 Aaron DiAntonio
Aaron DiAntonio Jeffrey Milbrandt2,3
Jeffrey Milbrandt2,3 Matthew D. Figley
Matthew D. Figley- 1Department of Developmental Biology, Washington University School of Medicine in Saint Louis, St. Louis, MO, United States
- 2Needleman Center for Neurometabolism and Axonal Therapeutics, Washington University School of Medicine in Saint Louis, St. Louis, MO, United States
- 3Department of Genetics, Washington University School of Medicine in Saint Louis, St. Louis, MO, United States
The Toll/interleukin-1 receptor (TIR) domain is the signature signalling motif of innate immunity, with essential roles in innate immune signalling in bacteria, plants, and animals. TIR domains canonically function as scaffolds, with stimulus-dependent multimerization generating binding sites for signalling molecules such as kinases and ligases that activate downstream immune mechanisms. Recent studies have dramatically expanded our understanding of the TIR domain, demonstrating that the primordial function of the TIR domain is to metabolize NAD+. Mammalian SARM1, the central executioner of pathological axon degeneration, is the founding member of the TIR-domain class of NAD+ hydrolases. This unexpected NADase activity of TIR domains is evolutionarily conserved, with archaeal, bacterial, and plant TIR domains all sharing this catalytic function. Moreover, this enzymatic activity is essential for the innate immune function of these proteins. These evolutionary relationships suggest a link between SARM1 and ancient self-defense mechanisms that has only been strengthened by the recent discovery of the SARM1 activation mechanism which, we will argue, is strikingly similar to bacterial toxin-antitoxin systems. In this brief review we will describe the regulation and function of SARM1 in programmed axon self-destruction, and highlight the parallels between the SARM1 axon degeneration pathway and bacterial innate immune mechanisms.
Introduction
Injured or diseased axons initiate a self-destruction program known as Wallerian degeneration. SARM1 triggers this pathological axon degeneration (1), and is a key driver of pathology in models of chemotherapy-induced peripheral neuropathy (2–5), traumatic brain injury (6–10), glaucoma (11), and retinal degeneration (12, 13). SARM1 also participates in antiviral defense. SARM1 triggers axon degeneration following rabies infection (14), presumably to halt the spread of the virus as it travels retrogradely down the axon to the neuronal cell body, and induces neuronal cell death in response to bunyavirus infection (15), killing infected cells and thereby reducing viral spread. Hence, the role of SARM1 in pathological axon degeneration is likely closely linked to its function in antiviral innate immunity.
SARM1 is a multi-domain protein comprised of an autoinhibitory ARM domain, tandem SAM domains mediating multimerization, and a C-terminal TIR domain NAD+ hydrolase (16, 17). In healthy neurons, SARM1 autoinhibition is maintained by multiple intra- and intermolecular interactions (18), including binding of the N-terminal ARM domain to the C-terminal TIR domain (19) (Figure 1A). SARM1 autoinhibition is regulated by an allosteric binding site within the autoinhibitory ARM domain that can bind either nicotinamide adenine dinucleotide (NAD+) (21, 22) or its precursor, nicotinamide mononucleotide (NMN) (23). NMN promotes SARM1-dependent axon degeneration (24–27). Axon injury leads to loss of the NAD+ biosynthetic enzyme NMNAT2 (28), resulting in an increased NMN/NAD+ ratio that promotes NMN binding to the allosteric site (23). The switch from NAD+ to NMN binding alters the conformation of the autoinhibitory ARM domain, thereby promoting TIR-TIR interactions and enzymatic activity (23) (Figure 1B). Below we will highlight commonalities between SARM1 activation and effector mechanisms with similar bacterial innate immune mechanisms.
