
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 03 November 2021
Sec. T Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.751138
This article is part of the Research TopicImplementing Logic Gates in Adoptive Cell TherapyView all 6 articles
 Loreen Sophie Rudek1†
Loreen Sophie Rudek1† Katharina Zimmermann1†
Katharina Zimmermann1† Melanie Galla1
Melanie Galla1 Johann Meyer1Johannes Kuehle2,3Andriana Stamopoulou1Daniel Brand1I. Erol Sandalcioglu4
Johann Meyer1Johannes Kuehle2,3Andriana Stamopoulou1Daniel Brand1I. Erol Sandalcioglu4 Belal Neyazi4Thomas Moritz1Claudia Rossig5
Belal Neyazi4Thomas Moritz1Claudia Rossig5 Bianca Altvater5
Bianca Altvater5 Christine S. Falk6
Christine S. Falk6 Hinrich Abken7
Hinrich Abken7 Michael Alexander Morgan1‡
Michael Alexander Morgan1‡ Axel Schambach1,8*‡
Axel Schambach1,8*‡Immune cell therapeutics are increasingly applied in oncology. Especially chimeric antigen receptor (CAR) T cells are successfully used to treat several B cell malignancies. Efforts to engineer CAR T cells for improved activity against solid tumors include co-delivery of pro-inflammatory cytokines in addition to CARs, via either constitutive cytokine expression or inducible cytokine expression triggered by CAR recognition of its target antigen—so-called “T cells redirected for universal cytokine-mediated killing” (TRUCKs) or fourth-generation CARs. Here, we tested the hypothesis that TRUCK principles could be expanded to improve anticancer functions of NK cells. A comparison of the functionality of inducible promoters responsive to NFAT or NFκB in NK cells showed that, in contrast to T cells, the inclusion of NFκB-responsive elements within the inducible promoter construct was essential for CAR-inducible expression of the transgene. We demonstrated that GD2CAR-specific activation induced a tight NFκB-promoter-driven cytokine release in NK-92 and primary NK cells together with an enhanced cytotoxic capacity against GD2+ target cells, also shown by increased secretion of cytolytic cytokines. The data demonstrate biologically relevant differences between T and NK cells that are important when clinically translating the TRUCK concept to NK cells for the treatment of solid malignancies.
Chimeric antigen receptors (CARs) as synthetic receptors combine antigen recognition and immune cell activation in one molecule and are designed to genetically modify immune cells, like T and NK cells, which find their application in clinical trials for adoptive anticancer cell therapy (1, 2). One of the most successful recent approaches in cancer immunotherapy was marked by the first US Food and Drug Administration (FDA) approvals of CAR T cell therapies against CD19+ hematological malignancies, including B cell lymphoma, acute lymphoblastic leukemia, and multiple myeloma (3–6). However, to date, adoptive cell-based CAR immunotherapies are limited in the treatment of solid tumors due to the immunosuppressive tumor microenvironment (TME), which results in insufficient tissue homing, restricted proliferation, and short-time persistence of modified immune cells within the tumor (7–9). Moreover, the antigenic heterogeneity of solid tumors limits the number of targetable antigens for CAR cell therapy (10, 11). Preclinical and clinical studies using T cells redirected for universal cytokine-mediated killing (TRUCKs) demonstrated improved antitumor activity due to additional CAR-mediated cytokine secretion after target antigen recognition in the tumor bed. Specific cytokine deposition promotes formation of an antitumoral and pro-inflammatory milieu in the TME, supporting the potential of such approaches to advance the treatment of solid tumors (12–14). As a further extension of the well-established TRUCK principle, we previously generated an “all-in-one” vector construct that combines constitutive GD2-specific CAR expression and inducible nuclear factor of activated T cells (NFAT)-driven cytokine secretion. T cells engineered with the “all-in-one” vector showed improved killing capacity against GD2+ target cells and enhanced migration capacities of macrophages in vitro (15).
However, there are some limitations that negatively impact CAR T cell success in the treatment of solid tumors; some of these limitations may not apply to CAR NK cells due to differences in cell function and signaling (16–18). Preclinical and clinical trials using CAR NK cells indicate important advantages of CAR NK cells over CAR T cells in adoptive cell therapy, including the option to prepare modified immune cells from healthy donors, potent antitumor effects, and therapeutic safety (19, 20). Thus, primary autologous and allogeneic NK cells including available NK cell lines, e.g., NK-92, are sources for CAR NK cell trials and simplify manufacturing of CAR NK cells with reduced treatment cost (21). First CAR NK cell studies showed antitumor effects in both hematologic and solid malignancies as well as low incidences of on-target/off-tumor toxicity and cytokine release syndrome (CRS) (22, 23). For instance, an early clinical study of CAR NK cells in hematological malignancies demonstrated their safe toxicity profile and efficiency to persist in peripheral blood of patients for more than 12 months (24). Additionally, Liu et al. reported a complete absence of graft-versus-host disease (GvHD) after treatment with CAR NK cells (24). For solid tumors, several preclinical studies targeting glioblastoma (GBM) (25), breast (26), ovarian (27), and pancreatic cancers (28), and Ewing sarcomas (29) demonstrated the potential of CAR NK cells and led to clinical trials, e.g., NCT03415100, NCT03940820, NCT03941457, and NCT03383978.
These beneficial CAR NK cell properties prompted us to transfer the “all-in-one” vector concept from T cells to NK cells with the aim to improve adoptive CAR cell therapy for solid tumors. As proof of concept, we focused on the target antigen disialoganglioside GD2, which is overexpressed in neuronal malignancies, such as GBM and neuroblastoma (NB) (30, 31). GBM and NB are two of the most common malignancies of the nervous system. GBMs account for 16% of all primary brain cancers and are the most frequent and aggressive malignancy of the central nervous system (32). NBs derive from the sympathetic nervous system and are the most frequent extracranial pediatric solid neoplasm accounting for 7% of all cancers in children under 15 years and 15% of all pediatric cancer-related deaths (33), indicating the clinical need for novel treatment strategies. As an additional treatment modality, CAR cell therapies may further increase the probabilities of disease-free survival.
To transfer the “all-in-one” vector concept to NK cells, we designed alpharetroviral “all-in-one” vector constructs with a GD2CAR and an inducible promoter-driven transgene based on the previously published lentiviral “all-in-one” vector constructs (15). The objective is based on recent studies indicating that alpharetroviral vectors modify NK cells more efficiently than lentiviral vectors (34, 35). Although it was previously described that NFAT signaling is relevant for human NK cell activation, other studies also showed that nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), which plays a key role in cytokine production and granule exocytosis in NK cells, is important for activation of NK cells and, thereby, potentially a superior inducible promoter element (36, 37). We generated alpharetroviral “all-in-one” vectors that combine constitutive GD2CAR expression and inducible cytokine expression under the control of an inducible NFκB promoter element. Here, we demonstrate the overall relevance of the NFκB signaling pathway in CAR-mediated activation in NK cells and show selective induction of the EGFP reporter as well as the anticancer cytokine human interleukin-12 (hIL-12) in modified NK-92 cells and primary NK cells after GD2 recognition. Moreover, we obtained an augmented in vitro cytotoxic capacity of engineered “all-in-one” CAR NK cells against GD2+ target cells assessed via lactate dehydrogenase (LDH) assays and against patient-derived primary GD2+ GBM cells shown by an increased release of NK cell-specific cytokines associated with enhanced lytic potential. We also observed that modified “all-in-one” NFκBmIL2.hIL12.GD2CAR NK-92 cells show a trend to recruit monocytic cells in vitro due to the induced IL-12 release after GD2CAR-specific stimulation.
Taken together, our data provide the basis to improve adoptive NK cell therapy approaches using the CAR and TRUCK concepts. Our engineered NFκB-driven “all-in-one” vector construct is a promising advance toward effective NK cell-based clinical strategies to treat solid tumors.
To generate novel alpharetroviral “all-in-one” self-inactivating (SIN) (24) vectors, multiple cloning steps were performed. In brief, previously described well-established codon-optimized lentiviral “all-in-one” SIN vectors (pCCL.PPT.NFATenhsyn.EGFP.PGK.GD2CAR.PRE, pCCL.PPT.NFATmIL2.EGFP.PGK.GD2CAR.PRE, pCCL.PPT.NFATenhsyn.hIL12.PGK.GD2CAR.PRE, pCCL.PPT.NFATmIL2.hIL12.PGK.GD2CAR.PRE) (15), which function well in primary T cells, provided the basis for our work. A second-generation GD2CAR including GD2-scFv (14.G2a), IgG1 hinge, CD28 transmembrane, CD137 (4-1BB) co-stimulatory, and CD3-ζ signaling domains was used for the “all-in-one” vector construct (38). As an additional CAR, a second-generation CAR targeting the carcinoembryonic antigen (CEA) BW431/26-scFv-Fc-CD28/CD3-ζ (607) was used (39). This CAR harbors an IgG1 (Fc) hinge region fused to a CD28 transmembrane and CD28-CD3-ζ signaling domain. As inducible genes of interest (iGOIs), EGFP and human IL-12 (single-chain p40–p35) were used. The “all-in-one” gene sequence was removed from the lentiviral backbone of CAR vectors and inserted into an alpharetroviral SIN vector backbone. An additional modification for suitable NK cell application was made by exchanging the inducible 6xNFAT response elements with two generated NFκB response elements (2xNFκB; each 52 bp). The cloned vectors were verified by Sanger sequencing (Microsynth Seqlab, Göttingen, Germany). Cloning details and sequences are available on request.
To produce alpharetroviral particles, 293T (#ACC 635; DSMZ, Brunswick, Germany) cells were seeded with a density of 5–6 × 106 cells in 10-cm dishes and cultivated overnight. The cells were transfected with vector plasmids using the calcium phosphate method in combination with 25 µM chloroquine. For RD114/TR-pseudotyped alpharetroviral vectors, 5 µg vector plasmid, 2 µg phCMV-RD114TR (40), and 2.5 µg pcDNA3.alpha gp.Co (34) were used. The packaging components were produced by PlasmidFactory (Bielefeld, Germany). Viral supernatants were harvested at 36 and 48 h after transfection, sterile-filtered, and concentrated via ultracentrifugation at 13,238 ×g overnight at 4°C and stored at -80°C until usage (41). Viral supernatants were titrated in HT1080 cells (#ACC 315; DSMZ) via spin infection-mediated transduction in the presence of 4 µg/ml protamine sulfate (Sigma-Aldrich, St. Louis, MO, USA). Transduction efficiencies were controlled at least 2 days after transduction by staining of GD2CAR expression and flow cytometric detection.
293T cells, fibrosarcoma cell line HT1080, genetically modified HT1080 cells expressing GD2 and described in Zimmermann et al. (15), genetically modified HT1080 expressing CEA, and the neuroblastoma cell line SH-SY5Y were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) (Biochrom, Berlin, Germany) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 µg/ml streptomycin, and 1 mM sodium pyruvate (all PAN-Biotech, Aidenbach, Germany). The human NK cell line NK-92 (kindly provided by Christine S. Falk, Hannover, Germany, and Eric Vivier, Marseille, France) (42) was maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Biochrom) supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, 1 mM sodium pyruvate, and 400 IU/ml hIL-2 (Proleukin®) (Novartis, Basel, Switzerland). The acute myeloid leukemia cell line KG-1a (#ACC 421; DSMZ), genetically modified to constitutively express the RFP657 reporter (APC+), was equipped with GD2 by retroviral transduction with a GD3/GD2S transgene to establish a suspension target cell line. Suspension target cells were cultivated in supplemented RPMI 1640 medium. All cells were cultured in humidified incubators at 37°C and 5% CO2.
Peripheral blood isolated from anonymous healthy donors was kindly provided by Transfusion Medicine (Hannover Medical School, Hannover, Germany). PBMCs were isolated via Leucosep™ (Greiner Bio-One, Kremsmünster, Austria) in combination with Bicoll Separating Solution (Ficoll Separating Solution, Biochrom) according to the manufacturer’s instructions. The separated lymphocytes were used to isolate NK cells or stored at -80°C until separation. Primary CD56+ NK cells were isolated from PBMCs with the human NK Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s protocol. In brief, a determined PBMC number was labeled firstly with NK Cell Biotin-Antibody Cocktail and secondly, after incubation and washing, with NK Cell MicroBead Cocktail. After a second incubation and washing, labeled cells were separated in equilibrated columns in the magnetic field of a MACS Separator. The collected flow-through consisted of enriched unlabeled CD56+ NK cells. The purity of isolated cells was quantified via flow cytometric staining for CD56 and CD3. Isolated CD56+ CD3- NK cells were cultivated in NK MACS Medium (MACS Miltenyi Biotec) with 5% human serum (c.c.pro, Oberdorla, Germany), 100 U/ml penicillin (PAN-Biotech), 100 µg/ml streptomycin (PAN-Biotech), 1% NK MACS supplement (MACS Miltenyi Biotec), 500 IU/ml of hIL-2 (Proleukin®, Novartis), and 70 ng/ml hIL-15 (PeproTech, Hamburg, Germany).
