
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 September 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.749659
This article is part of the Research Topic Extracellular Vesicles as Immunomodulatory Mediators in Inflammatory Processes View all 14 articles
 Guillaume Valade1
Guillaume Valade1 Nicolas Libert2
Nicolas Libert2 Christophe Martinaud3Eric Vicaut4
Christophe Martinaud3Eric Vicaut4 Sébastien Banzet1
Sébastien Banzet1 Juliette Peltzer1*
Juliette Peltzer1*Severe trauma is the principal cause of death among young people worldwide. Hemorrhagic shock is the leading cause of death after severe trauma. Traumatic hemorrhagic shock (THS) is a complex phenomenon associating an absolute hypovolemia secondary to a sudden and significant extravascular blood loss, tissue injury, and, eventually, hypoxemia. These phenomena are responsible of secondary injuries such as coagulopathy, endotheliopathy, microcirculation failure, inflammation, and immune activation. Collectively, these dysfunctions lead to secondary organ failures and multi-organ failure (MOF). The development of MOF after severe trauma is one of the leading causes of morbidity and mortality, where immunological dysfunction plays a central role. Damage-associated molecular patterns induce an early and exaggerated activation of innate immunity and a suppression of adaptive immunity. Severe complications are associated with a prolonged and dysregulated immune–inflammatory state. The current challenge in the management of THS patients is preventing organ injury, which currently has no etiological treatment available. Modulating the immune response is a potential therapeutic strategy for preventing the complications of THS. Mesenchymal stromal cells (MSCs) are multipotent cells found in a large number of adult tissues and used in clinical practice as therapeutic agents for immunomodulation and tissue repair. There is growing evidence that their efficiency is mainly attributed to the secretion of a wide range of bioactive molecules and extracellular vesicles (EVs). Indeed, different experimental studies revealed that MSC-derived EVs (MSC-EVs) could modulate local and systemic deleterious immune response. Therefore, these new cell-free therapeutic products, easily stored and available immediately, represent a tremendous opportunity in the emergency context of shock. In this review, the pathophysiological environment of THS and, in particular, the crosstalk between the immune system and organ function are described. The potential therapeutic benefits of MSCs or their EVs in treating THS are discussed based on the current knowledge. Understanding the key mechanisms of immune deregulation leading to organ damage is a crucial element in order to optimize the preparation of EVs and potentiate their therapeutic effect.
Severe trauma is the main cause of death among young people worldwide (1, 2), one-third being attributed to hemorrhage (3). In the military population, during modern conflicts, 90% of preventable deaths are of hemorrhagic origin (4).
Hemorrhage secondary to trauma is an emergency that can evolve into traumatic hemorrhagic shock (THS). Hemorrhagic shock in such condition is a complex association of tissue injuries and a severe hypovolemia due to blood loss. This leads to circulatory failure and inadequate tissue perfusion that induces a switch from aerobic to anaerobic metabolism (5). This phenomenon is responsible for secondary insults with tissue damage and inflammation, which can progress in the worst cases to organ dysfunction and multi-organ failure (MOF). The incidence of MOF is high in cases of severe trauma and remains a major cause of morbidity and mortality (≈33%) (6, 7).
Severe trauma is most often accompanied by significant tissue damage. Tissue attrition will rapidly lead to significant inflammation. The current challenge in the management of THS patients is preventing organ injuries, which currently have no etiological treatment available. Indeed, whereas post-hemorrhage resuscitation improves tissue perfusion, it does not treat the complex mechanisms that occur with reperfusion (ischemia/reperfusion, I/R) and activation of inflammatory and immune responses. Inflammatory and immune burst after trauma are major contributors of MOF (8, 9). The immune cells then become adherent to the vascular wall and decrease distal blood flow. These phenomena then induce tissue hypoperfusion, responsible for dysfunction of the microcirculation, hypoxia, and cellular acidosis, rapidly leading to organ failure and MOF. To improve the prognosis of patients, there is a critical need for new therapies to prevent and treat organ dysfunction and MOF after trauma.
Modulation of the immune and inflammatory response is a promising therapeutic strategy to treat complications of THS.
Mesenchymal stromal cells (MSCs) were discovered in the 1970s. Alexander Friedenstein, demonstrated the ability of culture-isolated fibroblast cells (now designated as MSCs) to recreate a hematopoietic environment in vivo after heterotopic grafting (10). These pioneering experiments provided the first clues to the existence of a cellular memory of the function they exerted in their original tissue. MSCs in the medullary microenvironment participate in the regulation of self-renewal and differentiation of hematopoietic stem cells (HSCs). More recently, clinical trials have shown that the co-graft of MSCs and HSCs allowed for better engraftment of HSCs while decreasing the risk of graft vs. host disease (GvHD) (11–14). Since then, many studies have shown the immunomodulatory capacities of MSCs in different contexts in vitro and in vivo and notably after trauma (15–17). MSCs exert their immunomodulation capacities by cell-to-cell contact or paracrine pathway via the secretion of various types of anti-inflammatory molecules and extracellular vesicles (EVs) (18).
In this review, we discuss the therapeutic potential and rationale for the application of EV-enriched MSC secretome for the prevention of organ injuries in an emergency context of THS.
Hemorrhagic shock is responsible for 1.9 million deaths per year worldwide, 79% of which are caused by physical trauma (1). According to the World Health Organization, 5.8 million deaths per year are due to trauma, which represents 10% of the causes of death (19). The majority of deaths occur at the site of the trauma or in the first hours of medical management, mainly as a result of brain injury or circulatory collapse following hemorrhage. Hospital deaths are the result of sepsis or MOF (20, 21). In modern conflicts, blast injuries have become predominant and account for nearly 75% of combat casualties in Iraq and Afghanistan (22). These injuries mainly concern poorly protected areas (limbs and the head and neck axis) in 34% of cases (23). Among soldiers killed in action, 87% died before reaching a medical facility, 24% of these deaths being considered to be potentially preventable. More than 90% of these potentially preventable deaths are associated with hemorrhage (4). During the last decade, the strategy to decrease the mortality rate was to prevent pre-hospital exsanguination. This has been partially achieved by the large diffusion of massive bleeding control strategies based on compressive devices such as tourniquets (24). However, the time of the pre-hospital phase has been considerably increased in recent conflicts (Sahel), promoting the duration of the shock and the onset of complications (25, 26).
The pathophysiology of THS is complex. We describe this phenomenon from the clinical to the cellular aspect, then discuss the 2021 guidelines for the management of critically ill patients without comorbidity factors.
THS associates tissue trauma and hemorrhagic shock, a form of hypovolemic shock in which sudden and severe blood loss leads to inadequate oxygen delivery at the cellular level (5). Hypovolemia causes a drop in venous return, blood pressure, and stroke volume. The clinical manifestations of shock include tachycardia, tachypnea, sweat, pallor, oliguria, and confusion. The clinical definition of shock associates one or several of these signs to a systolic blood pressure <90 mmHg. Metabolic cell activity is strongly dependent on the oxygen supply (DO2). The dioxygen artery concentration (CaO2) depends first on O2 binding to hemoglobin (Hb) and dioxygen saturation (SaO2) and, second, on dissolved (PaO2) (27). During hemorrhage, DO2 decreases because of a drop in Hb, cardiac output, or SaO2. Because of this drop, aerobic cell metabolism switches from aerobic to anaerobic metabolism, allowing the cell to maintain a minimal energy production (cf. Section 2.2.1). To maintain a sufficient DO2, the number of perfused capillaries increases (i.e., capillary recruitment) in proportion to the degree of tissue hypoxia, the oxygen extraction ratio increases, and regional vascular resistance is lowered to induce blood flow redistribution (28).
The adaptive mechanisms allowing the adaptation of the organism are neurological, renal, and hormonal. These can lead to the three phases of THS: compensated, decompensated and exceeded (29).
In the compensated shock phase, tissue hypoperfusion is counterbalanced by adaptive mechanisms.
The decrease in blood pressure is quickly detected by cardiopulmonary and arterial baroreceptors that induce an increase in sympathetic activity, resulting in arteriolar and venous vasoconstriction and an increase in heart rate to preserve vital organs such as the heart, lungs, and brain (30, 31). The renin–angiotensin–aldosterone system is also activated. Angiotensin promotes a ubiquitous vasoconstriction and stimulates aldosterone and anti-diuretic hormone production, sympathetic heart stimulation, thirst sensation, and decreased glomerular filtration rate (GFR) (32). Altogether, these compensatory mechanisms maintain the cardiac output, perfusion pressure, and circulating volume. All cellular functions are maintained as long as the combined yields of the aerobic and anaerobic sources of energy provide sufficient ATP (28). Nevertheless, these compensations can be overwhelmed.
When blood loss reaches a critical level (30%–40%) (29, 30), the compensatory mechanisms are overwhelmed: there is a massive decrease of reflex-activated sympathetic drive and an increase in cardiac vagal drive, resulting in reductions in heart rate and arterial blood pressure and loss of peripheral resistances (30). Uncompensated THS resulting in irreversible tissue damage occurs when the combined aerobic and anaerobic ATP supplies are not sufficient to maintain cellular function (28).
This last phase is associated with a “no reflow,” even if volemia is restored. Neutrophils adhere to the damaged endothelium, block capillaries, and aggravate local ischemic injuries. This worsens lesions such as coagulopathy, endotheliopathy, microcirculation failure, inflammation, and immune activation. All of these lead to secondary organ failure, MOF, and death (29).
The shift from aerobic to anaerobic metabolism results in the formation of lactate and protons and a decrease in ATP production. pH is maintained via H+/Na+ and Na+/Ca2+ pumps, causing an elevation of cytosolic Ca2+ (33). Moreover, ATP production is insufficient to maintain the function of these pumps. A disruption of the mitochondrial architecture also occurs, which destabilizes the mitochondrial membrane potential. This membrane potential is further affected by the opening of the mitochondrial permeability transition pore and inner membrane anion channels, finally impairing ATP production (34). The damaged mitochondria are no longer able to efficiently reduce O2 in H2O in the electron transport chain, leading to reactive oxygen species (ROS) formation (35). Oxidative stress is usually defined as an imbalance between the production of ROS and antioxidants. The ensuing pathophysiological consequences and oxidative damages correspond to protein nitrosylation, lipid peroxidation, or DNA damage and can lead to cell death. Necrotic cells and damage to the extracellular matrix release various intracellular and extracellular molecules, which act as “alarmins” triggering inflammatory cascades (36).
“Alarmins,” among which damage-associated molecular patterns (DAMPs) are released with tissue injuries, trigger both an intense pro-inflammatory systemic immune response syndrome (SIRS) and a counterbalancing anti-inflammatory response syndrome (CARS) within 30 min post-injury (37). Every DAMP proven to induce efferent pro-inflammatory pathways can be involved in the development of SIRS (38). This highlights the critical role of DAMPs in SIRS-associated MOF following THS. Moreover, it has been recently described that suppressing inducible DAMPs (SAMPs) (39), mainly produced by activated leukocytes and macrophages upon stress and injury (e.g., lipid mediators such as prostaglandin E2 or annexin A1) (40, 41), could trigger the pro-resolving pathways in CARS. An excessive CARS could lead to posttraumatic immunosuppression. In this review, we mainly focus on the mechanisms of THS-induced SIRS. DAMPs are passively released by necrotic cells, but also actively secreted by stressed or activated cells (e.g., high mobility group box protein 1, HMGB1). Elevated levels of HMGB1 (42–46), mtDNA (47–52), heat shock proteins (53–57), Ca2+-binding protein S100 (58), histones (59–63), ATP (64), interleukin 33 (IL-33) (65), or IL-1 (66) have been described after trauma in preclinical and clinical studies. DAMPs activate immune cells via their binding to pattern recognition receptors (PRRs), a group of receptors involved in the innate immune response, and induce the transcription of inflammatory factors (67, 68). Toll-like receptors (TLRs) form the most prominent group (69), and Nod-like receptors (NLRs) such as NLRP3 (70), receptor for advanced glycation end products (RAGE) (71), and purinergic (72) or complement receptors (73) also contribute to inflammation. The activation of these receptors triggers multiple pathways, notably the tumor necrosis factor alpha (TNF-α)/nuclear factor kappa B (NF-κB)/c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) signaling cascade (42, 66, 74–77), and the activation of NLRP3 inflammasome with production of IL-1β or IL-18 (78). In the case of major THS, the massive release of DAMPs may induce an excessive innate immune response, leading to coagulopathy, endothelial dysfunction, and an increase in vascular permeability, promoting the circulation of new DAMPs. This amplifies a vicious cycle of cell and tissue injuries that heightens the immunological response (73, 78). Resident inflammatory cells have the role of sentinels. They detect an increase in circulating DAMPs, and then they trigger the recruitment of circulating immune cells by releasing TNF-α, IL-6, IL-1β, etc. (34, 79, 80). DAMPs could also be secreted by activated immune cells such as neutrophils or monocytes and are also potent activators of the complement, leading to the generation of C3a, iC3b, and C5a (81, 82). Elevated plasma C3a, C5a, and C5b-9 levels correlate with trauma severity (83–85), and complement activation also contributes to neutrophil and monocyte recruitment (34).
Knowledge of the immune changes during the early phase is limited. A study on severe trauma patients revealed a massive leukocytosis, elevated serum pro- and anti-inflammatory cytokines, and evidence of innate cell activation within minutes of trauma (86).
The SIRS-primed circulating neutrophils home to the tissues and become activated by local inflammatory stimuli (87). Notably, data obtained in a cohort of trauma patients suggest that circulating platelet-activating factor (PAF) and IL-8 are potential mechanisms of circulating neutrophil priming. Indeed, the use of a PAF antagonist inhibits neutrophils priming 3 h after injury, and plasmatic levels of IL-8 increase between 6 and 12 h after injury. Moreover, at 12 h, IL-8 may also be an early predictive marker of the onset of MOF (88). Circulating neutrophil activation is associated with reduced surface expressions of CXCR2 (CD182) and C5aR (CD88) 3–4 h after injury, followed by gradual restoration (86, 89). Then, the expressions of CD62L (L-selectin) and CXCR1 (CD181) start decreasing at about 4–12 h (86), and CD62L remains low at 24 h (90). These phenotypic changes are directly related with inflammation (87) and phagocytosis (91). C5aR promotes phagocytosis, and its expression is downregulated by the binding of C5a (92). Conversely, the expression of CD11b is increased (93). Traumatic injury also leads to marked alterations in the phenotype, function, and life span of circulating neutrophils (94–96).
Circulating neutrophil counts increased sharply 3 h after injury and then decreased within 12 h, suggesting end organ sequestration. The drop in circulating neutrophils was significantly greater in MOF than that in non-MOF patients (93). Neutrophils reach the damaged tissues by diapedesis in the post-capillary venules. Neutrophil binding to the endothelium is first controlled by selectins (CD62L that binds to CD62E and CD62P), which promote the initial rolling or tethering. Then, integrins (the β2 integrins CD11a and CD11b) induce firm adhesion. Examination of autopsy specimens from patients with MOF revealed the presence of neutrophils that varies from renal blood vessels to large-scale tissue infiltration of the lung (97). Neutrophil apoptosis was profoundly delayed in severely injured patients, as well as their tissue clearance, correlating with a high risk of MOF (98, 99). When neutrophils are exposed to pro-inflammatory signals, they release not only ROS and proteases but also neutrophil extracellular traps (NETs), which induce injuries in healthy tissues. During NETosis, neutrophils release decondensed chromatin and proteins including neutrophil elastase, cathepsin G, and myeloperoxidase (MPO), as well as histones in NETs (100–102), which participate in the pathophysiology of trauma (103). The level of circulating cell-free DNA (used as a marker of NET formation) is higher in SIRS trauma patients than that in healthy subjects (104, 105).
Antigen-presenting cells (APCs), such as dendritic cells (DCs) and monocytes/macrophages, are important effector cells whose functional capacities are deeply influenced during tissue-induced injury (Figure 1). After THS, resident inflammatory cells serve as sentinels, then circulating neutrophil recruitment is rapidly followed by monocytes and macrophages. DAMPs bind to macrophage PRRs, leading to their pro-inflammatory activation, and can also trigger inflammasome formation, which does not support any direct transcriptional activity but allows the caspase-1-dependent cleavage of pro-IL-1β and pro-IL-18 into mature forms (37). It was recently demonstrated that inflammasomes, like TLRs, could trigger innate immune responses to aggression.
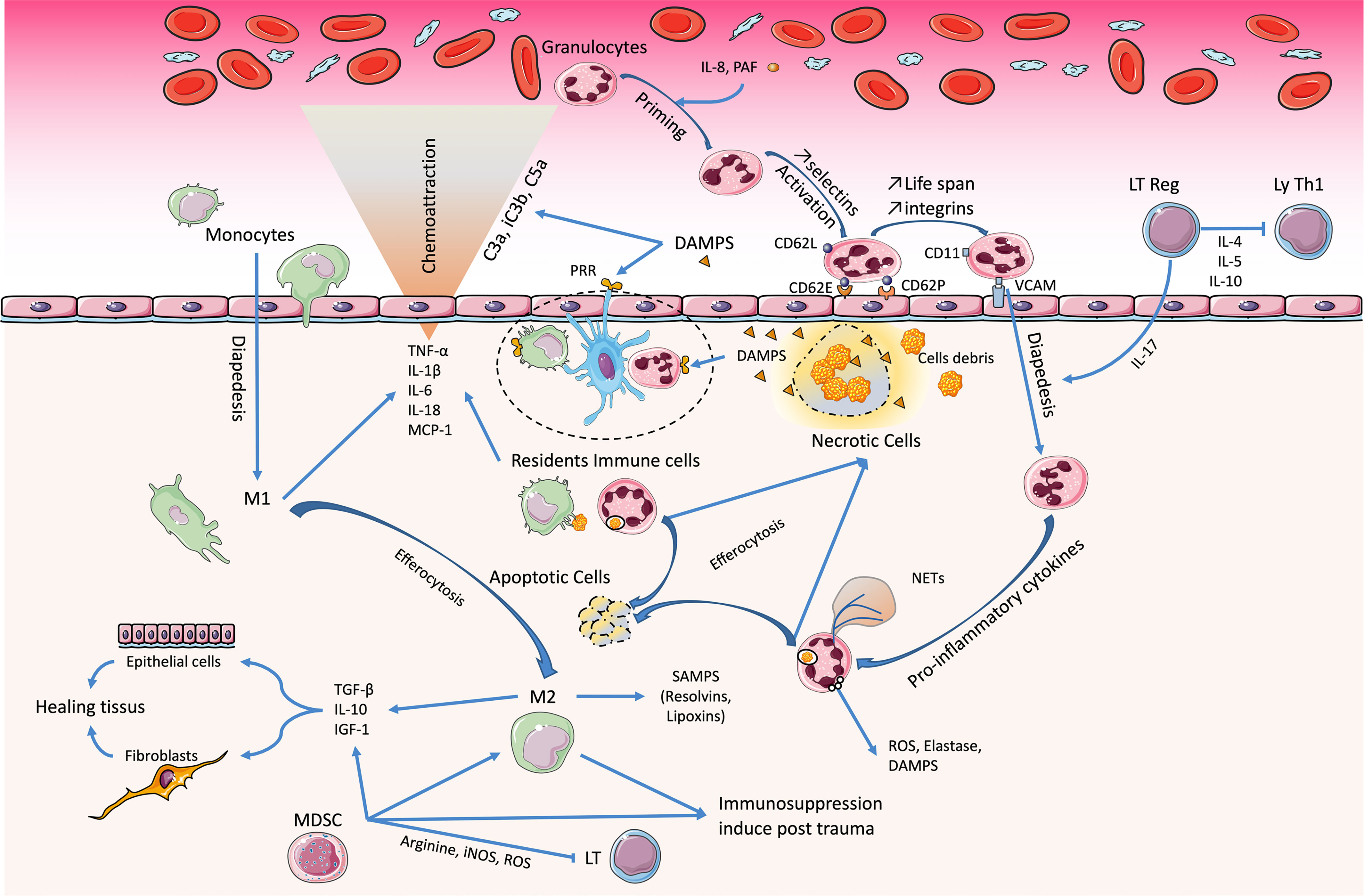
Figure 1 Immunological imbalance during traumatic hemorrhagic shock (THS). Damage-associated molecular patterns (DAMPs) play a key role in pathophysiology. The resident immune cells detect them and carry out the first reactions of phagocytosis and amplification of the inflammatory response. The circulating granulocytes infiltrate the tissue and maintain this reaction. Later-onset macrophages are pivotal in resolving this inflammatory phase (M1→M2) and initiate the healing phase. However, during THS, the abundance of DAMPs promotes the acquisition and maintenance of a pro-inflammatory phenotype. The trio of Treg, platelets, and endothelial cells co-stimulates and causes immunomodulation, with inhibition of Th1 lymphocytes. Bone marrow dysfunction induces an immunosuppression that favors the occurrence of sepsis.
Functional phenotypical changes of macrophages from pro-inflammatory (M1) to anti-inflammatory (M2) occur to support tissue repair at the damaged sites. The clearance of neutrophils in tissues by efferocytosis represents a central element in the induction of the M1-to-M2 switch (106). These M2 macrophages secrete growth factors and anti-inflammatory cytokines such as IL-10, transforming growth factor beta (TGF-β), and IGF-1, which enhance tissue remodeling (79, 107) mediators of resolution (e.g., lipoxins and resolvins) (108) and increase their expression of the receptors programmed cell death ligands 1 (PD-L1) and 2 (PD-L2) (109, 110). Within 2–4 h after injury, the activation of the p38 MAPK, ERK1/2, and JNK pathways triggers macrophage activation in the liver, which releases TNF-α, IL-6, and remarkably high levels of monocyte chemoattractant protein-1 (MCP-1) and keratinocyte-derived chemokine. Macrophages are the major producers of MCP-1 and IL-6 after trauma–hemorrhage and contribute, at least in part, to the trauma/hemorrhage-associated neutrophil infiltration (111, 112).