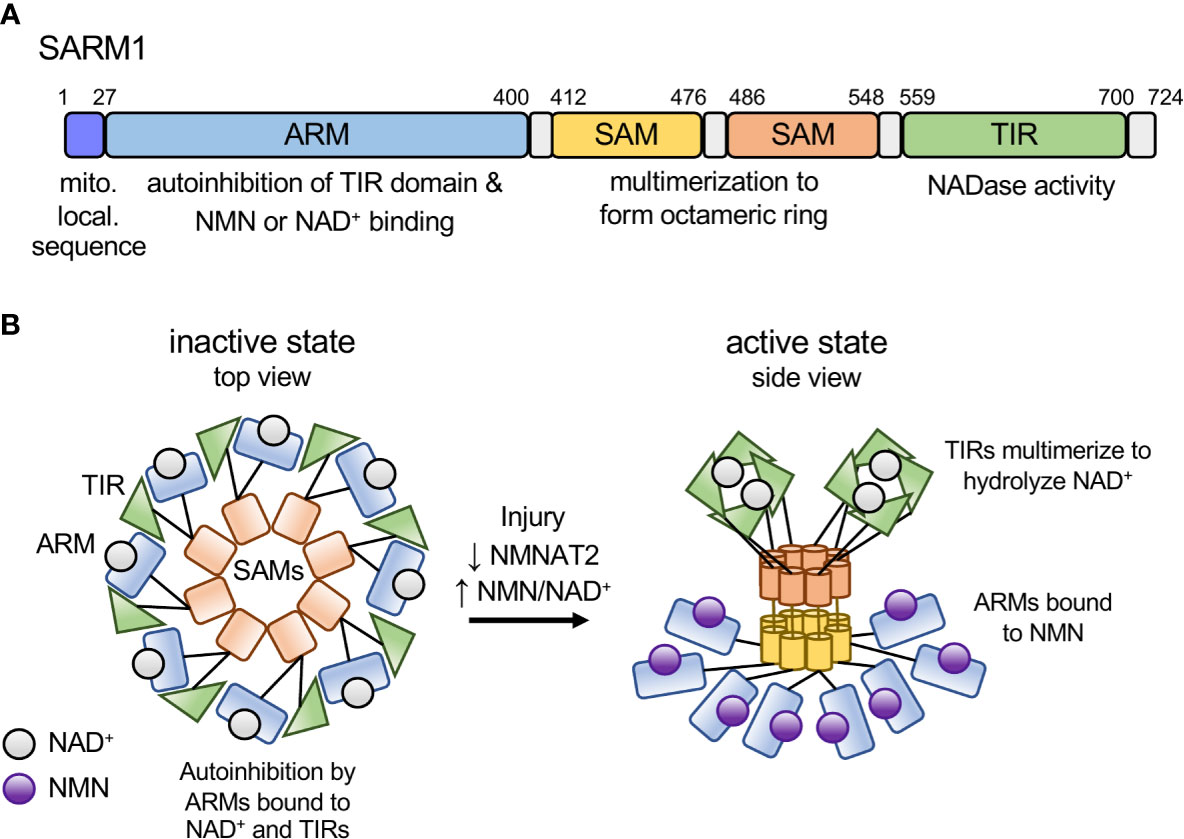
Figure 1 Model of SARM1 domain structure and activation mechanism. (A) Domain structure of the human SARM1 protein. SARM1 contains an N-terminal mitochondrial localization sequence and Armadillo-repeat containing domain (ARM), two tandem sterile alpha motif (SAM) domains, and a C-terminal Toll/interleukin-1 receptor (TIR) domain. Numbers denote the amino acid position of the domain boundaries. (B) Schematic depicting the activation mechanism of SARM1. In the inactive state SARM1’s ARM domains are bound to NAD+ at the allosteric site and bound to adjacent TIR domains both intra- and inter-molecularly, mediating autoinhibition of the TIR’s NADase activity. In response to an increase in the NMN/NAD+ ratio, NMN binds to the ARM domain allosteric site, resulting in a conformational change in the ARM domain, disengagement of the ARM-TIR interactions, multimerization of the TIR domains and NADase activity. Based on recent structural data from the RPP1 TIR domain, we depict active TIRs as a tetramer forming two active sites for NAD+ binding (20).
SARM1-NMNAT2 Is a Candidate Mammalian Toxin-Antitoxin Pair
Just as SARM1 can trigger axon self-destruction in response to rabies infection, so too can a bacterial population acquire immunity to bacteriophage infection through an altruistic suicide mechanism known as abortive infection. When infected, the bacterial cells activate a toxin-antitoxin (TA) system prior to phage replication, killing the infected cells and thereby protecting the community by preventing further phage expansion (29). In a TA system, bacteria express both a lethal toxin and its antagonist, the antitoxin. Upon infection, the antitoxin is degraded, unleashing the degenerative activity of the toxin. Genetic deletion of the toxin-encoding gene yields no phenotype in the absence of the inciting stimuli, whereas deletion of the antitoxin-encoding gene results in cell death due to unchecked toxin activity. This lethality can be rescued by concurrent deletion of the toxin-encoding gene. This TA relationship is strikingly reminiscent of the relationship between NMNAT2 and SARM1, with NMNAT2 serving as the antitoxin and SARM1 as the toxin (Figure 2). First, NMNAT2 inhibits the prodegenerative activity of SARM1, as antitoxins inhibit the functions of toxins. Second, the classic TA genetic relationship holds for SARM1 and NMNAT2. Loss of SARM1 has no obvious phenotype in mice until an appropriate stimulus, such as neuronal injury, occurs (17, 30). By contrast, genetic loss of NMNAT2 (the antitoxin) is embryonic lethal in mice (31, 32). Double mutants lacking both NMNAT2 and SARM1 fully rescue this lethality (33). Hence, the essential function of NMNAT2 is to inhibit SARM1. Third, similar to a type II TA system in bacteria (29), the antitoxin NMNAT2 is highly labile (28) and the levels of NMNAT2 are a key determinant of SARM1 activation (34). However, in contrast to the bacterial type II TA system, in which the antitoxin inactivates the toxin through direct binding, NMNAT2 inhibits SARM1 indirectly, by modulating the flow of metabolites that bind and regulate SARM1 activity.
