
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 05 November 2021
Sec. Viral Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.748417
This article is part of the Research TopicImmunometabolic Mechanisms Underlying the Severity of COVID-19View all 25 articles
 Camila Hartmann1,2
Camila Hartmann1,2 Anna Flavia Ribeiro dos Santos Miggiolaro1,2
Anna Flavia Ribeiro dos Santos Miggiolaro1,2 Jarbas da Silva Motta Junior2
Jarbas da Silva Motta Junior2 Lucas Baena Carstens1
Lucas Baena Carstens1 Caroline Busatta Vaz De Paula1
Caroline Busatta Vaz De Paula1 Sarah Fagundes Grobe1,2
Sarah Fagundes Grobe1,2 Larissa Hermann de Souza Nunes1,2
Larissa Hermann de Souza Nunes1,2 Gustavo Lenci Marques1,2
Gustavo Lenci Marques1,2 Peter Libby3Lidia Zytynski Moura1,2
Peter Libby3Lidia Zytynski Moura1,2 Lucia de Noronha1*
Lucia de Noronha1* Cristina Pellegrino Baena1,2
Cristina Pellegrino Baena1,2Rationale: Myocardial injury associates significantly and independently with mortality in COVID-19 patients. However, the pathogenesis of myocardial injury in COVID-19 remains unclear, and cardiac involvement by SARS-CoV-2 presents a major challenge worldwide.
Objective: This histological and immunohistochemical study sought to clarify the pathogenesis and propose a mechanism with pathways involved in COVID-19 myocardial injury.
Methods and Results: Postmortem minimally invasive autopsies were performed in six patients who died from COVID-19, and the myocardium samples were compared to a control group (n=11). Histological analysis was performed using hematoxylin-eosin and toluidine blue staining. Immunohistochemical (IHC) staining was performed using monoclonal antibodies against targets: caspase-1, caspase-9, gasdermin-d, ICAM-1, IL-1β, IL-4, IL-6, CD163, TNF-α, TGF-β, MMP-9, type 1 and type 3 collagen. The samples were also assessed for apoptotic cells by TUNEL. Histological analysis showed severe pericardiocyte interstitial edema and higher mast cells counts per high-power field in all COVID-19 myocardium samples. The IHC analysis showed increased expression of caspase-1, ICAM-1, IL-1β, IL-6, MMP-9, TNF-α, and other markers in the hearts of COVID-19 patients. Expression of caspase-9 did not differ from the controls, while gasdermin-d expression was less. The TUNEL assay was positive in all the COVID-19 samples supporting endothelial apoptosis.
Conclusions: The pathogenesis of COVID-19 myocardial injury does not seem to relate to primary myocardiocyte involvement but to local inflammation with associated interstitial edema. We found heightened TGF-β and interstitial collagen expression in COVID-affected hearts, a potential harbinger of chronic myocardial fibrosis. These results suggest a need for continued clinical surveillance of patients for myocardial dysfunction and arrythmias after recovery from the acute phase of COVID-19.
Since its emergence in December 2019 (1), the coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), continues to grow despite unprecedented worldwide efforts in the search of treatments and vaccines. COVID-19 is initially a respiratory disease, causing viral pneumonia and adult respiratory distress syndrome. However, cardiovascular manifestations occur commonly and relate to poor outcomes (2).
Myocardial injury (defined by troponin blood levels above the 99th-percentile upper reference) was observed in 7 to 17% (3) of patients and associates significantly and independently with mortality (4). Common cardiac complications among hospitalized patients with COVID-19 include arrhythmias and acute heart failure. Heart failure may contribute up to 40% of deaths, and circulatory failure may cause death even without respiratory failure (3). A prothrombotic coagulopathy can occur in 25% of patients resulting in venous and arterial thromboembolic events (5).
SARS-CoV-2 genome detection in endomyocardial biopsies (6) and autopsy specimens (7) has localized SARS-CoV-2 in the heart. However, evidence for infection of cardiocytes themselves remains controversial and the mechanism of cardiac damage by SARS-CoV-2 remains incompletely understood. Some autopsies of patients with COVID-19 revealed accumulation in the myocardial interstitium of mononuclear inflammatory cells (8) while others showed no increase in inflammatory cells despite the presence of the viral genome (7). SARS-CoV-2 particles have already been observed in myocardial interstitial cells (9) and endothelial cells (10) by electron microscopy, and it has been proposed that pyroptosis may have an important role in endothelial cell injury in patients with COVID-19 (10). Pyroptosis is a specific type of programmed pro-inflammatory cell death that culminates in caspase-1 activation, interleukin-6 (IL-6) secretion and endothelial disfunction (11). This could be the initial pathway for myocardial injury, and could also explain the involvement of various organs and tissues that has been described in COVID-19.
Interstitial myocardial fibrosis has been described as a possible consequence of myocardial injury (6, 12) by the expression of the pro-fibrotic mediator TGF-β, which can stimulate interstitial collagen production (13). Myocardial fibrosis may predispose to both systolic and diastolic dysfunction, as well as arrhythmias (14).
Given that the cardiac manifestations play a major role in adverse outcomes and discordant pathological studies regarding the mechanism of myocardial injury in COVID-19, we investigated myocardium samples in a histological and immunohistochemical study to help clarify its pathogenesis in lethal cases. Our findings provide new insight into the mechanisms involved in COVID-19-related myocardial injury.
Postmortem minimally invasive autopsies were performed in six patients who died from COVID-19 in Marcelino Champagnat Hospital, Brazil. All patients were symptomatic and tested positive for SARS-CoV-2 on nasopharyngeal swabs (RT-PCR) and had the chest computed tomography at admission compatible with pulmonary infection by COVID-19. This study was approved by the National Research Ethics Committee (CONEP), protocol number 3.944.734/2020. Patients’ families authorized the autopsies and provided informed consent form before the procedures. All methods were carried out following relevant guidelines and regulations. Clinical data were obtained from medical records during hospitalization in the Intensive Care Unit (ICU).
