 Zachary J. Morse
Zachary J. Morse Marc S. Horwitz
Marc S. Horwitz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 October 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.751337
This article is part of the Research TopicImmunopathology of Type 1 DiabetesView all 21 articles
In addition to genetic predisposition, environmental determinants contribute to a complex etiology leading to onset of type 1 diabetes (T1D). Multiple studies have established the gut as an important site for immune modulation that can directly impact development of autoreactive cell populations against pancreatic self-antigens. Significant efforts have been made to unravel how changes in the microbiome function as a contributor to autoimmune responses and can serve as a biomarker for diabetes development. Large-scale longitudinal studies reveal that common environmental exposures precede diabetes pathology. Virus infections, particularly those associated with the gut, have been prominently identified as risk factors for T1D development. Evidence suggests recent-onset T1D patients experience pre-existing subclinical enteropathy and dysbiosis leading up to development of diabetes. The start of these dysbiotic events coincide with detection of virus infections. Thus viral infection may be a contributing driver for microbiome dysbiosis and disruption of intestinal homeostasis prior to T1D onset. Ultimately, understanding the cross-talk between viral infection, the microbiome, and the immune system is key for the development of preventative measures against T1D.
Type 1 diabetes (T1D) is a persistent autoimmune disorder where immune cells attack and destroy the insulin-producing beta cells of the pancreas. Eventually, once enough beta cell mass is lost, individuals will begin to experience loss of natural blood glucose regulation and become reliant on exogenous administration of insulin. Numerous studies have characterized genetic variance and single nucleotide polymorphisms associated with T1D, which can explain why some individuals are more predisposed than others (1–6). Genome-wide association studies have found that Human leukocyte antigen (HLA) and insulin genes are responsible for a significant portion of the genetic risk for T1D. Additionally, many polymorphisms have been identified within immune-related genes including PTPN22, IFIH1, CTLA4, and IL2RA (5, 6). However, genetic make-up only accounts for part of the equation. After all, the immune system is shaped to an incredible extent by non-heritable forces and instead moulded largely by environmental exposures (7).
An array of exogenous stressors have been associated with precipitating autoimmunity (8). However, understanding exactly how environmental factors contribute to disease pathogenesis is a messy ordeal. Dysbiosis, infection, exposure to dietary antigen, and vitamin D deficiency have all been significantly implicated in altering susceptibility to T1D (9, 10). With such complicated etiology, incorporation of multi-faceted approaches, which take into account the extensive amount of cross-talk that occurs between each of these influences on the host, should be strongly considered in future studies.
Virus infections may be an instigating factor for the gut pathology and dysbiosis that is observed in patients leading up to islet autoimmunity and/or T1D onset (Figure 1). Clinical evidence suggests that diabetic patients experience prolonged enterovirus infections associated with the gut mucosa, resulting in persistent inflammation. Furthermore, patients with islet autoimmunity have increased intestinal permeability, low-grade enteropathy, and a dysbiotic microbiome. Seasonal patterns observed in T1D and other autoimmune disease diagnosis could, at least partially, be explained by seasonal variations in infection (11, 12). In this review, we will examine the known effects of virus infection on the microbiome and gastrointestinal (GI) physiology, and how this modulation may relate to T1D pathogenesis.
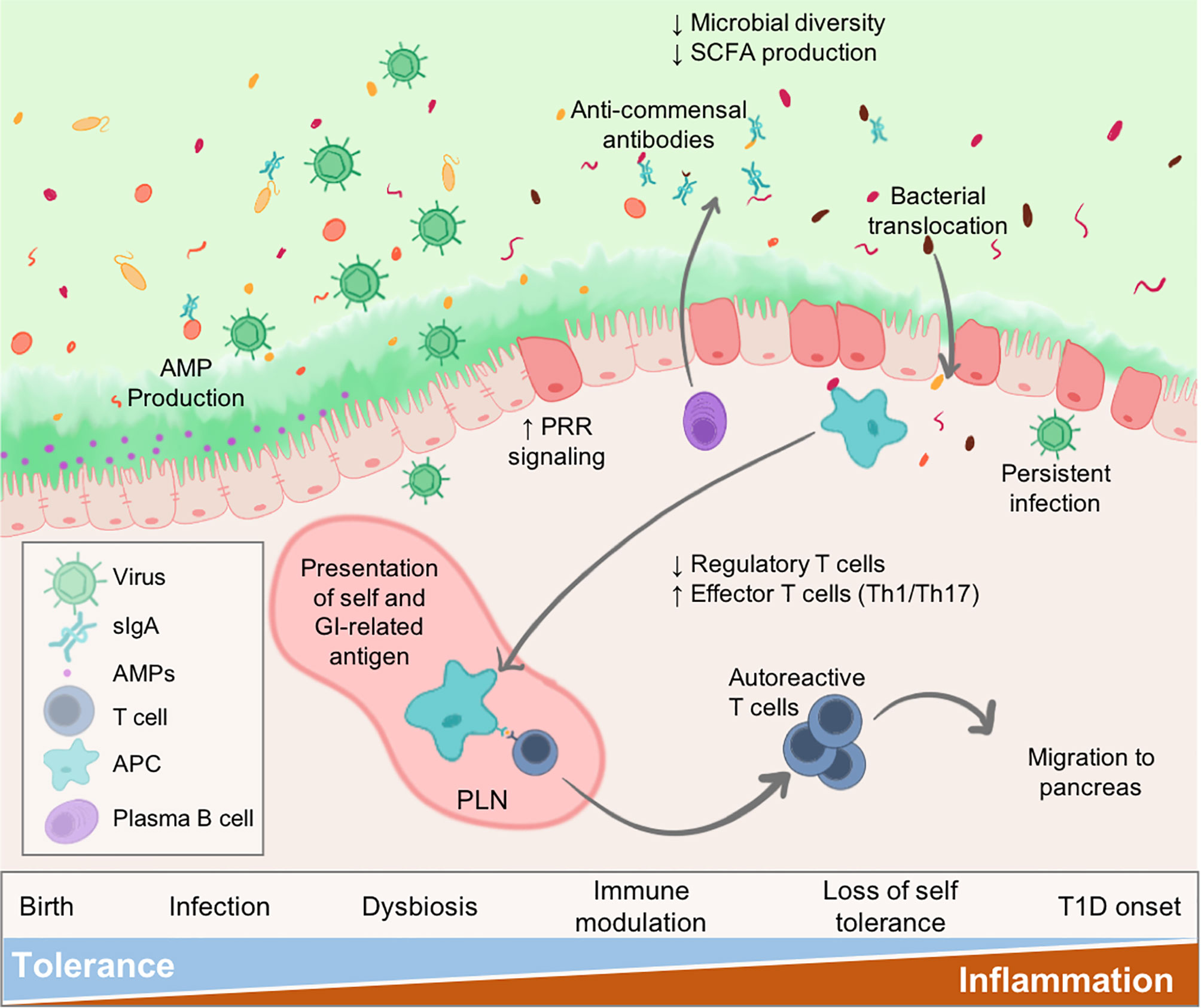
Figure 1 Virus infections alter intestinal homeostasis to contribute to T1D. The GI environment is tightly regulated by numerous mechanisms. Perturbations such as virus infection results in dysbiosis and disruption to the enteric environment. Microbial dysbiosis is characterized by loss of species diversity and production of SCFAs including butyrate and acetate. As a result of dysbiosis and inflammation, the epithelial barrier becomes more permeable due to loss of tight junctions between epithelial cells, alteration of secreted IgA (sIgA) antibodies, and diminished mucus production. Some persistent infections may be maintained contributing to sustained inflammatory signalling within the gut. Both pancreatic self-antigens and commensal microbial antigens are taken up by APCs and presented to T cells in the pLN causing loss of self-tolerance. These autoreactive T cells migrate to the pancreas to contribute to anti-islet responses and destruction of insulin-secreting beta cells. Individuals would progress to T1D once sufficient beta cell mass is lost resulting in loss of blood glucose regulation.
Numerous viruses, particularly those associated with the gut, have been connected with T1D pathogenesis including enterovirus, rotavirus, cytomegalovirus, and norovirus (13–17). The enterovirus, coxsackievirus B (CVB), has been the virus most frequently associated with T1D. So much so, that recently there has been movement and discussion towards the necessity to develop a vaccine specific for coxsackievirus to help mitigate the globally increasing rates of T1D (18–21). CVB binds to the coxsackie and adenovirus receptor (CAR), which is highly expressed on the insulin-secreting beta cells in the pancreatic islets (22). Variance in CAR expression has been correlated with increased predisposition for T1D (23). In both human populations and experimental mouse models, infection with enteroviruses has been identified to precede onset of islet autoimmunity (24–26). A recent large-scale study looking at virus shedding in the stool of children found that while those with islet-autoantibodies had no difference in total incidence of infection, they did experience a higher rate of sustained enterovirus B (particularly of CVB serotype) infections, which may be contributing to islet autoimmunity (23). Interestingly, this study also found association of other mammalian viruses including human masadenovirus-C (HAdV-C), which actually correlates with reduced incidence of auto-reactivity. The authors suggest that this may be due to HAdV-C competitively inhibiting CAR engagement or through sustained activation of innate immunity resulting in protection from other strains of virus including enterovirus. Children who developed T1D and islet-specific autoantibodies also have a history of increased incidence of respiratory infections in early adolescence (27). It is unclear, however, if there may be underlying immune differences that cause these populations to have increased susceptibility to both these types of infections and T1D autoimmunity. But, children who experience early loss of B cell tolerance to insulin exhibit weak humoral protection against CVB, whereas those with autoantibodies to the T1D biomarker, glutamic acid decarboxylase (GAD), have competent CVB responses - signifying viral clearance may be altered in individuals with T1D-related autoimmunity (28).
Rotavirus infection in children with a genetic predisposition to T1D is associated with increased islet autoimmunity, signifying that infection may exacerbate autoimmunity and diabetes (14). In non-obese diabetic (NOD) mice, rotavirus infection has also been shown to accelerate onset of T1D (29). However, pre-existing autoimmunity is necessary to accelerate disease onset (29). Thus, rotavirus may likely promote pathogenic events rather than serving as a trigger of diabetes.
Antiviral responses to viruses including CVB can likely have direct effects within the pancreas in precipitating T1D (30–32). While CVB has been shown to impair beta cell function in vitro, evidence suggests that the virus itself does not destroy beta cells through cytopathy (30, 33). Antiviral responses are largely mediated through expression of the three classes of interferon (IFN): type I (IFN-α and IFN-β), type II (IFN-γ) and type III (IFN-λ). Innate viral receptor engagement and ensuing immune pathway activation can have a significant role in T1D initiation and pathogenesis (34, 35). A transient type I IFN signature has been observed preceding islet autoantibody development in genetically-susceptible children, but is lost by the time of diabetes diagnosis (36–38). This IFN signalling may be a significant contributor to the hyperexpression of major histocompatibility complex (MHC) class I, endoplasmic reticulum stress, epigenetic and transcriptional/translational modifications observed in the islet microenvironment prior to T1D development. Recently, researchers were able to detect viral signatures (enteroviral protein and dsRNA) in the islets of autoantibody-positive and recent-onset T1D donors along with increased interferon and microbial stress markers (39). There has also been some suggestion that terminally-deleted viral genomes are able to persist in the islet microenvironment causing inflammation and increased immune cell recruitment (40). While there are existing direct links to virus-causing inflammation and modulation of the immune system within the islet microenvironment, there are likely secondary effects of infection, which are also long-term contributors to disease pathogenesis including microbial dysbiosis.
Comprising of a rich diversity of bacteria, archaea, viruses, fungi, and helminths – the microbiome is a dynamic environment that is constantly shifting. This review primarily focuses on the impact of commensal bacterial communities and later the collective virome. The microbiome has a substantial role in shaping peripheral immune tolerance, activation, migration, and differentiation, as well as local inflammatory responses (41). In response, the immune system is in constant communication to respond to these fluctuations in stimuli (42). Alterations in the microbiome have been heavily implicated in the pathogenesis of T1D (43–45) and genetic risk for T1D autoimmunity even confers differences in the bacterial microbiome (46–48). The intestinal microbiota can exert potent influence on immune homeostasis through the production of various metabolites and particularly short chain fatty acids (SCFAs). Both clinical studies and mouse models have established SCFAs including butyrate, propionate, and acetate as significant factors affecting immune regulation in T1D pathogenesis (43, 49). Metabolite-related dietary patterns have been shown to influence T1D susceptibility and metabolomic alterations precede the development of islet autoantibodies in children (50, 51).
