
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 16 September 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.747370
 Ruiqing Liu1Yashan Liu1
Ruiqing Liu1Yashan Liu1 Chang Liu1Aijiao Gao1Lin Wang1Huixin Tang1Qiang Wu2Xia Wang3
Chang Liu1Aijiao Gao1Lin Wang1Huixin Tang1Qiang Wu2Xia Wang3 Derun Tian1*
Derun Tian1* Zhi Qi4*
Zhi Qi4* Yanna Shen1,2*
Yanna Shen1,2*Staphylococcus aureus (S. aureus) is a foodborne pathogen that causes severe diseases, such as endocarditis, sepsis, and bacteremia. As an important component of innate immune system, the NLR family pyrin domain-containing 3 (NLRP3) inflammasome plays a critical role in defense against pathogen infection. However, the cellular mechanism of NLRP3 inflammasome activation during S. aureus infection remains unknown. In the present study, we found that spleen tyrosine kinase (Syk) and c-Jun N-terminal kinase (JNK) were rapidly phosphorylated during S. aureus infection. Moreover, a Syk/JNK inhibitor and Syk/JNK siRNA not only reduced NLRP3 inflammasome-associated molecule expression at the protein and mRNA levels, apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) speck formation, and interleukin-1β (IL-1β), and IL-18 release but also rescued the decreased NIMA-related kinase 7 (NEK7) expression level following suppression of the NEK7-NLRP3 interaction in macrophages. Interestingly, Syk/JNK phosphorylation levels and NLRP3 inflammasome-associated molecule expression were decreased by blockade of K+ efflux. Furthermore, activation of the NLRP3 inflammasome and a lower NEK7 protein level were found in vivo upon S. aureus infection. Taken together, our data indicated that S. aureus infection induces a K+ efflux/Syk/JNK/NEK7-NLRP3 signaling pathway and the subsequent activation of the NLRP3 inflammasome for the release of proinflammatory cytokines. This study expands our understanding of the basic molecular mechanism regulating inflammation and provides potential value for anti-infective drug development against S. aureus infection.
Staphylococcus aureus (S. aureus), a gram-positive foodborne pathogen, is carried by approximately 30% of the world population (1). As one of the most common nosocomial pathogens, S. aureus causes a variety of invasive infections (2), such as pneumonia, endocarditis, bacteremia, and sepsis (3), which can lead to death (4). Moreover, with the increasing rates of antibiotic resistance, morbidity and mortality are increasing (5). Therefore, studying the mechanism induced by S. aureus infection is extremely important for resisting S. aureus infection.
NLRP3 inflammasomes, which are multiprotein complexes, play a key role in the innate immune system against various pathogens. NLRP3 inflammasome activation is triggered by many stimuli, such as pathogen-associated molecular patterns (PAMPs), adenosine triphosphate (ATP), and toxins (6, 7), which lead to the aggregation of ASC and the proteolytic activation of pro-caspase-1 in macrophages, following IL-1β and IL-18 maturation and secretion (8, 9). In contrast, dietary selenium can attenuate S. aureus-induced mastitis by inhibiting the NLRP3 inflammasome (10). Despite these advances, the exact molecular mechanism of S. aureus infection-induced NLRP3 inflammasome activation remains uncertain.
NIMA-related kinase 7 (NEK7), which plays a crucial role in mitosis entry, cell cycle progression, cell division, and mitotic processes, has been proven to be a vital mediator during inflammasome activation in hepatocellular carcinoma (11). Moreover, potassium efflux is a common event in NLRP3 inflammasome activation (12), and NEK7-mediated assembly and activation of the NLRP3 inflammasome are downstream of potassium efflux in ventilator-induced lung injury (VILI) (13). However, whether S. aureus infection induces potassium efflux and the formation of the NEK7-NLRP3 complex needs to be fully elucidated.
In our previous studies, we found that Syk/JNK activated the NLRP3 inflammasome in diabetic cardiomyopathy (DCM) and diabetic nephropathy (DN) (14, 15). However, the role of Syk and JNK in infectious diseases remains unknown. Interestingly, a recent study found that sulforaphane reduces intracellular survival of S. aureus in macrophages through inhibition of JNK (16). Therefore, we hypothesized that Syk/JNK/NLRP3 plays an indispensable role upon S. aureus infection.
In the present study, we demonstrated that NEK7 and NLRP3 inflammasome activation was involved in S. aureus infection in vivo. Moreover, we are the first to provide evidence that Syk/JNK promotes NLRP3 inflammasome activation by regulating the formation of NEK7-NLRP3 in J774A.1 cells and peritoneal exudate cells (PECs), which was mediated by potassium efflux. In summary, the K+ efflux/Syk/JNK/NEK7-NLRP3 signaling pathway plays a critical role in defending against S. aureus-infection.
To test the mechanism by which S. aureus infection activates the NLRP3 inflammasome, we first established an S. aureus infectious pneumonia model and determined the colony-forming units (CFUs) of S. aureus in bronchoalveolar lavage fluid (BALF) and lung tissues. The average bacterial load in BALF was approximately 4.7×105 CFU/ml and the average bacterial load in the lung was approximately 1.7×106 CFU/ml, which demonstrated that the S. aureus infection pneumonia model had been built successfully (Figure 1A) (17). Additionally, H&E stain of the lungs in the infection group showed that the alveolar wall was thickened, the structure of the lung was severely damaged and lymphocytes were attached to the vessel wall (Figure 1B, black arrow), in addition, lymphocytes also accumulated outside of blood vessels and bronchi (Figure 1B, green arrow), and erythrocytes accumulated in the bronchi (Figure 1B, yellow arrow). However, these phenomena were not observed in the control group, suggesting that the lungs in the S. aureus infection group underwent a series of inflammatory pathological changes. Moreover, we quantify the pathological score in the lungs based on these pathological phenomena (Table 1).
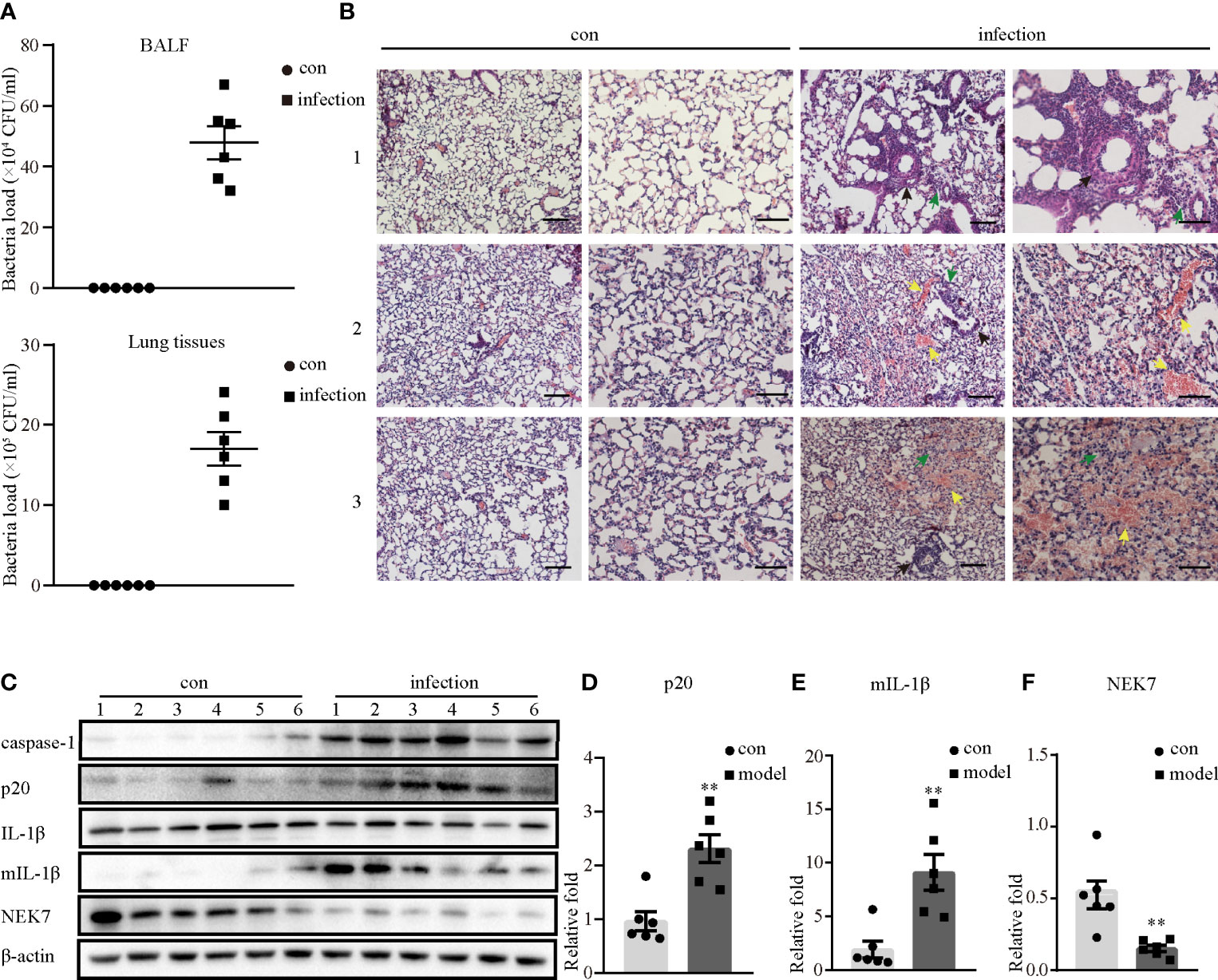
Figure 1 NLRP3 inflammasome was activated in S. aureus-induced mouse pneumonia. 6-8 weeks old female mice (C57BL/6) were randomly divided into the control group and the infection group, groups were both challenged with indicate stimulate groups. The infection group mice were challenged with 25 µl of a 109 CFU/mL suspension of S. aureus by intranasal administration, while the mice in the control group were given an equal volume of sterile PBS solution. (A) Bacterial load of BALF or lungs were determined in the control group and the infection group by CFU assay. (B) Histological examination for lungs were visualized by H&E stain (×100 magnification, first column and third column; ×200 magnification, second column and forth column). The arrows represent different inflammatory pathological changes in the infection group. (Scale bar: 100 µm.) (C) Western blot showing the protein levels of caspase-1, p20, pro-IL-1β, mIL-1β and NEK7. (D–F) Densitometry analysis of p20, mIL-1β and NEK7. Control group n=6, infection group n=6. Data shown are means ± SEMs; **P < 0.01 vs. control group.
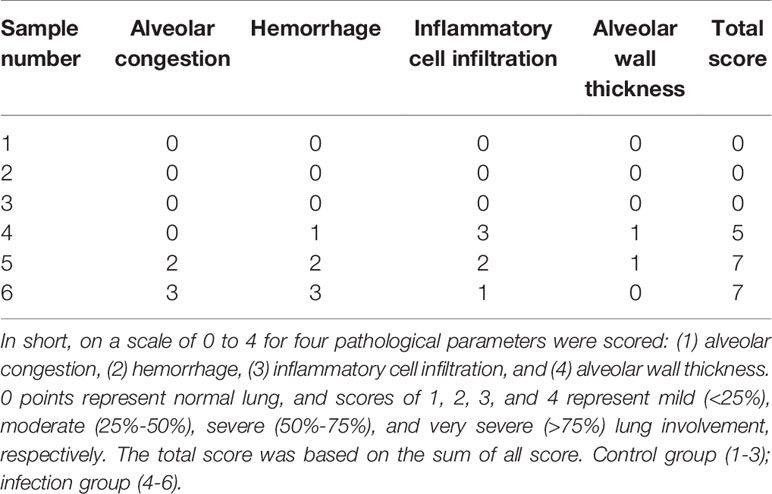
Table 1 Pathological score in the lung.
Next, we investigated whether inflammatory pathological changes were correlated with the NLRP3 inflammasome, and determined the NLRP3 inflammasome-related protein expression levels in the lungs. As shown in Figures 1C–F, the protein expression levels of activated caspase-1 (p20) and mature IL-1β (mIL-1β) in the infection group were significantly higher than those in the control group. Moreover, NEK7 expression at the protein level was markedly decreased in the infection group. Taken together, these data demonstrated that the NLRP3 inflammasome was activated in S. aureus-induced pneumonia, accompanied by decreased NEK7 protein levels.