Myocardial tissue was collected within 4 hours after death by left anterior mini thoracotomy for direct access to the left ventricle. The pericardium was sectioned and a fragment of myocardial tissue approximately 2 x 2 cm was obtained. The tissues from the myocardial biopsies were fixed in neutral buffered formalin for over 24 hours, and then processed under conventional histological technique. The myocardium biopsies of patients with COVID-19 were then independently compared to myocardium samples from control patients 6 hours after death from other. All the results were analyzed and integrated to the previous pathological knowledge.
The formalin fixed paraffin embedded (FFPE) sections were subjected to hematoxylin-eosin (H&E) and toluidine blue (TB) staining. Histological features (H&E) were observed and described by using Olympus BX40. Mast cells (only nucleated cells with granules) were scored (TB) by counting cells per high-power field (HPF – 40x objective – 0.26mm2) by screening 20 randomized HPFs (total area of 5.2mm2 per case).
Immunohistochemical (IHC) staining was performed in the myocardium samples using monoclonal and polyclonal antibodies against the following targets: caspase-1, caspase-9, gasdermin-d and interleukin-1β (IL-1β) to detect pyroptosis; intercellular adhesion molecule-1 (ICAM-1), tumor necrosis factor alpha (TNF-α), interleukin-4 (IL-4), interleukin-6 (IL-6), CD163 (macrophage-specific protein) and matrix metalloproteinase-9 (MMP-9) to detect inflammatory activation and response pathways; transforming growth factor (TGF-β), type 1 and type 3 collagen to detect myocardial fibrosis. The table in the Supplementary Material summarizes the specifications of the antibodies used to investigate the FFPE myocardial tissues. In order to detect apoptosis, the samples were also subjected to a TUNEL assay (Terminal deoxynucleotidyl transferase dUTP nick end labeling), using the ‘In Situ Cell Death Detection Kit, POD’ by Roche.
Scores of biomarker expression according to the IHC staining were given by an experienced pathologist and confirmed by two trained technicians. Biomarkers were analyzed using Allred scoring system (15, 16): score 0-5 depending on the proportion of cells which are stained (proportion score [PS]: score 0 = none stained cells, score 1 = 1%, score 2 = 1-10%, score 3 = 11-33%, score 4 = 34-66% and score 5 = 66-100%) and score 0-3 depending on the intensity of staining (intensity score [IS]: score 0 = none stained cells, score 1 = weak, score 2 = moderate, score 3 = strong). The semiquantitative analysis was obtained by summing the two scores (proportion and intensity of positivity), ranging from 0 to 8.
Clinical data and biomarkers score expression were evaluated independently in COVID-19 patients and controls. The comparison of the quantitative variables of the two groups was performed using the non-parametric Mann-Whitney test and the comparison of the proportions between groups was performed with Chi-Square. Values of p < 0.05 indicated statistical significance. The data were analyzed using the GraphPad Prism v9.0.2 software.
Clinical data from the baseline of COVID-19 patients (n=6) and control group (n=11) are presented in Table 1. Our sample was mainly male, with median age of 73.5 years, hypertensive, diabetic, with history of coronary artery disease and with no significant difference between groups (p>0.05 for all). Causes of death in the control group include bronchopneumonia (n=3), pulmonary thromboembolism (n=3), mesenteric ischemia (n=2), myocardial infarction (n=1), aortic dissection (n=1) and upper gastrointestinal bleeding (n=1).
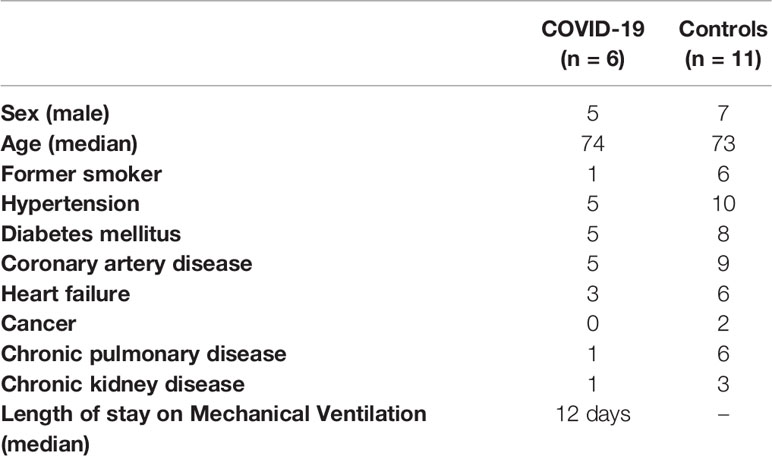
Table 1 Clinical data from the baseline of COVID-19 and control patients.
All the COVID-19 patients had symptoms of dyspnea with progressive worsening and had the chest computed tomography at admission suggestive of viral pulmonary infection for COVID-19. They were all admitted into the ICU and developed respiratory failure requiring mechanical ventilation. The median duration of mechanical ventilation was 12 days. During hospitalization, three patients developed acute kidney failure and patient 1 had pre-existing dialysis-dependent chronic kidney disease; two patients had incident acute atrial fibrillation; and one patient had an acute pulmonary embolism. Laboratory evaluation showed high levels of D-dimer and troponin in all COVID-19 patients. Transthoracic echocardiographic findings were heterogeneous as described in the Supplementary Material. Patients 2 and 5 had normal echocardiographic results with normal ejection fraction.