While the human microbiome can be quite heterogenous and studies examining the relationship between the microbiome and diabetes have produced highly variable results, there are some notable microbial hallmarks which have been often identified in individuals with T1D and islet-autoimmunity including: a marked decrease in the diversity of bacteria colonizing the gut, increased abundance of bacteria within the Bacteroides phylum, the loss of Firmicutes, and decreased production of SCFAs among other variances (52–55). However, understanding the effects of perturbations in humans while also controlling for extraneous factors is incredibly difficult. The use of NOD mice as a model for spontaneously developing diabetes has given significant insight into understanding the disease pathogenesis of T1D. While the autoimmunity experienced by NOD mice is not the same as that experienced by humans, it allows the use of environmental and genetic interventions in order to understand how they may impact diabetes development (56). Dysbiosis occurs in both humans and NOD mice prior to disease onset and diabetes incidence can even be predicted in these mice based on sampling from various mucosal microbiomes (43, 57, 58). A “diabetogenic” microbiome from a diabetes-prone NOD mouse is sufficient to promote insulitis when transferred to a non-obese diabetes-resistant (NOR) mouse (59). Typically, female NOD mice are more susceptible to developing autoimmune diabetes than male mice; however, this difference is not observed in germ-free mice (60). This discrepancy in sex bias can at least partially be explained by microbial stimulation of testosterone (60).
Pattern recognition receptors (PRRs) including toll-like receptors (TLRs), RIG-I-like receptors, and caspase activation and recruitment domains (CARDs) are innate sensors that can detect viral and microbial pathogen-associated molecular patterns (PAMPs). Signalling through these receptors can be detrimental for modifying susceptibility to T1D development (34). There are 13 total types of TLRs, each of which is specific for various bacterial (LPS, flagellin, peptidoglycan, etc.) and/or viral (dsRNA, CpG DNA, viral protein, etc.) antigens. Microbiota can regulate T1D through TLR signaling differences (61, 62). For instance, imbalance between TLR2 vs. TLR4 stimulation can determine T1D susceptibility where TLR2 provides a pro-diabetic signal whereas TLR4 provides microbiota-induced tolerization (61). This overlap in bacterial and virus infection immune signaling may signify a role between commensal microbes and virus infection in host immune regulation.
In the first few years of life, colonization of the GI tract plays an indispensable role in shaping host immune development, regulation, and maintenance (63, 64). With age, the microbiome experiences decreasing plasticity and tolerance for new antigen exposure and environmental disruptions (63, 65). Following infancy, the microbiome seems to stabilize with relatively established communities that continue to shape mucosal and systemic immune homeostasis into adulthood (66). Thus, timing of environmental perturbations is likely an important factor for producing dysbiosis, which impacts disease susceptibility. The “Hygiene Hypothesis” proposes that exposure to antigens in early life during immune development can have profound effects for the development of autoimmune and allergic disease later on. Evidence suggests that instigating factors leading to T1D occur early in life – especially since a majority of early-onset individuals who progress to overt T1D before adolescence develop autoantibodies by 3 years of age (1, 67). However, most individuals are diagnosed with T1D in adulthood, hinting that tolerance for environmental stressors may not necessarily be limited to a defined age or that triggering events can occur long before disease onset (68).
Infections that are relatively mild later in life, may have the ability to be quite detrimental early in life at promoting T1D, as the immune system is not yet fully developed and may lack the ability to properly defend the host (69). Viruses cause dysbiosis (70–72), potentially signifying lasting consequences whereby individuals may develop a more autoimmune-skewed microbiome that might be characterized by decreased diversity and less beneficial bacteria (e.g., less butyrate producers). Studies in NOD mice have shown early life exposure to a “diabetogenic microbiome” through fecal microbiome transfers (FMT) can regulate B cell activation and promote T1D onset later on (69). However, when mice are given this same microbiome composition post-adolescence they do not experience the same modulation of the immune system and increased incidence of diabetes autoimmunity. Thus, there may be a unique window, particularly early in life, whereby disruptions in the microbiome from exogenous stressors like infection can have much larger implications on future health.
Studies have found that detection of enteric infection precedes islet autoimmunity by 6 months or more (24, 73, 74). The existing confluence between intestinal inflammation and T1D maintain the gut as an important site for immune modulation that has implications for islet autoreactivity. While some viruses may have deleterious effects on the microbiome, others may actually promote tolerance. For example, norovirus infection was shown to protect from T1D through modulation of the microbiome (75). In this study, Pearson et al. found that NOD mice infected with murine norovirus (MNV) had significantly lower diabetes incidence, less immune infiltration into the islets, increased bacterial diversity, and an increased regulatory rather than inflammatory T cell profile.
Islet-autoreactive CD8+ T cells circulate in the blood in approximately the same quantities between healthy and diabetic patients – suggesting that these cells are a normal part of the T cell repertoire (76). However these cells are more abundant in the pancreata of T1D patients, indicating that they must home to the pancreas due to altered immunoregulatory signalling, proinflammatory islet environment, and/or peripheral activation (76). The GI tract plays a fundamental part in communicating between the host and microbiota. Even at healthy steady-state conditions, there is significant T cell trafficking between the gut and pancreatic tissues (77). Existing within this gut-pancreas axis, the pancreatic lymph nodes (pLN), which drain from the duodenum and pancreas, are sampling antigen heavily from both organs. The pLN resides at a critical and significant confluence whereby intestinal stress can alter the presentation of pancreatic self-antigens (77). It has even been suggested that this may be the portal connecting celiac disease with T1D, where GI inflammation due to gluten-sensitivity potentially stimulates anti-islet immune activation in the pancreas (77, 78). Diabetic patients experience prolonged enterovirus infections associated with the gut mucosa, resulting in persistent inflammation (79). This sustained inflammation may be sufficient to result in loss of self-tolerance and T1D development.
Resident T and B cells hold specificity for commensal microbes even under healthy homeostatic conditions (80). T cell polarization into T helper 1 (Th1), Th2, Th17, or regulatory (Treg) cell phenotypes can be driven in the gut by presence and abundance of specific microbes in autoimmunity (42, 81, 82). For instance, Bifidobacteria species can drive Th17 cell responses (83) – while Akkermansia, Bacteroides, and most notably Clostridium species, have been shown to promote Treg populations (82, 84). Produced in large quantities, particularly by Clostridium bacteria, the SCFA butyrate is a potent inducer of Treg delineation through histone modification promoting Foxp3 expression and, by eliciting high levels of transforming growth factor β (TGF-β), expression in gut-related CD103+ dendritic cells (85, 86).
Insight into the pathogenesis of T1D has revealed that Treg cells can be potent mediators for the suppression of autoreactive T cells and promotion of tolerance to islet antigen (87). Inflammasome-deficient mice have a microbiome that is protective for T1D (88). When NOD mice are co-housed with these protected mice they experience a corresponding reduction in diabetes incidence (89). This is attributed to an expansion of type 1 regulatory T cells in the gut, which home to the pancreas and secrete IL-10 to reduce inflammation in the pancreatic microenvironment. This microbiome-driven alteration in Treg populations is likely due to production of bacterial SCFA metabolites since administration of butyrate to NOD mice also causes initial expansion of Tregs in the colon, mesenteric lymph nodes (mLN), and Peyer’s patches with a subsequent migration to the pancreas and pLN to reduce T1D onset (90). Expansion of Ruminococcus species of bacteria can also promote CD8+ Treg cells to prevent diabetes in NOD mice and a streptozotocin-induced model. Furthermore, healthy human donors have increased CD8+ Tregs along with increased Ruminococcus when compared to T1D patients (91). These gut-primed Tregs may have a profound impact on maintaining pancreatic tolerance and may be limited in infection since enterovirus detection in young children is associated with ensuing depression of Treg responses and increased inflammatory Th1/Th17 responses (92).
Mucosa-associated invariant T cells (MAIT) are innate-like T cells expressing MHC class 1-related protein (MR1) that specifically binds microbial metabolites originating from riboflavin metabolite biosynthesis in bacteria. These cells are present in several tissues, and like their name suggests, they are important at mucosal sites (93). MAIT cells exist at an interesting interface and may be a key mediator between microbes, virus infection, and T1D. Germ-free mice lack MAIT cells, thus indicating that they likely rely on commensal bacteria for their development and maintenance (93). In fact, differences in bacterial metabolism can regulate MAIT cell activation (94, 95). Typically, MAIT cells are thought to have a protective phenotype whereby they promote intestinal homeostasis and have a role in supporting the gut epithelial barrier via secretion of IL-22, IL-12, and IL-17a (96). However, MAIT cells can also take on a more pathogenic nature in certain circumstances.
Rouxel et al. found that both recent-onset and established T1D patients have altered MAIT cell populations circulating in their blood whereby they are less abundant, express more activation/exhaustion markers, Th1-skewed, and are more cytotoxic (97). In NOD mice, MAIT cells seem to show a dimorphic phenotype depending on tissue specificity where MAIT cells in the lamina propria express IL-22 and IL-17a in non-diabetic mice; however, cells that infiltrate the pancreatic islets express IFN-γ and granzyme B to participate in beta cell destruction. Furthermore, the authors showed that MAIT cell-deficient (MR1-restricted) NOD mice have increased rates of diabetes and have a modified gut mucosal environment – suggesting that they can be protective (97). Beyond their role in sensing bacterial products, MAIT cells have also been identified to hold potent inflammatory responses in both acute and chronic virus infections (98). This is due to activation, which is independent of MR1 stimulation and instead due to cytokine signaling largely through type-1 interferon, IL-12, and IL-18 (98, 99). Ultimately, collective signalling from bacterial metabolites and cytokine profiles in infection may be detrimental in skewing MAIT populations toward either a protective or pathogenic nature in T1D pathogenesis.
Mariño et al. found that providing diets to NOD mice that yield increased production of acetate and/or butyrate are largely protected from autoimmune diabetes (49). These two SCFAs accomplish this through their own distinct mechanisms. While butyrate primarily boosted Tregs, acetate decreased frequency of islet-specific autoreactive T cells by modulating antigen presentation in B cell populations residing in the spleen and intestinal Peyer’s patches. Cross presentation of islet antigen by B cells in the pLN has been previously been shown to activate self-reactive CD8+ T cells (100).
Plasmacytoid dendritic cells (pDC) play an important part in mediating antiviral intestinal immunity. These cells extend their dendrites across the epithelial cell barrier to sample microbial antigen in the GI tract to present to resident adaptive immune cells and can produce a significant amount of IFN in infection. pDCs infected with rotavirus can induce bystander activation of islet-reactive T cells via type I interferon signalling (17). Mucosa-associated pDCs likely detect virus infection and travel to the mesenteric and/or pLN to promote B cell expression of MHC-I and proinflammatory T cell cytokine secretion to aid in inflammation (101, 102). Phagocytosis of Lactococcus lactis bacteria by pDCs can stimulate robust IFN-α secretion via TLR9 and MyD88 signalling (103). Oral administration of the L. lactis colonization factor antigen I fimbriae can also prevent T1D in NOD mice by promoting expansion of IL-10 and IFNγ while decreasing Th1 T cells (104).
MNV infection alters recruitment of macrophages in the pLN where they are deficient in CD86, signifying a decreased capacity to activate T cells leading to protection from T1D (75). Furthermore the offspring of antibiotic-treated pregnant NOD mice also experience reduced T1D incidence by instigating tolerized APCs (105). These APCs have a diminished ability to activate cytotoxic CD8+ T cells and thus represent the importance for microbiome-specific education of developing immune self-tolerance. Macrophages which lack previous exposure to bacteria in antibiotic-treated mice have reduced responses to LPS antigens (106). Decreased inflammatory responses by these APC populations due to microbiome differences may be sufficient to prevent autoreactivity – especially since islet resident macrophages are detrimental for the instigation of T1D autoimmunity in NOD mice (107).
Containment of commensal bacteria and dietary antigens within the intestinal lumen relies on several physiological and molecular barriers. The first line of defense is a layer of mucus created by O-linked glycoproteins (mucins) secreted from intestinal goblet cells combined with luminal saccharides. In the colon, a double layer of mucus serves as a physical barrier. The apical layer is typically colonized with various mucus-degrading microbes including those within the Akkermansia family. The innermost mucus layer, however, is predominately uncolonized and creates a largely impenetrable barrier for bacteria. A single cell layer of epithelial cells (IEB) is joined through tight junctions to create a continuous cellular barrier throughout the GI tract. This IEB can be maintained by cytokines including IL-22 produced by group 3 innate lymphoid cells and IL-17A from Th17 lamina propria T cells. Epithelial cells and resident lamina propria immune cells constantly sample the mucosal environment and respond to changes in microbial and viral stimuli. Commensal bacteria populations are regulated through production of antimicrobial peptides (AMPs) and by secreted IgA antibodies. AMPs are bactericidal for specific bugs, particularly within the small intestine where the mucus barrier can be more discontinuous. Colonization of bacteria within the GI tract is also highly regulated by IgA antibodies, which can coat bacteria for neutralization and opsonization. Most secreted IgA is polyreactive and holds an innate specificity to multiple strains of bacteria, but can also undergo somatic hypermutation to produce highly specific IgA against particular bacteria (91, 108).