Primary human GBM samples were provided from GBM resections after obtaining informed patient consent (Nordstadt Cerebral Cancer Study (NoCCA-Study), Register-Nr. 6864). After resection, the GBM cells were isolated and cultivated according to the protocol of Hasselbach et al. (43) with modifications of Zimmermann et al. (15). Briefly, isolated tumor spheroids were maintained in DMEM/F12 (Gibco, Karlsruhe, Germany) supplemented with 100 U/ml penicillin, 100 µg/ml streptomycin, N2 supplement (1×) (Miltenyi Biotec), 0.5 mg/ml bovine serum albumin (BSA) (PAN-Biotech), 25 µg/ml gentamicin (Gibco), 20 ng/ml human basic fibroblast growth factor (bFGF), and 20 ng/ml human epidermal growth factor (EGF) (both PeproTech). All primary cells were cultured in humidified incubators at 37°C and 5% CO2.
HT1080 cells and KG-1a cells were genetically modified via spinoculation-mediated transduction using protamine sulfate (Sigma-Aldrich). Therefore, 1 × 105 cells were seeded in a 24-well plate and generated viral vector supernatants were added with 4 µg/ml protamine sulfate (Sigma-Aldrich). For spinoculation, cells were centrifuged for 1 h at 800 ×g and 37°C. NK-92 cells and primary NK cells were transduced in the presence of RetroNectin® (TaKaRa Bio, Otsu, Shiga, Japan). In brief, 48-well plates were coated with 137 µl of 48 µg/ml RetroNectin® (TaKaRa) overnight at 4°C. The coated wells were blocked with sterile-filtered phosphate-buffered saline (PBS) (Biochrom) containing 2% BSA (PAN-Biotech) for 30 min at room temperature and washed with HBSS/HEPES (Gibco; PAN- Biotech). Viral vector supernatants were loaded into the wells, and plates were centrifuged for 30 min at 400 ×g and 4°C. After removing supernatants, NK-92 cells (1 × 105) or primary NK cells (2 × 105) were added to each well and incubated for 24 h.
The amount of used viral vector supernatants was calculated regarding the desired multiplicity of infection (MOI) (44). The transduction efficiency was determined via transgene staining and flow cytometric analysis at least 2 days after transduction.
Genomic DNA (gDNA) was isolated from unmodified and genetically modified NK-92 and primary NK cells with the QIAamp DNA Blood Mini Kit (Qiagen N.V., Venlo, Netherlands) according to the manufacturer’s instructions. For vector copy number (VCN) determination, gDNA was analyzed by real-time quantitative polymerase chain reaction (qPCR) with the help of TaqMan probe by using TaqMan Fast Advanced Master Mix (Applied Biosystems—ABI, Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). The primers were specific for the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and the genomic polypyrimidine tract binding protein 2 (PTPB2), as previously described (45). The qPCR was accomplished on a StepOnePlus device (Applied Biosystems).
Collected cell pellets were lysed in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 100 mM NaF, 1% Triton X-100 enriched with 1 mM DTT, 1 mM Na3VO4, and 1× cOmplete Mini Protease Inhibitor Cocktail (Roche Diagnostics, Mannheim, Germany)), incubated on ice for 20 min, and centrifuged for 15 min at high speed and 4°C. The protein concentration was determined via Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Protein sizes were identified with the help of the loaded Color Prestained Protein Standard ladder (New England Biolabs, Frankfurt am Main, Germany). After blotting separated proteins on a nitrocellulose membrane, membranes were blocked in Tris-buffer containing 5% milk powder and incubated with an anti-CD3-ζ-HRP-antibody (6B10.2, Santa Cruz Biotechnology, Dallas, TX, USA) to detect the endogenous CD3-ζ and the CD3-ζ domain of the CAR. As an additional control, membranes were also incubated with an antibody to detect the common housekeeping protein glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) (GeneTex, Eching, Germany). To detect the antibody protein binding, SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) was used according to the manufacturer’s instructions. Protein bands were visualized via a FusionFX instrument (Peqlab GmbH, Erlangen, Germany).
Flow cytometry was used for the analysis of surface marker or transgene expression. The following antibodies were used: CD3-APC (OKT3, BioLegend, San Diego, CA, USA), CD16-PE (REA423, Miltenyi), CD56-APC (AF12-7H3, BioLegend; HCD56, Miltenyi Biotec), CD56-FITC (REA196 Miltenyi), CD56-PE (B159, Becton Dickinson, BD, Heidelberg, Germany), Ganglidiomab-PE (Gangliomab-phycoerythrin (PE) monoclonal antibody was kindly provided by H. Lode and N. Siebert (46) and PE-conjugated), IgG, F(ab′)2 (Biotin-SP-conjugated, Jackson ImmunoResearch, West Grove, PA, USA), Ganglioside GD2-APC (14G2a, BioLegend), Ganglioside GD2-PE (14G2a, BioLegend), and Annexin V-PE (BD Biosciences, Franklin Lakes, USA) and the secondary antibodies Streptavidin-APC (BioLegend) and Streptavidin-PE (eBioscience, San Diego, CA, USA). For staining of the degranulation marker CD107a, cocultures were incubated with CD107a-APC (Miltenyi) for 1 h at 37°C and 5% CO2 with shaking. Afterward, Brefeldin A (1:1,000; BioLegend) and Monensin (1:1,000; BioLegend) were added for an additional incubation for 4 h at 37°C and 5% CO2 with shaking. Stained cells were measured via FACSCalibur cytometer (Becton Dickinson, BD) or CytoFLEX (Beckman Coulter) and analyzed via FlowJo software (Tree Star Inc., Ashland, OR, USA).
Modified and unmodified NK cells were cocultured with GD2+ or GD2- adherent or suspension target cells in the desired effector to target cell ratios (E:T) to investigate the specific inducible gene expression after GD2 antigen recognition. NFAT- or NFκB-driven EGFP induction was detected in addition to GD2CAR expression by flow cytometric analysis at defined time points. Inducible cytokine secretion was detected via ELISA, Bio-Plex assay, or LEGENDplexTM.
Cell culture supernatants of genetically modified and unmodified NK cell monocultures and NK cell cocultures with GD2+ or GD2- target cells were collected at desired time points to measure induced cytokine secretion after antigen recognition. Supernatants were analyzed by enzyme-linked immunosorbent assay (ELISA). For NFAT- or NFκB-driven human IL-12 secretion, the Human IL-12 (p70) ELISA MAX™ Deluxe Sets (BioLegend) was used according to the manufacturer’s instructions. The absorbance of the samples was measured with the SpectraMax® 340PC384 Microplate Reader (Molecular Devices, San Jose, CA, USA). The cytokine amount was calculated regarding measured absorbance and according to the manufacturer’s protocol.
In addition, coculture supernatants were analyzed via Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad Laboratories) or LEGENDplex™ Human CD8/NK Panel (BioLegend) according to the manufacturer’s protocols. For Bio-Plex assay, the provided plates were coated with magnetic beads and washed. Standards, blanks, and samples were added and incubated while shaking. After washing, the detection antibody was applied and incubated with shaking to bind desired biomarkers. After repeating washing, the wells were incubated with streptavidin. Then, the wells were washed and incubated with assay buffer while shaking. Afterward, the fluorescence of samples was measured by Bio-Plex 200 System (Bio-Rad Laboratories) and analyzed by Bio-Plex Manager 6.0 (Bio-Rad Laboratories). For LEGENDplex™ assay, standards, and diluted samples were incubated with premixed beads in a V-bottom plate overnight at 4°C. Afterward, the samples were incubated with detection antibody for 1 h and streptavidin-PE for 30 min at room temperature. Analysis was performed on a CytoFLEX (Beckman Coulter) and analyzed on legendplex.qognit.com (BioLegend).
The cytolytic capacity of modified NK-92 cells against GD2+/GD2- target cell lines (HT1080, HT1080 GD2, and SH-SY5Y) was analyzed by determination of the lactate dehydrogenase (LDH) release using the Pierce™ LDH Cytotoxicity Assay Kit (Thermo Scientific, Bremen, Germany). Cocultures were performed in 96-well plates with an E:T of 6:1, and 50 µl of cell coculture supernatants were collected after 24 h and used for the assay according to the manufacturer’s instructions. Fifty microliters of the provided reaction mix were added, and incubated for 30 min, and the reaction was stopped by adding 50 µl of the provided stop solution. Absorbance was determined at 490 nm and 680 nm (SPECTRAmax 340PC, Molecular Devices, Software SoftMax Pro 4.0) and cytototoxicity was calculated according to the manufacturer’s protocol.
Coculture supernatants were tested for their capacity to induce the migration of human monocytic cells through specific pore polycarbonate membranes using a modified Boyden chamber (Neuro Probe, MBB96). In more detail, a 96-well flat bottom plate was filled with 300 μl/well of 0.6% agarose and was placed inside the modified Boyden chamber after inserting the appropriate spacer. Then, 120 μl of sample was added into each well and a membrane with 8-μm pore size (Neuro Probe) was placed over the plate in direct contact with the cell culture supernatants. The chamber was closed, and 5 × 105 MONO-MAC-6 cells/well (in 200 μl RPMI medium) were added to the top wells of the chamber. The entire chamber was incubated at 37°C with 5% CO2 for 4 h. After the incubation time, the membrane was removed from the chamber and a scraper was used in order to remove the remaining cells from the top of the membrane. Migrated and membrane-attached cells were fixed with methanol for 10 min and stained with Giemsa’s solution for 1 h. Non-specific staining was removed via a washing step with water, and the membrane was dried overnight. Representative pictures for each sample were taken using an Olympus ιX71 microscope, and all stained cells were counted. Different concentrations of the IL-12 cytokine (0–500 pg/ml) diluted with the same coculture medium were used as positive controls.
The Axio Observer fluorescence microscope (ZEISS, Oberkochen, Germany) was used to take bright field cell images and to observe the fluorescence of target and effector cells in coculture experiments. An inducible GD2CAR-dependent EGFP expression in modified NK cells was detected in 24-h coculture experiments with GD2+ adherent and suspension target cells and patient-derived GBM spheroids. Additionally, the dynamic NFκB-dependent EGFP upregulation over time was visualized by live-cell imaging for modified NK cells cocultured with GD2+ RFP657+ KG-1a cells. RFP657+ KG-1a cells were distinguished from effector cells in cocultures by their red fluorescence. Microscope images were analyzed by AxioVision Software Rel. 4.8.
Presented data were statistically analyzed by GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). One-way ANOVA and two-way ANOVA tests with Tukey’s multiple-comparison tests and paired t-tests were used as indicated. Significance is indicated in all figures and legends (****p ≤ 0.0001; ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; ns p > 0.05). For technical and biological replicates, mean values ± SD (standard deviation) are shown.
Previous data of lentiviral “all-in-one” vector-modified T cells demonstrated the potent NFAT-driven upregulation of the inducible gene of interest (iGOI) upon GD2CAR-specific activation (15). The lentiviral “all-in-one” vector constructs for targeted T cell gene therapy were accordingly transferred to an alpharetroviral SIN vector system to create an effective platform for targeted NK cell therapy (Figure 1A). Analogously to the lentiviral vector architecture, alpharetroviral “all-in-one” SIN vector constructs combine constitutive second-generation CAR expression under the control of the human phosphoglycerate kinase (hPGK) promoter and NFAT-driven inducible expression of an iGOI, e.g., EGFP or hIL-12. Two different NFAT-driven inducible promoters, namely, NFATmIL2 or NFATsyn, both containing six NFAT consensus response elements (6xNFAT) fused to either a minimal IL2 promoter (48) or a synthetic promoter element and a synthetic TATA box (49) were used. Modification of NK-92 cells with these alpharetroviral vectors, which were pseudotyped with the RD114/TR envelope glycoprotein, showed average transduction rates from 50% to 75% (Figure 1B). The vector copy number (VCN) of modified NK-92 cells was analyzed by quantitative PCR to detect the wPRE sequence of the alpharetroviral vector construct and showed an average of one to two vector integrations per cell (Figure 1C). Immunoblot analyses confirmed the successful CAR expression of the “all-in-one” vectors in NK-92 cells on protein level as detected by expression of the CD3-ζ domain of the GD2CAR (about 53 kDa) (Figure 1D). The expression of endogenous CD3-ζ in NK-92 cells (about 17 kDa) and the housekeeping protein GAPDH were used as internal controls.
