 Jasmine George
Jasmine George Yuanyuan Zhang1†
Yuanyuan Zhang1† John D. Imig
John D. Imig Xueying Zhao
Xueying Zhao- 1Department of Physiology, Morehouse School of Medicine, Atlanta, GA, United States
- 2Drug Discovery Center, Medical College of Wisconsin, Milwaukee, WI, United States
Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH) is commonly associated with obesity and characterized by excessive lipid accumulation and liver inflammation. The T cell immunoglobulin and mucin domain 1 (Tim-1), also known as hepatitis A virus cellular receptor 1 (Havcr-1) and kidney injury molecule 1 (Kim-1), has been shown to affect innate immunity-driven proinflammatory cascade in liver ischemia-reperfusion injury. However, its contribution to obesity-related NAFLD/NASH remains unknown. Thus, this study was designed to evaluate the role of Tim-1 in obesity-related liver inflammation and injury in wild-type (WT) and Tim-1-deficient (Tim-1-/-) C57BL/6J mice fed a high-fat diet (HFD) for 5-6 months. HFD feeding induced steatosis and upregulated Tim-1 gene expression in the liver of WT mice. Surprisingly, Tim-1-/- mice on HFD diet exhibited an exacerbation of hepatic steatosis, accompanied with an elevation of protein levels of fatty acid translocase CD36 and sterol regulatory element binding protein 1 (SREBP1). Tim-1 deficiency also enhanced HFD-induced liver inflammation and injury, as evidenced by augmented increase in hepatic expression of pro-inflammatory factor lipocalin 2 and elevated serum alanine transaminase (ALT). In addition, gene expression of type I, III and IV collagens and liver fibrosis were greatly enhanced in HFD Tim-1-/- mice compared with HFD WT mice. HFD-induced hepatic expression of YM-1, a specific mouse M2 macrophage marker, was further upregulated by deletion of Tim-1. Together, these results show that Tim-1 deficiency aggravates the effects of HFD diet on lipid accumulation and liver fibrosis, most likely through enhanced infiltration and activation of inflammatory cells.
Introduction
Non-alcoholic fatty liver disease (NAFLD) has become the most common cause of chronic liver diseases in western countries (1). The prevalence of NAFLD in the U.S. adult population is approximately 25%, and about one-fourth of the patients with NAFLD progress to nonalcoholic steatohepatitis (NASH) (2). Through histological examinations, NASH is characterized by steatosis, hepatocyte ballooning, lobular inflammation, and varying degrees of liver fibrosis, which can lead to scarring of the liver (3). NASH has become a major health concern; however, there are no FDA-approved drugs for the treatment of this disease. Thus, improving our understanding of the functions of genes/mediators that contribute to the susceptibility to and severity of NAFLD/NASH is of great consequence to both the treatment and prevention of chronic liver disease.
The liver is recognized as an innate immunity organ and the hallmark of NASH is markedly enhanced infiltration of various immune cells, including hepatic macrophages (liver-resident Kupffer cells and recruited monocyte-derived macrophages), neutrophils, monocytes, and natural killer T cells (4–6). Given the functional role of hepatic macrophages as a master regulator of immune homeostasis and a pivotal coordinator of liver inflammation, it has been postulated that macrophage activation and polarization are involved in the progression of NAFLD to NASH. Nevertheless, it remains largely unknown how M1 versus M2 macrophages contribute to liver inflammation and fibrosis progression in advanced NASH.
T cell immunoglobulin and mucin domain-containing molecule 1 (Tim-1), also known as hepatitis A virus cellular receptor 1 (Havcr-1) and kidney injury molecule 1 (Kim-1), was first identified in kidney cells of African green monkeys in 1996 (7). Tim-1 protein, identified in activated T cells, B cells and dendritic cells, functions as co-stimulators and co-inhibitors of immune responses. For example, studies using blocking or activating antibodies have shown that the interaction between Tim-1 and ligand can enhance the activation of T cells and increase the production of Th2 type cytokines while blocking this interaction can greatly inhibit the activity of Th2 cells, thus regulating the immune response mediated by Th2 cells (8–15). Notably, a study performed in in vivo allergic airway disease revealed enhanced production of the Th2 cytokines (e.g., IL-4, IL-5, and IL-13) and inflammatory responses in the absence of Tim-1, suggesting that its primary role is to dampen, rather than promote, Th2-type immune responses (16). This concept is also supported by the findings that blockade of Tim-1 in low-density lipoprotein receptor (ldlr)-deficient mice aggravates atherosclerosis, which is likely related to the change in Th1/Th2 balance and reduced circulating regulatory T cells (17). Interestingly, Tim-1 signaling has been confirmed to be necessary for liver ischemia-reperfusion injury via increasing T cell, neutrophil, and macrophage sequestration (18, 19). High Tim-1 expression was observed in liver graft during ischemia-reperfusion injury development, and inhibition of Tim-1 abolished neutrophil and macrophage infiltration/activation in liver transplantation (18, 20) and subsequently ameliorated hepatocellular damage and improved liver function (18). Based on these observations, we hypothesized that Tim-1 plays a role in diet-induced NAFLD/NASH via its modulation of the inflammatory response to metabolic stress.
In this study, we generated mice deficient in Tim-1 and evaluated their responses to chronic high-fat diet (HFD) treatment. We found that the Tim-1 gene was significantly upregulated in HFD-fed wild-type (WT) livers. Tim-1 deficiency led to an exacerbation of HFD-induced steatosis concomitant with enhanced liver infiltration of neutrophils as well as macrophage activation and M2 polarization.
Materials And Methods
Animal Experiments
The Tim-1/Havcr-1/Kim-1 knockout (Tim-1-/-) mouse strain was generated using CRISPR/Cas9 technology on a C57BL/6J genetic background by the Gene Edit Biolab (Atlanta, GA, USA). Tim-1-/- mice were produced through heterozygous breeding. Tail samples were collected and genotyped. The animals were housed in a temperature-controlled room with 12 h light/dark regimen. All animal experiments were approved by the Institutional Animal Care and Use Committee at the Morehouse School of Medicine and were performed according to strict government and international guidelines on animal experimentation.
Male WT and Tim-1-/- mice at the age of 6 - 8 weeks were fed with standard chow or HFD diet for 5 - 6 months. The HFD, containing 42% fat, was purchased from the Envigo (TD.88137; Madison, WI, USA). Mouse body weight was measured every week, and blood glucose was monitored biweekly. The mice were fasted for 5-6 hours before euthanasia. Blood was collected by heart puncture. Liver and kidney tissues were harvested and stored in 4% paraformaldehyde or quickly frozen in liquid nitrogen and then stored at -80°C until further use.
Biochemical Analysis
Blood samples were clotted at room temperature for 2 hours and then centrifuged at 1200xg for 20 minutes. Serum samples were assayed immediately or split and stored at -80°C, avoiding repeated freeze-thaw cycles. Serum insulin concentration was measured using a kit from Thermo Fisher Scientific (Carlsbad, CA, USA). The serum lipid levels of cholesterol and triglyceride were measured to evaluate the relative lipid content changes via commercially available kits provided by Wako Chemicals USA Inc. (Richmond, VA, USA). Serum alanine aminotransferase (ALT) concentration was determined to evaluate liver function using an ALT activity assay kit (Thermo Fisher Scientific).
Quantitative Reverse Transcription PCR Analysis
Total RNA from mouse liver and kidney cortex was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Quantitative reverse transcription PCR (RT-qPCR) was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and primers for mouse genes (Table 1). GAPDH was used as endogenous control. Each sample was run in triplicate, and the comparative threshold cycle (Ct) method was used to quantify fold increase (2-ΔΔCt) compared with controls.

Table 1 Primer sequences for qPCR.
Light Microscopy of Liver Sections
Ten percent formalin-fixed paraffin sections (5 µm) were stained with hematoxylin and eosin (H&E) for histological evaluation. Another set of paraffin-embedded liver sections were stained with Sirius Red to identify collagen. Neutral lipid accumulation was determined by oil red O (ORO) staining of frozen hepatic tissue sections, which allows detection of triglyceride and cholesterol esters. The slides were observed and imaged by the Olympus microscope. All histological analysis was conducted by two observers in a blinded fashion. Sirius Red or ORO-stained areas were quantified by ImageJ software (21).
Western Blot Analysis
Liver tissue samples were lysed with RIPA lysis buffer (Sigma Aldrich Inc., St. Louis, MO, USA) and a protease inhibitor cocktail (Sigma-Aldrich). Protein (30 µg) samples were separated by 4-20% SDS-PAGE and transferred electrophoretically to nitrocellulose membranes (GE Healthcare, Piscataway, NJ, USA). The blots were incubated with primary antibodies for lipocalin-2 (LCN2, 1:1000, R&D, Minneapolis, MN, USA), fatty acid transport protein (FATP)2 (1:1000, Novus Biologicals, Centennial, CO, USA), FATP5 (1:1000, Novus Biologicals), CD36 (1:1000, Abcam, Cambridge, MA, USA), sterol regulatory element binding protein (SREBP)1 (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Ly6G/6C (1:1000; BD Diagnostic Systems, Sparks, MD, USA), F4/80 (1:1000; Bio-Rad Laboratories Inc., Irvine, CA, USA), and GAPDH (1:6000, Sigma-Aldrich). After incubation with HRP-conjugated secondary antibody, signals were detected using enhanced chemiluminescence ECL reagents (GE Healthcare). Relative band intensity was measured densitometrically by ImageJ software with GAPDH, a housekeeping protein, as an internal control.
Immunostaining
To examine the expression and distribution of LCN2, 5-µm cryostat liver sections were incubated with one or two primary antibodies overnight: goat anti-LCN2 (1:100; R&D) and rat anti-Ly6G/6C (a marker of neutrophils; 1:100; BD Diagnostic Systems) or goat anti-LCN2 and rat anti-F4/80 (a marker of macrophages; 1:100; Bio-Rad Laboratories Inc.). The secondary antibodies were Alexa Fluor 488-conjugated donkey anti goat or rat IgG (1:200) or Alexa Fluor 555-conjugated donkey anti-goat or rat IgG (1:200) from the Jackson ImmunoResearch Laboratories (West Grove, PA, USA). As a negative control, the sections were exposed to nonimmune IgG (in replacement of primary antibodies) with the same secondary antibodies, and no specific staining was observed. After nuclear staining with DAPI, the slides were mounted with ProLong gold antifade reagent (Thermo Fisher Scientific). The sections were observed and imaged by a Leica confocal microscope (Wetzlar, Germany).