Autoimmune disorders including T1D, rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus (SLE) have all been associated with increases in intestinal permeability - or a so-called “leaky gut” (109–111). Clinical studies have found that individuals with islet autoimmunity experience increased intestinal permeability and low-grade enteropathy (112–116). Loss of integrity occurs prior to T1D development in both human and mouse models, indicating that it may be a significant trigger – rather than a result – of autoimmunity (112, 117). In fact, Sorini et al. found that breaking the intestinal barrier using low-dose dextran-sulfate sodium (DSS) treatments in NOD mice was sufficient to increase onset of autoimmune diabetes (109). This subsequent loss of intestinal integrity can induce activation of islet-specific immune cells in the gut to travel to the pancreas and promote onset of diabetes in T cell receptor-transgenic BDC2.5 crossed NOD mice. Activation of these T cells also appeared to be dependent on the presence of the gut microbiome; however, microbial dysbiosis caused by the DSS-treatment alone was not sufficient to promote autoimmunity.
Breakage of the tight junctions, which glue together the intestinal epithelial barrier, may be a contributing factor in allowing permeability and contribute to T1D pathogenesis (118). As a result of reduced intestinal integrity, bacteria can cross mucosal barriers and leak into systemic circulation and various tissues. When disseminated systemically, commensal bacteria antigens can rapidly promote diabetes autoimmunity in NOD mice (119). Translocation of bacteria can contribute to autoreactivity in the following ways: 1) by directly damaging the beta cells (120) 2) through presentation of bacterial antigen to autoreactive T cells (109) 3) in promoting inflammation through innate receptor stimulation (121) 4) through bacterial molecular mimicry of self-antigens (122). In fact, translocation to the pLN has been observed in NOD mice prior to diabetes onset (123). It can also trigger activation of the innate bacterial peptidoglycan receptor, NOD2, to contribute to T1D development in a streptozotocin-induced mouse model (121). Islets exposed to translocated bacteria can directly mount anti-bacterial responses and promote inflammation (124). These responses may ultimately aid in recruitment and activation of autoreactive cells within the pancreatic environment.
Miranda et al. performed an extensive analysis looking at alterations at the mucosal immune environment in NOD mice prior to diabetes development (123). This study found that the mice developed impaired mucin production, dysbiosis, modified secretion of bacteria-specific IgA, and alterations in lamina propria dendritic and T cell populations – which skew toward an inflammatory rather than regulatory profile. Some of these changes were shown to be microbiome-driven since cross-fostering NOD pups with NOR mothers can restore mucus production. The intestinal mucus layers represent the first line of defense against intestinal microbes and can be modulated by the presence of specific bacteria. Specifically, butyrate-producing and mucin-degrading bacteria can improve intestinal integrity through regulation of epithelial tight junctions and stimulate production of mucin synthesis, respectively (49, 125, 126). Acetate- and butyrate-yielding diets correspond to a reduced concentration of bacterial lipopolysaccharide (LPS) antigens detected in the serum of mice - indicating reduced bacterial dissemination (49). The mucin-degrading bacteria, Akkermansia muciniphilia, can reduce intestinal permeability through fortification of epithelial tight junctions (126). Administration of A. muciniphilia can reduce diabetes incidence in NOD mice by modulating mucus production and expression of antimicrobial peptides (127). Furthermore, the colonization of A. muciniphilia decreased islet expression of TLRs and promoted regulatory T cells (127). This potentially signifies a change in the host’s ability to respond to subsequent infections and susceptibility to infection-induced diabetogenic responses.
Alterations in the abundance of certain bacterial antigens have been previously observed to elicit specific IgG antibody responses to commensal bacteria suggesting that B cell receptor and TLR stimulation can alter GI-related B cell profiles (128). Furthermore, SCFA metabolite concentration can drive production of bacteria strain-specific IgA in a T cell-dependent mechanism involving TLR recognition – resulting in altered bacterial colonization of mucosal environments (108). The presence of T1D and/or autoimmune risk alleles confers alterations in IgG and IgA anti-commensal microbial responses in HLA haplotype-dependent and -independent mechanisms (129). For example, Huang et al. observed that newly-diagnosed T1D patients have increased secretory IgA responses along with dysbiosis and decreased SCFA production (130). Performing FMTs to transfer the microbiota from these T1D patients to germ-free NOD mice results in similar alteration in IgA-mediated immunity in these mice. However, administration of the SCFA acetate is able to recover this modulation and restore IgA responses. It has yet to be determined if dysregulation of IgA-mediated control of commensal bacteria communities and intestinal homeostasis has role in contributing to T1D autoimmunity or if it is a by-product of dysbiosis and/or metabolic pathogenesis. Some evidence has indicated that changes in the anti-commensal antibody milieu occurs after seroconversion, but prior to T1D onset (48).
Collectively, research included in this review suggests that a “leaky gut” is a natural part of T1D pathophysiology that likely triggers and/or progresses disease (Table 1). Virus infections may be a causative agent to aid in microbiome-related promotion of autoreactivity. Increased gut inflammation invariably leads to loss of epithelial integrity and a breakdown of the barriers – thereby allowing dissemination of bacteria from the gut and increasing immune accessibility to antigens within the GI tract. Chronic viral infection is sufficient to drive sustained intestinal permeability (133). This infection-induced epithelial damage can be mitigated through blockade of type I IFN or depletion of CD8+ T cells (133). Infection with Citrobacter rodentium is able to produce barrier disruption along with increased insulitis in NOD mice (134). Respiratory infections are known to cause gastrointestinal distress, dysbiosis, and increased intestinal permeability despite an absence of virus in the GI environment (135). SARS-CoV2 patients experience noted dysbiosis and loss of intestinal integrity corresponding with more severe systemic inflammation, bacteremia, and higher mortality rate – potentially signifying a leaky gut as a contributor to worsening disease outcomes (136, 137). Additionally, human immunodeficiency virus (HIV) infection has been shown to cause systemic immune activation and AIDs-related morbidity due to translocation of bacteria from the intestinal lumen (138). In fact, HIV positive individuals can experience systemically disseminated bacteria resulting in stimulation of anti-CD4+ T cell autoantibody production (139).
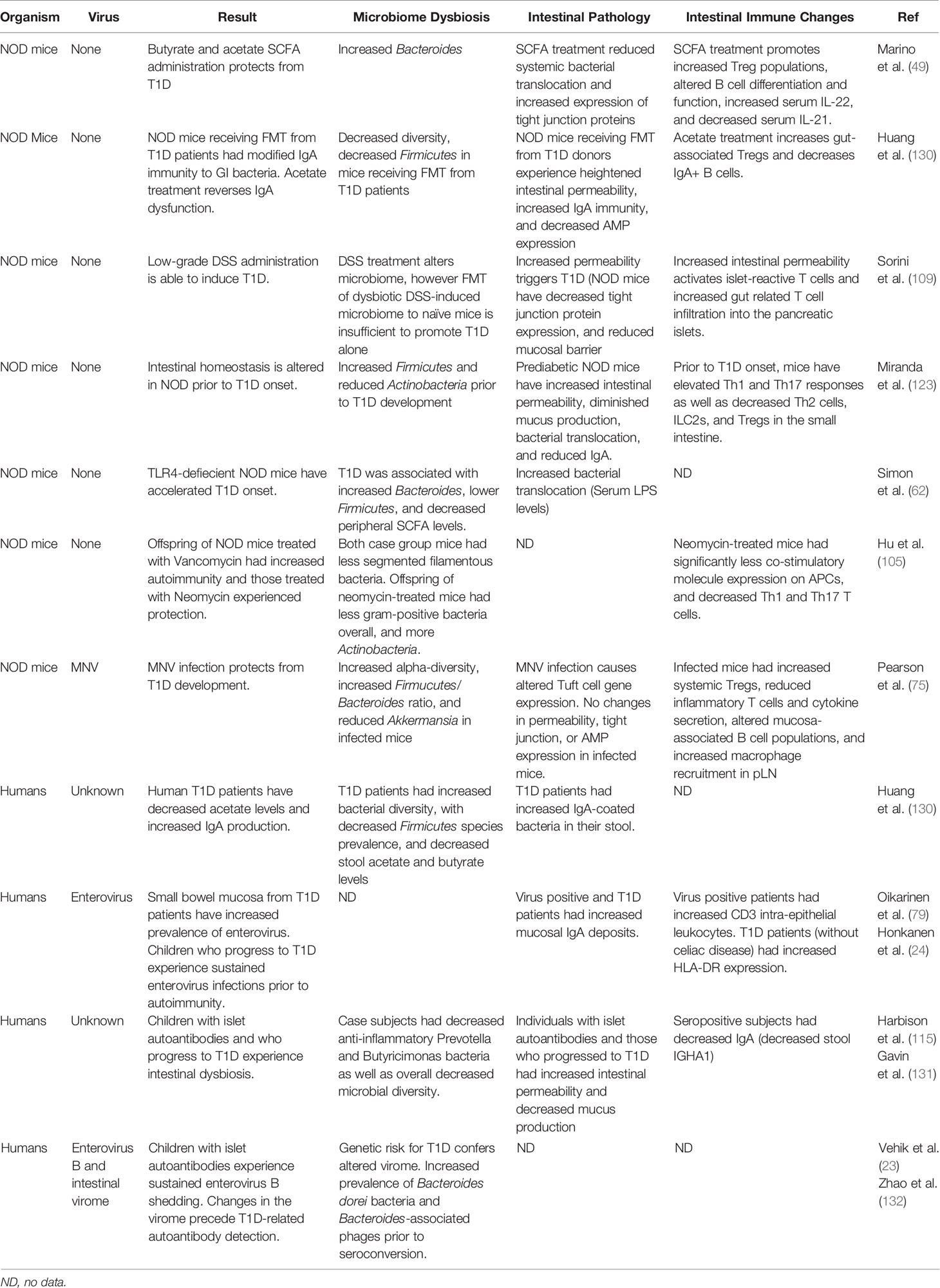
Table 1 Highlighted recent studies depicting intestinal changes associated with T1D.
The intestinal virome is made up of rich and diverse prokaryotic and eukaryotic viral communities, which are shaped by numerous factors including diet, genetics, disease, and geography (140). While a vast majority of the viruses in the body are bacteria-infecting phages, the human virome is also made up of: genomically-integrated human endogenous retroviruses (HERVs); latently-infecting viruses, such as human herpes viruses (HHVs); and potentially persistent/chronic infections – including common enteric viruses previously discussed in this review (CVB, norovirus, rotavirus, etc.) (23, 141). With the GI tract being the most abundant site of viral colonization, the intestinal virome is crucial for maintaining homeostasis and regulating disease pathogenesis through interaction with both commensal bacteria as well as the host (142). Typically germ-free and antibiotic-treated mice face immune dysfunction and altered intestinal morphology. However, infecting these mice with MNV mitigates these aberrations in the intestinal environment (143). Norovirus is therefore sufficient to preserve gut homeostasis and intestinal immunity in a manner that is typically served by microbiota. With potential for such an influential impact, it should be no surprise that alterations and dysbiosis in the viral composition have been associated with several diseases and can alter host immune homeostasis, particularly within mucosal environments (142, 144).
Using metagenomic analyses, researchers have observed the intestinal virome dramatically shifting prior to onset of T1D (23, 132, 145). Zhao et al., for instance, found that healthy donors had significantly higher viral diversity and increased abundance of Circoviridae-related sequences when compared to children who developed autoantibodies and T1D (132). These differences were observed prior to seroconversion and were also reflected in coinciding dysbiosis in bacterial communities. This suggests that there is a viral-bacterial relationship in precipitating autoimmunity. While modulation of commensal bacteria through phage bactericidal predation is not well understood, the ability of certain phages to affect bacterial abundance and modify bacterial fitness is particularly exemplified by the success of phage therapies in treating antibiotic-resistant bacterial infections (146). A study by Hsu et al. showed how phage-mediated killing has cascading effects within the microbiome, resulting in expansion or attrition of non-target bacterial populations and causing altered gut metabolomic profiles (147). These results suggest that lytic bacteriophages and the induction of prophages can be potent modulators of the bacterial microbiome and their effects can be amplified between molecular and cellular signals in the GI environment.
