 Sohee Lee1†
Sohee Lee1† Jong-Seok Kim
Jong-Seok Kim Jongdae Shin
Jongdae Shin Sung Ki Lee
Sung Ki Lee Hwan-Woo Park
Hwan-Woo Park
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 09 November 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.743700
Pathological maternal inflammation and abnormal placentation contribute to several pregnancy-related disorders, including preterm birth, intrauterine growth restriction, and preeclampsia. TANK-binding kinase 1 (TBK1), a serine/threonine kinase, has been implicated in the regulation of various physiological processes, including innate immune response, autophagy, and cell growth. However, the relevance of TBK1 in the placental pro-inflammatory environment has not been investigated. In this study, we assessed the effect of TBK1 inhibition on lipopolysaccharide (LPS)-induced NLRP3 inflammasome activation and its underlying mechanisms in human trophoblast cell lines and mouse placenta. TBK1 phosphorylation was upregulated in the trophoblasts and placenta in response to LPS. Pharmacological and genetic inhibition of TBK1 in trophoblasts ameliorated LPS-induced NLRP3 inflammasome activation, placental inflammation, and subsequent interleukin (IL)-1 production. Moreover, maternal administration of amlexanox, a TBK1 inhibitor, reversed LPS-induced adverse pregnancy outcomes. Notably, TBK1 inhibition prevented LPS-induced NLRP3 inflammasome activation by targeting the mammalian target of rapamycin complex 1 (mTORC1). Thus, this study provides evidence for the biological significance of TBK1 in placental inflammation, suggesting that amlexanox may be a potential therapeutic candidate for treating inflammation-associated pregnancy-related complications.
During pregnancy, at the maternal-fetal interface, physiological inflammation is beneficial for the implantation and the preliminary stages of placentation. An adequate response of the immune system at the placenta is vital to protect the fetus from various stimuli, such as infection and pathogens (1, 2). However, pathological inflammatory conditions can cause placental insufficiency, thereby contributing to several pregnancy-related disorders, including preterm birth, intrauterine growth restriction, and preeclampsia (3–5). Trophoblasts in the placenta recognize and respond to pathogen-associated molecular patterns such as bacterial cell wall components through the expression of toll-like receptors (TLRs) (6). Activation of TLRs stimulates intracellular signaling cascade, resulting in the activation of nuclear factor-kappa B (NF-κB/p65) and expression of pro-inflammatory cytokines, including interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α, which contribute to trophoblast dysfunction and placental abnormalities (7–9).
The NLR family pyrin domain-containing 3 (NLRP3) inflammasome is a large cytosolic multiprotein complex composed of an NLRP3 scaffold, an apoptosis-associated speck-like protein (ASC) adaptor, and caspase-1 as an IL-1β-converting enzyme (10). They are activated by a wide range of pathogen-associated molecular patterns, such as a TLR4 ligand, lipopolysaccharide (LPS), or damage-associated molecular patterns such as ATP, and play a role in inflammation (11). In response to stimuli, NLRP3 inflammasomes mediate the activation of caspase-1 and cleavage of inactive pro-IL-1β into its active form (12, 13) and are related to pregnancy dysfunction, including preterm birth, fetal growth restriction, and preeclampsia (14–16). Aberrant IL-1β production in the placenta and human trophoblasts has been associated with NLRP3 inflammasome activation, contributing to placental inflammation (17).
TANK-binding kinase 1 (TBK1) is a non-canonical IKK‐related innate immune kinase that plays a pivotal role in the innate immune system (18, 19). TBK1 can be activated by phosphorylation in response to stimulation by TLR3, TLR4, and pathogens (20); however, the molecular mechanisms involved in the phosphorylation of TBK1 are not entirely understood. Our previous study showed that activated TBK1 induces obesity-induced ubiquitinated protein inclusions in liver tissue and that inhibiting the TBK1 pathway protects against fibrotic liver pathologies in mouse models of obesity and nonalcoholic steatohepatitis (21). TBK1 has been associated with stimulus-dependent NF-κB activation, affecting several diseases, including inflammatory disorders and cancer (22–24). However, the relevance of TBK1 in LPS-induced NLRP3 inflammasome activation in the placenta and trophoblasts remains unexplored.
In this study, the potential effect of TBK1 inhibition on NLRP3 inflammasome activation in LPS-treated pregnant mice and human trophoblast cell lines was investigated for the first time. Further, the molecular mechanisms by which TBK1 regulates LPS-induced NLRP3 inflammasome activation was also studied. Our results suggest that TBK1 phosphorylation is induced in the placental tissues of LPS-treated mice and LPS-treated trophoblasts. Both pharmacological inhibition and genetic knockdown of TBK1 ameliorated LPS-induced NLRP3 inflammasome activation and placental inflammation. Moreover, we found that TBK1 deficiency suppresses LPS-induced NLRP3 inflammasome activation via a mTORC1-dependent mechanism. Therefore, TBK1 inhibition may act as a potential therapeutic target for placental dysfunction associated with NLRP3 inflammasome and inflammation.
Immunoblotting was performed using antibodies against phospho-TBK1, TBK1, NLRP3, Caspase-1, phospho-p65, p65, phospho-IκB, IκB, phospho-S6K, S6K (Cell Signaling Technology, Danvers, MA, USA), Caspase-1 (Santa Cruz Biotechnology, Dallas, TX, USA), and α-tubulin (Developmental Studies Hybridoma Bank, Iowa City, IA, USA). LPS (Escherichia coli LPS, serotype 0127: B8), ATP, and rapamycin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Amlexanox and Torin 1 were purchased from the Cayman Chemical Company, Ann Arbor, MI, USA. The sources of other reagents are indicated in the specified methods.