Statistical Analysis
Data are expressed as means ± SEM. Student’s t-test was used for comparison between two groups. Comparisons among multiple groups were performed by one-way ANOVA and Tukey post hoc test. Differences were considered statistically significant at P < 0.05.
Results
Tim-1 Gene Expression Was Upregulated in HFD-Fed C57BL/6J Mouse Livers
Previous studies have shown that hepatic expression of Tim-1 is increased in liver ischemia-reperfusion injury (18–20). Here, we examined the effect of metabolic stress on Tim-1 expression in the liver of C57BL/6J WT mice. Compared to chow control group, HFD feeding for 5-6 months significantly increased body weight (Figure 1A) and liver weight (Figure 1B). In addition, real-time qPCR analysis revealed an upregulation of hepatic Tim-1 transcript in HFD-induced obese mice compared with chow controls (Figures 1C, D), whereas its mRNA levels were not different in kidney tissues of chow (1.1 ± 0.2, n=5) and HFD (0.9 ± 0.1, n=4) WT mice.
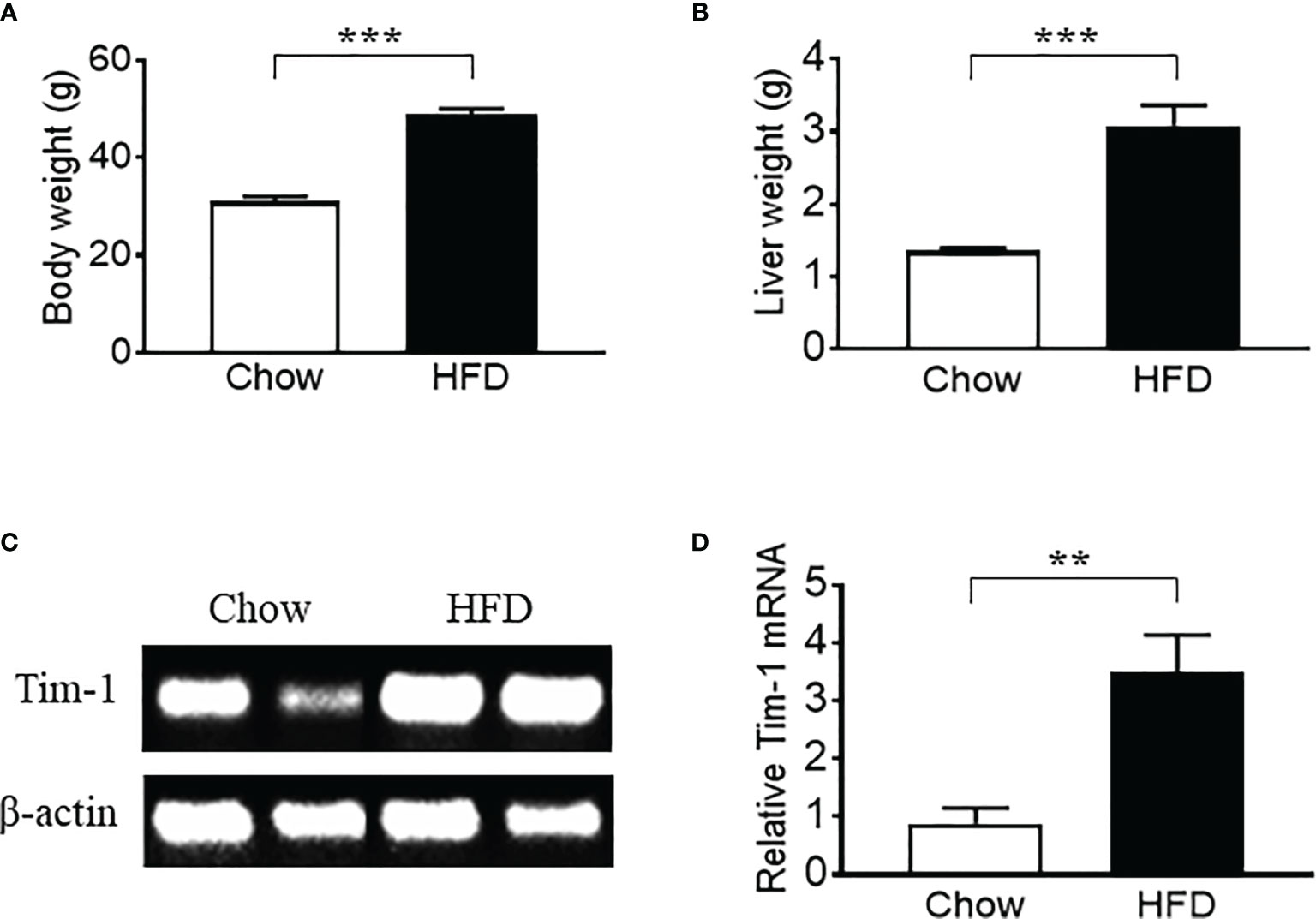
Figure 1 High-fat diet (HFD) feeding for 6 months led to an upregulation of hepatic Tim-1 in wild-type C57BL/6 mice. (A, B) Increased body weight and liver weight in HFD-fed mice compared to chow controls. (C, D) Representative gel and quantitative analysis after 35 cycles RT-PCR revealed an upregulation of Tim-1 gene expression in HFD mouse liver. Values are mean ± SEM. n = 5-6 mice; **P<0.01, ***P<0.001 vs. chow control group.
Tim-1 Deficiency Enhanced HFD-Induced Hepatic Steatosis
To further explore the function of Tim-1 in diet-induced hepatic steatosis, Tim-1-/- mice on C57BL/6J background were generated by inserting a Lox-Stop-Lox cassette into intron 2 of Tim-1 using CRISPR/Cas9 technology (Figures 2A, B). As expected, Tim-1 mRNA was not detectable in the liver (Figure 2C) of Tim-1-/- mice. The Tim-1-/- mice on chow diet had normal size and displayed normal physical behavior and activity. Moreover, deletion of Tim-1 had no effects on body weight gain and blood glucose in mice fed with chow or HFD diet. Serum insulin levels were similarly increased in HFD-fed WT (5.51 ± 1.29 ng/ml, n=6) and Tim-1-/- (6.58 ± 2.24 ng/ml, n=4) compared to chow-fed WT (1.73 ± 0.18 ng/ml, n=5) and Tim-1-/- (2.29 ± 0.37 ng/ml, n=6) mice.
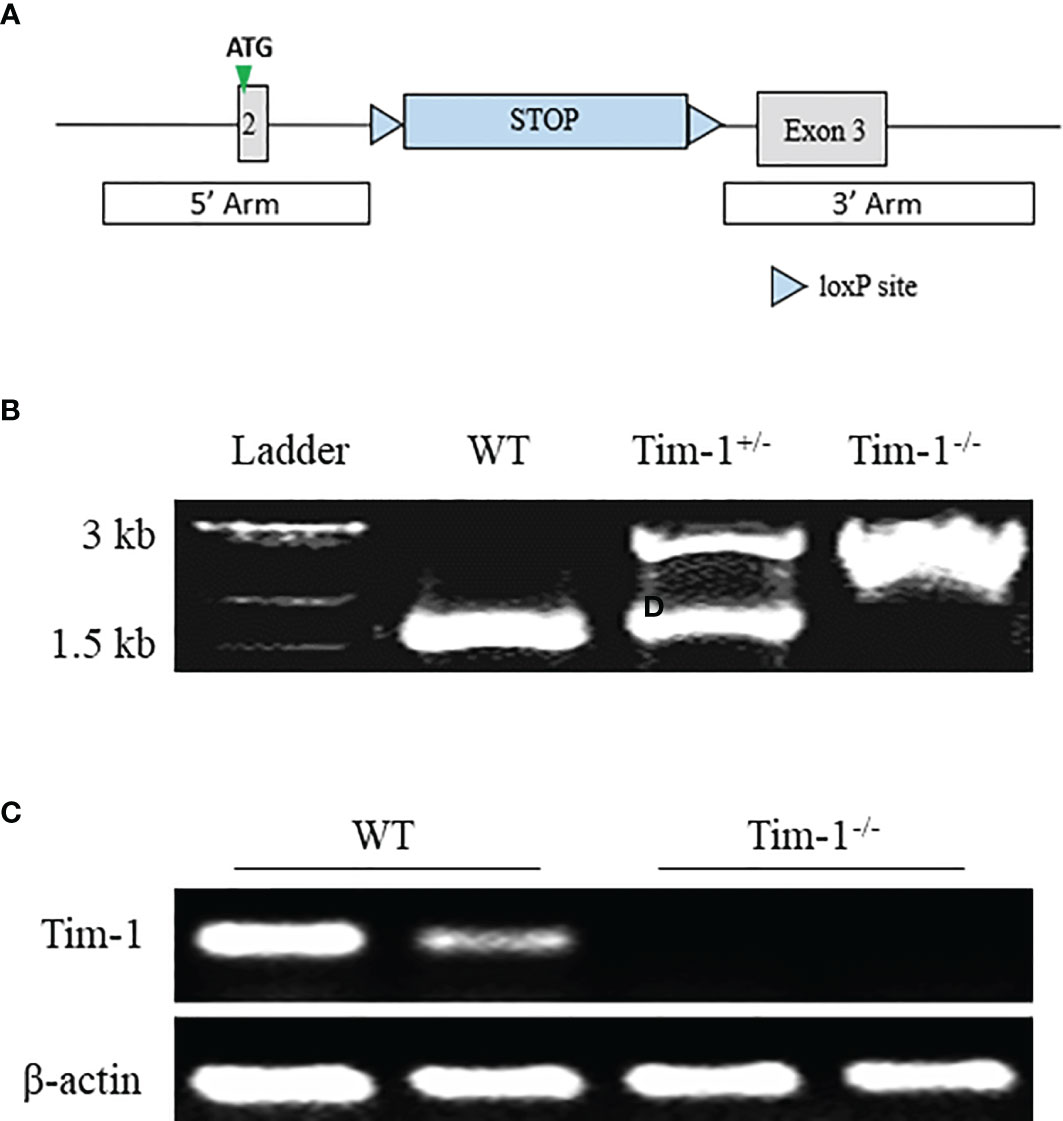
Figure 2 Generation of global Tim-1 knockout mice. (A) Schematic diagram of generation of Tim-1 Lox-Stop-Lox knock-in (Tim-1-/-) mice. (B) Genotyping PCR of tail DNA from wild-type (WT), heterozygous (Tim-1+/-), and homozygous (Tim-1-/-) mice. (C) Representative image of hepatic Tim-1 gene after 35 cycles RT-PCR in WT and Tim-1-/- mice.
