
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 October 2021
Sec. Vaccines and Molecular Therapeutics
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.753467
This article is part of the Research Topic AAV Gene Therapy: Immunology and Immunotherapeutics View all 14 articles
 Kleopatra Rapti1,2*
Kleopatra Rapti1,2* Dirk Grimm1,2,3*
Dirk Grimm1,2,3*Adeno-associated viruses (AAV) have emerged as the lead vector in clinical trials and form the basis for several approved gene therapies for human diseases, mainly owing to their ability to sustain robust and long-term in vivo transgene expression, their amenability to genetic engineering of cargo and capsid, as well as their moderate toxicity and immunogenicity. Still, recent reports of fatalities in a clinical trial for a neuromuscular disease, although linked to an exceptionally high vector dose, have raised new caution about the safety of recombinant AAVs. Moreover, concerns linger about the presence of pre-existing anti-AAV antibodies in the human population, which precludes a significant percentage of patients from receiving, and benefitting from, AAV gene therapies. These concerns are exacerbated by observations of cellular immune responses and other adverse events, including detrimental off-target transgene expression in dorsal root ganglia. Here, we provide an update on our knowledge of the immunological and molecular race between AAV (the “hedgehog”) and its human host (the “hare”), together with a compendium of state-of-the-art technologies which provide an advantage to AAV and which, thus, promise safer and more broadly applicable AAV gene therapies in the future.
The hallmark of gene therapy is the delivery of exogenous nucleic acids to cells with the aim to replace missing or defective genes, or to suppress (RNA interference technology) or correct (genome editing) deleterious ones, in order to ultimately ameliorate genetic causes of disease. The ideal delivery vehicle or vector should safely, specifically and efficiently transport the therapeutic cargo and allow expression for the desired duration. Although the delivery of “naked DNA” has progressed all the way to clinical trials, the use of non-viral and viral vectors continues to dominate the field [reviewed in (1–3)]. Viral vectors rely on natural, evolutionary evolved properties of viruses to efficiently evade an organism’s immune surveillance while delivering their cargo to specific cells. Several types of viral vectors are used in gene therapy today, mostly adenoviruses, retroviruses and adeno-associated viruses (AAVs), of which the latter have emerged over the past 20 years as the leading platform for a myriad of applications (1, 2).
AAVs are small, non-enveloped, non-pathogenic viruses endemic in humans and multiple vertebrate species. They belong to the genus Dependoparvovirus within the family Parvoviridae and are amongst the smallest animal DNA viruses [(4), reviewed in (5–7)]. They carry a ~4.7 kb single-stranded genome that is flanked by two 145 bp ITRs (inverted terminal repeats) forming a characteristic T-shaped hairpin and is packaged in a capsid of T=1 icosahedral symmetry and ~26 nm diameter. Their genome consists of two main open reading frames (ORFs), rep and cap [reviewed in (7)], and two additional ones encoding the assembly-activating protein (AAP) (8) and the recently discovered membrane-associated accessory protein (MAAP) (9). While the three viral capsid proteins VP1-3 share their C-terminal region, it is VP3, the shortest and most abundant of the three, that determines tissue tropism through receptor binding and interaction with factors in the circulation and interstitial tissue, including but not limited to antibodies [(10–12), reviewed in (2)]. These properties are mainly attributed to nine variable regions (VRI-IX) within VP3 (13).
So far, at least 13 naturally occurring primate serotypes and hundreds of variants have been identified, and countless engineered AAVs with specialized properties are constantly generated [(14–18), reviewed in (1, 2, 19)]. AAVs infect cells by binding to cell surface molecules, identified either as receptors, attachment or viral entry factors, and typically glycans or proteinaceous in nature. Some of these are serotype-specific while others are not, such as AAVR [(20–23), reviewed in (19)]. Binding to these factors is followed by receptor-mediated endocytosis, intracellular trafficking, endosomal escape, nuclear transport, capsid uncoating and finally second-strand genome conversion in the nucleus [reviewed in (19, 24)].
Reasons for the attraction of AAVs as vectors include their broad tropism, low immunogenicity as compared to other vectors, and apathogenicity. Moreover, they are easily engineered as gene delivery vector, by replacing the viral genome with a therapeutic expression cassette, yielding a recombinant AAV (rAAV) with the ITRs as the only essential cis elements. Recombinant AAVs transduce cells akin to an infection with their parental wild-type (wt)AAVs, but they cannot integrate into the host cell chromosome in a site-specific manner or integrate at very low frequency, due to the lack of the rep gene (25). Still, they can establish long-term transgene expression in both, animals and humans (26–29). Encouraging data in preclinical animal models and in clinical trials [reviewed in (30)] have led to the approval of several gene therapies in recent years, starting with Glybera, a rAAV1 carrying the lipoprotein lipase gene, whose intramuscular delivery aimed at the treatment of lipoprotein lipase deficiency. However, due to the high cost, the scarcity of the disease and the lack of approval in the US, it was withdrawn five years later (2017), despite its therapeutic efficacy (1, 2, 19, 31, 32). The first AAV gene therapy approved by the US Food and Drug Administration (FDA) in 2017 was Luxturna™ or voretigene neparvovec, followed by its approval in Europe end of 2018. Luxturna is an AAV2 vector carrying the RPE65 gene, which is delivered to the retina to treat an inherited form of blindness caused by a mutation in this gene (1, 2, 19, 31). The second gene therapy approved in the US in 2019 was ZOLGENSMA® (onasemnogene abeparvovec-xioi), i.e., an AAV9 vector carrying the human survival motor neuron (SMN) gene and used for the one-time treatment of children under the age of 2 who suffer from spinal muscular atrophy (SMA) (1, 2, 19, 31). Overall, the future of the field is bright, with a 2019 FDA report estimating the yearly approval of 10-20 new cell and gene therapy products by 2025 (2, 33).
Despite the three approved AAV gene therapies and the success of numerous clinical trials with AAVs (1, 2, 30, 31), challenges or impediments remain. This is exemplified by the first clinical trials for the treatment of severe hemophilia B, using an AAV2 vector carrying the gene for coagulation Factor IX (FIX) that was delivered either intramuscularly (34) or infused through the hepatic artery (35). While the first trial was hampered by low-level, short-term expression of <1% of FIX (34), immune responses against the AAV vector were noted in a second trial (35), which were not predicted by any of the preclinical studies in small or large animals (36). No long-lasting systemic toxicity was observed and therapeutic levels of FIX were obtained, but they rapidly declined to background levels, accompanied by a transient increase in liver transaminases. It was later determined that this was due to cytotoxic T-cell (CTL) responses from memory CD8+ T-cells against hepatocytes presenting AAV epitopes via major histocompatibility complex (MHC) class I (36–39). In another clinical trial using AAV8 to deliver a codon-optimized fIX gene, a short-term supply of immunosuppressants sufficed to block cellular immune responses and enabled long-term expression (27). In additional clinical trials, the presence of pre-existing anti-AAV antibodies governed the efficiency of the gene transfer (40, 41), as will be discussed in more detail below.
The experience and knowledge from these initial clinical trials shaped subsequent efforts to create new generations of AAV vectors that would perform better in humans. In particular, it quickly became evident that immune responses are a major roadblock and require thorough investigation, in order to develop novel, urgently needed strategies to evade or alleviate them. This review will first explore the mechanisms of anti-AAV immune responses and methods to measure them, before focusing on the multifaceted approaches to escape them in a (pre)clinical setting.
Immunity is the ability of higher organisms to protect themselves from pathogenic invaders, such as viruses or bacteria. Although AAVs are non-pathogenic and vectors derived thereof no longer express any viral proteins, their viral nature renders them a target for the immune system. On top, the fact that AAVs were discovered in human tissues explains why humans carry immunologic memory against them (3, 30, 42, 43).
Generally, the immune system consists of two major arms, the innate and adaptive immunity, which are intertwined and closely regulate one another (44). Adaptive immunity, also called acquired immunity, includes humoral immunity (B-cells, neutralizing antibodies) and cell-mediated immunity (T-lymphocytes, macrophages, natural killer cells).
The following chapters briefly summarize the current knowledge of the role of these arms in anti-AAV immune response in humans before we focus on clinically relevant countermeasures.
The innate system is the first line of defense against invaders and, therefore, is relatively non-specific. Professional antigen-presenting cells (APCs), which are present in most tissues, express pattern recognition receptors (PRRs). These recognize structural features on molecules, such as glycans and viral nucleic acids, which are shared between microorganisms and named pathogen-associated molecular patterns (PAMPs) (45). Toll-like receptors (TLRs) are PRRs that are critically involved in immune responses against AAVs together with myeloid differentiation primary response 88 (MyD88), i.e., their universal adaptor [reviewed in detail in (42)]. They are type I transmembrane proteins, which contain leucine-rich repeats and which are located on the cell surface (TLR1, 2, 4-6 and 10) or the endosome (TLR3, 7-9) (46), where they recognize the AAV capsid or the viral nucleic acid (CpG-containing viral genomes and double-stranded (ds)RNA), respectively [reviewed in (42, 45, 47)]. Triggering of PRRs results in nuclear translocation of nuclear factor κB (NF-κB) and interferon regulatory transcription factors (IRFs), which subsequently induce expression of pro-inflammatory cytokines and type I interferons [IFN, reviewed in (3, 42)]. Type I IFNs are the essential link between innate and adaptive immunity [(48–51), reviewed in (3, 47)].
The importance of innate immune responses in AAV gene transfer is subject to intense ongoing investigation. Early studies in mice showed low levels of chemokine induction, at least compared to adenoviral vectors, and the duration was also transient and did not lead to liver necrosis (52). One of the first factors identified to play a role in inhibiting AAV transduction is apolipoprotein B mRNA editing complex 3A (APOBEC3A) (53), a component of the intrinsic immunity. However, the link to innate immunity remains unclear (54). Further studies, mostly performed in the liver, revealed important roles for TLR2 and, most prominently, TLR9 receptors in the early-phase activation of the innate immune system, involving different cell types. TLR2 can sense the capsid of rAAVs (serotypes 2 and 8) on the surface of human non-parenchymal liver cells, such as Kupffer and liver sinusoidal endothelial cells (LSECs). This results in NFκB-mediated immune responses and activation of several interleukins and tumor necrosis factor α (TNFα), but not type I IFN (55). Extensive studies support the role of the viral genome and in particular of the presence of unmethylated CpGs in triggering the innate immune system through the TLR9-MyD88 pathway in different cell types (56, 57), not only in Kupffer cells (58), but also in dendritic cells (DCs) including plasmacytoid (59–61), conventional (59) and monocyte-derived DCs (59, 62). Different TLRs appear to be activated and have distinct effects on different DCs, all of which participate in linking innate and adaptive immunity (45). Still, TLR9 seems to be the most efficient in perpetrating downstream events, such as humoral responses (59) (see below for the link to the adaptive immunity).
Unmethylated CpGs, which are present in the ITRs but also in vector expression cassettes, were shown early on to play a central and enhancing role in the aforementioned TLR9-MyD88 activation (48, 52, 57, 61, 63) [reviewed in (56)]. The detrimental role of CpGs in expression cassettes became evident in clinical trials by the stronger immune responses triggered by codon-optimized transgenes that contained higher CpG levels compared to the wild-type sequences (57), which highlights the importance of the encapsidated nucleic acid. Similarly, the presence of self-complementary rather than single-stranded AAV vector DNA (scAAV vs ssAAV) also results in stronger induction of the innate immune system through TLR9 (58).
In addition to the role of the DNA, the role of double-stranded (ds)RNA produced from AAV vectors has most recently been identified as a factor governing the success of AAV gene therapy (64). Late rather than early innate immune responses are likely to explain the decline in transgene expression that is often observed in patients weeks after AAV delivery. In a recent study, it was hypothesized that the dsRNA produced by the inherent promoter activity of the AAV ITRs was sensed by the PRR melanoma differentiation-associated protein 5 (MDA5). Together with the signaling adaptor mitochondrial antiviral signaling protein (MAVS), MDA5 induces expression of IFN-β, a type I IFN. This induction was seen in different cell lines but, more importantly, also in vivo in the humanized liver of mice (64). However, another dsRNA PRR, TLR7, did not show a similar effect (59). Hence, the role and the mechanisms of dsRNA sensing in the innate immune responses require further study (42).
The aforementioned studies of innate immune responses against AAVs yield insights into the connection to the adaptive immunity arm. Indeed, the innate system is considered the key player in the induction of the adaptive responses, which is further corroborated by the fact that transient immune suppression of inflammatory cytokines in clinical trials also suppressed the adaptive immune responses (30). The TLR9-MyD88 pathway is central in this association with both, humoral (51, 59, 62, 65) and cellular immunity (48, 51, 56, 60, 61, 63, 65). In particular, the role of MyD88 in neutralizing antibody induction against the capsid was underlined (51), as well as its role in B-cell induction (59, 65), T helper 1 induction (3, 51, 65), or the shift from Th1 to Th2 (51). In contrast, TLRs are involved in the induction of CD8+ T-cells against the transgene product (51). B-cell induction can also be mediated by cytokines from monocyte-derived DCs (moDCs) (66). The role of TLRs in cell-mediated responses is strongly corroborated by multiple studies. CpG DNA in AAV vectors can induce CD8+ T-cell responses (63), and TLR9 was implicated in capsid antigen presentation through MHCI (51, 60, 63). This process was also shown to require type I IFN (60, 67), next to TLR9, which is secreted by plasmacytoid DCs (pDCs) and binds to its receptor on conventional DCs (cDCs). Subsequently, licensing of the latter activates CD8+ T-cells. Inhibition of this pathway reduced antibody production against the capsid (47, 48). Intramuscular AAV injection, with or without a TLR9 agonist depending on the mouse strain, elicited T-cell responses against the transgene product (59, 62). Similar effects were also observed following systemic administration (68). Finally, at lower doses, AAVs can interact directly with members of the complement system, especially iC3b, which can enhance humoral responses through the classical pathway. Yet, they also bind the complement regulatory protein factor H, which hinders the onset and intensity of antibody formation. At higher doses, AAVs can activate the complement and macrophages, in an antibody-dependent manner (69) [reviewed in (70)].
Together, a wealth of data supports the role of the innate immune system in animals or humans and especially in the induction of adaptive immune responses, as discussed in more detail below.
Adaptive immune responses follow, and are activated by, the innate system. The adaptive immune system is sophisticated and highly specific to the pathogens. The main actors of the two major branches of adaptive immunity, humoral and cellular, are the B- and T-cells. During development, they produce a vast amount of receptors by rearranging their DNA that recognize the pathogens in an initial encounter. Subsequently, this system generates the so-called immunological memory, which is more robust and is maintained for years after the first invasion (45). The key steps towards immunity include antigen capture and presentation by APCs to lymphocytes, which are in turn activated, clonally expanded and differentiated to effector cells. The effector functions include (1) activation of B-cells and production of antibodies against the pathogen, (2) activation of inflammation, of macrophages as well as of B- and T-cells by helper T-cells, (3) CTL responses to eliminate the pathogen, and (4) induction of regulatory T-lymphocytes to suppress immune responses. The effector phase is followed by contraction of lymphocytes by apoptosis that restores homeostasis and by survival of antigen-specific cells to yield immunologic memory (44, 45).
AAV vectors that are presently in clinical evaluation or used as basis for gene therapeutics are typically derived from wild-type AAVs with no or minimal modifications to the capsid, such as peptide insertions or point mutations. As humans are exposed to these viruses early in life, it comes as no surprise that adaptive immunity is a major challenge for gene therapy, as discussed below. Adaptive immune responses against AAVs (overview in Figure 1, Table 1) have been well documented in clinical trials and investigated in animal models [reviewed in (3, 30, 43, 95, 113)]. The next chapter will explore both, humoral and cellular immune responses against the capsid or the transgene product, as well as methodologies for their detection.
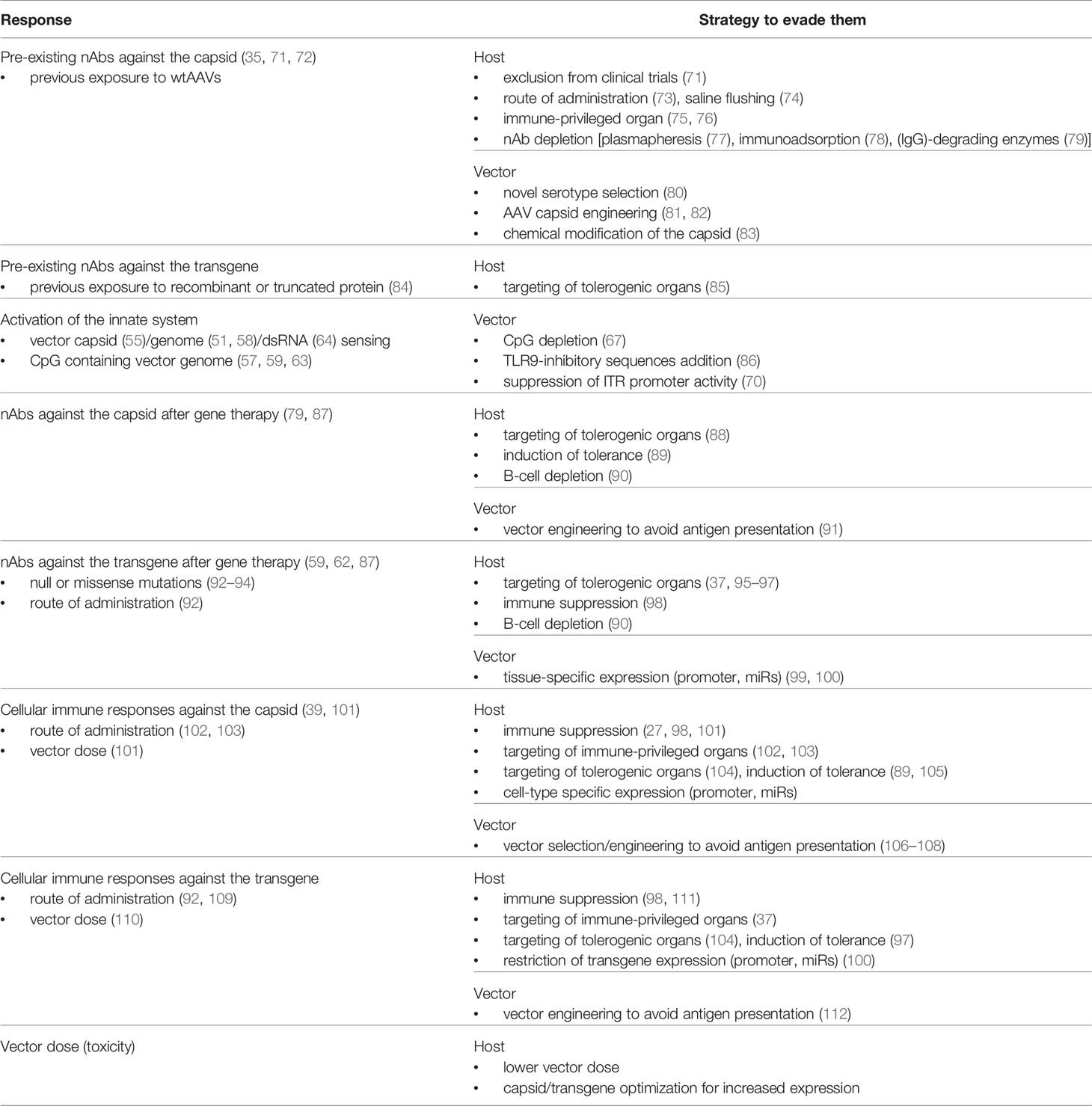
Table 1 Summary of immune responses to AAV gene therapy and evasion/prevention strategies.