Female C57BL/6 mice (6–8-weeks-old) were purchased from Samtako (Osan, Korea). All mice were maintained under a controlled temperature (20–25°C) and humidity (50 ± 5%) with a 12 h light/dark cycle and free access to food and drinking water. Female mice were mated with male mice, and the presence of vaginal plugs the following day confirmed successful mating. The timed pregnant mice received an intraperitoneal injection of LPS (200 µg/kg) or saline on gestation day 17.5. In the amlexanox + LPS group, the pregnant mice were intraperitoneally injected with amlexanox (2.5 mg/kg) at 27 h and 3 h before the intraperitoneal administration of LPS. The pregnant mice were sacrificed at various time points (4, 6, 8, 10, or 12 h) after LPS treatment, and placental samples were collected for molecular analysis. All animal studies were conducted following the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Animal Ethics Committee of Konyang University (P-21-19-A-01).
Human first-trimester trophoblast cell lines, HTR-8/SVneo (American Type Culture Collection, Rockville, MD, USA) and Sw.71 (gifted by Dr. Gil Mor, Yale University School of Medicine, New Haven, CT, USA), were used in our experiments. HTR-8/SVneo cells were cultured in RPMI 1640 medium (Welgene, Gyeongsan, Korea) supplemented with 10% fetal bovine serum (Welgene), 4500 mg/L D-glucose, 10 mM HEPES, 2mM L-glutamine, 1 mM sodium pyruvate, and 100 U/mL penicillin-streptomycin (Welgene). The Sw.71 cells were cultured in Dulbecco’s modified Eagle’s medium (Welgene) containing 10% fetal bovine serum, 4500 mg/L D-glucose with L-glutamine, and 100 U/mL penicillin-streptomycin. All cultures were maintained in a humidified 5% CO2 atmosphere at 37°C. For LPS treatment, cells were incubated in the presence of LPS or treated with LPS and then treated with 5 mM ATP for 45 min, as described previously (17). Identical volumes of phosphate-buffered saline (PBS) were used as the vehicle controls. When indicated, the cells were incubated with the TBK1 inhibitor amlexanox.
The HTR-8/SVneo cell survival was measured using the WST-1 assay (EZ-CytoX Enhanced Cell Viability Assay Kit, Daeil Lab Service, Seoul, Korea) according to the manufacturer’s protocol (25). Briefly, the HTR-8/SVneo cells (1 × 104 cells/well) were plated into 96-well plates and treated with the indicated concentrations of amlexanox for 24 h. Subsequently, 10 μL of WST-1 reagent was added to each well and incubated for 30 min at 37°C in a 5% CO2 incubator (Thermo Scientific, Waltham, MA, USA). The absorbance was measured at 450 nm using an Epoch 2 microplate reader (Bio-Tek Instruments, Winooski, VT, USA), and the absorbance values of the treated cells were expressed as a percentage of the absorbance values of the control.
Using a polyethylenimine reagent, HEK293T cells were transfected with the following lentiviral constructs with the packaging plasmids: sh-Luciferase (sh-Luc) and sh-TBK1 constructs (obtained from the RNAi Consortium collection of the Broad Institute). Lentiviral supernatants were collected and filtered 48 and 72 h after transfection. HTR-8/SVneo cells and Sw.71 cells were incubated 2–3 d with the lentiviral medium in the presence of 4 μg/mL polybrene.
Total RNA was extracted from placental tissues and HTR-8/SVneo cells using TRIzol reagent (Takara, Shiga, Japan) according to the manufacturer’s instructions. Complementary DNA was synthesized from RNA using a cDNA synthesis kit (BioFact, Daejeon, Korea). Quantitative real-time reverse transcription PCR (qRT-PCR) was performed in triplicate with the SYBR Green Real-Time PCR Master Mix Reagent (BioFact) using the QuantStudio 3 Real-time PCR System (Life Technologies, Carlsbad, CA, USA). Relative mRNA expression was calculated using comparative threshold cycle (Ct) values normalized to those of mouse or human cyclophilin. The following primers were used: mouse Nlrp3: forward 5′- AGCCTTCCAGGATCCTCTTC-3′, reverse 5′-CTTGGGCAGCAGTTTCTTTC-3′; mouse Tnf-α: forward 5′-TCCCAGGTTCTCTTCAAGGGA-3′, reverse 5′-GGTGAGGAGCACGTAGTCGG-3′; mouse Tgf-β1: forward 5′-CTCCCGTGGCTTCTAGTGC-3′, reverse 5′-GCCTTAGTTTGGACAGGATCTG-3′; mouse Il-6: forward 5′- TAGTCCTTCCTACCCCAATTTCC-3′, reverse 5′-TTGGTCCTTAGCCACTCCTTC-3′; mouse Il-10: forward 5′-GCTCTTACTGACTGGCATGAG-3′, reverse 5′-CGCAGCTCTAGGAGCATGTG-3′; mouse Mcp-1: forward 5′-CATCCACGTGTTGGCTCA-3′, reverse 5′-GATCATCTTGCTGGTGAATGAGT-3′; mouse Il-1β: forward 5′-TCTTTGAAGTTGACGGACCC-3′, reverse 5′-TGAGTGATACTGCCTGCCTG-3′; mouse IFN-β: forward 5′-GCACTGGGTGGAATGAGACT-3′, reverse 5′-AGTGGAGAGCAGTTGAGGAC-3′; mouse Cyclophilin A: forward 5′-GAGCTGTTTGCAGACAAAGTTC-3′, reverse 5′-CCCTGGCACATGAATCCTGG-3′; human NLRP3: forward 5′-GATCTTCGCTGCGATCAACAG-3′, reverse 5′-CGTGCATTATCTGAACCCCAC-3′; human IL-1β: forward 5′-AAAGAGGCACTGGCAGAA-3′, reverse 5′-AGCTCTGGCTTGTTCCTCAC-3′; and human Cyclophilin A: forward 5′-GCAAAGTGAAAGAAGGCATGAA-3′, reverse 5′-CCATTCCTGGACCCAAAGC-3′.