To understand the role of NEK7 more deeply, we assessed the protein and mRNA expression levels of NEK7 in macrophages infected with S. aureus. The results showed that the protein and mRNA expression of NEK7 was reduced in the infection group of J774A.1 cells (Figures 2A, B). Notably, the NLRP3-NEK7 compound was obviously increased upon S. aureus infection, as detected by coimmunoprecipitation (Figure 2C). Moreover, NEK7-siRNA attenuated both the mRNA levels of caspase-1 and Il-1β (Figure 2D) and the protein expression of activated caspase-1 (p20) and mature-IL-1β (mIL-1β) (Figure 2E) in J774A.1 cells infected with S. aureus. Similar results were observed in PECs (Figure 2F). These findings indicated that NEK7 was necessary for NLRP3 inflammasome activation.
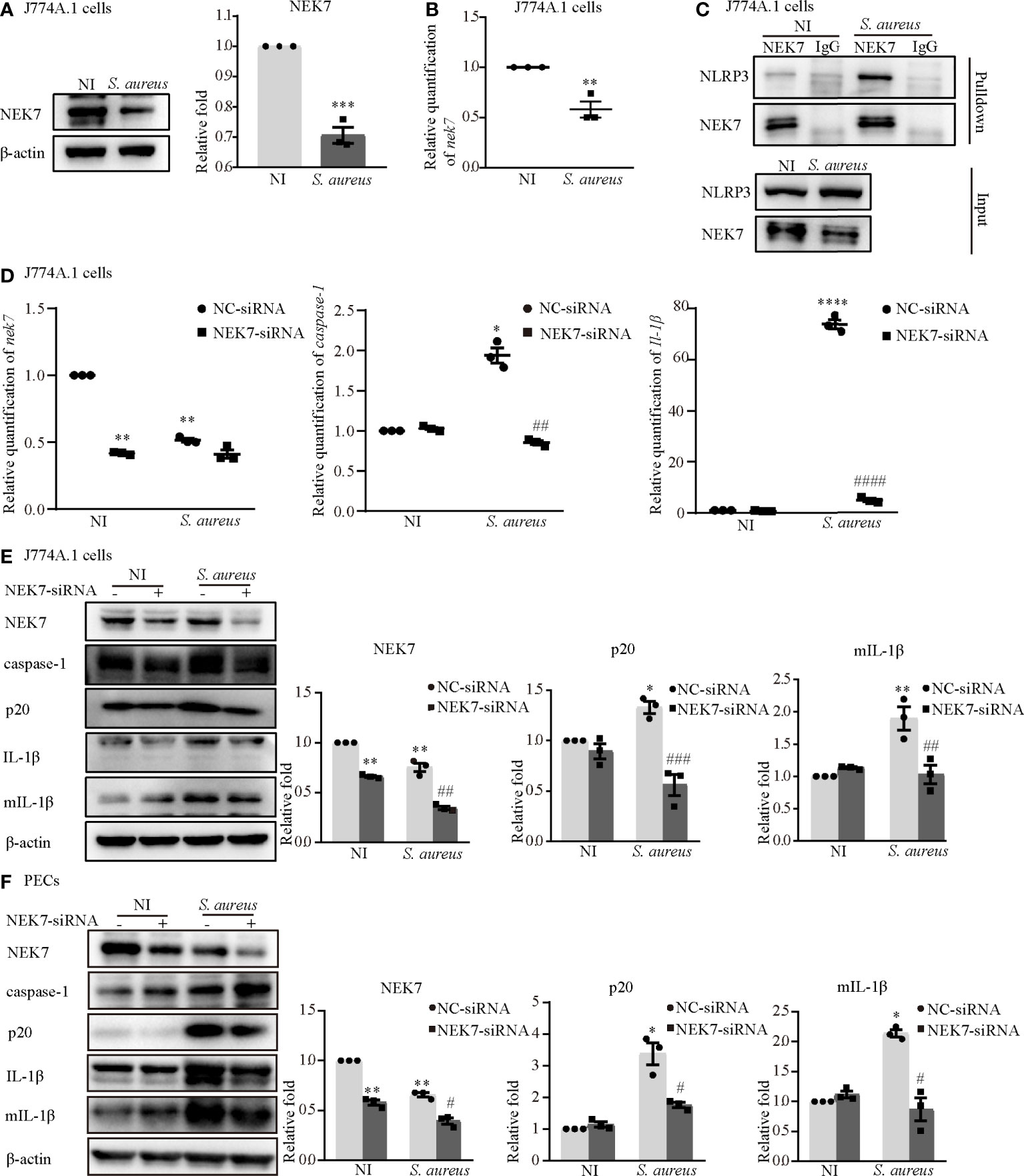
Figure 2 NEK7 was vital in S. aureus infection-induced NLRP3 inflammasome activation. The J774A.1 cells were infected with S. aureus (MOI of 1) for 24 h. (A) The protein expression level of NEK7 were detected by western blot and analyzed by relative densitometric quantification. (B) The mRNA expression level of nek7 were detected by RT-PCR. (C) The NEK7-NLRP3 ineraction were detected by coimmunoprecipitation. (D) Cells were pretreated with or without NEK7-siRNA for 48 h. The mRNA expression levels of caspase-1, Il-1β and nek7 were examined by RT-PCR. (E, F) The protein expression levels of caspase-1, p20, pro-IL-1β, mIL-1β and NEK7 were detected by western blot in J774A.1 cells (E) and PECs (F) and analyzed by relative densitometric quantification. Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. non-infection control group, #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 vs. infection control group.
The Syk/JNK/NLRP3 signaling pathway was involved in diabetic cardiomyopathy and diabetic nephropathy in our previous study (14, 15). Hence, we confirmed whether Syk and JNK mediated NLRP3 inflammasome activation during S. aureus infection. Interestingly, the phosphorylation levels of JNK and Syk were increased in a time-dependent manner in both J774A.1 cells (Figures 3A–D) and PECs (Figures 3G–J) during S. aureus infection. Moreover, the mRNA levels of syk and jnk were also increased starting at 15 minutes and 30 minutes after S. aureus infection (Figures 3E, F). These results indicated that Syk and JNK were involved in S. aureus infection.
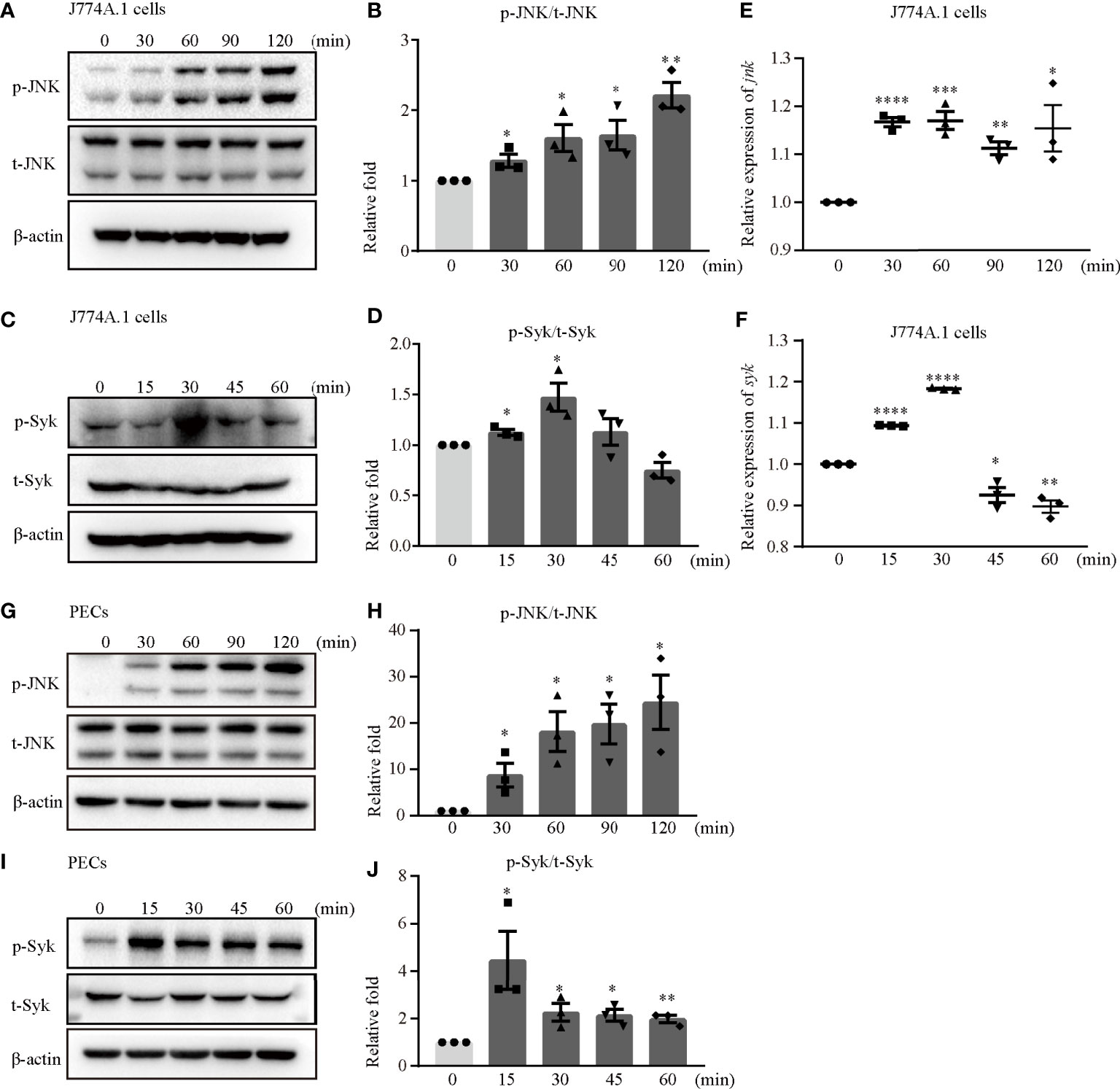
Figure 3 JNK and Syk were phosphorylated upon S. aureus infection. J774A.1 cells (A–F) and PECs (G–J) were infected with S. aureus at an MOI of 5 for different time. The cell lysates and total cellular RNA were collected. (A–D) Total and phosphorylation levels of JNK (A) and Syk (C) were detected by western blot. p-JNK/t-JNK (B) and p-Syk/t-Syk (D) were analyzed by relative densitometric quantification. (E, F) The mRNA expression levels of jnk (E) and syk (F) were quantified by RT-PCR. (G–J) The protein levels of t-JNK, p-JNK (G) and t-Syk, p-Syk (I) were detected by western blot. p-JNK/t-JNK (H) and p-Syk/t-Syk (J) were analyzed by relative densitometric quantification. Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. non-infection group.
Furthermore, JNK-siRNA or Syk-siRNA attenuated the S. aureus infection induced mRNA expression levels of caspase-1 and Il-1β (Figures 4A, C). The protein expression of p20 and mIL-1β was significantly decreased in cells exposed to S. aureus following JNK-siRNA (Figure 4B) or Syk-siRNA (Figure 4D) treatment. These data strongly indicated that JNK and Syk played a key role in NLRP3 inflammasome activation.