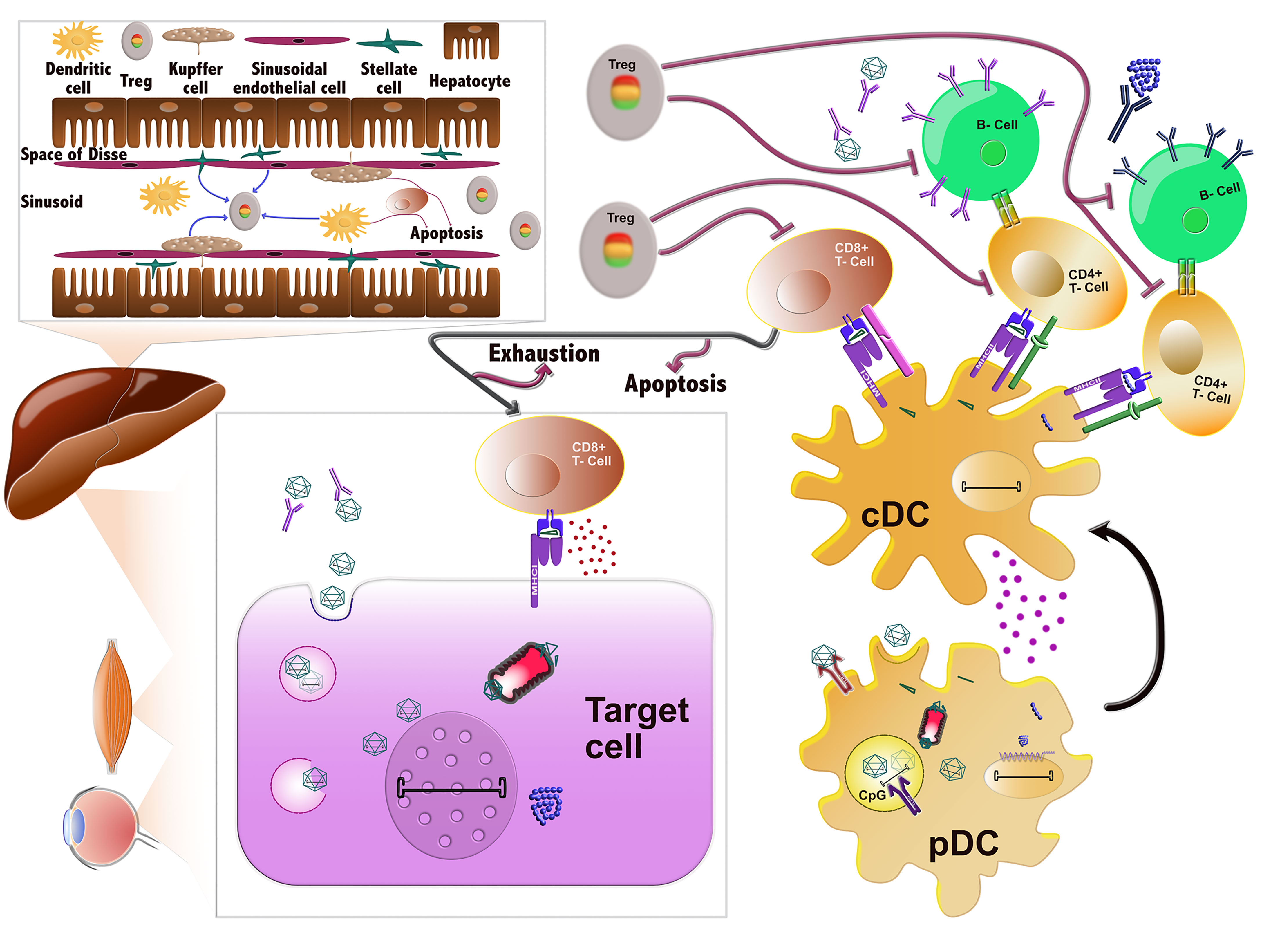
Figure 1 Immune responses against AAVs. AAVs delivered systemically can be neutralized by pre-existing antibodies prior to entering the cells. If they evade nAb binding, they enter the cells through endocytosis and can then be degraded in the endosomes. In certain cell types, such as DCs, their genome or capsid can be sensed by TLR9 and TLR2, respectively, which induces the innate immune response. Alternatively, AAVs can successfully escape the endosome and traffic in the cytoplasm, where they can be ubiquitinated, resulting in capsid degradation. The ensuing peptides can next be loaded to MHC class I molecules and presented on the surface of the cells, which are then targeted and possibly eliminated by CD8+ T-cells (CTL response). After endosomal escape, AAVs can also successfully transduce the cell and deliver their viral genome to the nucleus, where the transgene is expressed. Any misfolded protein encoded by the transgene can be degraded by the proteasome and the ensuing peptides can be loaded onto MHCI and also provoke a CTL response. Sensing of AAV vector components in plasmacytoid DCs (pDCs) by the innate immune system leads to the activation of conventional DCs (cDCs). cDCs employ antigen presentation to cross-prime CD8+ T-cells towards an effector type (Teff) and to activate CD4+ T-cells. The latter can activate B cells, which in turn produce the nAbs against the capsid and transgene product. After prolonged or inadequate stimulation, the CD8+ T-cells can be eliminated by different mechanisms including T-cell exhaustion, anergy or apoptosis. The tolerogenic environment in the liver can also stimulate the production of regulatory T-cells (Tregs), which can suppress the aforementioned immune responses at different stages.
Humoral immunity against AAVs, either exhibited by the prevalence of anti-AAV antibodies in the human population (71, 114, 115) but also in animals (116–118), or triggered by AAV vector administration, has been investigated since the early days of AAV vector engineering (119), and it has since been viewed and intensively discussed as a major impediment and exclusion criterion in AAV gene therapy clinical trials. Exacerbating this challenge is that not only up to 90% of individuals in certain areas of the world are seropositive for AAV and that 30-70% are believed to carry neutralizing anti-AAV antibodies (nAbs), but there is also significant cross-reactivity among the known naturally occurring AAV serotypes as well as their synthetic derivatives (115, 120, 121). As the topic of human seroprevalence against AAVs, induced by natural infection or by gene therapy, has already been covered extensively in a flurry of previous reviews including an excellent recent article by Weber in this journal (122) (also references therein), we kindly refer the reader to this literature for more background information. Below, we will instead focus on methodologies for the detection of humoral immune responses against AAVs and then later (chapter 3) discuss experimental strategies to circumvent these.
There are two major in vitro methodologies, i.e., cell-based assays and ELISAs (enzyme-linked immunosorbent assay), which are used for screening of anti-AAV nAbs and each exhibiting distinctive advantages (123–125). Cell-based assays are more widely used as they are robust and fast. Furthermore, they can distinguish between neutralizing and non-neutralizing, binding antibodies (123, 126). Recently, a variation of these assays has been reported, i.e., a cell-binding assay. This assay, albeit being fast, cannot make this distinction and can only detect neutralization at the level of receptor binding (127). ELISAs, on the other hand, are easy, relatively sensitive and highly useful for determining the immunoglobulin subclasses (114). However, they are typically used to measure binding, not necessarily neutralizing antibodies. There is a high but not absolute degree of correlation between the two assays (121, 128). In cell-based assays, serial dilutions of blood serum or plasma are mixed with equal amounts of an AAV vector, preincubated and then transferred to cells. Transduction efficiency is determined at a distinct time point and expressed as percent to a no-serum/plasma control. The titer is determined as the first dilution at which inhibition exceeds 50% (123, 124, 129, 130), which makes this assay similar to a half-maximal inhibitory concentration (IC50) assay. These assays have been performed in numerous variations, using different transgenes (green fluorescent protein (GFP), LacZ and luciferase), cell types (HEK293T or Huh7), serum or plasma, heat-inactivation or not, with adenovirus superinfection or not etc. The sensitivity of the assay was found to decrease with lower cell densities or with GFP [reviewed in (123, 131)]. Of these options, luciferase and HEK293T cells are the most widely used (123, 124, 131), although a need for optimization remains (132). It should also be noted that different AAV purification methods produce different full/empty capsid ratios, which could also impact data transferability efforts (133). Several studies have also attempted to determine the correlation between in vitro and in vivo assays, but this proved to be challenging and tedious (129, 131, 134). Despite the fact that humoral immunogenicity remains a major impediment for gene therapy in humans and in animal models (3, 30, 47, 116, 135, 136), an international standard assay that takes into consideration key parameters, such as sensitivity and specificity, has yet to be established (135). This underscores the importance of reporting these assays in sufficient detail to allow comparisons between studies and provides the opportunity for a call to the community for additional standardization efforts (78, 122, 137).
Intriguingly, even though the challenges posed by humoral immunity, predominantly against the AAV capsid, were well established in larger animal models and screened for in patients in clinical trials, the detected cellular immune responses were not as anticipated [reviewed in (3, 30, 43, 137, 138)]. In the first liver-directed clinical trial to treat hemophilia B, hepatic intra-arterial delivery of a recombinant AAV2 vector expressing the human blood-coagulation factor IX resulted in a limited duration of transgene expression and a transient, asymptomatic elevation of liver transaminases in the high-dose group. This was attributed to cellular immune responses towards the capsid that were targeting transduced hepatocytes, as the rise of transaminases was accompanied by a rise in capsid-specific CD8+ T-cells (35). In a subsequent trial using rAAV8 to systemically deliver a self-complementary genome encoding a codon-optimized FIX variant (scAAV2/8-LP1-hFIXco), an asymptomatic increase in serum transaminases or liver-enzyme levels was also observed in the medium- and high-dose groups, together with an increase in capsid-specific CD8+ T-cells in peripheral blood. Still, in patients treated with glucocorticoids, FIX expression was maintained years after vector application (27, 101). It was further established that humans carry capsid-specific T-cells against AAVs (38, 139), and that the elicited immune response is dose-dependent (35, 101). However, immune suppression strategies seem capable of obviating this obstacle, at least to some extent (27, 38, 98, 140–142) [reviewed in (43, 72, 143, 144)]. Besides dose, the route of administration may also play a role in the induction of T-cell immune responses. Intramuscular delivery typically induces stronger cellular responses, although these seem to only account for a reduction of transgene expression, but not for vector elimination (72, 102, 103). They are concomitant with the infiltration of T-cells that do not have a cytolytic (CTLs) but rather a regulatory phenotype, Tregs (145). The latter are formerly known as suppressor T-cells, which are responsible for tolerance to self-antigens. These results once again highlight the complexities of immune responses against AAVs.
The experience gained by the early clinical trials has motivated significant research on the characterization of these immune cells and the mechanisms underlying their induction, stimulation and regulation. APCs are professional, such as DCs, or non-professional, with the former expressing MHC class II molecules and the latter MHCI. Class I MHC molecules, expressed by almost all nucleated cells, mostly present antigens to cytotoxic T-cells, as opposed to MHCII that trigger helper (CD4+) and regulatory T-cells (45). After AAV administration, APCs intracellularly process transgene peptides or proteolytic products through proteasomal degradation of the AAV capsid (146) in the cytosol and cross-present them onto MHCI (60, 147, 148). This, in turn, flags the transduced cells for destruction (39, 149–151). Presentation on MHCII molecules facilitates humoral and cellular immune responses (48, 87, 105). The epitopes on the AAV capsid that are recognized by CD8+ cells are conserved across serotypes (38, 147). These cells are limited in peripheral blood mononuclear cells (PBMCs) (41, 147), which are typically screened during clinical trials, but are more abundant in lymphoid organs, such as the spleen, and recognize epitopes presented via major MHCI (147). Capsid-specific T-cells have been found in splenocytes from children (38, 147), which points to the induction of not only humoral, but also cellular immunity early in life after AAV infection and to the maintenance of memory T-cells in secondary lymphoid organs [reviewed in (3, 43)]. These cells express IFNγ, TNFα, IL-2, perforin and the degranulation marker CD107α (66, 147, 152), which equips them with a T-effector phenotype and the ability to exhibit cytolytic activity [reviewed in (3, 43)]. From the aforementioned research, it was long thought that the cellular immune responses against the capsid are mediated by memory CD8+ T-cells generated during childhood after natural infections. Recently, however, the presence of CpG motifs has been linked to the expansion of naïve T-cells directed against epitopes on the capsid. In contrast, memory T-cells react more vigorously to AAV vectors largely depleted of the CpG motifs as well as to empty capsids (63).
Another key aspect is the linkage of humoral to cellular immunity (48, 66). Typically, T-cells are identified by their ability to produce IFNγ upon stimulation with capsid peptides. One notable study that went a step further provided a link between seroprevalence and T-cell reactivity. Seropositive individuals had TNFα-secreting memory CD8+ cells, whereas seronegative individuals showed transient activation not of naïve T-cells, but of natural killer (NK) cells that secrete both, IFNγ and TNFα (66). Additionally, antibody formation requires the CD40-CD40L axis in CD4+ cells (48) and IL-1β and IL-6 in moDCs (66).
Finally, a contribution of the capsid itself in the induction of the cellular immune responses has been reported (87, 153, 154). In more detail, it was shown that AAVrh32.33 can induce stronger humoral and cellular immune responses than AAV8 (87) or other commonly used serotypes. This can be attributed to structural differences and, in particular, to the surface-exposed variable regions, mainly IV (153). This serotype is of particular interest for vaccine applications, due to its low seroprevalence. AAVs as a vaccine have multiple advantages: a single intramuscular application suffices for long-term expression, immunogenicity and protection, and their high thermal stability reduces the thermal-chain requirements, as shown recently for the AAVCOVID vaccine. However, even though large-scale production is feasible, it is challenging to meet the needs of a pandemic (155, 156). The capsid tropism, trafficking and transduction efficiency of APCs also seem to be factors contributing to vector immunogenicity (106–108, 154).
Several studies explored the cellular immune mechanisms in non-human primates (NHPs) and revealed that natural infections with AAVs also produce cellular and humoral responses. Capsid-specific CD4+ and CD8+ cells in rhesus macaques display distinctive differentiation status and function, as well as cell-subset frequencies, with higher proportions of T-effector (Teff) cells as compared to humans (139). Furthermore, unlike chimpanzees, human immune cells do not express CD33-related Siglecs (sialic acid-binding immunoglobulin-type lectins), which are inhibitory signaling molecules thought to downregulate immune cell activation (157).
Cellular immunity against AAV is predominantly evaluated by determining the frequency of capsid-specific T-cells. Two major assays are currently in use, namely, IFNγ enzyme-linked immune absorbent spot (ELISpot) assay and, more recently, flow cytometry combined with intracellular cytokine staining (ICS) (3, 158). ELISpot measures the frequency of T-cells that produce a cytokine, such as IFNγ or, as recently suggested, also TNFα (66), upon stimulation with the appropriate antigen. As a first step, peripheral blood mononuclear cells or splenocytes (usually from humans or from animals, respectively) are isolated, cultured and plated on ELISpot plates that contain membrane bottom wells pre-coated with antibodies against the target cytokine. Afterwards, the cells are stimulated with peptide pools from different AAV capsids. Upon stimulation, the immune cells, granted they have receptors recognizing the antigen, produce cytokines that are captured by the underlying antibodies. Cells are then removed and the cytokine is detected with another antibody, producing spots on the membranes corresponding to each cell that produced cytokines. Use of serial dilutions allows to determine the number of positive cells in a population (139, 147) [reviewed in (3, 113)]. For higher sensitivity, ICS of these immune cells after stimulation with AAV capsid peptide pools can be quantified using flow cytometry (66, 147) [reviewed in (3, 113)], which also allows for multifunctional analysis of T-cells (147). Additionally, because AAV-specific circulating T-cells are rare, an enrichment step based on MHCI tetramers or pentamers and magnetic beads can enhance the detection sensitivity for both assays (41, 66, 147).
As noted initially, the cellular immune responses observed in the first clinical trials, which led to rejection of AAV-transduced cells, were not predicted in any of the small or preclinical animal studies (36). Over time, multiple explanations have been proposed, such as the primate/human source of AAVs, immunological memory in humans, or differences in the immune system [reviewed in (3, 36, 43, 47, 113)]. Extended efforts were then dedicated to develop suitable animal models [reviewed in (113)], including incorporation of a highly immunogenic peptide (SIINFEKL) of ovalbumin in the AAV capsid. To study primary or secondary responses, AAV capsid-specific CD8+ T-cells, derived either from OT-1-transgenic animals (they carry the T-cell receptor for this peptide), or from mice immunized using adenoviral gene transfer of this peptide, are adoptively transferred to recipient mice that are injected with AAVs (60, 63, 159). The adoptive transfer can also be performed without the presence of the peptide or by including an in vitro expansion step and further stimulation of the immune system. However, these methods still fail to fully recapitulate the CTL responses [reviewed in (42, 113)].
Immune responses to the transgene products are influenced by multiple factors, which can be divided into (1) host-specific, such as the underlying mutations (missense, stop codon), the genetic background, disease-related inflammation and pre-existing immunity and (2) vector-specific, such as the AAV capsid and genome, delivery route, tissue-restricted promoters, vector dose, or transgene [reviewed in (30, 42, 95)]. Gene therapy in patients that lack a given protein, due to e.g., a stop codon, is likely to induce a transgene product-specific response. However, underlying mutations (92), even single-amino acid substitutions or transgene-derived cryptic epitopes (84, 93) can induce transgene product-specific cellular responses restricted to tissue-resident T-cells (109). Another determining factor is the genetic background of the patient (37, 94, 160). Disease-specific conditions, such as the dystrophic environment characterized by inflammation, contribute to transgene rejection (161). Pre-existing immunity against the transgene due to protein replacement therapy is also a limiting factor (84). Regarding vector-specific responses, the contribution of the capsid (107, 162, 163) and the vector genome (64, 67, 164) has already been elaborated on in the previous sections. Additionally, the route of administration and promoter-restricted expression are major determinants of immune responses to transgene products. Intravenous delivery results in concurrent expression in the liver and induction of immune tolerance (37, 95), as detailed in the next section. However, intramuscular injection or restriction to this tissue via muscle-specific promoters typically results in a stronger immune response (37, 88, 92, 162, 165). This example again highlights the critical role of the promoter in AAV vector constructs and concurrently illustrates the possibilities to reduce immune responses through a meticulous selection of promoters and other regulatory elements. Ideally, this results in the detargeting of vector gene expression from APCs and, thus, avoids presentation of transgene peptides on MHCI and subsequent CTL-mediated clearance of the transduced cells.
Such regulatory elements also comprise binding sites for tissue- or cell-specific mi(cro)RNAs, which can be easily included in the 3’ untranslated region of an AAV vector expression cassette and which will shut down unwanted gene expression in cells expressing the selected miRNA(s). For instance, this strategy has been exploited in the past to purposely detarget AAV transgene expression from the liver, by incorporating binding sites for the liver-specific miR-122 into the recombinant AAV genome (166–168). Most relevant in the context of anti-AAV immune responses are miRNAs that are abundantly expressed in professional APCs, especially miR-142 (169) or, as reported most recently, miR-652-5p (99). As shown repeatedly, inclusion of binding sites for these miRNAs can diminish both, antibody formation and CTL responses, in turn boosting transgene expression and extending its persistence in mice. Impressively, combination of binding sites for miR142 and miR652-5p even enabled robust expression of the highly immunogenic ovalbumin in vivo, through detargeting from APCs, inhibition of CTL activation and suppression of Th17 responses (99). The fact that incorporation of miRNA binding sites into AAV vectors is technically simple and that saturation of the endogenous miRNA/RNAi pathway is unlikely (due to the artifical design of these sites that bind miRNAs with perfect complementarity) makes this strategy very appealing and versatile. Finally, vector dose also plays a significant role in defining the T-cell immune response (110) against the capsid and the transgene (84, 170, 171).
Despite the possibility of immune responses against the transgene product, there are few reports of clinical trials encountering this limitation. This could be attributed to residual natural protein expression, to the type of application (e.g., gene replacement therapy), to the preconditions of the individuals, to vector delivery to immune-privileged organs, to the induction of immune tolerance and/or exhaustion, or to the application of immune suppression [reviewed in (3)]. Nonetheless, some transgene product-specific immune responses were observed in vector-treated individuals. In a phase I/II clinical trial, six Duchenne muscular dystrophy patients received the mini-dystrophin transgene intramuscularly. Dystrophin-specific cytolytic CD8+ T-cells were observed in all patients after treatment and in two before (84). T-cell responses were also observed in a separate clinical trial to treat another monogenic disorder, α-1-antitrypsin (AAT) deficiency. Intramuscular delivery resulted in AAT-specific T-cell responses in two participants as well as in a reduction in expression in one of them (172). Finally, in a phase I/II clinical trial to treat mucopolysaccharidosis type IIIB syndrome (Sanfilippo type B syndrome), an rAAV2/5 vector carrying the human α-N-acetylglucosaminidase (NAGLU) was delivered intracerebrally. In three of the four patients, circulating T-cells that produced TNFα upon stimulation with NAGLU peptides were detected but later subsided, indicative of the development of tolerance (173).
Immune tolerance is a vital part of the immune system, which encompasses a broad spectrum of processes that result in a state of non-reactivity towards antigens or immune homeostasis. This ensures protection from harmful, excessive immune responses inside the host, such as against self-antigens or against chronic infections and the ensuing inflammation that can cause significant tissue damage. The major mediators of immune tolerance are the regulatory T-cells, which include the natural and the induced, also called adaptive, Tregs (nTreg and iTreg), located in the thymus and in the periphery, respectively [reviewed in (143, 174)]. Tregs are the major actors involved in inducing systemic tolerance through liver-directed gene transfer, with the liver being a long recognized tolerogenic organ (175). Different markers are used to identify Tregs, most frequently CD4 and CD25 extracellularly and FoxP3 (forkhead box P3) intracellularly (CD4+CD25+FoxP3+ T-cells) (143, 175). The transcription factor FoxP3 is central to establishing the regulatory lineage. iTregs have a transient expression of FoxP3, whereas it is stable in nTregs (176). Tregs mediate tolerance via interaction with CD8+ T-effector cells, whereby they can either inhibit proliferation and IFNγ secretion, or induce cell death through granzyme or perforin (177–179). Tolerance through Tregs is also mediated in the liver draining lymph nodes via secretion of immunosuppressive cytokines, such as IL2 or IL10 that cause Teff anergy, exhaustion or suppression (88, 180–182), or differentiation of naïve CD4+CD25- T-cells into Tregs (182) [reviewed in (138)].