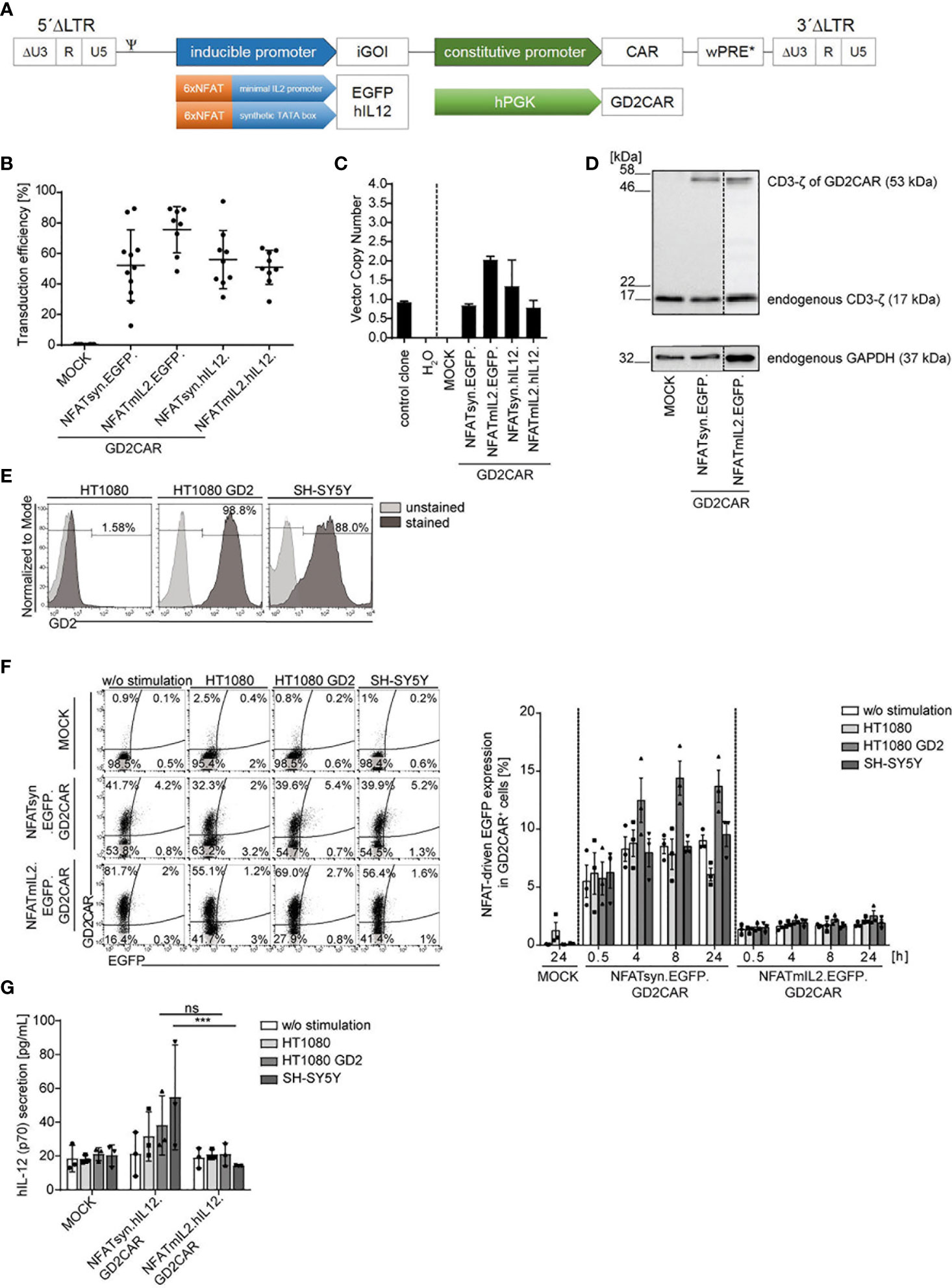
Figure 1 Generation and validation of the alpharetroviral NFAT-driven “all-in-one” SIN vector constructs. (A) Scheme of the alpharetroviral self-inactivating “all-in-one” provirus constructs encoding for a constitutively expressed GD2CAR and an iGOI, namely, EGFP and hIL-12. The constitutive GD2CAR expression is driven by a human phosphoglycerate kinase (hPGK) promoter. The inducible gene cassette includes six consensus NFAT (6xNFAT) elements in combination with either a minimal IL2 promoter (NFATmIL2) or a synthetic TATA box (NFATsyn). (B) Transduction efficiencies of NK-92 cells modified with the indicated alpharetroviral “all-in-one” SIN vector constructs and pseudotyped with RD114/TR (MOI 10). Transduction efficiencies were quantified by flow cytometric analyses for GD2CAR expression (n = 8–11). (C) VCN determination of unsorted modified (MOI 10) NK-92 cells. As controls, the VCNs of a water sample and an induced pluripotent stem cell (iPSC) clone HD2 (47) with a known VCN of 1 were calculated. Technical triplicates are shown. (D) CAR protein expression in unsorted transduced (MOI 10) NK-92 cells exemplarily shown for NFATmIL2.EGFP and NFATsyn.EGFP constructs via Western blot analysis. The CD3-ζ domain of the GD2CAR (expected size approximately 53 kDa) and the endogenous CD3-ζ (17 kDa) as an internal control were detected. The housekeeping protein glyceraldehyde-3-phosphate-dehydrogenase (GAPDH, 37 kDa) served as a second control. (E) Flow cytometric analysis of GD2 expression on target cell lines used for functional testing via coculture experiments. (F) Representative flow cytometric analysis of inducible EGFP upregulation of unsorted modified (MOI 10) NK-92 cells after 24 h of coculture with indicated target cells at an effector to target ratio (E:T) of 10:1. The bar graph summarizes the inducible GD2CAR-specific EGFP upregulation of modified NK-92 cells after coculture experiments (n = 3). (G) Inducible human IL-12 secretion of modified NK-92 cells after antigen recognition in 24-h coculture experiments at an E:T ratio of 10:1. Human IL-12 levels were determined by ELISA (n = 3). Indicated significance was determined by two-way analysis of variance (ANOVA) with Tukey´s multiple-comparison test; ***p ≤ 0.001. Mean values ± SD are shown. ns, not significant. Untransduced NK-92 cells were termed MOCK.
To assess the functionality and feasibility of the NFAT-driven “all-in-one” vector constructs for NK cell therapy, modified NK-92 cells were analyzed for GD2CAR-specific activation in a proof-of-concept cocultivation experiment with GD2+ target cell lines (HT1080 GD2 and SH-SY5Y). HT1080 cells, determined to be GD2-, served as a control target cell line (Figure 1E). NK-92 cells were modified with “all-in-one” alpharetroviral SIN vectors harboring an inducible EGFP or IL-12 expression cassette under the control of the different NFAT-driven promoters, NFATmIL2 and NFATsyn, and cocultured with GD2+ or GD2- target cells for 24 h. Specific GD2CAR activation upon recognition of the target antigen GD2 resulted in an NFAT-driven transgene upregulation as detected by flow cytometry for modular EGFP expression or by ELISA for IL-12 secretion in coculture supernatants. NK-92 cells transduced with constructs harboring an EGFP expression cassette under the control of the NFATsyn-responsive promoter led to a constant background expression in unstimulated modified NK-92 cells and modified NK-92 cells cocultured with GD2- HT1080 cells (Figure 1F). Moderate EGFP expression was observed in NK-92 cells modified with the NFATsyn.EGFP.GD2CAR construct after GD2CAR-specific activation upon coculture with GD2+ HT1080 GD2 or SH-SY5Y cells. The highest EGFP expression was reached after 4 to 8 h of coculture (Figure 1F). NK-92 cells transduced with constructs harboring an NFATmIL2-driven inducible EGFP expression cassette did not show increased GD2CAR-specific EGFP expression after cocultivation with the GD2+ target cells (Figure 1F). Comparably, NK-92 cells modified with “all-in-one” vector constructs harboring an NFAT-driven IL-12 cytokine cassette did not show an enhancement of GD2CAR-specific IL-12 secretion after cocultivation with GD2+ target cells, but did show a high background IL-12 secretion with GD2- target cells (Figure 1G). In addition to the background expression, NK-92 cells modified with NFATsyn-driven “all-in-one” constructs showed a slightly better IL-12 secretion after stimulation with GD2+ target cells compared to NK-92 cells expressing the NFATmIL2 promoter, but the level of secreted IL-12 was low, compared to previous results observed in T cells (15) (Figure 1G).
In conclusion, NFAT-driven inducible expression cassettes coupled to constitutive GD2CAR-specific signaling were far less efficient in NK-92 cells compared to T cells, which necessitated further optimization of the vector architecture to provoke “all-in-one” vector feasibility in CAR NK cell therapy.
NFκB is described as a key element in NK signaling pathways (50). Moreover, it was previously shown that NFκB was efficiently used for transgene induction in NK cells (37). Thus, to improve GD2CAR-specific inducible transgene expression in NK-92 cells, the six NFAT response elements within the inducible expression cassette were replaced by two NFκB response elements, as two NFκB consensus elements (2xNFκB) were linked to the minimal IL2 promoter (NFκBmIL2) or the synthetic promoter fused to the TATA box (NFκBsyn) (Figure 2A).
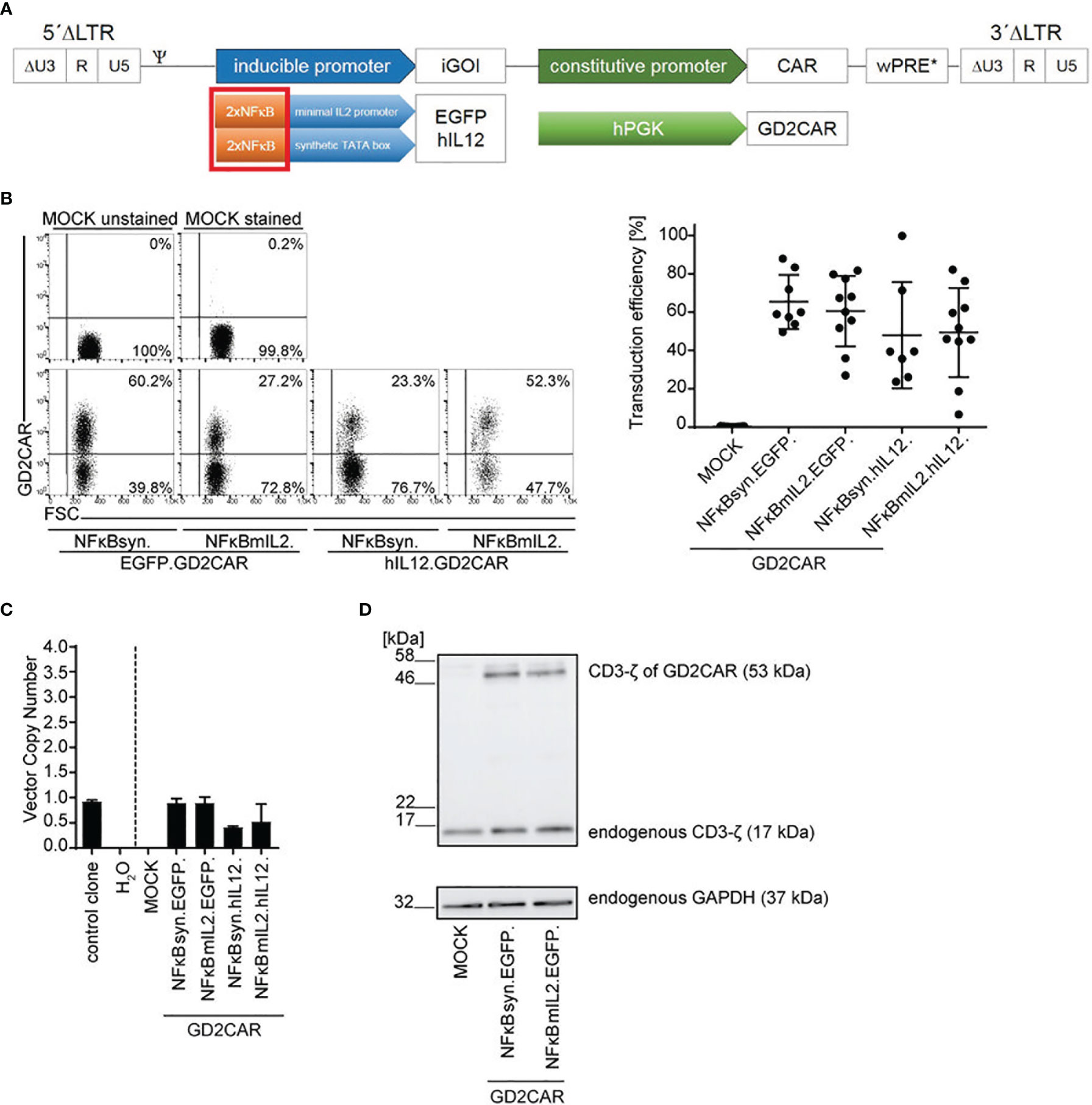
Figure 2 Optimization of alpharetroviral “all-in-one” SIN vectors with inducible NFκB-promoter elements for NK cell application. (A) Scheme of modified alpharetroviral SIN “all-in-one” provirus constructs encoding a constitutively expressed GD2CAR and an iGOI, namely, EGFP or hIL-12. The iGOI expression cassette included two consensus NFκB (2xNFκB) elements in combination with either a minimal IL2 promoter (NFκBmIL2) or a synthetic TATA box (NFκBsyn). (B) Representative flow cytometric analysis of unsorted modified NK-92 cells post transduction (RD114/TR-pseudotyped; MOI 10). Scatter plots summarize transduction efficiencies of unsorted modified (MOI 10) NK-92 cells (n = 7–10). (C) VCN of unsorted modified (MOI 10) NK-92 cells. As controls, the VCNs of a water sample and an induced pluripotent stem cell (iPSC) clone HD2 (47) with a known VCN of 1 were calculated. Technical triplicates are shown. (D) CAR expression in unsorted transduced (MOI 10) NK-92 cells was exemplarily shown for NFκBmIL2.EGFP and NFκBsyn.EGFP constructs on the protein level via Western blot analysis. The CD3-ζ domain of the GD2CAR (expected size approximately 53 kDa) and the endogenous CD3-ζ (17 kDa) as an internal control were detected. The housekeeping protein glyceraldehyde-3-phosphate-dehydrogenase (GAPDH, 37 kDa) served as a second loading control. Mean values ± SD are shown in Figure 2. Untransduced NK-92 cells were termed MOCK.