Serum triglyceride and total cholesterol levels were measured to evaluate the effect of Tim-1 deficiency on mouse lipid profile in serum. Compared to chow controls, HFD feeding led to a similar increase in cholesterol levels in WT and Tim-1-/- mice (Figure 3A), whereas triglyceride levels were not different among all four groups (Figure 3B). Although Tim-1 deficiency did not alter the liver weight and liver index (liver weight to body weight ratio) in chow-fed mice, there was a greater increase in liver weight and liver index in HFD-fed Tim-1-/- mice compared with HFD WT animals (Figures 3C, D). To further confirm the effect of Tim-1 deficiency on hepatic steatosis, we next evaluated lipid accumulation using H&E and ORO staining with liver sections. As shown in Figure 3E, there was no significant difference in liver histology and lipid storage between WT and Tim-1-/- mice on chow diet. However, HFD-induced hepatocyte ballooning and neutral lipid accumulation were more prominent in mice lacking Tim-1. ORO-positive area averaged 5.8 ± 1.1% in HFD-fed WT livers, which was further increased to 10.2 ± 1.3% (P<0.05) in HFD Tim-1-/- ones.
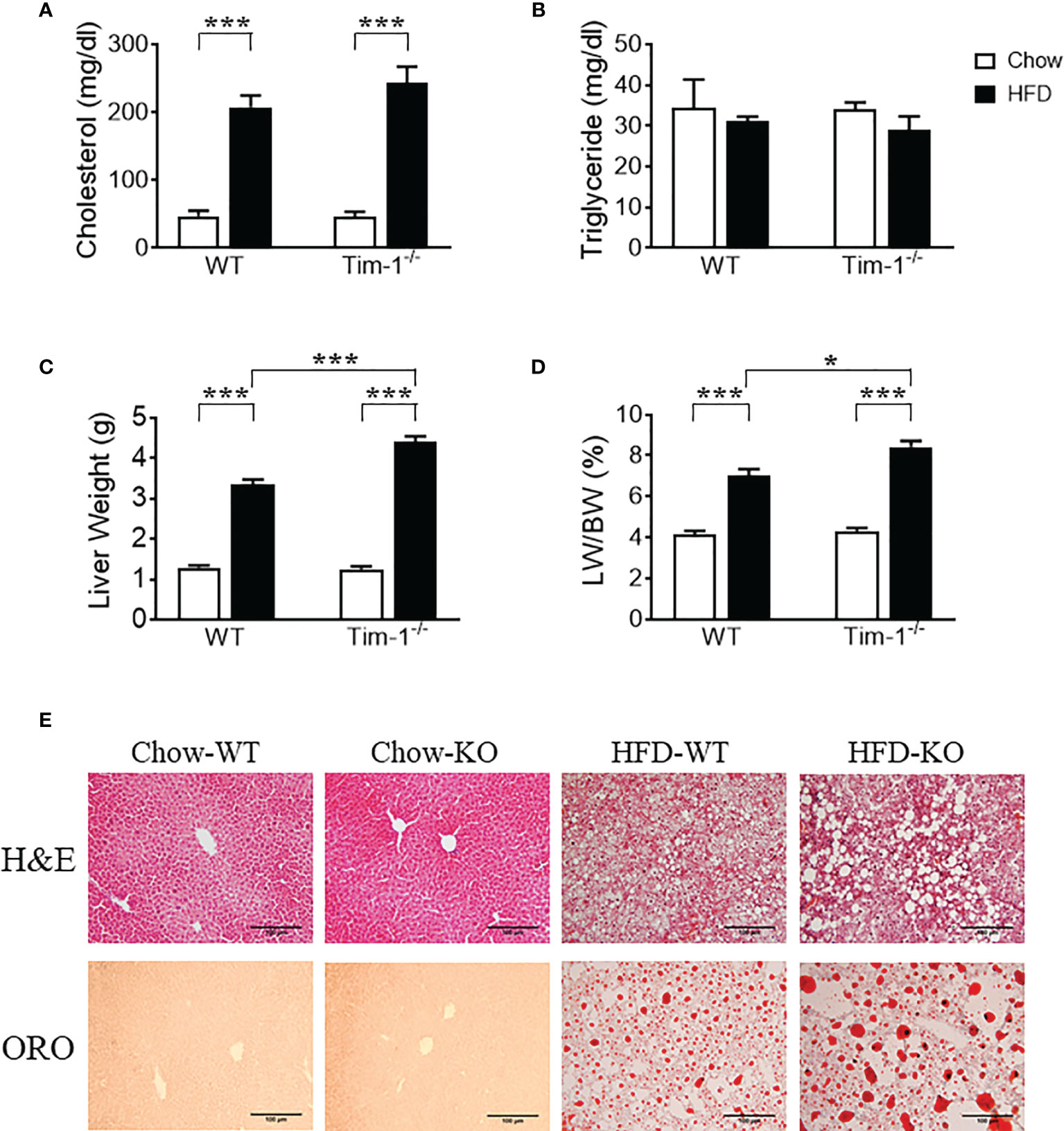
Figure 3 Tim-1 deficiency accelerated diet-induced hepatic steatosis. Serum levels of cholesterol (A) and triglyceride (B), liver weight (C), and liver weight to body weight (LW/BW) ratio (D) in chow or high-fat diet (HFD) WT and Tim-1-dificient (Tim-1-/-) mice. (E) Representative images of H&E and Oil red O (ORO) staining of WT and Tim-1-deficient (KO) livers (scale bar = 100 µm). Values are mean ± SEM. n=4-6 mice. Statistical differences were assessed by one-way ANOVA with Tukey’s multiple comparisons test; *P<0.05, ***P<0.001.
Tim-1 Deletion Enhanced HFD-Induced Hepatic Expression of Proteins Involved in Lipid Uptake and Biogenesis
To understand the mechanisms by which Tim-1 deficiency accelerates diet-induced hepatic steatosis, we next determined the proteins involved in the transport and biogenesis of fatty acids (FAs). Hepatic FA uptake is mainly mediated by SLC27A/FATPs and FA translocase CD36 (22, 23). Two members of the FATP family, FATP2 and FATP5, are robustly expressed in liver (24) and are thought to be involved in the early steps of long-chain FA uptake/activation (25, 26). Thus, we first compared the protein levels of FATP2 and FATP5 in the liver of chow or HFD-fed WT and Tim-1-/- mice. As shown in (Figures 4A, B), there was no significant difference in hepatic FATP2 protein among the four groups. Compared to chow controls, an upregulation of FATP5 protein expression was observed in HFD Tim-1-/- mice but not in HFD WT mice (Figures 4A, C). The FA translocase protein CD36 has been shown to accelerate FA uptake and extensive incorporation into triglycerides (27). Compared to chow controls, CD36 protein was significantly increased in liver tissues of HFD-fed WT mice, which was further enhanced by Tim-1 deletion (Figures 4A, D).
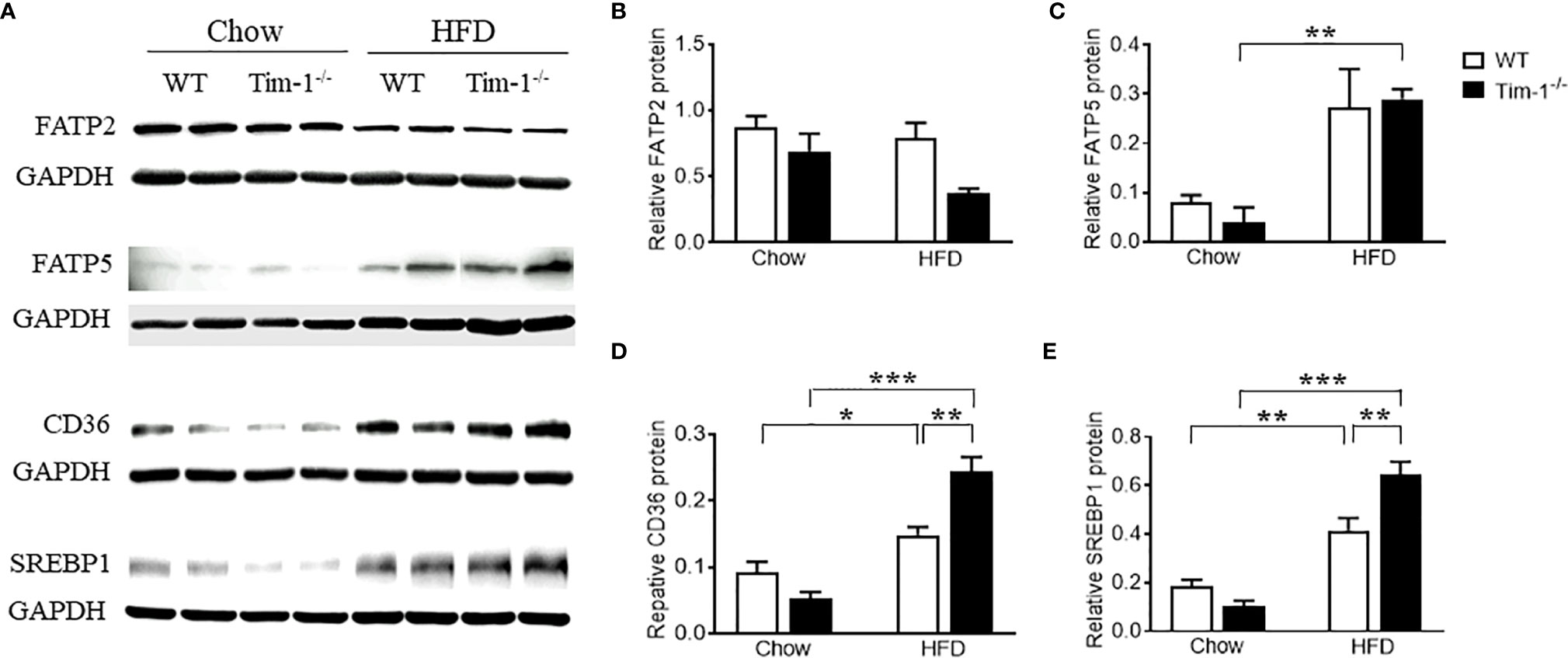
Figure 4 Tim-1 deficiency increased protein levels of FATP5, CD36 and SREBP1 in HFD mouse livers. (A) Representative Western blot images of FATP2, FATP5, CD36, and SREBP1 in the liver of chow or HFD-fed WT and Tim-1-/- mice. (B–E) Quantitative analysis of each protein. Values are mean ± SEM. n=4-6 mice. Statistical differences were assessed by one-way ANOVA with Tukey’s multiple comparisons test; *P<0.05, **P<0.01, ***P<0.001.