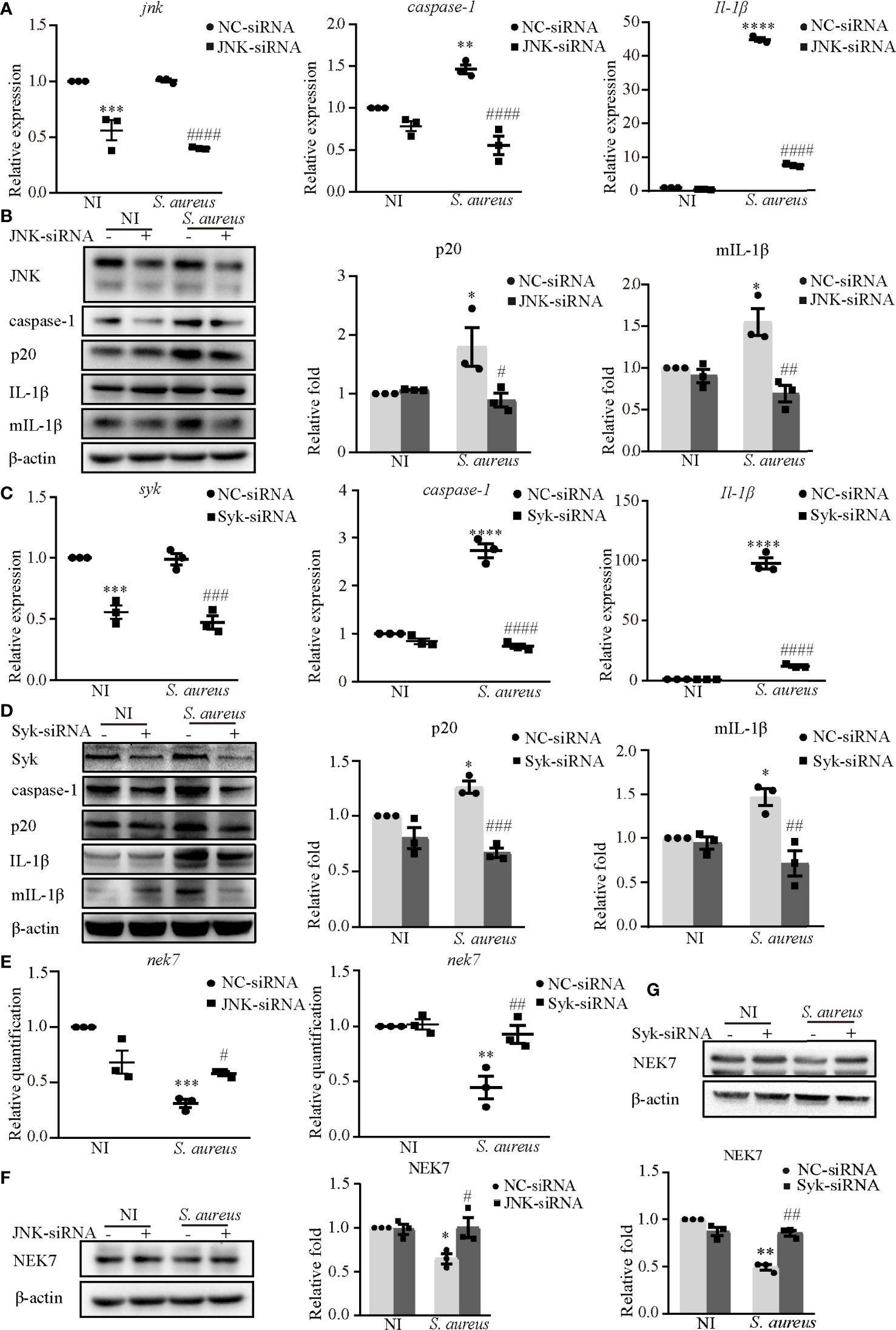
Figure 4 Syk and JNK promoted the NLRP3 inflammasome activation upon S. aureus infection. J774A.1 cells were infected with S. aureus (MOI of 1) for 24 h with or without JNK or Syk-siRNA pretreatment for 48 h. (A) The mRNA expression levels of caspase-1, Il-1β in the cells pretreated with or without JNK-siRNA were quantified by RT-PCR. (B) The protein expression levels of caspase-1, p20, pro-IL-1β and mIL-1β were detected by western blot and analyzed using densitometry. (C) The mRNA levels of caspase-1 and Il-1β in the cells pretreated with or without Syk-siRNA were quantified by RT-PCR. (D) The protein expression levels of caspase-1, p20, pro-IL-1β and mIL-1β were detected by western blot and analyzed using densitometry. (E) The mRNA expression level of nek7 was examined by RT-PCR with or without JNK/Syk-siRNA transfected. (F, G) The protein expression level of NEK7 was detected by western blot and analyzed using densitometry by transfected with or without JNK-siRNA (F) or Syk-siRNA (G). Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. non-infection control group, #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 vs. infection control group.
In our present study, NEK7 was involved in NLRP3 inflammasome activation upon S. aureus infection. Hence, we hypothesize that Syk and JNK may be linked to NEK7. Interestingly, the decreased mRNA expression level (Figure 4E) and protein expression level of NEK7 (Figures 4F, G) were rescued by JNK-siRNA or Syk-siRNA during S. aureus infection, indicating that NEK7 was downregulated by JNK and Syk in NLRP3 inflammasome activation.
To further confirm the role of p-JNK in NLRP3 inflammasome activation, both J774A.1 cells and PECs were pretreated with the JNK inhibitor (SP) or a DMSO control. SP significantly reduced the phosphorylation degree of JNK in both J774A.1 cells (Figure 5A) and PECs (Figure 5K). The mRNA levels of caspase-1 and Il-1β were decreased following SP pretreatment during S. aureus infection (Figure 5B and Figure S1A). Moreover, SP also attenuated the S. aureus-induced increase of p20 and mIL-1β in J774A.1 cells and PECs (Figure 5C and Figure S1B). In addition, the proinflammatory cytokines IL-1β and IL-18 in the supernatants of J774A.1 cells (Figure 5D) and PECs (Figures 5N, O) were substantially increased compared with those in the noninfected control group, whereas SP reduced the concentrations of IL-1β and IL-18 in the supernatant.
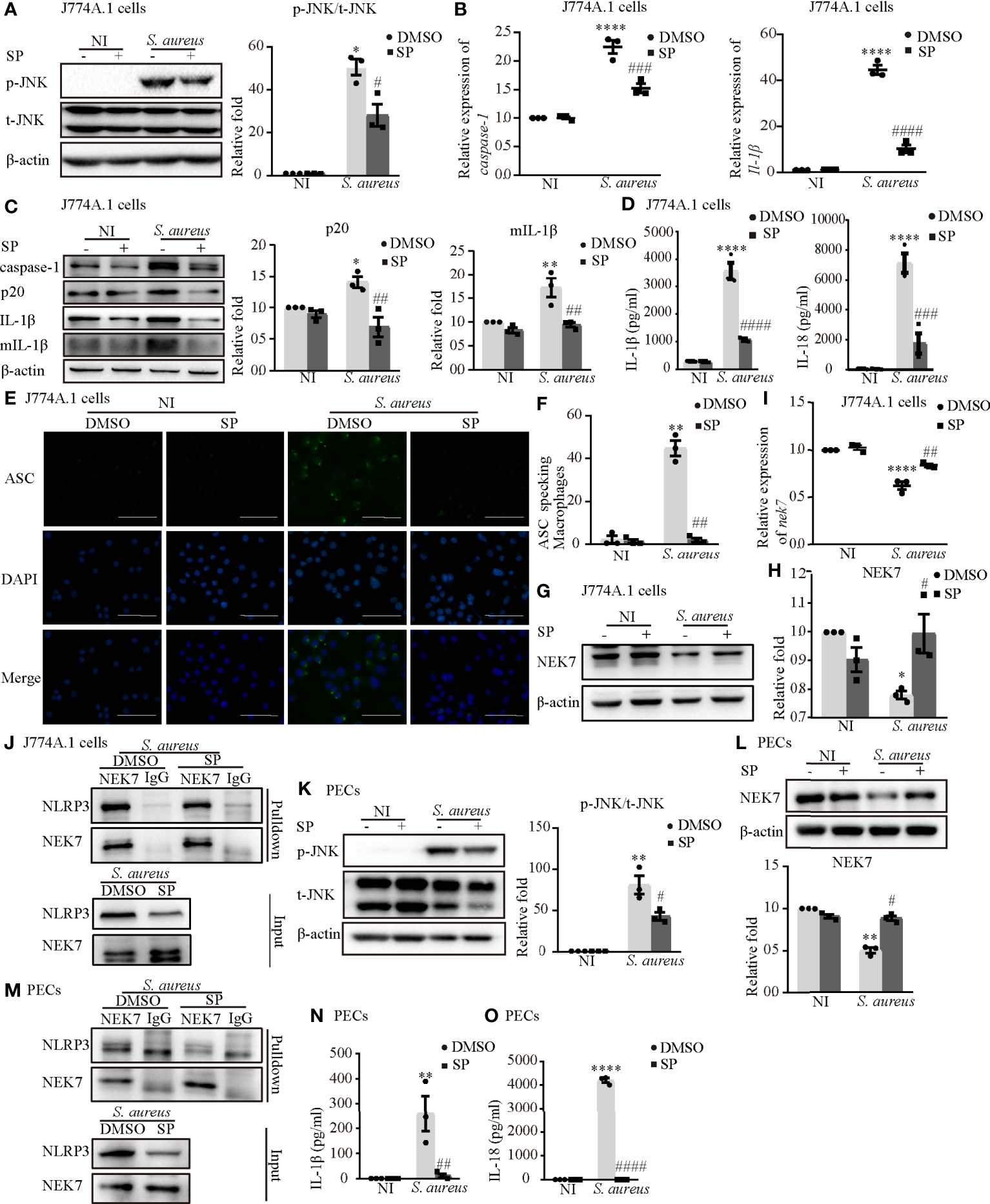
Figure 5 Phosphorylated JNK activated the NLRP3 inflammasome by accelarating NEK7-NLRP3 compound formation upon S. aureus infection. J774A.1 cells (A–J) and PECs (K–O) pretreated with or without JNK inhibitor (SP 600125) at the final concentration of 20 μmol/L for 2 h followed by S. aureus infecton (MOI of 1 or 5) for 24 h or 2 h. (A) The protein levels of t-JNK and p-JNK were detected by western blot and analyzed by relative densitometric quantification in J774A.1 cells and PECs (K). (B) The mRNA expression levels of caspase-1, Il-1β were quantified by RT-PCR. (C) The protein expression levels of caspase-1, p20, pro-IL-1β and mIL-1β were examined by western blot and analyzed by relative densitometric quantification. (D) The supernatants were collected at 48 h after S. aureus infection. The levels of IL-1β and IL-18 in supernatants were detected by ELISA in J774A.1 cells and PECs (N, O). (E) Confocal immunofluorescence imaged endogenous ASC specks. Cells were fixed and stained for ASC (Green), DAPI (Blue). (Scale bar: 100 µm.) (F) The percentage of J774A.1 cells with ASC specks were quantificated. More than 200 cells from at least 5 different fields were counted for ASC speck formation. (G, H) The protein expression level of NEK7 was examined by western blot and analyzed by relative densitometric quantification in J774A.1 cells and PECs (L). (I) The mRNA expression level of nek7 was examined by RT-PCR. (J) Coimmunoprecipitation analysis of NLRP3 interaction with NEK7 in J774.A cells and PECs (M). Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; *P < 0.05, **P < 0.01, ****P < 0.0001 vs. non-infection control group, #P < 0.05, ##P < 0.01, ###P < 0.001, ####P< 0.0001 vs. infection control group.
Upon NLRP3 inflammasome activation, ASC is recruited to the cytosol where it self-aggregates and forms specks, subsequently leading to caspase-1 activation (18). In Figures 5E, F and Figure S1C, we found that the number of ASC speck-positive cells in the infection group was much greater than that in the noninfection group through fluorescence microscopy. Inhibition of JNK phosphorylation by SP resulted in a significant reduction in S. aureus induced ASC speck formation in J774A.1 cells and PECs. Taken together, these results suggested that S. aureus-induced activation of the NLRP3 inflammasome and secretion of the proinflammatory cytokines IL-1β and IL-18 were dependent on p-JNK.
Whether p-JNK regulates NLRP3 inflammasome activation via NEK7 is poorly understood. In J774A.1 cells, the protein and mRNA expression of NEK7 was upregulated in the presence of SP during S. aureus infection (Figures 5G–I). Furthermore, the S. aureus induced NLRP3-NEK7 compound was obviously decreased by SP pretreatment (Figure 5J). Consistently, SP greatly increased the protein level of NEK7 and reduced NLRP3-NEK7 expression in PECs (Figures 5L, M). These results indicated that p-JNK activated the NLRP3 inflammasome by accelerating NEK7-NLRP3 compound formation upon S. aureus infection.
First, we confirmed that the inhibitor of Syk (BAY) reduced Syk phosphorylation in both J774A.1 cells (Figure 6A) and PECs (Figure 6J). The increased mRNA expression of caspase-1 and Il-1β induced by S. aureus infection in J774A.1 cells and PECs was significantly reduced following pretreatment with an inhibitor of Syk (BAY) (Figure 6B and Figure S1D). Moreover, the protein expression of p20 and mIL-1β showed similar tendencies (Figure 6C and Figure S1E). Furthermore, the secretion of IL-1β and IL-18 from J774A.1 cells (Figure 6D) and PECs (Figure 6M) in the S. aureus infection group was significantly increased compared with that in the noninfection group, while in comparison, the expression level of IL-1β and IL-18 in BAY pretreatment infection group was significantly decreased. In addition, BAY also reduced the formation of ASC specks in J774A.1 cells and PECs upon S. aureus infection (Figures 6E, F and Figure S1F). Taken together, these results suggested that the S. aureus-induced activation of the NLRP3 inflammasome and secretion of proinflammatory cytokines were dependent on p-Syk.
