 Ana Valle-Noguera
Ana Valle-Noguera Anne Ochoa-Ramos†
Anne Ochoa-Ramos† Maria José Gomez-Sánchez
Maria José Gomez-Sánchez Aranzazu Cruz-Adalia
Aranzazu Cruz-Adalia- Department of Immunology, School of Medicine, Universidad Complutense de Madrid; 12 de Octubre Health Research Institute (imas12), Madrid, Spain
Type 3 Innate lymphoid cells (ILC3s) have been described as tissue-resident cells and characterized throughout the body, especially in mucosal sites and classical first barrier organs such as skin, gut and lungs, among others. A significant part of the research has focused on their role in combating pathogens, mainly extracellular pathogens, with the gut as the principal organ. However, some recent discoveries in the field have unveiled their activity in other organs, combating intracellular pathogens and as part of the response to viruses. In this review we have compiled the latest studies on the role of ILC3s and the molecular mechanisms involved in defending against different microbes at the mucosal surface, most of these studies have made use of conditional transgenic mice. The present review therefore attempts to provide an overview of the function of ILC3s in infections throughout the body, focusing on their specific activity in different organs.
Introduction
Innate lymphoid cells (ILCs) are a subset of lymphocytes lacking the rearranged antigen receptors expressed by adaptive cells, and residing in peripheral tissues, particularly at barrier surfaces. It is now recognized that their location in peripheral tissues enables ILCs to respond promptly to tissue perturbation. Indeed, as a result of their location and effector phenotype, ILCs produce cytokines within hours of activation, as opposed to the number of days required for naive adaptive lymphocytes to be primed, expand, differentiate and enter tissues (1).
ILCs are found in both lymphoid and non-lymphoid tissues, although they are particularly abundant at mucosal surfaces as tissue-resident cells. The nomenclature for ILCs proposed in 2013 classified these cells into three groups based on their function, but recently the International Union of Immunological Societies proposed another classification, considering not only function but also development. Therefore, ILCs are divided into 5 groups: Natural Killer cells (NKs), ILC1, ILC2, ILC3 and Lymphoid tissue inducers (LTi) and they can be distinguished by different biomarkers expressed in each subset (2). Despite being identified as non-T, non-B lymphocytes, they can be considered as the innate counterparts of T lymphocytes. Therefore, regarding their function, ILC1s, ILC2s, and ILC3s mirror CD4+ T helper (Th)1, Th2, and Th17 cells, respectively, whereas natural killer (NK) cells mirror CD8+ cytotoxic T cells. Both ILCs and T cells work together to organize the most appropriate immune response to pathogens. ILC1s and Th1 cells react to intracellular pathogens, such as viruses, and to tumors; ILC2s and Th2 cells respond to large extracellular parasites and allergens; and ILC3s and Th17 cells mainly combat extracellular microbes, such as bacteria and fungi (1).
ILC3s express the nuclear factor retinoic acid–related orphan receptor γ t (RORγt) and the aryl hydrocarbon receptor (AhR) both of which are essential for their development and function (3, 4). Two subsets of ILC3s can be distinguished on the basis of the cell surface expression of Natural cytotoxicity receptors (NCR) and have been termed as NCR+ILC3s cells (Nkp46+ILC3s in mice and NKp44+ ILC3s) or NCR- ILC3s (3, 5). As the innate counterparts of T helper 17 (Th17), ILC3s are the early source of interleukin 17 (IL-17) and IL-22 during infections. They can also produce granulocyte-macrophage colony-stimulating factor GM-CSF, which plays a role in gut tolerance by inducing IL-10 secretion in macrophages and dendritic cells (DCs) that maintain the regulatory T (Treg) population (6). In mice, NKp46+ ILC3s are also dependent on T-box transcription factor TBX21 (T-bet) and can also produce interferon-gamma (IFN-γ) (7, 8). Ltis, previously included in ILC3s subset, are also strictly dependent on RORγt. Consequently, in many studies with conditional transgenic mice, which have depleted genes in RORγt-expressing cells, the function of the protein under study might also play a role in Ltis, rather than only in ILC3s. LTi cells were discovered in 1997 and identified as a discrete subset of lymphoid cells that are essential for the development of peripheral lymph nodes (LN) and Peyer’s patches during embryonic life (9). Embryonic LTis, which are involved in embryonic LN formation, are replaced in the adult by LTi cells derived from the bone marrow. In the adult mouse, LTi-like cells are present in high numbers within the adult gut mucosa; they express c-Kit and C-C-C chemokine receptor (CCR)6, but not NCRs. In recent years, some research has revealed that CCR6+ILC3s express MHC-II which, through antigen presentation, can induce apoptosis to commensal-specific CD4+T cells contributing to maintaining microbiome tolerance (10).
Cytokine release not only constitutes a defence mechanism but also helps maintain homeostasis and the mucosal barrier. IL-22 and IL-17 signaling promotes the production of antimicrobial peptides and regulates the expression of tight junction components in endothelial cells. Therefore, loss of IL-22 production gives rise to dissemination of intestinal bacteria, producing chronic body inflammation and resulting in susceptibility to infections, such as Citrobacter rodentium (4, 11, 12). IL-22, produced by ILC3s, is essential in the early phase of infection (11, 13). Furthermore, IL-17 production by ILC3s is also involved in combating fungal pathogens such as Candida albicans (14). The molecular mechanisms underlying ILC3 cytokine release for defence against pathogens have been well researched, and in the last few years ILC3s have been found to be regulated by different factors such as diet, the nervous system, circadian rhythms, microbiota and other immune cells (3, 4, 15, 16). Although ILC3 defence against pathogens has mostly been described in the gut, where they combat mostly extracellular bacteria, they also play an important role in other organs like lungs or skin. Therefore, the present paper discusses recent research on the role of ILC3s in infections by different pathogens, summarizing the cell signaling pathways involved in the activation of ILC3s, and analyzing each pathogen-specific tissue.
Defensive Function of ILC3s in the Skin
IL-17-Producing ILC3s Together With γδ T Cells Are Essential in the Defence Against the Epidermal Infection of Staphylococcus aureus
Staphylococcus aureus, a gram-positive bacterium, resides in 10–20% of healthy individuals, with the skin surface as its major infection site (17). The cutaneous immune response to S. aureus involves both the innate and adaptive immunity. Neutrophils represent the first-responder phagocytic cells that are recruited to the site of infection to help kill pathogens. IL-17A and IL-17F have been shown to promote neutrophil recruitment and previous research found that mice deficient in both cytokines developed spontaneous S. aureus skin infections but did not have an increased susceptibility to a systemic S. aureus challenge (18). Regarding the role of IL-17 against S. aureus cutaneous infections, mice deficient in γδ T cells (T cell receptor δ deficient; TCRδ−/−) developed larger skin lesions with higher bacterial counts, as well as an impaired neutrophil infiltration and induction of IL-17 compared with WT mice (19). Interestingly, when these TCRδ−/− mice were treated with an anti-CD90 monoclonal antibody (Mab), which depleted their ILCs, their disease scores were seen to be significantly lower in comparison with mice treated with control Mab, thus indicating that both ILCs and γδ T cells contributed to skin inflammation in response to S. aureus. Regarding the mechanism involved, the critical role of Myd88 for pathogen colonization was determined by means of Myd88−/− and Myd88fl/fl K14Cre mice [conditional deletion of Myd88 within keratinocytes (KCs)]. Remarkably, the induction of IL-1α and IL-36α produced by S. aureus in in vitro primary KCs from Myd88−/− mice was abolished compared with wild type (WT) cells. This provoked reduced IL-17A-producing cells (ILC3s and γδ T cells) infiltrating into the skin of Myd88−/− mice following epicutaneous infection. In conclusion, in order to combat the epidermal infection caused by S.aureus, there is an induction of IL-17-producing cells (ILC3s and γδ T cells) because of the Myd88-dependent IL-1α and IL-36α production by KCs (Figure 1) but further investigations are needed to determine the specific contribution of ILC3s in this model (20).
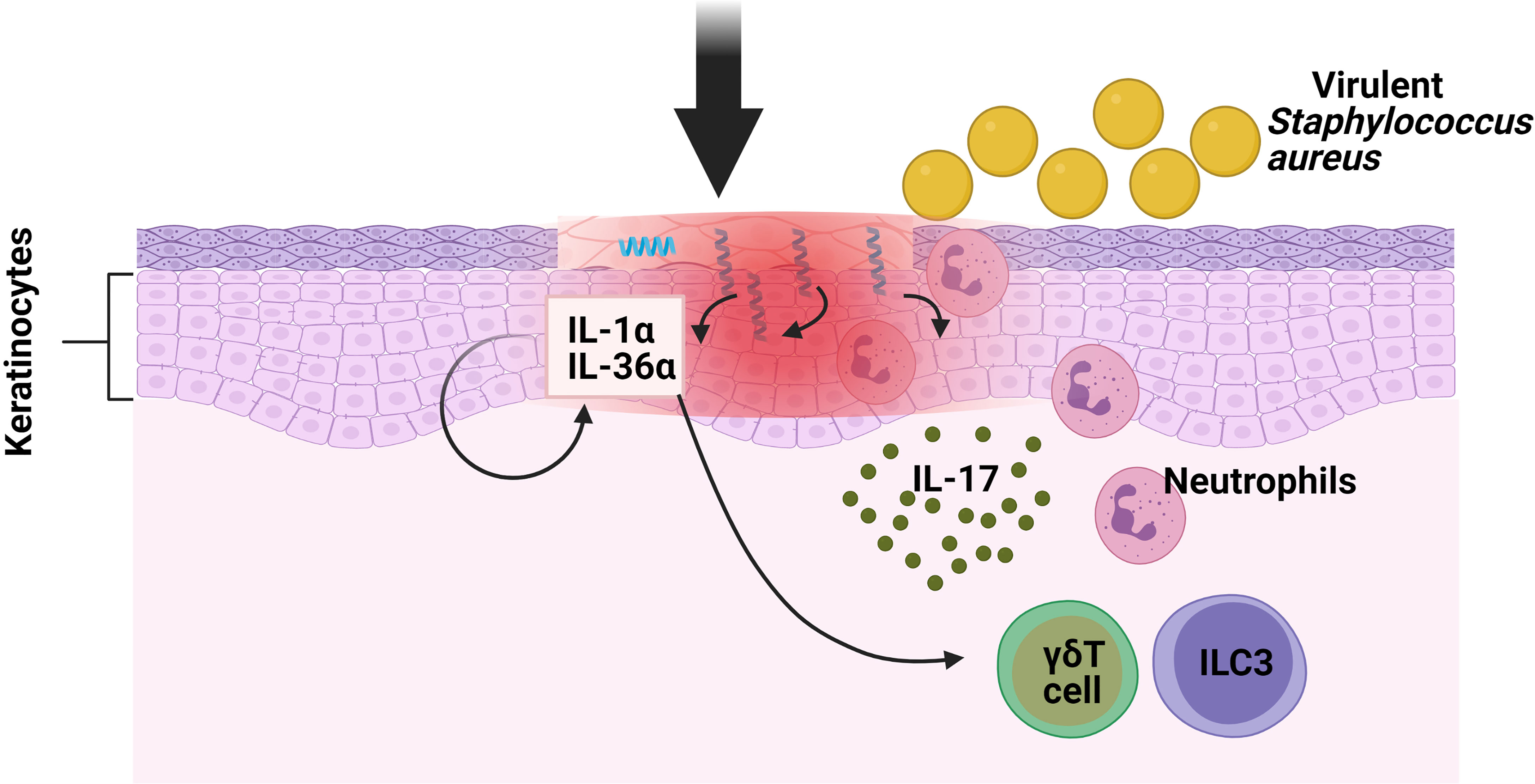
Figure 1 Function of ILC3s against pathogens in oral mucosa. S. aureus causes the keratinocytes of the skin to produce IL-1α and IL-36α. These interleukins bind to their receptors in ILC3s and γδ T cells, inducing the production of IL-17, which promotes neutrophil infiltration.
Role of ILC3s in Oral Mucosa
Complementary IL-17 Response by ILC3s and T Cells Is Required to Combat Infection by Candida albicans
C. albicans is an opportunistic pathogenic fungus considered to be a common member of the human microflora. One of the tissues most frequently affected by C. albicans is the oral cavity (21). Previous data had proposed ILCs as the IL-17A secretory cells at the onset of oropharyngeal candidiasis (OPC) in C. albicans-infected mice, sufficing for protection in Rag1−/− mice but not in Rag1−/− mice treated with anti-CD90 Mab. Indeed, IL-17A mRNA expression in the tongue at 24 h post-infection did not differ between the WT mice and the mice lacking the MHC-II, γδ T cells or NKT cells; however it did decrease in the in IL-23–deficient mice, suggesting that the fungal control was due to ILC function (14). However, the susceptibility in the early response was not analyzed. These results were challenged when mouse models that could not rearrange the TCR were susceptible to OPC, a fact that highlights the importance of T cells in the defence against C. albicans. Nonetheless, both of these studies analyzed indirect IL-17 production by antibodies. This has recently been demonstrated by direct visualization of IL-17A and IL-17F cytokines in the infected tongue of three IL-17-producing cell types: αβ T cells, γδ T cells and ILC3s (22) with the use of reporter transgenic mice. Interestingly, selective lack of nTh17 (unprimed population of CD4+ T cells) or γδ T cells did not affect fungal control (23), but elimination of both T cells and ILC3s was seen to be necessary to produce the high degree of susceptibility to OPC as with the IL-17RA or IL-17RC-deficient mice. Finally, the mechanism proposed for the production of innate IL-17 in the oral mucosa was through the induction of IL-1β, IL-6 and IL-23 by Langerin+ DCs, which are tissue-resident mononuclear phagocytes (MNPs) (22). To conclude, the coexistence of these three different but complementary cell types (nTh17, ILC3s and γδ T cells) is essential for the IL-17 response to the fungus in oral mucosa.
Function of the Tissue-Resident ILC3s in Liver
ILC3s Play a Critical Role in Liver Fibrosis
A wide range of viruses can cause acute or chronic hepatitis and liver infections, for example, Adenovirus (Ad) or lymphocytic choriomeningitis virus (LCMV) which is a prototypical virus found throughout the world and used in animal models of acute and persistent hepatitis (24). It has been demonstrated that the source of the early IL-17A/F production in liver after Ad and LCMV infection in mice was γδ T cells and ILCs, mostly the NKp46− ILC3 population. Importantly, IL-17 source was mostly from ILC3s because Ad-induced hepatitis in γδ−/− (TCRδ−/−) mice did not affect early IL-17A production or Th1/cytotoxic T-lymphocyte responses (25). However, there is a need to use knockout (KO) mice depleting RORγt+ cells or NKp46+ cells in this model in order to confirm that specifically IL-17-producing ILC3s are critical for the defence against adenovirus infection.