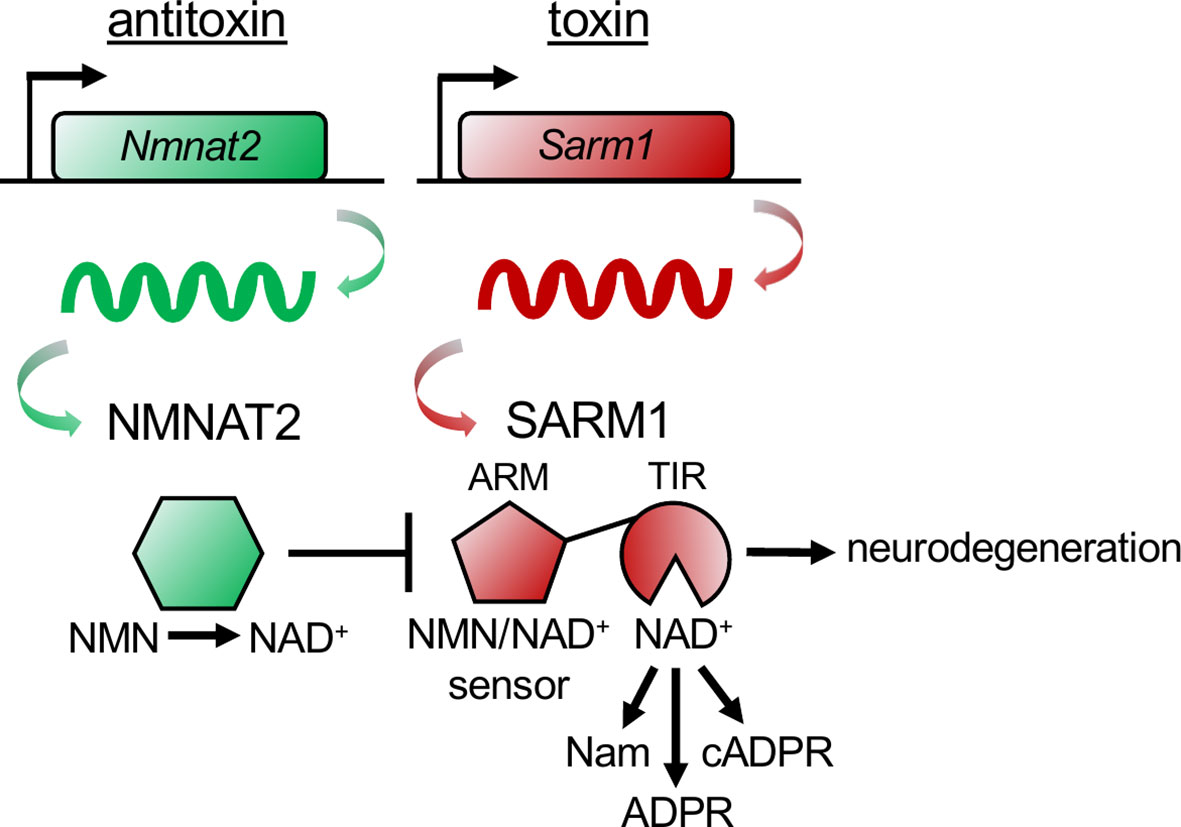
Figure 2 The NMNAT2 antitoxin inhibits the SARM1 toxin to prevent axon degeneration. The antitoxin NMNAT2 converts nicotinamide mononucleotide (NMN) to nicotinamide adenine dinucleotide (NAD+) and thus maintains a healthy NMN/NAD+ ratio in axons. SARM1’s ARM domain senses the ratio of NMN/NAD+ by binding to either metabolite. When the antitoxin NMNAT2 is lost, the NMN/NAD+ ratio rises, NMN binds to the toxin SARM1’s ARM domain, activating SARM1’s TIR domains to hydrolyze NAD+, producing nicotinamide (Nam) and adenosine diphosphate ribose (ADPR), or cyclizing ADPR into cyclic ADPR (cADPR). Activation of the toxin SARM1 drives pathological axon degeneration.