As a family of transcription factors involved in the biogenesis of cholesterol, FAs and triglycerides, SREBPs are implicated in the pathogenesis of NAFLD/NASH and SREBP1 is the primary subtype expressed in the liver of mice and humans (28, 29). Here, we confirmed that HFD feeding increased SREBP1 protein in WT livers (Figures 4A, E). Although Tim-1 deficiency had no effect on SREBP1 expression in the liver of chow-fed mice, HFD-induced upregulation of SREBP1 was greatly augmented by deletion of Tim-1 (P < 0.001 versus HFD WT group).
Tim-1 Deletion Accelerated HFD-Induced Liver Injury and Inflammation
In accordance with the more severe hepatomegaly and NAFLD liver phenotype shown by H&E and ORO staining in HFD-fed Tim-1-/- mice, HFD induced more hepatic secretion of ALT, a useful biomarker of liver injury, in mice lacking Tim-1. As shown in Figure 5A, HFD-fed Tim-1-/- mice showed significantly higher serum ALT level compared to the same diet-treated WT mice, supporting that deletion of Tim-1 promotes HFD-induced liver damage.
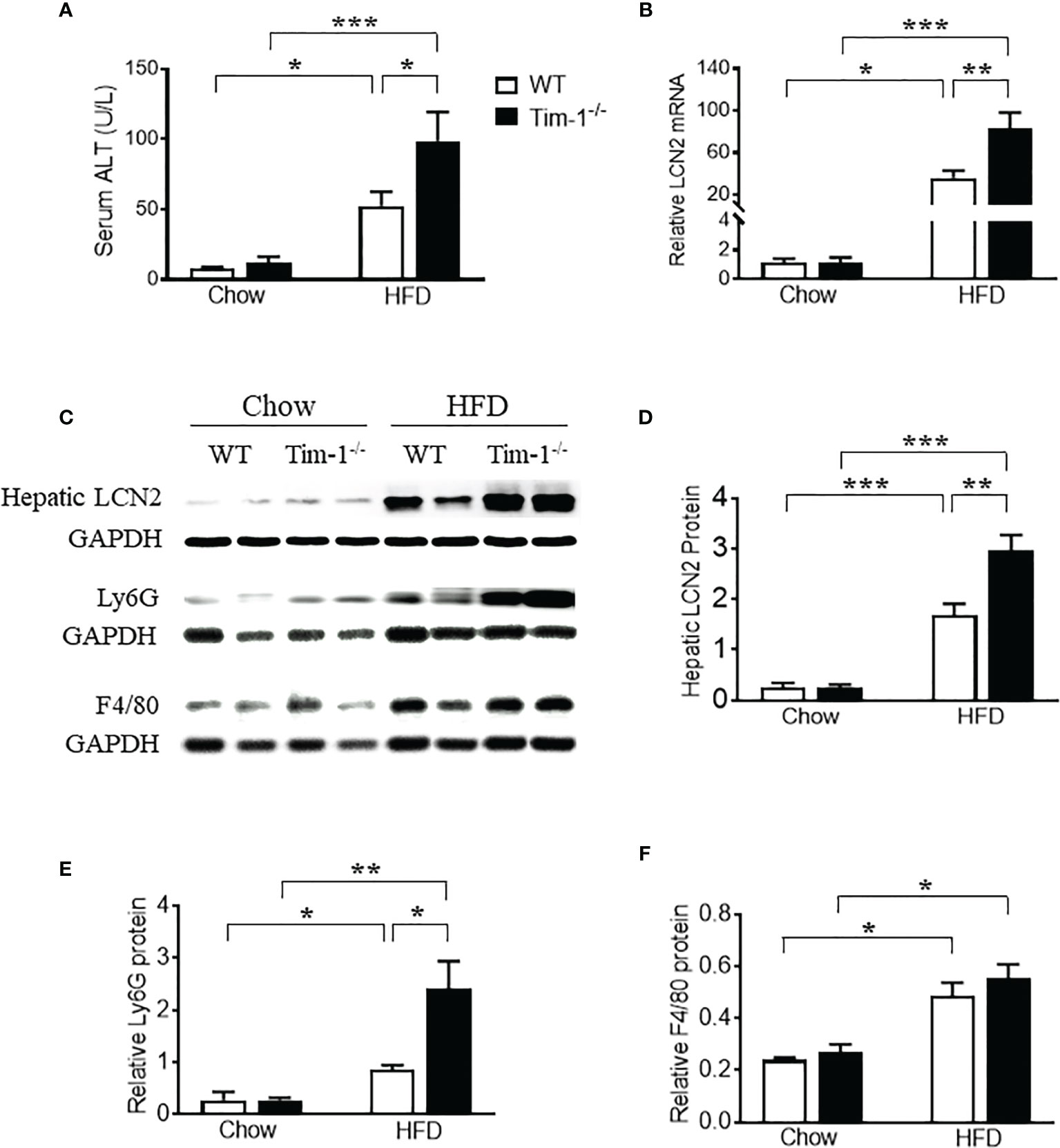
Figure 5 Tim-1 deficiency enhanced HFD-induced liver injury. (A) Serum alanine aminotransferase (ALT) concentration in chow or HFD-treated WT and Tim-1-/- mice. (B) Real-time qPCR analysis of lipocalin 2 (LCN2) transcript in chow or HFD-fed WT and Tim-1-/- mouse livers. (C–F) Representative Western blot image and quantitative analysis of hepatic LCN2, Ly6G/C, and F4/80 proteins. Values are mean ± SEM. n=4-6 mice. Statistical differences were assessed by one-way ANOVA with Tukey’s multiple comparisons test; *P<0.05, **P<0.01, ***P<0.001.
Lipocalin-2 [LCN2, also known as neutrophil gelatinase-associated lipocalin (NGAL)] is known to be expressed by a variety of cells including neutrophils, macrophages, epithelial cells like hepatocytes and its upregulation in liver is a reliable indicator of liver inflammation and damage. To further confirm the effects of Tim-1 deficiency on liver injury and inflammation, we examined the mRNA and protein levels of LCN2 in diet-induced fatty livers. As shown in Figure 5B, HFD feeding increased hepatic LCN2 mRNA by 34-fold in WT and 83-fold in Tim-1-/- mice (P < 0.01 versus HFD WT group). Accordingly, Western blot analysis revealed that HFD-induced upregulation of LCN2 protein in liver tissues was substantially enhanced in mice lacking Tim-1 (Figures 5C, D). Moreover, Tim-1 deletion accelerated HFD-induced neutrophil infiltration. As shown in Figures 5C, E, Ly6G protein was greatly higher in the liver of HFD Tim-1-/- mice compared to HFD WT mice, whereas F4/80 was similarly increased in HFD-fed WT and Tim-1-/- livers (Figures 5C, F).
Next, we determined if neutrophils and/or macrophages are the primary cell sources of elevated LCN2 in the obese liver by dual-labeling of the liver sections for LCN2 with Ly6G or with F4/80. In addition to barely detectable LCN2 staining in normal mouse livers, there were no apparent Ly6G or F4/80 signals in chow-fed WT and Tim-1-/- mouse livers. As depicted in Figures 6A, B, increased LCN2, Ly6G, and F4/80 were observed in HFD WT livers, which was further enhanced by Tim-1 deletion. Some but not all LCN2-positive cells were stained positive for Ly6G (Figure 6A). Similarly, a subset but not all LNC2-positive cells expressed macrophage marker F4/80. Our results support that Tim-1 deficiency enhanced LCN2 expression in both infiltrated neutrophils and macrophages in HFD mouse livers.
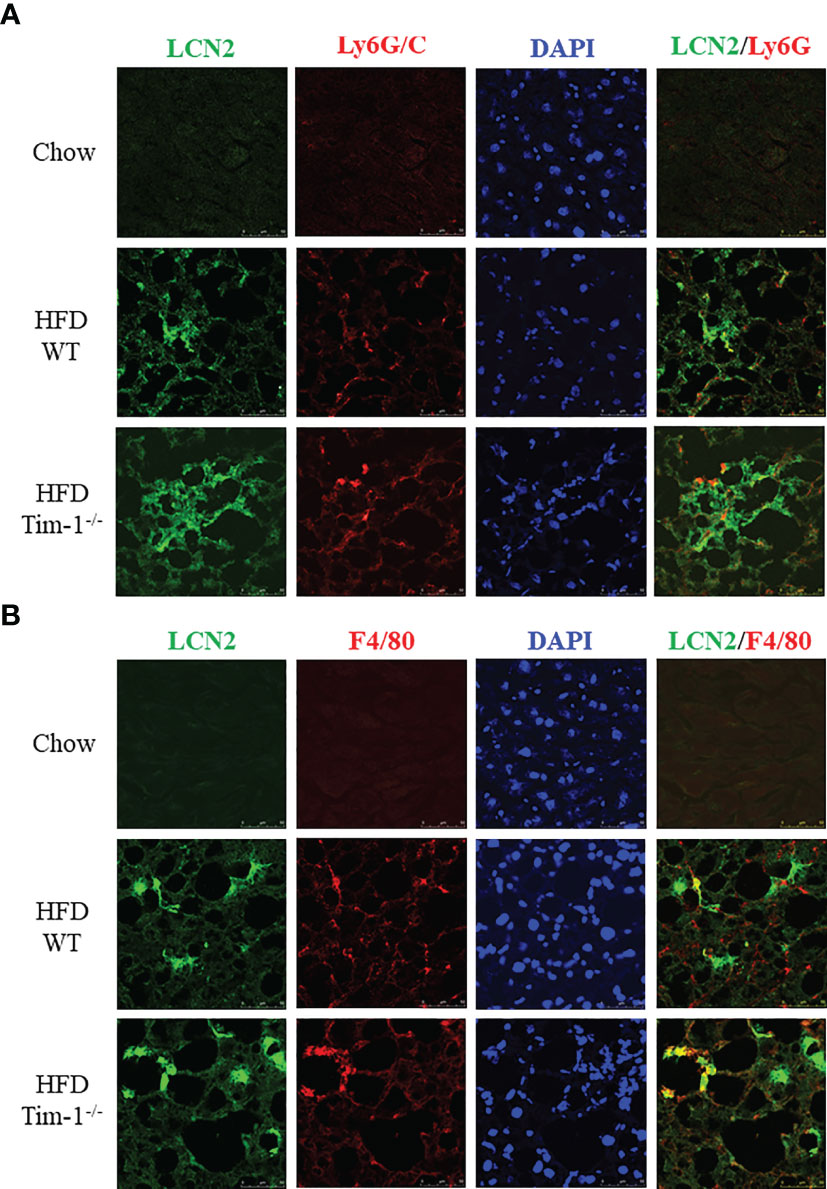
Figure 6 Tim-1 deficiency increased LCN2 expression in infiltrated liver neutrophils and macrophages. Representative immunofluorescence images of double staining for LCN2 with neutrophil marker Ly6G/C (A) or macrophage marker F4/80 (B) in chow or HFD-fed WT and Tim-1-/- livers.