An exhaustive study by Dallari et al. characterized host immune responses to several asymptomatic virus infections (acute and persistent strains of MNV, mastadenoviruses, astrovirus, parvoviruses, and reoviruses) in conventional and germ-free mice (144). The authors identified both distinct and common immune modulation contributed by viral and bacterial microbes. Viruses were generally responsible for eliciting Th1- and IL-22-mediated immunity as well as B cell and bacterial response pathway activation. While each virus exposure promoted profound immunomodulation, there was little consistency in immune pathways activated by each virus examined. Viral genome type, virus persistence, and viral load were only modestly attributed to the observed immune variance suggesting there is a largely individualistic and strain-specific contribution to intestinal immunity. While bacterial members of the microbiome have been the major focus of research in respect to their ability to shape mucosal immunity, this highlights importance and impact virus exposure also has within both GI-related and systemic immune homeostasis.
Despite eukaryotic cells not being a natural target for bacteriophages, their presence can alter host immune profiles. This is most often accomplished by bacteriophage stimulation of viral PRRs, including TLRs or RIG-I-like receptors. One study showed how phage taken up in antigen-presenting cells activates TLR3 signalling and subsequently type I IFN expression (148). Another study demonstrated Lactobacillus, Bacteroides, and Escherichia phages can promote IFN-γ-producing T cells along with IL-6, IL-10, and IL-12 secretion via TLR9 activation in germ-free mice (149). These changes can alter susceptibility to ensuing bacterial and viral infection. For instance, the presence of murine astrovirus has been shown to protect against MNV and rotavirus infection via stimulation of type III interferon signalling in the gut epithelium (150). Type III IFN expression in epithelial cells may also be detrimental in determining persistence of CVB in enteric environments (151). Phage-mediated cell lysis of bacteria would also result in increased release of antigenic bacterial PAMPS that go on to initiate inflammation through PRR activation. Bacteriophage induced amyloid production in E. coli has been associated with subsequent seroconversion and development of T1D (152). This effect is hypothesized to be caused by the release of E. coli amyloid-DNA PAMPs, which are known inducers of TLR2 and TLR9 and have been previously shown to trigger SLE autoimmunity in mice (153). However, more evidence is needed to determine if this mechanism can be directly contributing to T1D.
Ancestral viruses have integrated into the mammalian genome over millions of years of evolution, resulting in human endogenous retroviruses (HERVs). Some estimates attribute approximately 8% of the human genome to a viral origin (154). These genomic viral remnants largely go unexpressed. However, they can be induced by exogenous stressors including CVB and other viral infections (155–157). Expression of HERV antigen, particularly from the HERV-W family, has been associated with both T1D and multiple sclerosis autoimmunity in humans and mouse models (158–161). Mycobacterial infection can stimulate expression of the HERV-W envelope antigen, resulting in increased cross-reactive autoantibody expression in children at higher risk of T1D (162). Murine ERV antigens can be detected in the islets of NOD mice as disease progresses and anti-ERV immunity correlates with anti-islet reactivity (158). Furthermore, inducing expression of HERV-W-Env protein in mice causes hyperglycemia, reduced insulin production, and increased immune infiltration into the pancreas (159). This indicates a potential role in promoting inflammatory events within the islet microenvironment. While the exact role is to be determined, HERV-W-Env involvement in autoimmunity has been at least partially attributed to its signalling via CD14 and TLR4 PRR stimulation in APCs resulting in activation of Th1 and antimicrobial immune pathways (163).
Antigenic similarity between viral and host proteins can also potentially contribute to autoimmune responses. Antibodies against CVB4 viral protein can positively recognize beta cell antigen and induce cell apoptosis (164). Commensal viruses including Poxviruses, HHVs and other dsDNA viruses have been shown to exhibit sequence homology with multiple human peptide hormones such as insulin, insulin-like growth factors (IGFs), adiponectin, and resistin (165). Viruses in the Iridoviridae family express viral insulin/insulin-like growth-1-like peptides (VILPs), which share a significant homology with human insulin/IGF-1. These VILPS are able to adequately bind to, and cause activation of, their respective hormone receptors in both humans and mice (165). Whether this similarity can contribute to antigenic cross-reactivity against endogenous insulin in T1D has yet to be seen.
There is a significant degree of bidirectional influence between the microbiome and antiviral response. Not only does infection alter the microbial homeostasis, but the microbiome can also have a significant impact on the outcome of virus infection and the ensuing immunological responses (144, 166–168). The microbiome has been shown to determine severity of viral infection and promote resistance to enteric infection (169–171). Certain species of commensal bacteria can colonize intestinal lymphoid tissues including the Peyer’s patches and mLNs to modify antigen-presenting cell cytokine expression even under healthy homeostatic conditions (172). There is some evidence that microbial antigens may even share sufficient homology to induce cross-reactive T cells against pancreatic targets (76, 122). For instance, an integrase protein expressed by many bacteria within the Bacteroides genus is capable of serving as a low-avidity mimotope of pancreatic autoantigen (173).
Commensal bacteria can aid or limit virus infection through enhanced viral genetic recombination, stabilization of virus particles, promotion of virion dissemination to permissive cells, and modification to immune homeostasis (174). Surface bacterial polysaccharides, such as peptidoglycan and LPS, have been shown to promote virion stability and receptor engagement to increase poliovirus and reovirus infectivity in mice (170). Certain Bifidobacteria and Lactobacillus species have even exhibited an inhibitory potential of CVB4 in vitro (175, 176). Additionally, depleting microbiota through use of antibiotics is able to reduce rotavirus infection by promoting virus-specific humoral responses (177). Infection with H3N2 and H1N1 influenza strains in mice causes intestinal dysbiosis and results in reduced SCFA production and diminished immune responses to secondary infections (135). Conversely, commensal bacteria LPS and extracellular matrix-binding proteins have also been shown to destabilize influenza virions and block infection at mucosal sites, respectively (178, 179).
Intestinal bacteria can elicit prolonged steady-state activation of the innate and adaptive immune system to modify susceptibility to subsequent infection (180–182). For instance, commensal microbes can limit persistence of MNV infection in mice through stimulation of interferon signalling (183). Ultimately, bacterial stimulation of immune pathways may play an important role in setting the thermostat for ensuing pathogenic infections particularly in the intestinal environment (184). Antibiotic-treated mice have compromised innate and adaptive antiviral immune responses resulting in impaired ability to clear virus infection (181). This is likely because the sustained immunological stimulation from commensal microbiota lowers the activation threshold in order to establish a robust immune response against an invading pathogen. In fact, intestinal bacteria can send signals to lung stromal cells to maintain a primed baseline IFN signature to prepare against subsequent influenza infection and limit early viral replication (185). The antiviral thermostat may be altered in some individuals due to genetic variance and/or environmental stimulation. This may allow the establishment of persistent infections which have been observed prior to disease onset in individuals with islet-autoimmunity and T1D (23).
Understanding how enteric viruses contribute to homeostatic regulation of immunity and may contribute to autoimmune disorders is of great importance. Consequences of virus exposure within the intestinal environment are difficult to determine due to a lack of established animal models and confounding variables including commensal microbes commonly found in murine colonies (e.g., SFB, astrovirus), which may limit viral infection and skew results (150, 186). Ultimately, mice also exhibit differences in viral susceptibility, tropism, and pathogenesis when compared to humans.
Changes in the microbiota have been observed to occur prior to autoimmunity development, which suggests that dysbiosis has a causative role in T1D rather than a result of autoreactive or metabolic pathophysiological responses (58). Intervention studies in humans modulating the microbiome through dietary means or FMT have shown some success in improving T1D outcomes and prevention (187). However, conclusive results in these studies may be limited and require further work.
While there remains much controversy with regards to the precise role and importance of virus infection and the microbiome in determining whether a genetically susceptible individual will lose self-tolerance, significant efforts are being made to understand the patterns and commonalities that are able break through the heterogeneity of human data, background noise, and experimental limitations currently impeding understanding of these issues. Mouse models certainly provide a great deal of potential mechanistic insight into T1D; however, longitudinal human studies integrating clinical data for microbiome differences, infection history, and susceptibility to T1D-related autoimmunity are absolutely necessary to dissect the complicated etiology leading to diabetes development. Blood and stool samples from these large cohort studies can shed light on changes in the microbial, viral, and immunological landscape prior to disease onset. Furthermore, intestinal inflammation and potential increases in gut permeability can be identified by determining abundance of blood markers, clinical tests, and presence of translocated bacterial antigen (188).
Communication between the intestinal microbiota and resident immune populations likely have a profound role in dictating susceptibility and immune system response to virus infection. The intimate inter-relatedness of genetic susceptibility, viral responses, dysbiosis, and host immune state produces an incredibly complex web whereby perturbation can cause a myriad of effects. Understanding the experimental complexity in host-virus-microbe interactions is a monumental challenge. It is difficult to determine which factors and pathways are active contributors to, rather than incidental by-products of, disease. Though challenging, exploring this relationship further is necessary to inform the ultimate prevention, detection, and treatment of autoimmunity.
ZM and MH conceptualized, wrote, and edited the manuscript. ZM created the figures. All authors contributed to the article and approved the submitted version.
The work was funded by grants from the Juvenile Diabetes Research Foundation and GenomeBC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Isobel C. Mouat and Dr. Maya Lefkowich for their help with editing and reviewing the manuscript. Our work and research take place on the traditional, ancestral, and unceded territory of the xʷməθkʷəy̓əm (Musqueam), səlil̓ilw̓ətaʔɬ (Tsleil Waututh), and Sk;wx̱wú7mesh (Squamish) First Nations.
AMP, antimicrobial peptide; APC, antigen-presenting cell; CAR, coxsackie and adenovirus receptor; CARD, caspase activation and recruitment domains; CTLA4, cytotoxic T-lymphocyte associated protein 4; CVB, coxsackievirus B; dsRNA, double stranded RNA; DSS, dextran-sulfate sodium; EBV, Epstein-Barr virus; FMT, fecal microbiome transfer; GAD, glutamic acid decarboxylase; GI, gastrointestinal; HAdV-C, human masadenovirus-C; HERV, human endogenous retroviruses; HHV, human herpes viruses; HLA, Human leukocyte antigen; IEB, intestinal epithelial barrier; IFIH1, interferon induced with helicase C domain 1; IFN, interferon; IGF, insulin-like growth factors; IL2RA, interleukin-2 Receptor alpha; LPS, lipopolysaccharide; MAIT, Mucosa-associated invariant T cells; MHC, major histocompatibility complex; mLN, mesenteric lymph node; MNV, murine norovirus; MR1, MHC class 1-related protein; MyD88, Myeloid differentiation primary response 88; NOD, non-obese diabetic; NOD2, nucleotide-binding oligomerization domain-containing protein 2; NOR, non-obese diabetes resistant; PAMP, pathogen-associated molecular patterns; pDC, plasmacytoid dendritic cells; PRR, pattern recognition receptor; PTPN22, protein tyrosine phosphatase 22; SCFA, short chain fatty acid; SLE, systemic lupus erythematosus; T1D, type 1 diabetes; Th, T helper cell; TLR, toll-like receptor; Treg, regulatory T cell; VILP, viral insulin/insulin-like growth-1-like peptides.
