
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 26 October 2021
Sec. Antigen Presenting Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.746469
This article is part of the Research TopicLocal Immune Modulation of Macrophages and Dendritic CellsView all 14 articles
 David Hongo1
David Hongo1 Pingping Zheng2
Pingping Zheng2 Suparna Dutt1
Suparna Dutt1 Rahul D. Pawar1
Rahul D. Pawar1 Everett Meyer2
Everett Meyer2 Edgar G. Engleman3
Edgar G. Engleman3 Samuel Strober1*
Samuel Strober1*Classical dendritic cells (cDCs) in mice have been divided into 2 major subsets based on the expression of nuclear transcription factors: a CD8+Irf8+Batf3 dependent (DC1) subset, and a CD8-Irf4+ (DC2) subset. We found that the CD8+DC1 subset can be further divided into CD8+DC1a and CD8+DC1b subsets by differences in surface receptors, gene expression, and function. Whereas all 3 DC subsets can act alone to induce potent Th1 cytokine responses to class I and II MHC restricted peptides derived from ovalbumin (OVA) by OT-I and OT-II transgenic T cells, only the DC1b subset could effectively present glycolipid antigens to natural killer T (NKT) cells. Vaccination with OVA protein pulsed DC1b and DC2 cells were more effective in reducing the growth of the B16-OVA melanoma as compared to pulsed DC1a cells in wild type mice. In conclusion, the Batf3-/- dependent DC1 cells can be further divided into two subsets with different immune functional profiles in vitro and in vivo.
Two subsets of DC1 dendritic cells differ by surface phenotype, gene expression, and function.
The subset of CD8+dendritic cells (DCs) that is dependent on the Batf3 and Irf8 nuclear transcription factors for development and maturation in mouse lymphoid tissues has been extensively studied (1–5). This subset, that has been identified as DC1 by Murphy and co-workers (6, 7) plays a required role in the induction of CD8+ T cell immunity to tumors and viruses (8–15). DC1 cells express the surface markers CD11c and MHCII associated with all classical myeloid DCs, and express high levels of CD24 and XCR1 and low levels of CD172 (SIRPα) surface markers (5–16). This surface phenotype has been used to distinguish the DC1 subset from the CD8- DC2 subset that expresses low levels of CD24 and XCR1 and high levels of CD172a (6, 7). Inactivation of the gene encoding Batf-3 results in the selective elimination of CD8+ and CD103+ DC1 DCs (17, 18). Both subsets express high levels of XCR1, low levels of CD172, and can stimulate CD8+ T cell immunity (19–21). In contrast to the DC1 subset, the DC2 subset is dependent on the Irf4 nuclear transcription factor and has been subdivided further into Notch2-dependent and KIf4-dependent populations (22, 23). Batf3 is expressed in both the Irf8+ and Irf4+ DCs (5). Nevertheless, the DC2 cells are predominantly Batf3 independent (20).
It is not clear whether the CD8+ Batf-3 dependent DC1 subset is homogeneous or whether it can be divided further into additional subsets. Phenotypic and functional heterogeneity of the subset has been described previously with regard to the expression of CD103 in the spleen, skin draining lymph nodes, tumors, and intestines (24–28). DC1 cells in the skin draining lymph nodes, tumors, and intestines express high levels of CD103 whereas DC1 cells in the spleen have low or undetectable levels (24–28). The CD8+ subset of DCs has been reported to be more potent than the CD8- subset in cross-presenting cell associated protein antigens to conventional CD8+ T cells, and selective depletion of the CD8+ subset markedly attenuates CD8+ T cell immunity to tumors and viruses by virtue of cross antigen presentation (29–32). CD8+ DCs take up proteins from exogenous cell sources that are the source of the antigens, such as tumor cells and viral infected cells, process the proteins such that the derived peptides are cross-presented to CD8+ T cells after association with MHC receptors on the cell surface (1–11). In accordance with these observations, the ability of radiation therapy to induce T cell mediated durable complete remissions of solid and lymphoid tumors was dependent on the presence of CD8+Batf3 dependent DCs, since remissions observed in wild type mice were abrogated in Batf3-/- mice (33, 34). Remissions were restored by injecting CD8+ DCs into the tumors (33, 34).
Previous studies showed that after uptake of the soluble ovalbumin protein injected in vivo, the CD8+ DCs were effective at presenting the antigen to ovalbumin specific TCR transgenic CD8+T cells but not CD4+ T cells, and CD8-DCs were effective at presenting to transgenic CD4+T cell (32). These observations are consistent with the dominant role of CD8+DCs in the induction of CD8+T cell immunity to cell bound viral and tumor antigens. However, vaccination with soluble ovalbumin protein linked to antibodies directed to the DCIR2 surface receptor of the CD8- subset of DCs induced effective CD8+T cell immunity against the B16-OVA melanoma tumor after injection of the conjugate in vivo (35). Thus, CD8-DC2 cells can also induce CD8+T cell immunity to an ovalbumin tumor antigen depending on the nature of the vaccination. However, it was not determined whether CD8+DCs in the tumor microenvironment were also required for the CD8+ T cell anti-tumor effect in addition to the systemic immune response induced by the DCIR2 targeted CD8- DCs.
The CD8+DC1 and CD8-DC2 subsets have also been compared for their efficacy in presenting glycolipid antigens to NKT cells (36, 37). The latter T cells recognize glycolipids in association with the CD1d antigen presenting molecule (38–40). The DC1 subset was effective and the DC2 subset was ineffective in the induction of NKT cell anti-glycolipid immune responses. It is not clear whether the ability of the CD8+ DC1 subset to present soluble and cell associated protein antigens to CD8+ T cells, on one hand, and glycolipid antigens to NKT cells, on the other, is a function of a single subset or of at least two subsets of CD8+ DC1 cells.
The current study identified two major subsets of Batf3 dependent CD8+ DCs (DC1a and DC1b) that differ from each other and from the CD8-DC2 subset on the basis of clear differences in their surface receptors, and gene expression. The ability of all of the latter subsets to induce T cell immune responses to a variety of antigens was compared, and key differences were elucidated.
Single cells were harvested from the spleen of adult wild type (WT) C57BL/6 mice, stained for surface receptors, and CD3-CD56-CD3-CD19-Gr-1- (Lin-) cells were analyzed by flow cytometry for CD11c vs MHC class II expression. As shown in (Figure 1A), 4.5% were contained in the box enclosing a discrete population of CD11c+MHCII+ cells that were identified as dendritic cells (DCs). Further study of the gated DCs in the spleen of WT mice for expression of CD8 vs CD172a showed that 3 populations of cells with discrete concentric contours were present (enclosed in ellipses in Figure 1A). Population 1 (identified as the DC1a subset in all subsequent Figures and Tables) contained CD8hiCD172lo cells, population 2 (identified as the DC1b subset) contained CD8intCD172int cells, and population 3 (identified as DC2 subset) contained CD8-CD172hi cells. In order to determine whether populations 1 and 2 expressed the Irf8+/Irf4- nuclear factor expression pattern that characterizes DC1cells, all three populations were gated separately, and stained for intracellular Irf8 and Irf4. As shown in the histograms in Figure 1A, both the DC1a and DC1b populations expressed the Irf8+/Irf4- pattern. In contrast, the DC2 population, expressed the Irf8-Irf4+ pattern that has been shown previously to characterize the DC2 subset. The mean percentages of the 3 populations among gated CD11c+MHC+ cells enclosed in the ellipses in the spleen of C57BL/6 WT mice are shown in the panel of Figure 1B. DC1a, DC1b, and DC2 subsets made up about 12%,48%, and 40% of total DCs respectively. In additional experiments, the mean percentages of the 3 subsets among splenic DCs in C57BL/6 and BALB/c WT mice were compared. The DC1b cells represented the majority of DCs in both strains, and the mean percentage of DC2 cells was significantly higher in the BALB/c mice.