Placental tissues, HTR-8/SVneo cells, and Sw.71 cells were lysed in an ice-cold radioimmunoprecipitation assay buffer containing cOmplete protease inhibitor cocktail (Roche, Basel, Switzerland). Lysates were incubated for 20 min on ice and centrifuged at 18,000 × g for 15 min at 4°C. Protein concentrations were measured using a bicinchoninic acid protein assay (Pierce, Rockford, IL, USA). Lysates were boiled in 1× sodium dodecyl sulfate (SDS) and Laemmli sample buffer for 5 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA, USA), which were probed with primary antibodies against p-TBK1, TBK1, NLRP3, caspase-1, p-p65, p65, p-IκBα, IκBα, p-S6K, S6K, and α-tubulin. After incubation with secondary antibodies conjugated with horseradish peroxidase (Bio-Rad, Hercules, CA, USA), chemiluminescence was detected using a Fusion Solo System (Vilber Lourmat, Marne-la-Vallée, France). Densitometric analysis of the blots was performed using ImageJ (National Institutes of Health, Bethesda, MD, USA), in which the background was removed for each band.
Mouse placental tissues were fixed in 10% neutral buffered formalin for 24 h, dehydrated, and embedded in paraffin. Sections (5 μm) of the embedded tissue were prepared and stained with hematoxylin and eosin dyes or periodic acid-Schiff dyes. For immunohistochemistry, the paraffin-embedded sections were deparaffinized, rehydrated, and treated for antigen retrieval. Endogenous peroxidase was quenched using 3% hydrogen peroxide. After blocking the nonspecific antigens, the sections were incubated with anti-NLRP3 antibody or anti-IL-1β antibody overnight at 4°C, followed by incubation with biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA). Antibodies were visualized with streptavidin-HRP (BD Pharmingen, San Diego, CA, USA) using diaminobenzidine (Sigma-Aldrich). Hematoxylin was used to visualize nuclei. The samples were analyzed under a light microscope (DM2500; Leica, Wetzlar, Germany).
Cell supernatants collected from the HTR-8/SVneo cells were assayed for the levels of the secreted inflammatory cytokine IL-1β using the DuoSet ELISA development kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. The optical density of the final colored reaction product was determined at 450 nm using an Epoch 2 microplate reader (Bio-Tek Instruments).
Results are presented as the mean ± standard error of the mean (SEM). Data presented in the figures are representative of at least three independent experiments unless stated otherwise. The significance of differences between the two experimental groups was determined using a two-tailed Student’s t-test. Multiple comparisons were conducted using one-way ANOVA followed by Tukey’s or Fisher’s least significant difference (LSD) post hoc test. Differences were considered statistically significant at p < 0.05, *p < 0.05, **p < 0.01, ***p < 0.001).
