 Hayder M. Al-kuraishy1Ali Ismail Al-Gareeb1Gomaa Mostafa-Hedeab2*Keneth Iceland Kasozi3,4*Gerald Zirintunda5Akhmed Aslam6Mamdouh Allahyani7Susan Christina Welburn3,8Gaber El-Saber Batiha9
Hayder M. Al-kuraishy1Ali Ismail Al-Gareeb1Gomaa Mostafa-Hedeab2*Keneth Iceland Kasozi3,4*Gerald Zirintunda5Akhmed Aslam6Mamdouh Allahyani7Susan Christina Welburn3,8Gaber El-Saber Batiha9- 1Department of Clinical Pharmacology and Medicine, College of Medicine, ALmustansiriyia University, Baghdad, Iraq
- 2Pharmacology Department, Health Sciences Research Unit, Medical College, Jouf University, Sakaka, Saudi Arabia
- 3Infection Medicine, Deanery of Biomedical Sciences, College of Medicine and Veterinary Medicine, The University of Edinburgh, Edinburgh, United Kingdom
- 4School of Medicine, Kabale Unviersity, Kabale, Uganda
- 5Department of Animal Production and Management, Faculty of Agriculture and Animal Sciences, Busitema University, Tororo, Uganda
- 6Laboratory Medicine, Faculty of Applied Medical Sciences, Umm Al-Qura University, Makkah, Saudi Arabia
- 7Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Taif University, Taif, Saudi Arabia
- 8Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, Zhejiang University, Haining, China
- 9Department of Pharmacology and Therapeutics, Faculty of Veterinary Medicine, Damanhour University, Damanhour, Egypt
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a causative virus in the development of coronavirus disease 2019 (Covid-19) pandemic. Respiratory manifestations of SARS-CoV-2 infection such as acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) leads to hypoxia, oxidative stress, and sympatho-activation and in severe cases leads to sympathetic storm (SS). On the other hand, an exaggerated immune response to the SARS-CoV-2 invasion may lead to uncontrolled release of pro-inflammatory cytokine development of cytokine storm (CS). In Covid-19, there are interactive interactions between CS and SS in the development of multi-organ failure (MOF). Interestingly, cutting the bridge between CS and SS by anti-inflammatory and anti-adrenergic agents may mitigate complications that are induced by SARS-CoV-2 infection in severely affected Covid-19 patients. The potential mechanisms of SS in Covid-19 are through different pathways such as hypoxia, which activate the central sympathetic center through carotid bodies chemosensory input and induced pro-inflammatory cytokines, which cross the blood-brain barrier and activation of the sympathetic center. β2-receptors signaling pathway play a crucial role in the production of pro-inflammatory cytokines, macrophage activation, and B-cells for the production of antibodies with inflammation exacerbation. β-blockers have anti-inflammatory effects through reduction release of pro-inflammatory cytokines with inhibition of NF-κB. In conclusion, β-blockers interrupt this interaction through inhibition of several mediators of CS and SS with prevention development of neural-cytokine loop in SARS-CoV-2 infection. Evidence from this study triggers an idea for future prospective studies to confirm the potential role of β-blockers in the management of Covid-19.
Introduction
It is well-known in recent times that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a causative virus in the development of coronavirus disease 2019 (Covid-19) pandemic (1). This disease was initially documented in the Wuhan province of China (2). The SARS-COV-2 virus is highly infective with about 15% of the patients require hospitalization and 5% may need intensive care (3). Approximately half of the Covid-19 patients taken to intensive care units (ICU) die due to various complications associated with acute respiratory distress syndrome (ARDS) (4). The severe Covid-19 complications include respiratory failure, cardiac arrhythmias, acute kidney injury, and stroke (5). Respiratory failure is a result of acute lung injury (ALI) and ARDS (6). The respiratory system signs lead to hypoxia, oxidative stress, and sympatho-activation and in severe cases lead to sympathetic storm (SS) (7). SS is characterized by recurrent episodes of hyperhidrosis, hypertension, tachycardia, tachypnea, and hyperthermia (8).
On the other hand, exaggerated immune response to the SARS-CoV-2 invasion may lead to the production of various inflammatory substances (9). There may be an uncontrolled release of pro-inflammatory cytokines such as interleukins (IL-6, IL-1β, IL-8), tumor necrosis factor-alpha (TNF-α), and chemokines that together lead to the development of cytokine storm (CS) (10).
In Covid-19, there is interactive interaction between CS and SS in the development of multi-organ failure (MOF) and life-threatening complications (11). However, cutting the bridge between CS and SS by anti-inflammatory and anti-adrenergic agents may mitigate complications that are induced by SARS-CoV-2 infection in severely affected Covid-19 patients (12).
Anti-adrenergic β-blockers are a class of medications used in the management of cardiovascular disorders such as arrhythmia, acute coronary syndrome, and hypertension as well as other disorders like tremor and anxiety (13). β-blockers are either selective (block β1 or β2) or non-selective (block both β1 and β2). β-blockers reduce sympathetic stimulation-mediated by adrenalin and noradrenalin on β receptors (13). β1 receptors are located mainly on the heart and kidney while, β2 receptors are expressed primarily in the lungs, vascular smooth muscles, and gastrointestinal tract (14).
The objective of the present study was to summarize the current updates in the discussed topic and demonstrates the potential guidance for the usage of beta-blocker on the SS and CS in Covid-19.
β-Blockers and Sympathetic Storm in Covid-19
It has been reported that β-blockers such as propranolol, metoprolol, and labetalol are effective in the management of SS by mitigation of autonomic dysregulation and sympathetic spells in patients with thalamic injury (15). SS is due to increased activity of the sympathetic nervous system (SNS) at the expense of the parasympathetic nervous system (PSNS) due to brain injury (16). The severity of traumatic brain injury (TBI) correlates with the level of sympathetic activation. The implication is that early use of β-blockers in TBI may attenuate the development of SS (17). Luostarinen et al., a retrospective study showed that TBI in Covid-19 patients did not affect disease severity (18). About 55% of hospitalized Covid-19 patients develop neurological signs (19). These signs may remain for about three months following SARS-CoV-2 infection, suggesting the development of latent brain injury (20).
Invasion of the central nervous system (CNS) by SARS-CoV-2 has remained speculative (21). However, brain injury in Covid-19 patients might be due to the direct effect of SARS-CoV-2. Covid-19 may lead to brain injury because it manifests with hypoxemia, autoimmune response, thrombosis, and CS (22). Notably, involvement of the peripheral nervous system (PNS) and autonomic nervous system (ANS) results in an imbalance between SNS and PSNS with the development of SS (23). The imbalance of the SNS/PSNS axis of ANS may affect the release of pro-inflammatory cytokines and immune-inflammatory response during Covid-19 (24). In this context, high circulating catecholamine levels may reflect sympathetic-mediated neutrophilia and T cell dysfunction in Covid-19 due to SS (25). Thus, the development SS in Covid-19 is through central and peripheral effects of SARS-CoV-2 that increase sympathetic outflow (Figure 1).
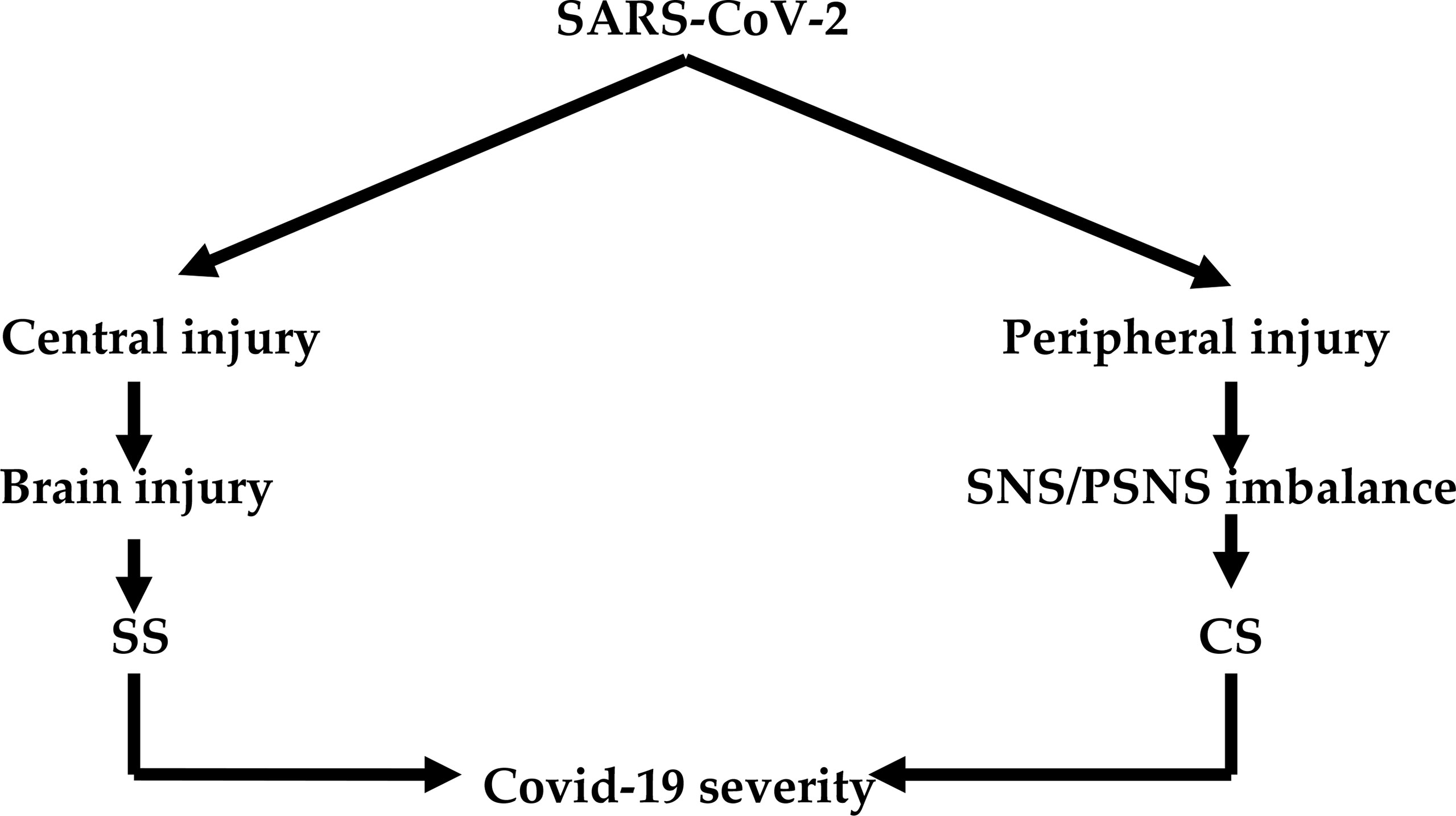
Figure 1 SARS-CoV-2 infection and development of sympathetic and cytokine storms: Central effect of SARS-CoV-2 leads to brain injury and development of sympathetic storm (SS). The peripheral effect of SARS-CoV-2 leads to induction imbalance between the sympathetic nervous system (SNS) and parasympathetic nervous system (PSNS) and development of cytokine storm (CS). Both SS and CS lead to Covid-19 severity.
The potential mechanisms of SS in Covid-19 are through the three pathways including; ALI/ARDS-induced hypoxia, which activate the central sympathetic center through carotid bodies chemosensory input (26). SARS-CoV-2-induced neuroinflammation directly activates sympathetic centers like locus coeruleus (LC), rostral ventrolateral medulla (RVLM), and hypothalamic paraventricular nucleus (HPVN) (27). SARS-CoV-2-induced pro-inflammatory cytokines, which cross the blood-brain barrier and activate sympathetic center (Figure 2) (28).
