
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 12 November 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.755961
This article is part of the Research TopicNegative Regulators of Innate Immunity and their Role in Host Responses to Injury and InfectionView all 13 articles
 Debabrata Chowdhury1Jason C. Gardner2Abhijit Satpati3
Debabrata Chowdhury1Jason C. Gardner2Abhijit Satpati3 Suba Nookala3
Suba Nookala3 Santhosh Mukundan3
Santhosh Mukundan3 Aleksey Porollo4,5,6Julio A. Landero Figueroa7
Aleksey Porollo4,5,6Julio A. Landero Figueroa7 Kavitha Subramanian Vignesh1*
Kavitha Subramanian Vignesh1*Non-canonical inflammasome activation by mouse caspase-11 (or human CASPASE-4/5) is crucial for the clearance of certain gram-negative bacterial infections, but can lead to severe inflammatory damage. Factors that promote non-canonical inflammasome activation are well recognized, but less is known about the mechanisms underlying its negative regulation. Herein, we identify that the caspase-11 inflammasome in mouse and human macrophages (Mϕ) is negatively controlled by the zinc (Zn2+) regulating protein, metallothionein 3 (MT3). Upon challenge with intracellular lipopolysaccharide (iLPS), Mϕ increased MT3 expression that curtailed the activation of caspase-11 and its downstream targets caspase-1 and interleukin (IL)-1β. Mechanistically, MT3 increased intramacrophage Zn2+ to downmodulate the TRIF-IRF3-STAT1 axis that is prerequisite for caspase-11 effector function. In vivo, MT3 suppressed activation of the caspase-11 inflammasome, while caspase-11 and MT3 synergized in impairing antibacterial immunity. The present study identifies an important yin-yang relationship between the non-canonical inflammasome and MT3 in controlling inflammation and immunity to gram-negative bacteria.
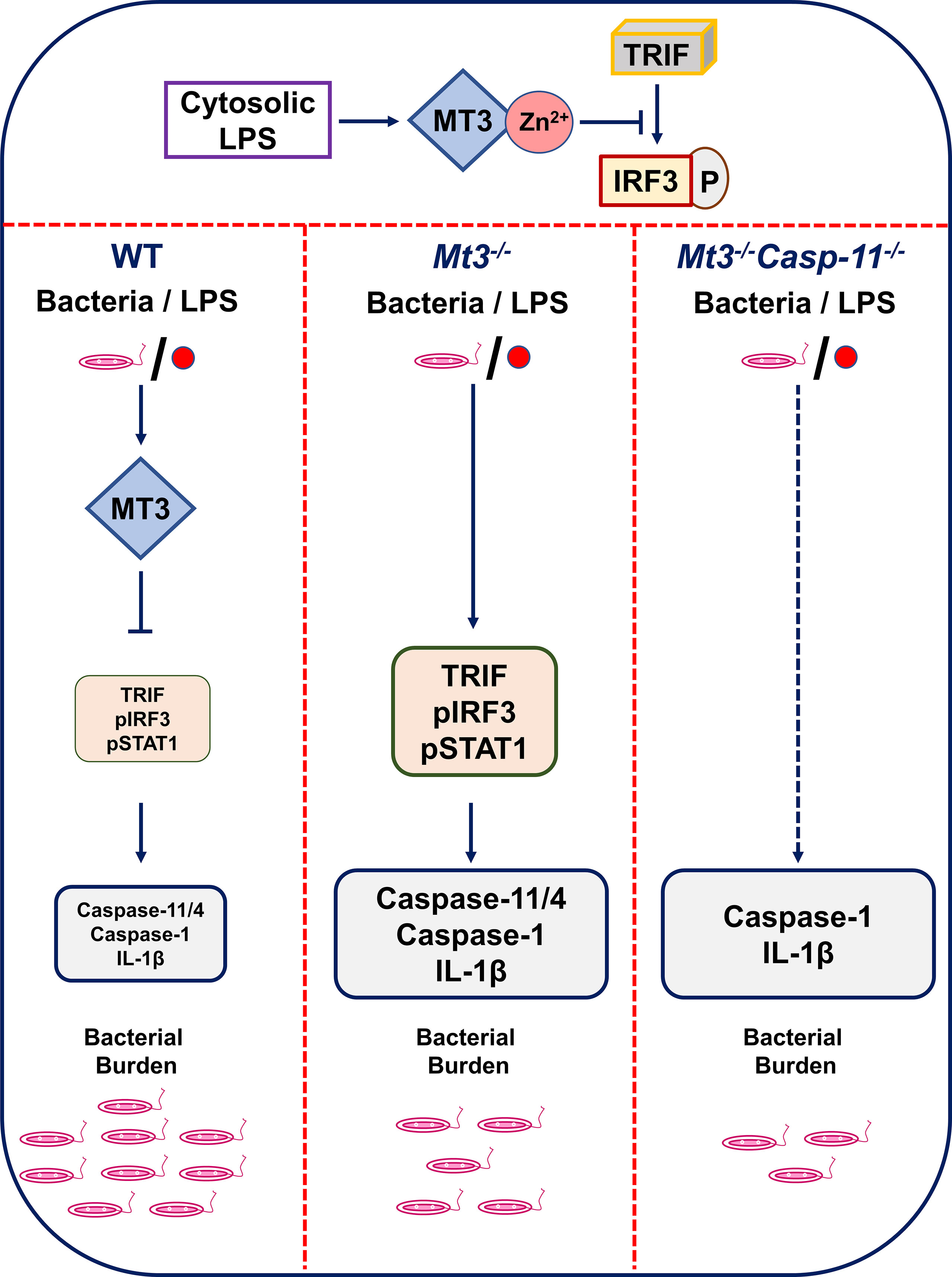
Graphical Abstract The MT3-Zn2+ axis suppresses TRIF signaling resulting in decreased IRF3 phosphorylation. When MT3 is absent, TRIF-IRF3-STAT1 signaling and non-canonical inflammasome activation are exaggerated. A lack of MT3 augments immunity to gram-negative bacteria, an effect, that is further enhanced by the combined absence of MT3 and caspase-11 in vivo. Thus, while MT3 curtails caspase-11 activation, the two molecules act together in compromising antibacterial immunity.
Gram-negative bacteria that cause more than 30% of the total healthcare-associated infections worldwide remain a global concern of morbidity and mortality (1). Assembly of inflammasome complexes during bacterial pathogenesis drives robust inflammation and shapes antibacterial immune responses. The non-canonical inflammasome is activated when innate immune cells such as Mϕ sense bacterial ligands in the cytosol (2). LPS from bacterial cell walls enters the cytosol during bacterial escape from vacuoles or via rupture of outer membrane vesicles (OMV) (3). iLPS directly binds caspase-11, triggering the non-canonical inflammasome cascade, followed by activation of pro-caspase-1 to caspase-1, processing of pro-IL-1β to mature IL-1β and pyroptosis, a lytic form of programmed cell death. Activation of gasdermin D (GSDMD) by caspase-11 and caspase-1, leads to pore formation on the cell membrane facilitating the exit of IL-1β from Mϕ (4). Unrestricted activation of this inflammatory cascade is a major underlying cause of tissue damage and sepsis-associated mortality (2). Thus, it is crucial to thoroughly understand the molecular cues that guard against excessive activation of the caspase-11 inflammasome. Although factors that promote non-canonical inflammasome activation have been extensively studied, less is known about the mechanisms that negatively regulate it.
MTs are Zn2+ regulating proteins induced by endogenous and exogenous stimuli including cytokines, infection, oxidative stress and heavy metals (5, 6). Intracellular availability of total Zn2+, exchangeable Zn2+ and Zn2+ redistribution among proteins is tightly regulated by MTs (7). Mice have 4 MT isoforms (MT1-4), whereas more than 16 MT isoforms are present in humans (8). Our knowledge on the role of the MT family in immune responses largely emerges from studies on MT1 and MT2. We and others have shown that MT1 and MT2 promote antifungal and antibacterial immunity in Mϕ primarily via Zn2+ sequestration (9, 10). MT3, on the other hand, suppresses manifestation of proinflammatory phenotypic and metabolic changes and impairs antifungal immunity in vitro in Mϕ and in vivo. Mϕ expression of MT3 is inducible by IL-4 stimulation. In these cells, MT3 increases intracellular free-Zn2+, promotes Zn2+ uptake by a prototypic intracellular pathogen and favors microbial survival (11). Thus, MTs and their ability to regulate Zn2+ homeostasis intricately ties Mϕ inflammatory responses to antimicrobial defense.
Zn2+ is indispensable in many biochemical processes due to its role in structural and catalytic functions of enzymes and macromolecules. Zn2+ excess or deficiency compromises the development and function of immune cells including monocytes and Mϕ, leading to increased risk of infection (12). Zn2+ also has a profound role as a signaling ion attributable to the transient changes in intracellular exchangeable Zn2+. LPS triggers increased Zn2+ import in leukocytes, monocytes and Mϕ (13, 14). In peripheral blood mononuclear cells (PBMCs) and human Mϕ (hMϕ), LPS-induced Zn2+ influx promotes IL-1β production (14). In contrast, exogenous exposure to Zn2+ in human monocytes may reduce IL-1β production due to inhibition of nucleotide phosphodiesterases (15). Thus, the effects of Zn2+ on signaling and cytokine production are context and Zn2+ concentration-dependent. Changes in ion flux, specifically K+ and Cl- egress and intracellular mobilization of Ca2+ underlie canonical nod-like receptor pyrin domain containing-3 (NLRP3) inflammasome activation (16). Intriguingly, Zn2+ exerts disparate effects on activation of this cascade. Long-term Zn2+ depletion disrupts lysosomal integrity leading to increased activation of the canonical NLRP3 inflammasome (17). On the other hand, short-term chelation of Zn2+ attenuates the canonical pathway due to impaired function of the pannexin-1 receptor (18).
The importance of Zn2+ regulation by MTs in the non-canonical inflammasome pathway remain unexplored. Given the suppressive role of MT3 in Mϕ inflammatory responses (19), we hypothesized that MT3 negatively regulates the highly inflammatory caspase-11 activation cascade. Bioinformatics analysis predicted the involvement of MT3 in regulating non-canonical inflammasome-associated pathways. Using a combination of protein-protein interaction network analysis, immunological and mass-spectrometric approaches, we demonstrate that triggering caspase-11 activation results in a profound, gradual increase in the Mϕ Zn2+ pool mediated by MT3. The increase in Zn2+ attenuates signaling via toll/interleukin-1 receptor (TIR) domain containing adaptor-inducing interferon (IFN)β - interferon regulatory factor 3 - signal transducer and activator of transcription factor 1 (TRIF-IRF3-STAT1), a pathway that is prerequisite for caspase-11 inflammasome activation (20). Zn2+ deficiency augments, whereas Zn2+ supplementation suppresses the non-canonical inflammasome in Mϕ. Using whole-body Mt3-/- and myeloid-MT3-deficient mice, we elucidate that MT3 blunts non-canonical inflammasome activation in vitro and in vivo upon challenge with iLPS or gram-negative bacteria but not gram-positive bacteria. Importantly, this function of MT3 is conserved in hMϕ. Although caspase-11 and MT3 form a negative regulatory loop, we find that these two molecules synergize in compromising antibacterial immunity. Our data uncover a previously unknown yin-yang relationship whereby the MT3-Zn2+ axis exerts a brake on non-canonical inflammasome activation but the functions of MT3 and caspase-11 converge in crippling immunity to invading bacteria (see Graphical Abstract).
MT3 attenuates cell death in neuronal and glial cells, but the precise underlying mechanisms are not fully understood (21). As non-canonical inflammasome activation leads to pyroptotic cell death, we investigated if MT3 effector function is related to caspase-11 activation in Mϕ. We explored whether MT3 is involved in inflammatory and cell-death processes using functional enrichment analysis. We assessed protein-protein interaction networks of MT3 in Mus musculus and Homo sapiens using the STRING database (22). The MT3 interaction partners significantly enriched 15 mouse and 43 human gene ontology categories for biological processes (GO BP) related to programmed cell death (PCD), LPS responses, signaling via TRIF, IL-1 and type-I IFN, cytokine responses and several immune processes. A complete network of MT3 interactions and GO BP categories is in Table 1, Supplementary Table S1, Supplementary Files S1 and S2. PCD and LPS responses are linked to inflammasome activation and more specifically, TRIF and type-I IFN signaling are tied to the non-canonical inflammasome pathway. A lack of TRIF signaling ablates non-canonical inflammasome activation in response to iLPS without impacting Mϕ response to canonical NLRP3 triggers such as ATP and nigericin (20). Thus, our bioinformatics analysis together with our previously reported role for MT3 in suppressing proinflammatory responses in Mϕ led us to investigate whether MT3 negatively regulates the non-canonical inflammasome pathway.
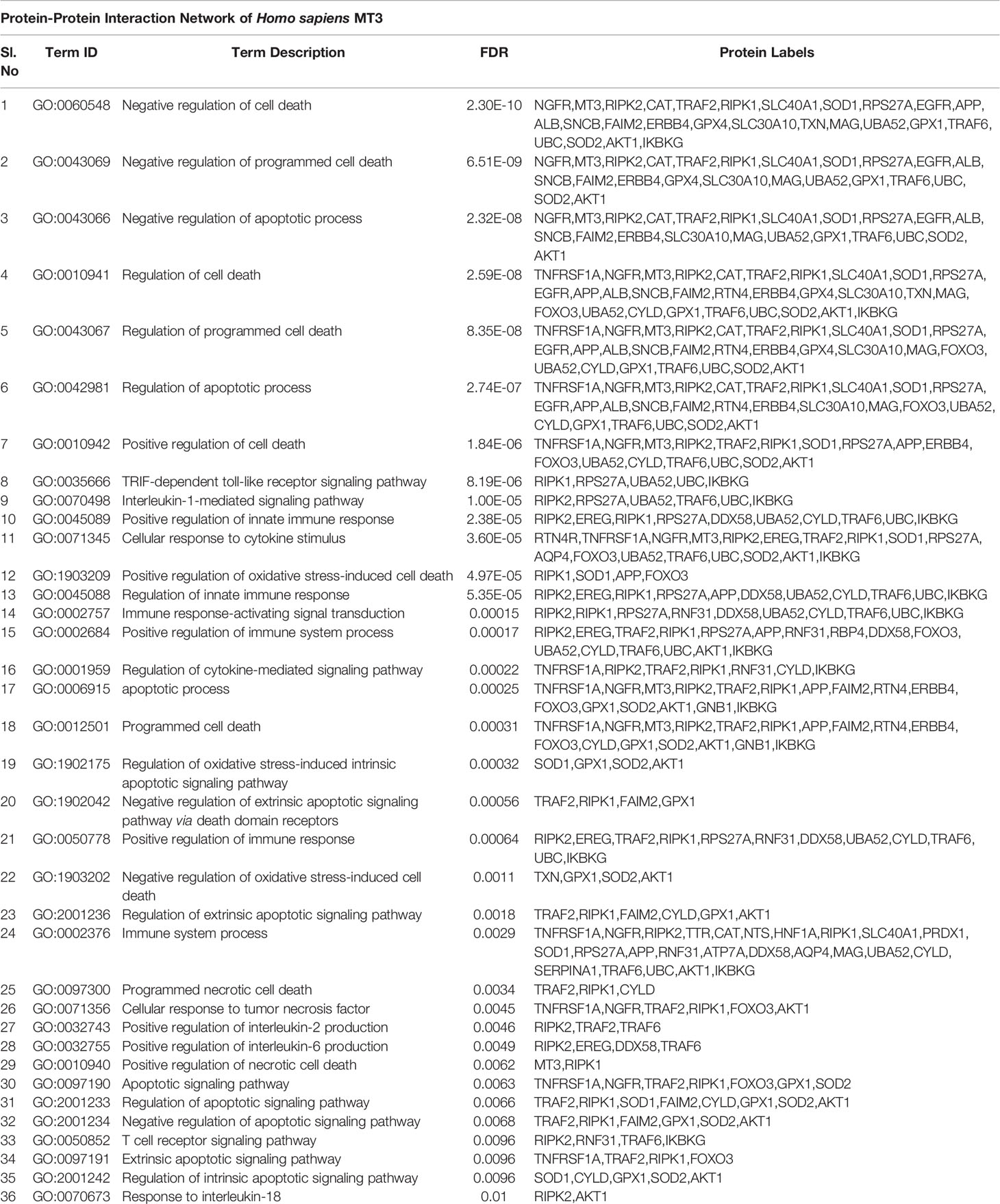
Table 1 See also Supplementary Table S1 and Files S1, S2 |Protein interaction network of Mus musculus MT3 to determine functionally enriched GO BP categories using the STRING database.