As observed in sepsis, suppression of the function of monocytes/macrophages is directly associated with the severity of trauma (113). SIRS and CARS occur concomitantly, but when the CARS is excessive or persistent, it promotes immunosuppression, secondary infections, and late or persisting organ dysfunctions (114). Macrophage dysfunction is a significant contributor to both innate and adaptive immune suppression (115). This suppressive function is related to a decrease in human leukocyte antigen DR (HLA-DR) and CD86 expression (116, 117). This impairment in the antigen presentation of macrophages appears early after injury and is maintained for several days (118–121). In addition, DCs, which represent the most potent APCs for the induction of primary T-cell responses, show a reduced responsiveness to bacterial components within a few hours after trauma–hemorrhage, secrete reduced levels of TNF-α and IL-6, as well as INF-γ, IL12, and IL-12p40, and are less potent to induce T-cell proliferation (122).
Maintaining the immune response following trauma requires the mobilization of bone marrow progenitors.
However, the formation of bone marrow granulocyte–macrophage colony-forming units (CFU-GM), erythroid burst-forming units (BFU-E), and erythrocyte colony-forming units (CFU-E) was significantly reduced, while peripheral blood CFU-GM, BFU-E, and CFU-E were increased in trauma patients. Bone marrow stroma failed to grow to confluence by day 14 in >90% of trauma patients. These data indicate that trauma induces a bone marrow dysfunction that releases immature white blood cells into circulation and may also contribute to a failure to clear infection and an increased propensity to organ failure (123, 124). Moreover, in pathophysiological conditions such as trauma, a partial blockade in the differentiation of immature myeloid cells into mature myeloid cells results in an expansion of this population called myeloid-derived suppressor cells (MDSCs), which have remarkable ability to suppress T-cell responses and to modulate macrophage cytokines (125). Moreover, MDSCs, like all APCs, interact and modulate the behavior of the adaptive immune system, notably T helper (Th) lymphocytes via major histocompatibility complex class II (MHCII), CD40, CD80, or CD86. MDSCs express low concentrations of MHCII and CD80/CD86 (126). The expansion of MDSC populations is proportional to the severity of the inflammatory insult (126, 127). Therefore, MDSCs could contribute to the post-trauma immunosuppression leading to the development of late sepsis and MOF (128).
The persistence of high levels of pro- and anti-inflammatory cytokines promotes T-cell exhaustion. There is a progressive decrease in the ability of T cells to produce cytokines (IFN-γ and TNF-α), higher expressions of CD28 and PD1 on CD4+ and a lower expression of CD127 on T cells, a loss of proliferative capacity, and a decreased cytotoxicity, which can lead to apoptotic cell death (129). Lymphocyte apoptosis occurs early after severe trauma and usually peaks at day 3 after the injury. There is a correlation between the injury severity score (ISS) and lymphopenia, aggravating the risk of subsequent major infection and sepsis (130). Apoptosis affects more the CD4+ and natural killer (NK) T lymphocytes than the CD8+. In contrast, the CD4+/CD25+ lymphocyte populations, regulatory T cells (Tregs), are more resistant to sepsis or burn-induced apoptosis (129, 131). Tregs are important mediators of the suppression of T-cell activation and the reduction in Th1 cytokine production after injury (132). Tregs also play a role in regulating neutrophils during I/R by modulating, for example, their sequestration diapedesis (133).
A leukocyte “genomic storm” occurs in critically injured patients, in which up to 80% of the leukocyte transcripts were altered in the first 12 h. It activates a large number of inflammatory mediators or pattern recognition receptors, but also suppresses genes involved in antigen presentation, T-cell proliferation and apoptosis, T-cell receptor function, or NK cell function (Figure 2). The unfavorable clinical course of the patients correlates with a higher and longer duration of expression of these genes (28 days, against 7–14 days for a favorable course), but not with the expression pattern (134). These results are consistent with another study describing an increase in blood Th17 CD4+ T cells and peripheral monocytes, as well as changes in the NK profile, and the plasma increase in IL-17F and IL-22, TNF-α, IFN-γ, and MCP-1 at 5 days of trauma (135). This suggests that it is illusory to imagine finding a specific marker or a single therapeutic agent that allows avoiding complicated outcomes of patients.
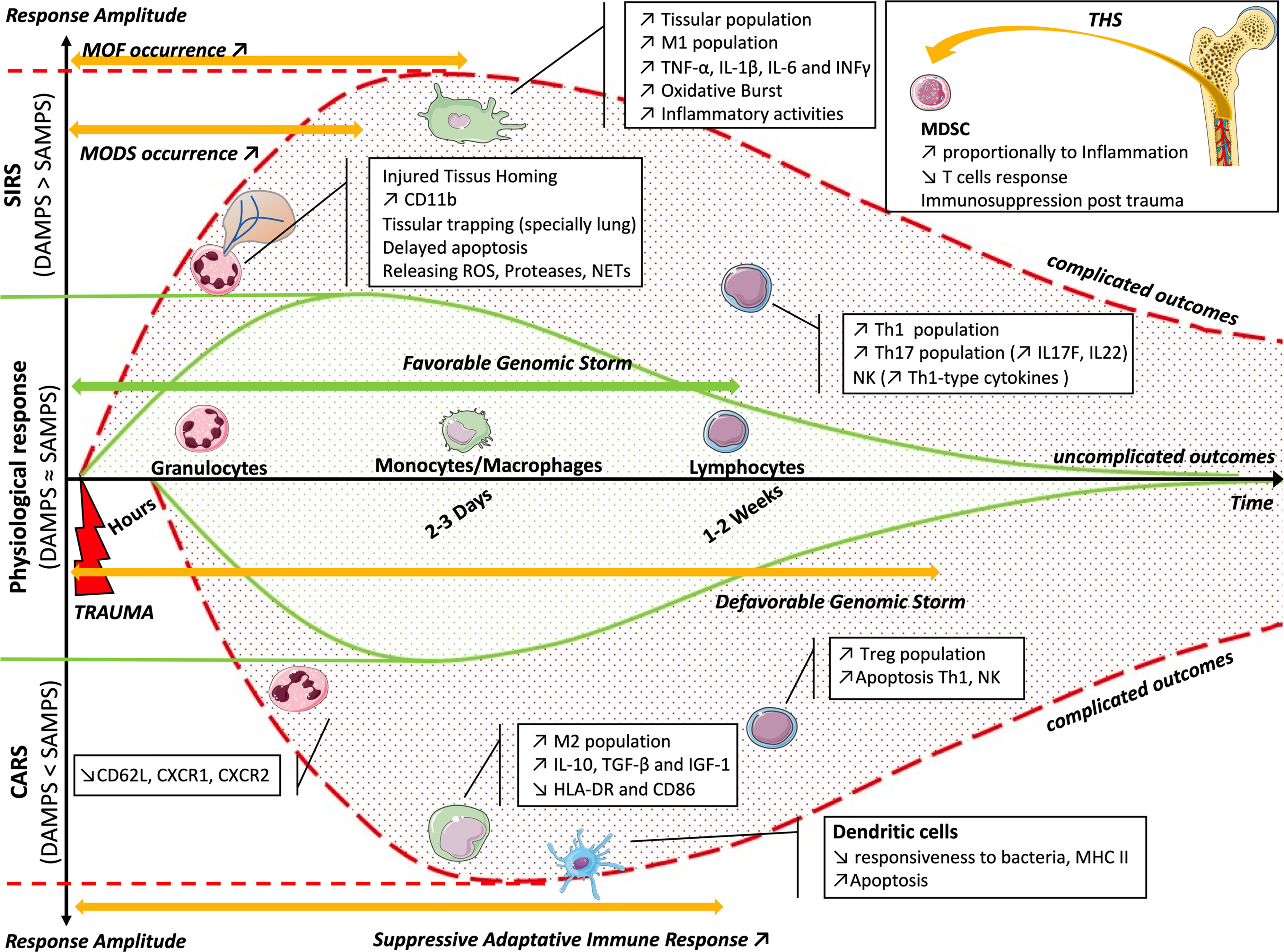
Figure 2 Balance of the inflammatory reaction (CARS or SIRS) as a function of time. The solid green curves represent the physiological response, following the favorable genomic storm and the balance between the effects of DAMPs and SAMPs. In the case of imbalance, the genomic storm becomes unfavorable. The upper dotted red curve represents the imbalance toward SIRS, with an increased effect of DAMPs, appearance of MODS and MOF, and cellular modifications. The lower dotted red curve represents the imbalance toward CARS, with an increased effect of SAMPs, appearance of suppressive adaptive immune response, and cellular changes. The box summarizes bone marrow dysfunction during THS. CARS, counterbalancing anti-inflammatory response syndrome; SIRS, systemic immune response syndrome; DAMPs, damage-associated molecular patterns; SAMPs, suppressing inducible DAMPs; MODS, multi-organ dysfunction syndrome; MOF, multi-organ failure; THS, traumatic hemorrhagic shock.
There is a concomitant and synchronous evolution of SIRS and CARS. To restore homeostasis, their evolution must be mirrored. If not, there is an imbalance on the SIRS side and, therefore, the appearance of deregulated inflammation, and even an MOF, or there is an imbalance on the CARS side and the occurrence of infection or delayed healing.
This balance could be the target of therapeutic strategies and help improve the prognosis of patients in the medium and long term after THS (Figure 2). Cell therapy or therapy by EVs could therefore be an interesting future strategy in this field.
Microcirculation is made up of three levels: arterioles, capillaries, and venules. All three are affected during THS. The vasoconstriction induced by epinephrine maintains local hypoxia and limits tissue exchange and, therefore, the clearance of lactic acid, for example. This association—coagulopathy, inflammation, anaerobiosis, and oxidation—promotes endotheliopathy (3). In this case, the arteriolar endothelium exhibits a dysfunction in relaxation linked to the local overproduction of ROS by CD11/CD18+ cells. In the capillaries, there is an adhesion of activated leukocytes to damaged endothelial cells. There is also a local exudate (Figure 3).
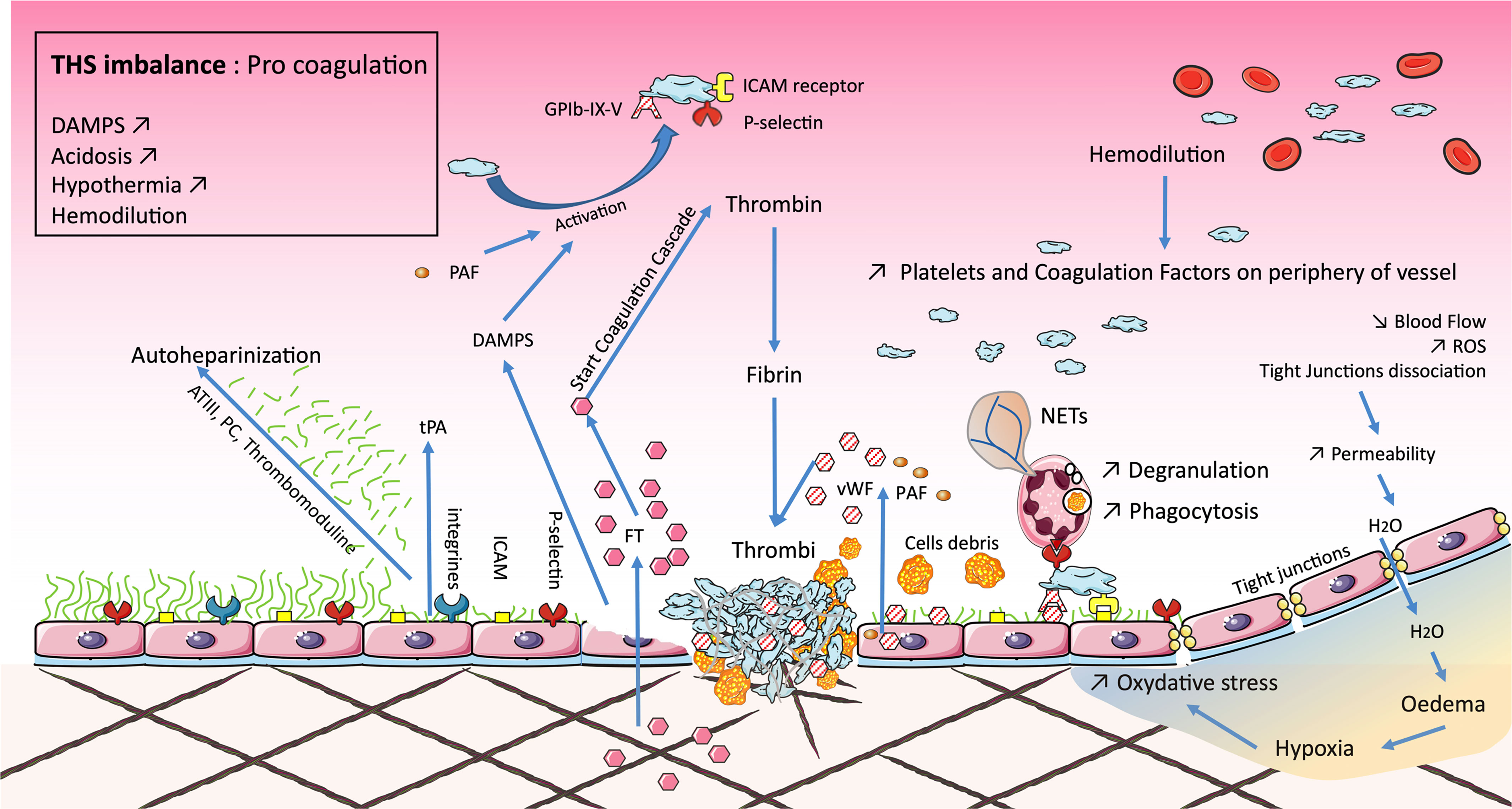
Figure 3 Microvascular dysfunction occurring during traumatic hemorrhagic shock (THS) induces the permeability of tight junctions, responsible for edema, increased oxidative stress, and, ultimately, local inflammation. Endotheliopathy is either direct from tissue damage or secondary to microvascular dysfunction. Endothelial damage degrades the glycocalyx resulting in local autoheparinization. The shedding of the glycocalyx exposes integrins and selectins, promoting the adhesion of platelets and polynuclear neutrophils. Their association stimulates endothelial cells, which release factors such as sCD40L, von Willebrand factor (vWF), and platelet-activating factor (PAF). Endothelial damage is also associated with the release of damage-associated molecular patterns (DAMPs) and tissue factor (TF). This activates the coagulation cascade reducing the downstream blood flow, forming the bed of coagulopathy in 15% of cases, the latter leading to disseminated intravascular coagulopathy (DIC). These phenomena are associated with the lethal triad: coagulopathy, acidosis, and hypothermia.
The endothelium of post-capillary venules plays a key role in the onset of complications secondary to THS. Firstly, ROS cause complement (C5) activation and the production of several factors (PAF and leukotriene B4), which are able to induce the adhesion and activation of leukocytes on the endothelium. ROS also induce the release of Weibel–Palade bodies, which are large endothelial vesicles that stock von Willebrand factor (vWF) and P-selectin. Then, ROS lead to the production, via the NFκB and AP-1 pathways, of E-selectin, intercellular adhesion molecules (ICAM), or even vascular cell adhesion molecules (VCAM). These elements allow the adhesion and diapedesis of CD11/CD18+ activated cells such as neutrophils. The inflammatory response is amplified by mast cells and macrophages, which release inflammatory mediators like TNF-α, nitric oxide (NO), histamine, or ROS. All these elements limit downstream blood flow, called microcirculation failure (136, 137).
Ischemia and inflammation often result in the disruption of endothelial tight junctions, adherent junctions and glycocalyx components (138–140). The decrease in blood flow is a mechanical stimulus inducing the activation of the adhesion molecule PECAM, vascular endothelial growth factor (VEGF) receptors, and VE-cadherin, which results in the depolarization of the endothelial cell membrane and subsequent ROS generation. These events finally disrupt the integrity of the endothelial cell–cell junction and compromise the endothelial barrier, leading to hyperpermeability (141).
The glycocalyx is an intravascular coat composed of glycosaminoglycans (e.g., heparan sulfate) and proteoglycans (e.g., syndecan) (142). The thickness of the glycocalyx decreases during hemorrhagic shock, in proportion to the reduction in blood flow (143). During I/R, glycocalyx shedding increases the circulating blood concentrations of syndecan-1 (which is highly associated with mortality) (144) and heparan sulfate (145). This results in the exposure of the injured endothelium to pro-inflammatory leukocytes, leading to the alteration of its structural integrity and hyperpermeability (146). Activated neutrophils cause glycocalyx disruption during trauma because they release proteolytic enzymes such as neutrophil elastase and degranulation, which promotes local inducible nitric oxide synthase (iNOS) or ROS synthesis (143).
Coagulopathy occurs in up to 15% of THS patients. It worsens the bleeding and is associated with excess mortality (139, 147). Tissue factor (TF) is the key element in initiating the coagulation cascade. Tissue damage exposes both TF and collagen, capable of binding factor VII and vWF (148), respectively, and initiating coagulation. At the damaged vascular site, the platelets come into contact with the thrombin formed during the initiation phase of the coagulation cascade and are then massively activated. Activated endothelial cells become procoagulant by secretion of plasminogen activator inhibitor-1 (PAI-1). Moreover, the damaged glycocalyx exposes P-selectin or ICAM-1, favoring platelet and neutrophil adhesion, respectively. In turn, neutrophils promote local fibrin activation and platelet adhesion (143). Observational data suggest a correlation between high levels of circulating syndecan-1 and higher catecholamines, IL-6, IL-10, histone-complexed DNA fragments, HMGB1, thrombomodulin, D-dimer, tissue plasminogen activator (tPA), and urokinase plasminogen activator and a threefold increased mortality (139). In addition, hypotension and hypovolemia during THS cause the release of tPA by endothelial cells (3). This could limit the procoagulant effects of the activated endothelium proteins (e.g., protein C and protein S), which inhibit the coagulation pathways and prevent an inappropriate extension of coagulation beyond the damaged vascular site. Nevertheless, this equilibrium may be broken and trigger trauma-associated coagulopathy. As previously described in the literature (139), there is a continuum between local and initial coagulopathy and disseminated intravascular coagulopathy (DIC), which appears later (hours/day). This DIC is the consequence of extensive trauma, overwhelmed anticoagulant capacity, and major inflammation. Coagulopathy is aggravated as part of the lethal triad via acidosis and hypothermia, which are traumatic or iatrogenic, but also by hypovolemia (149–151).
MOF is defined as alterations in the function of at least two organ systems, ranging from mild dysfunction to irreversible failure. Risk factors are related to the severity, type, and distribution of injuries (thoracic trauma), as well as the duration of hemorrhagic shock (152). As described previously, the pathogenesis of MOF is complex, with interrelated mechanisms involving neurohumoral and cellular cascades leading to generalized inflammatory reaction, capillary damage and permeability, interstitial edema, and, finally, organ dysfunction/failure (153). MOF should be distinguished from multi-organ dysfunction syndrome (MODS), which occurs frequently during resuscitation. Much of these early organ dysfunctions return to normal within 48 h of injury. The peak of MOF occurs within the first 3 days after injury. Disparate patterns were described: early MOF occurring within the first 3 days post-injury depending on shock severity, carrying high mortality, or late MOF whose incidence increases with age (154, 155). A retrospective study showed that lung failure was the most common organ failure, whereas cardiac and pulmonary system dysfunction decreased and renal and liver failures persisted at similar levels (155). Liver, kidney, or gastrointestinal tract injuries are directly linked to blood flow redistribution to vital organs such as the brain and heart after THS (155, 156).
A large number of scoring systems have been proposed to define MOF, without gold standard. All scoring systems [Denver, Marshall, and Sequential Organ Failure Assessment (SOFA)] include at least the monitoring of cardiac (e.g., mean arterial pressure), respiratory (e.g., PaO2/FiO2), hepatic (e.g., bilirubin), and renal (e.g., creatinine) functions (157, 158). Serum cytokine expression evaluated each 4 h during 24 h on 48 trauma patients revealed six candidate predictors of MOF occurrence: CXCL10, macrophage inflammatory protein-1 (MIP-1), IL-10, IL-6, IL-1Ra, and eotaxin (159), and the IL-4, IL-6, IL-8, and TNF-α levels are predictors of unfavorable outcomes (160).
However, depending on the type of scoring used and the classification of patients, retrospective studies can have very different conclusions. For example, over the years from around 2000 to 2010, some indicate a decrease in the incidence of MOFs with a MOF-related death rate that did not change. In contrast, others observed a significant increase of MOF prevalence and a decrease of mortality after multiple trauma and notably in the subgroup with MOF (84, 155).
The gastrointestinal tract and the tissues vascularized by the superior mesenteric artery are particularly sensitive to reduced blood perfusion (156). The loss of gut barrier integrity is hypothesized to be the “motor” of MOF by allowing the translocation of organisms from the external environment (including not only bacteria but also proteolytic enzymes) and by limiting systemic access for necessary nutrients (161, 162). Therefore, prevention of gut injury associated with intestinal ischemia could be a key therapeutic strategy. The decrease of mesenteric perfusion after THS leads to hypoxia of the villi (73). DAMPs govern the activation of resident leukocytes, the recruitment of circulating leukocytes, and also the activation of local and systemic complement (45, 82, 163). Inflammatory response, ROS production, and intraluminal pancreatic proteases also lead to mucus layer injury (164–166). Loss of the mucus layer was associated with increased gut permeability (164, 165, 167). Critical illness has a profound effect on the number of cells in the mucosal immune system (161). The lamina propria contains enteric glial cells (EGCs), and DCs; they can both recognize DAMPs and pathogen-associated molecular pattern molecules (PAMPs). EGCs are central in the homeostasis of the intestinal epithelium (168). Moreover, tissue damage can drive the dysregulation of pro-inflammatory group 3 innate lymphoid cell (ILC3s) response, which can contribute to immunopathology (169). In contrast, successful integration of environmental cues by ILC3s allows homeostasis of the gut–blood barrier by the production of IL-22. This route allows the restoration of local intestinal homeostasis after trauma (73, 169). It was demonstrated that severe THS caused an increase in bacterial translocation from the gut to the blood and organs, such as the liver and spleen. Moreover, it induces a modification toward a naive Th2 phenotype of CD4+ and a tolerogenic phenotype of DC in mesenteric lymphatic nodes, which is consistent with the clinical forms of immunosuppression observed in severe patients (170). Interestingly, a recent study of gut I/R showed that mice displayed a significant inflammatory response with neutrophil infiltration into mucosal areas, but also in the lung. Mesenteric lymph duct ligation, which had no effect on gut injury, attenuated lung injury following gut I/R. This study highlights the central role of the gut in the development of systemic inflammation and MOF, including acute lung injury (ALI) (171). Thus, the digestive tract can be both an instigator and a victim of MOF (172).
