 Julien M. P. Grenier
Julien M. P. Grenier Céline Testut
Céline Testut Cyril Fauriat
Cyril Fauriat Stéphane J. C. Mancini
Stéphane J. C. Mancini Michel Aurrand-Lions
Michel Aurrand-Lions- Aix-Marseille Université, Centre National de la Recherche Scientifique (CNRS), Institut National de la Santé et de la Recherche Médicale (INSERM), Institut Paoli Calmettes, Centre de Recherche en Cancérologie de Marseille (CRCM), Equipe Labellisée Ligue Nationale Contre le Cancer 2020, Marseille, France
In the bone marrow (BM) of adult mammals, haematopoietic stem cells (HSCs) are retained in micro-anatomical structures by adhesion molecules that regulate HSC quiescence, proliferation and commitment. During decades, researchers have used engraftment to study the function of adhesion molecules in HSC’s homeostasis regulation. Since the 90’s, progress in genetically engineered mouse models has allowed a better understanding of adhesion molecules involved in HSCs regulation by BM niches and raised questions about the role of adhesion mechanisms in conferring drug resistance to cancer cells nested in the BM. This has been especially studied in acute myeloid leukaemia (AML) which was the first disease in which the concept of cancer stem cell (CSC) or leukemic stem cells (LSCs) was demonstrated. In AML, it has been proposed that LSCs propagate the disease and are able to replenish the leukemic bulk after complete remission suggesting that LSC may be endowed with drug resistance properties. However, whether such properties are due to extrinsic or intrinsic molecular mechanisms, fully or partially supported by molecular crosstalk between LSCs and surrounding BM micro-environment is still matter of debate. In this review, we focus on adhesion molecules that have been involved in HSCs or LSCs anchoring to BM niches and discuss if inhibition of such mechanism may represent new therapeutic avenues to eradicate LSCs.
Introduction
Haematopoiesis takes place in the bone marrow of adult mammals and is the process leading to the formation of blood components throughout life. Haematopoietic stem cells (HSCs) are at the apex of the haematopoietic hierarchy and are able to self-renew and to differentiate into all blood cell types. The balance between differentiation and self-renewal is controlled by intrinsic properties of HSC and extrinsic cues delivered by the bone marrow microenvironment in micro-anatomical sites called “niches”. The concept of niche has been formulated by R. Schofield in 1978 who proposed that stem cell association with other cells prevents maturation while its progeny proliferate and differentiate, unless they can occupy a similar ‘niche’ (1). Although this working hypothesis turned to be true, its formal proof has long time been hampered by the lack of methods allowing precise localization of un-manipulated HSC within their niche (2, 3). In addition, because HSC activity has been essentially studied in transplantation assays, it has been difficult to decipher whether experimental assays were measuring intrinsic HSC stemness of engrafted cells or their ability to find a supportive niche in which they can self-renew (4, 5). The development of constitutive knock-out mouse models in the early 90’s, and conditional or inducible models later on, has represented a breakthrough to study the contribution of niche components to mammalian haematopoiesis (6, 7). Accordingly, a bibliographic search using combination of the words “haematopoiesis, adhesion and niche” reveals that only seven publications combine such words between 1989 and 2000, while more than hundred papers have been published thereafter. This likely indicates that adhesion was initially considered as an intrinsic property of HSC, while it has been integrated to the niche concept later on. This review is focused on adhesion molecules implicated in HSC or acute myeloid LSC interaction with the BM microenvironment (Figure 1).
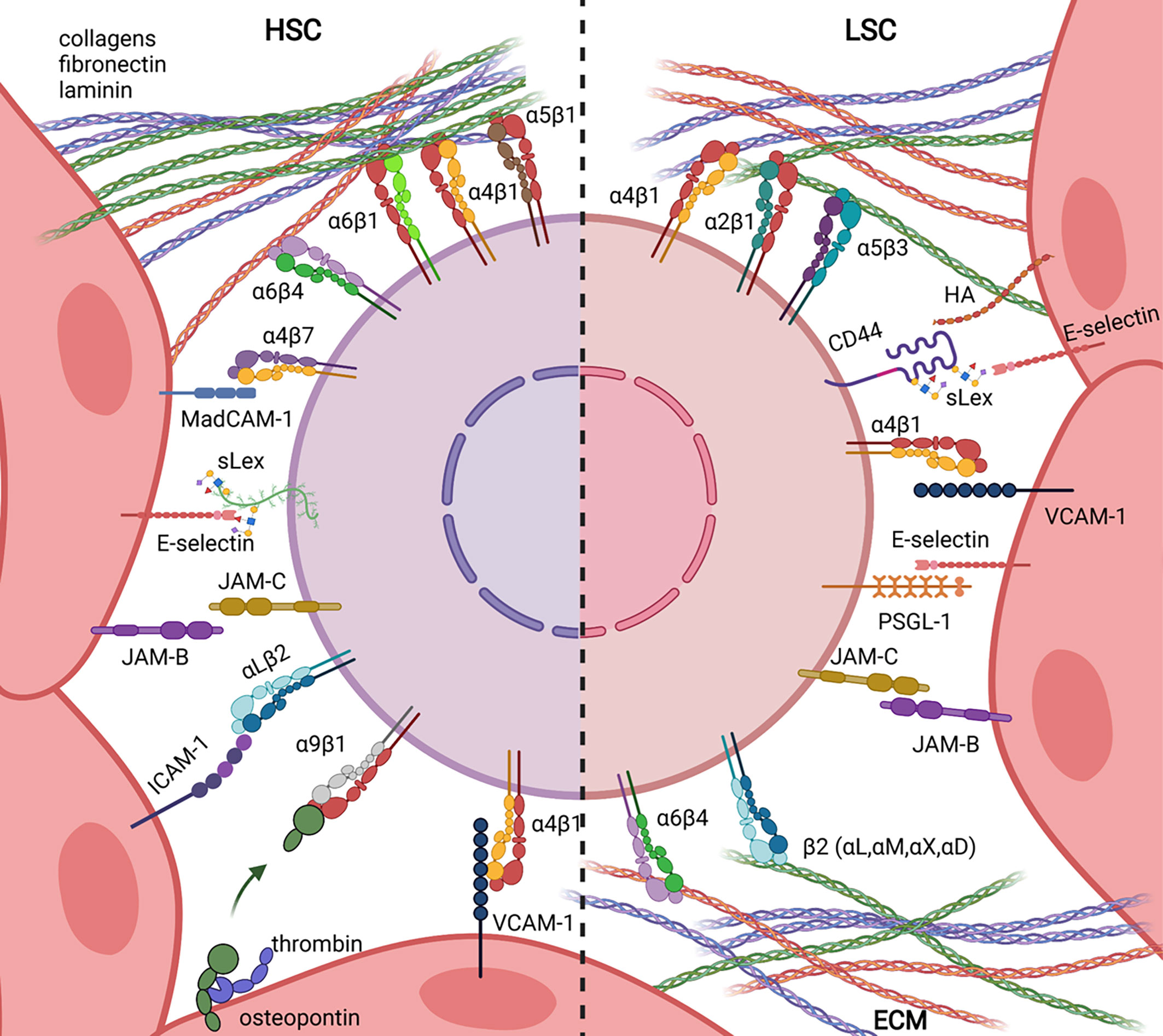
Figure 1 Ligand/Receptor adhesion pairs involved in Haematopoietic Stem Cell (HSC, left) and Leukemic Stem Cell (LSC, right) retention in bone marrow niches.
Adhesion Molecules Involved in HSC Retention in the Bone Marrow
With the exception of CD44, haematopoietic adhesion molecules belong to the immunoglobulin superfamily (Ig Sf), the cadherin family, the selectin family or the integrin family. Adhesion molecules promote cell/cell or cell/extracellular-matrix (ECM) interactions and deliver survival signals to haematopoietic cells. Reciprocally, stromal and endothelial cells express adhesion molecules interacting with haematopoietic cells or ECM contributing to the maintenance of bone marrow architecture.