The sample tissues from the COVID-19 patients were compared to the control group samples and histological assessment showed severe pericellular interstitial edema surrounding each cardiomyocyte in all of the COVID-19 patients. Histological analysis also showed neutrophilic myocarditis according to the Dallas criteria (17) in patient 1. All the other COVID-19 samples showed neither massive inflammatory cellular infiltration nor necrosis, indicating the absence of typical histological myocarditis. In contrast, toluidine blue staining revealed a higher interstitial and perivascular mast cell score in all COVID-19 myocardium samples compared to control (p=0.0023). Most mast cells appeared to be degranulating. Lipofuscin pigment and mild signs of cardiomyocyte hypertrophy were seen in COVID-19 and control patients.
Images of H&E, TB and IHC staining for COVID-19 patients and controls are shown in Figures 1A, B. The full scores of biomarker expression according to the IHC analysis are shown in the Supplementary Material.
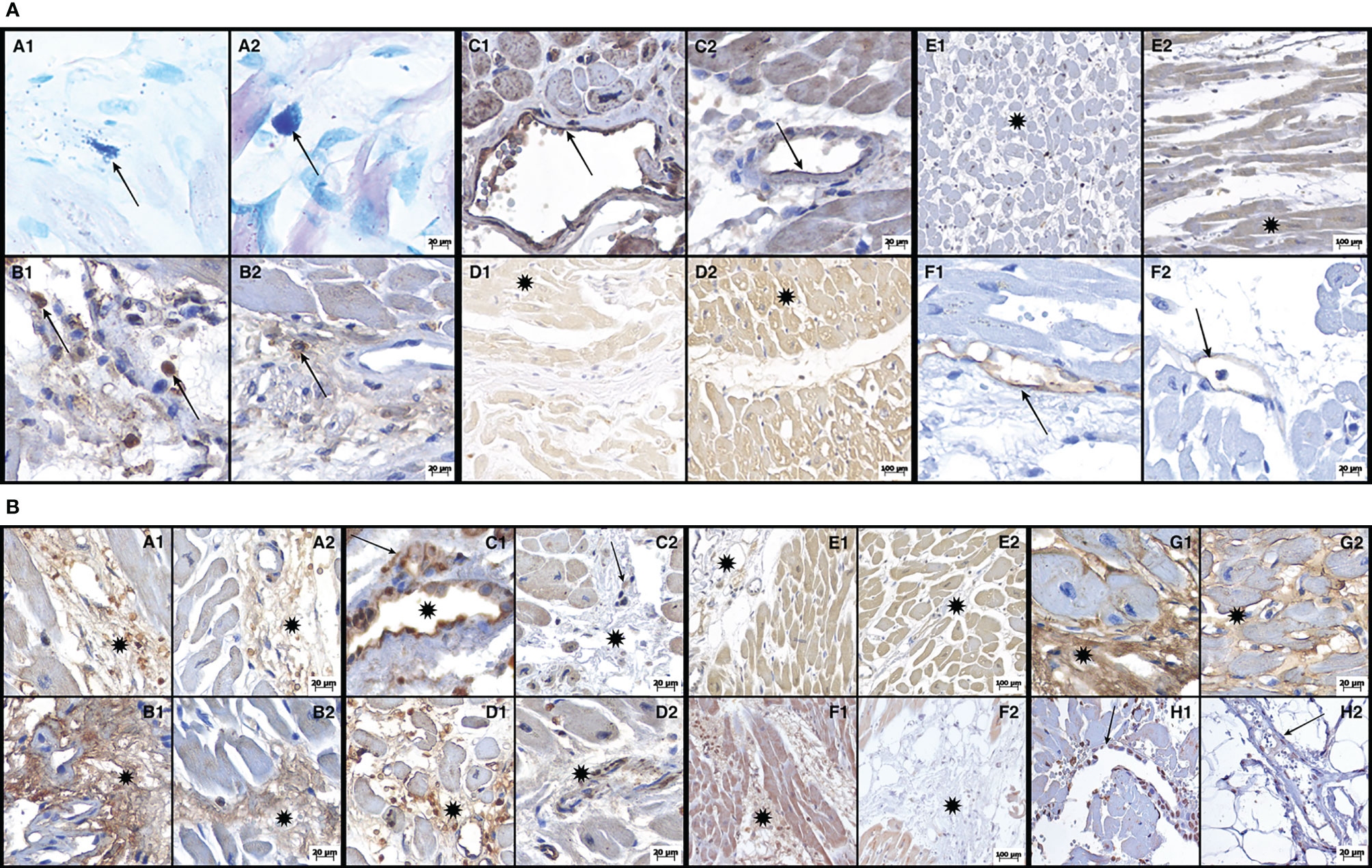
Figure 1 (A) Photomicrographs demonstrating histochemical (A1/2 - Toluidine Blue) and immunohistochemical (B = CD163; C = Casp-1; D = Casp-9; E = GSDM-D; F = ICAM-1) reactions of both groups: cases of COVID-19 (B1-F1) and cases of their respective controls (B2-F2). A1 (COVID-19) and A2 (control) show a mast cell in the perivascular space (arrows): in A1 the mast cell is degranulated, whereas in A2, the mast cell is intact. B1 (COVID-19) shows CD163-immunostained macrophages (arrows) in the myocardial interstitium, whereas in B2 (control) these CD163 macrophages (arrow) are less numerous. C1 (COVID-19) shows endothelial cells (arrow) with strong expression of Casp-1, whereas in C2 (control), Casp-1 expression in endothelial cells (arrow) is much more discrete. D1 (COVID-19) shows a weak expression of Casp-9 in the myocardium (asterisk), whereas in D2 (control), this expression is strong (asterisk). E1 (COVID-19) shows a weak expression of GSDM-D in the myocardium (asterisk), with E2 (control) showing a strong expression (asterisk). F1 (COVID-19) shows a strong expression of ICAM-1 in endothelial cells (arrow), whereas, in F2 (control), this expression is discrete (arrow). (B) Photomicrographs demonstrating immunohistochemical reactions (A = TNF-α; B = MMP-9; C = IL-4; D = IL-6; E = IL-1β; F = TGF-β; G = collagen 3; H = TUNEL) from both groups: cases of COVID-19 (A1-H1) and their respective controls (A2-H2). A1 (COVID-19) shows myocardial interstitium expressing TNF-α (asterisk), whereas, in A2 (control), we can observe that this expression is discrete (asterisk). B1(COVID-19) shows myocardial interstitium expressing MMP-9 (asterisk), whereas, in B2 (control), this expression is discrete (asterisk). C1 (COVID-19) shows interstitial macrophages (arrow) and endothelial cells (asterisk) with strong expression of IL-4, whereas in C2 (control), the expression of IL-4 in interstitial macrophages (arrow) and endothelial cells (asterisk) is discrete. D1 (COVID-19) shows a strong expression of IL-6 in the interstitium (asterisk), whereas, in D2 (control), this expression is discrete (asterisk). E1 (COVID-19) shows moderate expression of IL-1β in the myocardial interstitium (asterisk), and E2 (control) has a more discrete expression (asterisk). F1 (COVID-19) shows a strong expression of TGF-β in the interstitium of the myocardium (asterisk), whereas, in F2 (control), this expression is much more discrete or absent (asterisk). G1 (COVID-19) shows a strong expression of collagen 3 in the interstitium of the myocardium (asterisk), and this expression is much more delicate (asterisk) in G2 (control). H1 (COVID-19) demonstrates elevated DNA fragmentation in the TUNEL assay (arrow) compared to H2 (control), where few nuclei (arrow) are labeled.