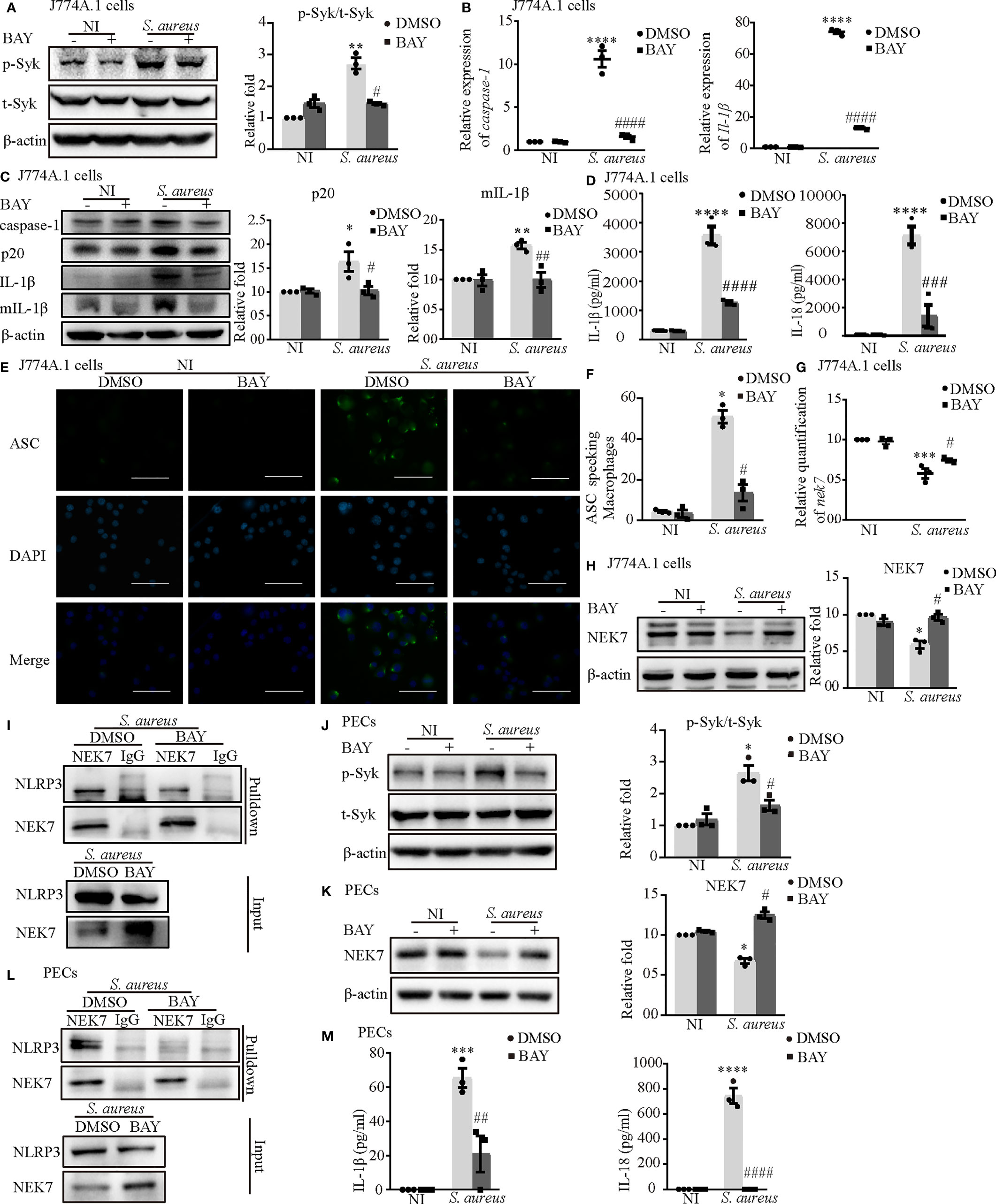
Figure 6 Phosphorylated Syk was involved in S. aureus infection-induced NEK7-NLRP3 signaling pathway activation. J774A.1 cells (A–I) and PECs (J–M) pretreated with or without Syk inhibitor (BAY 613606) at the final concentration of 1 μmol/L for 2 h followed by S. aureus infecton (MOI of 1 or 5) for different time. (A) The protein levels of t-Syk and p-Syk were detected by western blot and analyzed by relative densitometric quantification followed by S. aureus infection (MOI of 5) for 30 min in J774A.1 cells and PECs (J). (B) The mRNA expression levels of caspase-1 and Il-1β were quantified by RT-PCR. (C) The protein expression levels of caspase-1, p20, pro-IL-1β and mIL-1β were detected by western blot and analyzed by relative densitometric quantification. (D) The supernatants were collected at 48 h after S aureus infection. IL-1β and IL-18 in the supernatants were determined by ELISA in J774A.1 cells and PECs (M). (E) The ASC specks formation were imaged via fluorescence microscopy. (Scale bar: 100 µm.). (F) ASC specks were quantitied in more than 200 cells from at least 5 different fields. (G) The mRNA expression level of nek7 was examined by RT-PCR. (H) The protein expression level of NEK7 was detected by western blot and analyzed by relative densitometric quantification in J774A.1 cells and PECs (K). (I) The NEK7-NLRP3 compound were analysed by coimmunoprecipitation in J774A.1 cells and PECs (L). Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. non-infection control group, #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 vs. infection control group.
Consistent with our hypothesis, BAY obviously upregulated the mRNA and protein levels of NEK7 (Figures 6G, H) and decreased the NLRP3-NEK7 complex in J774A.1 cells (Figure 6I) during S. aureus infection. Likewise, BAY also greatly increased the protein level of NEK7 and reduced NLRP3-NEK7 compound in PECs (Figures 6K, L). These results confirmed that p-Syk was involved in the NEK7-NLRP3 signaling pathway during S. aureus infection.
Notably, the phosphorylation time of Syk was earlier than that of JNK and the expression of p-Syk gradually decreased when JNK was phosphorylated (Figure 3), so we hypothesized that the process of JNK phosphorylation may consume the amount of p-Syk.
To clarify the relationship between JNK and Syk during S. aureus infection, we assessed the expression level of JNK with or without Syk-siRNA or Syk inhibitor pretreatment in J774A.1 cells or PECs. As shown in Figures 7A, B, the phosphorylation level of JNK and the mRNA expression of jnk induced by S. aureus infection were obviously reduced in J774A.1 cells pretreated with Syk-siRNA. Moreover, the Syk inhibitor also reduced phosphorylation level of JNK in J774A.1 cells and PECs (Figures 7C, E) and the mRNA expression of jnk (Figure 7D) upon S. aureus infection. Combined with the results of Figure 3, it was clear that Syk was an important regulator of JNK phosphorylation during S. aureus infection.
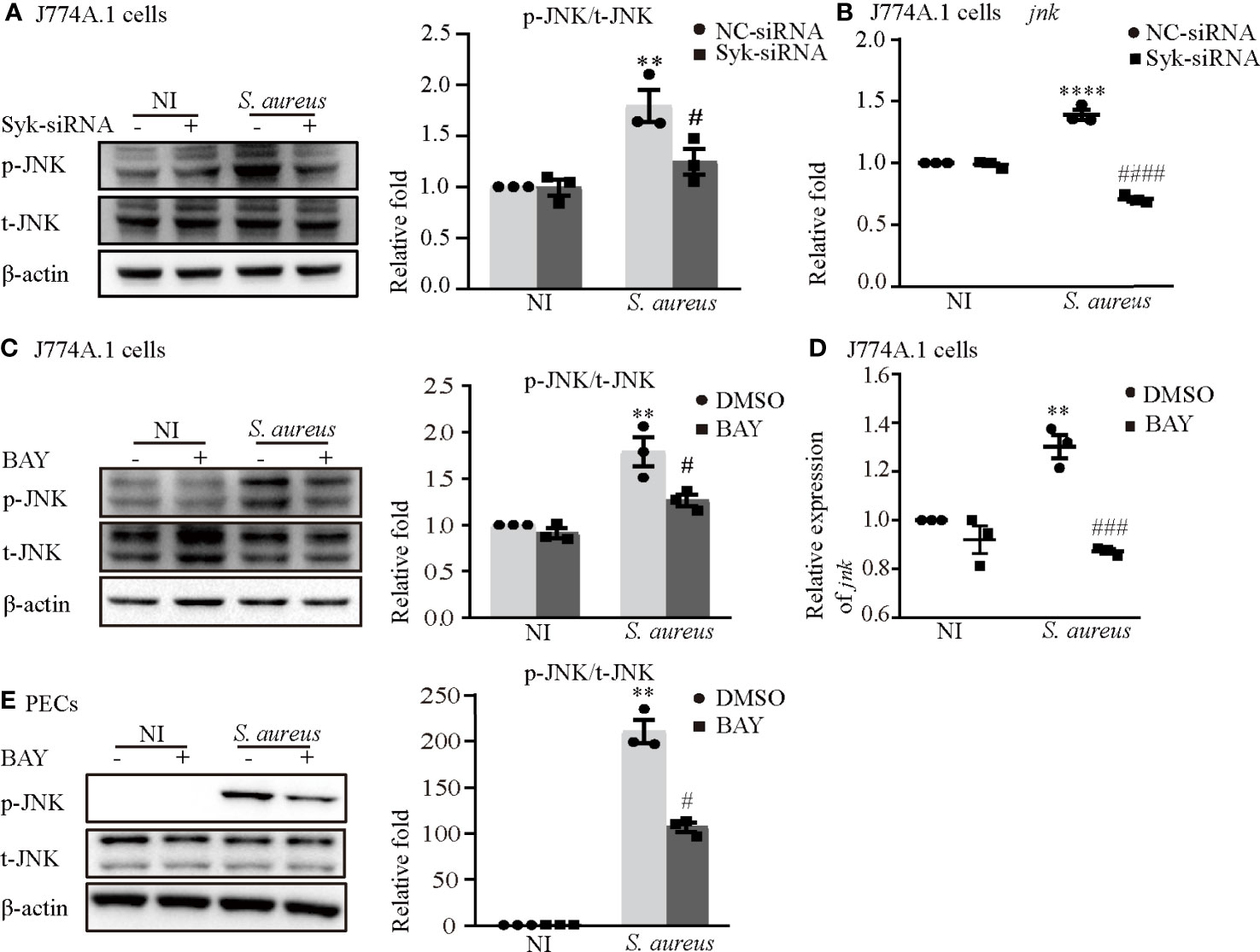
Figure 7 Syk/JNK signaling was critical for activation of the NLRP3 inflammasome upon S. aureus infection. Macrophages were infected with S. aureus (MOI of 5) for 2 h by pretreated with or without Syk-siRNA (A, B) for 48 h or Syk inhibitor (BAY) (C–E) for 2h. (A) The protein expression level of p-JNK was detected by western blot and analyzed by relative densitometric quantification in J774A.1 cells. (B) The mRNA expression level of jnk was quantified by RT-PCR. (C)The protein expression level of p-JNK was detected by western blot and analyzed by relative densitometric quantification in J774A.1 cells and PECs (E). (D) The mRNA expression level of jnk was quantified by RT-PCR. Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; **P < 0.01, ****P < 0.0001 vs. non-infection control group, #P < 0.05, ###P < 0.001, ####P < 0.0001 vs. infection control group.
Syk/JNK signaling was critical for activation of the NLRP3 inflammasome by regulating NEK7-NLRP3 compound formation upon S. aureus infection
It has been generally accepted that K+ efflux triggers NLRP3 activation in response to some stimuli (12). To assess whether activation of the NLRP3 inflammasome was dependent on K+ efflux and to explore the molecular mechanism during S. aureus infection, J774A.1 cells and PECs were pretreated with increasing concentrations of potassium chloride (KCl). First, the results showed that the K+ concentration in the supernatant in the infection group was much higher than that in the noninfection group, whereas the K+ concentration in cells in the infection group was much lower in both J774A.1 cells and PECs (Figure 8A). Moreover, a high extracellular concentration of KCl inhibited the S. aureus-induced phosphorylation of Syk and JNK in a dose-dependent manner in both J774A.1 cells (Figures 8B–E) and PECs (Figures 8F–I). Notably, the phosphorylation levels of Syk and JNK were decreased in the 40 mM group and 10 mM group, respectively. These results suggested that K+ efflux might activate the NLRP3 inflammasome through phosphorylation of Syk and JNK.