1. Krischer JP, Lynch KF, Lernmark Å, Hagopian WA, Rewers MJ, She J-X, et al. Genetic and Environmental Interactions Modify the Risk of Diabetes-Related Autoimmunity by 6 Years of Age: The TEDDY Study. Diabetes Care (2017) 40(9):1194–202. doi: 10.2337/dc17-0238
2. Bakay M, Pandey R, Grant SFA, Hakonarson H. The Genetic Contribution to Type 1 Diabetes. Curr Diabetes Rep (2019) 19(11):116. doi: 10.1007/s11892-019-1235-1
3. Pociot F, Kaur S, Nielsen LB. Effects of the Genome on Immune Regulation in Type 1 Diabetes. Pediatr Diabetes (2016) 17:37–42. doi: 10.1111/pedi.12336
4. Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, et al. A Genome-Wide Association Study of Nonsynonymous SNPs Identifies a Type 1 Diabetes Locus in the Interferon-Induced Helicase (IFIH1) Region. Nat Genet (2006) 38(6):617–9. doi: 10.1038/ng1800
5. Barrett JC, Clayton D, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-Wide Association Study and Meta-Analysis Finds Over 40 Loci Affect Risk of Type 1 Diabetes. Nat Genet (2009) 41(6):703–7. doi: 10.1038/ng.381
6. Westra H-J, Martínez-Bonet M, Onengut-Gumuscu S, Lee A, Luo Y, Teslovich N, et al. Fine-Mapping and Functional Studies Highlight Potential Causal Variants for Rheumatoid Arthritis and Type 1 Diabetes. Nat Genet (2018) 50(10):1366–74. doi: 10.1038/s41588-018-0216-7
7. Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJL, Furman D, et al. Variation in the Human Immune System Is Largely Driven by Non-Heritable Influences. Cell (2015) 160(1):37–47. doi: 10.1016/j.cell.2014.12.020
8. Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, Norris JM, et al. Epidemiology of Environmental Exposures and Human Autoimmune Diseases: Findings From a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun (2012) 39(4):259–71. doi: 10.1016/j.jaut.2012.05.002
9. Rewers M, Ludvigsson J. Environmental Risk Factors for Type 1 Diabetes. Lancet Lond Engl (2016) 387(10035):2340–8. doi: 10.1016/S0140-6736(16)30507-4
10. Esposito S, Toni G, Tascini G, Santi E, Berioli MG, Principi N. Environmental Factors Associated With Type 1 Diabetes. Front Endocrinol (2019) 10:592). doi: 10.3389/fendo.2019.00592
11. Watad A, Azrielant S, Bragazzi NL, Sharif K, David P, Katz I, et al. Seasonality and Autoimmune Diseases: The Contribution of the Four Seasons to the Mosaic of Autoimmunity. J Autoimmun (2017) 82:13–30. doi: 10.1016/j.jaut.2017.06.001
12. Moltchanova EV, Schreier N, Lammi N, Karvonen M. Seasonal Variation of Diagnosis of Type 1 Diabetes Mellitus in Children Worldwide. Diabetes Med (2009) 26(7):673–8. doi: 10.1111/j.1464-5491.2009.02743.x
13. Roep BO. A Viral Link for Type 1 Diabetes. Nat Med (2019) 25(12):1816–8. doi: 10.1038/s41591-019-0689-7
14. Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, et al. Association Between Rotavirus Infection and Pancreatic Islet Autoimmunity in Children at Risk of Developing Type 1 Diabetes. Diabetes (2000) 49(8):1319–24. doi: 10.2337/diabetes.49.8.1319
15. Perrett KP, Jachno K, Nolan TM, Harrison LC. Association of Rotavirus Vaccination With the Incidence of Type 1 Diabetes in Children. JAMA Pediatr (2019) 173(3):280–2. doi: 10.1001/jamapediatrics.2018.4578
16. Hiemstra HS, Schloot NC, van Veelen PA, Willemen SJM, Franken KLMC, van Rood JJ, et al. Cytomegalovirus in Autoimmunity: T Cell Crossreactivity to Viral Antigen and Autoantigen Glutamic Acid Decarboxylase. Proc Natl Acad Sci (2001) 98(7):3988–91. doi: 10.1073/pnas.071050898
17. Pane JA, Fleming FE, Graham KL, Thomas HE, Kay TWH, Coulson BS. Rotavirus Acceleration of Type 1 Diabetes in Non-Obese Diabetic Mice Depends on Type I Interferon Signalling. Sci Rep (2016) 6:29697. doi: 10.1038/srep29697
18. Stone VM, Hankaniemi MM, Svedin E, Sioofy-Khojine A, Oikarinen S, Hyöty H, et al. A Coxsackievirus B Vaccine Protects Against Virus-Induced Diabetes in an Experimental Mouse Model of Type 1 Diabetes. Diabetologia (2018) 61(2):476–81. doi: 10.1007/s00125-017-4492-z
19. Drescher KM, von Herrath M, Tracy S. Enteroviruses, Hygiene and Type 1 Diabetes: Toward a Preventive Vaccine. Rev Med Virol (2015) 25(1):19–32. doi: 10.1002/rmv.1815
20. Stone VM, Hankaniemi MM, Laitinen OH, Sioofy-Khojine AB, Lin A, Lozano IMD, et al. A Hexavalent Coxsackievirus B Vaccine Is Highly Immunogenic and Has a Strong Protective Capacity in Mice and Nonhuman Primates. Sci Adv (2020) 6(19):eaaz2433. doi: 10.1126/sciadv.aaz2433
21. Dunne JL, Richardson SJ, Atkinson MA, Craig ME, Dahl-Jørgensen K, Flodström-Tullberg M, et al. Rationale for Enteroviral Vaccination and Antiviral Therapies in Human Type 1 Diabetes. Diabetologia (2019) 62(5):744–53. doi: 10.1007/s00125-019-4811-7
22. Ifie E, Russell MA, Dhayal S, Leete P, Sebastiani G, Nigi L, et al. Unexpected Subcellular Distribution of a Specific Isoform of the Coxsackie and Adenovirus Receptor, CAR-SIV, in Human Pancreatic Beta Cells. Diabetologia (2018) 61(11):2344–55. doi: 10.1007/s00125-018-4704-1
23. Vehik K, Lynch KF, Wong MC, Tian X, Ross MC, Gibbs RA, et al. Prospective Virome Analyses in Young Children at Increased Genetic Risk for Type 1 Diabetes. Nat Med (2019) 25(12):1865–72. doi: 10.1038/s41591-019-0667-0
24. Honkanen H, Oikarinen S, Nurminen N, Laitinen OH, Huhtala H, Lehtonen J, et al. Detection of Enteroviruses in Stools Precedes Islet Autoimmunity by Several Months: Possible Evidence for Slowly Operating Mechanisms in Virus-Induced Autoimmunity. Diabetologia (2017) 60(3):424–31. doi: 10.1007/s00125-016-4177-z
25. Salminen K, Sadeharju K, Lönnrot M, Vähäsalo P, Kupila A, Korhonen S, et al. Enterovirus Infections Are Associated With the Induction of Beta-Cell Autoimmunity in a Prospective Birth Cohort Study. J Med Virol (2003) 69(1):91–8. doi: 10.1002/jmv.10260
26. Sioofy-Khojine A-B, Lehtonen J, Nurminen N, Laitinen OH, Oikarinen S, Huhtala H, et al. Coxsackievirus B1 Infections Are Associated With the Initiation of Insulin-Driven Autoimmunity That Progresses to Type 1 Diabetes. Diabetologia (2018) 61(5):1193–202. doi: 10.1007/s00125-018-4561-y
27. Lönnrot M, Lynch KF, Elding Larsson H, Lernmark Å, Rewers MJ, Törn C, et al. Respiratory Infections Are Temporally Associated With Initiation of Type 1 Diabetes Autoimmunity: The TEDDY Study. Diabetologia (2017) 60(10):1931–40. doi: 10.1007/s00125-017-4365-5
28. Ashton MP, Eugster A, Walther D, Daehling N, Riethausen S, Kuehn D, et al. Incomplete Immune Response to Coxsackie B Viruses Associates With Early Autoimmunity Against Insulin. Sci Rep (2016) 6:32899. doi: 10.1038/srep32899
29. Graham KL, Sanders N, Tan Y, Allison J, Kay TWH, Coulson BS. Rotavirus Infection Accelerates Type 1 Diabetes in Mice With Established Insulitis. J Virol (2008) 82(13):6139–49. doi: 10.1128/JVI.00597-08
30. Dotta F, Censini S, van Halteren AGS, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 Virus Infection of Beta Cells and Natural Killer Cell Insulitis in Recent-Onset Type 1 Diabetic Patients. Proc Natl Acad Sci USA (2007) 104(12):5115–20. doi: 10.1073/pnas.0700442104
31. Lietzén N, Hirvonen K, Honkimaa A, Buchacher T, Laiho JE, Oikarinen S, et al. Coxsackievirus B Persistence Modifies the Proteome and the Secretome of Pancreatic Ductal Cells. iScience (2019) 19:340–57. doi: 10.1016/j.isci.2019.07.040
32. Krogvold L, Edwin B, Buanes T, Frisk G, Skog O, Anagandula M, et al. Detection of a Low-Grade Enteroviral Infection in the Islets of Langerhans of Living Patients Newly Diagnosed With Type 1 Diabetes. Diabetes (2015) 64(5):1682–7. doi: 10.2337/db14-1370
33. Frisk G, Diderholm H. Tissue Culture of Isolated Human Pancreatic Islets Infected With Different Strains of Coxsackievirus B4: Assessment of Virus Replication and Effects on Islet Morphology and Insulin Release. Int J Exp Diabetes Res (2000) 1(3):165–75. doi: 10.1155/EDR.2000.165
34. Morse ZJ, Horwitz MS. Innate Viral Receptor Signaling Determines Type 1 Diabetes Onset. Front Endocrinol (2017) 8:249. doi: 10.3389/fendo.2017.00249
35. Szymczak F, Colli ML, Mamula MJ, Evans-Molina C, Eizirik DL. Gene Expression Signatures of Target Tissues in Type 1 Diabetes, Lupus Erythematosus, Multiple Sclerosis, and Rheumatoid Arthritis. Sci Adv (2021) 7(2):eabd7600. doi: 10.1126/sciadv.abd7600
36. Ferreira RC, Guo H, Coulson RMR, Smyth DJ, Pekalski ML, Burren OS, et al. A Type I Interferon Transcriptional Signature Precedes Autoimmunity in Children Genetically At-Risk of Type 1 Diabetes. Diabetes (2014) 63(7):2538–50. doi: 10.2337/db13-1777
37. Xhonneux L-P, Knight O, Lernmark Å, Bonifacio E, Hagopian WA, Rewers MJ, et al. Transcriptional Networks in at-Risk Individuals Identify Signatures of Type 1 Diabetes Progression. Sci Transl Med (2021) 13(587):eabd5666. doi: 10.1126/scitranslmed.abd5666
38. Kallionpää H, Elo LL, Laajala E, Mykkänen J, Ricaño-Ponce I, Vaarma M, et al. Innate Immune Activity Is Detected Prior to Seroconversion in Children With HLA-Conferred Type 1 Diabetes Susceptibility. Diabetes (2014) 63(7):2402–14. doi: 10.2337/db13-1775
39. Apaolaza PS, Balcacean D, Zapardiel-Gonzalo J, Nelson G, Lenchik N, Akhbari P, et al. Islet Expression of Type I Interferon Response Sensors Is Associated With Immune Infiltration and Viral Infection in Type 1 Diabetes. Sci Adv (2021) 7(9):eabd6527. doi: 10.1126/sciadv.abd6527
40. Tracy S, Smithee S, Alhazmi A, Chapman N. Coxsackievirus can Persist in Murine Pancreas by Deletion of 5’ Terminal Genomic Sequences. J Med Virol (2015) 87(2):240–7. doi: 10.1002/jmv.24039
41. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the Immune System. Nat Rev Immunol (2017) 17(4):219–32. doi: 10.1038/nri.2017.7
42. Lee N, Kim W-U. Microbiota in T-Cell Homeostasis and Inflammatory Diseases. Exp Mol Med (2017) 49(5):e340. doi: 10.1038/emm.2017.36
43. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes From the TEDDY Study. Nature (2018) 562(7728):589–94. doi: 10.1038/s41586-018-0620-2
44. Zhou H, Sun L, Zhang S, Zhao X, Gang X, Wang G. Evaluating the Causal Role of Gut Microbiota in Type 1 Diabetes and Its Possible Pathogenic Mechanisms. Front Endocrinol (2020) 11:125. doi: 10.3389/fendo.2020.00125
45. Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes (2015) 64(10):3510–20. doi: 10.2337/db14-1847
46. Russell JT, Roesch LFW, Ördberg M, Ilonen J, Atkinson MA, Schatz DA, et al. Genetic Risk for Autoimmunity Is Associated With Distinct Changes in the Human Gut Microbiome. Nat Commun (2019) 10(1):3621. doi: 10.1038/s41467-019-11460-x
47. Mullaney JA, Stephens JE, Costello M-E, Fong C, Geeling BE, Gavin PG, et al. Type 1 Diabetes Susceptibility Alleles Are Associated With Distinct Alterations in the Gut Microbiota. Microbiome (2018) 6:35. doi: 10.1186/s40168-018-0417-4