The IHC analysis showed increased expression for caspase-1 (p=0.0010), ICAM (p<0.0001), CD163 (p=0.0021), IL-1β (p<0.0001), IL-4 (p<0.0001), IL-6 (p<0.0001), MMP-9 (p=0.0002), TNF-α (p<0.0001), TGF-β (p=0.0063), type 1 collagen (p<0.0001) and type 3 collagen (p<0.0001) in the COVID-19 patients compared to the control. The expression for gadesmin-d, on the other hand, was decreased (p=0.0419). The graphical representation of these analyses are shown in Figure 2. No substantial differences from the control were observed in expression of caspase-9 (p>0.9999).
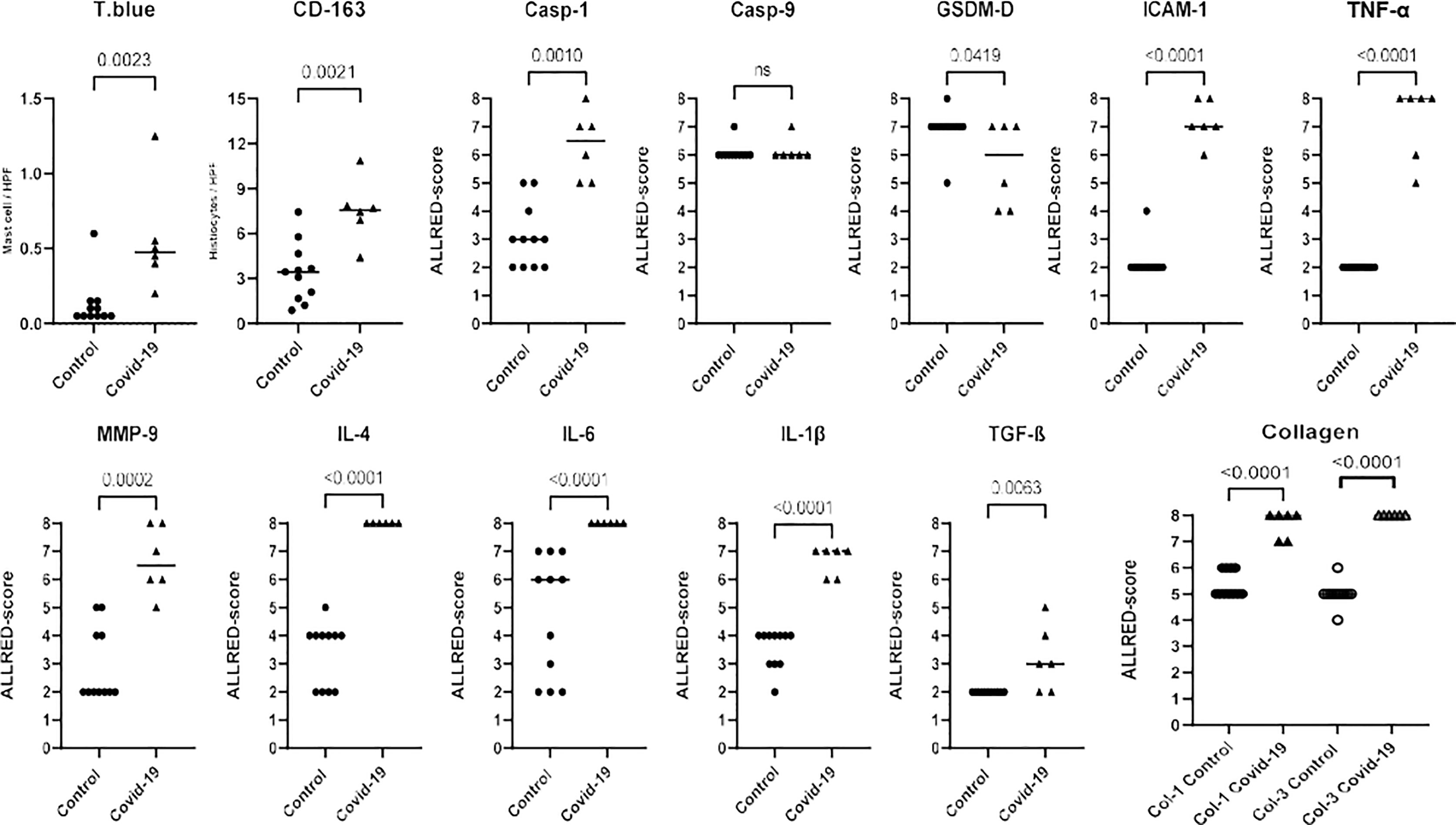
Figure 2 Graphical representation of the immunohistochemical analysis according to the biomarker expression (Toluidine Blue; CD-163; Casp-1; Casp-9; GSDM-D; ICAM-1; TNF-α; MMP-9; IL-4; IL-6; IL-1β; TGF-β; Collagen 1 and 3) in COVID 19 cases and controls. Biomarkers were analyzed using Allred scoring system: score 0-5 depending on the proportion of cells which are stained (proportion score [PS]: score 0 = none stained cells, score 1 = 1%, score 2 = 1-10%, score 3 = 11-33%, score 4 = 34-66% and score 5 = 66-100%) and score 0-3 depending on the intensity of staining (intensity score [IS]: score 0 = none stained cells, score 1 = weak, score 2 = moderate, score 3 = strong). The semiquantitative analysis was obtained by summing the two scores (proportion and intensity of positivity), ranging from 0 to 8.