In Mϕ, exposure to iLPS triggers caspase-11 activation and cell death by pyroptosis (2). We directly assessed if MT3 regulates non-canonical inflammasome activation using WT and Mt3-/- mice. BMDMϕ were exposed to iLPS or vehicle control and time-dependent changes in the gene expression of Mt1, Mt2 and Mt3 were examined. Mt3 expression increased gradually from 1 hour (h) and peaked at 48h in WT BMDMϕ challenged with iLPS (Figure 1A). The expression of Mt1 and Mt2 peaked at 6h, but receded over time in both WT and Mt3-/- BMDMϕ (Supplementary Figures S1A, B). To determine whether exogenous LPS had an effect on MT3, we stimulated WT BMDMϕ with extracellular LPS (exLPS) for 48h. While iLPS increased Mt3 expression by 10-fold, the fold increase observed with exLPS challenge was much lower (Figure 1B).
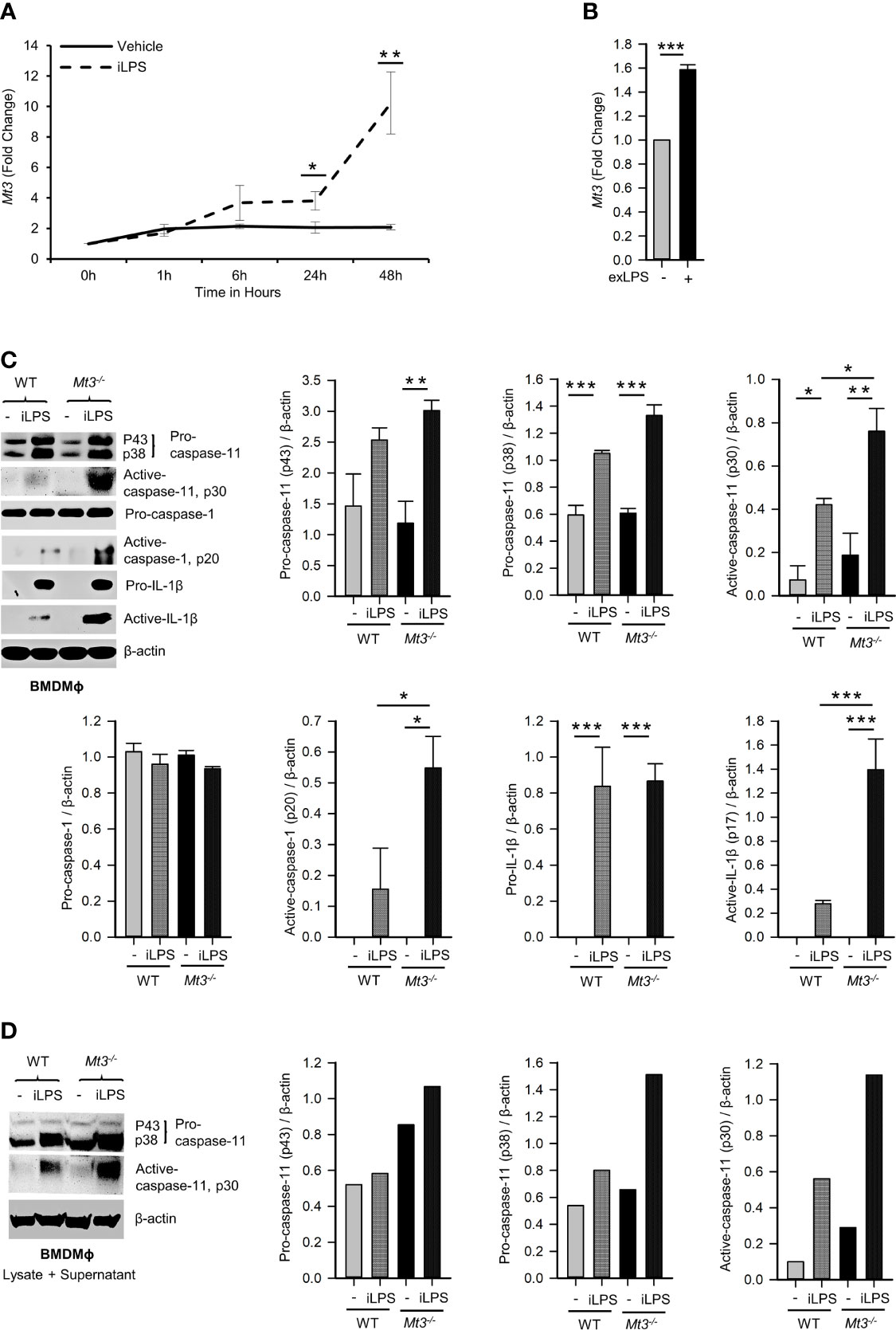
Figure 1 See also Supplementary Figure S1 MT3 suppresses caspase-11 inflammasome activation in BMDMϕ. qRT-PCR analysis of Mt3 expression in WT BMDMϕ stimulated with (A) iLPS (2 μg/ml) or vehicle control, 3-5 independent experiments and (B) exLPS (10 μg/ml) for 48h, 3 independent experiments, two-tailed t-test. (C) Western Blots of pro- and active-caspase-11, pro-caspase-1, pro-IL1β and β-actin in cell lysates and active-caspase-1 and active-IL-1β in supernatants of WT and Mt3-/- BMDMϕ stimulated with iLPS (10 μg/ml) or vehicle for 48h. Bar graphs are densitometric analysis of targets normalized to β-actin, 3-4 independent experiments, one-way ANOVA, data are mean ± SEM. (D) Western Blots of pro- and active-caspase-11 and β-actin in lysate + supernatant samples from WT and Mt3-/- BMDMϕ stimulated with iLPS (2 μg/ml) or vehicle for 48h. Bar graphs are densitometric analysis of targets normalized to β-actin. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we examined if MT3 regulated non-canonical inflammasome activation by assessing pro- and active forms of caspase-11, caspase-1 and IL-1β in cell lysates and supernatants of WT and Mt3-/- BMDMϕ challenged with iLPS. A lack of MT3 exacerbated activation of caspase-11 in cell lysates, and caspase-1 and IL-1β in culture supernatants. Pro-caspase-11, pro-caspase-1 and pro-IL-1β proteins in cell lysates were similar between iLPS treated WT and Mt3-/- BMDMϕ (Figure 1C). We further examined combined lysate and supernatant samples and found increased caspase-11 activation in Mt3-/- BMDMϕ challenged with iLPS (Figure 1D). Thus, the differences observed were not merely a result of reduced release of the active forms from cells, but an increase in non-canonical inflammasome activation in Mt3-/- BMDMϕ. Intracellular transfection of LPS at two different concentrations (2 and 10 μg/ml) yielded similar results (Figures 1C, D). We then examined the levels of IL-1α in supernatants of WT and Mt3-/- BMDMϕ challenged with iLPS. In contrast to IL-1β, IL-1α was moderately reduced in the supernatants of Mt3-/- BMDMϕ (Supplementary Figures S1C, D). We note that although IL-1α is activated by caspase-11, it is characteristically different from IL-1β, as it exists in both membrane-bound and secreted forms that are differentially regulated, and both pro- and cleaved IL-1α are bioactive (23–25).
Gram-negative bacteria activate the non-canonical inflammasome via CASPASE-4 in hMϕ (26). We investigated whether human MT3, similar to mouse MT3, suppressed non-canonical inflammasome activation and antibacterial immunity. Human monocyte-derived Mϕ obtained from PBMCs were transfected with scramble siRNA or MT3 siRNA followed by transfection with iLPS. To assess siRNA specificity, we analyzed the expression of MT3 and MT2A genes. MT3, but not MT2A expression was silenced in MT3 siRNA transfected hMϕ (Figure 2A). MT3 deficiency resulted in elevated activation of CASPASE-4 and heightened release of IL-1β from hMϕ (Figures 2B, C). We previously showed that a lack of MT3 increased resistance of mouse BMDMϕ to Escherichia coli (19). We therefore queried whether silencing MT3 in hMϕ impaired bacterial clearance in vitro. hMϕ treated with scramble siRNA or MT3 siRNA were infected with E. coli K12 for 24h. MT3-deficient hMϕ exerted a sharp decline in intracellular bacterial survival compared to control hMϕ (Figure 2D).
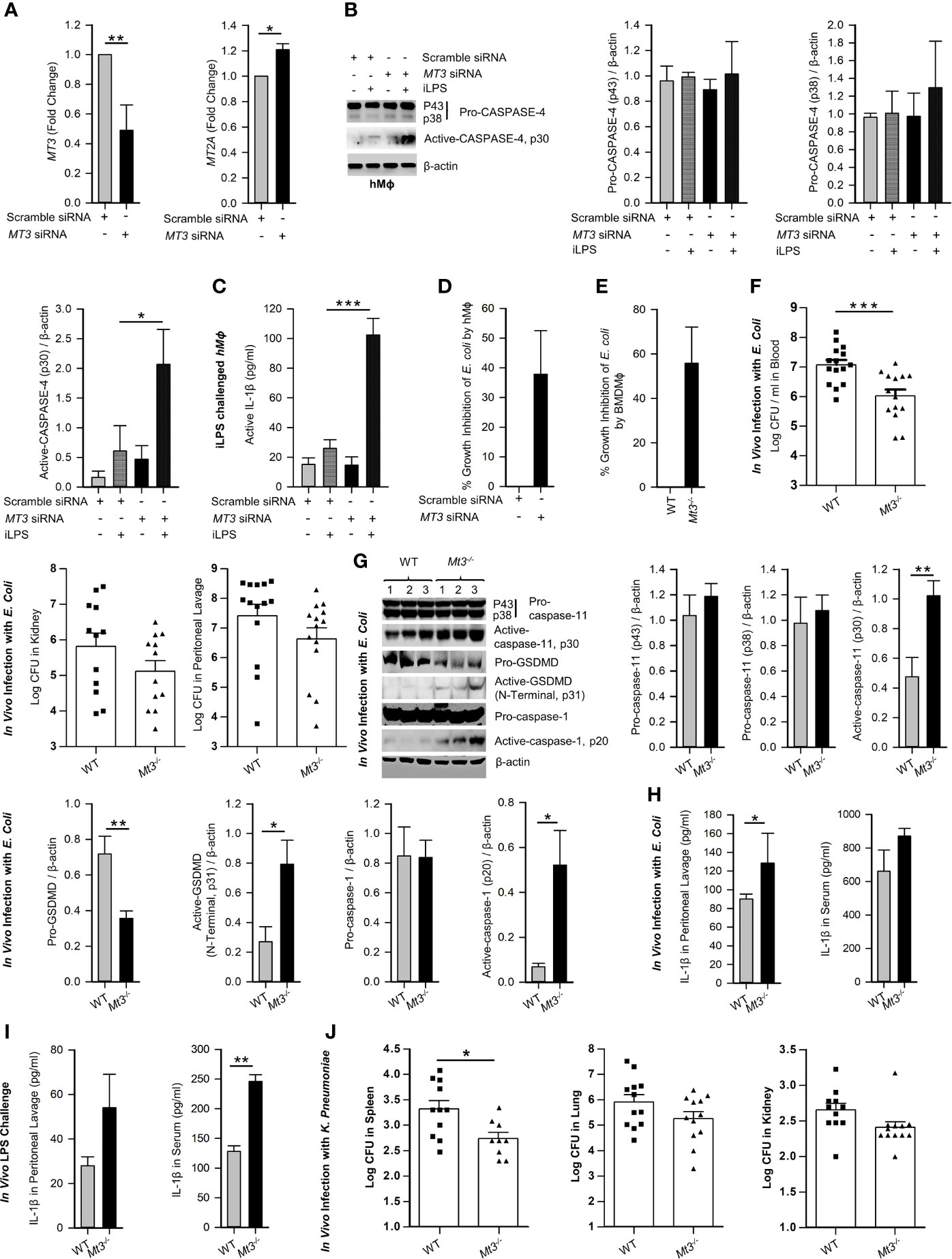
Figure 2 See also Supplementary Figure S2. MT3 curtails CASPASE-4 and caspase-11 signaling and antibacterial immunity in hMϕ and in vivo. (A) MT3 and MT2A expression analyzed by qRT-PCR in hMϕ transfected with scramble siRNA or MT3 siRNA for 24h, 3 independent experiments, two-tailed t-test. (B) Scramble siRNA or MT3 siRNA treated hMϕ stimulated with iLPS (10 μg/ml) or vehicle for 48h. Immunoblots of pro-CASPASE-4 and active-CASPASE-4 in cell extracts, 3 independent experiments, one-way ANOVA. (C) Active-IL-1β measured by ELISA in supernatants of hMϕ treated as above, 3 independent experiments, one-way ANOVA. (D) E. coli growth inhibition in hMϕ transfected with MT3 siRNA and infected with 25 E. coli (K12): 1 hMϕ for 24h compared to scramble siRNA treated hMϕ, 3 independent experiments, two-tailed t-test. (E) E. coli growth inhibition in WT and Mt3-/- BMDMϕ infected with 25 E. coli (K12):1 hMϕ for 24h, 4 independent experiments, two-tailed t-test. (F) WT and Mt3-/- mice infected i.p. with 1X109 E. coli for 6h, log CFUs of E. coli in blood, kidney and peritoneal lavage samples, n = 12-15 per group, two-tailed t-test. (G) Western blots of inflammasome mediators in kidney homogenates of WT and Mt3-/- mice infected as above, n = 6 per group, two-tailed t-test. (H) WT and Mt3-/- mice infected i.p. with 1 X109 E. coli for 1h and IL-1β measured in peritoneal lavage and serum by ELISA. n = 3 per group, two-tailed t-test. (I) WT and Mt3-/- mice primed i.p. with poly(I:C) (10 mg/kg) for 6h and challenged with LPS (2 mg/kg) i.p. After 18h, IL-1β was measured in peritoneal lavage and serum by ELISA, n = 3/group, two-tailed t-test. (J) Bacterial growth in spleen, lung and kidney of WT and Mt3-/- mice infected i.n. with K. pneumoniae (4 X104 CFUs/mouse) for 48h, n = 8-12 per group, two-tailed t-test, data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
We examined whether MT3 regulated antibacterial immunity and non-canonical inflammasome activation in vitro and in vivo. WT and Mt3-/- BMDMϕ were infected with E. coli. After 24h, bacterial survival was reduced in Mt3-/- BMDMϕ (Figure 2E). Next, we infected WT and Mt3-/- mice in vivo intraperitoneally (i.p.) with E. coli for 6h. Compared to WT mice, MT3 deficiency bolstered bacterial elimination from the blood and moderately improved bacterial clearance in the kidney and peritoneal lavage (Figure 2F). Caspase-11, GSDMD (N-terminal) and caspase-1 activation were heightened in kidney homogenates of infected Mt3-/- mice compared to infected WT mice (Figure 2G). The decrease in pro-GSDMD of Mt3-/- mice may be explained by increased conversion of pro- to active-GSDMD form (Figure 2G). IL-1β in the peritoneal lavage was significantly elevated (p<0.01) and serum IL-1β exhibited a trend towards increase in infected Mt3-/- mice compared to WT controls (Figure 2H). To determine if this response is consistent upon LPS challenge in vivo, we primed mice i.p. with poly(I:C) for 6h and challenged them i.p. with ultrapure LPS. After 18h, IL-1β was elevated in the peritoneal lavage and serum of Mt3-/- mice compared to WT mice (Figure 2I). We further queried the impact of MT3 on LPS-induced sepsis. WT and Mt3-/- mice were challenged with ultrapure LPS (20 mg/kg) and assayed for weight loss, murine sepsis scores (MSS) as reported previously (27) and survival. MT3 deficiency resulted in greater weight loss and increased sepsis scores, but both genotypes similarly succumbed to septic shock (Supplementary Figures S2A-C).