We evaluated the phosphorylation of TBK1 protein in the mouse placenta by LPS-induced maternal inflammatory response. Immunoblot analysis showed that the placental TBK1 phosphorylation was significantly increased at 4, 6, 8, and 10 h after LPS treatment (Figures 1A, B). To measure the phosphorylated levels of TBK1 in trophoblasts during maternal inflammation, human first-trimester extravillous trophoblasts (HTR-8/SVneo and Sw.71) were stimulated with various LPS concentrations at various time points. Immunoblot analysis showed that LPS increased TBK1 phosphorylation in HTR-8/SVneo after both short- (Figures 1C, D) and long-term treatments (Figures 1E, F) in a dose-dependent manner. The time course of TBK1 phosphorylation was further investigated by treating the cells with LPS (1 μg/mL), and the results showed that short- and long-term exposure to LPS significantly increased phosphorylated TBK1 levels in HTR-8/SVneo cells in a time-dependent manner (Figures 1G–J). Similar to that in HTR-8/SVneo cells, the expression of phosphorylated TBK1 was stimulated by LPS treatment in Sw.71 cells (Supplemental Figure 1).
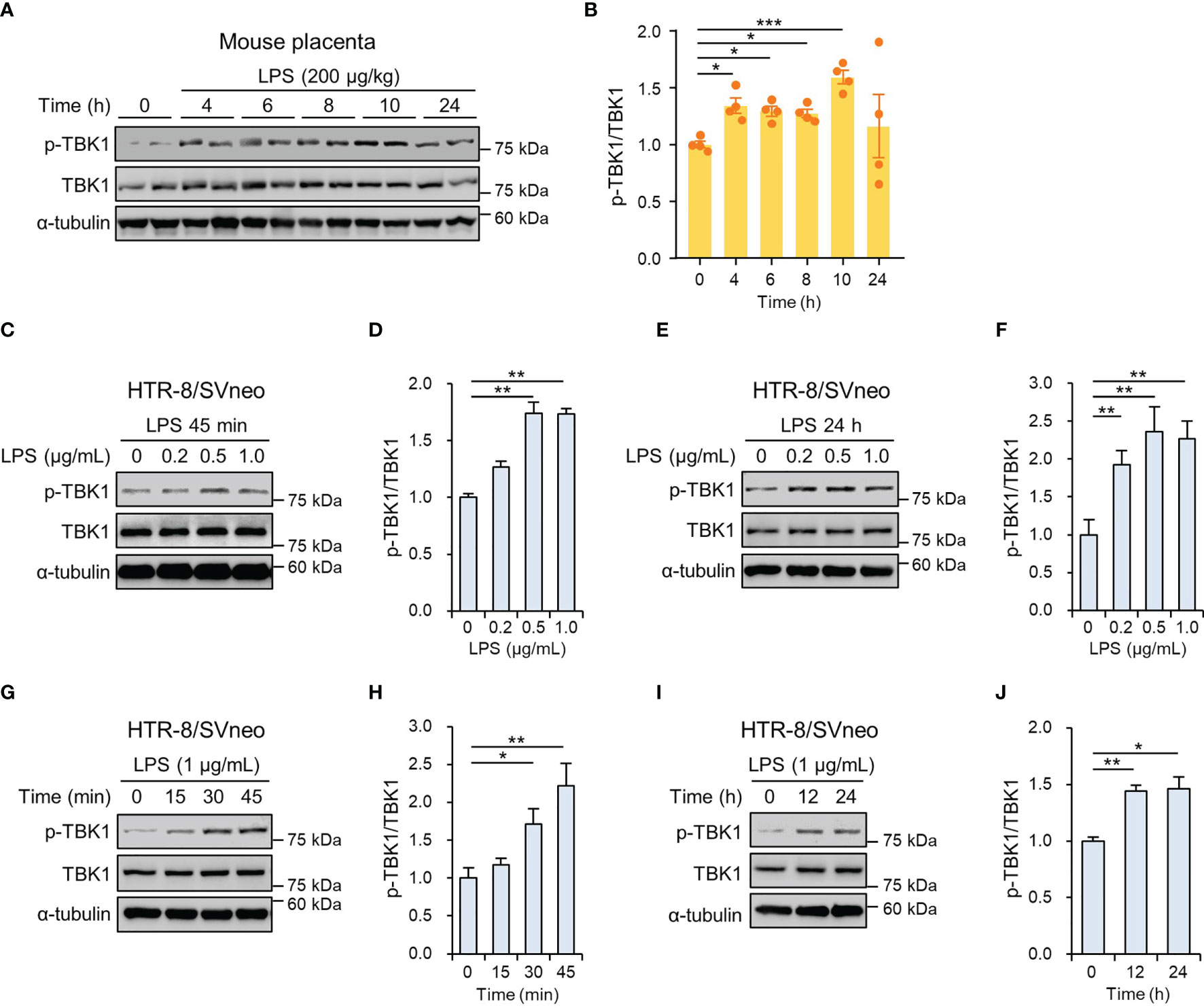
Figure 1 TBK1 phosphorylation in the placenta of mice and human trophoblasts exposed to lipopolysaccharide (LPS). (A, B) Pregnant mice were injected intraperitoneally with LPS (200 μg/kg) on gestation day 17.5. After the indicated number of hours, placental tissues were collected from mice and analyzed by immunoblotting with anti-p-TBK1 and TBK1 antibodies (n = 4 per group). (C–F) HTR-8/SVneo cells were treated with the indicated concentration of LPS for 45 min (C, D) and 24 h (E, F). Cell lysates were analyzed by immunoblotting with anti-p-TBK1 and anti-TBK1 antibodies. (G–J) HTR-8/SVneo cells were treated with LPS (1 μg/mL) for the indicated periods. Cell lysates were analyzed by immunoblotting with anti-p-TBK1 and anti-TBK1 antibodies. α-tubulin served as a loading control. Band intensities were quantified and normalized to total protein intensities. Data are shown as means ± SEM. Results are representative of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 (Student’s t-test).
NLRP3 inflammasome activation in trophoblasts is implicated in the pathogenesis of placental inflammation (17, 26). To investigate the effects of LPS on placental NLRP3 protein expression, placentas from mice exposed to LPS were analyzed by immunohistochemistry. The results showed that NLRP3 protein expression was increased in trophoblasts in the junctional zone and the labyrinth of the placentas of the LPS-treated mice compared with that of the control placentas of saline-treated mice (Supplemental Figure 2). Consistent with the above results, the NLRP3 inflammasome was activated in HTR-8/SVneo and Sw.71 cells upon stimulation with LPS and ATP (Supplemental Figures 3A, B). Furthermore, we investigated whether TBK1 is involved in LPS-induced NLRP3 inflammasome activation. Amlexanox, a TBK1 inhibitor, was used to explore the effect of TBK1 deficiency on LPS-induced NLRP3 inflammasome. We measured the cell viability of HTR-8/SVneo and Sw.71 cells treated with different concentrations of amlexanox. In vitro cytotoxicity assays revealed that amlexanox exhibited low cytotoxicity against cells up to 200 µM (Supplemental Figures 4A, B). Immunoblot analysis showed that amlexanox markedly decreased LPS-induced NLRP3 mRNA levels and NLRP3 and cleaved caspase-1 protein levels (Figures 2A–C and Supplemental Figure 3C). To confirm these results, IL-1β levels were analyzed using ELISA. Pretreatment of HTR-8/SVneo cells with amlexanox resulted in significantly lower levels of IL-1β compared to that in LPS-treated cells (Figure 2D).