The TUNEL assay in COVID-19 myocardium samples indicated endothelial cell apoptosis, distinct from the control samples, which tested negative. Cardiomyocytes did not display this marker of apoptosis. Other morphologic findings merit noting. First, caspase-1 and IL-6 were present in the cytoplasm, whereas ICAM-1 localized on the membrane of endothelial cells. Secondly, MMP-9 and both types of collagen were observed in large quantities in the interstitial and perivascular spaces. Our proposed mechanism of COVID-19 myocardial injury is shown in Figure 3.
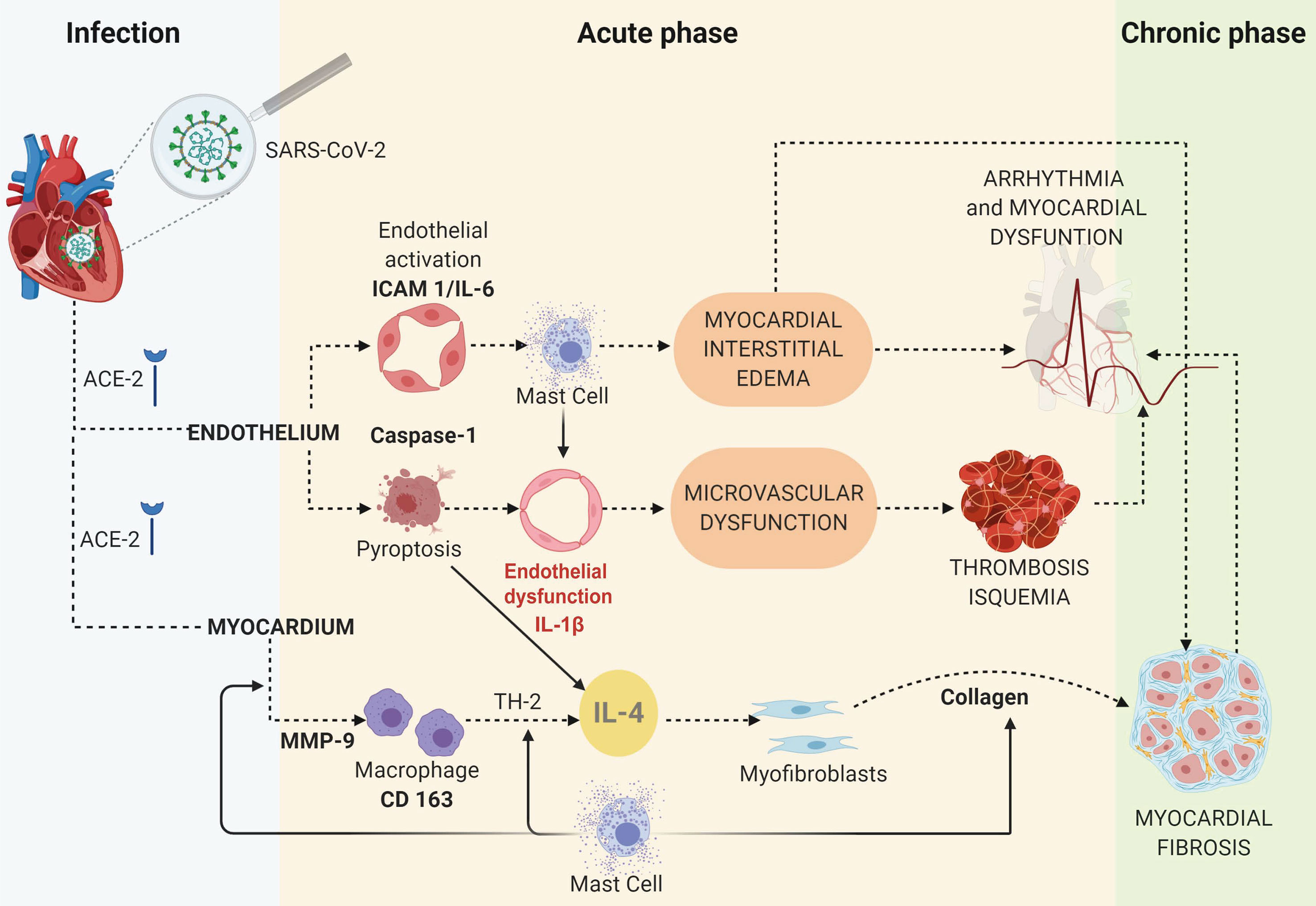
Figure 3 The proposed mechanisms of myocardial injury based on an IHC study of COVID-19 cases and controls.