NK-92 cells were modified with the newly designed vector constructs and analyzed for GD2CAR surface expression by staining the scFv region of the GD2CAR with a specific anti-idiotype antibody, Ganglidiomab. Average transduction efficiencies achieved with an MOI of 10 ranged between 40% and 70% (Figure 2B). Similarly to the NFAT-driven “all-in-one” vector constructs, the VCNs within the modified NK-92 cells were approximately 1 for most of the constructs (Figure 2C). Western blot analysis confirmed GD2CAR expression in NK-92 cells as shown exemplarily for the NFκB-driven “all-in-one” inducible EGFP vectors and visualized by the expression of the CD3-ζ domain of the CAR (Figure 2D). Expression of endogenous CD3-ζ and the housekeeping protein GAPDH served as controls.
The NFκB-driven “all-in-one” constructs pseudotyped with RD114/TR were also tested in primary NK cells that were enriched from human peripheral blood mononuclear cells (PBMCs) by magnetic separation (Figure S1A). Transduction efficiencies of approximately 30% on average for NFκB-driven constructs harboring the EGFP expression cassette and of <20% on average for NFκB-driven constructs harboring the IL-12 expression cassette were reached (Figure S1B). GD2CAR integration in primary NK cells was verified by VCN determination with measured VCNs of approximately 1 per cell (Figure S1C) and the CAR expression by immunoblot detection of the CD3-ζ domain of the GD2CAR (Figure S1D).
To analyze the functionality and feasibility of the alpharetroviral NFκB-driven inducible “all-in-one” vector constructs, coculture experiments of modified NK-92 cells with GD2- (HT1080, KG-1a) or GD2+ (HT1080 GD2, KG-1a GD2, and SH-SY5Y) target cells as well as with primary patient-derived GD2+ GBM cells were performed.
As a proof of concept of NFκB-driven transgene induction in modified NK cells after target stimulation, NK-92 cells modified with the modular EGFP “all-in-one” constructs were investigated. EGFP expression was induced in NK-92 cells transduced with vector constructs harboring an EGFP expression cassette under the control of the NFκB-responsive elements, independent of the promoter choice—NFκBmIL2 or NFκBsyn—after GD2CAR-mediated activation upon coculture with GD2+ target cells (Figure 3A). GD2CAR-specific NFκB-driven EGFP expression was also investigated in modified NK-92 cells over time (0.5, 4, 8, 24 h), showing that both NFκBsyn and NFκBmIL2 promoters reached their highest EGFP expression at 24 h after cocultivation with GD2+ target cells (Figure S2A). After a 24-h coculture with HT1080 GD2 target cells, the NFκBsyn promoter led to on average 60% EGFP upregulation in modified NK-92 cells compared with on average 50% EGFP upregulation driven by the NFκBmIL2 promoter. The same trend was observed with KG-1a cells expressing GD2 (Figure 3A). Coculture experiments with GD2+ SH-SY5Y cells showed 60% EGFP expression in modified NFκBsyn NK-92 cells compared to 40% NFκBmIL2-driven EGFP induction after GD2CAR-specific activation. The GD2CAR-specific stimulation of NFκBsyn.EGFP.GD2CAR-modified NK-92 cells by GD2+ target cells showed a twofold or fivefold EGFP induction compared to non-specific activated modified NK-92 cells (w/o stimulation; HT1080; KG-1a). NFκBmIL2.EGFP.GD2CAR-modified NK-92 cells showed a sevenfold or 10-fold EGFP induction after antigen recognition (Figure 3A). For all tested constructs, the density of EGFP expression recorded by the mean fluorescence intensity (MFI) was similar (Figures S2B, C). Regarding off-target leakiness and background expression of the inducible transgene, the NFκBmIL2 promoter element led to less background EGFP expression of unstimulated modified NK-92 cells and modified NK-92 cells cocultured with GD2- target cells compared to the NFκBsyn promoter. The NFκBsyn promoter showed an EGFP upregulation of more than 20% in unstimulated NK-92 cells and transduced NK-92 cells cocultured with HT1080 target cells, indicating that this vector configuration was less tightly controlled as compared to that with the NFκBmIL2 promoter element (Figure 3A).
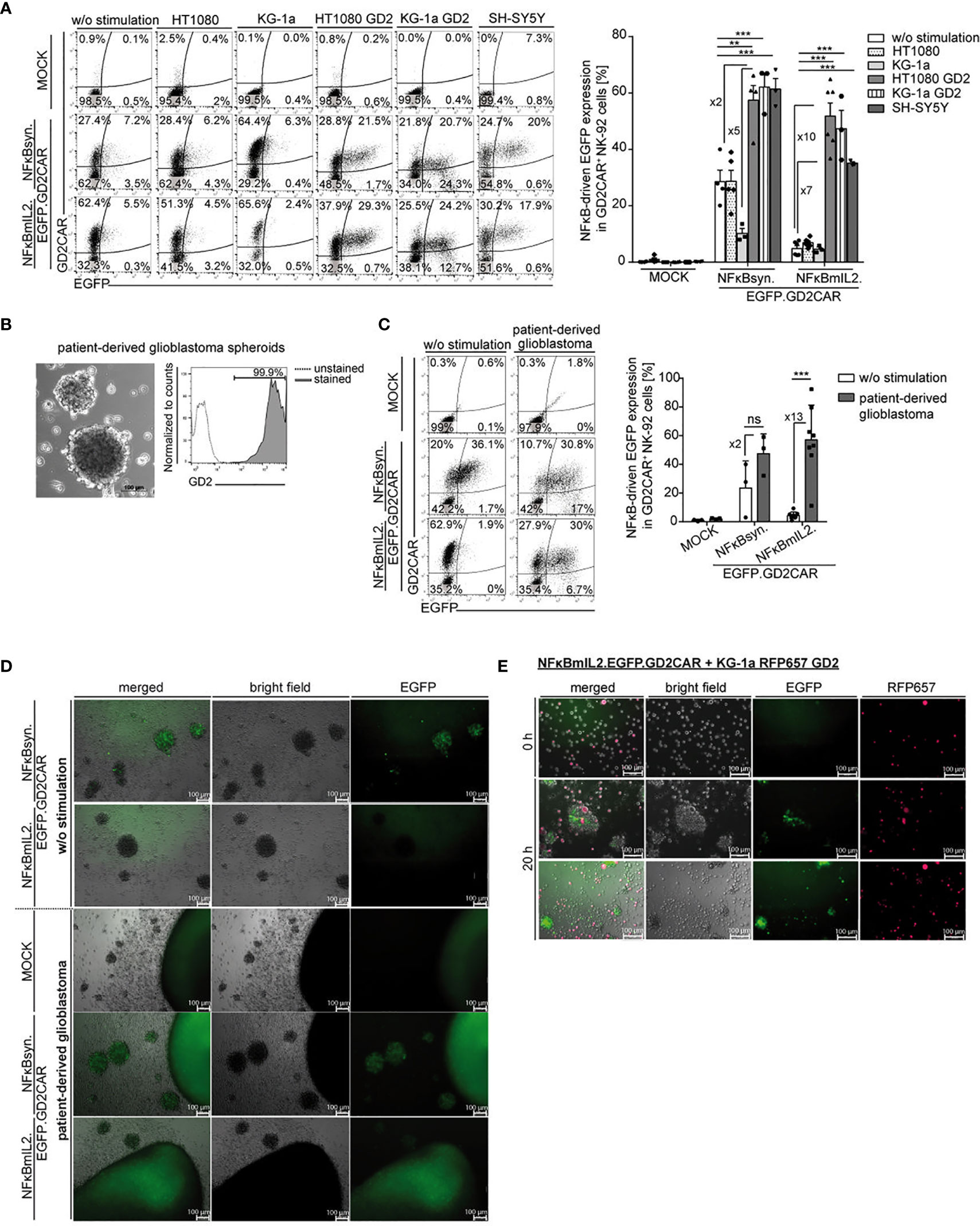
Figure 3 Modification of NK-92 cells with alpharetroviral SIN NFκB-driven “all-in-one” vector constructs led to GD2CAR-mediated EGFP induction. (A) Increased EGFP expression after target-specific stimulation. Exemplary flow cytometric analysis of unsorted modified (MOI 10) NK-92 cells after 24-h of coculture with indicated target cells (E:T 10:1). The bar graph summarizes the inducible NFκB-driven EGFP expression of unsorted modified (MOI 10) NK-92 cells after 24-h cocultures with indicated target cells (n = 3–6). Indicated significance was determined by one-way ANOVA with Tukey´s multiple-comparison test; ***p ≤ 0.001; **p ≤ 0.01. (B) Bright field picture of patient-derived primary glioblastoma (GBM) cells and flow cytometry analysis show GD2 expression of primary GBM cells. (C) Flow cytometric analysis of a 24-h coculture of unsorted modified (MOI 10) NK-92 cells in the presence or absence of primary GBM cells. The bar graph shows specific NFκB-driven EGFP expression in modified NK-92 cells (n = 3-8). Indicated significance was determined by paired t-test; ***p ≤ 0.001. ns, not significant. (D, E) Immunofluorescence microscopy analysis indicated GD2CAR-specific EGFP upregulation after incubation with (D) primary GBM cells or (E) KG-1a RFP657 GD2 cells. Shown are (D) 24-h or (E) 0- and 20-h (two representative pictures with identical culture conditions) cocultures of sorted modified NK-92 cells and tumor spheroids or suspension target cells. KG-1a RFP657 GD2 cells are indicated in red; NFκB-driven EGFP induction is indicated in green. Mean values +/- SEM are shown in 3A and mean values +/- SD are shown in 3C. Untransduced NK-92 cells were termed MOCK.
Similar results were observed in 24-h co-cultures of unsorted modified primary NK cells with GD2+- or GD2− adherent target cells at an E:T ratio of 10:1 with maximum average EGFP induction of 30% in NFκBsyn-modified cells compared to 25% in NFκBmIL2-engineered cells after GD2CAR-specific activation by HT1080 GD2 cells. The background EGFP expression of around 5% was also lower in the NFκBmIL2 vector configuration compared to the NFκBsyn vector configuration with 10% (Figure S3A).
The function of NK-92 cells modified with the alpharetroviral NFκBmIL2- or NFκBsyn-driven “all-in-one” EGFP vector constructs was further characterized by co-incubation with primary patient-derived GBM cells that highly express the GD2 antigen (Figure 3B). Modified NK-92 cells were cocultivated with patient-derived primary GBM cells. Specific GD2CAR-mediated induction of EGFP was assessed via flow cytometric analysis (Figure 3C) and fluorescence microscopy (Figure 3D), indicating the capability of modified NK-92 cells to recognize the GD2 antigen on primary GBM cells. In contrast to unmodified NK-92 cells, which did not show any NFκB-driven EGFP upregulation after cocultivation with primary GBM cells, a 13-fold and twofold higher EGFP expression was induced by NFκBmIL2.EGFP.GD2CAR- and NFκBsyn.EGFP.GD2CAR-modified NK92 cells after antigen recognition and GD2CAR signal transduction (Figure 3C). Similar results were observed for engineered primary NK cells in cocultures with patient-derived GBM spheroids with CAR-mediated EGFP induction of approximately 40% for NFκBsyn-responsive constructs and 20% for NFκBmIL2-responsive constructs (Figure S3B). However, NFκBsyn.EGFP.GD2CAR-modified NK-92 cells showed a CAR-independently elevated EGFP expression in up to 40% of modified cells in the absence of GD2+ target cells (Figure 3C), indicating leakiness of the NFκBsyn promoter element as described for cocultures with other target cell lines (Figure 3A). The MFI of EGFP confirmed the leakiness of the NFκBsyn promoter vector construct as seen with high fluorescence intensity values already in the non-induced state in comparison to the NFκBmIL2 promoter vector construct in NK-92 cells (Figures S2B–D).
NFκB-driven EGFP induction upon GD2CAR-specific activation was additionally detected by fluorescence microscopy after cocultivation of modified NK-92 cells with primary GBM cells. Activated modified NK-92 cells that induced EGFP expression became visible as green fluorescent cells (Figure 3D). Moreover, modified NK-92 cells migrated toward the GBM cells and were discernable as single green fluorescent cells on the outer layer and within tumor spheroids (Figure 3D). A CAR-signaling-independent EGFP expression was again detected in NFκBsyn.EGFP.GD2CAR-modified NK-92 cells in monoculture without GD2+ GBM cells, confirming the overall leakiness of the NFκBsyn promoter vector construct, which was less observed for the NFκBmIL2 construct (Figure 3D).