We then investigated whether MT3 increased susceptibility to other gram-negative bacteria. WT and Mt3-/- mice were infected intranasally (i.n.) with a virulent, heavily encapsulated strain of Klebsiella pneumoniae (KP2 2-70). MT3 deficiency significantly (p<0.05) improved K. pneumoniae clearance in the spleen, but no changes were observed in the lung and kidney (Figure 2J). Gram-positive bacteria activate caspase-11 via the NLRP6 inflammasome (28). We determined whether the increased non-canonical inflammasome activation and antibacterial resistance observed in Mt3-/- mice extended to gram-positive bacterial infection. WT and Mt3-/- mice were challenged subcutaneously (s.q.) with a clinical isolate of Group-A-Streptococcus GAS5448 (29). After 72h, Mt3-/- mice manifested significantly (p<0.05) reduced GAS burden in the kidney and spleen compared to WT mice. Bacterial CFUs in the blood exhibited a similar trend (Supplementary Figure S2D). Importantly, although Mt3-/- mice exhibited higher activation of caspase-1 and IL-1β, the levels of active caspase-11 and pro-caspase-11 were diminished in GAS-infected Mt3-/- mice compared to WT mice (Supplementary Figure S2E). Thus, although MT3 compromises resistance to both gram-negative and gram-positive bacteria, it specifically suppresses non-canonical inflammasome signaling in response to gram-negative microbial triggers.
Caspase-11 is crucial in antibacterial defenses particularly against gram-negative bacteria, although some studies have suggested a detrimental role for caspase-11 in bacterial elimination (30–35). MT3 suppressed antibacterial immunity as well as non-canonical inflammasome activation. Thus, we investigated whether the heightened immunity to E. coli in Mt3-/- mice was due to increased non-canonical inflammasome activation. We infected WT, Casp-11-/-, Mt3-/-, and Mt3-/-Casp-11-/- mice (Supplementary Figure S3A) i.p. with E. coli and assessed bacterial burden 6h post-infection. Compared to WT mice, bacterial elimination was enhanced in Mt3-/- and Casp-11-/- mice but this response was further exacerbated in Mt3-/- mice lacking caspase-11 (Figure 3A). These data indicate that the combined absence of MT3 and caspase-11 improves resistance to gram-negative bacterial infection. We then analyzed caspase-11 inflammasome mediators in kidney homogenates harvested 6h post-infection. Mice lacking MT3, or caspase-11, exhibited elevated activation of GSDMD (N-terminal), caspase-1 and IL-1β compared to WT mice (Figure 3B). These changes were also observed in the Mt3-/-Casp-11-/- mice. Since GSDMD is a target of caspase-11 as well as caspase-1 (36), an elevation in active GSDMD may result from higher caspase-1 activation observed in mice lacking MT3, caspase-11 or both. Caspase-8, a pro-apoptotic caspase, collaborates with caspase-11 to mediate systemic inflammation and septic shock (37, 38). Moreover, caspase-8, in addition to caspase-1 can directly cleave IL-1β. We therefore analyzed caspase-8 in kidney homogenates from E. coli infected WT, Mt3-/-, Casp-11-/- and Mt3-/-Casp-11-/- mice. Caspase-11 negatively influenced the activation of caspase-8 (Supplementary Figure S3B).
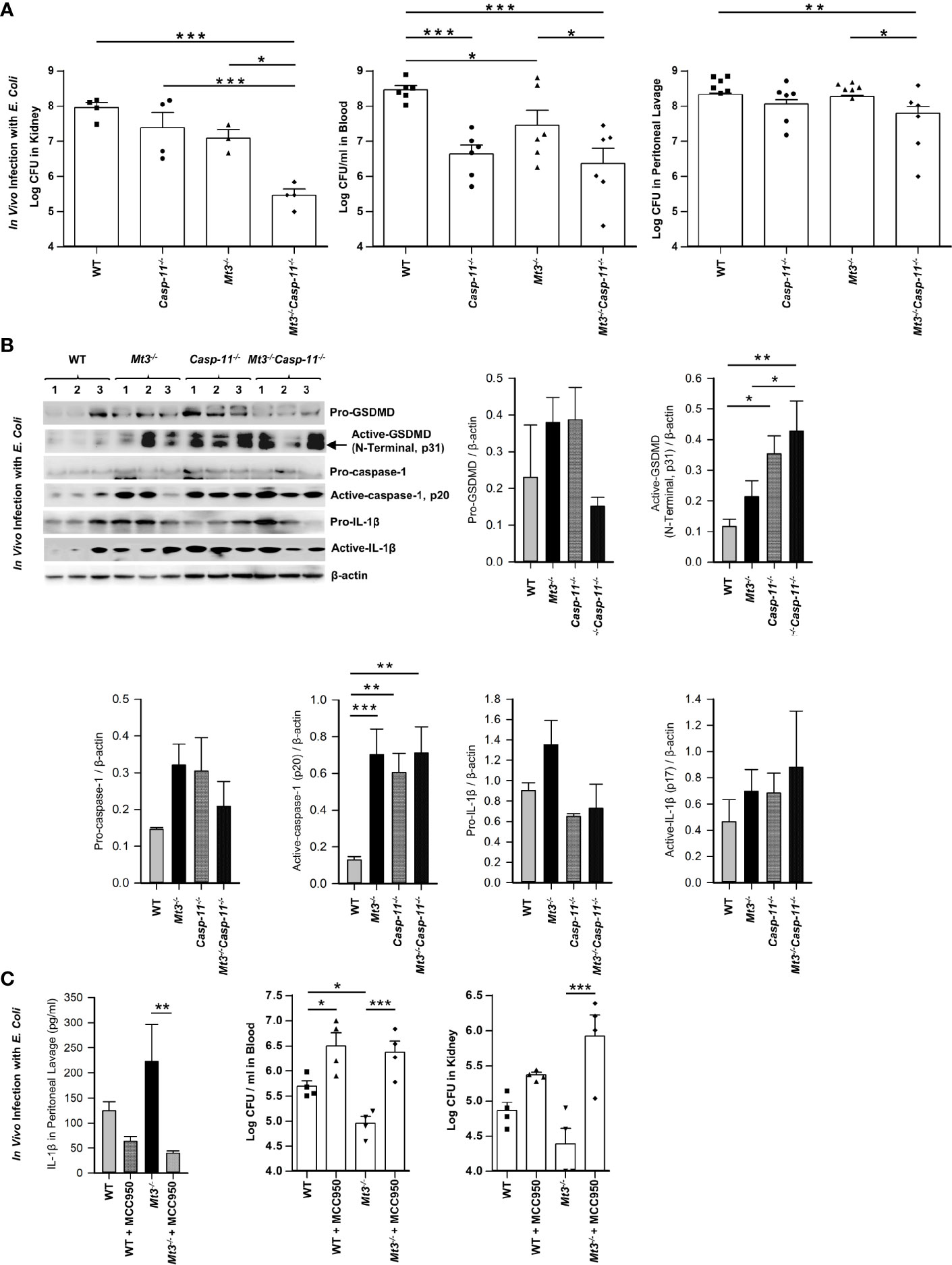
Figure 3 See also Supplementary Figure S3 Caspase-11 synergizes with MT3 in impairing bacterial clearance. WT, Casp-11-/-, Mt3-/- and Casp-11-/-Mt3-/- mice were infected i.p. with E. coli (1 X109 CFUs/mouse) for 6h. (A) Bacterial CFUs measured in kidney, blood and peritoneal lavage, n = 3-6 per group, one-way ANOVA. (B) Western blots of pro-GSDMD, active-GSDMD (p31), pro-caspase-1, active-caspase-1, pro-IL1β and active-IL-1β in kidney homogenates, n = 3-6 per group, one-way ANOVA, data are mean ± SEM. (C) WT and Mt3-/- mice treated i.p. with MCC950 (1 mg/mouse) or PBS and infected i.p. with E. coli (1 X109 CFUs/mouse) for 6h. IL1β was measured by ELISA in peritoneal lavage, n = 6 per group, one-way ANOVA, data are mean ± SEM. Bacterial CFUs in whole blood and kidney, n = 4 per group, one-way ANOVA, data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
The above data demonstrate that even when caspase-11 is absent, Mt3-/- mice exert heightened levels of active caspase-1, active IL-1β and antibacterial immunity. We therefore queried whether improved bacterial elimination in the absence of MT3 was facilitated by the canonical NLRP3 inflammasome. WT and Mt3-/- mice were treated i.p. with MCC950 (NLRP3 inhibitor) followed by infection i.p. with E. coli. Treatment with MCC950 reduced IL-1β levels and sharply blunted antibacterial resistance in the blood and kidney of Mt3-/- mice compared to vehicle-treated controls. Bacterial burdens were also elevated in WT mice by NLRP3 inhibition (Figure 3C). Collectively, these data demonstrate that MT3 negatively controls activation of the non-canonical inflammasome and that both MT3 and caspase-11 cripple resistance to bacterial infection.
To affirm that the effects on caspase-11 inflammasome activation observed in the Mt3-/- mice were dependent on myeloid-MT3, we generated mice specifically lacking MT3 in myeloid cells (Lys2Cre Mt3fl/fl) (Figure 4A and Supplementary Figures S4A, B). Genotyping analysis of BMDMϕ and peritoneal Mϕ (PMϕ) from Lys2Cre, Mt3fl/fl and Lys2Cre Mt3fl/fl mice demonstrated efficient removal of the Mt3 gene from BMDMϕ and PMϕ only in Lys2Cre Mt3fl/fl mice (Figure 4B). To determine if myeloid MT3 deficiency augmented non-canonical inflammasome activation in vivo, we infected Lys2Cre and Lys2Cre Mt3fl/fl mice i.p. with E. coli. After 6h, caspase-11 inflammasome targets and bacterial burden were examined in kidney and blood. Lys2Cre Mt3fl/fl mice exerted increased activation of caspase-11, GSDMD (N-terminal), caspase-1, and IL-1β in kidney homogenates and improved bacterial elimination compared to Lys2Cre control mice (Figures 4C, D). Thus, myeloid MT3 facilitates subversion of non-canonical inflammasome activation and contributes to antibacterial immunity in vivo.
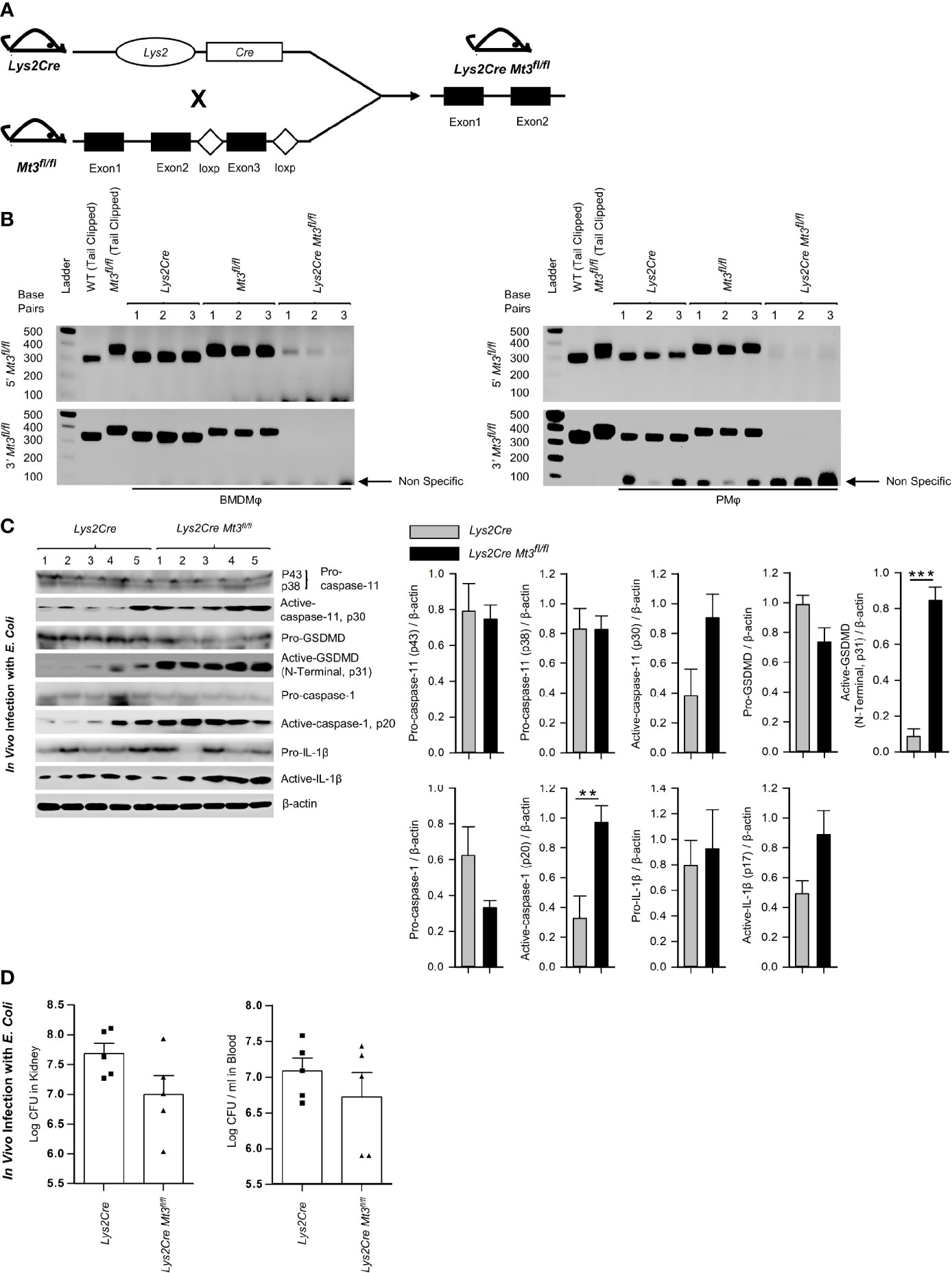
Figure 4 See also Supplementary Figure S4 Myeloid-MT3 suppresses non-canonical inflammasome activation and blunts gram-negative bacterial clearance in vivo. (A) Generation of Mt3fl/fl mice by inserting loxp sites flanking exon 3 of the Mt3 gene using the CRISPR-Cas9 gene targeting approach. Mt3fl/fl mice crossed with Lys2Cre mice to obtain Lys2Cre Mt3fl/fl mice. (B) Efficacy of myeloid Mt3 deletion assessed by genotyping peritoneal Mϕ (PMϕ) and BMDMϕ from Lys2Cre, Mt3fl/fl and Lys2Cre Mt3fl/fl mice. Gel electrophoresis analysis demonstrating efficient deletion of the Mt3 gene from BMDMϕ and PMϕ of Lys2Cre Mt3fl/fl mice. (C) Western blots of pro-caspase-11, active-caspase-11, pro-GSDMD, active-GSDMD (p31), pro-caspase-1, active-caspase-1, pro-IL1β and active-IL-1β in whole kidney homogenates of mice infected as above, n = 3-5 per group, two-tailed t-test. (D) Bacterial CFUs in kidney and whole blood of Lys2Cre and Lys2Cre Mt3fl/fl mice infected i.p. with E. coli (1 X109 CFUs/mouse) for 6h, n = 3-5 per group, two-tailed t-test, data are mean ± SEM. **p < 0.01, ***p < 0.001.