Hepatitis can develop into fibrosis and subsequently to cirrhosis, the latter being responsible for liver morbidity and mortality. In this regard, it has previously been demonstrated that ILC3s can contact directly with the hepatic stellate cells (HSCs) (26), which might affect the progression of liver fibrosis. The frequencies of ILC3s, as well as their production of IL-17A and IL-22, were considerably higher in chronic hepatitis B (CHB) and Hepatitis B Virus (HBV)-related liver cirrhosis (LC) patient subgroups compared to the healthy control group (HC), correlating to a more severe phenotype. In order to study the mechanism at play, pre-stimulated sorted ILC3s were co-cultured with LX-2 cells (human HSC line) revealing a greater proliferation and a higher expression of fibrogenic genes in the LX-2 cells, both in direct and indirect contact, suggesting the critical role of secreted cytokines rather than direct cell contact. Indeed, neutralizing either IL-17A or IL-22 in the cell culture reverted the results, thus revealing a regulation of the expression of the TGF-β receptor and a STAT3 activation in the HSC cell line. In addition, in vitro experiments demonstrated that ILC3s had also an indirect mechanism towards fibrosis by producing IL-22 which in turn suppressed IFN-γ (a well-known anti-fibrotic cytokine) production by other immune cells. Finally, to determine the in vivo effects of ILC3s in liver fibrogenesis, the carbon tetrachloride (CCl4) murine model was studied in Rag1−/− mice treated with anti-CD90.2 Mab, showing significantly lower HSC activation and less accumulation of the extracellular matrix (ECM) in liver compared with the non-ILC-depleted mice. Remarkably, adoptive transfer of ILC3s from WT to ILC-depleted Rag1−/− mice augmented HSC activation and ECM accumulation in liver, thus providing the in vivo pro-fibrotic effects of ILC3s in CCl4-induced mouse liver fibrosis (27) (Figure 2). In summary ILC3-produced cytokines influence the HSC cells, which contributes to liver fibrosis.
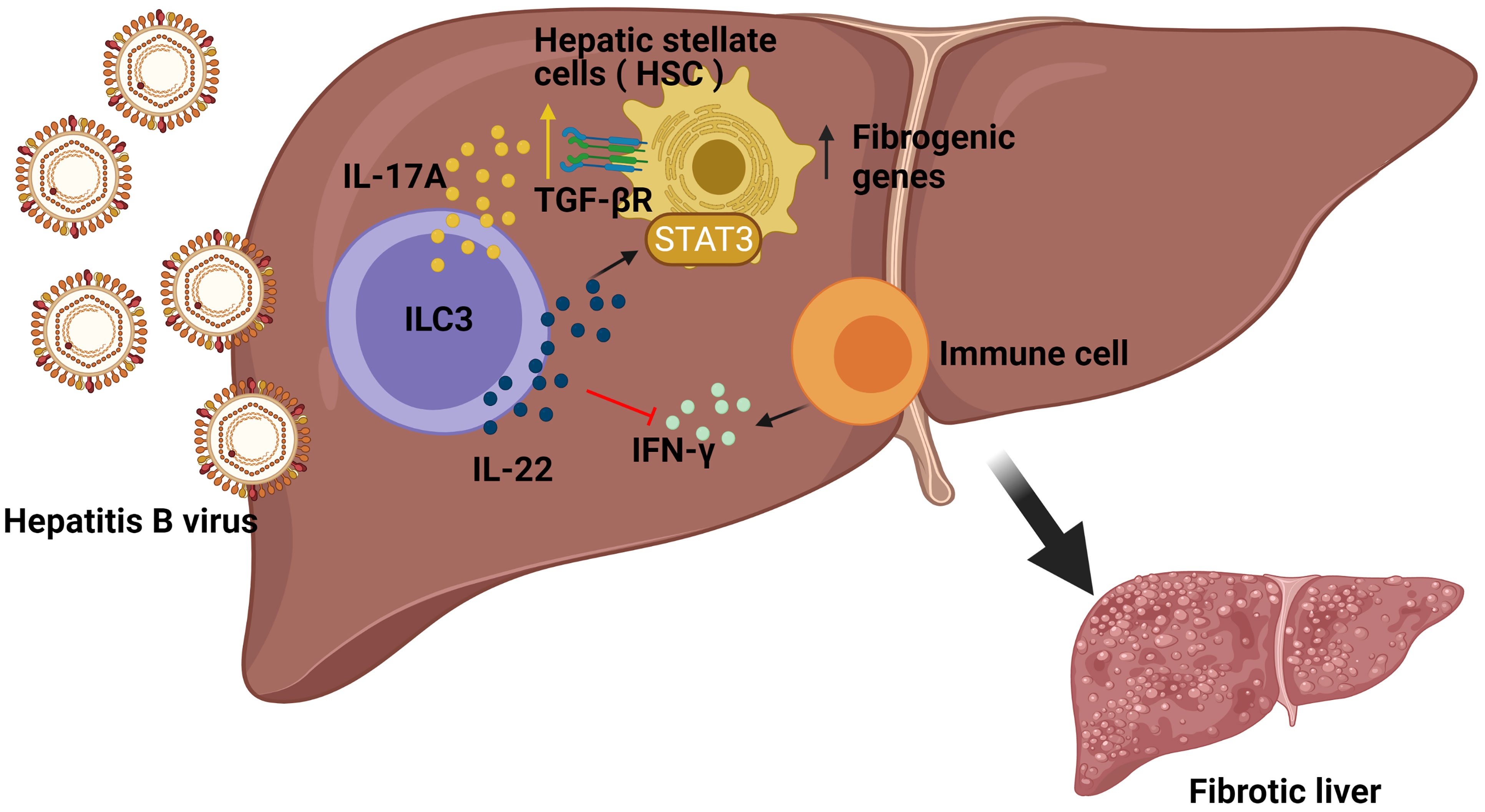
Figure 2 Role of ILC3s in the development of fibrosis induced by Hepatitis B (HBV) infection. In HBV infection, ILC3s can directly promote the expression of fibrogenic genes in hepatic stellate cells (HSC) in non-contact manners by producing IL-17A and IL-22. Additionally, ILC3s also have indirect fibrogenic effects by producing IL-22 to suppress interferon IFN-γ (an anti-fibrotic cytokine) production by other immune cells.
Innate Protection of ILC3s in Lung
ILC3s provide innate protection against pathogens within the lung, specifically against different primary or opportunistic pathogens (28) producing IL-17 and IL-22 upon stimulation (29).
IL-22 Producing Cells, Including ILC3s, Minimize Lung Inflammation During Influenza A Virus and Protect Against Secondary Bacterial Infections
The influenza viruses constitute some of the most predominant human respiratory pathogens, causing substantial seasonal and pandemic morbidity and mortality. ILC3s may play a role in the immune response to pulmonary viral infection due to the importance of the secretion of IL-22 and IL-17. Although IL-17-deficient mice present less lung injury following a viral infection (30). IL-17 has been shown to help to prevent secondary bacterial infections. Co-infection experiments with influenza and S. aureus showed that influenza triggered IFN-β production, which inhibited IL-17 production by T cells (31) and suppressed the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kβ) activation, thus increasing the chances of contracting a secondary pneumonia with S. aureus (32). However, there is a need for further research to establish the function of ILC3s, due to the fact that the role of only Th17 was demonstrated in this model. As for IL-22, a murine model of influenza A (IAV) viral infection followed by a secondary Streptococcus pneumoniae bacterial infection provoked an increase in RORγt+ cells and IL-22+ ILC3s in the lung. In addition, while IL-22-deficient mice exhibited no change in viral clearance, their survival was seen to be severely impaired after S. pneumoniae secondary infection (33). Indeed, a more recent study showed that infected transgenic IL-22 binding protein KO mice with influenza followed by S. aureus or S. pneumoniae infection exhibited greater bacterial clearance, a lower mortality rate from secondary bacterial infection, and enhanced airway epithelial integrity (34). In summary, IL-22 has been shown to reduce lung damage following an infection by influenza A infection and to protect against secondary bacterial infections. However, the specific contribution by ILC3s in this model should be demonstrated depleting CD90+ or RORγt+ cells in Rag−/− mice or NKp46+ cells.
ILC3s Produce IL-22 After Acute Aspergillus fumigatus Exposure
A. fumigatus is an environmental filamentous fungus which threatens the life of immunocompromised individuals, causing invasive aspergillosis and allergic disease (35). Genetic deficiency in or neutralization of IL-22 results in impaired clearance of A. fumigatus, indicating the critical role of IL-22 in pathogen elimination during acute infection (36); neutralization of IL-22 also improves lung function after chronic fungal exposure illustrating that IL-22 drives lung inflammatory responses that have a negative impact on lung function (37). Interestingly, it has been observed that innate and innate-like lymphocytes were involved in the production of IL-22 following 48h of A. fumigatus exposure in Il-22 Cre R26ReYFP reporter mice, in which yellow fluorescent protein (YFP) expression marks IL-22-producing cells. Moreover, lung IL-22 production was completely dependent on IL-7, partially dependent on IL-21, and negatively regulated by IL-15. Employing deficient mice for these interleukins, it was observed that IL-7 appeared to be essential with regard to maintaining invariant natural killer T (iNKT) cells and γδ T cells in the lung after fungal exposure; IL-21 for maintaining optimal numbers of iNKT cells and ILC3s and as a negative regulator of γδT cells; moreover, IL-15R signaling did not affect any of the absolute numbers of innate cells but iNKT cells and γδ T cells displayed higher levels of intracellular IL-22 in deficient mice of IL-15R but not in ILC3s (38) (Figure 3). In conclusion, the exposure to A. fumigatus can induce IL-22 in ILC3s and innate-like lymphocytes, which could drive lung inflammation; however, this should be demonstrated in mouse models depleting specifically RORγt+ cells, iNKT or γδ T cells in order to show the contribution of each population.
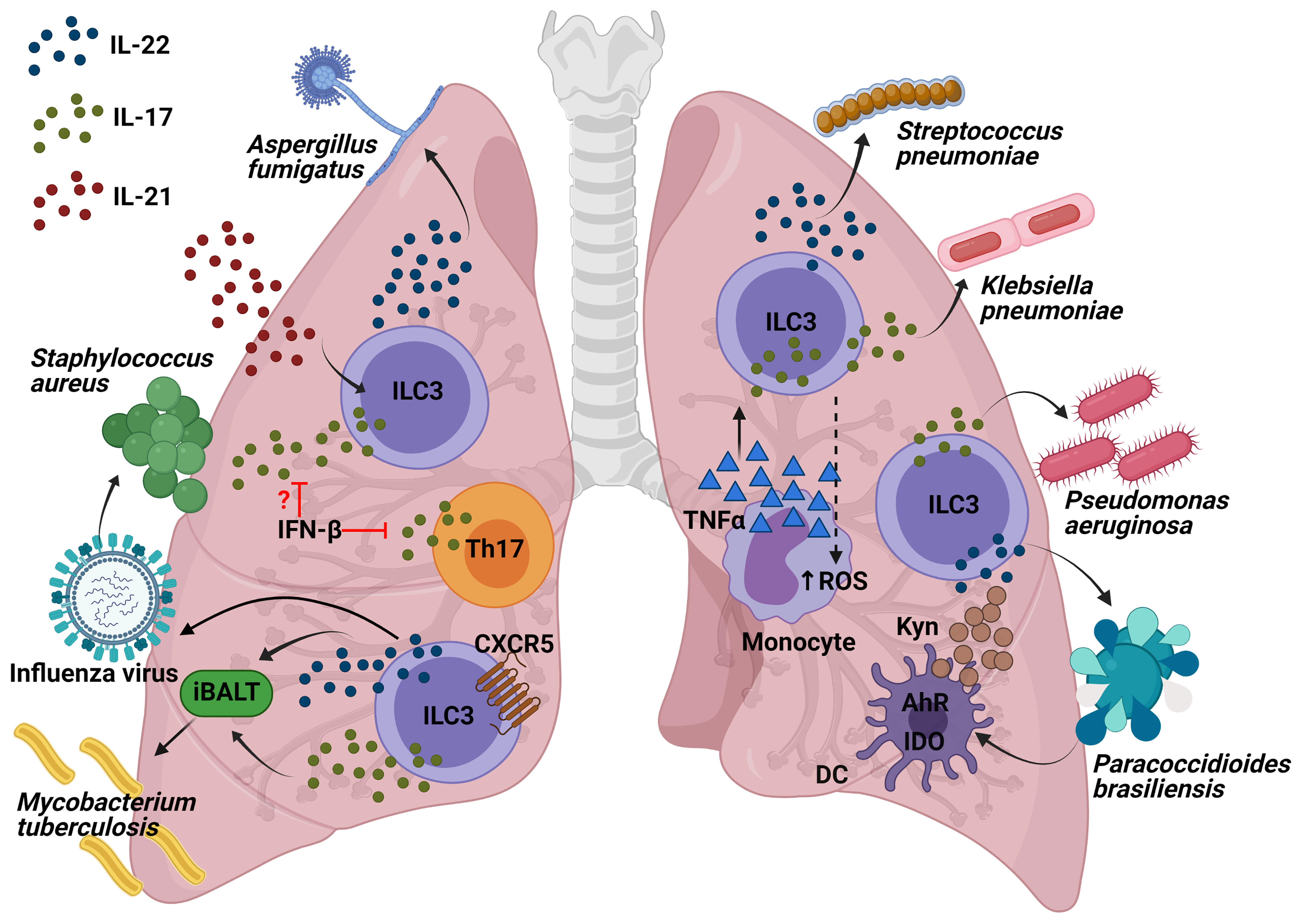
Figure 3 Interleukins produced by ILC3s and the molecular mechanisms of their role to protect against lung pathogens. Production of IL-17 and IL-22 by ILC3s following influenza infection might decrease the susceptibility to Staphylococcus infections. Innate-like lymphocytes are involved in the production of IL-22 following A. fumigatus exposure, being IL-21 the interleukin required for the maintaining of optimal numbers of iNKT cells and ILC3s. Both IL-17 and IL-22 from ILC3s are involved in the formation of iBALT which helps fight against M. tuberculosis. Recruitment of IL-22-producing ILC3s to the lung is required for protection from S. pneumoniae infection. IL-17 production in P. aeruginosa infection might come from ILC3s and it is essential in the defense against this pathogen. The recruitment of monocytes producing TNF-α increases IL-17-secreting ILCs which has been demonstrated to be important in the clearance of K. pneumoniae. AhR is important for IDO expression within the dendritic cells (DCs) as well as their production of kynurenines (Kyn). One of Kyn’s roles is to promote the IL-22 and IL-17-producing ILC3 function in order to fight against P. brasiliensis.