Our main findings from the myocardial postmortem biopsies of COVID-19 patients show myocardial interstitial edema, mast cell accumulation, and increased indicators of inflammation, apoptosis, and fibrosis compared to controls. The increased expression of ICAM-1 and IL-6 indicates inflammatory activation (18), which along with higher mast cell scores can explain the increased capillary permeability, microvascular leakage and the consequent formation of myocardial interstitial edema (19, 20). The increased expression of MMP-9, CD163, IL-4 and IL-6 demonstrates the presence of myocardial inflammatory response in the myocardial tissue (18, 21). More specifically, CD163 indicates macrophage recruitment (21) and MMP-9 promotes Th2 cell recruitment and matrix remodeling (22). The Th2 cytokine IL-4 and TGF-β can drive myocardial fibrosis (23) and provide a mechanism of elevated interstitial collagens type 1 and type 3 (24–27).
The TUNEL positivity in all the COVID-19 samples shows that this disease promotes endothelial cell apoptosis (programmed cell death). The probable mechanism is by pyroptosis, a specific inflammatory form of apoptosis that occurs most frequently upon infection by intracellular pathogens (like SARS-CoV-2) and requires the enzyme caspase-1 (28). Caspase-1 is activated as part of a multiprotein signaling platform, the inflammasome complex, and subsequently mediates the activation and secretion of interleukins, including IL-1β, as well as the rupture of the cell membrane (29). The lack of change in caspase-9 expression and the decrease in gasdermin-d expression corroborate the inflammasome complex, since gasdermin-d is activated and cleaved in this pathway, which has interaction with caspase-1 but usually no interaction with caspase-9 (30–32).
We also observed higher levels of caspase-1 adjacent to endothelial cells in the COVID-19 samples demonstrating endothelial infection, pyroptosis and injury in these patients. Moreover, SARS-CoV-2 particles have been described in endothelial cells by electron microscopy (10) and the caspase-1 identification is in accordance with Varga et al. (10), who suggested that pyroptosis might have an important role in endothelial cell injury in patients with COVID-19. These findings are also in line with previous biopsy studies which had already shown that the inflammatory process in cardiac tissue permeates the vascular wall (6, 11). SARS-CoV-2 potentially causes endotheliitis (10), which is determinant of microvascular dysfunction by shifting the vascular equilibrium towards more vasoconstriction with subsequent organ ischemia, inflammation with associated tissue edema, and a procoagulant state (33).
The expression of IL-6 and ICAM-1 increased in the endothelial cells and indicates endothelial activation as well as immune cell recruitment and response. Activated endothelial cells translocate NF-κB p50/p65 dimers to the nucleus and induce the expression of IL-6, TNF-α, and adhesion molecules such as ICAM-1. These molecules recruit leukocytes to the infected region of COVID-19 myocardial injury and propagate inflammation (18).
Endothelial cell functions regulate local vascular permeability and blood flow (19, 33). At rest, the endothelium is highly impermeable to large molecules. However, acute changes in vascular permeability result in loss of fluid and plasma proteins from the intravascular space into the interstitium, leading to edema (19, 20, 33). The abundance of mast cells rich in histamine suggests a mechanism for vascular leakage and interstitial myocardial edema observed here in specimens from COVID-19 patients. In addition to histamine, mast cell release TNF-α and proteases, which contribute to increased vascular permeability and local inflammation (19, 20). Mast cell activation occurs not only in the context of allergy, but also in viral infection (34).
A previous autopsy study showed that the presence of SARS-CoV-2 genome in the myocardial tissue was not associated with increased infiltration of mononuclear cells compared with the virus negative group (7). Although most of our COVID-19 myocardium samples also showed neither inflammatory cellular infiltration nor necrosis, which would be expected in typical histological myocarditis, the high levels of MMP-9, CD163, IL-4 and IL-6 demonstrate the presence of myocardial inflammatory response in this tissue.
We observed elevated type 1 and type 3 collagen in the interstitial and perivascular spaces in the COVID-19 samples, suggesting myocardial fibrosis, since synthesis of both types of collagen is markedly increased in the remodeling fibrotic heart, regardless of the etiology of fibrosis (23). TGF-β has major roles in cardiac fibrogenesis (23, 27, 35) and acts by activating SMAD2/3 pathways. IL-6 and IL-4 are also profibrotic cytokines, as they induce MMP-9 expression and collagen synthesis through gene transcription modulation (24–26). Mast cell degranulation may also be involved in fibrogenesis (23), since mast cell tryptase can directly induce fibroblast activation, myofibroblast differentiation and collagen synthesis independently of TGF-β (34)
Taken together our findings indicate that the microvascular dysfunction may lead to thrombosis and underscores the need for rigorous randomized evaluation of anticoagulant and anti-aggregating therapies in various stages of COVID-19 (36). The myocardial interstitial edema observed here may contribute to the high prevalence of cardiac arrhythmia in COVID-19 patients by loss of structure of the syncytium (19). Furthermore, our findings suggest that COVID-19 myocardial injury may cause myocardial fibrosis in the long term. Therapies which act on cardiac remodeling, such as angiotensin-converting enzyme inhibitors or mineralocorticoid receptor antagonists merit evaluation for myocardial protection in patients with or recovered from the acute phase of COVID-19 (37, 38).
Some limitations should be considered in our study. Our sample size was limited, since ethical issues, labor shortages and hospital overcrowding, among many other reasons, prevented larger samples of postmortem myocardial biopsies from being collected in the COVID-19 Intensive Care Units. Therefore, it is important to interpret our findings with caution and validate them in other samples to replicate our results. Additionally, interpretation of our findings should take into account that data based on FFPE postmortem samples only provides information at the time of death, and cannot reconstruct the entire disease process.