Integrins
Integrins are non-covalent heterodimers of α and β chains. In mammals, 18 α and 8 β subunits form 24 different integrin heterodimers involved in embryonic development and maintenance of tissue homeostasis. α/β chain pairing and integrin interaction with ECM, cell surface molecules or soluble factors have been extensively reviewed in the past and will not be described in further details here (8–11).
One key property of integrins is that they can be expressed in inactive, activated or clustered state on the surface. The switch between inactive and active state results in increased ligand affinity as a consequence of inside-out or outside-in signalling. Integrin clustering further induces cytoskeleton rearrangement and enhanced cell signalling (Figure 2).
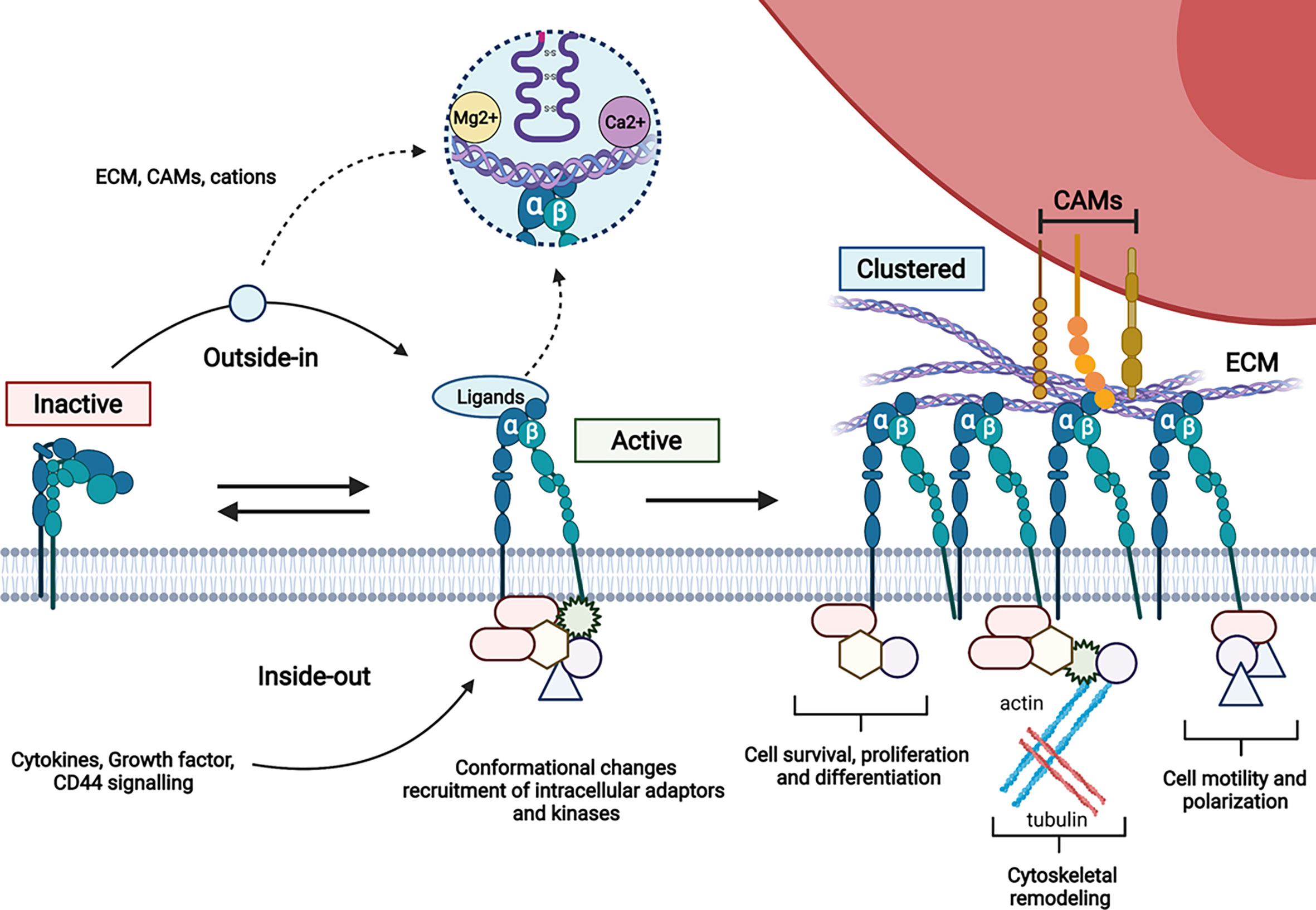
Figure 2 Schematic representation of integrin activation. The variety of intracellular protein complexes involved in integrin signalling (kinases, adaptors…) is depicted by forms recruited to the cytoplasmic tails of integrins.
Among α4β1, α5β1, α6β1, α6β4and α9β1 integrins that have been involved in interaction of HSC with bone marrow microenvironment (12–18), α4β1 is the most studied. The integrins α4β1 and α5β1 are activated by inside-out signalling that involves cytokines and divalent cations present in the bone marrow microenvironment, suggesting that they are essential for HSC retention in the bone marrow (19, 20). Accordingly, HSPC mobilization using G-CSF is correlated to decreased α4 integrin expression (21) and deletion or inhibition of α4β1 integrin result in accumulation of HSC in the blood circulation (22–25). Similar results were obtained using antibody against VCAM-1, suggesting a central role of α4β1/VCAM-1 axis in HSC retention in the bone marrow (26). This is consistent with the finding that β1 null HSC fail to engraft in irradiated recipient and that β1 null HSC from chimeric embryos are unable to seed foetal liver (27, 28).
Along this line, β7-deficient mice do not have defects in HSCs function (29), while interaction between α4β7 and MadCAM-1 (mucosal addressin cell adhesion molecule-1) accounts for half of the α4-integrin mediated homing activity to the bone marrow (30, 31). Therefore, it seems that β1 integrin heterodimers play a prominent role in bone marrow HSC retention as further supported by the fact that the dual α9β1/ α4β1 inhibitor BOP ((N-(benzenesulfonyl)-L-prolyl-L-O-(1-pyrrolidinylcarbonyl)tyrosine) induces a rapid mobilization of HSCs including those that are located in the endosteal region which bind thrombin-cleaved osteopontin with high affinity (32). This is also supported by the finding that patients treated with natalizumab, an anti-α4 integrin antibody, present increased levels of circulating CD34+ progenitor cells associated with an higher migratory profile as compared to GM-CSF mobilization (33, 34).
Finally, it has recently been reported in zebrafish that VCAM-1+ patrolling macrophages can interact with HSCs in an α4β1 dependent manner and contribute to their retention in the niche (35). This study confirms earlier findings in mouse models showing that macrophages contribute to HSC retention within niches through integrin-mediated interactions (36–38).
Selectins
The selectin family encompasses three members: E- (Endothelial), P- (Platelets) and L- (Leukocyte) selectins expressed by endothelial cells (E- and P- selectins), platelets (P-Selectin) and leukocytes (L-Selectin). They have been initially involved in the rolling of haematopoietic cells along vessels in flowing blood (39–41).
The minimal requirements for Ca2+-dependent ligand binding to selectins are the tetra-saccharides Sialyl Lewis X (Slex) and Sialyl Lewis A (SleA) (42, 43). As reviewed elsewhere (44), Slex and SleA synthesis requires several enzymes including α (1–3)-fucosyltransferase activities as illustrated by defective selectin-dependent leukocyte trafficking in FucT-VII deficient mice (45). This is reminiscent of the phenotype of P-Selectin deficient mice that harbour elevated number of circulating neutrophils, loss of leukocyte rolling in mesenteric venules and delayed leukocyte recruitment in peritonitis model (46). In contrast, E-selectin deficient mice have no defect in neutrophils trafficking suggesting a compensatory mechanism mediated by P-selectin (47).
The study of double knockout mice for E- and P-selectin has revealed defect in haematopoiesis with increased extramedullary erythropoiesis and reduced haematopoietic progenitor cell homing in irradiated deficient mice upon transplantation (41, 48). However, such functions were mostly attributed to HSPC homing and it is only in 2012 that E-selectin was shown to mediate HSC proliferation at the expense of self-renewal (49). In contrast to E- and P- Selectin, early haematopoietic defects in L-Selectin-deficient mice have not been reported so far (50).
