 Christina B. Schroeter1
Christina B. Schroeter1 Niklas Huntemann1Stefanie Bock2Christopher Nelke1
Niklas Huntemann1Stefanie Bock2Christopher Nelke1 David Kremer1Klaus Pfeffer3Sven G. Meuth1
David Kremer1Klaus Pfeffer3Sven G. Meuth1 Tobias Ruck1*
Tobias Ruck1*- 1Department of Neurology, Medical Faculty, Heinrich Heine University Düsseldorf, Düsseldorf, Germany
- 2Department of Neurology With Institute of Translational Neurology, University of Münster, Münster, Germany
- 3Institute of Medical Microbiology and Hospital Hygiene, Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany
Regulatory T cells (Tregs) are the major determinant of peripheral immune tolerance. Many Treg subsets have been described, however thymus-derived and peripherally induced Tregs remain the most important subpopulations. In multiple sclerosis, a prototypical autoimmune disorder of the central nervous system, Treg dysfunction is a pathogenic hallmark. In contrast, induction of Treg proliferation and enhancement of their function are central immune evasion mechanisms of infectious pathogens. In accordance, Treg expansion is compartmentalized to tissues with high viral replication and prolonged in chronic infections. In friend retrovirus infection, Treg expansion is mainly based on excessive interleukin-2 production by infected effector T cells. Moreover, pathogens seem also to enhance Treg functions as shown in human immunodeficiency virus infection, where Tregs express higher levels of effector molecules such as cytotoxic T-lymphocyte-associated protein 4, CD39 and cAMP and show increased suppressive capacity. Thus, insights into the molecular mechanisms by which intracellular pathogens alter Treg functions might aid to find new therapeutic approaches to target central nervous system autoimmunity. In this review, we summarize the current knowledge of the role of pathogens for Treg function in the context of autoimmune neuroinflammation. We discuss the mechanistic implications for future therapies and provide an outlook for new research directions.
1 Introduction
In the context of infections, Tregs mediate beneficial and detrimental effects on short- and long-term disease outcomes. Although many Treg subsets have been described, thymus-derived (tTregs) and peripherally induced Tregs (pTregs) remain the most important subpopulations (1, 2). Tregs are generally found to express CD4 and either or both the high-affinity receptor for interleukin (IL)-2 CD25+ as well as the forkhead box protein P3 (Foxp3) (3). Their expression of intracellular and surface markers, such as CD25, glucocorticoid-induced tumor necrosis factor receptor (GITR) and the inhibitory cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) define their phenotype and function (4–6). tTregs emerge with a CD4+CD25+Foxp3+ phenotype directly from the thymus. They are specific for self-antigens requiring continuous antigenic stimulation for their survival and preservation of self-tolerance, the lack of which may lead to autoimmune disorders (7–11). pTregs on the other hand adopt a regulatory function upon expression of Foxp3 and are therefore likely to be specific to an exogenous antigen (3, 12–16). In the context of infection, Tregs can ameliorate excessive immune responses by interaction with and suppression of immune cells. However, Treg expansion and enhanced Treg function are central mechanisms of pathogen-related immune evasion. Yet, the contribution of Tregs to the pathophysiology of pathogen-mediated diseases as well as the underlying molecular mechanisms remain largely elusive.
In the context of therapeutic interventions, it is important to consider the Janus-faced functions of Tregs in infections potentially providing beneficial or detrimental effects (Figure 1). Defining the mechanisms by which intracellular pathogens alter Treg function might pave the way toward new therapeutic approaches not only in the settings of infections, but also in autoimmune neuroinflammation.
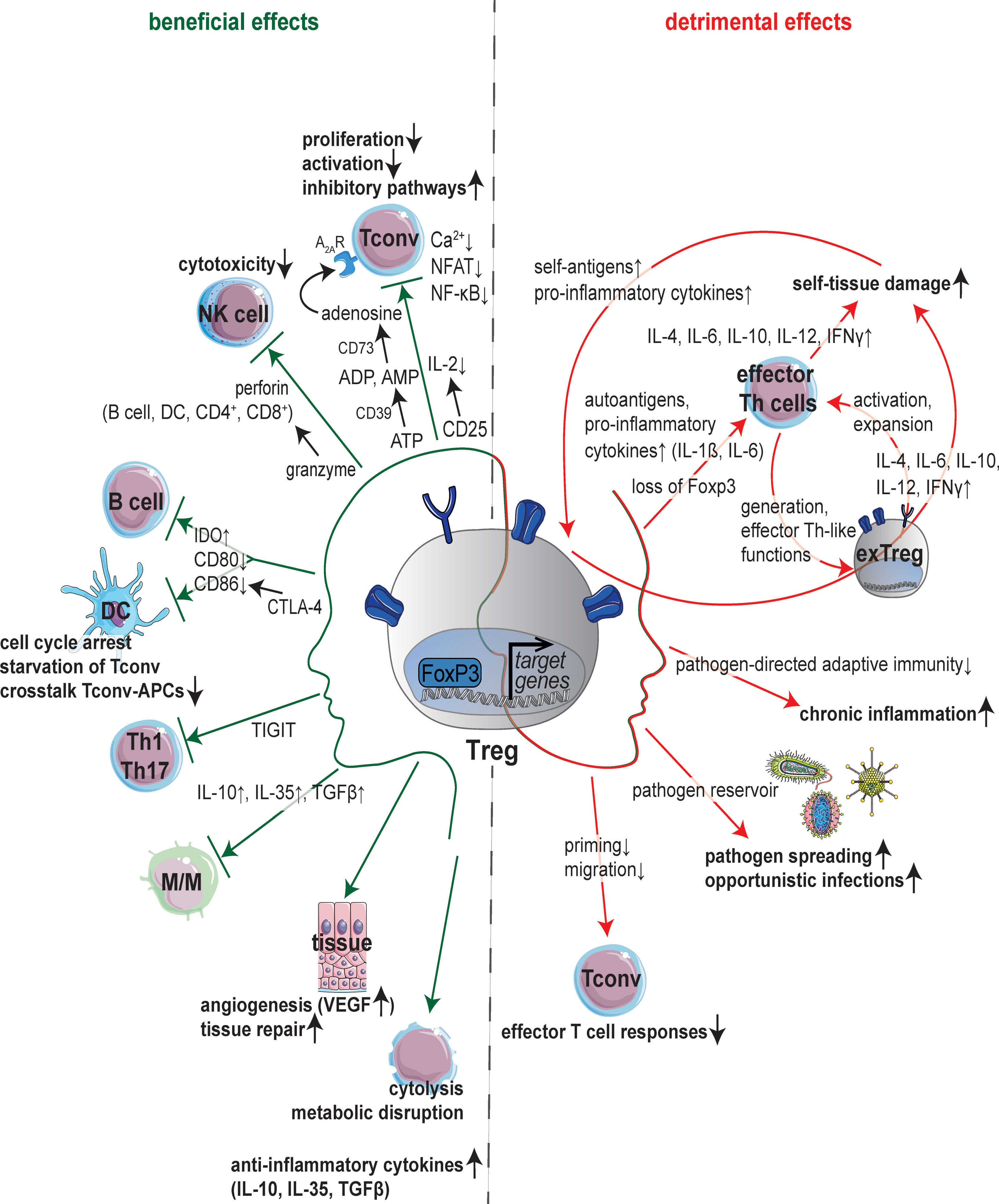
Figure 1 Janus-faced nature of Tregs. Schematic overview of molecular mechanisms underlying the regulation of immune cells and immune responses by regulatory T cells (Tregs). Anti-infective functions are mainly mediated by suppression of immune cells (left): Expression of CD25 leads to consumption of interleukin (IL)-2 inhibiting activation and proliferation of conventional T cells (Tconv). Suppression of Tconv can also be mediated by adenosine production via the ectoenzymes CD39 and CD73. Besides, Tregs are able to suppress T cell receptor (TCR)-induced Ca2+, NFAT and NF-κB signaling. Dendritic cells (DCs) and B cells are influenced by cytotoxic T-lymphocyte antigen 4 (CTLA-4) which binds CD80/CD86 and increases the expression of indoleamine 2,3-dioxygenase (IDO) resulting in starvation of Tconv next to cell cycle arrest and decrease in crosstalk between Tconv and antigen-presenting cells (APCs). Tregs can induce the death of effector cells (B cells, DCs, CD4+ and CD8+ cells) in a granzyme-perforin-dependent manner. The co-inhibitory molecule T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT) suppresses T helper (Th) 1 and Th17 cell responses. Tregs can also induce angiogenesis via vascular endothelial growth factor (VEGF) or target tissue cells directly. Further immunosuppressive effects of Tregs are mediated by cytokines (IL-10, IL-35, TGFβ), cytolysis or metabolic disruption. By contrast, Tregs can support inflammation (right) by a multi-layered feed-forward loop promoting the generation of ‘exTreg’ cells adapting Th-like functions, which in turn stimulate activation and expansion of autoreactive Th effector cells. Loss of immunosuppressive capacity adapting phenotype and functionality of Th cells is also reported upon loss of forkhead box protein 3 (Foxp3) in Tregs. Also, Tregs inhibit effector T cell responses thereby promoting chronic inflammation, pathogen spreading and opportunistic infections acting as pathogen reservoir. APCs, antigen-presenting cells; CTLA-4, cytotoxic T-lymphocyte antigen 4; DCs, dendritic cells; Foxp3, forkhead box protein 3; IDO, indoleamine 2,3-dioxygenase; IL, interleukin; M/M, monocytes and macrophages; TIGIT, T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif domains; Tconv, conventional T cells; TCR, T cell receptor; TGFβ, tumor growth factor β; Tregs, regulatory T cells; VEGF, vascular endothelial growth factor.
With this review, we intend to give a detailed overview of molecular mechanisms underlying altered Treg function in models of acute and chronic infections as well as in autoimmune neuroinflammation with a focus on multiple sclerosis (MS) (Table 1). We investigate the impact of pathogens on immune cell distributions, profiles, and functionality - particularly Treg functions - in the setting of neuroinflammation. We discuss how the complex changes in Tregs lead to altered function and that the underlying mechanisms could contribute to better understand the pathophysiology of neuroinflammatory diseases and their treatments. We further review the interplay of infection with pathogens and autoimmune processes (Figure 2) and, of particular interest, the clinical targets that result from these interactions (Table 2). In addition, we highlight the interplay between commensal bacteria and the function/plasticity of Tregs. In doing so, we particularly consider the implications for the phenotype of autoimmune phenomena. We point out the need for multi-omic approaches (functional analyses, transcriptomics, proteomics, and metabolomics) to illuminate the complex changes in Tregs leading to altered function (Figure 3).
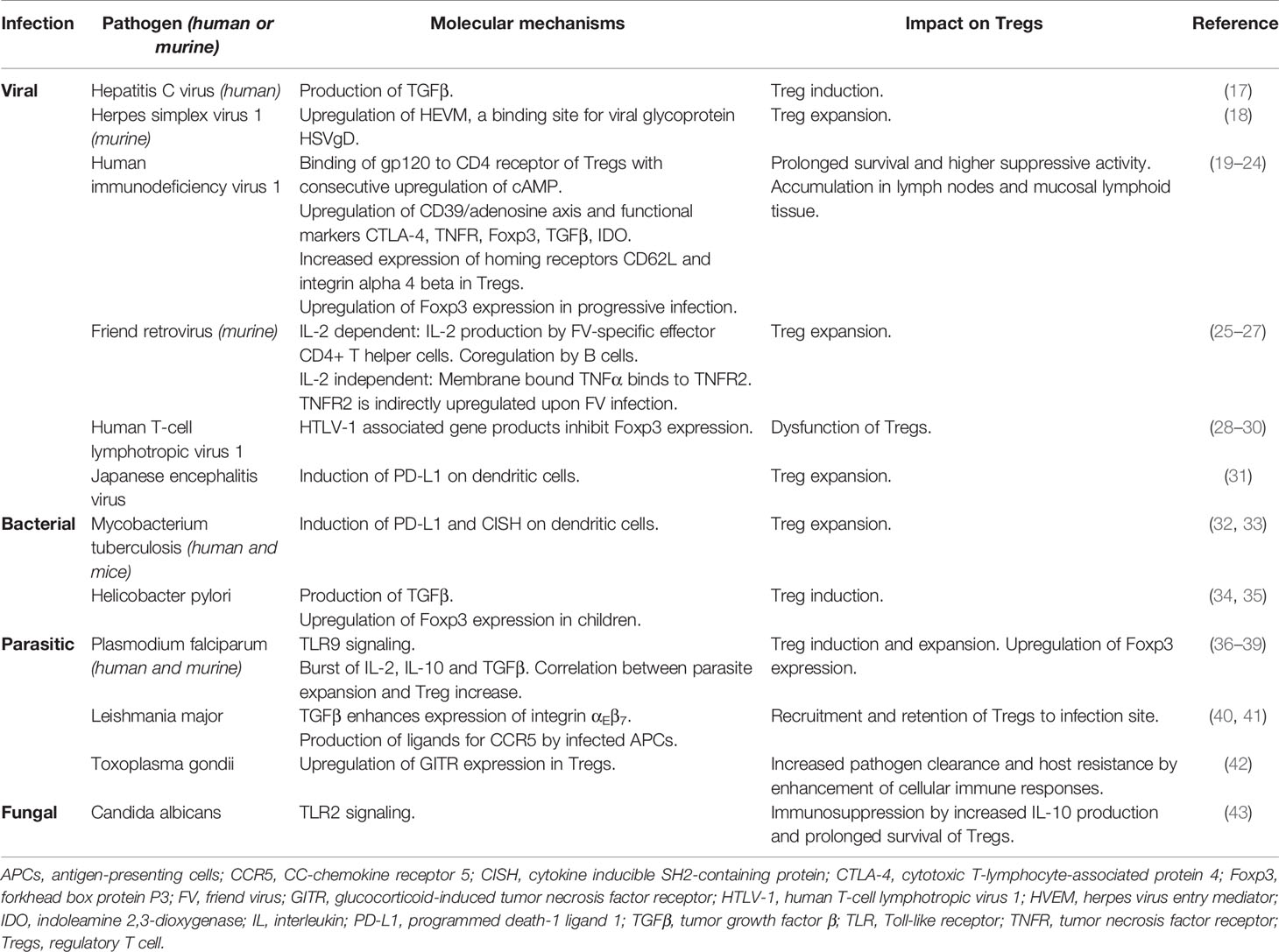
Table 1 Impact of pathogens on Tregs and the underlying molecular mechanisms.
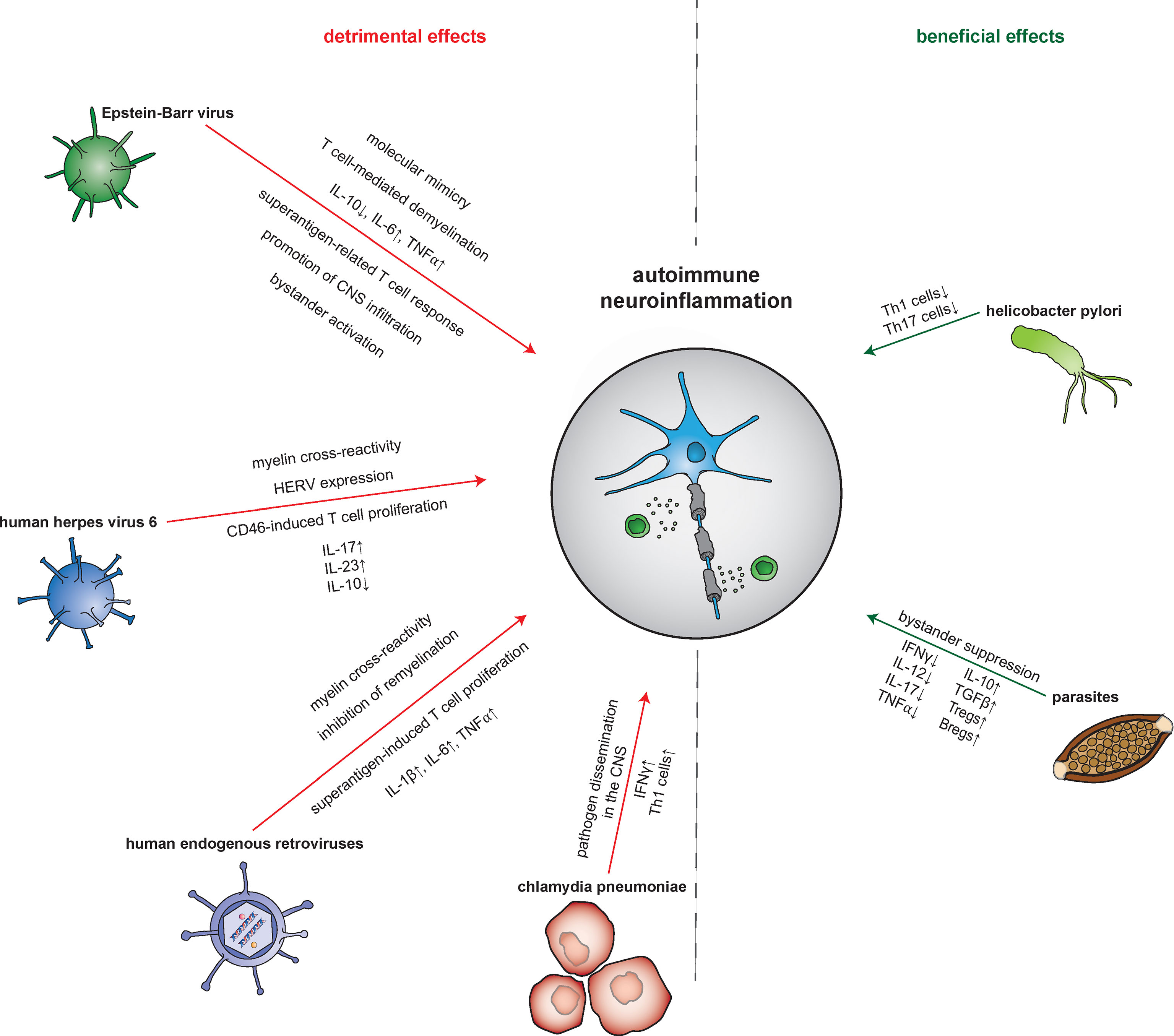
Figure 2 Pathogen-mediated impact on autoimmune neuroinflammation. The mechanisms by which infectious pathogens influence the processes of autoimmune neuroinflammation are diverse. Both detrimental and beneficial effects are reported. Epstein-Barr virus (EBV), for example, leads to an increase in neuronal damage via molecular mimicry, demyelination, an increase in pro- and decrease in anti-inflammatory molecules, and an augmented T cell response. Other pathways by which EBV induces amplification of the neuroinflammatory response include promotion of central nervous system (CNS) infiltration by autoreactive T and B cells next to bystander activation. Meanwhile, human herpes virus 6 (HHV-6) leads to a detrimental impact via CD8+ T cell-mediated cross-reactivity with myelin peptides and CD46-induced promotion of T cell proliferation. Furthermore, HHV-6 also triggers the expression of human endogenous retroviruses (HERVs) proteins. These in turn induce further damage via cross-reactivity with myelin antigens but also through acting as superantigens. Contributing to this is as well, HERVs trigger CD14- as well as Toll-like receptor (TLR) 4-mediated induction of proinflammatory cytokines. Interestingly, by suppression of oligodendrocyte precusor cells, HERVs also interfere with neurodegenerative processes. Finally, Chlamydia pneumoniae was shown to aggravate neuroinflammation in an animal model through pathogen dissemination into the CNS accompanied by an increase of pro-inflammatory Th1 cells. In contrast, a beneficial impact on the neuroinflammatory response was found for H. pylori and parasites. H. pylori improves the outcome in animal models of MS by reducing the proliferation of Th1 and Th17 cells. Parasites such as helminths attenuate the neuroinflammatory response by inducing bystander suppression via upregulation of regulatory B and T cells as well as anti-inflammatory cytokines. Bregs, regulatory B cells; CNS, central nervous system; HERV, human endogenous retrovirus; IFN, interferon; TGFβ, tumor growth factor β; Th cell, T helper cell; TLR, Toll-like receptor; TNFα, tumor necrosis factor α; Tregs, regulatory T cells.
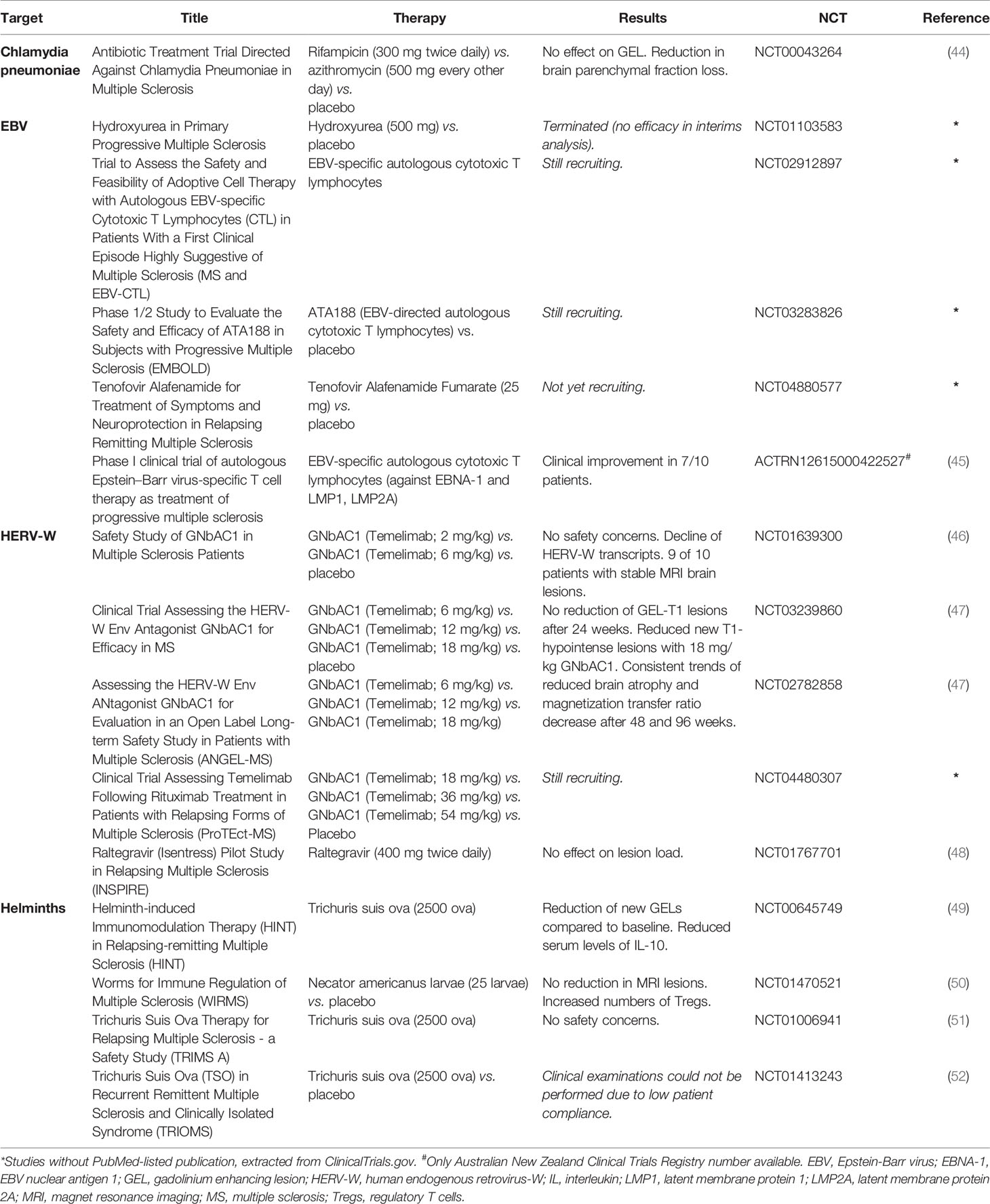
Table 2 Clinical studies on pathogen-derived targets in multiple sclerosis.