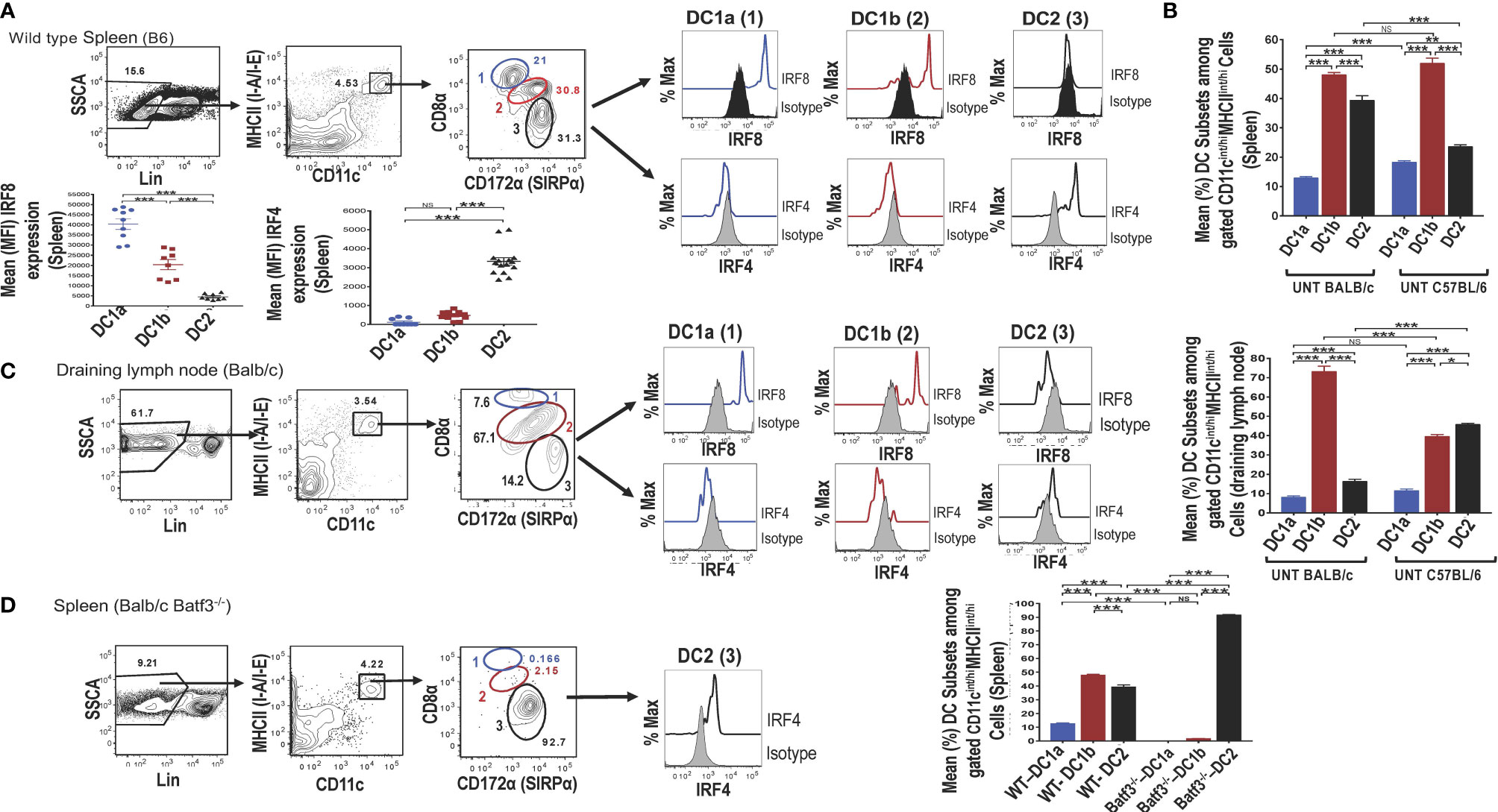
Figure 1 Identification of 2 subsets of CD8+ Batf3 dependent DCs. Spleen cells from untreated wild-type (WT) C57BL/6 or BALB/c mice were stained and analyzed by flow cytometry for DC susbsets. (A) Show representative two color FACS gating strategy to identify MHCII(I-A/I-E)+ CD11+ total DCs after gating on lineage negative (CD3, B220, CD19, CD56, Gr-1) spleen cells. Boxes in panels show the percentages of cells enclosed. Gated total DCs cells were further analyzed for CD8 versus CD172a, and 3 discrete populations were observed; populations 1 (DC1a), 2 (DC1b), and 3 (DC2) (indicated by blue, red, and black ovals) respectively. The 3 populations were analyzed for intracellular staining of the Irf8 and Irf4 nuclear factors compared to isotype control staining. Representative histograms are shown as well as the mean (MFI) for Irf8 and Irf4 staining intensity among the gated DC populations (n = 8-10). One-way ANOVA (Holm-Sidak’s multiple comparison test) was used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001, NS-not significant (p > 0.05). (B) Compares the mean (± SEM) percentages of populations 1 (DC1a), 2 (DC1b), and 3 (DC2) among total DCs from the spleens of untreated (UNT) WT C57BL/6 and BALB/c mice (n =18-20). (C) Shows staining for CD8α versus CD172 on gated CD11c+MHCII+ draining lymph node cells from WT BALB/c mice. The 3 gated populations of DCs were stained for Irf8 and Irf4, and representative histograms are shown. The bar graphs compare the mean percentages of DC1a, DC1b, and DC2 cells from draining lymph nodes of WT BALB/c and C57BL/6 mice (D) Shows representative two color FACS patterns of CD11c+ MHCII+ cells among spleen cells from Batf3-/- BALB/c mice. Gated cells from each box were further analyzed for CD8 versus CD172a. Too few populations 1 and 2 cells were available to stain for nuclear factors, and expression of Irf4 by population 3 is shown in a representative histogram. The mean percentages of the 3 DC populations are compared from WT versus Batf3-/- BALB/c mice in the bar graphs.
The mean fluorescence intensity (MFI) of staining for Irf8 and Irf4 among the 3 populations of gated splenic DCs is shown in Figure 1A. Mean intensity for Irf8 was highest in the DC1a cells, intermediate in DC1b cells and at background levels in DC2 cells. Differences in means were statistically significant for all 3 populations (p<0.001). As expected, the staining pattern for Irf4 showed the opposite pattern with the mean for Irf4 significantly increased (p<0.001) as compared to background staining levels for DC1a and DC1b cells.
Similar analysis of expression of CD8 vs CD172 on gated DCs from the draining lymph nodes of WT BALB/c mice showed that the same discrete contours for 3 populations was observed, and histograms of Irf8 and Irf4 staining followed the pattern observed in the spleen (Figure 1C). Although the mean percentage of DC1b cells in the draining nodes of BALB/c mice was about 70%, the DC2 cells constituted the majority of DCs in the draining nodes of C57BL/6 mice (Figure 1C).
Additional analysis of gated DCs from the spleen of Batf3-/- BALB/c mice showed a dramatic reduction of the DC1a and DC1b cells in representative histograms, and the DC2 cells with the Irf8-Irf4+ pattern accounted for about 93% of total DCs (Figure 1D). The DC1a and DC1b population each made up means of less than 3% of the gated DCs, and the DC2 cells made up a mean of over 90% (of Figure 1D). Mean percentages of each subset are compared for BALB/c WT versus Batf3-/- mice in the bar graph in Figure 1D. Mean percentages for DC1a and DC1b cells in the latter mice were below 2% and the mean for DC2 cells was above 90%. The loss of DC1a and DC1b subsets indicated that these were CD8+Batf3 dependent DCs that have been previously identified as DC1 cells (6, 7). The DC2 subset was similar to the CD8-CD172hi subset previously identified as the DC2 subset (6, 7).
In an effort to further distinguish the DC populations from one another, we stained the 3 gated subsets of DCs in the WT BALB/c spleen for a variety of additional surface receptors including those previously shown to identify DC1 cells such as XCR1 and CD24 (5–7). Figure 2A shows that XCR1, CD24, DEC205, Tim-3, Clec9A, BTLA, MHCII, and CD8 were highly expressed on DC1a cells as compared to other DC populations (MFI vs DC1b and DC2 cells p<0.001). In contrast, the MFI for CD1d, CD103, and Tim4 on DC1b cells was significantly increased as compared to that of DC1a and DC2 cells (Figure 2B). Surface receptors highly expressed on DC2 cells as compared to DC1a and DC1b cells were CD11b, CD40, CD172, and CD4 (p<0.001) (Figure 2C). Figure 2D summarizes the patterns of high levels of receptor expression that distinguishes each of the 3DC populations.
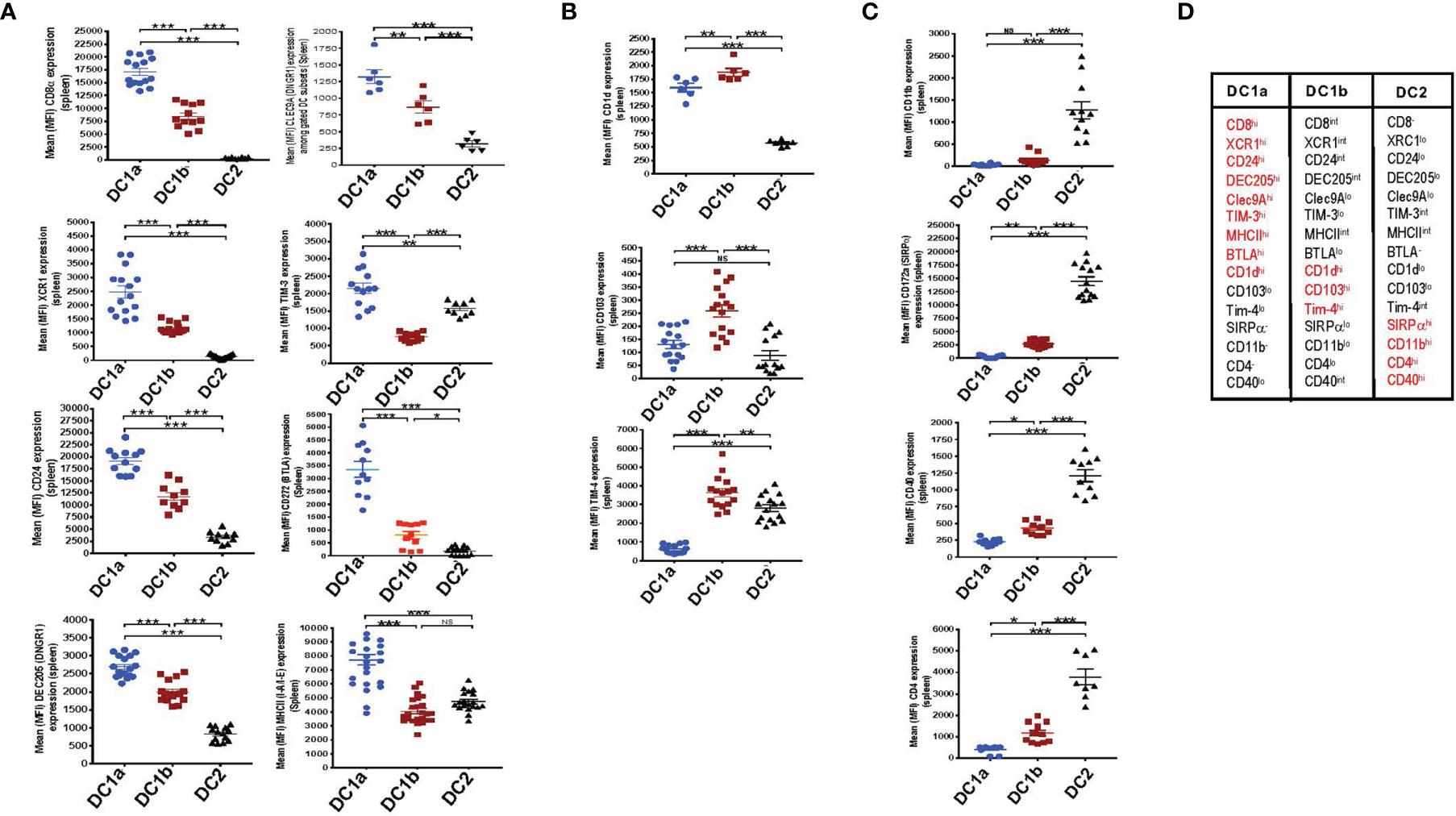
Figure 2 DC1a and DC1b cells in the spleen differ in their expression of multiple surface markers that also distinguish them from DC2 cells. (A) Shows individual and mean (MFI) levels of staining intensity of receptors that are most highly expressed on gated population 1(DC1a) cells including CD8a, XCR1, CD24, DEC205, Clec9A, TIM-3, MHCII, and BTLA (CD272) in WT BALB/c spleen cell samples. (B) Shows MFI levels of receptors most highly expressed on population 2 (DC1b) cells including CD1d, CD103, (DNGR1), and TIM-4. (C) Shows MFI levels of receptors most highly expressed on population 3 (DC2) cells including CD11b, CD172a (SIRP), CD40, and CD4. (D) Table shows the summary patterns of high expression of surface receptors on 3 DC subsets in the spleen. Receptors in red are those with MFIs that were significantly increased as compared to that of the two other DC populations. (N = 10-20). One-way ANOVA (Holm-Sidak’s multiple comparison test) was used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant (p > 0.05). MFI, mean fluorescence intensity.