48. Paun A, Yau C, Meshkibaf S, Daigneault MC, Marandi L, Mortin-Toth S, et al. Association of HLA-Dependent Islet Autoimmunity With Systemic Antibody Responses to Intestinal Commensal Bacteria in Children. Sci Immunol (2019) 4(32):eaau8125. doi: 10.1126/sciimmunol.aau8125
49. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect Against Type 1 Diabetes. Nat Immunol (2017) 18(5):552–62. doi: 10.1038/ni.3713
50. Johnson RK, Vanderlinden L, DeFelice BC, Kechris K, Uusitalo U, Fiehn O, et al. Metabolite-Related Dietary Patterns and the Development of Islet Autoimmunity. Sci Rep (2019) 9(1):14819. doi: 10.1038/s41598-019-51251-4
51. Li Q, Parikh H, Butterworth MD, Lernmark Å, Hagopian W, Rewers M, et al. Longitudinal Metabolome-Wide Signals Prior to the Appearance of a First Islet Autoantibody in Children Participating in the TEDDY Study. Diabetes (2020) 69(3):465–76. doi: 10.2337/db19-0756
52. Knip M, Siljander H. The Role of the Intestinal Microbiota in Type 1 Diabetes Mellitus. Nat Rev Endocrinol (2016) 12(3):154–67. doi: 10.1038/nrendo.2015.218
53. de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, et al. Aberrant Gut Microbiota Composition at the Onset of Type 1 Diabetes in Young Children. Diabetologia (2014) 57(8):1569–77. doi: 10.1007/s00125-014-3274-0
54. Endesfelder D, Engel M, Davis-Richardson AG, Ardissone AN, Achenbach P, Hummel S, et al. Towards a Functional Hypothesis Relating Anti-Islet Cell Autoimmunity to the Dietary Impact on Microbial Communities and Butyrate Production. Microbiome (2016) 4:17. doi: 10.1186/s40168-016-0163-4
55. Mejía-León ME, Petrosino JF, Ajami NJ, Domínguez-Bello MG, de la Barca AMC. Fecal Microbiota Imbalance in Mexican Children With Type 1 Diabetes. Sci Rep (2014) 4(1):3814. doi: 10.1038/srep03814
56. Pearson JA, Wong FS, Wen L. The Importance of the Non Obese Diabetic (NOD) Mouse Model in Autoimmune Diabetes. J Autoimmun (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
57. Hu Y, Peng J, Li F, Wong FS, Wen L. Evaluation of Different Mucosal Microbiota Leads to Gut Microbiota-Based Prediction of Type 1 Diabetes in NOD Mice. Sci Rep (2018) 8(1):1–13. doi: 10.1038/s41598-018-33571-z
58. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen A-M, et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression Toward Type 1 Diabetes. Cell Host Microbe (2015) 17(2):260–73. doi: 10.1016/j.chom.2015.01.001
59. Brown K, Godovannyi A, Ma C, Zhang Y, Ahmadi-Vand Z, Dai C, et al. Prolonged Antibiotic Treatment Induces a Diabetogenic Intestinal Microbiome That Accelerates Diabetes in NOD Mice. ISME J (2016) 10(2):321–32. doi: 10.1038/ismej.2015.114
60. Markle JGM, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex Differences in the Gut Microbiome Drive Hormone-Dependent Regulation of Autoimmunity. Science (2013) 339(6123):1084–8. doi: 10.1126/science.1233521
61. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota Regulates Type 1 Diabetes Through Toll-Like Receptors. Proc Natl Acad Sci USA (2015) 112(32):9973–7. doi: 10.1073/pnas.1508740112
62. Simon M-C, Reinbeck AL, Wessel C, Heindirk J, Jelenik T, Kaul K, et al. Distinct Alterations of Gut Morphology and Microbiota Characterize Accelerated Diabetes Onset in Nonobese Diabetic Mice. J Biol Chem (2020) 295(4):969–80. doi: 10.1016/S0021-9258(17)49908-X
63. Robertson RC, Manges AR, Finlay BB, Prendergast AJ. The Human Microbiome and Child Growth – First 1000 Days and Beyond. Trends Microbiol (2019) 27(2):131–47. doi: 10.1016/j.tim.2018.09.008
64. Sommer F, Bäckhed F. The Gut Microbiota — Masters of Host Development and Physiology. Nat Rev Microbiol (2013) 11(4):227–38. doi: 10.1038/nrmicro2974
65. Derrien M, Alvarez A-S, de Vos WM. The Gut Microbiota in the First Decade of Life. Trends Microbiol (2019) 27(12):997–1010. doi: 10.1016/j.tim.2019.08.001
66. Belkaid Y, Hand T. Role of the Microbiota in Immunity and Inflammation. Cell (2014) 157(1):121–41. doi: 10.1016/j.cell.2014.03.011
67. Parikka V, Näntö-Salonen K, Saarinen M, Simell T, Ilonen J, Hyöty H, et al. Early Seroconversion and Rapidly Increasing Autoantibody Concentrations Predict Prepubertal Manifestation of Type 1 Diabetes in Children at Genetic Risk. Diabetologia (2012) 55(7):1926–36. doi: 10.1007/s00125-012-2523-3
68. Thomas NJ, Jones SE, Weedon MN, Shields BM, Oram RA, Hattersley AT. Frequency and Phenotype of Type 1 Diabetes in the First Six Decades of Life: A Cross-Sectional, Genetically Stratified Survival Analysis From UK Biobank. Lancet Diabetes Endocrinol (2018) 6(2):122–9. doi: 10.1016/S2213-8587(17)30362-5
69. De Riva A, Wållberg M, Ronchi F, Coulson R, Sage A, Thorne L, et al. Regulation of Type 1 Diabetes Development and B-Cell Activation in Nonobese Diabetic Mice by Early Life Exposure to a Diabetogenic Environment. PloS One (2017) 12(8):e0181964. doi: 10.1371/journal.pone.0181964
70. Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the Gut Microbiota Is Associated With HIV Disease Progression and Tryptophan Catabolism. Sci Transl Med (2013) 5(193):193ra91. doi: 10.1126/scitranslmed.3006438
71. Inoue T, Nakayama J, Moriya K, Kawaratani H, Momoda R, Ito K, et al. Gut Dysbiosis Associated With Hepatitis C Virus Infection. Clin Infect Dis Off Publ Infect Dis Soc Am (2018) 67(6):869–77. doi: 10.1093/cid/ciy205
72. Yildiz S, Mazel-Sanchez B, Kandasamy M, Manicassamy B, Schmolke M. Influenza A Virus Infection Impacts Systemic Microbiota Dynamics and Causes Quantitative Enteric Dysbiosis. Microbiome (2018) 6(1):9. doi: 10.1186/s40168-017-0386-z
73. Oikarinen S, Martiskainen M, Tauriainen S, Huhtala H, Ilonen J, Veijola R, et al. Enterovirus RNA in Blood Is Linked to the Development of Type 1 Diabetes. Diabetes (2011) 60(1):276–9. doi: 10.2337/db10-0186
74. Stene LC, Oikarinen S, Hyöty H, Barriga KJ, Norris JM, Klingensmith G, et al. Enterovirus Infection and Progression From Islet Autoimmunity to Type 1 Diabetes: The Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes (2010) 59(12):3174–80. doi: 10.2337/db10-0866
75. Pearson JA, Tai N, Ekanayake-Alper DK, Peng J, Hu Y, Hager K, et al. Norovirus Changes Susceptibility to Type 1 Diabetes by Altering Intestinal Microbiota and Immune Cell Functions. Front Immunol (2019) 10:2654. doi: 10.3389/fimmu.2019.02654
76. Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, et al. Islet-Reactive CD8+ T Cell Frequencies in the Pancreas, But Not in Blood, Distinguish Type 1 Diabetic Patients From Healthy Donors. Sci Immunol (2018) 3(20):eaao4013. doi: 10.1126/sciimmunol.aao4013
77. Turley SJ, Lee J-W, Dutton-Swain N, Mathis D, Benoist C. Endocrine Self and Gut Non-Self Intersect in the Pancreatic Lymph Nodes. Proc Natl Acad Sci USA (2005) 102(49):17729–33. doi: 10.1073/pnas.0509006102
78. Hagopian W, Lee H-S, Liu E, Rewers M, She J-X, Ziegler A-G, et al. Co-Occurrence of Type 1 Diabetes and Celiac Disease Autoimmunity. Pediatrics (2017) 140(5):e20171305. doi: 10.1542/peds.2017-1305
79. Oikarinen M, Tauriainen S, Oikarinen S, Honkanen T, Collin P, Rantala I, et al. Type 1 Diabetes Is Associated With Enterovirus Infection in Gut Mucosa. Diabetes (2012) 61(3):687–91. doi: 10.2337/db11-1157
80. Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, et al. Circulating and Tissue-Resident CD4+ T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology (2017) 153(5):1320–37.e16. doi: 10.1053/j.gastro.2017.07.047
81. Brown EM, Kenny DJ, Xavier RJ. Gut Microbiota Regulation of T Cells During Inflammation and Autoimmunity. Annu Rev Immunol (2019) 37(1):599–624. doi: 10.1146/annurev-immunol-042718-041841
82. Krych Ł, Nielsen DS, Hansen AK, Hansen CHF. Gut Microbial Markers Are Associated With Diabetes Onset, Regulatory Imbalance, and IFN-γ Level in NOD Mice. Gut Microbes (2015) 6(2):101–9. doi: 10.1080/19490976.2015.1011876
83. Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng F, et al. Identifying Species of Symbiont Bacteria From the Human Gut That, Alone, Can Induce Intestinal Th17 Cells in Mice. Proc Natl Acad Sci USA (2016) 113(50):E8141–50. doi: 10.1073/pnas.1617460113
84. Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell (2017) 168(5):928–43.e11. doi: 10.1016/j.cell.2017.01.022
85. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
86. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
87. Yu H, Paiva R, Flavell RA. Harnessing the Power of Regulatory T-Cells to Control Autoimmune Diabetes: Overview and Perspective. Immunology (2018) 153(2):161–70. doi: 10.1111/imm.12867
88. de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol (2014) 6(12):a016287. doi: 10.1101/cshperspect.a016287
89. Yu H, Gagliani N, Ishigame H, Huber S, Zhu S, Esplugues E, et al. Intestinal Type 1 Regulatory T Cells Migrate to Periphery to Suppress Diabetogenic T Cells and Prevent Diabetes Development. Proc Natl Acad Sci USA (2017) 114(39):10443–8. doi: 10.1073/pnas.1705599114
90. Jacob N, Jaiswal S, Maheshwari D, Nallabelli N, Khatri N, Bhatia A, et al. Butyrate Induced Tregs Are Capable of Migration From the GALT to the Pancreas to Restore Immunological Tolerance During Type-1 Diabetes. Sci Rep (2020) 10(1):19120. doi: 10.1038/s41598-020-76109-y
91. Shimokawa C, Kato T, Takeuchi T, Ohshima N, Furuki T, Ohtsu Y, et al. CD8 + Regulatory T Cells Are Critical in Prevention of Autoimmune-Mediated Diabetes. Nat Commun (2020) 11(1):1922. doi: 10.1038/s41467-020-15857-x
92. Ruohtula T, Kondrashova A, Lehtonen J, Oikarinen S, Hämäläinen A-M, Niemelä O, et al. Immunomodulatory Effects of Rhinovirus and Enterovirus Infections During the First Year of Life. Front Immunol (2020) 11:567046. doi: 10.3389/fimmu.2020.567046
93. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of Evolutionarily Conserved Mucosal-Associated Invariant T Cells by MR1. Nature (2003) 422(6928):164–9. doi: 10.1038/nature01433
94. Schmaler M, Colone A, Spagnuolo J, Zimmermann M, Lepore M, Kalinichenko A, et al. Modulation of Bacterial Metabolism by the Microenvironment Controls MAIT Cell Stimulation. Mucosal Immunol (2018) 11(4):1060–70. doi: 10.1038/s41385-018-0020-9
95. Dias J, Leeansyah E, Sandberg JK. Multiple Layers of Heterogeneity and Subset Diversity in Human MAIT Cell Responses to Distinct Microorganisms and to Innate Cytokines. Proc Natl Acad Sci (2017) 114(27):E5434–43. doi: 10.1073/pnas.1705759114
96. Napier RJ, Adams EJ, Gold MC, Lewinsohn DM. The Role of Mucosal Associated Invariant T Cells in Antimicrobial Immunity. Front Immunol (2015) 6:344. doi: 10.3389/fimmu.2015.00344
97. Rouxel O, Da Silva J, Beaudoin L, Nel I, Tard C, Cagninacci L, et al. Cytotoxic and Regulatory Roles of Mucosal-Associated Invariant T Cells in Type 1 Diabetes. Nat Immunol (2017) 18(12):1321–31. doi: 10.1038/ni.3854