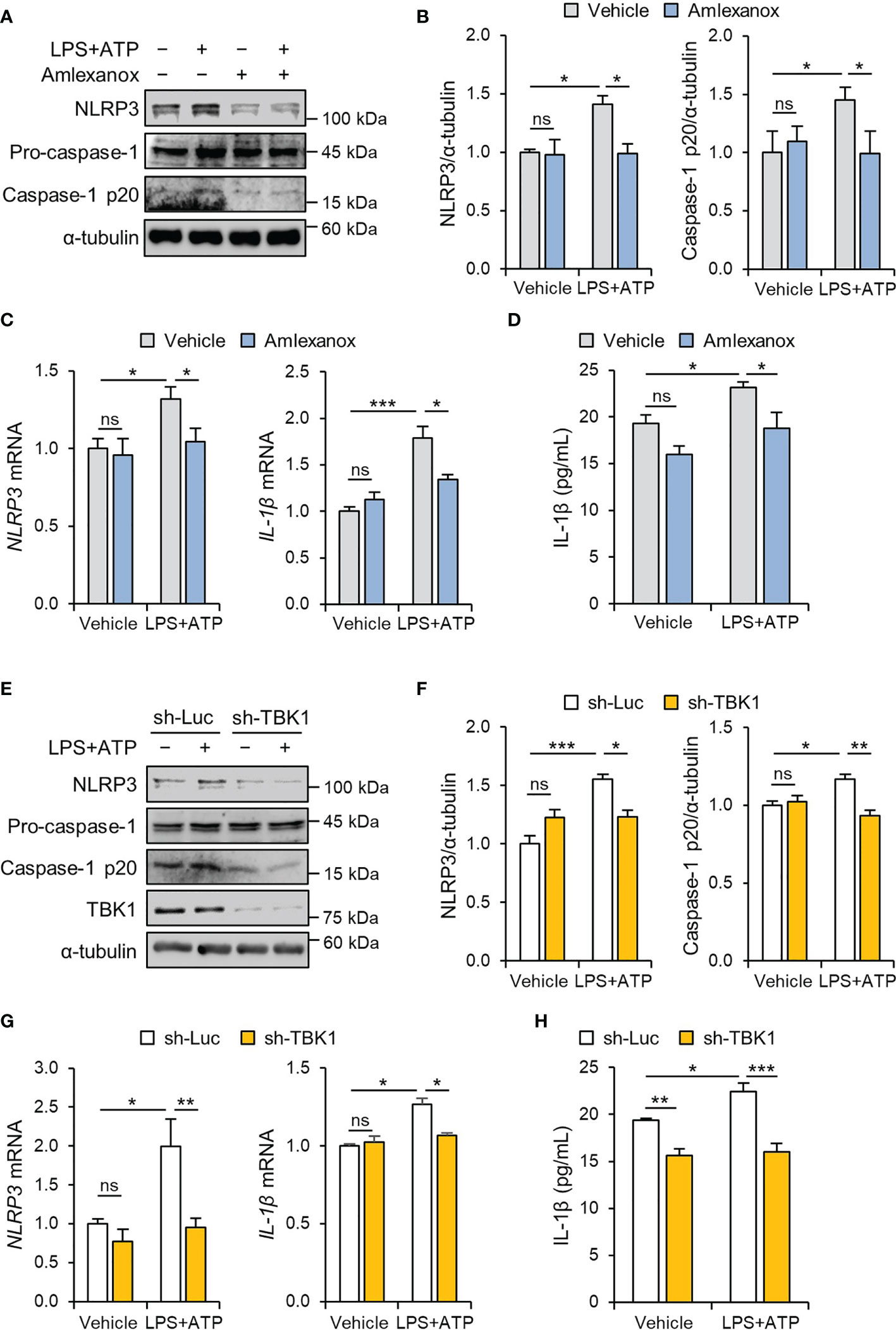
Figure 2 TBK1 deficiency abolishes LPS-induced NLRP3 inflammasome activation in trophoblasts. (A–C) HTR-8/SVneo cells were treated with 1 μg/mL LPS or 20 µM amlexanox for 24 h (A, B) or 12 h (C) followed by treatment with 5 mM ATP for 45 min, as indicated in the figure. (A, B) Cell lysates were analyzed by immunoblotting with anti-NLRP3 and anti-caspase-1 antibodies. (C) Relative mRNA expression of NLRP3 and IL-1β was quantified by qRT-PCR. (D) Conditioned media were collected from HTR-8/SVneo cells treated with 1 μg/mL LPS or 10 µM amlexanox for 36 h followed by treatment with 5 mM ATP for 45 min, as indicated in the figure. IL-1β levels were quantified by ELISA. (E–G) HTR-8/SVneo cells were infected with lentiviruses expressing shRNAs targeting luciferase (sh-Luc) or TBK1 (sh-TBK1) and treated with 1 μg/mL LPS for 24 h (E, F) or 12 h (G) followed by treatment with 5 mM ATP for 45 min, as indicated in the figure. (E–G) Cell lysates were analyzed by immunoblotting with anti-NLRP3, anti-caspase-1, and anti-TBK1 antibodies. (G) Relative mRNA expression of NLRP3 and IL-1β was quantified by qRT-PCR. α-tubulin served as a loading control. Band intensities were quantified and normalized to control band intensities. (H) Conditioned media were collected from HTR-8/SVneo cells infected with sh-Luc or sh-TBK1 lentiviruses and treated with 1 μg/mL LPS for 24 h followed by treatment with 5 mM ATP for 45 min, as indicated in the figure. IL-1β levels were quantified by ELISA. Data are shown as means ± SEM. Results are representative of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant (One-way ANOVA, followed by Tukey’s test).
To clarify the specific signaling mechanism by which amlexanox suppresses LPS-induced NLRP3 inflammasome activation in trophoblasts, lentiviral vectors expressing small hairpin RNAs (shRNAs) targeting human TBK1 were transduced to HTR-8/SVneo and Sw.71 cells. Knockdown of TBK1 was sufficient to block the upregulation of NLRP3 and cleaved caspase-1 in LPS-treated cells (Figures 2E–G and Supplemental Figure 4D), consistent with the inhibitory effect of amlexanox on NLRP3 inflammasome activation. ELISA showed that the lentiviral knockdown of TBK1 also decreased IL-1β production (Figure 2H), suggesting that it is an inhibitory effect of TBK1, not a nonspecific effect of amlexanox.
NF-κB signaling mediates the priming signal of NLRP3 inflammasome activation and upregulates the expression levels of NLRP3 and pro-IL-1β (27, 28). LPS increased the phosphorylation of NF-κB/p65 in a time-dependent manner, and the maximal effect was observed at 1 h following 1 μg/mL LPS treatment (Supplemental Figure 5A). To investigate the effect of amlexanox on the NF-κB pathway, we measured the phosphorylation levels of IκBα and NF-κB/p65 in HTR-8/SVneo and Sw.71 cells. Pretreatment with amlexanox decreased LPS-induced phosphorylation of IκBα and NF-κB/p65 (Figures 3A, B, and Supplemental Figure 5B). Further, we investigated the nuclear translocation of NF-κB/p65. LPS treatment resulted in a significant increase in NF-κB/p65 in the nucleus of HTR-8/SVneo cells, whereas the nuclear translocation of NF-κB/p65 was suppressed by amlexanox (Figure 3C).