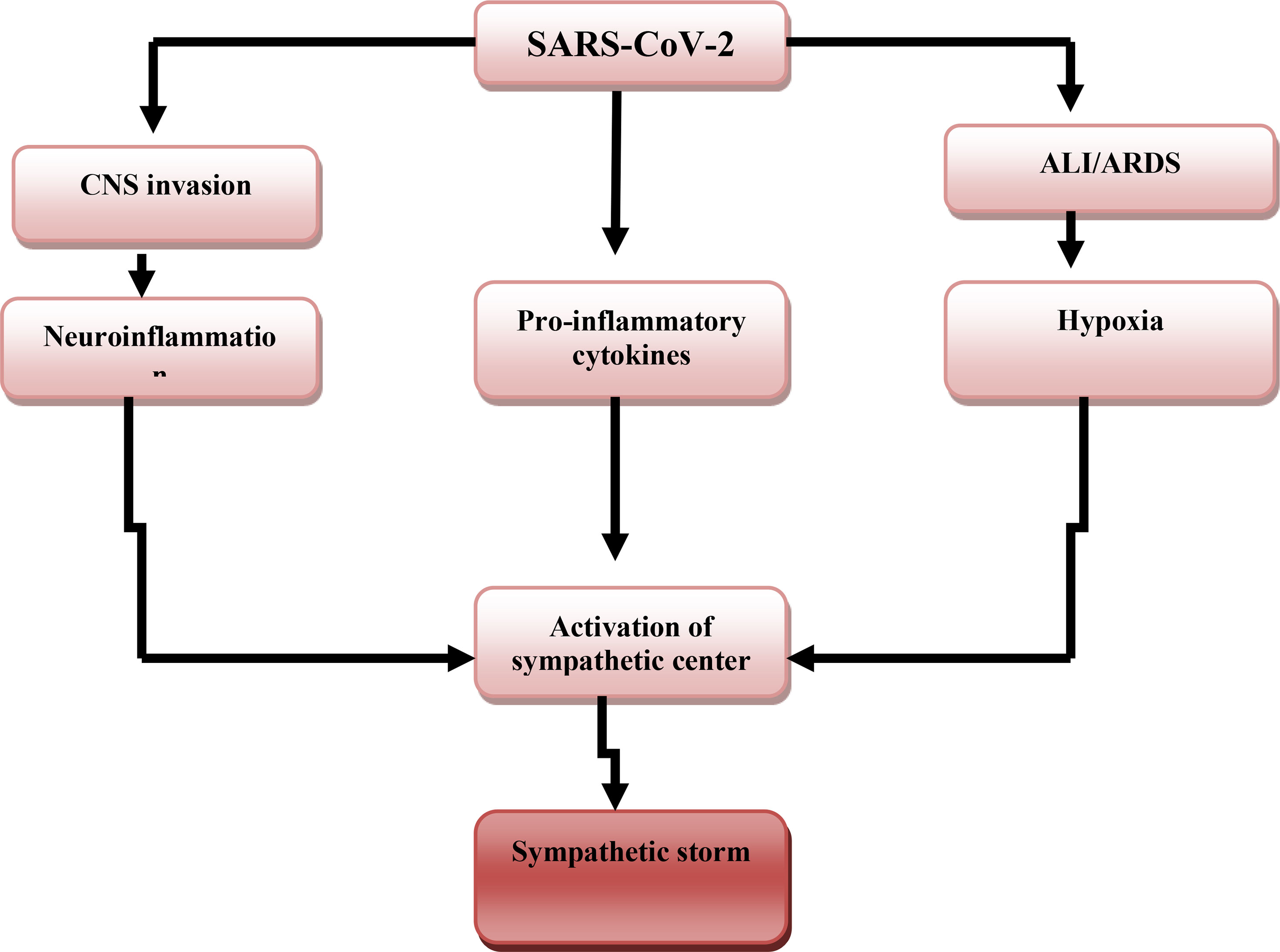
Figure 2 SARS-CoV-2-induced sympathetic storm. SARS-CoV-2 acute lung injury (ALI) and acute respiratory distress syndrome (ARDS)-induced hypoxia, SARS-CoV-2-induced neuroinflammation, and release of pro-inflammatory cytokines activate the central sympathetic center with the development of the sympathetic storm.
Moreover, comorbidities that induce a high sympathetic activity such as diabetes mellitus and hypertension may exacerbate cardiac arrhythmia, cardiac arrest, and acute myocardial infarction (29). Development of Covid-19 severity is linked with SS and vagal suppression that culminates into the CS (30). It is thus suggested that vagal stimulation might be valuable in Covid-19 patients through modulation of SS and release of pro-inflammatory cytokines (31). It has been shown that cholinergic agonists inhibit inflammation via suppression of inflammatory signals such as high mobility group protein 1 (HMGB1) (32). Furthermore, a molecular docking study observed that the nicotinic acetylcholine receptor (nAChR) may be a potential binding receptor for SARS-CoV-2 (32). Inhibition of nAChR by SARS-CoV-2 leads to inhibition of PSNS and exaggeration of SNS with subsequent progression of CS due to inhibition of vagal anti-inflammatory mediated by diminution of nAChR activity (33). Likewise, α-1 and β-receptor antagonists have valuable effects in Covid-19 via lessening of SS and development of CS (34). For that reason, β-blockers reduce sympathetic stimulation and inhibit the interaction between SARS-CoV-2 and receptor binding sites of angiotensin-converting enzyme 2 (ACE2) and CD147 (35) (Figure 3).
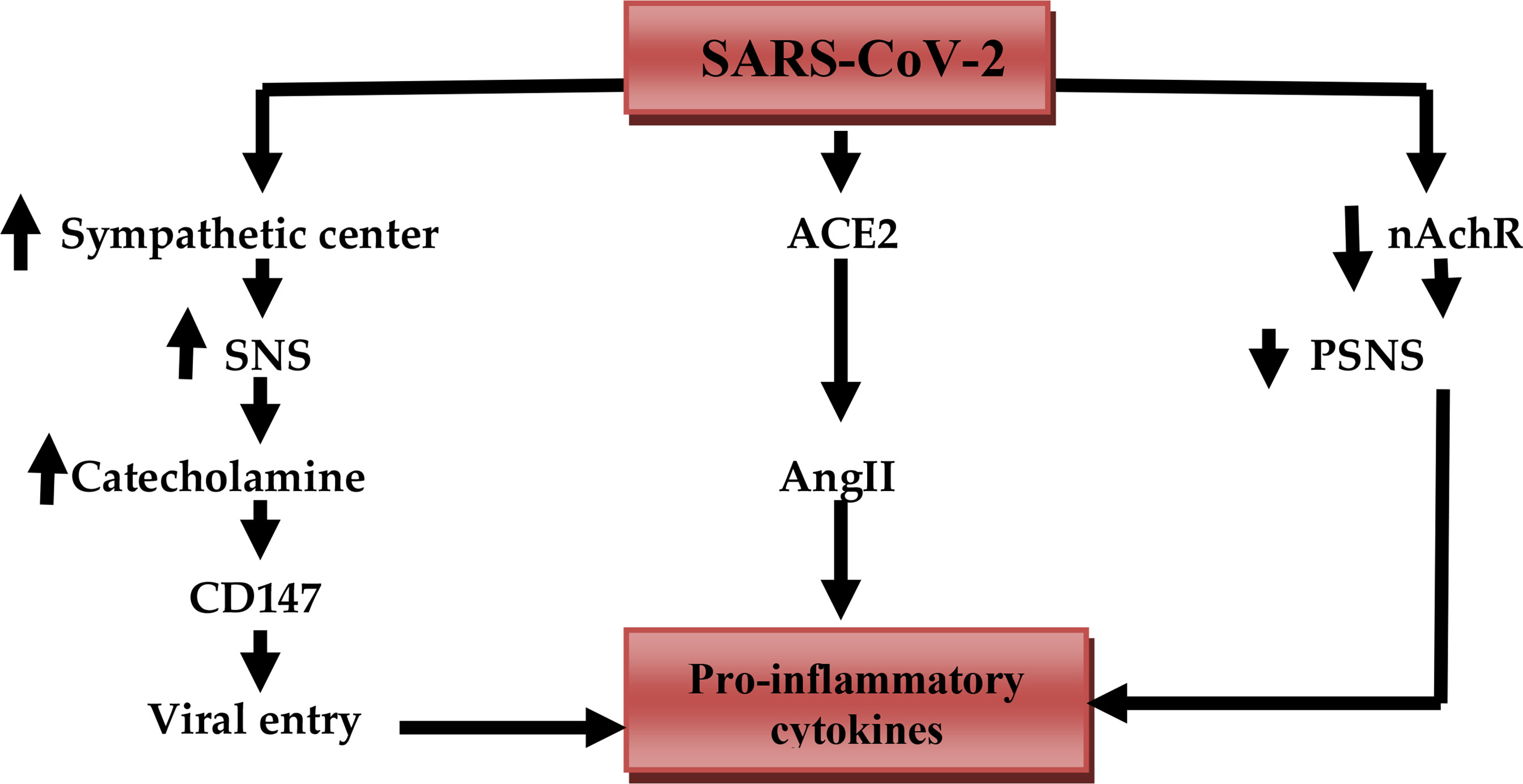
Figure 3 SARS-CoV-2 and release of pro-inflammatory cytokines: SARS-CoV-2 activates the sympathetic center, increases activity of the sympathetic nervous system (SNS), the release of catecholamine, which activates the expression of CD147 that increase viral entry. SARS-CoV-2 downregulates ACE2 which increases circulating angiotensin II (AngII). SARS-CoV-2 inhibits anti-inflammatory nicotinic acetylcholine receptor (nAChR) with reduced activity of the parasympathetic nervous system (PSNS). These changes together trigger the release of pro-inflammatory cytokines.
β-blockers reduce SS-induced cardiac arrhythmia, and destabilization of coronary plaques due to high circulating catecholamine, which cause positive inotropic and chronotropic effects through β1 receptor (36). As well, β-blockers reduce cardiac injury caused by sympathetic over-activation. Cardiomyocyte inflammation results from induction of local TNF-α and IL-6 expression (37).
Moreover, binding of SARS-CoV-2 to the ACE2 leads to deregulation of the renin-angiotensin system (RAS) with upregulation of vasoconstrictor angiotensin II (AngII). There is a co-current down-regulation of vasodilator Ang 1-7 leading to hypertension, sympathetic stimulation, and development of ALI and ARDS (38). β-blockers, therefore, reduce the activity of RAS by inhibiting the release of renin from renal juxtaglomerular cells, and protects the lungs and heart from exaggerated RAS and SS (39). An experimental study by Danukalo et al. illustrated that AngII increases firing and activity of LC with the propagation of sympathetic activation and hypertension in rats (40). Besides, β-blockers like propranolol modulate the activity and sensitivity of LC and prevent sympathetic stimulation in patients with migraines (41). Indeed, non-selective and lipophilic β-blockers like propranolol have a potent effect in the suppression of catecholamine from presynaptic adrenergic neurons through inhibition of excitatory presynaptic β2 autoreceptor (42). Taken together, β-blockers reduce the development of SS directly or indirectly through suppression of the central effect of AngII.
β-blockers prevent SS-induced ALI as high circulating catecholamines are linked with the development of ALI/ARDS (43). In addition, β-blockers prevent ALI through modulation of neutrophilia, lymphopenia, and the release of pro-inflammatory cytokines (44). In a retrospective study that involved 651 patients in ICU with sepsis, the patients on chronic β-blockers therapy had a lower risk of sepsis-induced ARDS. The patients required less mechanical ventilation due to upregulation of protective alveolar β2 adrenoceptors (45). Likewise in a randomized controlled clinical trial of 314 patients with acute respiratory failure in the ICU showed that patients on β-blockers therapy had a lower in-hospital mortality rate (46). Contrastingly, Mutlu et al., observed that β2-agonists improve alveolar fluid clearance in patients with pulmonary edema through up-regulation of alveolar epithelial sodium active transport (47). A study that involved 79 patients with ALI is associated with impairment of pulmonary alveolar clearance rate (48). These findings imply that selective β1-blockers are safer than non-selective ones in prevention of β2 adrenoceptors’ beneficial effect.