Cadherins
Cadherins are transmembrane glycoproteins characterized by tandemly repeated sequence motifs in their extracellular segments that allow homophilic interactions in a Ca2+ dependent manner (51). N-cadherin is not only expressed by neural cells but also by HSCs and spindle shaped osteoblastic cells lining the bones, called “Spindle-shaped N-cadherin+CD45– Osteoblastic” (SNO) in the original publication. Because conditional inactivation of BMP receptor type IA (BMPRIA) led to expansion of both SNO and HSC, with asymmetric N-Cadherin distribution between SNO and HSC adjacent cells, it has been proposed that N-cadherin-mediated adhesion contributes to HSCs maintenance in endosteal niche (52). This concept was further supported by the fact that the knock-out of N-cadherin in LSK cells impairs long term engraftment in the bone marrow but not in the spleen (53). However, the latter demonstration used LSK cells, a compartment in which less than 20% of the cells are HSCs. Therefore, the function of N-cadherin mediated adhesion in HSC maintenance has been challenged in several studies. First, it was demonstrated that N-cadherin is not expressed on purified HSCs and that osteoblasts are dispensable for HSC maintenance (54). Second, the conditional deletion of N-cadherin in HSC using Mx1-Cre did not affect haematopoiesis, nor did its specific deletion in osteoblasts (55–57). Therefore, the controversial function of N-cadherin in HSC maintenance has been revisited in the light of the methodology used to study its function (engraftment versus knock-out) and with respect to heterogeneous expression of N-cadherin by HSC subsets (58, 59). This led to the most recent concept that N-cadherin mediated adhesion of HSC to BM stromal progenitor cells (BMSPC) may only be revealed during emergency haematopoiesis such as the one needed by “reserve” HSC to survive chemotherapy (60).
Ig Sf Adhesion Molecules
Several Ig Sf adhesion molecules such as ALCAM (CD166), ESAM, JAM-A or JAM-C are expressed by HSPCs and BM stromal or endothelial cells (61–64). Some others such as ICAM-1 or VCAM-1 are expressed in the BM microenvironment and interact with integrins expressed by HSPCs or contribute to more complex adhesive networks involving IgSf/Integrin as well as IgSf/IgSf interactions such as the JAM family members (65–68). Therefore, early haematopoietic defects reported for IgSf deficient animals have to be interpreted with caution unless specific conditional knock-out mouse models are combined with orthogonal methods such as long-term engraftment. Defects in early haematopoiesis following knockout have been reported for ALCAM, ESAM, VCAM-1, JAM-C, JAM-B and ICAM-1 (Table 1).
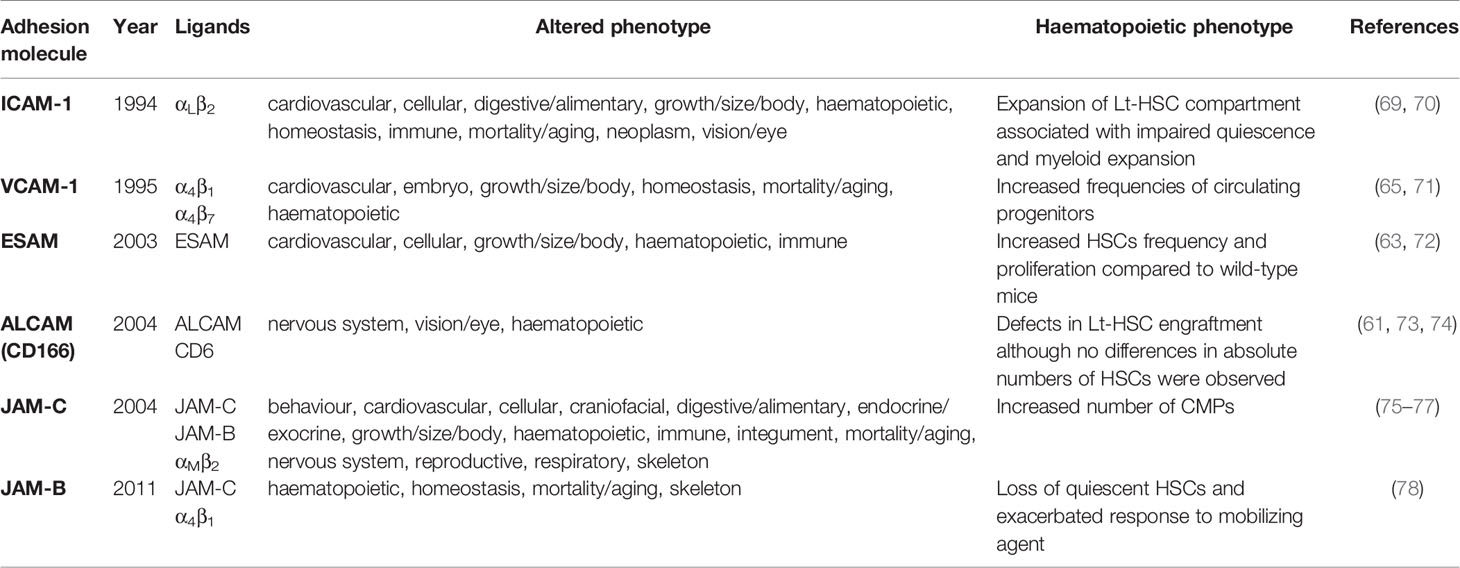
Table 1 Knock-out mice of Ig Sf molecules presenting haematopoietic defects.
Adhesion Molecules Involved in LSC Retention in the Bone Marrow
Similar to HSCs, LSCs are retained into specialized microanatomical sites by adhesive interactions. Indeed, AML development originates from LSC which share with HSCs the ability to self-renew (79, 80). After disease initiation, acute myeloid leukemic burst is accompanied by a remodelling of bone marrow niches that alters the physiological adhesive network of HSC (81–83). Whether adhesive remodelling occurs already at disease initiation in immunocompetent context remains to be addressed, but several adhesive Ligand/Receptor pairs have been involved in AML development in mouse models. Among them, only a limited number of Ligand/Receptor pairs that cross barrier species have been validated as putative therapeutic targets in preclinical setting using patient derived xenograft (PDX) models. This has encouraged some clinical trials targeting LSC adhesion to the niche in order to sensitize these cells to chemotherapy as recently reviewed by A. Villatoro et al. (84). In the next section, we will discuss the adhesion molecules known to contribute to LSC stemness maintenance that belong to the emerging class of adjuvant therapies for LSC eradication in AML.
CD44
CD44 is a class I transmembrane glycoprotein that does not belong to an adhesion molecular family and that interacts with ECM ligands such a as osteopontin, fibronectin or hyaluronan (HA). When CD44 is sialo-fucosylated and bears SleX glycan, it is called HCELL and interacts with E- and L-selectin (85, 86). In addition, several isoforms of CD44 are generated by alternative splicing and associated with different cellular processes (87). CD44 isoforms are widely expressed on AML cells and expression of the CD44-6v isoform has been associated with poor prognosis (88, 89). Functionally, CD44 has been involved in AML cell adhesion to bone marrow stromal cells (90, 91) and ligation of CD44 with HA or activating antibodies such as H90 has been shown to reverse differentiation blockage in AML cells (92). The same H90 activating antibody inhibited homing of AML-LSC to microenvironmental niches reducing the leukemic burden in a PDX setting. This was attributed to opposing effects of the H90 antibody which increases adhesion of normal CD34+CD38- cells to HA but inhibits adhesion of CD34+CD38- AML blasts to HA (93).
Integrins
Overexpression of the integrins αMβ2 (CD11b/Mac1), α2, α6 and α4β1 by AML cells has been associated with poor prognosis (94–96). Indeed, it has early been shown that both β1 and β2 integrin chains are necessary for AML blast adhesion to BM stromal cells (97).