In vitro live-cell imaging experiments were performed to record the behavior of the modified NK-92 cells while recognizing the target antigen GD2. KG-1a cells engineered to highly express GD2 (Figure S3C) in addition to an RFP657 reporter construct were used as target cells, as they were detectable due to their red fluorescence (Figure 3E). Modified NFκBmIL2.EGFP.GD2CAR-transduced NK-92 cells were cocultivated with the engineered KG-1a cells for the indicated time points. After 20 h of coculture, the NFκB-driven EGFP upregulation was clearly visible due to the green fluorescence signal detected in modified NK-92 cells, indicating engagement of the target antigen GD2 with consequent GD2CAR-mediated EGFP induction. This interaction was also seen by microscopic analysis as migration of the modified NK-92 cells to the GD2+ KG-1a cells and the integration of target cells into the NK cell cluster, which resulted in the additional detection of the merged yellow color (Figure 3E). Furthermore, comparable in vitro live-cell imaging experiments with NFκBmIL2.EGFP.GD2CAR-modified primary NK cells and GD2+ KG-1a cells confirmed the previously described observations after 16 h of coculture (Figure S3D).
The proof-of-concept experiments demonstrated that GD2CAR-mediated activation induced NFκB-driven EGFP transgene expression, confirming the feasibility of the transcription factor NFκB to translate GD2CAR-triggered signaling into transgene expression in the “all-in-one” alpharetroviral SIN vector system in NK cell subsets.
The modular design of the “all-in-one” vector system simplified the introduction of a different CAR construct to allow verification of the CAR-mediated induction of genes of interest (e.g., EGFP). Thus, GD2CAR was exchanged by a second-generation CEACAR. Modified NK-92 cells harboring the newly generated alpharetroviral “all-in-one” CEACAR vector constructs were compared regarding the promoter-dependent EGFP induction (NFκBsyn and NFκBmIL2) after CEA-specific CAR activation. Similar results as seen for the vector constructs with the GD2CAR were observed, indicating the adaptability of these principles to other applications and research questions (Figure S4).
Together, the presented data indicate the relevance of the choice of transcription factor-responsive elements within the inducible promoter cassette of the “all-in-one” vector construct that is likely cell-type dependent. As the NFκBsyn promoter-driven vector constructs led to high background expression in NK cells, the NFκBmIL2 promoter-driven “all-in-one” constructs were used in further analyses.
The cytotoxic activity of NK-92 cells modified with the alpharetroviral “all-in-one” vector constructs was assessed via lactate dehydrogenase (LDH) assays (Figure 4A). NK-92 cells harboring either the NFκBmIL2.EGFP.GD2CAR or the NFκBmIL2.hIL12.GD2CAR construct showed increased cytotoxicity after GD2CAR-specific activation with GD2+ target cells (HT1080 GD2 and SH-SY5Y). While NK-92 cells modified with the NFκBmIL2.EGFP.GD2CAR indicated enhanced killing capacity against GD2+ target cells compared to NK-92 modified with the NFκBmIL2.hIL12.GD2CAR vector construct, they also had higher background activity against GD2- target cells.
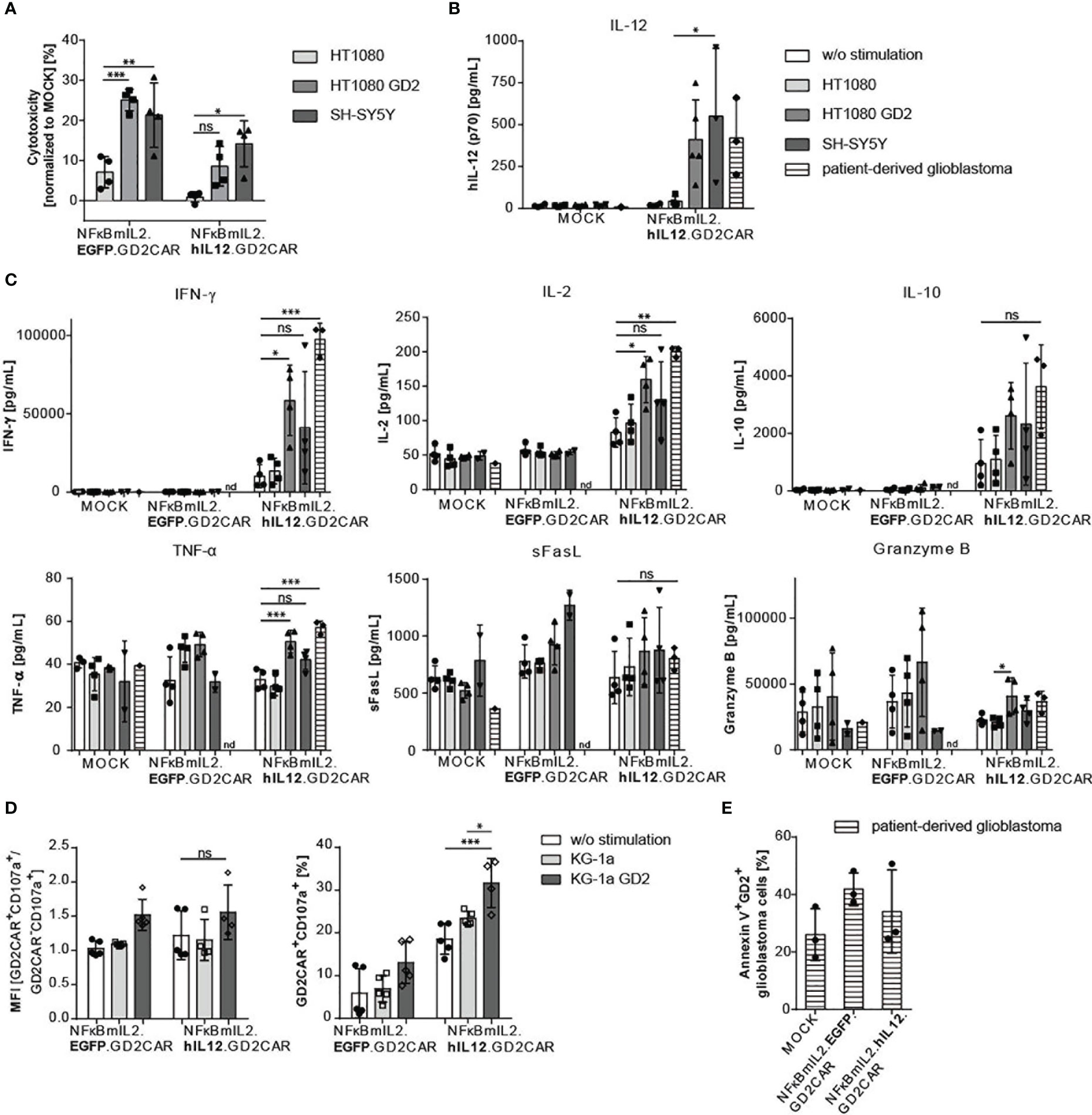
Figure 4 Increased cytotoxic potential of modified NK-92 cells with NFκB-driven “all-in-one” vectors after target gene stimulation. (A) Increased cytotoxic activity after GD2 antigen recognition. GD2CAR-mediated activation leads to antigen-specific killing. Lactate dehydrogenase (LDH) assays were performed with supernatants of coculture experiments. Cocultures were performed with “all-in-one” vector-modified, unsorted (MOI 10) NK-92 cells and indicated target cells for 24 h at an E:T of 6:1. Cytotoxicity was normalized to MOCK cells. Shown are mean values ± SD (n = 4 independent experiments done in triplicates). Indicated significance was determined by two-way ANOVA with Tukey’s multiple-comparison test; ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; ns, not significant. (B) Increased IL-12 secretion after GD2CAR-mediated stimulation. Bar graphs show inducible IL-12 secretion measured by ELISA of unsorted modified (MOI 10) NK-92 cells after 24 h of cocultivation with indicated target cell lines (E:T 10:1) or primary patient-derived GBM (n = 3–5). (C) Cytokine profile (IFN-γ, IL-2, IL-10, TNF-α, sFasL, and Granzyme B) of NK cell-specific activation markers after a 24-h coculture of unsorted modified (MOI 10) NK-92 cells and indicated target cell lines (E:T 10:1) or primary patient-derived GBM analyzed via LEGENDplex™ assay. Cytokines were determined in cell culture supernatants (n = 3–4). (D) Upregulation of the degranulation marker CD107a after GD2CAR-mediated stimulation in unsorted modified (MOI 10) NK cells cocultured for 24 h with KG-1a or KG-1a GD2 cells. Mean fluorescence intensity (MFI) of GD2CAR+ CD107a+/GD2CAR- CD107a+ ratio and percentages of CD107a expression in GD2CAR+ NK-92 cells are shown (n = 4–5). (E) Enhanced cytolytic potential of unsorted modified (MOI 10) NK-92 cells after coculture with primary GBM cells measured by Annexin V surface expression to detect apoptotic/necrotic GBM cells (n = 3). Shown are percentages of Annexin V+ GD2+ glioblastoma cells. Mean values ± SD are shown in Figure 4. Indicated significance was determined by one-way ANOVA with Tukey’s multiple-comparison test; ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; ns, not significant. Untransduced effector cells were termed MOCK.
Due to the modular alpharetroviral “all-in-one” vector architecture, the iGOI EGFP could be exchanged by a cDNA coding for human IL-12 (Figure 2A). The GD2CAR-mediated activation with GD2+ target cells resulted in NFκB-driven IL-12 secretion after 24 h of coculture (Figure 4B). A significant increase of IL-12 secretion was detected in modified NK-92 cells after GD2CAR-specific stimulation with HT1080 GD2, SH-SY5Y, or patient-derived GBM cells in comparison to no stimulation or activation by coculture with GD2- HT1080 cells as controls (Figure 4B).
To further test the cytolytic potential of the modified NK-92 cells, the secretion of antiviral, antitumoral, and NK cell activation-specific cytokines was determined in cell culture supernatants after cocultivation of unmodified and modified NK-92 cells with indicated target cells (Figure 4C). Unmodified NK-92 cells and NK-92 cells harboring the NFκBmIL2-driven “all-in-one” EGFP vector construct did not show increased IFN-γ expression after GD2CAR-specific activation. In contrast, NK-92 cells transduced with the NFκBmIL2-driven “all-in-one” IL-12 vector construct secreted elevated amounts of IFN-γ after cocultivation with GD2+ target cells (Figure 4C). Similar results were obtained for the secretion of the cytokines IL-2 and IL-10 (Figure 4C). Despite the absence of an overall leakiness resulting in non-specific IL-12 secretion (Figure 4B), NK-92 cells harboring the NFκBmIL2.hIL12.GD2CAR “all-in-one” vector already showed a high basal IFN-γ, IL-2, and IL-10 secretion even in the control setting—without stimulation or with cocultivation with GD2- target cells (Figure 4C). Furthermore, NFκBmIL2.hIL12.GD2CAR-modified NK-92 cells secreted higher levels of TNF-α, sFasL, and granzyme B after GD2CAR-mediated activation with GD2+ target cells. Increased secretion of these activation-specific cytokines was also observed after GD2CAR-specific activation of NFκBmIL2.EGFP.GD2CAR-modified NK-92 cells (Figure 4C). As all cocultures were performed under in vitro conditions without any additional immune cells that might be recruited and further help to eliminate the tumor, the experiment was not powered to detect significant differences between the different “all-in-one” vector constructs by a potential added value of IL-12 since the antitumor effect is mediated by the GD2CAR, which is included in all constructs.
In addition, several other cytokines known to indicate NK cell activation showed enhanced secretion levels after GD2CAR-mediated stimulation of modified NK-92 cells and primary NK cells with patient-derived primary GD2+ GBM cells (Figure S5). Here, a direct comparison of the cytokine expression profile in unmodified and modified NK-92 cells and primary NK cells stressed the differences between these NK cell subsets (Figure S5).
Furthermore, the enhanced secretion of cytolytic cytokines was supported by the increased expression of the degranulation marker CD107a in NFκBmIL2.hIL12.GD2CAR- and NFκBmIL2.EGFP.GD2CAR-modified NK-92 cells after antigen-specific CAR activation (Figure 4D). Again, high basal CD107a expression was observed in NFκBmIL2.hIL12.GD2CAR-modified NK-92 cells in monoculture and after CAR-unrelated activation (Figure 4D).
We further assessed the cytotoxic potential via Annexin V staining, which is commonly used to identify apoptotic cells. Flow cytometric analyses were performed after coculture of modified NK-92 cells with patient-derived GD2+ GBM cells (Figure 4E). Compared to cocultures of unmodified NK-92 cells with patient-derived GBM cells, Annexin V was upregulated in GD2+ GBM cells cocultured with modified NK-92 cells harboring either the NFκB-driven EGFP or NFκB-driven IL-12 “all-in-one” vector constructs. Annexin V expression, as an indicator of apoptotic cell death of GD2+ GBM cells, ranged between 30% and 40% (Figure 4E).
In summary, the increased killing capacity of GD2+ target cells, the enhanced secretion of NK cell-specific cytokines, and the upregulation of CD107a in modified NK-92 cells cocultivated with GD2+ target cells and the upregulation of Annexin V in GBM demonstrated an antigen-specific cytolytic activity of NK cells mediated by GD2CAR.
The functional assays shown above do not allow the investigation of effects of activation-specific cytokine secretion on additional immune cell types, e.g., recruitment of macrophages. To assess the potential of the GD2CAR-mediated secretion of IL-12, we performed migration assays with coculture supernatants using a modified Boyden chamber and the monocytic cell line MONO-MAC-6 (MM6). An IL-12-dependent trend to recruit MM6 cells was observed (Figure S6) in supernatants from cocultures with GD2+ HT1080 cells.