The findings support the favorable effects of β-blockers in the mitigation of SS-induced ALI/ARDS in severely affected Covid-19 patients (Figure 4).
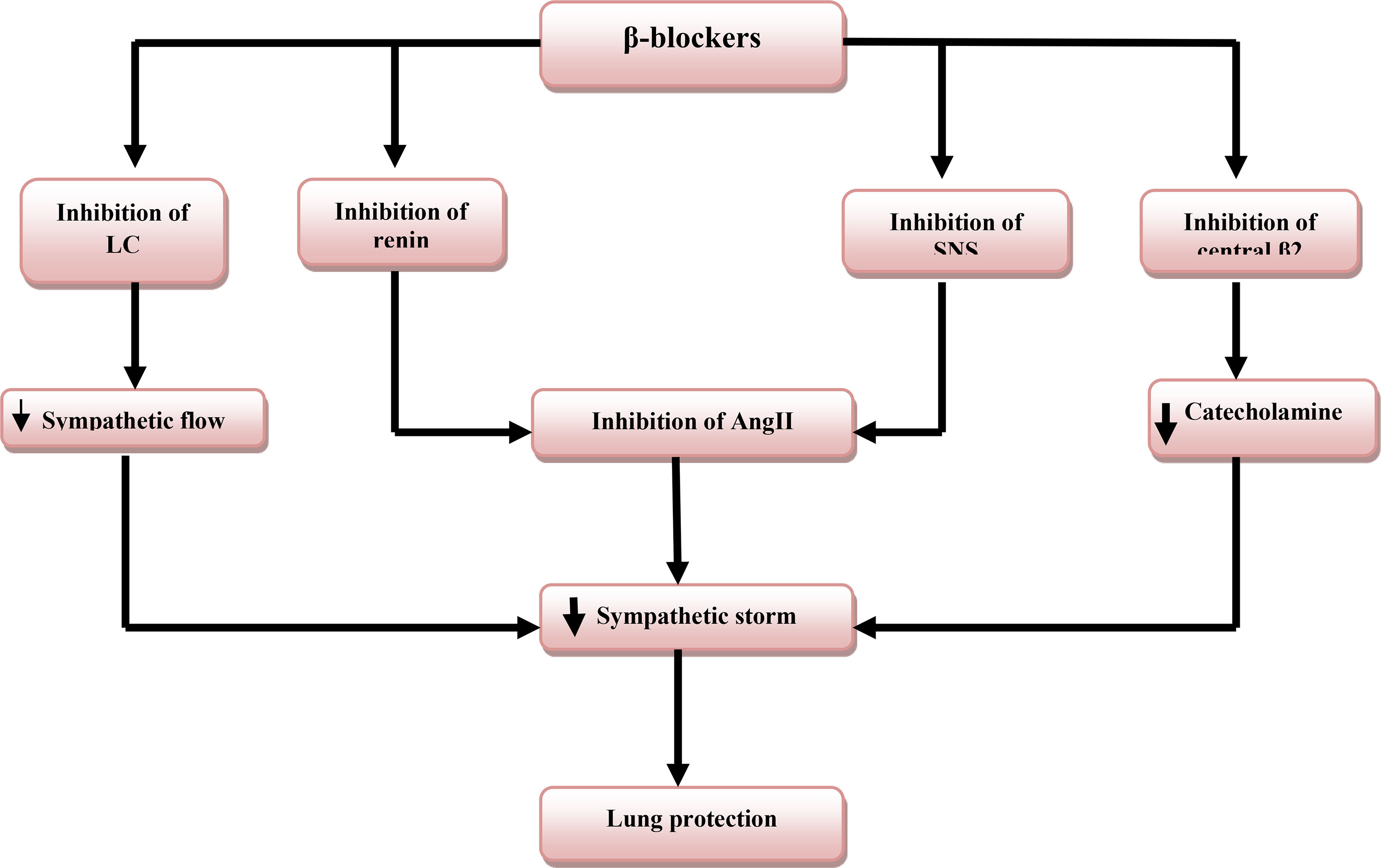
Figure 4 Role of β-blockers in lung protection: β-blockers inhibit sympathetic nervous system (SNS), renin release, locus coeruleus (LC) activity, and central presynaptic β2 receptors that decrease the release of catecholamine and angiotensin II (AngII) with subsequent inhibition of sympathetic storm and lung protection.
β-Blockers and Cytokine Storm in Covid-19
CS or cytokine releasing syndrome is a systemic inflammatory syndrome characterized by high circulating pro-inflammatory cytokines. CS also involves abnormal immunological hyperactivation involving pathogens, autoimmune reactions, and cancers (49). In Covid-19 pro-inflammatory cytokines including IL-6, TNF-α, IL-1β, and macrophage inflammatory protein (MIP) are elevated. Plasmablasts, CD4 and CD8, and other immune cells are also activated in CS (50). The interaction between SARS-CoV-2 and ACE2 on the affected cells induces cell damage. The interaction also causes the release of damage and inflammatory signals. The mentioned signals activate macrophages for the release of chemokines and pro-inflammatory cytokines that trigger T cells recruitment and activation (51). In addition, SARS-CoV-2 spike protein can activate CD147 and toll-like receptor 4 (TLR4) leading to stimulation of myeloid differentiation 88(MyD88) pathway. Myeloid differentiation provokes nuclear factor kappa B (NF-κB), which stimulates the release of pro-inflammatory cytokines and the development of CS (52) (Figure 5).
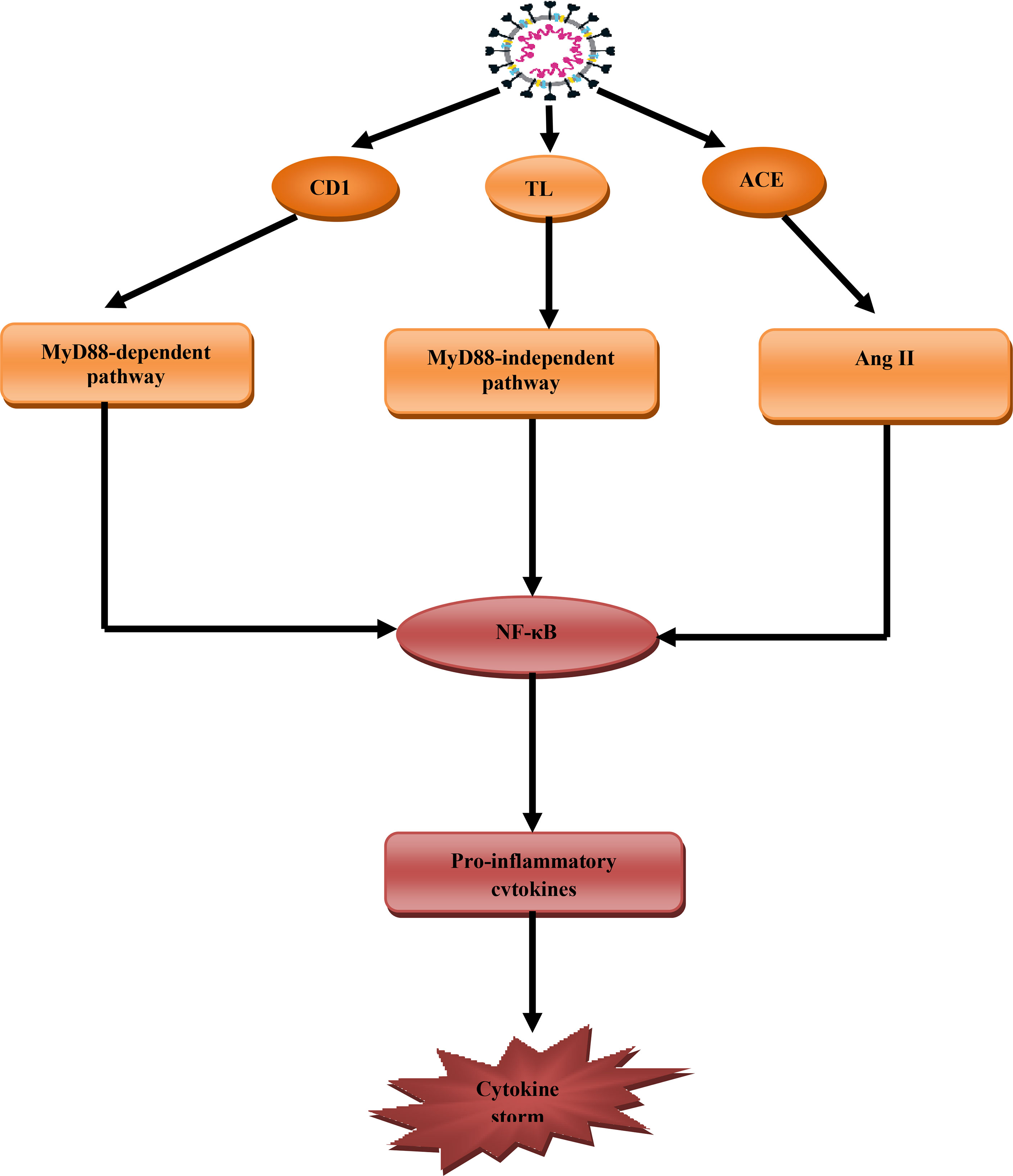
Figure 5 Role of SARS-CoV-2 in the development of cytokine storm (CS): SARS-CoV-2 through activation of CD147 activate myeloid differentiation 88 (MyD88), through toll-like receptor 4 (TLR4) and ACE2 activate angiotensin II (AngII) that together trigger NF-κB pathway, which stimulates the release of pro-inflammatory pathway and development of cytokine storm.
Interestingly, adrenergic receptors are linked with immunological disorders and the development of immune-mediated ALI since 90% of β-receptors are located in the lung alveoli with β2 predominant in 70% (53). β2 receptors are expressed by all immune cells especially macrophages, dendritic cells, and lymphocytes (54). Therefore, the β2-receptors signaling pathway play a crucial role in the production of pro-inflammatory cytokines, macrophage activation, and B-cells. The B-cells are involved in the production of antibodies which exacerbates the inflammation (55). Thus, β2-agonists may induce alveolar inflammation and pulmonary microvascular thrombosis via the accelerated release of IL-6 (56). Nossent et al. observed that β2-agonists increase the risk of venous thrombosis through activation of von Willebrand factor and factor VIII (57). In addition, activation of β2 receptors leads to the generation of reactive oxygen species (ROS) and induction of oxidative stress. Oxidative stress activates the release of IL-6, promotion of Th2 immune response, and inhibition of interferon-gamma (INF-γ) (58).
β-blockers have anti-inflammatory effects through reduction release of IL-6 and TNF-α, with inhibition of NF-κB and signal transduction and activator of transcription 3 (STAT3) (59). These pro-inflammatory cytokines and inflammatory signaling pathways are highly activated in Covid-19 in the progression of CS (60). Therefore, β-blockers may attenuate the development of CS in patients with severe Covid-19 (61). Additionally, β-blockers may reduce SARS-CoV-2-induced coagulopathy and pro-thrombotic complications through inhibition of platelet aggregations and factor VIII (62). β-blockers alleviate endothelial dysfunction and microvascular dysfunction linked with coagulopathy in Covid-19 through suppression of vascular endothelial growth factor (63).