Different APCs in the liver are critical for the induction of tolerance (96, 110). Kupffer cells (KCs), i.e., liver-resident macrophages, have a less mature phenotype and can induce expansion of Tregs or the conversion of Teff to Tregs, via programmed death ligand-1 (PD-L1) expression and IL-10 production (180, 182, 183). Liver sinusoidal endothelial cells (LSECs) are one of the first liver cells to recruit lymphocytes. Yet, due to high levels of IL-10, priming of T-cells is inefficient, thereby promoting Tregs (175, 184) [reviewed in (3, 95, 143, 175)]. Hepatic DCs also have the capacity to induce and maintain tolerance [reviewed in (175)]. Defective antigen presentation in the liver lymph nodes also results in T-cell exhaustion (110). T-cell exhaustion, that is CD8+ T-cells without effector functions, can be caused by interactions with Tregs, cytokines or by activation of inhibitory receptors, such as PD-1, and can also mediate long-term transgene expression (88, 185, 186). Remarkably, these cells persisted in human muscle biopsies even five years post-vector delivery (185).
AAV vectors have been used successfully in numerous clinical trials and in gene therapies. Still, as detailed above, the host immune system poses a substantial barrier to their broad and effective application. A major but unsatisfactory solution thus far has been the identification of patients with pre-existing, typically humoral immunity and their exclusion from participation. Importantly, several additional approaches were also widely explored to include more patients for whom gene therapy might be a preferred, if not the only therapy available. These can be classified into two major categories, i.e., modulation/suppression of the immune system or engineering of the AAV vector on the level of capsid and/or transgene (Figure 2, Table 1).
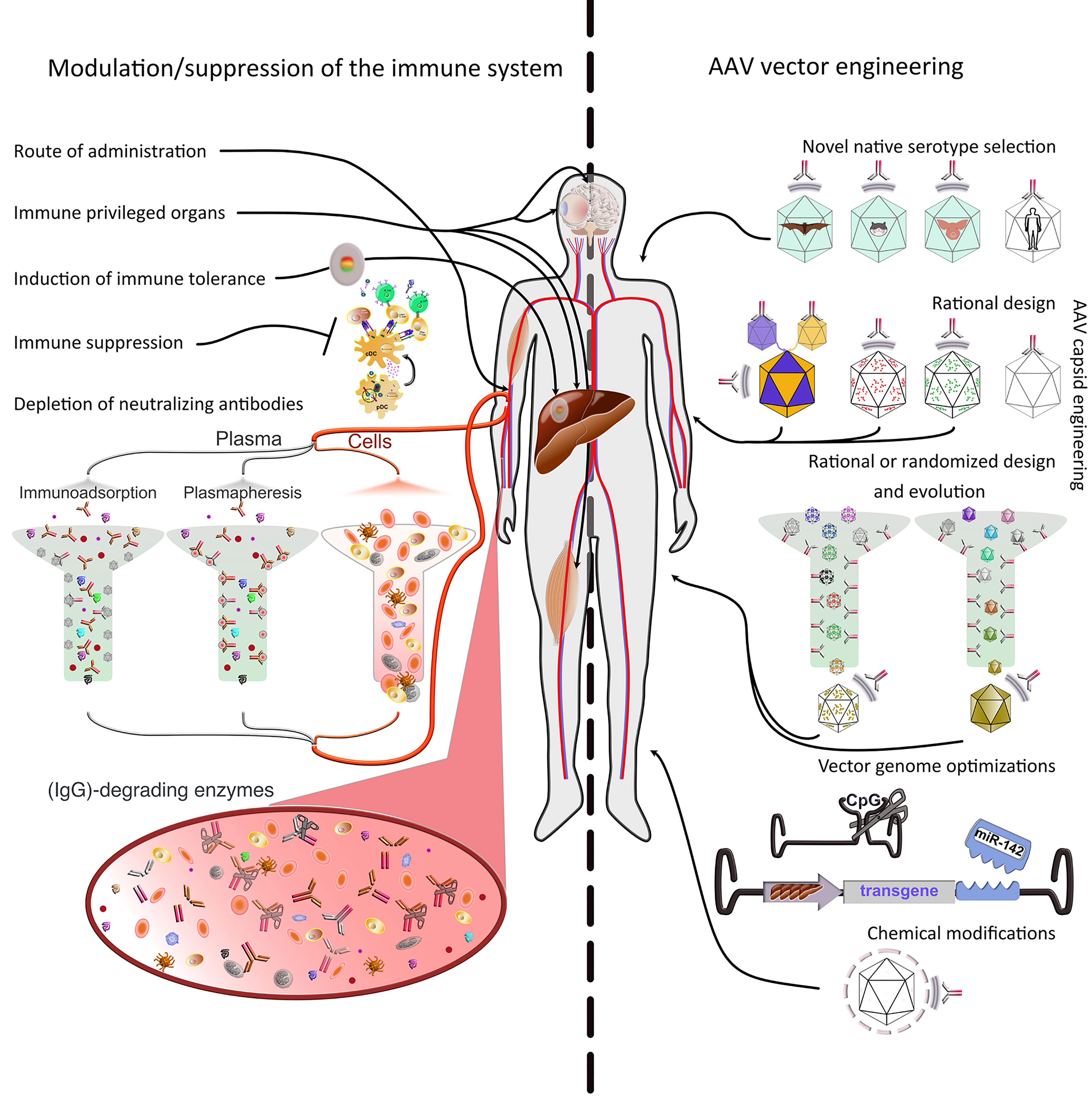
Figure 2 Strategies to evade immune responses. Several approaches to evade immune responses have been applied in the clinic or explored on a basic research level. They can be roughly divided into two main classes, i.e., 1) modulation or suppression of the subject’s immune system and 2) AAV vector engineering. 1) Modulation/suppression of the immune system. The route of administration is decisive in evading pre-existing immunity. Targeting immune-privileged organs, when possible, provides protection not only by evading immune responses, but also by inducing regulatory responses. The latter can also be induced through multiple exogenous interventions. Additionally, pharmacological immune suppression has been used extensively in the clinic. Pre-existing immunity (nAbs) is particularly difficult to evade or suppress. Promising approaches include plasmapheresis or the use of immunoadsorption columns ex vivo after separation from the cellular parts of the blood. Furthermore, recent studies have used IgG-degrading enzymes in vivo. 2) AAV vector engineering. The use of native serotypes from species other than humans or NHPs, or engineering the capsid of existing serotypes (predominantly from primate species) are advantageous strategies to evade immunity. The current serotypes can be modified in defined positions (rational design) based on acquired knowledge about sequence-structure-function relationships. To increase variability, the rational design strategy can be used to generate libraries, which then can be evolved/selected ex or in vivo. Libraries can also be fully randomized and interrogated for multiple properties, not only immune evasion. Moreover, the viral genome can be optimized to minimize antigen presentation (e.g., via cell-type specific promoters, miRNAs to prevent expression in APCs or peptides to inhibit antigen presentation) or immune system induction (CpG depletion). Finally, the capsid can be protected from the immune system by coating with molecules, such as PEG, engulfment in exosomes, or display of immune evasion peptides on its surface.
Below, we will discuss a selection of these approaches that have been published in a large body of work and that range from basic research all the way to the clinic.
The route of administration has been strongly implicated in the inhibition by, and the induction of, immune responses. The exact choice is usually determined by the type of disease. Examples for administration routes include intravenous for hemophilia or liver/heart diseases, intramuscular or intravenous for muscle diseases, or intracerebral, intraparenchymal, intrathecal, or intravenous for neurological diseases [reviewed in (1)]. Direct injection into the target organ, such as intramuscular injection (73), is the most straightforward method to avoid circulating antibodies. Additionally, saline flushing to avoid contact with nAbs in combination with direct or balloon catheter-guided vector injection has shown promise (74). Nonetheless, this does not obviate the generation of immune responses after gene therapy (187), which could eliminate transgene expression later on, as explained in detail before. Fortuitously, several delivery methods exist today that facilitate the evasion of these responses (immune-privileged organs) or their manipulation (immune tolerance or T-cell exhaustion).
The intravascular or intravenous route of administration has been predominantly associated with inactivation of AAVs by nAbs in the circulation and interstitial tissues (35, 40, 116, 117, 135, 188). Thus, it is encouraging, albeit not a panacea, that certain tissues provide shelter from the immune system. Three organs are considered immune-privileged, namely, the brain, the eye and the liver.
The CNS, and in particular the brain, have long been considered to be isolated from the immune system by the physical blood-brain barrier (BBB). Moreover, the brain lacks classical draining lymph nodes and APCs in the parenchyma. While these concepts have been challenged (189), CNS gene transfer, either to the cerebrospinal fluid (CSF) (75, 190) and, to a lesser extent, after intraparenchymal injection (191–193), remains comparatively successful notwithstanding the presence of circulating nAbs. Still, in patients with high titers of circulating antibodies, even CNS gene transfer is inhibited (134, 194). This was exemplified in a study in which preimmunization of mice after intramuscular injection hindered subsequent brain delivery. On the other hand, passive transfer of NHP serum containing nAb to mice did not impact gene transfer to the hippocampus or the thalamus (134).
The ocular immune privilege, akin to the one in the brain, was first described in the middle of last century. It is mediated by a blood-retina barrier (195), absence of efferent lymphatics, presence of elevated concentrations of immunomodulatory molecules and immunoinhibitory factors, and finally the ACAID system (anterior chamber-associated immune deviation), which is typically stimulated after perturbation of the ocular integrity. ACAID induction by intraocular antigens results in induction of Tregs, immunomodulators in the aqueous humor, anti-inflammatory cytokines and F4/80 macrophages that present the antigen to clusters of immune cells in the spleen (196, 197). The induction of a deviant immune response has already been reported for adenoviral and AAV gene transfer to the subretinal space (198). In combination with other mitigating factors, such as the low effective dosage and hence minimal toxicity and low manufacturing burden, this well-characterized immune privilege has catapulted ocular gene therapy to the forefront of the field [reviewed in (30)], with multiple clinical trials concluded or underway, culminating in the approval of Luxturna™ (199). There are two major delivery methods, subretinal and intravitreal, of which the latter is slightly more immunogenic (76). Although pre-existing immunity can pose a challenge to intravitreal delivery (200, 201), several clinical trials were successful due to the immune-privileged status of the eye (202) [reviewed in (30)].
In contrast to pre-existing immunity, induction of the immune system after gene therapy in the eye was observed more frequently (203), albeit not to the same extent reported for other delivery routes (detailed in previous sections). Induction of a dose-dependent inflammatory response was observed in animal models (204–207) and in humans (208–212). In several clinical trials using either of the two delivery methods (subretinal or intravitreal), inflammation occurred but could be treated with immune suppression regimes. Likewise, transient antibody response against the capsid and cellular immune responses were also noted [reviewed in (30, 76, 213)]. The low to mild and transient immune responses allowed repeated administration of AAV vectors, either contralateral or in one eye (214, 215) or the delivery of two boluses in the same eye (216). Finally, long-term expression could be achieved in most clinical trials, with a decline observed in some after a few years (202, 210) [reviewed in (30, 76, 213)].
Liver is the largest organ in the body, whose sinusoids filter an antigen-rich blood. In order to protect itself from the antigenic overload of nutritional components, chemicals and drugs, liver promotes immune tolerance rather than reaction (217). Even though systemically-delivered liver-targeted gene therapy cannot evade pre-existing immunity which challenges the immune-privileged status of the liver, the ability to harness the tolerogenic hepatic microenvironment has encouraged substantial research in the gene therapy field (145), especially regarding the expression of transgenes that are absent in subjects prior to the injection (187) [reviewed in (95, 175)]. Tolerance is dose-dependent, with higher dosage ensuring immune tolerance through Tregs, IL-10 expression, Fas-L and depletion of Teff cells as enabling factors (88, 110, 218). Another important consideration is whether the transgene product is secreted or not. Secreted proteins are presented in multiple organs and thus need a much lower threshold to produce Tregs. Interestingly, intracellularly restricted expression, e.g., using proteins located in the cytoplasm, results in antigen presentation by liver-draining lymph nodes (celiac and portal) and production of Tregs that are then disseminated to the periphery (96). This tolerance can be harnessed even with gene therapy targeted to other organs. Simultaneous expression of the transgene in the liver can induce expression of Tregs and tolerance to the transgene product expressed in muscle (104, 186). Most interestingly, the induction of immune tolerance through liver can be achieved despite the established presence of inhibitors (219) or even after gene delivery to another organ (88), hence reversing existing immunity [reviewed in (3, 30, 85, 138)].
Induction of immune tolerance can be achieved by targeting the liver, even at a later timepoint, as elaborated on in previous sections. Alternatively, Tregs can be ex vivo reprogrammed and adoptively transferred (97), or they can be induced through molecules (Tregitopes) or chemically (rapamycin). Tregitopes (Treg epitopes) are peptides found within human IgGs and exhibit high-affinity binding to MHCII. They were initially identified in humans (220) and are conserved in mammalian species (221). Via a mechanism requiring cell-to-cell contact, Tregitopes can trigger the proliferation of Tregs. Fusion of Tregitopes to the capsid proteins could also reduce CD8+ T-cell reactivity by fostering proliferation of Tregs in vivo (105). Alternatively, immunomodulatory drugs, such as rapamycin, can be used to induce Tregs. The phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K-Akt-mTOR) pathway regulates thymic and peripheral Treg generation (222). Rapamycin, an immunosuppressing compound used in graft rejection, can expand CD4+CD25+FoxP3+ Tregs (223). A hallmark study showed that simultaneous administration of synthetic vaccine particles encapsulating rapamycin [SVP(Rapa)] with AAV vectors alleviated anti-capsid humoral and cellular immune responses, thus enabling vector re-administration. Adoptive transfer of splenocytes to naïve mice transferred this immunomodulatory property while depletion of CD25+ cells neutralized this effect (89). Gene transfer to seropositive rhesus macaques after rapamycin treatment was only successful using subcutaneous, but not intravenous delivery (224), implying that route and dosing schedule may be key to success (225). Further studies in preclinical models are necessary to assess the efficacy of these treatments in the clinic as well as their safety, as they compromise the immune system and make the host vulnerable to infections [reviewed in (85, 138)].
Immune modulation involves a broad range of approaches, of which the earliest and most widely applied in human clinical trials is transient immune suppression predominantly of T-cells with corticosteroids, such as prednisolone (188) [reviewed in (30, 85, 98, 226)]. Immune suppression can also be mediated using alternatives to steroids, such as mycophenolate mofetil (MMF) and tacrolimus (227), or MMF and rapamycin (98). Immune suppression has had varying effects in studies and clinical trials. Despite positive results (27, 101, 142, 228, 229), it often did not sustain long-term expression of the transgene (88, 230). Of note, sustained immune suppression with daclizumab is associated with a reduction in Tregs (111). Therefore, a recent study proposed new protocols using a meticulously timed T-cell-directed IS, with an early administration of MMF and rapamycin and a delayed delivery of anti-thymocyte globulin (ATG), to determine the balance between immunogenicity and tolerance (98). Immune suppression regimens were also used to deplete B-cells and thereby nAbs, such as rituximab with cyclosporine (231), rituximab with sirolimus (90), or rituximab alone (232), rapamycin without or with prednisolone (89, 233), and anti-CD20 with rapamycin (234). However, one should be aware that such regimens target the immune system of the host as a whole and are not specifically tailored to the gene therapy. Finally, another intriguing approach that has shown great promise but requires further studies is inhibition of proteasomal processing of internalized capsids, which is necessary for antigen presentation and part of the immune reaction process, using a variety of molecules (146, 148) some of which are also approved for use in humans (146) [reviewed in (138)].
Immune suppression offers only moderate capabilities to evade pre-existing immunity, which would otherwise disqualify 15-50% of the population from clinical trials. Accordingly, additional approaches are urgently needed (235). Recently, a hallmark study illustrated the potential of Imlifidase (IdeS), a cysteine endopeptidase derived from the immunoglobulin G (IgG)-degrading enzyme of Streptococcus pyogenes. IdeS cleaves human IgG into F(ab’)2 and Fc fragments, thus eliminating its Fc-dependent effector functions. In this study, the authors successfully showed the cleavage of human intravenous immunoglobulin (IVIg, a cocktail of serum from thousands of human individuals) in vitro, as well as in pre-immunized mice and seropositive NHPs in vivo. IdeS administration prior to AAV gene transfer in vivo reduces the levels of IgGs, thereby allowing efficient transduction. Treatment with IdeS also enabled readministration of AAVs. Finally, the efficiency of IdeS was also successfully validated with human sera (236). In a similar study, IdeZ, an IdeS homolog isolated from S. equi ssp. zooepidemicus, showed efficient IgG cleavage of dog, monkey and human sera and facilitated in vivo gene transfer in passively immunized mice and macaques (237). Moreover, a large protein, protein M, was identified in Mycoplasma that binds Igs with broad reactivity by using a different mechanism. In vitro and in vivo studies showed the efficiency of this approach to protect AAVs from nAbs and to allow readministration (235, 238).
Removal of anti-AAV nAbs has also been attempted through plasmapheresis, an extracorporeal method in which a device separates plasma from the cellular component of the blood. Afterwards, the plasma is filtered through various techniques, remixed with the blood cells and returned to the subject. Alternatively, the plasma can be substituted by a replacement solution. Plasmapheresis was used to remove immunoglobulins against the capsid in humans (239) and in NHPs (77). In the latter study, AAV gene transfer after plasmapheresis resulted in efficient transduction (77). While promising, this approach has several limitations including that multiple rounds are needed. A further concern is the so-called “antibody rebound” effect, which means that the antibody pool is readily replaced. Also, certain patients with weak physical conditions might be more adversely affected by the procedure, and the complete removal of immunoglobulins leaves the patients vulnerable to infections (77, 239) [reviewed in (47, 138)]. Recently, elegant studies have thus combined plasmapheresis with immunoadsorption columns loaded with empty AAV capsids, in order to selectively deplete nAbs against the AAV capsid (78, 79). In a similar manner, a prior study exploited empty AAV capsids that were injected to mice together with the AAV vectors to act as decoys for AAV-specific nAb (240). However, a concern is that excess AAV capsids could elicit stronger cellular immune responses or immunotoxicities (151) [reviewed in (42)]. It should also be noted that these procedures non-discriminately remove all AAV-binding antibodies, some of which do not neutralize but actually enhance transduction or alter distribution (126, 241).
The majority of the AAV serotypes that are currently in clinical use are human or NHP isolates from liver or spleen [reviewed in (1, 2)]. However, a concern with their use is that epidemiological studies show a high seroprevalence of those serotypes in the human population (114, 115, 121, 242, 243). A rational approach to overcome this concern is to isolate novel natural AAV serotypes from other species, which are expected to exhibit lower seroprevalence in humans but may also display lower transduction efficiencies. To this end, novel native AAVs have already been isolated from avian (244), rat and mouse (14), caprine (245), porcine (246, 247) and bat (80, 248) species. Some of these showed promise in in vivo biodistribution studies where they were found capable of targeting multiple tissues (246), such as heart (246), muscle (246, 247, 249), lung (245) or retina (247). Despite these promising results, further studies are necessary in larger, preclinical animal models to better characterize and ideally validate their potential for clinical application.
There is a growing amount of information on immunogenic epitopes on the AAV capsid, based on cryo-EM structural mapping studies (250–255) [reviewed in (253)], single point mutants (256–258), or barcoded (9, 259) or not (260, 261) libraries of pooled vectors carrying point mutations. For the cryo-EM studies, monoclonal antibodies (mAbs) predominantly from hybridoma screens have been used, although recently, the isolation of human mAbs that are more clinically relevant has also been pursued (262). These studies facilitate the rational design of vector capsids or the engineering of libraries directed at the immunogenic epitopes, in order to render the complexity of the library technically attainable and to concurrently circumvent unwarranted changes to essential properties, such as tropism. Such information was applied to enhance nAb evasion by mutagenizing single [V719M (263), S671A (264), 265T (256)] or multiple positions (265, 266). A similar approach was used to modify surface tyrosines to avoid ubiquitination (267), proteasomal degradation and antigen presentation (149, 268, 269). This can be extended to other amino acids (serine, threonine, and lysine) (270, 271) and serotypes (272). Polyploid vectors, also called mosaic, combine capsid subunits from different serotypes and are formed by simply mixing the production plasmids at different ratios. This methodology identified a triploid AAV2/8/9 vector that could evade immunity more than the parental vectors (273).
A conceptually different approach is to display peptides on the surface of the capsid to evade or quench the immune system. Display on the AAV capsid of a self-peptide (SP), a 21-amino-acid long truncated bioactive form of CD47, whose binding to SIRPα (Signal regulatory protein α) on macrophages acts as a “don’t-eat-me” signal for macrophages, was shown to reduce macrophage uptake (91). Likewise, fusion of Tregitopes on the AAV capsid protein VP1 reduced CD8+ T-cell responses and increased Tregs (105).