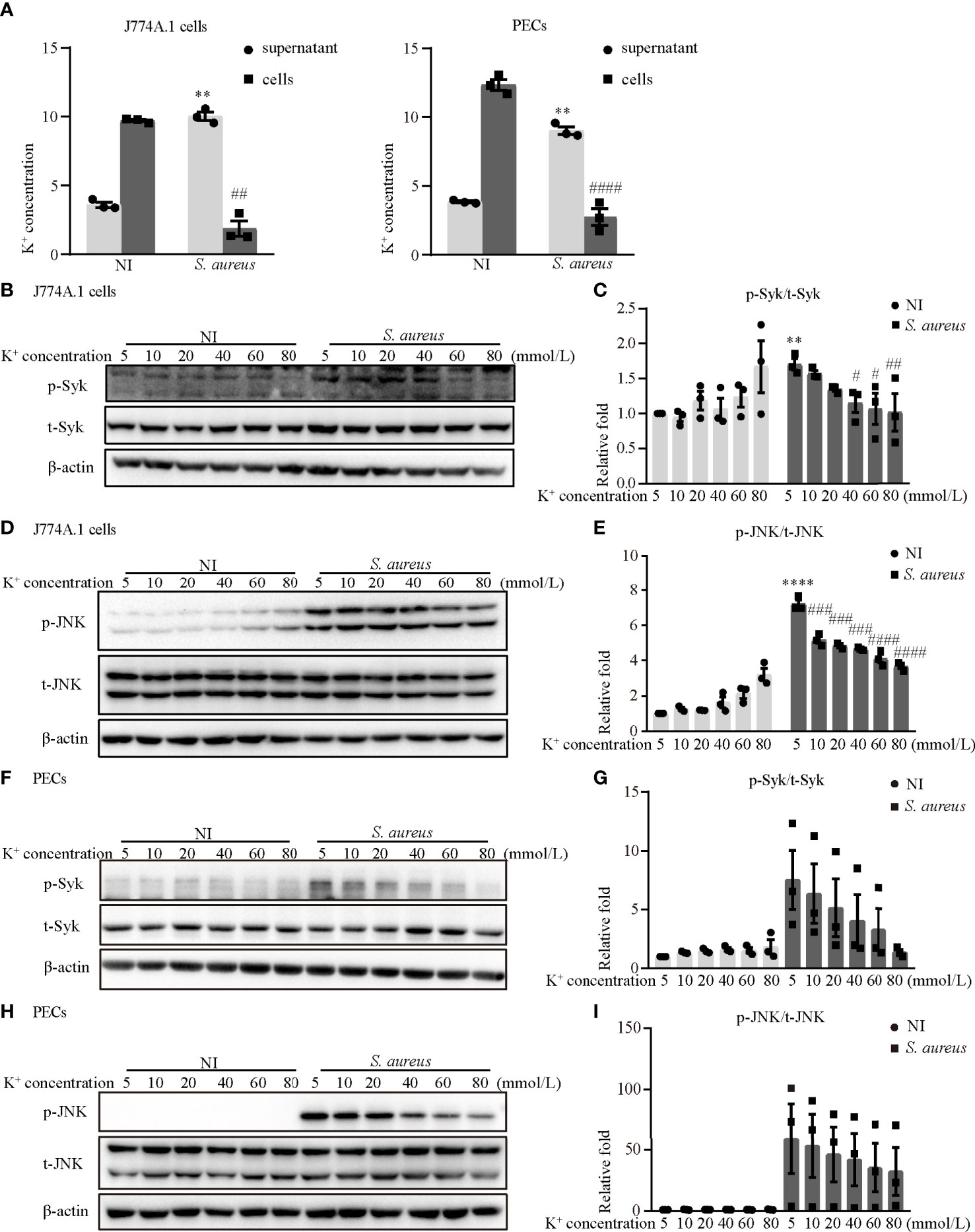
Figure 8 Blockade of potassium efflux decreased the phosphorylation levels of Syk and JNK upon S. aureus infection. (A) K+ concentration in the supernatant and cells of J774A.1 cells and PECs infected with S. aureus (MOI of 1) for 24 h (B–I) J774A.1 cells and PECs were infected with S. aureus (MOI of 5) for different time in the presence of following treating with increasing concentration of KCl (5 mM-80 mM). The cell lysates were collected at 0.5 h and 2 h. (B) The protein levels of t-Syk and p-Syk were detected by western blot in J774A.1 cells and PECs (F). (C) Relative densitometric quantification analysis of p-Syk/t-Syk in J774A.1 cells and PECs (G). (D) The protein levels of t-JNK and p-JNK were detected by western blot in J774A.1 cells and PECs (H). (E) Relative densitometric quantification analysis of p-JNK/t-JNK in J774A.1 cells and PECs (I). Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; **P < 0.01, ****P < 0.0001 vs. non-infection 5 mM group, #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 vs. infection 5 mM group.
To further determine the role of K+ efflux in NLRP3 inflammasome activation upon S. aureus infection, J774A.1 cells and PECs were cultured in medium with or without 40 mM KCl for 24 h. There was no significant difference in cell viability between the 5 mM group and the 40 mM group in the J774A.1 cells, while the cell viability was decreased upon S. aureus infection (Figure 9A). Compared to the 5 mM group, the mRNA expression levels of caspase-1, Il-1β and nek7 in the 40 mM group were obviously decreased in J774A.1 cells upon S. aureus infection (Figure 9B). Furthermore, 40 mM KCl also significantly reduced the protein expression level of p20, mIL-1β and NEK7 in both J774A.1 cells (Figure 9C) and PECs (Figure 9D). In summary, these results indicated that the K+ efflux/Syk/JNK signaling pathway was involved in NLRP3 inflammasome activation by regulating NLRP3-NEK7 complex formation during S. aureus infection.
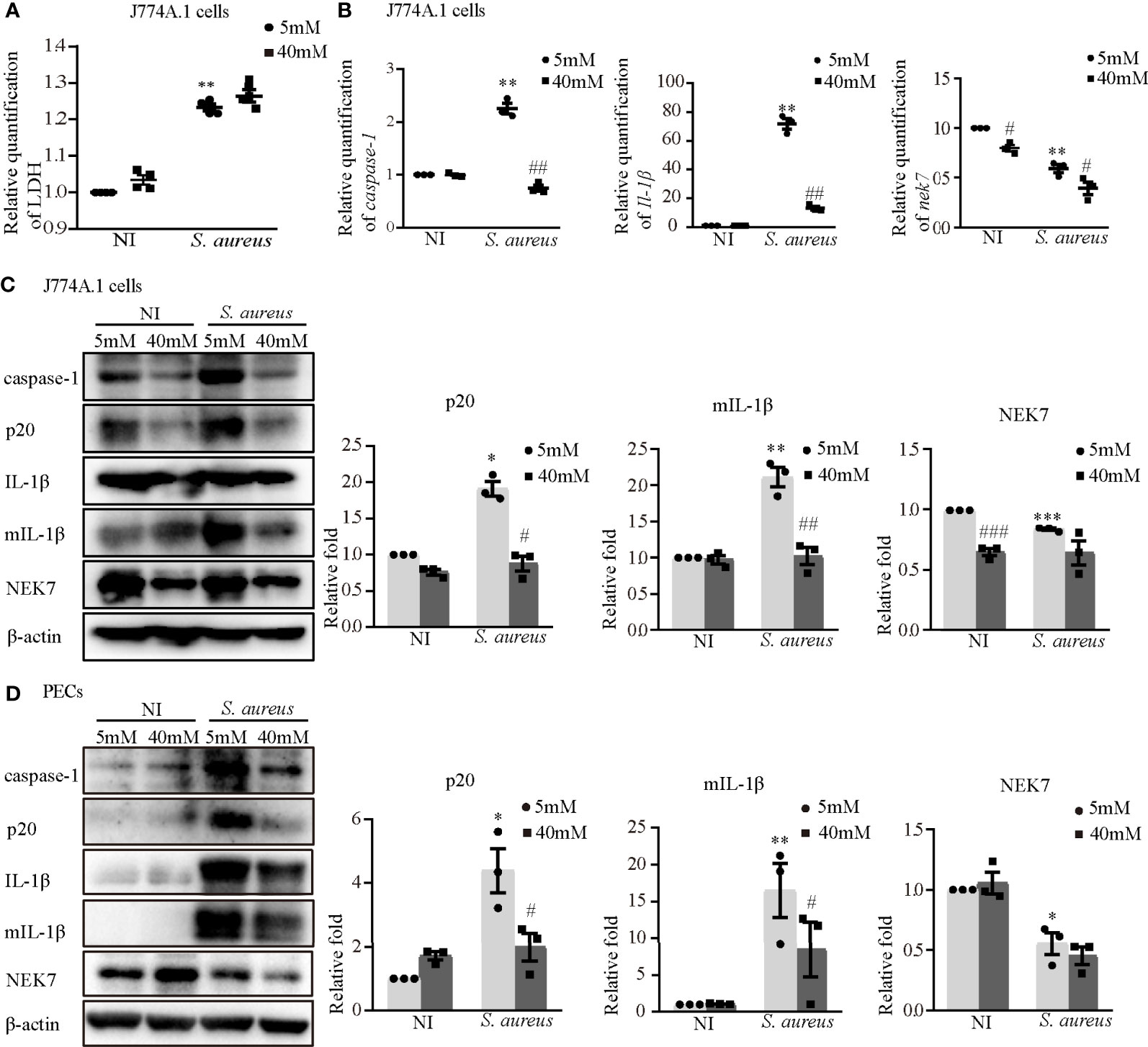
Figure 9 Blockade of potassium efflux downregulated NLRP3 inflammasome activation and NEK7 expression. J774A.1 cells (A–C) and PECs (D) were infected with S. aureus infection (MOI of 1) for 24 h in the presence of different concentration of KCl (5 mM or 40 mM). (A) The level of LDH in the supernatants were detected. (B) The mRNA expression levels of caspase-1, Il-1β and nek7 were examined by RT-PCR. (C) The protein expression levels of caspase-1, p20, pro-IL-1β, mIL-1β and NEK7 were detected by western blot and analyzed by relative densitometric quantification in J774A.1 cells and PECs (D). Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; *P < 0.05, **P < 0.01, ***P < 0.001 vs. non-infection control group, #P <0.05, ##P < 0.01, ###P < 0.001 vs. infection control group.
As an opportunistic pathogen, S. aureus infection is a main cause of invasive infections, which can range from superficial skin infections to potentially life-threatening systemic diseases. A series of inflammatory processes can be observed during S. aureus infection, such as cytokine release, tissue fibrosis, and neutrophil aggregation (19). Herein we showed that many inflammatory pathological phenomena appeared in the lung tissue of mice in the S. aureus infection group following NLRP3 inflammasome-associated molecules activation (Figure 1 and Table 1), suggesting that inflammation may be induced by NLRP3 inflammasome activation. Thus, the exact mechanism of NLRP3 inflammasome activation needs to be studied more deeply at the cellular level.
To gain a better understanding of the molecular mechanism of NLRP3 inflammasome activation, we focused on Syk and JNK because they particapate in NLRP3 inflammasome activation in DCM and DN (14, 15). In addition, a recent report found that the Dectin-2/Syk/JNK/NF-κB pathway induces NLRP3 and pro-IL-1β expression in Kawasaki disease (20), which is similar to our conclusion. Furthermore, Syk is known to be critical for IL-1β released during Toxoplasma gondii infection (21) and important in TLR4-mediated signaling upon LPS stimulation. Herein, we demonstrated that Syk and JNK were phosphorylated in S. aureus-infected macrophages (Figure 3), and that genetic knockdown of Syk or JNK in J774A.1 cells not only significantly reduced the transcripts of caspase-1 and IL-1β but also decreased the protein expression of p20 and mIL-1β (Figures 4A–D). In addition, similar results were obtained using phosphorylation inhibitors of Syk or JNK (Figures 5B, C; Figures 6B, C and Figures S1A, B, D, E). Moreover, our data showed that p-Syk/p-JNK promoted the formation of ASC specks and mediated caspase-1 activation, IL-1β maturation and secretion (Figures 5D–F, N, O; 6D–F, M and Figures S1C, F), indicating that the phosphorylation levels of Syk and JNK play a key role in NLRP3 inflammation activation upon S. aureus infection. Similarly, a previous study revealed that Syk enhances ASC oligomerization and regulates NLRP3 inflammasome activation via ASC phosphorylation at the Y146 and Y187 residues (22), although we have not confirmed the specific activation domain of ASC during S. aureus infection. Moreover, we demonstrated that Syk participated in this process by regulating JNK phosphorylation (Figure 7), indicating that Syk/JNK signaling is critical for activation of the NLRP3 inflammasome upon S. aureus infection.
NEK7, a serine/threonine kinase implicated in mitosis, shows lower protein and mRNA levels in systemic lupus erythematosus (SLE) patients (23). NEK7 is also necessary for NLRP3 inflammasome assembly and activation in bone marrow cells treated with LPS, followed by nigericin or ATP stimulation (24). Interestingly, our study found that the protein level of NEK7 was decreased in S. aureus induced mouse pneumonia (Figures 1C, F). However, whether NEK7 participates in S. aureus infection-induced NLRP3 inflammasome activation remains unknown.