In further studies, we elucidated the distribution of the DC populations in the mesenteric lymph nodes (msLNs), intestines (gut), liver, and peripheral blood. Mononuclear cells were collected from the four tissue sources from WT BALB/c mice, and the percentages of these cells among total DCs were determined and compared to that in the spleen and draining lymph nodes. MHCII+CD11c+ total DCs from each tissue were gated as before and analyzed for the expression of CD8 versus CD172 as shown in Figure 3A. Three DC populations were clearly identified among the the msLN cells and peripheral blood mononuclear cells (PBMC) as shown in the representative two color stainings. In contrast, only one population of DCs that had the staining characteristics of DC1b cells was found among the gut mononuclear cells, and neither the DC1a nor the DC2 populations were identified (Figure 3A). Two DC populations with characteristics of the DC1a and DC1b cells were found among liver mononuclear cells, and the DC2 population was not observed (Figure 3A).
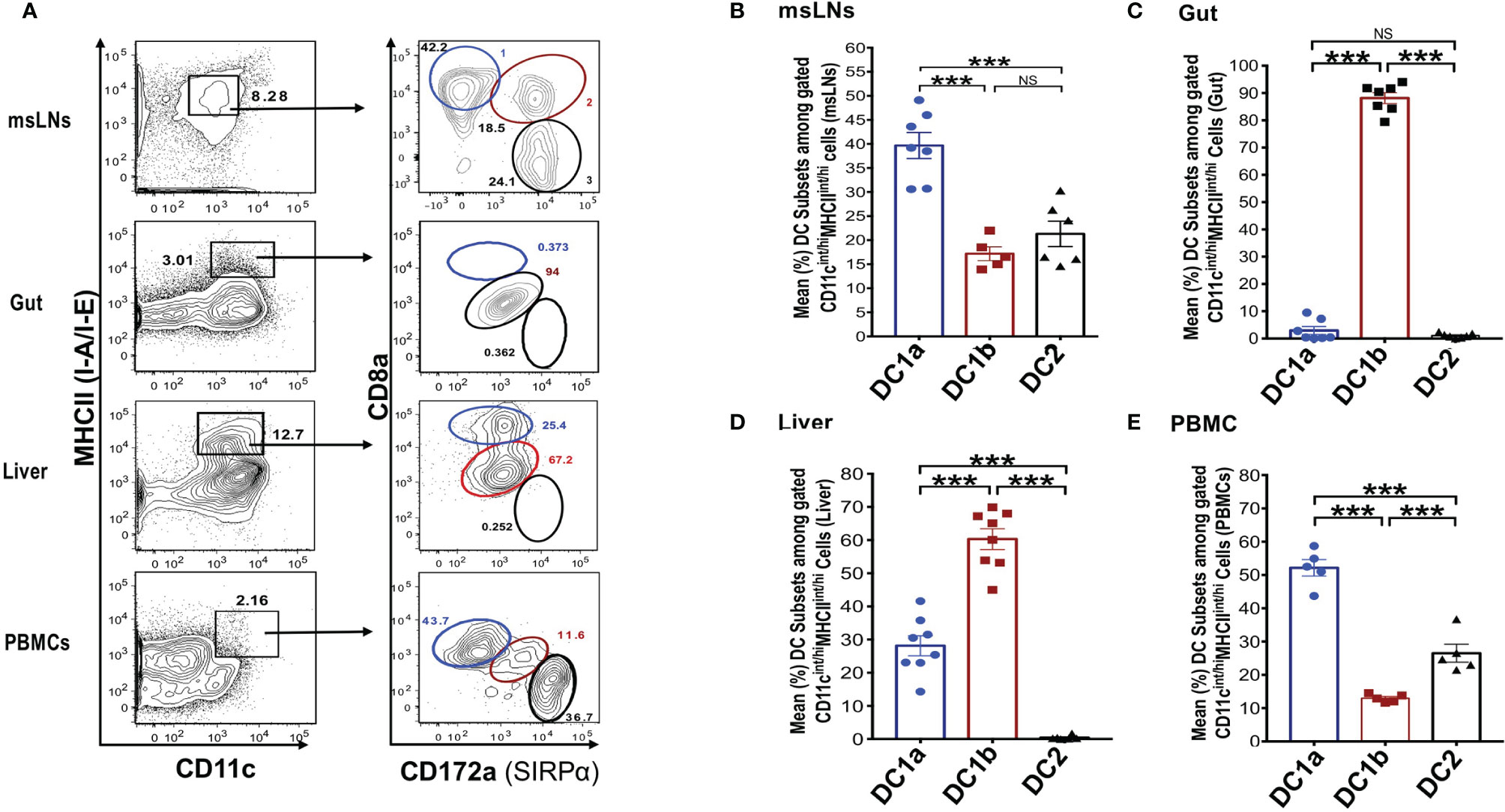
Figure 3 Distribution of 3 DC subsets in different tissues (mesenteric LNs, gut, liver and PBMCs). (A) Shows representative FACS staining of gated MHCII+CD11c+ total DCs from mesenteric lymph nodes (msLNs), intestines (gut), liver and PBMCs of WT BALB/c mice. Total DCs were further analyzed for CD172a versus CD8a staining, and ellipses outline populations 1,2, and 3(DC1a, DC1b, DC2). Mean (SEM) percentages of 3 DC populations among total DCs in (B) msLNs, (C) gut, (D) liver, and (E) PBMCs cells. (N = 6-10). One-way ANOVA (Holm-Sidak’s multiple comparison test) was used for statistical analysis. ***p < 0.001, NS, not significant.
Figures 3B-E shows the mean percentages of each DC population among total DCs from the 4 tissues. Each population accounted for a mean of at least 10% among total DCs in the msLNs and PBMC, and the mean percentage of DC1a cells were significantly increased as compared to the other populations in these tissues (p<0.001) (Figures 3B, E). The mean percentage of DC1a and DC2 cells among total DCs in the gut accounted for less that 2%, and almost all were DC1b cells (Figure 3C). The mean percentage of DC2 cells was below 1% in the liver, and about 60% were DC1b cells and 30% DC1a (Figure 3D). In summary, three populations of DCs were found in the msLNs and PBMC as was observed in the spleen and draining lymph nodes. Whereas the dominant population in the latter two tissues was DC1b, the dominant population among the msLNs and the PBMC was DC1a. DC2 cells were not identified in the two non-lymphoid tissues (gut and liver), and were easily identified in the four lymphoid tissues.
In order to compare the gene expression pattern of the 3 DC populations in the spleen of WT BALB/c mice, the cells were sorted by flow cytometry according to the gates shown in Figure 1A for populations 1, 2, and 3, RNA was extracted from the sorted cells, and the gene expression profiles were compared using the RNAseq. Figure 4A shows the heat map comparison of gene expression for sorted DC1a vs DC1b cells, Figure 4B shows the comparison for DC1a vs DC2 cells, and Figure 4C shows the comparison for DC1b vs DC2 cells. The 3 heat maps clearly distinguished the gene expression patterns of the 3 sorted populations. Further quantitative analysis of differences in the immune response and signaling pathways encoded by the upregulated genes of the DC1b versus DC1a cells are shown in Supplemental Figure 1.
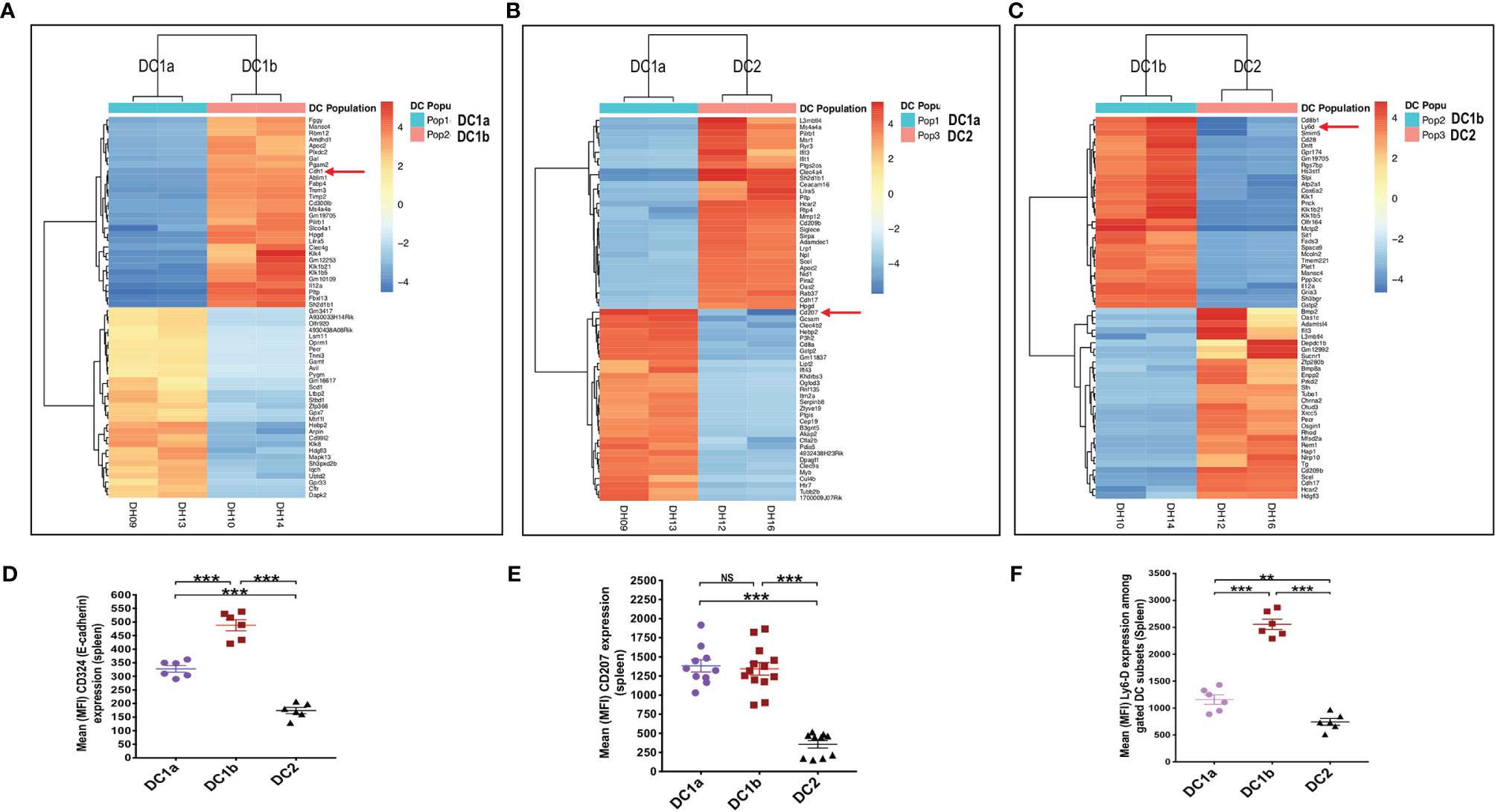
Figure 4 Gene expression profiling reveals additional differences between the 3 DC subsets. (A) Heat map of genes with significantly increased or decreased expression levels (as determined by RNA-seq analysis) in sorted DC1a (Pop 1) versus DC1b (Pop 2) or (B) DC1a (Pop 1) versus DC2 (Pop 3) or (C) DC1b (Pop 2) versus DC2 (Pop 3) from the WT BALB/c spleen. Each row represents a gene identified on the right side of the map, and each column represents a result from a single sample. The color scale shown on the map shows the relative expression levels of the genes across all the samples. Red and yellow shades represent higher expression levels, while blue shades represent lower expression levels. (D–F) Representative staining intensities and MFI of upregulated receptors CD207 (langerin), CD324 (E-cadherin; Cdh1), and Ly6-D cells on DC subsets (n = 10-20). The genes of these three markers were identified by the RNAseq analysis and were stained to confirm their expression at the protein level. One-way ANOVA (Holm-Sidak’s multiple comparison test) was used for statistical analysis of MFI. *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant (p > 0.05).