CS is also developed due to activation of nod-like receptor pyrin 3 (NLRP3) inflammasome by SARS-CoV-2 viroporin (64). Gao et al. found that β-blocker nebivolol inhibits NLRP3 inflammasome in obesity-induced vascular remodeling in experimental animals (65). So, β-blockers could have potential benefits in mitigating the progression of SARS-CoV-2-mediated CS. SS with high catecholamine levels activates RAS with induction of AngII-mediated ALI and release of pro-inflammatory cytokines. Therefore β-blockers through inhibition of renin release and suppression of RAS may weaken the release of pro-inflammatory cytokines and the development of CS (66). Furthermore, macrophage activation syndrome (MAS) like disease is developed in severely affected Covid-19 leading to ALI, ARDS, and MOF (67). Xia et al. illustrated that high circulating catecholamine levels are associated with macrophages activation and release of pro-inflammatory cytokines (68). A prospective study involving 32 patients with immune-mediated dilated cardiomyopathy showed that β-blockers therapy reduces pro-inflammatory TNF-α. β-blockers increase anti-inflammatory IL-10 through inhibition of macrophage activation (69). Thus, β-blockers may reduce the development of MAS through inhibition of macrophage activation and release of pro-inflammatory cytokines (70). Nateasan preprinted study summarized the beneficial effects of β-blockers in Covid-19 in some points including that β-blockers improve oxygenation, reduce bronchial secretion, inhibit entry of SARS-CoV-2 through ACE2 and CD147, inhibit the release of pro-inflammatory cytokines, reduce the development of pulmonary edema and ARDS, inhibit the development of endothelial dysfunction and coagulopathy, block proliferation of SARS-CoV-2, and finally suppression of NLRP3 inflammasome and NF-κB signaling (62).
Taken together, according to these findings, β-blockers might have a potential therapeutic modality in the prevention development of CS in Covid-19 (Figure 6).
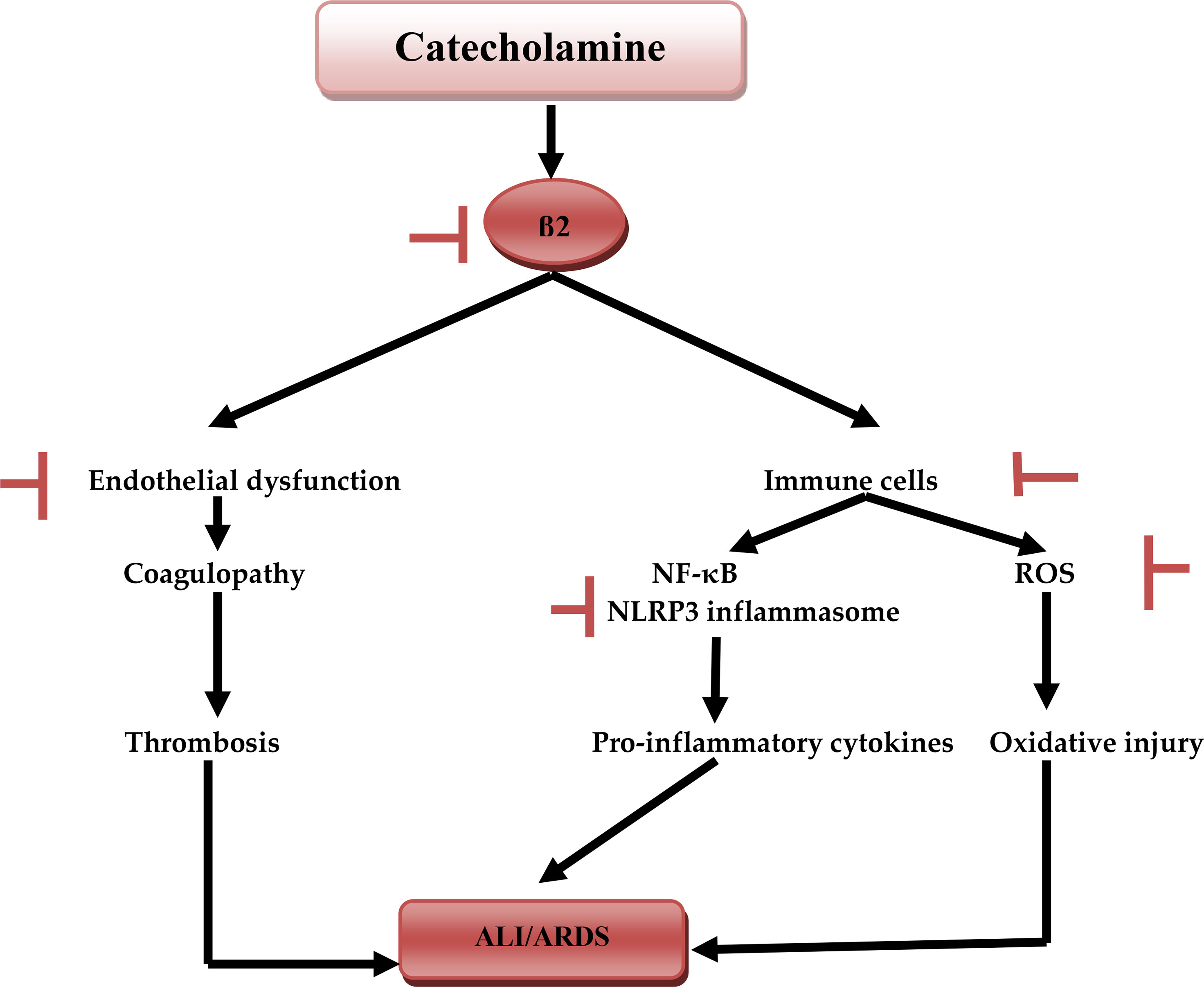
Figure 6 Catecholamine and acute lung injury: catecholamine during SARS-CoV-2-induced sympathetic storm, activates β1, which activates NF-κB and NLRP3 inflammasome of immune cells macrophages and neutrophils induces the release of pro-inflammatory cytokines. Activation of immune cells triggers the generation of reactive oxygen species (ROS), activation of β2 leads to the development of endothelial dysfunction, coagulopathy, and thrombosis. Together these changes cause acute lung injury (ALI) and acute respiratory syndrome (ARDS). Inhibitory effects of β-blockers.
Neuroinflammation, Sympathetic and Cytokine Storms in Covid-19
Covid-19 causes systemic as well as different neurological disorders characterized by anosmia, headache, seizure, cerebrovascular complications, impaired consciousness, psychiatric disorders and dysautonomia (71). These neurological manifestations in Covid-19 are due to direct SARS-CoV-2 cytopathic effects or indirectly due to cerebral endothelial dysfunction and neuroinflammation caused by the cytokine storm (71). In addition, Covid-19 can aggravate neuroinflammation through activation of neurons, microglia, astrocytes and endothelial cells with subsequent increase of cytokines and chemokines in the CNS (72). Similarly, activations of dendritic cells, monocytes/macrophages, mast cells, endothelial and epithelial cells contribute to induction of neuroinflammation by releasing of pro-inflammatory cytokines and development of cytokine storm-induced neuroinflammation (73). Furthermore, Covid-19-induced psychological stress and sympathetic activation can exacerbate neuroinflammation via induction activation of mast cells and other immune cells to release pro-inflammatory cytokines (73). Therefore, there is a neuro-immunopathological mechanistic linkage between neuroinflammation with SS and CS in Covid-19 patients. It has been shown that neuroinflammation together with activated AngII and oxidative stress can provoke activation of sympathetic outflow with development of hypertension (74). Winklewski et al., observed that neuroinflammation plays a crucial role in the neuronal injury and activation of SNS. In turn, overactivated SNS induces pulmonary complications with increased risk of pneumonia, which causes a bystander autoimmune response against the CNS, thereby initiating a vicious cycle (75). Therefore, SARS-CoV-2-induced neuroinflammation could be the potential cause of sympathetic overactivity in severely affected Covid-19 patients (76). Sympathetic overactivity and development of SS may exert noteworthy detrimental effects on Covid-19 patients through action on the CNS and other vital organs (76). In turn, SARS-CoV-2 infection may increase sympathetic discharge through induction of CS, hypoxia and RAS imbalance (76). β-blockers mainly propranolol are effective in reduction of paroxysmal sympathetic overactivity following TBI (77). Furthermore, selective β-blockers can mitigate sympathetic overactivity and SS-induced neuroinflammation following ischemic stroke since up-regulated β2-adrenoceptor anti-inflammatory activity acts against post-stroke neuroinflammation (78). However, Evans et al., experimental study illustrated that β-blockers have pro-inflammatory effects so may increase neuroinflammation by inducing synaptic phagocytosis (79). The chronic use of β-blockers reduces systemic inflammation in patients with chronic liver disorders (80). Also, prolonged use of β-blockers in hypertensive Covid-19 patients does not affect the mortality but decreases risk for development of CS and SS (81). Taken together, these findings give a clue that β-blockers could have potential role in attenuation of neuroinflammation, SS and CS in Covid-19 patients.
Cross talk Between Sympathetic and Cytokine Storms in Covid-19
It is proposed that cortical inhibitory GABAerigic neurons inhibit pre-sympathetic hypothalamic PVN neurons (82). These GABAerigic neurons have a high expression of ACE2 receptors. Therefore down-regulation of ACE2 receptors during SARS-CoV-2 infection may suppress these inhibitory inter-neurons with activation of hypothalamic PVN sympathetic neurons (83). Down-regulation of ACE2 during SARS-CoV-2 infection also augments AngII level, which has a potent stimulatory effect on the central hypothalamic PVN sympathetic neurons (84). Notably, central sympathetic stimulation due to SARS-CoV-2 infection increases circulating catecholamine. Catecholamines activate macrophages and neutrophils for the release of pro-inflammatory cytokines. Activated macrophages and neutrophils also release catecholamines, which act in a paracrine manner for augmentation of pro-inflammatory cytokines release (85). Rlddell speculated that catecholamine acts as a fuel for the activation and boosting of macrophages and neutrophils and the development of CS (86). Indeed, high catecholamine levels interact with pro-inflammatory cytokines in the progression of capillary leak syndrome and the development of MOF (87). These findings confirm the potential nexus between SS and CS in the development of MOF in patients with severe Covid-19 (88). An experimental study showed that interruption of catecholamine synthesis and release by metyrosine inhibits the development of CS in mice induced by T cell targeting antibodies (89).
Furthermore, high catecholamine levels during the development of SS in Covid-19 patients facilitate entry of SARS-CoV-2 via induction expression of CD147. Expressed CD147 causes damage to the lung alveolar basement membrane through activation of matrix metalloproteinase (MMPs) (90). In turn, alveolar membrane injury triggers the release of catecholamine from activated macrophages and neutrophils with the generation of the vicious cycle of injury (91). Thus, inhibition of CD147 may alleviate ALI through disruption of catecholamine-mediated acute inflammatory reactions (92). Hence, β-blockers may reduce pulmonary inflammation and alveolar dysfunction through inhibition of CD147 and MMPs in SARS-CoV-2 infection (93). Inhibition of CD147 leads to significant down-regulation of NF-κB signaling, which is the central pathway for the activation release of pro-inflammatory cytokines (94). Therefore, β-blockers through inhibition of CD147, NF-κB signaling and other inflammatory molecules (95) are potentially considered anti-inflammatory agents and may mitigate Covid-19 severity.