In conclusion, our observations provide new insight into the multifactorial mechanisms that provoke myocardial injury in COVID-19 including interstitial edema associated with mast cells, local inflammation, and fibrogenesis. These findings help to understand the acute cardiac complications of COVID-19, and also raise concerns about possible chronic cardiac sequelae of SARS-CoV-2 infection which will require further study in survivors of this pandemic illness.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
This study was reviewed and approved by the National Research Ethics Committee (CONEP), Pontifícia Universidade Católica do Paraná (PUCPR), protocol number 3.944.734/2020. Patients’ families authorized the autopsies and provided written informed consent to participate in the study before the procedures. All methods were carried out following relevant guidelines and regulations.
Study design, data analysis, and critical review: CB, CH, GL, LN, and LZ. Data analysis and critical review: PL. Data collection, data analysis, and critical review: AM, CB, JM, LB, LH, and SF. All authors contributed to the article and approved the submitted version.
CH is a recipient of a scholarship of PUCPR and AFSM is recipient of a scholarship from CAPES. PL receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund.
PL is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Cartesian, Esperion, Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Novo Nordisk, Merck, Novartis, Pfizer, Sanofi-Regeneron. PL is a member of scientific advisory board for Amgen, Corvidia Therapeutics, Caristo, CSL Behring, DalCor Pharmaceuticals, Dewpoint, PlaqueTec, Kancera, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech, Inc.
PL’s laboratory has received research funding in the last 2 years from Novartis. PL is on the Board of Directors of XBiotech, Inc. PL has a financial interest in Xbiotech, a company developing therapeutic human antibodies. PL’s interests were reviewed and are managed by Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to the patients and their families that made this study possible. Figure 3 was created with BioRender.com.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.748417/full#supplementary-material
Supplementary Material | Photomicrograph demonstrating the positive and negative controls of the biomarkers studied (CD163, Casp-1, Casp-9, GSDM-D, ICAM-1, TNF-alpha, MMP-9, IL-4, IL-6, IL-1beta, TGF -beta, TUNEL). Placenta with CD163 positive histiocytes (arrow), Casp-1 and Casp-9 positive skeletal muscle fibers (arrow), GSDM-D positive enterocytes (arrow), ICAM-1 positive pulmonary capillaries (arrow) can be seen; cutaneous inflammatory process positive for TNF-alpha, IL-4, IL-6, and TGF-beta (arrow) and fibrous alveolar septum positive for MMP-9 and IL-1beta (arrow) can also be observed.
1. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical Course and Outcomes of Critically Ill Patients With SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir Med (2020) 8(5)::475–81. doi: 10.1016/S2213-2600(20)30079-5
2. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary Manifestations of COVID-19. Nat Med (2020) 26(7):1017–32. doi: 10.1038/s41591-020-0968-3
3. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical Predictors of Mortality Due to COVID-19 Based on an Analysis of Data of 150 Patients From Wuhan, China. Intensive Care Med (2020) 46(5):846–8. doi: 10.1007/s00134-020-05991-x
4. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol (2020) 5:802–10. doi: 10.1001/jamacardio.2020.0950
5. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA - J Am Med Assoc (2020) 324:782–93. doi: 10.1001/jama.2020.12839
6. Escher F, Pietsch H, Aleshcheva G, Bock T, Baumeier C, Elsaesser A, et al. Detection of Viral SARS-CoV-2 Genomes and Histopathological Changes in Endomyocardial Biopsies. ESC Heart Fail (2020) 7:2440–7. doi: 10.1002/ehf2.12805
7. Lindner D, Fitzek A, Bräuninger H, Aleshcheva G, Edler C, Meissner K, et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol (2020) 5(11):1281–5. doi: 10.1001/jamacardio.2020.3551
8. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of COVID-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
9. Tavazzi G, Pellegrini C, Maurelli M, Belliato M, Sciutti F, Bottazzi A, et al. Myocardial Localization of Coronavirus in COVID-19 Cardiogenic Shock. Eur J Heart Fail (2020) 22:911–5. doi: 10.1002/ejhf.1828
10. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
11. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host Cell Death and Inflammation. Nat Rev Microbiol (2009) 7:99–109. doi: 10.1038/nrmicro2070
12. Babapoor-Farrokhran S, Gill D, Walker J, Rasekhi RT, Bozorgnia B, Amanullah A. Myocardial Injury and COVID-19: Possible Mechanisms. Life Sci (2020) 253:117723. doi: 10.1016/j.lfs.2020.117723
13. Wang Y, Xu F, Chen J, Shen X, Deng Y, Xu L, et al. Matrix Metalloproteinase-9 Induces Cardiac Fibroblast Migration, Collagen and Cytokine Secretion: Inhibition by Salvianolic Acid B From Salvia Miltiorrhiza. Phytomedicine (2011) 19:13–9. doi: 10.1016/j.phymed.2011.06.024
14. González A, Schelbert EB, Díez J, Butler J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J Am Coll Cardiol (2018) 71:1696–706. doi: 10.1016/j.jacc.2018.02.021
15. Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen Receptor Status by Immunohistochemistry is Superior to the Ligand-Binding Assay for Predicting Response to Adjuvant Endocrine Therapy in Breast Cancer. J Clin Oncol (1999) 17:1474–81. doi: 10.1200/JCO.1999.17.5.1474
16. Qureshi A, Pervez S. Allred Scoring for ER Reporting and It’s Impact in Clearly Distinguishing ER Negative From ER Positive Breast Cancers. J Pakistan Med Assoc (2010) 60(5):350–3.