ALI and acute respiratory distress syndrome (ARDS) are serious complications of traumatic injury. ALI/ARDS constitute a pathophysiological continuum that is defined by a lung disease with acute onset, non-cardiac, diffuse bilateral pulmonary infiltrates and a PaO2/FiO2 ≤300 for ALI or ≤200 for ARDS (173).
In a recent study, 30% of patients developed ARDS as a result of trauma, with a death rate three times higher. Lung damage can be caused by pulmonary contusion, shock, administration of blood products including platelets, and an element of volume overload that can occur in the presence of increased pulmonary vascular permeability (174). Following hemorrhagic shock, neutrophils (activated via NF-kB and NLRP3 signaling) and macrophages (via HMGB1/TLR4 pathway) induce pulmonary inflammation (175). Moreover, in a model of THS lymph-induced ALI, the lung injury was totally abrogated in neutrophil-depleted animals (176). This inflammation locally damaged tight junctions and endothelial cells and ultimately led to the production of edema and the deterioration of capillary alveolar exchanges (107). Furthermore, pulmonary edema is aggravated by the decreased production of surfactant by injured endothelial cells (177).
During hemorrhage, the spongy hepatic structure and vascular response, modulated by hepatic sympathetic nerves, could temporarily compensate for the volume of blood lost. Moreover, hepatic glycogenosis could also compensate for hypovolemia by an osmotic effect toward the vessels (178). Nevertheless, THS inducing liver ischemia rapidly leads to endothelial and hepatocyte cell death (179). The diagnostic evaluation includes a combination of biochemical tests with, for example, the determination of serum hepatobiliary enzymatic activities [alanine aminotransferase (ALT) and aspartate aminotransferase (AST)] and gamma-glutamyl transpeptidase (GGT). Histopathologic changes include cellular swelling, vacuolization, endothelial cell disruption, neutrophil infiltration, and hepatocellular necrosis (180, 181). Following THS, the number of activated Ito cells (perisinusoidal fat-storing cells, stellate cells, and lipocytes) and Kupffer cells (KCs, resident hepatic macrophages) are increased. Activated KCs migrate from hepatic sinusoids into the injury areas, increase phagocytosis, and release ROS and various cytokines such as TNF-α, IL-1, IL-6, or IFN-γ (34, 181). This results notably in neutrophil activation and their sequestration in different vascular beds of the liver (156, 180). Neutrophil release NETs, proteases, and ROS, inducing hepatocyte injury and their release of DAMPs (133, 182). DAMPs (the most described in the liver is HMGB1) and also the complement pathway can activate KCs. The pro-inflammatory cytokines and ROS released by activated KCs also exert cytotoxic effects by inducing changes in the cell membrane receptors of hepatocytes and endothelial cells. They also activate other KCs and produce chemotactic factors for neutrophils and CD4+ lymphocytes (181, 183–187), which aggravate microvascular/hepatocellular injury by the formation of cellular thrombi (133).
The incidence of acute kidney injury (AKI) is indicated at 13% in trauma and increases to 42.5% in THS; 96% of AKI appear within the first 5 days (188). AKI is the clinical endpoint of multiple processes and results in a decrease in the GFR, which is a measure of global renal function. The injury mechanisms identified are I/R, inflammation, and rhabdomyolysis. In the nephron, the glomerulus is exposed to vasoconstriction of the afferent glomerulus artery, resulting in a decrease in the GFR by injury of the glomerular–blood barrier. Cellular debris can precipitate in the tubule, further decreasing renal filtration and reabsorption. I/R injury is among the most common causes of AKI, and the underlying pathogenesis involves injury to the nephron by both ischemia and oxidative stress survival/death mechanisms. Proximal renal tubular cells along the nephron segments are particularly sensitive to hypoxia. One of the early events in renal I/R is the activation of the endothelium (increased expressions of E-selectin, ICAM-1, and CX3CL1), increasing vascular permeability and promoting leukocyte extravasation. Moreover, tubular epithelial cells increase complement binding and upregulate TLRs, leading to cytokine/chemokine production. A study in patients suffering from AKI post-blunt trauma showed a rapid increase in concentrations on D0 (time of measurements after injury within the first 12 h = D0, 24–96 h = D1–D4, and ≥96 h ≥ D4) in inflammatory factors [e.g., IL-8, MCP-1 (alias CCL2), and IL-6] and anti-inflammatory factor (e.g., IL-1ra), followed by a drop on D4, in IL-1ra, IL-4, and IL-6 (189). In the tubules, neutrophils are observed between 3 and 24 h, followed by an ascending plateau up to 72 h after I/R injury (190). Macrophages are recruited at D1, with a peak at D5. The M1 is the dominant population from D1 to D3, then the M2 from D5 to D7. The authors demonstrated the role of M1 in the onset of tissue lesions and more of M2 in tubular repair (191). The I/R model also induces a maturation of the DC phenotypes and their production of TNF-α, IL-6, or MCP-1 in the first 24 h (192). Moreover, the injured epithelium releases fraktaline, which recruits more DCs (193). Finally, the kidney disposes type 2 innate lymphoid cells (ILC-2 cells), which appear to be involved in the anti-inflammatory phase. ILC-2 releases IL-4 and IL-13, allowing the polarization of macrophages and lymphocytes to M2 and Th2/Treg phenotypes, respectively (194). Finally, rhabdomyolysis is a classic complication of severe trauma ranging from the elevation of serum myoglobin and creatinine kinase (CK) activity to AKI and disseminated intravascular coagulation. It induces disturbances in intracellular ionic gradients, leading to increased concentrations of intracellular Ca2+. The pathogenesis of AKI by rhabdomyolysis involves myoglobin-induced intrarenal vasoconstriction, direct ischemic injury, and tubular obstruction (195). Moreover, in a model of rhabdomyolysis-induced AKI, the heme-activated platelets enhanced the production of macrophage extracellular traps (METs) by increasing intracellular ROS generation and histone citrullination (196). There is a need today to find new therapies to prevent/treat kidney damage in order to avoid the clinical consequences associated with AKI and progress to chronic renal failure (197).
The current management of hemorrhagic shock is based on two main pillars: stopping the bleeding and damage control resuscitation. This is applied during the pre-hospital and intrahospital phases (198). Bleeding control is first achieved by local compression, placing tourniquets or hemostatic dressings. The definitive management of these wounds requires surgical hemostasis (150, 198). The aim of damage control resuscitation is to maintain permissive hypotension (80–90 mmHg) as long as surgical hemostasis is not achieved; it is a compromise between tissue perfusion and aggressive resuscitation with high doses of fluids (199). Moreover, this limits hemodilution by overfilling, which helps maintain DO2 above the critical limit (<8/10 ml min−1 kg−1) (28, 198).
Preserving blood pressure begins with vascular filling. It is recommended to use a plasma first (200). Treatment with plasma during massive bleeding allows restoration of the glycocalyx (145). The use of vasopressors or sympathomimetics is only recommended as a second-line treatment (198, 201–203). Hemoglobinemia is not the only criterion for optimal transfusion. It is recommended to start the plasma transfusion at the same time, with a plasma/blood cell ratio between 1:2 and 1:1. Platelet infusion should be administered to maintain a minimum count, depending on the clinical situation (150, 198). To finish, tranexamic acid must be used before the third hour after THS for anti-fibrinolytic action. Other treatments such as coagulation factor concentrate, fibrinogen supplementation, or calcium supplementation could be used against coagulopathy (150, 198).
The complexity and heterogeneity of the multiple factors involved in the pathophysiology of THS can give rise to MOF despite constant improvement in patient care. Deregulations of the immune system are at the heart of systemic deregulations after injury; therefore, modulating the immune response is a promising therapeutic strategy for preventing the complications of THS.
Preclinical and clinical proof-of-concept studies have analyzed the efficacy of new and emerging therapeutic candidates in the context of individual organ failure. Although informative, these studies do not address the full complexity of THS. Hence, hypothesis-driven research studies targeting the multi-organ dysfunction of THS are urgently needed. The therapeutic potential of MSC therapy has been well characterized and demonstrated to improve tissue function and regeneration. The established immunomodulation capacity and ability to restore tissue damage may also be applied in the treatment of THS-induced MOF. THS is a life-threatening emergency requiring immediate medical intervention. While cell-based therapy carries multiple advantages, the drawback is the delay of the supply of MSCs that require in vitro expansion and the complex storage and transport before administration. EVs, on the other hand, are secretory products of MCS. The major advantage of using cell derivatives rather than cells is the immediate availability of the product, which may be prepared, amplified, characterized, and easily stored for future use in the emergency context of THS patients. Existing evidence indicates that MSC-derived EVs are able to prevent immunological disturbances that lead to organ failure.
MSCs have been described since 1970 (204). These cells of mesenchymal origin have been found in both perinatal tissues and numerous adult tissues (205). Although isolated from various tissues, they share common properties described in 2006 by the International Society for Cellular Therapy (ISCT), which proposed minimal criteria to define MSCs. These plastic-adherent fibroblastic-like cells express a panel of antigenic surface markers (positive for CD73, CD90, and CD105 and negative for hematopoietic markers) and have an in vitro multipotency capacity in the three canonical pathways: osteoblastic, chondroblastic, and adipocytic (206). They have many capacities: trophic support and immunomodulation, as described above, but also anti-apoptosis, pro-angiogenesis, or even antioxidation (207). MSCs were first described as key regulators of the HSC niche homeostasis. Later, in the 2000s, it has been described that allogeneic MSC transplantation given intravenously is well tolerated (11), can promote hematopoietic engraftment (208), accelerate lymphocyte recovery (209), reduce the risk of graft failure, and reduce the incidence of GvHD (12, 210). MSCs can modulate innate immunity by promoting the repolarization of monocytes and macrophages from a type 1 (pro-inflammatory) to a type 2 (anti-inflammatory) (211), by suppressing the proliferation, cytokine secretion, and NK cell cytotoxicity (212), and by inhibiting the maturation and migration of DCs (213), as well as modulate polymorphonuclear cell apoptosis and activity (214). MSCs can also modulate both adaptive immune effector activity by inhibition of T-cell (215) and B-cell (216) functions. These data open the way to their utilization as cell therapy products in degenerative and/or inflammatory diseases lacking appropriate treatments (217). Presently, hundreds of clinical trials are using MSCs to evaluate their therapeutic effects in numerous severe diseases (217). The first clinical trial in this context, using the systemic administration of allogeneic MSCs, did not exacerbate the elevated cytokine levels in the plasma of septic shock patients, consistent with a safe response. This cohort also revealed patient-specific and dose-dependent perturbations in cytokines, including an early but transient dampening of pro-inflammatory cytokines (218).
This immunomodulation potential has been extensively documented. Caplan and Dennis (219), in 2006, postulated that MSCs could mediate their therapeutic activity via the secretion of soluble factors such as prostaglandin E2 (40), IL-1 receptor antagonist (IL-1RA) (220), TGF-β (221), hepatocyte growth factor (HGF) (222), indoleamine 2,3-dioxygenase (IDO) (223), or tumor necrosis factor-stimulated gene 6 (TSG-6) (224–226) rather than by direct cellular interactions. In 2007, it was then demonstrated that MSC-conditioned media rich in small EVs could exert cardioprotective effects in a myocardial infarction model (227). Another team described the beneficial effects of MSC-conditioned media enriched with larger EVs in a mouse model of AKI (228). Consequently, today, there is a growing interest in MSC-derived EVs (MSC-EVs). More recently, it was also demonstrated that MSC-EVs can be a promising therapy for preventing chronic GvHD by exhibiting potent immunomodulatory effects (229, 230). Moreover, in several preclinical studies, it was shown that MSC-EV therapy reduced inflammation in kidney injury animal models (231) and decreased the inflammatory cell influx, altering alveolar macrophages toward an anti-inflammatory phenotype in lung injury models (232, 233) or the pro-inflammatory cytokine messenger RNA (mRNA) levels in liver injury (234). Finally, it is important to understand that MSCs are sensitive to their environment. Their properties and those of their by-products may vary depending on growing conditions, known as the concept of “priming” (235, 236). Stimulating MSCs with pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-1α, or IL-1β induces the secretion of soluble or EV-encapsulated anti-inflammatory factors (237–240). Interestingly, the secretome of MSCs primed with IL-1β and the sera of polytrauma patients share important characteristics. IL-1β priming enhances the secretion of pro-inflammatory and pro-angiogenic factors (IL-6 and VEGFA) and chemokines (CXCL1 and CCL2). Moreover, MSC-IL-1β priming may improve their therapeutic effects by inducting cell adhesion molecules and anti-inflammatory and anti-fibrotic molecules (241).
The few studies using MSCs in THS showed that their administration early after hemorrhagic +/− traumatic shock limited vascular permeability by preserving the barrier junction proteins (VE-cadherin, claudin-1, and occludin-1), inhibiting the expressions of leukocyte adhesion molecules (VCAM-1 and ICAM-1) on endothelial cells, and decreasing both serum concentrations of inflammatory molecules and CD68- and MPO-positive cell tissue infiltration (17). We recently showed that IL-1β-primed MSCs attenuated hemorrhagic shock-induced early hepatic and kidney injury and dysfunction and reduced the SIRS/CARS syndrome, as shown by the decreases in the plasma cytokine concentrations and the phenotypic activation of circulating CD11bc+ cells (242). MSCs would also prevent the decrease in hematopoietic progenitors induced by THS in the bone marrow (15, 17, 243). Whether the use of MSCs could alleviate or potentially exacerbate THS-induced coagulopathy is unclear. MSCs express TF (244, 245) and phosphatidylserine (246), which are thrombogenic and tend to increase the clotting rates. MSCs can therefore behave as beneficial hemostatic agents, but can also be excessively procoagulant, depending on the dose, the time of administration, and the method of preparation, which, in this case, may require the concomitant administration of anticoagulants to prevent venous thromboembolism or disseminated coagulopathy (247). Moreover, prothrombotic factors on their surface could trigger the instant blood-mediated inflammatory reaction (IBMIR) after blood exposure. IBMIR is characterized by the activation of complement/coagulation cascades, the binding of activated platelets to the MSCs, and clot infiltration by neutrophils and monocytes, which could lead to cell destruction. It is important to note that the induction of IBMIR depends on the MSC source and the dose administered and increases when their in vitro expansion has been high (passages 5 to 8) (245). This means that the choice of the modes of preparation and administration of MSCs can modulate their thrombotic activity. In contrast, cultured fibrin-embedded human MSCs can dissolve the surrounding fibrin mesh. This fibrinolytic capacity may be related to the transcriptional expression of the urokinase plasminogen activator (uPA) and its receptor (uPAR), the tPA, and the PAI (248). In conclusion, MSCs are being employed as an experimental therapy in a variety of human diseases and represent an important hope in the context of lesions induced by THS. They act on several biological processes including inflammation and reprogramming of immune cells, but also by the activation of endogenous repair pathways. Current dogma indicates that they improve disease through the secretion of paracrine-acting factors and, more recently, via the production of EVs.
In the emergency context of THS, requiring very quick availability of the therapeutic product, a ready-to-use EV solution, appears to be a particularly interesting innovative strategy.
Cells use multiple and sophisticated modes of communication. Besides direct cellular communication through the expression of cell surface markers, they communicate not only by the secretion of soluble molecules but also via the production of EVs. The term “extracellular vesicle” corresponds to a generic term that refers to particles naturally released from the cells, delineated by a lipid bilayer, and are devoid of replicative activity (i.e., without functional nucleus). The three main EV subtypes found in the literature include microvesicles (MVs; also known as microparticles or ectosomes), exosomes (exo), and apoptotic bodies. They are characterized by their size (small vesicles, <100–200 nm; medium/large, >200 nm), density, cellular origin, and their biochemical composition (tetraspanin, annexin V, etc.) or according to their biogenesis process (249). The biogenesis of small vesicles (exosomes) occurs in early endosomes; then, during the process of maturation, the early endosomes become endosomes or multivesicular bodies and accumulate intraluminal vesicles, which can either be degraded by lysosomes or released as exosomes in the extracellular space. The biogenesis of medium/large vesicles (MVs) occurs via the direct budding of the cell membrane and are released into the extracellular space (250). Apoptotic bodies are large-sized vesicles that specifically originate from apoptotic cells (251). These EVs contain bioactive soluble molecules (mRNA, miRNA, proteins, lipids, etc.) and membrane-bound molecules (CDs and enzymes). The most currently available EV isolation methods [ultracentrifugation, tangential filtration, immunocapture, or precipitation (252), including those used for clinical grade isolation] do not allow the precise isolation or purification of a specific EV subpopulation (exo or MVs) (253–255). Therefore, the International Society for Extracellular Vesicles (ISEV) has suggested minimal information for studies of extracellular vesicles (MISEV). These guidelines indicate that “EV” remains a collective term describing a complex continuum of vesicles of different sizes and composition and resulting from various mechanisms of formation and release (249). Moreover, in most cases, EV preparations are composed of different vesicles and a greater or a lesser amount of soluble proteins that may participate in the biological activity of the final product. We must therefore also take into consideration the heterogeneity of the final preparations used in the different studies, which mostly include soluble factors. The most suitable term would ultimately be “EV-enriched secretome” rather than “EVs.”
Intercellular communication via extracellular cargo is highly conserved across species (from bacteria to human); therefore, EVs are likely to be a highly efficient, robust, and economic manner of exchanging information between cells (256).
The specific combinations of molecules in EVs generally reflect the unique characteristics of their original cells and influence their functional properties; therefore, these EVs could recapitulate most effects of the cells from which they originate from and be used as substitutes of those cells in therapeutic objectives (18). EVs can be harvested from all body fluids and take part in many physiological and pathophysiological processes (18). Indeed, EVs are frequently produced in greater abundance in stressed than in unstressed cells; therefore, they can promote the activation of immune cells such as macrophages, which can, in turn, also release EVs and soluble factors and promote stress cell and tissue inflammation and injury (257).
The most extensive studies on EV-mediated communication have been performed between tumor and immune cells and between different types of immune cells. Currently, dendritic cells and mesenchymal stromal cells are the sources for which the prospects for clinical use in humans are most advanced. Since the first descriptions of the therapeutic potential of MSC-EVs in AKI and MI models (227, 228), many studies have addressed the therapeutic functions of MSC-EVs. They could provide new therapeutics and have to be better described and understood (249, 258).
As described above, MSCs release a unique signature of proteins (259), lipids (260), and membrane receptors or various types of nucleic acids through EVs (258), which participate in the protection and the regeneration process of damaged cells notably by mitigating the immune response (261, 262). Proteome analysis of MSC-EVs provided by the ExoCarta database showed that the MSC-EV proteome is rich in IL-10, HGF, and leukemia inhibitory factor (LIF) anti-inflammatory cytokines. Moreover, some cytokines, chemokines, and chemokine receptors involved in immune cell recruitment, cell migration, immunosuppression, or neutrophil degranulation, such as CCL2, VEGFC, CCL20, as well as chemokine ligand 2 (CXCL2), CXCL8, CXCL16, defensin α1, HERC5, and IFITM2, are also expressed (261). Similarly, they carry microRNAs (miRNAs) involved in immune function, like miR-146b, identified as an IL-10 effector on macrophages by targeting the TLR4 pathway (263), or miR-181c, which also decreases the expression of TLR4 and the activation of the NF-κB pathway (264). In addition, the pro-inflammatory priming of MSCs, for example by TNF-α and IFN-γ, generates modifications of the protein content and the transcripts of MSC-EVs, notably a greater expression of COX2 leading to an increased release of PGE2, which could promote their anti-inflammatory activity (260). Hypoxia also modulates the MSC-EV miRNA expression profile with notably significant overexpressions of miR-223 and miR-146b, which are implicated in the inflammatory phase of the healing process (265).
Concerning the anti-inflammatory effects on DCs, the authors described 49 miRNAs enriched in MSC-EVs, including miR-21-5p, miR-142-3p, miR-223-3p, and miR-126-3p, known for their role in DC maturation and function (266).
Macrophages have an important role in the inflammatory phase firstly by their pro-inflammatory phenotype and then by their switch to a pro-resolving, anti-inflammatory phenotype. A study in which unfractionated PBMCs were co-cultured with PKH26+-MSC-derived EVs showed that EVs were mostly internalized by monocytes and scarcely by lymphocytes after 24 h or 4 days, but inflammatory priming of MSCs increases EV internalization by lymphocytes (267). It was already described that PBMC or macrophage co-culture with adipose-derived MSC-Exo (MSC-derived exosomes) could induce M2 macrophage polarization (265, 268). MSC-EVs also inhibited TNF-α and IL-6 production by inflammatory glial cells and limited their activation (loss of CD45 and CD11b expressions) and induction of CCL2, one of the membrane markers of M2 polarization (269). Finally, bone marrow MSC-EVs could also downregulate the production of IL-23 and IL-22 by macrophages and pro-inflammatory cytokines, inducing Th17 effector T cells. Therefore, MSC-EV-educated macrophages could promote resolution via the decrease of Th17 pathogenicity (270).