Moreover, CD147 play an important role in the progression of inflammatory and thrombotic pathways by triggering the interaction between platelets, immune and endothelial cells (96). Jin et al., experimental study demonstrated that thrombotic-induced acute ischemic stroke triggers expression and upregulation of CD147 in the brain micro-vessels and circulating platelets and leukocytes causing secondary microthrombosis (97). Therefore, inhibition of CD147 improves acute ischemic stroke by inhibiting thrombo-inflammation (97). CD147 could represent a novel therapeutic target against thrombo-inflammatory disorders. In addition, cyclophilin A stimulates platelets adhesion and thrombus formation through activation of CD147 (98). Thus, inhibition of the cyclophilin A-CD147 interaction attenuates acute pulmonary thrombosis in experimental rats (99). In SARS-CoV-2 infection both cyclophilin A and CD147 are activated with substantial development of pulmonary micro-thrombosis (100, 101). In addition, activation of cyclophilin A and CD147 is associated with cardiac inflammation and fibrosis in patients with heart failure (102). β-blockers improve cardiac function in patients with heart failure via inhibiting expression of cyclophilin A and CD147 (102). Furthermore, CD147 induces expression of vascular endothelial growth factor (VEGF) in different inflammatory disorders (103). Yin et al., found that high VEGF serum levels are linked with neuroinflammation through recruitment of inflammatory cells and activation of brain AngII in Covid-19 patients (104), while Barbieri et al., showed that high endothelial nitric oxide synthase levels associated with VEGF activity in stressed mice (105). It has been reported that β-blockers can reduce expression of VEGF in patients with lung cancer (71). Therefore, β-blockers are able to inhibit development of pulmonary thrombo-embolism and SARS-CoV-2 infection-induced heart failure by inhibiting cyclophilin A-CD147 expression in the platelets and cardiomyocytes respectively with attenuation of neuroinflammation via inhibition of VEGF.
It has also been proposed that α1-blockers like prazosin are effective in the mitigation of CS in Covid-19 through inhibition release of IL-6 (106). Therefore, dual β and α1-blocker like labetalol might be more effective in suppressing the development of CS through complete blocking of catecholamine effects on the immune cells during SS in Covid-19 (107).
Interestingly, β-blockers mainly carvedilol has anti-oxidant effects that are induced by high catecholamine level in patients with heart failure (108). Therefore, β-blockers block the development of oxidative stress during the development of SS and CS in Covid-19 that is associated with various complications like endothelial dysfunction and coagulopathy (109). It has been shown that toxic gas-induced pulmonary alveolar membrane injury triggers cascades for the development of oxidative stress. Oxidative stress then provokes neutrophils and macrophages to release pro-inflammatory cytokines and the development of ALI (110). Similarly, oxidative stress injury in SARS-CoV-2 infection escalates the release of pro-inflammatory cytokines in an oxidative-dependent manner in the development of CS (111). Notably, myeoloperoxidase (MPO) is regarded as a linking marker between oxidative stress and inflammation. High MPO activity is linked with the development of cardiovascular complications (112). MPO is regarded as a natural immune response, which releases hypochloruous acid (HOCI). HOCL competes for oxygen binding at heme molecule of hemoglobin causing heme destruction and release of free iron that cause acute tissue injury through the generation of ROS in Covid-19 (113). This confirms that MPO-induced oxidative stress is regarded as the chief central pathway linking the development of CS and SS in Covid19. β-blockers mostly metoprolol block the activity of MPO and mitigate the development of oxidative stress and further development of sympoatho-cytokine storm (108). Therefore, there is considerable crosstalk between SS and CS in Covid-19 (Figure 7).
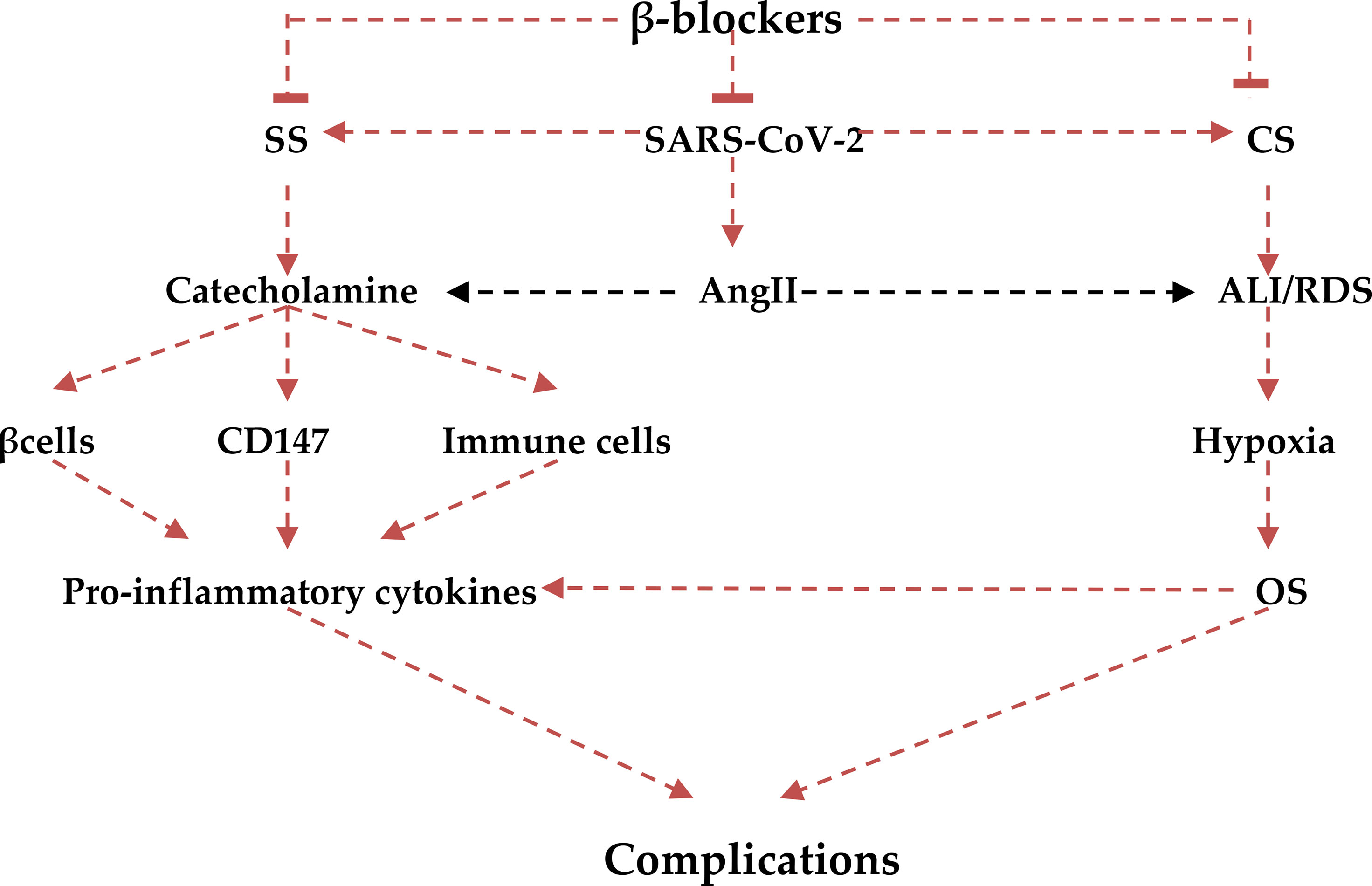
Figure 7 Role of β-blockers in the irruptions of the crosstalk between cytokine (CS) and sympathetic storms(SS): β-blockers block reduces the release of catecholamine and decreases its stimulatory effect on β2, CD147, and immune cells with reduction release of pro-inflammatory cytokines. The anti-inflammatory effects of β-blockers also attenuate CS-induced acute lung injury (ALI) and acute respiratory syndrome (ARDS), development of oxidative stress (OS), and final systemic complications.
The present study revealed β-blockers are effective mitigators of both SS and CS through interruption of catecholamine-β receptors interaction and inhibition release of pro-inflammatory cytokine and development of CS in Covid-19. Despite these beneficial effects of β-blockers in Covid-19 patients, β-blockers have some adverse effects including bradycardia, bronchospasm, peripheral vasoconstrictions, insomnia and depression (114). These adverse effects of β-blockers mainly bronchospasm and peripheral vasoconstrictions may adversely affect the pulmonary function in aged critically ill Covid-19 patients. However, selective β-blockers like celiprolol, nebivolol and carvedilol have less adverse effects due to their selectivity against β1-adrenoceptors (114). Moreover, celiprolol, which is β1-adrenoceptors antagonist and β2-adrenoceptors agonist, is safe in asthmatic patients as it induces bronodilation (115). However, β2-adrenoceptors which expressed on airways and immune cells contribute to exaggerated immune response in the early phase of SARS-CoV-2 infection (61). β2-adrenoceptors on the alveolar macrophages promote secretion of IL-6 and induction of immuno-thrombosis with development of pulmonary thromboembolism (61). Thus, non-selective β-blockers counteract pulmonary thromboembolism by inhibiting prothrombotic response, secretion of VEGF and vascular tone (116). Futhermore, selective inhibitors of β2-adrenoceptors (ICI) blocks the fosforilation of NfKB principal orchestrator of inflammatory response (117).
Therefore, use of non-selective β-blockers in Covid-19 patients in the early phase could be beneficial to attenuate SARS-CoV-2 infection-induced hyperinflammation through inhibition pro-inflammatory cytokines release (61). Likewise, non-selective β-blockers block exaggerated immune response and pulmonary thrombosis by suppressing Th17 activation and procoagulant status respectively. Therefore, despite the adverse effects of β-blockers, non-selective β-blockers appear to be more effective than selective β-blockers in the clinical setting of Covid-19.
We lacked access to clinical data to enforce the concept, however, this study proposes a mechanism of cross-talk between CS and SS in Covid-19 regarding the potential role of β-blockers to guide further studies.
Conclusion
The anti-inflammatory effect of β-blockers through the inhibition release of pro-inflammatory cytokines contributes to the mitigation of CS progression. In addition, β-blockers attenuate the development of SS due to SARS-CoV-2 infection-induced catecholamine release and sympatho-excitation. CS and SS interact at various levels to cause lethal complications in patients with severe COVID-19 like ALI, ARDS, and MOF. However, β-blockers interrupt this interaction through inhibition of several mediators of CS and SS. β-blockers also prevent the development of the neural-cytokine loop in SARS-CoV-2 infection. Evidence from this study triggers an idea for prospective studies to confirm the potential role of β-blockers in the management of Covid-19 in clinical trials.