Tim-1 Deletion Accelerated HFD-Induced Liver Fibrosis
To further determine if increased hepatic injury and inflammation in HFD Tim-1-/- mice would enhance collagen deposition, we next performed Sirius Red staining on paraffin-embedded liver sections. As depicted in (Figures 7A, B), Sirius red staining revealed that HFD feeding for 6 months induced minor to moderate accumulation of collagen fibers in liver tissue of WT mice. Tim-1 deficiency greatly enhanced HFD-induced fibrosis, as shown by abundant fibrillar collagen deposition in HFD Tim-1-/- livers. The liver expresses type I (Col1), III (Col3) and IV (Col4) collagens with type I being the major collagen associated with hepatic fibrosis in Western diet-fed mice and humans with NASH. Compared to chow controls, we found that HFD substantially increased the expression of Col1a1, Col3a1, and Col4a1 by 24-, 10-, and 2.6-fold, respectively in Tim-1-/- livers and 7-, 4-, and 2-fold, respectively in WT livers (Figure 7C), while fibronectin 1 (Fn1) transcript levels were not different among the groups.
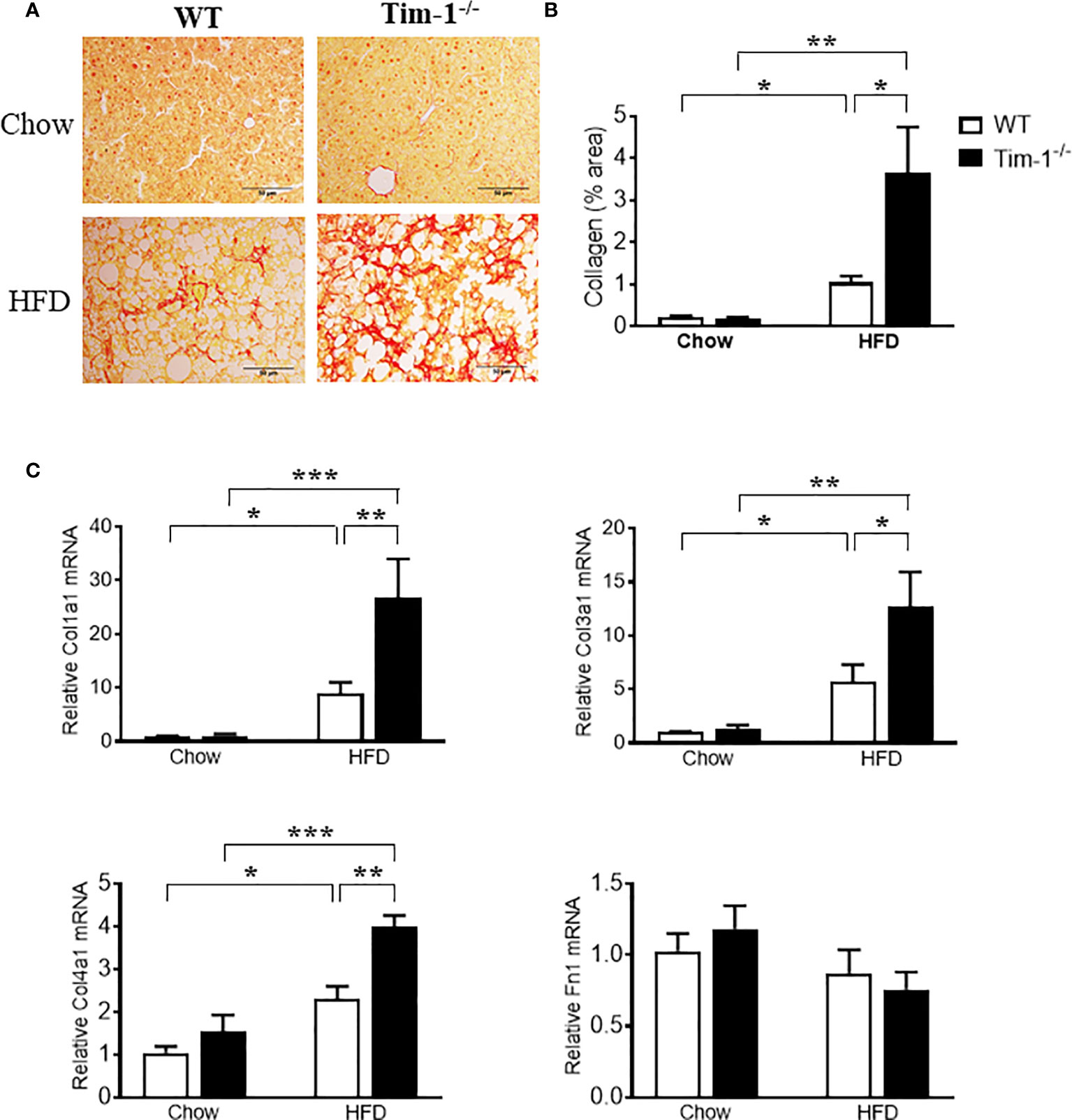
Figure 7 Tim-1 deficiency accelerated HFD-induced liver fibrosis. (A) Representative images of Sirius Red staining to identify collagen deposition in liver sections from chow or HFD-fed WT and Tim-1-/- mice. (B) Quantitative analysis of Sirius Red-stained liver tissue sections. (C) Real-time qPCR analysis of hepatic gene expression of type I (Col1a1), type III (Col3a1), and type IV (Col4a1) collagen as well as fibronectin (Fn1) in chow or HFD-treated WT and Tim-1-/- mice. Values are mean ± SEM. n=4-6 mice. Statistical differences were assessed by one-way ANOVA with Tukey’s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001.
Tim-1 Deletion Enhanced M2 Macrophage Polarization
Macrophages have been found to both promote liver fibrosis and contribute to its resolution by reprogramming the two different polarization states, pro-inflammatory M1 macrophages (classically activated) and anti-inflammatory M2 macrophages (alternatively activated), which was driven by micro-environmental cues. To further evaluate the effect of Tim-1 deficiency on macrophage activation and polarization, we next determined the expression levels of markers for M1 and M2 macrophages in liver tissues. Compared to chow controls, HFD feeding caused a similar increase in mRNA levels of M1 marker CD11c (Figure 8A) and pro-inflammatory cytokine TNF-α (Figure 8B) in both WT and Tim-1-/- mice. As indicated in Figure 8C, hepatic expression of CD206, a well-known M2 marker for both mouse and human, was not affected by HFD diet or Tim-1 deletion. In contrast, HFD feeding substantially increased hepatic expression of YM-1, a specific mouse M2 marker, by 18-fold in WT and 42-fold in Tim-1-/- mice (P < 0.05 vs. HFD-WT) compared with chow controls (Figure 8D). mRNA levels of anti-inflammatory cytokine TGF-β1 were elevated to a similar extent in HFD-fed WT and Tim-1-/- mice (Figure 8E). Additionally, hepatic expression of IL-10 was not significantly modified by diet or Tim-1 deletion (Figure 8F).