Signaling via the TRIF pathway is crucial for caspase-11 activation and synergistic engagement of the NLRP3 inflammasome leading to activation of caspase-1 and IL-1β (20). Downstream of TRIF, IRF3 and IRF7 induce IFNβ production that activates STAT1 signaling and promotes transcription of inflammasome components including caspase-11 and guanylate binding proteins (GBPs). GBP2 and GBP5 facilitate LPS release into the cytosol from intracellular vacuoles containing bacteria (39, 40). Our functional enrichment data based on protein-protein interaction network analyses revealed a potential involvement of MT3 in LPS, TRIF, type-I IFN and IL-1 signaling (Table 1, Supplementary Table S1 and Supplementary Files S1, S2). We further examined our published RNA-seq data (NCBI SRA: PRJNA533616) to determine differentially expressed genes by comparing resting WT and Mt3-/- BMDMϕ (19). The derived list of differentially expressed genes significantly enriched 12 GO BP categories directly related to cytokine and chemokine signaling and regulation of inflammatory responses based on DAVID functional enrichment analysis (Figure 5A) (41). These analyses suggested that MT3 deficiency perturbed the expression of immune-related genes even at the resting state. We reported that a lack of MT3 augments IFNγ responsiveness (19). Herein, from our RNA-seq analysis, we identified 20 genes related to IFN-signaling that were upregulated in resting Mt3-/- BMDMϕ compared to resting WT BMDMϕ (p adj <0.05) (Figures 5B, C). Among these, Isg15, Mx1 and Ifit (Ifit1bl1, Ifit3, Ifit3b, Ifit2, Ifit1, Ifit1bl2) family of genes are known targets of type-I IFNs (42–45). These observations led us to posit that MT3 regulated the cellular response to LPS challenge by modulating the TRIF-IRF3-STAT1 axis upstream of non-canonical inflammasome activation. LPS engages the TRIF-IRF3-STAT1 axis via toll-like receptor 4 (TLR4) signaling in Mϕ. To address this hypothesis, we challenged WT and Mt3-/- BMDMϕ with iLPS or vehicle and examined activation of the TRIF-IRF3-STAT1 pathway. Mϕ lacking MT3 exerted increased activation of phospho-IRF3 (pIRF3), pSTAT1, GBP2 and GBP5 (Figure 5D). TRIF protein levels were unaltered by MT3 deficiency (Figure 5E). Type-I IFN signaling is required for activation of the caspase-11 inflammasome cascade by gram-negative bacteria (20). As MT3 deficiency augmented the expression of genes involved in IFN signaling (Figures 5B, C), we blocked the interferon-α/β receptor (IFNAR)1 using a monoclonal antibody prior to iLPS challenge in WT and Mt3-/- Mϕ. IFNAR1 blockade resulted in decreased pro-caspase-11 (p43 subunit). Total STAT1 and pro-caspase-1 (p38 subunit) were not greatly affected (Supplementary Figure S5). We found robust attenuation of pSTAT1, active-caspase-11 and active-caspase-1, but secretion of active-IL-1β in both WT and Mt3-/- Mϕ was increased upon blockade of IFNAR1 signaling. This finding corresponded with high pro-IL-1β levels in Mϕ treated with the IFNAR1 antibody (Supplementary Figure S5). These data indicate that although IFNAR1 signaling is required for fueling the non-canonical inflammasome cascade and activation of caspase-1, pro-IL-1β and its activation are suppressed by IFNAR1.
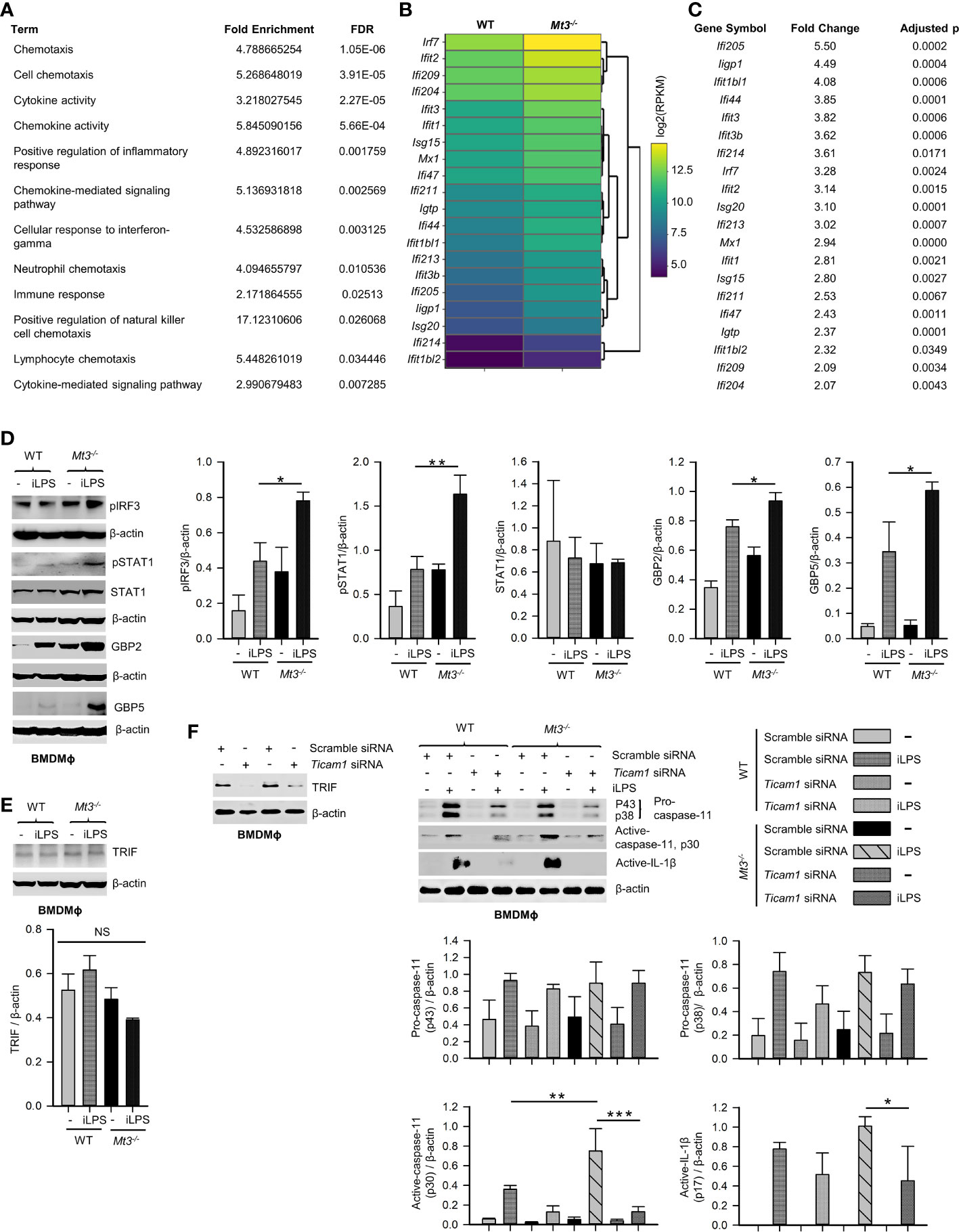
Figure 5 See also Supplementary Figure S5 MT3 thwarts TRIF-IRF3-STAT1 signaling to suppress non-canonical inflammasome activation. (A) Functional enrichment analysis of differentially expressed genes using RNA-seq data from resting WT and Mt3-/- BMDMϕ (NCBI SRA: PRJNA533616) (19) FDR, false detection rates. (B, C) Heat map (left) and table (right) show differentially expressed IFN-related genes in resting Mt3-/- BMDMϕ compared to resting WT BMDMϕ obtained from RNA-seq analysis. (D) Western blots of pIRF3, pSTAT1, STAT1, GBP2 and GBP5 in vehicle or iLPS (10 μg/ml)-treated WT and Mt3-/- BMDMϕ lysates, 3-4 independent experiments, one-way ANOVA. (E) Western blots of TRIF in lysates from WT and Mt3-/- BMDMϕ stimulated as above, 3 independent experiments, one-way ANOVA. (F) Scramble and Ticam1 siRNA treated WT and Mt3-/- BMDMϕ treated with iLPS (10 μg/ml) or vehicle for 48h. Immunoblots of TRIF (2 independent experiments), pro-caspase-11, and active-caspase-11 in lysates and active-IL-1β in supernatants, 3 independent experiments, one-way ANOVA, data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001; NS, not significant.
We queried if MT3 exerted a brake on TRIF signaling to downmodulate non-canonical inflammasome activation. WT and Mt3-/- BMDMϕ were treated with scramble or Ticam1 (gene encoding TRIF) siRNA, and challenged with iLPS (Figure 5F). Ticam1 silencing reversed the effects of MT3 deficiency resulting in a sharp reduction in caspase-11 and IL-1β activation in Mϕ (Figure 5F). These data reveal a central role for MT3 in attenuating the crosstalk between TRIF signaling and the caspase-11 activation cascade.
MTs are master regulators of intracellular Zn2+ availability and distribution (46, 47). We determined if negative control of the non-canonical inflammasome by MT3 was Zn2+-dependent. First, we systematically assessed Zn2+ changes in WT and Mt3-/- BMDMϕ upon challenge with iLPS over time using SEC-ICP-MS. Activation of the non-canonical inflammasome in WT Mϕ was associated with profound changes in the intracellular Zn2+ pool. iLPS exposure led to a gradual increase in total Zn2+ largely associated with the chromatogram peak(s) between 18-21 min. that we previously identified as MTs (Figures 6A, B) (9, 11). The time-dependent elevation in Zn2+ corresponded with kinetics of Mt3 induction (Figures 1A and 6A, B). Mϕ lacking MT3 failed to elevate total Zn2+ and MT-associated Zn2+ in response to iLPS (Figure 6B). In contrast to the increase in Zn2+ pool observed in WT Mϕ challenged with iLPS, resting Mt3-/- Mϕ harbored higher Zn2+ content that reduced over time post iLPS challenge (Figure 6B). These data indicate that MT3 drives an elevation in intracellular Zn2+ in Mϕ during non-canonical inflammasome activation.
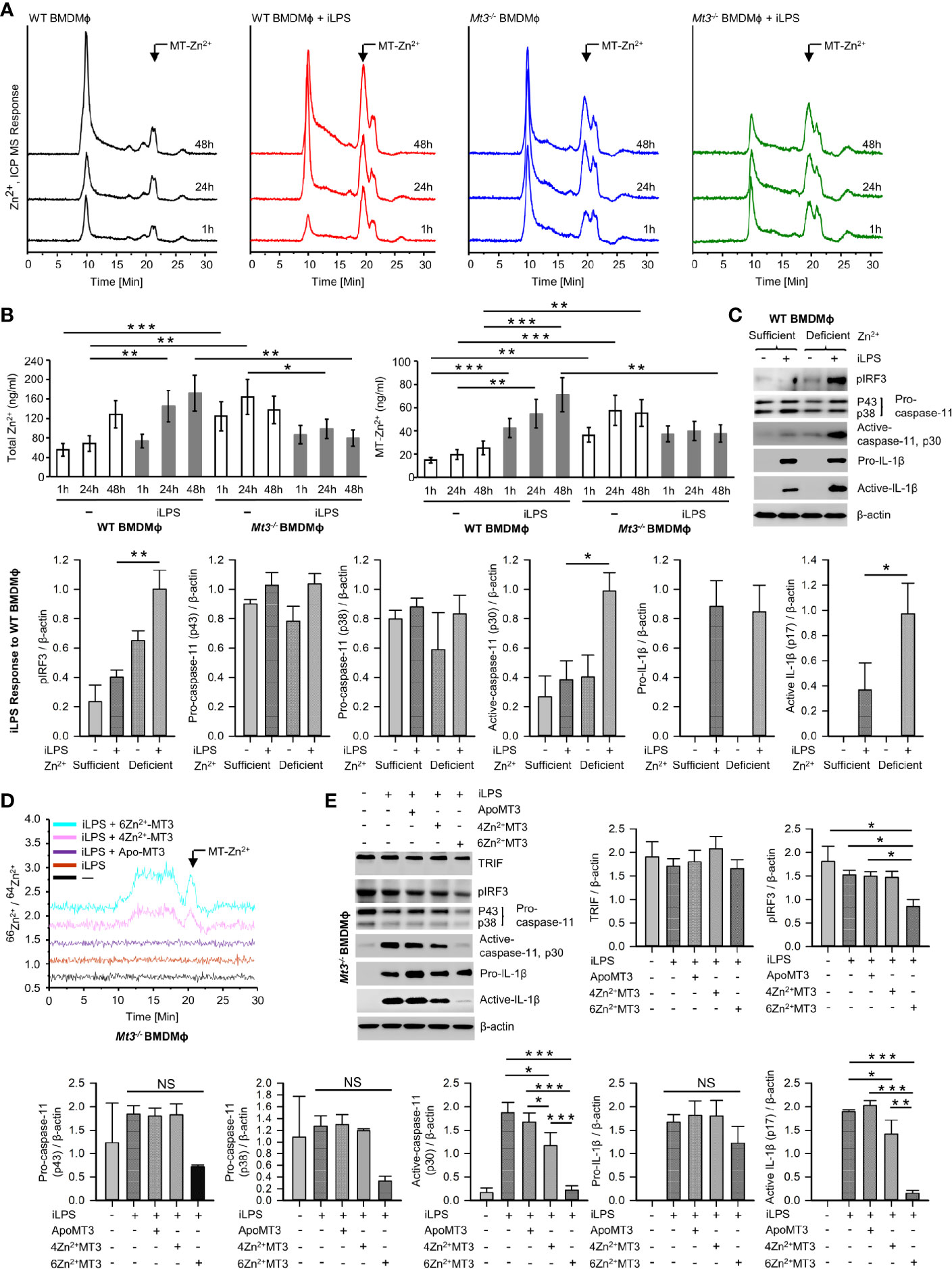
Figure 6 See also Supplementary Figures S6, 7 MT3-Zn2+ axis drives negative regulation of the non-canonical inflammasome. (A) SEC-ICP-MS of WT and Mt3-/- BMDMϕ exposed to vehicle or iLPS (10 ug/ml) for the indicated time points, chromatograms depict Zn2+ distribution in cell lysates across various molecular masses, arrow indicates Zn2+ associated with the MT-peak (18-21 min.) on the chromatogram, Y axis is off-set to allow easy comparison under the same scale. (B) Bar graphs of total Zn2+ and MT-Zn2+ in WT and Mt3-/- BMDMϕ post iLPS (10 μg/ml) or vehicle exposure. Two-way t-test against respective BMDMϕ controls at each time point, 3 independent experiments, data are mean ± SD. (C) WT BMDMϕ treated with iLPS (10 μg/ml) or vehicle for 24h in Zn2+ sufficient or Zn2+ deficient Opti-MEM media, immunoblots of pIRF3, pro-caspase-11, active-caspase-11 and pro-IL-1β in lysates and active-IL-1β in media supernatants, one-way ANOVA, data are mean ± SEM. (D, E) Mt3-/- BMDMϕ transfected with Pro-Ject™ or Pro-Ject™ complexed with apo-MT3, 4Zn2+MT3 or 6Zn2+MT3 and treated with iLPS (10 μg/ml) or vehicle for 24h in Zn2+ deficient Opti-MEM media. (D) Chromatograms depict Zn2+ distribution in cell lysates across various molecular masses, arrow indicates Zn2+ signal associated with the MT-peak (18-21 min.) on the chromatogram, Y axis is off-set to allow easy comparison under the same scale. (E) Western blots of pIRF3, pro-caspase-11, active-caspase-11 and pro-IL1β in lysates and active-IL-1β in supernatants, 3 independent experiments, one-way ANOVA, data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001; NS, not significant.
Zn2+ chelation in human monocytes increases IRF3 activation (48). We reasoned that if the effects of MT3 were Zn2+ dependent, altering the intracellular Zn2+ concentration will at least in part reverse the heightened non-canonical inflammasome signaling observed in Mt3-/- cells. To test this postulate, we exposed WT and Mt3-/- BMDMϕ to increasing amounts of ZnSO4 and challenged them with iLPS in vitro. Exogenous ZnSO4 supplementation remarkably reduced the ability of Mt3-/- Mϕ to respond to iLPS. pIRF3, pSTAT1 and activation of caspase-11 were reduced in ZnSO4-supplemented Mt3-/- Mϕ (Supplementary Figure S6A). A similar effect of Zn2+ was also observed in WT Mϕ (Supplementary Figure S6A). We investigated if exposing WT Mϕ to a Zn2+-deficient environment would mimic the effects MT3 deficiency on the non-canonical inflammasome. WT BMDMϕ were cultured in Zn2+-sufficient or Zn2+-deficient Opti-MEM media prior to iLPS exposure. WT Mϕ exposed to a Zn2+-deficient milieu manifested higher pIRF3 and caspase-11 activation accompanied by increased activation and release of IL-1β (Figure 6C). The amount of TRIF, pro-caspase-11 and pro-IL-1β were not affected by Zn2+ deficiency (Figures 6C and Supplementary Figure S6B).