In particular, genes encoding pro-inflammatory and allograft rejection immune response pathways including upregulation of signaling pathways for TNFalpha and IL-2 were significantly increased in the DC1b versus DC1a cells as judged by normalized enrichment scores (NES)(Supplementary Figure 1A). Similar upregulation of genes encoding pro-inflammatory response pathways and cytokine signaling including interferon alpha and gamma, TNFalpha, or IL-2 were observed as judged by NES when DC2 cells were compared to DC1a cells or when DC2 cells were compared to DC1b cells (Supplementary Figures 1B, C). In contrast, expression of gene pathways encoding Myc targets and oxidative phosphorylation were reduced when comparing DC1b versus DC1a cells, and DC2 versus DC1a cells. Principal component analysis (Supplementary Figure 1D) also showed marked differences in gene expression patterns of the 3 DC populations with variances of 40% and 29% for PC1 and PC2 respectively. Duplicate experiments for purification of each of the 3 populations showed marked concordance of gene expression patterns within each population as compared to marked differences between populations (Supplemental Figure 1E).
Analysis of the upregulated genes shown in Figures 4A-C identified three surface proteins that were predicted to be upregulated in DC1b vs DC1a cells: CD324 (E-cadherin), DC1a vs DC2 cells CD207 (langerin), and DC1b vs DC2 cells (Ly6-D). Staining of the splenic DCs showed that the MFIs of the 3 receptors were increased the appropriate cell populations as predicted by the gene expression patterns (Figures 4D–F).
We tested the ability of the 3 DC subsets to present a class I MHC restricted ovalbumin (OVA) peptide antigen to OT-1 CD8+T cells harvested from C57BL/6 mice bearing a transgenic TCR that recognizes this peptide antigen (41, 42). In addition, we tested the ability of the 3 DC subsets to present a class II MHC restricted peptide to OT-II CD4+ T cells with the appropriate transgenic TCR (42). Since the 3 DC subsets described above expressed both class I and class II MHC on their cell surfaces, we hypothesized that each DC subset would be able to present both the class I and class II restricted peptides to the OT-I and OT-II T cells respectively when the peptides were added to cultures of each DC subset with the appropriate T cells. However, the lower levels of class II MHC on DC1b cells as compared to the DC1a and DC2 cells, could reduce the activity of the former as compared to the latter subsets with regard to presentation of the class II restricted peptide.
In order to test the antigen presenting activity of the 3 DC subsets independent of the need for antigen processing, OT-1 CD8+ T cells were harvested from the spleens of C57BL/6 transgenic mice and cultured in the presence or absence of sorted C57BL/6 DC1a, DC1b, or DC2 cells in the presence or absence of the class I MHC restricted SIINFEKL peptide derived from ovalbumin. Culture supernatants were assayed for IL-4, IFN, IL-13, and IL-2. Figure 5A shows that all 3 subsets of DCs stimulated the robust secretion of IFN and IL-2 with little or no secretion of IL-4 (<100pg/ml) and modest secretion of IL-13 (<600pg/ml). The mean concentrations of IFN (about 9,000 pg/mL) in the cultures containing the combination of CD8+T cells with each DC subset, and peptide were increased (p<0.001) as compared to the means in cultures containing the combination of CD8+ T cells and peptide without the DCs. DC1a cells stimulated a mean concentration that was a few hundred pg/mL higher than that of the DC1b and DC2 cells (p<0.05). In contrast, the DC2 subset stimulated the highest secretion of IL-2 (mean about 12,5000pg/mL) that was about 10,000 pg/mL higher than that stimulated by the DC1a subset (p<0.001).
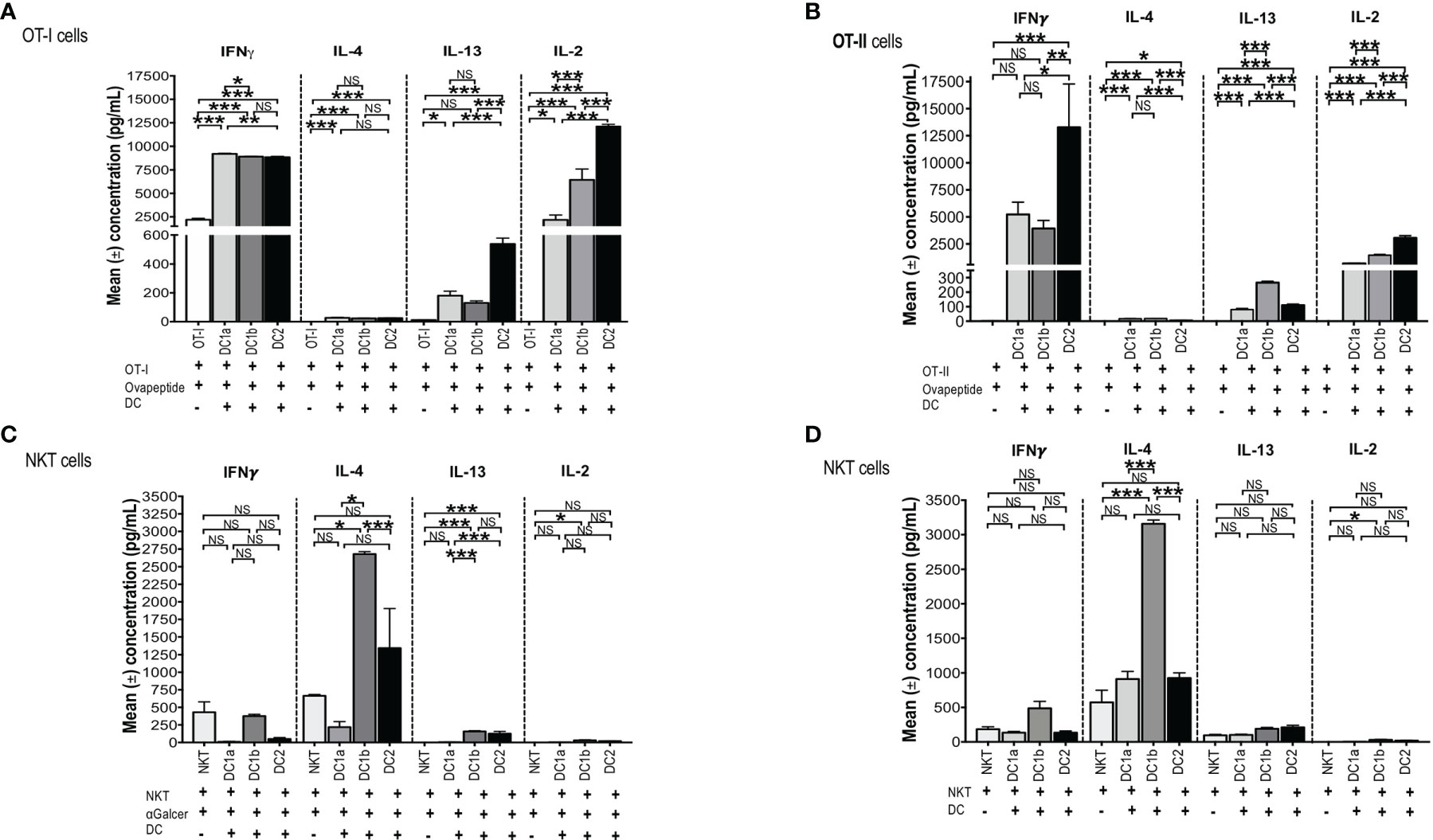
Figure 5 All 3 DC subsets can present Class I, and II MHC restricted OVA peptide antigens, but only DC1b cells present glycolipid antigens. (A) Mean concentrations of IL-2 (Right), IL-13 (Right Middle), IL-4 (Left Middle), and IFNγ (Left) in culture supernatants of FACS-sorted DC subsets (DC1a, DC1b and DC2) (1x104 per well) from wild type untreated C57BL/6 mice that were cultured with OT-I restricted OVA257-264 peptide (1g/mL) and with CFSE-labelled OT-I CD8+ T-cells (1x105 cells per well). In control experiments, CFSE-labelled OT-I CD8+ T-cells (1x105 cells per well) were incubated with OVA257-264 peptide (1g/mL) but without DCs for 5 days. The concentrations of IL-4, IL-13, IL-2, IFN in culture supernatants was measured by Luminex. (B) Mean concentrations of IL-2 (Right), IL-13 (Right Middle), IL-4 (Left Middle), and IFNγ (Lef) in culture supernatants of FACS-sorted DC subsets (DC1a, DC1b and DC2) (1x104 per well) from wild type untreated C57BL/6 mice that were cultured with OT-II restricted OVA323-339 peptide (10g/mL) with CFSE-labelled OT-II CD4+ T-cells (1x105 cells per well). In control experiments, CFSE-labelled OT-II CD4+ T-cells (1x105 cells per well) were incubated with OVA323-339 peptide (10g/mL) without DCs for 5 days. The concentrations of IL-4, IL-13, IL-2, IFN in culture supernatants was measured by Luminex. All data are representative of three independent experiments (6 mice per experiment). (C, D) Mean concentrations of cytokines in cultures of sorted DC1a (Pop 1), DC1b (Pop 2) and DC2 (Pop 3) (15x103 cells/well) subsets incubated with sorted NKT cells (5x105 cells/well) from untreated wild type BALB/c spleen cells. AlphaGalcer (α-galactosylceramide) (100ng/mL) was added to all cultures in (C) but not in (D). Control cultures contained NKT cells with (C) or without (D) glycolipid but without DCs. Supernatants were harvested after 5 days, and concentrations were measured by Luminex. Data pooled from 2 or more independent experiments for a total of at least 10-12 mice per group, represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, NS, not significant (p > 0.05). One-way ANOVA (Holm-Sidak’s multiple comparison test) was used for statistical analysis.