ILC3s Regulate the Mycobacterium tuberculosis Burden During Infection Through the Formation of Inducible Bronchus-Associated Lymphoid Tissue in Lungs
M. tuberculosis (Mtb) is an aerobic bacillus belonging to the Mtb complex, which constitutes the group of mycobacterial pathogens that cause tuberculosis in mammalian species (39). A recently published article has highlighted for the first time the importance of ILC3s in the control of Mtb infection. Ardain et al. demonstrated that ILC3s accumulated rapidly in the lungs of Mtb-infected mice, coinciding with alveolar macrophage accumulation, which has been confirmed in humans through examination of lung tissue from Mtb patients. Importantly, mice lacking ILCs (Rag2−/− cγc−/− mice) exhibited less early alveolar macrophage accumulation and a lower level of Mtb than the control mice. Notably, an increased load of early Mtb in Rag2−/− cγc−/− mice could be reverted by adoptive transfer of sorted lung ILCs from M. tuberculosis-infected control mice that expressed CCR6, RORγt and AhR. These results were further confirmed in mice with specific depletion of ILC3s (Ahrfl/fl Rorc Cre and Cbfbfl/fl Nkp46 Cre mice), which exhibited higher, early and late, Mtb burdens. In order to investigate the possible mechanisms, the migration of mouse ILC3s in response to C-X-C Motif Chemokine Ligand 13 (CXCL13) was confirmed by means of in vitro assays. The receptor C-X-C motif chemokine receptor 5 (CXCR5) expressed in ILC3s is important for the formation of inducible bronchus-associated lymphoid tissue (iBALT), the formation of which showed a decrease in ILC3-depleted, Il-17−/− Il-22−/− and IL-23-depleted M. tuberculosis-infected mice. However, iBALT formation was not entirely via ILC3s, as it was also observed in neonatal Rorc−/− mice. Similarly, Cxcr5−/− mice also exhibited an increase in lung Mtb colony forming units (CFUs) and decreased accumulation of ILC3s within lymphoid follicles, as well as reduced formation of iBALT structures. All these data support a protective role for ILC3s in regulating early Mtb control through the production of IL-17 and IL-22 with the formation of iBALT structures in a CXCR5-dependent manner (40) (Figure 3). Other published studies have confirmed the importance of both NKp46+ ILC3s and Ltis in Mtb infection, which were found to have accumulated in the lungs of mice challenged with aerosolized Mtb (41). In short, ILC3s are important not only for the defence against extracellular pathogens, as has previously been shown, they are also essential for controlling infection by the intracellular bacterium M. tuberculosis.
IL-22-Producing ILC3s in the Gut Are Essential to Fight Against Streptococcus pneumoniae
S. pneumoniae is a gram-positive bacterium that is a common cause of pneumonia, septicaemia, and meningitis. As for IL-22, during S. pneumoniae infection, it has been seen to be produced by CCR6+ ILC3s in a MyD88 dependent manner and triggered by DCs (42). Interestingly, IL-22 has recently been found to be essential in neonate mice, because recruitment of IL-22-producing ILC3s to the lung, by commensal microbes in the gut, was required for protection from S. pneumoniae infection. Treatment of RORγt diphtheria toxin receptor (DTR) newborn mice (Rorc Cre mice crossed with Rosa26-iDTR mice; RorγtiDTR) with DT diminished the number of ILC3s in the lungs and reduced IL-22 in bronchial lavage fluid (BAL), making them more susceptible to pneumonia. Importantly, it was reversed by transferring lung ILC3s. Indeed, reduced concentrations of IL-22 as well as lower numbers of lung IL-22+ ILC3s in BAL were found in human newborns exposed to prolonged durations of antibiotics, thus contributing to increased susceptibility to pneumonia, which was reversed by transfer of commensal bacteria after birth. Intestinal CD103+CD11b+ DCs were able to capture antigens from commensal bacteria to induce the expression of the lung homing signal CCR4 in ILC3s (43). Later on, transgenic Rorc GFP/+ newborn mice were used to confirm the need for pulmonary IL-22-producing ILC3s as a defence against S. pneumoniae infection. Furthermore, part of the development of this population in the lungs is generated by ILC precursors expressing the transcription factor promyelocytic leukemia zinc finger (ZBTB16), because their absence reduced ILC3 numbers from birth and throughout adulthood. The expansion and maturation of pulmonary ILC precursors was mediated by the insulin-like growth factor 1 (IGF-1) from alveolar fibroblasts; consequently, co-transplanting the common lymphoid precursors from newborns lacking IGF1R on ZBTB16+ ILC precursors in Rag2−/− cγc−/− mice (lacking all ILC subsets) made them susceptible to S. pneumoniae intratracheal challenge (44) (Figure 3). More recently, prophylactic intranasal administration of IL-7, an important factor for RORγt+ cell survival and homeostasis, has been found to increase the number of RORγt+ innate T cells in the lung (NKT, γδT cells and mucosal-associated invariant T (MAIT) cells) and to enhance expression of IL-17A, resulting in a reduction of bacterial burdens upon S. pneumoniae challenge (45). In general terms, this brings us to conclude that some of the factors for the generation of pulmonary IL-22-producing ILC3s involve: the commensal microbes in the gut, ZBTB16+ ILC precursors, and the expression of IGF-1 by the alveolar fibroblasts. IL-22-producing ILC3s are important both in the newbornhood and adulthood to control a S. pneumoniae infection and prevent pneumonia.
IL-17 and IL-22 From ILC3s Are Essential for Combating Against Klebsiella pneumoniae Infection
K. pneumoniae is a gram-negative bacterium, found in the normal flora. It can cause different clinical diseases including pneumonia. In recent years, Klebsiella species have become relevant pathogens in nosocomial infections (46). A mouse model of K. pneumoniae infection was used to describe how the recruitment of monocytes producing TNF-α increased IL-17-secreting ILCs which potentiated reactive oxygen species (ROS) production and, therefore enhanced bacteria killing by monocytes. Indeed, both T cell deficient and WT mice exhibited a similar bacterial clearance, but Rag2−/−cγc−/− mice and Rag2−/− depleted of ILCs by means of an anti-CD90 MAb led to increased bacterial burden and mortality. Exogenous IL-17 in K. pneumoniae-infected Rag2−/−cγc−/− mice were able to clear the pathogen. Remarkably, an adoptive transfer of ILCs from Rag2−/− (but not Il-17a−/−Rag2−/−) mice reduced the bacterial burden in the lungs of recipient Rag2−/− cγc−/− mice, thus demonstrating the importance of innate IL-17-producing cells in the defence against K. pneumoniae (Figure 3). Moreover, IL-22 did not seem to be significant in the early defence against K. pneumoniae since its clearance was similar both in Il-22−/− and in the WT mice (47). However, the lung burdens of infected Rag2−/− cγc−/− mice were significantly reduced through addition of exogenous IL-22. Indeed, a distinct group of IL-17+, IL-22+ and inducible T cell costimulatory molecule–positive (ICOS+) ILC3s was detected to be essential for host resistance against K. pneumoniae in lungs from infected Rag2−/− mice, using single cell RNA sequencing (48). In conclusion, IL-17 from ILC3s potentiates the bacterial clearance of K. pneumoniae; furthermore, production of IL-22 by ILC3s could also help to control the infection.
IL-17 from ILC3s Could Be Important in the Fight Against Pseudomonas aeruginosa Infection
P. aeruginosa is a common gram-negative bacterium capable of colonizing a wide range of ecological niches (49). It was previously discovered that exposing mice to C. albicans reduced lung injury and bacterial burden from a posterior P. aeruginosa infection (50), mainly because C. albicans increased the production of IL-22 from ILCs, promoting production of antimicrobial peptides and thus, P. aeruginosa protection (51). Moreover, it has recently been demonstrated in a murine P. aeruginosa pneumonia model that IL-22 upregulated IFN-λ expression and reduced recruitment of neutrophils, which provided a better outcome in the lung pathology of the mice compared with the exacerbated lung inflammation and pathology in mice with IFN-λ or IL-22 neutralization (52). In addition, further data showed that 90% of IL-17 production in P. aeruginosa infection appeared to come from ILC3s rather than CD3+ lineages, including γδ T cells (Figure 3). IL-17 was shown to be important for this infection since Il-17ra−/− mice presented greater pulmonary bacterial loads at 2 weeks following infection by YH5 strain, and with the NH57388A strain, all of which died. However, there is a need for further characterization with CD127 and RORγt as ILC3 markers, in order to be sure of the identity of these IL-17+ CD3low-int CD19- CD11c- NK1.1- cells (53). To conclude, in a P. aeruginosa infection, IL-22 is important for a controlled response and IL-17 is necessary in order to reduce the bacterial load. However, there is still a need to characterize ILC3s in this regard.
Reduced Numbers of ILC3s Provoke a Greater Paracoccidioides brasiliensis Load in Ahr−/− Mice
The genus paracoccidioides (P. brasiliensis and P. lutzii) is a dimorphic fungus that causes paracoccidioidomycosis (PCM), which is a systemic granulomatous mycosis (54). Recent studies of P. brasiliensis infection in Ahr−/− mice have revealed a reduction of the cytokines, indoleamine 2,3-dioxygenase 1 (IDO-1) expression and kyneurine (Kyn) synthesis by pulmonary DCs. This altered pulmonary microenvironment provided a poorer outcome, a greater fungal load and a decrease in the number of NK and ILC3s, with no differences in the ILC1 or ILC2 populations (55) (Figure 3). However, there exists a need to address the potential role of ILC3s in defending against P. brasiliensis with the use of more specific transgenic mice.
The Repercussion of Blood or Systemic Infections in ILC3 Population
Immunodeficiency Viruses Reduce ILC3s in Infected Subjects
Human immunodeficiency virus (HIV) is a retroviral RNA virus that attacks the immune system; it is characterized by a systemic chronic inflammation and if not treated, it can lead to the development of acquired immunodeficiency syndrome (AIDS) (56). Studies published to date on HIV and Simian Immunodeficiency Virus (SIV) infections suggest that there is an early and sustained reduction of ILC numbers, as well as diminished function thereof, most notably ILC3s in the gut; this causes disruption of the mucosal barrier and immune dysregulation. A better understanding of the mechanisms of loss and dysfunction is relevant with regard to designing new immunotherapies for restoring these cells. Different mucosal NK-cell subpopulations were studied in rectal tissue of macaques chronically infected with SIV, finding a reduction of the NKp44+ NK (ILC3s) population and alteration of its functional profile resulting from diminished IL-17 secretion due to IDO1 catabolites, which were secreted by DCs (57). In a subsequent investigation with an HIV model of humanized mice, ILC3s became depleted as a result of CD95 induction in ILC3s by HIV, via a plasmacytoid DC- and IFN-I-dependent mechanism that sensitized ILC3s to undergo CD95/FasL-mediated apoptosis. It was therefore hypothesized that the lack of IL-22 was one of the reasons why the intestinal barrier function was impaired in HIV patients (58–60). On the other hand, other studies suggest that the loss of ILC3s in lymphoid tissues during SIV infection might be due to the induction of apoptosis by microbial products through the toll-like receptor 2 (TLR2) (lipoteichoic acid) and TLR4 (LPS) pathways (61). More recently a study of simian-human immunodeficiency viruses (SHIV), which explored breastfeeding-related HIV infection in newborns, found that infection caused depletion of ILC3s in the gut. The infection altered the trafficking and chemokine receptors in ILC3s, reducing the expression of α4β7 integrin in colonic ILC3s and consequently minimizing IL-22 secretion in colon and oral mucosa. The aforementioned study corroborated the premise that the lentivirus infection could have significant effects on the frequency, phenotype, and function of innate cells in the oral and gut mucosa such as the reduction of ILC3 IL-22+ which might be particularly relevant in infants infected through breastfeeding, because their immune system is not fully functional (62). Importantly, it has recently been demonstrated that NKp44+ ILCs play a protective role in the progression of the disease, as their higher frequency in the mucosa was associated with delayed SIV acquisition and decreased viremia in vaccinated macaques (63), thus confirming previous published data (64).
In human subjects it was found that all ILCs in blood were depleted during infection, but surprisingly a reduction of ILC numbers in the gut was not detected (65). Recently, however, depletion of ILCs has been found in the blood, but this has also been detected in the gut of people with HIV-1, even with effective antiretroviral therapy (ART). It was demonstrated ex vivo that inflammatory cytokines associated with HIV-1 infection irreversibly disrupted ILCs and expanded Transcription Factor 7 (TCF7)-dependent memory NK cells, thus explaining the chronic inflammation in people with HIV-1 (66). Interestingly, preterm birth was also associated with lower levels of all three peripheral ILC subsets in pregnant HIV positive women (67). Moreover, vertically HIV-infected children also exhibited a loss of all circulating ILCs which, unlike CD4+ T cells, were not restored by long-term ART if not initiated at birth (68). Further research is required in order to study the specific consequences of the lack of ILC3s in HIV-infected patients. In summary, ILC3s are depleted in the blood and gut of HIV and SIV infected human and macaque subjects potentially via the CD95 induction or TLRs pathway.
Defensive Role of ILC3s in Gut
Several Transcription Factors Expressed in ILC3s Are Essential in the Fight Against Salmonella and the Possible Subsequent Development of Intestinal Fibrosis
Salmonella enterica serovar Typhimurium (S. typhimurium) is an enteropathogenic bacterium that causes inflammation in both the small and large intestine (69). Salmonellosis comprises major food-borne gastrointestinal diseases caused by the Salmonella species, and most infectious serovars cause enterocolitis or diarrhea. Mice lacking hematopoietic expression of RORα were protected from intestinal fibrosis in a Crohn’s disease model of S. typhimurium because ILC3s produced less IL-17A. Reconstituting lethally irradiated mice with equal numbers of bone marrow (BM) cells from Rag1−/− mice combined with either WT or Rora sg/sg BM cells, showed that RORα-expressing ILC3s were sufficient to cause fibrosis (70) (Figure 4). In addition, diminished expression of a number of ILC3-defining transcripts in the cecum and mLN was observed in Salmonella-infected Rora sg/sg BM transplant chimeric mice using single-cell RNA sequence analysis compared with their WT counterparts. These cells, known as ex-RORγt ILC3s, were similar to ILC1s and promoted Salmonella and IFN-γ–mediated enterocolitis. Therefore, even after chronic Salmonella infection, RORα is still essential with regard to conserving the role of ILC3s in peripheral tissues and their lineage fate (71). In summary, these results show that expression of RORα promotes susceptibility to intestinal fibrosis due to an increase in ILC3 responses.