Next, we directly addressed whether Zn2+ is required for the suppressive function of MT3 on the caspase-11 inflammasome. We first overexpressed the MT3 gene in Mt1-/-Mt2-/- Mϕ and isolated the protein. Mt1-/-Mt2-/- BMDMϕ were transfected with the Mt3 overexpressing vector (pCMV6-Ac-MT3-GFP) or an empty vector (pCMV6-Ac-GFP) control. Mt1-/-Mt2-/- Mϕ were used so as to exclude any contribution of these MTs in the MT3 purification process. The MT-associated peak from MT3-overexpressed Mϕ was identified by SEC-ICP-MS (Supplementary Figure S7) and collected. We complexed MT3 with the 66Zn2+ isotope to acquire an MT3-Zn2+ saturation of 4 Zn2+ ions per MT3 (MT3-4Zn2+) and 6 Zn2+ ions per MT3 (MT3-6Zn2+). The 66Zn2+ isotope was used to monitor changes in the ratio of 66Zn2+/64Zn2+ post-transfection of the MT3-66Zn2+ complexes in Mϕ. We transfected apo-MT3, MT3-4Zn2+ or MT3-6Zn2+ into Mt3-/- BMDMϕ in Zn2+-deficient media. The use of Mt3-/- BMDMϕ and Zn2+-deficient media enabled exclusion of any possible contribution from endogenous MT3 and exogenous Zn2+ in our analysis. Post-transfection of apo-MT3 or MT3-Zn2+ complexes, Mϕ were challenged with iLPS to activate the non-canonical inflammasome. To confirm that intracellular Zn2+ changes occurred upon transfection of the MT3- 66Zn2+ complexes, we analyzed BMDMϕ lysates by SEC-ICP-MS. Mt3-/- cells transfected with MT3-4Zn2+ and MT3-6Zn2+ but not apo-MT3 exhibited an increase in the 66Zn2+/64Zn2+ ratio in the MT-peak region at 18-21 mins. in the chromatogram indicating an elevation in the intracellular 66Zn2+ isotope (Figure 6D). These data confirm that transfection of the MT3-Zn2+ complexes resulted in an increase in intracellular 66Zn2+ in Mϕ. In parallel, we isolated cell lysates and supernatants proteins from these Mϕ to determine whether apo-MT3 or the MT3-Zn2+ complexes modulated the non-canonical inflammasome pathway. Transfection of MT3-4Zn2+ and MT3-6Zn2+ but not apo-MT3, dampened pIRF3, active caspase-11 and active IL-1β in response to iLPS (Figure 6E). TRIF levels were unaffected by MT3 transfection (Figure 6E). Pro-caspase-11 and pro-IL-1β were modestly diminished by MT3-4Zn2+ and MT3-6Zn2+ exposure, but these changes were not significant (Figure 6E). The effect of MT3-6Zn2+ was more profound than that of MT3-4Zn2+ indicating that a higher Zn2+ saturation on MT3 corresponded with a stronger suppressive effect on the non-canonical inflammasome (Figure 6E).
Taken together, these findings reveal a previously undescribed interplay between the non-canonical inflammasome and its negative regulator, whereby the MT3-Zn2+ axis suppresses caspase-11 inflammasome, but the two molecules concur in compromising immunological fitness of the host during bacterial pathogenesis.
Human CASPASE-4 or mouse caspase-11 are inflammatory caspases that drive cell death via pyroptosis. These caspases directly recognize bacterial LPS in the cytosol resulting in CASPASE-4 or caspase-11 auto-processing and synergistic activation of the NLRP3 inflammasome that culminates in caspase-1 activation, processing and release of IL-1β and IL-18 (2, 32). While the non-canonical inflammasome boosts host immunological fitness to some bacterial infections, heightened activation of this cascade poses the danger of tissue injury and organ failure. Thus far, negative regulation of IFNβ production by prostaglandin E2, immunity-related GTPases M clade, cyclic-adenosine monophosphate, and low dose oxidized phospholipid oxPAPC have been shown to thwart activation of the non-canonical inflammasome (49–52). The role of MTs in regulating inflammasome activation pathways has largely been unknown. Herein, we identify a previously undescribed function of MT3 in curtailing the highly inflammatory non-canonical inflammasome activation cascade via Zn2+ regulation. We demonstrate that while MT3 orchestrates negative regulation of the caspase-11 inflammasome, the combined presence of MT3 and caspase-11 blunts resistance to E. coli infection in vivo. These studies illuminate a central role for the MT3-Zn2+ axis in shaping the intricate balance between host antibacterial immunity and unrestrained inflammation.
MT1 and MT2 are ubiquitously expressed and can be induced by infection (53–56). Initial studies on MT3 revealed tissue-restricted expression with high levels predominantly found in the brain tissue where it inhibits neuronal cell death (21, 57). The immunological functions of MT3, particularly in the innate arm have only recently been investigated (11, 19, 21). We reported that Mt3 is inducible by the pro-resolving cytokines IL-4 and IL-13 in Mϕ. One inducer of Mt3 expression is STAT6 signaling, and this MT is crucial in shaping the phenotypic and metabolic attributes of Mϕ stimulated with type-2 cytokines (11, 19). Studies on MTs in response to exogenous LPS stimulation have largely focused on MT1 and MT2. Monocytes and Mϕ induce MT1 and MT2 upon extracellular LPS exposure (58, 59). We found that iLPS challenge also induced Mt1 and Mt2 in Mϕ, although their expression receded to baseline over time. In contrast, Mt3 expression gradually increased as non-canonical inflammasome activation progressed. Subversion of inflammatory responses in Mϕ by MT3 and the delayed expression pattern in response to a non-canonical inflammasome trigger are reminiscent of waning inflammation after the initial peak of inflammasome activation has subsided (19). In line with this hypothesis, protein interaction network analysis predicted the involvement of MT3 in cellular responses related to non-canonical inflammasome activation. Mϕ lacking MT3 exerted robust activation of caspase-11, caspase-1 and IL-1β. Similar to our observations with mouse MT3, a lack of human MT3 exacerbated the activation of CASPASE4 and IL-1β in hMϕ. Human and mouse MT3 proteins that share 86% identity thus have consistent roles that culminate in negative regulation of the non-canonical inflammasome cascade in Mϕ (60). As non-canonical inflammasome activation progressed, MT3 guarded against its unrestrained activation to avert potential inflammatory damage. Together, these observations reveal a pivotal role for MT3 in curtailing the vigor of the caspase-11 activation cascade. Although Mt3-/- mice exerted higher sepsis scores and weight loss, they succumbed to septic shock similar to WT controls, suggesting that a threshold level of caspase-11 activation may be sufficient to promote sepsis-associated mortality.
In vivo, LPS released from OMV of gram-negative bacteria triggers caspase-11 activation (2, 3). Myeloid-MT3 contributed to averting excessive activation of caspase-11 and synergistic activation of the canonical inflammasome in response to gram-negative microbial triggers. MT3 compromised antibacterial resistance to E. coli and K. pneumoniae, but this was not due to its suppressive action on the caspase-11 inflammasome in vivo. Instead, Mt3-/- and Casp-11-/- mice manifested improved antibacterial immunity, an effect that was further augmented when Mt3-/- mice lacked caspase-11. The synergism between MT3 and caspase-11 may result from independent or combined effects of MT3 and caspase-11 in vivo. Of note, the activation of caspase-1 and caspase-8 in the absence of MT3 and caspase-11 reveal that canonical inflammasome activation was operational and both caspase-1 and caspase-8 may contribute to IL-1β activation in vivo (38).
Mϕ utilize Zn2+ deprivation and Zn2+ intoxication mechanisms as strategies for antimicrobial defense (56, 61, 62). We previously showed that ablation of MT3 in Mϕ augments immunity to Histoplasma capsulatum as well as E. coli. The increased antimicrobial resistance in Mt3-/- Mϕ is at least partially attributable to a decrease in the Mϕ exchangeable Zn2+ pool and exaggerated IFNγ responsiveness (11, 19). Myeloid and non-myeloid cells may together contribute to bacterial elimination in whole-body MT3-deficient mice. Nonetheless, the augmented bacterial elimination observed in Lys2Cre Mt3fl/fl mice indicates that myeloid-MT3 contributes to the suppression of antibacterial defenses in vivo. The finding that MT3 deficiency dually bolstered inflammasome activation and antibacterial immunity underpins a role for this protein in suppressing the emergence of a proinflammatory phenotype in Mϕ. Our data unveil a unique crosstalk between caspase-11 and its negative regulator, whereby although MT3 keeps caspase-11 activation under control, the two synergistically compromise host immunological fitness to gram-negative bacterial infection.
Caspase-11 activation can have opposing effects on clearance of different bacteria. It improves resistance to Burkholderia thailandensis, B. pseudomallei, Brucella abortus, and Legionella pneumophila but may compromise immunity to B. cenocepacia, Salmonella typhimurium, E. coli, Shigella flexneri, K. pneumoniae and gram-positive infections including Streptococcus pyogenes, Staphylococcus aureus and Listeria monocytogenes (28, 31–33, 35, 63–69). Lipoteichoic acid from gram-positive bacteria engages the caspase-11 inflammasome via NLRP6 (28). Likewise, GAS infection led to caspase-11 activation in vivo. Although MT3 exerted disparate effects on caspase-11 activation in gram-positive and gram-negative infections, caspase-1 and IL-1β activation was suppressed by MT3 in both infection settings. In the context of GAS infection, both the host and the pathogen contribute to canonical inflammasome activation. Surface and secreted GAS virulence factor emm, and the streptococcal pyrogenic exotoxin B (SpeB) proteins act as second signals to activate caspase-1 signaling (70–72). Our data do not exclude the role of pathogen-derived factors in contributing to the increased canonical inflammasome activation observed in Mt3-/- mice infected with GAS. Nonetheless, our findings indicate that MT3 exerted a suppressive effect on the canonical caspase-1 pathway activated by gram-positive bacteria, while sparing negative regulation of the upstream non-canonical inflammasome activation in vivo.
The TRIF pathway is a central node in activation of the caspase-11 inflammasome in response to gram-negative infection (20). Targeting TRIF, but not IFNAR1, completely reversed the inflammatory cascade, including IL-1β activation. Blockade of IFNAR1 attenuated downstream activation of STAT1, caspase-11 and caspase-1, but both pro-IL-1β and active IL-1β levels were dramatically enhanced. This finding contrasts with the previously reported requirement of both TRIF and IFNAR1 in this pathway (20). Although that study utilized BMDMϕ from IFNAR1-/- mice and we used an anti-IFNAR1 monoclonal antibody, both approaches resulted in attenuation of targets downstream of IFNAR1. Emerging evidence points to an indirect inhibitory effect of type-I IFNs on inflammasome activation by decreasing pro-IL-1β transcription via IL-10 or 25-hydroxycholesterol (73, 74). The subdued activation of caspase-1 and heightened active IL-1β levels suggests that IL-1β activation occurs via a caspase-1 independent pathway when IFNAR is blocked. Interfering with IFNAR1 signaling can therefore subdue activation of critical inflammasome components including caspase-11 and caspase-1 but sustain IL-1β production and activation.
The crucial function of Zn2+ as a signaling molecule is well documented (13, 75–77). Changes in plasma and cellular Zn2+ levels regulate the production of various cytokines via NF-κB signaling (78, 79). Specifically, Zn2+ deficiency in humans increases the production of tumor necrosis factor (TNF)α and IL-1β by LPS-treated PBMCs ex vivo, whereas Zn2+ supplementation reduces it (80, 81). Therefore, a shift from physiological Zn2+ concentrations at the systemic or cellular level can impact proinflammatory cytokine responses and inflammation. To our knowledge, modulation of Mϕ Zn2+ homeostasis during non-canonical inflammasome activation has not been previously demonstrated. Our data show that caspase-11 activation is accompanied by a gradual expansion of the intracellular Zn2+ pool driven by MT3. Zn2+ deficiency did not augment pro- forms of caspase-11 (p43 and p38), but specifically increased their activation. Zn2+ diminishes signaling via IRF3 by limiting its nuclear localization (48). Accordingly, MT3 interfered with signaling via the TRIF-IRF3-STAT1 axis by shaping the Mϕ Zn2+ pool. The MT3-Zn2+ axis dampened IRF3 phosphorylation and downstream mediators without impacting TRIF levels. Although we cannot rule out the direct effect of Zn2+ on inflammasome components downstream of IRF3, the suppressive action of MT3 on the non-canonical inflammasome was Zn2+ dependent. Our data demonstrate that by manipulating the Mϕ Zn2+ milieu, caspase-11 activation can either be triggered or averted. This finding has important implications in defining a role for Zn2+ in subverting caspase-11 driven hyperinflammation. Developing therapeutic strategies that temper activation of the caspase-11/4 inflammasome have garnered tremendous interest to alleviate endotoxemia. In light of this, the MT3-Zn axis emerges as a fresh and vital candidate that guards the vigor of a caspase-11 fueled inflammatory response. In the context of gram-negative bacterial pathogenesis, our data indicate that strategies aimed at combined targeting of MT3 and the caspase-11 inflammasome may be more beneficial in infection control than targeting caspase-11 alone.
Taken together, our studies illuminate a double-edged phenomenon in inflammasome regulation whereby the MT3-Zn2+ axis is a sentinel of the caspase-11 inflammasome but MT3 and the non-canonical inflammasome function in concert to compromise host antibacterial resistance.
Reagents and resources can be found in Table 2.
Table 2 Reagents and resources.
E. coli (K12) and K. pneumoniae were kindly provided by Dr. Jason Gardner at the University of Cincinnati. Group A Streptococcus (GAS 5448) was kindly provided by Dr. Suba Nookala at the University of North Dakota.
All mice used in this study were on the C57BL/6 background. WT and Casp4tm1Yuan/J (Casp-11-/-) mice were acquired from the Jackson Laboratory. Lys2Cre mice were kindly provided by Dr. George S. Deepe. Jr. (University of Cincinnati). Mt3-/- (exon 3 deleted) and Mt3fl/fl mice were generated using clustered regularly interspaced short palindromic repeats (CRISPR) by the Transgenic Animal and Genome Editing Core facility at the Cincinnati Children’s Hospital Medical Center (CCHMC). In mice, the Mt gene cluster is located on chromosome 8. The Mt3 gene consists of 3 exons and is preceded by Mt1 and Mt2 genes. We targeted exon 3 of Mt3 by flanking it with loxp sites (Mt3fl/fl) using the CRISPR-Cas9 gene targeting approach. Lys2Cre Mt3fl/fl mice that exhibit myeloid Mt3 deficiency were generated by crossing Lys2Cre mice to Mt3fl/fl mice. Deletion of the Mt3 gene was confirmed in BMDMϕ and PMϕ of Lys2Cre Mt3fl/fl mice by genotyping as detailed in the CRISPR/Cas9 generation of Mt3fl/fl mice section. To confirm 3’ loxp site insertion or deletion, genomic DNA amplified using forward (5' TAG GCT TCC CAC CTG TTT GG 3') and reverse (5' GCC AAG ATA AAG TCC GGG GT 3') primers. To confirm 5’ loxp site insertion or deletion, genomic DNA amplified using forward (5' TCG AAC TAC CTC CAA ACA GAG AAC 3') and reverse (5' TCA GTT TGG TCC AAA CGG GAT G 3') primers. To confirm Lys2Cre gene insertion, genomic DNA amplified using mutant (5' CCC AGA AAT GCC AGA TTA CG 3'), common (5' CTT GGG CTG CCA GAA TTT CTC 3') and WT (5' TTA CAG TCG GCC AGG CTG AC 3') primers. Casp-11-/-Mt3-/- mice were generated by crossing Casp-11-/- mice to Mt3-/- mice. Casp-11 was amplified using tail genomic DNA by mutant reverse (5' CGC TTC CTC GTG CTT TAC GGT AT 3'), common forward (5' ACA ATT GCC ACT GTC CAG GT 3') and WT reverse (5' CAT TGC TGA CCT TAT TTC TGT ATG G 3') primers. Mt3 was amplified using tail genomic DNA by forward (5' TTG GGG TGA GGT GTA GAG GT 3') and reverse (5 GCC AAG ATA AAG TCC GGG GT ' 3') primers. Mice used in this study had ad-libitum access to food and water. All mice were housed in the Department of Laboratory Animal Medicine, University of Cincinnati, accredited by American Association for Accreditation of Laboratory Animal Care (Frederick, MD) and experiments were conducted in accordance with Animal Welfare Act guidelines of the National Institutes of Health.