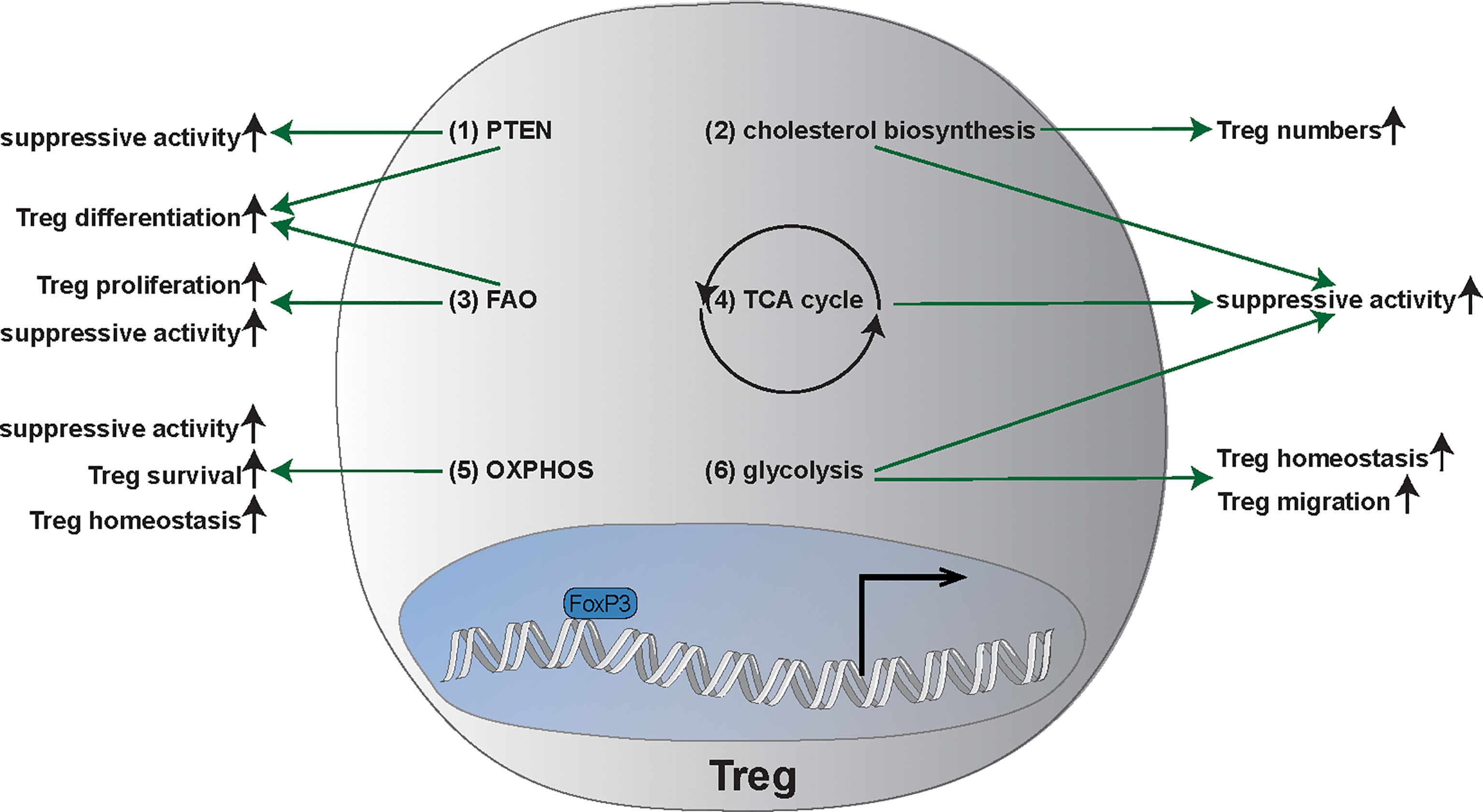
Figure 3 Main metabolic pathways and metabolic plasticity of Tregs. (1) Phosphatase and tensin homolog (PTEN) enhances regulatory T cell (Treg) differentiation as well as the suppressive activity of Tregs. (2) Cholesterol biosynthesis is required for suppressive functionality of Tregs and increases Treg frequency. (3) Fatty axid oxidation (FAO) is important for Treg generation, proliferation, as well as the suppressive activity. (4) Tricarboxylic acid cycle (TCA): TCA promotes the suppressive activity of Tregs. (5) Oxidative Phosphorylation (OXPHOS) is not only important for survival of Tregs but also for the suppressive capacity as well as the homeostasis of Tregs. (6) Glycolysis promotes suppressive function, the migration, and the homeostasis of Tregs. FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation; PTEN, Phosphatase and tensin homolog; TCA, Tricarboxylic acid cycle; Tregs, regulatory T cells.
Overall, our review focuses on past, present and future insights into the role of Tregs in the pathophysiology of pathogen-mediated diseases and in autoimmune neuroinflammation. We discuss established and potential therapeutic strategies in MS resulting from this new molecular understanding.
2 Manuscript/Subsections
2.1 Janus-Faced Nature: Duality of Tregs in Pathogen-Mediated Diseases
In the context of pathogen-mediated diseases, Tregs are Janus-faced (Figure 1): On the one hand, they are the major determinant of peripheral immune tolerance not only controlling immune responsiveness to intrinsic antigens and infective pathogens but also modulating immune capacity (9, 11–13). On the other hand, Tregs dampen favorable pathogen-directed adaptive immunity counter acting complete pathogen clearance and giving rise to chronic infections (14, 53, 54). Their phenotype and functions are, among others, dependent on their localization and the tissue type (55–62).
2.1.1 One Janus Face: Beneficial Treg Effects in Infections
Aside from their immunosuppressive capacity forestalling pathology, Tregs have been shown to facilitate appropriate effector mechanisms. Furthermore, Tregs control immunopathology detrimental to the host body arising from excessive effector immune responses.
Tregs utilize diverse immunosuppressive mechanisms depending on their microenvironment: Expression of CD25, the α-chain of IL-2, leads to consumption of IL-2 inhibiting activation and proliferation of conventional T cells (Tconv) (63–65). Interestingly, Chinen et al. showed that IL-2 expression activates signal transducer and activator of transcription (STAT) 5 further boosting the suppressor function of Tregs (66). Suppression of Tconv next to macrophages can also be triggered by cAMP-mediated protein kinase A (PKA) activation. Here, expression of the ectonucleotidase CD39 by Tregs leads to hydrolyzation of ATP followed by further cleavage of AMP to adenosine by CD73 (67–69). Subsequently, activation of the adenosine receptor A2A causes intracellular accumulation of cAMP which in turn stimulates PKA and associated downstream signaling (70–73). Tregs can induce the death of natural killer cells (NK cells) and other effector cells, such as B cells, dendritic cells (DCs), CD4+ and CD8+ cells, by releasing granzyme resulting in perforin-dependent apoptosis of target cells (74–76). B cells and DCs are regulated by Tregs via CTLA-4 which binds CD80/CD86 and increases the expression of indoleamine 2,3-dioxygenase (IDO) (77–79). Consecutively, binding of CD28 to CD80/CD86 is limited hampering the crosstalk between Tconv and antigen-presenting cells (APCs). Also, accumulation of IDO can lead to starvation of Tconv and cell cycle arrest, amongst others via degradation of the essential amino acid tryptophan (80, 81). Besides that, T cells can suppress T cell receptor (TCR)-induced Ca2+, NFAT and NF-κB downstream signaling (77). The co-inhibitory molecule T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT) suppresses pro-inflammatory T helper (Th) 1 and Th17 cells, but not Th2 cell responses (82, 83). Tregs can directly induce angiogenesis via vascular endothelial growth factor or target tissue cells (58). Further immunosuppressive effects of Tregs, e.g. on monocytes and macrophages, are mediated via anti-inflammatory cytokines such as IL-10, IL-35 or tumor growth factor β (TGFβ), cytolysis or metabolic disruption (62, 77, 84).
During acute and chronic (retro-) viral infections, Tregs have been shown to promote the in- and efflux of pro-inflammatory cells into lymph nodes (85, 86). Also, they suppress the proliferation and entry of infected cells (87, 88) and contribute to a memory formation via antigen persistence (89). In mice, Treg response is locally defined controlling magnitude and duration of virus-specific cytotoxic T cell responses (90, 91). For example, in human and murine cytomegalovirus, vaccinia virus and influenza virus, CD8+ T cell responses are controlled by Tregs by suppression of the immune response to immunodominant epitopes (92, 93). In human immunodeficiency virus (HIV) low Treg frequencies are strongly associated with increased immune activation, accelerated atherosclerosis and other morbidities linked to inflammation (94–103). A negative correlation between the relative amount of Tregs and inflammation has also been observed for hepatitis C virus (HCV) in humans and mice (104, 105). Here, Tregs suppress not only the production of interferon gamma (IFNγ) but also the expansion and activation-induced cell death of HCV-specific T cells resulting in reduced CD4+ T cell reactivity and mitigation of T cell-mediated liver disease (105–109).
In bacterial infections, Tregs show a predominant regulatory function controlling and limiting adaptive and innate immune responses as shown in different mouse studies (110): Immunosuppressive functions of Tregs have been already described for helicobacter hepaticus (111, 112), helicobacter pylori (H. pylori) (113–115), listeria monocytogenes (116), pneumocystis carinii (117, 118).
In toxoplasmosis as a prototypical parasitic infection, Treg depleted mice showed 50-60% mortality during acute infection (119).
2.1.2 Other Janus Face: Detrimental Treg Effects in Infections
Immune responses to pathogens can be impaired by an overly strong suppressive effect of Tregs interfering with early protective immunity (84): Tregs can inhibit effector T cell responses thereby promoting chronic inflammation. In turn, lack of complete eradication of pathogens leads to a reservoir function of human and murine Tregs acting as carriers for respective pathogens promoting their expansion in the environment resulting in spread of infections (120–122). Accordingly, pathogen clearance during the disease course correlates with a decrease in the suppressive capacity of Tregs (123). Vice versa, states of chronic inflammation are often characterized in humans by resistance to immune regulation by Tregs (124–126).
In friend retrovirus (FV) (127–130) and herpes simplex virus (HSV) infection in mice (54, 131, 132), Tregs limit CD8+ effector T cell proliferation and functions resulting in viral persistence. Treg expansion is mainly based on excessive IL-2 production by murine, FV-infected effector T cells (90, 91, 133, 134). In HIV, human Tregs inhibit effector T cell responses thereby promoting viral chronicity and opportunistic infections acting as a viral reservoir (Figure 1) (100, 135, 136).
In human and murine mycobacterium tuberculosis (Mtb) infection, expansion of Mtb-specific Tregs interferes with priming and migration of T cells to the infected lung resulting in deficient clearance of Mtb (Figure 1) (137–140). Here, Treg numbers inversely correlate with local mycobacteria-specific immunity. Three human studies have shown an increase in T reg numbers in the blood and at sites of infection during active disease (140–142). In human hepatitis B virus (HBV), the expansion of antigen-specific Tregs suggests their contribution to the liver pathology (143–145). Here, the frequency of Tregs correlates with viral load.
In murine fungal infections such as Candida albicans, the absence of Toll-like receptors (TLRs) and Tregs lead to an increase in immunity to candida via secretion of anti-inflammatory cytokines and improved leukocyte recruitment to infection sites (146, 147).
In parasitic infections [e.g., schistosoma in mice (148, 149), leishmania in humans and mice (150–155), plasmodium, and helminths (156, 157)], Tregs are reported to favor parasite expansion and persistence by limiting effector responses, especially of Th1 and Th2 cells, in an IL-10-dependent manner and by suppression of antigen-specific T cell proliferation (36, 158). Nevertheless, while Treg frequency correlated with parasite pathogen load, it also accounted for reduced liver pathology and improved host survival rates. Also, in murine chronic infections of the protozoan parasite Toxoplasma gondii, a nonresolving Th1 myositis occurs where Treg ablation during chronic infection rescues macrophage homeostasis and skeletal muscle fiber regeneration (159).
2.1.3 Treg/Th17 Ratio in Pathogen-Mediated Diseases
In general, the balance between Th17 and Tregs is crucial for the maintenance of immune homeostasis during pathogen-mediated infections (160–162). By presentation of antigens via major histocompatibility complex II molecules on APCs and certain cytokine environments, naïve Th cells are activated and polarized into either peripherally-induced Tregs or Th17 cells to maintain homeostasis. Among APCs, macrophages are known to promote Treg responses, while DCs mainly activate Th17 cell responses (163). In mice, Th17 differentiation is mainly dependent on the cytokines IL-6 and TGFβ which induce the transcription factor retinoic acid-related orphan receptor gamma t (RORγt) in a STAT3-dependent manner (164, 165). In humans, Th17 differentiation mainly depends on IL-23 and IL-1ß (166–168). Th17 cell differentiation is further stimulated by TGFβ, TNF, IL-6, and IL-21. Maintenance and expansion of Th17 cells is regulated by IL-23 (168). Differentiation towards the Treg subset is stimulated via induction of the transcription factor STAT5 by TGFβ and in the absence of IL-6 (169–172). IL-2 and IL-10 also play important roles in the differentiation of Tregs (173). Th17 cells show pro-inflammatory effects during disease progression, which can result in autoimmunity. Tregs on the other hand, serve as immunoregulatory cells and maintain self-tolerance. These opposing effects are also represented on cytokine level: While Th17 cells secrete mainly IL17, IL-21 and IL-22, Tregs produce IL-10, IL-35 and TGFβ (160). Via IL-17 Th17 cells attract other immune cells, such as macrophages and neutrophils resulting in a state of chronic inflammation. In contrast, Tregs regulate differentiation and activity of Th17 and other T cells (8, 174). The Treg/Th17 ratio can contribute to a wide range of immune responses ranging from predominantly regulatory to stimulatory function. Its balance is crucial for immune homeostasis.
In recently infected SARS-CoV-2 patients, the number of Th17 cells was significantly increased compared to healthy controls causing inflammatory responses due to the production of pro-inflammatory cytokines (175, 176). In contrast, Treg numbers were decreased and even further downregulated in the disease course (177–179). Interestingly, the Treg/Th17 cell ratio and expression levels of their related cytokines were significantly higher in deceased patients than during remission (175).
Likewise, in respiratory syncytial virus infection but also in pulmonary infections in general, Tregs and Th17 cells have opposing features determining clinical severity (136, 177, 180, 181): Tregs promote viral clearance by recruiting CD8+ cytotoxic T cells to the lungs and limiting inefficient or excessive T cell responses (Th2, CD4+ and CD8+) (182–186). In addition, they control the innate immune response by neutrophils and NK cells. In contrast, Th17 cells hamper viral clearance by limiting CD8+ T cell responses and enhance Th2 immune responses resulting in a more severe clinical picture (187, 188).
In peripheral blood analysis of HBV patients, the Treg/Th17 ratio was decreased with reduced Treg levels and increased Th17 cell numbers (189). The latter correlated with TGFβ and IL-21 levels. Interestingly, here, the Treg/Th17 ratio was the best marker for predicting the stage of HBV-associated liver cirrhosis (190). In HCV patients, Th17 cells were associated with early infection and repair processes leading to liver fibrosis. Here, TGFβ and IL-10 suppressed Th17 cells (191).
For Mtb, the balance between Tregs and Th17 cells regulates encapsulation and control of lung lesions (192). If Tregs become predominant over Th17, Mtb disseminates more easily and recruitment of neutrophils to the infection sites gets delayed (192).
In gastrointestinal infectious diseases, Th17 are predominant in the acute phase producing the cytokines IL-17A, IL-17F and IL-22. In contrast, Treg/Th17 ratio increases in the chronic phase and infection progress because of the suppressive function of Tregs (193).
2.1.4 Impact of Pathogens on Tregs
Several pathogens impact the immune status exploiting the regulatory T cell compartment to enhance their replication and become persistent (95, 194–196). While tTregs are usually specific for self-antigens requiring continuous antigen stimulation for their survival (8–11), pTregs are converted in the periphery and therefore more likely to be influenced by and specific for a microorganism-derived antigen (13, 14). Induction of Treg proliferation and enhancement of Treg function might be central to immune evasion mechanisms of intracellular pathogens (123, 197). In accordance, Treg expansion is compartmentalized to tissues with high viral replication (here Treg frequency often correlates with viral load) and prolonged in chronic infections (19, 25, 90). However, the molecular mechanisms by which intracellular pathogens alter Treg functions as well as the origin of these Tregs remain incompletely understood (for detailed overview see Table 1).
Some pathogens directly contribute to Treg induction. In humans, for example, hepatocytes infected with HCV or gastric epithelial cells infected with H. pylori induce Tregs via TGFβ production (Table 1) (17, 34). Upon infection with plasmodium falciparum there is a burst of IL-2, IL-10 and TGFβ associated with Treg induction and expansion. Here, Tregs were, among others, induced by TLR9 signaling in mice and expressed high levels of Foxp3 suppressing inflammatory processes and immunity-driving mediators of effector T cells (36–39).
Likewise, viral pathogens such as HSV-1, FV and Japanese encephalitis virus enhance Treg expansion (Table 1). In murine HSV-1, the viral binding site herpes virus entry mediator is upregulated (18). For FV infection, there are two possible mechanisms underlying Treg expansion: IL-2-dependent stimulation versus IL-2-independent, tumor necrosis factor (TNF) receptor 2-dependent upregulation (Table 1) (25–27). In both, Japanese encephalitis virus and Mtb, Treg expansion is a result of programmed death-1 ligand 1 (PD-L1) induction (31–33).
Further, pathogens can enhance Treg functions as shown in HIV infection. Here, upon binding of HIV glycoprotein 120 to the CD4 receptor, Tregs express higher levels of effector molecules such as CTLA-4, CD39 and cAMP and show increased suppressive capacity next to prolonged survival rates (Table 1) (19–23). Of note, expression of Foxp3 by patients with a progressive HIV-1 infection seems to be upregulated by individual T cells due to antigen stimulation. Moreover, Foxp3 expression in CD4+ T cells was shown to be a marker of HIV infection and potentially even a prognostic marker of HIV progression (24). Also, in the context of COVID-19 disease, an alteration in the expression of Foxp3 could be detected. More precisely, in patients with a severe disease course, a downregulation of Foxp3 could be detected in T cells indicating an impaired Foxp3-mediated feedback on T cell activation as potential mechanisms underlying disease progression (198). Similarly, human T cell lymphotropic virus 1 associated gene products are reported to inhibit Foxp3 gene expression thereby causing Treg dysfunction (Table 1) (28–30).
Interestingly, women infected with Chlamydia trachomatis displayed increased expression levels of Foxp3 in vaginal swab samples following the clearance of infection due to antibiotic treatment (199). In Candida albicans, prolonged Treg survival rates were achieved by TLR2 signaling and IL-10 production (Table 1) (43). Altered Foxp3 expression could also be detected in the context of parasitic infections: During chronic infection with Leishmania of the Viannia subgenus, a decreased Foxp3 expression was detected (200). Recruitment of Tregs to infection sites of Leishmania major was improved by expression of integrin αEβ7 and CC-chemokine receptor 5 (40, 41). Excitingly, infection with helminth parasites mediated by parasite-secreted proteins could also induce de novo T cell Foxp3 expression (201). This, in turn, may be a way through which parasites can evade the host immune response. Regarding H. pylori infection, it is worth noting that increased expression of Foxp3 on Tregs was observed in Tregs isolated from infected children, possibly contributing to an inverse association between H. pylori infection and allergic disease through changes in Treg functionality (35).
2.2 Role of Tregs in Multiple Sclerosis
Immune homeostasis and self-tolerance are regulated by the development, stability and function of Tregs (202). Tregs control immune capacity thereby influencing bystander immune responses such as allergies or autoimmune diseases (11, 14). Tregs prevent the activation and infiltration of T cells into the central nervous system (CNS) and maintain the homeostasis of the immune system (203–205). By suppressing CD8+ T effector cell responses, they limit parenchymal damage during CNS inflammation (206).
Tregs can contribute to the pathogenesis of autoimmune diseases by a multi-layered feed-forward loop (Figure 1) (84): Autoantigens and pro-inflammatory cytokines (IL-1β, IL-6 etc.) activate effector Th cells which further aggravate self-tissue damage by the expression of IL-4, IL-6, IL-10, IL-12 and IFNγ (207). Antigens and cytokines from damaged tissue promote the generation of ‘exTreg’ cells adapting Th-like functions which, in turn, stimulate the activation and expansion of autoreactive Th effector cells. These effector Th cell-like functions of ‘exTreg’ cells also directly stimulate pathogenic immune responses in local tissues and promote the pathogenesis of autoimmune diseases by participating in a feed-forward loop (Figure 1). Strikingly, dysfunctional, instable or insufficient Foxp3 expression can also trigger autoimmunity (15, 208–212): Upon loss of Foxp3, Tregs lose their immunosuppressive capacity adapting phenotype and functionality of Th cells (Th1, Th2, Th17), such as production of IFNγ and IL-17 (Figure 1) (213–215).
In contrast, CD4+ HLA-G+ tTregs cells were shown to ameliorate polyclonal adaptive immune response suppressing graft-versus-host disease in vivo (216). Likewise, Foxp3+ Tregs limit muscle destruction by cytotoxic T cells in dermatomyositis, polymyositis and inclusion body myositis (217).
Treg/Th17 imbalance is associated with autoimmune diseases such as MS, myasthenia gravis, psoriasis, inflammatory bowel diseases and rheumatoid arthritis (165, 218–223). Here, Th17 cells are regarded as the main driver of autoimmune inflammation activating other immune cells and secreting pro-inflammatory cytokines (224, 225). A decrease of Tregs in autoimmune and inflammatory diseases is reported to cause disease progression (226). Therapeutic approaches targeting the described Treg/Th17 axis are promising (227) and mainly aim at neutralizing Th17-secreted cytokines, reducing Th17 cell counts, increasing Treg cell levels and regulating transcription factors such as RORγt, STAT3 and Foxp3 (228–231).
2.2.1 Qualitative Treg Alterations in MS
In MS pathogenesis, T cells acquire an autoreactive phenotype against CNS autoantigens followed by migration into the CNS causing inflammatory lesions. Activation of T cells is induced by molecular mimicry in the periphery or by autoreactive T cells in the CNS (232). Control mechanisms that should prevent autoimmunity, such as selection processes during tTreg development or peripheral suppression by Tregs, are often circumvented by autoreactive T cells (233, 234).
Tregs acquire a phenotype and expression profile resembling Th1 cells, thereby contributing to disease progression (235): Lower expression levels of Foxp3, TGF, CTLA-4 and CD39 were accompanied by an increase in IFN secretion in relapsing-remitting MS (RRMS) patients (236–238). Myelin-reactive T cells secreted high levels of IL-17, IFNγ and granulocyte-macrophage colony-stimulating factor compared to healthy controls (239). Next to an upregulation of markers associated with Th1 identity, Tregs expressed higher levels of the migration markers CD103 and CD49d enhancing transmigration of ‘exTregs’ into the CNS (238). High IL-17 levels have also been detected in the cerebrospinal fluid (CSF) of MS patients during relapse as well as in chronic lesions (222, 240), suggesting that both, the upregulation of IL-17 and down-regulation of Treg-mediated immunity, contribute to MS pathogenesis (241, 242).
Likewise, in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, an altered phenotype and impaired suppressive capacity of Tregs have been associated with clinical deterioration (235, 243, 244). Transfer of Th1-like ‘exTregs’ even lead to induction of EAE in naïve recipient mice. Interestingly, Othy et al. (245) showed that Tregs can suppress Th17 cells by inhibition of intracellular Ca2+ signaling and their contact to APCs.
Therefore, therapeutic induction of Tregs as well as modulation of Treg/Th17-related pathways could attenuate the inflammatory immune response resulting in mitigation of disease symptoms (246–250). Interestingly, Haas et al. showed that the immunosuppressive effect of Tregs after alemtuzumab treatment of MS patients was mainly due to an altered composition and reactivity of conventional CD4+ cells after immunodepletion (251).
2.2.2 Quantitative Treg Alterations in MS
Treg frequency and the Treg/Th17 ratio were negatively correlated with disease severity in MS patients (252, 253). In RRMS patients, reduced Treg numbers were observed. In EAE, Treg plasticity was studied in detail showing an increase of ‘exTreg’ counts during the preclinical phase until disease maximum (212, 254). In line, remission is linked to an increase in Treg numbers representing a recovery of Treg identity (254). Interestingly, clinically isolated syndrome often preceding the diagnosis of MS was associated with alterations of the Treg compartment in peripheral blood (255) Here, patients displayed lower levels of immunosuppressive CD45RA+ Foxp3low Tregs while levels of non-immunosuppressive CD45RA- Foxp3low Tregs and Th17-like Tregs increased. These observations suggest that progression to MS might be preceded by changes to the Treg compartment. A recent study investigating the mechanisms driving Treg dysfunction reported an inhibitory effect of circulating exosomes from MS on Tregs (256). This effect is thought to be mediated by let-7i miRNA interacting with insulin like growth factor 1 receptor and TGFβ receptor 1 expressed by CD4+ T cells. Thus, miRNA profiles from MS patients might directly inhibit Treg expansion (256).
2.3 The Interplay of Infections and Autoimmunity - Translation in the Setting of Multiple Sclerosis
It is commonly accepted that the interaction of genetic susceptibility and the exposure to certain environmental factors is crucial for the occurrence of autoimmunity. A major environmental factor contributing to the pathogenesis of autoimmune diseases and, more specifically, autoimmune neuroinflammation is pathogen-mediated infection. One of the underlying mechanisms is so-called molecular mimicry. Here, due to structural similarity of pathogen-derived peptides with host molecules, autoreactive B and T cells become cross-activated leading to an immune response directed against self-antigens (257). Likewise, epitope spreading is involved in the interplay of autoimmunity and infections. In this context, a new infection in an ongoing autoimmune disease leads to tissue damage with exposure of further self-antigens (258). APC-mediated presentation of these antigens to autoreactive lymphocytes then accelerates inflammatory processes (258). Furthermore, infections can facilitate inflammatory processes through bystander activation leading to a general immune response with activation of immune cells such as NK cells or macrophages and thus release of pro-inflammatory cytokines (259). This inflammatory milieu induces an antigen-independent activation of primed B and T lymphocytes at the inflammation site and thereby enhances autoimmune damage. Lastly, pathogen-mediated amplification of autoimmune events involves bacterial or viral superantigens leading to an extremely potent activation of polyclonal autoreactive T cells by binding to major histocompatibility complex II (258). These superantigens lead to a massive proliferation of T lymphocytes with excessive cytokine production, especially of IL-2 and IFNγ, resulting in an exacerbation of autoimmune processes (258).