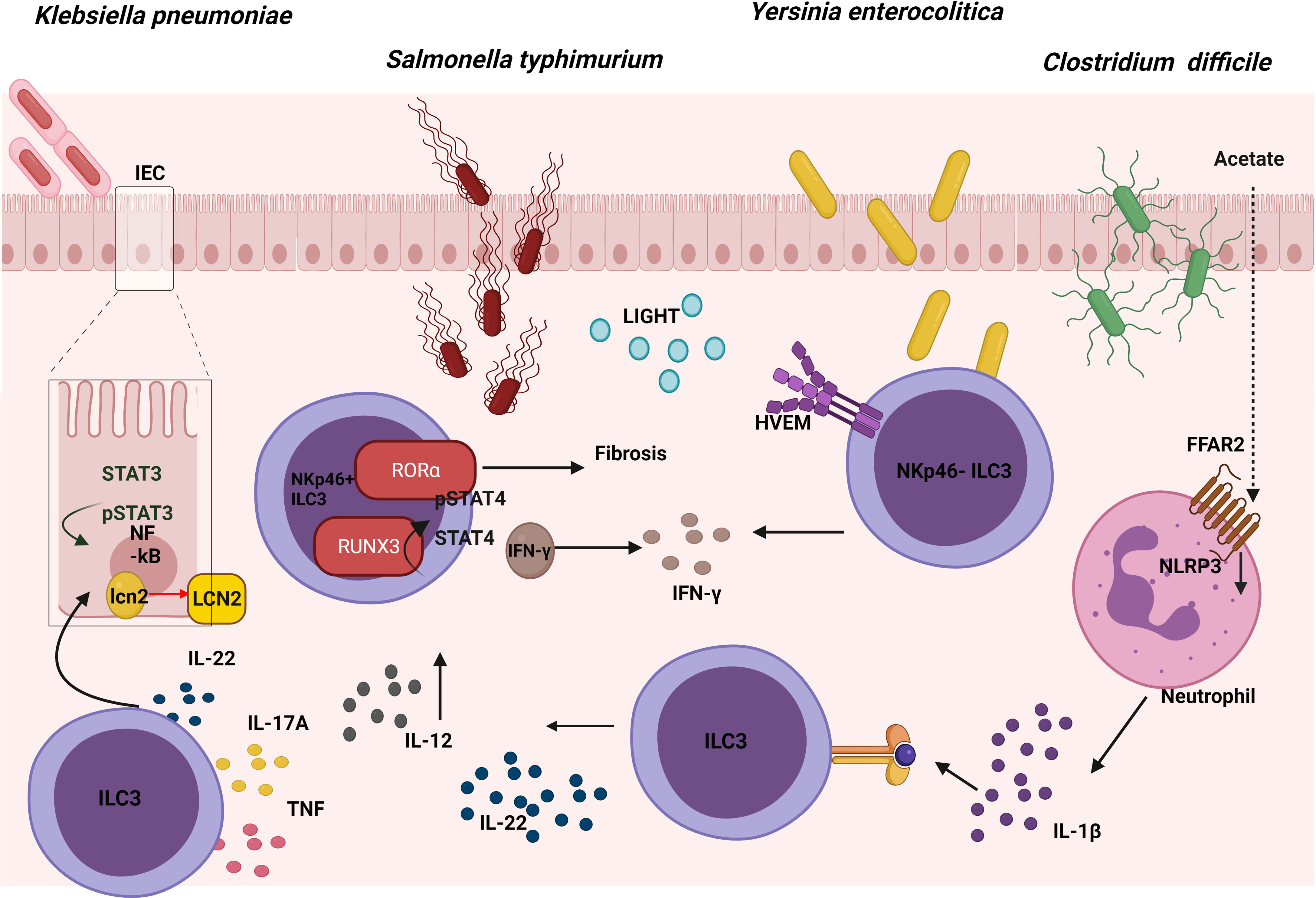
Figure 4 Role of ILC3s in the response against gut pathogens. The expression of RORα in ILC3s leads to intestinal fibrosis in a Crohn’s disease model of S. Typhimurium. At the same time, an up-regulation of RUNX3 expression in ILC3s promotes the IL-12/STAT4/IFN-γ signaling pathway, which is also involved in the fight against this pathogen. Cytokines produced by ILC3s promote the NFkB and STAT3 pathway, both of which are involved in the production of lipocalin-2 (LCN-2) by intestinal epithelial cells (IECs), inhibiting the growth of K. pneumoniae. The binding between LIGHT and its receptor HVEM in ILC3s promotes IFN-γ production which is involved in the defence against Y. enterocolitica. In the defence against C. difficile, acetate promotes the production of IL-1β in neutrophils via the FFAR2-NLRP3 inflammasome axis, activating ILC3s via IL-1R to produce IL-22.
Following oral infection with S. typhimurium a significant increase was observed in the number and percentage of Nkp46+ILC3 cells in the lamina propria (LP) of the murine small intestine, with elevated levels of Runx3 (Runt-related transcription factor 3). To study the role of this transcription factor during infection in these cells, Runx3fl/fl Plzf Cre (Runx3 cKO) mice were generated; these proved to be much more susceptible to S. typhimurium intracellular bacterial infection. Promyelocytic leukemia zinc finger protein (PLZf) is expressed by ILCs during their development, as well as in Th1, CD8+ T cells and several invariant T cells. Flow cytometry was employed to demonstrate that part of the inability to control the infection was because IFN-γ secretion decreased in Nkp46+ILC3s. Previously, it was reported that after binding IL-12 to IL-12R, STAT4 proteins were phosphorylated and accumulated in the promoter areas of IFN-γ to induce its expression. Thus, to analyze the role of this axis, sorted Nkp46+ILC3s from Runx3 cKO mice were stimulated with IL-12; by means of flow cytometry a decrease was seen in pSTAT4 compared with WT, which demonstrated that these cells had impaired the IL-12/STAT4 signaling. Moreover, in order to understand the underlying mechanism, chromatin immunoprecipitation (CHIP) analysis was used to show that RUNX3 was directly bound to the promoter region and intron 8 of the IL12Rβ2 gene (72). In conclusion, upon infection with S. typhimurium, Nkp46+ILC3s upregulated Runx3 expression, which promotes the IL-12/STAT4/IFN-γ signaling pathway, and which would in turn limit intracellular bacterial infection (Figure 4).
Giardia Lamblia Infection Produces IL-17-Expressing ILC3s in Gut
Giardia lamblia is an extracellular protozoan pathogen that inhabits the human small intestine, causing diarrhea (73), which is mainly due to food contamination; it can cause foodborne diseases including giardiasis (74). It has been found that G. lamblia increased the secretion of IL-17A, IL-17F, IL-1β of isolated ILCs from the LP of mouse small intestine in vitro. Moreover, mice inoculated with G. lamblia trophozoites showed more IL-17-expressing ILC3s in the LP of mouse small intestine. Therefore, IL-17-producing ILC3s could be key players upon infection with G. lamblia (75) but further research studying this model in Rag−/− mice with the depletion of Il-17 gene in the RORγt+ cells is required to demonstrate this hypothesis.
ILC3-Derived Cytokines Induce Lipocalin-2 Expression in Epithelial Cells Which Inhibits the Growth of Klebsiella pneumoniae
K. pneumoniae is an opportunistic pathogen commonly related to lung infections, and studies in recent years are beginning to associate it with gastrointestinal tract-related diseases (76). As a response to this infection in the gut, a release occurs of epithelial-derived antimicrobial factors produced by intestinal epithelial cells (IECs). In particular, lipocalin-2 (LCN-2) provides a specific protection against the Enterobacteriaceae family. Furthermore, one of the cell types playing an important role in the regulation of antimicrobial responses in IECs involves ILC3s; in particular, these act by secreting IL-22 during enterobacteriaceae infections. It was demonstrated that IL-22 regulated the expression and production of LCN-2 in IECs which was capable of inhibiting the growth of K. pneumoniae in vitro. The supernatant of NKp44+ and NKp44- ILC3s culture isolated from human tonsil samples and stimulated with IL-2, IL-1β, and IL-23, was able to increase expression of LCN-2 in an epithelial cell line in vitro. The importance of IL-22 has been confirmed by the use of anti-IL-22 Mab, which reverted LCN-2 expression and protein release in IECs in vitro. All these data supported the premise that ILC3-derived IL-22 could enhance the expression of LCN-2 in human IECs (77) (Figure 4). The role of other cells in this mechanism remains to be established, and the next step should involve demonstrating this mechanism in an in vivo model. To this end, neonatal mice exposed to a short (SE) or long (LE) exposure of broad-spectrum antibiotics were examined, thus observing that the LE mice were more susceptible to K. pneumoniae-induced sepsis at 2 weeks of life. Appreciably, the percentage of ILC3s and their capacity to produce IL-17A in the LP showed a decrease in the LE mice and were capable of partially rescuing the physiological phenotype by means of reconstitution with mature microbiota (78). This study supported a role for microbiota dependent suppression of IL-17A-producing ILC3s in increasing susceptibility to late-onset sepsis. In conclusion, cytokines produced by ILC3s can regulate the intestinal epithelial cells in order to defend against K. pneumoniae.
ILC3-Derived IFN-γ Production Protects Against Yersinia enterocolitica Infection
Y. enterocolitica is a gram-negative bacterium that produces enterocolitis and can cause infection by ingestion of contaminated food, particularly raw or undercooked pork products, and of unpasteurized milk (79). Remarkably, Ahrfl/flRorc Cre mice were used to demonstrate that ILC3s play a significant role in the defence against Y. enterocolitica. In order to investigate which ILC3 subset was necessary, Rorcfl/flNkp46 Cre mice were infected. This showed that the NKp46+ ILC3 subset was not required to mediate early host defence. Interestingly, the survival, weight loss and areas of necrosis were rescued by performing an adoptive transfer of NKp46- ILC3 into Rag2−/−cγc−/− infected with Y. enterocolitica. However, the same result was obtained transferring equal numbers of NKp46+ ILC3s, indicating that these cells also had a protective capacity in immunosuppressive mice but were redundant in immuno-competent mice [also demonstrated in C. rodentium infection model (80)]. By contrast, recipient mice injected with CCR6+ ILC3s sorted from LP of the small intestine (LTi-like cells) were not protected. In addition, bacterial translocation in Ifng−/−, IL-17Rra−/−, and IL-22−/− mice after oral Y. enterocolitica infection showed that IFN-γ was required for host defence, especially those produced by NKp46- ILC3. An adoptive transfer of CCR6− ILC3s from Ifng−/− or WT mice in Rag2−/− cγc−/− mice confirmed the importance of IFN-γ production by ILC3s, but, surprisingly, IL-17 and IL-22 cytokines had little effect on the outcome. Previous work suggested that the herpesvirus entry mediator (HVEM) expression was important as a regulator of the mucosal immune system in multiple cell types (81); it was expressed by all small intestine ILC subsets in mice and all ILCs in human peripheral blood. Therefore, Hvem−/−, Hvemfl/fl Rorc Cre, and Hvemfl/fl Cd4 Cre mice were infected, showing that ILC3s (but not CD4+T cells expressing HVEM) were essential for the early protection of Y. enterocolitica. Indeed, RORγt-mediated deletion of HVEM only decreased IFN-γ secretion from ILC3s. The essential role of HVEM in ILC3s was confirmed by the poorer outcome of an adoptive transfer of Hvem−/− ILC3s into Rag2−/− cγc−/− mice infected with Y. enterocolitica, in comparison with the adoptive transfer of WT ILC3s. IFN-γ production by ILC3s increased through a HVEM ligand known as LIGHT, at both the transcript and protein levels. All these data showed that HVEM signaling mediated by LIGHT plays a critical role in regulating ILC3-derived IFN-γ production for protection against Y. enterocolitica following infection (82) (Figure 4). In summary, the signature ILC3 cytokines, IL-22 and IL-17A, are not important for protection against Y. enterocolitica, but the IFN-γ produced by these cells does play an important role.
Acetate-Induced IL-22 Secretion by ILC3s Could Help to Defend Against Clostridium difficile
C. difficile is an opportunistic anaerobic gram-positive bacterium that causes most nosocomial antibiotic–associated diarrheas and colitis. During colonization, pathogenic strains of C. difficile produce 2 exotoxins that induce acute inflammation, cell necrosis, and fluid secretion in/into the gastrointestinal tract (83). The role of ILC3s in the defence against C. difficile was studied in the murine model and although upregulation of the expression of ILC1- or ILC3-associated proteins in the large intestine was detected following C. difficile infection; selective loss of ILC3s (Rorc−/− mice) or ILC3 effector molecules (Il-22−/− and Il-17−/− mice) did not affect the survival or the severity of the infection. This would appear to suggest that ILC3s made a minor contribution to resistance, while the loss of IFN-γ or T-bet-expressing ILC1s in Rag1−/− mice increased susceptibility to C. difficile (84). However, according to the research conducted by J. L. Fachi, who investigated whether hypoxia could regulate the activation of ILC3s, hypoxia inducible factor-1α (HIF-1α)-deficient RORγt mice infected with C. difficile presented more severe infection compared with their WT counterparts, although they displayed significantly lower numbers of gut ILC3 and higher numbers of ILC1s (85). The aforementioned group previously investigated previously the mechanism through which oral administration of acetate enhanced the host’s resistance to C. difficile. Acetate-treated mice showed a moderate increase in the number of ILC3s (but not that of other ILCs) in the gut and increased expression of RORγt and IL-22 specifically in colon. In addition, free fatty acid receptor (Ffar)2−/− and Rag2−/− cγc−/− transgenic mice presented a more severe infection, although acetate was not triggering the expression of IL-22 through FFAR2 directly in ILC3s in vitro. As neutrophils also expressed FFAR2, acetate actually stimulated production of IL-1β through the FFAR2-NLRP3 inflammasome axis in these cells, thus activating ILC3s through IL-1R (86) (Figure 4). Therefore, acetate supplementation improves protection against C. difficile in colon by inducing the production of IL-22 in ILC3s via FFAR2/IL1-β axis in neutrophils. Considering the controversy of the possible role of ILC3s in the defense against C. difficile, there is a need for further study with other transgenic mice in order to elucidate the role play by ILC3s in this infection model.