In agreement with the in vivo results, our in vitro data showed that the expression of NEK7 in both types of macrophages was decreased during S. aureus infection, simultaneously, the formation of NEK7 and NLRP3 complexes was increased (Figures 2A–C). Consistently, some studies reported that the expression of NEK7 was decreased with the enhanced interaction of NEK7-NLRP3 under the stimulation of LPS and ATP (25), LPS and cytotoxic necrosis factor (CNF1) (26) or LPS and IgA ICs (27). Furthermore, S. aureus infection-induced expression of NLRP3 inflammasome-associated molecules were significantly decreased by NEK7-siRNA pretreatment (Figures 2D–F), suggesting that NEK7 participated in NLRP3 inflammasome activation. In addition, a previous study reported that NEK7 may be a switch between mitosis and the NLRP3 inflammasome, indicating that the interaction between NEK7 and NLRP3 is restricted to interphase of the cell cycle and that cells with activated NLRP3 inflammasomes cannot initiate mitosis (24). Based on above all, we speculated that the decreased expression of NEK7 may be due to the cell damage. As shown in Figure 9A, the LDH level in the culture supernatant was effectively increased. Unfortunately, we did not assess the growth cycle of the cells in our study.
To study the role of Syk/JNK in regulating NEK7-NLRP3 compound formation more deeply, we assessed the expression level of NEK7 with JNK/Syk-siRNA or JNK/Syk inhibitor pretreatment. Notably, our study demonstrated, for the first time, that the decreased mRNA and protein expression levels of NEK7 were rescued by JNK or Syk-siRNA (Figures 4E–G) and JNK or Syk inhibitors (Figures 5G–I, L; Figures 6G, H, K), indicating a role for Syk/JNK in regulating NEK7 expression in S. aureus-infected cells. At the same time, the inhibition of JNK or Syk in J774A.1 cells and PECs significantly reduced the interaction of NEK7-NLRP3 in S. aureus infection (Figures 5J, M; Figures 6I, L), suggesting that p-Syk/p-JNK accelerates NEK7-NLRP3 complex formation by downregulating NEK7 expression. In contrast, our recent study in Listeria monocytogenes infection found that the protein expression of NEK7 was increased (28). Taken together our findings in the present research suggest that NEK7 might play different roles in the infection of intracellular bacteria and extracellular bacteria, but we also need to pay attention to the cell cycle during infection.
To date, there are four main modes involved in activating NLRP3 inflammasome. One of them is the ion flux model. In this model, K+ efflux is widely accepted as an essential second step in the activation of NLRP3 inflammasomes, and this process is mediated by NEK7 during LPS and ATP costimulation (29, 30). Our data showed that K+ efflux increased upon S. aureus infection (Figure 8A), in addition, blockade of K+ efflux decreased the phosphorylation levels of Syk and JNK in both J774A.1 cells and PECs (Figures 8B–I). Moreover, the protein and mRNA level of NLRP3 inflammasome-associated molecules were all decreased when K+ efflux was inhibited (Figure 9), further suggesting that the NLRP3 inflammasome activation regulated by the Syk/JNK/NEK7 pathway was downstream of K+ efflux. Another mode of NLRP3 inflammasome activation is the reactive oxygen species (ROS) activation model. Mitochondrial ROSs are mediators of multiple activators of the NLRP3 inflammasome (31, 32), and the interaction between NEK7 and NLRP3 by ROS is K+ efflux-independent (21). However, macrophages are unable to effectively kill S. aureus internalized in mitochondria (33). Furthermore, cell lysosomal destruction and metabolites can also activate the NLRP3 inflammasome (34, 35). However, we did not consider the mechanism of S. aureus infection induced NLRP3 inflammasome activation from these aspects in the present study, and further studies are needed to verify this view.
NLRP3 inflammasome activation triggers caspase-1-dependent pyroptosis, which is a lytic form of programmed cell death that leads to the release of inflammatory cytokines (36), but there are very few studies about pyroptosis induced by S. aureus infection. Taken together with our present study, whether infection triggers pyroptosis to defend against bacteria needs further study. Furthermore, excessive inflammation may cause serious damage in normal tissue, so the anti-inflammatory mechanism needs to be activated to prevent it (37). Our previous study found that resveratrol (RSV) inhibits tumor progression by downregulating of NLRP3 (38), but whether RSV plays an anti-inflammatory role in S. aureus pneumonia to protect normal lung tissues needs further investigation.
Although our study of S. aureus infection induced NLRP3 inflammasome activation is more detailed, there are still some questions that need to be answered. It is well established that S. aureus employs a wide array of virulence factors, including surface proteins, pore-forming toxins, and alpha toxin (39). Moreover, alpha toxin potentiates opportunistic bacterial lung infections (40). Therefore, whether alpha toxin plays a key role in this process needs to be addressed in future studies. Furthermore, with the increasing mortality induced by antibiotic-resistant bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA), whose mortality is similar to that of acquired immune deficiency syndrome (AIDS) (41), whether the K+ efflux/Syk/JNK/NEK7-NLRP3 signaling pathway can be activated to defend against MRSA infection remains unknown.
In conclusion, as shown in Figure 10, our work provides evidence of a novel signaling pathway. Phosphorylation of Syk and JNK was dependent on potassium efflux during S. aureus infection, thereafter promoting NEK7-NLRP3 interaction and NLRP3 inflammasome activation through downregulation of NEK7, leading to the cleavage of pro-caspase-1 and maturation of IL-1β, and finally contributing to S. aureus induced-pneumonia. Our study provides a new dimension to understand the inflammation induced by S. aureus infection and may help to find potential therapeutic targets for drugs against S. aureus infection. However, future work should include more in vivo experiments to verify this conclusion.
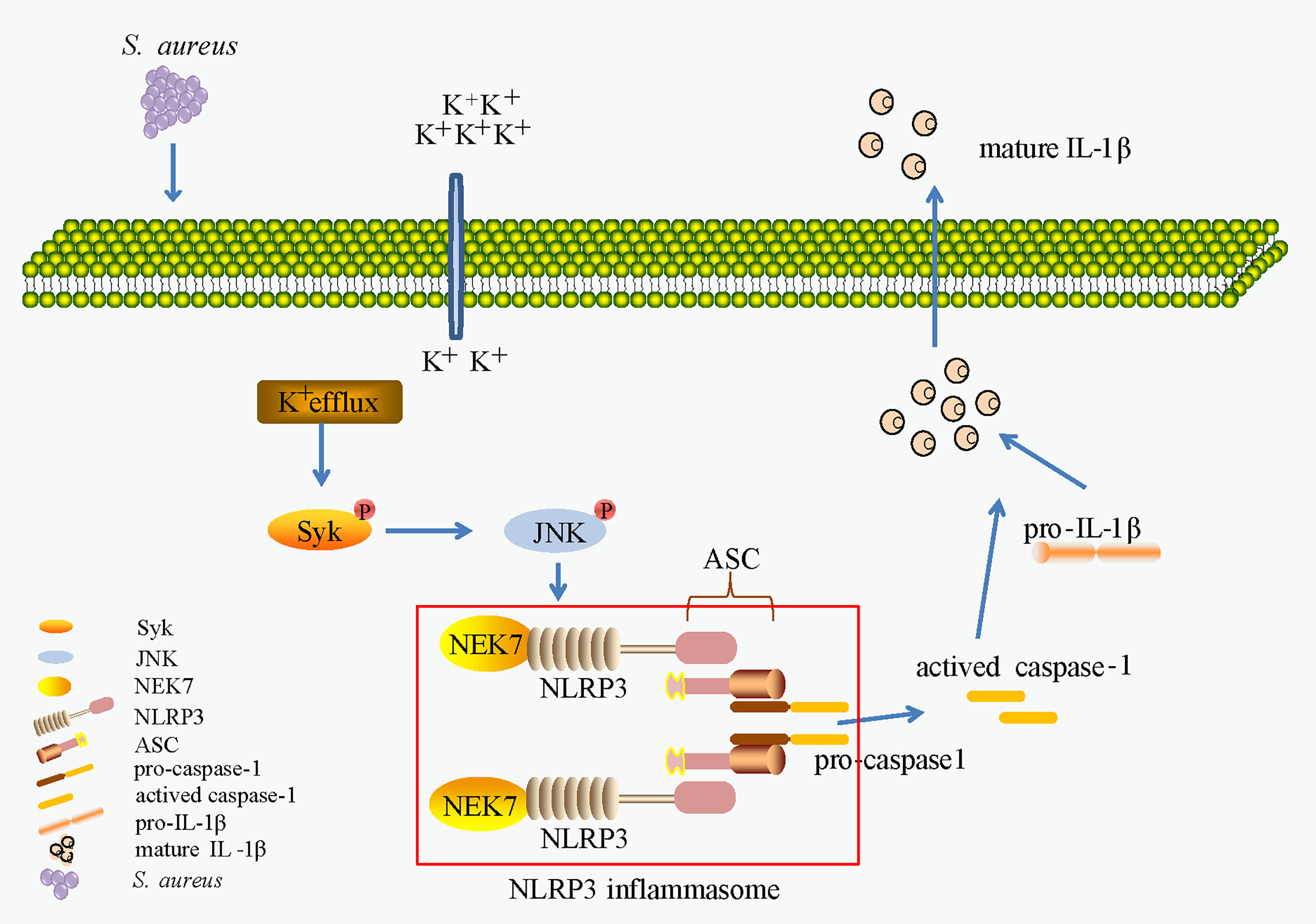
Figure 10 K+ efflux/Syk/JNK was important in NEK7-NLRP3 interaction and NLRP3 inflammasome activtion upon S. aureus infection.
The JNK inhibitor II SP600125 (SP) was purchased from Merck KGaA, Inc. Syk inhibitor IV BAY 61-3606 was purchased from Santa Cruz Biotechnology, Inc. TRIzol® reagent was purchased from Invitrogen Thermo Fisher Scientific, Inc. The H&E stain kit (cat. no. G1120), RIPA buffer (cat. no. R0020), DAPI solution (cat. no. C0060) and 4% paraformaldehyde (cat. no. P1110) were purchased from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). Antibodies for phospho-JNK (cat. no. 9255s, 1:1,000), JNK (cat. no. 9252, 1:1,000), p-Syk (cat. no. 2710, 1:1,000), Syk (cat. no. 2712, 1:1,000), NLRP3 (cat. no. 15101, 1:1,000), rabbit IgG (cat. no. 2729) and caspase-1 (cat. no. 4199, 1:1,000) were purchased from Cell Signaling Technology, Inc. Antibodies for IL-1β (cat. no. ab9722, 1:2,000) and NEK7 (cat. no. ab133514, 1:2,000) were purchased from Abcam (Cambridge, UK). The antibody for β-actin (cat. no. sc-47778, 1:1,000) was purchased from Santa Cruz Biotechnology, Inc. An anti-ASC antibody (cat. no. A1170, 1:500) was obtained from Abclonal (Wuhan, China). HRP-conjugated anti-mouse immunoglobulin G (IgG) (cat. no. W4021, 1:5,000) and HRP-conjugated anti-rabbit IgG (cat. no. W4011, 1:5,000) were from Promega (Madison, WI, USA). Antibodies for FITC goat anti-rabbit IgG (cat. no. GR200G-02C, 1:100) were obtained from Tianjin Sungene Biotech Co., Ltd (Tianjin, China). The primers for syk (cat# MQP030294), jnk (cat# MQP099059), caspase1(cat# MQP028800), Il-1β (cat# MQP092978), nek7 (cat# MQP101958) and gapdh (cat# MQP027158) were purchased from GeneCopoeia, Inc. (Beijing, China).
C57BL/6 WT female mice (age, 6-8 weeks old) were purchased from The Laboratory Animal Center of the Academy of Military Medical Sciences (Beijing, China, SYXK (Jin), 2019-0004). The mice were maintained under specific pathogen-free conditions of temperature (23 ± 5°C) and humidity (60 ± 5%) with alternating 12 h light/dark cycles. Clean drinking water and food were available freely. Mice were randomly divided into an infection group and a control group, with 6 mice per group. Infection group mice were challenged with 25 µl of a 109 CFU/mL suspension of S. aureus (ATCC 25923) by intranasal administration, while the mice in the control group were given an equal volume of sterile PBS solution. After 24 hours, the number of viable S. aureus in both lung tissue and bronchoalveolar lavage fluid (BVLF) was counted by CFU counting assay. In addition, part of the lung tissue was homogenized for western blot and H&E stain.