The wealth of knowledge regarding the immunogenic epitopes on the AAV capsid has been mostly implemented in rational engineering and directed evolution of viral libraries, to create novel, engineered immune evading AAVs. In an early study, five immunogenic amino acid positions (449, 458, 459, 493, 551) were randomized on AAV and the library was evolved under negative selection pressure using IVIg, producing immune-escaping variants (260). By studying the AAV1 complexed with four different Fabs of mouse anti-AAV1 mAbs, the cryo-reconstructed structures revealed three capsid antigenic footprints: region IV (456-AQNK-459), region V (492-TKTDNNNS-499), and region VIII (588-STDPATGDVH-597). Each region was separately randomized, and the libraries subjected to iterative rounds of evolution on vascular endothelial cells that are highly permissible to AAV1, in order to evolve the library for properties other than efficient transduction. The top variants were then combined albeit some could not be juxtaposed, as observed previously (274). In this case, the variant was combined with a library based on another footprint and a new evolution was applied. One of the evolved CAMs (Capsid Antigenic Motifs), CAM130, was significantly enhanced compared to the parent vector and, whilst maintaining tropism, showed an advantageous, immune-evading serological profile (81). A similar study evolved an AAV8-based variant, AAVhum.8, that exhibits mouse and human hepatocyte tropism and sera-evading properties (275). Another group harnessed a mAb PAV9.1 selected from a panel of hybridoma clones to identify a conformational epitope on AAV9 for further mutagenesis that comprises 494-TQNNN-498 and 586-SAQAQ-590. These Fab complementary determining regions (CDRs) were mutated using serotype swapping, alanine replacement, and additional point mutations. The resulting CDRs were efficient at evading the mAb binding, but they could not evade binding or neutralization by polyclonal serum or plasma from mice, macaques, or human donors (276). Another recent rational approach focused on residues identified to be different among 150 AAV3B variants, selected the surface-exposed ones and mutagenized them only to naturally occurring residues in this position. The library was evolved in human hepatocarcinoma spheroid cultures and the top variant, AAV3B-DE5, was further evaluated. It exhibited reduced seroreactivity against IVIg and individual human samples as well as tropism towards human, but not mouse hepatocytes in chimeric livers, similar to the parental serotype (277). Rational design strategies have contributed significantly to the field. However, screening of completely randomized libraries without a priori knowledge of immunogenic epitopes also offers valuable solutions, as discussed next.
Complementing the efforts outlined in the prior chapter that rely on limited antecedent knowledge to facilitate some degree of rational design followed by further AAV evolution, many groups have devised and applied experimental forward-oriented strategies in order to identify immunoevasive AAV variants. To this end, comprehensive libraries of synthetically engineered AAV capsid variants are first generated and then subjected to a negative selection pressure, which ideally eliminates all candidates that cross-react with, and are neutralized by, anti-AAV antibodies. The methodologies to create such diverse capsids libraries are manifold and have been extensively reviewed in the past by us and others (2, 278), hence it may suffice below to name some of the most widely used approaches including DNA family shuffling, peptide display, error-prone PCR and ancestral reconstruction.
One of the first studies to illustrate the power of directed AAV library screens and antibody-mediated selection was reported by the Kay lab in 2008 (279), which exploited the fact that IVIg contains a mixture of anti-AAV antibodies that is a good proxy for the human population. Accordingly, the group first created a library of ~7×105 shuffled AAV capsid variants from eight parental viral serotypes and then iteratively amplified this capsid pool on human liver cells in the presence of IVIg, with the aim to eliminate all variants that were recognized by the anti-AAV antibodies. Indeed, this strategy enabled the isolation of a single chimeric AAV variant, called AAV-DJ, that at least partially resisted antibody neutralization in vitro and in vivo to a much greater extent than AAV2, which is one of its dominant parental serotypes and which has been used extensively in humans to date. The ability of shuffled AAV capsids to partially evade IVIg neutralization was later also confirmed by several other groups including notable work from Koerber et al., albeit this group only used IVIg during the stratification of already isolated chimeric AAVs and not for selection (280).
Similarly, in another representative example from the Schaffer lab (261), negative selection via a neutralizing anti-AAV2 rabbit serum was harnessed to enrich antibody-resistant AAV capsid variants from libraries that had been created through error-prone PCR amplification of the entire AAV2 cap gene. Interestingly, their lead candidate had not only become more antibody-resistant than wild-type AAV2 but had also acquired additional properties, such as altered DNA packaging efficiency, heparin affinity or cell specificity, likely explained by the well-known pleiotropic roles of many residues within the AAV capsid proteins.
These conclusions were confirmed in a flurry of more recent work from several labs, which cannot be covered comprehensively here; hence we will only highlight examples below that are representative for numerous other studies. One such example is another pivotal study by the Kay lab (82) in which Paulk and co-workers performed a multiplexed AAV library screen combining iterative in vivo selection of a shuffled library in “humanized” mice (i.e., mice xenotransplanted with human hepatocytes), followed by two rounds of ex vivo AAV capsid depletion on IVIg-coated beads. Of the shuffled capsid variants enriched by this procedure, NP59 is remarkable as it combines robust and specific in vivo transduction of human hepatocytes with good performance in seroreactivity and transduction neutralization assays, including the use of individual serum samples from macaques or humans (healthy or hemophilia B patients), or again IVIg. A very similar approach has also been reported more recently by the Li lab (281), using a different starting library and resulting in unique shuffled AAV variants. As a last example in this category, a study by the Samulski lab should be pointed out, which is remarkable for the fact that Li et al. selected a shuffled AAV capsid library in the presence of neutralizing sera not only in cultured cells, but directly in the muscle of mice (282, 283). Furthermore, rather than using an IVIg pool for selection, the group harnessed individual sera from human patients who had participated in a clinical trial for Duchenne muscular dystrophy with an AAV2.5 vector. A particularly notable conclusion in this work was such a stringent selection strategy may be beneficial, since pools such as IVIg are a mixture comprising sera without neutralizing anti-AAV antibodies, hence AAV capsid variants emerging from IVIg selection may only escape neutralization in subjects with high individual antibody titers, whereas those selected with a stringent serum may be more broadly resistant.
Similar conclusions were also drawn in parallel work in which capsids were diversified not via DNA shuffling, but rather via insertion of short peptides (typically 7 to 14 amino acids long) on the capsid surface. While the primary purpose of these peptides is binding to (usually unknown) receptors on target cells, several groups have reported that display of these short additional peptides on the AAV shell can also modulate its recognition by neutralizing antibodies. As a representative example, one of the first studies reporting this finding should be highlighted (284), in which Huttner and colleagues found that insertion of peptides selected in previous screens in AAV2 amino acid positions 534 and 573 substantially reduced capsid affinity for neutralizing anti-AAV2 antibodies in human sera.
In general, it should be an interesting goal for future work to compare the lead candidates from these and other studies side-by-side in genuine or humanized mouse livers in the presence of anti-AAV antibodies, ideally using a vector DNA/RNA barcoding strategy (285) to enable a fair comparative analysis in the same animal(s). Besides, the aforementioned research, along with the rational design studies, further corroborates the notion that antigenic, tropism and potency determinants overlap in the structural context of the AAV capsid and thus perfectly complements the alternative strategies noted above, not only on a technical but also on the biological level.
Rather than engineering the capsid itself, either via directed evolution or rational design, numerous groups have pursued an alternative and complementary strategy to mask AAV from neutralizing antibodies, by either chemically modifying the shell or encapsulating the particles in extracellular vesicles (exosome). As with the capsid engineering approaches, the diversity of strategies is so substantial and the literature so complex that we can only highlight a few representative examples below, and we apologize to all colleagues whose work we had to omit for space reasons.
One particular strategy that has been reported frequently is AAV modification via chemical conjugation with polyethylene glycol (PEG), which is a simple, cheap and effective means to cover and protect the capsid from neutralization, but which may also come at the cost of (steric) interference with AAV transduction and tropism. This dilemma was exemplified, for instance, in one of the first reports of AAV PEGylation by Lee and co-workers (286) who found that there is only a small window of PEGylation, i.e., PEG:lysine conjugation ratio and PEG molecular weight, that enables effective antibody protection while maintaining infectivity. Subsequently, other used various alternative strategies for AAV PEGylation, such as AAV2 modification with a series of activated PEGs, some of which yielded protection from neutralization without severely impeding transduction efficiency (287), or use of genetic code expansion for insertion of a lysine mimic into the AAV2 capsid that enabled site-specific PEGylation and escape from neutralization (288).
Instead of coating the AAV particle via chemical modification, other groups, most notably the one of Casey Maguire, rather encapsulate the vector particles in naturally occurring cellular vesicles, resulting in what was originally called “vexosomes” and later exo-AAVs. As, for instance, demonstrated by György et al. in 2014 (83), exosome-encapsulated AAV9 vectors were significantly more resistant to both, pooled human serum as well as IVIg, and they also performed better than naked AAV9 in passively IVIg-immunized mice. Importantly, this method is not restricted to a particular serotype, since escape from neutralization in cultured cells was also observed for AAV1 and AAV2. These encouraging results were corroborated and extended in a series of more recent studies, including a notable piece of work from Meliani et al. who showed that exo-AAV8 vectors (and also exo-AAV5) performed better than the wild-type counterpart at liver-directed human factor IX expression in mice, perhaps owing to a faster nuclear translocation rate and autophagy-independent trafficking. Remarkably, in turn, the higher expression was also correlated with an increased frequency of Tregs in lymph nodes of exo-AAV8-treated mice, suggesting that improved induction of immunological tolerance may be an additional benefit of exosome-encapsulated AAVs. Moreover, exo-AAV8 was also more resistant to neutralizing anti-AAV8 antibodies in human sera and in a passive immunization mouse model of liver gene transfer, further illustrating the potential of this strategy to expand the proportion of human subjects who are eligible for AAV gene therapies.
Finally, another intriguing approach worth mentioning has recently been reported by Katrekar and colleagues (289), who combined genetic code expansion and click-labeling to precisely tether oligonucleotides to the surface of the chimeric AAV-DJ capsid (279). When incubated with lipofectamine, this resulted in so-called “cloaked” AAVs that were much more resistant to neutralizing anti-AAV antibodies and concurrently yielded higher CRISPR gene editing efficiencies than an unconjugated AAV-DJ control, reminiscent of the dual benefit observed with the exo-AAV strategy (see above).
While these and other, equally compelling studies that could unfortunately not be mentioned here are very promising, it is also clear that additional, meticulous work is needed and that a number of challenges have to be overcome before chemically modified AAVs can be used more broadly and even clinically. This includes the need to optimize and standardize large-scale production and purification protocols, as well as the requirement of a thorough characterization of possible impurities and contaminations especially in cell-derived exo-AAVs. In addition, immune responses against PEG or lipofectamine will have to be studied and, if detected, may limit the widespread application of certain formulations. Last but not least, cloaking or coating the viral shell may inadvertently and negatively impact in vivo features such as biodistribution, kinetics and blood clearance, which would be particularly detrimental for synthetic capsids that have been genetically engineered to have a more defined tropism or other advantageous properties.
Next to the viral capsid, also the cargo, i.e., the recombinant genome consisting of the transgene expression cassette flanked by the ITRs, offers multiple opportunities for engineering and alleviation of immune responses, including its structure and components.
Regarding structure, a favorable design with respect to efficiency are self-complementary (sc) or double-stranded AAV vector genomes, which―after replication and encapsidation―carry two inverted copies of a transgene that rapidly and effectively self-anneal in the transduced cell. Thereby, scAAV genomes alleviate the rate-limiting step of second-strand DNA conversion that normally restricts transduction with conventional single-stranded AAV vectors and that explains their slow kinetics of transgene expression that is typically observed in vivo. While beneficial in this aspect, a drawback of scAAV vector genomes that has become apparent over the last decade is their higher propensity to trigger an innate immune response via the endosomal DNA receptor TLR9, as observed in several tissues including the liver and the muscle. This includes early data in mice where scAAV vectors led to increased expression of various innate immune-related genes and induced innate responses in a dose-dependent manner via TLR9 signaling. This, in turn, enhanced adaptive immune responses to the capsid but not the transgene product, probably due to the short-lived and self-limiting nature of the innate response (58).
Still, in independent work, the Ertl lab showed that scAAV vectors, as a result of their faster transgene expression kinetics, are also more prone than ssAAV to induce transgene product-specific CD8+ T-cell and B-cell/antibody responses in mice (164). The extent of these responses depended on the capsid serotype, with AAV7 yielding much stronger effects than AAV2, probably owing to the higher efficiency of AAV7 in the muscle.
While both studies unanimously concluded that AAV genome configuration governs the immunogenicity of AAV particles, the authors also concurred that lowering doses may allow scAAV vectors to dodge the immune system. Alternatively, or in addition, as demonstrated consistently by the Wilson, Ertl and Church labs, innate immune responses can also be blunted by directly engineering the vector genome. First, Faust and colleagues reported that vector genomes depleted of CpG islands, which are typically sensed by TLR9, evade the innate and adaptive immune response and establish persistent transgene expression in skeletal muscle in mice in the absence of T-cell infiltrates, even when delivered by a highly immunogenic AAVrh32.33 capsid (67). Concurrent with this, and as already noted above in chapter 2.2.2.1, the Ertl lab found that CpG depletion in AAV vector genomes can diminish a primary, de novo T-cell response, by reducing expansion of naïve CD8+ T-cells against AAV capsid epitopes (63). In contrast, these engineered vectors triggered a secondary response, by driving proliferation of anti-AAV capsid-specific memory CD8+ T-cells, a phenomenon that was also observed with empty AAV capsids. The latter is particularly relevant in view of the fact that spiking in empty capsids into AAV vector preparations has previously been shown to dampen the humoral immune response to the viral particles, implying that the empty capsids acted as antibody sponges. Hence, the findings by Xiang et al. (63), that empty capsids do not stimulate a primary T-cell response to antigenic capsid epitopes and also do not boost T-cell activation triggered by full AAV particles, are pivotal for our understanding of the role of full versus empty capsids and for the future optimization of strategies to circumvent humoral and cellular anti-AAV immune responses.
Most recently, Chan and co-workers moreover showed that TLR9 activation can also be dampened through incorporation of short oligonucleotides that antagonize TLR9 activation, dubbed TLR9i (i for inhibitory) (86). Such TLR9i sequences were found to cloak the AAV vectors in multiple, but not all tested animal models and tissues, confirming the pivotal role of TLR9 sensing but concurrently implying the existence of other, TLR9-independent immune mechanisms. The latter may also have contributed to the results of clinical trials using AAV8 for expression of the human blood coagulation factor IX in hemophilia B patients, in which capsid-specific CD8+ T-cell responses were observed despite the use of CpG-reduced vector genomes (101). In this context, another original hypothesis that is noteworthy and that was presented by Li and Samulski (1) suggests that the intrinsic promoter activity of the AAV ITRs might drive the generation of dsRNA at later stages of transduction, in turn activating cellular RNA sensors and thus stimulating an innate immune response. If true, this would imply a solution whereby this promoter activity is diminished or blocked, by deliberately engineering the ITRs and/or insulating the expression cassette from the ITRs (1).
Interestingly, the authors of the latest work on TLR9i postulated that these sequences may act by outcompeting CpG islands in the vector genome for TLR9 binding, potentially via their higher TLR9 affinity and their ability to prevent TLR9 dimerization and activation upon binding (86). While not tested in this work, this raises the intriguing question whether combining TLR9i with the aforementioned depletion of CpG islands may synergize and further dampen innate immune responses to the AAV vector genome. Additional topics for future work should include the persistence of the effect and the impact on vector transgene expression, as well as on clinically relevant immune responses, including humoral or cellular immunity to the capsid proteins or the transgene product. Furthermore, it will be prudent and informative to assess this strategy in more animal models and with other targets, using different clinically applicable administration routes. Until then, the fact that Chan et al. noted beneficial effects on the anti-AAV immune response in a variety of experimental settings and largely independent of the other vector components, such as capsid or promoter, is already highly encouraging as it suggests a large degree of versatility, modularity and translatability.
Despite the success of AAV-based gene therapeutics and the prevailing view that AAV is less immunogenic than other recombinant virus platforms, rapidly mounting evidence from preclinical work in large animals and clinical studies in humans implies that AAV vectors can cause inflammatory and immune responses as well as other dose-dependent toxicities and pathologies. This includes observations of severe adverse events, possibly related to innate and cellular immune responses, in patients suffering from Duchenne muscular dystrophy (DMD) or spinal muscular atrophy type 1 who had been treated with high AAV vector doses (290, 291). In the DMD trial (IGNITE), treatment of a boy with a high dose (2x1014 vg/kg) of the therapeutic vector (SGT-001, encoding micro-dystrophin) resulted in lower red blood cell and platelet counts (thrombocytopenia), caused kidney damage, and activated the complement system. Fortunately, these complications were all resolved and the sponsoring company (SOLID) improved their manufacturing protocol (to reduce the number of empty capsids), eventually allowing the FDA to lift their initial ban on this trial.
Similar findings were reported in a phase I trial (sponsored by Rocket Pharmaceuticals) for treatment of Danon disease, a devastating and rare X-linked autophagic vacuolar myopathy that results from mutations in the LAMP-2 gene and that can cause dysfunction of the musculature and other organs, frequently triggering early mortality. In this trial, one patient treated with the high dose (1x1014 vg/kg) and who had a pre-existing anti-AAV9 immunity, experienced adverse events that were presumably immune-related and that also comprised thrombocytopenia and acute kidney injury. Luckily, also this patient ultimately recovered and regained normal organ function. Still, the serious and consistent adverse events in these two trials clearly raise a warning flag about possible toxicity from in vivo application of excessive AAV vector doses.
In addition, AAV dose-dependent pathology was observed in dorsal root ganglia (DRG) in vector-treated non-human primates that seemed to be largely independent of capsid or cargo (292, 293). The effects were only mild to moderate and not associated with fatalities; moreover, a possible mitigation has recently been proposed in which the inclusion of binding sites for miR-183 a miRNA largely restricted to DRG neurons, can alleviate DRG toxicity (294). Nonetheless, the possible link between AAV delivery and sensory neuropathies in DRGs requires further investigation and careful monitoring in ongoing or future clinical studies.
However, most dire and most alarming is the outcome of the recent ASPIRO trial, in which three children affected by X-linked myotubular myopathy (XLMTM) and given a very high dose of 3x1014 AAV8 vg/kg bodyweight developed progressive liver dysfunction, bacterial infection and sepsis (two of the three patients), eventually resulting in death of all three individuals (295–298). Also here, a critical role of the immune system has been suspected, including the presence of anti-AAV antibodies in these patients that could have triggered an innate response or activated the classical arm of the complement system. Notably, none of the patients in this trial who had received a lower vector dose developed liver-related adverse events. Instead, several children regained the ability to sit, stand or walk and no longer needed ventilator support, clearly illustrating the great potential of this gene therapy approach and of the vector used. Of note is that no such toxicities had previously been observed with the same vector in mice or non-human primates, even using higher doses of 8x1014 AAV8 vg/kg, and that very encouraging efficacy data had been obtained in murine and canine XLMTM models (299, 300). This highlights the species-specific differences in AAV-host interactions and the urgent need for better characterization including a possible contribution by immune mechanisms, with the clinically highly relevant aim to control and overcome, or ideally altogether prevent, such adverse events in patients.
Last but not least, we note another very recent, serious adverse event in an AAV gene therapy clinical trial (INFINITY, company sponsor Adverum), this time the loss of sight in a patient with diabetic macular edema who was treated with a high dose (6x1011 vg/eye) of a vector based on a synthetic AAV capsid (AAV2.7m8). The fact that the eye is more immunoprivileged than other organs (see also chapter 3.12.2.) and that the effect occurred long (30 weeks) after dosing may argue at least against an acute anti-AAV immune response, but the mechanisms are still unclear.
The historic fable “The race between the hare and the hedgehog”, originally published by the Brothers Grimm in 1843, describes the race of two animals, one fast (hare) and one slow (hedgehog), of which the latter has no chance of winning the contest. Still, the hedgehog seemingly outcompetes the hare through cheating, as he places his lookalike wife at the finish line and thereby ultimately frustrates the hare to an extent that it dies. For many reasons, one can readily apply the same image and draw comparisons to the incessant arms race between AAV and its human host, starting with natural infections and nowadays significantly accelerated by the growing clinical use of recombinant AAV vectors. Akin to the fable, there is substantial hope that the hedgehog in this analogy, i.e., the AAVs, will eventually win this race against the host (the hare), assisted by a little cheating in the form of our ever expanding understanding of AAV-host interactions and the concurrent advent of powerful new technologies to blunt or alleviate anti-AAV immune responses, a collection of which has been highlighted and discussed in this article. It is also evident that this looming happy ending requires a considerable body of additional, concerted work from the field, aimed at a better characterization of the new, genetically engineered, evolved or designed, and/or chemically modified AAV particles along with improvements in the technology for their clinical-grade manufacturing including production, purification, quality control and batch release. Still, in view of the remarkable pace which the race between AAV and the host has picked up in recent years, there is every reason to hope and believe that the fairy tale of AAV gene therapies in the absence of adverse immune responses will eventually become a reality.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
KR was supported by a Marie Curie International Incoming Fellowship PIIF-GA-2013-627329. DG greatly appreciates support by the German Research Foundation (DFG) through the DFG Collaborative Research Centers SFB1129 (Projektnummer 240245660) and TRR179 (Projektnummer 272983813), as well as by the German Center for Infection Research (DZIF, BMBF; TTU-HIV 04.819). DG is moreover grateful for support from the ANR/DFG-funded project MATRIXNASH and the DLR-funded project KARTLE.