An Increase in IL-22-Producing ILC3 Cells Following Exposure of Dectin-3−/− Mice to Candida albicans Promotes Colitis-Associated Colon Cancer
Among the fungi that resides in the gastrointestinal tract of healthy individuals, Candida is the dominant one. In order to sense a fungi infection and induce an activation of downstream signaling, on their surface pattern innate immune cells have recognition receptors such as mammalian C-type lectin receptors. In particular, Dectin-3 receptors recognize α-mannans on the surface of multiple fungi such as C.albicans, Paracoccidioides brasiliensis, and Cryptococcus. Interestingly, Dectin-3−/− mice have recently been found to present enhanced colitis-associated colon cancer (CAC) correlating with an increase in the C. albicans burden. The Dectin-3−/− mice recruited more macrophages and ILC3s to mesenteric LNs and LP tissues than the WT tumor-bearing mice. Regarding the mechanism, it was shown that an elevated C. albicans load triggered glycolysis and IL-7 production through a HIF-1α-dependent pathway in macrophages from LP in Dectin-3−/− mice. Importantly, IL-7 induced IL-22 secretion in sorted ILC3s in vitro via AhR and STAT3. In conclusion, an increase in IL-22 production by ILC3s through STAT3 and AhR promotes CAC progression as a result of a Dectin-3 deficiency in macrophages, which increases IL-7 production through HIF-1α-dependent glycolysis (87) (Figure 5).
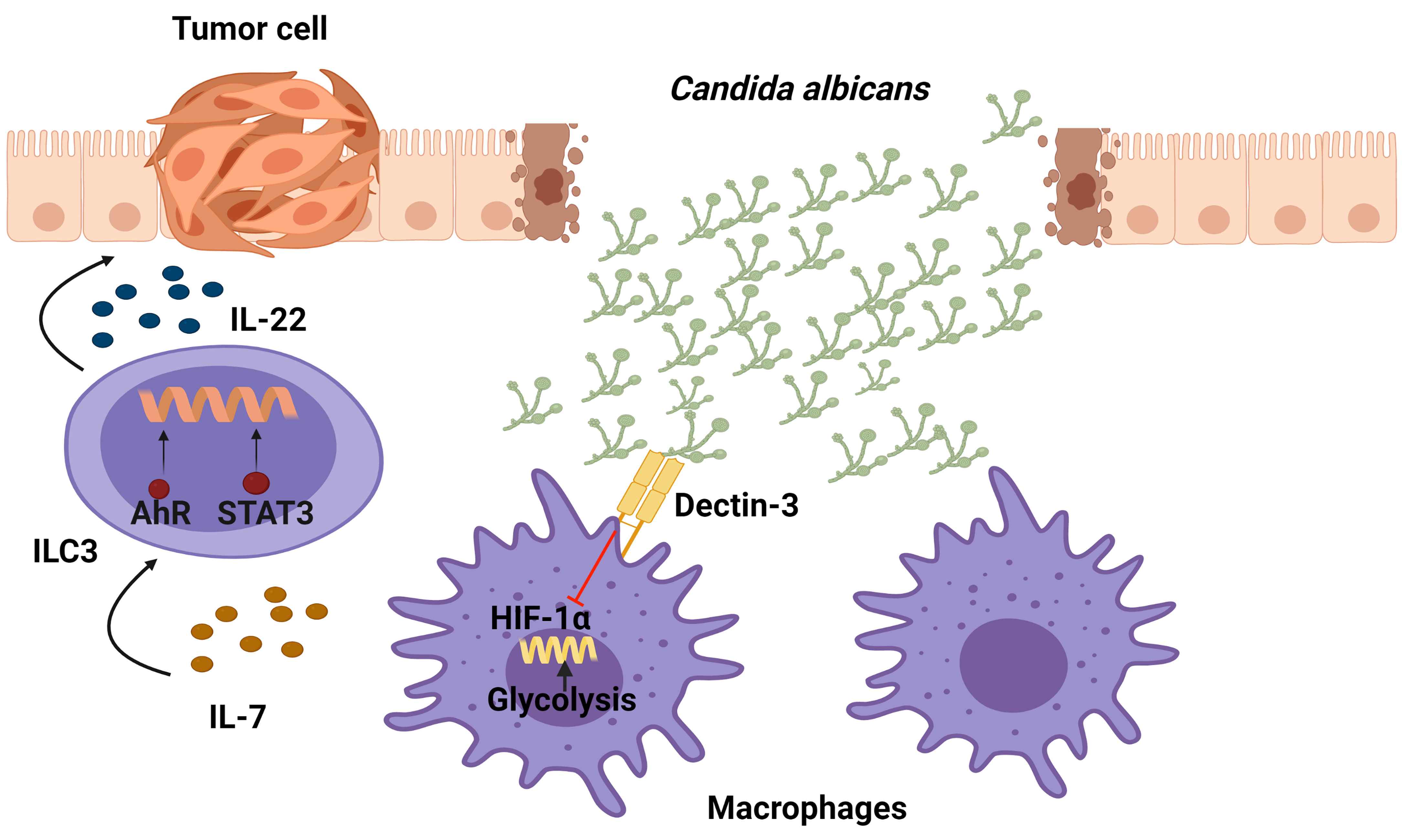
Figure 5 Function of IL-22-producing ILC3 cells in the development of colitis-associated colon cancer (CAC) following exposure of Candida albicans. Dectin-3, expressed by myeloid cells, recognizes this fungus and down-regulates the production of IL-7. In Dectin-3-deficient mice, macrophages display a HIF-1α-dependent glycolysis which brings ILC3s to produce IL-22 (through STAT3 and AhR), leading to the progression of CAC.
Several Signals Can Regulate ILC3 Responses Against Citrobacter rodentium
C. rodentium is a gram-negative mucosal pathogen that shares several pathogenic mechanisms with enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic E. coli (EHEC); it therefore constitutes a useful murine model for investigating infectious colitis as well as intestinal bowel disease (IBD), enabling an assessment of the mucosal immune responses, host-pathogen interactions and wound healing, among others (88). Previous field studies have demonstrated that ILC3s plays a key role in the control of C. rodentium infection by secreting large amounts of IL-22 and IL-17 (11, 13, 89). However, some studies support the idea that Nkp46+ ILC3s prove to be redundant for control of C. rodentium infection in the presence of T cells; these studies involved the use of transgenic mice with conditional deletion of the key ILC3 genes Stat3, Il-22, Tbx21 and Mcl1 in Nkp46-expressing cells (80). In recent years, different signals have been found to regulate ILC3 responses against C. rodentium.
Hypoxia: ILC3s have been observed to undergo metabolic reprogramming combining glycolysis with mitochondrial ROS production to fight C. rodentium infection. Rag1−/− mice lacking the gene for the mTOR complex 1 (mTORC1) subunit Raptor in RORγt-expressing cells, or WT mice treated with rapamycin, showed greater susceptibility to C. rodentium infection. Significantly, Raptorfl/fl Rorc Cre mice presented much lower numbers of ILC3s at steady state suggesting a possible role in haematopoiesis although this possibility was not addressed in the aforementioned research. To study whether mTORC1-HIF-1α could drive an impact in ILC3 function, ILC3 cell line MNK3 and primary intestinal ILC3s were activated in vitro and incubated with mTORC1 and HIF-1α inhibitors, showing a decrease in IL-17A and IL-22 production, as well as proliferation. Therefore, the mTORC1 pathway could sustain ILC3s activation, cytokine production, and proliferation, but there is a need to conduct further studies in order establish the function of mTORC1-HIF-1α pathway in C. rodentium infection in vivo (90).
Microbiota: Microbiota-derived short-chain fatty acids (SCFAs) promoted IL-22 production by CD4+ T cells and ILC3s both in vitro and in vivo, resulting in a significant inhibition of C. rodentium-colitis. In addition, in vitro assays with G protein-coupled receptor (GPR)41(FFAR3)-specific agonist and other inhibitors showed that butyrate promoted IL-22 production through the FFAR3, activating HIF-1α, AhR, STAT3 and mTOR proteins. In contrast, IL-22 was induced by butyrate at similar levels in WT and GPR109a-deficient or GPR43(FFAR2)-deficient CD4+ T cells, suggesting that it was particularly dependent on GPR41 (91). However, the deficiency of FFAR2 in RORγt-expressing cells caused a decrease in in situ proliferation and IL-22 production, specifically in colonic CCR6+ILC3s (and not in Nkp46+ ILC3s) which led to increased susceptibility to C. rodentium infection in Ffar2fl/fl Rorc Cre mice. Additionally, FFAR2 agonists increased ILC3-derived IL-22 via an AKT and STAT3 axis in vitro (92). Interestingly, neither alterations in cell frequency nor IL-22 nor IL-17A production in colonic RORγt+ CD4+ T cells from Ffar2fl/fl Rorc Cre mice were observed, as suggested the data published by Yang in CD4+ T cells (91). In this regard, it has recently been published that SCFAs, produced by the commensal microbiota from dietary fibres (DF), supported optimal expansion of all ILCs via both FFAR2 and FFAR3 receptors. Mice fed with a DF diet increased the number of ILC3s in the gut but not in the lymphoid organs, improving intestinal immunity to C. rodentium infection. In addition, the signaling triggered by SCFAs on ILCs was analyzed in vitro, showing an activation of Phosphoinositide 3-kinase (PI3K), STAT3, STAT5, and mTOR, which is important for ILC proliferation. Furthermore, FFAR2 was required for normal expansion of ILC1s and ILC3s both in the intestine and in systemic tissues following C. rodentium infection; this was demonstrated with the use of Ffar2−/− mice (93) (Figure 6). To summarize, the protection against C. rodentium provided by IL-22 secreting-ILC3 cells can be activated by FFAR2 and FFAR3 triggered by microbiota-derived SCFAs.
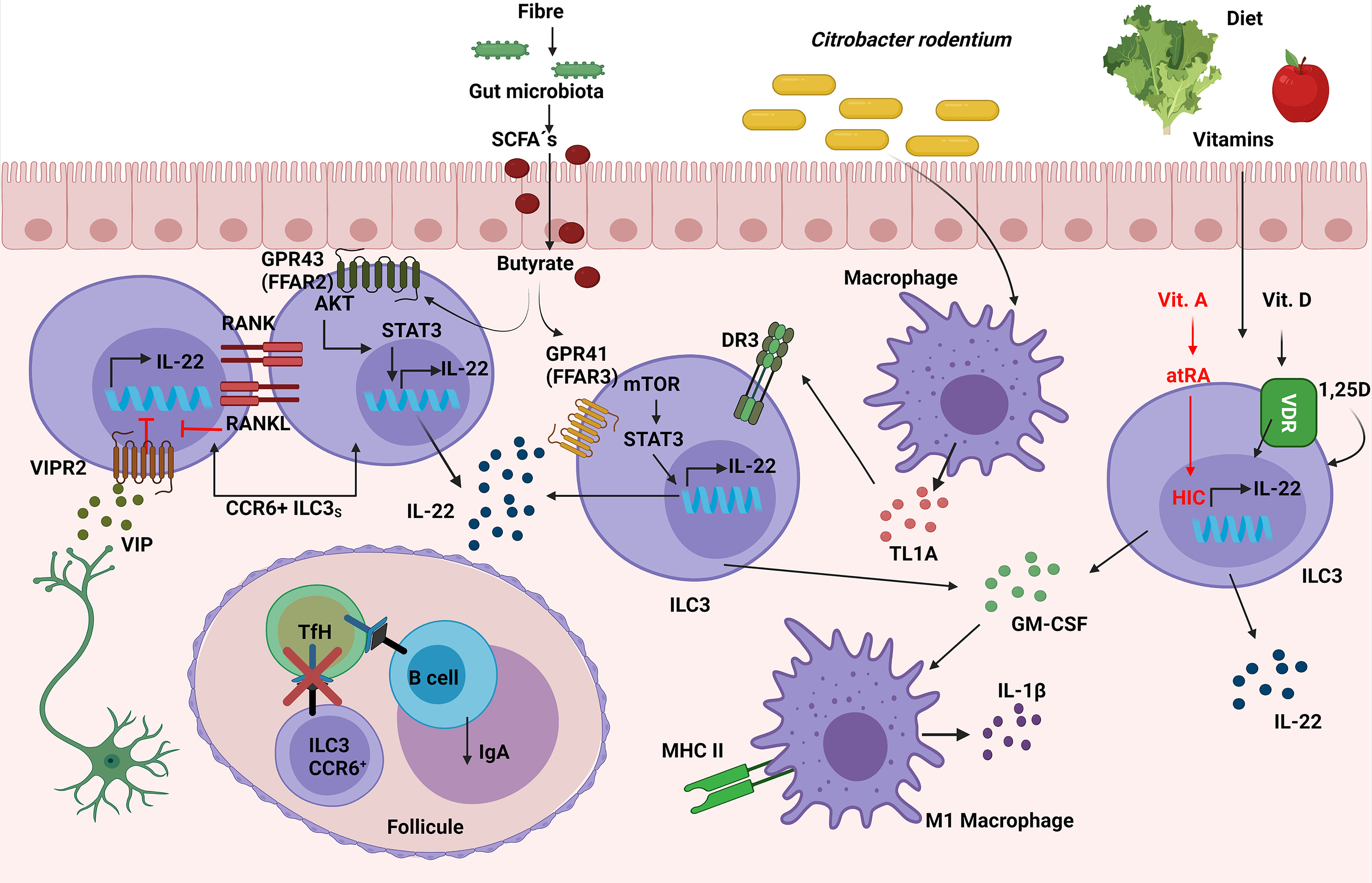
Figure 6 Role of ILC3s in the response against C. rodentium. Vasoactive intestinal peptide (VIP), released by neurons in lymphoid patches in close contact with CCR6+ ILC3s, inhibits their IL-22 secretion. Simultaneously, ILC3s have a negative intrinsic modulation, expressing both RANK and RANKL, which suppresses the induction of IL-22 and IL-17. Susceptibility to C. rodentium infection is caused by these inhibitory mechanisms. The interaction between CCR6+ ILC3 and TfH cells through MHC-II in the mesenteric lymph nodes limits the TfH responses as well as pathogen specific-IgA. Simultaneously, ILC3s expression of TL1A receptor DR3 confers protection against C. rodentium. SCFAs produced by the commensal microbiota from dietary fibers increase IL-22 secretion in ILC3s via the FFAR2/FFAR3-AKT/mTOR/STAT3 molecular pathway, thus contributing to the elimination of the infection. Moreover, ILC3-derived GM-CSF production can improve antimicrobial responses by increasing the expression of IL-1β and MHC-II in macrophages (M1-like macrophages). Furthermore, Vitamins D and A in the diet increase ILC3’s synthesis of IL-22, which protects against C. rodentium; vitamin A’s molecular mechanism is via HIC (Hypermethylated in cancer 1).