The J774A.1 cell line (cat. no. ZQ0565) was purchased from Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (Shanghai, China). J774A.1 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA.) supplemented with 15% FBS (FBS, LONSERA, cat. no. s711-001s, Shanghai, China) and 1% streptomycin/penicillin. Cells were seeded at various densities in 6/12/96−well plates. After 2.5 h, the adherent cells were pretreated with or without Syk inhibitor (1 μM) or JNK inhibitor (20 μM) for 2 h, or siRNA targeting Syk, JNK or NEK7 (50 nM) for 48 h or KCl (5-80 mM), after which they were subsequently exposed to a noninfectious environment or an S. aureus infectious environment (MOI 1/MOI 5) for different times, followed by harvesting of supernatant and cell lysates for analysis.
Mice were given a single intraperitoneal injection of 3 ml of 4% thioglycollate medium (Eiken Chemical, Tokyo, Japan). Three and a half days later, peritoneal exudate cells were collected and washed with RPMI 1640 medium (Gibco, USA). Cells were seeded in cell culture plates with RPMI 1640 medium containing 10% fetal bovine serum at 37°C and 5% CO2. After 2.5 h, adherent macrophages were used for experiments.
Staphylococcus aureus (ATCC 25923) was grown overnight in brain heart infusion broth (BHI, Eiken Chemical Co., Ltd., Tokyo, Japan) at 37°C with shaking. One volume of the overnight culture (2 ml) was added to 100 volumes of fresh BHI broth and cultured for 4 to 5 h. The bacteria were washed, suspended in PBS containing 10% glycerol and stored in a -80°C refrigerator. Three and a half days later, we selected three tubes of S. aureus randomly, determined the concentration of bacteria by plating a 10-fold serial dilution of bacterial suspension on tryptic soy agar (TSA) plates and counted the colonies at 24 h after incubation at 37°C. For infections of the cultured cells, an MOI of 1 or 5 was used. After 1 h, gentamicin was added to the culture at a final concentration of 10 mg/L.
J774A.1 cells were transfected with Syk, JNK or NEK7-siRNA (50 nM) using Lipofectamine™ 3000 (Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol to knockdown syk, jnk, and nek7, respectively; a scrambled siRNA, the negative control (NC)-siRNA, was used as a control in the experiment. The sequences for mouse Syk-siRNA were 5’-CCAGGUGGAAUAAUCUCAATT-3’ (sense) and 5’-UUGAGAUUAUUCCACCUGGTT-3’ (antisense); the sequences for mouse JNK-siRNA were 5’-GGAGGAACGAACUAAGAAUTT-3’ (sense) and 5’-AUUCUUAGUUCGUUCCUCCTT-3’ (antisense); and the sequences for mouse NEK7-siRNA were 5’-GCAACUCAACCAUCCAAAUTT-3’ (sense) and 5’- AUUUGGAUGGUUGAGUUGCTT-3’ (antisense). Sequences for the NC-siRNA were 5’-UUCUCCGAACGUGUCACGUTT-3’ (sense) and 5’-ACGUGACACGUUC GGAGAATT-3’ (antisense). Following 48 h of transfection, the cells were infected with S. aureus and harvested for western blot or RT-PCR. The efficiency of Syk, JNK or NEK7 silencing was assessed by western blot and RT-PCR.
Macrophages at a concentration of 1x106 cells/well were infected with S. aureus at an MOI of 1 or 5. At the harvest time, cells were lysed by SDS lysis buffer and lung tissues were homogenized in RIPA buffer. Total protein concentration was assessed using a bicinchoninic acid protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). Samples were separated by SDS-PAGE, and transferred to polyvinylidene difluoride (PVDF) membranes (Merck. Millipore). PVDF membranes were blocked with 5% nonfat milk or 5% bovine serum albumin (BSA) for 2 h at room temperature (RT). Membranes were then incubated with primary antibody overnight at 4°C. Following three washes with Tris-buffered saline/Tween 20 (TBST), the membranes were incubated with secondary antibodies at RT for 1 h. After being washed 3 times with TBST, the corresponding protein bands were detected using enhanced chemiluminescent HRP substrate (Millipore). β-actin was employed as a loading control. The results were analyzed using ImageJ software 6.0 (National Institutes of Health, Bethesda, MD, USA).
Mouse IL-1β and IL-18 proteins released into the supernatants were measured using an ELISA. The ELISA kits for IL-1β (cat.no. EK201B/3-96) and IL-18 (cat no. EK2182-96T) were purchased from Multi Sciences (Lianke) Biotech, Co., Ltd. (Hangzhou, China).
Macrophages at a concentration of 9x106 cells/well were seeded in 10 cm plates and infected with S. aureus at an MOI of 1 for 24 h. Cells were lysed in cold lysis buffer (40 mM Tris base, 120 mM NaCl, 1% Triton X-100, 0.5 mM Na3VO4, 1 mM PMSF, 0.5 mM NaF, 1 × EDTA-free Roche protease inhibitor cocktail, pH 7.4) with shaking at a high speed for 30 min. Cell lysates were collected by centrifugation (12,000 × rpm) at 4°C for 30 min. Total protein concentrations were determined using a BCA protein assay. Some of the samples were used to detect the expression level of intracellular proteins. Dynabeads™ Protein A were incubated with anti-NEK7 (4 μg) or control IgG (4 μg) antibodies on a rotary shaker at 4°C for 6 h. The other samples were equalized into two tubes and incubated with Dynabeads carrying different antibodies by shaking at 4°C overnight. The beads were washed more than 5 times. Proteins were denatured at 97°C for 5 min following the addition of twofold loading buffer and then detected by western blot.
J774A.1 cells (at a concentration of 2×105/well) were seeded in 12-well plates on slides. Adherent cells were treated according to the experimental conditions. Slides were washed with PBS three times and fixed in 4% paraformaldehyde at RT for 15 minutes. Slides were blocked in 5% BSA buffer at 37°C for 1 h and incubated with anti-ASC antibody at 4°C overnight. After being washed three times with PBS, the cells were incubated with secondary antibody for 1 h at RT. DAPI was used to stain nuclei. Cell images were taken using a Nikon DS-Ri1 fluorescence microscope. The ASC specks in at least 5 random fields of one slide were counted, and the percentage of ASC-speck-positive cells was calculated.
The lung tissue of mice at 24 h postinfection was fixed in 4% paraformaldehyde and routinely processed into paraffin. Sections 5 µm in thickness were cut for hematoxylin and eosin staining according to the manufacturer’s protocol for histopathologic evaluation. The histopathological samples were blindly assessed according to a histological scoring system as previously described (42). In short, on a scale of 0 to 4 for four pathological parameters were scored: (1) alveolar congestion, (2) hemorrhage, (3) inflammatory cell infiltration, and (4) alveolar wall thickness. 0 points represent normal lung, and scores of 1, 2, 3, and 4 represent mild (<25%), moderate (25%-50%), severe (50%-75%), and very severe (>75%) lung involvement, respectively. The total score was based on the sum of all score. Control group (1-3); infection group (4-6).
Total RNA was harvested using TRIzol reagent according to the manufacturer’s protocol, and first-strand cDNA was synthesized using the FastQuant RT kit (Tiangen Biotech Co., Ltd., Beijing, China). The concentration of the isolated RNA was measured using a NanoDrop (Thermo Fisher Scientific, Inc.). The mRNA levels were analyzed by RT-PCR using SYBR Green. The PCRs were performed with an initial denaturation at 95°C for 10 min, followed by 40 cycles at 95°C for 10 sec, 60°C for 20 sec, and 72°C for 20 sec, with a final extension at 72°C for 5 min. The PCR products were analyzed by melting curve analysis using MxPro-Mx3005P software v4.10 (Agilent Technologies, Inc.). The relative quantification of mRNA was performed using the comparative quantification cycle (Cq) method. The mRNA expression level was measured using the 2−ΔΔCq method.
Macrophages were seeded in 6-well-plates for 2.5 h and infected at an MOI of 1 for 24 h by culturing with or without KCl (40 mM). LDH release in supernatants was measured according to the manufacturer’s protocol using an LDH assay kit (cat. no. BC0685, Beijing Solarbio Science & Technology Co., Ltd.).
Macrophages were seeded in 6-well-plates for 2.5 h and infected at an MOI of 1 for 24 h. The concentrations of intracellular and extracellular potassium were measured following the manufacturer’s protocol via a biochemical assay kit (cat. no. E-BC-K279-M, Elabscience Biotechnology Co., Ltd. Wuhan, China).
Data are presented as the means ± SEM deviation of independent experiments. All the statistical results are representative of three independent experiments, and differences between groups were analyzed by GraphPad Prism 8 software (GraphPad Software, Inc., La Jolla, CA, USA). Student’s t-test, one-way ANOVA with Tukey’s post hoc test, and two-way ANOVA with Tukey’s multiple comparisons test were used for analysis. A statistically significant difference was defined as p<0.05 (*p< 0.05, **p< 0.01, ***p< 0.001).
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the Laboratory Animal Ethical Committee of Tianjin Medical University.
RL: data curation, project administration and writing-original draft. RL, YS, DT: conceptualization. RL, YL, AG, LW, HT: investigation. YS, ZQ, CL, XW: funding acquisition. CL, QW: supervision. All authors contributed to the article and approved the submitted version.
This work was supported by grants of the National Natural Science Foundation of China to YS (81772252, 32071263) and ZQ (81971887, 82172170); the Tianjin Natural Science Foundation to ZQ (20JCYBJC01260, 20YDTPJC00250); the Key Laboratory of Emergency and Trauma (Hainan Medical University), Ministry of Education to YS (KLET-201906) and ZQ (KLET-201906); the Fundamental Research Funds for the Central Universities, Nankai University to ZQ (63211140); the Scientific Research Project of Tianjin Education Commission to CL (2020KJ206); the Science & Technology Department of Sichuan Province to XW (2020YFS0103).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to express our gratitude to Lei Yang (Tianjin Institute of Acute Abdominal Diseases of Integrated Traditional Chinese and Western Medicine, Tianjin Nankai Hospital, Tianjin, China) for technical support in vivo experiment.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.747370/full#supplementary-material
Supplementary Figure 1 | JNK and Syk activated the NLRP3 inflammasome upon S. aureus infection in PECs. PECs pretreated with or without JNK inhibitor at the final concentration of 20 μmol/L or Syk inhibitor at the final concentration of 1 μmol/L for 2 h followed by S. aureus infecton (MOI of 1) for 24 h. (A) The mRNA expression levels of caspase-1, Il-1β were quantified by RT-PCR. (B) The protein levels of caspase-1, p20, pro-IL-1β and mIL-1β were examined by western blot and analyzed by relative densitometric quantification. (C) Immunofluorescence imaged endogenous ASC specks. Cells were fixed and stained for ASC (Green), DAPI (Blue). (Scale bar: 100 µm.) (D) The mRNA expression levels of caspase-1, Il-1β were examined by RT-PCR. (E) The protein expression levels of caspase-1, p20, pro-IL-1β and mIL-1β were examined by western blot and analyzed by relative densitometric quantification. (F) The ASC specks formation were imaged via fluorescence microscopy. (Scale bar: 100 µm.). Similar results were obtained in three independent experiments in data. Data shown are means ± SEMs; **P < 0.01, ****P < 0.0001 vs. non-infection control group, #P < 0.05, ##P < 0.01, ####P < 0.0001 vs. infection control group.
AIDS, acquired immune deficiency syndrome; ATP, adenosine triphosphate; ASC, apoptosis-associated speck-like protein containing a caspase-recruitment domain; BALF, bronchoalveolar lavage fluid; CFU, colony-forming units; DCM, diabetic cardiomyopathy; DN, diabetic nephropathy; H&E, hematoxylin and eosin; JNK, c-Jun N-terminal kinase; KCl, potassium chloride; IL-1β, interleukin-1β; IL-18, interleukin-18; mIL-1β, mature IL-1β; MRSA, methicillin-resistant Staphylococcus aureus; NEK7, NIMA-related kinase 7; NLRP3, NOD-like receptor family, pyrin domain containing 3; PAMPs, pathogen-associated molecular patterns; PECs, peritoneal exudate cells; p20, activated caspase-1; ROS, reactive oxygen species; RSV, resveratrol; SLE, systemic lupus erythematosus; Staphylococcus aureus, S. aureus; Syk, spleen tyrosine kinase.