To our knowledge, all known TA systems are found in bacteria. Here we posit that via convergent evolution SARM1 and NMNAT2 have developed into an analogous TA system to control axonal fate in mammalian neurons. In an injured or unhealthy axon the transport of NMNAT2 is disrupted (3, 28), and neuronal stress pathways promote NMNAT2 turnover (34, 35), leading to the loss of the labile NMNAT2 antitoxin and the subsequent activation of the toxin SARM1, resulting in rapid and efficient axonal self-destruction. This may be a physiological function of SARM1, enabling the phagocytosis and clearance of damaged axons before their contents leak and potentially harm adjacent axons or cells or induce inflammation. Indeed, this scenario was recently demonstrated in a mouse model of ulcerative colitis, in which SARM1 promotes axon degeneration in the enteric nervous system and thereby limits inflammation in the colon (36). It will be interesting to determine whether NMNAT2 is also lost in this colitis model, or in response to rabies infection, when SARM1 is activated and axons degenerate as an antiviral defense (14).
SARM1 Is the Founding Member of the TIR Domain Family of Innate Immune NADases
The role of SARM1 and its connection to ancient surveillance mechanisms extends beyond its TA relationship with NMNAT2 to its mechanism of degeneration, NAD+ cleavage. In many bacterial TA relationships, the toxins are, like SARM1, NAD+ glycohydrolases (37). Examples include the toxins SPN (S. pyogenes), TNT and MbcT (M. tuberculosis), Tne2 (P. protegens), and RES (P. luminescens), all of which deplete NAD+ to induce cellular dysfunction or death and are neutralized by their respective antitoxins (38–42). The newly discovered bacterial TIR domain NADases also participate in phage defense as they are essential components of the Thoeris phage defense mechanism (43, 44). Moreover, bacteria not only use TIR NADases to defend against phage invasion, but also to disrupt mammalian innate immune mechanisms via metabolic disruption of the host cell (45). In addition, plant TIR domain innate immune receptors are active NADases and this enzymatic function is essential for the cell death that confers disease resistance (20, 46–48). The conservation of TIR NADase function (Table 1) in bacterial, plant, and animal response to infection suggests that TIR-mediated NAD+ cleavage is a primordial innate immune function.