MSC-EVs limit the proliferation and differentiation of activated CD4+ and CD8+ lymphocytes (271). They induce CD3+ and CD4+ lymphocyte apoptosis and increase the Treg/T effector balance (272) by promoting the passage from Th1/Th17 to Th2 (273–275). Otherwise, in co-culture with activated PBMCs, MSC-EVs inhibit the secretion of TNF-α and IL-1β, but increase the concentrations of TGF-β (276) and IL-10 in the co-culture medium (272). As described above, monocytes and, to a lesser extent, lymphocytes were able to internalize PKH26+-MSC-derived EVs. Interestingly, the uptake of MSC-derived EVs occurred in resting but mostly in activated immune effector cells, allowing presumption of a possible role of EVs in immunosuppression, and the inhibition of EV secretion impairs the immunosuppressive capacities of MSCs. Moreover, EV uptake by stimulated B lymphocytes and NK cells is more important than that by T lymphocytes and correlates with the immunosuppressive activity of EV, observed only for B lymphocytes and NK cells, but not for T lymphocytes. Finally, pro-inflammatory priming of MSCs induced an increase in the levels of the anti-inflammatory miRNA-155 and miRNA-146 in both MSCs and their EVs (267, 277, 278). Another study also reported a concentration-dependent immunosuppressive effect of MSC-derived exosomes on B lymphocytes (263).
All these elements show MSC-EVs representing a promising therapy for inflammatory diseases.
There is significant expanded literature concerning the use of MSC-EVs in multiple preclinical models, in particular on isolated organ damage (Table 1). However, no data are currently available on their use in the context of THS. In the following paragraphs, the beneficial effects of MSC-EVs on immunological deregulations and the endothelial dysfunctions of several critical organs injured in MOF are exposed. Otherwise, although inflammation and coagulation are interdependent processes that can initiate a vicious cycle in which each process intensifies the other, the potential benefit of MSC-EVs on coagulopathy has been poorly explored. However, as discussed previously in the section on MSCs, EVs express phosphatidylserine and TF on their surfaces, which were functionally thrombogenic and tended to increase the clotting rates (246) or IBMIR.

Table 1 Overview of the applications of mesenchymal stromal cell-derived extracellular vesicles (MSC-EVs) in preclinical experimental studies.
To our knowledge, no study has investigated the role of EV administration in THS-induced intestinal ischemia. Most studies have investigated the role of EVs in inflammatory bowel diseases (IBDs), mainly ulcerative colitis and Crohn’s disease. A number of rodent models of colitis have been developed; among them, chemical models, notably dextran sulfate sodium (DSS), are widely used (308). The data listed below will relate to the results obtained in this context.
The intravenous injection of MSC-EVs from different sources [bone marrow, umbilical cord, and adipose-derived stromal cells (ADSCs)] attenuated the severity of colitis. Indeed, they exert antioxidative and anti-apoptotic effects, they also reduce the mRNA and protein levels of NF-kB, numerous cytokines, chemokines (TNF-α, IFN-γ, IL-12, IL-1β, IL-6, IL-7, CCL-24, and CCL-17) and enzymes (iNOS and COX2) and they increase IL-10 and TGF-β in the injured colon (279, 280, 282). However, it was observed that TSG-6 depletion in EVs reduced their immunomodulatory efficacy. TSG-6 in EVs plays a key role in increasing the population of Tregs and for macrophage polarization from M1 to M2 in the colon (269). Moreover, intraperitoneal injection of MSC-Exo in a mouse model of inflammatory bowel disease indicated a protective role in the intestinal barrier not only by preventing the destruction of tight junctions, therefore decreasing permeability, but also by modulating the responses of Th2 and Th17 cells in the mesenteric lymph nodes. Again, the knockdown of TSG-6 abrogated the therapeutic effects of MSC-Exo; conversely, administration of a recombinant of TSG-6 showed beneficial effects similar to those of MSC-Exo (286). Therefore, TSG-6 appears to play a major role in the anti-inflammatory effects of MSC-EVs in inflammatory bowel pathologies. Moreover, another study revealed that bone marrow MSC-EVs could inhibit the differentiation of Th17 cells in ulcerative colitis by regulating histone H3 lysine-27 trimethylation (H3K27me3) that is closely associated with the differentiation of Th17 cells. Therefore, MSC-EVs, which regulate H3K27me3, could be promising agents for inflammatory immune diseases associated with abnormal Th17 cell differentiation (283). Administration of MSC-EVs also increases the percentages of CD4+ CD25+Foxp3+ Tregs in lymph nodes and the spleen (281).
Moreover, as is described in other pathologies, TNF-α and IFN-γ MSC priming increased the immunosuppression of MSC-EVs (270). Finally, Yu et al. evaluated the effect of EphB2-overexpressing bone marrow MSC-EVs. EphB2 is a signaling receptor involved in the regulation of inflammatory response, immune homeostasis, and cell migration. They showed that the overexpression of EphB2 improved the colonic targeting ability of EVs and demonstrated a robust immunomodulatory effect by the modulation of the Th17/Treg balance (278).
Regarding the potential beneficial effect of the miRNA content in EVs in this pathology, a crucial role of exosomal miR200b has been described by using heme oxygenase-1 (HO-1)-modified bone marrow MSC-Exo, which overexpresses miR200b. This miRNA targets the HMGB3 gene involved in intestinal inflammation (284). Moreover, EVs overexpressing miR-146a seem to exert better anti-inflammatory effects in an experimental rat model of colitis (285). Furthermore, the study of Wang et al. demonstrated a stronger therapeutic effect of human umbilical cord (hUC)-MSC-derived exosomes that highly expressed miR-326. Indeed, this miRNA played an important role in the inhibition of the neddylation process that indirectly activated NF-κB pro-inflammatory transcription factor (277).
MSC-EVs have been extensively studied in septic ALI, including in clinical trials (309) and, more recently, in COVID-19 patients (Table 2). In the few models of ALI induced by THS, it was demonstrated that bone marrow MSC-EVs can modulate cytoskeletal signaling and attenuate lung vascular permeability (290). Moreover, ADSC-MSC-EVs could decrease endothelial damage via the PI3K/Akt signaling pathway (289). In a mouse model of lung I/R injury, EV treatment significantly attenuated lung dysfunction and injury by decreasing edema, neutrophil infiltration, and myeloperoxidase levels. Moreover, significant decreases in pro-inflammatory cytokines (IL-17, TNF-α, and CXCL1) and HMGB1 were observed. An upregulation of KGF, PGE2, and IL-10 in the bronchoalveolar fluid was also shown. Finally, MSC-EVs significantly downregulated the iNKT-produced IL-17 and the macrophage-produced HMGB1 and TNF-α in an in vitro model of hypoxia/reoxygenation (291). Moreover, as described previously, intestinal I/R is a common clinical occurrence caused by a number of pathophysiological contexts, including THS. ALI is a primary component of MOF triggered by intestinal I/R. In a rat model of ALI induced by occlusion/reperfusion of the superior mesenteric artery, intravenous treatment by rat bone marrow MSC-derived exosomes attenuated lung damage by decreasing apoptosis and the pulmonary levels of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, accompanied by a downregulation of the expressions of TLR4 and NF-κB (288).

Table 2 Overview of the applications of mesenchymal stromal cell-derived extracellular vesicles (MSC-EVs) in clinical studies.
EV miRNAs also play a role. miR-124-3p, abundantly expressed in rat MSC-derived exosomes, inhibits the expression of the purinergic receptor P2X ligand gated ion channel 7 (P2X7). Inhibition of P2X7, which is overexpressed in traumatic ALI rats, improves oxidative stress and decreases the levels of inflammatory factors, including TNF-α, IL-6, and IL-8, and increases IL-10 (287). Furthermore, the transfer of MSC-EV miR-451 to macrophages in vitro not only inhibits TNF-α and macrophage migration inhibitory factor secretion but also represses their TLR signaling. This repression allowed the mitochondrial transfer of MSC-EVs to macrophages. All these immunomodulatory effects on macrophages were exerted by different MSC-EV populations (310). Altogether, these data indicate that MSC-EVs, by limiting oxidative stress and vascular permeability and by downregulating the activity of immune cells in the lungs, represent a novel therapeutic option in the treatment of traumatic ALI.
As for ALI, many studies have evaluated MSC-EVs in models of hepatic injury induced by the administration of D-galactosamine/TNF-α, various toxic drugs, or LPS. Systemic administration of human MSC-EVs on hepatic I/R injury suppressed not only hepatocyte necrosis and sinusoidal congestion but also AST and ALT injury markers (294, 298, 311). Moreover, in a model of I/R-induced hepatic apoptosis, hUC-MSC-EVs reduced neutrophil infiltration and, therefore, their respiratory burst. This alleviated oxidative stress in hepatic tissue (293). This suggests that MSC-EVs could reduce hepatic injury by suppressing inflammatory responses (of TNF-α, IL-6, and HMGB1) and attenuating the oxidative stress response [by increasing glutathione, glutathione peroxidase, and superoxide dismutase (SOD)] and apoptosis (by decreasing caspase-3 and Bax) (292, 294). hUC-MSC-EVs could induce anti-apoptotic and pro-survival effects in a human liver cell line and ameliorated the I/R injury-induced hepatic dysfunction in mice. This study highlighted the crucial role of miR-1246 via the regulation of the GSK3β-Wnt/β-catenin pathway to mediate these effects (295). Subsequently, exosomes expressing miR-1246 had protective effects against hepatic I/R by regulating Th17/Treg imbalance via the interaction of miR-1246 and IL-6-gp130-STAT3 (296). Another team described in an I/R mouse model that hUC-MSC-EVs significantly modulated the membranous expression of CD154 of intra-hepatic CD4+ T cells, which initiated the inflammatory response in the liver and can aggravate liver I/R (297). As shown in the few studies exploring the effects of treatment with MSC-EVs after I/R, their capacity to inhibit immune cell activation (mainly neutrophils) and pro-inflammatory cytokine release, as well as their capacity to attenuate oxidative stress and to inhibit hepatic cell apoptosis, makes MSC-EVs a promising therapy to treat liver injury following THS.
Many studies have shown the beneficial effects of the administration of MSC-EVs in AKI (312). As in the previous sections, only studies using models of I/R or rhabdomyolysis were discussed since toxicity studies (cisplatin) are not relevant to THS. The therapeutic effects of EVs are mediated by different biological processes, including anti-apoptosis, anti-inflammation, angiogenesis, and anti-fibrosis (303, 306, 307). After systemic injection, labeled MSC-EVs accumulated specifically in the kidneys of mice with AKI, but not in healthy controls (299). This suggests a homing capacity of EV-derived MSCs on the site of injury.
In an I/R-induced AKI mouse model, exosomes from human amnion epithelial cells (hAEC-Exo) could improve animal survival and renal function and induce M2 macrophage polarization. This M1/M2 shift was associated with increases in the IL-4 and IL-13 levels and decreases in the TNF-α and IFN-γ levels, which helped reduce the inflammatory response (301). Similarly, EVs from ADSCs decreased the protein levels of NF-κB, TNF-α, IL-1β, and MIF, as well as PAI-1 and COX-2 in the kidney parenchyma, 72 h after I/R (302). Moreover, administration of human Wharton jelly MSC-EVs also alleviated inflammation (decreased TNF-α and increased IL-10 expressions in the kidney) in the first 48 h, but also suppressed the expression of CX3CL1 (a potent chemo-attractant factor for macrophages) and decreased the number of CD68+ macrophages in the kidney (231). Several studies suggest that the therapeutic effects of EVs can be mediated by functional mRNAs and miRNAs (228, 300, 303). MSC-EVs express high levels of miR-15a, miR-15b, and miR-16 that may modulate CX3CL1 expression (231). The same team also described that the number of NK cells increased in the kidney after I/R injury. EVs also decreased the percentage of NK cells in ischemic kidney, suggesting that MSC-EVs could alleviate kidney injury by regulating NK cells (304). Several proteins expressed by both naive and IFN-γ-primed EV-MSCs, such as galectin-1 and galectin-3 described as mediators of MSC T-cell immunosuppression, or the membrane markers CD90 and CD73 are also associated with MSC-immunosuppressive capacity (305). Finally, EV-MSCs contain anti-inflammatory and anti-oxidative apolipoprotein A1 (ApoA1). ApoA1 is described to have therapeutic effects in kidney injury, leading to the reduction of serum creatinine levels, serum TNF-α and IL-1β levels, and tissue MPO activity. Moreover, ApoA1 can suppress the expressions of ICAM-1 and P-selectin in the endothelium, thus diminishing neutrophil adherence (313). This literature, reduced here to I/R and rhabdomyolysis injuries, indicates the benefit of treatment with MSC-EVs of AKI by limiting the leukocyte chemoattraction and activation through inducing a shift from M1 to M2 macrophages or by decreasing pro-inflammatory and increasing anti-inflammatory cytokine production. All these encouraging arguments suggest that there is a potential interest in the use of MSC-EVs in the context of THS.
The pathophysiology of THS-induced MOF is complex and still not fully understood. The aim of most treatments currently used in the clinic is to compensate for the function of the affected organ with, for example, dialysis, parenteral nutrition, or controlled ventilation and oxygenation. Limited options are available to prevent the occurrence or limit the extent of organ failure in THS. The imbalance between SIRS and CARS is a key mechanism in MOF, but because it is at the crossroad of multiple system dysfunctions, no unique physiological or molecular therapeutic target can be identified. As shown in previous sections, MSCs and their EVs have an important potential to treat isolated organ failure through multiple intricate molecular mechanisms that target notably inflammation and oxidative stress. This is the reason why we believe that taking advantage of the pleiotropic effects of MSC-EVs could be a precious new approach in a pathophysiological situation as complex and multifactorial as that of THS leading to MOF.
MSC-EVs represent a great hope for the treatment of THS. Their use can have important advantages, but unknowns persist. Although a number of preclinical studies have explored the biology of MSC-EVs, only a few clinical trials have been listed concerning acute injuries of isolated organs, systemic immune dysfunctions, I/R injuries, or trauma and MOF (Table 2). A significant increase has occurred with the SARS-CoV-2 pandemic, and complete studies indicated that the administration of MSC-EVs decreases systemic inflammation and allows restoration of pulmonary oxygenation; most other studies are in progress.
Regarding the systemic administration of EVs, which seems the most relevant in the context of THS-induced MOF, although the biodistribution/homing of MSCs has been explored, it is still poorly understood for EVs. However, it was demonstrated that 70% of near-infrared lipophilic dye-labeled human MSC-EVs accumulated in the liver after systemic administration in healthy mice. Interestingly, dendritic cell-derived EVs showed an increased accumulation in the spleen, suggesting that the homing pattern of EVs reflects those of their original cells (314). On the other hand, the unpredictable nature of THS, as well as the need for emergency administration of the therapeutic product, requires the use of EVs from allogeneic MSCs. It is known that the survival of allogeneic MSCs is limited after administration, but during this time, they continuously secrete soluble factors/EVs, adapted to the pathophysiological context that they encounter. This is not the case with EVs, but iterative administrations can be more easily considered. Indeed, it will be a product already prepared/qualified, immediately available, and easily stored and transportable, allowing patients to be treated anywhere without the need for nearby production facilities. In addition, we hope that the lack of adaptability of EVs to the pathophysiological context could be compensated by the use of optimized priming upstream.
In addition, as has been the case with MSCs, there may be a mismatch between the hope raised by exciting preclinical publications and the ability to enter daily clinical practice. These difficulties could result not only from differences between human and animal species but also from the heterogeneity of the products used. Variability in MSC-EVs is associated with the variability of the cells from which they are produced. The variability of MSCs arises from several key factors such as the tissue origin (bone marrow, adipose tissue, perinatal tissues, etc.), donor, culture condition media/support (platelet lysate, fetal bovine serum, bioreactor, priming by hypoxia, or cytokines), age (age of donor and culture passage), or cryopreservation. Moreover, depending on the therapeutic target, a strategic choice between primary MSCs or cell line, native or modified, must be carefully considered. Therefore, EVs could be selected based on the advantages of MSC sourcing/efficacy. A study comparing the protein profiles of MSC-EVs with the proteome profiles of EVs from other cells showed a specific protein signature of MSC-EVs, despite the huge diversity in the sources of MSCs or the preparation methods of MSC-EVs. However, 22 proteins were exclusively found in the bone marrow-derived MSC-EV profiles. Identification of the functional markers of potency and the development of easily deployable and standardized methods of evaluation would benefit the field of EVs, as it did for cell therapy, and in this study, it was also suggested that several membrane and extracellular proteins (i.e., COL6A2 or COL6A3 and THY1) could be used as a standard for the quality control of production either in research or in clinical settings (259). EVs can also be transformed/loaded (without prior transformation of the producing MSCs) to improve their targeting or their therapeutic properties. All these provide a large field of possibilities for the clinical use of EVs (315). Moreover, in most EV manufacturing processes, the therapeutic product is composed of a continuum of different types of vesicles (size and origin) and certain amounts of soluble proteins that may participate in the biological and therapeutic activity of the final product (Figure 4). In fact, most of the studies described in the literature are based on products that do not consist solely of EVs (due very often to isolation by ultracentrifugation), but which contain a greater or a lesser proportion of soluble proteins. The most effective product could therefore be an “EV-enriched secretome.” In preclinical studies, EVs are isolated using different techniques: ultracentrifugation, tangential filtration, immunocapture, or precipitation (252). However, not all of them are easy to consider when moving to clinical grade production. Indeed, although ultracentrifugation is the most widely used, it is time-consuming and additional stages of purification (washing and microfiltration) are generally necessary to increase the purity of the EV products. Tangential flow filtration, for example, already validated for industrial-scale productions, seems more suitable. On the other hand, specifically concerning MSCs, the culture media used for the expansion phases are enriched with fetal calf serum or platelet lysate, which contain large amounts of EVs that cannot be distinguished or separated from MSC-derived EVs. To overcome this problem, in many studies, the culture medium is removed after the expansion of MSCs, rinsed, and replaced by a medium without these additives during the entire period of MSC secretion. The cellular stress generated by starving must, however, be taken into consideration since it generates modifications in the state of the cells and, therefore, in the quality/functionality of the EVs produced. On the other hand, the secretion times of the EVs, and therefore the potential quantity of EVs recovered, are limited. The alternatives for the clinic consist in the use of serum-free or platelet lysate-free media (containing specific cocktails of growth factors and additives), but these media are very specific of a cell type and very expensive, which is problematic for the large-scale production of conditioned medium. Commercial “exosome-free” media also exist. Depletions are performed by ultracentrifugation; however, the levels of depletion are not optimal, with variations depending on the centrifugation conditions and durations. Likewise, tangential flow filtration appears to be a possible solution concerning the purification of culture media for large-scale clinical productions.
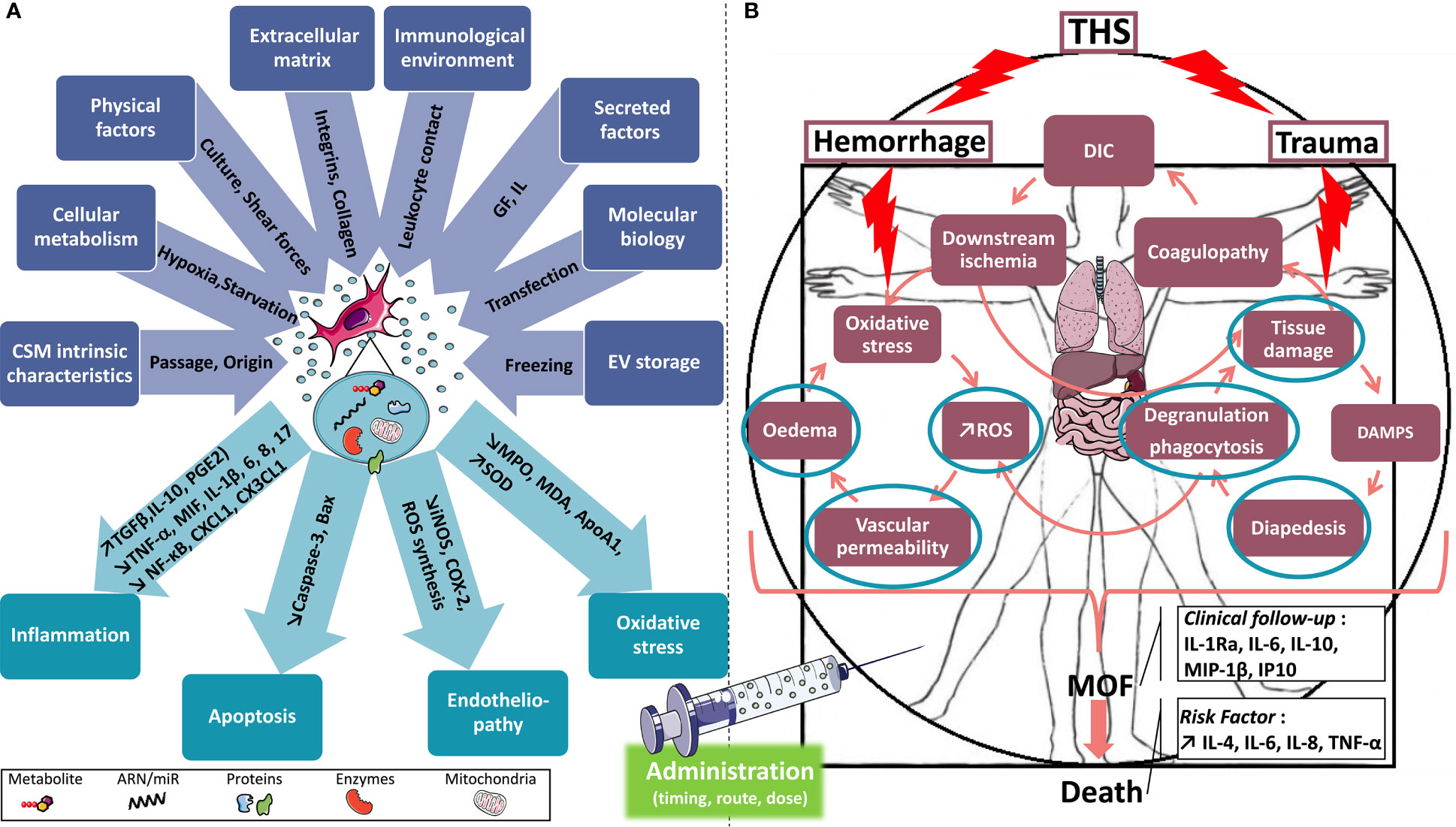
Figure 4 MSC-EVs in THS, or how to apply recent knowledge at the service of those seriously injured. (A) In blue are the main factors modulating the production of EVs. EVs in the center contain, depending on the priming, organelles, proteins, enzymes, RNA, and miR in variable quantities. In aquamarine are the possible main pathways of the potential beneficial effects of EVs in THS models. The administration methods vary by model and must be explored. (B) Simplified consequences of THS. Three loops (coagulopathy, inflammation, and endotheliopathy) are involved in the vicious circle leading to MOF. Cytokines of clinical interest are predictors of the onset of MOF. IL-4, IL-6, IL-8, and TNF-α are significantly increased in trauma patients with MOF and not surviving it. Items circled in aquamarine are potential targets for EV action. EVs, extracellular vesicles; MSC-EVs, mesenchymal stromal cell-derived extracellular vesicles; THS, traumatic hemorrhagic shock; MOF, multi-organ failure; MPO, myeloperoxidase; MDA, malondialdehyde; SOD, superoxide dismutase; GF, growth factor; IL, interleukin.