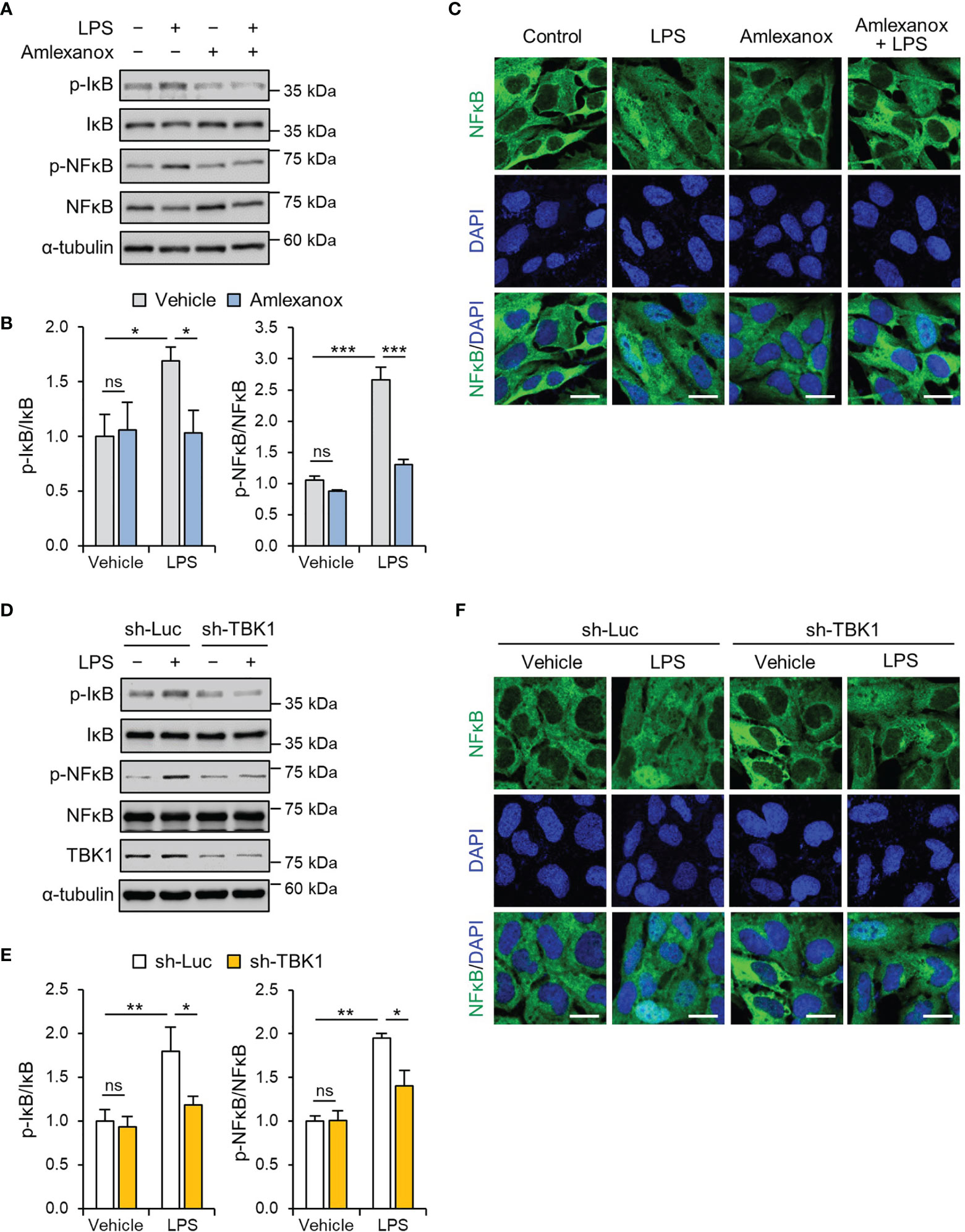
Figure 3 TBK1 deficiency abolishes LPS-induced NF-κB/p65 activation in trophoblasts. (A, B) HTR-8/SVneo cells were treated with 1 μg/mL LPS or 100 µM amlexanox for 1 h, as indicated in the figure. Cell lysates were analyzed by immunoblotting with anti-p-IκBα, anti-IκBα, anti-p-p65, and anti-p65 antibodies. α-tubulin served as a loading control. (C) Immunofluorescence staining of p65 (green) in HTR-8/SVneo cells treated with 1 μg/mL LPS or vehicle for 45 min in the presence or absence of 100 µM amlexanox. Nuclei were stained with DAPI (blue). Scale bars, 20 μm. (D, F) HTR-8/SVneo cells were infected with lentiviruses expressing shRNAs targeting luciferase (sh-Luc) or TBK1 (sh-TBK1) and treated with 1 μg/mL LPS for 1 h, as indicated in the figure. Cell lysates were analyzed by immunoblotting with anti-p-IκBα, anti-IκBα, anti-p-p65, and anti-p65 antibodies. α-tubulin served as a loading control. (E) Immunofluorescence staining of p65 (green) in HTR-8/SVneo cells infected with sh-Luc or sh-TBK1 lentiviruses and treated with 1 μg/mL LPS or vehicle for 45 min. Nuclei were stained with DAPI (blue). Scale bars, 20 μm. Data are shown as means ± SEM. Results are representative of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant (One-way ANOVA, followed by Tukey’s test).
We also examined whether the knockdown of TBK1 reversed the effect of LPS on the NF-κB/p65 pathway. Lentiviral knockdown of TBK1 reduced LPS-induced phosphorylation of IκBα and NF-κB/p65 (Figures 3D, E, and Supplemental Figure 4C). Immunofluorescence staining showed that the knockdown of TBK1 was sufficient to inhibit the nuclear translocation of NF-κB/p65 in LPS-treated cells (Figure 3F). These results collectively indicate that TBK1 deficiency prevents the LPS-induced nuclear translocation of NF-κB/p65 by phosphorylation of IκBα.