The majority of CAR-based therapeutic approaches employ CAR T cells, which are the most commonly applied immune cell type in the treatment of hematological malignancies (3, 4, 17, 51–53). A major interest in the field of CAR therapy for cancer treatment is improvements to effectively address solid tumors. One approach is the extension of CAR engineering to alternative immune cell types, such as macrophages, NKT cells, and NK cells. Natural anticancer properties of NK cells might potentiate CAR-mediated cytotoxic activity against heterogeneous tumor populations as CAR NK cells still possess their natural killing capacities, which are not dependent on antigen recognition but rather are regulated via a balanced stimulation of activating and inhibitory receptors (1, 18). This may provide more complete elimination of tumors and could be especially advantageous in the case of target antigen loss. Compared to T cells, NK cells have a reduced risk of GvHD in allogeneic settings due to the lack of endogenous antigen-specific receptors, which engage allo-restricted peptides presented on the major histocompatibility complex (MHC) (19). Moreover, NK cells showed minimal cytokine release syndrome and neurotoxicity in first clinical studies, attributed to lower association of their cytokine activation profile with these disease patterns (24). Another benefit of CAR NK cells is the feasibility for an “off-the-shelf” product, e.g., by using the NK-92 cell line, for clinical application, thus reducing cost and time limitations (54).
These beneficial CAR NK cell properties, together with the TRUCK strategy to increase local anticancer efficacy in solid tumors via additional cytokine delivery (12, 14), have motivated us to the aim of this study to apply the TRUCK-based “all-in-one” vector concept to NK cells. Our group recently showed the applicability of an “all-in-one” lentiviral vector system designed to engineer primary human T cells to target GD2+ GBM (15). In line with this, other recent studies took advantage of the strategy to co-deliver an additional cytokine and showed the potential of the armored CARs (14, 55). Although the feasibility, efficacy, and selectivity to induce NFAT-driven cytokine secretion in a CAR-dependent fashion was successfully demonstrated (15), we encountered some challenges and problems when applying the system to NK cells. The switch from lentiviral to alpharetroviral SIN vectors was accomplished to obtain a more efficient transduction of NK cells. This is in line with earlier studies that demonstrated increased transduction rates with RD114/TR-pseudotyped alpharetroviral vectors in NK cells (35). Furthermore, alpharetroviral vectors may provide an additional level of safety due to the more neutral genomic integration patterns as compared to gammaretroviral and lentiviral vectors (44, 56). The established one-vector system might further decrease adverse therapy effects and reduce the risk of insertional mutagenesis of retroviral gene transfer because only a single transduction is required for transfer of the inducible cassette and constitutive CAR expression cassette (57).
Thus, despite obtaining adequate and stable transduction efficiencies with alpharetroviral “all-in-one” vector gene transfer in NK-92 cells and in primary NK cells, we observed no significant NFATmIL2-driven gene induction and only low-level CAR-mediated gene induction with the NFATsyn promoter element accompanied with high background expression levels. These results demonstrate the challenge to directly transfer established CAR T cell vector concepts to alternative types of immune cells and highlight the importance of the interplay between the engineered signaling cascade of the vector system and the overall natural properties of the individual immune cell to be genetically modified for cell therapy.
Due to the phenotypic heterogeneity of NK cells, further investigations regarding NK cell activation mechanisms are needed to fully understand the role of the different transcription factors. While NFAT is a key transcription factor in T and B cell development and activation, NFAT might also influence NK cell reactivity and function (36, 58). However, previous studies identified NFAT as a negative regulator of NK cell function and demonstrated its dispensability for NK cell activation (37, 50, 58). In line with this, Kulemzin et al. recently showed that activation-induced NFAT-driven promoters showed different activation patterns in T cells and NK cells. NFAT-driven promoters, which worked in T cells, were not efficient in NK-92 cells, whereas NFκB promoters were able to induce the activation of inducible gene cassettes in both cell types (37). NFκB is a key regulator in T and B cell differentiation as well as in transcription, immune response regulation, and feedback mechanisms (50, 59), but it also plays a role in cytokine production and granule exocytosis in NK cells. Therefore, NFκB is a promising inducible promoter element for the development of NK cell therapeutics. Indeed, after vector modification by the exchange of NFAT to NFκB in the inducible promoter element, comparable transduction efficiencies of the novel NFκB-driven “all-in-one” vectors in NK-92 cells and in primary NK cells were observed and, more importantly, GD2CAR-dependent gene induction was shown in modified NK cell subsets after cocultivation with all tested GD2+ target cell lines and patient-derived GD2+ GBM spheroids. Appropriate GD2CAR-mediated transgene induction was detected for both the EGFP control vector modules and the hIL-12 vector constructs. Here, the NFκBmIL2 promoter combination performed more efficaciously than the NFκBsyn promoter element as evidenced by greater responsiveness to GD2CAR signaling and lower background expression of the inducible transgene. Furthermore, even in coculture conditions with a mix of CAR-modified and unmodified NK cells, used to more closely resemble the clinical application approach, the potency of CAR-mediated gene induction and cell activation was clearly visible.
Kulemzin et al., who tested NFκB-driven lentiviral prostate-specific membrane antigen (PSMA)-CAR vectors for cytokine induction with different NFκB response elements, showed the highest fold activation with promoters containing 10xNFκB and 30xNFκB response elements, but also clear leakiness in non-activated cells. In contrast, we achieved efficient NFκB-promoter induction with two NFκB response elements, which were in comparison larger in bp size and thus correspond to eight NFκB response elements used by Kulemzin et al. (37, 60). Moreover, low background expression of the inducible transgene was obtained by the fusion of the NFκB response elements to the minimal IL2 promoter. This observation might indicate that not only the number of response repeats, but also the choice of the fused promoter elements (mIL2 and syn) is a critical design feature that impacts the successful implementation of the CAR-controlled therapeutic gene delivery.
Although the background expression of the inducible transgene was very low in NK cells modified with the NFκBmIL2-driven “all-in-one” vector construct harboring an IL-12 gene cassette, unexpectedly high IFN-γ and CD107a expression levels were detected in non-activated and CAR-independently activated modified NK cells. The observation that EGFP and IL-12 levels were specifically increased after GD2CAR-mediated activation with GD2+ target cells argues against the assumption that this is a result of the leakiness of the designed vector construct. This observation might rather be explained by a high auto-activation of the modified NK cells due to the insertion of the construct and a resulting GD2CAR-mediated positive autocrine feedback loop, which did not occur in the NFκB-driven “all-in-one” construct containing EGFP as an inducible transgene. While high auto-activation may be suboptimal in certain settings and might cause severe side effects as well as non-specific off-target cytotoxicity, higher baseline NK cell activation, further amplified by a specific GD2CAR-mediated stimulation, might be favorable with respect to increasing anticancer potency of applied NK cells. Nevertheless, further investigations are needed to explore possibilities to improve the CAR-construct design with the aim to diminish the non-specific activation profile.
For CAR T cell application, CARs are usually designed with the signaling domains found in their natural activation pathways (2). In the present study, we used a second generation CAR with a 4-1BB costimulatory and CD3-ζ signaling domain. This combination was shown to work adequately in both T and NK cells (38, 41, 61, 62). Despite this CAR configuration that was designed for T cell studies, we showed the functionality of GD2CAR also in NK cells, which is in accordance with the general importance of CD3-ζ for NK cell proliferation and activation (63, 64). After GD2CAR-specific stimulation, upregulation of the iGOIs (EGFP and IL-12) as well as specific cytokines such as IFN-γ, TNF-α, and IL-10 was observed to indicate a clear link from CAR signaling to iGOI and cytokine release. Moreover, expression of the degranulation marker CD107a further supports the functional initiation of cytolytic activity of GD2CAR NK cells, although there was a high basal activation in non-stimulated cells. This high basal activation in non-modified NK cells may be expected, as these cells can recognize tumor cell markers other than the GD2 antigen targeted by the CAR used in this study. This seems to also be reflected in the high levels of Annexin V+GD2+ tumor cells found upon coculture with unmodified or GD2CAR NK cells (Figure 4E). Although the CD107a expression level as measured by MFI was only slightly increased in GD2CAR NK cells, the percentage of CD107a+ GD2CAR NK cells was significantly increased upon coculture with GD2+ target cells (Figure 4D). The general activation of NK cells upon coculture with tumor cells may also partially explain the minimal, non-significant differences in secretion levels observed for some cytokines (e.g., IL-10, sFas-L, and granzyme B) upon comparison of MOCK and CAR-expressing NK-92 cells. The high effector-to-target ratio (10:1) used for these experiments, which allowed quantification of secretion and expression levels of the iGOI (IL-12 and EGFP), might be inappropriate for the analysis of other cytokines. Since untransduced NK-92 cells secrete cytokines when activated, the high effector-to-target ratio may have obscured potential differences between MOCK and modified NK-92 cells. Thus, lower effector-to-target ratios might increase the possibility to assess differences in cytokine secretion levels between the unmodified and modified NK-92 cells, similar to what was observed in the cytotoxicity analyses. Furthermore, the efficiency of the “all-in-one” vector constructs, including CAR-mediated transgene induction, was lower in modified primary NK cells than in engineered NK-92 cells. This might be due to donor-specific differences or may indicate inherent differences in components of signal transduction cascades in these NK cell sources. Better delineation of signaling pathways used in NK cells may help direct further strategies to improve CAR-directed cytokine induction in these settings. For example, remodeling the CAR with more NK cell-specific signaling domains, such as DAP-10, DAP-12, and 2B4, might further improve the antitumor cytotoxicity of these cells (65–67).
Prospectively, the specific “all-in-one” alpharetroviral NFκBmIL2-driven GD2CAR vectors can be further optimized by additional or different cytokine transgenes. Especially, the pro-inflammatory cytokines hIL-15 and hIL-21 are promising candidates for CAR NK cell therapy as previously described (68–70). These cytokines can create an antitumoral milieu in the tumor bed by immune cell activation and recruitment of other immune cells, thus enhancing CAR-dependent cytotoxicity. Especially IL-15 is known to improve NK cell proliferation and in vivo persistence.
Another possibility to improve CAR NK cell function is to overexpress the chemokine receptor CXCR4 to promote migration to the tumor site. Preclinical studies on CAR NK cells in solid tumors show that CXCR4 overexpression led to complete remissions in mouse models and increased in vivo survival of CAR NK cells (71). The examined cytokine profile of modified NK-92 cells after cocultivation with primary GBM further highlights the importance of chemokine receptors and shows an increased upregulation of the chemokine ligand CXCL10, which might be induced by IL-12 and is associated with an increased migration potential toward tumor cells (72). While in vitro migration assays supported the capacity of “all-in-one” CAR NK cells to recruit monocytic cells (i.e., MM6 cells) upon antigen stimulation of CAR NK cells, further in vivo studies would more completely show the potential of secreted IL-12 to remodel the tumor microenvironment. Noteworthily, the additive effect of cytokine (e.g., IL-12, IL-15, IL-18) secretion by CAR T cells was previously shown in in vivo mouse studies and this concept is being evaluated in clinical trials (e.g., NCT03721068) (13, 73–75).
CAR NK cell therapy might also be expanded to address further solid malignancies or even diseases beyond cancer, for example, autoimmune and inflammatory diseases as well as chronic infections. Such strategies could be developed by exchange of the targeting scFv region of the CAR or even the entire CAR cassette as demonstrated for the CEACAR (Figure S4) within the described vectors, which can be easily accomplished due to the modular design.
In conclusion, the presented data demonstrate the in vitro proof-of-concept of the transferability of the designed “all-in-one” vector constructs from T cells to NK cells via incorporation of the inducible NFκB-driven transgene cassettes. The data especially highlight that biological differences in the respective immune cells substantially impact the outcome of functional reprogramming. Linking CAR signaling to responsive, cell-specific enhancer/promoter elements can be exploited to improve the delivery of gene cargos, such as cytokines. Our developed NFκBmIL2.iGOI.GD2CAR construct demonstrated tightly linked antigen-specific transgene induction and effective cytolytic potential shown by increased NK cell activation-specific cytokine release in NK cell subsets, even after the recognition of primary patient-derived tumor cells. One limitation of our study is the lack of in vivo testing in humanized mouse models that allow tumor formation and investigation of immune responses. Still, the data presented here indicate that engineered alpharetroviral NFκBmIL2-driven vectors represent potentially useful candidates for application in targeted adoptive cell therapy with genetically engineered NK cells and provide a basis for clinical translation of CAR NK cell therapy to treat malignancies.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Nordstadt Cerebral Cancer Study (NoCCA-Study), Register-Nr. 6864. The patients/participants provided their written informed consent to participate in this study.
Conceptualization and design, KZ, MM, ASc. Methodology, LR, KZ, MG, ASt, DB, MM, CF. Analysis and interpretation of data, KZ, LR, JM, HA, MM, ASc. Resources, HA, JK, IS, BN, BA, CR, TM, MG, MM, ASc. Writing—original draft preparation, KZ, LR, MM, ASc. Writing—review and editing, all authors. Supervision, ASc, MM. Funding acquisition, ASc.
This work was supported by DFG-funded SFB738 (projects C4 and C9), from CARs to TRUCKs (Krebshilfe/German Cancer Aid-Priority Program in Translational Oncology) and REBIRTH Center for Translational Regenerative Medicine (funded by MWK Lower Saxony) as well as BMBF [01EN2007A (GD2-IL18 CART)].