The Treg compartment is needed to control immunopathology throughout life. However, while Tregs are indispensable for immune regulation, exuberant Treg function might prove detrimental for host defense. For example, in Mtb as a model for chronic bacterial infection, Tregs can delay leukocyte migration from lymph nodes to sites of ongoing infection (107, 260). In line, Treg ablation reduces accumulation of Mtb in lungs of infected mice (139). These observations underpin the potential of Tregs to exert detrimental effects in immune-mediated diseases. The continuum of dysfunctional Treg action is shared between infectious conditions, characterized by exuberant immunosuppression, and autoimmune conditions, characterized by promotion of immunogenicity (84). Common to Treg dysfunction is the instability of Foxp3 (213). Foxp3 is pivotal for Treg homeostasis. However, lineage tracing studies revealed that Foxp3 is frequently lost under autoimmune conditions (213). Loss of Foxp3 leads to generation of the so called ‘exTreg’ phenotype characterized by functions shared with effector Th cells, such as secretion of pro-inflammatory cytokines e.g. IFN-γ and IL-17 (84). Moreover, continuous IL-2 signaling is needed to prevent loss of Foxp3 (261). Intriguingly, inflammatory conditions promote loss of Foxp3 and, therefore, contribute to maintaining autoimmune states (212). Inflamed tissue constitutes a complex micro-environment characterized by immune cell infiltration, pro-inflammatory cytokine secretion and increased (self-)antigen presentation. Treg instability might therefore contribute to sustaining inflammatory conditions whereas inflammation promotes loss of Foxp3 and generation of Tregs more closely resembling effector Th cells further contributing to pro-inflammatory stimuli in a feed-forward loop (84). Intriguingly, Tregs from patients who resolved an HCV infection reacted to a virus-encoded peptide with substantial human homology while Tregs from non-infected patients did not (260). Taken together, the pathogenic potential and lineage instability of Tregs make them suspects for mediating autoimmunity following chronic infections.
One of the best studied pathogens involved in MS pathophysiology is Epstein-Barr virus (EBV). Based on epidemiological similarities, an association of EBV infection and MS was suspected early on (262). Further research subsequently not only proved that virtually all MS patients exhibit an EBV infection (263), but also that prior infectious mononucleosis is associated with a 2-3 times higher risk of developing MS (264). Conversely, this risk is significantly reduced for individuals with a negative EBV serology (265). Particularly interesting in this context are data showing that initially seronegative patients experience seroconversion shortly before the onset of MS symptoms (266). Even in pre-symptomatic patients with EBV, a significant increase in anti-EBV antibodies was found over five years before disease onset suggesting involvement of EBV in early disease stages (267). Interestingly, since an association between EBV serology and early conversion of clinically isolated syndrome into clinically definite MS has been demonstrated (268), EBV serology also appears to correlate with disease activity. Consistently, a correlation between anti-EBV nuclear antigen 1 (EBNA-1) titers, disease progression, lesion load, brain atrophy, and the extent of demyelination in MS patients has been demonstrated (269–271). Further support for an involvement of EBV in MS pathophysiology derives from histological studies revealing an accumulation of EBV-infected B- and plasma cells in MS brain meninges, in cortical as well as in white matter lesions (272–274).
Despite this overwhelming evidence, the molecular mechanisms underlying the role of EBV in the immunopathophysiology of MS are still not properly understood. However, a number of hypotheses exist involving discussion of both modulation of peripheral and CNS-localized immune responses. In the periphery, EBV might lead to a cross-activation of pathogenic T cells via molecular mimicry as described above (275). This theory is supported by the fact that EBNA-1-specific T cells react to myelin antigens more frequently than to other auto-antigens causing a release of pro-inflammatory IFNγ (276). Furthermore, EBV-reactive T cells were isolated from the CSF of MS patients also recognizing myelin basic protein (MBP) (277). In addition, a very recent study found cross-reactivity between EBNA-1 and the recently identified MS autoantigen called anoctamin 2 further supporting EBV-induced molecular mimicry (278). Another hypothesis proposes that EBV infection of peripheral B cells induces the expression of aB-crystallin. As it is also expressed in oligodendrocytes, an aB-crystallin-directed T cell response might ultimately lead to demyelination (279). Furthermore, there is evidence for EBV infection of B cells leading to a release of predominantly pro-inflammatory cytokines such as IL-6 or TNFα and simultaneously impeding immunoregulatory processes by reducing IL-10 levels (280, 281). Further evidence derives from recent data providing another pathophysiological link between EBV infections and MS. Wang et al. reported an autoreactive CD4+ T cell clone showing cross-reactivity between HLA-DR-derived self-proteins, EBV antigens, as well as autoantigens presented by HLA-DR allomorphs DR2a and DR2b (282). Thus, EBV antigens could be actively involved in the activity of autoreactive CD4+ T cells. Since HLA-DR15 is one of the genetic factors most strongly associated with MS, this link highlights the relevance of EBV infection in the pathogenesis of MS (282). A theory with regard to the modulation of peripheral immune processes describes that the invasion of autoreactive T- and EBV-infected B cells into the CNS is forced by expression induction of EBV-induced G protein-coupled receptor 2 thereby fostering the neuroinflammatory response (Figure 2) (275, 283–285).
In the CNS, the accumulation of infected B cells within the meninges and perivascular cuffs suggests that these B cells may elicit a CD8+ T cell response, leading to a multiplication of the inflammatory response via bystander activation (272, 286). Expression of superantigens by EBV-infected B cells could further lead to an excessive T cell response (287). Finally, it is hypothesized that EBV-induced immortalization of infected B cells and exhaustion-induced defective elimination lead to an accumulation of EBV-infected autoreactive B cells causing a permanent exposure to CNS antigens (Figure 2) (288–290). This exposure might considerably aggravate CNS damage in the context of autoimmune neuronal inflammation by antigen expression, autoantibody production, as well as by providing survival signals to autoreactive T cells.
Besides EBV, human endogenous retroviruses (HERVs) seem to be significantly involved in MS pathophysiology. These proviruses which account for circa 8% of the genome originate from exogenous infection of primate germ line cells millions of years ago and are today part of the human DNA (291). While they are functionally inactive under physiological conditions, pathological triggers such as viral infections can induce reactivation and thus production of viral proteins (292). First evidence of an involvement in MS dates back more than 30 years when retrovirus transcription was found in the supernatants of meningeal cell cultures of MS patients (293). Different HERVs such as HERV-H, HERV-K and HERV-W were subsequently associated with MS (294, 295). In addition to an increased HERV-W expression in MS patients (296), observations of higher antibody reactivity to certain HERV-W sequences in MS patients (297) and HERV-W upregulation in MS plaques correlating with disease activity support the involvement of HERVs in MS (298). There is also clinical evidence of a relationship of HERVs and MS since patients expressing high levels of HERV-W show a poorer prognosis in early disease stages and increased disease progression (299). Accordingly, HERV-W load also correlates with disease activity and the occurrence of relapses (300). Once more, the possible underlying mechanisms are diverse. For example, reactivation of HERV-W proteins leads to an activation of both innate and adaptive immune responses in MS (301). Thus, dysregulated expression of HERVs may contribute to CNS damage such as observed in a severe combined immunodeficiency mouse model (302). This dysregulated HERV-W activity is likely to involve binding of its envelope protein (ENV) to TLR4 and its co-receptor CD14 which triggers the release of pro-inflammatory cytokines such as TNFα, IL-1β or IL-6 fostering the autoimmune response (303–305). Furthermore, HERV-W-derived proteins such as ENV show cross-reactivity with myelin antigens amplifying the neuroinflammatory response (306). Aside from immune-mediated mechanisms, HERV-W ENV also interferes with remyelination via the inhibition of oligodendrocyte precursor cell (OPC) differentiation (307). Moreover, it also induces a pro-inflammatory activation of myeloid cells which, in turn, contributes to axonal damage and thus neurodegeneration even in long-standing MS cases (308). As there is a monoclonal antibody available that neutralizes HERV-W-induced detrimental effects, endogenous retroviruses constitute an attractive target for future MS therapies (Figure 2) (47, 309).
There is an exciting connection between EBV and HERVs. Exposure of EBV-derived glycoprotein 350 to B cells, monocytes, macrophages, as well as astrocytes leads to a significant increase in the expression of HERV-W and syncytin-1 and is thus also associated with unfavorable processes (310). Similar to EBV, human herpes virus 6 (HHV-6) infection can trigger the expression of HERV-W as well as a HERV-related superantigen (Figure 2) (311, 312). HHV-6 is a neurotropic virus that is divided into two subtypes, of which subtype A can be found in oligodendrocytes of MS white matter lesions (313). In addition to the expression of HHV-6 antigens in MS plaques (314), further evidence for an involvement of HHV-6 in MS pathogenesis derives from elevated anti-HHV-6 antibodies in the CSF of MS patients, especially in patients with an exacerbated disease indicating HHV-6 as a trigger for disease aggravation (315–317). Interestingly, in a non-human primate MS-like animal model, prior infection with HHV-6 resulted in a worse outcome further supporting a detrimental impact of HHV-6 on MS (318). One of the HHV-6-mediated mechanisms contributing to MS pathophysiology involves molecular mimicry since cross-reactivity between HHV-6 and MBP was shown to induce cytotoxic T cell-mediated oligodendrocyte death (319). This idea is further supported by a close sequence homology between MBP and the HHV-6-derived U24 protein (320). Furthermore, it is suggested that HHV-6 binding to the CD46 receptor leads to a T cell-mediated autoimmune reaction (321). Also, increased IL-23 release by DCs and IL-17 production by T cells with a concomitant decreased secretion of the immunoregulatory IL-10 provide potential mechanisms of how also HHV-6 might exacerbate neuroinflammatory processes (Figure 2) (321).
Apart from viral infections, there is also evidence for the involvement of bacterial pathogens in MS pathophysiology. A large meta-analysis, for instance, has shown that MS patients have a significantly higher incidence of Chlamydia pneumoniae (C. pneumoniae) DNA and intrathecally synthesized immunoglobulins in their CSF compared to patients with other neurological diseases (322). In EAE, systemic infection of mice with C. pneumoniae led to dissemination of the pathogen into the CNS accompanied by an aggravation of autoimmune neuroinflammation through reduced Th1 cell proliferation as well as IFNγ production (Figure 2) (323). Nevertheless, the available data is still unclear and controversially discussed.
Whereas the pathogens mentioned so far all have a negative impact on the processes in MS, this is different for H. pylori. In MS cohorts for example, a reduced prevalence of the pathogen compared to controls was demonstrated (324). Even more, MS patients being seropositive for H. pylori showed reduced disability scores (325). In EAE, infection with H. pylori resulted in reduced disease progression, milder proliferation of autoreactive cells, and lower infiltration of pro-inflammatory effector Th1 and Th17 cells into the CNS (Figure 2) (326). A protective role of H. pylori is also assumed in other autoimmune diseases such as asthma (327) or inflammatory bowel disease (328).
Likewise, strong evidence for a protective role in MS disease development has been demonstrated for some parasites. First indications derive from epidemiological investigations, which showed an inverse relationship between parasites like Trichuris trichiura and the occurrence of MS (329). In fact, the prevalence of MS seemed to decrease when the contamination level exceeded 10% (329). Of note, parasite-infected MS patients showed a significantly decreased number of relapses, a minor decline in disability scores, and reduced magnetic resonance imaging (MRI) disease activity compared to patients without helminthic infection (144). Parasitic infections exert an anti-inflammatory effects both on the parasite-specific response and the inflammatory response directed against other antigens in the sense of bystander suppression (330). In mice, helminth infection significantly attenuated both the incidence and clinical symptoms of EAE (331, 332). This amelioration was accompanied by a decrease in pro-inflammatory IFNγ, TNFα, IL-17, and IL-12 with a simultaneous increase in the release of immunoregulatory IL-10 and TGFβ (330–332). Also in humans, helminth infection was associated with induction of CD4+CD25+Foxp3+ T cells suppressing the inflammatory response (333). Beyond Tregs, regulatory B cells secreting IL-10 were detected in greater numbers in helminth-infected individuals suffering from MS (334). Further, the MBP-specific immune response was characterized by a decreased release of pro- next to an enhanced release of anti-inflammatory cytokines in patients with a parasitic infection (Figure 2) (333). Interestingly, the protective effects of helminths infection were shown to be reversed following an anthelmintic treatment concerning both the clinical as well as radiological MS activity and the immunosuppressive effects in terms of the Treg activity (335, 336).
In summary, for many pathogens there is versatile evidence for modulation of autoimmune processes in the context of neuroinflammation in MS. Nevertheless, it has not yet been possible to conclusively define the underlying molecular mechanisms.
2.3.1 Therapeutic Targets
The above findings on detrimental but also beneficial effects of pathogenic infections have led to therapeutic approaches - in some cases despite continuing doubts about the mechanistic background.
In the case of EBV, attempts have been made to prevent an acute EBV infection by prophylactic vaccination thereby reducing the risk for development of MS (274). However, there is currently no appropriate vaccination available. In general, antibodies directed against certain EBV proteins expressed during latency to increase anti-EBV immunity would be a promising strategy. Once again, however, no study results are available to date (274). In contrast, cell-based immunotherapies appear to be a more promising approach. In particular, the application of autologous or allogenic T cells targeting EBV-infected B cells came into focus. A first successful application of this therapy was already demonstrated in a patient suffering from secondary progressive MS (337). Subsequently, a study on the effects of a EBV-specific autologous T cell therapy using in vitro expanded T lymphocytes interfering with EBNA-1 and latent membrane proteins 1 and 2A was initiated (Table 2) (45). Clinical improvement occurred in 7 out of 10 included MS patients. Of note, this was only a safety trial therefore lacking a placebo group. Further studies elucidating the impact of autologous or allogenic T cells attacking EBV-infected B cells are underway (Table 2).
HERV-targeted therapies, on the other hand, have reached a more advanced stage. As already pointed out, HERV and HERV-related proteins such as ENV exert an unfavorable effect on OPCs and myeloid cells and thus on remyelination and neurodegeneration (307–309). In view of the persistent lack of remyelinating therapies (338), it is particularly interesting that this inhibition can be reversed by the anti-HERV-W IgG4 monoclonal antibody GNbAC1 (temelimab) (309). Besides promotion of remyelination, GNbAC1 impedes the release of pro-inflammatory cytokines (339). Application of GNbAC1 led to favorable effects in numerous early-phase studies (46, 340–342). Given these pre-clinical results, two phase IIa and IIb trials were initiated (Table 2) (47). Therapy with 18 mg/kg resulted in a significant reduction of the number of T1-hypointense lesions after 48 hours. Moreover, there was a consistent trend of reduced brain atrophy and a magnetization transfer ratio decrease indicating a positive impact on remyelination. However, GNbAC1 failed to achieve the primary endpoint of the study, i.e. reduction of gadolinium-enhancing lesions (GELs), possibly because of underdosing. However, the beneficial MRI effects on neurodegeneration raised hope and led to the initiation of a new phase II study (Table 2) (47). Another HERV-related approach is based on the theory that antiretroviral therapies can also induce inhibition of HERVs in MS (343). In a baseline-versus-treatment phase IIb study, 20 patients with active RRMS were treated with the integrase inhibitor raltegravir for 3 months (Table 2) (48). However, the primary study endpoint reduction in lesion load or development of new lesions during the treatment period compared with baseline was not met.
Although to date the evidence regarding C. pneumoniae is quite sparse, therapy with rifampicin or azithromycin for 6 months were compared with placebo in newly diagnosed RRMS patients with evidence of C. pneumoniae infection in the CSF (Table 2) (44). The primary endpoint, reduction of GELs, was not reached. Only a decrease in brain atrophy was found under antibiotic therapy. However, given the very small number of subjects, these results should be interpreted with great caution.
Recently, there are also first therapeutic approaches that exploit the protective effects of helminth infection on MS. In a small phase I study, MRI activity in five treatment-naïve RRMS patients was compared between baseline and after probiotic treatment with 2500 Trichuris suis ova every two weeks for three months (Table 2) (49). Under treatment, there was a reduction of new GELs by 70% compared to baseline with a return to baseline after two months of follow-up. The reduction of lesions was also associated with increased serum levels of of IL-4 and IL-10 in 80% of the participants (336). The follow-up study including 16 RRMS patients also showed a trend of reduction of active MRI lesions compared to baseline (344). Furthermore, there was an increase of Tregs observed during this trial. A safety study evaluating the effect of orally administered 2500 Trichuris suis eggs in 10 MS patients could not observe Trichuris suis ova-induced effects on disease progression (Table 2) (51, 336). In line, in this study there were no significant alterations detected regarding cytokine expression and T cell-specific transcriptions factors (336). A 9-month double-blind, randomized, placebo-controlled study enrolling 71 RRMS patients investigated the effect of transcutaneous application of hookworm larvae on lesion burden (Table 2) (50). Of note, treatment with hookworm larvae increased the proportion of Tregs in the peripheral blood. Furthermore, the study showed a tendency of reduced new or enlarging lesions as well as an ameliorated MRI activity in the treatment group. However, these differences were not significant (50). Given these inconsistent results together with methodological limitations such as small sample sizes, further studies are required to sufficiently address the therapeutic potential of helminth infections in MS (336).
2.4 Microbiome - the Missing Link Between Biomolecular Treg Signatures and Clinical Phenotype?
In recent years, the important role of the gut microbiome has been recognized in autoimmune diseases and pathogen-induced immune responses (345, 346). The interplay of the gut microbiome and the immune system may explain its seemingly universal impact on a great variety of diseases including autoimmune diseases, cancer, vascular disease, and even psychiatric disorders (347–350).
Importantly, a variety of factors modulate the composition of the microbiome. Hence, the relationship between the host and the microbiome needs to be understood as a dynamic rather than static process (351). One of the most influential factors of the microbiome is the diet, which under unfavorable conditions induces dysregulation in the form of dysbiosis (351). This dysbiosis, in turn, contributes to an increased incidence of gut-distal autoimmune phenomena such as autoimmune arthritis (352) or type 1 diabetes (353) through alterations in Treg/Th17 balance. In general, the dynamic balance or dysbalance of Tregs and Th17 is suggested to be a main effector mechanism by which the gut microbiome influences systemic immunity (354). Furthermore, antibiotic therapy has an enormous impact on the microbiome and thus on the function of CD4+ T cells. For example, antibiotic therapy not only leads to an altered colonic but also tTreg TCR repertoire (355). Likewise, antibiotic-treated mice show a significant reduction of Tregs in the colonic lamina propria (356, 357). Similarly, also germ-free mice exhibited a reduced Treg population (356, 358). However, restoration of the microbiome caused a strong re-expansion of Tregs in these mice (358). Importantly, the effects of an altered microbiome and thus altered Treg/Th17 balance expanded from the local milieu into the whole body. Consistent with this, it has been shown that gut-resident T cells traffic between different organs thereby exerting a systemic effect on the immune system (359, 360). In addition, a reduction of CD4+ memory T cells expressing the gut-homing chemokine receptor CC-chemokine 9 was detected in secondary progressive MS patients (361). These gut tropic CC-chemokine 9+ memory T cells were shown to exhibit a regulatory phenotype potentially contributing to the conversion of RRMS to secondary progressive MS (361).
Importantly, with respect to microbiome-mediated effects on immunoregulatory mechanisms, tTregs and pTregs need to be distinguished. Unlike tTregs, pTregs exhibit a TCR repertoire directed against microbiota and dietary antigens (362–364). In addition to pTregs and tTregs, other Treg populations such as latency-associated peptide-expressing Tregs, type 1 Tregs, RORγt-expressing Tregs, or GATA binding protein 3+ Tregs have been shown to play an essential role in mediating intestinal and extra-intestinal immune homeostasis (365).
This knowledge on the interplay of microbiome, Tregs and autoimmunity provides the rationale to investigate possible microbiome-associated therapeutic strategies such as a modified diet or use of probiotics to change Treg plasticity and function in a favorable way to treat autoimmunity. Therefore, the identification of the involved pathogens and signaling pathways is essential. One of the underlying mechanisms involves intestinal epithelial cell- and microbiota-mediated modulation of DCs leading to an immunoregulatory phenotype through upregulation of IL-10, TGFβ and retinoic acid (365). The potent induction of colonic Tregs by Clostridium species has been well studied. Colonization of germ-free mice with a composition of 46 Clostridium strains resulted in a marked expansion of Tregs (356). Matrix metalloproteinases induced by Clostridium were involved in this expansion and promoted the differentiation and survival of Tregs via TGFβ. Furthermore, Clostridia are known to enhance levels of short-chain fatty acids (SCFAs) (366) which effectively stimulate Tregs. For example, in contrast to long-chain fatty acids that promote a Th1 as well as Th17 response, SCFA administration induces a reduction in the number of Th1 cells but an increased proliferation of gut Tregs (367, 368). Accordingly, application of SCFAs led to a significantly reduced disease progression in EAE and inflammation in ulcerative colitis (367, 369). Protective effects on type 1 diabetes in pre-diabetic mice as well as in a model of collagen-induced arthritis have also been described (368, 370). Interestingly, the SCFAs-induced effects appear to be mediated in part by epigenetic modifications via a suppression of histone deacetylases (371, 372). Despite these protective effects in several models of autoimmune diseases, SCFA application led to disease aggravation in an inflammatory model of rheumatoid arthritis (368). Therefore, a better understanding of the role of SCFAs and other microbiota metabolites is essential in order to exploit these findings therapeutically in clinical practice. In line with a significant impact of dysbiosis in the pathophysiology of MS, a Japanese study observed moderate dysbiosis in the microbiome of RRMS patients compared to healthy controls (373). Primarily, this involved a reduction of clostridial species as well as Bacteroidetes. Interestingly, however, the reduction did not affect the clostridial species, which is commonly known to have a protective effect on autoimmune processes by inducing colonic Tregs. This, in turn, underlines that deeper insights, especially into the influence of distinct species, are essential for translational approaches aiming to modulate the microbiome.
Additionally, several studies have demonstrated the relevance of Bacteroides fragilis, a human symbiont, for the function of Tregs (374, 375). By binding to TLR2, Bacteroides fragilis-derived capsular polysaccharide A as well as outer-membrane vesicles cause an immunosuppressive phenotype in terms of a significant expansion of IL-10-releasing intestinal Tregs (376, 377).
There is also evidence for a role of Akkermansia calcoaceticus and Akkermansia muciniphila in the context of autoimmune neuroinflammation. These bacteria induce impaired Treg conversion in peripheral blood mononuclear cells with concomitant stimulation of Th1 cell differentiation (378). Interestingly, in MS patients, a considerably increased occurrence of Acinetobacter and Akkermansia could be found, which, however, was decreased under MS therapy (351, 378, 379).
Furthermore, there is evidence of a DC-mediated Treg-promoting effect of Lactobacillus, Streptococcus, and Bifidobacterium (380, 381). Regarding potential therapeutic opportunities, application of probiotic mixtures consisting of these bacteria resulted in an expansion of Tregs (382). Therapy with Lactobacilli strains was also able to prevent disease progression in EAE by suppression of autoreactive T cells (383). In addition to these significant pre-clinical results derived from EAE, there is also human data underlining the impact as well as therapeutic potential of probiotic therapeutic approaches: In a small study including 9 MS patients and 13 healthy controls, probiotic therapy with Lactobacillus, Bifidobacterium and Streptococcus over two months led to a modulation of the microbiome as well as of the immune response (384). Thus, this therapy led to an enhancement of immunoregulatory processes with a reduction in the number of inflammatory monocytes as well as the expression of the costimulatory marker CD80 on monocytes. At the same time, probiotic therapy led to a decreased expression of the MS risk alleles HLA.DQA1 and HLA.DPB1 in control patients. However, no significant effects on peripheral Tregs were observed due to the intake of the probiotic treatment. On the microbiome level, several taxa known to be depleted in MS were increased whereas taxa associated with dysbiosis in MS were reduced (384).
Other interesting players influencing the activity and phenotype of bowel tropic Tregs are enteric neurons. Very recent data, for example, show that microbe-responsive Tregs in the colon lamina propria are localized in close proximity to nitrergic and peptidergic nerve fibers (385). Enteric neurons, in turn, prevent differentiation as well as the activity of Tregs in vitro by modulating IL-6. These processes, in turn, are decidedly influenced by microbial signals (385). Recent data describing a vago-vagal liver-brain-gut neuronal arc that induces proliferation and maintenance of pTregs further highlight the relevance of enteric neurons in the context of gut-tropic Tregs (386). Underlying mechanisms involve hepatic vagal sensory afferents that sense the gut microenvironment and on the other hand give input to vagal parasympathetic nerves as well as enteric neurons. Perturbation of the aforementioned afferent neurons led to a reduction in the frequency of pTregs. In vivo, this subsequently led to an increased symptomatology in animal models of colitis (386).
All these considerations have led to therapeutic approaches, such as antigen-based oral immunotherapies, which aim to achieve amelioration of autoimmune diseases via an expansion of Tregs induced by oral antigen administration (365). However, further studies on the critically responsible bacteria of the microbiome as well as the signaling pathways involved are required for a more widespread translational application.
2.5 Tregs in the Advent of the Era of Immunometabolomics
The large variety of pathogens, different transmission routes and complex underlying molecular mechanisms necessitate new comprehensive diagnostic approaches such as immunometabolomics. Several clinical studies addressing autoimmune inflammatory diseases like systemic lupus erythematosus, rheumatoid arthritis, Crohn’s disease and colitis ulcerosa, suggested that disease activity and outcome correlate with immunometabolic signatures (e.g. metabolite levels) (387–389). Moreover, Treg levels were found to be inversely correlated with body weight and body mass index (390). Altered Treg function is not restricted to adipose tissue but can be retrieved in flow cytometry analysis from circulating blood cells pointing to systemic effects (390). Thus, tissue-derived metabolic signals can exert systemic effects via local modulation of function and expansion of Tregs (391). This is particularly true for the adipose tissue. In the visceral adipose tissue of healthy mice, there is a significant enrichment of Tregs, whose deficiency leads to systemic consequences such as metabolic inflammation (60, 60, 392). In obese mice, for example, there is a reduction in the number of Tregs which, in turn, is associated with a decreased lipid content (60). Interestingly, at least in mice, there seems to be an inverse relationship between external lipid accumulation and intrinsic Treg lipid contents. In obese humans, on the other hand, a slight increase in visceral adipose tissue Tregs could be detected (393). Therefore, findings from murine experiments should not be carelessly extrapolated to humans, since underlying mechanisms may vary between species.