The methods for the design of sgRNAs, donor oligos and the production of Mt3fl/fl (loxp sites surrounding exon 3 of the murine Mt3 gene) animals were as described previously (82). The sgRNAs were selected according to the on- and off-target scores from the CRISPR design web tool (http://genome-engineering.org) as well as CRISPOR (http://crispor.tefor.net) (83). The selected sgRNA target sequences were cloned, according to the published method (84), into the pX458 vector (addgene #48138) that was modified by us to contain an optimized sgRNA scaffold (85) and a Cas9-2A-GFP. Their editing activity were validated by the T7E1 assay in mouse mK4 cells (86), compared side-by-side with Tet2 sgRNA that was known to work in mouse embryos efficiently (87). Validated sgRNA was transcribed in vitro using the MEGAshorscript T7 kit (ThermoFisher) and purified by the MEGAclear Kit (ThermoFisher), and stored at -80˚C. To prepare the injection mix, we incubated sgRNA and Cas9 protein (ThermoFisher) at 37˚C for 5 mins. to form the ribonucleoprotein complex and then added the donor oligos to it. The initial attempt was to insert both loxP sites simultaneously via piezo-driven cytoplasmic injection (88) of 100 ng/ul Cas9 protein, 50 ng/ul 5’ sgRNA, 50 ng/ul 3’ sgRNA, 50 ng/ul 5’ donor, and 50 ng/ul 3’ donor into fertilized eggs. Injected eggs were transferred into the oviductal ampulla of pseudo-pregnant CD-1 females on the same day. Pups were born and genotyped by PCR and Sanger sequencing. However, only 3’ loxP-containing mice were obtained from this attempt. After breeding them to homozygosity for the 3’ loxP, a new set of 5’ sgRNA and the donor oligo was designed and injected into the zygotes with the mix containing 150 ng/ul Cas9 protein, 75 ng/ul 5’ sgRNA, and 100 ng/ul ssDNA donor oligo. Injected eggs were transferred into the oviductal ampulla of pseudopregnant CD-1 females on the same day. Pups were born and genotyped by PCR and Sanger sequencing. Founder mice carrying both 5’ and 3’ loxP sites in cis were finally obtained. Animals were housed in a controlled environment with a 12-h light/12-h dark cycle, with free access to water and a standard chow diet. All animal procedures were carried out in accordance with the Institutional Animal Care and Use Committee-approved protocol of Cincinnati Children’s Hospital and Medical Center.
hMϕ were prepared from peripheral blood mononuclear cells (PBMCs). Briefly, human blood obtained from the Hoxworth Blood Center, University of Cincinnati was diluted (1:2) with calcium- and magnesium-free 1X Dulbecco’s phosphate-buffered saline (DPBS) and inverted gently to mix. Ficoll-Paque (10 ml for the total volume of 40 ml diluted blood) was layered at the bottom of the tube. The tubes were centrifuged at 400 X g for 30 mins. without break at 20˚C. The PBMC interface was transferred to sterile tubes and washed three times using 40 ml of DPBS containing 2mM EDTA and centrifuged at 120 X g for 10 mins. without break at 4˚C. A final wash was performed with DPBS (without EDTA). Isolated PBMCs were resuspended in complete RPMI 1640 medium. PBMCs (5 X 106) were plated in 24 well plates containing complete RPMI medium. After 24h, adherent monocytes were washed three times with DPBS and plated in complete RPMI 1640 medium (Corning®) containing 10 ng/ml macrophage-colony stimulating factor (M-CSF), 10%FBS, 10 μg/ml gentamycin sulfate (Alkali Scientific Inc.) and 2-mercaptoethanol. Cells were differentiated by exposure to human recombinant M-CSF on days 0, 2 and 4. After 6 days, hMϕ were washed with DPBS prior to use for the experiment.
Mouse BMDMϕ were prepared by differentiating bone marrow cells in complete RPMI 1640 medium containing 10 ng/ml mouse M-CSF, 10% fetal bovine serum (FBS) (HyClone Laboratories, Utah), gentamycin sulfate (10 μg/ml) and 2-mercaptoethanol. BMDMϕ were fed on days 0, 2 with complete RPMI 1640 medium containing 10 ng/ml M-CSF and were supplemented on day 4 with M-CSF. After 6 days, adherent Mϕ were harvested by washing with DPBS followed by trypsinization and centrifuged at 1600 rpm for 5 mins. at 4˚C. Mϕ were washed again with DPBS at 1600 rpm for 5 mins. at 4˚C and counted under the microscope. Dead cells were excluded from enumeration using Trypan Blue stain. BMDMϕ (0.5 X 106 in 24 well or 1 X 106 in 12 well plate) were seeded.
The STRING database was used to review the protein-protein interaction networks of MT3 in Mus musculus and Homo sapiens (22). A full network analysis was conducted based on text mining, experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence data with a minimum required interaction score of 0.4 and a maximum of 50 interactors in the first shell and 50 interactors in the second shell. Statistical significance of the enriched biological processes (GO BP categories) in the MT3 network was set with a false discovery rate (FDR) <0.05.
Identification of differentially expressed genes in resting WT compared to resting Mt3-/- Mϕ was based on our previously published RNA-seq data (NCBI SRA: PRJNA533616) (19). The number of biological replicates used in the analysis was 3 per group. Genes differentially expressed with a fold change FC>2 and adjusted p value q<0.05 were considered significant. The Benjamini-Hochberg correction was used to adjust p values for multiple hypothesis testing. Differentially expressed genes in Mt3-/- BMDMϕ compared to WT BMDMϕ with q<0.05 were queried using the functional annotation clustering tool DAVID to identify statistically enriched GO categories (using the GO terms, BP direct, CC direct and MF direct) in Mt3-/- BMDMϕ compared to WT BMDMϕ (41). Significance of enrichment was set to FDR <0.05.
For gene silencing, Mϕ were transfected with the transfection complex (50 μl) of siRNA and TransIT-TKO® (0.5%) transfection reagent (Mirus Bio™) in 500 μl of complete RPMI 1640 medium without antibiotics as per the manufacturer’s instructions. Concentration of siRNAs used for gene silencing were 100 nM each of the non-targeting pool (ON-TARGETplus™ scramble siRNA), human MT3 (MT3 Silencer® Pre-designed siRNA) and mouse Ticam1 (ON-TARGETplus SMARTpool). All siRNAs were purchased from Dharmacon (GE Healthcare). Both BMDMϕ and hMϕ were incubated with the siRNA containing transfection complexes for 24h in RPMI medium and washed prior to transfection with LPS in Opti-MEM medium. TRIF silencing was assessed by protein expression using Western blots. Human MT3 silencing was assessed by gene expression using qRT-PCR.
IFNAR1 on WT and Mt3-/- BMDMϕ was neutralized using 10 μg/ml monoclonal anti-IFNAR1 antibody (BioLegend; Clone: MAR1-5A3) 1h prior and 24h after iLPS (10 μg/ml) stimulation. Negative control groups were treated with the same dose of isotype control IgG antibody (BioLegend; Clone: MOPC-21). After a total 48h, cell lysates and supernatants were harvested for the molecular analysis.
To activate the non-canonical inflammasome, 1 X 106 Mϕ were transfected with a transfection complex (50 μl) of 0.3% TransIT™-LT1 (Mirus Bio™) transfection reagent (vehicle) and 2 μg/ml or 10 μg/ml ultrapure LPS-B5 (In vivo Gen) prepared from E. coli 055:K59(B5) in 500 μl Opti-MEM medium (Thermo Fisher Scientific-US) as per manufacturer’s instructions for 24h.
Molecular biology grade chelex-100 resin (BioRad) was washed three times with metal free ddiH2O prior to use. To prepare Zn2+-deficient Opti-MEM medium, washed Chelex-100 resin (3 g per 100 ml) was added to Opti-MEM media and vigorously shaken for 1h on an orbital shaker at room temperature. After this time, media was filtered using a 0.22 μm filter and mixed with fresh washed chelex-100 resin and the same procedure was repeated for a total of 3 times to eliminate metals from Opti-MEM media. Chelex was removed from the media by a final filtration step. During each of these stages, an aliquot of the media was saved to monitor the efficiency of Ca2+, Mg2+, Mn2+, Co2+, Zn2+, Cu2+, Ni2+ and Fe2+ elimination by ICP-MS. The amount of Zn2+ in Opti-MEM media was decreased by 95% by the above chelation method. To prepare Zn2+-sufficient media, chelexed Opti-MEM was reconstituted with Ca2+, Mg2+, Mn2+, Co2+, Cu2+ and Zn2+ at the original concentrations as measured by ICP-MS. To prepare Zn2+-deficient media, all measured elements except Zn2+ were added to the chelexed Opti-MEM media at the original measured concentrations. Finally, the pH of Zn2+-sufficient and Zn2+-deficient Opti-MEM media was adjusted to 7.4 and filtered prior to use.
The pCMV6-Ac-GFP vector containing the mouse Mt3 gene (pCMV6-Ac-MT3-GFP) and empty pCMV6-Ac-GFP vectors were acquired from Origene and dissolved in nuclease-free sterile H2O. Plasmid DNA (5 ng) was added to 50 μl of thawed Novablue competent E. coli cells (EMD Millipore) and transformation was performed as per manufacturer’s instructions. E. coli cells were serially diluted in S. O. C media (ThermoFisher Scientific) and plated onto Luria-Bertani (LB) plates with 50 μg/ml carbenicillin and grown for 24h at 37˚C. A single colony was isolated and inoculated in LB media containing carbenicillin and grown at 37˚C for 5h in a shaker. The culture was further amplified by passaging for another 24h. E. coli cells were then harvested by centrifugation at 2000 rpm for 10 mins. and plasmid was extracted using the EndoFree plasmid MAXI kit (Qiagen) as per the manufacturer’s instructions. The plasmid was reconstituted in endotoxin-free TE buffer and OD readings obtained were in the range of 1.8-1.9. The resulting endotoxin-free plasmid DNA was set to a concentration of 1 mg/ml in filter-sterilized EndoFree TE buffer and frozen into aliquots until further use.
Mt1-/-Mt2-/- BMDMϕ were transfected with pCMV6-Ac-GFP control vector or pCMV6-Ac-MT3-GFP vector using the LT1 transfection reagent (Mirus Bio) in RPMI media containing 10% serum without antibiotics as per the manufacturer’s instructions. After 48h, BMDMϕ cultures were lysed with 250 μl of 0.1% SDS prepared in double-deionized (ddi) H2O for 20 min. on ice with intermittent mixing. Cell lysates were transferred to 0.22 μm filter tubes and centrifuged at 13000 rpm for 5 mins. Filtered cell lysates were subjected to SEC-ICP-MS to isolate the MT3 protein as described below.
Cell lysates from above were analyzed by SEC-ICP-MS to detect the MT3-associated peak, followed by collection of the fraction of interest (18-21 mins.). The collected fraction was concentrated by freeze drying in Millrock lyophilizer (Millrock, NY). The concentrated fraction was treated with 1 g of Chelex X-100 resin to remove the divalent metals associated to the protein. The total MT concentration was calculated by the total sulfur concentration in the sample with a 1:20 stoichiometry. The sample was divided into 3 fractions, and each fraction was incubated for 2 h in 50 mM Tris-HCl with the appropriate concentration of 66Zn2+ nitrate to obtain an MT3-Zn2+ saturation of 0, 4 or 6 Zn2+ ions per MT3 molecule. After incubation, samples were filtered using a 3 kDa MWCO filter to remove the unbound Zn2+ and reconstituted in 1X PBS.
In a 24 well plate, 5 X 105 Mt3-/- BMDMϕ were transfected with the transfection complex (10 μl) containing 500 ng of apo-MT3, 4Zn2+-MT3 or 6Zn2+-MT3 and Pro-Ject™ (1.75 μl) protein transfection reagent (ThermoScientific-US) in 250 μl of 2% FBS containing antibiotic free RPMI 1640 medium. Control Mt3-/- BMDMϕ were treated with Pro-Ject™ alone. Cells were incubated with the transfection complexes for 3.5h and washed two times using HBSS prior to challenge with iLPS in Zn2+ free Opti-MEM medium. At the experiment end point, cell lysates were either prepared for SEC-ICP-MS analysis or both cell lysates and supernatants were collected for the analysis of non-canonical inflammasome activation.
SEC-ICP-MS-MS analysis of WT and Mt3−/− BMDMϕ was performed as described previously (11). Mϕ were plated in Opti-MEM media, and either left untreated or transfected with 10 μg/ml LPS for 1, 24 and 48h. After this time, Mϕ were washed twice in HBSS and cells were lysed with 0.1% SDS on ice for 20 mins. Cell lysates were then centrifuged in 0.22 mm filter tubes at 13000 rpm for 10 mins. Filtered cell lysates were frozen at -80°C until further analysis by SEC-ICP-MS. 50-80 µl of cell lysates were injected to the HPLC-ICP-MS system according to protein concentration. To normalize the response of ICP-MS-MS signal from SEC separations on different days, 50 μl of 0.5 mg/ml carbonic anhydrase was injected into the liquid chromatography system, and area of Zn2+ signals from samples was normalized to area of the carbonic anhydrase peak from each day. The absorbance of carbonic anhydrase at 280 nm was followed to ensure protein integrity.
The instrumentation consisted of an Agilent 1100 HPLC equipped with a degasser, a binary pump, a thermostated auto sampler, a column oven compartment and a diode array detector. For the Mϕ lysates, a TSK gel 3000SW gel filtration column (TSK Tokyo Japan) 7.8 × 300 mm, 10 mm particle size was used. The mobile phase was ammonium acetate pH 7.4, 0.05% MeOH at 0.5 ml/minute. The HPLC system was coupled to the ICP-MS-MS nebulizer via a short polyether ether ketone capillary of 0.17 mm internal diameter. An Agilent 7500ce ICP-MS system equipped with a micromist quartz nebulizer, a chilled double pass Scott spray chamber and a standard 2 mm insert quartz torch with shield torch was used for all experiments. The ICP-MS was operated by the Agilent Mass Hunter integrated chromatographic software in the helium collision mode as reported previously (11). The isotope dilution experiments were processed by exporting the chromatographic data to Origin (Origin labs, CA) and the signal, in the form of counts per second, was used to calculate the ratio of 66Zn2+/64Zn2+ at every point in the chromatograms. This was used to generate a new chromatogram that reflected the input of 66Zn2+ from the MT3-Zn2+ complex at every molecular mass region in the chromatogram. The total area under the chromatograms was used to calculate the concentration of total Zn2+ (64Zn2+) and 66Zn2+ from the MT3-66Zn2+ complexes (66Zn2+/64Zn2+) against a calibration curve of Zn2+ based on carbonic anhydrase.
All metal analysis experiments were performed using trace metal grade reagents with acid washed plastic vials. Reagent blanks were used to correct the background signal. The analysis was performed through a metal free encased auto sampler. The concentration of Zn2+ in the blanks was always below 100 parts per trillion (ppt), the blank estimate concentration on the calibration curves was always below 50 ppt, while the detection limits were below 30 ppt.
For chromatographic analysis, the mobile phase was cleaned using a Chelex 100 resin, using the batch method. In brief, 3 g of Chelex-100 was added to a liter of mobile phase, stirred for 30 mins. and passed through a 0.45 mm membrane. This decreased the Zn2+ concentration below 200 ppt (measured as total). By this method, the base line ICP-MS-MS 66Zn2+ signal was below 1000 counts per second, which represents sub-ppb levels. The SEC column was cleaned using 10 volumes of 0.2 M NaCl and equilibrated with the mobile phase, followed by injection of 50 μl of 2% HNO3 three times to remove any accumulated Zn2+ in the column. With this procedure, Zn2+ distribution in the samples never deviated more than 10% compared to the theoretical natural Zn2+ isotope distribution in the control Mϕ samples. Four blanks and four carbonic anhydrase standards were injected after the cleaning procedure for monitoring Zn2+ signal by ICP-MS-MS to ensure optimal column performance. Typically, the column was cleaned every 30-40 samples.
Thirteen-week-old WT and Mt3-/- mice were primed with i.p. injection of 10 mg/kg poly(I:C) for 6h followed by 2 mg/kg ultrapure LPS-B5 (In vivoGen) prepared from E. coli 055:K59(B5) (i.p. injection) for 18h. At the experiment end point, blood was collected by cardiac puncture, allowed to clot, and centrifuged at 2000 rpm for 30 mins. at 4˚C to isolate serum. Serum was used to measure cytokines by enzyme-linked immunosorbent assay (ELISA).