Author Contributions
HA-k, KK, GM-h, and GB conceptualized the study. HA-k, AA-G, KK, GZ, SW, and GB developed the study design. HA-k, AA-G, GM-h, KK, GZ, AA, MA, SW, and GB collected the data and analyzed it for scientific content. HA-k, KK, GM-h, SW, GB drafted the initial manuscript and revised it for intellectual content while HA-k, AA-G, GM-h, KK, GZ, AA, MA, SW, and GB approved the final version for publication and remain accountable on all aspects of the work therein. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Deanship of Scientific Research at Jouf University under grant Number DSR-2021-01-0374.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Moubarak M, Kasozi KI, Hetta HF, Shaheen HM, Rauf A, Al-Kuraishy HM, et al. The Rise of SARS-CoV-2 Variants and the Role of Convalescent Plasma Therapy for Management of Infections. Life (2021) 11(8):734. doi: 10.3390/life11080734
2. Al-Kuraishy HM, Al-Gareeb AI, Alblihed M, Cruz-Martins N, Batiha GE. COVID-19 and Risk of Acute Ischemic Stroke and Acute Lung Injury in Patients With Type II Diabetes Mellitus: The Anti-Inflammatory Role of Metformin. Front Med (2021) 8:110. doi: 10.3389/fmed.2021.644295
3. Al-Kuraishy HM, Al-Gareeb AI, Almulaiky YQ, Cruz-Martins N, Batiha GE. Role of Leukotriene Pathway and Montelukast in Pulmonary and Extrapulmonary Manifestations of Covid-19: The Enigmatic Entity. Eur J Pharmacol (2021) 15:174196. doi: 10.1016/j.ejphar.2021.174196
4. Al-Kuraishy HM, Al-Gareeb AI, Alqarni M, Cruz-Martins N, El-Saber Batiha G. Pleiotropic Effects of Tetracyclines in the Management of COVID-19: Emerging Perspectives. Front Pharmacol (2021) 12:136. doi: 10.3389/fphar.2021.642822
5. Al-Kuraishy HM, Al-Gareeb AI, Qusty N, Cruz-Martins N, Batiha GE. Sequential Doxycycline and Colchicine Combination Therapy in Covid-19: The Salutary Effects. Pulm Pharmacol Ther (2021) 67:102008. doi: 10.1016/j.pupt.2021.102008
6. Al-Kuraishy HM, Al-Gareeb AI, Qusty N, Alexiou A, Batiha GE. Impact of Sitagliptin in Non-Diabetic Covid-19 Patients. Curr Molec Pharmacol (2021) 1. doi: 10.2174/1874467214666210902115650
7. Lugnier C, Al-Kuraishy HM, Rousseau E. PDE4 Inhibition as a Therapeutic Strategy for Improvement of Pulmonary Dysfunctions in Covid-19 and Cigarette Smoking. Biochem Pharmacol (2021) 185:114431. doi: 10.1016/j.bcp.2021.114431
8. Levy ER, McVeigh U, Ramsay AM. Paroxysmal Sympathetic Hyperactivity (Sympathetic Storm) in a Patient With Permanent Vegetative State. J Palliat Med (2011) 14(12):1355–7. doi: 10.1089/jpm.2010.0444
9. Tufan A, Avanoğlu Güler A, Matucci-Cerinic M. COVID-19, Immune System Response, Hyperinflammation and Repurposing Antirheumatic Drugs. Turk J Med Sci (2020) 50(SI-1):620–32. doi: 10.3906/sag-2004-168
10. Al-Kuraishy HM, Al-Gareeb AI, Cruz-Martins N, Batiha GE. Hyperbilirubinemia in Gilbert Syndrome Attenuates Covid-19 Induced-Metabolic Disturbances: A Case-Report Study. Front Cardiovasc Med (2021) 8:71. doi: 10.3389/fcvm.2021.642181
11. Zaim S, Chong JH, Sankaranarayanan V, Harky A. COVID-19 and Multiorgan Response. Curr Probl Cardiol (2020) 45(8):100618. doi: 10.1016/j.cpcardiol.2020.100618
12. Al-kuraishy H, Al-Gareeb AI, Guerreiro SG, Cruz-Martins N, Batiha GE. COVID-19 in Relation to Hyperglycemia and Diabetes Mellitus. Front Cardiovasc Med (2021) 8:335. doi: 10.3389/fcvm.2021.644095
13. Villanueva C, Albillos A, Genescà J, Garcia-Pagan JC, Calleja JL, Aracil C, et al. β Blockers to Prevent Decompensation of Cirrhosis in Patients With Clinically Significant Portal Hypertension (PREDESCI): A Randomised, Double-Blind, Placebo-Controlled, Multicentre Trial. Lancet (2019) 393(10181):1597–608. doi: 10.1016/S0140-6736(18)31875-0
14. Vilar S, Sobarzo-Sanchez E, Santana L, Uriarte E. Molecular Docking and Drug Discovery in β-Adrenergic Receptors. Curr Med Chem (2017) 24(39):4340–59. doi: 10.2174/0929867324666170724101448
15. Do D, Sheen VL, Bromfield E. Treatment of Paroxysmal Sympathetic Storm With Labetalol. J Neurol Neurosurg Psychiatry (2000) 69(6):832–3. doi: 10.1136/jnnp.69.6.832
16. Bratton SL, Chestnut RM, Ghajar J, McConnell Hammond FF, Harris OA, Hartl R, et al. Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons; Joint Section on Neurotrauma and Critical Care, AANS/CNS. Guidelines for the Management of Severe Traumatic Brain Injury. J Neurotrauma (2007) 24(Suppl 1):S59–64. doi: 10.1089/neu.2007.9987
17. Ammar MA, Hussein NS. Using Propranolol in Traumatic Brain Injury to Reduce Sympathetic Storm Phenomenon: A Prospective Randomized Clinical Trial. Saudi J Anaesth (2018) 12(4):514. doi: 10.4103/sja.SJA_33_18
18. Luostarinen T, Virta J, Satopää J, Bäcklund M, Kivisaari R, Korja M, et al. Intensive Care of Traumatic Brain Injury and Aneurysmal Subarachnoid Hemorrhage in Helsinki During the Covid-19 Pandemic. Acta Neurochir (2020) 162(11):2715–24. doi: 10.1007/s00701-020-04583-4
19. Chou SH, Beghi E, Helbok R. Global Incidence of Neurological Manifestations Among Patients Hospitalized With COVID-19-A Report for the GCS-NeuroCOVID Consortium and the ENERGY Consortium. JAMA Netw Open (2021) 4(5):e2112131. doi: 10.1001/jamanetworkopen.2021.12131
20. DeKosky ST, Kochanek PM, Valadka AB, Clark RS, Chou SH, Au AK, et al. Blood Biomarkers for Detection of Brain Injury in COVID-19 Patients. J Neurotrauma (2021) 38(1):1–43. doi: 10.1089/neu.2020.7332
21. Li H, Xue Q, Xu X. Involvement of the Nervous System in SARS-CoV-2 Infection. Neurotoxicity Res (2020) 38(1):1–7. doi: 10.1007/s12640-020-00219-8
22. Al-Kuraishy HM, Al-Gareeb AI, Qusti S, Alshammari EM, Atanu FO, Batiha GE. Arginine Vasopressin and Pathophysiology of COVID-19: An Innovative Perspective. BioMed Pharmacother (2021) 15:112193. doi: 10.1016/j.biopha.2021.112193
23. Nersesjan V, Amiri M, Lebech AM, Roed C, Mens H, Russell L, et al. Central and Peripheral Nervous System Complications of COVID-19: A Prospective Tertiary Center Cohort With 3-Month Follow-Up. J Neurol (2021) 13:1–9. doi: 10.1007/s00415-020-10380-x
24. Perlmutter A. Immunological Interfaces: The COVID-19 Pandemic and Depression. Front Neurol (2021) 12:657004. doi: 10.3389/fneur.2021.657004
25. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells (2020) 9(6):1383. doi: 10.3390/cells9061383
26. Porzionato A, Emmi A, Stocco E, Barbon S, Boscolo-Berto R, Macchi V, et al. The Potential Role of the Carotid Body in COVID-19. Am J Physiol Lung Cell Mol Physiol (2020) 319(4):L620–6. doi: 10.1152/ajplung.00309.2020
27. Giorgi FS, Biagioni F, Galgani A, Pavese N, Lazzeri G, Fornai F. Locus Coeruleus Modulates Neuroinflammation in Parkinsonism and Dementia. Int J Mol Sci (2020) 21(22):8630. doi: 10.3390/ijms21228630
28. Jin S, Dai J, Teng X, Wu YM. Adverse Effects of Sympathetic Activation Should Not be Neglected During the Coronavirus Disease 2019 Pandemic. Chin Med J (2021) 134(4):413. doi: 10.1097/CM9.0000000000001106
29. Díaz HS, Toledo C, Andrade DC, Marcus NJ, Del Rio R. Neuroinflammation in Heart Failure: New Insights for an Old Disease. J Physiol (2020) 598(1):33–59. doi: 10.1113/JP278864
30. Del Rio R, Marcus NJ, Inestrosa NC. Potential Role of Autonomic Dysfunction in Covid-19 Morbidity and Mortality. Front Physiol (2020) 11:56174. doi: 10.3389/fphys.2020.561749
31. Alexandris N, Lagoumintzis G, Chasapis CT, Leonidas DD, Papadopoulos GE, Tzartos SJ, et al. Nicotinic Cholinergic System and COVID-19: In Silico Evaluation of Nicotinic Acetylcholine Receptor Agonists as Potential Therapeutic Interventions. Toxicol Rep (2021) 8:73–83. doi: 10.1016/j.toxrep.2020.12.013
32. Konig MF, Powell M, Staedtke V, Bai RY, Thomas DL, Fischer N, et al. Preventing Cytokine Storm Syndrome in COVID-19 Using α-1 Adrenergic Receptor Antagonists. J Clin Invest (2020) 130(7):3345–47. doi: 10.1172/JCI139642
33. Rose L, Graham L, Koenecke A, Powell M, Xiong R, Shen Z, et al. The Association Between Alpha-1 Adrenergic Receptor Antagonists and In-Hospital Mortality From COVID-19. Front Med (2021) 8:637647. doi: 10.3389/fmed.2021.637647
34. Vasanthakumar N. Beta-Adrenergic Blockers as a Potential Treatment for COVID-19 Patients. BioEssays (2020) 42(11):2000094. doi: 10.1002/bies.202000094
35. Lampert R, Burg MM, Jamner LD, Dziura J, Brandt C, Li F, et al. Effect of β-Blockers on Triggering of Symptomatic Atrial Fibrillation by Anger or Stress. Heart Rhythm (2019) 16(8):1167–73. doi: 10.1016/j.hrthm.2019.03.004
36. Panico K, Abrahão MV, Trentin-Sonoda M, Muzi-Filho H, Vieyra A, Carneiro-Ramos MS. Cardiac Inflammation After Ischemia-Reperfusion of the Kidney: Role of the Sympathetic Nervous System and the Renin-Angiotensin System. Cell Physiol Biochem (2019) 53(4):587–605. doi: 10.33594/000000159
37. Al-Kuraishy HM, Hussien NR, Al-Naimi MS, Al-Buhadily AK, Al-Gareeb AI, Lungnier C. Renin–Angiotensin System and Fibrinolytic Pathway in COVID-19: One-Way Skepticism. Biom Biotech Rese J (BBRJ) (2020) 4(SuppS1):33–40.
38. Heriansyah T, Nur Chomsy I, Febrianda L, Farahiya Hadi T, Andri Wihastuti T. The Potential Benefit of Beta-Blockers for the Management of Covid-19 Protocol Therapy-Induced QT Prolongation: A Literature Review. Scientia Pharm (2020) 88(4):55. doi: 10.3390/scipharm88040055
39. Verbrugge FH, Duchenne J, Bertrand PB, Dupont M, Tang WW, Mullens W. Uptitration of Renin-Angiotensin System Blocker and Beta-Blocker Therapy in Patients Hospitalized for Heart Failure With Reduced Versus Preserved Left Ventricular Ejection Fractions. Am J Cardiol (2013) 112(12):1913–20. doi: 10.1016/j.amjcard.2013.08.013
40. Boyer N, Signoret-Genest J, Artola A, Dallel R, Monconduit L. Propranolol Treatment Prevents Chronic Central Sensitization Induced by Repeated Dural Stimulation. Pain (2017) 158(10):2025–34. doi: 10.1097/j.pain.0000000000001007
41. Dakhale GN, Sharma VS, Thakre MN, Kalikar M. Low-Dose Sodium Valproate Versus Low-Dose Propranolol in Prophylaxis of Common Migraine Headache: A Randomized, Prospective, Parallel, Open-Label Study. Indian J Pharmacol (2019) 51(4):255. doi: 10.4103/ijp.IJP_457_18
42. Sweeney RM, Griffiths M, McAuley D. Treatment of Acute Lung Injury: Current and Emerging Pharmacological Therapies. In: Seminars in Respiratory and Critical Care Medicine, vol. 34. Thieme Medical Publishers (2013) 34(4):487–98. doi: 10.1055/s-0033-1351119
43. Henriquez AR, Snow SJ, Schladweiler MC, Miller CN, Dye JA, Ledbetter AD, et al. Adrenergic and Glucocorticoid Receptor Antagonists Reduce Ozone-Induced Lung Injury and Inflammation. Toxicol Appl Pharmacol (2018) 339:161–71. doi: 10.1016/j.taap.2017.12.006
44. Al-Qadi MO, Kashyap R. A42 ARDS: Risk, Treatment, and Outcomes: Effect of Chronic Beta Blockers Use on Sepsis-Related Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2015) 191:1.