In further experiments, we tested the ability of the 3 DC subsets to present a class II MHC restricted peptide to CD4+ OT-II T cells. Figure 5B shows that all 3 DC subsets stimulated robust secretion of IFN and IL-2 at levels that were comparable to those observed with OT-I cells (2,500-12,500pg/ml), and minimal secretion (<100pg/ml) of IL-4 or IL-13 (<300/pg/ml) consistent with the Th1 pattern observed with OT-I cells. The experiments with OT-II cells were repeated using ovalbumin protein instead of ovalbumin peptide. As shown in Supplemental Figures 2A-D, the Th1 pattern of OT-II cell immune response was similar when the protein was used instead of the peptide. In particular, the 3 DC subsets stimulated robust secretion of IFN gamma and IL-22 (>2,500pg/ml), and considerably less IL-2, IL-13, TNF alpha, and IL-17 (<1,000 pg/ml). The results indicate that all 3 DC subsets can process the protein for presentation to the OT-II T cells. Stimulation of the OT-I and OT-II cells by DCs pulsed with peptide or protein as measured by proliferation (CFSE dilution), indicated that all 3 subsets were effective inducers of proliferation (data not shown).
A previous study, showed that Batf-3 dependent CD8+ DCs were able to present a variety of glycolipids, including the potent activating Galcer glycolipid, to NKT cells in vitro and in vivo, but the Batf-3 independent DC2 cells could not (36). We hypothesized that the DC1b subset described above would be more potent than the DC1a and DC2 subsets in stimulating an immune response to the Galcer glycolipid after co-culture with purified NKT cells, since the DC1b subset expressed significantly higher levels of glycolipid antigen presenting molecule, CD1d, as compared to the DC1a and DC2 DC subsets (Figure 2).
To test this hypothesis, CD1dtetramer+ NKT cells were harvested from the spleens of BALB/c mice and cultured in the presence or absence of sorted BALB/c DC1a, DC1b, or DC2 cells in the presence of Galcer. Figure 5C compares the mean concentrations of IFN, IL-4, IL-13 and IL-2 from cultures of NKT cells and Galcer without or without the purified DC subsets. Only DC1b cells induced IL-4 (about 2,500 pg/mL) as compared to cultures lacking DCs. DC1b cells induced significantly higher levels of IL-4 as compared to DC1a and DC2 cells. DC2 cells failed to stimulate significantly increased production of IL-4 as compared to control cultures without DCs. Figure 5C shows that DC1b cells (but not DC1a) stimulated low levels of secretion of IL-13 (mean <100pg/ml) and IL-2 (mean <50pg/ml in vitro that were increased as compared to the background controls, but 25-50-fold less than IL-4.
Since, DCs are able to present constitutively expressed endogenous glycolipid antigens to NKT cells (48), the above experiments that tested the ability of DC subsets to stimulate cytokine secretion of purified NKT cells were repeated in the absence of Galcer. Figure 5D shows that the secretion of IL-4 induced by DC1b cells in the absence of Galcer was at least as robust as in the presence of Galcer. There was a highly significant increase in the mean concentration of IL-4 in NKT cell cultures with DC1b cells versus cultures with NKT cells alone (p<0.001). In addition, DC1a and DC2 cells failed to significantly increase IL-4 concentrations as compared to control cultures with NK T cells alone. A similar pattern was observed with IL-13 secretion; however, the concentrations were all below 200pg/ml. None of the 3 DC subsets induced robust NKT cell secretion of IFN or IL-2 (Figures 5C, D). In summary, only DC1b cells induced robust Th2 cytokine secretion by NKT cells that was predominantly polarized toward IL-4 in contrast to the TH1 pattern induced in OT-I and OT-II by all 3 DC subsets. The response of NKT cells induced by DC1b cells is likely due to endogenous glycolipid antigens.
Batf3 dependent CD8+CD103+ DC1 cells, present in the microenvironment of tumors, cross-present tumor antigens to CD8+ T cells in the regional lymph nodes, and are required to initiate an immune response to the tumor (9–11). It is unclear, whether the DC1 cells are also required to initiate an anti-tumor immune response when the tumor antigens are incorporated into a subcutaneous vaccination to treat a tumor. In order to determine whether tumor antigen pulsed DC1a, DC1b, and/or DC2 cells can be used as a vaccination to initiate an immune response and slow the growth of the subcutaneous B16-OVA melanoma tumor, the purified subsets were incubated with ovalbumin whole protein and injected subcutaneously twice into wild type BALB/c mice 4 and 2 weeks before injection of the tumor cells. Figure 6 shows the growth of the subcutaneous tumors as measured by tumor volume on days 11, 15, 17 and 22 after tumor cell injection. Control wild type mice had no vaccination.
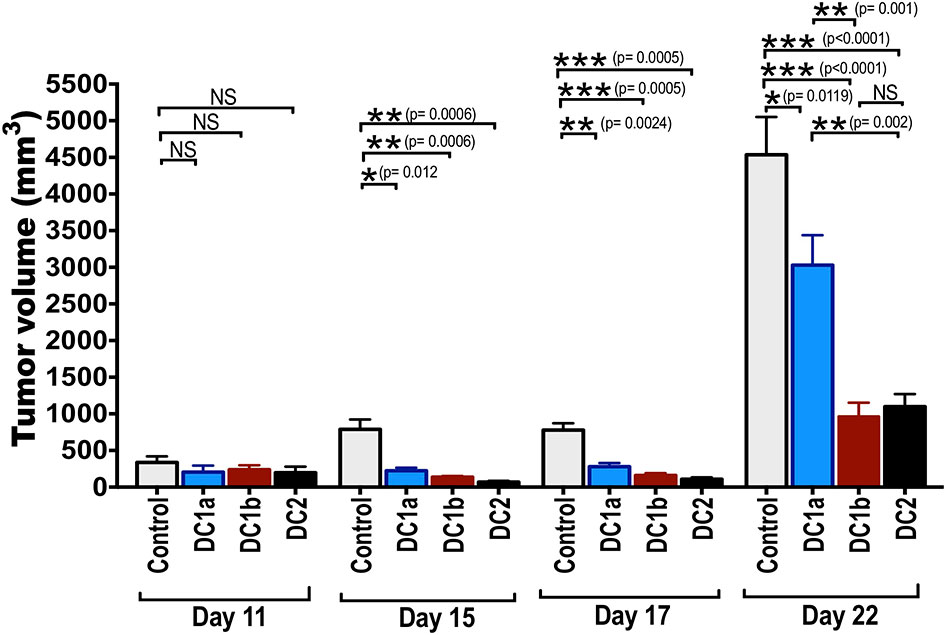
Figure 6 Tumor antigen-pulsed DC1b and DC2 cells are more effective than tumor antigen-pulsed DC1a cells at reducing tumor growth. Bar graphs depicting mean (+/-SE) tumor volumes in wild type C57BL/6 hosts monitored for 22 days. FACS-sorted DC subsets from wild type C57BL/6 mice were pulsed with whole OVA protein (500g/mL) in vitro for 1 hour, and then injected in vivo into wild type C57BL/6 hosts twice subcutaneously with a 2-week interval between injections. Hosts were then challenged with B16F10-ova tumor cells (5x104 cells/mouse) injected subcutaneously on the flank of mouse 2 weeks after the second injection of OVA pulsed DCs, and the tumor volumes monitored over time. Control mice received tumor cells with no vaccination. Data are representative of two independent experiments. Each data point reflects data from 6-10 mice injected with FACS-sorted DC subsets pooled from 8-10 mice. One-way ANOVA (Holm-Sidak’s multiple comparison test) results are indicated as *P < 0.05, **p < 0.01, ***p < 0.001, NS, not significant (p > 0.05).
The tumor growth in control mice was rapid, and the mean tumor volume at day 22 was about 4500mm3. Experimental mice that were vaccinated with the 3 different subsets of DCs showed significant slowing of tumor growth with all 3 subsets as compared to control mice. When DC1a cells were used as the vaccination, the day 22 mean volume was reduced to 2500 mm3 (p<0.05), and when DC1b and DC2 cells were used the volumes were further reduced to about 1,200 and 1,000 mm3 respectively (p<0.001). The DC1b and DC2 cells were significantly (p<0.01) more effective in slowing tumor growth than the DC1a cells.
Our findings show that Batf3 dependent CD8+DC1 cells, which had been previously distinguished from DC2 cells by the expression of surface markers such as CD24 and XCR1 (6, 20) and the nuclear transcription factor Irf8 (4, 5), can be further divided by their surface phenotype, gene expression profile and function into two subsets that we have designated DC1a and DC1b. Both DC1a and DC1b cells expressed CD24, XCR1, CD8, and Irf8 but the levels of expression were significantly different, and the DC1b cells expressed significantly lower levels. DC1b cells expressed significantly higher levels of CD1d, CD103, and Tim-4 as compared to the DC1a cells. In contrast, the DC2 cells failed to express CD24, XCR1, CD8, and Irf8, but did express Irf4. The DC1a, DC1b, and DC2 subsets were clearly identified by flow cytometry in the lymphoid tissues including the spleen, skin draining lymph nodes, msLNs, and PBMC when MHCII+CD11c+ total DCs were further analyzed for the expression of CD8 versus CD172.