17. Aretz HT. Myocarditis: The Dallas Criteria. Hum Pathol (1987) 18:619–24. doi: 10.1016/S0046-8177(87)80363-5
18. Pober JS, Sessa WC. Evolving Functions of Endothelial Cells in Inflammation. Nat Rev Immunol (2007) 7:803–15. doi: 10.1038/nri2171
19. Morimoto SI, Kato S, Hiramitsu S, Uemura A, Ohtsuki M, Kato Y, et al. Role of Myocardial Interstitial Edema in Conduction Disturbances in Acute Myocarditis. Heart Vessels (2006) 21:356–60. doi: 10.1007/s00380-006-0922-4
20. Kunder CA, St John AL, Abraham SN. Mast Cell Modulation of the Vascular and Lymphatic Endothelium. Blood (2011) 118:5383–93. doi: 10.1182/blood-2011-07-358432
21. Etzerodt A, Moestrup SK. CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid Redox Signal (2013) 18:2352–63. doi: 10.1089/ars.2012.4834
22. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix Metalloproteinase-9: Many Shades of Function in Cardiovascular Disease. Physiology (2013) 28:391–403. doi: 10.1152/physiol.00029.2013
23. Kong P, Christia P, Frangogiannis NG. The Pathogenesis of Cardiac Fibrosis. Cell Mol Life Sci (2014) 71:549–74. doi: 10.1007/s00018-013-1349-6
24. Peng H, Sarwar Z, Yang XP, Peterson EL, Xu J, Janic B, et al. Profibrotic Role for Interleukin-4 in Cardiac Remodeling and Dysfunction. Hypertension (2015) 66:582–9. doi: 10.1161/HYPERTENSIONAHA.115.05627
25. Fang L, Ellims AH, Beale AL, Taylor AJ, Murphy A, Dart AM. Systemic Inflammation is Associated With Myocardial Fibrosis, Diastolic Dysfunction, and Cardiac Hypertrophy in Patients With Hypertrophic Cardiomyopathy. Am J Transl Res (2017) 9:5063–73.
26. Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 Mediates Myocardial Fibrosis, Concentric Hypertrophy, and Diastolic Dysfunction in Rats. Hypertension (2010) 56:225–31. doi: 10.1161/HYPERTENSIONAHA.109.148635
27. Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, et al. Myocardial Fibrosis: Biomedical Research From Bench to Bedside. Eur J Heart Fail (2017) 19:177–91. doi: 10.1002/ejhf.696
28. Le HT, Harton JA. Pyrin- and CARD-Only Proteins as Regulators of NLR Functions. Front Immunol (2013) 4:275. doi: 10.3389/fimmu.2013.00275
29. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ (2007) 14:1590–604. doi: 10.1038/sj.cdd.4402194
30. Dubois H, Sorgeloos F, Sarvestani ST, Martens L, Saeys Y, Mackenzie JM, et al. Nlrp3 Inflammasome Activation and Gasdermin D-Driven Pyroptosis are Immunopathogenic Upon Gastrointestinal Norovirus Infection. PloS Pathog (2019) 15:e1007709. doi: 10.1371/journal.ppat.1007709
31. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
32. Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
33. Bonetti PO, Lerman LO, Lerman A. Endothelial Dysfunction: A Marker of Atherosclerotic Risk. Arterioscler Thromb Vasc Biol (2003) 23:168–75. doi: 10.1161/01.ATV.0000051384.43104.FC
34. Legere SA, Haidl ID, Légaré JF, Marshall JS. Mast Cells in Cardiac Fibrosis: New Insights Suggest Opportunities for Intervention. Front Immunol (2019) 10:580. doi: 10.3389/fimmu.2019.00580
35. Cojan-Minzat BO, Zlibut A, Agoston-Coldea L. Non-Ischemic Dilated Cardiomyopathy and Cardiac Fibrosis. Heart Fail Rev (2020), 1–21. doi: 10.1007/s10741-020-09940-0
36. Kollias A, Kyriakoulis KG, Dimakakos E, Poulakou G, Stergiou GS, Syrigos K. Thromboembolic Risk and Anticoagulant Therapy in COVID-19 Patients: Emerging Evidence and Call for Action. Br J Haematol (2020) 189(5):846–7. doi: 10.1111/bjh.16727
37. Lacolley P, Safar ME, Lucet B, Ledudal K, Labat C, Benetos A. Prevention of Aortic and Cardiac Fibrosis by Spironolactone in Old Normotensive Rats. J Am Coll Cardiol (2001) 37:662–7. doi: 10.1016/S0735-1097(00)01129-3
38. Kosmala W, Przewlocka-Kosmala M, Szczepanik-Osadnik H, Mysiak A, O'Moore-Sullivan T, Marwick TH. A Randomized Study of the Beneficial Effects of Aldosterone Antagonism on Lv Function, Structure, and Fibrosis Markers in Metabolic Syndrome. JACC Cardiovasc Imaging (2011) 4:1239–49. doi: 10.1016/j.jcmg.2011.08.014
Keywords: heart, myocardial injury, COVID-19, SARS-CoV-2, endothelium, immunohistochemistry, pathology, fibrosis
Citation: Hartmann C, Miggiolaro AFRdS, Motta JdS Junior, Baena Carstens L, Busatta Vaz De Paula C, Fagundes Grobe S, Hermann de Souza Nunes L, Lenci Marques G, Libby P, Zytynski Moura L, de Noronha L and Pellegrino Baena C (2021) The Pathogenesis of COVID-19 Myocardial Injury: An Immunohistochemical Study of Postmortem Biopsies. Front. Immunol. 12:748417. doi: 10.3389/fimmu.2021.748417
Received: 27 July 2021; Accepted: 18 October 2021;
Published: 05 November 2021.
Edited by:
Julia Kzhyshkowska, Heidelberg University, GermanyReviewed by:
Qi Wang, Southern Medical University, ChinaCopyright © 2021 Hartmann, Miggiolaro, Motta, Baena Carstens, Busatta Vaz De Paula, Fagundes Grobe, Hermann de Souza Nunes, Lenci Marques, Libby, Zytynski Moura, de Noronha and Pellegrino Baena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucia de Noronha, bHVjaWEubm9yb25oYUBwdWNwci5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.