To evaluate whether maternal administration of amlexanox affects the fetal-placental development of LPS-treated mice, we first measured the fetal and placental weight and length in each group. Fetal and placental weight and length were significantly reduced in the LPS-treated mice compared to that in the control mice, whereas amlexanox treatment prevented the decrease in fetal and placental weight and length associated with LPS treatment (Figures 4A–C). We also evaluated whether amlexanox affects placental growth and morphology in LPS-treated mice. Periodic acid-Schiff staining revealed that LPS-exposed placenta had significantly reduced length of decidual layers and junctional zones; however, no appreciable change was observed in the length of the labyrinth layer. Interestingly, amlexanox treatment abrogated the LPS-induced placental morphological changes (Figures 4D, E). These findings suggest that maternal administration of amlexanox alleviates LPS-induced morphological changes in the placenta.
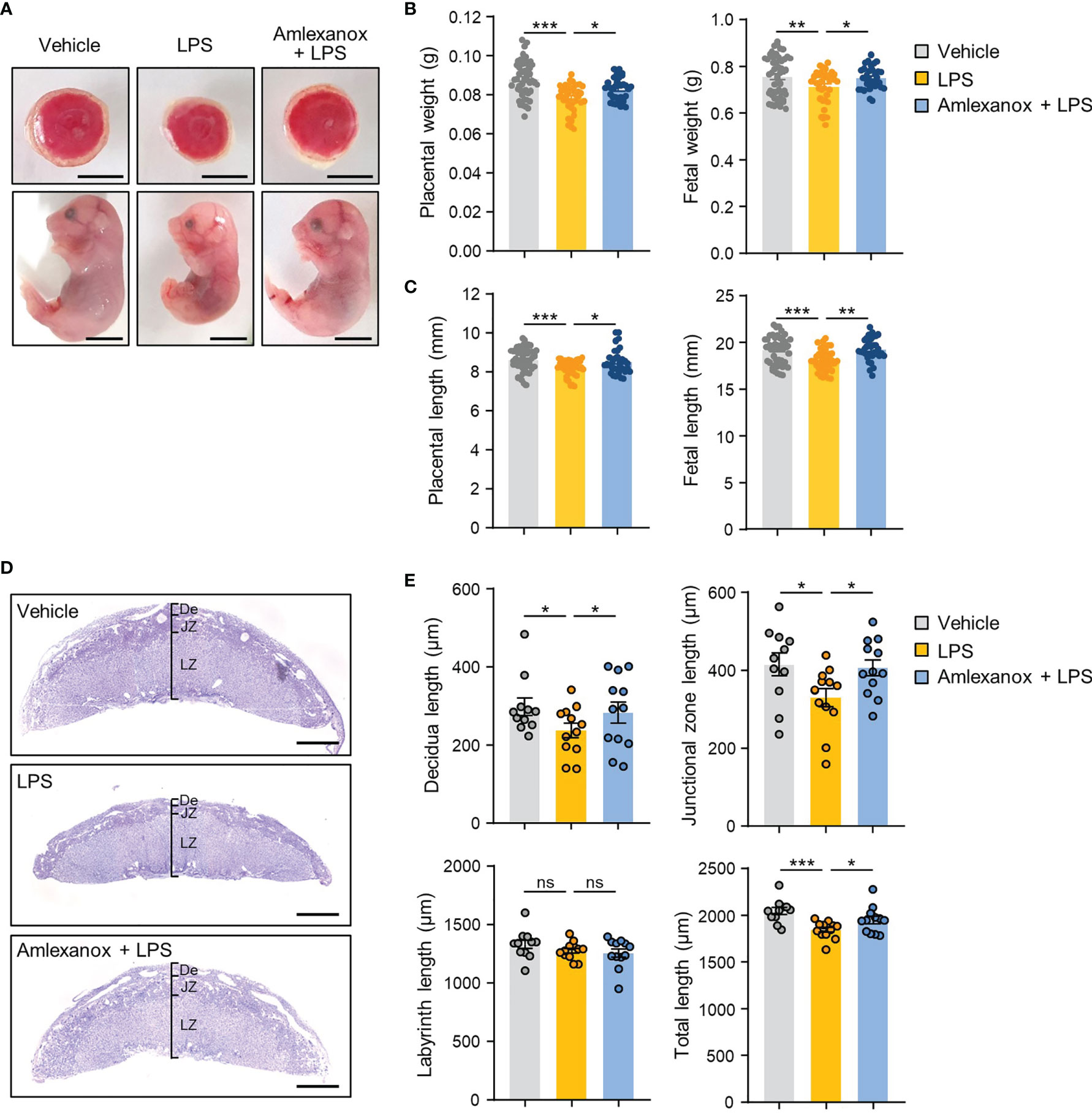
Figure 4 The effect of amlexanox on the gross placental morphology in the LPS-treated pregnant mice. (A–E) Pregnant mice were intraperitoneally injected with LPS (200 μg/kg) or amlexanox (2.5 mg/kg) on gestation day 17.5, as indicated in the figure. (A) Representative images of fetus and placenta for each maternal group. Scale bars, 0.5 cm (B, C) Average fetal and placental weights and lengths for each group (n = 33-50 per group). (D, E) Periodic acid-Schiff (PAS) staining of placental tissues from vehicle-, LPS-, LPS+amlexanox-treated pregnant mice at gestation day 17.5. Decidua (De), labyrinth (LZ), and junctional zone (JZ) length on the midline section was quantified (n = 11 per group). Scale bars, 1 mm. Data are shown as means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant (One-way ANOVA, followed by Fisher’s LSD test).
To further confirm whether maternal administration of amlexanox influences placental NLRP3 inflammasome activation in vivo, we measured the expression levels of NLRP3 inflammasome markers in the placentas of LPS-treated mice. Immunoblot analysis showed that NLRP3 and cleaved caspase-1 protein levels were elevated in response to LPS treatment; this increase was suppressed in amlexanox-treated mice exposed to LPS (Figures 5A, B). Correspondingly, amlexanox treatment decreased NLRP3 mRNA levels in the LPS-treated mouse placenta (Figure 5C). Immunohistochemical analysis showed that NLRP3 and IL-1β staining was significantly increased in the cytoplasm of trophoblasts in the junctional zone and labyrinth layer in LPS-treated mouse placenta; however, their expression was decreased with amlexanox treatment (Figure 5D).