Diet: Vitamins can also regulate innate responses to infections. Vitamin A metabolite all-trans-retinoic acid (atRA) has been implicated in the regulation of immune tolerance to food antigens, partially by regulating ILC responses in the intestine. The role of the transcription factor Hypermethylated in cancer 1 (HIC1), previously identified as an all-trans retinoic acid (atRA) responsive gene in intestinal Th cells, was essential for the regulation of ILC3s in the defence against C. rodentium, because Hic1flflRorc Cre mice treated with depleted anti-CD4 Mab were more susceptible to the infection and showed less RORγt+ T-bet+ ILC3s in LP (94). The role of vitamin D in the clearance of C. rodentium infection was tested in Cyp27B1 (vitamin D 1α-hydroxylase mimicking Vitamin D deficiency) KO mice previously fed with a low vitamin D diet (D-), making them more susceptible due to lower levels of IL-22 from both ILC3s and Th17 than mice with a vitamin D supplemented diet (D+). ILC3s were important with regard to maintaining the defence against C. rodentium, because Rag−/− mice on a D- diet presented lower levels of IL-22 from ILC3, and therefore had more severe colitis and a higher mortality than their D+ diet littermates. Furthermore, this phenotype was rescued by administering 1,25D which raise ILC3s and IL-22 levels, or by administering IL-22Fc fusion protein IL-22 in vivo, thus proving that vitamin D was required for healthy regulation of ILC3 response against C. rodentium (95) (Figure 6). In short, vitamins A and D are are needed to maintain suitable levels of IL-22 secretion from ILC3s in order to enhance the protection against C. rodentium.
Nervous system: Previous studies have associated ILC3s with the mucosal neural system (16). It was demonstrated that ILC3s expressed circadian clock genes and IL-17/IL-22-producing ILC3s diminished significantly in Arntlfl/fl Vav1 Cre and the Arntlfl/fl Rorc Cre mice (conditional deletion of the master circadian activator aryl hydrocarbon receptor nuclear translocator-like gene Arntl in hematopoietic cells and RORγt-expressing cells respectively), showing greater susceptibility to C. rodentium. This ILC3 reduction in the gut was not due to an alteration of the numbers of common lymphoid progenitors but rather involved an impairment of the migration of ILC3s to LP, because ILC3s from Arntlfl/fl Rorc Cre mice displayed reduced expression of CCR9, an essential chemokine receptor for intestinal LP migration. In general terms, circadian clock genes play a significant role in the migration and regulation of ILC3s for fighting against C. rodentium (96).
As for the modulation of ILC3s by the nervous system, CCR6+ ILC3 which expressed neural-related genes such as the vasoactive intestinal peptide receptor 2 (VIPR2) were in close proximity with VIP+ neurons in the cryptopatches and lymphoid patches of ileum and colon. VIPR2−/− CCR6+ ILC3s generated more IL-22, which was inhibited by using VIPR2 agonists, suggesting that VIP inhibited the production of IL-22 in ILC3s. Additionally, with the use of DREADD mice (designer receptors exclusively activated by designer drugs in VIP+ cells), VIP neurons were shown to modulate CCR6+ ILC3 cytokine secretion through VIPR2, which is key in the protection against C. rodentium. The DREADD inhibition of VIP+ neurons provided protection against C. rodentium; this is in contrast with DREADD activation in the presence of C. rodentium, which led to severe disease that could be reversed with the administration of recombinant mouse IL-22, indicating that activation of VIPergic neurons may be contributing to intestinal barrier malfunction (97) (Figure 6). Thus, the neuron system regulates ILC3 response to C. rodentium by the modulation of Vipergic neurons.
Cytokines or chemoattractant: The primary source of tumor necrosis factor-like cytokine 1A (TL1A) in the gut are CX3CR1+ MNPs and interestingly, Rag2−/− Tnfsf25fl/fl Rorc Cre (RORγt+ cells deficient for TL1A receptor DR3) mice infected with C. rodentium lost more weight and presented lower survival rates compared to their littermate controls, thus revealing a protective mechanism of TL1A in ILC3s during acute colitis (98) (Figure 6). As for chemotactic receptors, it has been shown that GPR183, which is normally expressed by follicular B cells, DCs and CD4+ T cells, was also highly expressed in ILC3s. Additionally, GPR183 was found to be an important receptor for the migration of ILC3s towards its ligand 7a,25-OHC (produced by stromal cells); this was revealed by means of a transwell migration assay in vitro with sorted GPR183−/− ILC3s. Moreover, Gpr183LacZ/+ (KO reporter mice) mice exhibited lower numbers of ILC3s and fewer cryptopatches, a fact which made them more susceptible to colitis by C. rodentium infection, demonstrating that GPR183 and 7a,25-OHC controlled the distribution and accumulation of ILC3s in the mLNs and Peyer’s patches (99). Therefore, GPR183 plays an essential role in terms of migration and distribution of ILC3 and consequently in a C. rodentium infection. As for TL1A, it would be of general interest to study in greater depth the molecular pathway involved in the protection against C. rodentium.
Membrane proteins: NKR-P1A, expressed in human NKs, is a type II transmembrane protein belonging to the C-type lectin superfamily and which can recognize C-type lectin-related (Clr) proteins. E. Abou-Samra et al. found that ILC3s in the gut also expressed NKR-P1B (homologue of NKR-1PA in mice). Although NKR-P1B-deficient mice showed higher numbers of ILCs and γδT cells in steady state, lower levels of IL-22 were observed. Moreover, upon C. rodentium infection NKR-P1B-deficient mice had more CFUs showing lower levels of IL-22 production from CCR6+ ILC3s than their control WT mice. Similar results were obtained in Rag1−/−Nkr-p1b−/− mice demonstrating the importance of NKR-P1B in ILC3s in the context of infection (100). As a result, the NKR-P1B ILC3 membrane protein is required to regulate ILC3 numbers and function, although further research on the molecular mechanism is needed.
Another molecular regulation of ILC3s is based on the receptor activator of nuclear factor κ B (RANK) interaction with RANK Ligand (RANKL). A novel negative intrinsic regulation of production of IL-22 and IL-17 by ILC3s was discovered by generating Tnfrsf11afl/fl Rorc Cre (RANK ligand deficient mice in RORγt-expressing cells) and Tnfrsf11fl/fl Rorc Cre (RANK deficient mice in RORγt+ cells). Tnfrsf11fl/fl Rorc Cre mice exhibited hyperresponsive CCR6+ ILC3s and increased IL-17/IL-22 production, presenting less severe colitis upon C. rodentium infection. Rag1−/− Tnfrsf11fl/fl Rorc Cre mice had a similar outcome of the disease demonstrating that the regulation of RANK-RANKL was in ILC3s and not in T cells (101) (Figure 6). Several years ago, AhR was shown to constitute a relevant transcription factor relevant for combating C. rodentium, as knocking out AhR resulted in a loss of IL-22+ ILC3s (4, 102, 103). It has recently been determined that AhR expression in RORγt+ cells alone was sufficient with regard to maintaining a functional ILC3 compartment and controlling C. rodentium infection; this was demonstrated by the complete protection of AhrCAIR/CAIR Rorc Cre mice, an AhR-knockin mouse model that expressed a constitutively active form of AhR (CA-AhR) only in RORγt+ cells (104). Conclusively, AhR is required for ILC3 to produce IL-22 in the presence of C. rodentium, and ILC3s possess intrinsic self-regulation of their immune response, which is mediated via the RANK-RANKL interaction
ILC3s can regulate other immune cells during C. rodentium infection through GM-CSF secretion. Mice lacking ILCs and T cells (Rag1/2−/− treated with CD90.2 Mab) were more susceptible to C. rodentium infection; they presented an accumulation of immature monocyte in gut following dextran sulfate sodium colitis and a reduction of pro-IL-1β expression in intestinal mononuclear phagocytes. As ILC3s were the major GM-CSF-producing cell type in the colon during colitis, sorted RORγt+ ILC3s or ILC3-conditioned media were co-cultured with BMDM, resulting in an enhancement of the IL-1β, MHC-II expression in BMDM and suppression of IL-10 (characteristics of M1 macrophage). Importantly, these effects were inhibited by the addition of a GM-CSF-neutralizing antibody. In addition, an increase was also observed in the expression of genes associated with M2 macrophage activation in colonic macrophages of ILC-depleted mice in vivo. Consequently, these data reveal that GM-CSF-mediated ILC3-macrophage crosstalk play a vital role in calibrating the intestinal macrophage phenotype to enhance anti-bacterial responses (105) (Figure 6).
It has been shown that Lti-like ILC3s might also be interacting with follicular T helper cells (TfH) in the interfollicular region of mesenteric lymph nodes, thus limiting the TfH response and B cell-derived IgA production against mucosal microbiota as well as B cell class switching through MHC-II antigen presentation in steady state. This interaction between Lti-like ILC3s and TfH cells was also relevant in C. rodentium infection, which was demonstrated with the use of mice lacking MHC-II in ILC3s, observing greater numbers of TfH cells and pathogen-specific IgA after infection with C. rodentium. Further studies are required to determine the function of Lti-like ILC3 interactions with TfH and the consequences on TfH function in infection (106) (Figure 6).
Discussion
The importance of cytokine release for combatting pathogens has long been extensively described. The present review addresses the latest studies on ILC3s in the early release of cytokines in various organs of the body in mice and humans - in particular the secretion of IL-17 and IL-22, and their role in the early response to a wide range of pathogens. Although ILC3s have previously been described to be important for defending against extracellular bacteria, recent evidence suggests they can also provide defence against parasites or even viruses. And apart from playing an essential role in the gut, they also have an important function in other organs. Furthermore, the present review highlights the new molecular pathways that have recently been described for the different roles ILC3s play in each organ (Table 1); this renders our review a useful tool for further investigation in the field. It is important to highlight that the methodology employed to study the molecular mechanisms involved in the function of ILC3s during infections in vivo, entails generating conditional transgenic mice, usually in RORγt-expressing cells, and in order to avoid the contribution of T cells, these mice are crossed with Rag−/− mice. Consequently, the function of RORγt-expressing cells, which include not only Nkp46+ ILC3s or Nkp46- but also Lti cells, has been studied in immunosuppressive mice. Other experimental approaches involve depleting genes in Nkp46+ cells; this has led to some groups discovering that Nkp46+ ILC3s are redundant during infection in the presence of T cells (immunocompetent mice). In general terms, there is a need for further study of ILC3s and their molecular mechanisms at play in the different diseases of each organ in order to develop new therapies or to make earlier diagnoses.
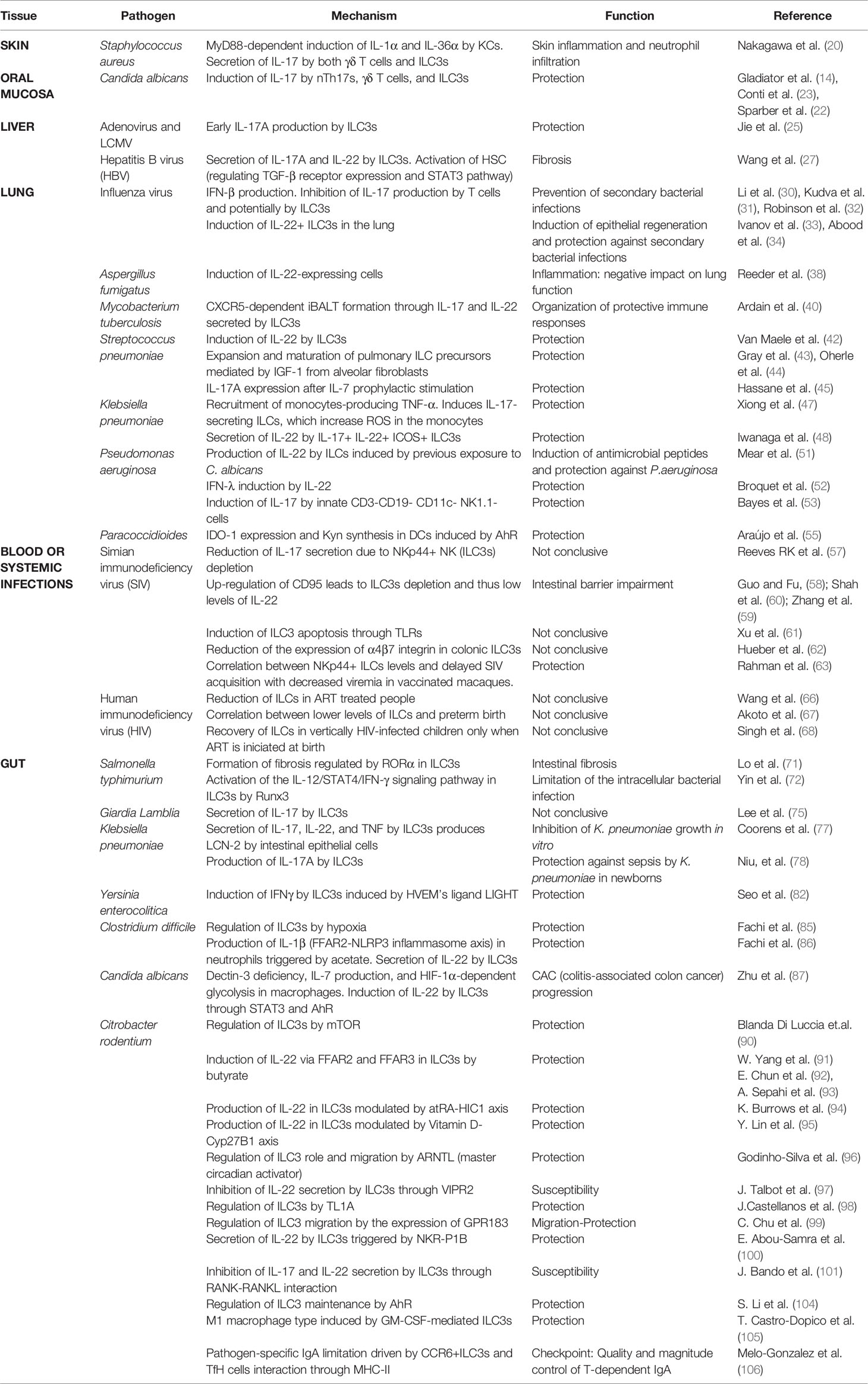
Table 1 Main results of the function and molecular mechanism described in each research article divided by tissue and pathogen.
Author Contributions
AV-N, AO-R and AC-A wrote and prepared the manuscript. MG-S designed the figures. All authors contributed to the article and approved the submitted version.
Funding
The present review was supported by Ramon y Cajal Program (RYC-2017-21837) and the grant N° RTI2018-093647-B-I00 to AC-A from the Ministerio de Ciencia, Innovación e Universidades (MCIU), Agenda Estatal de Investigación and Fondo Europeo de Desarrollo Regional (FEDER). A.V-N is a recipient of an FPI fellowship (PRE2019-090341) from the Spanish Ministry of Science, Innovation, and Universities.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Cormac de Brun, a native English-speaking science editor, who copyedited the manuscript text. Figures were created using BioRender.