45. Noveanu M, Breidthardt T, Reichlin T, Gayat E, Potocki M, Pargger H, et al. Effect of Oral Beta-Blocker on Short and Long-Term Mortality in Patients With Acute Respiratory Failure: Results From the BASEL-II-ICU Study. J Crit Care (2010) 14(6):1–0. doi: 10.1186/cc9317
46. Mutlu GM, Factor P. Alveolar Epithelial β2-Adrenergic Receptors. Am J Respir Cell Mol Biol (2008) 38(2):127–34. doi: 10.1165/rcmb.2007-0198TR
47. Ware LB, Matthay MA. Alveolar Fluid Clearance is Impaired in the Majority of Patients With Acute Lung Injury and the Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2001) 163(6):1376–83. doi: 10.1164/ajrccm.163.6.2004035
48. McCarthy C, Kokosi M, Bonella F. Shaping the Future of an Ultra-Rare Disease: Unmet Needs in the Diagnosis and Treatment of Pulmonary Alveolar Proteinosis. Curr Opin Pulm Med (2019) 25(5):450–8. doi: 10.1097/MCP.0000000000000601
49. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
50. Al-Kuraishy HM, Al-Gareeb AI, Alkazmi L, Alexiou A, Batiha GE. Levamisole Therapy in COVID-19. Viral Immunol (2021) 12. doi: 10.1089/vim.2021.0042
51. Moradian N, Gouravani M, Salehi MA, Heidari A, Shafeghat M, Hamblin MR, et al. Cytokine Release Syndrome: Inhibition of Pro-Inflammatory Cytokines as a Solution for Reducing COVID-19 Mortality. Eur Cytokine Netw (2020) 31(3):81–93. doi: 10.1684/ecn.2020.0451
52. Al-Kuraishy HM, Al-Gareeb AI, Alzahrani KJ, Cruz-Martins N, Batiha GE. The Potential Role of Neopterin in Covid-19: A New Perspective. Molecul Cellul Bioch (2021) 476(11):4161–6. doi: 10.1007/s11010-021-04232-z
53. Lamyel F, Warnken-Uhlich M, Seemann WK, Mohr K, Kostenis E, Ahmedat AS, et al. The β 2-Subtype of Adrenoceptors Mediates Inhibition of Pro-Fibrotic Events in Human Lung Fibroblasts. Naunyn Schmiedebergs Arch Pharmacol (2011) 384(2):133. doi: 10.1007/s00210-011-0655-5
54. Gálvez I, Martín-Cordero L, Hinchado MD, Álvarez-Barrientos A, Ortega E. Obesity Affects β2 Adrenergic Regulation of the Inflammatory Profile and Phenotype of Circulating Monocytes From Exercised Animals. Nutrients (2019) 11(11):2630. doi: 10.3390/nu11112630
55. Wu L, Tai Y, Hu S, Zhang M, Wang R, Zhou W, et al. Bidirectional Role of β2-Adrenergic Receptor in Autoimmune Diseases. Front Pharmacol (2018) 9:1313. doi: 10.3389/fphar.2018.01313
56. Budinger GS, Mutlu GM. Reply: β2-Agonists and Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2014) 189(11):1448–. doi: 10.1164/rccm.201404-0644LE
57. Nossent AY, Dai L, Rosendaal FR, Vos HL, Eikenboom JC. Beta 2 Adrenergic Receptor Polymorphisms: Association With Factor VIII and Von Willebrand Factor Levels and the Risk of Venous Thrombosis. J Thromb Haemost (2005) 3(2):405–7. doi: 10.1111/j.1538-7836.2005.01109.x
58. Changotra H, Jia Y, Moore TN, Liu G, Kahan SM, Sosnovtsev SV, et al. Type I and Type II Interferons Inhibit the Translation of Murine Norovirus Proteins. J Virol (2009) 83(11):5683–92. doi: 10.1128/JVI.00231-09
59. Zhou L, Li Y, Li X, Chen G, Liang H, Wu Y, et al. Propranolol Attenuates Surgical Stress–Induced Elevation of the Regulatory T Cell Response in Patients Undergoing Radical Mastectomy. J Immunol (2016) 196(8):3460–9. doi: 10.4049/jimmunol.1501677
60. Kim KH, Park YJ, Jang HJ, Lee SJ, Lee S, Yun BS, et al. Rugosic Acid A, Derived From Rosa Rugosa Thunb., Is Novel Inhibitory Agent for NF-κb and IL-6/STAT3 Axis in Acute Lung Injury Model. Phytother Res (2020) 34(12):3200–10. doi: 10.1002/ptr.6767
61. Barbieri A, Robinson N, Palma G, Maurea N, Desiderio V, Botti G. Can Beta-2-Adrenergic Pathway Be a New Target to Combat SARS-CoV-2 Hyperinflammatory Syndrome?—Lessons Learned From Cancer. Front Immunol (2020) 11:2615. doi: 10.3389/fimmu.2020.588724
62. Vasanthakumar N. Beta-Adrenergic Blocker Treatment for COVID-19 Patients. BioEssays : News and Reviews in Molecular, Cellular and Developmental Biology (2020) 42(11):e2000094. doi: 10.1002/bies.202000094.
63. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant Treatment is Associated With Decreased Mortality in Severe Coronavirus Disease 2019 Patients With Coagulopathy. J Thromb Haemost (2020) 18(5):1094–9. doi: 10.1111/jth.14817
64. Freeman TL, Swartz TH. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front Immunol (2020) 11:1518. doi: 10.3389/fimmu.2020.01518
65. Gao J, Xie Q, Wei T, Huang C, Zhou W, Shen W. Nebivolol Improves Obesity-Induced Vascular Remodeling by Suppressing NLRP3 Activation. J Cardiovasc Pharmacol (2019) 73(5):326–33. doi: 10.1097/FJC.0000000000000667
66. Jin P, Zhao T, Wei Y, Zhao F. Efficacy of Beta-Blockers in the Treatment of Sepsis. Banglad J Pharmacol (2021) 316(1):1–8. doi: 10.3329/bjp.v16i1.46001
67. McGonagle D, Ramanan AV, Bridgewood C. Immune Cartography of Macrophage Activation Syndrome in the COVID-19 Era. Nat Rev Rheumatol (2021) 5:1–3. doi: 10.1038/s41584-020-00571-1
68. Xia Y, Wei Y, Li ZY, Cai XY, Zhang LL, Dong XR, et al. Catecholamines Contribute to the Neovascularization of Lung Cancer via Tumor-Associated Macrophages. Brain Behav Immun (2019) 81:111–21. doi: 10.1016/j.bbi.2019.06.004
69. Ohtsuka T, Hamada M, Hiasa G, Sasaki O, Suzuki M, Hara Y, et al. Effect of Beta-Blockers on Circulating Levels of Inflammatory and Anti-Inflammatory Cytokines in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol (2001) 37(2):412–7. doi: 10.1016/S0735-1097(00)01121-9
70. Upadhyayula S, Kasliwal RR. Covid Cardiology: A Neologism for an Evolving Subspecialty. J Clin Prev Cardiol (2020) 9(2):40–4.
71. Yang P, Deng W, Han Y, Shi Y, Xu T, Shi J, et al. Analysis of the Correlation Among Hypertension, the Intake of β-Blockers, and Overall Survival Outcome in Patients Undergoing Chemoradiotherapy With Inoperable Stage III Non-Small Cell Lung Cancer. Am J Cancer Res (2017) 7(4):946954.
72. Dewanjee S, Vallamkondu J, Kalra RS, Puvvada N, Kandimalla R, Reddy PH. Emerging COVID-19 Neurological Manifestations: Present Outlook and Potential Neurological Challenges in COVID-19 Pandemic. Mol Neurobiol (2021) 58(9):4694–715. doi: 10.1007/s12035-021-02450-6
73. Amruta N, Chastain WH, Paz M, Solch RJ, Murray-Brown IC, Befeler JB, et al. SARS-CoV-2 Mediated Neuroinflammation and the Impact of COVID-19 in Neurological Disorders. Cytokine Growth Factor Rev (2021) 58:1–15. doi: 10.1016/j.cytogfr.2021.02.002
74. Kempuraj D, Selvakumar GP, Ahmed ME, Raikwar SP, Thangavel R, Khan A, et al. COVID-19, Mast Cells, Cytokine Storm, Psychological Stress, and Neuroinflammation. Neuroscientist (2020) 26(5-6):402–14. doi: 10.1177/1073858420941476
75. Haspula D, Clark MA. Neuroinflammation and Sympathetic Overactivity: Mechanisms and Implications in Hypertension. Auton Neurosci (2018) 210:10–7. doi: 10.1016/j.autneu.2018.01.002
76. Winklewski PJ, Radkowski M, Demkow U. Cross-Talk Between the Inflammatory Response, Sympathetic Activation and Pulmonary Infection in the Ischemic Stroke. J Neuroinflamm (2014) 11:213. doi: 10.1186/s12974-014-0213-4
77. Porzionato A, Emmi A, Barbon S, Boscolo-Berto R, Stecco C, Stocco E, et al. Sympathetic Activation: A Potential Link Between Comorbidities and COVID-19. FEBS J (2020) 287(17):3681–8. doi: 10.1111/febs.15481
78. Nguembu S, Meloni M, Endalle G, Dokponou H, Dada OE, Senyuy WP, et al. Paroxysmal Sympathetic Hyperactivity in Moderate-To-Severe Traumatic Brain Injury and the Role of Beta-Blockers: A Scoping Review. Emerg Med Int (2021) 2021:5589239. doi: 10.1155/2021/5589239
79. Lechtenberg KJ, Meyer ST, Doyle JB, Peterson TC, Buckwalter MS. Augmented β2-Adrenergic Signaling Dampens the Neuroinflammatory Response Following Ischemic Stroke and Increases Stroke Size. J Neuroinflamm (2019) 16(1):1–8. doi: 10.1186/s12974-019-1506-4
80. Evans AK, Ardestani PM, Yi B, Park HH, Lam RK, Shamloo M. Beta-Adrenergic Receptor Antagonism Is Proinflammatory and Exacerbates Neuroinflammation in a Mouse Model of Alzheimer's Disease. Neurobiol Disease (2020) 146:105089. doi: 10.1016/j.nbd.2020.105089
81. Mookerjee RP, Pavesi M, Thomsen KL, Mehta G, Macnaughtan J, Bendtsen F, et al. Treatment With non-Selective Beta Blockers Is Associated With Reduced Severity of Systemic Inflammation and Improved Survival of Patients With Acute-on-Chronic Liver Failure. J Hepatol (2016) 64(3):574–82. doi: 10.1016/j.jhep.2015.10.018
82. Park JB, Jo JY, Zheng H, Patel KP, Stern JE. Regulation of Tonic GABA Inhibitory Function, Presympathetic Neuronal Activity and Sympathetic Outflow From the Paraventricular Nucleus by Astroglial GABA Transporters. J Physiol (2009) 587(19):4645–60. doi: 10.1113/jphysiol.2009.173435
83. Mukerjee S, Gao H, Xu J, Sato R, Zsombok A, Lazartigues E. ACE2 and ADAM17 Interaction Regulates the Activity of Presympathetic Neurons. Hypertension (2019) 74(5):1181–91. doi: 10.1161/HYPERTENSIONAHA.119.13133