Finally, from a regulatory point of view, EV-derived products are classified as medicinal products. Within the framework of medicinal products, EV-derived products are categorized as “biological medicinal products” (Directive 2003/63/EC of June 25, 2003, amending Directive 2001/83/EC). However, MSC-EVs could be subcategorized. When they originate from unmodified primary cells or from genetically modified cells that do not contain a transgene product (immortalized cells), they belong to the biological medicinal product category, without any further subcategory. In contrast, MSC-EVs containing a transgene considered as a gene therapy product (e.g., recombinant mRNA and miRNA) are classified as gene therapy products, a subclass of advanced therapy medicinal products (ATMP). This means that the active substance and mode of action of MSC-EVs are decisive for their regulatory classification and can have significant repercussions on the manufacturing process. The use of primary MSCs may have some limitations for large clinical-scale manufacturing due to their limited life span and the donor-to-donor or batch-to-batch heterogeneity. Therefore, EVs produced from immortalized MSCs could be the most promising strategy to prevent MOF.
In recent years, a considerable number of studies have contributed to a better understanding of the biology of EVs and paved the way for their therapeutic use. In cases of isolated organ injuries, MSC-EVs can help restore local homeostasis by decreasing inflammation and oxidative stress, by having an anti-apoptotic effect, or even inhibiting endotheliopathy. Locally protecting the onset of organ damage is a means to prevent the onset of SIRS and the depression of CARS at the systemic level, which promote the development MOF.
This new therapeutic tool could revolutionize the field of cell therapy because it opens the way to treatments that can be administered as early as possible for the care of patients, not only in civilian life but also in hostile contexts such as those encountered in theaters of military operations.
GV and JP wrote the manuscript with input from all authors. All authors (NL, CM, EV, and SB) provided critical feedback and helped shape the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and Regional Mortality From 235 Causes of Death for 20 Age Groups in 1990 and 2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380:2095–128. doi: 10.1016/S0140-6736(12)61728-0
2. Krug EG, Mercy JA, Dahlberg LL, Zwi AB. The World Report on Violence and Health. Lancet (2002) 360:1083–8. doi: 10.1016/S0140-6736(02)11133-0
3. White NJ, Ward KR, Pati S, Strandenes G, Cap AP. Hemorrhagic Blood Failure: Oxygen Debt, Coagulopathy, and Endothelial Damage. J Trauma Acute Care Surg (2017) 82:S41–9. doi: 10.1097/TA.0000000000001436
4. Eastridge BJ, Mabry RL, Seguin P, Cantrell J, Tops T, Uribe P, et al. Death on the Battlefield (2001-2011): Implications for the Future of Combat Casualty Care. J Trauma Acute Care Surg (2012) 73:S431–437. doi: 10.1097/TA.0b013e3182755dcc
6. Negoi I, Paun S, Hostiuc S, Stoica B, Tanase I, Negoi RI, et al. Mortality After Acute Trauma: Progressive Decreasing Rather Than a Trimodal Distribution. J Acute Dis (2015) 4:205–9. doi: 10.1016/j.joad.2015.03.001
7. Teixeira PGR, Inaba K, Hadjizacharia P, Brown C, Salim A, Rhee P, et al. Preventable or Potentially Preventable Mortality at a Mature Trauma Center. J Trauma (2007) 63:1338–47. doi: 10.1097/TA.0b013e31815078ae
8. Jarrar D, Chaudry IH, Wang P. Organ Dysfunction Following Hemorrhage and Sepsis: Mechanisms and Therapeutic Approaches. Int J Mol Med (1999) 4:575–83. doi: 10.3892/ijmm.4.6.575
9. McGhan LJ, Jaroszewski DE. The Role of Toll-Like Receptor-4 in the Development of Multi-Organ Failure Following Traumatic Haemorrhagic Shock and Resuscitation. Injury (2012) 43:129–36. doi: 10.1016/j.injury.2011.05.032
10. Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV. Stromal Cells Responsible for Transferring the Microenvironment of the Hemopoietic Tissues. Cloning In Vitro and Retransplantation In Vivo. Transplantation (1974) 17:331–40. doi: 10.1097/00007890-197404000-00001
11. Koç ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI, et al. Rapid Hematopoietic Recovery After Coinfusion of Autologous-Blood Stem Cells and Culture-Expanded Marrow Mesenchymal Stem Cells in Advanced Breast Cancer Patients Receiving High-Dose Chemotherapy. J Clin Oncol (2000) 18:307–16. doi: 10.1200/JCO.2000.18.2.307
12. Lazarus HM, Koc ON, Devine SM, Curtin P, Maziarz RT, Holland HK, et al. Cotransplantation of HLA-Identical Sibling Culture-Expanded Mesenchymal Stem Cells and Hematopoietic Stem Cells in Hematologic Malignancy Patients. Biol Blood Marrow Transplant (2005) 11:389–98. doi: 10.1016/j.bbmt.2005.02.001
13. Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, et al. Treatment of Severe Acute Graft-Versus-Host Disease With Third Party Haploidentical Mesenchymal Stem Cells. Lancet (2004) 363:1439–41. doi: 10.1016/S0140-6736(04)16104-7
14. Lee ST, Jang JH, Cheong J-W, Kim JS, Maemg H-Y, Hahn JS, et al. Treatment of High-Risk Acute Myelogenous Leukaemia by Myeloablative Chemoradiotherapy Followed by Co-Infusion of T Cell-Depleted Haematopoietic Stem Cells and Culture-Expanded Marrow Mesenchymal Stem Cells From a Related Donor With One Fully Mismatched Human Leucocyte Antigen Haplotype. Br J Haematol (2002) 118:1128–31. doi: 10.1046/j.1365-2141.2002.03767.x
15. Gore AV, Bible LE, Livingston DH, Mohr AM, Sifri ZC. Mesenchymal Stem Cells Reverse Trauma and Hemorrhagic Shock-Induced Bone Marrow Dysfunction. J Surg Res (2015) 199:615–21. doi: 10.1016/j.jss.2015.06.023
16. Nandra KK, Takahashi K, Collino M, Benetti E, Wong WSF, Goh FY, et al. Acute Treatment With Bone Marrow-Derived Mononuclear Cells Attenuates the Organ Injury/Dysfunction Induced by Hemorrhagic Shock in the Rat. Shock (2012) 37:592–8. doi: 10.1097/SHK.0b013e31824e4c0d
17. Pati S, Gerber MH, Menge TD, Wataha KA, Zhao Y, Baumgartner JA, et al. Bone Marrow Derived Mesenchymal Stem Cells Inhibit Inflammation and Preserve Vascular Endothelial Integrity in the Lungs After Hemorrhagic Shock. PloS One (2011) 6:e25171. doi: 10.1371/journal.pone.0025171
18. Yáñez-Mó M, Siljander PR-M, Andreu Z, Zavec AB, Borràs FE, Buzas EI, et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J Extracell Vesicles (2015) 4:27066. doi: 10.3402/jev.v4.27066
19. WHO. Injuries and Violence: The Facts. World Health Organization (2020). Available at: https://www.who.int/news-room/fact-sheets/detail/injuries-and-violence
20. Sobrino J, Shafi S. Timing and Causes of Death After Injuries. Proc (Bayl Univ Med Cent) (2013) 26:120–3. doi: 10.1080/08998280.2013.11928934
21. Pfeifer R, Teuben M, Andruszkow H, Barkatali BM, Pape H-C. Mortality Patterns in Patients With Multiple Trauma: A Systematic Review of Autopsy Studies. PloS One (2016) 11:e0148844. doi: 10.1371/journal.pone.0148844
22. Belmont PJ, McCriskin BJ, Sieg RN, Burks R, Schoenfeld AJ. Combat Wounds in Iraq and Afghanistan From 2005 to 2009. J Trauma Acute Care Surg (2012) 73:3–12. doi: 10.1097/TA.0b013e318250bfb4
23. Schoenfeld AJ, Dunn JC, Bader JO, Belmont PJ. The Nature and Extent of War Injuries Sustained by Combat Specialty Personnel Killed and Wounded in Afghanistan and Iraq, 2003-2011. J Trauma Acute Care Surg (2013) 75:287–91. doi: 10.1097/TA.0b013e31829a0970
24. Cannon JW, Hofmann LJ, Glasgow SC, Potter BK, Rodriguez CJ, Cancio LC, et al. Dismounted Complex Blast Injuries: A Comprehensive Review of the Modern Combat Experience. JACS (2016) 223:652–664.e8. doi: 10.1016/j.jamcollsurg.2016.07.009
25. Dubost C, Goudard Y, Landevoisin ESD, Contargyris C, Evans D, Pauleau G. Combat Casualties From Two Current Conflicts With the Seventh French Forward Surgical Team in Mali and Central African Republic in 2014. BMJ Mil Health (2016) 25. doi: 10.1136/jramc-2015-000557
26. Travers S, Carfantan C, Luft A, Aigle L, Pasquier P, Martinaud C, et al. Five Years of Prolonged Field Care: Prehospital Challenges During Recent French Military Operations. Transfusion (2019) 59:1459–66. doi: 10.1111/trf.15262
27. Longrois D, Mertes P-M. Choc Hémorragique. EMC - Anesth-Réanim (2010) 7:1–19. doi: 10.1016/S0246-0289(10)44705-2
28. Gutierrez G, Reines HD, Wulf-Gutierrez ME. Clinical Review: Hemorrhagic Shock. Crit Care (2004) 8:373–81. doi: 10.1186/cc2851
29. Copotoiu R, Cinca E, Collange O, Levy F, Mertes P-M. Pathophysiology of Hemorragic Shock. Transfus Clin Biol (2016) 23:222–8. doi: 10.1016/j.tracli.2016.07.004
30. Evans RG, Ventura S, Dampney RA, Ludbrook J. Neural Mechanisms in the Cardiovascular Responses to Acute Central Hypovolaemia. Clin Exp Pharmacol Physiol (2001) 28:479–87. doi: 10.1046/j.1440-1681.2001.03473.x
31. Serratrice J, Verschueren A, Serratrice G. Système Nerveux Autonome. EMC - Neurol (2013) 10:1–18. doi: 10.1016/S0246-0378(12)60767-2
32. Albrecht E, Haberer J-P, Buchser E, Moret V. Manuel Pratique D’anesthésie. Elsevier Masson (2015) (978-2294731891):864. doi: 10.1016/j.pratan.2016.01.003
33. Baines CP. The Mitochondrial Permeability Transition Pore and Ischemia-Reperfusion Injury. Basic Res Cardiol (2009) 104:181–8. doi: 10.1007/s00395-009-0004-8
34. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/Reperfusion. Compr Physiol (2016) 7:113–70. doi: 10.1002/cphy.c160006
35. Hervera A, Santos CX, De Virgiliis F, Shah AM, Di Giovanni S. Paracrine Mechanisms of Redox Signalling for Postmitotic Cell and Tissue Regeneration. Trends Cell Biol (2019) 29:514–30. doi: 10.1016/j.tcb.2019.01.006
36. Lugrin J, Rosenblatt-Velin N, Parapanov R, Liaudet L. The Role of Oxidative Stress During Inflammatory Processes. Biol Chem (2014) 395:203–30. doi: 10.1515/hsz-2013-0241
37. Bortolotti P, Faure E, Kipnis E. Inflammasomes in Tissue Damages and Immune Disorders After Trauma. Front Immunol (2018) 9:1900. doi: 10.3389/fimmu.2018.01900
38. Relja B, Land WG. Damage-Associated Molecular Patterns in Trauma. Eur J Trauma Emerg Surg (2020) 46:751–75. doi: 10.1007/s00068-019-01235-w
39. Land WG. Use of DAMPs and SAMPs as Therapeutic Targets or Therapeutics: A Note of Caution. Mol Diagn Ther (2020) 24:251–62. doi: 10.1007/s40291-020-00460-z
40. Németh K, Leelahavanichkul A, Yuen PST, Mayer B, Parmelee A, Doi K, et al. Bone Marrow Stromal Cells Attenuate Sepsis via Prostaglandin E(2)-Dependent Reprogramming of Host Macrophages to Increase Their Interleukin-10 Production. Nat Med (2009) 15:42–9. doi: 10.1038/nm.1905
41. Tsai W-H, Li I-T, Yu Y-B, Hsu H-C, Shih C-H. Serial Changes in Plasma Annexin A1 and Cortisol Levels in Sepsis Patients. Chin J Physiol (2014) 57:1–7. doi: 10.4077/CJP.2014.BAB193
42. Kim JY, Park JS, Strassheim D, Douglas I, Diaz del Valle F, Asehnoune K, et al. HMGB1 Contributes to the Development of Acute Lung Injury After Hemorrhage. Am J Physiol Lung Cell Mol Physiol (2005) 288:L958–965. doi: 10.1152/ajplung.00359.2004
43. Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, et al. Systemic Inflammation and Remote Organ Injury Following Trauma Require HMGB1. Am J Physiol Regul Integr Comp Physiol (2007) 293:R1538–1544. doi: 10.1152/ajpregu.00272.2007
44. Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early Release of High Mobility Group Box Nuclear Protein 1 After Severe Trauma in Humans: Role of Injury Severity and Tissue Hypoperfusion. Crit Care (2009) 13:R174. doi: 10.1186/cc8152
45. Sodhi CP, Jia H, Yamaguchi Y, Lu P, Good M, Egan C, et al. Intestinal Epithelial TLR-4 Activation Is Required for the Development of Acute Lung Injury After Trauma/Hemorrhagic Shock via the Release of HMGB1 From the Gut. J Immunol (2015) 194:4931–9. doi: 10.4049/jimmunol.1402490
46. Ottestad W, Rognes IN, Pischke SE, Mollnes TE, Andersson U, Eken T. Biphasic Release of the Alarmin High Mobility Group Box 1 Protein Early After Trauma Predicts Poor Clinical Outcome. Crit Care Med (2019) 47:e614–22. doi: 10.1097/CCM.0000000000003800
47. Lam NYL, Rainer TH, Chiu RWK, Joynt GM, Lo YMD. Plasma Mitochondrial DNA Concentrations After Trauma. Clin Chem (2004) 50:213–6. doi: 10.1373/clinchem.2003.025783
48. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
49. Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The Plasma Mitochondrial DNA is an Independent Predictor for Post-Traumatic Systemic Inflammatory Response Syndrome. PloS One (2013) 8:e72834. doi: 10.1371/journal.pone.0072834
50. Simmons JD, Lee Y-L, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated Levels of Plasma Mitochondrial DNA DAMPs are Linked to Clinical Outcome in Severely Injured Human Subjects. Ann Surg (2013) 258:591–596; discussion 596-598. doi: 10.1097/SLA.0b013e3182a4ea46
51. Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, et al. Mitochondrial DNA Released by Trauma Induces Neutrophil Extracellular Traps. PloS One (2015) 10:e0120549. doi: 10.1371/journal.pone.0120549
52. Lubkin DT, Bishawi M, Barbas AS, Brennan TV, Kirk AD. Extracellular Mitochondrial DNA and N-Formyl Peptides in Trauma and Critical Illness: A Systematic Review. Crit Care Med (2018) 46:2018–28. doi: 10.1097/CCM.0000000000003381
53. Hashiguchi N, Ogura H, Tanaka H, Koh T, Aoki M, Shiozaki T, et al. Enhanced Expression of Heat Shock Proteins in Leukocytes From Trauma Patients. J Trauma (2001) 50:102–7. doi: 10.1097/00005373-200101000-00018
54. Ogura H, Hashiguchi N, Tanaka H, Koh T, Noborio M, Nakamori Y, et al. Long-Term Enhanced Expression of Heat Shock Proteins and Decelerated Apoptosis in Polymorphonuclear Leukocytes From Major Burn Patients. J Burn Care Rehabil (2002) 23:103–9. doi: 10.1097/00004630-200203000-00006
55. Pespeni M, Mackersie RC, Lee H, Morabito D, Hodnett M, Howard M, et al. Serum Levels of Hsp60 Correlate With the Development of Acute Lung Injury After Trauma. J Surg Res (2005) 126:41–7. doi: 10.1016/j.jss.2005.01.012
56. Guisasola MC, Ortiz A, Chana F, Alonso B, Vaquero J. Early Inflammatory Response in Polytraumatized Patients: Cytokines and Heat Shock Proteins. A Pilot Study. Orthop Traumatol Surg Res (2015) 101:607–11. doi: 10.1016/j.otsr.2015.03.014
57. Ren B, Zou G, Huang Y, Xu G, Xu F, He J, et al. Serum Levels of HSP70 and Other DAMP Proteins can Aid in Patient Diagnosis After Traumatic Injury. Cell Stress Chaperones (2016) 21:677–86. doi: 10.1007/s12192-016-0694-4
58. Joly P, Marshall JC, Tessier PA, Massé C, Page N, Frenette AJ, et al. S100A8/A9 and sRAGE Kinetic After Polytrauma; an Explorative Observational Study. Scand J Trauma Resusc Emerg Med (2017) 25:114. doi: 10.1186/s13049-017-0455-0
59. Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous Histones Function as Alarmins in Sterile Inflammatory Liver Injury Through Toll-Like Receptor 9 in Mice. Hepatology (2011) 54:999–1008. doi: 10.1002/hep.24501
60. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular Histones Promote Thrombin Generation Through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
61. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones From Dying Renal Cells Aggravate Kidney Injury via TLR2 and TLR4. J Am Soc Nephrol (2012) 23:1375–88. doi: 10.1681/ASN.2011111077
62. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating Histones are Mediators of Trauma-Associated Lung Injury. Am J Respir Crit Care Med (2013) 187:160–9. doi: 10.1164/rccm.201206-1037OC
63. Kawai C, Kotani H, Miyao M, Ishida T, Jemail L, Abiru H, et al. Circulating Extracellular Histones Are Clinically Relevant Mediators of Multiple Organ Injury. Am J Pathol (2016) 186:829–43. doi: 10.1016/j.ajpath.2015.11.025
64. Hasan D, Blankman P, Nieman GF. Purinergic Signalling Links Mechanical Breath Profile and Alveolar Mechanics With the Pro-Inflammatory Innate Immune Response Causing Ventilation-Induced Lung Injury. Purinergic Signal (2017) 13:363–86. doi: 10.1007/s11302-017-9564-5
65. Sundnes O, Ottestad W, Schjalm C, Lundbäck P, la Cour Poulsen L, Mollnes TE, et al. Rapid Systemic Surge of IL-33 After Severe Human Trauma: A Prospective Observational Study. Mol Med (2021) 27:29. doi: 10.1186/s10020-021-00288-1
66. Ehrnthaller C, Flierl M, Perl M, Denk S, Unnewehr H, Ward PA, et al. The Molecular Fingerprint of Lung Inflammation After Blunt Chest Trauma. Eur J Med Res (2015) 20:70. doi: 10.1186/s40001-015-0164-y
67. Relja B, Mörs K, Marzi I. Danger Signals in Trauma. Eur J Trauma Emerg Surg (2018) 44:301–16. doi: 10.1007/s00068-018-0962-3
68. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu Rev Immunol (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
69. Darrabie MD, Cheeseman J, Limkakeng AT, Borawski J, Sullenger BA, Elster EA, et al. Toll-Like Receptor Activation as a Biomarker in Traumatically Injured Patients. J Surg Res (2018) 231:270–7. doi: 10.1016/j.jss.2018.05.059
70. Martinon F. Detection of Immune Danger Signals by NALP3. J Leukoc Biol (2008) 83:507–11. doi: 10.1189/jlb.0607362
71. Teissier T, Boulanger É. The Receptor for Advanced Glycation End-Products (RAGE) is an Important Pattern Recognition Receptor (PRR) for Inflammaging. Biogerontology (2019) 20:279–301. doi: 10.1007/s10522-019-09808-3
72. Giuliani AL, Sarti AC, Falzoni S, Di Virgilio F. The P2X7 Receptor-Interleukin-1 Liaison. Front Pharmacol (2017) 8:123. doi: 10.3389/fphar.2017.00123
73. Huber-Lang M, Lambris JD, Ward PA. Innate Immune Responses to Trauma. Nat Immunol (2018) 19:327–41. doi: 10.1038/s41590-018-0064-8
74. Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim J-Y, Strassheim D, et al. High Mobility Group Box 1 Protein Interacts With Multiple Toll-Like Receptors. Am J Physiol Cell Physiol (2006) 290:C917–924. doi: 10.1152/ajpcell.00401.2005
75. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A Critical Cysteine is Required for HMGB1 Binding to Toll-Like Receptor 4 and Activation of Macrophage Cytokine Release. Proc Natl Acad Sci USA (2010) 107:11942–7. doi: 10.1073/pnas.1003893107
76. Yu L, Wang L, Chen S. Endogenous Toll-Like Receptor Ligands and Their Biological Significance. J Cell Mol Med (2010) 14:2592–603. doi: 10.1111/j.1582-4934.2010.01127.x
77. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) Pathway. Sci Signal (2010) 3:cm1. doi: 10.1126/scisignal.3105cm1
78. Ma KC, Schenck EJ, Pabon MA, Choi AMK. The Role of Danger Signals in the Pathogenesis and Perpetuation of Critical Illness. Am J Respir Crit Care Med (2018) 197:300–9. doi: 10.1164/rccm.201612-2460PP
79. Denk S, Weckbach S, Eisele P, Braun CK, Wiegner R, Ohmann JJ, et al. Role of Hemorrhagic Shock in Experimental Polytrauma. Shock (2018) 49:154–63. doi: 10.1097/SHK.0000000000000925
80. Lu S, Aguilar A, Subramani K, Poulose N, Ayub A, Raju R. Alteration of Cytokine Profile Following Hemorrhagic Shock. Cytokine (2016) 81:35–8. doi: 10.1016/j.cyto.2016.01.022