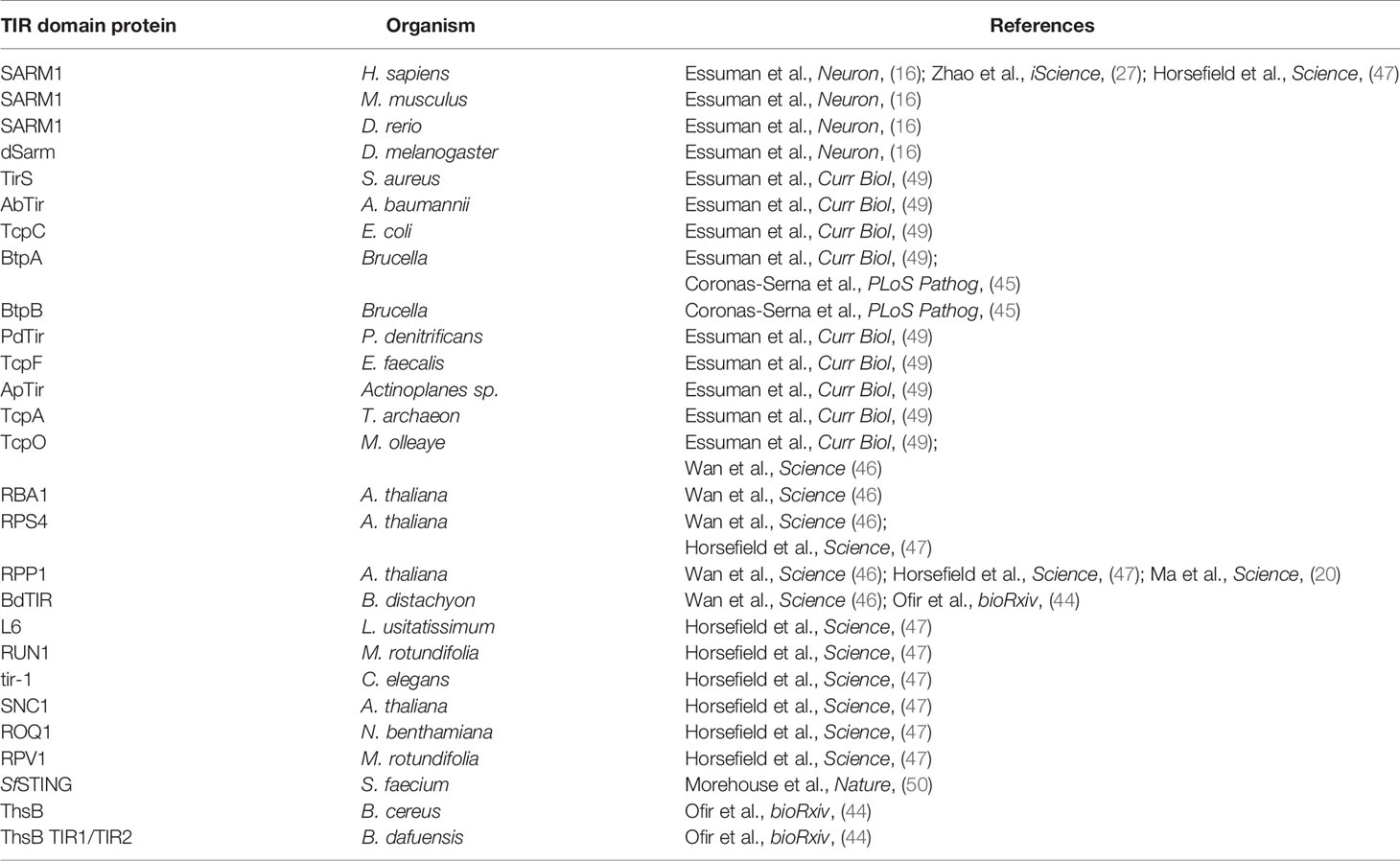
Table 1 TIR domain-containing proteins with demonstrated intrinsic NADase activity.
Finally, SARM1 and evolutionarily diverse TIR domain proteins not only share NADase function, but can also possess regulatory domains controlled via allosteric binding to cellular metabolites. The SARM1 TIR domain NADase is fused to a metabolic sensing ARM domain that acts to inhibit the NADase activity until specific environmental signals are present. This is likely a general regulatory mechanism for TIR NADase activation, as organisms from all kingdoms of life encode proteins with TIR domains fused to a variety of other motifs, such as leucine-rich repeat (LRR), tetratricopeptide repeat (TPR), WD repeat, and coiled coil (CC) domains (51–53), that may function as environmental sensors to tune the NADase activity of the fused effector TIR domains. Indeed, this precise regulatory relationship occurs in ancient STING (stimulator of interferon genes) receptor proteins. In both prokaryotes and lower eukaryotes, STING domains are fused with TIR NADase domains. This effectively couples STING domain sensing of cyclic dinucleotides produced by the cyclic AMP-GMP synthase (cGAS) cellular surveillance system to TIR domain NADase activity (50). We suggest that additional multi-domain proteins encoding TIR NADases are likely regulated via metabolite binding, sensing changes in cellular metabolism and responding via NAD+ hydrolysis.
Discussion
This brief survey of SARM1 and the family of TIR domain NADases demonstrates that mechanistic insights into SARM1 regulation and function have enabled major breakthroughs in our understanding of TIR domain proteins across the domains of life. Key insights from these studies are the identification of SARM1/NMNAT2 as the first candidate mammalian toxin/antitoxin pair, the recognition of multidomain TIR containing proteins as coordinated metabolic sensors and effectors, and the realization that there is a striking commonality between mechanisms of neurodegeneration and the primordial battle between bacteria and bacteriophages.
Author Contributions
Writing – original draft, all authors. Writing – review and editing, all authors. Funding acquisition, AD and JM. Supervision, AD and JM. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by National Institutes of Health grants R37NS065053 to AD and RO1NS087632 to AD and JM.