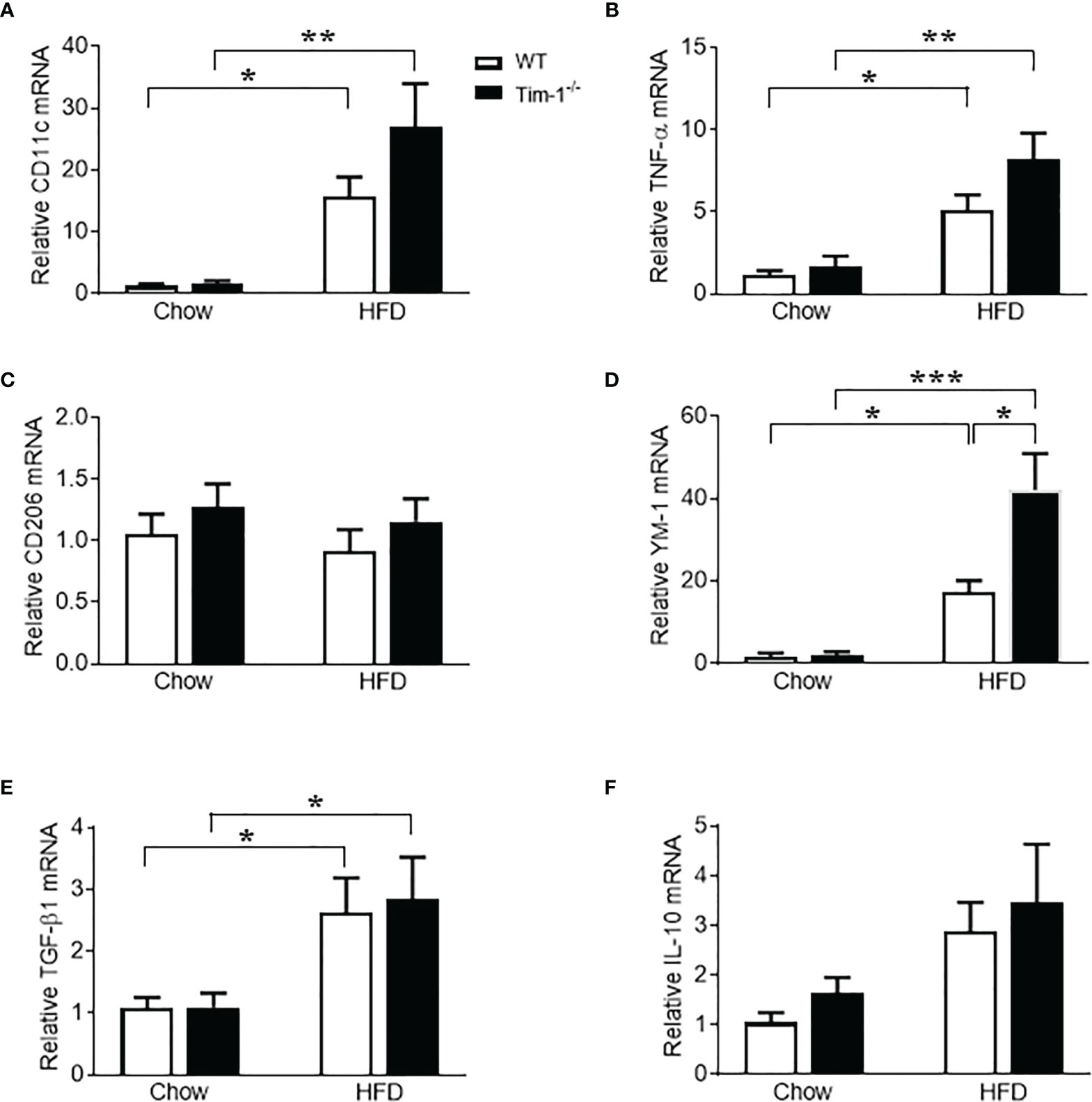
Figure 8 Effects of Tim-1 deficiency on M1 and M2 macrophages. Real-time PCR analysis of hepatic gene expression of M1 macrophage marker CD11c (A), proinflammatory cytokine TNF-α (B), M2 markers CD206 (C) and YM-1 (D), and anti-inflammatory cytokine TGF-β1 (E) and IL-10 (F). Values are mean ± SEM. n=4-6 mice. Statistical differences were assessed by one-way ANOVA with Tukey’s multiple comparisons test; *P<0.05, **P<0.01, ***P<0.001.
Discussion
This is the first study that documents the functional significance of Tim-1 in diet-induced NAFLD/NASH. We found that Tim-1 expression was significantly upregulated in the liver of HFD-fed WT mice. Tim-1 deficiency resulted in an exacerbation of HFD-induced hepatic steatosis and inflammation, as evidenced by increased hepatocyte ballooning and excessive lipid accumulation concomitant with augmented infiltration of inflammatory cells as well as pronounced hepatic expression of LCN2 in HFD Tim-1-/- mice. Moreover, HFD-induced collagen deposition and hepatic expression of fibrotic and M2 macrophage markers were significantly enhanced by genetic deletion of Tim-1. Our results suggest that Tim-1 functions in pathways that suppress the recruitment and activation of inflammatory cells in the liver and protect against NASH progression.
In agreement with previous reports (30–35), we found that HFD-fed C57BL/6J mice gained excessive weight and developed NAFLD/NASH as assessed by liver histology within 6 months of HFD feeding. Interestingly, diet-induced liver steatosis was significantly exacerbated by Tim-1 deletion, as evidenced by higher liver weight, more severe hepatocyte ballooning and excessive lipid accumulation in HFD Tim-1-/- mice. These results support a functional role for Tim-1 in lipid metabolism and/or hepatic distribution in response to metabolic stress.
Hepatic steatosis develops when lipid uptake and de novo synthesis surpass lipid oxidation and export. In NAFLD, hepatic FA uptake and de novo lipogenesis are increased. Long-chain FAs can be either transported directly by FATPs across the plasma membrane or, alternatively, are first accumulated on the plasma membrane by binding to CD36, which subsequently transfers FAs to transport proteins. As a member of glycoprotein, FA translocation enzyme CD36 is weakly expressed in hepatocytes and liver tissue under physiological conditions but significantly upregulated in animal models and NAFLD patients (36, 37). Moreover, previous studies confirmed that overexpression of CD36 led to steatosis in mice, and liver-specific knockout of CD36 reduced lipid content in mice fed with HFD (23, 38). Our finding that Tim-1 deletion greatly enhanced HFD-induced upregulation of hepatic CD36 protein expression suggests that Tim-1 deficiency may disrupt lipid homeostasis by increasing FA uptake via its modulation of CD36. In addition, hepatic expression of SREBP1 protein, an important transcriptional promoter of lipogenesis activated by insulin signaling in the fed state, was induced by HFD treatment in WT mice, which was further upregulated by Tim-1 deletion. Together, our results strongly support that Tim-1 deficiency may accelerate the severity of hepatic steatosis by increasing both FA uptake and de novo lipogenesis.
Next, we confirmed that Tim-1 deficiency aggravated HFD-induced liver injury and inflammation, as evidenced by elevated serum ALT, and increased LCN2 mRNA and protein in liver tissues. One of the striking effects of Tim-1 deficiency in diet-induced NAFLD was substantially increased hepatic expression of LCN2 concomitant with increased infiltration of inflammatory cells including neutrophils and macrophages. Moreover, confocal immunofluorescence images revealed a partial colocalization of LCN2 with neutrophil marker Ly6G or with macrophage marker F4/80, demonstrating both neutrophils and macrophages as the major cell populations contributing to Tim-1-enhanced LCN2 upregulation in diet-induced NAFLD/NASH mouse model. LCN2, an acute protein induced in response to bacterial infection, metabolic stress, or injury, has been proven to be a reliable biomarker of liver injury and inflammation. Previous studies also demonstrated that injury-induced upregulation of hepatic LCN2 has a significant hepatoprotective effect in acute liver injury and that hepatocytes are the major source for hepatic LCN2 (39–41). Furthermore, these data suggest that LCN2 might act as an intrinsic “help-me” sensor upon injury to recruit inflammatory cells. In line with this assumption, Asimakopoulou et al. observed that Lcn2-deficient mice showed a significantly lower recruitment of neutrophils and leukocytes, compared to WT animals, when fed with a methionine-choline-deficient (MCD) diet that induces hepatic inflammation and injury (42). A recent report also provides evidence that LCN2 is implicated in the progression of simple steatosis to NASH by promoting neutrophil-macrophage crosstalk (43). The authors found that Lcn2-deficient mice on high-fat high-cholesterol (HFHC) diet had reduced infiltration of both neutrophils and macrophages, and chronic LCN2 administration-induced elevation of hepatic macrophages was abrogated by Ly6G antibody-mediated depletion of neutrophils (43). Likewise, mice treated with ethanol exhibited elevated LCN2 expression in neutrophils, and Lcn2-deficient mice are protected from alcoholic steatohepatitis (ASH) as demonstrated by reduced neutrophil infiltration and liver injury (44). Thus, Tim-1 deficiency may accelerate obesity-related liver inflammation and injury by upregulating LCN2, which warrants further investigation.
Upon chronic inflammation and injury, hepatic stellate cells (HSCs) are activated and turn into the primary source of extracellular matrix in ASH and NASH. A recent study provides evidence that hepatic LCN2 is also involved in the activation of HSCs in ASH (45). The authors found that hepatic expression of Col1a1 was elevated in ASH patients and correlated with hepatic LCN2 expression, and Lcn2-deficient mice were protected from liver fibrosis caused by either ethanol or CCl4 exposure. The causal role of LCN2 in tissue fibrosis is also supported by an in vitro study that recombinant LCN2 induced type 1 collagen protein expression in human fibroblasts in a dose-dependent fashion (46). In the current study, we found that mice lacking Tim-1 showed an enhanced fibrogenic response associated with an upregulation of LCN2 expression in the liver of HFD-fed mice.
The development of fibrosing steatohepatitis is a complex process that involves multicellular responses other than HSCs. For example, activated macrophages can differentiate into diverse phenotypes contributing to both the progression and regression of the fibrotic process (47–49). Deletion of the macrophage population either during injury or during repair/resolution has dramatically different effects on the overall fibrotic response (49, 50). Specifically, macrophage depletion in progressive inflammatory injury results in amelioration of fibrosis; in contrast, depletion during recovery results in a failure of resolution with the persistence of cellular and matrix components of the fibrotic response (49, 50). Using a set of established markers, Belijaars et al. further localized and quantified M1 (classically activated)- and M2 (alternatively activated)-dominant macrophages in CCl4-damaged mouse livers as well as human end-stage cirrhotic livers (47). They found that M2 markers were present in liver fibrotic lesions but nearly absent during the resolution of fibrosis, suggesting a more pro-fibrotic character of M2-dominant macrophages in human and mouse livers (47). In this study, we also quantified hepatic gene expression of M1 and M2 markers to further determine if Tim-1 is involved in macrophage polarization and activation, A similar upregulation of CD11c and TNF-α gene expression was observed in HFD-fed WT and Tim-1-/- livers, suggesting that Tim-1 may not be required for M1 macrophage polarization. We next determined CD206 (mannose receptor, MCR-1) and YM-1, M2 macrophage markers, in mouse NAFLD/NASH livers. Hepatic mRNA levels of CD206, a well-known marker for both mouse and human M2 macrophages, was unaltered by either HFD diet or Tim-1 deletion. While in many organs M2 macrophages specifically express CD206, in livers its expression was found in macrophages as well as in sinusoidal endothelial cells (47), which complicate the quantitative interpretation of mRNA expression analysis of whole liver homogenates. Of note, YM-1 does not have this disadvantage and acts as a specific and useful marker for M2 macrophages in mouse liver (47, 51). Therefore, a substantial enhancement of HFD-induced hepatic expression of YM-1 transcript by loss of Tim-1 supports an important role for Tim-1 in M2 macrophage polarization.