For in vitro infection, E. coli (K12) was grown in LB broth at 37˚C overnight in an orbital shaker. The culture was pelleted, washed and resuspended with ice-cold DPBS. Optical Density (OD) of the culture was measured at 600nm using a spectrophotometer. To analyze E. coli burden in in vitro, hMϕ were transfected with scramble siRNA or MT3 siRNA as described above. hMϕ, WT and Mt3-/- BMDMϕ were infected with a multiplicity of infection (MOI) of 25 E. coli: 1 hMϕ for 3.5h in Opti-MEM media. Mϕ were washed 3 times with 10 μg/ml gentamycin sulfate containing DPBS to kill extracellular bacteria and incubated in Opti-MEM media with antibiotic for 24h. Mϕ were again washed 3 times with antibiotic-free DPBS, diH2O was added and cells were incubated for 30 min. to induce osmotic lysis. Cells were scraped and lysates were diluted in DPBS followed by plating on LB agar plates and incubated at 37˚C for 24h. Colonies were enumerated as above. Intracellular bacterial burden was represented as percent inhibition of bacterial growth in MT3-silenced hMϕ compared to scramble siRNA treated hMϕ and in Mt3-/- BMDMϕ compared to WT BMDMϕ.
To analyze antibacterial immunity in vivo, 10 to12 week-old mice were used. Mice were infected with E. coli 1 X 109 CFUs via i.p. injection (300 µl/mouse) for 1h or 6h. To investigate the role of NLRP3 inflammasome in antibacterial immunity, mice were treated with 1 mg/mouse MCC950 (an inhibitor of NLRP3 inflammasome) via i.p. injection (100 µl/mouse) for 1h followed by E. coli 1 X 109 CFUs via i.p. injection (300 µl/mouse) for 6h. K. pneumoniae KP2 2-70, a virulent, heavily encapsulated gram-negative bacterial strain (89, 90), was grown overnight in brain heart infusion (BHI) broth. The following morning bacteria were washed with DPBS, and administered at 4 x 104 CFUs in 50 µl per mouse by the i.n. to isoflurane-anesthetized mice for 48h. At the infection end point, blood was collected by cardiac puncture. A portion of the blood sample was acquired in anticoagulant (3% Na-citrate or EDTA) containing tubes to determine bacterial CFUs in blood. The remaining blood was allowed to clot, and centrifuged at 2000 rpm for 30 mins. at 4˚C to isolate serum. Peritoneal lavage was collected using ice-cold 10 ml DPBS. Kidney, lung, and spleen were collected after perfusion with 3 ml of DPBS, indicated organs and skin was rinsed in DPBS and ground with 5 ml DPBS using a glass grinder. Bacterial growth was measured in blood, peritoneal lavage, kidney, lung, skin and spleen samples. Serum and peritoneal lavage were used to measure cytokines by enzyme-linked immunosorbent assay (ELISA).
Septic shock was induced in mice via i.p. injection with ultrapure LPS-B5 (20 mg/kg) (In vivoGen) prepared from E. coli 055:K59(B5). Mice were weighed and sepsis scores were determined at various intervals. The MSS scoring method assesses sepsis severity with scores ranging from 1-4 based on 7 parameters (appearance, consciousness, activity, stimulus (sounds/touch), eyes aspect, respiration rate and respiration quality) (27). Survival analysis was conducted using the Kaplan-Meier analysis method and log-rank (Mantel-Cox test) was used to determine statistical differences in survival.
A representative M1T1 clonal Group-A-Streptococcus GAS5448 was used for subcutaneous infections (91). GAS was grown at 37°C under static conditions in Todd-Hewitt broth (BD, MD, USA) supplemented with 1.5% yeast extract (BD Biosciences, MD, USA) and in vivo infections were performed as described previously (29, 92). WT and Mt3-/- mice (n=8/group) were used in this study as a model for subcutaneous GAS infections. One day prior to infection, the hair on the back of the mice was depilated (using Nair cream) and mice were infected subcutaneously with 0.1 ml of GAS suspension prepared in sterile DPBS (Ca2+/Mg2+ free, low endotoxin, Mediatech, VA, USA, DPBS) (OD600 adjusted to yield ~1-5x10 (8) CFUs). Actual inoculum was determined by plating on trypticase soy agar containing 5% sheep blood (BD Biosciences, MD, USA). Mice were monitored twice daily for body weight, lesions, and mortality. To determine GAS dissemination and load, mice were humanely euthanized 72h post-infection. Blood was drawn through cardiac puncture; necrotic skin, kidney, and spleen were recovered aseptically and weighed. One ml of DPBS was added per 100 mg tissue and homogenized (Omni International, Marietta, GA) followed by plating of ten-fold dilutions on blood agar plates. GAS burden was calculated as colony-forming units (CFUs) per ml (blood) or per mg of tissue. The remaining homogenates were centrifuged for 15 mins. at 12,000 x g at 4°C, and supernatants were stored at -80°C for western blot analysis.
RNA was isolated from Mϕ after elimination of genomic DNA using RNeasy Plus Mini kit (Qiagen) or QUICK-RNA™ MINIPREP KIT (Thomas Scientific). cDNA was prepared using Reverse Transcription Systems Kit (Promega, WI) or rAmp First Strand cDNA Synthesis Flex Kit (Thomas Scientific). Taqman primer/probe sets (Applied Biosystems, CA) were used for real-time gene expression analysis using ABI Prism 7500. For time course analysis of expression of murine Mt genes, Mϕ were left unstimulated or stimulated with 2 μg/ml iLPS for 0h, 1h, 6h, 24h and 48h. Data are presented as fold change in gene expression normalized to unstimulated Mϕ at the 0h time point. To examine the effects of extracellular ultrapure LPS (exLPS) on murine Mt3 gene expression, BMDMϕ were left unstimulated or stimulated with 10 μg/ml exLPS. Data are presented as fold change in gene expression normalized to unstimulated Mϕ at the 48h time point. For MT2A gene expression analysis in MT3 silenced hMϕ, cells were treated with either MT3 siRNA or scramble siRNA as mentioned above. Data are presented as fold change in gene expression normalized to control siRNA treated hMϕ. Hypoxanthine guanine phosphoribosyl transferase (Hprt) was used as an internal control to compare target gene expression.
TRIF (Proteintech), pIRF3 (BIOSS), STAT1, pSTAT1 (Abcam), GBP2, GBP5 (Proteintech), caspase-11 (Abcam and eBioscience™), CASPASE-4 (MBL), Gasdermin D (Proteintech and Cell Signaling Technologies), caspase-1 (AdipoGen Life Sciences), IL-1β (R&D Systems) and caspase-8 (Enzo Life Sciences) were assessed in kidney homogenates of E. coli infected mice. Cell lysates were prepared using Denaturing Cell Extraction Buffer (Invitrogen) containing protease & phosphatase inhibitor cocktail (ThermoScientific). Culture supernatants were frozen at -80˚C until use and processed using methanol-chloroform protein extraction method. Briefly, supernatants were mixed with equal volume of 100% ice-cold methanol and 0.25 times of the total volume of chloroform followed by gentle vortexing and centrifugation at 20,000 X g at 4˚C for 10 min. Upper-phase was discarded without disturbing inter-phase proteins. Ice-cold methanol (500 μl) was added to the tube, gently vortexed and centrifuged at 20,000 X g at 4˚C for 10 mins. Supernatants were discarded and pellet was dried at 37˚C for 3-5 mins. Urea (8 M, pH-8.0) was used to dissolve the pellet and extracted proteins were stored at -80˚C. Total cell lysates, supernatants proteins, cell lysates + supernatants and kidney homogenates were boiled in SDS-PAGE 1X sample buffer at 95˚C for 5 mins. Kidney homogenates were centrifuged at 20,000 X g at 4˚C for 15 mins. Supernatants were collected for protein analysis. Reduced proteins were run on 8%,10% or 12% SDS-PAGE gels and transferred on to 0.22 μm nitrocellulose membranes (GE Healthcare Life Sciences). Membranes were blocked using 5% skim milk in 1X Tris-buffered saline and 0.1% Tween 20 (1XTBST) and probed overnight with primary antibodies at 4˚C. Membranes were washed 3 times for 10 mins. each with 1XTBST and probed with corresponding HRP conjugated or IRDyes (LI-COR) secondary antibodies, washed and developed using BrightStar™ Femto HRP Chemiluminescent 2-Component Substrate Kit (Alkali Scientific Inc. β-actin was used as an internal loading control. Western blots were analyzed using ImageJ software and densitometry data were normalized to β-actin.
Human and mouse IL-1β (BioLegend) concentration in media supernatants, serum and in peritoneal lavage and mouse IL-1α (BioLegend) in media supernatants were determined using commercial ELISA kits according to the manufacturer’s instructions.
Data were analyzed using Sigma plot or GraphPad Prism by one-way ANOVA for multiple comparisons using the indicated ad-hoc methods with at least 3 or more independent biological replicates. Where two groups were compared, two-tailed t-test was used. For in vivo infection, bacterial CFUs were log-transformed for statistical analysis. p values were calculated, *p < 0.05, **p < 0.01, ***p< 0.001; NS, not significant, ND, not detected.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Cincinnati and were conducted within the Department of Laboratory Animal Medicine accredited by American Association for Accreditation of Laboratory Animal Care (Frederick, MD). All animal experiments were conducted in accordance with Animal Welfare Act guidelines of the National Institutes of Health.
DC and KSV planned and conducted molecular and biochemical in vitro and in vivo experiments, generated Casp-11-/-Mt3-/- and Lys2Cre Mt3fl/fl mice, analyzed data, and wrote the manuscript. JG conducted in vivo infections with K. pneumoniae. AS, SN, and SM conducted in vivo experiments with GAS infection and analyzed data. AP assisted with bioinformatics analysis. JL conducted chromatographic and mass spectrometric analysis using ICP-MS and SEC-ICP-MS and MT3-Zn2+ complex preparations, and analyzed data. KSV designed and supervised the project. All authors contributed to the article and approved the submitted version.
This work was supported by a Junior Faculty Pilot Project Award, American Heart Association 19CDA34770022 Award, American Association of Immunology Careers in Immunology Fellowship Awards to KSV and NIAIDR01 AI106269-06 awarded in part to KSV. GAS studies were supported by the UND CoBRE Host–Pathogen Interactions Pilot Award - NIH/NIGMS award P20GM113123, UND SMHS funds (SN), and UND VPRED Postdoctoral funding support (AS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Transgenic Animal and Genome Editing Core at Cincinnati Children’s Hospital Medical (CCHMC) Center for production of Mt3fl/fl mice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.755961/full#supplementary-material
1. Haque M, Sartelli M, McKimm J, Abu Bakar M. Health Care-Associated Infections - an Overview. Infect Drug Resist (2018) 11:2321–33. doi: 10.2147/IDR.S177247
2. Kayagaki N, Kagawa S, Hamada H, Ariyoshi T. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
3. Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, et al. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell (2016) 165:1106–19. doi: 10.1016/j.cell.2016.04.015
4. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
5. Davis SR, Cousins RJ. Metallothionein Expression in Animals: A Physiological Perspective on Function. J Nutr (2000) 130:1085–8. doi: 10.1093/jn/130.5.1085
6. Kimura T, Kambe T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int J Mol Sci (2016) 17:336. doi: 10.3390/ijms17030336
7. Krezel A, Maret W. The Functions of Metamorphic Metallothioneins in Zinc and Copper Metabolism. Int J Mol Sci (2017) 18:1237. doi: 10.3390/ijms18061237.
8. Palmiter RD. The Elusive Function of Metallothioneins. Proc Natl Acad Sci U S A (1998) 95:8428–30. doi: 10.1073/pnas.95.15.8428
9. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS Jr. Granulocyte Macrophage-Colony Stimulating Factor Induced Zn Sequestration Enhances Macrophage Superoxide and Limits Intracellular Pathogen Survival. Immunity (2013) 39:697–710. doi: 10.1016/j.immuni.2013.09.006
10. Wu A, Tymoszuk P, Haschka D, Heeke S, Dichtl S, Petzer V, et al. Salmonella Utilizes Zinc To Subvert Antimicrobial Host Defense of Macrophages via Modulation of NF-kappaB Signaling. Infect Immun (2017) 85:e00418–17. doi: 10.1128/IAI.00418-17.
11. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Divanovic S, Caruso JA, Deepe GS Jr. IL-4 Induces Metallothionein 3- and SLC30A4-Dependent Increase in Intracellular Zn(2+) That Promotes Pathogen Persistence in Macrophages. Cell Rep (2016) 16:3232–46. doi: 10.1016/j.celrep.2016.08.057
12. Shankar AH, Prasad AS. Zinc and Immune Function: The Biological Basis of Altered Resistance to Infection. Am J Clin Nutr (1998) 68:447S–63S. doi: 10.1093/ajcn/68.2.447S
13. Pyle CJ, Akhter S, Bao S, Dodd CE, Schlesinger LS, Knoell DL. Zinc Modulates Endotoxin-Induced Human Macrophage Inflammation Through ZIP8 Induction and C/EBPbeta Inhibition. PloS One (2017) 12:e0169531. doi: 10.1371/journal.pone.0169531
14. Haase H, Ober-Blöbaum JL, Engelhardt G, Hebel S, Heit A, Heine H, et al. Zinc Signals are Essential for Lipopolysaccharide-Induced Signal Transduction in Monocytes. J Immunol (2008) 181:6491–502. doi: 10.4049/jimmunol.181.9.6491
15. von Bulow V, Rink L, Haase H. Zinc-Mediated Inhibition of Cyclic Nucleotide Phosphodiesterase Activity and Expression Suppresses TNF-Alpha and IL-1 Beta Production in Monocytes by Elevation of Guanosine 3’,5’-Cyclic Monophosphate. J Immunol (2005) 175:4697–705. doi: 10.4049/jimmunol.175.7.4697
16. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol (2018) 39:393–406. doi: 10.1016/j.it.2018.01.009
17. Summersgill H, England H, Lopez-Castejon G, Lawrence CB, Luheshi NM, Pahle J, et al. Zinc Depletion Regulates the Processing and Secretion of IL-1beta. Cell Death Dis (2014) 5:e1040. doi: 10.1038/cddis.2013.547
18. Brough D, Pelegrin P, Rothwell NJ. Pannexin-1-Dependent Caspase-1 Activation and Secretion of IL-1beta is Regulated by Zinc. Eur J Immunol (2009) 39:352–8. doi: 10.1002/eji.200838843
19. Chowdhury D, Alrefai H, Landero Figueroa JA, Candor K, Porollo A, Fecher R, et al. Metallothionein 3 Controls the Phenotype and Metabolic Programming of Alternatively Activated Macrophages. Cell Rep (2019) 27:3873–86.e3877. doi: 10.1016/j.celrep.2019.05.093
20. Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
21. Lee SJ, Koh JY. Roles of Zinc and Metallothionein-3 in Oxidative Stress-Induced Lysosomal Dysfunction, Cell Death, and Autophagy in Neurons and Astrocytes. Mol Brain (2010) 3:30. doi: 10.1186/1756-6606-3-30
22. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING V11: Protein-Protein Association Networks With Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res (2019) 47:D607–13. doi: 10.1093/nar/gky1131
23. Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, et al. The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front Immunol (2013) 4:391. doi: 10.3389/fimmu.2013.00391
24. Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER. Identification of a Membrane-Associated Interleukin 1 in Macrophages. Proc Natl Acad Sci U S A (1985) 82:1204–8. doi: 10.1073/pnas.82.4.1204
25. Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, et al. Inflammasome Activation and IL-1beta Target IL-1alpha for Secretion as Opposed to Surface Expression. Proc Natl Acad Sci U S A (2011) 108:18055–60. doi: 10.1073/pnas.1109176108
26. Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, et al. Human Caspase-4 Mediates Noncanonical Inflammasome Activation Against Gram-Negative Bacterial Pathogens. Proc Natl Acad Sci U S A (2015) 112:6688–93. doi: 10.1073/pnas.1421699112
27. Shrum B, Anantha RV, Xu SX, Donnelly M, Haeryfar SM, McCormick JK, et al. A Robust Scoring System to Evaluate Sepsis Severity in an Animal Model. BMC Res Notes (2014) 7:1–11. doi: 10.1186/1756-0500-7-233
28. Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, et al. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell (2018) 175:1651–1664 e1614. doi: 10.1016/j.cell.2018.09.047
29. Chella Krishnan K, Mukundan S, Alagarsamy J, Hur J, Nookala S, Siemens N, et al. Genetic Architecture of Group A Streptococcal Necrotizing Soft Tissue Infections in the Mouse. PloS Pathog (2016) 12:e1005732. doi: 10.1371/journal.ppat.1005732
30. Wang J, Shao Y, Wang W, Li S, Xin N, Xie F, et al. Caspase-11 Deficiency Impairs Neutrophil Recruitment and Bacterial Clearance in the Early Stage of Pulmonary Klebsiella Pneumoniae Infection. Int J Med Microbiol (2017) 307:490–6. doi: 10.1016/j.ijmm.2017.09.012
31. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science (2013) 339:975–8. doi: 10.1126/science.1230751
32. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 Increases Susceptibility to Salmonella Infection in the Absence of Caspase-1. Nature (2012) 490:288–91. doi: 10.1038/nature11419
33. Krause K, Daily K, Estfanous S, Hamilton K, Badr A, Abu Khweek A, et al. Caspase-11 Counteracts Mitochondrial ROS-Mediated Clearance of Staphylococcus Aureus in Macrophages. EMBO Rep (2019) 20:e48109. doi: 10.15252/embr.201948109
34. Kovacs SB, Oh C, Maltez VI, McGlaughon BD, Verma A, Miao EA, et al. Neutrophil Caspase-11 Is Essential to Defend Against a Cytosol-Invasive Bacterium. Cell Rep (2020) 32:107967. doi: 10.1016/j.celrep.2020.107967
35. Cerqueira DM, Gomes MTR, Silva ALN, Rungue M, Assis NRG, Guimarães ES, et al. Guanylate-Binding Protein 5 Licenses Caspase-11 for Gasdermin-D Mediated Host Resistance to Brucella Abortus Infection. PloS Pathog (2018) 14:e1007519. doi: 10.1371/journal.ppat.1007519
36. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell (2020) 180:941–955 e920. doi: 10.1016/j.cell.2020.02.002
37. Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, et al. Caspase-8 Collaborates With Caspase-11 to Drive Tissue Damage and Execution of Endotoxic Shock. Immunity (2018) 49:42–55 e46. doi: 10.1016/j.immuni.2018.06.011
38. Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, et al. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem (2015) 290:20167–84. doi: 10.1074/jbc.M115.652321
39. Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, et al. LPS Targets Host Guanylate-Binding Proteins to the Bacterial Outer Membrane for non-Canonical Inflammasome Activation. EMBO J (2018) 37:e98089. doi: 10.15252/embj.201798089
40. Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, et al. Human Caspase-4 Detects Tetra-Acylated LPS and Cytosolic Francisella and Functions Differently From Murine Caspase-11. Nat Commun (2018) 9:242. doi: 10.1038/s41467-017-02682-y
41. Huang da W, Sherman BT, Lempicki RA. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat Protoc (2009) 4:44–57. doi: 10.1038/nprot.2008.211
42. Durfee LA, Huibregtse JM. Identification and Validation of ISG15 Target Proteins. Subcell Biochem (2010) 54:228–37. doi: 10.1007/978-1-4419-6676-6_18
43. Pletneva LM, Haller O, Porter DD, Prince GA, Blanco JCG. Induction of Type I Interferons and Interferon-Inducible Mx Genes During Respiratory Syncytial Virus Infection and Reinfection in Cotton Rats. J Gen Virol (2008) 89:261–70. doi: 10.1099/vir.0.83294-0
44. Varela M, Diaz-Rosales P, Pereiro P, Forn-Cuní G, Costa MM, Dios S, et al. Interferon-Induced Genes of the Expanded IFIT Family Show Conserved Antiviral Activities in non-Mammalian Species. PloS One (2014) 9:e100015. doi: 10.1371/journal.pone.0100015
45. Tirumurugaan KG, Pawar RM, Dhinakar Raj G, Thangavelu A, Hammond JA, Parida S. RNAseq Reveals the Contribution of Interferon Stimulated Genes to the Increased Host Defense and Decreased PPR Viral Replication in Cattle. Viruses (2020) 12:463. doi: 10.3390/v12040463
46. Hojyo S, Fukada T. Roles of Zinc Signaling in the Immune System. J Immunol Res (2016) 2016:6762343. doi: 10.1155/2016/6762343
47. Maret W. Metals on the Move: Zinc Ions in Cellular Regulation and in the Coordination Dynamics of Zinc Proteins. BioMetals (2011) 24:411–8. doi: 10.1007/s10534-010-9406-1
48. Brieger A, Rink L, Haase H. Differential Regulation of TLR-Dependent MyD88 and TRIF Signaling Pathways by Free Zinc Ions. J Immunol (2013) 191:1808–17. doi: 10.4049/jimmunol.1301261
49. Zaslona Z, Flis E, Wilk MM, Carroll RG, Palsson-McDermott EM, Hughes MM, et al. Caspase-11 Promotes Allergic Airway Inflammation. Nat Commun (2020) 11:1055. doi: 10.1038/s41467-020-14945-2
50. Finethy R, Dockterman J, Kutsch M, Orench-Rivera N, Wallace GD, Piro AS, et al. Dynamin-Related Irgm Proteins Modulate LPS-Induced Caspase-11 Activation and Septic Shock. EMBO Rep (2020) 21:e50830. doi: 10.15252/embr.202050830
51. Chen R, Zeng L, Zhu S, Liu J, Zeh HJ, Kroemer G, et al. cAMP Metabolism Controls Caspase-11 Inflammasome Activation and Pyroptosis in Sepsis. Sci Adv (2019) 5:eaav5562. doi: 10.1126/sciadv.aav5562
52. Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, Monack DM, et al. The Oxidized Phospholipid oxPAPC Protects From Septic Shock by Targeting the non-Canonical Inflammasome in Macrophages. Nat Commun (2018) 9:996. doi: 10.1038/s41467-018-03409-3
53. Subramanian Vignesh K, Deepe GS Jr. Metallothioneins: Emerging Modulators in Immunity and Infection. Int J Mol Sci (2017) 18:2197. doi: 10.3390/ijms18102197.
54. Dalton T, Palmiter RD, Andrews GK. Transcriptional Induction of the Mouse Metallothionein-I Gene in Hydrogen Peroxide-Treated Hepa Cells Involves a Composite Major Late Transcription Factor/Antioxidant Response Element and Metal Response Promoter Elements. Nucleic Acids Res (1994) 22:5016–23. doi: 10.1093/nar/22.23.5016
55. Chowdhury D, Deepe GS, Subramanian Vignesh K. The Metallothionein-Zinc Landscape: How It Shapes Antimicrobial Immunity. In: Zinc Signaling. Singapore: Springer (2019). p. 57–77.
56. Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I, et al. Mycobacterial P(1)-Type ATPases Mediate Resistance to Zinc Poisoning in Human Macrophages. Cell Host Microbe (2011) 10:248–59. doi: 10.1016/j.chom.2011.08.006
57. Palmiter RD, Findley SD, Whitmore TE, Durnam DM. MT-III, a Brain-Specific Member of the Metallothionein Gene Family. Proc Natl Acad Sci U S A (1992) 89:6333–7. doi: 10.1073/pnas.89.14.6333
58. Leibbrandt ME, Koropatnick J. Activation of Human Monocytes With Lipopolysaccharide Induces Metallothionein Expression and is Diminished by Zinc. Toxicol Appl Pharmacol (1994) 124:72–81. doi: 10.1006/taap.1994.1010
59. Arizono K, Kagawa S, Hamada H, Ariyoshi T. Nitric Oxide Mediated Metallothionein Induction by Lipopolysaccharide. Res Commun Mol Pathol Pharmacol (1995) 90:49–58.
60. Coordinators NR. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res (2016) 44:D7–19. doi: 10.1093/nar/gkx1095
61. Stocks CJ, von Pein JB, Curson JEB, Rae J, Phan MD, Foo D, et al. Frontline Science: LPS-Inducible SLC30A1 Drives Human Macrophage-Mediated Zinc Toxicity Against Intracellular Escherichia Coli. J Leukoc Biol (2021) 109:287–97. doi: 10.1002/JLB.2HI0420-160R
62. Stocks CJ, Phan MD, Achard MES, Nhu NTK, Condon ND, Gawthorne JA, et al. Uropathogenic Escherichia Coli Employs Both Evasion and Resistance to Subvert Innate Immune-Mediated Zinc Toxicity for Dissemination. Proc Natl Acad Sci U S A (2019) 116:6341–50. doi: 10.1073/pnas.1820870116
63. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, et al. Caspase-11 Stimulates Rapid Flagellin-Independent Pyroptosis in Response to Legionella Pneumophila. Proc Natl Acad Sci U S A (2013) 110:1851–6. doi: 10.1073/pnas.1211521110
64. Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, et al. Caspase-11 Promotes the Fusion of Phagosomes Harboring Pathogenic Bacteria With Lysosomes by Modulating Actin Polymerization. Immunity (2012) 37:35–47. doi: 10.1016/j.immuni.2012.05.001
65. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses Against Enteric Bacterial Pathogens. Cell Host Microbe (2014) 16:249–56. doi: 10.1016/j.chom.2014.07.002
66. Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, et al. The Shigella OspC3 Effector Inhibits Caspase-4, Antagonizes Inflammatory Cell Death, and Promotes Epithelial Infection. Cell Host Microbe (2013) 13:570–83. doi: 10.1016/j.chom.2013.04.012
67. Codo AC, Saraiva AC, Dos Santos LL, Visconde MF, Gales AC, Zamboni DS, et al. Inhibition of Inflammasome Activation by a Clinical Strain of Klebsiella Pneumoniae Impairs Efferocytosis and Leads to Bacterial Dissemination. Cell Death Dis (2018) 9:1182. doi: 10.1038/s41419-018-1214-5
68. Krause K, Caution K, Badr A, Hamilton K, Saleh A, Patel K, et al. CASP4/caspase-11 Promotes Autophagosome Formation in Response to Bacterial Infection. Autophagy (2018) 14:1928–42. doi: 10.1080/15548627.2018.1491494
69. Mueller NJ, Wilkinson RA, Fishman JA. Listeria Monocytogenes Infection in Caspase-11-Deficient Mice. Infect Immun (2002) 70:2657–64. doi: 10.1128/IAI.70.5.2657-2664.2002
70. LaRock CN, Nizet V. Inflammasome/IL-1beta Responses to Streptococcal Pathogens. Front Immunol (2015) 6:518. doi: 10.3389/fimmu.2015.00518
71. Hancz D, Westerlund E, Bastiat-Sempe B, Sharma O, Valfridsson C, Meyer L, et al. Inhibition of Inflammasome-Dependent Interleukin 1beta Production by Streptococcal NAD(+)-Glycohydrolase: Evidence for Extracellular Activity. mBio (2017) 8:e00756–17. doi: 10.1128/mBio.00756-17
72. Valderrama JA, Riestra AM, Gao NJ, LaRock CN, Gupta N, Ali SR, et al. Group A Streptococcal M Protein Activates the NLRP3 Inflammasome. Nat Microbiol (2017) 2:1425–34. doi: 10.1038/s41564-017-0005-6
73. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-Hydroxycholesterol Suppresses Interleukin-1-Driven Inflammation Downstream of Type I Interferon. Science (2014) 345:679–84. doi: 10.1126/science.1254790
74. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity (2011) 34:213–23. doi: 10.1016/j.immuni.2011.02.006
75. Sayadi A, Nguyen AT, Bard FA, Bard-Chapeau EA. Zip14 Expression Induced by Lipopolysaccharides in Macrophages Attenuates Inflammatory Response. Inflamm Res (2013) 62:133–43. doi: 10.1007/s00011-012-0559-y
76. Wessels I, Maywald M, Rink L. Zinc as a Gatekeeper of Immune Function. Nutrients (2017) 9:1286. doi: 10.3390/nu9121286
77. Maywald M, Wessels I, Rink L. Zinc Signals and Immunity. Int J Mol Sci (2017) 18:2222. doi: 10.3390/ijms18102222
78. Foster M, Samman S. Zinc and Regulation of Inflammatory Cytokines: Implications for Cardiometabolic Disease. Nutrients (2012) 4:676–94. doi: 10.3390/nu4070676
79. Prasad AS, Beck FW, Grabowski SM, Kaplan J, Mathog RH. Zinc Deficiency: Changes in Cytokine Production and T-Cell Subpopulations in Patients With Head and Neck Cancer and in Noncancer Subjects. Proc Assoc Am Physicians (1997) 109:68–77.
80. Beck FW, Li Y, Bao B, Prasad AS, Sarkar FH. Evidence for Reprogramming Global Gene Expression During Zinc Deficiency in the HUT-78 Cell Line. Nutrition (2006) 22:1045–56. doi: 10.1016/j.nut.2006.08.001
81. Prasad AS. Zinc: Mechanisms of Host Defense. J Nutr (2007) 137:1345–9. doi: 10.1093/jn/137.5.1345
82. Yuan CL, Hu YC. A Transgenic Core Facility’s Experience in Genome Editing Revolution. Adv Exp Med Biol (2017) 1016:75–90. doi: 10.1007/978-3-319-63904-8_4
83. Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud JB, et al. Evaluation of Off-Target and on-Target Scoring Algorithms and Integration Into the Guide RNA Selection Tool CRISPOR. Genome Biol (2016) 17:148. doi: 10.1186/s13059-016-1012-2
84. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome Engineering Using the CRISPR-Cas9 System. Nat Protoc (2013) 8:2281–308. doi: 10.1038/nprot.2013.143
85. Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, et al. Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell (2013) 155:1479–91. doi: 10.1016/j.cell.2013.12.001
86. Valerius MT, Patterson LT, Witte DP, Potter SS. Microarray Analysis of Novel Cell Lines Representing Two Stages of Metanephric Mesenchyme Differentiation. Mech Dev (2002) 112:219–32. doi: 10.1016/S0925-4773(02)00008-4
87. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell (2013) 153:910–8. doi: 10.1016/j.cell.2013.04.025
88. Yang H, Wang H, Jaenisch R. Generating Genetically Modified Mice Using CRISPR/Cas-Mediated Genome Engineering. Nat Protoc (2014) 9:1956–68. doi: 10.1038/nprot.2014.134
89. Domenico P, Diedrich DL, Straus DC. Extracellular Polysaccharide Production by Klebsiella Pneumoniae and its Relationship to Virulence. Can J Microbiol (1985) 31:472–8. doi: 10.1139/m85-088
90. Wei RQ, Yee JB, Straus DC, Hutson JC. Bactericidal Activity of Testicular Macrophages. Biol Reprod (1988) 38:830–5. doi: 10.1095/biolreprod38.4.830
91. Chatellier S, Ihendyane N, Kansal RG, Khambaty F, Basma H, Norrby-Teglund A, et al. Genetic Relatedness and Superantigen Expression in Group A Streptococcus Serotype M1 Isolates From Patients With Severe and Nonsevere Invasive Diseases. Infect Immun (2000) 68:3523–34. doi: 10.1128/IAI.68.6.3523-3534.2000
Keywords: macrophage, non-canonical inflammasome, zinc, metallothionein, innate immunity, caspase-11 non-canonical inflammasome, MT3
Citation: Chowdhury D, Gardner JC, Satpati A, Nookala S, Mukundan S, Porollo A, Landero Figueroa JA and Subramanian Vignesh K (2021) Metallothionein 3-Zinc Axis Suppresses Caspase-11 Inflammasome Activation and Impairs Antibacterial Immunity. Front. Immunol. 12:755961. doi: 10.3389/fimmu.2021.755961
Received: 09 August 2021; Accepted: 15 October 2021;
Published: 12 November 2021.
Edited by:
Maciej Lech, LMU Munich University Hospital, GermanyReviewed by:
Amal O. Amer, The Ohio State University, United StatesCopyright © 2021 Chowdhury, Gardner, Satpati, Nookala, Mukundan, Porollo, Landero Figueroa and Subramanian Vignesh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kavitha Subramanian Vignesh, S2F2aXRoYS5TdWJyYW1hbmlhbkB1Yy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.