Conflict of Interest
AD and JM are co-founders, scientific advisory board members, and shareholders of Disarm Therapeutics, Inc. (a wholly-owned subsidiary of Eli Lilly and Company). The editor declared a past co-authorship with all the authors.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Figley MD, DiAntonio A. The SARM1 Axon Degeneration Pathway: Control of the NAD+ Metabolome Regulates Axon Survival in Health and Disease. Curr Opin Neurobiol (2020) 63:59–66. doi: 10.1016/j.conb.2020.02.012
2. Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A. Prevention of Vincristine-Induced Peripheral Neuropathy by Genetic Deletion of SARM1 in Mice. Brain (2016) 139:3092–108. doi: 10.1093/brain/aww251
3. Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, et al. Vincristine and Bortezomib Use Distinct Upstream Mechanisms to Activate a Common SARM1-Dependent Axon Degeneration Program. JCI Insight (2019) 4(17):e129920. doi: 10.1172/jci.insight.129920
4. Cetinkaya-Fisgin A, Luan X, Reed N, Jeong YE, Oh BC, Hoke A. Cisplatin Induced Neurotoxicity is Mediated by Sarm1 and Calpain Activation. Sci Rep (2020) 10:21889. doi: 10.1038/s41598-020-78896-w
5. Gould SA, White M, Wilbrey AL, Pór E, Coleman MP, Adalbert R. Protection Against Oxaliplatin-Induced Mechanical and Thermal Hypersensitivity in Sarm1-/- Mice. Exp Neurol (2021) 338:113607. doi: 10.1016/j.expneurol.2021.113607
6. Maynard ME, Redell JB, Zhao J, Hood KN, Vita SM, Kobori N, et al. Sarm1 Loss Reduces Axonal Damage and Improves Cognitive Outcome After Repetitive Mild Closed Head Injury. Exp Neurol (2020) 327:113207. doi: 10.1016/j.expneurol.2020.113207
7. Marion CM, McDaniel DP, Armstrong RC. Sarm1 Deletion Reduces Axon Damage, Demyelination, and White Matter Atrophy After Experimental Traumatic Brain Injury. Exp Neurol (2019) 321:113040. doi: 10.1016/j.expneurol.2019.113040
8. Bradshaw DV Jr, Knutsen AK, Korotcov A, Sullivan GM, Radomski KL, Dardzinski BJ, et al. Genetic Inactivation of SARM1 Axon Degeneration Pathway Improves Outcome Trajectory After Experimental Traumatic Brain Injury Based on Pathological, Radiological, and Functional Measures. Acta Neuropathol Commun (2021) 9:89. doi: 10.1186/s40478-021-01193-8
9. Ziogas NK, Koliatsos VE. Primary Traumatic Axonopathy in Mice Subjected to Impact Acceleration: A Reappraisal of Pathology and Mechanisms With High-Resolution Anatomical Methods. J Neurosci (2018) 38:4031–47. doi: 10.1523/JNEUROSCI.2343-17.2018
10. Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, et al. Attenuated Traumatic Axonal Injury and Improved Functional Outcome After Traumatic Brain Injury in Mice Lacking Sarm1. Brain (2016) 139:1094–105. doi: 10.1093/brain/aww001
11. Ko KW, Milbrandt J, DiAntonio A. SARM1 Acts Downstream of Neuroinflammatory and Necroptotic Signaling to Induce Axon Degeneration. J Cell Biol (2020) 219(8):e201912047. doi: 10.1083/jcb.201912047
12. Ozaki E, Gibbons L, Neto NG, Kenna P, Carty M, Humphries M, et al. SARM1 Deficiency Promotes Rod and Cone Photoreceptor Cell Survival in a Model of Retinal Degeneration. Life Sci Alliance (2020) 3(5):e201900618. doi: 10.26508/lsa.201900618
13. Sasaki Y, Kakita H, Kubota S, Sene A, Lee TJ, Ban N, et al. SARM1 Depletion Rescues NMNAT1-Dependent Photoreceptor Cell Death and Retinal Degeneration. Elife (2020) 9:e62027. doi: 10.7554/eLife.62027
14. Sundaramoorthy V, Green D, Locke K, O’Brien CM, Dearnley M, Bingham J. Novel Role of SARM1 Mediated Axonal Degeneration in the Pathogenesis of Rabies. PloS Pathog (2020) 16:e1008343. doi: 10.1371/journal.ppat.1008343
15. Mukherjee P, Woods TA, Moore RA, Peterson KE. Activation of the Innate Signaling Molecule MAVS by Bunyavirus Infection Upregulates the Adaptor Protein SARM1, Leading to Neuronal Death. Immunity (2013) 38:705–16. doi: 10.1016/j.immuni.2013.02.013
16. Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity That Promotes Pathological Axonal Degeneration. Neuron (2017) 93:1334–1343.e5. doi: 10.1016/j.neuron.2017.02.022
17. Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1-Mediated Axon Degeneration Requires Both SAM and TIR Interactions. J Neurosci (2013) 33:13569–80. doi: 10.1523/JNEUROSCI.1197-13.2013
18. Shen C, Vohra M, Zhang P, Mao X, Figley MD, Zhu J, et al. Multiple Domain Interfaces Mediate SARM1 Autoinhibition. Proc Natl Acad Sci USA (2021) 118(4):e2023151118. doi: 10.1073/pnas.2023151118
19. Summers DW, Gibson DA, DiAntonio A, Milbrandt J. SARM1-Specific Motifs in the TIR Domain Enable NAD+ Loss and Regulate Injury-Induced SARM1 Activation. Proc Natl Acad Sci USA (2016) 113:E6271–80. doi: 10.1073/pnas.1601506113
20. Ma S, Lapin D, Liu L, Sun Y, Song W, Zhang X, et al. Direct Pathogen-Induced Assembly of an NLR Immune Receptor Complex to Form a Holoenzyme. Science (2020) 370(6521):eabe3069. doi: 10.1126/science.abe3069
21. Jiang Y, Liu T, Lee C-H, Chang Q, Yang J, Zhang Z. The NAD+-Mediated Self-Inhibition Mechanism of Pro-Neurodegenerative SARM1. Nature (2020) 588:658–63. doi: 10.1038/s41586-020-2862-z
22. Sporny M, Guez-Haddad J, Khazma T, Yaron A, Dessau M, Shkolnisky Y, et al. Structural Basis for SARM1 Inhibition and Activation Under Energetic Stress. Elife (2020) 9:e62021. doi: 10.7554/eLife.62021
23. Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, et al. SARM1 is a Metabolic Sensor Activated by an Increased NMN/NAD+ Ratio to Trigger Axon Degeneration. Neuron (2021) 109:1118–36.e11. doi: 10.1016/j.neuron.2021.02.009
24. Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, et al. A Rise in NAD Precursor Nicotinamide Mononucleotide (NMN) After Injury Promotes Axon Degeneration. Cell Death Differ (2015) 22:731–42. doi: 10.1038/cdd.2014.164
25. Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J. NMNAT1 Inhibits Axon Degeneration via Blockade of SARM1-Mediated NAD+ Depletion. Elife (2016) 5:1010. doi: 10.7554/eLife.19749
26. Di Stefano M, Loreto A, Orsomando G, Mori V, Zamporlini F, Hulse RP, et al. NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Curr Biol (2017) 27:784–94. doi: 10.1016/j.cub.2017.01.070
27. Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, et al. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-Apoptotic Cell Death. iScience (2019) 15:452–66. doi: 10.1016/j.isci.2019.05.001
28. Gilley J, Coleman MP. Endogenous Nmnat2 is an Essential Survival Factor for Maintenance of Healthy Axons. PloS Biol (2010) 8:e1000300. doi: 10.1371/journal.pbio.1000300
29. Harms A, Brodersen DE, Mitarai N, Gerdes K. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol Cell (2018) 70:768–84. doi: 10.1016/j.molcel.2018.01.003
30. Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, et al. Dsarm/Sarm1 is Required for Activation of an Injury-Induced Axon Death Pathway. Science (2012) 337:481–4. doi: 10.1126/science.1223899
31. Hicks AN, Lorenzetti D, Gilley J, Lu B, Andersson K-E, Miligan C, et al. Nicotinamide Mononucleotide Adenylyltransferase 2 (Nmnat2) Regulates Axon Integrity in the Mouse Embryo. PloS One (2012) 7:e47869–10. doi: 10.1371/journal.pone.0047869
32. Gilley J, Adalbert R, Yu G, Coleman MP. Rescue of Peripheral and CNS Axon Defects in Mice Lacking NMNAT2. J Neurosci (2013) 33:13410–24. doi: 10.1523/JNEUROSCI.1534-13.2013
33. Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP. Absence of SARM1 Rescues Development and Survival of NMNAT2-Deficient Axons. Cell Rep (2015) 10:1974–81. doi: 10.1016/j.celrep.2015.02.060
34. Summers DW, Frey E, Walker LJ, Milbrandt J, DiAntonio A. DLK Activation Synergizes With Mitochondrial Dysfunction to Downregulate Axon Survival Factors and Promote SARM1-Dependent Axon Degeneration. Mol Neurobiol (2020) 57:1146–58. doi: 10.1007/s12035-019-01796-2
35. Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, DiAntonio A. MAPK Signaling Promotes Axonal Degeneration by Speeding the Turnover of the Axonal Maintenance Factor NMNAT2. Elife (2017) 6:1–20. doi: 10.7554/eLife.22540
36. Sun Y, Wang Q, Wang Y, Ren W, Cao Y, Li J, et al. Sarm1-Mediated Neurodegeneration Within the Enteric Nervous System Protects Against Local Inflammation of the Colon. Protein Cell (2021) 12:621–38. doi: 10.1007/s13238-021-00835-w