Next to the adipose tissue, liver-resident Tregs could also exert systemic effects on metabolic inflammation and systemic metabolism, depending on local metabolism and leading to the development of steatohepatitis or exacerbation of atherosclerosis upon deprivation (394, 395). Furthermore, hyperglycemia in diabetes poses a substantial risk for systemic infections disrupting lymphoid tissue integrity and affecting leukocyte development, phenotype and activity (396). Obesity and type 2 diabetes mellitus drive immune dysfunction, deteriorate infections, and increase sepsis mortality (397). Beyond the direct permissive effects of hyperglycemia on bacterial survival, our knowledge about molecular mechanism of immune deviation in obesity and diabetes remains fragmentary. T cells support coordinated immune responses with their metabolic flexibility (Figure 3) (398–401). Development, activation, function and maintenance of T cells are linked to their metabolic household and, depending on the phase, are associated with an altered metabolic state (399, 402–410).
Basal metabolism of Tregs is characterized by fatty acid oxidation (FAO), metabolism of acetyl CoA, and subsequently tricarboxylic acid cycle activity and oxidative phosphorylation (OXPHOS) (391). In contrast, during the induction of Tregs, there is a switch to a more glycolytic metabolism. This switch is characterized by a displacement of the glycolytic enzyme enolase-1 from the Foxp3 locus allowing the transcription of Foxp3 (411). Inhibition of glycolysis, e.g. using 2-deoxy-D-glucose, results in impeded Treg development due to lack of displacement of enolase-1 and ongoing Foxp3 inhibition (411). In contrast, multiple metabolic pathways are relevant for both homeostasis and proliferation of Tregs. Thus, Tregs show a constitutive activity of mechanistic target of rapamycin (mTOR), which, just like glucose transporter 1, showed an increased expression in proliferating Tregs (412). Interestingly, Procaccini et al. demonstrated that freshly extracted human Tregs show a very high level of glycolysis (413). Furthermore, Li et al. could show that both naturally occurring and tumor-derived Tregs cells exhibit significantly enhanced glucose metabolism as well as increased expression of glycolysis-associated genes further highlighting the role of glycolysis in this state (414). Besides glycolysis, oxidative metabolism has been shown to be critically involved in Treg homeostasis and expansion. Treg proliferation and survival, for instance, influence the function of liver kinase B 1, which, in turn, is an essential inducer of these pathways (415, 416). Further evidence for an involvement of mitochondrial metabolomic pathways derives from a study demonstrating an impaired Treg fitness due to an ablation of the mitochondrial complex III (417). Last, AMP activated protein kinase (AMPK)-dependent FAO is not only crucial for Treg generation, but also for their proliferation (Figure 3) (418–420). While the metabolism of Tregs during homeostasis and proliferation is characterized by a balance of glycolysis and OXPHOS, a predominance of OXPHOS seems to be essential for the suppressive activity of Tregs. Thus, Tregs deficient for proteins that are crucial for mitochondrial metabolism, show significantly reduced suppressive functions and impaired allograft survival (421). Conversely, increased expression of genes that are important to OXPHOS leads to improved Treg suppressive function (421). Further support for a critical role of OXPHOS in the function of Tregs derives from the finding that liver kinase B 1, an enzyme involved in OXPHOS, is essential for the suppressive capacity of Tregs (417). Consequently, deficiency of liver kinase B 1 in mice was associated with a fatal, early-onset autoimmune disease (415). Moreover, also mTOR appears to be involved in the immunoregulatory activity of Tregs via induction of OXPHOS (422). Thus, for example, the suppressive activity of Tregs is associated with increased mTOR signaling. Furthermore, inhibition of mTOR reduced Treg-mediated suppression of T cell proliferation, while deficiency of mTOR in mice triggered autoimmune phenomena (422). In addition, the adequate immunosuppressive function of Tregs may also be associated with both enhanced FAO and fatty acid synthesis (423, 424). For example, stimulation of Tregs leads to increased FAO (418). On the other hand, inhibition of the mevalonate pathway, i.e. fatty acid synthesis, causes a restriction of the suppressive capacity of Tregs (425, 426). In contrast to OXPHOS, FAO, and fatty acid synthesis, enhancement of glycolysis could possibly even induce an impairment of the suppressive capacity of Tregs. For example, Treg-specific deficiency of phosphatase and tensin homolog (PTEN) in mice leads to spontaneous lupus-like disease accompanied by a reduction in the suppressive function of Tregs (427). Of note, increased glycolytic activity was detected in these Tregs, which in turn resulted in insufficient resolution of inflammatory activity EAE and subsequently to augmented disease progression (427). Further evidence for glycolysis-associated insufficiency of immunosuppressive activities derives from Tregs with an enhanced expression of glucose transporter 1, which showed an increased extent of glycolysis but impaired suppression (412). In human Tregs, however, maintenance of glycolytic activity was required for optimal suppressive properties of Tregs further highlighting the need for differential evaluation of immunometabolic processes in human and murine Tregs (411, 428).
In contrast, during migration of Tregs, there appears to be a shift towards glycolytic pathways. For example, hypoxia-inducible factor 1 α (HIF-1α)-deficient Tregs, which exhibit low levels of both glycolysis and enhanced OXPHOS, show increased suppressive properties but impaired migration (429). In mice, this subsequently led to reduced tumor growth and increased survival, likely due to enhanced anti-tumor immunity (429). Consistent with a prominent role of glycolytic processes during migration, stimuli such as CD28 triggering migration of Tregs induced an increased uptake of glucose and markedly enhanced glycolysis (430). Underlying mechanisms involve a PI3K-mTORC2-mediated pathway leading to an enhanced expression of glucokinase. In turn, Tregs deficient for this pathway showed diminished migration but preserved suppressive function. Consistently, Tregs from individuals with a mutation leading to enhanced glucokinase activity showed enhanced migration (430). In summary, a complex interplay and balance of different metabolic pathways underlie the different functional stages of Tregs activity.
Treg dysfunctionality is suggested to be the link between dysmetabolism or dysimmune reactions and pathogens. In fact, hyperglycemia suppressed HIF-1α effects on Tregs in aspergillus fumigatus infection (431). Together with defective NLRP3/IL-1β signal pathway, this led to a more widespread disbalance of Th1/Th2 ratio and Th17 cells (431). The HIF-1α-dependent glycolytic pathway stimulates Th17 differentiation and limits Treg development by promoting the function of RORγt (432, 433).
Targeting immunometabolism as an anti-inflammatory strategy has become an established method in clinical therapies (250, 434, 435). Recently, Palma et al. (436) reported on caloric restriction in mice promoting elimination of Mtb. By means of a multi-omic approach they defined changes in glycolysis next to reduction in fatty acid metabolism and mTOR activity as crucial effects of metabolic reprogramming in this condition (436). In addition to anti-inflammatory therapy, these findings on the metabolic properties of Tregs could also be used in tumor therapy to improve anti-tumor immunity via metabolic interventions. Thus, several aspects of the tumor microenvironment offer an advantage to Tregs, especially in comparison to Th1 or Th17 cells. For example, despite hypoxia, glucose shortness and acidosis, Tregs can compete with other T cells by increased expression of glucose transporters, enhanced glycolytic metabolism, and resistance to lactate-induced acidosis (391).
In the future, omic approaches might enable us to appreciate the complex interplay of immunorelevant pathological conditions and respective Treg functions. To this end, we hypothesize that the metabolic state is capable of reprograming immune responses, with Tregs in a key position, and Treg/Th17 ratio as a valuable judge for T cell function from blood-derived T cell analysis (437). Netea et al. (147) described the chance of so-called ‘innate immune training’: Metabolic shifts can cause epigenetic reprogramming in activated immune cells resulting in an innate immune memory resembling the adaptive immune system.
Strikingly, Arpaia et al. (438) showed that several metabolites produced by commensal bacteria, such as butyrate, short fatty acid and propionate (PA), represent a missing link between microbiota and the immune system promoting generation of pTregs. Interestingly, PA in particular led to a substantial increase in differentiation and proliferation of murine and human Tregs as well as a reduction in the differentiation of Th17 cells (367). Consistently, PA-treated mice showed an ameliorated EAE disease course accompanied by an increase in Treg frequency and IL-10 levels compared to controls. Of note, even application of Tregs derived from mice pre-treated with PA attenuated the clinical symptoms of EAE (367). In addition to these findings obtained from EAE, clinical data from MS patients also pointed in this direction. Not only decreased PA serum and feces levels were found in all disease entities of MS, especially after the first relapse (439). Rather, the add-on administration of PA in MS patients led to a reversal of the Treg/Th17 imbalance, i.e. to an induction of Tregs and a reduction of pro-inflammatory Th1 and Th17 cells. Furthermore, these patients even showed a lower annual relapse rate, a reduced disability progression, and a minor brain atrophy. In line, the microbiome of these patients also showed an upregulation of Treg-inducing genes following PA treatment. These findings indicate a large potential benefit of PA therapy (439).
3 Discussion
In conclusion, microorganisms and immune responses to pathogens are closely interrelated: Programming of the immune system determines the effectiveness of pathogen elimination or their persistence, respectively. Vice versa, pathogens can affect immune responses. Specifically, the balance between T cell subpopulations, in particular the Treg/Th17 ratio, influences the defense against pathogens. In this context, Tregs exhibit a Janus-faced nature: Pronounced Treg activation correlates with a restricted and tissue-protective immune response but can result in pathogen persistence. In contrast, impaired Treg activity, together with Th1 and Th17 responses, can effectively eliminate pathogens but can also result in substantial collateral tissue damage. Consistent with this, mechanisms potentially reprogramming and modulating Treg responses, and their pathogenic conversion are currently intensely investigated. The pathogen itself can impact frequency and function of Tregs by a variety of mechanisms. It can alter immune responses to escape immune activation or even infiltrate immune cells using them as Trojan horses to infect the environment. Metabolic conditions like hyperglycemia or diabetes correlating with high fat diet in animal experiments can reprogram those immune responses by a variety of complex mechanisms not yet fully understood and in urgent need for further investigation. In this context, certain commensal microbiota alter the immune balance, especially the Treg/Th17 ratio.
There is substantial evidence that the interplay of pathogenic infections and immune processes are crucial in the pathogenesis of autoimmune disorders. Several pathogens were shown to be critically involved in MS immunopathophysiology by modulating the immune response. The best evidence is available for host-detrimental effects of EBV and HERVs which seem to fuel neuroinflammation. These effects are induced by different mechanisms such as bystander activation, molecular mimicry, superantigen-induced promotion of the T cell response, and epitope spreading. However, apart from these deleterious effects, protective effects have been demonstrated for numerous parasites, especially helminths, and also H. pylori. Such insights into the interplay of infection and MS have subsequently led to numerous translational approaches aimed at inhibiting deleterious and promoting favorable interactions. In addition to cell-based immunotherapies targeting EBV-infected B cells, the application of the monoclonal antibody GNbAC1 inhibiting HERV-W ENV seems particularly promising. Interestingly, GNbAC1 not only leads to a suppression of pro-inflammatory signaling pathways but also to possible critically desired neuroregenerative effects. The underlying mechanisms probably include improved OPC differentiation and a neutralization of pro-inflammatory myeloid cells. Accordingly, results of the ongoing phase IIb ProTect-MS study are awaited with great interest.
Further therapeutic opportunities might be provided by a closer understanding of the interaction of the intestinal microbiome with Tregs thereby influencing the Treg/Th17 balance. Dysregulation of this balance or of the microbiome in the sense of dysbiosis promotes the occurrence of autoimmune diseases. Furthermore, the composition of the microbiome seems to have enormous effects on the phenotype of Tregs. However, these effects are not only local, but lead to a systemic modulation of the immune response with both potentially unfavorable and favorable consequences. Nevertheless, deeper insights into this interplay are necessary to develop therapeutic strategies to take advantage of this knowledge. Possible candidates for this are Clostridium species or Bacteroides fragilis, which provide favorable immune modulatory properties with regard to autoimmunity by promoting regulatory capacities of Tregs.
Impacting both innate and adaptive immune responses, future research should identify the most relevant pathogen species as well as define molecular mechanisms and substrates underlying reprogramming of the immune balance. Such new insights could be used to therapeutically induce or modify Tregs to treat autoimmune diseases and immune responses in general. Hence, multi-omic approaches may unravel the interplay between microorganism-modulated Treg function and autoimmune neuroinflammation. Understanding the underlying molecular mechanisms could further contribute to the development of targeted therapies.
Author Contributions
CS and TR conceptualized the outline of the manuscript. CS and NH wrote the original draft of the manuscript. CS and NH were responsible for graphical illustrations. SB, CN, DK, KP, SM, and TR reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Thornton AM, Lu J, Korty PE, Kim YC, Martens C, Sun PD, et al. Helios+ and Helios- Treg Subpopulations Are Phenotypically and Functionally Distinct and Express Dissimilar TCR Repertoires. Eur J Immunol (2019) 49:398–412. doi: 10.1002/eji.201847935
2. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T Cells: Recommendations to Simplify the Nomenclature. Nat Immunol (2013) 14:307–8. doi: 10.1038/ni.2554
3. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-Dependent Programme of Regulatory T-Cell Differentiation. Nature (2007) 445:771–5. doi: 10.1038/nature05543
4. Rudensky AY. Regulatory T Cells and Foxp3. Immunol Rev (2011) 241:260–8. doi: 10.1111/j.1600-065X.2011.01018.x
5. Tone M, Tone Y, Adams E, Yates SF, Frewin MR, Cobbold SP, et al. Mouse Glucocorticoid-Induced Tumor Necrosis Factor Receptor Ligand Is Costimulatory for T Cells. Proc Natl Acad Sci USA (2003) 100:15059–64. doi: 10.1073/pnas.2334901100
6. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic Self-Tolerance Maintained by CD25(+)CD4(+) Regulatory T Cells Constitutively Expressing Cytotoxic T Lymphocyte-Associated Antigen 4. J Exp Med (2000) 192:303–10. doi: 10.1084/jem.192.2.303
7. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the Regulatory T Cell Lineage In Vivo. Science (2010) 329:1667–71. doi: 10.1126/science.1191996
8. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155:1151–64.
9. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous Requirement for the TCR in Regulatory T Cell Function. Nat Immunol (2014) 15:1070–8. doi: 10.1038/ni.3004
10. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T Cell Receptor Signals Maintain a Functional Regulatory T Cell Pool. Immunity (2014) 41:722–36. doi: 10.1016/j.immuni.2014.10.012
11. Legoux FP, Lim J-B, Cauley AW, Dikiy S, Ertelt J, Mariani TJ, et al. CD4+ T Cell Tolerance to Tissue-Restricted Self Antigens Is Mediated by Antigen-Specific Regulatory T Cells Rather Than Deletion. Immunity (2015) 43:896–908. doi: 10.1016/j.immuni.2015.10.011
12. Bluestone JA, Abbas AK. Natural Versus Adaptive Regulatory T Cells. Nat Rev Immunol (2003) 3:253–7. doi: 10.1038/nri1032
13. Maizels RM, Smith KA. Regulatory T Cells in Infection. In: Advances in Immunology. Elsevier. (2011) 112:73–136. doi: 10.1016/B978-0-12-387827-4.00003-6
14. Arce-Sillas A, Álvarez-Luquín DD, Tamaya-Domínguez B, Gomez-Fuentes S, Trejo-García A, Melo-Salas M, et al. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J Immunol Res (2016) 2016:1720827. doi: 10.1155/2016/1720827
15. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
16. Williams LM, Rudensky AY. Maintenance of the Foxp3-Dependent Developmental Program in Mature Regulatory T Cells Requires Continued Expression of Foxp3. Nat Immunol (2007) 8:277–84. doi: 10.1038/ni1437
17. Hall CHT, Kassel R, Tacke RS, Hahn YS. HCV+ Hepatocytes Induce Human Regulatory CD4+ T Cells Through the Production of TGF-Beta. PloS One (2010) 5:e12154. doi: 10.1371/journal.pone.0012154
18. Sharma S, Rajasagi NK, Veiga-Parga T, Rouse BT. Herpes Virus Entry Mediator (HVEM) Modulates Proliferation and Activation of Regulatory T Cells Following HSV-1 Infection. Microbes Infect (2014) 16:648–60. doi: 10.1016/j.micinf.2014.06.005
19. Schulze Zur Wiesch J, Thomssen A, Hartjen P, Tóth I, Lehmann C, Meyer-Olson D, et al. Comprehensive Analysis of Frequency and Phenotype of T Regulatory Cells in HIV Infection: CD39 Expression of FoxP3+ T Regulatory Cells Correlates With Progressive Disease. J Virol (2011) 85:1287–97. doi: 10.1128/JVI.01758-10
20. Nikolova M, Carriere M, Jenabian M-A, Limou S, Younas M, Kök A, et al. CD39/adenosine Pathway Is Involved in AIDS Progression. PloS Pathog (2011) 7:e1002110. doi: 10.1371/journal.ppat.1002110
21. Becker C, Taube C, Bopp T, Becker C, Michel K, Kubach J, et al. Protection From Graft-Versus-Host Disease by HIV-1 Envelope Protein Gp120-Mediated Activation of Human CD4+CD25+ Regulatory T Cells. Blood (2009) 114:1263–9. doi: 10.1182/blood-2009-02-206730
22. Ji J, Cloyd MW. HIV-1 Binding to CD4 on CD4+CD25+ Regulatory T Cells Enhances Their Suppressive Function and Induces Them to Home to, and Accumulate in, Peripheral and Mucosal Lymphoid Tissues: An Additional Mechanism of Immunosuppression. Int Immunol (2009) 21:283–94. doi: 10.1093/intimm/dxn146
23. Nilsson J, Boasso A, Velilla PA, Zhang R, Vaccari M, Franchini G, et al. HIV-1-Driven Regulatory T-Cell Accumulation in Lymphoid Tissues Is Associated With Disease Progression in HIV/AIDS. Blood (2006) 108:3808–17. doi: 10.1182/blood-2006-05-021576
24. Suchard MS, Mayne E, Green VA, Shalekoff S, Donninger SL, Stevens WS, et al. FOXP3 Expression Is Upregulated in CD4T Cells in Progressive HIV-1 Infection and Is a Marker of Disease Severity. PloS One (2010) 5:e11762. doi: 10.1371/journal.pone.0011762
25. Joedicke JJ, Dietze KK, Zelinskyy G, Dittmer U. The Phenotype and Activation Status of Regulatory T Cells During Friend Retrovirus Infection. Virol Sin (2014) 29:48–60. doi: 10.1007/s12250-014-3396-z
26. Moore TC, Gonzaga LM, Mather JM, Messer RJ, Hasenkrug KJ. B Cell Requirement for Robust Regulatory T Cell Responses to Friend Retrovirus Infection. mBio (2017) 8:e01122–17. doi: 10.1128/mBio.01122-17
27. Myers L, Joedicke JJ, Carmody AB, Messer RJ, Kassiotis G, Dudley JP, et al. IL-2-Independent and TNF-α-Dependent Expansion of Vβ5+ Natural Regulatory T Cells During Retrovirus Infection. J Immunol (2013) 190:5485–95. doi: 10.4049/jimmunol.1202951
28. Yamano Y, Takenouchi N, Li H-C, Tomaru U, Yao K, Grant CW, et al. Virus-Induced Dysfunction of CD4+CD25+ T Cells in Patients With HTLV-I-Associated Neuroimmunological Disease. J Clin Invest (2005) 115:1361–8. doi: 10.1172/JCI23913
29. Walsh PT, Benoit BM, Wysocka M, Dalton NM, Turka LA, Rook AH. A Role for Regulatory T Cells in Cutaneous T-Cell Lymphoma; Induction of a CD4 + CD25 + Foxp3+ T-Cell Phenotype Associated With HTLV-1 Infection. J Invest Dermatol (2006) 126:690–2. doi: 10.1038/sj.jid.5700121
30. Oh U, Grant C, Griffith C, Fugo K, Takenouchi N, Jacobson S. Reduced Foxp3 Protein Expression Is Associated With Inflammatory Disease During Human T Lymphotropic Virus Type 1 Infection. J Infect Dis (2006) 193:1557–66. doi: 10.1086/503874
31. Gupta N, Hegde P, Lecerf M, Nain M, Kaur M, Kalia M, et al. Japanese Encephalitis Virus Expands Regulatory T Cells by Increasing the Expression of PD-L1 on Dendritic Cells: Immunity to Infection. Eur J Immunol (2014) 44:1363–74. doi: 10.1002/eji.201343701
32. Periasamy S, Dhiman R, Barnes PF, Paidipally P, Tvinnereim A, Bandaru A, et al. Programmed Death 1 and Cytokine Inducible SH2-Containing Protein Dependent Expansion of Regulatory T Cells Upon Stimulation With Mycobacterium Tuberculosis. J Infect Dis (2011) 203:1256–63. doi: 10.1093/infdis/jir011
33. Trinath J, Maddur MS, Kaveri SV, Balaji KN, Bayry J. Mycobacterium Tuberculosis Promotes Regulatory T-Cell Expansion via Induction of Programmed Death-1 Ligand 1 (PD-L1, CD274) on Dendritic Cells. J Infect Dis (2012) 205:694–6. doi: 10.1093/infdis/jir820