References
1. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
2. Valle-Noguera A, Gómez-Sánchez MJ, Girard-Madoux MJH, Cruz-Adalia A. Optimized Protocol for Characterization of Mouse Gut Innate Lymphoid Cells. Front Immunol (2020) 11:563414. doi: 10.3389/fimmu.2020.563414
3. Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. Rorγt and Commensal Microflora Are Required for the Differentiation of Mucosal Interleukin 22-Producing NKp46+ Cells. Nat Immunol (2009) 10:83–91. doi: 10.1038/ni.1684
4. Qiu J, Heller JJ, Guo X, Chen ZME, Fish K, Fu YX, et al. The Aryl Hydrocarbon Receptor Regulates Gut Immunity Through Modulation of Innate Lymphoid Cells. Immunity (2012) 36:92–104. doi: 10.1016/j.immuni.2011.11.011
5. Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, et al. Human Fetal Lymphoid Tissue–Inducer Cells Are Interleukin 17–Producing Precursors to RORC + CD127 + Natural Killer–Like Cells. Nat Immunol 2008 101 (2009) 10:66–74. doi: 10.1038/ni.1668
6. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-Dependent Crosstalk Between Macrophages and ILC3 Promotes Intestinal Homeostasis. Science (2014) 343:1249288. doi: 10.1126/science.1249288
7. Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-Bet Gradient Controls the Fate and Function of CCR6 – Rorγt + Innate Lymphoid Cells. Nat (2013) 494:261–5. doi: 10.1038/nature11813
8. Rankin L, Groom JR, Chopin M, Herold M, Walker JA, Mielke LA, et al. T-Bet Is Essential for NKp46+ Innate Lymphocyte Development Through the Notch Pathway. Nat Immunol (2013) 14:389. doi: 10.1038/NI.2545
9. Mebius RE, Rennert P, Weissman IL. Developing Lymph Nodes Collect CD4+CD3– Ltβ+ Cells That Can Differentiate to APC, NK Cells, and Follicular Cells But Not T or B Cells. Immunity (1997) 7:493–504. doi: 10.1016/S1074-7613(00)80371-4
10. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Group 3 Innate Lymphoid Cells Mediate Intestinal Selection of Commensal Bacteria-Specific CD4+ T Cells. Science (2015) 348:1031–5. doi: 10.1126/science.aaa4812
11. Satoh-Takayama N, Vosshenrich CAJ, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial Flora Drives Interleukin 22 Production in Intestinal NKp46+ Cells That Provide Innate Mucosal Immune Defense. Immunity (2008) 29:958–70. doi: 10.1016/j.immuni.2008.11.001
12. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate Lymphoid Cells Promote Anatomical Containment of Lymphoid-Resident Commensal Bacteria. Science (2012) 336:1321–5. doi: 10.1126/science.1222551
13. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) Lymphoid Tissue-Inducer Cells Promote Innate Immunity in the Gut. Immunity (2011) 34:122–34. doi: 10.1016/j.immuni.2010.12.009
14. Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting Edge: IL-17–Secreting Innate Lymphoid Cells Are Essential for Host Defense Against Fungal Infection. J Immunol (2013) 190:521–5. doi: 10.4049/jimmunol.1202924
15. Julliard W, De Wolfe ÃTJ, Fechner JH, Safdar ÃN, Agni R, Mezrich JD. Amelioration of Clostridium Difficile Infection in Mice by Dietary. Ann Surg (2017) 265:1183–91. doi: 10.1097/SLA.0000000000001830
16. Ibiza S, García-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, et al. Glial-Cell-Derived Neuroregulators Control Type 3 Innate Lymphoid Cells and Gut Defence. Nature (2016) 535:440–3. doi: 10.1038/nature18644
17. Balasubramanian D, Harper L, Shopsin B, Torres VJ. Staphylococcus Aureus Pathogenesis in Diverse Host Environments. Pathog Dis (2017) 75:5. doi: 10.1093/FEMSPD/FTX005
18. Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential Roles of Interleukin-17A and -17F in Host Defense Against Mucoepithelial Bacterial Infection and Allergic Responses. Immunity (2009) 30:108–19. doi: 10.1016/J.IMMUNI.2008.11.009
19. Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, et al. IL-17 Is Essential for Host Defense Against Cutaneous Staphylococcus Aureus Infection in Mice. J Clin Invest (2010) 120:1762. doi: 10.1172/JCI40891
20. Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus Aureus Virulent Psmα Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe (2017) 22:667–77.e5. doi: 10.1016/j.chom.2017.10.008
21. Pavlova A, Sharafutdinov I. Recognition of Candida Albicans and Role of Innate Type 17 Immunity in Oral Candidiasis. Microorganisms (2020) 8:1–18hep. doi: 10.3390/microorganisms8091340
22. Sparber F, Dolowschiak T, Mertens S, Lauener L, Clausen BE, Joller N, et al. Langerin+DCs Regulate Innate IL-17 Production in the Oral Mucosa During Candida Albicans-Mediated Infection. PloS Pathog (2018) 14:e1007069. doi: 10.1371/journal.ppat.1007069
23. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 Cells and IL-17 Receptor Signaling Are Essential for Mucosal Host Defense Against Oral Candidiasis. J Exp Med (2009) 206:299–311. doi: 10.1084/jem.20081463
24. Mrzljak A, Tabain I, Premac H, Bogdanic M, Barbic L, Savic V, et al. The Role of Emerging and Neglected Viruses in the Etiology of Hepatitis. Curr Infect Dis Rep (2019) 21:1–10. doi: 10.1007/S11908-019-0709-2
25. Jie Z, Liang Y, Hou L, Dong C, Iwakura Y, Soong L, et al. Intrahepatic Innate Lymphoid Cells Secrete IL-17A and IL-17f That Are Crucial for T Cell Priming in Viral Infection. J Immunol (2014) 192:3289–300. doi: 10.4049/jimmunol.1303281
26. Muhanna N, Horani A, Doron S, Safadi R. Lymphocyte–hepatic Stellate Cell Proximity Suggests a Direct Interaction. Clin Exp Immunol (2007) 148:338–47. doi: 10.1111/J.1365-2249.2007.03353.X
27. Wang S, Li J, Wu S, Cheng L, Shen Y, Ma W, et al. Type 3 Innate Lymphoid Cell: A New Player in Liver Fibrosis Progression. Clin Sci (2018) 132:2565–82. doi: 10.1042/CS20180482
28. Ardain A, Porterfield JZ, Kløverpris HN, Leslie A. Type 3 ILCs in Lung Disease. Front Immunol (2019) 10:92. doi: 10.3389/fimmu.2019.00092
29. Hoffmann JP, Kolls JK, McCombs JE. Regulation and Function of ILC3s in Pulmonary Infections. Front Immunol (2021) 12:672523. doi: 10.3389/fimmu.2021.672523
30. Li C, Yang P, Sun Y, Li T, Wang C, Wang Z, et al. IL-17 Response Mediates Acute Lung Injury Induced by the 2009 Pandemic Influenza A (H1N1) Virus. Cell Res (2012) 22:528–38. doi: 10.1038/cr.2011.165
31. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A Inhibits Th17-Mediated Host Defense Against Bacterial Pneumonia in Mice. J Immunol (2011) 186:1666–74. doi: 10.4049/jimmunol.1002194
32. Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, et al. Influenza A Exacerbates Staphylococcus Aureus Pneumonia by Attenuating IL-1β Production in Mice. J Immunol (2013) 191:5153–9. doi: 10.4049/jimmunol.1301237
33. Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, et al. Interleukin-22 Reduces Lung Inflammation During Influenza A Virus Infection and Protects Against Secondary Bacterial Infection. J Virol (2013) 87:6911. doi: 10.1128/JVI.02943-12
34. Abood RN, McHugh KJ, Rich HE, Ortiz MA, Tobin JM, Ramanan K, et al. IL-22 Binding Protein Exacerbates Influenza, Bacterial Super-Infection. Mucosal Immunol (2019) 12:1231. doi: 10.1038/S41385-019-0188-7
35. van de Veerdonk FL, Gresnigt MS, Romani L, Netea MG, Latgé J-P. Aspergillus Fumigatus Morphology and Dynamic Host Interactions. Nat Rev Microbiol (2017) 15:661–74. doi: 10.1038/nrmicro.2017.90
36. Gessner MA, Werner JL, Lilly LM, Nelson MP, Metz AE, Dunaway CW, et al. Dectin-1-Dependent Interleukin-22 Contributes to Early Innate Lung Defense Against Aspergillus Fumigatus. Infect Immun (2012) 80:410. doi: 10.1128/IAI.05939-11
37. Lilly LM, Gessner MA, Dunaway CW, Metz AE, Schwiebert L, Weaver CT, et al. The β-Glucan Receptor Dectin-1 Promotes Lung Immunopathology During Fungal Allergy via IL-22. J Immunol (2012) 189:3653–60. doi: 10.4049/JIMMUNOL.1201797
38. Reeder KM, Mackel JJ, Godwin MS, Dunaway CW, Blackburn JP, Patel RP, et al. Role of Common γ-Chain Cytokines in Lung Interleukin-22 Regulation After Acute Exposure to Aspergillus Fumigatus. Infect Immun (2018) 86(10):e00157-18. doi: 10.1128/IAI.00157-18
39. Gordon SV, Parish T. Microbe Profile: Mycobacterium Tuberculosis: Humanity’s Deadly Microbial Foe. Microbiology (2018) 164:437–9. doi: 10.1099/MIC.0.000601
40. Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, et al. Group 3 Innate Lymphoid Cells Mediate Early Protective Immunity Against Tuberculosis. Nature (2019) 570:528–32. doi: 10.1038/s41586-019-1276-2
41. Tripathi D, Radhakrishnan RK, Thandi RS, Paidipally P, Devalraju KP, Neela VSK, et al. IL-22 Produced by Type 3 Innate Lymphoid Cells (ILC3s) Reduces the Mortality of Type 2 Diabetes Mellitus (T2DM) Mice Infected With Mycobacterium Tuberculosis. PloS Pathog (2019) 15:e1008140. doi: 10.1371/journal.ppat.1008140
42. Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, et al. Activation of Type 3 Innate Lymphoid Cells and Interleukin 22 Secretion in the Lungs During Streptococcus Pneumoniae Infection. J Infect Dis (2014) 210:493–503. doi: 10.1093/infdis/jiu106
43. Gray J, Oehrle K, Worthen G, Alenghat T, Whitsett J, Deshmukh H. Intestinal Commensal Bacteria Mediate Lung Mucosal Immunity and Promote Resistance of Newborn Mice to Infection. Sci Transl Med (2017) 9:eaaf9412. doi: 10.1126/scitranslmed.aaf9412
44. Oherle K, Acker E, Bonfield M, Wang T, Gray J, Lang I, et al. Insulin-Like Growth Factor 1 Supports a Pulmonary Niche That Promotes Type 3 Innate Lymphoid Cell Development in Newborn Lungs. Immunity (2020) 52:275–94.e9. doi: 10.1016/j.immuni.2020.01.005
45. Hassane M, Jouan Y, Creusat F, Soulard D, Boisseau C, Gonzalez L, et al. Interleukin-7 Protects Against Bacterial Respiratory Infection by Promoting IL-17A-Producing Innate T-Cell Response. Mucosal Immunol (2020) 13:128–39. doi: 10.1038/s41385-019-0212-y
46. Li B, Zhao Y, Liu C, Chen Z, Zhou D. Molecular Pathogenesis of Klebsiella Pneumoniae. Future Microbiol (2014) 9:1071–81. doi: 10.2217/fmb.14.48
47. Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, Pamer EG. Innate Lymphocyte/Ly6Chi Monocyte Crosstalk Promotes Klebsiella Pneumoniae Clearance. Cell (2016) 165:679–89. doi: 10.1016/j.cell.2016.03.017
48. Iwanaga N, Sandquist I, Wanek A, McCombs J, Song K, Kolls JK. Host Immunology and Rational Immunotherapy for Carbapenem-Resistant Klebsiella Pneumoniae Infection. JCI Insight (2020) 5:e135591. doi: 10.1172/jci.insight.135591
49. Chevalier S, Bouffartigues E, Bodilis J, Maillot O, Lesouhaitier O, Feuilloley MGJ, et al. Structure, Function and Regulation of Pseudomonas Aeruginosa Porins. FEMS Microbiol Rev (2017) 41:698–722. doi: 10.1093/FEMSRE/FUX020
50. Ader F, Jawhara S, Nseir S, Kipnis E, Faure K, Vuotto F, et al. Short Term Candida Albicans Colonization Reduces Pseudomonas Aeruginosa-Related Lung Injury and Bacterial Burden in a Murine Model. Crit Care (2011) 15:R150. doi: 10.1186/CC10276
51. Mear JB, Gosset P, Kipnis E, Faure E, Dessein R, Jawhara S, et al. Candida Albicans Airway Exposure Primes the Lung Innate Immune Response Against Pseudomonas Aeruginosa Infection Through Innate Lymphoid Cell Recruitment and Interleukin-22-Associated Mucosal Response. Infect Immun (2014) 82:306–15. doi: 10.1128/IAI.01085-13
52. Broquet A, Besbes A, Martin J, Jacqueline C, Vourc’h M, Roquilly A, et al. Interleukin-22 Regulates Interferon Lambda Expression in a Mice Model of Pseudomonas Aeruginosa Pneumonia. Mol Immunol (2020) 118:52–9. doi: 10.1016/j.molimm.2019.12.003
53. Bayes HK, Ritchie ND, Evans TJ. Interleukin-17 Is Required for Control of Chronic Lung Infection Caused by Pseudomonas Aeruginosa. Infect Immun (2016) 84:3507–16. doi: 10.1128/IAI.00717-16
54. Borges-Walmsley MI, Chen D, Shu X, Walmsley AR. The Pathobiology of Paracoccidioides Brasiliensis. Trends Microbiol (2002) 10:80–7. doi: 10.1016/S0966-842X(01)02292-2
55. de Araújo EF, Preite NW, Veldhoen M, Loures FV, Calich VLG. Pulmonary Paracoccidioidomycosis in AhR Deficient Hosts Is Severe and Associated With Defective Treg and Th22 Responses. Sci Rep (2020) 10:1–16. doi: 10.1038/s41598-020-68322-6
56. Chinen J, Shearer WT. Molecular Virology and Immunology of HIV Infection. J Allergy Clin Immunol (2002) 110:189–98. doi: 10.1067/mai.2002.126226