A patent application has been submitted by ASc, HA, JK and KZ.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Holger Lode and Nikolai Siebert from the Department of Pediatric Oncology and Hematology, University Medicine Greifswald (Germany), as well as Ulrike Köhl and Wolfgang Glienke from the Institute of Cellular Therapeutics, Integrated Research and Treatment Center Transplantation, Hannover Medical School (MHH, Germany), for providing us the Ganglidiomab-PE antibody for GD2CAR-specific staining as well as helpful suggestions and advice. Additionally, the authors thank Karina Müller-Wallat from the Department of Neurosurgery, KRH Clinical Center Nordstadt (Hannover, Germany), for the excellent cooperation in providing us the patient-derived GBM cells. The authors also thank Kerstin Beushausen and Jana Keil from the Institute of Transplant Immunology, Integrated Research and Treatment Center Transplantation, MHH (Germany), for the excellent support with the Bio-Plex experiment. Moreover, the authors thank Daniela Lenz, Larissa Nassauer, Julia Dahlke, and Thomas Neumann from the Institute of Experimental Hematology, MHH (Germany), for the technical support and Matthias Ballmeier from the core facility cell sorting, MHH (Germany), for the FACS-based cell sorting of transduced NK cells.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.751138/full#supplementary-material
Supplementary Figure 1 | Feasibility of genetic engineering of primary NK cells with alpharetroviral NFκB-driven “all-in-one” GD2CAR vectors. (A) Representative flow cytometric analysis of primary peripheral blood mononuclear cells (PBMCs) pre and post magnetic separation of CD56+ CD3- primary NK cells for cell modification. (B) Exemplary flow cytometric analysis of unsorted modified primary NK cells post transduction (RD114/TR-pseudotyped; MOI 10). The scatter plot shows stable transduction efficiencies (MOI 10) in primary NK cells. (n=7-9) (C) Vector copy number (VCN) determination of unsorted modified (MOI 10) primary NK cells, exemplarily shown for NFκBsyn.EGFP.GD2CAR and NFκBmIL2.EGFP.GD2CAR constructs. The VCNs of a water sample and an induced pluripotent stem cell (iPSC) clone HD2 (49) with a known VCN of 1 were calculated as controls. Technical triplicates are shown. (D) GD2CAR expression in modified (MOI 10) primary NK cells was exemplarily investigated for NFκBsyn.EGFP.GD2CAR and NFκBmIL2.EGFP.GD2CAR constructs on protein level via Western blot analysis. The CD3-ζ domain of the GD2CAR (expected size approximately 53 kDa) was detected in modified (MOI 10) primary NK cells in addition to the endogenous CD3-ζ (17 kDa) and the housekeeping gene glyceraldehyde-3-phosphate-dehydrogenase (GAPDH, 37 kDa) as internal controls. Untransduced primary NK cells were termed MOCK.
Supplementary Figure 2 | (A) Time-dependent increase of inducible EGFP expression after target antigen stimulation. Inducible EGFP upregulation of NK-92 cells modified (MOI 10) with NFκB-driven “all-in-one” vector constructs after target antigen recognition over time. Unsorted modified (MOI 10) NK-92 cells were cocultivated with indicated target cells in an E:T ratio of 10:1 and analyzed after 0.5, 4, 8 and 24 h via flow cytometry. The bar graph summarizes inducible GD2CAR-mediated EGFP expression over time (n=3). Untransduced NK-92 cells were termed MOCK. (B–D) Mean fluorescent intensity (MFI) of EGFP in GD2CAR+ NK92 cells is exemplarily shown after coculture of modified NK-92 cells with (B) HT1080, HT1080 GD2 and SH-SY5Y (n=1-3), (C) KG-1a and KG-1a GD2 (n=3) and (D) patient-derived glioblastoma cells (n=1-3). Mean values ± SD are shown for all graphs.
Supplementary Figure 3 | GD2CAR-mediated induction of EGFP expression after target-specific stimulation. (A) Summary of flow cytometric analysis of inducible NFκB-driven EGFP expression of unsorted modified (MOI 10) primary NK cells after 24 h of coculture with indicated target cells in an effector to target ratio of 10:1 (E:T 10:1) (n=4-6). Additionally, mean fluorescent intensity (MFI) of EGFP upon GD2CAR+ NK cells is exemplarily shown after coculture with indicated target cells (n=3). (B) Representative flow cytometric analysis of a 24 h coculture of unsorted modified (MOI 10) primary NK cells cultivated with or without GD2+ patient-derived GBM spheroids. The bar graph summarizes the induced NFκB-driven EGFP expression of primary “all-in-one” CAR NK cells after target-specific stimulation. MFI of EGFP upon GD2CAR+ NK cells is exemplarily shown after coculture with patient-derived glioblastoma cells (n=3). (C) GD2 expression of tested suspension target cells detected via flow cytometry. (D) Immunofluorescent microscopy analysis indicated GD2CAR-mediated EGFP induction of “all-in-one” CAR-modified primary NK cells (MOI 10) after 0 h and 16 h of stimulation with KG-1α RFP657 GD2 cells. KG-1α RFP657 GD2 cells are distinguished from primary NK cells by their red fluorescence. NFκB-driven EGFP expression of primary modified NK cells is indicated in green. Untransduced primary NK cells were termed MOCK. Mean values ± SD are shown.
Supplementary Figure 4 | Generation of alpharetroviral “all-in-one” SIN vectors with inducible NFκB-promoter elements to control EGFP expression and a constitutive promoter to express a CEACAR. (A) Scheme of modified alpharetroviral SIN “all-in-one” provirus vector constructs harboring a constitutively expressed CEACAR and an inducible gene of interest (iGOI), EGFP. The EGFP expression cassette included two consensus NFκB (2xNFκB) elements in combination with either a minimal IL2 promoter (NFκBmIL2) or a synthetic TATA box (NFκBsyn). (B) Flow cytometric analysis of unsorted modified NK-92 cells 4 days post transduction (RD114/TR-pseudotyped). The CEACAR was detected via the anti-IgG antibody binding to the IgG hinge domain of the CAR. (C) Flow cytometric analysis of CEA expression on generated HT1080 CEA target cells used for functional testing in coculture experiments. (D) CEA-specific stimulation of modified NK-92 cells (IgG positive) led to CEACAR-mediated EGFP induction. Exemplary flow cytometric analysis of unsorted modified NK-92 cells after 24 h of coculture with indicated target cells (E:T 5:1). The bar graph summarizes the inducible NFκB-driven EGFP expression of modified NK-92 cells after 24 h cocultures with indicated target cells (n=3). Indicated significance was determined by two-way ANOVA with Tukey’s multiple comparison test; ***p ≤ 0.001.
Supplementary Figure 5 | Differences in NK cell-specific cytokine release profiles of NK-92 cells and primary NK cells. NK-92 cells and primary NK cells were modified (MOI 10) with the indicated “all-in-one” vector constructs and cocultured without (-) or with (+) patient-derived glioblastoma cells (GBM) for 24 h. Cell culture supernatant was collected for cytokine analysis via Bio-Plex assay. Cytokine release of (A) IL-2, (B) IL-8 (CXCL8), (C) IL-10, (D) IFN-γ, (E) IP-10 (CXCL10), (F) MCP-1 (CCL2) and (G) TNF-α is shown in a side-by-side comparison of NK-92 cells (n=3) and primary NK cells (n=4).
Supplementary Figure 6 | Antigen-induced IL-12 secretion from NK cells modified with the alpharetroviral “all-in-one” NFκBmIL2.hIL12.GD2CAR vector construct recruited monocytic cells MONO-MAC-6 (MM6). Cell culture supernatants (also used for experiments in Figure 4C) from 24 h coculture experiments with modified (MOI 10) NK-92 cells and indicated target cells (E:T 10:1) were used in a cell migration assay with a modified Boyden chamber and the monocytic cell line MM6. (A) Graph summarizes the recruitment of MM6 cells and shows the migrated cells assessed by Giemsa staining of the membrane after 4 h incubation time (n=3). (B) Exemplary pictures of the fixed and Giemsa stained membranes from cell culture supernatants of NK-92 cells modified with the “all-in-one” NFκBmIL2.hIL12.GD2CAR vector construct and cocultured with the indicated target cells showing enhanced IL-12-mediated recruitment.
1. Basar R, Daher M, Rezvani K. Next-Generation Cell Therapies: The Emerging Role of CAR-NK Cells. Blood Adv (2020) 4(22):5868–76. doi: 10.1182/bloodadvances.2020002547
2. Larson RC, Maus MV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
3. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
4. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
5. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med (2011) 365(8):725–33. doi: 10.1056/NEJMoa1103849
6. Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med (2021) 384(8):705–16. doi: 10.1056/NEJMoa2024850
7. Mata M, Gottschalk S. Engineering for Success: Approaches to Improve Chimeric Antigen Receptor T Cell Therapy for Solid Tumors. Drugs (2019) 79(4):401–15. doi: 10.1007/s40265-019-01071-7
8. Morgan MA, Schambach A. Engineering CAR-T Cells for Improved Function Against Solid Tumors. Front Immunol (2018) 9:2493. doi: 10.3389/fimmu.2018.02493
9. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther J Am Soc Gene Ther (2010) 18(4):843–51. doi: 10.1038/mt.2010.24
10. Doucette T, Rao G, Rao A, Shen L, Aldape K, Wei J, et al. Immune Heterogeneity of Glioblastoma Subtypes: Extrapolation From the Cancer Genome Atlas. Cancer Immunol Res (2013) 1(2):112–22. doi: 10.1158/2326-6066.CIR-13-0028
11. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Sci (New York NY) (2014) 344(6190):1396–401. doi: 10.1126/science.1254257
12. Chmielewski M, Abken H. CAR T Cells Transform to Trucks: Chimeric Antigen Receptor-Redirected T Cells Engineered to Deliver Inducible IL-12 Modulate the Tumour Stroma to Combat Cancer. Cancer Immunol Immunother CII (2012) 61(8):1269–77. doi: 10.1007/s00262-012-1202-z
13. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res (2011) 71(17):5697–706. doi: 10.1158/0008-5472.CAN-11-0103
14. Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J Immunol (Baltimore Md 1950) (2019) 203(1):198–207. doi: 10.4049/jimmunol.1800033
15. Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, et al. Design and Characterization of an "All-In-One" Lentiviral Vector System Combining Constitutive Anti-G(D2) CAR Expression and Inducible Cytokines. Cancers (2020) 12(2):375. doi: 10.3390/cancers12020375
16. Roddie C, O'Reilly M, Dias Alves Pinto J, Vispute K, Lowdell M. Manufacturing Chimeric Antigen Receptor T Cells: Issues and Challenges. Cytotherapy (2019) 21(3):327–40. doi: 10.1016/j.jcyt.2018.11.009
17. Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(10):1112–21. doi: 10.1200/JCO.2015.64.5929
18. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01869
19. Lin C, Zhang J. Reformation in Chimeric Antigen Receptor Based Cancer Immunotherapy: Redirecting Natural Killer Cell. Biochim Biophys Acta Rev Cancer (2018) 1869(2):200–15. doi: 10.1016/j.bbcan.2018.01.005
20. Wrona E, Borowiec M, Potemski P. CAR-NK Cells in the Treatment of Solid Tumors. Int J Mol Sci (2021) 22(11):5899. doi: 10.3390/ijms22115899
21. Morgan MA, Büning H, Sauer M, Schambach A. Use of Cell and Genome Modification Technologies to Generate Improved "Off-The-Shelf" CAR T and CAR NK Cells. Front Immunol (2020) 11:1965. doi: 10.3389/fimmu.2020.01965
22. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK Cells: A Promising Cellular Immunotherapy for Cancer. EBioMedicine (2020) 59:102975. doi: 10.1016/j.ebiom.2020.102975
23. Lupo KB, Matosevic S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers (2019) 11(6):769. doi: 10.3390/cancers11060769
24. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
25. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci Rep (2015) 5(1):11483. doi: 10.1038/srep11483
26. Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, et al. Engineering Antigen-Specific Primary Human NK Cells Against HER-2 Positive Carcinomas. Proc Natl Acad Sci USA (2008) 105(45):17481–6. doi: 10.1073/pnas.0804788105
27. Klapdor R, Wang S, Morgan M, Dork T, Hacker U, Hillemanns P, et al. Characterization of a Novel Third-Generation Anti-CD24-CAR Against Ovarian Cancer. Int J Mol Sci (2019) 20(3):660. doi: 10.3390/ijms20030660
28. Lee YE, Ju A, Choi HW, Kim JC, Kim EE, Kim TS, et al. Rationally Designed Redirection of Natural Killer Cells Anchoring a Cytotoxic Ligand for Pancreatic Cancer Treatment. J Control Release Off J Control Release Soc (2020) 326:310–23. doi: 10.1016/j.jconrel.2020.07.016
29. Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing Sarcoma With Activated and GD2-Specific Chimeric Antigen Receptor-Engineered Human NK Cells Induces Upregulation of Immune-Inhibitory HLA-G. Oncoimmunology (2017) 6(1):e1250050. doi: 10.1080/2162402X.2016.1250050
30. Fleurence J, Cochonneau D, Fougeray S, Oliver L, Geraldo F, Terme M, et al. Targeting and Killing Glioblastoma With Monoclonal Antibody to O-Acetyl GD2 Ganglioside. Oncotarget (2016) 7(27):41172–85. doi: 10.18632/oncotarget.9226
31. Wu ZL, Schwartz E, Seeger R, Ladisch S. Expression of GD2 Ganglioside by Untreated Primary Human Neuroblastomas. Cancer Res (1986) 46(1):440–3.
32. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored By Am Soc Prev Oncol (2014) 23(10):1985–96. doi: 10.1158/1055-9965.EPI-14-0275
33. Kholodenko IV, Kalinovsky DV, Doronin II, Deyev SM, Kholodenko RV. Neuroblastoma Origin and Therapeutic Targets for Immunotherapy. J Immunol Res (2018) 2018:7394268. doi: 10.1155/2018/7394268
34. Suerth JD, Maetzig T, Galla M, Baum C, Schambach A. Self-Inactivating Alpharetroviral Vectors With a Split-Packaging Design. J Virol (2010) 84(13):6626–35. doi: 10.1128/JVI.00182-10
35. Suerth JD, Morgan MA, Kloess S, Heckl D, Neudörfl C, Falk CS, et al. Efficient Generation of Gene-Modified Human Natural Killer Cells via Alpharetroviral Vectors. J Mol Med (Berlin Germany) (2016) 94(1):83–93. doi: 10.1007/s00109-015-1327-6
36. Aramburu J, Azzoni L, Rao A, Perussia B. Activation and Expression of the Nuclear Factors of Activated T Cells, NFATp and NFATc, in Human Natural Killer Cells: Regulation Upon CD16 Ligand Binding. J Exp Med (1995) 182(3):801–10. doi: 10.1084/jem.182.3.801
37. Kulemzin SV, Matvienko DA, Sabirov AH, Sokratyan AM, Chernikova DS, Belovezhets TN, et al. Design and Analysis of Stably Integrated Reporters for Inducible Transgene Expression in Human T Cells and CAR NK-Cell Lines. BMC Med Genomics (2019) 12(Suppl 2):44. doi: 10.1186/s12920-019-0489-4
38. Kailayangiri S, Altvater B, Lesch S, Balbach S, Göttlich C, Kühnemundt J, et al. EZH2 Inhibition in Ewing Sarcoma Upregulates G(D2) Expression for Targeting With Gene-Modified T Cells. Mol Ther J Am Soc Gene Ther (2019) 27(5):933–46. doi: 10.1016/j.ymthe.2019.02.014
39. Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, et al. Tumor-Specific T Cell Activation by Recombinant Immunoreceptors: CD3 Zeta Signaling and CD28 Costimulation Are Simultaneously Required for Efficient IL-2 Secretion and can be Integrated Into One Combined CD28/CD3 Zeta Signaling Receptor Molecule. J Immunol (Baltimore Md 1950) (2001) 167(11):6123–31. doi: 10.4049/jimmunol.167.11.6123
40. Sandrin V, Boson B, Salmon P, Gay W, Nègre D, Le Grand R, et al. Lentiviral Vectors Pseudotyped With a Modified RD114 Envelope Glycoprotein Show Increased Stability in Sera and Augmented Transduction of Primary Lymphocytes and CD34+ Cells Derived From Human and Nonhuman Primates. Blood (2002) 100(3):823–32. doi: 10.1182/blood-2001-11-0042
41. Klapdor R, Wang S, Hacker U, Büning H, Morgan M, Dörk T, et al. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor-Based Immunotherapy and Chemotherapy. Hum Gene Ther (2017) 28(10):886–96. doi: 10.1089/hum.2017.168
42. Kurschus FC, Bruno R, Fellows E, Falk CS, Jenne DE. Membrane Receptors Are Not Required to Deliver Granzyme B During Killer Cell Attack. Blood (2005) 105(5):2049–58. doi: 10.1182/blood-2004-06-2180
43. Hasselbach LA, Irtenkauf SM, Lemke NW, Nelson KK, Berezovsky AD, Carlton ET, et al. Optimization of High Grade Glioma Cell Culture From Surgical Specimens for Use in Clinically Relevant Animal Models and 3D Immunochemistry. J Vis Exp JoVE (2014) 7(83):e51088. doi: 10.3791/51088
44. Moiani A, Suerth JD, Gandolfi F, Rizzi E, Severgnini M, De Bellis G, et al. Genome-Wide Analysis of Alpharetroviral Integration in Human Hematopoietic Stem/Progenitor Cells. Genes (2014) 5(2):415–29. doi: 10.3390/genes5020415
45. Maetzig T, Kuehle J, Schwarzer A, Turan S, Rothe M, Chaturvedi A, et al. All-In-One Inducible Lentiviral Vector Systems Based on Drug Controlled FLP Recombinase. Biomaterials (2014) 35(14):4345–56. doi: 10.1016/j.biomaterials.2014.01.057
46. Lode HN, Schmidt M, Seidel D, Huebener N, Brackrock D, Bleeke M, et al. Vaccination With Anti-Idiotype Antibody Ganglidiomab Mediates a GD(2)-Specific Anti-Neuroblastoma Immune Response. Cancer Immunol Immunother CII (2013) 62(6):999–1010. doi: 10.1007/s00262-013-1413-y
47. Hoffmann D, Schott JW, Geis FK, Lange L, Muller FJ, Lenz D, et al. Detailed Comparison of Retroviral Vectors and Promoter Configurations for Stable and High Transgene Expression in Human Induced Pluripotent Stem Cells. Gene Ther (2017) 24(5):298–307. doi: 10.1038/gt.2017.20
48. Hooijberg E, Bakker AQ, Ruizendaal JJ, Spits H. NFAT-Controlled Expression of GFP Permits Visualization and Isolation of Antigen-Stimulated Primary Human T Cells. Blood (2000) 96(2):459–66. doi: 10.1182/blood.V96.2.459
49. Merlet E, Lipskaia L, Marchand A, Hadri L, Mougenot N, Atassi F, et al. A Calcium-Sensitive Promoter Construct for Gene Therapy. Gene Ther (2013) 20(3):248–54. doi: 10.1038/gt.2012.30
50. Kwon HJ, Choi GE, Ryu S, Kwon SJ, Kim SC, Booth C, et al. Stepwise Phosphorylation of P65 Promotes NF-κb Activation and NK Cell Responses During Target Cell Recognition. Nat Commun (2016) 7:11686. doi: 10.1038/ncomms11686
51. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma. Clin Cancer Res An Off J Am Assoc Cancer Res (2019) 25(6):1702–8. doi: 10.1158/1078-0432.CCR-18-2743
52. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults With Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci Trans Med (2013) 5(177):177ra38. doi: 10.1126/scitranslmed.3005930
53. Kochenderfer JN, Somerville RPT, Lu T, Yang JC, Sherry RM, Feldman SA, et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma After Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol Ther J Am Soc Gene Ther (2017) 25(10):2245–53. doi: 10.1016/j.ymthe.2017.07.004
54. Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, et al. NK-92: An 'Off-the-Shelf Therapeutic' for Adoptive Natural Killer Cell-Based Cancer Immunotherapy. Cancer Immunol Immunother CII (2016) 65(4):485–92. doi: 10.1007/s00262-015-1761-x
55. Lange S, Sand LGL, Bell M, Patil SL, Langfitt D, Gottschalk S. A Chimeric GM-CSF/IL18 Receptor to Sustain CAR T-Cell Function. Cancer Discov (2021) 11(7):1661–71. doi: 10.1158/2159-8290.CD-20-0896
56. Suerth JD, Maetzig T, Brugman MH, Heinz N, Appelt JU, Kaufmann KB, et al. Alpharetroviral Self-Inactivating Vectors: Long-Term Transgene Expression in Murine Hematopoietic Cells and Low Genotoxicity. Mol Ther J Am Soc Gene Ther (2012) 20(5):1022–32. doi: 10.1038/mt.2011.309
57. Biasco L, Rothe M, Schott JW, Schambach A. Integrating Vectors for Gene Therapy and Clonal Tracking of Engineered Hematopoiesis. Hematol/Oncol Clin N Am (2017) 31(5):737–52. doi: 10.1016/j.hoc.2017.06.009
58. Rothfelder K, Märklin M, Wild J, Dörfel D, Kanz L, Müller MR, et al. Involvement of NFAT Transcription Factors in NK Cell Reactivity. Blood (2016) 128(22):1344. doi: 10.1182/blood.V128.22.1344.1344
59. Mitchell S, Vargas J, Hoffmann A. Signaling via the Nfκb System. Wiley Interdiscip Rev Syst Biol Med (2016) 8(3):227–41. doi: 10.1002/wsbm.1331
60. Badr C, Niers JM, Tjon-Kon-Fat L-A, Noske DP, Wurdinger T, Tannous BA. Real-Time Monitoring of NF-kappaB Activity in Cultured Cells and in Animal Models. Mol Imaging (2009) 8(5):278–90.
61. Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, et al. Advantages and Applications of CAR-Expressing Natural Killer Cells. Front Pharmacol (2015) 6:21. doi: 10.3389/fphar.2015.00021
62. Kloess S, Kretschmer A, Stahl L, Fricke S, Koehl U. CAR-Expressing Natural Killer Cells for Cancer Retargeting. Transfusion Med Hemother (2019) 46(1):4–13. doi: 10.1159/000495771
63. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating Receptors and Coreceptors Involved in Human Natural Killer Cell-Mediated Cytolysis. Annu Rev Immunol (2001) 19:197–223. doi: 10.1146/annurev.immunol.19.1.197
64. Pitcher LA, van Oers NS. T-Cell Receptor Signal Transmission: Who Gives an ITAM? Trends Immunol (2003) 24(10):554–60. doi: 10.1016/j.it.2003.08.003
65. Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, et al. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J Immunol (Baltimore Md 1950) (2015) 194(7):3201–12. doi: 10.4049/jimmunol.1400330
66. Imai C, Iwamoto S, Campana D. Genetic Modification of Primary Natural Killer Cells Overcomes Inhibitory Signals and Induces Specific Killing of Leukemic Cells. Blood (2005) 106(1):376–83. doi: 10.1182/blood-2004-12-4797
67. Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2b4 (CD244) Signaling by Recombinant Antigen-Specific Chimeric Receptors Costimulates Natural Killer Cell Activation to Leukemia and Neuroblastoma Cells. Clin Cancer Res An Off J Am Assoc Cancer Res (2009) 15(15):4857–66. doi: 10.1158/1078-0432.CCR-08-2810
68. Ojo EO, Sharma AA, Liu R, Moreton S, Checkley-Luttge MA, Gupta K, et al. Membrane Bound IL-21 Based NK Cell Feeder Cells Drive Robust Expansion and Metabolic Activation of NK Cells. Sci Rep (2019) 9(1):14916. doi: 10.1038/s41598-019-51287-6
69. Klöß S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, Improved Ex Vivo Expansion Using IL-21 With Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum Gene Ther (2017) 28(10):897–913. doi: 10.1089/hum.2017.157
70. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord Blood NK Cells Engineered to Express IL-15 and a CD19-Targeted CAR Show Long-Term Persistence and Potent Antitumor Activity. Leukemia (2018) 32(2):520–31. doi: 10.1038/leu.2017.226
71. Muller N, Michen S, Tietze S, Topfer K, Schulte A, Lamszus K, et al. Engineering NK Cells Modified With an EGFRvIII-Specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1alpha-Secreting Glioblastoma. J Immunother (2015) 38(5):197–210. doi: 10.1097/CJI.0000000000000082
72. Karin N, Razon H. Chemokines Beyond Chemo-Attraction: CXCL10 and Its Significant Role in Cancer and Autoimmunity. Cytokine (2018) 109:24–8. doi: 10.1016/j.cyto.2018.02.012
73. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors That Exhibit Augmented Activity Against Advanced Solid Tumors. Cell Rep (2017) 21(11):3205–19. doi: 10.1016/j.celrep.2017.11.063
74. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep (2017) 20(13):3025–33. doi: 10.1016/j.celrep.2017.09.002
Keywords: immunotherapy, NK cells, chimeric antigen receptor (CAR), tumor microenvironment, immunomodulatory cytokines, alpharetroviral vectors, “all-in-one” TRUCK, NFκB
Citation: Rudek LS, Zimmermann K, Galla M, Meyer J, Kuehle J, Stamopoulou A, Brand D, Sandalcioglu IE, Neyazi B, Moritz T, Rossig C, Altvater B, Falk CS, Abken H, Morgan MA and Schambach A (2021) Generation of an NFκB-Driven Alpharetroviral “All-in-One” Vector Construct as a Potent Tool for CAR NK Cell Therapy. Front. Immunol. 12:751138. doi: 10.3389/fimmu.2021.751138
Received: 31 July 2021; Accepted: 12 October 2021;
Published: 03 November 2021.
Edited by:
Loretta Tuosto, Sapienza University of Rome, ItalyReviewed by:
Antonio Rosato, University of Padua, ItalyCopyright © 2021 Rudek, Zimmermann, Galla, Meyer, Kuehle, Stamopoulou, Brand, Sandalcioglu, Neyazi, Moritz, Rossig, Altvater, Falk, Abken, Morgan and Schambach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Axel Schambach, c2NoYW1iYWNoLmF4ZWxAbWgtaGFubm92ZXIuZGU=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.