34. Beswick EJ, Pinchuk IV, Earley RB, Schmitt DA, Reyes VE. Role of Gastric Epithelial Cell-Derived Transforming Growth Factor Beta in Reduced CD4+ T Cell Proliferation and Development of Regulatory T Cells During Helicobacter Pylori Infection. Infect Immun (2011) 79:2737–45. doi: 10.1128/IAI.01146-10
35. León MA, Palma C, Hernández C, Sandoval M, Cofre C, Perez-Mateluna G, et al. Helicobacter Pylori Pediatric Infection Changes FcϵRI Expression in Dendritic Cells and Treg Profile In Vivo and In Vitro. Microbes Infect (2019) 21:449–55. doi: 10.1016/j.micinf.2019.05.001
36. Belkaid Y, Blank RB, Suffia I. Natural Regulatory T Cells and Parasites: A Common Quest for Host Homeostasis. Immunol Rev (2006) 212:287–300. doi: 10.1111/j.0105-2896.2006.00409.x
37. Hisaeda H, Tetsutani K, Imai T, Moriya C, Tu L, Hamano S, et al. Malaria Parasites Require TLR9 Signaling for Immune Evasion by Activating Regulatory T Cells. J Immunol (2008) 180:2496–503. doi: 10.4049/jimmunol.180.4.2496
38. Hisaeda H, Maekawa Y, Iwakawa D, Okada H, Himeno K, Kishihara K, et al. Escape of Malaria Parasites From Host Immunity Requires CD4+ CD25+ Regulatory T Cells. Nat Med (2004) 10:29–30. doi: 10.1038/nm975
39. Scholzen A, Mittag D, Rogerson SJ, Cooke BM, Plebanski M. Plasmodium Falciparum-Mediated Induction of Human CD25Foxp3 CD4 T Cells Is Independent of Direct TCR Stimulation and Requires IL-2, IL-10 and TGFbeta. PloS Pathog (2009) 5:e1000543. doi: 10.1371/journal.ppat.1000543
40. Suffia I, Reckling SK, Salay G, Belkaid Y. A Role for CD103 in the Retention of CD4+CD25+ Treg and Control of Leishmania Major Infection. J Immunol (2005) 174:5444–55. doi: 10.4049/jimmunol.174.9.5444
41. Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5-Dependent Homing of Naturally Occurring CD4+ Regulatory T Cells to Sites of Leishmania Major Infection Favors Pathogen Persistence. J Exp Med (2006) 203:2451–60. doi: 10.1084/jem.20060956
42. Costa FRC, Mota CM, Santiago FM, Silva MV, Ferreira MD, Fonseca DM, et al. GITR Activation Positively Regulates Immune Responses Against Toxoplasma Gondii. PloS One (2016) 11:e0152622. doi: 10.1371/journal.pone.0152622
43. Netea MG, Sutmuller R, Hermann C, van der Graaf CAA, van der Meer JWM, van Krieken JH, et al. Toll-Like Receptor 2 Suppresses Immunity Against Candida Albicans Through Induction of IL-10 and Regulatory T Cells. J Immunol (2004) 172:3712–8. doi: 10.4049/jimmunol.172.6.3712
44. Sriram S, Yao SY, Stratton C, Moses H, Narayana PA, Wolinsky JS. Pilot Study to Examine the Effect of Antibiotic Therapy on MRI Outcomes in RRMS. J Neurol Sci (2005) 234:87–91. doi: 10.1016/j.jns.2005.03.042
45. Pender MP, Csurhes PA, Smith C, Douglas NL, Neller MA, Matthews KK, et al. Epstein-Barr Virus-Specific T Cell Therapy for Progressive Multiple Sclerosis. JCI Insight (2018) 3:124714. doi: 10.1172/jci.insight.124714
46. Derfuss T, Curtin F, Guebelin C, Bridel C, Rasenack M, Matthey A, et al. A Phase IIa Randomised Clinical Study of GNbAC1, a Humanised Monoclonal Antibody Against the Envelope Protein of Multiple Sclerosis-Associated Endogenous Retrovirus in Multiple Sclerosis Patients. Mult Scler (2015) 21:885–93. doi: 10.1177/1352458514554052
47. Hartung H-P, Derfuss T, Cree BA, Sormani MP, Selmaj K, Stutters J, et al. Efficacy and Safety of Temelimab in Multiple Sclerosis: Results of a Randomized Phase 2b and Extension Study. Mult Scler (2021) 13524585211024996. doi: 10.1177/13524585211024997
48. Gold J, Marta M, Meier UC, Christensen T, Miller D, Altmann D, et al. A Phase II Baseline Versus Treatment Study to Determine the Efficacy of Raltegravir (Isentress) in Preventing Progression of Relapsing Remitting Multiple Sclerosis as Determined by Gadolinium-Enhanced MRI: The INSPIRE Study. Mult Scler Relat Disord (2018) 24:123–8. doi: 10.1016/j.msard.2018.06.002
49. Fleming JO, Isaak A, Lee JE, Luzzio CC, Carrithers MD, Cook TD, et al. Probiotic Helminth Administration in Relapsing-Remitting Multiple Sclerosis: A Phase 1 Study. Mult Scler (2011) 17:743–54. doi: 10.1177/1352458511398054
50. Tanasescu R, Tench CR, Constantinescu CS, Telford G, Singh S, Frakich N, et al. Hookworm Treatment for Relapsing Multiple Sclerosis: A Randomized Double-Blinded Placebo-Controlled Trial. JAMA Neurol (2020) 77:1089–98. doi: 10.1001/jamaneurol.2020.1118
51. Voldsgaard A, Bager P, Garde E, Åkeson P, Leffers AM, Madsen CG, et al. Trichuris Suis Ova Therapy in Relapsing Multiple Sclerosis Is Safe But Without Signals of Beneficial Effect. Mult Scler (2015) 21:1723–9. doi: 10.1177/1352458514568173
52. Yordanova IA, Ebner F, Schulz AR, Steinfelder S, Rosche B, Bolze A, et al. The Worm-Specific Immune Response in Multiple Sclerosis Patients Receiving Controlled Trichuris Suis Ova Immunotherapy. Life (Basel) (2021) 11:101. doi: 10.3390/life11020101
53. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T Cells Prevent Catastrophic Autoimmunity Throughout the Lifespan of Mice. Nat Immunol (2007) 8:191–7. doi: 10.1038/ni1428
54. Belkaid Y, Tarbell K. Regulatory T Cells in the Control of Host-Microorganism Interactions (*). Annu Rev Immunol (2009) 27:551–89. doi: 10.1146/annurev.immunol.021908.132723
55. Xia M, Hu S, Fu Y, Jin W, Yi Q, Matsui Y, et al. CCR10 Regulates Balanced Maintenance and Function of Resident Regulatory and Effector T Cells to Promote Immune Homeostasis in the Skin. J Allergy Clin Immunol (2014) 134:634–44.e10. doi: 10.1016/j.jaci.2014.03.010
56. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, But Not Obese, Fat Is Enriched for a Unique Population of Regulatory T Cells That Affect Metabolic Parameters. Nat Med (2009) 15:930–9. doi: 10.1038/nm.2002
57. Burzyn D, Benoist C, Mathis D. Regulatory T Cells in Nonlymphoid Tissues. Nat Immunol (2013) 14:1007–13. doi: 10.1038/ni.2683
58. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang L-P, et al. Tumour Hypoxia Promotes Tolerance and Angiogenesis via CCL28 and Treg Cells. Nature (2011) 475:226–30. doi: 10.1038/nature10169
59. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-Infiltrating Regulatory T Cells Stimulate Mammary Cancer Metastasis Through RANKL–RANK Signalling. Nature (2011) 470:548–53. doi: 10.1038/nature09707
60. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ Is a Major Driver of the Accumulation and Phenotype of Adipose Tissue Treg Cells. Nature (2012) 486:549–53. doi: 10.1038/nature11132
61. Sharma A, Rudra D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front Immunol (2018) 9:883. doi: 10.3389/fimmu.2018.00883
62. Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk H-D. Regulatory T Cell-Mediated Anti-Inflammatory Effects Promote Successful Tissue Repair in Both Indirect and Direct Manners. Front Pharmacol (2015) 6:184. doi: 10.3389/fphar.2015.00184
63. Ghelani A, Bates D, Conner K, Wu M-Z, Lu J, Hu Y-L, et al. Defining the Threshold IL-2 Signal Required for Induction of Selective Treg Cell Responses Using Engineered IL-2 Muteins. Front Immunol (2020) 11:1106. doi: 10.3389/fimmu.2020.01106
64. de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 Is Essential for CD4+CD25+ Regulatory T Cell Function. Eur J Immunol (2004) 34:2480–8. doi: 10.1002/eji.200425274
65. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4 + CD25 High Regulatory Cells in Human Peripheral Blood. J Immunol (2001) 167:1245–53. doi: 10.4049/jimmunol.167.3.1245
66. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An Essential Role for the IL-2 Receptor in Treg Cell Function. Nat Immunol (2016) 17:1322–33. doi: 10.1038/ni.3540
67. Zhang HY, Yan KX, Huang Q, Ma Y, Fang X, Han L. Target Tissue Ectoenzyme CD39/CD73-Expressing Foxp3 + Regulatory T Cells in Patients With Psoriasis. Clin Exp Dermatol (2015) 40:182–91. doi: 10.1111/ced.12497
68. Schuler PJ, Saze Z, Hong C-S, Muller L, Gillespie DG, Cheng D, et al. Human CD4 + CD39 + Regulatory T Cells Produce Adenosine Upon Co-Expression of Surface CD73 or Contact With CD73 + Exosomes or CD73 + Cells: CD73 Co-Expression in Human CD39 + T Reg. Clin Exp Immunol (2014) 177:531–43. doi: 10.1111/cei.12354
69. Bopp T, Becker C, Klein M, Klein-Heßling S, Palmetshofer A, Serfling E, et al. Cyclic Adenosine Monophosphate Is a Key Component of Regulatory T Cell–Mediated Suppression. J Exp Med (2007) 204:1303–10. doi: 10.1084/jem.20062129
70. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med (2007) 204:1257–65. doi: 10.1084/jem.20062512
71. Rueda CM, Jackson CM, Chougnet CA. Regulatory T-Cell-Mediated Suppression of Conventional T-Cells and Dendritic Cells by Different cAMP Intracellular Pathways. Front Immunol (2016) 7:216. doi: 10.3389/fimmu.2016.00216
72. Klein M, Bopp T. Cyclic AMP Represents a Crucial Component of Treg Cell-Mediated Immune Regulation. Front Immunol (2016) 7:315. doi: 10.3389/fimmu.2016.00315
73. Su W, Chen X, Zhu W, Yu J, Li W, Li Y, et al. The cAMP–Adenosine Feedback Loop Maintains the Suppressive Function of Regulatory T Cells. J Immunol (2019) 203:1436–46. doi: 10.4049/jimmunol.1801306
74. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and Perforin Are Important for Regulatory T Cell-Mediated Suppression of Tumor Clearance. Immunity (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
75. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T Regulatory Cells Can Use the Perforin Pathway to Cause Autologous Target Cell Death. Immunity (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
76. Gondek DC, DeVries V, Nowak EC, Lu L-F, Bennett KA, Scott ZA, et al. Transplantation Survival Is Maintained by Granzyme B + Regulatory Cells and Adaptive Regulatory T Cells. J Immunol (2008) 181:4752–60. doi: 10.4049/jimmunol.181.7.4752
77. Schmidt A, Oberle N, Krammer PH. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front Immun (2012) 3:51. doi: 10.3389/fimmu.2012.00051
78. Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T Lymphocyte Antigen-4-Dependent Down-Modulation of Costimulatory Molecules on Dendritic Cells in CD4+ CD25+ Regulatory T-Cell-Mediated Suppression. Immunology (2006) 118:240–9. doi: 10.1111/j.1365-2567.2006.02362.x
79. Lippens C, Duraes FV, Dubrot J, Brighouse D, Lacroix M, Irla M, et al. IDO-Orchestrated Crosstalk Between pDCs and Tregs Inhibits Autoimmunity. J Autoimmun (2016) 75:39–49. doi: 10.1016/j.jaut.2016.07.004
80. Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of Tryptophan Catabolism by Human Leukemic Cells Results in the Conversion of CD25- Into CD25+ T Regulatory Cells. Blood (2007) 109:2871–7. doi: 10.1182/blood-2006-07-036863
81. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of Tryptophan Catabolism by Regulatory T Cells. Nat Immunol (2003) 4:1206–12. doi: 10.1038/ni1003
82. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
83. Kurtulus S, Sakuishi K, Ngiow S-F, Joller N, Tan DJ, Teng MWL, et al. TIGIT Predominantly Regulates the Immune Response via Regulatory T Cells. J Clin Invest (2015) 125:4053–62. doi: 10.1172/JCI81187
84. Guo J, Zhou X. Regulatory T Cells Turn Pathogenic. Cell Mol Immunol (2015) 12:525–32. doi: 10.1038/cmi.2015.12
85. Kassiotis G, O’Garra A. Immunology. Immunity Benefits From a Little Suppression. Science (2008) 320:1168–9. doi: 10.1126/science.1159090
86. Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of Early Protective Immunity to Viral Infection by Regulatory T Cells. Science (2008) 320:1220–4. doi: 10.1126/science.1155209
87. Moreno-Fernandez ME, Rueda CM, Rusie LK, Chougnet CA. Regulatory T Cells Control HIV Replication in Activated T Cells Through a cAMP-Dependent Mechanism. Blood (2011) 117:5372–80. doi: 10.1182/blood-2010-12-323162
88. Haase AT. Perils at Mucosal Front Lines for HIV and SIV and Their Hosts. Nat Rev Immunol (2005) 5:783–92. doi: 10.1038/nri1706
89. Graham JB, Da Costa A, Lund JM. Regulatory T Cells Shape the Resident Memory T Cell Response to Virus Infection in the Tissues. J Immunol (2014) 192:683–90. doi: 10.4049/jimmunol.1202153
90. Zelinskyy G, Dietze KK, Hüsecken YP, Schimmer S, Nair S, Werner T, et al. The Regulatory T-Cell Response During Acute Retroviral Infection Is Locally Defined and Controls the Magnitude and Duration of the Virus-Specific Cytotoxic T-Cell Response. Blood (2009) 114:3199–207. doi: 10.1182/blood-2009-03-208736
91. Iwashiro M, Messer RJ, Peterson KE, Stromnes IM, Sugie T, Hasenkrug KJ. Immunosuppression by CD4+ Regulatory T Cells Induced by Chronic Retroviral Infection. Proc Natl Acad Sci (2001) 98:9226–30. doi: 10.1073/pnas.151174198
92. Haeryfar SMM, DiPaolo RJ, Tscharke DC, Bennink JR, Yewdell JW. Regulatory T Cells Suppress CD8+ T Cell Responses Induced by Direct Priming and Cross-Priming and Moderate Immunodominance Disparities. J Immunol (2005) 174:3344–51. doi: 10.4049/jimmunol.174.6.3344
93. Aandahl EM, Michaëlsson J, Moretto WJ, Hecht FM, Nixon DF. Human CD4+ CD25+ Regulatory T Cells Control T-Cell Responses to Human Immunodeficiency Virus and Cytomegalovirus Antigens. J Virol (2004) 78:2454–9. doi: 10.1128/jvi.78.5.2454-2459.2004
94. Hunt PW, Landay AL, Sinclair E, Martinson JA, Hatano H, Emu B, et al. A Low T Regulatory Cell Response May Contribute to Both Viral Control and Generalized Immune Activation in HIV Controllers. PloS One (2011) 6:e15924. doi: 10.1371/journal.pone.0015924
95. Hasenkrug KJ, Chougnet CA, Dittmer U. Regulatory T Cells in Retroviral Infections. PloS Pathog (2018) 14:e1006776. doi: 10.1371/journal.ppat.1006776
96. Eggena MP, Barugahare B, Jones N, Okello M, Mutalya S, Kityo C, et al. Depletion of Regulatory T Cells in HIV Infection Is Associated With Immune Activation. J Immunol (2005) 174:4407–14. doi: 10.4049/jimmunol.174.7.4407
97. Oswald-Richter K, Grill SM, Leelawong M, Unutmaz D. HIV Infection of Primary Human T Cells Is Determined by Tunable Thresholds of T Cell Activation. Eur J Immunol (2004) 34:1705–14. doi: 10.1002/eji.200424892
98. Chase AJ, Yang H-C, Zhang H, Blankson JN, Siliciano RF. Preservation of FoxP3+ Regulatory T Cells in the Peripheral Blood of Human Immunodeficiency Virus Type 1-Infected Elite Suppressors Correlates With Low CD4+ T-Cell Activation. J Virol (2008) 82:8307–15. doi: 10.1128/JVI.00520-08
99. Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, et al. The Prevalence of Regulatory T Cells in Lymphoid Tissue Is Correlated With Viral Load in HIV-Infected Patients. J Immunol (2005) 174:3143–7. doi: 10.4049/jimmunol.174.6.3143
100. Kinter AL, Hennessey M, Bell A, Kern S, Lin Y, Daucher M, et al. CD25(+)CD4(+) Regulatory T Cells From the Peripheral Blood of Asymptomatic HIV-Infected Individuals Regulate CD4(+) and CD8(+) HIV-Specific T Cell Immune Responses In Vitro and Are Associated With Favorable Clinical Markers of Disease Status. J Exp Med (2004) 200:331–43. doi: 10.1084/jem.20032069
101. Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human Immunodeficiency Virus–Driven Expansion of CD4+CD25+ Regulatory T Cells, Which Suppress HIV-Specific CD4 T-Cell Responses in HIV-Infected Patients. Blood (2004) 104:3249–56. doi: 10.1182/blood-2004-01-0365
102. Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, et al. Cutting Edge: The Prevalence of Regulatory T Cells in Lymphoid Tissue Is Correlated With Viral Load in HIV-Infected Patients. J Immunol (2005) 174:3143–7. doi: 10.4049/jimmunol.174.6.3143
103. Oswald-Richter K, Grill SM, Shariat N, Leelawong M, Sundrud MS, Haas DW, et al. HIV Infection of Naturally Occurring and Genetically Reprogrammed Human Regulatory T-Cells. PloS Biol (2004) 2:e198. doi: 10.1371/journal.pbio.0020198
104. Bolacchi F, Sinistro A, Ciaprini C, Demin F, Capozzi M, Carducci FC, et al. Increased Hepatitis C Virus (HCV)-Specific CD4+CD25+ Regulatory T Lymphocytes and Reduced HCV-Specific CD4+ T Cell Response in HCV-Infected Patients With Normal Versus Abnormal Alanine Aminotransferase Levels. Clin Exp Immunol (2006) 144:188–96. doi: 10.1111/j.1365-2249.2006.03048.x
105. Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang K-M. Suppression of HCV-Specific T Cells Without Differential Hierarchy Demonstrated Ex Vivo in Persistent HCV Infection. Hepatology (2003) 38:1437–48. doi: 10.1016/j.hep.2003.09.026
106. Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, Ferrari C, et al. T Cells With a CD4+CD25+ Regulatory Phenotype Suppress In Vitro Proliferation of Virus-Specific CD8+ T Cells During Chronic Hepatitis C Virus Infection. J Virol (2005) 79:7860–7. doi: 10.1128/JVI.79.12.7860-7867.2005
107. Li S, Jones KL, Woollard DJ, Dromey J, Paukovics G, Plebanski M, et al. Defining Target Antigens for CD25+ FOXP3 + IFN-Gamma- Regulatory T Cells in Chronic Hepatitis C Virus Infection. Immunol Cell Biol (2007) 85:197–204. doi: 10.1038/sj.icb.7100020
108. Boyer O, Saadoun D, Abriol J, Dodille M, Piette J-C, Cacoub P, et al. CD4+CD25+ Regulatory T-Cell Deficiency in Patients With Hepatitis C-Mixed Cryoglobulinemia Vasculitis. Blood (2004) 103:3428–30. doi: 10.1182/blood-2003-07-2598
109. Cabrera R, Tu Z, Xu Y, Firpi RJ, Rosen HR, Liu C, et al. An Immunomodulatory Role for CD4(+)CD25(+) Regulatory T Lymphocytes in Hepatitis C Virus Infection. Hepatology (2004) 40:1062–71. doi: 10.1002/hep.20454
110. Belkaid Y, Rouse BT. Natural Regulatory T Cells in Infectious Disease. Nat Immunol (2005) 6:353–60. doi: 10.1038/ni1181
111. Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ TR Cells Suppress Innate Immune Pathology Through Cytokine-Dependent Mechanisms. J Exp Med (2003) 197:111–9. doi: 10.1084/jem.20021345
112. Kullberg MC, Jankovic D, Gorelick PL, Caspar P, Letterio JJ, Cheever AW, et al. Bacteria-Triggered CD4+ T Regulatory Cells Suppress Helicobacter Hepaticus–Induced Colitis. J Exp Med (2002) 196:505–15. doi: 10.1084/jem.20020556
113. Kaparakis M, Laurie KL, Wijburg O, Pedersen J, Pearse M, van Driel IR, et al. CD4+ CD25+ Regulatory T Cells Modulate the T-Cell and Antibody Responses in Helicobacter-Infected BALB/c Mice. Infect Immun (2006) 74:3519–29. doi: 10.1128/IAI.01314-05
114. Raghavan S, Suri-Payer E, Holmgren J. Antigen-Specific In Vitro Suppression of Murine Helicobacter Pylori-Reactive Immunopathological T Cells by CD4+CD25+ Regulatory T Cells. Scand J Immunol (2004) 60:82–8. doi: 10.1111/j.0300-9475.2004.01447.x
115. Lundgren A, Suri-Payer E, Enarsson K, Svennerholm A-M, Lundin BS. Helicobacter Pylori- Specific CD4 + CD25 High Regulatory T Cells Suppress Memory T-Cell Responses to H . Pylori in Infected Individuals. Infect Immun (2003) 71:1755–62. doi: 10.1128/IAI.71.4.1755-1762.2003
116. Kursar M, Bonhagen K, Fensterle J, Köhler A, Hurwitz R, Kamradt T, et al. Regulatory CD4+CD25+ T Cells Restrict Memory CD8+ T Cell Responses. J Exp Med (2002) 196:1585–92. doi: 10.1084/jem.20011347
117. McKinley L, Logar AJ, McAllister F, Zheng M, Steele C, Kolls JK. Regulatory T Cells Dampen Pulmonary Inflammation and Lung Injury in an Animal Model of Pneumocystis Pneumonia. J Immunol (2006) 177:6215–26. doi: 10.4049/jimmunol.177.9.6215
118. Hori S, Carvalho TL, Demengeot J. CD25+CD4+ Regulatory T Cells Suppress CD4+ T Cell-Mediated Pulmonary Hyperinflammation Driven by Pneumocystis Carinii in Immunodeficient Mice. Eur J Immunol (2002) 32:1282–91. doi: 10.1002/1521-4141(200205)32:5<1282::AID-IMMU1282>3.0.CO;2-
119. Tenorio EP, Olguín JE, Fernández J, Vieyra P, Saavedra R. Reduction of Foxp 3 + Cells by Depletion With the PC61 mAb Induces Mortality in Resistant BALB/c Mice Infected With Toxoplasma Gondii. J Biomed Biotechnol (2010) 2010:1–9. doi: 10.1155/2010/786078
120. Johanns TM, Ertelt JM, Rowe JH, Way SS. Regulatory T Cell Suppressive Potency Dictates the Balance Between Bacterial Proliferation and Clearance During Persistent Salmonella Infection. PloS Pathog (2010) 6:e1001043. doi: 10.1371/journal.ppat.1001043
121. Monack DM. Helicobacter and Salmonella Persistent Infection Strategies. Cold Spring Harb Perspect Med (2013) 3:a010348. doi: 10.1101/cshperspect.a010348
122. Neill DR, Coward WR, Gritzfeld JF, Richards L, Garcia-Garcia FJ, Dotor J, et al. Density and Duration of Pneumococcal Carriage Is Maintained by Transforming Growth Factor β1 and T Regulatory Cells. Am J Respir Crit Care Med (2014) 189:1250–9. doi: 10.1164/rccm.201401-0128OC
123. Boer MC, Joosten SA, Ottenhoff THM. Regulatory T-Cells at the Interface Between Human Host and Pathogens in Infectious Diseases and Vaccination. Front Immunol (2015) 6:217. doi: 10.3389/fimmu.2015.00217
124. Trinschek B, Lüssi F, Haas J, Wildemann B, Zipp F, Wiendl H, et al. Kinetics of IL-6 Production Defines T Effector Cell Responsiveness to Regulatory T Cells in Multiple Sclerosis. PloS One (2013) 8:e77634. doi: 10.1371/journal.pone.0077634
125. Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, et al. In Active Relapsing-Remitting Multiple Sclerosis, Effector T Cell Resistance to Adaptive Tregs Involves IL-6-Mediated Signaling. Sci Trans Med (2013) 5:170ra15–170ra15. doi: 10.1126/scitranslmed.3004970
126. Gross CC, Meyer C, Bhatia U, Yshii L, Kleffner I, Bauer J, et al. CD8+ T Cell-Mediated Endotheliopathy Is a Targetable Mechanism of Neuro-Inflammation in Susac Syndrome. Nat Commun (2019) 10:5779. doi: 10.1038/s41467-019-13593-5
127. Robertson SJ, Messer RJ, Carmody AB, Hasenkrug KJ. In Vitro Suppression of CD8+ T Cell Function by Friend Virus-Induced Regulatory T Cells. J Immunol (2006) 176:3342–9. doi: 10.4049/jimmunol.176.6.3342
128. Zelinskyy G, Kraft ARM, Schimmer S, Arndt T, Dittmer U. Kinetics of CD8+ Effector T Cell Responses and Induced CD4+ Regulatory T Cell Responses During Friend Retrovirus Infection. Eur J Immunol (2006) 36:2658–70. doi: 10.1002/eji.200636059
129. He H, Messer RJ, Sakaguchi S, Yang G, Robertson SJ, Hasenkrug KJ. Reduction of Retrovirus-Induced Immunosuppression by In Vivo Modulation of T Cells During Acute Infection. J Virol (2004) 78:11641–7. doi: 10.1128/JVI.78.21.11641-11647.2004
130. Dittmer U, He H, Messer RJ, Schimmer S, Olbrich ARM, Ohlen C, et al. Functional Impairment of CD8+ T Cells by Regulatory T Cells During Persistent Retroviral Infection. Immunity (2004) 20:293–303. doi: 10.1016/S1074-7613(04)00054-8
131. Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4 + CD25 + Regulatory T Cells Control the Severity of Viral Immunoinflammatory Lesions. J Immunol (2004) 172:4123–32. doi: 10.4049/jimmunol.172.7.4123
132. Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T Cells Regulate Virus-Specific Primary and Memory CD8+ T Cell Responses. J Exp Med (2003) 198:889–901. doi: 10.1084/jem.20030171
133. Myers L, Messer RJ, Carmody AB, Hasenkrug KJ. Tissue-Specific Abundance of Regulatory T Cells Correlates With CD8+ T Cell Dysfunction and Chronic Retrovirus Loads. J Immunol (2009) 183:1636–43. doi: 10.4049/jimmunol.0900350
134. Myers L, Hasenkrug KJ. Retroviral Immunology: Lessons From a Mouse Model. Immunol Res (2009) 43:160–6. doi: 10.1007/s12026-008-8061-x
135. Kinter A, McNally J, Riggin L, Jackson R, Roby G, Fauci AS. Suppression of HIV-Specific T Cell Activity by Lymph Node CD25+ Regulatory T Cells From HIV-Infected Individuals. Proc Natl Acad Sci USA (2007) 104:3390–5. doi: 10.1073/pnas.0611423104
136. Wan Z, Zhou Z, Liu Y, Lai Y, Luo Y, Peng X, et al. Regulatory T Cells and T Helper 17 Cells in Viral Infection. Scand J Immunol (2020) 91:e12873. doi: 10.1111/sji.12873
137. Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB. Pathogen-Specific Regulatory T Cells Delay the Arrival of Effector T Cells in the Lung During Early Tuberculosis. J Exp Med (2010) 207:1409–20. doi: 10.1084/jem.20091885
138. Shafiani S, Dinh C, Ertelt JM, Moguche AO, Siddiqui I, Smigiel KS, et al. Pathogen-Specific Treg Cells Expand Early During Mycobacterium Tuberculosis Infection But Are Later Eliminated in Response to Interleukin-12. Immunity (2013) 38:1261–70. doi: 10.1016/j.immuni.2013.06.003
139. Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, et al. Expansion and Function of Foxp3-Expressing T Regulatory Cells During Tuberculosis. J Exp Med (2007) 204:2159–69. doi: 10.1084/jem.20062105
140. Chen X, Zhou B, Li M, Deng Q, Wu X, Le X, et al. CD4(+)CD25(+)FoxP3(+) Regulatory T Cells Suppress Mycobacterium Tuberculosis Immunity in Patients With Active Disease. Clin Immunol (2007) 123:50–9. doi: 10.1016/j.clim.2006.11.009
141. Ribeiro-Rodrigues R, Resende Co T, Rojas R, Toossi Z, Dietze R, Boom WH, et al. A Role for CD4+CD25+ T Cells in Regulation of the Immune Response During Human Tuberculosis. Clin Exp Immunol (2006) 144:25–34. doi: 10.1111/j.1365-2249.2006.03027.x
142. Guyot-Revol V, Innes JA, Hackforth S, Hinks T, Lalvani A. Regulatory T Cells Are Expanded in Blood and Disease Sites in Patients With Tuberculosis. Am J Respir Crit Care Med (2006) 173:803–10. doi: 10.1164/rccm.200508-1294OC
143. Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, et al. Circulating and Liver Resident CD4+CD25+ Regulatory T Cells Actively Influence the Antiviral Immune Response and Disease Progression in Patients With Hepatitis B. J Immunol (2006) 177:739–47. doi: 10.4049/jimmunol.177.1.739
144. Yang G, Liu A, Xie Q, Guo TB, Wan B, Zhou B, et al. Association of CD4+CD25+Foxp3+ Regulatory T Cells With Chronic Activity and Viral Clearance in Patients With Hepatitis B. Int Immunol (2007) 19:133–40. doi: 10.1093/intimm/dxl130
145. Stoop JN, van der Molen RG, Kuipers EJ, Kusters JG, Janssen HLA. Inhibition of Viral Replication Reduces Regulatory T Cells and Enhances the Antiviral Immune Response in Chronic Hepatitis B. Virology (2007) 361:141–8. doi: 10.1016/j.virol.2006.11.018
146. Montagnoli C, Bacci A, Bozza S, Gaziano R, Mosci P, Sharpe AH, et al. B7/CD28-Dependent CD4 + CD25 + Regulatory T Cells Are Essential Components of the Memory-Protective Immunity to Candida albicans. J Immunol (2002) 169:6298–308. doi: 10.4049/jimmunol.169.11.6298
147. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
148. Cai X-P, Zhang H, Zhang Y-C, Wang Y, Su C, Ji M-J, et al. Dynamics of CD4+CD25+ T Cells in Spleens and Mesenteric Lymph Nodes of Mice Infected With Schistosoma Japonicum. Acta Biochim Biophys Sin (Shanghai) (2006) 38:299–304. doi: 10.1111/j.1745-7270.2006.00168.x
149. Hesse M, Piccirillo CA, Belkaid Y, Prufer J, Mentink-Kane M, Leusink M, et al. The Pathogenesis of Schistosomiasis Is Controlled by Cooperating IL-10-Producing Innate Effector and Regulatory T Cells. J Immunol (2004) 172:3157–66. doi: 10.4049/jimmunol.172.5.3157
150. Campanelli AP, Roselino AM, Cavassani KA, Pereira MSF, Mortara RA, Brodskyn CI, et al. CD4+CD25+ T Cells in Skin Lesions of Patients With Cutaneous Leishmaniasis Exhibit Phenotypic and Functional Characteristics of Natural Regulatory T Cells. J Infect Dis (2006) 193:1313–22. doi: 10.1086/502980
151. Aseffa A, Gumy A, Launois P, MacDonald HR, Louis JA, Tacchini-Cottier F. The Early IL-4 Response to Leishmania Major and the Resulting Th2 Cell Maturation Steering Progressive Disease in BALB/c Mice Are Subject to the Control of Regulatory CD4 + CD25 + T Cells. J Immunol (2002) 169:3232–41. doi: 10.4049/jimmunol.169.6.3232
152. Xu D, Liu H, Komai-Koma M, Campbell C, McSharry C, Alexander J, et al. CD4 + CD25 + Regulatory T Cells Suppress Differentiation and Functions of Th1 and Th2 Cells, Leishmania Major Infection, and Colitis in Mice. J Immunol (2003) 170:394–9. doi: 10.4049/jimmunol.170.1.394
153. Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4+ CD25+ Regulatory T Cells in Reactivation of Persistent Leishmaniasis and Control of Concomitant Immunity. J Exp Med (2004) 200:201–10. doi: 10.1084/jem.20040298
154. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ Regulatory T Cells Control Leishmania Major Persistence and Immunity. Nature (2002) 420:502–7. doi: 10.1038/nature01152
155. Liu H, Hu B, Xu D, Liew FY. CD4 + CD25 + Regulatory T Cells Cure Murine Colitis: The Role of IL-10, TGF-β, and CTLA4. J Immunol (2003) 171:5012–7. doi: 10.4049/jimmunol.171.10.5012
156. Kitagaki K, Businga TR, Racila D, Elliott DE, Weinstock JV, Kline JN. Intestinal Helminths Protect in a Murine Model of Asthma. J Immunol (2006) 177:1628–35. doi: 10.4049/jimmunol.177.3.1628
157. McKee AS, Pearce EJ. CD25 + CD4 + Cells Contribute to Th2 Polarization During Helminth Infection by Suppressing Th1 Response Development. J Immunol (2004) 173:1224–31. doi: 10.4049/jimmunol.173.2.1224
158. Taylor JJ, Mohrs M, Pearce EJ. Regulatory T Cell Responses Develop in Parallel to Th Responses and Control the Magnitude and Phenotype of the Th Effector Population. J Immunol (2006) 176:5839–47. doi: 10.4049/jimmunol.176.10.5839
159. Jin RM, Blair SJ, Warunek J, Heffner RR, Blader IJ, Wohlfert EA. Regulatory T Cells Promote Myositis and Muscle Damage in Toxoplasma Gondii Infection. J Immunol (2017) 198:352–62. doi: 10.4049/jimmunol.1600914
160. Sehrawat S, Rouse BT. Interplay of Regulatory T Cell and Th17 Cells During Infectious Diseases in Humans and Animals. Front Immunol (2017) 8:341. doi: 10.3389/fimmu.2017.00341
161. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 Cells in Human Disease. Immunol Rev (2008) 223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x
162. Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: An Effector CD4 T Cell Lineage With Regulatory T Cell Ties. Immunity (2006) 24:677–88. doi: 10.1016/j.immuni.2006.06.002
163. Denning TL, Wang Y, Patel SR, Williams IR, Pulendran B. Lamina Propria Macrophages and Dendritic Cells Differentially Induce Regulatory and Interleukin 17-Producing T Cell Responses. Nat Immunol (2007) 8:1086–94. doi: 10.1038/ni1511
164. Awasthi A, Kuchroo VK. Th17 Cells: From Precursors to Players in Inflammation and Infection. Int Immunol (2009) 21:489–98. doi: 10.1093/intimm/dxp021
165. Kimura A, Kishimoto T. IL-6: Regulator of Treg/Th17 Balance. Eur J Immunol (2010) 40:1830–5. doi: 10.1002/eji.201040391
166. Wu B, Wan Y. Molecular Control of Pathogenic Th17 Cells in Autoimmune Diseases. Int Immunopharmacol (2020) 80:106187. doi: 10.1016/j.intimp.2020.106187
167. Revu S, Wu J, Henkel M, Rittenhouse N, Menk A, Delgoffe GM, et al. IL-23 and IL-1β Drive Human Th17 Cell Differentiation and Metabolic Reprogramming in Absence of CD28 Costimulation. Cell Rep (2018) 22:2642–53. doi: 10.1016/j.celrep.2018.02.044
168. Toussirot E. The IL23/Th17 Pathway as a Therapeutic Target in Chronic Inflammatory Diseases. Inflamm Allergy Drug Targets (2012) 11:159–68. doi: 10.2174/187152812800392805
169. Bi Y, Yang R. Direct and Indirect Regulatory Mechanisms in TH17 Cell Differentiation and Functions: Direct and Indirect Regulatory Mechanisms in TH17 Cell. Scand J Immunol (2012) 75:543–52. doi: 10.1111/j.1365-3083.2012.02686.x
170. Tran DQ. TGF-β: The Sword, the Wand, and the Shield of FOXP3(+) Regulatory T Cells. J Mol Cell Biol (2012) 4:29–37. doi: 10.1093/jmcb/mjr033
171. Li MO, Flavell RA. TGF-β: A Master of All T Cell Trades. Cell (2008) 134:392–404. doi: 10.1016/j.cell.2008.07.025
172. Zhou L, Chong MMW, Littman DR. Plasticity of CD4+ T Cell Lineage Differentiation. Immunity (2009) 30:646–55. doi: 10.1016/j.immuni.2009.05.001
173. Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 Axis in the Immunopathogenesis of Psoriasis. J Invest Dermatol (2009) 129:1339–50. doi: 10.1038/jid.2009.59
174. Suri-Payer E, Amar AZ, Thornton AM, Shevach EM. CD4+CD25+ T Cells Inhibit Both the Induction and Effector Function of Autoreactive T Cells and Represent a Unique Lineage of Immunoregulatory Cells. J Immunol (1998) 160:1212–8.
175. Sadeghi A, Tahmasebi S, Mahmood A, Kuznetsova M, Valizadeh H, Taghizadieh A, et al. Th17 and Treg Cells Function in SARS-CoV2 Patients Compared With Healthy Controls. J Cell Physiol (2021) 236:2829–39. doi: 10.1002/jcp.30047
176. Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and Functional Features of Human Th17 Cells. J Exp Med (2007) 204:1849–61. doi: 10.1084/jem.20070663
177. Qin L, Hu C, Feng J, Xia Q. Activation of Lymphocytes Induced by Bronchial Epithelial Cells With Prolonged RSV Infection. PloS One (2011) 6:e27113. doi: 10.1371/journal.pone.0027113
178. Hoe E, Anderson J, Nathanielsz J, Toh ZQ, Marimla R, Balloch A, et al. The Contrasting Roles of Th17 Immunity in Human Health and Disease. Microbiol Immunol (2017) 61:49–56. doi: 10.1111/1348-0421.12471
179. Wu Y-C, Chen C-S, Chan Y-J. The Outbreak of COVID-19: An Overview. J Chin Med Assoc (2020) 83:217–20. doi: 10.1097/JCMA.0000000000000270
180. Mangodt TC, Van Herck MA, Nullens S, Ramet J, De Dooy JJ, Jorens PG, et al. The Role of Th17 and Treg Responses in the Pathogenesis of RSV Infection. Pediatr Res (2015) 78:483–91. doi: 10.1038/pr.2015.143
181. Lu X, McCoy KS, Xu J, Hu W, Chen H, Jiang K, et al. Galectin-9 Ameliorates Respiratory Syncytial Virus-Induced Pulmonary Immunopathology Through Regulating the Balance Between Th17 and Regulatory T Cells. Virus Res (2015) 195:162–71. doi: 10.1016/j.virusres.2014.10.011
182. Fulton RB, Meyerholz DK, Varga SM. Foxp3 + CD4 Regulatory T Cells Limit Pulmonary Immunopathology by Modulating the CD8 T Cell Response During Respiratory Syncytial Virus Infection. J Immunol (2010) 185:2382–92. doi: 10.4049/jimmunol.1000423
183. Lee DCP, Harker JAE, Tregoning JS, Atabani SF, Johansson C, Schwarze J, et al. CD25 + Natural Regulatory T Cells Are Critical in Limiting Innate and Adaptive Immunity and Resolving Disease Following Respiratory Syncytial Virus Infection. J Virol (2010) 84:8790–8. doi: 10.1128/JVI.00796-10
184. Ruckwardt TJ, Bonaparte KL, Nason MC, Graham BS. Regulatory T Cells Promote Early Influx of CD8 + T Cells in the Lungs of Respiratory Syncytial Virus-Infected Mice and Diminish Immunodominance Disparities. J Virol (2009) 83:3019–28. doi: 10.1128/JVI.00036-09
185. Loebbermann J, Thornton H, Durant L, Sparwasser T, Webster KE, Sprent J, et al. Regulatory T Cells Expressing Granzyme B Play a Critical Role in Controlling Lung Inflammation During Acute Viral Infection. Mucosal Immunol (2012) 5:161–72. doi: 10.1038/mi.2011.62
186. Durant LR, Makris S, Voorburg CM, Loebbermann J, Johansson C, Openshaw PJM. Regulatory T Cells Prevent Th2 Immune Responses and Pulmonary Eosinophilia During Respiratory Syncytial Virus Infection in Mice. J Virol (2013) 87:10946–54. doi: 10.1128/JVI.01295-13
187. Mukherjee S, Lindell DM, Berlin AA, Morris SB, Shanley TP, Hershenson MB, et al. IL-17–Induced Pulmonary Pathogenesis During Respiratory Viral Infection and Exacerbation of Allergic Disease. Am J Pathol (2011) 179:248–58. doi: 10.1016/j.ajpath.2011.03.003
188. Bystrom J, Al-Adhoubi N, Al-Bogami M, Jawad A, Mageed R. Th17 Lymphocytes in Respiratory Syncytial Virus Infection. Viruses (2013) 5:777–91. doi: 10.3390/v5030777
189. Chen Y, Fang J, Chen X, Pan C, Liu X, Liu J. Effects of the Treg/Th17 Cell Balance and Their Associated Cytokines in Patients With Hepatitis B Infection. Exp Ther Med (2015) 9:573–8. doi: 10.3892/etm.2014.2104
190. Lan Y-T, Wang Z, Tian P, Gong X-N, Fan Y-C, Wang K. Treg/Th17 Imbalance and Its Clinical Significance in Patients With Hepatitis B-Associated Liver Cirrhosis. Diagn Pathol (2019) 14:114. doi: 10.1186/s13000-019-0891-4
191. Rowan AG, Fletcher JM, Ryan EJ, Moran B, Hegarty JE, O’Farrelly C, et al. Hepatitis C Virus-Specific Th17 Cells Are Suppressed by Virus-Induced TGF-Beta. J Immunol (2008) 181:4485–94. doi: 10.4049/jimmunol.181.7.4485
192. Cardona P, Cardona P-J. Regulatory T Cells in Mycobacterium Tuberculosis Infection. Front Immunol (2019) 10:2139. doi: 10.3389/fimmu.2019.02139
193. Adibzadeh Sereshgi MM, Abdollahpour-Alitappeh M, Mahdavi M, Ranjbar R, Ahmadi K, Taheri RA, et al. Immunologic Balance of Regulatory T Cell/T Helper 17 Responses in Gastrointestinal Infectious Diseases: Role of miRNAs. Microb Pathog (2019) 131:135–43. doi: 10.1016/j.micpath.2019.03.029
194. Silva-Campa E, Flores-Mendoza L, Reséndiz M, Pinelli-Saavedra A, Mata-Haro V, Mwangi W, et al. Induction of T Helper 3 Regulatory Cells by Dendritic Cells Infected With Porcine Reproductive and Respiratory Syndrome Virus. Virology (2009) 387:373–9. doi: 10.1016/j.virol.2009.02.033
195. Cecere TE, Todd SM, Leroith T. Regulatory T Cells in Arterivirus and Coronavirus Infections: Do They Protect Against Disease or Enhance It? Viruses (2012) 4:833–46. doi: 10.3390/v4050833
196. Li S, Gowans EJ, Chougnet C, Plebanski M, Dittmer U. Natural Regulatory T Cells and Persistent Viral Infection. J Virol (2008) 82:21–30. doi: 10.1128/JVI.01768-07
197. Belkaid Y. Regulatory T Cells and Infection: A Dangerous Necessity. Nat Rev Immunol (2007) 7:875–88. doi: 10.1038/nri2189
198. Kalfaoglu B, Almeida-Santos J, Tye CA, Satou Y, Ono M. T-Cell Hyperactivation and Paralysis in Severe COVID-19 Infection Revealed by Single-Cell Analysis. Front Immunol (2020) 11:589380. doi: 10.3389/fimmu.2020.589380
199. Ziklo N, Huston WM, Taing K, Timms P. High Expression of IDO1 and TGF-β1 During Recurrence and Post Infection Clearance With Chlamydia Trachomatis, Are Independent of Host IFN-γ Response. BMC Infect Dis (2019) 19:218. doi: 10.1186/s12879-019-3843-4
200. Díaz YR, Rojas R, Valderrama L, Saravia NG. T-Bet, GATA-3, and Foxp3 Expression and Th1/Th2 Cytokine Production in the Clinical Outcome of Human Infection With Leishmania (Viannia) Species. J Infect Dis (2010) 202:406–15. doi: 10.1086/653829
201. Grainger JR, Smith KA, Hewitson JP, McSorley HJ, Harcus Y, Filbey KJ, et al. Helminth Secretions Induce De Novo T Cell Foxp3 Expression and Regulatory Function Through the TGF-β Pathway. J Exp Med (2010) 207:2331–41. doi: 10.1084/jem.20101074
202. Kleinewietfeld M, Hafler DA. Regulatory T Cells in Autoimmune Neuroinflammation. Immunol Rev (2014) 259:231–44. doi: 10.1111/imr.12169
203. Pette M, Fujita K, Kitze B, Whitaker JN, Albert E, Kappos L, et al. Myelin Basic Protein-Specific T Lymphocyte Lines From MS Patients and Healthy Individuals. Neurology (1990) 40:1770–6. doi: 10.1212/wnl.40.11.1770
204. Mykicki N, Herrmann AM, Schwab N, Deenen R, Sparwasser T, Limmer A, et al. Melanocortin-1 Receptor Activation Is Neuroprotective in Mouse Models of Neuroinflammatory Disease. Sci Transl Med (2016) 8:362ra146. doi: 10.1126/scitranslmed.aaf8732
205. Koutrolos M, Berer K, Kawakami N, Wekerle H, Krishnamoorthy G. Treg Cells Mediate Recovery From EAE by Controlling Effector T Cell Proliferation and Motility in the CNS. Acta Neuropathol Commun (2014) 2:163. doi: 10.1186/s40478-014-0163-1
206. Göbel K, Bittner S, Melzer N, Pankratz S, Dreykluft A, Schuhmann MK, et al. CD4(+) CD25(+) FoxP3(+) Regulatory T Cells Suppress Cytotoxicity of CD8(+) Effector T Cells: Implications for Their Capacity to Limit Inflammatory Central Nervous System Damage at the Parenchymal Level. J Neuroinflammation (2012) 9:41. doi: 10.1186/1742-2094-9-41
207. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the Inheritance of Regulatory T Cell Identity by a Cis Element in the Foxp3 Locus. Cell (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
208. Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, et al. Cutting Edge: Depletion of Foxp3+ Cells Leads to Induction of Autoimmunity by Specific Ablation of Regulatory T Cells in Genetically Targeted Mice. J Immunol (2009) 183:7631–4. doi: 10.4049/jimmunol.0804308
209. Huehn J, Polansky JK, Hamann A. Epigenetic Control of FOXP3 Expression: The Key to a Stable Regulatory T-Cell Lineage? Nat Rev Immunol (2009) 9:83–9. doi: 10.1038/nri2474
210. Li X, Liang Y, LeBlanc M, Benner C, Zheng Y. Function of a Foxp3 Cis-Element in Protecting Regulatory T Cell Identity. Cell (2014) 158:734–48. doi: 10.1016/j.cell.2014.07.030
211. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) Is Caused by Mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
212. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-Antigen-Driven Activation Induces Instability of Regulatory T Cells During an Inflammatory Autoimmune Response. Immunity (2013) 39:949–62. doi: 10.1016/j.immuni.2013.10.016
213. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the Transcription Factor Foxp3 Leads to the Generation of Pathogenic Memory T Cells In Vivo. Nat Immunol (2009) 10:1000–7. doi: 10.1038/ni.1774
214. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular Antagonism and Plasticity of Regulatory and Inflammatory T Cell Programs. Immunity (2008) 29:44–56. doi: 10.1016/j.immuni.2008.05.007
215. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic Maintenance of Natural Foxp3(+) CD25(+) CD4(+) Regulatory T Cells by Interleukin (IL)-2 and Induction of Autoimmune Disease by IL-2 Neutralization. J Exp Med (2005) 201:723–35. doi: 10.1084/jem.20041982
216. Pankratz S, Bittner S, Herrmann AM, Schuhmann MK, Ruck T, Meuth SG, et al. Human CD4+ HLA-G+ Regulatory T Cells Are Potent Suppressors of Graft-Versus-Host Disease In Vivo. FASEB J (2014) 28:3435–45. doi: 10.1096/fj.14-251074
217. Waschbisch A, Schwab N, Ruck T, Stenner M-P, Wiendl H. FOXP3+ T Regulatory Cells in Idiopathic Inflammatory Myopathies. J Neuroimmunol (2010) 225:137–42. doi: 10.1016/j.jneuroim.2010.03.013
218. Göbel K, Pankratz S, Asaridou C-M, Herrmann AM, Bittner S, Merker M, et al. Blood Coagulation Factor XII Drives Adaptive Immunity During Neuroinflammation via CD87-Mediated Modulation of Dendritic Cells. Nat Commun (2016) 7:11626. doi: 10.1038/ncomms11626
219. Heylen M, Ruyssers NE, Gielis EM, Vanhomwegen E, Pelckmans PA, Moreels TG, et al. Of Worms, Mice and Man: An Overview of Experimental and Clinical Helminth-Based Therapy for Inflammatory Bowel Disease. Pharmacol Ther (2014) 143:153–67. doi: 10.1016/j.pharmthera.2014.02.011
220. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and Effector Functions of TH17 Cells. Nature (2008) 453:1051–7. doi: 10.1038/nature07036
221. Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, et al. Human Interleukin-17: A T Cell-Derived Proinflammatory Cytokine Produced by the Rheumatoid Synovium. Arthritis Rheum (1999) 42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E
222. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-Microarray Analysis of Multiple Sclerosis Lesions Yields New Targets Validated in Autoimmune Encephalomyelitis. Nat Med (2002) 8:500–8. doi: 10.1038/nm0502-500
223. Kleinewietfeld M, Hafler DA. The Plasticity of Human Treg and Th17 Cells and Its Role in Autoimmunity. Semin Immunol (2013) 25:305–12. doi: 10.1016/j.smim.2013.10.009
224. Noack M, Miossec P. Th17 and Regulatory T Cell Balance in Autoimmune and Inflammatory Diseases. Autoimmun Rev (2014) 13:668–77. doi: 10.1016/j.autrev.2013.12.004
225. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When Worlds Collide: Th17 and Treg Cells in Cancer and Autoimmunity. Cell Mol Immunol (2018) 15:458–69. doi: 10.1038/s41423-018-0004-4
226. Riou J, Althaus CL. Pattern of Early Human-to-Human Transmission of Wuhan 2019 Novel Coronavirus (2019-nCoV), December 2019 to January 2020. Eurosurveillance (2020) 25:7–11. doi: 10.2807/1560-7917.ES.2020.25.4.2000058
227. Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules (2017) 22:134. doi: 10.3390/molecules22010134
228. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief Report: Inhibition of Interleukin-6 Function Corrects Th17/Treg Cell Imbalance in Patients With Rheumatoid Arthritis. Arthritis Rheum (2012) 64:2499–503. doi: 10.1002/art.34477
229. Kikuchi J, Hashizume M, Kaneko Y, Yoshimoto K, Nishina N, Takeuchi T. Peripheral Blood CD4+CD25+CD127low Regulatory T Cells Are Significantly Increased by Tocilizumab Treatment in Patients With Rheumatoid Arthritis: Increase in Regulatory T Cells Correlates With Clinical Response. Arthritis Res Ther (2015) 17:10. doi: 10.1186/s13075-015-0526-4
230. Li S, Wu Z, Li L, Liu X. Interleukin-6 (IL-6) Receptor Antagonist Protects Against Rheumatoid Arthritis. Med Sci Monit (2016) 22:2113–8. doi: 10.12659/MSM.896355
231. Lee J, Baek S, Lee J, Lee J, Lee D-G, Park M-K, et al. Digoxin Ameliorates Autoimmune Arthritis via Suppression of Th17 Differentiation. Int Immunopharmacol (2015) 26:103–11. doi: 10.1016/j.intimp.2015.03.017
232. Waisman A, Johann L. Antigen-Presenting Cell Diversity for T Cell Reactivation in Central Nervous System Autoimmunity. J Mol Med (2018) 96:1279–92. doi: 10.1007/s00109-018-1709-7
233. Prinz M, Priller J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat Neurosci (2017) 20:136–44. doi: 10.1038/nn.4475