37. Roussin M, Salcedo SP. NAD+-Targeting by Bacteria: An Emerging Weapon in Pathogenesis. FEMS Microbiol Rev (2021) fuab037. doi: 10.1093/femsre/fuab037
38. Freire DM, Gutierrez C, Garza-Garcia A, Grabowska AD, Sala AJ, Ariyachaokun K, et al. An NAD+ Phosphorylase Toxin Triggers Mycobacterium Tuberculosis Cell Death. Mol Cell (2019) 73:1282–91.e8. doi: 10.1016/j.molcel.2019.01.028
39. Skjerning RB, Senissar M, Winther KS, Gerdes K, Brodersen DE. The RES Domain Toxins of RES-Xre Toxin-Antitoxin Modules Induce Cell Stasis by Degrading NAD+. Mol Microbiol (2019) 111:221–36. doi: 10.1111/mmi.14150
40. Smith CL, Ghosh J, Elam JS, Pinkner JS, Hultgren SJ, Caparon MG, et al. Structural Basis of Streptococcus Pyogenes Immunity to Its NAD+ Glycohydrolase Toxin. Structure (2011) 19:192–202. doi: 10.1016/j.str.2010.12.013
41. Sun J, Siroy A, Lokareddy RK, Speer A, Doornbos KS, Cingolani G, et al. The Tuberculosis Necrotizing Toxin Kills Macrophages by Hydrolyzing NAD. Nat Struct Mol Biol (2015) 22:672–8. doi: 10.1038/nsmb.3064
42. Tang JY, Bullen NP, Ahmad S, Whitney JC. Diverse NADase Effector Families Mediate Interbacterial Antagonism via the Type VI Secretion System. J Biol Chem (2018) 293:1504–14. doi: 10.1074/jbc.RA117.000178
43. Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, et al. Systematic Discovery of Antiphage Defense Systems in the Microbial Pangenome. Science (2018) 3:eaar4120–18. doi: 10.1126/science.aar4120
44. Ofir G, Herbst E, Baroz M, Cohen D, Millman A, Doron S, et al. Antiviral Activity of Bacterial TIR Domains via Signaling Molecules That Trigger Cell Death. bioRxiv (2021). doi: 10.1101/2021.01.06.425286
45. Coronas-Serna JM, Louche A, Rodríguez-Escudero M, Roussin M, Imbert PRC, Rodríguez-Escudero I, et al. The TIR-Domain Containing Effectors BtpA and BtpB From Brucella Abortus Impact NAD Metabolism. PloS Pathog (2020) 16:e1007979. doi: 10.1371/journal.ppat.1007979
46. Wan L, Essuman K, Anderson RG, Sasaki Y, Monteiro F, Chung E-H, et al. TIR Domains of Plant Immune Receptors Are NAD+-Cleaving Enzymes That Promote Cell Death. Science (2019) 365:799–803. doi: 10.1126/science.aax1771
47. Horsefield S, Burdett H, Zhang X, Manik MK, Shi Y, Chen J, et al. NAD+ Cleavage Activity by Animal and Plant TIR Domains in Cell Death Pathways. Science (2019) 365:793–9. doi: 10.1126/science.aax1911
48. Swiderski MR, Birker D, Jones JDG. The TIR Domain of TIR-NB-LRR Resistance Proteins Is a Signaling Domain Involved in Cell Death Induction. Mol Plant Microbe Interact (2009) 22:157–65. doi: 10.1094/MPMI-22-2-0157
49. Essuman K, Summers DW, Sasaki Y, Mao X, Yim AKY, DiAntonio A, et al. TIR Domain Proteins Are an Ancient Family of NAD+-Consuming Enzymes. Curr Biol (2018) 28:421–30.e4. doi: 10.1016/j.cub.2017.12.024
50. Morehouse BR, Govande AA, Millman A, Keszei AFA, Lowey B, Ofir G, et al. STING Cyclic Dinucleotide Sensing Originated in Bacteria. Nature (2020) 586:429–33. doi: 10.1038/s41586-020-2719-5
51. Zhang Q, Zmasek CM, Cai X, Godzik A. TIR Domain-Containing Adaptor SARM Is a Late Addition to the Ongoing Microbe-Host Dialog. Dev Comp Immunol (2011) 35:461–8. doi: 10.1016/j.dci.2010.11.013
52. Nanson JD, Kobe B, Ve T. Death, TIR, and RHIM: Self-Assembling Domains Involved in Innate Immunity and Cell-Death Signaling. J Leukoc Biol (2019) 105:363–75. doi: 10.1002/JLB.MR0318-123R
Keywords: NAD+, innate immunity, NMNAT2, axon degeneration, plant, metabolism, TIR domain, abortive infection
Citation: DiAntonio A, Milbrandt J and Figley MD (2021) The SARM1 TIR NADase: Mechanistic Similarities to Bacterial Phage Defense and Toxin-Antitoxin Systems. Front. Immunol. 12:752898. doi: 10.3389/fimmu.2021.752898
Received: 03 August 2021; Accepted: 26 August 2021;
Published: 23 September 2021.
Edited by:
Bostjan Kobe, The University of Queensland, AustraliaReviewed by:
Hyun Ho Park, Chung-Ang University, South KoreaJonathan Gilley, University of Cambridge, United Kingdom
Michael Carty, Trinity College Dublin, Ireland
Copyright © 2021 DiAntonio, Milbrandt and Figley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron DiAntonio, ZGlhbnRvbmlvQHd1c3RsLmVkdQ==