DG is a co-founder and shareholder (CSO) of AaviGen GmbH. DG and KR are inventors on a pending patent application related to the generation of immune-evading AAV capsid variants.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Li C, Samulski RJ. Engineering Adeno-Associated Virus Vectors for Gene Therapy. Nat Rev Genet (2020) 21:255–72. doi: 10.1038/s41576-019-0205-4
2. Wang D, Tai PWL, Gao G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat Rev Drug Discov (2019) 18:358–78. doi: 10.1038/s41573-019-0012-9
3. Ronzitti G, Gross DA, Mingozzi F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front Immunol (2020) 11:670. doi: 10.3389/fimmu.2020.00670
4. Blacklow NR, Hoggan MD, Kapikian AZ, Austin JB, Rowe WP. Epidemiology of Adenovirus-Associated Virus Infection in a Nursery Population. Am J Epidemiol (1968) 88:368–78. doi: 10.1093/oxfordjournals.aje.a120897
5. Carter BJ. Adeno-Associated Virus and the Development of Adeno-Associated Virus Vectors: A Historical Perspective. Mol Ther (2004) 10:981–9. doi: 10.1016/j.ymthe.2004.09.011
6. Hastie E, Samulski RJ. Adeno-Associated Virus at 50: A Golden Anniversary of Discovery, Research, and Gene Therapy Success–A Personal Perspective. Hum Gene Ther (2015) 26:257–65. doi: 10.1089/hum.2015.025
7. Balakrishnan B, Jayandharan GR. Basic Biology of Adeno-Associated Virus (AAV) Vectors Used in Gene Therapy. Curr Gene Ther (2014) 14:86–100. doi: 10.2174/1566523214666140302193709
8. Sonntag F, Kother K, Schmidt K, Weghofer M, Raupp C, Nieto K, et al. The Assembly-Activating Protein Promotes Capsid Assembly of Different Adeno-Associated Virus Serotypes. J Virol (2011) 85:12686–97. doi: 10.1128/JVI.05359-11
9. Ogden PJ, Kelsic ED, Sinai S, Church GM. Comprehensive AAV Capsid Fitness Landscape Reveals a Viral Gene and Enables Machine-Guided Design. Science (2019) 366:1139–43. doi: 10.1126/science.aaw2900
10. Denard J, Beley C, Kotin R, Lai-Kuen R, Blot S, Leh H, et al. Human Galectin 3 Binding Protein Interacts With Recombinant Adeno-Associated Virus Type 6. J Virol (2012) 86:6620–31. doi: 10.1128/JVI.00297-12
11. Denard J, Marolleau B, Jenny C, Rao TN, Fehling HJ, Voit T, et al. C-Reactive Protein (CRP) Is Essential for Efficient Systemic Transduction of Recombinant Adeno-Associated Virus Vector 1 (rAAV-1) and rAAV-6 in Mice. J Virol (2013) 87:10784–91. doi: 10.1128/JVI.01813-13
12. Denard J, Rouillon J, Leger T, Garcia C, Lambert MP, Griffith G, et al. AAV-8 and AAV-9 Vectors Cooperate With Serum Proteins Differently Than AAV-1 and AAV-6. Mol Ther Methods Clin Dev (2018) 10:291–302. doi: 10.1016/j.omtm.2018.08.001
13. Agbandje-McKenna M, Kleinschmidt J. AAV Capsid Structure and Cell Interactions. Methods Mol Biol (2011) 807:47–92. doi: 10.1007/978-1-61779-370-7_3
14. Lochrie MA, Tatsuno GP, Arbetman AE, Jones K, Pater C, Smith PH, et al. Adeno-Associated Virus (AAV) Capsid Genes Isolated From Rat and Mouse Liver Genomic DNA Define Two New AAV Species Distantly Related to AAV-5. Virology (2006) 353:68–82. doi: 10.1016/j.virol.2006.05.023
15. Schmidt M, Grot E, Cervenka P, Wainer S, Buck C, Chiorini JA. Identification and Characterization of Novel Adeno-Associated Virus Isolates in ATCC Virus Stocks. J Virol (2006) 80:5082–5. doi: 10.1128/JVI.80.10.5082-5085.2006
16. Schmidt M, Voutetakis A, Afione S, Zheng C, Mandikian D, Chiorini JA. Adeno-Associated Virus Type 12 (AAV12): A Novel AAV Serotype With Sialic Acid- and Heparan Sulfate Proteoglycan-Independent Transduction Activity. J Virol (2008) 82:1399–406. doi: 10.1128/JVI.02012-07
17. Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, et al. Clades of Adeno-Associated Viruses Are Widely Disseminated in Human Tissues. J Virol (2004) 78:6381–8. doi: 10.1128/JVI.78.12.6381-6388.2004
18. Cabanes-Creus M, Hallwirth CV, Westhaus A, Ng BH, Liao SHY, Zhu E, et al. Restoring the Natural Tropism of AAV2 Vectors for Human Liver. Sci Transl Med (2020) 12:eaba3312. doi: 10.1126/scitranslmed.aba3312
19. Zengel J, Carette JE. Structural and Cellular Biology of Adeno-Associated Virus Attachment and Entry. Adv Virus Res (2020) 106:39–84. doi: 10.1016/bs.aivir.2020.01.002
20. Dudek AM, Pillay S, Puschnik AS, Nagamine CM, Cheng F, Qiu J, et al. An Alternate Route for Adeno-Associated Virus (AAV) Entry Independent of AAV Receptor. J Virol (2018) 92:e02213–17. doi: 10.1128/JVI.02213-17
21. Dudek AM, Zabaleta N, Zinn E, Pillay S, Zengel J, Porter C, et al. GPR108 Is a Highly Conserved AAV Entry Factor. Mol Ther (2020) 28:367–81. doi: 10.1016/j.ymthe.2019.11.005
22. Meisen WH, Nejad ZB, Hardy M, Zhao H, Oliverio O, Wang S, et al. Pooled Screens Identify GPR108 and TM9SF2 as Host Cell Factors Critical for AAV Transduction. Mol Ther Methods Clin Dev (2020) 17:601–11. doi: 10.1016/j.omtm.2020.03.012
23. Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, et al. An Essential Receptor for Adeno-Associated Virus Infection. Nature (2016) 530:108–12. doi: 10.1038/nature16465
24. Nonnenmacher M, Weber T. Intracellular Transport of Recombinant Adeno-Associated Virus Vectors. Gene Ther (2012) 19:649–58. doi: 10.1038/gt.2012.6
25. Colella P, Ronzitti G, Mingozzi F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol Ther Methods Clin Dev (2018) 8:87–104. doi: 10.1016/j.omtm.2017.11.007
26. Buchlis G, Podsakoff GM, Radu A, Hawk SM, Flake AW, Mingozzi F, et al. Factor IX Expression in Skeletal Muscle of a Severe Hemophilia B Patient 10 Years After AAV-Mediated Gene Transfer. Blood (2012) 119:3038–41. doi: 10.1182/blood-2011-09-382317
27. Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med (2014) 371:1994–2004. doi: 10.1056/NEJMoa1407309
28. Niemeyer GP, Herzog RW, Mount J, Arruda VR, Tillson DM, Hathcock J, et al. Long-Term Correction of Inhibitor-Prone Hemophilia B Dogs Treated With Liver-Directed AAV2-Mediated Factor IX Gene Therapy. Blood (2009) 113:797–806. doi: 10.1182/blood-2008-10-181479
29. Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, et al. Multiyear Follow-Up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia a. N Engl J Med (2020) 382:29–40. doi: 10.1056/NEJMoa1908490
30. Verdera HC, Kuranda K, Mingozzi F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol Ther (2020) 28:723–46. doi: 10.1016/j.ymthe.2019.12.010
31. Keeler AM, Flotte TR. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu Rev Virol (2019) 6:601–21. doi: 10.1146/annurev-virology-092818-015530
32. Crowe K. The Million-Dollar Drug. (2018). Available at: https://newsinteractives.cbc.ca/longform/glybera (Mar. 2021).
33. Gottlieb S. Statement From FDA Commissioner Scott Gottlieb, M.D. and Peter Marks M.D., Ph.D., Director of the Center for Biologics Evaluation and Research on New Policies to Advance Development of Safe and Effective Cell and Gene Therapies, Commissioner of Food and Drugs - Food and Drug Administration Scott Gottlieb M.D (2019). Available at: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics.
34. Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, et al. AAV-Mediated Factor IX Gene Transfer to Skeletal Muscle in Patients With Severe Hemophilia B. Blood (2003) 101:2963–72. doi: 10.1182/blood-2002-10-3296
35. Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful Transduction of Liver in Hemophilia by AAV-Factor IX and Limitations Imposed by the Host Immune Response. Nat Med (2006) 12:342–7. doi: 10.1038/nm1358
36. Herzog RW. Immune Responses to AAV Capsid: Are Mice Not Humans After All? Mol Ther (2007) 15:649–50. doi: 10.1038/sj.mt.6300123
37. Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of Immune Tolerance to Coagulation Factor IX Antigen by In Vivo Hepatic Gene Transfer. J Clin Invest (2003) 111:1347–56. doi: 10.1172/JCI200316887
38. Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-Cell Responses to Adeno-Associated Virus Capsid in Humans. Nat Med (2007) 13:419–22. doi: 10.1038/nm1549
39. Pien GC, Basner-Tschakarjan E, Hui DJ, Mentlik AN, Finn JD, Hasbrouck NC, et al. Capsid Antigen Presentation Flags Human Hepatocytes for Destruction After Transduction by Adeno-Associated Viral Vectors. J Clin Invest (2009) 119:1688–95. doi: 10.1172/JCI36891
40. Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, et al. Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease Trial, Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID Trial), a First-in-Human Phase 1/2 Clinical Trial. J Card Fail (2009) 15:171–81. doi: 10.1016/j.cardfail.2009.01.013
41. Vandamme C, Xicluna R, Hesnard L, Devaux M, Jaulin N, Guilbaud M, et al. Tetramer-Based Enrichment of Preexisting Anti-AAV8 CD8(+) T Cells in Human Donors Allows the Detection of a TEMRA Subpopulation. Front Immunol (2019) 10:3110. doi: 10.3389/fimmu.2019.03110
42. Martino AT, Markusic DM. Immune Response Mechanisms Against AAV Vectors in Animal Models. Mol Ther Methods Clin Dev (2020) 17:198–208. doi: 10.1016/j.omtm.2019.12.008
43. Vandamme C, Adjali O, Mingozzi F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum Gene Ther (2017) 28:1061–74. doi: 10.1089/hum.2017.150
44. Abbas AK, Lichtman AH, Pillai S. Basic Immunology: Functions and Disorders of the Immune System. Sixth edition ed. Elsevier, Philadelphia (2019). p. vii.
46. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
47. Shirley JL, de Jong YP, Terhorst C, Herzog RW. Immune Responses to Viral Gene Therapy Vectors. Mol Ther (2020) 28:709–22. doi: 10.1016/j.ymthe.2020.01.001
48. Shirley JL, Keeler GD, Sherman A, Zolotukhin I, Markusic DM, Hoffman BE, et al. Type I IFN Sensing by cDCs and CD4(+) T Cell Help Are Both Requisite for Cross-Priming of AAV Capsid-Specific CD8(+) T Cells. Mol Ther (2020) 28:758–70. doi: 10.1016/j.ymthe.2019.11.011
49. Sun J, Shao W, Chen X, Merricks EP, Wimsey L, Abajas YL, et al. An Observational Study From Long-Term AAV Re-Administration in Two Hemophilia Dogs. Mol Ther Methods Clin Dev (2018) 10:257–67. doi: 10.1016/j.omtm.2018.07.011
50. Suzuki M, Bertin TK, Rogers GL, Cela RG, Zolotukhin I, Palmer DJ, et al. Differential Type I Interferon-Dependent Transgene Silencing of Helper-Dependent Adenoviral vs. adeno-associated viral vectors in vivo. Mol Ther (2013) 21:796–805. doi: 10.1038/mt.2012.277
51. Rogers GL, Suzuki M, Zolotukhin I, Markusic DM, Morel LM, Lee B, et al. Unique Roles of TLR9- and MyD88-Dependent and -Independent Pathways in Adaptive Immune Responses to AAV-Mediated Gene Transfer. J Innate Immun (2015) 7:302–14. doi: 10.1159/000369273
52. Zaiss AK, Liu Q, Bowen GP, Wong NC, Bartlett JS, Muruve DA. Differential Activation of Innate Immune Responses by Adenovirus and Adeno-Associated Virus Vectors. J Virol (2002) 76:4580–90. doi: 10.1128/JVI.76.9.4580-4590.2002
53. Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, et al. APOBEC3A is a Potent Inhibitor of Adeno-Associated Virus and Retrotransposons. Curr Biol (2006) 16:480–5. doi: 10.1016/j.cub.2006.01.031
54. Stavrou S, Ross SR. APOBEC3 Proteins in Viral Immunity. J Immunol (2015) 195:4565–70. doi: 10.4049/jimmunol.1501504
55. Hosel M, Broxtermann M, Janicki H, Esser K, Arzberger S, Hartmann P, et al. Toll-Like Receptor 2-Mediated Innate Immune Response in Human Nonparenchymal Liver Cells Toward Adeno-Associated Viral Vectors. Hepatology (2012) 55:287–97. doi: 10.1002/hep.24625
56. Wright JF. Codon Modification and PAMPs in Clinical AAV Vectors: The Tortoise or the Hare? Mol Ther (2020) 28:701–3. doi: 10.1016/j.ymthe.2020.01.026
57. Wright JF. Quantification of CpG Motifs in rAAV Genomes: Avoiding the Toll. Mol Ther (2020) 28:1756–8. doi: 10.1016/j.ymthe.2020.07.006
58. Martino AT, Suzuki M, Markusic DM, Zolotukhin I, Ryals RC, Moghimi B, et al. The Genome of Self-Complementary Adeno-Associated Viral Vectors Increases Toll-Like Receptor 9-Dependent Innate Immune Responses in the Liver. Blood (2011) 117:6459–68. doi: 10.1182/blood-2010-10-314518
59. Butterfield JSS, Biswas M, Shirley JL, Kumar SRP, Sherman A, Terhorst C, et al. TLR9-Activating CpG-B ODN But Not TLR7 Agonists Triggers Antibody Formation to Factor IX in Muscle Gene Transfer. Hum Gene Ther Methods (2019) 30:81–92. doi: 10.1089/hgtb.2019.013
60. Rogers GL, Shirley JL, Zolotukhin I, Kumar SRP, Sherman A, Perrin GQ, et al. Plasmacytoid and Conventional Dendritic Cells Cooperate in Crosspriming AAV Capsid-Specific CD8(+) T Cells. Blood (2017) 129:3184–95. doi: 10.1182/blood-2016-11-751040
61. Zhu J, Huang X, Yang Y. The TLR9-MyD88 Pathway is Critical for Adaptive Immune Responses to Adeno-Associated Virus Gene Therapy Vectors in Mice. J Clin Invest (2009) 119:2388–98. doi: 10.1172/JCI37607
62. Herzog RW, Cooper M, Perrin GQ, Biswas M, Martino AT, Morel L, et al. Regulatory T Cells and TLR9 Activation Shape Antibody Formation to a Secreted Transgene Product in AAV Muscle Gene Transfer. Cell Immunol (2019) 342:103682. doi: 10.1016/j.cellimm.2017.07.012
63. Xiang Z, Kurupati RK, Li Y, Kuranda K, Zhou X, Mingozzi F, et al. The Effect of CpG Sequences on Capsid-Specific CD8(+) T Cell Responses to AAV Vector Gene Transfer. Mol Ther (2020) 28:771–83. doi: 10.1016/j.ymthe.2019.11.014
64. Shao W, Earley LF, Chai Z, Chen X, Sun J, He T, et al. Double-Stranded RNA Innate Immune Response Activation From Long-Term Adeno-Associated Virus Vector Transduction. JCI Insight (2018) 3:e120474. doi: 10.1172/jci.insight.120474
65. Sudres M, Cire S, Vasseur V, Brault L, Da Rocha S, Boisgerault F, et al. MyD88 Signaling in B Cells Regulates the Production of Th1-Dependent Antibodies to AAV. Mol Ther (2012) 20:1571–81. doi: 10.1038/mt.2012.101
66. Kuranda K, Jean-Alphonse P, Leborgne C, Hardet R, Collaud F, Marmier S, et al. Exposure to Wild-Type AAV Drives Distinct Capsid Immunity Profiles in Humans. J Clin Invest (2018) 128:5267–79. doi: 10.1172/JCI122372
67. Faust SM, Bell P, Cutler BJ, Ashley SN, Zhu Y, Rabinowitz JE, et al. CpG-Depleted Adeno-Associated Virus Vectors Evade Immune Detection. J Clin Invest (2013) 123:2994–3001. doi: 10.1172/JCI68205
68. Ashley SN, Somanathan S, Giles AR, Wilson JM. TLR9 Signaling Mediates Adaptive Immunity Following Systemic AAV Gene Therapy. Cell Immunol (2019) 346:103997. doi: 10.1016/j.cellimm.2019.103997
69. Zaiss AK, Cotter MJ, White LR, Clark SA, Wong NC, Holers VM, et al. Complement Is an Essential Component of the Immune Response to Adeno-Associated Virus Vectors. J Virol (2008) 82:2727–40. doi: 10.1128/JVI.01990-07
70. Muhuri M, Maeda Y, Ma H, Ram S, Fitzgerald KA, Tai PW, et al. Overcoming Innate Immune Barriers That Impede AAV Gene Therapy Vectors. J Clin Invest (2021) 131:e143780. doi: 10.1172/JCI143780
71. Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Pogoda JM, et al. Prevalence of AAV1 Neutralizing Antibodies and Consequences for a Clinical Trial of Gene Transfer for Advanced Heart Failure. Gene Ther (2016) 23:313–9. doi: 10.1038/gt.2015.109
72. Ferreira V, Petry H, Salmon F. Immune Responses to AAV-Vectors, the Glybera Example From Bench to Bedside. Front Immunol (2014) 5:82. doi: 10.3389/fimmu.2014.00082
73. Greig JA, Calcedo R, Grant RL, Peng H, Medina-Jaszek CA, Ahonkhai O, et al. Intramuscular Administration of AAV Overcomes Pre-Existing Neutralizing Antibodies in Rhesus Macaques. Vaccine (2016) 34:6323–9. doi: 10.1016/j.vaccine.2016.10.053
74. Mimuro J, Mizukami H, Hishikawa S, Ikemoto T, Ishiwata A, Sakata A, et al. Minimizing the Inhibitory Effect of Neutralizing Antibody for Efficient Gene Expression in the Liver With Adeno-Associated Virus 8 Vectors. Mol Ther (2013) 21:318–23. doi: 10.1038/mt.2012.258
75. Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Jude Samulski R. Global CNS Gene Delivery and Evasion of Anti-AAV-Neutralizing Antibodies by Intrathecal AAV Administration in non-Human Primates. Gene Ther (2013) 20:450–9. doi: 10.1038/gt.2012.101
76. Bucher K, Rodriguez-Bocanegra E, Dauletbekov D, Fischer MD. Immune Responses to Retinal Gene Therapy Using Adeno-Associated Viral Vectors - Implications for Treatment Success and Safety. Prog Retin Eye Res (2021) 83:100915. doi: 10.1016/j.preteyeres.2020.100915
77. Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN, et al. Plasmapheresis Eliminates the Negative Impact of AAV Antibodies on Microdystrophin Gene Expression Following Vascular Delivery. Mol Ther (2014) 22:338–47. doi: 10.1038/mt.2013.244
78. Orlowski A, Katz MG, Gubara SM, Fargnoli AS, Fish KM, Weber T. Successful Transduction With AAV Vectors After Selective Depletion of Anti-AAV Antibodies by Immunoadsorption. Mol Ther Methods Clin Dev (2020) 16:192–203. doi: 10.1016/j.omtm.2020.01.004
79. Bertin B, Veron P, Leborgne C, Deschamps JY, Moullec S, Fromes Y, et al. Capsid-Specific Removal of Circulating Antibodies to Adeno-Associated Virus Vectors. Sci Rep (2020) 10:864. doi: 10.1038/s41598-020-57893-z
80. Li Y, Li J, Liu Y, Shi Z, Liu H, Wei Y, et al. Bat Adeno-Associated Viruses as Gene Therapy Vectors With the Potential to Evade Human Neutralizing Antibodies. Gene Ther (2019) 26:264–76. doi: 10.1038/s41434-019-0081-8
81. Tse LV, Klinc KA, Madigan VJ, Castellanos Rivera RM, Wells LF, Havlik LP, et al. Structure-Guided Evolution of Antigenically Distinct Adeno-Associated Virus Variants for Immune Evasion. Proc Natl Acad Sci U S A (2017) 114:E4812–21. doi: 10.1073/pnas.1704766114
82. Paulk NK, Pekrun K, Zhu E, Nygaard S, Li B, Xu J, et al. Bioengineered AAV Capsids With Combined High Human Liver Transduction In Vivo and Unique Humoral Seroreactivity. Mol Ther (2018) 26:289–303. doi: 10.1016/j.ymthe.2017.09.021
83. Gyorgy B, Fitzpatrick Z, Crommentuijn MH, Mu D, Maguire CA. Naturally Enveloped AAV Vectors for Shielding Neutralizing Antibodies and Robust Gene Delivery In Vivo. Biomaterials (2014) 35:7598–609. doi: 10.1016/j.biomaterials.2014.05.032
84. Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin Immunity in Duchenne's Muscular Dystrophy. N Engl J Med (2010) 363:1429–37. doi: 10.1056/NEJMoa1000228
85. Samelson-Jones BJ, Arruda VR. Translational Potential of Immune Tolerance Induction by AAV Liver-Directed Factor VIII Gene Therapy for Hemophilia a. Front Immunol (2020) 11:618. doi: 10.3389/fimmu.2020.00618
86. Chan YK, Wang SK, Chu CJ, Copland DA, Letizia AJ, Costa Verdera H, et al. Engineering Adeno-Associated Viral Vectors to Evade Innate Immune and Inflammatory Responses. Sci Transl Med (2021) 13:eabd3438. doi: 10.1126/scitranslmed.abd3438
87. Mays LE, Vandenberghe LH, Xiao R, Bell P, Nam HJ, Agbandje-McKenna M, et al. Adeno-Associated Virus Capsid Structure Drives CD4-Dependent CD8+ T Cell Response to Vector Encoded Proteins. J Immunol (2009) 182:6051–60. doi: 10.4049/jimmunol.0803965
88. Poupiot J, Costa Verdera H, Hardet R, Colella P, Collaud F, Bartolo L, et al. Role of Regulatory T Cell and Effector T Cell Exhaustion in Liver-Mediated Transgene Tolerance in Muscle. Mol Ther Methods Clin Dev (2019) 15:83–100. doi: 10.1016/j.omtm.2019.08.012
89. Meliani A, Boisgerault F, Hardet R, Marmier S, Collaud F, Ronzitti G, et al. Antigen-Selective Modulation of AAV Immunogenicity With Tolerogenic Rapamycin Nanoparticles Enables Successful Vector Re-Administration. Nat Commun (2018) 9:4098. doi: 10.1038/s41467-018-06621-3
90. Corti M, Elder M, Falk D, Lawson L, Smith B, Nayak S, et al. B-Cell Depletion Is Protective Against Anti-AAV Capsid Immune Response: A Human Subject Case Study. Mol Ther Methods Clin Dev (2014) 1:14033. doi: 10.1038/mtm.2014.33
91. Robinson TM, Chen MY, Lam MT, Ykema MR, Suh J. Display of Self-Peptide on Adeno-Associated Virus Capsid Decreases Phagocytic Uptake In Vitro. ACS Synth Biol (2020) 9:2246–51. doi: 10.1021/acssynbio.0c00203
92. Cao O, Hoffman BE, Moghimi B, Nayak S, Cooper M, Zhou S, et al. Impact of the Underlying Mutation and the Route of Vector Administration on Immune Responses to Factor IX in Gene Therapy for Hemophilia B. Mol Ther (2009) 17:1733–42. doi: 10.1038/mt.2009.159
93. Li C, Goudy K, Hirsch M, Asokan A, Fan Y, Alexander J, et al. Cellular Immune Response to Cryptic Epitopes During Therapeutic Gene Transfer. Proc Natl Acad Sci U S A (2009) 106:10770–4. doi: 10.1073/pnas.0902269106
94. Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, et al. Cross-Reactive Immunologic Material Status Affects Treatment Outcomes in Pompe Disease Infants. Mol Genet Metab (2010) 99:26–33. doi: 10.1016/j.ymgme.2009.08.003
95. Herzog RW. Complexity of Immune Responses to AAV Transgene Products - Example of Factor IX. Cell Immunol (2019) 342:103658. doi: 10.1016/j.cellimm.2017.05.006
96. Perrin GQ, Zolotukhin I, Sherman A, Biswas M, de Jong YP, Terhorst C, et al. Dynamics of Antigen Presentation to Transgene Product-Specific CD4(+) T Cells and of Treg Induction Upon Hepatic AAV Gene Transfer. Mol Ther Methods Clin Dev (2016) 3:16083. doi: 10.1038/mtm.2016.83
97. Herzog RW, Kuteyeva V, Saboungi R, Terhorst C, Biswas M. Reprogrammed CD4(+) T Cells That Express FoxP3(+) Control Inhibitory Antibody Formation in Hemophilia A Mice. Front Immunol (2019) 10:274. doi: 10.3389/fimmu.2019.00274
98. Samelson-Jones BJ, Finn JD, Favaro P, Wright JF, Arruda VR. Timing of Intensive Immunosuppression Impacts Risk of Transgene Antibodies After AAV Gene Therapy in Nonhuman Primates. Mol Ther Methods Clin Dev (2020) 17:1129–38. doi: 10.1016/j.omtm.2020.05.001
99. Muhuri M, Zhan W, Maeda Y, Li J, Lotun A, Chen J, et al. Novel Combinatorial MicroRNA-Binding Sites in AAV Vectors Synergistically Diminish Antigen Presentation and Transgene Immunity for Efficient and Stable Transduction. Front Immunol (2021) 12:674242. doi: 10.3389/fimmu.2021.674242
100. Xiao Y, Muhuri M, Li S, Qin W, Xu G, Luo L, et al. Circumventing Cellular Immunity by Mir142-Mediated Regulation Sufficiently Supports rAAV-Delivered OVA Expression Without Activating Humoral Immunity. JCI Insight (2019) 5:e99052. doi: 10.1172/jci.insight.99052
101. Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-Associated Virus Vector-Mediated Gene Transfer in Hemophilia B. N Engl J Med (2011) 365:2357–65. doi: 10.1056/NEJMoa1108046
102. Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, Rouhani F, et al. Phase 2 Clinical Trial of a Recombinant Adeno-Associated Viral Vector Expressing Alpha1-Antitrypsin: Interim Results. Hum Gene Ther (2011) 22:1239–47. doi: 10.1089/hum.2011.053
103. Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT, et al. Sustained Transgene Expression Despite T Lymphocyte Responses in a Clinical Trial of Raav1-AAT Gene Therapy. Proc Natl Acad Sci USA (2009) 106:16363–8. doi: 10.1073/pnas.0904514106
104. Doerfler PA, Todd AG, Clement N, Falk DJ, Nayak S, Herzog RW, et al. Copackaged AAV9 Vectors Promote Simultaneous Immune Tolerance and Phenotypic Correction of Pompe Disease. Hum Gene Ther (2016) 27:43–59. doi: 10.1089/hum.2015.103
105. Hui DJ, Basner-Tschakarjan E, Chen Y, Davidson RJ, Buchlis G, Yazicioglu M, et al. Modulation of CD8+ T Cell Responses to AAV Vectors With IgG-Derived MHC Class II Epitopes. Mol Ther (2013) 21:1727–37. doi: 10.1038/mt.2013.166
106. Mays LE, Wang L, Lin J, Bell P, Crawford A, Wherry EJ, et al. AAV8 Induces Tolerance in Murine Muscle as a Result of Poor APC Transduction, T Cell Exhaustion, and Minimal MHCI Upregulation on Target Cells. Mol Ther (2014) 22:28–41. doi: 10.1038/mt.2013.134
107. Lu Y, Song S. Distinct Immune Responses to Transgene Products From Raav1 and Raav8 Vectors. Proc Natl Acad Sci USA (2009) 106:17158–62. doi: 10.1073/pnas.0909520106
108. Rossi A, Dupaty L, Aillot L, Zhang L, Gallien C, Hallek M, et al. Vector Uncoating Limits Adeno-Associated Viral Vector-Mediated Transduction of Human Dendritic Cells and Vector Immunogenicity. Sci Rep (2019) 9:3631. doi: 10.1038/s41598-019-40071-1
109. Greig JA, Calcedo R, Kuri-Cervantes L, Nordin JML, Albrecht J, Bote E, et al. AAV8 Gene Therapy for Crigler-Najjar Syndrome in Macaques Elicited Transgene T Cell Responses That Are Resident to the Liver. Mol Ther Methods Clin Dev (2018) 11:191–201. doi: 10.1016/j.omtm.2018.10.012
110. Kumar SRP, Hoffman BE, Terhorst C, de Jong YP, Herzog RW. The Balance Between CD8(+) T Cell-Mediated Clearance of AAV-Encoded Antigen in the Liver and Tolerance Is Dependent on the Vector Dose. Mol Ther (2017) 25:880–91. doi: 10.1016/j.ymthe.2017.02.014
111. Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, Edmonson SA, Hui DJ, Sabatino DE, et al. Modulation of Tolerance to the Transgene Product in a Nonhuman Primate Model of AAV-Mediated Gene Transfer to Liver. Blood (2007) 110:2334–41. doi: 10.1182/blood-2007-03-080093
112. Shao W, Chen X, Samulski RJ, Hirsch ML, Li C. Inhibition of Antigen Presentation During AAV Gene Therapy Using Virus Peptides. Hum Mol Genet (2018) 27:601–13. doi: 10.1093/hmg/ddx427
113. Ertl HCJ. Preclinical Models to Assess the Immunogenicity of AAV Vectors. Cell Immunol (2019) 342:103722. doi: 10.1016/j.cellimm.2017.11.006
114. Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum Gene Ther (2010) 21:704–12. doi: 10.1089/hum.2009.182
115. Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide Epidemiology of Neutralizing Antibodies to Adeno-Associated Viruses. J Infect Dis (2009) 199:381–90. doi: 10.1086/595830
116. Calcedo R, Franco J, Qin Q, Richardson DW, Mason JB, Boyd S, et al. Preexisting Neutralizing Antibodies to Adeno-Associated Virus Capsids in Large Animals Other Than Monkeys May Confound In Vivo Gene Therapy Studies. Hum Gene Ther Methods (2015) 26:103–5. doi: 10.1089/hgtb.2015.082
117. Wang L, Calcedo R, Bell P, Lin J, Grant RL, Siegel DL, et al. Impact of Pre-Existing Immunity on Gene Transfer to Nonhuman Primate Liver With Adeno-Associated Virus 8 Vectors. Hum Gene Ther (2011) 22:1389–401. doi: 10.1089/hum.2011.031
118. Rapti K, Louis-Jeune V, Kohlbrenner E, Ishikawa K, Ladage D, Zolotukhin S, et al. Neutralizing Antibodies Against AAV Serotypes 1, 2, 6, and 9 in Sera of Commonly Used Animal Models. Mol Ther (2012) 20:73–83. doi: 10.1038/mt.2011.177
119. Chirmule N, Propert K, Magosin S, Qian Y, Qian R, Wilson J. Immune Responses to Adenovirus and Adeno-Associated Virus in Humans. Gene Ther (1999) 6:1574–83. doi: 10.1038/sj.gt.3300994
120. Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T. Pre-Existing Anti-Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum Gene Ther Methods (2013) 24:59–67. doi: 10.1089/hgtb.2012.243
121. Kruzik A, Fetahagic D, Hartlieb B, Dorn S, Koppensteiner H, Horling FM, et al. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol Ther Methods Clin Dev (2019) 14:126–33. doi: 10.1016/j.omtm.2019.05.014
122. Weber T. Anti-AAV Antibodies in AAV Gene Therapy: Current Challenges and Possible Solutions. Front Immunol (2021) 12:658399. doi: 10.3389/fimmu.2021.658399
123. Gorovits B, Fiscella M, Havert M, Koren E, Long B, Milton M, et al. Recommendations for the Development of Cell-Based Anti-Viral Vector Neutralizing Antibody Assays. AAPS J (2020) 22:24. doi: 10.1208/s12248-019-0403-1
124. Jungmann A, Muller O, Rapti K. Cell-Based Measurement of Neutralizing Antibodies Against Adeno-Associated Virus (AAV). Methods Mol Biol (2017) 1521:109–26. doi: 10.1007/978-1-4939-6588-5_7
125. Basner-Tschakarjan E, Bijjiga E, Martino AT. Pre-Clinical Assessment of Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front Immunol (2014) 5:28. doi: 10.3389/fimmu.2014.00028
126. Fitzpatrick Z, Leborgne C, Barbon E, Masat E, Ronzitti G, van Wittenberghe L, et al. Influence of Pre-Existing Anti-Capsid Neutralizing and Binding Antibodies on AAV Vector Transduction. Mol Ther Methods Clin Dev (2018) 9:119–29. doi: 10.1016/j.omtm.2018.02.003
127. Guo P, Zhang J, Chrzanowski M, Huang J, Chew H, Firrman JA, et al. Rapid AAV-Neutralizing Antibody Determination With a Cell-Binding Assay. Mol Ther Methods Clin Dev (2019) 13:40–6. doi: 10.1016/j.omtm.2018.11.007
128. Falese L, Sandza K, Yates B, Triffault S, Gangar S, Long B, et al. Strategy to Detect Pre-Existing Immunity to AAV Gene Therapy. Gene Ther (2017) 24:768–78. doi: 10.1038/gt.2017.95
129. Kruzik A, Koppensteiner H, Fetahagic D, Hartlieb B, Dorn S, Romeder-Finger S, et al. Detection of Biologically Relevant Low-Titer Neutralizing Antibodies Against Adeno-Associated Virus Require Sensitive In Vitro Assays. Hum Gene Ther Methods (2019) 30:35–43. doi: 10.1089/hgtb.2018.263
130. Meliani A, Leborgne C, Triffault S, Jeanson-Leh L, Veron P, Mingozzi F. Determination of Anti-Adeno-Associated Virus Vector Neutralizing Antibody Titer With an In Vitro Reporter System. Hum Gene Ther Methods (2015) 26:45–53. doi: 10.1089/hgtb.2015.037
131. Wang M, Crosby A, Hastie E, Samulski JJ, McPhee S, Joshua G, et al. Prediction of Adeno-Associated Virus Neutralizing Antibody Activity for Clinical Application. Gene Ther (2015) 22:984–92. doi: 10.1038/gt.2015.69
132. Krotova K, Aslanidi G. Modifiers of Adeno-Associated Virus-Mediated Gene Expression in Implication for Serotype-Universal Neutralizing Antibody Assay. Hum Gene Ther (2020) 31:1124–31. doi: 10.1089/hum.2020.074
133. Hajba L, Guttman A. Recent Advances in the Analysis Full/Empty Capsid Ratio and Genome Integrity of Adeno-Associated Virus (AAV) Gene Delivery Vectors. Curr Mol Med (2020) 20:806–13. doi: 10.2174/1566524020999200730181042
134. Wang D, Zhong L, Li M, Li J, Tran K, Ren L, et al. Adeno-Associated Virus Neutralizing Antibodies in Large Animals and Their Impact on Brain Intraparenchymal Gene Transfer. Mol Ther Methods Clin Dev (2018) 11:65–72. doi: 10.1016/j.omtm.2018.09.003
135. Scallan CD, Jiang H, Liu T, Patarroyo-White S, Sommer JM, Zhou S, et al. Human Immunoglobulin Inhibits Liver Transduction by AAV Vectors at Low AAV2 Neutralizing Titers in SCID Mice. Blood (2006) 107:1810–7. doi: 10.1182/blood-2005-08-3229
136. Meadows AS, Pineda RJ, Goodchild L, Bobo TA, Fu H. Threshold for Pre-Existing Antibody Levels Limiting Transduction Efficiency of Systemic Raav9 Gene Delivery: Relevance for Translation. Mol Ther Methods Clin Dev (2019) 13:453–62. doi: 10.1016/j.omtm.2019.04.004
137. Mingozzi F. AAV Immunogenicity: A Matter of Sensitivity. Mol Ther (2018) 26:2335–6. doi: 10.1016/j.ymthe.2018.09.001
138. Nidetz NF, McGee MC, Tse LV, Li C, Cong L, Li Y, et al. Adeno-Associated Viral Vector-Mediated Immune Responses: Understanding Barriers to Gene Delivery. Pharmacol Ther (2020) 207:107453. doi: 10.1016/j.pharmthera.2019.107453
139. Martino AT, Herzog RW, Anegon I, Adjali O. Measuring Immune Responses to Recombinant AAV Gene Transfer. Methods Mol Biol (2011) 807:259–72. doi: 10.1007/978-1-61779-370-7_11
140. Gaudet D, Methot J, Dery S, Brisson D, Essiembre C, Tremblay G, et al. Efficacy and Long-Term Safety of Alipogene Tiparvovec (AAV1-LPLS447X) Gene Therapy for Lipoprotein Lipase Deficiency: An Open-Label Trial. Gene Ther (2013) 20:361–9. doi: 10.1038/gt.2012.43
141. George LA, Ragni MV, Rasko JEJ, Raffini LJ, Samelson-Jones BJ, Ozelo M, et al. Long-Term Follow-Up of the First in Human Intravascular Delivery of AAV for Gene Transfer: AAV2-Hfix16 for Severe Hemophilia B. Mol Ther (2020) 28:2073–82. doi: 10.1016/j.ymthe.2020.06.001
142. George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, et al. Hemophilia B Gene Therapy With a High-Specific-Activity Factor IX Variant. N Engl J Med (2017) 377:2215–27. doi: 10.1056/NEJMoa1708538
143. Gernoux G, Wilson JM, Mueller C. Regulatory and Exhausted T Cell Responses to AAV Capsid. Hum Gene Ther (2017) 28:338–49. doi: 10.1089/hum.2017.022
144. Mingozzi F, High KA. Immune Responses to AAV in Clinical Trials. Curr Gene Ther (2007) 7:316–24. doi: 10.2174/156652307782151425
145. Mueller C, Chulay JD, Trapnell BC, Humphries M, Carey B, Sandhaus RA, et al. Human Treg Responses Allow Sustained Recombinant Adeno-Associated Virus-Mediated Transgene Expression. J Clin Invest (2013) 123:5310–8. doi: 10.1172/JCI70314
146. Finn JD, Hui D, Downey HD, Dunn D, Pien GC, Mingozzi F, et al. Proteasome Inhibitors Decrease AAV2 Capsid Derived Peptide Epitope Presentation on MHC Class I Following Transduction. Mol Ther (2010) 18:135–42. doi: 10.1038/mt.2009.257
147. Hui DJ, Edmonson SC, Podsakoff GM, Pien GC, Ivanciu L, Camire RM, et al. AAV Capsid CD8+ T-Cell Epitopes Are Highly Conserved Across AAV Serotypes. Mol Ther Methods Clin Dev (2015) 2:15029. doi: 10.1038/mtm.2015.29
148. Li C, He Y, Nicolson S, Hirsch M, Weinberg MS, Zhang P, et al. Adeno-Associated Virus Capsid Antigen Presentation Is Dependent on Endosomal Escape. J Clin Invest (2013) 123:1390–401. doi: 10.1172/JCI66611
149. Martino AT, Basner-Tschakarjan E, Markusic DM, Finn JD, Hinderer C, Zhou S, et al. Engineered AAV Vector Minimizes In Vivo Targeting of Transduced Hepatocytes by Capsid-Specific CD8+ T Cells. Blood (2013) 121:2224–33. doi: 10.1182/blood-2012-10-460733
150. Li C, Hirsch M, DiPrimio N, Asokan A, Goudy K, Tisch R, et al. Cytotoxic-T-Lymphocyte-Mediated Elimination of Target Cells Transduced With Engineered Adeno-Associated Virus Type 2 Vector In Vivo. J Virol (2009) 83:6817–24. doi: 10.1128/JVI.00278-09
151. Pei X, Earley LF, He Y, Chen X, Hall NE, Samulski RJ, et al. Efficient Capsid Antigen Presentation From Adeno-Associated Virus Empty Virions In Vivo. Front Immunol (2018) 9:844. doi: 10.3389/fimmu.2018.00844
152. Li H, Lasaro MO, Jia B, Lin SW, Haut LH, High KA, et al. Capsid-Specific T-Cell Responses to Natural Infections With Adeno-Associated Viruses in Humans Differ From Those of Nonhuman Primates. Mol Ther (2011) 19:2021–30. doi: 10.1038/mt.2011.81
153. Mays LE, Wang L, Tenney R, Bell P, Nam HJ, Lin J, et al. Mapping the Structural Determinants Responsible for Enhanced T Cell Activation to the Immunogenic Adeno-Associated Virus Capsid From Isolate Rhesus 32.33. J Virol (2013) 87:9473–85. doi: 10.1128/JVI.00596-13
154. Vandenberghe LH, Wang L, Somanathan S, Zhi Y, Figueredo J, Calcedo R, et al. Heparin Binding Directs Activation of T Cells Against Adeno-Associated Virus Serotype 2 Capsid. Nat Med (2006) 12:967–71. doi: 10.1038/nm1445
155. Zhu F, Chen T, Zhang Y, Sun H, Cao H, Lu J, et al. A Novel Adeno-Associated Virus-Based Genetic Vaccine Encoding the Hepatitis C Virus NS3/4 Protein Exhibits Immunogenic Properties in Mice Superior to Those of an NS3-Protein-Based Vaccine. PloS One (2015) 10:e0142349. doi: 10.1371/journal.pone.0142349
156. Zabaleta N, Dai W, Bhatt U, Herate C, Maisonnasse P, Chichester JA, et al. An AAV-Based, Room-Temperature-Stable, Single-Dose COVID-19 Vaccine Provides Durable Immunogenicity and Protection in non-Human Primates. Cell Host Microbe (2021) 29:1437–53.e8. doi: 10.1016/j.chom.2021.08.002
157. Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of Siglec Expression on T Lymphocytes During Human Evolution. Proc Natl Acad Sci USA (2006) 103:7765–70. doi: 10.1073/pnas.0510484103
158. Calcedo R, Chichester JA, Wilson JM. Assessment of Humoral, Innate, and T-Cell Immune Responses to Adeno-Associated Virus Vectors. Hum Gene Ther Methods (2018) 29:86–95. doi: 10.1089/hgtb.2018.038
159. He Y, Weinberg MS, Hirsch M, Johnson MC, Tisch R, Samulski RJ, et al. Kinetics of Adeno-Associated Virus Serotype 2 (AAV2) and AAV8 Capsid Antigen Presentation In Vivo Are Identical. Hum Gene Ther (2013) 24:545–53. doi: 10.1089/hum.2013.065
160. Bilic I, Monahan P, Berg V, Scheiflinger F, Reipert B. Whole Exome Sequencing of Patients Treated With Adeno-Associated Virus Serotype 8-Factor IX (AAV8-FIX) Gene Therapy Reveals Potential Determinants of Persistent Transgene Expression. Res Pract Thromb Haemost (2019) 3(Suppl. 1):95.