234. Engelhardt B, Ransohoff RM. Capture, Crawl, Cross: The T Cell Code to Breach the Blood-Brain Barriers. Trends Immunol (2012) 33:579–89. doi: 10.1016/j.it.2012.07.004
235. Venken K, Hellings N, Broekmans T, Hensen K, Rummens J-L, Stinissen P. Natural Naive CD4+CD25+CD127low Regulatory T Cell (Treg) Development and Function Are Disturbed in Multiple Sclerosis Patients: Recovery of Memory Treg Homeostasis During Disease Progression. J Immunol (2008) 180:6411–20. doi: 10.4049/jimmunol.180.9.6411
236. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T Helper Type 1-Like, Foxp3+ Regulatory T Cells in Human Autoimmune Disease. Nat Med (2011) 17:673–5. doi: 10.1038/nm.2389
237. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of Ectonucleotidase CD39 by Foxp3+ Treg Cells: Hydrolysis of Extracellular ATP and Immune Suppression. Blood (2007) 110:1225–32. doi: 10.1182/blood-2006-12-064527
238. Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens J-L, et al. Compromised CD4+ CD25(high) Regulatory T-Cell Function in Patients With Relapsing-Remitting Multiple Sclerosis Is Correlated With a Reduced Frequency of FOXP3-Positive Cells and Reduced FOXP3 Expression at the Single-Cell Level. Immunology (2008) 123:79–89. doi: 10.1111/j.1365-2567.2007.02690.x
239. Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, et al. Functional Inflammatory Profiles Distinguish Myelin-Reactive T Cells From Patients With Multiple Sclerosis. Sci Transl Med (2015) 7:287ra74. doi: 10.1126/scitranslmed.aaa8038
240. Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, et al. Interleukin-17 mRNA Expression in Blood and CSF Mononuclear Cells Is Augmented in Multiple Sclerosis. Mult Scler (1999) 5:101–4. doi: 10.1177/135245859900500206
241. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of Functional Suppression by CD4+CD25+ Regulatory T Cells in Patients With Multiple Sclerosis. J Exp Med (2004) 199:971–9. doi: 10.1084/jem.20031579
242. Kimura K. Regulatory T Cells in Multiple Sclerosis. Clin Exp Neuroimmunol (2020) 11:148–55. doi: 10.1111/cen3.12591
243. Frisullo G, Nociti V, Iorio R, Patanella AK, Caggiula M, Marti A, et al. Regulatory T Cells Fail to Suppress CD4T+-Bet+ T Cells in Relapsing Multiple Sclerosis Patients. Immunology (2009) 127:418–28. doi: 10.1111/j.1365-2567.2008.02963.x
244. Chen X, Winkler-Pickett RT, Carbonetti NH, Ortaldo JR, Oppenheim JJ, Howard OMZ. Pertussis Toxin as an Adjuvant Suppresses the Number and Function of CD4+CD25+ T Regulatory Cells. Eur J Immunol (2006) 36:671–80. doi: 10.1002/eji.200535353
245. Othy S, Jairaman A, Dynes JL, Dong TX, Tune C, Yeromin AV, et al. Regulatory T Cells Suppress Th17 Cell Ca 2+ Signaling in the Spinal Cord During Murine Autoimmune Neuroinflammation. Proc Natl Acad Sci USA (2020) 117:20088–99. doi: 10.1073/pnas.2006895117
246. Breuer J, Schwab N, Schneider-Hohendorf T, Marziniak M, Mohan H, Bhatia U, et al. Ultraviolet B Light Attenuates the Systemic Immune Response in Central Nervous System Autoimmunity. Ann Neurol (2014) 75:739–58. doi: 10.1002/ana.24165
247. Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann B. Glatiramer Acetate Improves Regulatory T-Cell Function by Expansion of Naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-Cells in Patients With Multiple Sclerosis. J Neuroimmunol (2009) 216:113–7. doi: 10.1016/j.jneuroim.2009.06.011
248. Muls N, Dang HA, Sindic CJM, van Pesch V. Fingolimod Increases CD39-Expressing Regulatory T Cells in Multiple Sclerosis Patients. PloS One (2014) 9:e113025. doi: 10.1371/journal.pone.0113025
249. Korporal M, Haas J, Balint B, Fritzsching B, Schwarz A, Moeller S, et al. Interferon Beta-Induced Restoration of Regulatory T-Cell Function in Multiple Sclerosis Is Prompted by an Increase in Newly Generated Naive Regulatory T Cells. Arch Neurol (2008) 65:1434–9. doi: 10.1001/archneur.65.11.1434
250. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl Fumarate Targets GAPDH and Aerobic Glycolysis to Modulate Immunity. Science (2018) 360:449–53. doi: 10.1126/science.aan4665
251. Haas J, Würthwein C, Korporal-Kuhnke M, Viehoever A, Jarius S, Ruck T, et al. Alemtuzumab in Multiple Sclerosis: Short- and Long-Term Effects of Immunodepletion on the Peripheral Treg Compartment. Front Immunol (2019) 10:1204. doi: 10.3389/fimmu.2019.01204
252. Jamshidian A, Shaygannejad V, Pourazar A, Zarkesh-Esfahani S-H, Gharagozloo M. Biased Treg/Th17 Balance Away From Regulatory Toward Inflammatory Phenotype in Relapsed Multiple Sclerosis and Its Correlation With Severity of Symptoms. J Neuroimmunol (2013) 262:106–12. doi: 10.1016/j.jneuroim.2013.06.007
253. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39 + Foxp3 + Regulatory T Cells Suppress Pathogenic Th17 Cells and Are Impaired in Multiple Sclerosis. J Immunol (2009) 183:7602–10. doi: 10.4049/jimmunol.0901881
254. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-Specific Regulatory T Cells Accumulate in the CNS But Fail to Control Autoimmune Inflammation. Nat Med (2007) 13:423–31. doi: 10.1038/nm1564
255. Jones AP, Trend S, Byrne SN, Fabis-Pedrini MJ, Geldenhuys S, Nolan D, et al. Altered Regulatory T-Cell Fractions and Helios Expression in Clinically Isolated Syndrome: Clues to the Development of Multiple Sclerosis. Clin Transl Immunol (2017) 6:e143. doi: 10.1038/cti.2017.18
256. Kimura K, Hohjoh H, Fukuoka M, Sato W, Oki S, Tomi C, et al. Circulating Exosomes Suppress the Induction of Regulatory T Cells via Let-7i in Multiple Sclerosis. Nat Commun (2018) 9:17. doi: 10.1038/s41467-017-02406-2
257. Rojas M, Restrepo-Jiménez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, et al. Molecular Mimicry and Autoimmunity. J Autoimmun (2018) 95:100–23. doi: 10.1016/j.jaut.2018.10.012
258. Getts DR, Chastain EML, Terry RL, Miller SD. Virus Infection, Antiviral Immunity, and Autoimmunity. Immunol Rev (2013) 255:197–209. doi: 10.1111/imr.12091
259. Pacheco Y, Acosta-Ampudia Y, Monsalve DM, Chang C, Gershwin ME, Anaya J-M. Bystander Activation and Autoimmunity. J Autoimmun (2019) 103:102301. doi: 10.1016/j.jaut.2019.06.012
260. Losikoff PT, Mishra S, Terry F, Gutierrez A, Ardito MT, Fast L, et al. HCV Epitope, Homologous to Multiple Human Protein Sequences, Induces a Regulatory T Cell Response in Infected Patients. J Hepatol (2015) 62:48–55. doi: 10.1016/j.jhep.2014.08.026
261. Duarte JH, Zelenay S, Bergman M-L, Martins AC, Demengeot J. Natural Treg Cells Spontaneously Differentiate Into Pathogenic Helper Cells in Lymphopenic Conditions. Eur J Immunol (2009) 39:948–55. doi: 10.1002/eji.200839196
262. Warner HB, Carp RI. Multiple Sclerosis and Epstein-Barr Virus. Lancet (1981) 2:1290. doi: 10.1016/s0140-6736(81)91527-0
263. Dobson R, Kuhle J, Middeldorp J, Giovannoni G. Epstein-Barr-Negative MS: A True Phenomenon? Neurol Neuroimmunol Neuroinflamm (2017) 4:e318. doi: 10.1212/NXI.0000000000000318
264. Thacker EL, Mirzaei F, Ascherio A. Infectious Mononucleosis and Risk for Multiple Sclerosis: A Meta-Analysis. Ann Neurol (2006) 59:499–503. doi: 10.1002/ana.20820
265. Pakpoor J, Disanto G, Gerber JE, Dobson R, Meier UC, Giovannoni G, et al. The Risk of Developing Multiple Sclerosis in Individuals Seronegative for Epstein-Barr Virus: A Meta-Analysis. Mult Scler (2013) 19:162–6. doi: 10.1177/1352458512449682
266. Levin LI, Munger KL, O’Reilly EJ, Falk KI, Ascherio A. Primary Infection With the Epstein-Barr Virus and Risk of Multiple Sclerosis. Ann Neurol (2010) 67:824–30. doi: 10.1002/ana.21978
267. Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal Relationship Between Elevation of Epstein-Barr Virus Antibody Titers and Initial Onset of Neurological Symptoms in Multiple Sclerosis. JAMA (2005) 293:2496–500. doi: 10.1001/jama.293.20.2496
268. Lünemann JD, Tintoré M, Messmer B, Strowig T, Rovira A, Perkal H, et al. Elevated Epstein-Barr Virus-Encoded Nuclear Antigen-1 Immune Responses Predict Conversion to Multiple Sclerosis. Ann Neurol (2010) 67:159–69. doi: 10.1002/ana.21886
269. Farrell RA, Antony D, Wall GR, Clark DA, Fisniku L, Swanton J, et al. Humoral Immune Response to EBV in Multiple Sclerosis Is Associated With Disease Activity on MRI. Neurology (2009) 73:32–8. doi: 10.1212/WNL.0b013e3181aa29fe
270. Zivadinov R, Cerza N, Hagemeier J, Carl E, Badgett D, Ramasamy DP, et al. Humoral Response to EBV Is Associated With Cortical Atrophy and Lesion Burden in Patients With MS. Neurol Neuroimmunol Neuroinflamm (2016) 3:e190. doi: 10.1212/NXI.0000000000000190
271. Jakimovski D, Ramanathan M, Weinstock-Guttman B, Bergsland N, Ramasamay DP, Carl E, et al. Higher EBV Response Is Associated With More Severe Gray Matter and Lesion Pathology in Relapsing Multiple Sclerosis Patients: A Case-Controlled Magnetization Transfer Ratio Study. Mult Scler (2020) 26:322–32. doi: 10.1177/1352458519828667
272. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr Virus Infection in the Multiple Sclerosis Brain. J Exp Med (2007) 204:2899–912. doi: 10.1084/jem.20071030
273. Magliozzi R, Serafini B, Rosicarelli B, Chiappetta G, Veroni C, Reynolds R, et al. B-Cell Enrichment and Epstein-Barr Virus Infection in Inflammatory Cortical Lesions in Secondary Progressive Multiple Sclerosis. J Neuropathol Exp Neurol (2013) 72:29–41. doi: 10.1097/NEN.0b013e31827bfc62
274. Hassani A, Corboy JR, Al-Salam S, Khan G. Epstein-Barr Virus Is Present in the Brain of Most Cases of Multiple Sclerosis and May Engage More Than Just B Cells. PloS One (2018) 13:e0192109. doi: 10.1371/journal.pone.0192109
275. Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung H-P, Maniar T, et al. Epstein-Barr Virus in Multiple Sclerosis: Theory and Emerging Immunotherapies. Trends Mol Med (2020) 26:296–310. doi: 10.1016/j.molmed.2019.11.003
276. Lünemann JD, Jelcić I, Roberts S, Lutterotti A, Tackenberg B, Martin R, et al. EBNA1-Specific T Cells From Patients With Multiple Sclerosis Cross React With Myelin Antigens and Co-Produce IFN-Gamma and IL-2. J Exp Med (2008) 205:1763–73. doi: 10.1084/jem.20072397
277. Holmøy T, Kvale EØ, Vartdal F. Cerebrospinal Fluid CD4+ T Cells From a Multiple Sclerosis Patient Cross-Recognize Epstein-Barr Virus and Myelin Basic Protein. J Neurovirol (2004) 10:278–83. doi: 10.1080/13550280490499524
278. Tengvall K, Huang J, Hellström C, Kammer P, Biström M, Ayoglu B, et al. Molecular Mimicry Between Anoctamin 2 and Epstein-Barr Virus Nuclear Antigen 1 Associates With Multiple Sclerosis Risk. Proc Natl Acad Sci USA (2019) 116:16955–60. doi: 10.1073/pnas.1902623116
279. van Noort JM, Bajramovic JJ, Plomp AC, van Stipdonk MJ. Mistaken Self, a Novel Model That Links Microbial Infections With Myelin-Directed Autoimmunity in Multiple Sclerosis. J Neuroimmunol (2000) 105:46–57. doi: 10.1016/s0165-5728(00)00181-8
280. Li R, Rezk A, Healy LM, Muirhead G, Prat A, Gommerman JL, et al. MSSRF Canadian B Cells in MS Team. Cytokine-Defined B Cell Responses as Therapeutic Targets in Multiple Sclerosis. Front Immunol (2015) 6:626. doi: 10.3389/fimmu.2015.00626
281. D’Addario M, Libermann TA, Xu J, Ahmad A, Menezes J. Epstein-Barr Virus and Its Glycoprotein-350 Upregulate IL-6 in Human B-Lymphocytes via CD21, Involving Activation of NF-kappaB and Different Signaling Pathways. J Mol Biol (2001) 308:501–14. doi: 10.1006/jmbi.2001.4589
282. Wang J, Jelcic I, Mühlenbruch L, Haunerdinger V, Toussaint NC, Zhao Y, et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell (2020) 183:1264–81.e20. doi: 10.1016/j.cell.2020.09.054
283. Clottu AS, Mathias A, Sailer AW, Schluep M, Seebach JD, Du Pasquier R, et al. EBI2 Expression and Function: Robust in Memory Lymphocytes and Increased by Natalizumab in Multiple Sclerosis. Cell Rep (2017) 18:213–24. doi: 10.1016/j.celrep.2016.12.006
284. Wanke F, Moos S, Croxford AL, Heinen AP, Gräf S, Kalt B, et al. EBI2 Is Highly Expressed in Multiple Sclerosis Lesions and Promotes Early CNS Migration of Encephalitogenic CD4 T Cells. Cell Rep (2017) 18:1270–84. doi: 10.1016/j.celrep.2017.01.020
285. Rutkowska A, Shimshek DR, Sailer AW, Dev KK. EBI2 Regulates Pro-Inflammatory Signalling and Cytokine Release in Astrocytes. Neuropharmacology (2018) 133:121–8. doi: 10.1016/j.neuropharm.2018.01.029
286. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of Ectopic B-Cell Follicles With Germinal Centers in the Meninges of Patients With Secondary Progressive Multiple Sclerosis. Brain Pathol (2004) 14:164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x
287. Sutkowski N, Palkama T, Ciurli C, Sekaly RP, Thorley-Lawson DA, Huber BT. An Epstein-Barr Virus-Associated Superantigen. J Exp Med (1996) 184:971–80. doi: 10.1084/jem.184.3.971
288. Agarwal AK, Shah A. Menstrual-Linked Asthma. J Asthma (1997) 34:539–45. doi: 10.3109/02770909709055398
289. Humme S, Reisbach G, Feederle R, Delecluse H-J, Bousset K, Hammerschmidt W, et al. The EBV Nuclear Antigen 1 (EBNA1) Enhances B Cell Immortalization Several Thousandfold. Proc Natl Acad Sci USA (2003) 100:10989–94. doi: 10.1073/pnas.1832776100
290. Pender MP, Csurhes PA, Burrows JM, Burrows SR. Defective T-Cell Control of Epstein-Barr Virus Infection in Multiple Sclerosis. Clin Transl Immunol (2017) 6:e126. doi: 10.1038/cti.2016.87
291. Marrodan M, Alessandro L, Farez MF, Correale J. The Role of Infections in Multiple Sclerosis. Mult Scler (2019) 25:891–901. doi: 10.1177/1352458518823940
292. Brütting C, Emmer A, Kornhuber M, Staege MS. A Survey of Endogenous Retrovirus (ERV) Sequences in the Vicinity of Multiple Sclerosis (MS)-Associated Single Nucleotide Polymorphisms (SNPs). Mol Biol Rep (2016) 43:827–36. doi: 10.1007/s11033-016-4004-0
293. Perron H, Geny C, Laurent A, Mouriquand C, Pellat J, Perret J, et al. Leptomeningeal Cell Line From Multiple Sclerosis With Reverse Transcriptase Activity and Viral Particles. Res Virol (1989) 140:551–61. doi: 10.1016/s0923-2516(89)80141-4
294. Gifford R, Tristem M. The Evolution, Distribution and Diversity of Endogenous Retroviruses. Virus Genes (2003) 26:291–315. doi: 10.1023/a:1024455415443
295. Morandi E, Tanasescu R, Tarlinton RE, Constantinescu CS, Zhang W, Tench C, et al. The Association Between Human Endogenous Retroviruses and Multiple Sclerosis: A Systematic Review and Meta-Analysis. PloS One (2017) 12:e0172415. doi: 10.1371/journal.pone.0172415
296. Garcia-Montojo M, Rodriguez-Martin E, Ramos-Mozo P, Ortega-Madueño I, Dominguez-Mozo MI, Arias-Leal A, et al. Syncytin-1/HERV-W Envelope Is an Early Activation Marker of Leukocytes and Is Upregulated in Multiple Sclerosis Patients. Eur J Immunol (2020) 50:685–94. doi: 10.1002/eji.201948423
297. Christensen T, Petersen T, Thiel S, Brudek T, Ellermann-Eriksen S, Møller-Larsen A. Gene-Environment Interactions in Multiple Sclerosis: Innate and Adaptive Immune Responses to Human Endogenous Retrovirus and Herpesvirus Antigens and the Lectin Complement Activation Pathway. J Neuroimmunol (2007) 183:175–88. doi: 10.1016/j.jneuroim.2006.09.014
298. Mameli G, Astone V, Arru G, Marconi S, Lovato L, Serra C, et al. Brains and Peripheral Blood Mononuclear Cells of Multiple Sclerosis (MS) Patients Hyperexpress MS-Associated Retrovirus/HERV-W Endogenous Retrovirus, But Not Human Herpesvirus 6. J Gen Virol (2007) 88:264–74. doi: 10.1099/vir.0.81890-0
299. Sotgiu S, Mameli G, Serra C, Zarbo IR, Arru G, Dolei A. Multiple Sclerosis-Associated Retrovirus and Progressive Disability of Multiple Sclerosis. Mult Scler (2010) 16:1248–51. doi: 10.1177/1352458510376956
300. Dolei A, Serra C, Mameli G, Pugliatti M, Sechi G, Cirotto MC, et al. Multiple Sclerosis-Associated Retrovirus (MSRV) in Sardinian MS Patients. Neurology (2002) 58:471–3. doi: 10.1212/wnl.58.3.471
301. Grandi N, Tramontano E. HERV Envelope Proteins: Physiological Role and Pathogenic Potential in Cancer and Autoimmunity. Front Microbiol (2018) 9:462. doi: 10.3389/fmicb.2018.00462
302. Firouzi R, Rolland A, Michel M, Jouvin-Marche E, Hauw JJ, Malcus-Vocanson C, et al. Multiple Sclerosis-Associated Retrovirus Particles Cause T Lymphocyte-Dependent Death With Brain Hemorrhage in Humanized SCID Mice Model. J Neurovirol (2003) 9:79–93. doi: 10.1080/13550280390173328
303. Rolland A, Jouvin-Marche E, Viret C, Faure M, Perron H, Marche PN. The Envelope Protein of a Human Endogenous Retrovirus-W Family Activates Innate Immunity Through CD14/TLR4 and Promotes Th1-Like Responses. J Immunol (2006) 176:7636–44. doi: 10.4049/jimmunol.176.12.7636
304. Duperray A, Barbe D, Raguenez G, Weksler BB, Romero IA, Couraud P-O, et al. Inflammatory Response of Endothelial Cells to a Human Endogenous Retrovirus Associated With Multiple Sclerosis Is Mediated by TLR4. Int Immunol (2015) 27:545–53. doi: 10.1093/intimm/dxv025
305. Perron H, Dougier-Reynaud H-L, Lomparski C, Popa I, Firouzi R, Bertrand J-B, et al. Human Endogenous Retrovirus Protein Activates Innate Immunity and Promotes Experimental Allergic Encephalomyelitis in Mice. PloS One (2013) 8:e80128. doi: 10.1371/journal.pone.0080128
306. de Luca V, Martins Higa A, Malta Romano C, Pimenta Mambrini G, Peroni LA, Trivinho-Strixino F, et al. Cross-Reactivity Between Myelin Oligodendrocyte Glycoprotein and Human Endogenous Retrovirus W Protein: Nanotechnological Evidence for the Potential Trigger of Multiple Sclerosis. Micron (2019) 120:66–73. doi: 10.1016/j.micron.2019.02.005
307. Kremer D, Schichel T, Förster M, Tzekova N, Bernard C, van der Valk P, et al. Human Endogenous Retrovirus Type W Envelope Protein Inhibits Oligodendroglial Precursor Cell Differentiation. Ann Neurol (2013) 74:721–32. doi: 10.1002/ana.23970
308. Kremer D, Gruchot J, Weyers V, Oldemeier L, Göttle P, Healy L, et al. pHERV-W Envelope Protein Fuels Microglial Cell-Dependent Damage of Myelinated Axons in Multiple Sclerosis. Proc Natl Acad Sci USA (2019) 116:15216–25. doi: 10.1073/pnas.1901283116
309. Kremer D, Förster M, Schichel T, Göttle P, Hartung H-P, Perron H, et al. The Neutralizing Antibody GNbAC1 Abrogates HERV-W Envelope Protein-Mediated Oligodendroglial Maturation Blockade. Mult Scler (2015) 21:1200–3. doi: 10.1177/1352458514560926
310. Mameli G, Poddighe L, Mei A, Uleri E, Sotgiu S, Serra C, et al. Expression and Activation by Epstein Barr Virus of Human Endogenous Retroviruses-W in Blood Cells and Astrocytes: Inference for Multiple Sclerosis. PloS One (2012) 7:e44991. doi: 10.1371/journal.pone.0044991
311. Libbey JE, Cusick MF, Fujinami RS. Role of Pathogens in Multiple Sclerosis. Int Rev Immunol (2014) 33:266–83. doi: 10.3109/08830185.2013.823422
312. Tai AK, Luka J, Ablashi D, Huber BT. HHV-6A Infection Induces Expression of HERV-K18-Encoded Superantigen. J Clin Virol (2009) 46:47–8. doi: 10.1016/j.jcv.2009.05.019
313. Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, et al. Plaque-Associated Expression of Human Herpesvirus 6 in Multiple Sclerosis. Proc Natl Acad Sci USA (1995) 92:7440–4. doi: 10.1073/pnas.92.16.7440
314. Tarlinton RE, Martynova E, Rizvanov AA, Khaiboullina S, Verma S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses (2020) 12:E643. doi: 10.3390/v12060643
315. Soldan SS, Berti R, Salem N, Secchiero P, Flamand L, Calabresi PA, et al. Association of Human Herpes Virus 6 (HHV-6) With Multiple Sclerosis: Increased IgM Response to HHV-6 Early Antigen and Detection of Serum HHV-6 DNA. Nat Med (1997) 3:1394–7. doi: 10.1038/nm1297-1394
316. Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM. Human Herpesvirus 6 Genome and Antigen in Acute Multiple Sclerosis Lesions. J Infect Dis (2003) 187:1365–76. doi: 10.1086/368172
317. Sanders VJ, Felisan S, Waddell A, Tourtellotte WW. Detection of Herpesviridae in Postmortem Multiple Sclerosis Brain Tissue and Controls by Polymerase Chain Reaction. J Neurovirol (1996) 2:249–58. doi: 10.3109/13550289609146888
318. Leibovitch EC, Caruso B, Ha SK, Schindler MK, Lee NJ, Luciano NJ, et al. Herpesvirus Trigger Accelerates Neuroinflammation in a Nonhuman Primate Model of Multiple Sclerosis. Proc Natl Acad Sci USA (2018) 115:11292–7. doi: 10.1073/pnas.1811974115
319. Hogestyn JM, Mock DJ, Mayer-Proschel M. Contributions of Neurotropic Human Herpesviruses Herpes Simplex Virus 1 and Human Herpesvirus 6 to Neurodegenerative Disease Pathology. Neural Regen Res (2018) 13:211–21. doi: 10.4103/1673-5374.226380
320. Tejada-Simon MV, Zang YCQ, Hong J, Rivera VM, Zhang JZ. Cross-Reactivity With Myelin Basic Protein and Human Herpesvirus-6 in Multiple Sclerosis. Ann Neurol (2003) 53:189–97. doi: 10.1002/ana.10425
321. Cavallo S. Immune-Mediated Genesis of Multiple Sclerosis. J Transl Autoimmun (2020) 3:100039. doi: 10.1016/j.jtauto.2020.100039
322. Bagos PG, Nikolopoulos G, Ioannidis A. Chlamydia Pneumoniae Infection and the Risk of Multiple Sclerosis: A Meta-Analysis. Mult Scler (2006) 12:397–411. doi: 10.1191/1352458506ms1291oa
323. Du C, Yao S-Y, Ljunggren-Rose A, Sriram S. Chlamydia Pneumoniae Infection of the Central Nervous System Worsens Experimental Allergic Encephalitis. J Exp Med (2002) 196:1639–44. doi: 10.1084/jem.20020393
324. Jaruvongvanich V, Sanguankeo A, Jaruvongvanich S, Upala S. Association Between Helicobacter Pylori Infection and Multiple Sclerosis: A Systematic Review and Meta-Analysis. Mult Scler Relat Disord (2016) 7:92–7. doi: 10.1016/j.msard.2016.03.013
325. Pedrini MJF, Seewann A, Bennett KA, Wood AJT, James I, Burton J, et al. Helicobacter Pylori Infection as a Protective Factor Against Multiple Sclerosis Risk in Females. J Neurol Neurosurg Psychiatry (2015) 86:603–7. doi: 10.1136/jnnp-2014-309495
326. Cook KW, Crooks J, Hussain K, O’Brien K, Braitch M, Kareem H, et al. Helicobacter Pylori Infection Reduces Disease Severity in an Experimental Model of Multiple Sclerosis. Front Microbiol (2015) 6:52. doi: 10.3389/fmicb.2015.00052
327. McCune A, Lane A, Murray L, Harvey I, Nair P, Donovan J, et al. Reduced Risk of Atopic Disorders in Adults With Helicobacter Pylori Infection. Eur J Gastroenterol Hepatol (2003) 15:637–40. doi: 10.1097/00042737-200306000-00010
328. Luther J, Dave M, Higgins PDR, Kao JY. Association Between Helicobacter Pylori Infection and Inflammatory Bowel Disease: A Meta-Analysis and Systematic Review of the Literature. Inflamm Bowel Dis (2010) 16:1077–84. doi: 10.1002/ibd.21116