Among the β1 integrins, α4β1 seems to play the most prominent role through its interaction with fibronectin (FN) and VCAM-1. Interaction of integrin α4β1 with FN protects AML cells from chemotherapy and is associated with the maintenance of minimal residual disease (MRD). Treatment with a blocking antibody against α4β1 abrogates chemoresistance and MRD in mice (98). Similarly, integrin α4β1 interaction with VCAM-1 contributes to drug resistance by activating NF-κB pathway in BM stromal cells which is essential to promote chemoresistance in leukemic cells as demonstrated by inhibition of NF-kB signalling (99). This study illustrates the reciprocal crosstalk between LSC and stromal cells since NF-κB activation in stromal cells upregulates VCAM-1 which serves as a positive feedback loop for leukemic cell adhesion to stromal cells.
More recently, the interaction between the integrin α2β1 and collagen has been shown to confer doxorubicin chemoresistance via the inhibition of Rac-1 (100). This protective effect is reversed by anti-α2β1. Although these studies show the therapeutic potential of integrin inhibition in AML, they do not formally prove that LSC are more addicted to integrin-mediated adhesion than normal HSC. To find such differential adhesive cues, Ebert and collaborators have used results from pooled in vivo shRNA screens. They have found that the integrin αvβ3 is essential for leukemic initiation and maintenance but dispensable for normal HSPC activity (101). This was attributed to constitutive activation of Syk, a candidate therapeutic target in AML, that is phosphorylated upon engagement of surface receptors including not only αvβ3 integrin, but also β2 integrins (102, 103). In summary, integrin signalling converging toward specific activation pathway such as NF-kB or Syk may represent attractive therapeutic targets.
E-Selectin
E- and P-Selectins are constitutively expressed by bone marrow endothelial cells and play a role in HSPC rolling on micro vessels (39, 104, 105). However, they induce contrasting effects in HSPC upon interaction in vitro (86, 106–108). The study of early haematopoiesis in E-Selectin deficient mice (Sele-/-) has revealed that inhibition of E-Selectin in vivo increases dormancy and self-renewal of HSC (49). This is not mediated by the conventional ligands of E-Selectin since HSC isolated from mice deficient for P-selectin glycoprotein ligand-1 (Psgl-1 encoded by Selplg), HCELL (Cd44) or both do not present increased dormancy. In contrast, LSC of AML make a different selectin receptor usage that promotes AML cell survival. Indeed, leukemic cells present alterations in glycosylation which leads to expression of fucosylated ligands such as PSGL-1 (CD162) that activate PI3K/Akt survival pathway (109, 110). Even more interesting is the fact that inhibition of E-selectin interaction with its ligands using a glycomimetic stimulates proliferation of AML blast while dampening HSC cycling. Since these finding have been confirmed in preclinical mouse models, this led to the opening of phase II/III clinical trials combining inhibition of E-selectin with conventional chemotherapy in AML (NCT03616470, NCT03701308).
Ig Sf Adhesion Molecules
Most of the Ig Sf molecules expressed by normal HSC are also expressed by LSCs in AML, however only few of them allows enrichment of cells with leukemic initiating activity associated to poor prognosis. We have shown that JAM-C is expressed by a fraction of LSCs presenting high activation of Src kinase family and enriched for leukaemia initiating activity. Increased frequencies of JAM-C expressing cells identify AML patients with poor disease outcome (111). This has been confirmed in an independent study on a large cohort of AML patients (112, 113). The “CD34+ CD38low CD123+ CD41- JAM-C+” cells are enriched tenfold for LSCs as compared to cells lacking JAM-C expression within the same compartment suggesting that JAM-C may play a cell-autonomous signalling function at the transition between healthy HSC and LSC. This would be consistent with results showing that PDX or AML cell line engraftment of JAM-C-expressing cells is only partially dependent on JAM-B expression by recipient mice and with results showing that silencing JAM-C expression is sufficient to decrease Src family kinase activation (111). This could be due to promiscuous cis-interactions between JAM-C and the integrin α4β1 since JAM-B has been shown to bind α4β1 when interaction is facilitated by the simultaneous engagement with JAM-C (67).
NCAM1(CD56) is another Ig Sf molecules whose expression is correlated with poor overall survival in AML with t(8;21) (q22; q22) and highly expressed by LSC in mouse AML models using MLL-AF9 or Hoxa9-Meis1 as driver translocations (114). NCAM1 expression confers drug resistance to AML cells and knockdown of NCAM1 sensitizes blasts to genotoxic agents (115). This is likely due to constitutive activation of the MEK-ERK pathway, similar to what has been reported during neural development (116). These two examples pave the way for the use of Ig Sf molecule expression to stratify patients eligible to treatments targeting downstream signalling pathways such as Src or Mek/Erk.
Outlook
Recent studies have shown that HSC niches are altered during AML development with strong coordinated changes of the osteolineage and endothelial compartments, and alterations of the mesenchymal compartment occurring early during leukemic development. Whether such alterations depend on adhesive interaction of leukemic initiating cells with BM microenvironment resulting in localization of LSCs in specific sites remain to be defined, but it seems that LSC take advantage of pre-existing adhesive pathways in the niche to maintain survival signals and dormancy that protect them from chemotherapies. Therefore, the selective disruption of LSC from their niche by targeting single adhesion molecule remains a major limitation for current therapies. A better knowledge of the differences between LSC/Niche and HSC/Niche integrated adhesive networks will help refining specificity of therapeutic strategies directed against adhesive cues.
Author Contributions
JG wrote and revised the manuscript. CT, CF, and SM revised the manuscript and MA-L supervised the work. All authors contributed to the article and approved the submitted version.
Funding
JG was supported by a grant from the French Society of Haematology (SFH). CT was supported by a grant from the ligue nationale contre le cancer. The work was supported by the Ligue Nationale Contre le Cancer, ELN2020.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We apologize for colleagues whose work has not been cited due to space limitation. Figures 1, 2 were created by JG with BioRender.com.
References
1. Schofield R. The Relationship Between the Spleen Colony-Forming Cell and the Haemopoietic Stem Cell. Blood Cells (1978) 4:7–25.
2. Pinho S, Frenette PS. Haematopoietic Stem Cell Activity and Interactions With the Niche. Nat Rev Mol Cell Biol (2019) 20:303–20. doi: 10.1038/s41580-019-0103-9
3. Crane GM, Jeffery E, Morrison SJ. Adult Haematopoietic Stem Cell Niches. Nat Rev Immunol (2017) 17:573–90. doi: 10.1038/nri.2017.53