81. Lord JM, Midwinter MJ, Chen Y-F, Belli A, Brohi K, Kovacs EJ, et al. The Systemic Immune Response to Trauma: An Overview of Pathophysiology and Treatment. Lancet (2014) 384:1455–65. doi: 10.1016/S0140-6736(14)60687-5
82. Huber-Lang M, Gebhard F, Schmidt CQ, Palmer A, Denk S, Wiegner R. Complement Therapeutic Strategies in Trauma, Hemorrhagic Shock and Systemic Inflammation - Closing Pandora’s Box? Semin Immunol (2016) 28:278–84. doi: 10.1016/j.smim.2016.04.005
83. Burk A-M, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, et al. Early Complementopathy After Multiple Injuries in Humans. Shock (2012) 37:348–54. doi: 10.1097/SHK.0b013e3182471795
84. Fröhlich M, Lefering R, Probst C, Paffrath T, Schneider MM, Maegele M, et al. Epidemiology and Risk Factors of Multiple-Organ Failure After Multiple Trauma: An Analysis of 31,154 Patients From the TraumaRegister DGU. J Trauma Acute Care Surg (2014) 76:921–7. doi: 10.1097/TA.0000000000000199
85. Rittirsch D, Redl H, Huber-Lang M. Role of Complement in Multiorgan Failure. Clin Dev Immunol (2012) 2012:962927. doi: 10.1155/2012/962927
86. Hazeldine J, Naumann DN, Toman E, Davies D, Bishop JRB, Su Z, et al. Prehospital Immune Responses and Development of Multiple Organ Dysfunction Syndrome Following Traumatic Injury: A Prospective Cohort Study. PloS Med (2017) 14:e1002338. doi: 10.1371/journal.pmed.1002338
87. Pillay J, Hietbrink F, Koenderman L, Leenen LPH. The Systemic Inflammatory Response Induced by Trauma is Reflected by Multiple Phenotypes of Blood Neutrophils. Injury (2007) 38:1365–72. doi: 10.1016/j.injury.2007.09.016
88. Botha AJ, Moore FA, Moore EE, Peterson VM, Silliman CC, Goode AW. Sequential Systemic Platelet-Activating Factor and Interleukin 8 Primes Neutrophils in Patients With Trauma at Risk of Multiple Organ Failure. Br J Surg (1996) 83:1407–12. doi: 10.1002/bjs.1800831027
89. Visser T, Hietbrink F, Groeneveld KM, Koenderman L, Leenen LPH. Isolated Blunt Chest Injury Leads to Transient Activation of Circulating Neutrophils. Eur J Trauma Emerg Surg (2011) 37:177–84. doi: 10.1007/s00068-010-0041-x
90. Seekamp A, van Griensven M, Hildebrandt F, Brauer N, Jochum M, Martin M. The Effect of Trauma on Neutrophil L-Selectin Expression and sL-Selectin Serum Levels. Shock (2001) 15:254–60. doi: 10.1097/00024382-200115040-00002
91. Doroshenko T, Chaly Y, Savitskiy V, Maslakova O, Portyanko A, Gorudko I, et al. Phagocytosing Neutrophils Down-Regulate the Expression of Chemokine Receptors CXCR1 and CXCR2. Blood (2002) 100:2668–71. doi: 10.1182/blood.100.7.2668
92. Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, et al. C5a Mediates Peripheral Blood Neutrophil Dysfunction in Critically Ill Patients. Am J Respir Crit Care Med (2009) 180:19–28. doi: 10.1164/rccm.200812-1928OC
93. Botha AJ, Moore FA, Moore EE, Sauaia A, Banerjee A, Peterson VM. Early Neutrophil Sequestration After Injury: A Pathogenic Mechanism for Multiple Organ Failure. J Trauma (1995) 39:411–7. doi: 10.1097/00005373-199509000-00003
94. Paunel-Görgülü A, Flohé S, Scholz M, Windolf J, Lögters T. Increased Serum Soluble Fas After Major Trauma is Associated With Delayed Neutrophil Apoptosis and Development of Sepsis. Crit Care (2011) 15:R20. doi: 10.1186/cc9965
95. Paunel-Görgülü A, Kirichevska T, Lögters T, Windolf J, Flohé S. Molecular Mechanisms Underlying Delayed Apoptosis in Neutrophils From Multiple Trauma Patients With and Without Sepsis. Mol Med (2012) 18:325–35. doi: 10.2119/molmed.2011.00380
96. Hazeldine J, Hampson P, Lord JM. The Impact of Trauma on Neutrophil Function. Injury (2014) 45:1824–33. doi: 10.1016/j.injury.2014.06.021
97. Brown KA, Brain SD, Pearson JD, Edgeworth JD, Lewis SM, Treacher DF. Neutrophils in Development of Multiple Organ Failure in Sepsis. Lancet (2006) 368:157–69. doi: 10.1016/S0140-6736(06)69005-3
98. Biffl WL, Moore EE, Zallen G, Johnson JL, Gabriel J, Offner PJ, et al. Neutrophils are Primed for Cytotoxicity and Resist Apoptosis in Injured Patients at Risk for Multiple Organ Failure. Surgery (1999) 126:198–202. doi: 10.1016/S0039-6060(99)70155-8
99. Nolan B, Collette H, Baker S, Duffy A, De M, Miller C, et al. Inhibition of Neutrophil Apoptosis After Severe Trauma is NF Kappa Beta Dependent. J Trauma (2000) 48:599–604; discussion 604-605. doi: 10.1097/00005373-200004000-00004
100. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
101. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
102. Neeli I, Dwivedi N, Khan S, Radic M. Regulation of Extracellular Chromatin Release From Neutrophils. J Innate Immun (2009) 1:194–201. doi: 10.1159/000206974
103. Jorch SK, Kubes P. An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat Med (2017) 23:279–87. doi: 10.1038/nm.4294
104. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PloS One (2012) 7:e48111. doi: 10.1371/journal.pone.0048111
105. Meng W, Paunel-Görgülü A, Flohé S, Witte I, Schädel-Höpfner M, Windolf J, et al. Deoxyribonuclease is a Potential Counter Regulator of Aberrant Neutrophil Extracellular Traps Formation After Major Trauma. Mediators Inflamm (2012) 2012:149560. doi: 10.1155/2012/149560
106. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages That Have Ingested Apoptotic Cells In Vitro Inhibit Proinflammatory Cytokine Production Through Autocrine/Paracrine Mechanisms Involving TGF-Beta, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi: 10.1172/JCI1112
107. Herold S, Mayer K, Lohmeyer J. Acute Lung Injury: How Macrophages Orchestrate Resolution of Inflammation and Tissue Repair. Front Immunol (2011) 2:65. doi: 10.3389/fimmu.2011.00065
108. Stables MJ, Gilroy DW. Old and New Generation Lipid Mediators in Acute Inflammation and Resolution. Prog Lipid Res (2011) 50:35–51. doi: 10.1016/j.plipres.2010.07.005
109. Peiseler M, Kubes P. Macrophages Play an Essential Role in Trauma-Induced Sterile Inflammation and Tissue Repair. Eur J Trauma Emerg Surg (2018) 44:335–49. doi: 10.1007/s00068-018-0956-1
110. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: Defects in Immune Function in Patients With Sepsis are Associated With PD-1 or PD-L1 Expression and can be Restored by Antibodies Targeting PD-1 or PD-L1. J Leukoc Biol (2016) 100:1239–54. doi: 10.1189/jlb.4HI0616-255R
111. Hildebrand F, Hubbard WJ, Choudhry MA, Frink M, Pape H-C, Kunkel SL, et al. Kupffer Cells and Their Mediators: The Culprits in Producing Distant Organ Damage After Trauma-Hemorrhage. Am J Pathol (2006) 169:784–94. doi: 10.2353/ajpath.2006.060010
112. Thobe BM, Frink M, Hildebrand F, Schwacha MG, Hubbard WJ, Choudhry MA, et al. The Role of MAPK in Kupffer Cell Toll-Like Receptor (TLR) 2-, TLR4-, and TLR9-Mediated Signaling Following Trauma-Hemorrhage. J Cell Physiol (2007) 210:667–75. doi: 10.1002/jcp.20860
113. Wutzler S, Maier M, Lehnert M, Henrich D, Walcher F, Maegele M, et al. Suppression and Recovery of LPS-Stimulated Monocyte Activity After Trauma is Correlated With Increasing Injury Severity: A Prospective Clinical Study. J Trauma (2009) 66:1273–80. doi: 10.1097/TA.0b013e3181968054
114. Sauaia A, Moore FA, Moore EE. Postinjury Inflammation and Organ Dysfunction. Crit Care Clin (2017) 33:167–91. doi: 10.1016/j.ccc.2016.08.006
115. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent Inflammation and Immunosuppression: A Common Syndrome and New Horizon for Surgical Intensive Care. J Trauma Acute Care Surg (2012) 72:1491–501. doi: 10.1097/TA.0b013e318256e000
116. Cheron A, Floccard B, Allaouchiche B, Guignant C, Poitevin F, Malcus C, et al. Lack of Recovery in Monocyte Human Leukocyte Antigen-DR Expression is Independently Associated With the Development of Sepsis After Major Trauma. Crit Care (2010) 14:R208. doi: 10.1186/cc9331
117. Manjuck J, Saha DC, Astiz M, Eales LJ, Rackow EC. Decreased Response to Recall Antigens is Associated With Depressed Costimulatory Receptor Expression in Septic Critically Ill Patients. J Lab Clin Med (2000) 135:153–60. doi: 10.1067/mlc.2000.104306
118. Stephan RN, Saizawa M, Conrad PJ, Dean RE, Geha AS, Chaudry IH. Depressed Antigen Presentation Function and Membrane Interleukin-1 Activity of Peritoneal Macrophages After Laparotomy. Surgery (1987) 102:147–54. doi: 10.5555/uri:pii:0039606087902285
119. Ayala A, Perrin MM, Chaudry IH. Defective Macrophage Antigen Presentation Following Haemorrhage is Associated With the Loss of MHC Class II (Ia) Antigens. Immunology (1990) 70:33–9.
120. Redmond HP, Hofmann K, Shou J, Leon P, Kelly CJ, Daly JM. Effects of Laparotomy on Systemic Macrophage Function. Surgery (1992) 111:647–55.
121. McCarter MD, Mack VE, Daly JM, Naama HA, Calvano SE. Trauma-Induced Alterations in Macrophage Function. Surgery (1998) 123:96–101. doi: 10.1016/S0039-6060(98)70234-X
122. Kawasaki T, Fujimi S, Lederer JA, Hubbard WJ, Choudhry MA, Schwacha MG, et al. Trauma-Hemorrhage Induces Depressed Splenic Dendritic Cell Functions in Mice. J Immunol (2006) 177:4514–20. doi: 10.4049/jimmunol.177.7.4514
123. Livingston DH, Anjaria D, Wu J, Hauser CJ, Chang V, Deitch EA, et al. Bone Marrow Failure Following Severe Injury in Humans. Ann Surg (2003) 238:748–53. doi: 10.1097/01.sla.0000094441.38807.09
124. Francis WR, Ireland RE, Spear AM, Jenner D, Watts SA, Kirkman E, et al. Flow Cytometric Analysis of Hematopoietic Populations in Rat Bone Marrow. Impact of Trauma and Hemorrhagic Shock. Cytometry A (2019) 95:1167–77. doi: 10.1002/cyto.a.23903
125. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
126. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-Dependent Expansion of an Immature GR-1(+)CD11b(+) Population Induces T Cell Suppression and Th2 Polarization in Sepsis. J Exp Med (2007) 204:1463–74. doi: 10.1084/jem.20062602
127. Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ Myeloid Suppressor Cells Cause T Cell Dysfunction After Traumatic Stress. J Immunol (2006) 176:2085–94. doi: 10.4049/jimmunol.176.4.2085
128. Mannick JA, Rodrick ML, Lederer JA. The Immunologic Response to Injury. J Am Coll Surg (2001) 193:237–44. doi: 10.1016/s1072-7515(01)01011-0
129. Hotchkiss RS, Monneret G, Payen D. Sepsis-Induced Immunosuppression: From Cellular Dysfunctions to Immunotherapy. Nat Rev Immunol (2013) 13:862–74. doi: 10.1038/nri3552
130. Girardot T, Rimmelé T, Venet F, Monneret G. Apoptosis-Induced Lymphopenia in Sepsis and Other Severe Injuries. Apoptosis (2017) 22:295–305. doi: 10.1007/s10495-016-1325-3
131. Huang L-F, Yao Y-M, Dong N, Yu Y, He L-X, Sheng Z-Y. Association Between Regulatory T Cell Activity and Sepsis and Outcome of Severely Burned Patients: A Prospective, Observational Study. Crit Care (2010) 14:R3. doi: 10.1186/cc8232
132. MacConmara MP, Maung AA, Fujimi S, McKenna AM, Delisle A, Lapchak PH, et al. Increased CD4+ CD25+ T Regulatory Cell Activity in Trauma Patients Depresses Protective Th1 Immunity. Ann Surg (2006) 244:514–23. doi: 10.1097/01.sla.0000239031.06906.1f
133. Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T Cells Contribute to Postischemic Liver Injury in Mice by Interacting With Sinusoidal Endothelium and Platelets. Hepatology (2006) 43:306–15. doi: 10.1002/hep.21017
134. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A Genomic Storm in Critically Injured Humans. J Exp Med (2011) 208:2581–90. doi: 10.1084/jem.20111354
135. Seshadri A, Brat GA, Yorkgitis BK, Keegan J, Dolan J, Salim A, et al. Phenotyping the Immune Response to Trauma: A Multiparametric Systems Immunology Approach. Crit Care Med (2017) 45:1523–30. doi: 10.1097/CCM.0000000000002577
136. Carden DL, Granger DN. Pathophysiology of Ischaemia-Reperfusion Injury. J Pathol (2000) 190:255–66. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6
137. Flohé SB, Flohé S, Schade FU. Invited Review: Deterioration of the Immune System After Trauma: Signals and Cellular Mechanisms. Innate Immun (2008) 14:333–44. doi: 10.1177/1753425908100016
138. Denk S, Wiegner R, Hönes FM, Messerer DAC, Radermacher P, Weiss M, et al. Early Detection of Junctional Adhesion Molecule-1 (JAM-1) in the Circulation After Experimental and Clinical Polytrauma. Mediators Inflamm (2015) 2015:463950. doi: 10.1155/2015/463950
139. Johansson PI, Sørensen AM, Perner A, Welling KL, Wanscher M, Larsen CF, et al. Disseminated Intravascular Coagulation or Acute Coagulopathy of Trauma Shock Early After Trauma? An Observational Study. Crit Care (2011) 15:R272. doi: 10.1186/cc10553
140. Giannotta M, Trani M, Dejana E. VE-Cadherin and Endothelial Adherens Junctions: Active Guardians of Vascular Integrity. Dev Cell (2013) 26:441–54. doi: 10.1016/j.devcel.2013.08.020
141. Alves NG, Motawe ZY, Yuan SY, Breslin JW. Endothelial Protrusions in Junctional Integrity and Barrier Function. Curr Top Membr (2018) 82:93–140. doi: 10.1016/bs.ctm.2018.08.006
142. Chignalia AZ, Yetimakman F, Christiaans SC, Unal S, Bayrakci B, Wagener BM, et al. The Glycocalyx and Trauma: A Review. Shock (2016) 45:338–48. doi: 10.1097/SHK.0000000000000513
143. Tuma M, Canestrini S, Alwahab Z, Marshall J. Trauma and Endothelial Glycocalyx: The Microcirculation Helmet? Shock (2016) 46:352–7. doi: 10.1097/SHK.0000000000000635
144. Haywood-Watson RJ, Holcomb JB, Gonzalez EA, Peng Z, Pati S, Park PW, et al. Modulation of Syndecan-1 Shedding After Hemorrhagic Shock and Resuscitation. PloS One (2011) 6:e23530. doi: 10.1371/journal.pone.0023530
145. Jedlicka J, Becker BF, Chappell D. Endothelial Glycocalyx. Crit Care Clin (2020) 36:217–32. doi: 10.1016/j.ccc.2019.12.007
146. Peng Z, Pati S, Potter D, Brown R, Holcomb JB, Grill R, et al. Fresh Frozen Plasma Lessens Pulmonary Endothelial Inflammation and Hyperpermeability After Hemorrhagic Shock and is Associated With Loss of Syndecan-1. Shock (2013) 40:195–202. doi: 10.1097/SHK.0b013e31829f91fc
147. MacLeod JBA, Lynn M, McKenney MG, Cohn SM, Murtha M. Early Coagulopathy Predicts Mortality in Trauma. J Trauma (2003) 55:39–44. doi: 10.1097/01.TA.0000075338.21177.EF
148. Romani de Wit T, van Mourik JA. Biosynthesis, Processing and Secretion of Von Willebrand Factor: Biological Implications. Best Pract Res Clin Haematol (2001) 14:241–55. doi: 10.1053/beha.2001.0132
149. Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet J-F. Acute Traumatic Coagulopathy: Initiated by Hypoperfusion: Modulated Through the Protein C Pathway? Ann Surg (2007) 245:812–8. doi: 10.1097/01.sla.0000256862.79374.31
150. Spahn DR, Bouillon B, Cerny V, Duranteau J, Filipescu D, Hunt BJ, et al. The European Guideline on Management of Major Bleeding and Coagulopathy Following Trauma: Fifth Edition. Crit Care (2019) 23:98. doi: 10.1186/s13054-019-2347-3
151. Petros S. Trauma-Induced Coagulopathy. Hamostaseologie (2019) 39:20–7. doi: 10.1055/s-0039-1677853
152. Regel G, Grotz M, Weltner T, Sturm JA, Tscherne H. Pattern of Organ Failure Following Severe Trauma. World J Surg (1996) 20:422–9. doi: 10.1007/s002689900067
153. Messerer DAC, Halbgebauer R, Nilsson B, Pavenstädt H, Radermacher P, Huber-Lang M. Immunopathophysiology of Trauma-Related Acute Kidney Injury. Nat Rev Nephrol (2021) 17:91–111. doi: 10.1038/s41581-020-00344-9
154. Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury Multiple Organ Failure: A Bimodal Phenomenon. J Trauma (1996) 40:501–510; discussion 510-512. doi: 10.1097/00005373-199604000-00001
155. Sauaia A, Moore EE, Johnson JL, Chin TL, Banerjee A, Sperry JL, et al. Temporal Trends of Postinjury Multiple-Organ Failure: Still Resource Intensive, Morbid, and Lethal. J Trauma Acute Care Surg (2014) 76:582–93. doi: 10.1097/TA.0000000000000147
156. Veith NT, Histing T, Menger MD, Pohlemann T, Tschernig T. Helping Prometheus: Liver Protection in Acute Hemorrhagic Shock. Ann Transl Med (2017) 5:206–6. doi: 10.21037/atm.2017.03.109
157. Hutchings L, Watkinson P, Young JD, Willett K. Defining Multiple Organ Failure After Major Trauma: A Comparison of the Denver, Sequential Organ Failure Assessment, and Marshall Scoring Systems. J Trauma Acute Care Surg (2017) 82:534–41. doi: 10.1097/TA.0000000000001328
158. Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury Multiple Organ Failure. Injury (2009) 40:912–8. doi: 10.1016/j.injury.2009.05.024
159. Jastrow KM, Gonzalez EA, McGuire MF, Suliburk JW, Kozar RA, Iyengar S, et al. Early Cytokine Production Risk Stratifies Trauma Patients for Multiple Organ Failure. J Am Coll Surg (2009) 209:320–31. doi: 10.1016/j.jamcollsurg.2009.05.002
160. Hranjec T, Swenson BR, Dossett LA, Metzger R, Flohr TR, Popovsky KA, et al. Diagnosis-Dependent Relationships Between Cytokine Levels and Survival in Patients Admitted for Surgical Critical Care. J Am Coll Surg (2010) 210:833–844, 845–846. doi: 10.1016/j.jamcollsurg.2009.12.042
161. Clark JA, Coopersmith CM. Intestinal Crosstalk: A New Paradigm for Understanding the Gut as the “Motor” of Critical Illness. Shock (2007) 28:384–93. doi: 10.1097/shk.0b013e31805569df
162. Kistler EB, Alsaigh T, Chang M, Schmid-Schönbein GW. Impaired Small-Bowel Barrier Integrity in the Presence of Lumenal Pancreatic Digestive Enzymes Leads to Circulatory Shock. Shock (2012) 38:262–7. doi: 10.1097/SHK.0b013e31825b1717
163. Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM. The Role of the Complement System in Ischemia-Reperfusion Injury. Shock (2004) 21:401–9. doi: 10.1097/00024382-200405000-00002
164. Rupani B, Caputo FJ, Watkins AC, Vega D, Magnotti LJ, Lu Q, et al. Relationship Between Disruption of the Unstirred Mucus Layer and Intestinal Restitution in Loss of Gut Barrier Function After Trauma Hemorrhagic Shock. Surgery (2007) 141:481–9. doi: 10.1016/j.surg.2006.10.008
165. Fishman JE, Levy G, Alli V, Sheth S, Lu Q, Deitch EA. Oxidative Modification of the Intestinal Mucus Layer is a Critical But Unrecognized Component of Trauma Hemorrhagic Shock-Induced Gut Barrier Failure. Am J Physiol Gastrointest Liver Physiol (2013) 304:G57–63. doi: 10.1152/ajpgi.00170.2012
166. DeLano FA, Hoyt DB, Schmid-Schönbein GW. Pancreatic Digestive Enzyme Blockade in the Intestine Increases Survival After Experimental Shock. Sci Transl Med (2013) 5:169ra11. doi: 10.1126/scitranslmed.3005046
167. Lu Q, Xu D-Z, Sharpe S, Doucet D, Pisarenko V, Lee M, et al. The Anatomic Sites of Disruption of the Mucus Layer Directly Correlate With Areas of Trauma/Hemorrhagic Shock-Induced Gut Injury. J Trauma (2011) 70:630–5. doi: 10.1097/TA.0b013e3181e1221b
168. Yu Y-B, Li Y-Q. Enteric Glial Cells and Their Role in the Intestinal Epithelial Barrier. World J Gastroenterol (2014) 20:11273–80. doi: 10.3748/wjg.v20.i32.11273
169. Withers DR, Hepworth MR. Group 3 Innate Lymphoid Cells: Communications Hubs of the Intestinal Immune System. Front Immunol (2017) 8:1298. doi: 10.3389/fimmu.2017.01298
170. Zhang J, Zhang Y, Xu T, Pan S-J, Nie G, Miao X-Y, et al. Severe Traumatic Hemorrhagic Shock Induces Compromised Immune Barrier Function of the Mesenteric Lymph Node Leading to an Increase in Intestinal Bacterial Translocation. Am J Transl Res (2017) 9:2363–73.