Previous studies have demonstrated that M2-dominant macrophages, activated by Th2 cytokines (e.g., IL-4 and IL-13), are associated with increased fibrogenesis, tissue remodeling, and angiogenesis (52–54). When cultured with myofibroblasts, M2 macrophages promote complex matrix deposition (52, 53, 55, 56). Although Tim-1 has been proposed to have both activating and inhibitory effects in immune responses by studies using different monoclonal antibodies (8–15), a recent study performed in in vivo allergic airway disease revealed enhanced inflammatory responses and production of the Th2 cytokines IL-4, IL-5, and IL-13 in the absence of Tim-1, suggesting that its primary role is to dampen, rather than promote, Th2-type immune responses (16). Here, we found that Tim-1 deficiency enhanced HFD-induced macrophage accumulation and M2 polarization concomitant with accelerated fibrotic response in the mouse NAFLD/NASH model. It is likely that Tim-1 exerts an anti-fibrotic effect by inhibiting alternative activation of macrophages though its modulation of Th2-type immune response.
Our results suggest that Tim-1 deficiency may accelerate HFD-induced fibrosis by increasing macrophage infiltration and M2 polarization. However, we cannot answer where the M2-dominant macrophages come from, meaning are they derived from bone marrow monocytes, or do they develop from tissue-resident Kupffer cells. Previous studies showed that monocytes do infiltrate the liver during fibrogenesis and resolution. Moreover, during sustained Th2-type profiles, alternatively-activated Kupffer cells may be essential contributors in collagen synthesis, probably leading to an active fibrogenic state (53, 57, 58). Therefore, understanding the dynamics of all these different macrophages during fibrogenesis as well as the interactions between immune cells and macrophages is a subject of future research interest.
We are also aware that the use of global knockout mice, to define the role of Tim-1 in NAFLD/NASH, does not allow to distinguish between its direct hepatic and extrahepatic effects. It is well known that Tim-1/Kim-1 is massively induced in damaged renal proximal tubules after acute and chronic kidney injury (59, 60). Moreover, Tim-1/Kim-1 expression is anti-inflammatory and reduces acute kidney injury due to its mediation of phagocytic processes in renal tubular cells (59, 60). There is growing evidence that NAFLD and chronic kidney disease share common pathogenetic mechanisms and that the fatty liver per se may promote kidney injury and vice versa (61). In the present study, we used the C57BL/6J strain, one of the most susceptible to obesity but relatively resistant to kidney injury when fed high fat diets (62). Of note, we found that HFD feeding did not alter Tim-1/Kim-1 expression in the kidney of WT mice, suggesting that renal Tim-1 may play a minor role in diet-induced NAFLD/NASH. To better define the role of hepatic Tim-1, however, using tissue-specific Tim-1 knockout mouse model is warranted in the future.
In summary, this study revealed that Tim-1 deficiency led to an enhancement of lipid accumulation, and liver inflammation and injury in diet-induced NAFLD/NASH mouse model. In addition, we provide evidence that Tim-1 expression regulates lipid metabolism in hepatocytes by targeting CD36 and SREBP1. Our results indicate a regulatory role for Tim-1 in HFD-induced steatohepatitis and might be considered as a target for the prevention and treatment of chronic liver disease.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee at the Morehouse School of Medicine.
Author Contributions
JG and YZ have contributed equally to this work and share first authorship. JG, YZ, and XZ designed, performed and analyzed data from most of the experiments. JS and JMS performed histology and Western blot analysis. XZ conceived and supervised all studies. XZ wrote the manuscript. JI reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health Grants SC1DK112151 and S21MD000101.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Dr. Xiaoming Chen for her technical assistance with histology and immunostaining analysis.
References
1. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and Liver Cancer. Nat Rev Gastroenterol Hepatol (2013) 10:656–65. doi: 10.1038/nrgastro.2013.183
2. Diehl AM, Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med (2017) 377:2063–72. doi: 10.1056/NEJMra1503519
3. Aly FZ, Kleiner DE. Update on Fatty Liver Disease and Steatohepatitis. Adv Anat Pathol (2011) 18:294–300. doi: 10.1097/PAP.0b013e318220f59b
4. Brunt EM, Kleiner DE, Wilson LA, Unalp A, Behling CE, Lavine JE, et al. Portal Chronic Inflammation in Nonalcoholic Fatty Liver Disease (NAFLD): A Histologic Marker of Advanced NAFLD-Clinicopathologic Correlations From the Nonalcoholic Steatohepatitis Clinical Research Network. Hepatology (2009) 49:809–20. doi: 10.1002/hep.22724
5. Tajiri K, Shimizu Y, Tsuneyama K, Sugiyama T. Role of Liver-Infiltrating CD3+CD56+ Natural Killer T Cells in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Eur J Gastroenterol Hepatol (2009) 21:673–80. doi: 10.1097/MEG.0b013e32831bc3d6
6. Wu L, Gao X, Guo Q, Li J, Yao J, Yan K, et al. The Role of Neutrophils in Innate Immunity-Driven Nonalcoholic Steatohepatitis: Lessons Learned and Future Promise. Hepatol Int (2020) 14:652–66. doi: 10.1007/s12072-020-10081-7
7. Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone SM. Identification of a Surface Glycoprotein on African Green Monkey Kidney Cells as a Receptor for Hepatitis A Virus. EMBO J (1996) 15:4282–96. doi: 10.1002/j.1460-2075.1996.tb00803.x
8. Encinas JA, Janssen EM, Weiner DB, Calarota SA, Nieto D, Moll T, et al. Anti-T-Cell Ig and Mucin Domain-Containing Protein 1 Antibody Decreases TH2 Airway Inflammation in a Mouse Model of Asthma. J Allergy Clin Immunol (2005) 116:1343–9. doi: 10.1016/j.jaci.2005.08.031
9. Rennert PD. Novel Roles for TIM-1 in Immunity and Infection. Immunol Lett (2011) 141:28–35. doi: 10.1016/j.imlet.2011.08.003
10. Sizing ID, Bailly V, McCoon P, Chang W, Rao S, Pablo L, et al. Epitope-Dependent Effect of Anti-Murine TIM-1 Monoclonal Antibodies on T Cell Activity and Lung Immune Responses. J Immunol (2007) 178:2249–61. doi: 10.4049/jimmunol.178.4.2249
11. Sonar SS, Hsu YM, Conrad ML, Majeau GR, Kilic A, Garber E, et al. Antagonism of TIM-1 Blocks the Development of Disease in a Humanized Mouse Model of Allergic Asthma. J Clin Invest (2010) 120:2767–81. doi: 10.1172/JCI39543
12. Ueno T, Habicht A, Clarkson MR, Albin MJ, Yamaura K, Boenisch O, et al. The Emerging Role of T Cell Ig Mucin 1 in Alloimmune Responses in an Experimental Mouse Transplant Model. J Clin Invest (2008) 118:742–51. doi: 10.1172/JCI32451
13. Umetsu SE, Lee WL, McIntire JJ, Downey L, Sanjanwala B, Akbari O, et al. TIM-1 Induces T Cell Activation and Inhibits the Development of Peripheral Tolerance. Nat Immunol (2005) 6:447–54. doi: 10.1038/ni1186
14. Xiao S, Najafian N, Reddy J, Albin M, Zhu C, Jensen E, et al. Differential Engagement of Tim-1 During Activation Can Positively or Negatively Costimulate T Cell Expansion and Effector Function. J Exp Med (2007) 204:1691–702. doi: 10.1084/jem.20062498
15. Yuan X, Ansari MJ, D'Addio F, Paez-Cortez J, Schmitt I, Donnarumma M, et al. Targeting Tim-1 to Overcome Resistance to Transplantation Tolerance Mediated by CD8 T17 Cells. Proc Natl Acad Sci USA (2009) 106:10734–9. doi: 10.1073/pnas.0812538106
16. Curtiss ML, Gorman JV, Businga TR, Traver G, Singh M, Meyerholz DK, et al. Tim-1 Regulates Th2 Responses in an Airway Hypersensitivity Model. Eur J Immunol (2012) 42:651–61. doi: 10.1002/eji.201141581
17. Foks AC, Engelbertsen D, Kuperwaser F, Alberts-Grill N, Gonen A, Witztum JL, et al. Blockade of Tim-1 and Tim-4 Enhances Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice. Arterioscler Thromb Vasc Biol (2016) 36:456–65. doi: 10.1161/ATVBAHA.115.306860
18. Uchida Y, Ke B, Freitas MC, Ji H, Zhao D, Benjamin ER, et al. The Emerging Role of T Cell Immunoglobulin Mucin-1 in the Mechanism of Liver Ischemia and Reperfusion Injury in the Mouse. Hepatology (2010) 51:1363–72. doi: 10.1002/hep.23442
19. Zhang Y, Liu Y, Chen H, Zheng X, Xie S, Chen W, et al. TIM-1 Attenuates the Protection of Ischemic Preconditioning for Ischemia Reperfusion Injury in Liver Transplantation. Am J Transl Res (2017) 9:3665–75.