4. Lemischka IR. Clonal, In Vivo Behavior of the Totipotent Hematopoietic Stem Cell. Semin Immunol (1991) 3:349–55.
5. Mielcarek M, Reems J, Torok-Storb B. Extrinsic Control of Stem Cell Fate: Practical Considerations. Stem Cells (1997) 15(Suppl 1):229–232; discussion 233-236. doi: 10.1002/stem.5530150831
6. Chen KG, Johnson KR, Robey PG. Mouse Genetic Analysis of Bone Marrow Stem Cell Niches: Technological Pitfalls, Challenges, and Translational Considerations. Stem Cell Rep (2017) 9:1343–58. doi: 10.1016/j.stemcr.2017.09.014
7. Joseph C, Quach JM, Walkley CR, Lane SW, Lo Celso C, Purton LE. Deciphering Hematopoietic Stem Cells in Their Niches: A Critical Appraisal of Genetic Models, Lineage Tracing, and Imaging Strategies. Cell Stem Cell (2013) 13:520–33. doi: 10.1016/j.stem.2013.10.010
8. Samaržija I, Dekanić A, Humphries JD, Paradžik M, Stojanović N, Humphries MJ, et al. Integrin Crosstalk Contributes to the Complexity of Signalling and Unpredictable Cancer Cell Fates. Cancers (Basel) (2020) 12:E1910. doi: 10.3390/cancers12071910
9. Hynes RO. Integrins: Versatility, Modulation, and Signaling in Cell Adhesion. Cell (1992) 69:11–25. doi: 10.1016/0092-8674(92)90115-s
10. Humphries JD, Byron A, Humphries MJ. Integrin Ligands at a Glance. J Cell Sci (2006) 119:3901–3. doi: 10.1242/jcs.03098
11. Harburger DS, Calderwood DA. Integrin Signalling at a Glance. J Cell Sci (2009) 122:159–63. doi: 10.1242/jcs.018093
12. Grassinger J, Haylock DN, Storan MJ, Haines GO, Williams B, Whitty GA, et al. Thrombin-Cleaved Osteopontin Regulates Hemopoietic Stem and Progenitor Cell Functions Through Interactions With Alpha9beta1 and Alpha4beta1 Integrins. Blood (2009) 114:49–59. doi: 10.1182/blood-2009-01-197988
13. Hartz B, Volkmann T, Irle S, Loechelt C, Neubauer A, Brendel C. α4 Integrin Levels on Mobilized Peripheral Blood Stem Cells Predict Rapidity of Engraftment in Patients Receiving Autologous Stem Cell Transplantation. Blood (2011) 118:2362–5. doi: 10.1182/blood-2011-02-331918
14. Qian H, Tryggvason K, Jacobsen SE, Ekblom M. Contribution of Alpha6 Integrins to Hematopoietic Stem and Progenitor Cell Homing to Bone Marrow and Collaboration With Alpha4 Integrins. Blood (2006) 107:3503–10. doi: 10.1182/blood-2005-10-3932
15. Schreiber TD, Steinl C, Essl M, Abele H, Geiger K, Müller CA, et al. The Integrin Alpha9beta1 on Hematopoietic Stem and Progenitor Cells: Involvement in Cell Adhesion, Proliferation and Differentiation. Haematologica (2009) 94:1493–501. doi: 10.3324/haematol.2009.006072
16. Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of Single Human Hematopoietic Stem Cells Capable of Long-Term Multilineage Engraftment. Science (2011) 333:218–21. doi: 10.1126/science.1201219
17. Huntsman HD, Bat T, Cheng H, Cash A, Cheruku PS, Fu J-F, et al. Human Hematopoietic Stem Cells From Mobilized Peripheral Blood can be Purified Based on CD49f Integrin Expression. Blood (2015) 126:1631–3. doi: 10.1182/blood-2015-07-660670
18. Soligo D, Schiró R, Luksch R, Manara G, Quirici N, Parravicini C, et al. Expression of Integrins in Human Bone Marrow. Br J Haematol (1990) 76:323–32. doi: 10.1111/j.1365-2141.1990.tb06363.x
19. Takamatsu Y, Simmons PJ, Lévesque JP. Dual Control by Divalent Cations and Mitogenic Cytokines of Alpha 4 Beta 1 and Alpha 5 Beta 1 Integrin Avidity Expressed by Human Hemopoietic Cells. Cell Adhes Commun (1998) 5:349–66. doi: 10.3109/15419069809010781
20. Lévesque JP, Leavesley DI, Niutta S, Vadas M, Simmons PJ. Cytokines Increase Human Hemopoietic Cell Adhesiveness by Activation of Very Late Antigen (VLA)-4 and VLA-5 Integrins. J Exp Med (1995) 181:1805–15. doi: 10.1084/jem.181.5.1805
21. Prosper F, Stroncek D, McCarthy JB, Verfaillie CM. Mobilization and Homing of Peripheral Blood Progenitors Is Related to Reversible Downregulation of Alpha4 Beta1 Integrin Expression and Function. J Clin Invest (1998) 101:2456–67. doi: 10.1172/JCI188
22. Arroyo AG, Yang JT, Rayburn H, Hynes RO. Differential Requirements for Alpha4 Integrins During Fetal and Adult Hematopoiesis. Cell (1996) 85:997–1008. doi: 10.1016/s0092-8674(00)81301-x
23. Scott LM, Priestley GV, Papayannopoulou T. Deletion of Alpha4 Integrins From Adult Hematopoietic Cells Reveals Roles in Homeostasis, Regeneration, and Homing. Mol Cell Biol (2003) 23:9349–60. doi: 10.1128/MCB.23.24.9349-9360.2003
24. Papayannopoulou T, Nakamoto B. Peripheralization of Hemopoietic Progenitors in Primates Treated With Anti-VLA4 Integrin. Proc Natl Acad Sci U.S.A. (1993) 90:9374–8. doi: 10.1073/pnas.90.20.9374
25. Ramirez P, Rettig MP, Uy GL, Deych E, Holt MS, Ritchey JK, et al. BIO5192, a Small Molecule Inhibitor of VLA-4, Mobilizes Hematopoietic Stem and Progenitor Cells. Blood (2009) 114:1340–3. doi: 10.1182/blood-2008-10-184721
26. Papayannopoulou T, Craddock C, Nakamoto B, Priestley GV, Wolf NS. The VLA4/VCAM-1 Adhesion Pathway Defines Contrasting Mechanisms of Lodgement of Transplanted Murine Hemopoietic Progenitors Between Bone Marrow and Spleen. Proc Natl Acad Sci USA (1995) 92:9647–51. doi: 10.1073/pnas.92.21.9647
27. Potocnik AJ, Brakebusch C, Fässler R. Fetal and Adult Hematopoietic Stem Cells Require Beta1 Integrin Function for Colonizing Fetal Liver, Spleen, and Bone Marrow. Immunity (2000) 12:653–63. doi: 10.1016/s1074-7613(00)80216-2
28. Hirsch E, Iglesias A, Potocnik AJ, Hartmann U, Fässler R. Impaired Migration But Not Differentiation of Haematopoietic Stem Cells in the Absence of Beta1 Integrins. Nature (1996) 380:171–5. doi: 10.1038/380171a0
29. Bungartz G, Stiller S, Bauer M, Müller W, Schippers A, Wagner N, et al. Adult Murine Hematopoiesis can Proceed Without Beta1 and Beta7 Integrins. Blood (2006) 108:1857–64. doi: 10.1182/blood-2005-10-007658
30. Murakami JL, Xu B, Franco CB, Hu X, Galli SJ, Weissman IL, et al. Evidence That β7 Integrin Regulates Hematopoietic Stem Cell Homing and Engraftment Through Interaction With MAdCAM-1. Stem Cells Dev (2016) 25:18–26. doi: 10.1089/scd.2014.0551
31. Katayama Y, Hidalgo A, Peired A, Frenette PS. Integrin Alpha4beta7 and Its Counterreceptor MAdCAM-1 Contribute to Hematopoietic Progenitor Recruitment Into Bone Marrow Following Transplantation. Blood (2004) 104:2020–6. doi: 10.1182/blood-2003-12-4157
32. Cao B, Zhang Z, Grassinger J, Williams B, Heazlewood CK, Churches QI, et al. Therapeutic Targeting and Rapid Mobilization of Endosteal HSC Using a Small Molecule Integrin Antagonist. Nat Commun (2016) 7:11007. doi: 10.1038/ncomms11007