Interestingly, only the DC1b subset was clearly identified by flow cytometry among total DCs in the intestines. The results suggest a failure of the DC1a and DC2 subsets to migrate to the gut. Our continuing studies investigate the chemokine receptors and other trafficking molecules on all three subsets, and whether sorted DC1b cells from the intestines differ in their gene expression pattern and function from the sorted DC1b cells from the lymphoid tissues. It has been previously shown that almost all CD8+DCs in the intestines express CD103 (27). The latter is consistent with the current finding that only DC1b cells are present in the intestines, since DC1b cells expressed significantly higher levels of CD103 than the DC1a or DC2 cells. E-Cadherin was also highly expressed along with CD103 on the DC1b cells, and this is likely to promote interactions between these cells since E-Cadherin binds to CD103, and the heterodimers can modulate their immune functions (48).
Sorted DC1a, DC1b and DC2 cells from the spleen were compared for their gene expression patterns using RNAseq analysis. Differences in gene expression patterns predicted that the expression of E-cadherin and Ly6-D surface receptors also could be used to distinguish the subsets, and this was confirmed by surface staining. Despite the capacity of all 3 DC subsets to present peptide and protein antigens to OT-II CD4 T cells and OT-I CD8 T cells, only the DC1b subset with the highest level of expression of the CD1d was able to effectively present the Galcer or endogenous glycolipid antigens to NKT cells. This finding is consistent with the previously reported ability of CD8+DCs to present a variety of glycolipid antigens to NKT cells in vitro, whereas the CD8-DCs were ineffective (36, 37). Interestingly, the NKT cell response elicited by the DC1b cells showed a Th2 skewing made up predominantly of IL-4 with minimal IFN or IL-2 (27). This polarization is likely due to the low level of CD40 on the DC1b subset, since blocking of the CD40/CD40L interaction has been shown to result in a Th2 polarization of NKT cells (49).
The DC2 cells were also more effective than DC1 cells in eliciting immune responses after vaccination with OVA pulsed DC subsets to control the growth of the OVA-B16 melanoma tumor cells. DC1 cells have been studied extensively for their capacity to cross-present intracellular tumor antigens and viral antigens to CD8+ T cells in local lymph nodes to subsequently induce systemic immune responses (9). However, the role of DC1 versus DC2 cells in the context of the induction of immune responses after subcutaneous vaccination with tumor or viral antigens remains to be elucidated, since the antigens in vaccines can be taken up directly by DCs in local lymph nodes without the need for cross-presentation. In addition, DCs can be pulsed in vitro with these antigens. Previous studies showed that DC2 cells can be at least as effective as DC1 cells in the induction of anti-tumor immune responses after vaccination, and can generate anti-tumor CD4+ and CD8+ memory T cells (35).
In the current study, we found that vaccination with OVA pulsed DC1b and DC2 cells were more effective than the pulsed DC1a cells in controlling the growth of the OVA-B16 melanoma. The results suggest that the DC1b cells are more effective in generating memory T cells to OVA tumor antigen than DC1a cells in the context of vaccination. However, further studies are needed to compare the efficacy of the two subsets for antigen cross-presentation of tumor antigens in the tumor microenvironment. Tumor control is likely to involve DCs in the generation of memory T cells from the vaccination as well as cross-presentation of antigen by DCs in the tumor microenvironment. In conclusion, our studies showed that CD8+Batf3 dependent DC1 cells are comprised of DC1a and DC1b subsets that differ in their surface marker phenotype, gene expression patterns, and function from each other as well as from DC2 cells.
Adult 8- to 10-week-old male Batf3-/- BALB/c (H-2Kd)1 mice, wild type C57BL/6 (H-2Kb), and (C57BL/6-Transgenic (TcraTcrb)1100Mj) OT-I (CD8+ TCR-Tag specific for ovalbumin (OVA) derived SIINFEKL peptide) mice (41) were used for the studies. Transgenic C57BL/6-OT-II mice (B6. Cg (TcraTcrb) 425Cbn/J) with a CD4+TCR-Tag specific for OVA 323-339 peptide, were used also. All mice were obtained from the Jackson Laboratory (Bar Harbor, ME) (42). Ja18-/- BALB/c mice were originally obtained through a material transfer agreement with Dr. Taniguchi of RIKEN Research, Japan. Wild type BALB/c (H-2Kd), Cd1-/- BALB/c mice and Ja18-/- BALB/c mice (39) were bred in the Department of Comparative Medicine, Stanford University (Stanford, CA), and all mice were maintained in the Department according to institutional guidelines approved by the National Institutes of Health.
Staining procedures and flow cytometry analysis performed on FACS LSRII or Aria machines (Becton Dickinson, San Jose, CA) and have previously been described in detail (43). The Fluorochrome conjugated mAbs used for staining were purchased from Invitrogen (San Diego, CA), Biolegend (San Diego, CA), BD biosciences (San Jose, CA), Novus Biologicals (Littleton, CA), Bio-Rad (Hercules, CA) and R&D Systems, (Minneapolis, MN). The following reagents and antibodies were used for flow cytometry:
CD11c (clone: N418; HL3); CD8a (clone: 53-6.7); CD205 (DEC205)(clone: NLDC-145); MHCII (I-A/I-E)(clone: M5/144.15.2); TLR3 (clone: 40C1285.6); TLR3 (clone: 40C1285.6); TLR4 (clone: MTs510); CD16 (FcγRIIIA)(clone: 5B11); CD32B (FcγRIIB); CD11b (clone: M1/70); CD172a (SIRPa) (clone: P84); CD45R/B220 (clone: RA3-6B2); CD370 (CLEC9A, DNGR1, clone: 7H11), CD103 (clone: 2E7), Treml4 (clone: 16E5), TIM-4 (clone: RMT4-54); CD40 (3/23); Rae-1; CD69 (clone: H1.2F3); TCRb (clone: H57-597); CD24 (clone: M1/69); XCR1 (clone: ZET); TIM-3 (CD366, clone: RMT3-32); CD1d (clone: 1B1); Clec9A (DNGR1, clone: 7H11); CD4 (clone: GK1.5); CD207 (clone: 929F3.01); CD324 (E-cadherin, clone: DECMA); Ly6-D (clone: 49-H4); PDL-2 (clone: TY25) and PDL-1 (clone: MIH5). Cells were stained with monoclonal antibodies and aqua dye (Zombie Aqua, Biolegend, San Diego, CA), a dead cell exclusion dye prior to flow cytometry analysis as per the manufacturer’s protocol (eBioscience, BD biosciences). Phycoerythrin and allophycocyanin conjugated CD1d-tetramers were obtained from the National Institutes of Health (NIH) Tetramer Facility, Rockville, MD. FACS analysis used FlowJo Software.
The C57BL/6 spleen cells from either OT-I or OT-II transgenic mice (Jackson Laboratory) were harvested, and the single cell suspensions lysed with ACK lysis buffer (0.15M NH4Cl, 1mM KHCO3, and 0.1mM Na2EDTA {pH 7.4]) for 2 min at room temperature. Subsequently, cells were purified using conventional CD8+ T cell or CD4+ T cell isolation kit by negative selection on MACS LS columns, and then CFSE-labeled (labeled with 2.5μM CFSE) (Molecular Probe, Invitrogen, Eugene, OR). The DC subsets were co-cultured with enriched CFSE-labeled CD8+ T or CD4+ T cells from OT-I or OT-II mice spleen respectively with or without Ovalbumin peptides (OVA257-264 (1g/mL), OVA323-339 (10g/mL) or whole OVA protein (1g/mL) in 96-well round bottom plates at a ratio of 1:10 (DCs: T cells) at 37C, 5% CO2 in Complete (10% FCS) RPMI medium for 5 days. After co-culture the culture supernatants were collected for cytokine analysis using Luminex, and the cells were stained for OT-I or OT-II proliferation analysis by flow cytometry. OVA peptides and whole OVA protein were purchased from Sigma-Aldrich (St. Louis, MO).
Invariant NKT (iNKT) cells were isolated using a gating strategy as described in detail before (44). Briefly splenocytes from wild-type BALB/c mice were stained with glycolipid loaded PE-conjugated CD1d tetramers (National Institutes of Health, Tetramer Facility, Emory-Atlanta, GA), and PE-conjugated-Galcer loaded CD1d dimer-mouse IgG fusion protein (BD Biosciences, San Jose, CA). iNKT cells were enriched subsequently by incubating with anti-PE microbeads (Miltenyi Biotech, Auburn, CA), and passing through a magnetic column (Miltenyi Biotech). The cells were then stained with anti-TCR-FITC and sorted by FACS Aria II (Becton Dickinson). The purity of sorted iNKT cells (TCR+CD1d-tet+) was more than 97%. Sorted iNKT cells were then cocultured in vitro with DC subsets from untreated wild type BALB/c mice.
The C5BL/6 spleen cells from wild type untreated mice were cut into pieces with scalpel blades, and then digested with collagenase solution at 37C for 30 min on a shaker. The digested spleen cells were filtered with a cell strainer (70μm), and then RBCs were lysed with lysis buffer. For sorting, Miltenyi Mouse Pan-DC enrichment kit was used to enriched CD11c+ cells by magnetic beads labeling and purification on a LS column. For sorting DC cell subsets, the enriched CD11c+ cells were stained with the following monoclonal antibodies; CD11c, B220, MHCII, CD8a, and CD172a. The sorted DC subsets (DC1a (Pop 1), DC1b (Pop 2), DC2 (Pop 3) used for antigen (Ova) presentation to OT-I T cells, were irradiated (3,000 rads) from a 137Cs source (J.L. Shephard & Associates, San Fernando; CA).