57. Reeves RK, Rajakumar PA, Evans TI, Connole M, Gillis J, Wong FE, et al. Gut Inflammation and Indoleamine Deoxygenase Inhibit IL-17 Production and Promote Cytotoxic Potential in NKp44 Mucosal NK Cells During SIV Infection. Blood (2011) 118(12):3321–30. doi: 10.1182/blood-2011-04-347260
58. Guo X, Fu YX. The Tragic Fate of Group 3 Innate Lymphoid Cells During HIV-1 Infection. J Clin Invest (2015) 125:3430–2. doi: 10.1172/JCI83823
59. Zhang Z, Cheng L, Zhao J, Li G, Zhang L, Chen W, et al. Plasmacytoid Dendritic Cells Promote HIV-1-Induced Group 3 Innate Lymphoid Cell Depletion. J Clin Invest (2015) 125:3692–703. doi: 10.1172/JCI82124
60. Shah SV, Manickam C, Ram DR, Reeves RK. Innate Lymphoid Cells in Hiv/Siv Infections. Front Immunol (2017) 8:1818. doi: 10.3389/fimmu.2017.01818
61. Xu H, Wang X, Lackner AA, Veazey RS. Type 3 Innate Lymphoid Cell Depletion Is Mediated by TLRs in Lymphoid Tissues of Simian Immunodeficiency Virus-Infected Macaques. FASEB J (2015) 29:5072–80. doi: 10.1096/FJ.15-276477
62. Hueber B, Curtis AD, Kroll K, Varner V, Jones R, Pathak S, et al. Functional Perturbation of Mucosal Group 3 Innate Lymphoid and Natural Killer Cells in Simian-Human Immunodeficiency Virus/Simian Immunodeficiency Virus-Infected Infant Rhesus Macaques. J Virol (2020) 94:1–15. doi: 10.1128/jvi.01644-19
63. Rahman MA, Ko E-J, Enyindah-Asonye G, Hait SH, Hogge C, Hunegnaw R, et al. Differential Effect of Mucosal NKp44+ Innate Lymphoid Cells and Δγ Cells on SIV Infection Outcome in Rhesus Macaques. J Immunol (2019) 203:2459. doi: 10.4049/JIMMUNOL.1900572
64. Vaccari M, Gordon SN, Fourati S, Schifanella L, Liyanage NPM, Cameron M, et al. Adjuvant-Dependent Innate and Adaptive Immune Signatures of Risk of SIVmac251 Acquisition. Nat Med (2016) 22:762. doi: 10.1038/NM.4105
65. Kløverpris HN, Kazer SW, Mjösberg J, Mabuka JM, Wellmann A, Ndhlovu Z, et al. Innate Lymphoid Cells Are Depleted Irreversibly During Acute HIV-1 Infection in the Absence of Viral Suppression. Immunity (2016) 44:391. doi: 10.1016/J.IMMUNI.2016.01.006
66. Wang Y, Lifshitz L, Gellatly K, Vinton CL, Busman-Sahay K, McCauley S, et al. HIV-1-Induced Cytokines Deplete Homeostatic Innate Lymphoid Cells and Expand TCF7-Dependent Memory NK Cells. Nat Immunol (2020) 21:274. doi: 10.1038/S41590-020-0593-9
67. Akoto C, Chan CYS, Tshivuila-Matala COO, Ravi K, Zhang W, Vatish M, et al. Innate Lymphoid Cells Are Reduced in Pregnant HIV Positive Women and Are Associated With Preterm Birth. Sci Rep (2020) 10:13265. doi: 10.1038/s41598-020-69966-0
68. Singh A, Kazer SW, Roider J, Krista KC, Millar J, Asowata OE, et al. Innate Lymphoid Cell Activation and Sustained Depletion in Blood and Tissue of Children Infected With HIV From Birth Despite Antiretroviral Therapy. Cell Rep (2020) 32:108153. doi: 10.1016/j.celrep.2020.108153
69. Crump JA, Sjölund-Karlsson M, Gordon MA, Parry CM. Epidemiology, Clinical Presentation, Laboratory Diagnosis, Antimicrobial Resistance, and Antimicrobial Management of Invasive Salmonella Infections. Clin Microbiol Rev (2015) 28:901–37. doi: 10.1128/CMR.00002-15
70. Lo BC, Gold MJ, Hughes MR, Antignano F, Valdez Y, Zaph C, et al. The Orphan Nuclear Receptor RORa and Group 3 Innate Lymphoid Cells Drive Fibrosis in a Mouse Model of Crohn’s Disease. Sci Immunol (2016) 1:eaaf8864–eaaf8864. doi: 10.1126/sciimmunol.aaf8864
71. Lo BC, Canals Hernaez D, Scott RW, Hughes MR, Shin SB, Underhill TM, et al. The Transcription Factor Rorα Preserves ILC3 Lineage Identity and Function During Chronic Intestinal Infection. J Immunol (2019) 203:3209–15. doi: 10.4049/jimmunol.1900781
72. Yin S, Yu J, Hu B, Lu C, Liu X, Gao X, et al. Runx3 Mediates Resistance to Intracellular Bacterial Infection by Promoting IL12 Signaling in Group 1 ILC and NCR+Ilc3. Front Immunol (2018) 9:2101. doi: 10.3389/fimmu.2018.02101
73. Adam RD. Biology of Giardia Lamblia. Clin Microbiol Rev (2001) 14:447. doi: 10.1128/CMR.14.3.447-475.2001
74. Ryan U, Hijjawi N, Feng Y, Xiao L. Giardia: An Under-Reported Foodborne Parasite. Int J Parasitol (2019) 49:1–11. doi: 10.1016/J.IJPARA.2018.07.003
75. Lee HY, Park EA, Lee KJ, Lee KH, Park SJ. Increased Innate Lymphoid Cell 3 and IL-17 Production in Mouse Lamina Propria Stimulated With Giardia Lamblia. Korean J Parasitol (2019) 57:225–32. doi: 10.3347/kjp.2019.57.3.225
76. Kaur CP, Vadivelu J, Chandramathi S. Impact of Klebsiella Pneumoniae in Lower Gastrointestinal Tract Diseases. J Dig Dis (2018) 19:262–71. doi: 10.1111/1751-2980.12595
77. Coorens M, Rao A, Gräfe SK, Unelius D, Lindforss U, Agerberth B, et al. Innate Lymphoid Cell Type 3– Derived Interleukin-22 Boosts Lipocalin-2 Production in Intestinal Epithelial Cells via Synergy Between STAT3 and NF-B. J Biol Chem (2019) 294:6027–41. doi: 10.1074/jbc.RA118.007290
78. Niu X, Daniel S, Kumar D, Ding EY, Savani RC, Koh AY, et al. Transient Neonatal Antibiotic Exposure Increases Susceptibility to Late-Onset Sepsis Driven by Microbiota-Dependent Suppression of Type 3 Innate Lymphoid Cells. Sci Rep (2020) 10:12974. doi: 10.1038/s41598-020-69797-z
79. Bancerz-Kisiel A, Szweda W. Yersiniosis – A Zoonotic Foodborne Disease of Relevance to Public Health. Ann Agric Environ Med (2015) 22:397–402. doi: 10.5604/12321966.1167700
80. Rankin LC, Girard-Madoux MJH, Seillet C, Mielke LA, Kerdiles Y, Fenis A, et al. Complementarity and Redundancy of IL-22-Producing Innate Lymphoid Cells. Nat Immunol (2016) 17:179–86. doi: 10.1038/ni.3332
81. Shui J-W, Kronenberg M. HVEM: An Unusual TNF Receptor Family Member Important for Mucosal Innate Immune Responses to Microbes. Gut Microbes (2013) 4:146. doi: 10.4161/GMIC.23443
82. Seo GY, Shui JW, Takahashi D, Song C, Wang Q, Kim K, et al. LIGHT-HVEM Signaling in Innate Lymphoid Cell Subsets Protects Against Enteric Bacterial Infection. Cell Host Microbe (2018) 24:249–60.e4. doi: 10.1016/j.chom.2018.07.008
83. Saleh MM, Petri WA. Type 3 Immunity During Clostridioides Difficile Infection: Too Much of a Good Thing? Infect Immun (2020) 88:e00306-19. doi: 10.1128/IAI.00306-19
84. Abt MC, Lewis BB, Caballero S, Ling L, Leiner I, Pamer EG, et al. Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection Against Acute Clostridium Difficile Infection Article Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection Against Acute Clostridium Difficile Infection. Cell Host Microbe (2015) 18:27–37. doi: 10.1016/j.chom.2015.06.011
85. Fachi JL, Pral LP, dos Santos JAC, Codo AC, de Oliveira S, Felipe JS, et al. Hypoxia Enhances ILC3 Responses Through HIF-1α-Dependent Mechanism. Mucosal Immunol (2021) 14:828–41. doi: 10.1038/s41385-020-00371-6
86. Fachi JL, Sécca C, Rodrigues PB, de Mato FCP, Di Luccia B, Felipe J de S, et al. Acetate Coordinates Neutrophil and ILC3 Responses Against C. Difficile Through FFAR2. J Exp Med (2020) 217:jem.20190489. doi: 10.1084/jem.20190489
87. Zhu Y, Shi T, Lu X, Xu Z, Qu J, Zhang Z, et al. Fungal-Induced Glycolysis in Macrophages Promotes Colon Cancer by Enhancing Innate Lymphoid Cell Secretion of IL-22. EMBO J (2021) 40:e105320. doi: 10.15252/embj.2020105320
88. Bouladoux N, Harrison OJ, Belkaid Y. The Mouse Model of Infection With Citrobacter Rodentium. Curr Protoc Immunol (2017) 119:19.15.1–19.15.25. doi: 10.1002/cpim.34
89. Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, et al. Lineage Relationship Analysis of Rorγt+ Innate Lymphoid Cells. Science (2010) 330:665–9. doi: 10.1126/science.1194597
90. Di Luccia B, Gilfillan S, Cella M, Colonna M, Huang SCC. ILC3s Integrate Glycolysis and Mitochondrial Production of Reactive Oxygen Species to Fulfill Activation Demands. J Exp Med (2019) 216:2231–41. doi: 10.1084/jem.20180549
91. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal Microbiota-Derived Short-Chain Fatty Acids Regulation of Immune Cell IL-22 Production and Gut Immunity. Nat Commun (2020) 11:1–18. doi: 10.1038/s41467-020-18262-6
92. Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity (2019) 51:871–84.e6. doi: 10.1016/j.immuni.2019.09.014
93. Sepahi A, Liu QY, Friesen L, Kim CH. Dietary Fiber Metabolites Regulate Innate Lymphoid Cell Responses. Mucosal Immunol (2021) 14:317–30. doi: 10.1038/s41385-020-0312-8
94. Burrows K, Antignano F, Chenery A, Bramhall M, Korinek V, Underhill TM, et al. HIC1 Links Retinoic Acid Signalling to Group 3 Innate Lymphoid Cell-Dependent Regulation of Intestinal Immunity and Homeostasis. PloS Pathog (2018) 14:e1006869. doi: 10.1371/journal.ppat.1006869
95. Lin YD, Arora J, Diehl K, Bora SA, Cantorna MT. Vitamin D Is Required for ILC3 Derived IL-22 and Protection From Citrobacter Rodentium Infection. Front Immunol (2019) 10:1. doi: 10.3389/fimmu.2019.00001
96. Godinho-Silva C, Domingues RG, Rendas M, Raposo B, Ribeiro H, da Silva JA, et al. Light-Entrained and Brain-Tuned Circadian Circuits Regulate ILC3s and Gut Homeostasis. Nature (2019) 574:254–8. doi: 10.1038/s41586-019-1579-3
97. Talbot J, Hahn P, Kroehling L, Nguyen H, Li D, Littman DR. Feeding-Dependent VIP Neuron-ILC3 Circuit Regulates the Intestinal Barrier HHS Public Access. Nature (2020) 579:575–80. doi: 10.1038/s41586-020-2039-9
98. Castellanos JG, Woo V, Viladomiu M, Shih DQ, Scherl EJ, Longman RS. Microbiota-Induced TNF-Like Ligand 1a Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation During Colitis Correspondence. Immunity (2018) 49:1077–89.e5. doi: 10.1016/j.immuni.2018.10.014
99. Chu C, Moriyama S, Li Z, Zhou L, Flamar AL, Klose CSN, et al. Anti-Microbial Functions of Group 3 Innate Lymphoid Cells in Gut-Associated Lymphoid Tissues Are Regulated by G-Protein-Coupled Receptor 183. Cell Rep (2018) 23:3750–8. doi: 10.1016/j.celrep.2018.05.099
100. Abou-Samra E, Hickey Z, Aguilar OA, Scur M, Mahmoud AB, Pyatibrat S, et al. NKR-P1B Expression in Gut-Associated Innate Lymphoid Cells Is Required for the Control of Gastrointestinal Tract Infections. Cell Mol Immunol (2019) 16:868–77. doi: 10.1038/s41423-018-0169-x
101. Bando JK, Gilfillan S, Song C, McDonald KG, Huang SCC, Newberry RD, et al. The Tumor Necrosis Factor Superfamily Member RANKL Suppresses Effector Cytokine Production in Group 3 Innate Lymphoid Cells. Immunity (2018) 48:1208–219.e4. doi: 10.1016/j.immuni.2018.04.012
102. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural Aryl Hydrocarbon Receptor Ligands Control Organogenesis of Intestinal Lymphoid Follicles. Science (2011) 334:1561–5. doi: 10.1126/SCIENCE.1214914
103. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR Drives the Development of Gut ILC22 Cells and Postnatal Lymphoid Tissues via Pathways Dependent on and Independent of Notch. Nat Immunol (2012) 13:144–52. doi: 10.1038/ni.2187
104. Li S, Bostick JW, Ye J, Qiu J, Zhang B, Urban JF, et al. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity (2018) 49:915–28.e5. doi: 10.1016/j.immuni.2018.09.015
105. Castro-Dopico T, Fleming A, Dennison TW, Ferdinand JR, Harcourt K, Stewart BJ, et al. GM-CSF Calibrates Macrophage Defense and Wound Healing Programs During Intestinal Infection and Inflammation. Cell Rep (2020) 32:107857. doi: 10.1016/j.celrep.2020.107857
Keywords: type 3 innate lymphoid cells, infection, pathogens, mucosa, host-pathogen interaction, IL-22, IL-17, ILC3s
Citation: Valle-Noguera A, Ochoa-Ramos A, Gomez-Sánchez MJ and Cruz-Adalia A (2021) Type 3 Innate Lymphoid Cells as Regulators of the Host-Pathogen Interaction. Front. Immunol. 12:748851. doi: 10.3389/fimmu.2021.748851
Received: 28 July 2021; Accepted: 13 September 2021;
Published: 29 September 2021.
Edited by:
Stefan Jordan, Charité-Universitätsmedizin Berlin, GermanyReviewed by:
Naoko Satoh-Takayama, RIKEN Center for Integrative Medical Sciences, JapanSantosh Panda, Washington University in St. Louis, United States
Copyright © 2021 Valle-Noguera, Ochoa-Ramos, Gomez-Sánchez and Cruz-Adalia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aranzazu Cruz-Adalia, YXJhbmNydXpAdWNtLmVz
†These authors share first authorship