161. Ferrand M, Galy A, Boisgerault F. A Dystrophic Muscle Broadens the Contribution and Activation of Immune Cells Reacting to rAAV Gene Transfer. Gene Ther (2014) 21:828–39. doi: 10.1038/gt.2014.61
162. Arruda VR, Schuettrumpf J, Herzog RW, Nichols TC, Robinson N, Lotfi Y, et al. Safety and Efficacy of Factor IX Gene Transfer to Skeletal Muscle in Murine and Canine Hemophilia B Models by Adeno-Associated Viral Vector Serotype 1. Blood (2004) 103:85–92. doi: 10.1182/blood-2003-05-1446
163. Herzog RW, Yang EY, Couto LB, Hagstrom JN, Elwell D, Fields PA, et al. Long-Term Correction of Canine Hemophilia B by Gene Transfer of Blood Coagulation Factor IX Mediated by Adeno-Associated Viral Vector. Nat Med (1999) 5:56–63. doi: 10.1038/4743
164. Wu T, Topfer K, Lin SW, Li H, Bian A, Zhou XY, et al. Self-Complementary AAVs Induce More Potent Transgene Product-Specific Immune Responses Compared to a Single-Stranded Genome. Mol Ther (2012) 20:572–9. doi: 10.1038/mt.2011.280
165. Boisgerault F, Mingozzi F. The Skeletal Muscle Environment and Its Role in Immunity and Tolerance to AAV Vector-Mediated Gene Transfer. Curr Gene Ther (2015) 15:381–94. doi: 10.2174/1566523215666150630121750
166. Geisler A, Jungmann A, Kurreck J, Poller W, Katus HA, Vetter R, et al. Microrna122-Regulated Transgene Expression Increases Specificity of Cardiac Gene Transfer Upon Intravenous Delivery of AAV9 Vectors. Gene Ther (2011) 18:199–209. doi: 10.1038/gt.2010.141
167. Qiao C, Yuan Z, Li J, He B, Zheng H, Mayer C, et al. Liver-Specific microRNA-122 Target Sequences Incorporated in AAV Vectors Efficiently Inhibits Transgene Expression in the Liver. Gene Ther (2011) 18:403–10. doi: 10.1038/gt.2010.157
168. Xie J, Xie Q, Zhang H, Ameres SL, Hung JH, Su Q, et al. MicroRNA-Regulated, Systemically Delivered Raav9: A Step Closer to CNS-Restricted Transgene Expression. Mol Ther (2011) 19:526–35. doi: 10.1038/mt.2010.279
169. Brown BD, Cantore A, Annoni A, Sergi LS, Lombardo A, Della Valle P, et al. A microRNA-Regulated Lentiviral Vector Mediates Stable Correction of Hemophilia B Mice. Blood (2007) 110:4144–52. doi: 10.1182/blood-2007-03-078493
170. Dasgupta S, Navarrete AM, Bayry J, Delignat S, Wootla B, Andre S, et al. A Role for Exposed Mannosylations in Presentation of Human Therapeutic Self-Proteins to CD4+ T Lymphocytes. Proc Natl Acad Sci USA (2007) 104:8965–70. doi: 10.1073/pnas.0702120104
171. Puzzo F, Colella P, Biferi MG, Bali D, Paulk NK, Vidal P, et al. Rescue of Pompe Disease in Mice by AAV-Mediated Liver Delivery of Secretable Acid Alpha-Glucosidase. Sci Transl Med (2017) 9:eaam6375. doi: 10.1126/scitranslmed.aam6375
172. Calcedo R, Somanathan S, Qin Q, Betts MR, Rech AJ, Vonderheide RH, et al. Class I-Restricted T-Cell Responses to a Polymorphic Peptide in a Gene Therapy Clinical Trial for Alpha-1-Antitrypsin Deficiency. Proc Natl Acad Sci USA (2017) 114:1655–9. doi: 10.1073/pnas.1617726114
173. Tardieu M, Zerah M, Gougeon ML, Ausseil J, de Bournonville S, Husson B, et al. Intracerebral Gene Therapy in Children With Mucopolysaccharidosis Type IIIB Syndrome: An Uncontrolled Phase 1/2 Clinical Trial. Lancet Neurol (2017) 16:712–20. doi: 10.1016/S1474-4422(17)30169-2
174. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
175. Keeler GD, Markusic DM, Hoffman BE. Liver Induced Transgene Tolerance With AAV Vectors. Cell Immunol (2019) 342:103728. doi: 10.1016/j.cellimm.2017.12.002
176. Curotto de Lafaille MA, Lafaille JJ. Natural and Adaptive Foxp3+ Regulatory T Cells: More of the Same or a Division of Labor? Immunity (2009) 30:626–35. doi: 10.1016/j.immuni.2009.05.002
177. Arce-Sillas A, Alvarez-Luquin DD, Tamaya-Dominguez B, Gomez-Fuentes S, Trejo-Garcia A, Melo-Salas M, et al. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J Immunol Res (2016) 2016:1720827. doi: 10.1155/2016/1720827
178. Dowling MR, Kan A, Heinzel S, Marchingo JM, Hodgkin PD, Hawkins ED. Regulatory T Cells Suppress Effector T Cell Proliferation by Limiting Division Destiny. Front Immunol (2018) 9:2461. doi: 10.3389/fimmu.2018.02461
179. Piccirillo CA, Shevach EM. Cutting Edge: Control of CD8+ T Cell Activation by CD4+CD25+ Immunoregulatory Cells. J Immunol (2001) 167:1137–40. doi: 10.4049/jimmunol.167.3.1137
180. Hoffman BE, Martino AT, Sack BK, Cao O, Liao G, Terhorst C, et al. Nonredundant Roles of IL-10 and TGF-Beta in Suppression of Immune Responses to Hepatic AAV-Factor IX Gene Transfer. Mol Ther (2011) 19:1263–72. doi: 10.1038/mt.2011.33
181. Dobrzynski E, Mingozzi F, Liu YL, Bendo E, Cao O, Wang L, et al. Induction of Antigen-Specific CD4+ T-Cell Anergy and Deletion by In Vivo Viral Gene Transfer. Blood (2004) 104:969–77. doi: 10.1182/blood-2004-03-0847
182. Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic Regulatory T Cells and Kupffer Cells Are Crucial Mediators of Systemic T Cell Tolerance to Antigens Targeting Murine Liver. Hepatology (2009) 50:612–21. doi: 10.1002/hep.23043
183. Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and Role of Regulatory CD4+CD25+ T Cells in Tolerance to the Transgene Product Following Hepatic In Vivo Gene Transfer. Blood (2007) 110:1132–40. doi: 10.1182/blood-2007-02-073304
184. Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, et al. TGF-Beta-Dependent Induction of CD4(+)CD25(+)Foxp3(+) Tregs by Liver Sinusoidal Endothelial Cells. J Hepatol (2014) 61:594–9. doi: 10.1016/j.jhep.2014.04.027
185. Gernoux G, Gruntman AM, Blackwood M, Zieger M, Flotte TR, Mueller C. Muscle-Directed Delivery of an AAV1 Vector Leads to Capsid-Specific T Cell Exhaustion in Nonhuman Primates and Humans. Mol Ther (2020) 28:747–57. doi: 10.1016/j.ymthe.2020.01.004
186. Bartolo L, Li Chung Tong S, Chappert P, Urbain D, Collaud F, Colella P, et al. Dual Muscle-Liver Transduction Imposes Immune Tolerance for Muscle Transgene Engraftment Despite Preexisting Immunity. JCI Insight (2019) 4:e127008. doi: 10.1172/jci.insight.127008
187. Fuchs SP, Martinez-Navio JM, Rakasz EG, Gao G, Desrosiers RC. Liver-Directed But Not Muscle-Directed AAV-Antibody Gene Transfer Limits Humoral Immune Responses in Rhesus Monkeys. Mol Ther Methods Clin Dev (2020) 16:94–102. doi: 10.1016/j.omtm.2019.11.010
188. High KA. The Gene Therapy Journey for Hemophilia: Are We There Yet? Hematol Am Soc Hematol Educ Program (2012) 2012:375–81. doi: 10.1182/asheducation.V2012.1.375.3797885
189. Louveau A, Harris TH, Kipnis J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol (2015) 36:569–77. doi: 10.1016/j.it.2015.08.006
190. Samaranch L, Salegio EA, San Sebastian W, Kells AP, Foust KD, Bringas JR, et al. Adeno-Associated Virus Serotype 9 Transduction in the Central Nervous System of Nonhuman Primates. Hum Gene Ther (2012) 23:382–9. doi: 10.1089/hum.2011.200
191. Sanftner LM, Suzuki BM, Doroudchi MM, Feng L, McClelland A, Forsayeth JR, et al. Striatal Delivery of rAAV-hAADC to Rats With Preexisting Immunity to AAV. Mol Ther (2004) 9:403–9. doi: 10.1016/j.ymthe.2003.12.005
192. Peden CS, Manfredsson FP, Reimsnider SK, Poirier AE, Burger C, Muzyczka N, et al. Striatal Readministration of rAAV Vectors Reveals an Immune Response Against AAV2 Capsids That can be Circumvented. Mol Ther (2009) 17:524–37. doi: 10.1038/mt.2008.284
193. Haurigot V, Marco S, Ribera A, Garcia M, Ruzo A, Villacampa P, et al. Whole Body Correction of Mucopolysaccharidosis IIIA by Intracerebrospinal Fluid Gene Therapy. J Clin Invest (2013) 123:3254–71. doi: 10.1172/JCI66778
194. Janelidze S, Nordstrom U, Kugler S, Brundin P. Pre-Existing Immunity to Adeno-Associated Virus (AAV)2 Limits Transgene Expression Following Intracerebral AAV2-Based Gene Delivery in a 6-Hydroxydopamine Model of Parkinson's Disease. J Gene Med (2014) 16:300–8. doi: 10.1002/jgm.2779
195. Lee S, Kang IK, Kim JH, Jung BK, Park K, Chang H, et al. Relationship Between Neutralizing Antibodies Against Adeno-Associated Virus in the Vitreous and Serum: Effects on Retinal Gene Therapy. Transl Vis Sci Technol (2019) 8:14. doi: 10.1167/tvst.8.2.14
198. Anand V, Duffy B, Yang Z, Dejneka NS, Maguire AM, Bennett J. A Deviant Immune Response to Viral Proteins and Transgene Product Is Generated on Subretinal Administration of Adenovirus and Adeno-Associated Virus. Mol Ther (2002) 5:125–32. doi: 10.1006/mthe.2002.0525
199. High KA, Roncarolo MG. Gene Therapy. N Engl J Med (2019) 381:455–64. doi: 10.1056/NEJMra1706910
200. Kotterman MA, Yin L, Strazzeri JM, Flannery JG, Merigan WH, Schaffer DV. Antibody Neutralization Poses a Barrier to Intravitreal Adeno-Associated Viral Vector Gene Delivery to non-Human Primates. Gene Ther (2015) 22:116–26. doi: 10.1038/gt.2014.115
201. Andrzejewski S, Moyle PM, Stringer BW, Steel JC, Layton CJ. Neutralisation of Adeno-Associated Virus Transduction by Human Vitreous Humour. Gene Ther (2021) 28:242–55. doi: 10.1038/s41434-020-0162-8
202. Jacobson SG, Cideciyan AV, Roman AJ, Sumaroka A, Schwartz SB, Heon E, et al. Improvement and Decline in Vision With Gene Therapy in Childhood Blindness. N Engl J Med (2015) 372:1920–6. doi: 10.1056/NEJMoa1412965
203. Reichel FF, Dauletbekov DL, Klein R, Peters T, Ochakovski GA, Seitz IP, et al. AAV8 Can Induce Innate and Adaptive Immune Response in the Primate Eye. Mol Ther (2017) 25:2648–60. doi: 10.1016/j.ymthe.2017.08.018
204. Vandenberghe LH, Bell P, Maguire AM, Cearley CN, Xiao R, Calcedo R, et al. Dosage Thresholds for AAV2 and AAV8 Photoreceptor Gene Therapy in Monkey. Sci Transl Med (2011) 3:88ra54. doi: 10.1126/scitranslmed.3002103
205. Ye GJ, Budzynski E, Sonnentag P, Nork TM, Miller PE, Sharma AK, et al. Safety and Biodistribution Evaluation in Cynomolgus Macaques of Raav2tyf-PR1.7-Hcngb3, a Recombinant AAV Vector for Treatment of Achromatopsia. Hum Gene Ther Clin Dev (2016) 27:37–48. doi: 10.1089/humc.2015.164
206. Ramachandran PS, Lee V, Wei Z, Song JY, Casal G, Cronin T, et al. Evaluation of Dose and Safety of AAV7m8 and AAV8BP2 in the Non-Human Primate Retina. Hum Gene Ther (2017) 28:154–67. doi: 10.1089/hum.2016.111
207. Timmers AM, Newmark JA, Turunen HT, Farivar T, Liu J, Song C, et al. Ocular Inflammatory Response to Intravitreal Injection of Adeno-Associated Virus Vector: Relative Contribution of Genome and Capsid. Hum Gene Ther (2020) 31:80–9. doi: 10.1089/hum.2019.144
208. Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, Nanda A, Davies A, Wood LJ, et al. Initial Results From a First-in-Human Gene Therapy Trial on X-Linked Retinitis Pigmentosa Caused by Mutations in RPGR. Nat Med (2020) 26:354–9. doi: 10.1038/s41591-020-0763-1
209. Xue K, Jolly JK, Barnard AR, Rudenko A, Salvetti AP, Patricio MI, et al. Beneficial Effects on Vision in Patients Undergoing Retinal Gene Therapy for Choroideremia. Nat Med (2018) 24:1507–12. doi: 10.1038/s41591-018-0185-5
210. Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, et al. Long-Term Effect of Gene Therapy on Leber's Congenital Amaurosis. N Engl J Med (2015) 372:1887–97. doi: 10.1056/NEJMoa1414221
211. Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, et al. Effect of Gene Therapy on Visual Function in Leber's Congenital Amaurosis. N Engl J Med (2008) 358:2231–9. doi: 10.1056/NEJMoa0802268
212. Guy J, Feuer WJ, Davis JL, Porciatti V, Gonzalez PJ, Koilkonda RD, et al. Gene Therapy for Leber Hereditary Optic Neuropathy: Low- and Medium-Dose Visual Results. Ophthalmology (2017) 124:1621–34. doi: 10.1016/j.ophtha.2017.05.016
213. Buck TM, Wijnholds J. Recombinant Adeno-Associated Viral Vectors (rAAV)-Vector Elements in Ocular Gene Therapy Clinical Trials and Transgene Expression and Bioactivity Assays. Int J Mol Sci (2020) 21:4197. doi: 10.3390/ijms21124197
214. Bennett J, Ashtari M, Wellman J, Marshall KA, Cyckowski LL, Chung DC, et al. AAV2 Gene Therapy Readministration in Three Adults With Congenital Blindness. Sci Transl Med (2012) 4:120ra115. doi: 10.1126/scitranslmed.3002865
215. Weed L, Ammar MJ, Zhou S, Wei Z, Serrano LW, Sun J, et al. Safety of Same-Eye Subretinal Sequential Readministration of AAV2-Hrpe65v2 in Non-Human Primates. Mol Ther Methods Clin Dev (2019) 15:133–48. doi: 10.1016/j.omtm.2019.08.011
216. Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, et al. Gene Therapy for Leber Congenital Amaurosis Caused by RPE65 Mutations: Safety and Efficacy in 15 Children and Adults Followed Up to 3 Years. Arch Ophthalmol (2012) 130:9–24. doi: 10.1001/archophthalmol.2011.298
217. Bogdanos DP, Gao B, Gershwin ME. Liver Immunology. Compr Physiol (2013) 3:567–98. doi: 10.1002/cphy.c120011
218. Faust SM, Bell P, Zhu Y, Sanmiguel J, Wilson JM. The Role of Apoptosis in Immune Hyporesponsiveness Following AAV8 Liver Gene Transfer. Mol Ther (2013) 21:2227–35. doi: 10.1038/mt.2013.94
219. Keeler GD, Kumar S, Palaschak B, Silverberg EL, Markusic DM, Jones NT, et al. Gene Therapy-Induced Antigen-Specific Tregs Inhibit Neuro-Inflammation and Reverse Disease in a Mouse Model of Multiple Sclerosis. Mol Ther (2018) 26:173–83. doi: 10.1016/j.ymthe.2017.09.001
220. De Groot AS, Moise L, McMurry JA, Wambre E, Van Overtvelt L, Moingeon P, et al. Activation of Natural Regulatory T Cells by IgG Fc-Derived Peptide "Tregitopes". Blood (2008) 112:3303–11. doi: 10.1182/blood-2008-02-138073
221. Cousens L, Najafian N, Martin WD, De Groot AS. Tregitope: Immunomodulation Powerhouse. Hum Immunol (2014) 75:1139–46. doi: 10.1016/j.humimm.2014.10.012
222. Pompura SL, Dominguez-Villar M. The PI3K/AKT Signaling Pathway in Regulatory T-Cell Development, Stability, and Function. J Leukoc Biol (2018) 103:1065–76. doi: 10.1002/JLB.2MIR0817-349R
223. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin Selectively Expands CD4+CD25+FoxP3+ Regulatory T Cells. Blood (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
224. Stone D, Kenkel EJ, Loprieno MA, Tanaka M, De Silva Feelixge HS, Kumar AJ, et al. Gene Transfer in Adeno-Associated Virus Seropositive Rhesus Macaques Following Rapamycin Treatment and Subcutaneous Delivery of AAV6, But Not Retargeted AAV6 Vectors. Hum Gene Ther (2021) 32:96–112. doi: 10.1089/hum.2020.113
225. Nayak S, Sarkar D, Perrin GQ, Moghimi B, Hoffman BE, Zhou S, et al. Prevention and Reversal of Antibody Responses Against Factor IX in Gene Therapy for Hemophilia B. Front Microbiol (2011) 2:244. doi: 10.3389/fmicb.2011.00244
226. Mingozzi F, High KA. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race Between Clearance, Tolerance, Neutralization, and Escape. Annu Rev Virol (2017) 4:511–34. doi: 10.1146/annurev-virology-101416-041936
227. Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of Transient Immunosuppression on Adenoassociated, Virus-Mediated, Liver-Directed Gene Transfer in Rhesus Macaques and Implications for Human Gene Therapy. Blood (2006) 108:3321–8. doi: 10.1182/blood-2006-04-017913
228. Herzog RW, Mount JD, Arruda VR, High KA, Lothrop CD Jr. Muscle-Directed Gene Transfer and Transient Immune Suppression Result in Sustained Partial Correction of Canine Hemophilia B Caused by a Null Mutation. Mol Ther (2001) 4:192–200. doi: 10.1006/mthe.2001.0442
229. Miesbach W, Meijer K, Coppens M, Kampmann P, Klamroth R, Schutgens R, et al. Gene Therapy With Adeno-Associated Virus Vector 5-Human Factor IX in Adults With Hemophilia B. Blood (2018) 131:1022–31. doi: 10.1182/blood-2017-09-804419
230. Unzu C, Hervas-Stubbs S, Sampedro A, Mauleon I, Mancheno U, Alfaro C, et al. Transient and Intensive Pharmacological Immunosuppression Fails to Improve AAV-Based Liver Gene Transfer in non-Human Primates. J Transl Med (2012) 10:122. doi: 10.1186/1479-5876-10-122
231. Mingozzi F, Chen Y, Murphy SL, Edmonson SC, Tai A, Price SD, et al. Pharmacological Modulation of Humoral Immunity in a Nonhuman Primate Model of AAV Gene Transfer for Hemophilia B. Mol Ther (2012) 20:1410–6. doi: 10.1038/mt.2012.84
232. Mingozzi F, Chen Y, Edmonson SC, Zhou S, Thurlings RM, Tak PP, et al. Prevalence and Pharmacological Modulation of Humoral Immunity to AAV Vectors in Gene Transfer to Synovial Tissue. Gene Ther (2013) 20:417–24. doi: 10.1038/gt.2012.55
233. Velazquez VM, Meadows AS, Pineda RJ, Camboni M, McCarty DM, Fu H. Effective Depletion of Pre-Existing Anti-AAV Antibodies Requires Broad Immune Targeting. Mol Ther Methods Clin Dev (2017) 4:159–68. doi: 10.1016/j.omtm.2017.01.003
234. Biswas M, Palaschak B, Kumar SRP, Rana J, Markusic DM, Cell Depletion Eliminates FVIII Memory B Cells B. and Enhances AAV8-Cof8 Immune Tolerance Induction When Combined With Rapamycin. Front Immunol (2020) 11:1293. doi: 10.3389/fimmu.2020.01293
235. Herzog RW, Biswas M. Neutralizing the Neutralizers in AAV Gene Therapy. Mol Ther (2020) 28:1741–2. doi: 10.1016/j.ymthe.2020.07.015
236. Leborgne C, Barbon E, Alexander JM, Hanby H, Delignat S, Cohen DM, et al. IgG-Cleaving Endopeptidase Enables In Vivo Gene Therapy in the Presence of Anti-AAV Neutralizing Antibodies. Nat Med (2020) 26:1096–101. doi: 10.1038/s41591-020-0911-7
237. Elmore ZC, Oh DK, Simon KE, Fanous MM, Asokan A. Rescuing AAV Gene Transfer From Neutralizing Antibodies With an IgG-Degrading Enzyme. JCI Insight (2020) 5:e139881. doi: 10.1172/jci.insight.139881
238. Askew C, Li C. A Vector Independent Method of Neutralizing Antibody Evasion Potently Protects AAV for Efficient Gene Delivery, Molecular Therapy. 50 HAMPSHIRE ST, FLOOR 5, CAMBRIDGE, MA 02139 USA: Cell Press (2020). p. 22–2.