98. van Wilgenburg B, Scherwitzl I, Hutchinson EC, Leng T, Kurioka A, Kulicke C, et al. MAIT Cells Are Activated During Human Viral Infections. Nat Commun (2016) 7:11653. doi: 10.1038/ncomms11653
99. Ussher JE, Bilton M, Attwod E, Shadwell J, Richardson R, de Lara C, et al. CD161++ CD8+ T Cells, Including the MAIT Cell Subset, Are Specifically Activated by IL-12+IL-18 in a TCR-Independent Manner. Eur J Immunol (2014) 44(1):195–203. doi: 10.1002/eji.201343509
100. Mariño E, Tan B, Binge L, Mackay CR, Grey ST. B-Cell Cross-Presentation of Autologous Antigen Precipitates Diabetes. Diabetes (2012) 61(11):2893–905. doi: 10.2337/db12-0006
101. Pane JA, Webster NL, Graham KL, Holloway G, Zufferey C, Coulson BS. Rotavirus Acceleration of Murine Type 1 Diabetes Is Associated With a T Helper 1-Dependent Specific Serum Antibody Response and Virus Effects in Regional Lymph Nodes. Diabetologia (2013) 56(3):573–82. doi: 10.1007/s00125-012-2798-4
102. Pane JA, Webster NL, Zufferey C, Coulson BS. Rotavirus Acceleration of Murine Type 1 Diabetes Is Associated With Increased MHC Class I-Restricted Antigen Presentation by B Cells and Elevated Proinflammatory Cytokine Expression by T Cells. Virus Res (2014) 179:73–84. doi: 10.1016/j.virusres.2013.11.009
103. Jounai K, Ikado K, Sugimura T, Ano Y, Braun J, Fujiwara D. Spherical Lactic Acid Bacteria Activate Plasmacytoid Dendritic Cells Immunomodulatory Function via TLR9-Dependent Crosstalk With Myeloid Dendritic Cells. PloS One (2012) 7(4):e32588. doi: 10.1371/journal.pone.0032588
104. Nelson AS, Maddaloni M, Abbott JR, Hoffman C, Akgul A, Ohland C, et al. Oral Therapy With Colonization Factor Antigen I Prevents Development of Type 1 Diabetes in Non-Obese Diabetic Mice. Sci Rep (2020) 10(1):6156. doi: 10.1038/s41598-020-62881-4
105. Hu Y, Jin P, Peng J, Zhang X, Wong FS, Wen L. Different Immunological Responses to Early-Life Antibiotic Exposure Affecting Autoimmune Diabetes Development in NOD Mice. J Autoimmun (2016) 72:47–56. doi: 10.1016/j.jaut.2016.05.001
106. Umenai T, Hirai H, Shime N, Nakaya T, Asahara T, Nomoto K, et al. Eradication of the Commensal Intestinal Microflora by Oral Antimicrobials Interferes With the Host Response to Lipopolysaccharide. Eur J Clin Microbiol Infect Dis (2010) 29(6):633–41. doi: 10.1007/s10096-010-0905-3
107. Carrero JA, McCarthy DP, Ferris ST, Wan X, Hu H, Zinselmeyer BH, et al. Resident Macrophages of Pancreatic Islets Have a Seminal Role in the Initiation of Autoimmune Diabetes of NOD Mice. Proc Natl Acad Sci USA (2017) 114(48):E10418–27. doi: 10.1073/pnas.1713543114
108. Takeuchi T, Miyauchi E, Kanaya T, Kato T, Nakanishi Y, Watanabe T, et al. Acetate Differentially Regulates IgA Reactivity to Commensal Bacteria. Nature (2021) 595(7868):560–4. doi: 10.1038/s41586-021-03727-5
109. Sorini C, Cosorich I, Lo Conte M, De Giorgi L, Facciotti F, Lucianò R, et al. Loss of Gut Barrier Integrity Triggers Activation of Islet-Reactive T Cells and Autoimmune Diabetes. Proc Natl Acad Sci (2019) 116(30):15140–9. doi: 10.1073/pnas.1814558116
110. Tajik N, Frech M, Schulz O, Schälter F, Lucas S, Azizov V, et al. Targeting Zonulin and Intestinal Epithelial Barrier Function to Prevent Onset of Arthritis. Nat Commun (2020) 11(1):1995. doi: 10.1038/s41467-020-15831-7
111. Mu Q, Kirby J, Reilly CM, Luo XM. Leaky Gut As a Danger Signal for Autoimmune Diseases. Front Immunol (2017) 8:598. doi: 10.3389/fimmu.2017.00598
112. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased Intestinal Permeability Precedes Clinical Onset of Type 1 Diabetes. Diabetologia (2006) 49(12):2824–7. doi: 10.1007/s00125-006-0465-3
113. Sapone A, de Magistris L, Pietzak M, Clemente MG, Tripathi A, Cucca F, et al. Zonulin Upregulation Is Associated With Increased Gut Permeability in Subjects With Type 1 Diabetes and Their Relatives. Diabetes (2006) 55(5):1443–9. doi: 10.2337/db05-1593
114. Li X, Atkinson MA. The Role for Gut Permeability in the Pathogenesis of Type 1 Diabetes – A Solid or Leaky Concept? Pediatr Diabetes (2015) 16(7):485–92. doi: 10.1111/pedi.12305
115. Harbison JE, Roth-Schulze AJ, Giles LC, Tran CD, Ngui KM, Penno MA, et al. Gut Microbiome Dysbiosis and Increased Intestinal Permeability in Children With Islet Autoimmunity and Type 1 Diabetes: A Prospective Cohort Study. Pediatr Diabetes (2019) 20(5):574–83. doi: 10.1111/pedi.12865
116. You Q, He DM, Shu GF, Cao B, Xia YQ, Xing Y, et al. Increased Formation of Neutrophil Extracellular Traps Is Associated With Gut Leakage in Patients With Type 1 But Not Type 2 Diabetes. J Diabetes (2019) 11(8):665–73. doi: 10.1111/1753-0407.12892
117. Neu J, Reverte CM, Mackey AD, Liboni K, Tuhacek-Tenace LM, Hatch M, et al. Changes in Intestinal Morphology and Permeability in the Biobreeding Rat Before the Onset of Type 1 Diabetes. J Pediatr Gastroenterol Nutr (2005) 40(5):589–95. doi: 10.1097/01.MPG.0000159636.19346.C1
118. Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, et al. Role of the Intestinal Tight Junction Modulator Zonulin in the Pathogenesis of Type I Diabetes in BB Diabetic-Prone Rats. Proc Natl Acad Sci USA (2005) 102(8):2916–21. doi: 10.1073/pnas.0500178102
119. Sofi MH, Johnson BM, Gudi RR, Jolly A, Gaudreau M-C, Vasu C. Polysaccharide A-Dependent Opposing Effects of Mucosal and Systemic Exposures to Human Gut Commensal Bacteroides Fragilis in Type 1 Diabetes. Diabetes (2019) 68(10):1975–89. doi: 10.2337/db19-0211
120. Myers MA, Hettiarachchi KD, Ludeman JP, Wilson AJ, Wilson CR, Zimmet PZ. Dietary Microbial Toxins and Type 1 Diabetes. Ann NY Acad Sci (2003) 1005:418–22. doi: 10.1196/annals.1288.071
121. Costa FRC, Françozo MCS, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut Microbiota Translocation to the Pancreatic Lymph Nodes Triggers NOD2 Activation and Contributes to T1D Onset. J Exp Med (2016) 213(7):1223–39. doi: 10.1084/jem.20150744
122. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. Microbial Antigen Mimics Activate Diabetogenic CD8 T Cells in NOD Mice. J Exp Med (2016) 213(10):2129–46. doi: 10.1084/jem.20160526
123. Miranda MCG, Oliveira RP, Torres L, Aguiar SLF, Pinheiro-Rosa N, Lemos L, et al. Frontline Science: Abnormalities in the Gut Mucosa of Non-Obese Diabetic Mice Precede the Onset of Type 1 Diabetes. J Leukoc Biol (2019) 106(3):513–29. doi: 10.1002/JLB.3HI0119-024RR
124. Abdellatif AM, Jensen Smith H, Harms RZ, Sarvetnick NE. Human Islet Response to Selected Type 1 Diabetes-Associated Bacteria: A Transcriptome-Based Study. Front Immunol (2019) 10:2623. doi: 10.3389/fimmu.2019.02623
125. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut Microbiome Metagenomics Analysis Suggests a Functional Model for the Development of Autoimmunity for Type 1 Diabetes. PloS One (2011) 6(10):e25792. doi: 10.1371/journal.pone.0025792
126. Chelakkot C, Choi Y, Kim D-K, Park HT, Ghim J, Kwon Y, et al. Akkermansia Muciniphila -Derived Extracellular Vesicles Influence Gut Permeability Through the Regulation of Tight Junctions. Exp Mol Med (2018) 50(2):e450–0. doi: 10.1038/emm.2017.282
127. Hänninen A, Toivonen R, Pöysti S, Belzer C, Plovier H, Ouwerkerk JP, et al. Akkermansia Muciniphila Induces Gut Microbiota Remodelling and Controls Islet Autoimmunity in NOD Mice. Gut (2017) 67(8):1445–53. doi: 10.1136/gutjnl-2017-314508
128. Zeng MY, Cisalpino D, Varadarajan S, Hellman J, Warren HS, Cascalho M, et al. Gut Microbiota-Induced Immunoglobulin G Controls Systemic Infection by Symbiotic Bacteria and Pathogens. Immunity (2016) 44(3):647–58. doi: 10.1016/j.immuni.2016.02.006
129. Ferreira RC, Pan-Hammarström Q, Graham RR, Gateva V, Fontán G, Lee AT, et al. Association of IFIH1 and Other Autoimmunity Risk Alleles With Selective IgA Deficiency. Nat Genet (2010) 42(9):777–80. doi: 10.1038/ng.644
130. Huang J, Pearson JA, Peng J, Hu Y, Sha S, Xing Y, et al. Gut Microbial Metabolites Alter IgA Immunity in Type 1 Diabetes. JCI Insight (2020) 5(10):e135718. doi: 10.1172/jci.insight.135718
131. Gavin PG, Mullaney JA, Loo D, Cao K-AL, Gottlieb PA, Hill MM, et al. Intestinal Metaproteomics Reveals Host-Microbiota Interactions in Subjects at Risk for Type 1 Diabetes. Diabetes Care (2018) 41(10):2178–86. doi: 10.2337/dc18-0777
132. Zhao G, Vatanen T, Droit L, Park A, Kostic AD, Poon TW, et al. Intestinal Virome Changes Precede Autoimmunity in Type I Diabetes-Susceptible Children. Proc Natl Acad Sci (2017) 114(30):E6166–75. doi: 10.1073/pnas.1706359114
133. Labarta-Bajo L, Nilsen SP, Humphrey G, Schwartz T, Sanders K, Swafford A, et al. Type I IFNs and CD8 T Cells Increase Intestinal Barrier Permeability After Chronic Viral Infection. J Exp Med (2020) 217(12):e20192276. doi: 10.1084/jem.20192276
134. Lee AS, Gibson DL, Zhang Y, Sham HP, Vallance BA, Dutz JP. Gut Barrier Disruption by an Enteric Bacterial Pathogen Accelerates Insulitis in NOD Mice. Diabetologia (2010) 53(4):741–8. doi: 10.1007/s00125-009-1626-y
135. Sencio V, Barthelemy A, Tavares LP, Machado MG, Soulard D, Cuinat C, et al. Gut Dysbiosis During Influenza Contributes to Pulmonary Pneumococcal Superinfection Through Altered Short-Chain Fatty Acid Production. Cell Rep (2020) 30(9):2934–47.e6. doi: 10.1016/j.celrep.2020.02.013
136. Giron LB, Dweep H, Yin X, Wang H, Damra M, Goldman AR, et al. Plasma Markers of Disrupted Gut Permeability in Severe COVID-19 Patients. Front Immunol (2021) 12:686240. doi: 10.3389/fimmu.2021.686240
137. Prasad R, Patton MJ, Floyd JL, Vieira CP, Fortmann S, DuPont M, et al. Plasma Microbiome in COVID-19 Subjects: An Indicator of Gut Barrier Defects and Dysbiosis. bioRxiv (2021) 2021.04.06.438634. doi: 10.1101/2021.04.06.438634
138. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial Translocation Is a Cause of Systemic Immune Activation in Chronic HIV Infection. Nat Med (2006) 12(12):1365–71. doi: 10.1038/nm1511
139. Xu W, Luo Z, Alekseyenko AV, Martin L, Wan Z, Ling B, et al. Distinct Systemic Microbiome and Microbial Translocation Are Associated With Plasma Level of Anti-CD4 Autoantibody in HIV Infection. Sci Rep (2018) 8(1):12863. doi: 10.1038/s41598-018-31116-y
140. Liang G, Bushman FD. The Human Virome: Assembly, Composition and Host Interactions. Nat Rev Microbiol (2021) 19(8):514–27. doi: 10.1038/s41579-021-00536-5
141. Virgin HW. The Virome in Mammalian Physiology and Disease. Cell (2014) 157(1):142–50. doi: 10.1016/j.cell.2014.02.032
142. Li Y, Handley SA, Baldridge MT. The Dark Side of the Gut: Virome–host Interactions in Intestinal Homeostasis and Disease. J Exp Med (2021) 218:e20201044. doi: 10.1084/jem.20201044