84. Masi S, Uliana M, Virdis A. Angiotensin II and Vascular Damage in Hypertension: Role of Oxidative Stress and Sympathetic Activation. Vascul Pharmacol (2019) 115:13–7. doi: 10.1016/j.vph.2019.01.004
85. Gubbi S, Nazari MA, Taieb D, Klubo-Gwiezdzinska J, Pacak K. Catecholamine Physiology and Its Implications in Patients With COVID-19. Lancet Diabetes Endocrinol (2020) 8(12):978986. doi: 10.1016/S2213-8587(20)30342-9
86. RIddell SR. Adrenaline Fuels a Cytokine Storm During Immunotherapy. Nature (2018) 564:194. doi: 10.1038/d41586-018-07581-w
87. Siddall E, Radhakrishnan J. Capillary Leak Syndrome: A Cytokine and Catecholamine Storm? Kidney Int (2019) 95(5):1009–11. doi: 10.1016/j.kint.2019.03.001
88. Konig MF, Powell M, Staedtke V, Bai RY, Thomas DL, Fischer N, et al. Targeting the Catecholamine-Cytokine Axis to Prevent SARS-CoV-2 Cytokine Storm Syndrome. medRxiv (2020) 1. doi: 10.1101/2020.04.02.20051565
89. Staedtke V, Bai RY, Kim K, Darvas M, Davila ML, Riggins GJ, et al. Disruption of a Self-Amplifying Catecholamine Loop Reduces Cytokine Release Syndrome. Nature (2018) 564(7735):273–7. doi: 10.1038/s41586-018-0774-y
90. Radzikowska U, Ding M, Tan G, Zhakparov D, Peng Y, Wawrzyniak P, et al. Distribution of ACE2, CD147, CD26, and Other SARS-CoV-2 Associated Molecules in Tissues and Immune Cells in Health and in Asthma, COPD, Obesity, Hypertension, and COVID-19 Risk Factors. Allergy (2020) 75(11):2829–45. doi: 10.1111/all.14429
91. Flierl MA, Rittirsch D, Nadeau BA, Sarma JV, Day DE, Lentsch AB, et al. Upregulation of Phagocyte-Derived Catecholamines Augments the Acute Inflammatory Response. PloS One (2009) 4(2):e4414. doi: 10.1371/journal.pone.0004414
92. Jin R, Liu S, Wang M, Zhong W, Li G. Inhibition of CD147 Attenuates Stroke-Associated Pneumonia Through Modulating Lung Immune Response in Mice. Front Neurol (2019) 10:853. doi: 10.3389/fneur.2019.00853
93. Vasanthakumar N. Adrenergic Storm-Induced Warburg Effect in COVID-19: A Hypothesis. OSF Preprints (2020). doi: 10.31219/osf.io/z654v
94. Wang Q, Xu B, Fan K, Wu J, Wang T. Inflammation Suppression by Dexamethasone via Inhibition of CD147-Mediated NF-κb Pathway in Collagen-Induced Arthritis Rats. J Biol Chem (2020) 473(1):63–76. doi: 10.1007/s11010-020-03808-5
95. Zhou C, Chen X, Zeng W, Peng C, Huang G, Li XA, et al. Propranolol Induced G0/G1/S Phase Arrest and Apoptosis in Melanoma Cells via AKT/MAPK Pathway. Oncotarget (2016) 7(42):68314. doi: 10.18632/oncotarget.11599
96. Pennings GJ, Kritharides L. CD147 in Cardiovascular Disease and Thrombosis. In: Seminars in Thrombosis and Hemostasis, vol. 40. Thieme Medical Publishers (2014) 40(7):747–55. doi: 10.1055/s-0034-1390001
97. Jin R, Xiao AY, Chen R, Granger DN, Li G. Inhibition of CD147 (Cluster of Differentiation 147) Ameliorates Acute Ischemic Stroke in Mice by Reducing Thromboinflammation. Stroke (2017) 48(12):3356–65. doi: 10.1161/STROKEAHA.117.018839
98. Seizer P, Ungern-Sternberg SN, Schönberger T, Borst O, Münzer P, Schmidt EM, et al. Extracellular Cyclophilin A Activates Platelets via EMMPRIN (CD147) and PI3K/Akt Signaling, Which Promotes Platelet Adhesion and Thrombus Formation In Vitro and In Vivo. Arterioscler Thromb Vasc Biol (2015) 35(3):655–63. doi: 10.1161/ATVBAHA.114.305112
99. Lu G, Jia Z, Zu Q, Zhang J, Zhao L, Shi H. Inhibition of the Cyclophilin A–CD147 Interaction Attenuates Right Ventricular Injury and Dysfunction After Acute Pulmonary Embolism in Rats. J Biol Chem (2018) 293(31):12199–208. doi: 10.1074/jbc.RA118.002845
100. Liu C, von Brunn A, Zhu D. Cyclophilin A and CD147: Novel Therapeutic Targets for the Treatment of COVID-19. Med Drug Discovery (2020) 7:100056. doi: 10.1016/j.medidd.2020.100056
101. Mulder MM, Brandts L, Brüggemann RA, Koelmann M, Streng AS, Olie RH, et al. Serial Markers of Coagulation and Inflammation and the Occurrence of Clinical Pulmonary Thromboembolism in Mechanically Ventilated Patients With SARS-CoV-2 Infection; the Prospective Maastricht Intensive Care COVID Cohort. Thromb J (2021) 19(1):35. doi: 10.1186/s12959-021-00286-7
102. Satoh K, Suzuki K, Otsuki T, Sunamura S, Kudo S, Ikeda S, et al. Novel Biomarkers for Cardiac Hypertrophy, Fibrosis and Failure: Cyclophilin A and Basigin—Clinical Application of Basic Research. J Card Fail (2016) 22(9):S159. doi: 10.1016/j.cardfail.2016.07.045
103. Wang CH, Yao H, Chen LN, Jia JF, Wang L, Dai JY, et al. CD147 Induces Angiogenesis Through a Vascular Endothelial Growth Factor and Hypoxia-Inducible Transcription Factor 1α–Mediated Pathway in Rheumatoid Arthritis. Arthrit Rheuma (2012) 64(6):1818–27. doi: 10.1002/art.34341
104. Yin XX, Zheng XR, Peng W, Wu ML, Mao XY. Vascular Endothelial Growth Factor (VEGF) as a Vital Target for Brain Inflammation During the COVID-19 Outbreak. ACS Chem Neurosci (2020) 11(12):1704–5. doi: 10.1021/acschemneuro.0c00294
105. Barbieri A, Palma G, Rosati A, Giudice A, Falco A, Petrillo A, et al. Role of Endothelial Nitric Oxide Synthase (eNOS) in Chronic Stress-Promoted Tumour Growth. J Cell Mol Med (2012) 16(4):920–6. doi: 10.1111/j.1582-4934.2011.01375.x
106. Raza IM, Zaman Z, Waseem Y. Cytokine Storm Syndrome, a Potential Cause of Death in COVID-19 Patients. Pak J Surg Med (2021) 1(4):e139–. doi: 10.37978/pjsm.v1i4.139
107. Alexander SP, Armstrong JF, Davenport AP, Davies JA, Faccenda E, Harding SD, et al. A Rational Roadmap for SARS-CoV-2/COVID-19 Pharmacotherapeutic Research and Development: IUPHAR Review 29. Br J Pharmacol (2020) 177(21):4942–66. doi: 10.1111/bph.15094
108. Nakamura K, Murakami M, Miura D, Yunoki K, Enko K, Tanaka M, et al. Beta-Blockers and Oxidative Stress in Patients With Heart Failure. Pharmaceuticals (Basel Switzerland) (2011) 4(8):1088–100. doi: 10.3390/ph4081088
109. Pincemail J, Cavalier E, Charlier C, Cheramy–Bien JP, Brevers E, Courtois A, et al. Oxidative Stress Status in COVID-19 Patients Hospitalized in Intensive Care Unit for Severe Pneumonia. A Pilot Study. Antioxidants (2021) 10(2):257. doi: 10.3390/antiox10020257
110. Zhang Y, Fan L, Xi R, Mao Z, Shi D, Ding D, et al. Lethal Concentration of Perfluoroisobutylene Induces Acute Lung Injury in Mice Mediated via Cytokine Storm, Oxidative Stress and Apoptosis. Inhal Toxicol (2017) 29(6):255–65. doi: 10.1080/08958378.2017.1357772
111. Francisqueti-Ferron FV, Garcia JL, Ferron AJ, Nakandakare-Maia ET, Gregolin CS, das Chagas Silva JP, et al. Gamma-Oryzanol as a Potential Modulator of Oxidative Stress and Inflammation via PPAR-Y in Adipose Tissue: A Hypothetical Therapeutic for Cytokine Storm in COVID-19? Molecul Cellul Endocrinol (2021) 520:111095. doi: 10.1016/j.mce.2020.111095
112. Goud PT, Bai D, Abu-Soud HM. A Multiple-Hit Hypothesis Involving Reactive Oxygen Species and Myeloperoxidase Explains Clinical Deterioration and Fatality in COVID-19. Int J Biol Sci (2021) 17(1):62. doi: 10.7150/ijbs.51811
113. Gok HB, Solaroglu I, Okutan O, Cimen B, Kaptanoglu E, Palaoglu S. Metoprolol Treatment Decreases Tissue Myeloperoxidase Activity After Spinal Cord Injury in Rats. J Clin Neurosci (2007) 14(2):138–42. doi: 10.1016/j.jocn.2005.10.016
114. Cleland JGF, Bunting KV, Flather MD, Altman DG, Holmes J, Coats AJS, et al. Beta-Blockers for Heart Failure With Reduced, Mid-Range, and Preserved Ejection Fraction: An Individual Patient-Level Analysis of Double-Blind Randomized Trials. Eur Heart J (2018) 39(1):26–35. doi: 10.1093/eurheartj/ehx564
115. Nawarskas JJ, Cheng-Lai A, Frishman WH. Celiprolol: A Unique Selective Adrenoceptor Modulator. Cardiol Rev (2017) 25(5):247. doi: 10.1097/CRD.0000000000000159
116. Madden KS, Szpunar MJ, Brown EB. β-Adrenergic Receptors (β-AR) Regulate VEGF and IL-6 Production by Divergent Pathways in High β-AR-Expressing Breast Cancer Cell Lines. Breast Cancer Res Treat (2011) 130(3):747–58. doi: 10.1007/s10549-011-1348-y
Keywords: SARS-CoV-2, cytokine storm, sympathetic storm, pharmacology and Covid, Beta blockers
Citation: Al-kuraishy HM, Al-Gareeb AI, Mostafa-Hedeab G, Kasozi KI, Zirintunda G, Aslam A, Allahyani M, Welburn SC and Batiha GE-S (2021) Effects of β-Blockers on the Sympathetic and Cytokines Storms in Covid-19. Front. Immunol. 12:749291. doi: 10.3389/fimmu.2021.749291
Received: 29 July 2021; Accepted: 15 October 2021;
Published: 11 November 2021.
Edited by:
Lei Xi, Virginia Commonwealth University, United StatesReviewed by:
Antonio Barbieri, Istituto Nazionale Tumori Fondazione G. Pascale (IRCCS), ItalyLu Qin, Penn State Milton S. Hershey Medical Center, United States
Copyright © 2021 Al-kuraishy, Al-Gareeb, Mostafa-Hedeab, Kasozi, Zirintunda, Aslam, Allahyani, Welburn and Batiha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gomaa Mostafa-Hedeab, Gomaa@ju.edu.sa; Keneth Iceland Kasozi, kicelandy@gmail.com; kicelandy@kab.ac.ug