329. Fleming JO, Cook TD. Multiple Sclerosis and the Hygiene Hypothesis. Neurology (2006) 67:2085–6. doi: 10.1212/01.wnl.0000247663.40297.2d
330. Walsh KP, Brady MT, Finlay CM, Boon L, Mills KHG. Infection With a Helminth Parasite Attenuates Autoimmunity Through TGF-Beta-Mediated Suppression of Th17 and Th1 Responses. J Immunol (2009) 183:1577–86. doi: 10.4049/jimmunol.0803803
331. Sewell D, Qing Z, Reinke E, Elliot D, Weinstock J, Sandor M, et al. Immunomodulation of Experimental Autoimmune Encephalomyelitis by Helminth Ova Immunization. Int Immunol (2003) 15:59–69. doi: 10.1093/intimm/dxg012
332. La Flamme AC, Ruddenklau K, Bäckström BT. Schistosomiasis Decreases Central Nervous System Inflammation and Alters the Progression of Experimental Autoimmune Encephalomyelitis. Infect Immun (2003) 71:4996–5004. doi: 10.1128/IAI.71.9.4996-5004.2003
333. Correale J, Farez M. Association Between Parasite Infection and Immune Responses in Multiple Sclerosis. Ann Neurol (2007) 61:97–108. doi: 10.1002/ana.21067
334. Correale J, Farez M, Razzitte G. Helminth Infections Associated With Multiple Sclerosis Induce Regulatory B Cells. Ann Neurol (2008) 64:187–99. doi: 10.1002/ana.21438
335. Correale J, Farez MF. The Impact of Parasite Infections on the Course of Multiple Sclerosis. J Neuroimmunol (2011) 233:6–11. doi: 10.1016/j.jneuroim.2011.01.002
336. Douglas B, Oyesola O, Cooper MM, Posey A, Tait Wojno E, Giacomin PR, et al. Immune System Investigation Using Parasitic Helminths. Annu Rev Immunol (2021) 39:639–65. doi: 10.1146/annurev-immunol-093019-122827
337. Pender MP, Csurhes PA, Smith C, Beagley L, Hooper KD, Raj M, et al. Epstein-Barr Virus-Specific Adoptive Immunotherapy for Progressive Multiple Sclerosis. Mult Scler (2014) 20:1541–4. doi: 10.1177/1352458514521888
338. Huntemann N, Rolfes L, Pawlitzki M, Ruck T, Pfeuffer S, Wiendl H, et al. Failed, Interrupted, or Inconclusive Trials on Neuroprotective and Neuroregenerative Treatment Strategies in Multiple Sclerosis: Update 2015-2020. Drugs (2021) 81:1031–63. doi: 10.1007/s40265-021-01526-w
339. Madeira A, Burgelin I, Perron H, Curtin F, Lang AB, Faucard R. MSRV Envelope Protein Is a Potent, Endogenous and Pathogenic Agonist of Human Toll-Like Receptor 4: Relevance of GNbAC1 in Multiple Sclerosis Treatment. J Neuroimmunol (2016) 291:29–38. doi: 10.1016/j.jneuroim.2015.12.006
340. Curtin F, Perron H, Kromminga A, Porchet H, Lang AB. Preclinical and Early Clinical Development of GNbAC1, a Humanized IgG4 Monoclonal Antibody Targeting Endogenous Retroviral MSRV-Env Protein. MAbs (2015) 7:265–75. doi: 10.4161/19420862.2014.985021
341. Curtin F, Vidal V, Bernard C, Kromminga A, Lang AB, Porchet H. Serum Pharmacokinetics and Cerebrospinal Fluid Concentration Analysis of the New IgG4 Monoclonal Antibody GNbAC1 to Treat Multiple Sclerosis: A Phase 1 Study. MAbs (2016) 8:854–60. doi: 10.1080/19420862.2016.1168956
342. Curtin F, Lang AB, Perron H, Laumonier M, Vidal V, Porchet HC, et al. GNbAC1, a Humanized Monoclonal Antibody Against the Envelope Protein of Multiple Sclerosis-Associated Endogenous Retrovirus: A First-in-Humans Randomized Clinical Study. Clin Ther (2012) 34:2268–78. doi: 10.1016/j.clinthera.2012.11.006
343. Maruszak H, Brew BJ, Giovannoni G, Gold J. Could Antiretroviral Drugs be Effective in Multiple Sclerosis? A Case Report. Eur J Neurol (2011) 18:e110–111. doi: 10.1111/j.1468-1331.2011.03430.x
344. Fleming J, Hernandez G, Hartman L, Maksimovic J, Nace S, Lawler B, et al. Safety and Efficacy of Helminth Treatment in Relapsing-Remitting Multiple Sclerosis: Results of the HINT 2 Clinical Trial. Mult Scler (2019) 25:81–91. doi: 10.1177/1352458517736377
345. Rooks MG, Garrett WS. Gut Microbiota, Metabolites and Host Immunity. Nat Rev Immunol (2016) 16:341–52. doi: 10.1038/nri.2016.42
346. Thaiss CA, Zmora N, Levy M, Elinav E. The Microbiome and Innate Immunity. Nature (2016) 535:65–74. doi: 10.1038/nature18847
347. Li W, Deng Y, Chu Q, Zhang P. Gut Microbiome and Cancer Immunotherapy. Cancer Lett (2019) 447:41–7. doi: 10.1016/j.canlet.2019.01.015
348. De Luca F, Shoenfeld Y. The Microbiome in Autoimmune Diseases. Clin Exp Immunol (2019) 195:74–85. doi: 10.1111/cei.13158
349. Dickerson F, Severance E, Yolken R. The Microbiome, Immunity, and Schizophrenia and Bipolar Disorder. Brain Behav Immun (2017) 62:46–52. doi: 10.1016/j.bbi.2016.12.010
350. Barrington WT, Lusis AJ. Atherosclerosis: Association Between the Gut Microbiome and Atherosclerosis. Nat Rev Cardiol (2017) 14:699–700. doi: 10.1038/nrcardio.2017.169
351. Sprouse ML, Bates NA, Felix KM, Wu H-JJ. Impact of Gut Microbiota on Gut-Distal Autoimmunity: A Focus on T Cells. Immunology (2019) 156:305–18. doi: 10.1111/imm.13037
352. Scher JU, Abramson SB. The Microbiome and Rheumatoid Arthritis. Nat Rev Rheumatol (2011) 7:569–78. doi: 10.1038/nrrheum.2011.121
353. Mathis D, Benoist C. Microbiota and Autoimmune Disease: The Hosted Self. Cell Host Microbe (2011) 10:297–301. doi: 10.1016/j.chom.2011.09.007
354. Omenetti S, Pizarro TT. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front Immunol (2015) 6:639. doi: 10.3389/fimmu.2015.00639
355. Cebula A, Seweryn M, Rempala GA, Pabla SS, McIndoe RA, Denning TL, et al. Thymus-Derived Regulatory T Cells Contribute to Tolerance to Commensal Microbiota. Nature (2013) 497:258–62. doi: 10.1038/nature12079
356. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science (2011) 331:337–41. doi: 10.1126/science.1198469
357. Ochoa-Repáraz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, et al. Role of Gut Commensal Microflora in the Development of Experimental Autoimmune Encephalomyelitis. J Immunol (2009) 183:6041–50. doi: 10.4049/jimmunol.0900747
358. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg Induction by a Rationally Selected Mixture of Clostridia Strains From the Human Microbiota. Nature (2013) 500:232–6. doi: 10.1038/nature12331
359. Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. Endoscopic Photoconversion Reveals Unexpectedly Broad Leukocyte Trafficking to and From the Gut. Proc Natl Acad Sci USA (2014) 111:6696–701. doi: 10.1073/pnas.1405634111
360. Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, et al. Control of TH17 Cells Occurs in the Small Intestine. Nature (2011) 475:514–8. doi: 10.1038/nature10228
361. Kadowaki A, Saga R, Lin Y, Sato W, Yamamura T. Gut Microbiota-Dependent CCR9+CD4+ T Cells Are Altered in Secondary Progressive Multiple Sclerosis. Brain (2019) 142:916–31. doi: 10.1093/brain/awz012
362. Tanoue T, Atarashi K, Honda K. Development and Maintenance of Intestinal Regulatory T Cells. Nat Rev Immunol (2016) 16:295–309. doi: 10.1038/nri.2016.36
363. Chai JN, Peng Y, Rengarajan S, Solomon BD, Ai TL, Shen Z, et al. Helicobacter Species Are Potent Drivers of Colonic T Cell Responses in Homeostasis and Inflammation. Sci Immunol (2017) 2:eaal5068. doi: 10.1126/sciimmunol.aal5068
364. Whibley N, Tucci A, Powrie F. Regulatory T Cell Adaptation in the Intestine and Skin. Nat Immunol (2019) 20:386–96. doi: 10.1038/s41590-019-0351-z
365. Bertolini TB, Biswas M, Terhorst C, Daniell H, Herzog RW, Piñeros AR. Role of Orally Induced Regulatory T Cells in Immunotherapy and Tolerance. Cell Immunol (2021) 359:104251. doi: 10.1016/j.cellimm.2020.104251
366. Arroyo Hornero R, Hamad I, Côrte-Real B, Kleinewietfeld M. The Impact of Dietary Components on Regulatory T Cells and Disease. Front Immunol (2020) 11:253. doi: 10.3389/fimmu.2020.00253
367. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity (2015) 43:817–29. doi: 10.1016/j.immuni.2015.09.007
368. Mizuno M, Noto D, Kaga N, Chiba A, Miyake S. The Dual Role of Short Fatty Acid Chains in the Pathogenesis of Autoimmune Disease Models. PloS One (2017) 12:e0173032. doi: 10.1371/journal.pone.0173032
369. Scheppach W, Sommer H, Kirchner T, Paganelli GM, Bartram P, Christl S, et al. Effect of Butyrate Enemas on the Colonic Mucosa in Distal Ulcerative Colitis. Gastroenterology (1992) 103:51–6. doi: 10.1016/0016-5085(92)91094-k
370. Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, et al. Pancreatic β-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed Under the Influence of the Gut Microbiota. Immunity (2015) 43:304–17. doi: 10.1016/j.immuni.2015.07.013
371. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
372. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
373. Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, et al. Dysbiosis in the Gut Microbiota of Patients With Multiple Sclerosis, With a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PloS One (2015) 10:e0137429. doi: 10.1371/journal.pone.0137429
374. Mazmanian SK, Round JL, Kasper DL. A Microbial Symbiosis Factor Prevents Intestinal Inflammatory Disease. Nature (2008) 453:620–5. doi: 10.1038/nature07008
375. Round JL, Mazmanian SK. Inducible Foxp3+ Regulatory T-Cell Development by a Commensal Bacterium of the Intestinal Microbiota. Proc Natl Acad Sci USA (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
376. Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The Toll-Like Receptor 2 Pathway Establishes Colonization by a Commensal of the Human Microbiota. Science (2011) 332:974–7. doi: 10.1126/science.1206095
377. Chu H, Khosravi A, Kusumawardhani IP, Kwon AHK, Vasconcelos AC, Cunha LD, et al. Gene-Microbiota Interactions Contribute to the Pathogenesis of Inflammatory Bowel Disease. Science (2016) 352:1116–20. doi: 10.1126/science.aad9948
378. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut Bacteria From Multiple Sclerosis Patients Modulate Human T Cells and Exacerbate Symptoms in Mouse Models. Proc Natl Acad Sci USA (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
379. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the Human Gut Microbiome in Multiple Sclerosis. Nat Commun (2016) 7:12015. doi: 10.1038/ncomms12015
380. Smits HH, Engering A, van der Kleij D, de Jong EC, Schipper K, van Capel TMM, et al. Selective Probiotic Bacteria Induce IL-10-Producing Regulatory T Cells In Vitro by Modulating Dendritic Cell Function Through Dendritic Cell-Specific Intercellular Adhesion Molecule 3-Grabbing Nonintegrin. J Allergy Clin Immunol (2005) 115:1260–7. doi: 10.1016/j.jaci.2005.03.036
381. Kwon H-K, Lee C-G, So J-S, Chae C-S, Hwang J-S, Sahoo A, et al. Generation of Regulatory Dendritic Cells and CD4+Foxp3+ T Cells by Probiotics Administration Suppresses Immune Disorders. Proc Natl Acad Sci USA (2010) 107:2159–64. doi: 10.1073/pnas.0904055107
382. Di Giacinto C, Marinaro M, Sanchez M, Strober W, Boirivant M. Probiotics Ameliorate Recurrent Th1-Mediated Murine Colitis by Inducing IL-10 and IL-10-Dependent TGF-Beta-Bearing Regulatory Cells. J Immunol (2005) 174:3237–46. doi: 10.4049/jimmunol.174.6.3237
383. Lavasani S, Dzhambazov B, Nouri M, Fåk F, Buske S, Molin G, et al. A Novel Probiotic Mixture Exerts a Therapeutic Effect on Experimental Autoimmune Encephalomyelitis Mediated by IL-10 Producing Regulatory T Cells. PloS One (2010) 5:e9009. doi: 10.1371/journal.pone.0009009
384. Tankou SK, Regev K, Healy BC, Tjon E, Laghi L, Cox LM, et al. A Probiotic Modulates the Microbiome and Immunity in Multiple Sclerosis. Ann Neurol (2018) 83:1147–61. doi: 10.1002/ana.25244
385. Yan Y, Ramanan D, Rozenberg M, McGovern K, Rastelli D, Vijaykumar B, et al. Interleukin-6 Produced by Enteric Neurons Regulates the Number and Phenotype of Microbe-Responsive Regulatory T Cells in the Gut. Immunity (2021) 54:499–513.e5. doi: 10.1016/j.immuni.2021.02.002
386. Teratani T, Mikami Y, Nakamoto N, Suzuki T, Harada Y, Okabayashi K, et al. The Liver-Brain-Gut Neural Arc Maintains the Treg Cell Niche in the Gut. Nature (2020) 585:591–6. doi: 10.1038/s41586-020-2425-3
387. McKinney EF, Lyons PA, Carr EJ, Hollis JL, Jayne DRW, Willcocks LC, et al. A CD8+ T Cell Transcription Signature Predicts Prognosis in Autoimmune Disease. Nat Med (2010) 16:586–591, 1p following 591. doi: 10.1038/nm.2130
388. Lee JC, Lyons PA, McKinney EF, Sowerby JM, Carr EJ, Bredin F, et al. Gene Expression Profiling of CD8+ T Cells Predicts Prognosis in Patients With Crohn Disease and Ulcerative Colitis. J Clin Invest (2011) 121:4170–9. doi: 10.1172/JCI59255
389. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring Oxidant Signaling Suppresses Proarthritogenic T Cell Effector Functions in Rheumatoid Arthritis. Sci Transl Med (2016) 8:331ra38. doi: 10.1126/scitranslmed.aad7151
390. Wagner N-M, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S, et al. Circulating Regulatory T Cells Are Reduced in Obesity and May Identify Subjects at Increased Metabolic and Cardiovascular Risk: Regulatory T Cells and Obesity. Obesity (2013) 21:461–8. doi: 10.1002/oby.20087
391. Pacella I, Piconese S. Immunometabolic Checkpoints of Treg Dynamics: Adaptation to Microenvironmental Opportunities and Challenges. Front Immunol (2019) 10:1889. doi: 10.3389/fimmu.2019.01889
392. Schmidleithner L, Thabet Y, Schönfeld E, Köhne M, Sommer D, Abdullah Z, et al. Enzymatic Activity of HPGD in Treg Cells Suppresses Tconv Cells to Maintain Adipose Tissue Homeostasis and Prevent Metabolic Dysfunction. Immunity (2019) 50:1232–48.e14. doi: 10.1016/j.immuni.2019.03.014
393. Wu D, Han JM, Yu X, Lam AJ, Hoeppli RE, Pesenacker AM, et al. Characterization of Regulatory T Cells in Obese Omental Adipose Tissue in Humans. Eur J Immunol (2019) 49:336–47. doi: 10.1002/eji.201847570
394. Li M, Zhao W, Wang Y, Jin L, Jin G, Sun X, et al. A Wave of Foxp3+ Regulatory T Cell Accumulation in the Neonatal Liver Plays Unique Roles in Maintaining Self-Tolerance. Cell Mol Immunol (2020) 17:507–18. doi: 10.1038/s41423-019-0246-9
395. Klingenberg R, Gerdes N, Badeau RM, Gisterå A, Strodthoff D, Ketelhuth DFJ, et al. Depletion of FOXP3+ Regulatory T Cells Promotes Hypercholesterolemia and Atherosclerosis. J Clin Invest (2013) 123:1323–34. doi: 10.1172/JCI63891
396. Andersen CJ, Murphy KE, Fernandez ML. Impact of Obesity and Metabolic Syndrome on Immunity. Adv Nutr (2016) 7:66–75. doi: 10.3945/an.115.010207
397. Frydrych LM, Bian G, O’Lone DE, Ward PA, Delano MJ. Obesity and Type 2 Diabetes Mellitus Drive Immune Dysfunction, Infection Development, and Sepsis Mortality. J Leukoc Biol (2018) 104:525–34. doi: 10.1002/JLB.5VMR0118-021RR
398. Ganeshan K, Chawla A. Metabolic Regulation of Immune Responses. Annu Rev Immunol (2014) 32:609–34. doi: 10.1146/annurev-immunol-032713-120236
399. van der Windt GJW, Everts B, Chang C-H, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
400. Pearce EL, Pearce EJ. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
401. Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
402. Seth RB, Sun L, Chen ZJ. Antiviral Innate Immunity Pathways. Cell Res (2006) 16:141–7. doi: 10.1038/sj.cr.7310019
403. Waickman AT, Powell JD. mTOR, Metabolism, and the Regulation of T-Cell Differentiation and Function. Immunol Rev (2012) 249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x
404. Wang R, Green DR. Metabolic Checkpoints in Activated T Cells. Nat Immunol (2012) 13:907–15. doi: 10.1038/ni.2386
405. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria Are Required for Antigen-Specific T Cell Activation Through Reactive Oxygen Species Signaling. Immunity (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
406. Pearce EL. Metabolism in T Cell Activation and Differentiation. Curr Opin Immunol (2010) 22:314–20. doi: 10.1016/j.coi.2010.01.018
407. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature (2009) 460:103–7. doi: 10.1038/nature08097
408. van der Windt GJW, Pearce EL. Metabolic Switching and Fuel Choice During T-Cell Differentiation and Memory Development. Immunol Rev (2012) 249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x
409. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The Transcription Factor Myc Controls Metabolic Reprogramming Upon T Lymphocyte Activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
410. Lo Y-C, Lee C-F, Powell JD. Insight Into the Role of mTOR and Metabolism in T Cells Reveals New Potential Approaches to Preventing Graft Rejection. Curr Opin Organ Transplant (2014) 19:363–71. doi: 10.1097/MOT.0000000000000098
411. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis Controls the Induction of Human Regulatory T Cells by Modulating the Expression of FOXP3 Exon 2 Splicing Variants. Nat Immunol (2015) 16:1174–84. doi: 10.1038/ni.3269
412. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-Like Receptor Signaling Balance Treg Cell Anabolic Metabolism for Suppression. Nat Immunol (2016) 17:1459–66. doi: 10.1038/ni.3577
413. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity (2016) 44:406–21. doi: 10.1016/j.immuni.2016.01.028
414. Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab (2019) 29:103–23.e5. doi: 10.1016/j.cmet.2018.09.020
415. He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, et al. Metabolic Control of Regulatory T Cell (Treg) Survival and Function by Lkb1. Proc Natl Acad Sci USA (2017) 114:12542–7. doi: 10.1073/pnas.1715363114
416. Yang K, Blanco DB, Neale G, Vogel P, Avila J, Clish CB, et al. Homeostatic Control of Metabolic and Functional Fitness of Treg Cells by LKB1 Signalling. Nature (2017) 548:602–6. doi: 10.1038/nature23665
417. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, et al. Mitochondrial Complex III Is Essential for Suppressive Function of Regulatory T Cells. Nature (2019) 565:495–9. doi: 10.1038/s41586-018-0846-z
418. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
419. Xie M, Zhang D, Dyck JRB, Li Y, Zhang H, Morishima M, et al. A Pivotal Role for Endogenous TGF-Beta-Activated Kinase-1 in the LKB1/AMP-Activated Protein Kinase Energy-Sensor Pathway. Proc Natl Acad Sci USA (2006) 103:17378–83. doi: 10.1073/pnas.0604708103
420. Gualdoni GA, Mayer KA, Göschl L, Boucheron N, Ellmeier W, Zlabinger GJ. The AMP Analog AICAR Modulates the Treg/Th17 Axis Through Enhancement of Fatty Acid Oxidation. FASEB J (2016) 30:3800–9. doi: 10.1096/fj.201600522R
421. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential Role of Mitochondrial Energy Metabolism in Foxp3+ T-Regulatory Cell Function and Allograft Survival. FASEB J (2015) 29:2315–26. doi: 10.1096/fj.14-268409
422. Chapman NM, Zeng H, Nguyen T-LM, Wang Y, Vogel P, Dhungana Y, et al. mTOR Coordinates Transcriptional Programs and Mitochondrial Metabolism of Activated Treg Subsets to Protect Tissue Homeostasis. Nat Commun (2018) 9:2095. doi: 10.1038/s41467-018-04392-5
423. Kempkes RWM, Joosten I, Koenen HJPM, He X. Metabolic Pathways Involved in Regulatory T Cell Functionality. Front Immunol (2019) 10:2839. doi: 10.3389/fimmu.2019.02839
424. Cluxton D, Petrasca A, Moran B, Fletcher JM. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front Immunol (2019) 10:115. doi: 10.3389/fimmu.2019.00115
425. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 Couples Immune Signals and Metabolic Programming to Establish T(reg)-Cell Function. Nature (2013) 499:485–90. doi: 10.1038/nature12297
426. Thurnher M, Gruenbacher G. T Lymphocyte Regulation by Mevalonate Metabolism. Sci Signal (2015) 8:re4. doi: 10.1126/scisignal.2005970
427. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) Kinase in Treg Cells Maintains Homeostasis and Lineage Stability. Nat Immunol (2015) 16:188–96. doi: 10.1038/ni.3077
428. Turner MS, Kane LP, Morel PA. Dominant Role of Antigen Dose in CD4+Foxp3+ Regulatory T Cell Induction and Expansion. J Immunol (2009) 183:4895–903. doi: 10.4049/jimmunol.0901459
429. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1α Is a Metabolic Switch Between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep (2019) 27:226–37.e4. doi: 10.1016/j.celrep.2019.03.029
430. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity (2017) 47:875–89.e10. doi: 10.1016/j.immuni.2017.10.017
431. Ye Y, Chen Y, Sun J, Zhang H, Meng Y, Li W, et al. Hyperglycemia Suppresses the Regulatory Effect of Hypoxia-Inducible Factor-1α in Pulmonary Aspergillus Fumigatus Infection. Pathog Dis (2020) 78:ftaa038. doi: 10.1093/femspd/ftaa038
432. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med (2011) 208:1367–76. doi: 10.1084/jem.20110278
433. Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of TH17/Treg Balance by Hypoxia-Inducible Factor 1. Cell (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
434. Pålsson-McDermott EM, O’Neill LAJ. Targeting Immunometabolism as an Anti-Inflammatory Strategy. Cell Res (2020) 30:300–14. doi: 10.1038/s41422-020-0291-z
435. Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. Adenosine-Mono-Phosphate-Activated Protein Kinase-Independent Effects of Metformin in T Cells. PloS One (2014) 9:e106710. doi: 10.1371/journal.pone.0106710
436. Palma C, La Rocca C, Gigantino V, Aquino G, Piccaro G, Di Silvestre D, et al. Caloric Restriction Promotes Immunometabolic Reprogramming Leading to Protection From Tuberculosis. Cell Metab (2021) 33:300–18.e12. doi: 10.1016/j.cmet.2020.12.016
437. Li Y, Wei C, Xu H, Jia J, Wei Z, Guo R, et al. The Immunoregulation of Th17 in Host Against Intracellular Bacterial Infection. Mediators Inflamm (2018) 2018:6587296. doi: 10.1155/2018/6587296
438. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
Keywords: T cells, regulatory T cells, microorganism, pathogens, neuroinflammation, autoimmunity, microbiome, immunometabolomics
Citation: Schroeter CB, Huntemann N, Bock S, Nelke C, Kremer D, Pfeffer K, Meuth SG and Ruck T (2021) Crosstalk of Microorganisms and Immune Responses in Autoimmune Neuroinflammation: A Focus on Regulatory T Cells. Front. Immunol. 12:747143. doi: 10.3389/fimmu.2021.747143
Received: 25 July 2021; Accepted: 20 September 2021;
Published: 07 October 2021.
Edited by:
Cinthia Farina, San Raffaele Scientific Institute (IRCCS), ItalyReviewed by:
Claudio Procaccini, Istituto per l’Endocrinologia e l’oncologia “Gaetano Salvatore” (CNR), ItalyWakiro Sato, National Center of Neurology and Psychiatry, Japan
Copyright © 2021 Schroeter, Huntemann, Bock, Nelke, Kremer, Pfeffer, Meuth and Ruck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tobias Ruck, dG9iaWFzLnJ1Y2tAbWVkLnVuaS1kdWVzc2VsZG9yZi5kZQ==