To determine cytokine production in culture supernatants, NKT cells were cultured (at 5x104 cells/well) with DC subsets (at 1x104 cells/well), or alone. In another study of antigen presentation, OT-I cells (CD8+ T cells; 1x105 cells/well), or OT-II cells or were cocultured with DC subsets (at 1.5x104 cells/well) or alone for 4 days at 37 °C under 5% CO2 in 96-well flat-bottomed plates (Falcon, Becton Dickinson, Franklin Lakes, NJ) in a volume of 0.2 ml in Complete (10% FCS) RPMI medium for 4 days. Cell-free supernatants were collected after centrifugation for protein analysis, while cell pellets were resuspended and stained for flow cytometric analysis. Supernatants were assayed for IL-4, IL-13, IL-2 and IFN protein amounts using Multiplex magnetic Bead Array kit (ThermoFisher Scientific, Waltham, MA), quantified using a MAGPIX instrument (Luminex Corporation, TX).
To assess the efficacy of the 3 DC subsets to stimulate NKT cell cytokine production, sorted iNKT cells from untreated BALB/c mice, and sorted DC subsets (DC1a, DC1b, DC2) obtained from untreated wild type BALB/c spleen cells were cultured alone or together or in some cultures, α-galactosylceramide (100ng/mL) was added. iNKT cells (5x104 cells/well) and DC subsets (1.5x104 cells/well) were cultured at 37C, 5% CO2 in Complete (10% FCS) RPMI medium for 4 days. After culture, the supernatants were harvested, and the concentrations of cytokines in supernatants were quantified using a MAGPIX instrument (Luminex Corporation, TX).
To evaluate antigen presentation capabilities of sorted DCs, irradiated DC subsets (DC1a, DC1b, and DC2) were plated in a round bottom 96-well plate at concentration of 1x104 per/well. Purified OT-I CD8+ T cells, or OT-II+CD4+ T cells were mixed with DC subsets at a ratio of 1:10 and cocultured for 4 days at 37C in 5% CO2 in Complete (10% FCS) RPMI media. In some wells, MHCI restricted ovalbumin specific peptide SIINFEKL (OVA peptide 257-64), or MHCII restricted ISQAVHAAHAEINEAGR (OVA peptide 323-339, recognized by transgene encoded TCR of OT-II+CD4+ T cells), or whole OVA protein were added to the cell cultures. DC subsets were cultured either alone or with OVA or with OT-I T cells or OT-II cells. OT-I T, OT-II or were also either cultured alone or with OVA peptides or with whole OVA protein in control cultures.
The ability of DC subsets to process and present antigen to naïve T cells in vivo to control OVA-specific tumor (B16F10-Ova) was compared. Sorted DC subsets were pulsed in vitro with whole OVA protein (500g/mL) for 1hr in 37C, 5%CO2 incubator. Pulsed DCs were then used to vaccinate naïve wild type C57BL/6 hosts 2 times in a 2-week interval at a dose of 2x105 cells per mouse by intravenous injection of the tail vein.
Two weeks after the last vaccination with primed DC subsets, C57BL/6 hosts were injected subcutaneously into the flank with 5x104 B16F10-OVA cells/mouse and monitored for tumor growth. B16F10-OVA cells were kindly provided by Dr. Darrell Irvine (Massachusetts Institute of Technology MIT (Cambridge, MA). The equation used to calculate tumor volume was as follows: tumor volume = length x width2 x 0.52. Mice were euthanized when the tumor diameter of the tumor mass reached 2 cm.
Total RNA was prepared by sorting enriched CD11c+ subsets; DC1a (Pop 1), DC1b (Pop 2) and DC2 (Pop 3) from spleen cells (1,000 cells per sample) into RLT Plus lysis buffer (Qiagen) and stored at –80 °C, and then processed using RNeasy Micro Plus kit (Qiagen) per the manufacturer’s protocol. Two biological replicates were used for DC1a, DC1b and DC2 RNA-seq. The RNA quality was assessed by Agilent 2100 Bioanalyzer (Agilent Technologies). cDNA synthesis and amplification were prepared using SMARTseq v4 UltraLow Input RNA kit (Takara Inc.). The cDNA library was further processed for bulk RNASeq using a KAPA HyperPlus library kit (Roche Inc.) followed by ligation of Illumina Adaptor tags (Illumina Inc.). Library were quantified and sequenced at Human Immune monitoring Core Center and Genomics/Microarray Core Facility at Stanford University on Illumina Hiseq 4000.
Sequencing yielded 76 bp paired-end reads with a mean sequencing depth of 24.8 million paired-end reads/sample. Trimmomatic (version 0.36) (45) was used to remove adaptors from reads. Clean reads were then aligned to the Mus musculus transcriptome (GRCm38) using histat2(version 2.1.0) (46) and gene count matrix of those mapped uniquely to known mRNAs (GRCm38.84.gtf) were generated by stringtie (version 1.3.1c, prepDE.py function) (47). DESeq2 was used to carry out differential gene expression of pairwise comparisons between tissues with the same genotype and between genotypes in the same tissue.
Results from independent experiments were pooled, and all data were analyzed using Prism (GraphPad Software; VERSION 7) by comparison of means using unpaired two-tailed Student’s t tests. A difference of 0.05 was accepted as statistically significant. The data in all figures represent mean ± SEM to indicate variation within each experiment. One-way ANOVA (Holm-Sidak’s multiple comparison test) was used for statistical analysis. *-p<0.05; **-p<0.01; ***-p<0.001; NS-not significant (p>0.05). Statistical analysis for RNA-seq data is described above.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/GSE181475.
The animal study was reviewed and approved by Stanford University IACUC and APLAC committee according to institutional guidelines approved by the National Institutes of Health.
DH designed and performed research, contributed vital analytical methods, collected, analyzed and interpreted data, and wrote manuscript. PZ helped perform RNA sequencing and gene expression analysis. SD helped perform tumor experiment. RP helped with Luminex analysis. EM helped design experiments. EE helped design experiments, and helped write manuscript. SS provided overall research supervision and wrote manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Institutes of Allergy and Infectious Diseases (R01CA23395801), and National Heart, Lung, and Blood Institute (PO1HL- 075462).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.746469/full#supplementary-material
Supplementary Figure 1 | DC1a, DC1b and DC2 cells shows differences in expression of genes encoding immune response and cytokine signaling pathways. (A–C) GSEA (gene set enrichment analysis) of the most significantly (p < 0.1) upregulated and downregulated genes was compared using NES (normalized enrichment scores) of gene transcripts obtained by RNAseq analysis of sorted DC1a (Pop 1), DC1b (Pop 2) and DC2 (Pop 3) cells from WT BALB/c spleens. GSEA was used to determine NES as follows: - NES = actual ES/mean (ESs against all permutations of the data set). A small normal p value and a high FDR value indicates not significant. FDR is adjusted for gene set size and multiple hypothesis testing but not p value. (D, E). Shows the PCA of the three DC subsets, and the distance among the samples respectively. The information for gene set analysis were obtained from the following websites. GSEA website: https://www.gseamsigdb.org/gsea/doc/GSEAUserGuideTEXT.htm#_Enrichment_Score_(ES) Hallmark gene sets were downloaded from website: http://bioinf.wehi.edu.au/MSigDB/v7.1/Mm.h.all.v7.1.entrez.rds: H hallmark gene sets.
Supplementary Figure 2 | All 3 DC subsets pulsed with OVA protein stimulated robust secretion of Th1 cytokines by OT-II CD4+ T-cells. (A–D) Mean concentrations of IFN, IL-4, IL-13, IL-2, IL-22, TNF, and IL-17a in culture supernatants. FACS-sorted enriched DC subsets (DC1a, DC1b and DC2) (1x104 per well) from wild type untreated C57Bl/6 mice cultured alone or with CFSE-labelled OT-II CD4+ T-cells from (1x105 cells per well) in the presence of whole OVA protein (1g/mL). Cytokines in culture supernatants were measured by Luminex. Control cultures contained OT-II cells alone. Data was pooled from 2 or more independent experiments for a total of at least 10-12 mice per group. One-way ANOVA (Holm-Sidak’s multiple comparison test) results are indicated.
DCs, Dendritic cells; DC1, Type I dendritic cell; DC2, Type II dendritic cell; Batf3, basic leucine zipper transcription factor, ATF-3; NKT cell, Natural killer T cell; MLR, Mixed leukocyte reaction.
1. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 Deficiency Reveals a Critical Role for CD8alpha+ Dendritic Cells in Cytotoxic T Cell Immunity. Science (2008) 322:1097–100. doi: 10.1126/science.1164206
2. Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ Dendritic Cells Form a Unified Subset Developmentally Related to CD8alpha+ Conventional Dendritic Cells. J Exp Med (2010) 207:823–36. doi: 10.1084/jem.20091627
3. Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis e Sousa C, Ozato K, et al. Essential Role for ICSBP in the In Vivo Development of Murine CD8alpha + Dendritic Cells. Blood (2003) 101:305–10. doi: 10.1182/blood-2002-04-1088
4. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, et al. Batf3 Maintains Autoactivation of Irf8 for Commitment of a CD8alpha(+) Conventional DC Clonogenic Progenitor. Nat Immunol (2015) 16:708–17. doi: 10.1038/ni.3197
5. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, et al. Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
6. Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the Dendritic Cell Lineage. Nat Immunol (2012) 13:1145–54. doi: 10.1038/ni.2467
7. Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep (2016) 15:2462–74. doi: 10.1016/j.celrep.2016.05.025
8. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
9. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating Dendritic Cells of the Tumor Microenvironment Cross-Present Tumor Antigens and Stably Engage Tumor-Specific T Cells. Cancer Cell (2012) 21:402–17. doi: 10.1016/j.ccr.2012.01.008
10. Krueger PD, Kim TS, Sung SS, Braciale TJ, Hahn YS. Liver-Resident CD103+ Dendritic Cells Prime Antiviral CD8+ T Cells in Situ. J Immunol (2015) 194:3213–22. doi: 10.4049/jimmunol.1402622
11. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
12. Pascual DW, Wang X, Kochetkova I, Callis G, Riccardi C. The Absence of Lymphoid CD8+ Dendritic Cell Maturation in L-Selectin-/- Respiratory Compartment Attenuates Antiviral Immunity. J Immunol (2008) 181:1345–56. doi: 10.4049/jimmunol.181.2.1345
13. Sun T, Rojas OL, Li C, Ward LA, Philpott DJ, Gommerman JL. Intestinal Batf3-Dependent Dendritic Cells are Required for Optimal Antiviral T-Cell Responses in Adult and Neonatal Mice. Mucosal Immunol (2017) 10:775–88. doi: 10.1038/mi.2016.79
14. Desai P, Tahiliani V, Abboud G, Stanfield J, Salek-Ardakani S. Batf3-Dependent Dendritic Cells Promote Optimal CD8 T Cell Responses Against Respiratory Poxvirus Infection. J Virol (2018) 92. doi: 10.1128/JVI.00495-18
15. GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, et al. Clearance of Influenza Virus From the Lung Depends on Migratory Langerin+CD11b- But Not Plasmacytoid Dendritic Cells. J Exp Med (2008) 205:1621–34. doi: 10.1084/jem.20071365
16. Anderson DA 3rd, Murphy KM, Briseno CG. Development, Diversity, and Function of Dendritic Cells in Mouse and Human. Cold Spring Harb Perspect Biol (2018) 10. doi: 10.1101/cshperspect.a028613
17. Sutherland RM, Londrigan SL, Brady JL, Azher H, Carrington EM, Zhan Y, et al. Shutdown of Immunological Priming and Presentation After In Vivo Administration of Adenovirus. Gene Ther (2012) 19:1095–100. doi: 10.1038/gt.2011.187
18. Murillo O, Dubrot J, Palazon A, Arina A, Azpilikueta A, Alfaro C, et al. In Vivo Depletion of DC Impairs the Anti-Tumor Effect of Agonistic Anti-CD137 mAb. Eur J Immunol (2009) 39:2424–36. doi: 10.1002/eji.200838958
19. Kamphorst AO, Guermonprez P, Dudziak D, Nussenzweig MC. Route of Antigen Uptake Differentially Impacts Presentation by Dendritic Cells and Activated Monocytes. J Immunol (2010) 185:3426–35. doi: 10.4049/jimmunol.1001205
20. Gurka S, Hartung E, Becker M, Kroczek RA. Mouse Conventional Dendritic Cells Can be Universally Classified Based on the Mutually Exclusive Expression of XCR1 and SIRPalpha. Front Immunol (2015) 6:35. doi: 10.3389/fimmu.2015.00035
21. Vanders RL, Murphy VE, Gibson PG, Hansbro PM, Wark PA. CD8 T Cells and Dendritic Cells: Key Players in the Attenuated Maternal Immune Response to Influenza Infection. J Reprod Immunol (2015) 107:1–9. doi: 10.1016/j.jri.2014.09.051
22. Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-Dependent Classical Dendritic Cells Orchestrate Intestinal Immunity to Attaching-and-Effacing Bacterial Pathogens. Nat Immunol (2013) 14:937–48. doi: 10.1038/ni.2679
23. Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 Expression in Conventional Dendritic Cells is Required for T Helper 2 Cell Responses. Immunity (2015) 42:916–28. doi: 10.1016/j.immuni.2015.04.017
24. Bachem A, Hartung E, Guttler S, Mora A, Zhou X, Hegemann A, et al. Expression of XCR1 Characterizes the Batf3-Dependent Lineage of Dendritic Cells Capable of Antigen Cross-Presentation. Front Immunol (2012) 3:214. doi: 10.3389/fimmu.2012.00214
25. Beavis PA, Henderson MA, Giuffrida L, Davenport AJ, Petley EV, House IG, et al. Dual PD-1 and CTLA-4 Checkpoint Blockade Promotes Antitumor Immune Responses Through CD4(+)Foxp3(-) Cell-Mediated Modulation of CD103(+) Dendritic Cells. Cancer Immunol Res (2018) 6:1069–81. doi: 10.1158/2326-6066.CIR-18-0291
26. Crozat K, Tamoutounour S, Vu Manh TP, Fossum E, Luche H, Ardouin L, et al. Cutting Edge: Expression of XCR1 Defines Mouse Lymphoid-Tissue Resident and Migratory Dendritic Cells of the CD8alpha+ Type. J Immunol (2011) 187:4411–5. doi: 10.4049/jimmunol.1101717
27. Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, et al. Small Intestinal CD103+ Dendritic Cells Display Unique Functional Properties That are Conserved Between Mice and Humans. J Exp Med (2008) 205:2139–49. doi: 10.1084/jem.20080414
28. Tomura M, Hata A, Matsuoka S, Shand FH, Nakanishi Y, Ikebuchi R, et al. Tracking and Quantification of Dendritic Cell Migration and Antigen Trafficking Between the Skin and Lymph Nodes. Sci Rep (2014) 4:6030. doi: 10.1038/srep06030
29. Torti N, Walton SM, Murphy KM, Oxenius A. Batf3 Transcription Factor-Dependent DC Subsets in Murine CMV Infection: Differential Impact on T-Cell Priming and Memory Inflation. Eur J Immunol (2011) 41:2612–8. doi: 10.1002/eji.201041075
30. Grees M, Sharbi-Yunger A, Evangelou C, Baumann D, Cafri G, Tzehoval E, et al. Optimized Dendritic Cell Vaccination Induces Potent CD8 T Cell Responses and Anti-Tumor Effects in Transgenic Mouse Melanoma Models. Oncoimmunology (2018) 7:e1445457. doi: 10.1080/2162402X.2018.1445457
31. Behboudi S, Moore A, Hill AV. Splenic Dendritic Cell Subsets Prime and Boost CD8 T Cells and are Involved in the Generation of Effector CD8 T Cells. Cell Immunol (2004) 228:15–9. doi: 10.1016/j.cellimm.2004.03.010
32. Pooley JL, Heath WR, Shortman K. Cutting Edge: Intravenous Soluble Antigen is Presented to CD4 T Cells by CD8- Dendritic Cells, But Cross-Presented to CD8 T Cells by CD8+ Dendritic Cells. J Immunol (2001) 166:5327–30. doi: 10.4049/jimmunol.166.9.5327
33. Dutt S, Atallah MB, Minamida Y, Filatenkov A, Jensen KP, Iliopoulou BP, et al. Accelerated, But Not Conventional, Radiotherapy of Murine B-Cell Lymphoma Induces Potent T Cell-Mediated Remissions. Blood Adv (2018) 2:2568–80. doi: 10.1182/bloodadvances.2018023119
34. Filatenkov A, Baker J, Mueller AM, Kenkel J, Ahn GO, Dutt S, et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin Cancer Res (2015) 21:3727–39. doi: 10.1158/1078-0432.CCR-14-2824
35. Neubert K, Lehmann CH, Heger L, Baranska A, Staedtler AM, Buchholz VR, et al. Antigen Delivery to CD11c+CD8- Dendritic Cells Induces Protective Immune Responses Against Experimental Melanoma in Mice in Vivo. J Immunol (2014) 192:5830–8. doi: 10.4049/jimmunol.1300975
36. Arora P, Baena A, Yu KO, Saini NK, Kharkwal SS, Goldberg MF, et al. A Single Subset of Dendritic Cells Controls the Cytokine Bias of Natural Killer T Cell Responses to Diverse Glycolipid Antigens. Immunity (2014) 40:105–16. doi: 10.1016/j.immuni.2013.12.004
37. Venkataswamy MM, Porcelli SA. Lipid and Glycolipid Antigens of CD1d-Restricted Natural Killer T Cells. Semin Immunol (2010) 22:68–78. doi: 10.1016/j.smim.2009.10.003
38. Smiley ST, Kaplan MH, Grusby MJ. Immunoglobulin E Production in the Absence of Interleukin-4-Secreting CD1-Dependent Cells. Science (1997) 275:977–9. doi: 10.1126/science.275.5302.977
39. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT Cells in IL-12-Mediated Rejection of Tumors. Science (1997) 278:1623–6. doi: 10.1126/science.278.5343.1623
40. Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, et al. CD1d-Lipid-Antigen Recognition by the Semi-Invariant NKT T-Cell Receptor. Nature (2007) 448:44–9. doi: 10.1038/nature05907
41. Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T Cell Receptor Antagonist Peptides Induce Positive Selection. Cell (1994) 76:17–27. doi: 10.1016/0092-8674(94)90169-4
42. Robertson JM, Jensen PE, Evavold BD. DO11.10 and OT-II T Cells Recognize a C-Terminal Ovalbumin 323-339 Epitope. J Immunol (2000) 164:4706–12. doi: 10.4049/jimmunol.164.9.4706
43. Hongo D, Tang X, Dutt S, Nador RG, Strober S. Interactions Between NKT Cells and Tregs are Required for Tolerance to Combined Bone Marrow and Organ Transplants. Blood (2012) 119:1581–9. doi: 10.1182/blood-2011-08-371948
44. Tang X, Zhang B, Jarrell JA, Price JV, Dai H, Utz PJ, et al. Ly108 Expression Distinguishes Subsets of Invariant NKT Cells That Help Autoantibody Production and Secrete IL-21 From Those That Secrete IL-17 in Lupus Prone NZB/W Mice. J Autoimmun (2014) 50:87–98. doi: 10.1016/j.jaut.2014.01.002
45. Kim D, et al. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol (2013) 14:R36. doi: 10.1186/gb-2013-14-4-r36
46. Robinson PN, Krawitz P, Mundlos S. Strategies for Exome and Genome Sequence Data Analysis in Disease-Gene Discovery Projects. Clin Genet (2011) 80:127–32. doi: 10.1111/j.1399-0004.2011.01713.x
47. Anders S, Pyl PT, Huber W. HTSeq–a Python Framework to Work With High-Throughput Sequencing Data. Bioinformatics (2015) 31:166–9. doi: 10.1093/bioinformatics/btu638
48. Van den Bossche J, Laoui D, Naessens T, Smits HH, Hokke CH, Stijlemans B, et al. E-Cadherin Expression in Macrophages Dampens Their Inflammatory Responsiveness In Vitro, But Does Not Modulate M2-Regulated Pathologies In Vivo. Sci Rep (2015) 5:12599. doi: 10.1038/srep12599
Keywords: dendritic cells, type I dendritic cells, type II dendritic cell, CD4 T cell, CD8 T cell, tumor vaccination
Citation: Hongo D, Zheng P, Dutt S, Pawar RD, Meyer E, Engleman EG and Strober S (2021) Identification of Two Subsets of Murine DC1 Dendritic Cells That Differ by Surface Phenotype, Gene Expression, and Function. Front. Immunol. 12:746469. doi: 10.3389/fimmu.2021.746469
Received: 23 July 2021; Accepted: 17 September 2021;
Published: 26 October 2021.
Edited by:
Daniel Saban, Duke University, United StatesReviewed by:
Luc Van Kaer, Vanderbilt University, United StatesCopyright © 2021 Hongo, Zheng, Dutt, Pawar, Meyer, Engleman and Strober. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel Strober, c3N0cm9iZXJAc3RhbmZvcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.