171. Ma Y, Zabell T, Creasy A, Yang X, Chatterjee V, Villalba N, et al. Gut Ischemia Reperfusion Injury Induces Lung Inflammation via Mesenteric Lymph-Mediated Neutrophil Activation. Front Immunol (2020) 11:586685. doi: 10.3389/fimmu.2020.586685
172. Hassoun H, Kone B, Mercer D, Moody F, Weisbrodt N, Moore F. Post-Injury Multiple Organ Failure: The Role of the Gut. Shock (2001) 15:1–10. doi: 10.1097/00024382-200115010-00001
173. Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, Mechanisms, Relevant Outcomes, and Clinical Trial Coordination. Am J Respir Crit Care Med (1994) 149:818–24. doi: 10.1164/ajrccm.149.3.7509706
174. Matthay MA, Pati S, Lee J-W. Concise Review: Mesenchymal Stem (Stromal) Cells: Biology and Preclinical Evidence for Therapeutic Potential for Organ Dysfunction Following Trauma or Sepsis. Stem Cells (2017) 35:316–24. doi: 10.1002/stem.2551
175. Wen Z, Fan L, Li Y, Zou Z, Scott MJ, Xiao G, et al. Neutrophils Counteract Autophagy-Mediated Anti-Inflammatory Mechanisms in Alveolar Macrophage: Role in Posthemorrhagic Shock Acute Lung Inflammation. J Immunol (2014) 193(9):4623–33. doi: 10.4049/jimmunol.1400899
176. Reino DC, Palange D, Feketeova E, Bonitz RP, Xu DZ, Lu Q, et al. Activation of TLR4 is Necessary for Trauma Hemorrhagic Shock-Induced Gut Injury and PMN Priming. Shock (2012) 38:107–14. doi: 10.1097/SHK.0b013e318257123a
177. Grommes J, Soehnlein O. Contribution of Neutrophils to Acute Lung Injury. Mol Med (2011) 17:293–307. doi: 10.2119/molmed.2010.00138
178. Lautt WW. Chapter 14, Integrative Hepatic Response to Hemorrhage. In: Hepatic Circulation: Physiology and Pathophysiology. San Rafael (CA: Morgan & Claypool Life Sciences (2009).
179. Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of Cell Death During Warm Hepatic Ischemia-Reperfusion in Rats: Apoptosis or Necrosis? Hepatology (2001) 33:397–405. doi: 10.1053/jhep.2001.22002
180. Selzner N, Rudiger H, Graf R, Clavien P-A. Protective Strategies Against Ischemic Injury of the Liver. Gastroenterology (2003) 125:917–36. doi: 10.1016/s0016-5085(03)01048-5
181. Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic Ischemia/Reperfusion Injury–a Fresh Look. Exp Mol Pathol (2003) 74:86–93. doi: 10.1016/s0014-4800(03)00008-x
182. Nace GW, Huang H, Klune JR, Eid RE, Rosborough BR, Korff S, et al. Cellular-Specific Role of Toll-Like Receptor 4 in Hepatic Ischemia-Reperfusion Injury in Mice. Hepatology (2013) 58:374–87. doi: 10.1002/hep.26346
183. Jaeschke H, Farhood A. Neutrophil and Kupffer Cell-Induced Oxidant Stress and Ischemia-Reperfusion Injury in Rat Liver. Am J Physiol (1991) 260:G355–362. doi: 10.1152/ajpgi.1991.260.3.G355
184. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The Nuclear Factor HMGB1 Mediates Hepatic Injury After Murine Liver Ischemia-Reperfusion. J Exp Med (2005) 201:1135–43. doi: 10.1084/jem.20042614
185. Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic Ischemia/Reperfusion Injury Involves Functional TLR4 Signaling in Nonparenchymal Cells. J Immunol (2005) 175:7661–8. doi: 10.4049/jimmunol.175.11.7661
186. Hanschen M, Zahler S, Krombach F, Khandoga A. Reciprocal Activation Between CD4+ T Cells and Kupffer Cells During Hepatic Ischemia-Reperfusion. Transplantation (2008) 86:710–8. doi: 10.1097/TP.0b013e3181821aa7
187. Mihm S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int J Mol Sci (2018) 19:3104. doi: 10.3390/ijms19103104
188. Harrois A, Soyer B, Gauss T, Hamada S, Raux M, Duranteau J, et al. Prevalence and Risk Factors for Acute Kidney Injury Among Trauma Patients: A Multicenter Cohort Study. Crit Care (2018) 22:344. doi: 10.1186/s13054-018-2265-9
189. Bihorac A, Baslanti TO, Cuenca AG, Hobson CE, Ang D, Efron PA, et al. Acute Kidney Injury is Associated With Early Cytokine Changes After Trauma. J Trauma Acute Care Surg (2013) 74:1005–13. doi: 10.1097/TA.0b013e31828586ec
190. Awad AS, Rouse M, Huang L, Vergis AL, Reutershan J, Cathro HP, et al. Compartmentalization of Neutrophils in the Kidney and Lung Following Acute Ischemic Kidney Injury. Kidney Int (2009) 75:689–98. doi: 10.1038/ki.2008.648
191. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi B-S, et al. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. JASN (2011) 22:317–26. doi: 10.1681/ASN.2009060615
192. Dong X, Swaminathan S, Bachman L-A, Croatt A-J, Nath K-A, Griffin M-D. Resident Dendritic Cells are the Predominant TNF-Secreting Cell in Early Renal Ischemia–Reperfusion Injury. Kidney Int (2007) 71:619–28. doi: 10.1038/sj.ki.5002132
193. Gunay Y, Inal A, Yener N, Sinanoglu O, Selvi O. Bircan HY. A Novel Mechanism of Anti–T-Lymphocyte Globulin Mediated by Fractalkine in Renal Ischemia–Reperfusion Injury in Rats. Transplant Proc (2013) 45:2461–8. doi: 10.1016/j.transproceed.2013.02.118
194. Cao Q, Wang Y, Niu Z, Wang C, Wang R, Zhang Z, et al. Potentiating Tissue-Resident Type 2 Innate Lymphoid Cells by IL-33 to Prevent Renal Ischemia-Reperfusion Injury. JASN (2018) 29:961–76. doi: 10.1681/ASN.2017070774
195. Elterman J, Zonies D, Stewart I, Fang R, Schreiber M. Rhabdomyolysis and Acute Kidney Injury in the Injured War Fighter. J Trauma Acute Care Surg (2015) 79:S171. doi: 10.1097/TA.0000000000000572
196. Okubo K, Kurosawa M, Kamiya M, Urano Y, Suzuki A, Yamamoto K, et al. Macrophage Extracellular Trap Formation Promoted by Platelet Activation is a Key Mediator of Rhabdomyolysis-Induced Acute Kidney Injury. Nat Med (2018) 24:232–8. doi: 10.1038/nm.4462
197. Kaushal GP, Shah SV. Challenges and Advances in the Treatment of AKI. J Am Soc Nephrol (2014) 25:877–83. doi: 10.1681/ASN.2013070780
198. Duranteau J, Asehnoune K, Pierre S, Ozier Y, Leone M, Lefrant J-Y. Recommandations Sur La Réanimation Du Choc Hémorragique - Société Française D’Anesthésie Et De Réanimation (SFAR) 2014. Anesth Réanim (2015) 1:62–74. doi: 10.1016/j.anrea.2014.12.007
199. Rossaint R, Bouillon B, Cerny V, Coats TJ, Duranteau J, Fernández-Mondéjar E, et al. The European Guideline on Management of Major Bleeding and Coagulopathy Following Trauma: Fourth Edition. Crit Care (2016) 20:100. doi: 10.1186/s13054-016-1265-x
200. Sperry JL, Guyette FX, Brown JB, Yazer MH, Triulzi DJ, Early-Young BJ, et al. Prehospital Plasma During Air Medical Transport in Trauma Patients at Risk for Hemorrhagic Shock. N Eng J Med (2018) 379:315–26. doi: 10.1056/NEJMoa1802345
201. Plurad DS, Talving P, Lam L, Inaba K, Green D, Demetriades D. Early Vasopressor Use in Critical Injury Is Associated With Mortality Independent From Volume Status. J Trauma Acute Care Surg (2011) 71:565–72. doi: 10.1097/TA.0b013e3182213d52
202. Sperry JL, Minei JP, Frankel HL, West MA, Harbrecht BG, Moore EE, et al. Early Use of Vasopressors After Injury: Caution Before Constriction. J Trauma Acute Care Surg (2008) 64:9–14. doi: 10.1097/TA.0b013e31815dd029
203. Gauss T, Gayat E, Harrois A, Raux M, Follin A, Daban J-L, et al. Prehospital Traumabase Group Ile De France, SAMU=Service D’aide Médicale Urgente. Effect of Early Use of Noradrenaline on in-Hospital Mortality in Haemorrhagic Shock After Major Trauma: A Propensity-Score Analysis. Br J Anaesth (2018) 120:1237–44. doi: 10.1016/j.bja.2018.02.032
204. Friedenstein AJ, Chailakhjan RK, Lalykina KS. The Development of Fibroblast Colonies in Monolayer Cultures of Guinea-Pig Bone Marrow and Spleen Cells. Cell Prolif (1970) 3:393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x
205. Tavakoli S, Jafarbeigloo HRG, Shariati A, Jahangiryan A, Jadidi F, Kouhbanani MAJ, et al. Mesenchymal Stromal Cells; a New Horizon in Regenerative Medicine. J Cell Physiol (2020) 235:9185–210. doi: 10.1002/jcp.29803
206. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy (2006) 8:315–7. doi: 10.1080/14653240600855905
207. Liang X, Ding Y, Zhang Y, Tse H-F, Lian Q. Paracrine Mechanisms of Mesenchymal Stem Cell-Based Therapy: Current Status and Perspectives. Cell Transplant (2014) 23:1045–59. doi: 10.3727/096368913X667709
208. MacMillan ML, Blazar BR, DeFor TE, Wagner JE. Transplantation of Ex-Vivo Culture-Expanded Parental Haploidentical Mesenchymal Stem Cells to Promote Engraftment in Pediatric Recipients of Unrelated Donor Umbilical Cord Blood: Results of a Phase I–II Clinical Trial. Bone Marrow Transplant (2009) 43:447–54. doi: 10.1038/bmt.2008.348
209. Ball LM, Bernardo ME, Roelofs H, Lankester A, Cometa A, Egeler RM, et al. Cotransplantation of Ex Vivo Expanded Mesenchymal Stem Cells Accelerates Lymphocyte Recovery and may Reduce the Risk of Graft Failure in Haploidentical Hematopoietic Stem-Cell Transplantation. Blood (2007) 110:2764–7. doi: 10.1182/blood-2007-04-087056
210. Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringdén O. Mesenchymal Stem Cells Inhibit and Stimulate Mixed Lymphocyte Cultures and Mitogenic Responses Independently of the Major Histocompatibility Complex. Scand J Immunol (2003) 57:11–20. doi: 10.1046/j.1365-3083.2003.01176.x
211. Kim J, Hematti P. Mesenchymal Stem Cell-Educated Macrophages: A Novel Type of Alternatively Activated Macrophages. Exp Hematol (2009) 37:1445–53. doi: 10.1016/j.exphem.2009.09.004
212. Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. Interactions Between Human Mesenchymal Stem Cells and Natural Killer Cells. Stem Cells (2006) 24:74–85. doi: 10.1634/stemcells.2004-0359
213. Jung Y-J, Ju S-Y, Yoo E-S, Cho SJ, Cho K-A, Woo S-Y, et al. MSC-DC Interactions: MSC Inhibit Maturation and Migration of BM-Derived DC. Cytotherapy (2007) 9:451–8. doi: 10.1080/14653240701452057
214. Su VY-F, Lin C-S, Hung S-C, Yang K-Y. Mesenchymal Stem Cell-Conditioned Medium Induces Neutrophil Apoptosis Associated With Inhibition of the NF-κb Pathway in Endotoxin-Induced Acute Lung Injury. Int J Mol Sci (2019) 20(9):2208. doi: 10.3390/ijms20092208
215. Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringdén O. HLA Expression and Immunologic Properties of Differentiated and Undifferentiated Mesenchymal Stem Cells. Exp Hematol (2003) 31:890–6. doi: 10.1016/S0301-472X(03)00110-3
216. Rafei M, Hsieh J, Fortier S, Li M, Yuan S, Birman E, et al. Mesenchymal Stromal Cell-Derived CCL2 Suppresses Plasma Cell Immunoglobulin Production via STAT3 Inactivation and PAX5 Induction. Blood (2008) 112:4991–8. doi: 10.1182/blood-2008-07-166892
217. Naji A, Eitoku M, Favier B, Deschaseaux F, Rouas-Freiss N, Suganuma N. Biological Functions of Mesenchymal Stem Cells and Clinical Implications. Cell Mol Life Sci (2019) 76:3323–48. doi: 10.1007/s00018-019-03125-1
218. Schlosser K, Wang J-P, Dos Santos C, Walley KR, Marshall J, Fergusson DA, et al. Effects of Mesenchymal Stem Cell Treatment on Systemic Cytokine Levels in a Phase 1 Dose Escalation Safety Trial of Septic Shock Patients. Crit Care Med (2019) 47:918–25. doi: 10.1097/CCM.0000000000003657
219. Caplan AI, Dennis JE. Mesenchymal Stem Cells as Trophic Mediators. J Cell Biochem (2006) 98:1076–84. doi: 10.1002/jcb.20886
220. Luz-Crawford P, Djouad F, Toupet K, Bony C, Franquesa M, Hoogduijn MJ, et al. Mesenchymal Stem Cell-Derived Interleukin 1 Receptor Antagonist Promotes Macrophage Polarization and Inhibits B Cell Differentiation. Stem Cells (2016) 34:483–92. doi: 10.1002/stem.2254
221. Kosaric N, Srifa W, Bonham CA, Kiwanuka H, Chen K, Kuehlmann BA, et al. Macrophage Subpopulation Dynamics Shift Following Intravenous Infusion of Mesenchymal Stromal Cells. Mol Ther (2020) 28:2007–22. doi: 10.1016/j.ymthe.2020.05.022
222. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human Bone Marrow Stromal Cells Suppress T-Lymphocyte Proliferation Induced by Cellular or Nonspecific Mitogenic Stimuli. Blood (2002) 99:3838–43. doi: 10.1182/blood.v99.10.3838
223. Kim DS, Jang IK, Lee MW, Ko YJ, Lee D-H, Lee JW, et al. Enhanced Immunosuppressive Properties of Human Mesenchymal Stem Cells Primed by Interferon-γ. EBioMedicine (2018) 28:261–73. doi: 10.1016/j.ebiom.2018.01.002
224. Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, et al. Intravenous hMSCs Improve Myocardial Infarction in Mice Because Cells Embolized in Lung are Activated to Secrete the Anti-Inflammatory Protein TSG-6. Cell Stem Cell (2009) 5:54–63. doi: 10.1016/j.stem.2009.05.003
225. Sala E, Genua M, Petti L, Anselmo A, Arena V, Cibella J, et al. Mesenchymal Stem Cells Reduce Colitis in Mice via Release of TSG6, Independently of Their Localization to the Intestine. Gastroenterology (2015) 149:163–176.e20. doi: 10.1053/j.gastro.2015.03.013
226. Magaña-Guerrero FS, Domínguez-López A, Martínez-Aboytes P, Buentello-Volante B, Garfias Y. Human Amniotic Membrane Mesenchymal Stem Cells Inhibit Neutrophil Extracellular Traps Through TSG-6. Sci Rep (2017) 7:12426. doi: 10.1038/s41598-017-10962-2
227. Lai RC, Arslan F, Lee MM, Sze NSK, Choo A, Chen TS, et al. Exosome Secreted by MSC Reduces Myocardial Ischemia/Reperfusion Injury. Stem Cell Res (2010) 4:214–22. doi: 10.1016/j.scr.2009.12.003
228. Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, et al. Mesenchymal Stem Cell-Derived Microvesicles Protect Against Acute Tubular Injury. J Am Soc Nephrol (2009) 20:1053–67. doi: 10.1681/ASN.2008070798
229. Lai P, Chen X, Guo L, Wang Y, Liu X, Liu Y, et al. A Potent Immunomodulatory Role of Exosomes Derived From Mesenchymal Stromal Cells in Preventing cGVHD. J Hematol Onco (2018) 11:135. doi: 10.1186/s13045-018-0680-7
230. Wang L, Gu Z, Zhao X, Yang N, Wang F, Deng A, et al. Extracellular Vesicles Released From Human Umbilical Cord-Derived Mesenchymal Stromal Cells Prevent Life-Threatening Acute Graft-Versus-Host Disease in a Mouse Model of Allogeneic Hematopoietic Stem Cell Transplantation. Stem Cells Dev (2016) 25:1874–83. doi: 10.1089/scd.2016.0107
231. Zou X, Zhang G, Cheng Z, Yin D, Du T, Ju G, et al. Microvesicles Derived From Human Wharton’s Jelly Mesenchymal Stromal Cells Ameliorate Renal Ischemia-Reperfusion Injury in Rats by Suppressing CX3CL1. Stem Cell Res (2014) 5:40. doi: 10.1186/scrt428
232. Mansouri N, Willis GR, Fernandez-Gonzalez A, Reis M, Nassiri S, Mitsialis SA, et al. Mesenchymal Stromal Cell Exosomes Prevent and Revert Experimental Pulmonary Fibrosis Through Modulation of Monocyte Phenotypes. JCI Insight (2019) 4(21):e128060. doi: 10.1172/jci.insight.128060
233. Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, et al. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am J Respir Crit Care Med (2017) 196:1275–86. doi: 10.1164/rccm.201701-0170OC
234. Tamura R, Uemoto S, Tabata Y. Immunosuppressive Effect of Mesenchymal Stem Cell-Derived Exosomes on a Concanavalin A-Induced Liver Injury Model. Inflamm Regen (2016) 36:26. doi: 10.1186/s41232-016-0030-5
235. Lane SW, Williams DA, Watt FM. Modulating the Stem Cell Niche for Tissue Regeneration. Nat Biotechnol (2014) 32:795–803. doi: 10.1038/nbt.2978
236. Zhou Y, Tsai T-L, Li W-J. Strategies to Retain Properties of Bone Marrow–Derived Mesenchymal Stem Cells Ex Vivo. Ann N Y Acad Sci (2017) 1409:3–17. doi: 10.1111/nyas.13451
237. Le Blanc K, Mougiakakos D. Multipotent Mesenchymal Stromal Cells and the Innate Immune System. Nat Rev Immunol (2012) 12:383–96. doi: 10.1038/nri3209
238. Gao F, Chiu SM, Motan DAL, Zhang Z, Chen L, Ji H-L, et al. Mesenchymal Stem Cells and Immunomodulation: Current Status and Future Prospects. Cell Death Dis (2016) 7:e2062–2. doi: 10.1038/cddis.2015.327
239. Müller L, Tunger A, Wobus M, von Bonin M, Towers R, Bornhäuser M, et al. Immunomodulatory Properties of Mesenchymal Stromal Cells: An Update. Front Cell Dev Biol (2021) 9:637725. doi: 10.3389/fcell.2021.637725
240. Roura S, Monguió-Tortajada M, Munizaga-Larroudé M, Clos-Sansalvador M, Franquesa M, Rosell A, et al. Potential of Extracellular Vesicle-Associated TSG-6 From Adipose Mesenchymal Stromal Cells in Traumatic Brain Injury. Int J Mol Sci (2020) 21:6761. doi: 10.3390/ijms21186761
241. Amann EM, Groß A, Rojewski MT, Kestler HA, Kalbitz M, Brenner RE, et al. Inflammatory Response of Mesenchymal Stromal Cells After In Vivo Exposure With Selected Trauma-Related Factors and Polytrauma Serum. PloS One (2019) 14:e0216862. doi: 10.1371/journal.pone.0216862
242. Aussel C, Baudry N, Grosbot M, Caron C, Vicaut E, Banzet S, et al. IL-1β Primed Mesenchymal Stromal Cells Moderate Hemorrhagic Shock Induced Organ Injuries. Stem Cell Res Ther (2021) 12(1):438. doi: 10.1186/s13287-021-02505-4
243. Schweizer R, Kamat P, Schweizer D, Dennler C, Zhang S, Schnider JT, et al. Bone Marrow-Derived Mesenchymal Stromal Cells Improve Vascular Regeneration and Reduce Leukocyte-Endothelium Activation in Critical Ischemic Murine Skin in a Dose-Dependent Manner. Cytotherapy (2014) 16:1345–60. doi: 10.1016/j.jcyt.2014.05.008
244. Gleeson BM, Martin K, Ali MT, Kumar AHS, Pillai MG-K, Kumar SPG, et al. Bone Marrow-Derived Mesenchymal Stem Cells Have Innate Procoagulant Activity and Cause Microvascular Obstruction Following Intracoronary Delivery: Amelioration by Antithrombin Therapy. Stem Cells (2015) 33:2726–37. doi: 10.1002/stem.2050
245. Moll G, Rasmusson-Duprez I, von Bahr L, Connolly-Andersen A-M, Elgue G, Funke L, et al. Are Therapeutic Human Mesenchymal Stromal Cells Compatible With Human Blood? Stem Cells (2012) 30:1565–74. doi: 10.1002/stem.1111
246. Chance TC, Rathbone CR, Kamucheka RM, Peltier GC, Cap AP, Bynum JA. The Effects of Cell Type and Culture Condition on the Procoagulant Activity of Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles. J Trauma Acute Care Surg (2019) 87:S74–82. doi: 10.1097/TA.0000000000002225