33. Jing D, Oelschlaegel U, Ordemann R, Hölig K, Ehninger G, Reichmann H, et al. CD49d Blockade by Natalizumab in Patients With Multiple Sclerosis Affects Steady-State Hematopoiesis and Mobilizes Progenitors With a Distinct Phenotype and Function. Bone Marrow Transplant (2010) 45:1489–96. doi: 10.1038/bmt.2009.381
34. Zohren F, Toutzaris D, Klärner V, Hartung H-P, Kieseier B, Haas R. The Monoclonal Anti-VLA-4 Antibody Natalizumab Mobilizes CD34+ Hematopoietic Progenitor Cells in Humans. Blood (2008) 111:3893–5. doi: 10.1182/blood-2007-10-120329
35. Li D, Xue W, Li M, Dong M, Wang J, Wang X, et al. VCAM-1+ Macrophages Guide the Homing of HSPCs to a Vascular Niche. Nature (2018) 564:119–24. doi: 10.1038/s41586-018-0709-7
36. Jacobsen RN, Forristal CE, Raggatt LJ, Nowlan B, Barbier V, Kaur S, et al. Mobilization With Granulocyte Colony-Stimulating Factor Blocks Medullar Erythropoiesis by Depleting F4/80(+)VCAM1(+)CD169(+)ER-HR3(+)Ly6G(+) Erythroid Island Macrophages in the Mouse. Exp Hematol (2014) 42:547–561.e4. doi: 10.1016/j.exphem.2014.03.009
37. Dutta P, Hoyer FF, Grigoryeva LS, Sager HB, Leuschner F, Courties G, et al. Macrophages Retain Hematopoietic Stem Cells in the Spleen via VCAM-1. J Exp Med (2015) 212:497–512. doi: 10.1084/jem.20141642
38. Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone Marrow CD169+ Macrophages Promote the Retention of Hematopoietic Stem and Progenitor Cells in the Mesenchymal Stem Cell Niche. J Exp Med (2011) 208:261–71. doi: 10.1084/jem.20101688
39. Mazo IB, Gutierrez-Ramos JC, Frenette PS, Hynes RO, Wagner DD, von Andrian UH. Hematopoietic Progenitor Cell Rolling in Bone Marrow Microvessels: Parallel Contributions by Endothelial Selectins and Vascular Cell Adhesion Molecule 1. J Exp Med (1998) 188:465–74. doi: 10.1084/jem.188.3.465
40. Vestweber D, Blanks JE. Mechanisms That Regulate the Function of the Selectins and Their Ligands. Physiol Rev (1999) 79:181–213. doi: 10.1152/physrev.1999.79.1.181
41. Frenette PS, Subbarao S, Mazo IB, von Andrian UH, Wagner DD. Endothelial Selectins and Vascular Cell Adhesion Molecule-1 Promote Hematopoietic Progenitor Homing to Bone Marrow. Proc Natl Acad Sci U.S.A. (1998) 95:14423–8. doi: 10.1073/pnas.95.24.14423
42. Tvaroška I, Selvaraj C, Koča J. Selectins-The Two Dr. Jekyll and Mr. Hyde Faces of Adhesion Molecules-A Review. Molecules (2020) 25:E2835. doi: 10.3390/molecules25122835
43. Varki A. Selectin Ligands. Proc Natl Acad Sci USA (1994) 91:7390–7. doi: 10.1073/pnas.91.16.7390
44. Lowe JB. Glycosylation in the Control of Selectin Counter-Receptor Structure and Function. Immunol Rev (2002) 186:19–36. doi: 10.1034/j.1600-065x.2002.18603.x
45. Malý P, Thall A, Petryniak B, Rogers CE, Smith PL, Marks RM, et al. The Alpha(1,3)Fucosyltransferase Fuc-TVII Controls Leukocyte Trafficking Through an Essential Role in L-, E-, and P-Selectin Ligand Biosynthesis. Cell (1996) 86:643–53. doi: 10.1016/s0092-8674(00)80137-3
46. Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte Rolling and Extravasation Are Severely Compromised in P Selectin-Deficient Mice. Cell (1993) 74:541–54. doi: 10.1016/0092-8674(93)80055-j
47. Labow MA, Norton CR, Rumberger JM, Lombard-Gillooly KM, Shuster DJ, Hubbard J, et al. Characterization of E-Selectin-Deficient Mice: Demonstration of Overlapping Function of the Endothelial Selectins. Immunity (1994) 1:709–20. doi: 10.1016/1074-7613(94)90041-8
48. Frenette PS, Mayadas TN, Rayburn H, Hynes RO, Wagner DD. Susceptibility to Infection and Altered Hematopoiesis in Mice Deficient in Both P- and E-Selectins. Cell (1996) 84:563–74. doi: 10.1016/s0092-8674(00)81032-6
49. Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular Niche E-Selectin Regulates Hematopoietic Stem Cell Dormancy, Self Renewal and Chemoresistance. Nat Med (2012) 18:1651–7. doi: 10.1038/nm.2969
50. Arbonés ML, Ord DC, Ley K, Ratech H, Maynard-Curry C, Otten G, et al. Lymphocyte Homing and Leukocyte Rolling and Migration Are Impaired in L-Selectin-Deficient Mice. Immunity (1994) 1:247–60. doi: 10.1016/1074-7613(94)90076-0
51. Hatta K, Nose A, Nagafuchi A, Takeichi M. Cloning and Expression of cDNA Encoding a Neural Calcium-Dependent Cell Adhesion Molecule: Its Identity in the Cadherin Gene Family. J Cell Biol (1988) 106:873–81. doi: 10.1083/jcb.106.3.873
52. Zhang J, Niu C, Ye L, Huang H, He X, Tong W-G, et al. Identification of the Haematopoietic Stem Cell Niche and Control of the Niche Size. Nature (2003) 425:836–41. doi: 10.1038/nature02041
53. Hosokawa K, Arai F, Yoshihara H, Iwasaki H, Nakamura Y, Gomei Y, et al. Knockdown of N-Cadherin Suppresses the Long-Term Engraftment of Hematopoietic Stem Cells. Blood (2010) 116:554–63. doi: 10.1182/blood-2009-05-224857
54. Kiel MJ, Radice GL, Morrison SJ. Lack of Evidence That Hematopoietic Stem Cells Depend on N-Cadherin-Mediated Adhesion to Osteoblasts for Their Maintenance. Cell Stem Cell (2007) 1:204–17. doi: 10.1016/j.stem.2007.06.001
55. Kiel MJ, Acar M, Radice GL, Morrison SJ. Hematopoietic Stem Cells do Not Depend on N-Cadherin to Regulate Their Maintenance. Cell Stem Cell (2009) 4:170–9. doi: 10.1016/j.stem.2008.10.005
56. Bromberg O, Frisch BJ, Weber JM, Porter RL, Civitelli R, Calvi LM. Osteoblastic N-Cadherin Is Not Required for Microenvironmental Support and Regulation of Hematopoietic Stem and Progenitor Cells. Blood (2012) 120:303–13. doi: 10.1182/blood-2011-09-377853
57. Greenbaum AM, Revollo LD, Woloszynek JR, Civitelli R, Link DC. N-Cadherin in Osteolineage Cells Is Not Required for Maintenance of Hematopoietic Stem Cells. Blood (2012) 120:295–302. doi: 10.1182/blood-2011-09-377457
58. Arai F, Hosokawa K, Toyama H, Matsumoto Y, Suda T. Role of N-Cadherin in the Regulation of Hematopoietic Stem Cells in the Bone Marrow Niche. Ann N Y Acad Sci (2012) 1266:72–7. doi: 10.1111/j.1749-6632.2012.06576.x
59. Haug JS, He XC, Grindley JC, Wunderlich JP, Gaudenz K, Ross JT, et al. N-Cadherin Expression Level Distinguishes Reserved Versus Primed States of Hematopoietic Stem Cells. Cell Stem Cell (2008) 2:367–79. doi: 10.1016/j.stem.2008.01.017
60. Zhao M, Tao F, Venkatraman A, Li Z, Smith SE, Unruh J, et al. N-Cadherin-Expressing Bone and Marrow Stromal Progenitor Cells Maintain Reserve Hematopoietic Stem Cells. Cell Rep (2019) 26:652–669.e6. doi: 10.1016/j.celrep.2018.12.093
61. Chitteti BR, Kobayashi M, Cheng Y, Zhang H, Poteat BA, Broxmeyer HE, et al. CD166 Regulates Human and Murine Hematopoietic Stem Cells and the Hematopoietic Niche. Blood (2014) 124:519–29. doi: 10.1182/blood-2014-03-565721