20. Zhang Y, Ji H, Shen X, Cai J, Gao F, Koenig KM, et al. Targeting TIM-1 on CD4 T Cells Depresses Macrophage Activation and Overcomes Ischemia-Reperfusion Injury in Mouse Orthotopic Liver Transplantation. Am J Transplant (2013) 13:56–66. doi: 10.1111/j.1600-6143.2012.04316.x
21. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 Years of Image Analysis. Nat Methods (2012) 9:671–5. doi: 10.1038/nmeth.2089
22. Wang X, Ma Y, Yang LY, Zhao D. MicroRNA-20a-5p Ameliorates Non-Alcoholic Fatty Liver Disease via Inhibiting the Expression of CD36. Front Cell Dev Biol (2020) 8:596329. doi: 10.3389/fcell.2020.596329
23. Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology (2016) 157:570–85. doi: 10.1210/en.2015-1866
24. Hirsch D, Stahl A, Lodish HF. A Family of Fatty Acid Transporters Conserved From Mycobacterium to Man. Proc Natl Acad Sci USA (1998) 95:8625–9. doi: 10.1073/pnas.95.15.8625
25. Doege H, Stahl A. Protein-Mediated Fatty Acid Uptake: Novel Insights From In Vivo Models. Physiol (Bethesda ) (2006) 21:259–68. doi: 10.1152/physiol.00014.2006
26. Stahl A, Gimeno RE, Tartaglia LA, Lodish HF. Fatty Acid Transport Proteins: A Current View of a Growing Family. Trends Endocrinol Metab (2001) 12:266–73. doi: 10.1016/S1043-2760(01)00427-1
27. Xu S, Jay A, Brunaldi K, Huang N, Hamilton JA. CD36 Enhances Fatty Acid Uptake by Increasing the Rate of Intracellular Esterification But Not Transport Across the Plasma Membrane. Biochemistry (2013) 52:7254–61. doi: 10.1021/bi400914c
28. Deng X, Cagen LM, Wilcox HG, Park EA, Raghow R, Elam MB. Regulation of the Rat SREBP-1c Promoter in Primary Rat Hepatocytes. Biochem Biophys Res Commun (2002) 290:256–62. doi: 10.1006/bbrc.2001.6148
29. Shimano H. SREBPs: Physiology and Pathophysiology of the SREBP Family. FEBS J (2009) 276:616–21. doi: 10.1111/j.1742-4658.2008.06806.x
30. Fengler VH, Macheiner T, Kessler SM, Czepukojc B, Gemperlein K, Muller R, et al. Susceptibility of Different Mouse Wild Type Strains to Develop Diet-Induced NAFLD/AFLD-Associated Liver Disease. PloS One (2016) 11:e0155163. doi: 10.1371/journal.pone.0155163
31. Machado MV, Michelotti GA, Xie G, Almeida PT, Boursier J, Bohnic B, et al. Mouse Models of Diet-Induced Nonalcoholic Steatohepatitis Reproduce the Heterogeneity of the Human Disease. PloS One (2015) 10:e0127991. doi: 10.1371/journal.pone.0127991
32. Nandi A, Kitamura Y, Kahn CR, Accili D. Mouse Models of Insulin Resistance. Physiol Rev (2004) 84:623–47. doi: 10.1152/physrev.00032.2003
33. Roberts-Toler C, O'Neill BT, Cypess AM. Diet-Induced Obesity Causes Insulin Resistance in Mouse Brown Adipose Tissue. Obes (Silver Spring) (2015) 23:1765–70. doi: 10.1002/oby.21134
34. Wang CY, Liao JK. A Mouse Model of Diet-Induced Obesity and Insulin Resistance. Methods Mol Biol (2012) 821:421–33. doi: 10.1007/978-1-61779-430-8_27
35. Zhang S, Sousa A, Lin M, Iwano A, Jain R, Ma B, et al. Role of Chitinase 3-Like 1 Protein in the Pathogenesis of Hepatic Insulin Resistance in Nonalcoholic Fatty Liver Disease. Cells (2021) 10:201. doi: 10.3390/cells10020201
36. Garcia-Monzon C, Lo IO, Crespo J, Romero-Gomez M, Garcia-Samaniego J, Fernandez-Bermejo M, et al. Increased Soluble CD36 Is Linked to Advanced Steatosis in Nonalcoholic Fatty Liver Disease. Eur J Clin Invest (2014) 44:65–73. doi: 10.1111/eci.12192
37. Greco D, Kotronen A, Westerbacka J, Puig O, Arkkila P, Kiviluoto T, et al. Gene Expression in Human NAFLD. Am J Physiol Gastrointest Liver Physiol (2008) 294:G1281–7. doi: 10.1152/ajpgi.00074.2008
38. Koonen DP, Jacobs RL, Febbraio M, Young ME, Soltys CL, Ong H, et al. Increased Hepatic CD36 Expression Contributes to Dyslipidemia Associated With Diet-Induced Obesity. Diabetes (2007) 56:2863–71. doi: 10.2337/db07-0907
39. Borkham-Kamphorst E, Drews F, Weiskirchen R. Induction of Lipocalin-2 Expression in Acute and Chronic Experimental Liver Injury Moderated by Pro-Inflammatory Cytokines Interleukin-1beta Through Nuclear Factor-KappaB Activation. Liver Int (2011) 31:656–65. doi: 10.1111/j.1478-3231.2011.02495.x
40. Borkham-Kamphorst E, van de Leur E, Zimmermann HW, Karlmark KR, Tihaa L, Haas U, et al. Protective Effects of Lipocalin-2 (LCN2) in Acute Liver Injury Suggest a Novel Function in Liver Homeostasis. Biochim Biophys Acta (2013) 1832:660–73. doi: 10.1016/j.bbadis.2013.01.014
41. Xu MJ, Feng D, Wu H, Wang H, Chan Y, Kolls J, et al. Liver Is the Major Source of Elevated Serum Lipocalin-2 Levels After Bacterial Infection or Partial Hepatectomy: A Critical Role for IL-6/Stat3. Hepatology (2015) 61:692–702. doi: 10.1002/hep.27447
42. Asimakopoulou A, Borkham-Kamphorst E, Tacke F, Weiskirchen R. Lipocalin-2 (NGAL/LCN2), a "Help-Me" Signal in Organ Inflammation. Hepatology (2016) 63:669–71. doi: 10.1002/hep.27930
43. Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, et al. Lipocalin-2 Mediates Non-Alcoholic Steatohepatitis by Promoting Neutrophil-Macrophage Crosstalk via the Induction of CXCR2. J Hepatol (2016) 65:988–97. doi: 10.1016/j.jhep.2016.05.041
44. Wieser V, Tymoszuk P, Adolph TE, Grander C, Grabherr F, Enrich B, et al. Lipocalin 2 Drives Neutrophilic Inflammation in Alcoholic Liver Disease. J Hepatol (2016) 64:872–80. doi: 10.1016/j.jhep.2015.11.037
45. Chen J, Argemi J, Odena G, Xu MJ, Cai Y, Massey V, et al. Hepatic Lipocalin 2 Promotes Liver Fibrosis and Portal Hypertension. Sci Rep (2020) 10:15558. doi: 10.1038/s41598-020-72172-7
46. Tarjus A, Martinez-Martinez E, Amador C, Latouche C, El MS, Berger T, et al. Neutrophil Gelatinase-Associated Lipocalin, A Novel Mineralocorticoid Biotarget, Mediates Vascular Profibrotic Effects of Mineralocorticoids. Hypertension (2015) 66:158–66. doi: 10.1161/HYPERTENSIONAHA.115.05431
47. Beljaars L, Schippers M, Reker-Smit C, Martinez FO, Helming L, Poelstra K, et al. Hepatic Localization of Macrophage Phenotypes During Fibrogenesis and Resolution of Fibrosis in Mice and Humans. Front Immunol (2014) 5:430. doi: 10.3389/fimmu.2014.00430
48. Duffield JS. The Inflammatory Macrophage: A Story of Jekyll and Hyde. Clin Sci (Lond) (2003) 104:27–38. doi: 10.1042/CS20020240
49. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective Depletion of Macrophages Reveals Distinct, Opposing Roles During Liver Injury and Repair. J Clin Invest (2005) 115:56–65. doi: 10.1172/JCI200522675
50. Han J, Zhang X, Lau JK, Fu K, Lau HC, Xu W, et al. Bone Marrow-Derived Macrophage Contributes to Fibrosing Steatohepatitis Through Activating Hepatic Stellate Cells. J Pathol (2019) 248:488–500. doi: 10.1002/path.5275
51. Raes G, Van den Bergh R, De BP, Ghassabeh GH, Scotton C, Locati M, et al. Arginase-1 and Ym1 Are Markers for Murine, But Not Human, Alternatively Activated Myeloid Cells. J Immunol (2005) 174:6561–2. doi: 10.4049/jimmunol.174.11.6561
52. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi Monocytes Direct Alternatively Activated Profibrotic Macrophage Regulation of Lung Fibrosis. Am J Respir Crit Care Med (2011) 184:569–81. doi: 10.1164/rccm.201010-1719OC
53. Lopez-Navarrete G, Ramos-Martinez E, Suarez-Alvarez K, Aguirre-Garcia J, Ledezma-Soto Y, Leon-Cabrera S, et al. Th2-Associated Alternative Kupffer Cell Activation Promotes Liver Fibrosis Without Inducing Local Inflammation. Int J Biol Sci (2011) 7:1273–86. doi: 10.7150/ijbs.7.1273
54. Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of Alternatively and Classically Activated Macrophages on Fibrogenic Activities of Human Fibroblasts. Cell Immunol (2000) 204:19–28. doi: 10.1006/cimm.2000.1687
55. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
56. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol (2008) 8:958–69. doi: 10.1038/nri2448
57. Liaskou E, Zimmermann HW, Li KK, Oo YH, Suresh S, Stamataki Z, et al. Monocyte Subsets in Human Liver Disease Show Distinct Phenotypic and Functional Characteristics. Hepatology (2013) 57:385–98. doi: 10.1002/hep.26016
58. Tacke F, Zimmermann HW. Macrophage Heterogeneity in Liver Injury and Fibrosis. J Hepatol (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
59. Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, et al. KIM-1-Mediated Phagocytosis Reduces Acute Injury to the Kidney. J Clin Invest (2015) 125:1620–36. doi: 10.1172/JCI75417
60. Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney Injury Molecule-1 Is a Phosphatidylserine Receptor That Confers a Phagocytic Phenotype on Epithelial Cells. J Clin Invest (2008) 118:1657–68. doi: 10.1172/JCI34487
61. Musso G, Cassader M, Cohney S, De MF, Pinach S, Saba F, et al. Fatty Liver and Chronic Kidney Disease: Novel Mechanistic Insights and Therapeutic Opportunities. Diabetes Care (2016) 39:1830–45. doi: 10.2337/dc15-1182
Keywords: non-alcoholic steatohepatitis, high-fat diet, lipid metabolism, inflammation, Tim-1
Citation: George J, Zhang Y, Sloan J, Sims JM, Imig JD and Zhao X (2021) Tim-1 Deficiency Aggravates High-Fat Diet-Induced Steatohepatitis in Mice. Front. Immunol. 12:747794. doi: 10.3389/fimmu.2021.747794
Received: 26 July 2021; Accepted: 16 September 2021;
Published: 05 October 2021.
Edited by:
Saurabh Chatterjee, University of South Carolina, United StatesReviewed by:
Gianfranco Danilo Alpini, Indiana University, United StatesJuliane Beier, University of Pittsburgh, United States
Copyright © 2021 George, Zhang, Sloan, Sims, Imig and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueying Zhao, xzhao@msm.edu
†These authors have contributed equally to this work and share first authorship