239. Monteilhet V, Saheb S, Boutin S, Leborgne C, Veron P, Montus MF, et al. A 10 Patient Case Report on the Impact of Plasmapheresis Upon Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 6, and 8. Mol Ther (2011) 19:2084–91. doi: 10.1038/mt.2011.108
240. Mingozzi F, Anguela XM, Pavani G, Chen Y, Davidson RJ, Hui DJ, et al. Overcoming Preexisting Humoral Immunity to AAV Using Capsid Decoys. Sci Transl Med (2013) 5:194ra192. doi: 10.1126/scitranslmed.3005795
241. Adachi K, Dissen GA, Lomniczi A, Xie Q, Ojeda SR, Nakai H. Adeno-Associated Virus-Binding Antibodies Detected in Cats Living in the Northeastern United States Lack Neutralizing Activity. Sci Rep (2020) 10:10073. doi: 10.1038/s41598-020-66596-4
242. Leborgne C, Latournerie V, Boutin S, Desgue D, Quere A, Pignot E, et al. Prevalence and Long-Term Monitoring of Humoral Immunity Against Adeno-Associated Virus in Duchenne Muscular Dystrophy Patients. Cell Immunol (2019) 342:103780. doi: 10.1016/j.cellimm.2018.03.004
243. Stanford S, Pink R, Creagh D, Clark A, Lowe G, Curry N, et al. Adenovirus-Associated Antibodies in UK Cohort of Hemophilia Patients: A Seroprevalence Study of the Presence of Adenovirus-Associated Virus Vector-Serotypes AAV5 and AAV8 Neutralizing Activity and Antibodies in Patients With Hemophilia a. Res Pract Thromb Haemost (2019) 3:261–7. doi: 10.1002/rth2.12177
244. Yates VJ, el-Mishad AM, McCormick KJ, Trentin JJ. Isolation and Characterization of an Avian Adenovirus-Associated Virus. Infect Immun (1973) 7:973–80. doi: 10.1128/iai.7.6.973-980.1973
245. Arbetman AE, Lochrie M, Zhou S, Wellman J, Scallan C, Doroudchi MM, et al. Novel Caprine Adeno-Associated Virus (AAV) Capsid (AAV-Go.1) Is Closely Related to the Primate AAV-5 and has Unique Tropism and Neutralization Properties. J Virol (2005) 79:15238–45. doi: 10.1128/JVI.79.24.15238-15245.2005
246. Bello A, Chand A, Aviles J, Soule G, Auricchio A, Kobinger GP. Novel Adeno-Associated Viruses Derived From Pig Tissues Transduce Most Major Organs in Mice. Sci Rep (2014) 4:6644. doi: 10.1038/srep06644
247. Bello A, Tran K, Chand A, Doria M, Allocca M, Hildinger M, et al. Isolation and Evaluation of Novel Adeno-Associated Virus Sequences From Porcine Tissues. Gene Ther (2009) 16:1320–8. doi: 10.1038/gt.2009.82
248. Li Y, Ge X, Zhang H, Zhou P, Zhu Y, Zhang Y, et al. Host Range, Prevalence, and Genetic Diversity of Adenoviruses in Bats. J Virol (2010) 84:3889–97. doi: 10.1128/JVI.02497-09
249. Tulalamba W, Weinmann J, Pham QH, El Andari J, VandenDriessche T, Chuah MK, et al. Distinct Transduction of Muscle Tissue in Mice After Systemic Delivery of AAVpo1 Vectors. Gene Ther (2020) 27:170–9. doi: 10.1038/s41434-019-0106-3
250. Gurda BL, DiMattia MA, Miller EB, Bennett A, McKenna R, Weichert WS, et al. Capsid Antibodies to Different Adeno-Associated Virus Serotypes Bind Common Regions. J Virol (2013) 87:9111–24. doi: 10.1128/JVI.00622-13
251. Gurda BL, Raupp C, Popa-Wagner R, Naumer M, Olson NH, Ng R, et al. Mapping a Neutralizing Epitope Onto the Capsid of Adeno-Associated Virus Serotype 8. J Virol (2012) 86:7739–51. doi: 10.1128/JVI.00218-12
252. Jose A, Mietzsch M, Smith JK, Kurian J, Chipman P, McKenna R, et al. High-Resolution Structural Characterization of a New Adeno-Associated Virus Serotype 5 Antibody Epitope Toward Engineering Antibody-Resistant Recombinant Gene Delivery Vectors. J Virol (2019) 93:e01394–18. doi: 10.1128/JVI.01394-18
253. Smith JK, Agbandje-McKenna M. Creating an Arsenal of Adeno-Associated Virus (AAV) Gene Delivery Stealth Vehicles. PloS Pathog (2018) 14:e1006929. doi: 10.1371/journal.ppat.1006929
254. Tseng YS, Agbandje-McKenna M. Mapping the AAV Capsid Host Antibody Response Toward the Development of Second Generation Gene Delivery Vectors. Front Immunol (2014) 5:9. doi: 10.3389/fimmu.2014.00009
255. Tseng YS, Gurda BL, Chipman P, McKenna R, Afione S, Chiorini JA, et al. Adeno-Associated Virus Serotype 1 (AAV1)- and AAV5-Antibody Complex Structures Reveal Evolutionary Commonalities in Parvovirus Antigenic Reactivity. J Virol (2015) 89:1794–808. doi: 10.1128/JVI.02710-14
256. Li C, Diprimio N, Bowles DE, Hirsch ML, Monahan PE, Asokan A, et al. Single Amino Acid Modification of Adeno-Associated Virus Capsid Changes Transduction and Humoral Immune Profiles. J Virol (2012) 86:7752–9. doi: 10.1128/JVI.00675-12
257. Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R, et al. Mutations on the External Surfaces of Adeno-Associated Virus Type 2 Capsids That Affect Transduction and Neutralization. J Virol (2006) 80:821–34. doi: 10.1128/JVI.80.2.821-834.2006
258. Moskalenko M, Chen LL, van Roey M, Donahue BA, Snyder RO, McArthur JG, et al. Epitope Mapping of Human Anti-Adeno-Associated Virus Type 2 Neutralizing Antibodies: Implications for Gene Therapy and Virus Structure. J Virol (2000) 74:1761–6. doi: 10.1128/JVI.74.4.1761-1766.2000
259. Adachi K, Enoki T, Kawano Y, Veraz M, Nakai H. Drawing a High-Resolution Functional Map of Adeno-Associated Virus Capsid by Massively Parallel Sequencing. Nat Commun (2014) 5:3075. doi: 10.1038/ncomms4075
260. Maersch S, Huber A, Buning H, Hallek M, Perabo L. Optimization of Stealth Adeno-Associated Virus Vectors by Randomization of Immunogenic Epitopes. Virology (2010) 397:167–75. doi: 10.1016/j.virol.2009.10.021
261. Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed Evolution of Adeno-Associated Virus Yields Enhanced Gene Delivery Vectors. Nat Biotechnol (2006) 24:198–204. doi: 10.1038/nbt1182
262. Giles AR, Calcedo R, Tretiakova AP, Wilson JM. Isolating Human Monoclonal Antibodies Against Adeno-Associated Virus From Donors With Pre-Existing Immunity. Front Immunol (2020) 11:1135. doi: 10.3389/fimmu.2020.01135
263. Kim Y, Kim E, Oh S, Yoon YE, Jang JH. Mutagenic Analysis of an Adeno-Associated Virus Variant Capable of Simultaneously Promoting Immune Resistance and Robust Gene Delivery. Hum Gene Ther (2018) 29:25–41. doi: 10.1089/hum.2017.034
264. Selot R, Arumugam S, Mary B, Cheemadan S, Jayandharan GR. Optimized AAV Rh.10 Vectors That Partially Evade Neutralizing Antibodies During Hepatic Gene Transfer. Front Pharmacol (2017) 8:441. doi: 10.3389/fphar.2017.00441
265. Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, et al. Phase 1 Gene Therapy for Duchenne Muscular Dystrophy Using a Translational Optimized AAV Vector. Mol Ther (2012) 20:443–55. doi: 10.1038/mt.2011.237
266. Burg M, Rosebrough C, Drouin LM, Bennett A, Mietzsch M, Chipman P, et al. Atomic Structure of a Rationally Engineered Gene Delivery Vector, AAV2.5. J Struct Biol (2018) 203:236–41. doi: 10.1016/j.jsb.2018.05.004
267. Mary B, Maurya S, Arumugam S, Kumar V, Jayandharan GR. Post-Translational Modifications in Capsid Proteins of Recombinant Adeno-Associated Virus (AAV) 1-Rh10 Serotypes. FEBS J (2019) 286:4964–81. doi: 10.1111/febs.15013
268. Markusic DM, Herzog RW, Aslanidi GV, Hoffman BE, Li B, Li M, et al. High-Efficiency Transduction and Correction of Murine Hemophilia B Using AAV2 Vectors Devoid of Multiple Surface-Exposed Tyrosines. Mol Ther (2010) 18:2048–56. doi: 10.1038/mt.2010.172
269. Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next Generation of Adeno-Associated Virus 2 Vectors: Point Mutations in Tyrosines Lead to High-Efficiency Transduction at Lower Doses. Proc Natl Acad Sci USA (2008) 105:7827–32. doi: 10.1073/pnas.0802866105
270. Gabriel N, Hareendran S, Sen D, Gadkari RA, Sudha G, Selot R, et al. Bioengineering of AAV2 Capsid at Specific Serine, Threonine, or Lysine Residues Improves Its Transduction Efficiency In Vitro and In Vivo. Hum Gene Ther Methods (2013) 24:80–93. doi: 10.1089/hgtb.2012.194
271. Li B, Ma W, Ling C, Van Vliet K, Huang LY, Agbandje-McKenna M, et al. Site-Directed Mutagenesis of Surface-Exposed Lysine Residues Leads to Improved Transduction by AAV2, But Not AAV8, Vectors in Murine Hepatocytes In Vivo. Hum Gene Ther Methods (2015) 26:211–20. doi: 10.1089/hgtb.2015.115
272. Hakim CH, Yue Y, Shin JH, Williams RR, Zhang K, Smith BF, et al. Systemic Gene Transfer Reveals Distinctive Muscle Transduction Profile of Tyrosine Mutant AAV-1, -6, and -9 in Neonatal Dogs. Mol Ther Methods Clin Dev (2014) 1:14002. doi: 10.1038/mtm.2014.2
273. Chai Z, Sun J, Rigsbee KM, Wang M, Samulski RJ, Li C. Application of Polyploid Adeno-Associated Virus Vectors for Transduction Enhancement and Neutralizing Antibody Evasion. J Control Release (2017) 262:348–56. doi: 10.1016/j.jconrel.2017.08.005
274. Marsic D, Govindasamy L, Currlin S, Markusic DM, Tseng YS, Herzog RW, et al. Vector Design Tour De Force: Integrating Combinatorial and Rational Approaches to Derive Novel Adeno-Associated Virus Variants. Mol Ther (2014) 22:1900–9. doi: 10.1038/mt.2014.139
275. Havlik LP, Simon KE, Smith JK, Klinc KA, Tse LV, Oh DK, et al. Coevolution of Adeno-Associated Virus Capsid Antigenicity and Tropism Through a Structure-Guided Approach. J Virol (2020) 94:e00976–20. doi: 10.1128/JVI.00976-20
276. Giles AR, Govindasamy L, Somanathan S, Wilson JM. Mapping an Adeno-Associated Virus 9-Specific Neutralizing Epitope To Develop Next-Generation Gene Delivery Vectors. J Virol (2018) 92:e01011–18. doi: 10.1128/JVI.01011-18
277. Biswas M, Marsic D, Li N, Zou C, Gonzalez-Aseguinolaza G, Zolotukhin I, et al. Engineering and In Vitro Selection of a Novel AAV3B Variant With High Hepatocyte Tropism and Reduced Seroreactivity. Mol Ther Methods Clin Dev (2020) 19:347–61. doi: 10.1016/j.omtm.2020.09.019
278. Grimm D, Zolotukhin S. E Pluribus Unum: 50 Years of Research, Millions of Viruses, and One Goal–Tailored Acceleration of AAV Evolution. Mol Ther (2015) 23:1819–31. doi: 10.1038/mt.2015.173
279. Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, et al. In Vitro and In Vivo Gene Therapy Vector Evolution via Multispecies Interbreeding and Retargeting of Adeno-Associated Viruses. J Virol (2008) 82:5887–911. doi: 10.1128/JVI.00254-08
280. Koerber JT, Jang JH, Schaffer DV. DNA Shuffling of Adeno-Associated Virus Yields Functionally Diverse Viral Progeny. Mol Ther (2008) 16:1703–9. doi: 10.1038/mt.2008.167
281. Pei X, Shao W, Xing A, Askew C, Chen X, Cui C, et al. Development of AAV Variants With Human Hepatocyte Tropism and Neutralizing Antibody Escape Capacity. Mol Ther Methods Clin Dev (2020) 18:259–68. doi: 10.1016/j.omtm.2020.06.003
282. Li C, Wu S, Albright B, Hirsch M, Li W, Tseng YS, et al. Development of Patient-Specific AAV Vectors After Neutralizing Antibody Selection for Enhanced Muscle Gene Transfer. Mol Ther (2016) 24:53–65. doi: 10.1038/mt.2015.134
283. Chai Z, Samulski RJ, Li C. Nab Escaping AAV Mutants Isolated From Mouse Muscles. Bio Protoc (2018) 8:e2841. doi: 10.21769/BioProtoc.2841
284. Huttner NA, Girod A, Perabo L, Edbauer D, Kleinschmidt JA, Buning H, et al. Genetic Modifications of the Adeno-Associated Virus Type 2 Capsid Reduce the Affinity and the Neutralizing Effects of Human Serum Antibodies. Gene Ther (2003) 10:2139–47. doi: 10.1038/sj.gt.3302123
285. Weinmann J, Weis S, Sippel J, Tulalamba W, Remes A, El Andari J, et al. Identification of a Myotropic AAV by Massively Parallel In Vivo Evaluation of Barcoded Capsid Variants. Nat Commun (2020) 11:5432. doi: 10.1038/s41467-020-19230-w
286. Lee GK, Maheshri N, Kaspar B, Schaffer DV. PEG Conjugation Moderately Protects Adeno-Associated Viral Vectors Against Antibody Neutralization. Biotechnol Bioeng (2005) 92:24–34. doi: 10.1002/bit.20562
287. Le HT, Yu QC, Wilson JM, Croyle MA. Utility of PEGylated Recombinant Adeno-Associated Viruses for Gene Transfer. J Control Release (2005) 108:161–77. doi: 10.1016/j.jconrel.2005.07.019
288. Yao T, Zhou X, Zhang C, Yu X, Tian Z, Zhang L, et al. Site-Specific PEGylated Adeno-Associated Viruses With Increased Serum Stability and Reduced Immunogenicity. Molecules (2017) 22:1155. doi: 10.3390/molecules22071155
289. Katrekar D, Moreno AM, Chen G, Worlikar A, Mali P. Oligonucleotide Conjugated Multi-Functional Adeno-Associated Viruses. Sci Rep (2018) 8:3589. doi: 10.1038/s41598-018-21742-x
290. Flotte T-i. Revisiting the "New" Inflammatory Toxicities of Adeno-Associated Virus Vectors. Hum Gene Ther (2020) 31:398–9. doi: 10.1089/hum.2020.29117.trf
291. Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med (2017) 377:1713–22. doi: 10.1056/NEJMoa1706198
292. Hordeaux J, Buza EL, Dyer C, Goode T, Mitchell TW, Richman L, et al. Adeno-Associated Virus-Induced Dorsal Root Ganglion Pathology. Hum Gene Ther (2020) 31:808–18. doi: 10.1089/hum.2020.167
293. Hordeaux J, Hinderer C, Goode T, Katz N, Buza EL, Bell P, et al. Toxicology Study of Intra-Cisterna Magna Adeno-Associated Virus 9 Expressing Human Alpha-L-Iduronidase in Rhesus Macaques. Mol Ther Methods Clin Dev (2018) 10:79–88. doi: 10.1016/j.omtm.2018.06.003
294. Hordeaux J, Buza EL, Jeffrey B, Song C, Jahan T, Yuan Y, et al. MicroRNA-Mediated Inhibition of Transgene Expression Reduces Dorsal Root Ganglion Toxicity by AAV Vectors in Primates. Sci Transl Med (2020) 12:eaba9188. doi: 10.1126/scitranslmed.aba9188
295. Morales L, Gambhir Y, Bennett J, Stedman HH. Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys Following Systemic High-Dose AAV. Mol Ther (2020) 28:1753–5. doi: 10.1016/j.ymthe.2020.07.009
296. Shieh PB, Bonnemann CG, Muller-Felber W, Blaschek A, Dowling JJ, Kuntz NL, et al. Re: "Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy" by Wilson and Flotte. Hum Gene Ther (2020) 31:787. doi: 10.1089/hum.2020.217
297. Wilson JM, Flotte TR. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum Gene Ther (2020) 31:695–6. doi: 10.1089/hum.2020.182
298. Srivastava A. AAV Vectors: Are They Safe? Hum Gene Ther (2020) 31:697–9. doi: 10.1089/hum.2020.187
299. Childers MK, Joubert R, Poulard K, Moal C, Grange RW, Doering JA, et al. Gene Therapy Prolongs Survival and Restores Function in Murine and Canine Models of Myotubular Myopathy. Sci Transl Med (2014) 6:220ra210. doi: 10.1126/scitranslmed.3007523
Keywords: AAV, antibody response, cellular response, capsid, engineering, immune evasion, pre-existing immunity, neutralizing antibodies
Citation: Rapti K and Grimm D (2021) Adeno-Associated Viruses (AAV) and Host Immunity – A Race Between the Hare and the Hedgehog. Front. Immunol. 12:753467. doi: 10.3389/fimmu.2021.753467
Received: 04 August 2021; Accepted: 28 September 2021;
Published: 29 October 2021.
Edited by:
Nicole K. Paulk, University of California, San Francisco, United StatesReviewed by:
Eduard Ayuso, DiNAQOR (Switzerland), SwitzerlandCopyright © 2021 Rapti and Grimm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kleopatra Rapti, a2xlb3BhdHJhLnJhcHRpQGJpb3F1YW50LnVuaS1oZWlkZWxiZXJnLmRl; Dirk Grimm, ZGlyay5ncmltbUBiaW9xdWFudC51bmktaGVpZGVsYmVyZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.