247. Herzig MC, Cap AP. Challenges in Translating Mesenchymal Stem Cell Therapies for Trauma and Critical Care: TRANSLATING MSC THERAPIES FOR TRAUMA. Transfusion (2016) 56:20S–5S. doi: 10.1111/trf.13566
248. Neuss S, Schneider RKM, Tietze L, Knüchel R, Jahnen-Dechent W. Secretion of Fibrinolytic Enzymes Facilitates Human Mesenchymal Stem Cell Invasion Into Fibrin Clots. Cells Tissues Organs (2010) 191:36–46. doi: 10.1159/000215579
249. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J Extracell Vesicles (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
250. van Niel G, D’Angelo G, Raposo G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat Rev Mol Cell Biol (2018) 19:213–28. doi: 10.1038/nrm.2017.125
251. Caruso S, Poon IKH. Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Front Immunol (2018) 9:1486. doi: 10.3389/fimmu.2018.01486
252. Gardiner C, Vizio DD, Sahoo S, Théry C, Witwer KW, Wauben M, et al. Techniques Used for the Isolation and Characterization of Extracellular Vesicles: Results of a Worldwide Survey. J Extracell Vesicles (2016) 5:32945. doi: 10.3402/jev.v5.32945
253. Makridakis M, Roubelakis MG, Vlahou A. Stem Cells: Insights Into the Secretome. Biochim Biophys Acta (2013) 1834:2380–4. doi: 10.1016/j.bbapap.2013.01.032
254. Phinney DG, Pittenger MF. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells (2017) 35:851–8. doi: 10.1002/stem.2575
255. Doron G, Klontzas ME, Mantalaris A, Guldberg RE, Temenoff JS. Multiomics Characterization of Mesenchymal Stromal Cells Cultured in Monolayer and as Aggregates. Biotechnol Bioeng (2020) 117:1761–78. doi: 10.1002/bit.27317
256. van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R. Classification, Functions, and Clinical Relevance of Extracellular Vesicles. Pharmacol Rev (2012) 64:676–705. doi: 10.1124/pr.112.005983
257. Hu Q, Lyon CJ, Fletcher JK, Tang W, Wan M, Hu TY. Extracellular Vesicle Activities Regulating Macrophage- and Tissue-Mediated Injury and Repair Responses. Acta Pharm Sin B (2021) 11:1493–512. doi: 10.1016/j.apsb.2020.12.014
258. Park K-S, Bandeira E, Shelke GV, Lässer C, Lötvall J. Enhancement of Therapeutic Potential of Mesenchymal Stem Cell-Derived Extracellular Vesicles. Stem Cell Res Ther (2019) 10:288. doi: 10.1186/s13287-019-1398-3
259. van Balkom BWM, Gremmels H, Giebel B, Lim SK. Proteomic Signature of Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles. Proteomics (2019) 19:e1800163. doi: 10.1002/pmic.201800163
260. Harting MT, Srivastava AK, Zhaorigetu S, Bair H, Prabhakara KS, Furman NET, et al. Inflammation-Stimulated Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Inflammation. Stem Cells (2018) 36:79–90. doi: 10.1002/stem.2730
261. Mardpour S, Hamidieh AA, Taleahmad S, Sharifzad F, Taghikhani A, Baharvand H. Interaction Between Mesenchymal Stromal Cell-Derived Extracellular Vesicles and Immune Cells by Distinct Protein Content. J Cell Physiol (2019) 234:8249–58. doi: 10.1002/jcp.27669
262. Dabrowska S, Andrzejewska A, Janowski M, Lukomska B. Immunomodulatory and Regenerative Effects of Mesenchymal Stem Cells and Extracellular Vesicles: Therapeutic Outlook for Inflammatory and Degenerative Diseases. Front Immunol (2020) 11:591065. doi: 10.3389/fimmu.2020.591065
263. Budoni M, Fierabracci A, Luciano R, Petrini S, Di Ciommo V, Muraca M. The Immunosuppressive Effect of Mesenchymal Stromal Cells on B Lymphocytes is Mediated by Membrane Vesicles. Cell Transplant (2013) 22:369–79. doi: 10.3727/096368911X582769b
264. Li X, Liu L, Yang J, Yu Y, Chai J, Wang L, et al. Exosome Derived From Human Umbilical Cord Mesenchymal Stem Cell Mediates MiR-181c Attenuating Burn-Induced Excessive Inflammation. EBioMedicine (2016) 8:72–82. doi: 10.1016/j.ebiom.2016.04.030
265. Sicco CL, Reverberi D, Balbi C, Ulivi V, Principi E, Pascucci L, et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem Cells Transl Med (2017) 6:1018–28. doi: 10.1002/sctm.16-0363
266. Reis M, Mavin E, Nicholson L, Green K, Dickinson AM, Wang X. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Dendritic Cell Maturation and Function. Front Immunol (2018) 9:2538. doi: 10.3389/fimmu.2018.02538
267. Di Trapani M, Bassi G, Midolo M, Gatti A, Kamga PT, Cassaro A, et al. Differential and Transferable Modulatory Effects of Mesenchymal Stromal Cell-Derived Extracellular Vesicles on T, B and NK Cell Functions. Sci Rep (2016) 6:24120. doi: 10.1038/srep24120
268. Heo JS, Choi Y, Kim HO. Adipose-Derived Mesenchymal Stem Cells Promote M2 Macrophage Phenotype Through Exosomes. Stem Cells Int (2019) 2019:7921760. doi: 10.1155/2019/7921760
269. An J-H, Li Q, Ryu M-O, Nam A-R, Bhang D-H, Jung Y-C, et al. TSG-6 in Extracellular Vesicles From Canine Mesenchymal Stem/Stromal is a Major Factor in Relieving DSS-Induced Colitis. PloS One (2020) 15:e0220756. doi: 10.1371/journal.pone.0220756
270. An J-H, Li Q, Bhang D-H, Song W-J, Youn H-Y. TNF-α and INF-γ Primed Canine Stem Cell-Derived Extracellular Vesicles Alleviate Experimental Murine Colitis. Sci Rep (2020) 10:2115. doi: 10.1038/s41598-020-58909-4
271. Blazquez R, Sanchez-Margallo FM, de la Rosa O, Dalemans W, Álvarez V, Tarazona R, et al. Immunomodulatory Potential of Human Adipose Mesenchymal Stem Cells Derived Exosomes on In Vitro Stimulated T Cells. Front Immunol (2014) 5:556. doi: 10.3389/fimmu.2014.00556
272. Del Fattore A, Luciano R, Pascucci L, Goffredo BM, Giorda E, Scapaticci M, et al. Immunoregulatory Effects of Mesenchymal Stem Cell-Derived Extracellular Vesicles on T Lymphocytes. Cell Transplant (2015) 24:2615–27. doi: 10.3727/096368915X687543
273. Zhang B, Yin Y, Lai RC, Tan SS, Choo ABH, Lim SK. Mesenchymal Stem Cells Secrete Immunologically Active Exosomes. Stem Cells Dev (2014) 23:1233–44. doi: 10.1089/scd.2013.0479
274. Roura S, Monguió-Tortajada M, Munizaga-Larroudé M, Clos-Sansalvador M, Franquesa M, Rosell A, et al. Potential of Extracellular Vesicle-Associated TSG-6 From Adipose Mesenchymal Stromal Cells in Traumatic Brain Injury. Int J Mol Sci (2020) 21(18):6761. doi: 10.3390/ijms21186761
275. Li X, Liu L, Yang J, Yu Y, Chai J, Wang L, et al. Exosome Derived From Human Umbilical Cord Mesenchymal Stem Cell Mediates MiR-181c Attenuating Burn-Induced Excessive Inflammation. EBioMedicine (2016) 8:72–82. doi: 10.1016/j.ebiom.2016.04.030
276. Chen W, Huang Y, Han J, Yu L, Li Y, Lu Z, et al. Immunomodulatory Effects of Mesenchymal Stromal Cells-Derived Exosome. Immunol Res (2016) 64:831–40. doi: 10.1007/s12026-016-8798-6
277. Wang G, Yuan J, Cai X, Xu Z, Wang J, Kofi Wiredu Ocansey D, et al. HucMSC-Exosomes Carrying miR-326 Inhibit Neddylation to Relieve Inflammatory Bowel Disease in Mice. Clin Transl Med (2020) 10(2):e113. doi: 10.1002/ctm2.113
278. Yu T, Chu S, Liu X, Li J, Chen Q, Xu M, et al. Extracellular Vesicles Derived From EphB2-Overexpressing Bone Marrow Mesenchymal Stem Cells Ameliorate DSS-Induced Colitis by Modulating Immune Balance. Stem Cell Res Ther (2021) 12:181. doi: 10.1186/s13287-021-02232-w
279. Yang J, Liu X-X, Fan H, Tang Q, Shou Z-X, Zuo D-M, et al. Extracellular Vesicles Derived From Bone Marrow Mesenchymal Stem Cells Protect Against Experimental Colitis via Attenuating Colon Inflammation, Oxidative Stress and Apoptosis. PloS One (2015) 10:e0140551. doi: 10.1371/journal.pone.0140551
280. Cao L, Xu H, Wang G, Liu M, Tian D, Yuan Z. Extracellular Vesicles Derived From Bone Marrow Mesenchymal Stem Cells Attenuate Dextran Sodium Sulfate-Induced Ulcerative Colitis by Promoting M2 Macrophage Polarization. Int Immunopharmacol (2019) 72:264–74. doi: 10.1016/j.intimp.2019.04.020
281. Heidari N, Abbasi-Kenarsari H, Namaki S, Baghaei K, Zali MR, Khaligh SG, et al. Adipose-Derived Mesenchymal Stem Cell-Secreted Exosome Alleviates Dextran Sulfate Sodium-Induced Acute Colitis by Treg Cell Induction and Inflammatory Cytokine Reduction. J Cell Physiol (2021) 236:5906–20. doi: 10.1002/jcp.30275
282. Mao F, Wu Y, Tang X, Kang J, Zhang B, Yan Y, et al. Exosomes Derived From Human Umbilical Cord Mesenchymal Stem Cells Relieve Inflammatory Bowel Disease in Mice. BioMed Res Int (2017) 2017:e5356760. doi: 10.1155/2017/5356760
283. Chen Q, Duan X, Xu M, Fan H, Dong Y, Wu H, et al. BMSC-EVs Regulate Th17 Cell Differentiation in UC via H3k27me3. Mol Immunol (2020) 118:191–200. doi: 10.1016/j.molimm.2019.12.019
284. Sun D, Cao H, Yang L, Lin L, Hou B, Zheng W, et al. MiR-200b in Heme Oxygenase-1-Modified Bone Marrow Mesenchymal Stem Cell-Derived Exosomes Alleviates Inflammatory Injury of Intestinal Epithelial Cells by Targeting High Mobility Group Box 3. Cell Death Dis (2020) 11:1–18. doi: 10.1038/s41419-020-2685-8
285. Wu H, Fan H, Shou Z, Xu M, Chen Q, Ai C, et al. Extracellular Vesicles Containing miR-146a Attenuate Experimental Colitis by Targeting TRAF6 and IRAK1. Int Immunopharmacol (2019) 68:204–12. doi: 10.1016/j.intimp.2018.12.043
286. Yang S, Liang X, Song J, Li C, Liu A, Luo Y, et al. A Novel Therapeutic Approach for Inflammatory Bowel Disease by Exosomes Derived From Human Umbilical Cord Mesenchymal Stem Cells to Repair Intestinal Barrier via TSG-6. Stem Cell Res Ther (2021) 12:315. doi: 10.1186/s13287-021-02404-8
287. Li Q-C, Liang Y, Su Z-B. Prophylactic Treatment With MSC-Derived Exosomes Attenuates Traumatic Acute Lung Injury in Rats. Am J Physiol Lung Cell Mol Physiol (2019) 316:L1107–17. doi: 10.1152/ajplung.00391.2018
288. Liu J, Chen T, Lei P, Tang X, Huang P. Exosomes Released by Bone Marrow Mesenchymal Stem Cells Attenuate Lung Injury Induced by Intestinal Ischemia Reperfusion via the TLR4/NF-κb Pathway. Int J Med Sci (2019) 16:1238–44. doi: 10.7150/ijms.35369
289. Mizuta Y, Akahoshi T, Guo J, Zhang S, Narahara S, Kawano T, et al. Exosomes From Adipose Tissue-Derived Mesenchymal Stem Cells Ameliorate Histone-Induced Acute Lung Injury by Activating the PI3K/Akt Pathway in Endothelial Cells. Stem Cell Res Ther (2020) 11:508. doi: 10.1186/s13287-020-02015-9
290. Potter DR, Miyazawa BY, Gibb SL, Deng X, Togaratti PP, Croze RH, et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Pulmonary Vascular Permeability and Lung Injury Induced by Hemorrhagic Shock and Trauma. J Trauma Acute Care Surg (2018) 84:245–56. doi: 10.1097/TA.0000000000001744
291. Stone ML, Zhao Y, Robert Smith J, Weiss ML, Kron IL, Laubach VE, et al. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Lung Ischemia-Reperfusion Injury and Enhance Reconditioning of Donor Lungs After Circulatory Death. Respir Res (2017) 18:212. doi: 10.1186/s12931-017-0704-9
292. Haga H, Yan IK, Borrelli DA, Matsuda A, Parasramka M, Shukla N, et al. Extracellular Vesicles From Bone Marrow–Derived Mesenchymal Stem Cells Protect Against Murine Hepatic Ischemia/Reperfusion Injury. Liver Transpl (2017) 23:791–803. doi: 10.1002/lt.24770
293. Yao J, Zheng J, Cai J, Zeng K, Zhou C, Zhang J, et al. Extracellular Vesicles Derived From Human Umbilical Cord Mesenchymal Stem Cells Alleviate Rat Hepatic Ischemia-Reperfusion Injury by Suppressing Oxidative Stress and Neutrophil Inflammatory Response. FASEB J (2019) 33:1695–710. doi: 10.1096/fj.201800131RR
294. Nong K, Wang W, Niu X, Hu B, Ma C, Bai Y, et al. Hepatoprotective Effect of Exosomes From Human-Induced Pluripotent Stem Cell–Derived Mesenchymal Stromal Cells Against Hepatic Ischemia-Reperfusion Injury in Rats. Cytotherapy (2016) 18:1548–59. doi: 10.1016/j.jcyt.2016.08.002
295. Xie K, Liu L, Chen J, Liu F. Exosomes Derived From Human Umbilical Cord Blood Mesenchymal Stem Cells Improve Hepatic Ischemia Reperfusion Injury via Delivering miR-1246. Cell Cycle (2019) 18:3491–501. doi: 10.1080/15384101.2019.1689480
296. Xie K, Liu L, Chen J, Liu F. Exosomal miR-1246 Derived From Human Umbilical Cord Blood Mesenchymal Stem Cells Attenuates Hepatic Ischemia Reperfusion Injury by Modulating T Helper 17/Regulatory T Balance. IUBMB Life (2019) 71:2020–30. doi: 10.1002/iub.2147
297. Zheng J, Lu T, Zhou C, Cai J, Zhang X, Liang J, et al. Extracellular Vesicles Derived From Human Umbilical Cord Mesenchymal Stem Cells Protect Liver Ischemia/Reperfusion Injury by Reducing CD154 Expression on CD4+ T Cells via CCT2. Adv Sci (2020) 7:1903746. doi: 10.1002/advs.201903746
298. Anger F, Camara M, Ellinger E, Germer C-T, Schlegel N, Otto C, et al. Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles Improve Liver Regeneration After Ischemia Reperfusion Injury in Mice. Stem Cells Dev (2019) 28:1451–62. doi: 10.1089/scd.2019.0085
299. Grange C, Tapparo M, Bruno S, Chatterjee D, Quesenberry PJ, Tetta C, et al. Biodistribution of Mesenchymal Stem Cell-Derived Extracellular Vesicles in a Model of Acute Kidney Injury Monitored by Optical Imaging. Int J Mol Med (2014) 33:1055–63. doi: 10.3892/ijmm.2014.1663
300. Gatti S, Bruno S, Deregibus MC, Sordi A, Cantaluppi V, Tetta C, et al. Microvesicles Derived From Human Adult Mesenchymal Stem Cells Protect Against Ischaemia-Reperfusion-Induced Acute and Chronic Kidney Injury. Nephrol Dial Transplant (2011) 26:1474–83. doi: 10.1093/ndt/gfr015
301. Ren Y, Chen Y, Zheng X, Wang H, Kang X, Tang J, et al. Human Amniotic Epithelial Cells Ameliorate Kidney Damage in Ischemia-Reperfusion Mouse Model of Acute Kidney Injury. Stem Cell Res Ther (2020) 11:410. doi: 10.1186/s13287-020-01917-y
302. Lin K-C, Yip H-K, Shao P-L, Wu S-C, Chen K-H, Chen Y-T, et al. Combination of Adipose-Derived Mesenchymal Stem Cells (ADMSC) and ADMSC-Derived Exosomes for Protecting Kidney From Acute Ischemia–Reperfusion Injury. Int J Cardiol (2016) 216:173–85. doi: 10.1016/j.ijcard.2016.04.061
303. Collino F, Bruno S, Incarnato D, Dettori D, Neri F, Provero P, et al. AKI Recovery Induced by Mesenchymal Stromal Cell-Derived Extracellular Vesicles Carrying MicroRNAs. JASN (2015) 26:2349–60. doi: 10.1681/ASN.2014070710
304. Zou X, Gu D, Zhang G, Zhong L, Cheng Z, Liu G, et al. NK Cell Regulatory Property is Involved in the Protective Role of MSC-Derived Extracellular Vesicles in Renal Ischemic Reperfusion Injury. Hum Gene Ther (2016) 27:926–35. doi: 10.1089/hum.2016.057
305. Kilpinen L, Impola U, Sankkila L, Ritamo I, Aatonen M, Kilpinen S, et al. Extracellular Membrane Vesicles From Umbilical Cord Blood-Derived MSC Protect Against Ischemic Acute Kidney Injury, a Feature That is Lost After Inflammatory Conditioning. J Extracell Vesicles (2013) 2:21927. doi: 10.3402/jev.v2i0.21927
306. Bruno S, Tapparo M, Collino F, Chiabotto G, Deregibus MC, Soares Lindoso R, et al. Renal Regenerative Potential of Different Extracellular Vesicle Populations Derived From Bone Marrow Mesenchymal Stromal Cells. Tissue Eng Part A (2017) 23:1262–73. doi: 10.1089/ten.TEA.2017.0069
307. Zou X, Gu D, Xing X, Cheng Z, Gong D, Zhang G, et al. Human Mesenchymal Stromal Cell-Derived Extracellular Vesicles Alleviate Renal Ischemic Reperfusion Injury and Enhance Angiogenesis in Rats. Am J Transl Res (2016) 8:4289–99.
308. Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr Protoc Immunol (2014) 104:15.25.1–15.25.14. doi: 10.1002/0471142735.im1525s104
309. Matthay MA, Calfee CS, Zhuo H, Thompson BT, Wilson JG, Levitt JE, et al. Treatment With Allogeneic Mesenchymal Stromal Cells for Moderate to Severe Acute Respiratory Distress Syndrome (START Study): A Randomised Phase 2a Safety Trial. Lancet Respir Med (2019) 7:154–62. doi: 10.1016/S2213-2600(18)30418-1
310. Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM, et al. Mesenchymal Stem Cells Use Extracellular Vesicles to Outsource Mitophagy and Shuttle microRNAs. Nat Commun (2015) 6:8472. doi: 10.1038/ncomms9472
311. Damania A, Jaiman D, Teotia AK, Kumar A. Mesenchymal Stromal Cell-Derived Exosome-Rich Fractionated Secretome Confers a Hepatoprotective Effect in Liver Injury. Stem Cell Res Ther (2018) 9:31. doi: 10.1186/s13287-017-0752-6
312. Fazekas B, Griffin MD. Mesenchymal Stromal Cell–Based Therapies for Acute Kidney Injury: Progress in the Last Decade. Kidney Int (2020) 97:1130–40. doi: 10.1016/j.kint.2019.12.019
313. Shi N, Wu M-P. Apolipoprotein A-I Attenuates Renal Ischemia/Reperfusion Injury in Rats. J BioMed Sci (2008) 15:577–83. doi: 10.1007/s11373-008-9258-7
314. Wiklander OPB, Nordin JZ, O’Loughlin A, Gustafsson Y, Corso G, Mäger I, et al. Extracellular Vesicle In Vivo Biodistribution is Determined by Cell Source, Route of Administration and Targeting. J Extracell Vesicles (2015) 4:26316. doi: 10.3402/jev.v4.26316
Keywords: mesenchymal stromal cell, extracellular vesicles, inflammation, traumatic hemorrhagic shock, multi-organ failure, acute injury
Citation: Valade G, Libert N, Martinaud C, Vicaut E, Banzet S and Peltzer J (2021) Therapeutic Potential of Mesenchymal Stromal Cell-Derived Extracellular Vesicles in the Prevention of Organ Injuries Induced by Traumatic Hemorrhagic Shock. Front. Immunol. 12:749659. doi: 10.3389/fimmu.2021.749659
Received: 29 July 2021; Accepted: 06 September 2021;
Published: 29 September 2021.
Edited by:
Paula Barbim Donate, University of São Paulo, BrazilReviewed by:
Paulus Mrass, University of New Mexico, United StatesCopyright © 2021 Valade, Libert, Martinaud, Vicaut, Banzet and Peltzer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juliette Peltzer, anVsaWV0dGUucGVsdHplckB3YW5hZG9vLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.