62. Ooi AGL, Karsunky H, Majeti R, Butz S, Vestweber D, Ishida T, et al. The Adhesion Molecule Esam1 is a Novel Hematopoietic Stem Cell Marker. Stem Cells (2009) 27:653–61. doi: 10.1634/stemcells.2008-0824
63. Yokota T, Oritani K, Butz S, Kokame K, Kincade PW, Miyata T, et al. The Endothelial Antigen ESAM Marks Primitive Hematopoietic Progenitors Throughout Life in Mice. Blood (2009) 113:2914–23. doi: 10.1182/blood-2008-07-167106
64. Sugano Y, Takeuchi M, Hirata A, Matsushita H, Kitamura T, Tanaka M, et al. Junctional Adhesion Molecule-A, JAM-A, Is a Novel Cell-Surface Marker for Long-Term Repopulating Hematopoietic Stem Cells. Blood (2008) 111:1167–72. doi: 10.1182/blood-2007-03-081554
65. Ulyanova T, Scott LM, Priestley GV, Jiang Y, Nakamoto B, Koni PA, et al. VCAM-1 Expression in Adult Hematopoietic and Nonhematopoietic Cells Is Controlled by Tissue-Inductive Signals and Reflects Their Developmental Origin. Blood (2005) 106:86–94. doi: 10.1182/blood-2004-09-3417
66. Balzano M, De Grandis M, Vu Manh T-P, Chasson L, Bardin F, Farina A, et al. Nidogen-1 Contributes to the Interaction Network Involved in Pro-B Cell Retention in the Peri-Sinusoidal Hematopoietic Stem Cell Niche. Cell Rep (2019) 26:3257–3271.e8. doi: 10.1016/j.celrep.2019.02.065
67. Cunningham SA, Rodriguez JM, Arrate MP, Tran TM, Brock TA. JAM2 Interacts With Alpha4beta1. Facilitation by JAM3. J Biol Chem (2002) 277:27589–92. doi: 10.1074/jbc.C200331200
68. Lamagna C, Meda P, Mandicourt G, Brown J, Gilbert RJC, Jones EY, et al. Dual Interaction of JAM-C With JAM-B and Alpha(M)beta2 Integrin: Function in Junctional Complexes and Leukocyte Adhesion. Mol Biol Cell (2005) 16:4992–5003. doi: 10.1091/mbc.e05-04-0310
69. Liu Y-F, Zhang S-Y, Chen Y-Y, Shi K, Zou B, Liu J, et al. ICAM-1 Deficiency in the Bone Marrow Niche Impairs Quiescence and Repopulation of Hematopoietic Stem Cells. Stem Cell Rep (2018) 11:258–73. doi: 10.1016/j.stemcr.2018.05.016
70. Xu H, Gonzalo JA, St Pierre Y, Williams IR, Kupper TS, Cotran RS, et al. Leukocytosis and Resistance to Septic Shock in Intercellular Adhesion Molecule 1-Deficient Mice. J Exp Med (1994) 180:95–109. doi: 10.1084/jem.180.1.95
71. Kwee L, Burns DK, Rumberger JM, Norton C, Wolitzky B, Terry R, et al. Creation and Characterization of E-Selectin- and VCAM-1-Deficient Mice. Ciba Found Symp (1995) 189:17–28; discussion 28-34, 77–78. doi: 10.1002/9780470514719.ch3
72. Ishida T, Kundu RK, Yang E, Hirata K, Ho Y-D, Quertermous T. Targeted Disruption of Endothelial Cell-Selective Adhesion Molecule Inhibits Angiogenic Processes In Vitro and In Vivo. J Biol Chem (2003) 278:34598–604. doi: 10.1074/jbc.M304890200
73. Weiner JA, Koo SJ, Nicolas S, Fraboulet S, Pfaff SL, Pourquié O, et al. Axon Fasciculation Defects and Retinal Dysplasias in Mice Lacking the Immunoglobulin Superfamily Adhesion Molecule BEN/ALCAM/Sc1. Mol Cell Neurosci (2004) 27:59–69. doi: 10.1016/j.mcn.2004.06.005
74. Jeannet R, Cai Q, Liu H, Vu H, Kuo Y-H. Alcam Regulates Long-Term Hematopoietic Stem Cell Engraftment and Self-Renewal. Stem Cells (2013) 31:560–71. doi: 10.1002/stem.1309
75. Gliki G, Ebnet K, Aurrand-Lions M, Imhof BA, Adams RH. Spermatid Differentiation Requires the Assembly of a Cell Polarity Complex Downstream of Junctional Adhesion Molecule-C. Nature (2004) 431:320–4. doi: 10.1038/nature02877
76. Praetor A, McBride JM, Chiu H, Rangell L, Cabote L, Lee WP, et al. Genetic Deletion of JAM-C Reveals a Role in Myeloid Progenitor Generation. Blood (2009) 113:1919–28. doi: 10.1182/blood-2008-06-159574
77. Pellegrini M, Claps G, Orlova VV, Barrios F, Dolci S, Geremia R, et al. Targeted JAM-C Deletion in Germ Cells by Spo11-Controlled Cre Recombinase. J Cell Sci (2011) 124:91–9. doi: 10.1242/jcs.072959
78. Arcangeli M-L, Frontera V, Bardin F, Obrados E, Adams S, Chabannon C, et al. JAM-B Regulates Maintenance of Hematopoietic Stem Cells in the Bone Marrow. Blood (2011) 118:4609–19. doi: 10.1182/blood-2010-12-323972
79. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367:645–8. doi: 10.1038/367645a0
80. Bonnet D, Dick JE. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3:730–7. doi: 10.1038/nm0797-730
81. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell (2019) 177:1915–1932.e16. doi: 10.1016/j.cell.2019.04.040
82. Passaro D, Garcia-Albornoz M, Diana G, Chakravarty P, Ariza-McNaughton L, Batsivari A, et al. Integrated OMICs Unveil the Bone-Marrow Microenvironment in Human Leukemia. Cell Rep (2021) 35:109119. doi: 10.1016/j.celrep.2021.109119
83. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell (2018) 22:64–77.e6. doi: 10.1016/j.stem.2017.11.006
84. Villatoro A, Konieczny J, Cuminetti V, Arranz L. Leukemia Stem Cell Release From the Stem Cell Niche to Treat Acute Myeloid Leukemia. Front Cell Dev Biol (2020) 8:607. doi: 10.3389/fcell.2020.00607
85. Zöller M. CD44, Hyaluronan, the Hematopoietic Stem Cell, and Leukemia-Initiating Cells. Front Immunol (2015) 6:235. doi: 10.3389/fimmu.2015.00235
86. Dimitroff CJ, Lee JY, Rafii S, Fuhlbrigge RC, Sackstein R. CD44 is a Major E-Selectin Ligand on Human Hematopoietic Progenitor Cells. J Cell Biol (2001) 153:1277–86. doi: 10.1083/jcb.153.6.1277
87. Ponta H, Sherman L, Herrlich PA. CD44: From Adhesion Molecules to Signalling Regulators. Nat Rev Mol Cell Biol (2003) 4:33–45. doi: 10.1038/nrm1004
88. Bendall LJ, Bradstock KF, Gottlieb DJ. Expression of CD44 Variant Exons in Acute Myeloid Leukemia is More Common and More Complex Than That Observed in Normal Blood, Bone Marrow or CD34+ Cells. Leukemia (2000) 14:1239–46. doi: 10.1038/sj.leu.2401830
89. Legras S, Günthert U, Stauder R, Curt F, Oliferenko S, Kluin-Nelemans HC, et al. A Strong Expression of CD44-6v Correlates With Shorter Survival of Patients With Acute Myeloid Leukemia. Blood (1998) 91:3401–13. doi: 10.1182/blood.V91.9.3401
90. Legras S, Levesque JP, Charrad R, Morimoto K, Le Bousse C, Clay D, et al. CD44-Mediated Adhesiveness of Human Hematopoietic Progenitors to Hyaluronan is Modulated by Cytokines. Blood (1997) 89:1905–14. doi: 10.1182/blood.V89.6.1905
91. Gutjahr JC, Bayer E, Yu X, Laufer JM, Höpner JP, Tesanovic S, et al. CD44 Engagement Enhances Acute Myeloid Leukemia Cell Adhesion to the Bone Marrow Microenvironment by Increasing VLA-4 Avidity. Haematologica (2021) 106:2102–13. doi: 10.3324/haematol.2019.231944
92. Charrad RS, Li Y, Delpech B, Balitrand N, Clay D, Jasmin C, et al. Ligation of the CD44 Adhesion Molecule Reverses Blockage of Differentiation in Human Acute Myeloid Leukemia. Nat Med (1999) 5:669–76. doi: 10.1038/9518
93. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 Eradicates Human Acute Myeloid Leukemic Stem Cells. Nat Med (2006) 12:1167–74. doi: 10.1038/nm1483
94. Graf M, Reif S, Kröll T, Hecht K, Nuessler V, Schmetzer H. Expression of MAC-1 (CD11b) in Acute Myeloid Leukemia (AML) is Associated With an Unfavorable Prognosis. Am J Hematol (2006) 81:227–35. doi: 10.1002/ajh.20526
95. Lian X-Y, Zhang W, Wu D-H, Ma J-C, Zhou J-D, Zhang Z-H, et al. Methylation-Independent ITGA2 Overexpression Is Associated With Poor Prognosis in De Novo Acute Myeloid Leukemia. J Cell Physiol (2018) 233:9584–93. doi: 10.1002/jcp.26866
96. Yamakawa N, Kaneda K, Saito Y, Ichihara E, Morishita K. The Increased Expression of Integrin α6 (ITGA6) Enhances Drug Resistance in EVI1(high) Leukemia. PloS One (2012) 7:e30706. doi: 10.1371/journal.pone.0030706
97. Bendall LJ, Kortlepel K, Gottlieb DJ. Human Acute Myeloid Leukemia Cells Bind to Bone Marrow Stroma via a Combination of Beta-1 and Beta-2 Integrin Mechanisms. Blood (1993) 82:3125–32. doi: 10.1182/blood.V82.10.3125.3125
98. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction Between Leukemic-Cell VLA-4 and Stromal Fibronectin Is a Decisive Factor for Minimal Residual Disease of Acute Myelogenous Leukemia. Nat Med (2003) 9:1158–65. doi: 10.1038/nm909
99. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal Leukemia-Stroma VCAM-1/VLA-4-Dependent Activation of NF-κb Mediates Chemoresistance. Blood (2014) 123:2691–702. doi: 10.1182/blood-2013-06-511527
100. Naci D, Berrazouane S, Barabé F, Aoudjit F. Cell Adhesion to Collagen Promotes Leukemia Resistance to Doxorubicin by Reducing DNA Damage Through the Inhibition of Rac1 Activation. Sci Rep (2019) 9:19455. doi: 10.1038/s41598-019-55934-w
101. Miller PG, Al-Shahrour F, Hartwell KA, Chu LP, Järås M, Puram RV, et al. In Vivo RNAi Screening Identifies a Leukemia-Specific Dependence on Integrin Beta 3 Signaling. Cancer Cell (2013) 24:45–58. doi: 10.1016/j.ccr.2013.05.004
102. Hahn CK, Berchuck JE, Ross KN, Kakoza RM, Clauser K, Schinzel AC, et al. Proteomic and Genetic Approaches Identify Syk as an AML Target. Cancer Cell (2009) 16:281–94. doi: 10.1016/j.ccr.2009.08.018
103. Oellerich T, Oellerich MF, Engelke M, Münch S, Mohr S, Nimz M, et al. β2 Integrin-Derived Signals Induce Cell Survival and Proliferation of AML Blasts by Activating a Syk/STAT Signaling Axis. Blood (2013) 121:3889–3899, S1-66. doi: 10.1182/blood-2012-09-457887
104. Schweitzer KM, Dräger AM, van der Valk P, Thijsen SF, Zevenbergen A, Theijsmeijer AP, et al. Constitutive Expression of E-Selectin and Vascular Cell Adhesion Molecule-1 on Endothelial Cells of Hematopoietic Tissues. Am J Pathol (1996) 148:165–75.
105. Hidalgo A, Weiss LA, Frenette PS. Functional Selectin Ligands Mediating Human CD34(+) Cell Interactions With Bone Marrow Endothelium Are Enhanced Postnatally. J Clin Invest (2002) 110:559–69. doi: 10.1172/JCI14047
106. Lévesque JP, Zannettino AC, Pudney M, Niutta S, Haylock DN, Snapp KR, et al. PSGL-1-Mediated Adhesion of Human Hematopoietic Progenitors to P-Selectin Results in Suppression of Hematopoiesis. Immunity (1999) 11:369–78. doi: 10.1016/s1074-7613(00)80112-0
107. Winkler IG, Snapp KR, Simmons PJ, Lévesque J-P. Adhesion to E-Selectin Promotes Growth Inhibition and Apoptosis of Human and Murine Hematopoietic Progenitor Cells Independent of PSGL-1. Blood (2004) 103:1685–92. doi: 10.1182/blood-2003-06-1921
108. Eto T, Winkler I, Purton LE, Lévesque J-P. Contrasting Effects of P-Selectin and E-Selectin on the Differentiation of Murine Hematopoietic Progenitor Cells. Exp Hematol (2005) 33:232–42. doi: 10.1016/j.exphem.2004.10.018
109. Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, et al. Endothelial E-Selectin Inhibition Improves Acute Myeloid Leukaemia Therapy by Disrupting Vascular Niche-Mediated Chemoresistance. Nat Commun (2020) 11:2042. doi: 10.1038/s41467-020-15817-5
110. Erbani J, Tay J, Barbier V, Levesque J-P, Winkler IG. Acute Myeloid Leukemia Chemo-Resistance Is Mediated by E-Selectin Receptor CD162 in Bone Marrow Niches. Front Cell Dev Biol (2020) 0:668. doi: 10.3389/fcell.2020.00668
111. De Grandis M, Bardin F, Fauriat C, Zemmour C, El-Kaoutari A, Sergé A, et al. JAM-C Identifies Src Family Kinase-Activated Leukemia-Initiating Cells and Predicts Poor Prognosis in Acute Myeloid Leukemia. Cancer Res (2017) 77:6627–40. doi: 10.1158/0008-5472.CAN-17-1223
112. von Bonin M, Moll K, Kramer M, Oelschlägel U, Wermke M, Röllig C, et al. JAM-C Expression as a Biomarker to Predict Outcome of Patients With Acute Myeloid Leukemia-Letter. Cancer Res (2018) 78:6339–41. doi: 10.1158/0008-5472.CAN-18-0642
113. De Grandis M, Mancini SJC, Vey N, Aurrand-Lions M. JAM-C Expression as a Biomarker to Predict Outcome of Patients With Acute Myeloid Leukemia-Response. Cancer Res (2018) 78:6342–3. doi: 10.1158/0008-5472.CAN-18-0834
114. Baer MR, Stewart CC, Lawrence D, Arthur DC, Byrd JC, Davey FR, et al. Expression of the Neural Cell Adhesion Molecule CD56 Is Associated With Short Remission Duration and Survival in Acute Myeloid Leukemia With T(8;21)(Q22;Q22). Blood (1997) 90:1643–8. doi: 10.1182/blood.V90.4.1643
115. Sasca D, Szybinski J, Schüler A, Shah V, Heidelberger J, Haehnel PS, et al. NCAM1 (CD56) Promotes Leukemogenesis and Confers Drug Resistance in AML. Blood (2019) 133:2305–19. doi: 10.1182/blood-2018-12-889725
Keywords: adhesion, haematopoietic stem cell, leukemic stem cell, haematopoiesis, bone marrow, acute myeloid leukaemia
Citation: Grenier JMP, Testut C, Fauriat C, Mancini SJC and Aurrand-Lions M (2021) Adhesion Molecules Involved in Stem Cell Niche Retention During Normal Haematopoiesis and in Acute Myeloid Leukaemia. Front. Immunol. 12:756231. doi: 10.3389/fimmu.2021.756231
Received: 10 August 2021; Accepted: 27 October 2021;
Published: 12 November 2021.
Edited by:
Pedro Jose Esteves, Centro de Investigacao em Biodiversidade e Recursos Geneticos (CIBIO-InBIO), PortugalReviewed by:
Jean-Pierre Levesque, University of Queensland, AustraliaLouise Purton, University of Melbourne, Australia
Copyright © 2021 Grenier, Testut, Fauriat, Mancini and Aurrand-Lions. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel Aurrand-Lions, bWljaGVsLmF1cnJhbmQtbGlvbnNAaW5zZXJtLmZy
†Present Address: Stéphane J. C. Mancini, INSERM U1236, MIcroenvironment, Cell differentiation, iMmunology And Cancer (MICMAC), Rennes, France