1. Bowman L, Zeden MS, Schuster CF, Kaever V, Gründling A. New Insights Into the Cyclic Di-Adenosine Monophosphate (C-Di-AMP) Degradation Pathway and the Requirement of the Cyclic Dinucleotide for Acid Stress Resistance in Staphylococcus Aureus. J Biol Chem (2016) 291(53):26970–86. doi: 10.1074/jbc.M116.747709
2. Lowy FD. Staphylococcus Aureus Infections. N Engl J Med (1998) 339:520–32. doi: 10.1056/NEJM199808203390806
3. Faridi A, Kareshk AT, Fatahi-Bafghi M, Ziasistani M, Ghahraman MRK, Seyyed-Yousefi SZ, et al. Detection of Methicillin-Resistant Staphylococcus Aureus (MRSA) in Clinical Samples of Patients With External Ocular Infection. Iran J Microbiol (2018) 10:215–9.
4. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus Aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin Microbiol Rev (2015) 28(3):603–61. doi: 10.1128/CMR.00134-14
5. Parker D, Ahn D, Cohen T, Prince A. Innate Immune Signaling Activated by MDR Bacteria in the Airway. Physiol Rev (2016) 96:19–53. doi: 10.1152/physrev.00009.2015
6. Lin L, Xu L, Lv WH, Han L, Xiang YZ, Fu L, et al. An NLRP3 Inflammasome-Triggered Cytokine Storm Contributes to Streptococcal Toxic Shock-Like Syndrome (STSLS). PloS Pathog (2019) 15(6):e1007795. doi: 10.1371/journal.ppat.1007795
7. Elliott EI, Sutterwala FS. Initiation and Perpetuation of NLRP3 Inflammasome Activation and Assembly. Immunol Rev (2015) 265(1):35–52. doi: 10.1111/imr.12286
8. Chiu HW, Li LH, Hsieh CY, Rao YK, Chen FH, Chen A, et al. Glucosamine Inhibits IL-1β Expression by Preserving Mitochondrial Integrity and Disrupting Assembly of the NLRP3 Inflammasome. Sci Rep (2019) 9(1):5603. doi: 10.1038/s41598-019-42130-z
9. Hara H, Kohsuke T, Kawamura I, Fang RD, Cuellar EH, Shen YN, et al. Phosphorylation of the Adaptor ASC Acts as a Molecular Switch That Controls the Formation of Speck-Like Aggregates and Inflammasome Activity. Nat Immunol (2013) 14:1247−1255. doi: 10.1038/ni.2749
10. Bi CL, Zhang SJ, Shen YZ, Pauline M, Li H, Tang H. Selenium Plays an Anti-Inflammatory Role by Regulation NLRP3 Inflammasome in Staphylococcus Aureus-Infected Mouse Mammary Gland. Biol Trace Elem Res (2021) 199(2):604–10. doi: 10.1007/s12011-020-02166-z
11. Zhou L, Wang Z, Xu X, Wan Y, Qu K, Fan H, et al. Nek7 is Overexpressed in Hepatocellular Carcinoma and Promotes Hepatocellular Carcinoma Cell Proliferation In Vitro and In Vivo. Oncotarget (2016) 7:18620–30. doi: 10.18632/oncotarget.7620
12. Muñoz-Planillo R, Kuffa P, Martı´nez-Colo´n G, Smith BL, Rajendiran TM, Núñez G. K+Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
13. Liu H, Gu CP, Liu MJ, Liu G, Wang YL. NEK7 Mediated Assembly and Activation of NLRP3 Inflammasome Downstream of Potassium Efflux in Ventilator-Induced Lung Injury. Biochem Pharmacol (2020) 177:113998. doi: 10.1016/j.bcp.2020.113998
14. Li SY, Liu RQ, Xue MT, Qiao YC, Chen YF, Long GF, et al. Spleen Tyrosine Kinase−Induced JNK−dependent NLRP3 Activation is Involved in Diabetic Cardiomyopathy. Int J Mol Med (2019) 43(6):2481–90. doi: 10.3892/ijmm.2019.4148
15. Qiao YC, Tian XX, Li M, Li SY, Chen YF, Xue MT, et al. Spleen Tyrosine Kinase Promotes NLR Family Pyrin Domain Containing 3 Inflammasome−Mediated IL−1β Secretion via C−Jun N−terminal Kinase Activation and Cell Apoptosis During Diabetic Nephropathy. Mol Med Rep (2018) 18(2):1995–2008. doi: 10.3892/mmr.2018.9164
16. Therese B Deramaudt TB, Ali M, Vinit S, Bonay M. Sulforaphane Reduces Intracellular Survival of Staphylococcus Aureus in Macrophages Through Inhibition of JNK and P38 MAPK–induced Inflammation. Int J Mol Med (2020) 45(6):1927–41. doi: 10.3892/ijmm.2020.4563
17. Zhao G, Wu HC, Jiang KF, Rui GZ, Zhu Z, Qiu CW, et al. IFN-τ Inhibits S. Aureus-Induced Inflammation by Suppressing the Activation of NF-κb and MAPKs in RAW 264.7 Cells and Mice With Pneumonia. Int Immunopharmacol (2016) 35:332–40. doi: 10.1016/j.intimp.2016.02.016
18. Sun LB, Ma W, Gao WL, Xing YM, Chen LX, Xia ZY, et al. Propofol Directly Induces Caspase-1-Dependent Macrophage Pyroptosis Through the NLRP3-ASC Inflammasome. Cell Death Dis (2019) 10(8):542. doi: 10.1038/s41419-019-1761-4
19. Pollitt EJG, Szkuta PT, Burns N, Foster SJ. Staphylococcus Aureus Infection Dynamics. PloS Pathog (2018) 14(6):e1007112. doi: 10.1371/journal.ppat.1007112
20. Anzai F, Watanabe S, Kimura H, Kamata R, Karasawa T, Komada T, et al. Crucial Role of NLRP3 Inflammasome in a Murine Model of Kawasaki Disease. J Mol Cell Cardiol (2020) 138:185–96. doi: 10.1016/j.yjmcc.2019.11.158
21. Pandori WJ, Lima TS, Mallya S, Kao TH, Gov L, Lodoen MB. Toxoplasma Gondii Activates a Syk-CARD9-NF-κb Signaling Axis and Gasdermin D-Independent Release of IL-1β During Infection of Primary Human Monocytes. PloS Pathog (2019) 15(8):e1007923. doi: 10.1371/journal.ppat.1007923
22. Lin YC, Huang DY, Wang JS, Lin YL, Hsieh SL, Huang KC, et al. Syk is Involved in NLRP3 Inflammasome-Mediated Caspase-1 Activation Through Adaptor ASC Phosphorylation and Enhanced Oligomerization. J Leukoc Biol (2015) 97(5):825–35. doi: 10.1189/jlb.3HI0814-371RR
23. Ma ZZ, Sun HS, Lv JC, Guo L, Yang QR. Expression and Clinical Significance of the NEK7-NLRP3 Inflammasome Signaling Pathway in Patients With Systemic Lupus Erythematosus. J Inflammation (Lond) (2018) 15:16. doi: 10.1186/s12950-018-0192-9
24. Shi HX, Wang Y, Li XH, Zhan XM, Tang M, Fina M, et al. NLRP3 Activation and Mitosis are Mutually Exclusive Events Coordinated by NEK7, a New Inflammasome Component. Nat Immunol (2016) 17(3):250–8. doi: 10.1038/ni.3333
25. Wu CH, Gan CH, Li LH, Chang JC, Chen ST, Menon MP, et al. A Synthetic Small Molecule F240B Decreases NLRP3 Inflammasome Activation by Autophagy Induction. Front Immunol (2020) 11:607564. doi: 10.3389/fimmu.2020.607564
26. Dufies O, Doye A, Courjon J, Torre C, Michel G, Loubatier C, et al. Escherichia Coli RhoGTPase-Activating Toxin CNF1 Mediates NLRP3 Inflammasome Activation via P21 Activated Kinases-1/2 During Bacteremia in Mice. Nat Microbiol (2021) 6(3):401–12. doi: 10.1038/s41564-020-00832-5
27. Yang SR, Hua KF, Takahata A, Wu CY, Hsieh CY, Chiu HW, et al. LCC18, a Benzamide-Linked Small Molecule, Ameliorates IgA Nephropathy in Mice. J Pathol (2021) 253(4):427–41. doi: 10.1002/path.5609
28. Gao AJ, Tang HX, Zhang Q, Liu RQ, Wang L, Liu YS, et al. Mst1/2-ALK Promotes NLRP3 Inflammasome Activation and Cell Apoptosis During Listeria Monocytogenes Infection. J Microbiol (2021) 59(7):681–92. doi: 10.1007/s12275-021-0638-2
29. Groß CJ, Mishra R, Schneider KS, Médard G, Wettmarshausen J, Dittlein DC, et al. K+ Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity (2016) 45(4):761–73. doi: 10.1016/j.immuni.2016.08.010
30. He Y, Zeng MY, Yang DH, Motro B, Núñez G. NEK7 is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
31. Yabal M, Calleja DJ, Simpson DS, Lawlor KE. Stressing Out the Mitochondria: Mechanistic Insights Into NLRP3 Inflammasome Activation. J Leukoc Biol (2019) 105(2):377–99. doi: 10.1002/JLB.MR0318-124R
32. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity (2015) 42:406–17. doi: 10.1016/j.immuni.2015.02.002
33. Cohen TS, Boland ML, Boland BB, Takahashi V, Tovchigrechko A, Lee Y, et al. S. Aureus Evades Macrophage Killing Through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep (2018) 22(9):2431–41. doi: 10.1016/j.celrep.2018.02.027
34. Lee HM, Yuk JM, Kim KH, Jang J, Kang G, Park JB, et al. Mycobacterium Abscessus Activates the NLRP3 Inflammasome via Dectin-1-Syk and P62/SQSTM1. Immunol Cell Biol (2012) 90(6):601–10. doi: 10.1038/icb.2011.72
35. Yu HB, Finlay BB. The Caspase-1 Inflammasome: A Pilot of Innate Immune Responses. Cell Host Microbe (2008) 4(3):198–208. doi: 10.1016/j.chom.2008.08.007
36. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host Cell Death and Inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi: 10.1038/nrmicro2070
37. Wang JY, Sahoo M, Lantier1 L, Warawa J, Cordero H, Deobald K, et al. Caspase-11-Dependent Pyroptosis of Lung Epithelial Cells Protects From Melioidosis While Caspase-1 Mediates Macrophage Pyroptosis and Production of IL-18. PloS Pathog (2018) 14(5):e1007105. doi: 10.1371/journal.ppat.1007105
38. Tian XX, Zhang SZ, Zhang Q, Kang LC, Ma CZ, Feng LF, et al. Resveratrol Inhibits Tumor Progression by Down-Regulation of NLRP3 in Renal Cell Carcinoma. J Nutr Biochem (2020) 85:108489. doi: 10.1016/j.jnutbio.2020.108489
39. Seilie ES, Bubeck Wardenburg J. Staphylococcus Aureus Pore-Forming Toxins: The Interface of Pathogen and Host Complexity. Semin Cell Dev Biol (2017) 72:101–16. doi: 10.1016/j.semcdb.2017.04.003
40. Cohen TS, Hilliard JJ, Jones-Nelson O, Keller AE, O’Day T, Tkaczyk C, et al. Staphylococcus Aureus α Toxin Potentiates Opportunistic Bacterial Lung Infections. Sci Transl Med (2016) 8(329):329ra31. doi: 10.1126/scitranslmed.aad9922
41. Dadashi M, Nasiri MJ, Fallah F, Owlia P, Hajikhani B, Emaneini M, et al. Methicillin-Resistant Staphylococcus Aureus(MRSA) in Iran: A Systematic Review and Meta-Analysis. J Glob Antimicrob Resist (2018) 12:96–103. doi: 10.1016/j.jgar.2017.09.006
Keywords: Syk, JNK, NLRP3 inflammasome, S. aureus, K+ efflux, NEK7
Citation: Liu R, Liu Y, Liu C, Gao A, Wang L, Tang H, Wu Q, Wang X, Tian D, Qi Z and Shen Y (2021) NEK7-Mediated Activation of NLRP3 Inflammasome Is Coordinated by Potassium Efflux/Syk/JNK Signaling During Staphylococcus aureus Infection. Front. Immunol. 12:747370. doi: 10.3389/fimmu.2021.747370
Received: 26 July 2021; Accepted: 30 August 2021;
Published: 16 September 2021.
Edited by:
Chaofeng Han, Second Military Medical University, ChinaReviewed by:
Chen Wang, China Pharmaceutical University, ChinaCopyright © 2021 Liu, Liu, Liu, Gao, Wang, Tang, Wu, Wang, Tian, Qi and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derun Tian, dGlhbmRyQHRtdS5lZHUuY24=; Zhi Qi, cWl6aGlAbmFua2FpLmVkdS5jbg==; Yanna Shen, c2hlbnlhbm5hQHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.