143. Kernbauer E, Ding Y, Cadwell K. An Enteric Virus Can Replace the Beneficial Function of Commensal Bacteria. Nature (2014) 516(7529):94–8. doi: 10.1038/nature13960
144. Dallari S, Heaney T, Rosas-Villegas A, Neil JA, Wong S-Y, Brown JJ, et al. Enteric Viruses Evoke Broad Host Immune Responses Resembling Those Elicited by the Bacterial Microbiome. Cell Host Microbe (2021) 29(6):1014–29.e8. doi: 10.1016/j.chom.2021.03.015
145. Wook Kim K, Allen DW, Briese T, Couper JJ, Barry SC, Colman PG, et al. Distinct Gut Virome Profile of Pregnant Women With Type 1 Diabetes in the ENDIA Study. Open Forum Infect Dis (2019) 6(2):ofz025. doi: 10.1093/ofid/ofz025
146. Kortright KE, Chan BK, Koff JL, Turner PE. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe (2019) 25(2):219–32. doi: 10.1016/j.chom.2019.01.014
147. Hsu BB, Gibson TE, Yeliseyev V, Liu Q, Lyon L, Bry L, et al. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe (2019) 25(6):803–14.e5. doi: 10.1016/j.chom.2019.05.001
148. Sweere JM, Belleghem JDV, Ishak H, Bach MS, Popescu M, Sunkari V, et al. Bacteriophage Trigger Antiviral Immunity and Prevent Clearance of Bacterial Infection. Science (2019) 363(6434):eaat9691. doi: 10.1126/science.aat9691
149. Gogokhia L, Buhrke K, Bell R, Hoffman B, Brown DG, Hanke-Gogokhia C, et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe (2019) 25(2):285–99.e8. doi: 10.1016/j.chom.2019.01.008
150. Ingle H, Lee S, Ai T, Orvedahl A, Rodgers R, Zhao G, et al. Viral Complementation of Immunodeficiency Confers Protection Against Enteric Pathogens via Interferon-λ. Nat Microbiol (2019) 4(7):1120–8. doi: 10.1038/s41564-019-0416-7
151. Stone VM, Ringqvist EE, Larsson PG, Domsgen E, Holmlund U, Sverremark-Ekström E, et al. Inhibition of Type III Interferon Expression in Intestinal Epithelial Cells-A Strategy Used by Coxsackie B Virus to Evade the Host’s Innate Immune Response at the Primary Site of Infection? Microorganisms (2021) 9(1):105. doi: 10.3390/microorganisms9010105
152. Tetz G, Brown SM, Hao Y, Tetz V. Type 1 Diabetes: An Association Between Autoimmunity, the Dynamics of Gut Amyloid-Producing E. Coli and Their Phages. Sci Rep (2019) 9(1):9685. doi: 10.1038/s41598-019-46087-x
153. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity (2015) 42(6):1171–84. doi: 10.1016/j.immuni.2015.06.002
154. Bannert N, Kurth R. Retroelements and the Human Genome: New Perspectives on an Old Relation. Proc Natl Acad Sci USA (2004) 101(Suppl 2):14572–9. doi: 10.1073/pnas.0404838101
155. Feschotte C, Gilbert C. Endogenous Viruses: Insights Into Viral Evolution and Impact on Host Biology. Nat Rev Genet (2012) 13(4):283–96. doi: 10.1038/nrg3199
156. Mameli G, Poddighe L, Mei A, Uleri E, Sotgiu S, Serra C, et al. Expression and Activation by Epstein Barr Virus of Human Endogenous Retroviruses-W in Blood Cells and Astrocytes: Inference for Multiple Sclerosis. PloS One (2012) 7(9):e44991. doi: 10.1371/journal.pone.0044991
157. Dechaumes A, Bertin A, Sane F, Levet S, Varghese J, Charvet B, et al. Coxsackievirus-B4 Infection Can Induce the Expression of Human Endogenous Retrovirus W in Primary Cells. Microorganisms. Microorganisms (2020) 8(9):1335. doi: 10.3390/microorganisms8091335
158. Bashratyan R, Regn D, Rahman MJ, Marquardt K, Fink E, Hu W-Y, et al. Type 1 Diabetes Pathogenesis Is Modulated by Spontaneous Autoimmune Responses to Endogenous Retrovirus Antigens in NOD Mice. Eur J Immunol (2017) 47(3):575–84. doi: 10.1002/eji.201646755
159. Levet S, Medina J, Joanou J, Demolder A, Queruel N, Réant K, et al. An Ancestral Retroviral Protein Identified as a Therapeutic Target in Type-1 Diabetes. JCI Insight (2017) 2(17):e94387. doi: 10.1172/jci.insight.94387
160. Perron H, Germi R, Bernard C, Garcia-Montojo M, Deluen C, Farinelli L, et al. Human Endogenous Retrovirus Type W Envelope Expression in Blood and Brain Cells Provides New Insights Into Multiple Sclerosis Disease. Mult Scler Houndmills Basingstoke Engl (2012) 18(12):1721–36. doi: 10.1177/1352458512441381
161. Perron H, Garson JA, Bedin F, Beseme F, Paranhos-Baccala G, Komurian-Pradel F, et al. Molecular Identification of a Novel Retrovirus Repeatedly Isolated From Patients With Multiple Sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc Natl Acad Sci USA (1997) 94(14):7583–8. doi: 10.1073/pnas.94.14.7583
162. Niegowska M, Wajda-Cuszlag M, Stępień-Ptak G, Trojanek J, Michałkiewicz J, Szalecki M, et al. Anti-HERV-W Env Antibodies Are Correlated With Seroreactivity Against Mycobacterium Avium Subsp. Paratuberculosis in Children and Youths at T1D Risk. Sci Rep (2019) 9(1):6282. doi: 10.1038/s41598-019-42788-5
163. Rolland A, Jouvin-Marche E, Viret C, Faure M, Perron H, Marche PN. The Envelope Protein of a Human Endogenous Retrovirus-W Family Activates Innate Immunity Through CD14/TLR4 and Promotes Th1-Like Responses. J Immunol (2006) 176(12):7636–44. doi: 10.4049/jimmunol.176.12.7636
164. Bason C, Lorini R, Lunardi C, Dolcino M, Giannattasio A, d’Annunzio G, et al. In Type 1 Diabetes a Subset of Anti-Coxsackievirus B4 Antibodies Recognize Autoantigens and Induce Apoptosis of Pancreatic Beta Cells. PloS One (2013) 8(2):e57729. doi: 10.1371/journal.pone.0057729
165. Altindis E, Cai W, Sakaguchi M, Zhang F, GuoXiao W, Liu F, et al. Viral Insulin-Like Peptides Activate Human Insulin and IGF-1 Receptor Signaling: A Paradigm Shift for Host-Microbe Interactions. Proc Natl Acad Sci USA (2018) 115(10):2461–6. doi: 10.1073/pnas.1721117115
166. Pfeiffer JK, Virgin HW. Transkingdom Control of Viral Infection and Immunity in the Mammalian Intestine. Science (2016) 351(6270):aad5872. doi: 10.1126/science.aad5872
167. Harper A, Vijayakumar V, Ouwehand AC, Ter Haar J, Obis D, Espadaler J, et al. Viral Infections, the Microbiome, and Probiotics. Front Cell Infect Microbiol (2020) 10:596166. doi: 10.3389/fcimb.2020.596166
168. Berger AK, Mainou BA. Interactions Between Enteric Bacteria and Eukaryotic Viruses Impact the Outcome of Infection. Viruses (2018) 10(1):19. doi: 10.3390/v10010019
169. Pickard JM, Zeng MY, Caruso R, Núñez G. Gut Microbiota: Role in Pathogen Colonization, Immune Responses, and Inflammatory Disease. Immunol Rev (2017) 279(1):70–89. doi: 10.1111/imr.12567
170. Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, et al. Intestinal Microbiota Promote Enteric Virus Replication and Systemic Pathogenesis. Science (2011) 334(6053):249–52. doi: 10.1126/science.1211057
171. Karst SM. The Influence of Commensal Bacteria on Infection With Enteric Viruses. Nat Rev Microbiol (2016) 14(4):197–204. doi: 10.1038/nrmicro.2015.25
172. Fung TC, Bessman NJ, Hepworth MR, Kumar N, Shibata N, Kobuley D, et al. Lymphoid-Tissue-Resident Commensal Bacteria Promote Members of the IL-10 Cytokine Family to Establish Mutualism. Immunity (2016) 44(3):634–46. doi: 10.1016/j.immuni.2016.02.019
173. Hebbandi Nanjundappa R, Ronchi F, Wang J, Clemente-Casares X, Yamanouchi J, Sokke Umeshappa C, et al. A Gut Microbial Mimic That Hijacks Diabetogenic Autoreactivity to Suppress Colitis. Cell (2017) 171(3):655–67.e17. doi: 10.1016/j.cell.2017.09.022
174. Li N, Ma W-T, Pang M, Fan Q-L, Hua J-L. The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front Immunol (2019) 10:1551. doi: 10.3389/fimmu.2019.01551
175. Arena MP, Elmastour F, Sane F, Drider D, Fiocco D, Spano G, et al. Inhibition of Coxsackievirus B4 by Lactobacillus Plantarum. Microbiol Res (2018) 210:59–64. doi: 10.1016/j.micres.2018.03.008
176. El Kfoury KA, Romond M-B, Scuotto A, Alidjinou EK, Dabboussi F, Hamze M, et al. Bifidobacteria-Derived Lipoproteins Inhibit Infection With Coxsackievirus B4 In Vitro. Int J Antimicrob Agents (2017) 50(2):177–85. doi: 10.1016/j.ijantimicag.2017.03.010
177. Uchiyama R, Chassaing B, Zhang B, Gewirtz AT. Antibiotic Treatment Suppresses Rotavirus Infection and Enhances Specific Humoral Immunity. J Infect Dis (2014) 210(2):171–82. doi: 10.1093/infdis/jiu037
178. Bandoro C, Runstadler JA. Bacterial Lipopolysaccharide Destabilizes Influenza Viruses. mSphere (2017) 2(5):e00267–17. doi: 10.1128/mSphere.00267-17
179. Chen H-W, Liu P-F, Liu Y-T, Kuo S, Zhang X-Q, Schooley RT, et al. Nasal Commensal Staphylococcus Epidermidis Counteracts Influenza Virus. Sci Rep (2016) 6:27870. doi: 10.1038/srep27870
180. Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, et al. The Microbial Metabolite Desaminotyrosine Protects From Influenza Through Type I Interferon. Science (2017) 357(6350):498–502. doi: 10.1126/science.aam5336
181. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal Bacteria Calibrate the Activation Threshold of Innate Antiviral Immunity. Immunity (2012) 37(1):158–70. doi: 10.1016/j.immuni.2012.04.011
182. Gonzalez-Perez G, Lamousé-Smith ESN. Gastrointestinal Microbiome Dysbiosis in Infant Mice Alters Peripheral CD8+ T Cell Receptor Signaling. Front Immunol (2017) 8:265. doi: 10.3389/fimmu.2017.00265
183. Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, et al. Commensal Microbes and Interferon-λ Determine Persistence of Enteric Murine Norovirus Infection. Science (2015) 347(6219):266–9. doi: 10.1126/science.1258025
184. Koch BEV, Yang S, Lamers G, Stougaard J, Spaink HP. Intestinal Microbiome Adjusts the Innate Immune Setpoint During Colonization Through Negative Regulation of Myd88. Nat Commun (2018) 9(1):4099. doi: 10.1038/s41467-018-06658-4
185. Bradley KC, Finsterbusch K, Schnepf D, Crotta S, Llorian M, Davidson S, et al. Microbiota-Driven Tonic Interferon Signals in Lung Stromal Cells Protect From Influenza Virus Infection. Cell Rep (2019) 28(1):245–56.e4. doi: 10.1016/j.celrep.2019.05.105
186. Shi Z, Zou J, Zhang Z, Zhao X, Noriega J, Zhang B, et al. Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell (2019) 179(3):644–58.e13. doi: 10.1016/j.cell.2019.09.028
187. de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut (2021) 70(1):92–105. doi: 10.1136/gutjnl-2020-322630
Keywords: type 1 diabetes, microbiome, autoimmunity, coxsackievirus, dysbiosis, gut
Citation: Morse ZJ and Horwitz MS (2021) Virus Infection Is an Instigator of Intestinal Dysbiosis Leading to Type 1 Diabetes. Front. Immunol. 12:751337. doi: 10.3389/fimmu.2021.751337
Received: 31 July 2021; Accepted: 28 September 2021;
Published: 15 October 2021.
Edited by:
F. Susan Wong, Cardiff University, United KingdomReviewed by:
Matthias G. Von Herrath, La Jolla Institute for Immunology (LJI), United StatesCopyright © 2021 Morse and Horwitz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc S. Horwitz, bWhvcndpdHpAbWFpbC51YmMuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.