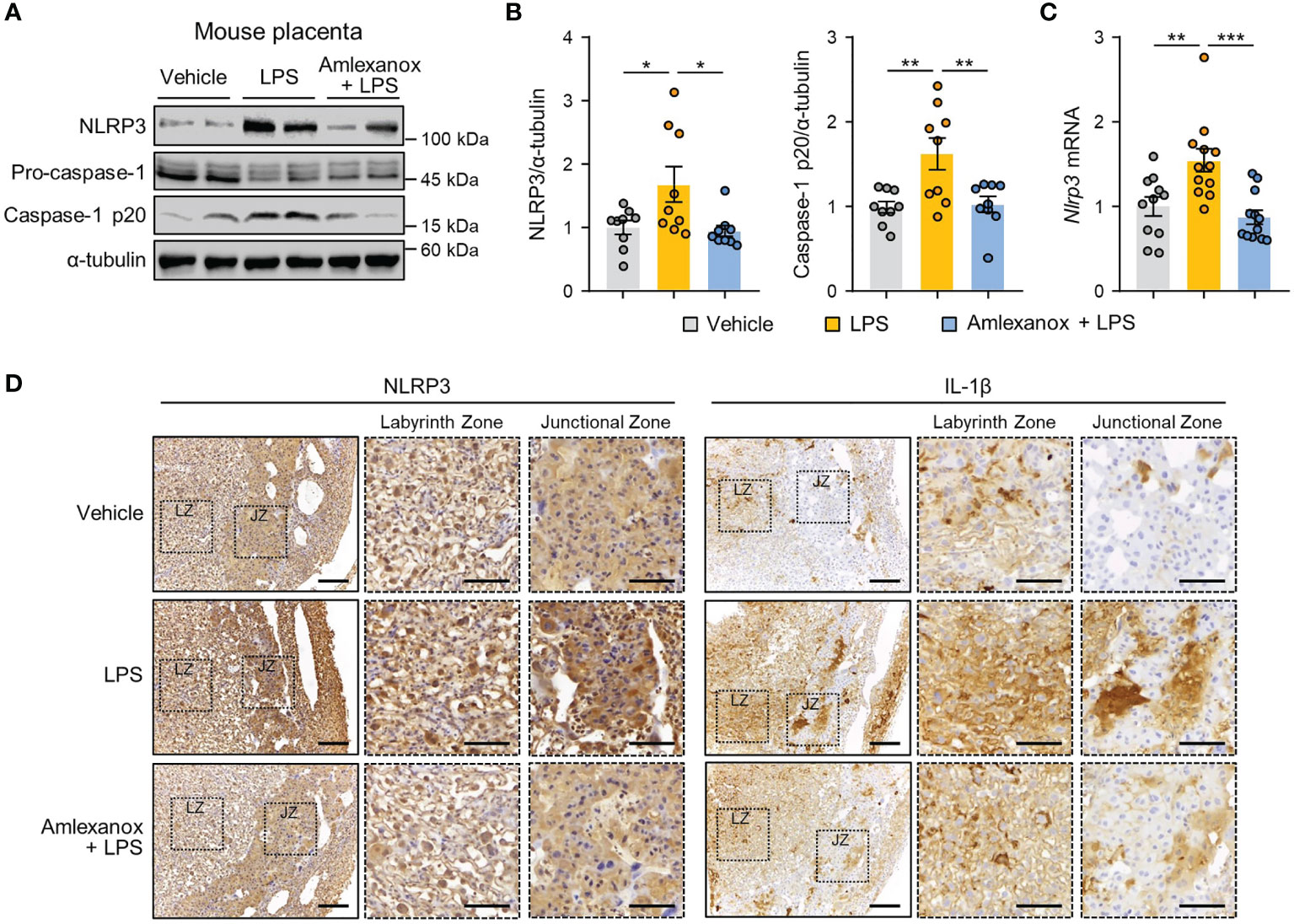
Figure 5 Amlexanox ameliorates placental NLRP3 inflammasome activation in LPS-treated pregnant mice. (A–D) Pregnant mice were intraperitoneally injected with LPS (200 μg/kg) or amlexanox (2.5 mg/kg) on gestation day 17.5, as indicated in the figure. (A, B) Placental tissues were collected from mice and analyzed by immunoblotting with anti-NLRP3 and anti-caspase-1 antibodies (n = 9 per group). α-tubulin served as a loading control. Band intensities were quantified and normalized to control band intensities. (C) Placental tissues were collected from mice and analyzed by qRT-PCR for the expression of NLRP3 (n = 11–12 per group). (D) Immunohistochemical analysis of NLRP3 and IL-1β in placental tissues from vehicle-, LPS-, and LPS+amlexanox-treated pregnant mice at gestation day 17.5. Nuclei were stained with hematoxylin. Boxed areas are magnified in the adjacent panels. LZ and JZ represent the labyrinth zone and the junctional zone, respectively. Scale bars, 200 μm; inset, 100 μm. Data are shown as means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (One-way ANOVA, followed by Tukey’s test).
To determine whether the effect of amlexanox on the NF-κB pathway could be observed in vivo, we measured the phosphorylation levels of NF-κB/p65 in mouse placental tissues. Immunoblot analysis revealed that phosphorylation of NF-κB/p65 was increased, whereas amlexanox treatment prevented the increase in phosphorylation levels of NF-κB/p65 (Figures 6A, B). We also evaluated whether maternal administration of amlexanox affected gene expression of inflammatory cytokines and chemokines using qRT-PCR. The expression of IL-1β, IL-6, IL-10, TNF-α, TGF-β1, MCP-1, and IFN-β genes was elevated in the placenta of LPS-treated mice as compared to that of control mice; this increase was largely attenuated in amlexanox-treated mice exposed to LPS (Figure 6C). Taken together, these data indicate that TBK1 inhibition by amlexanox alleviates placental inflammation and NLRP3 inflammasome activation.
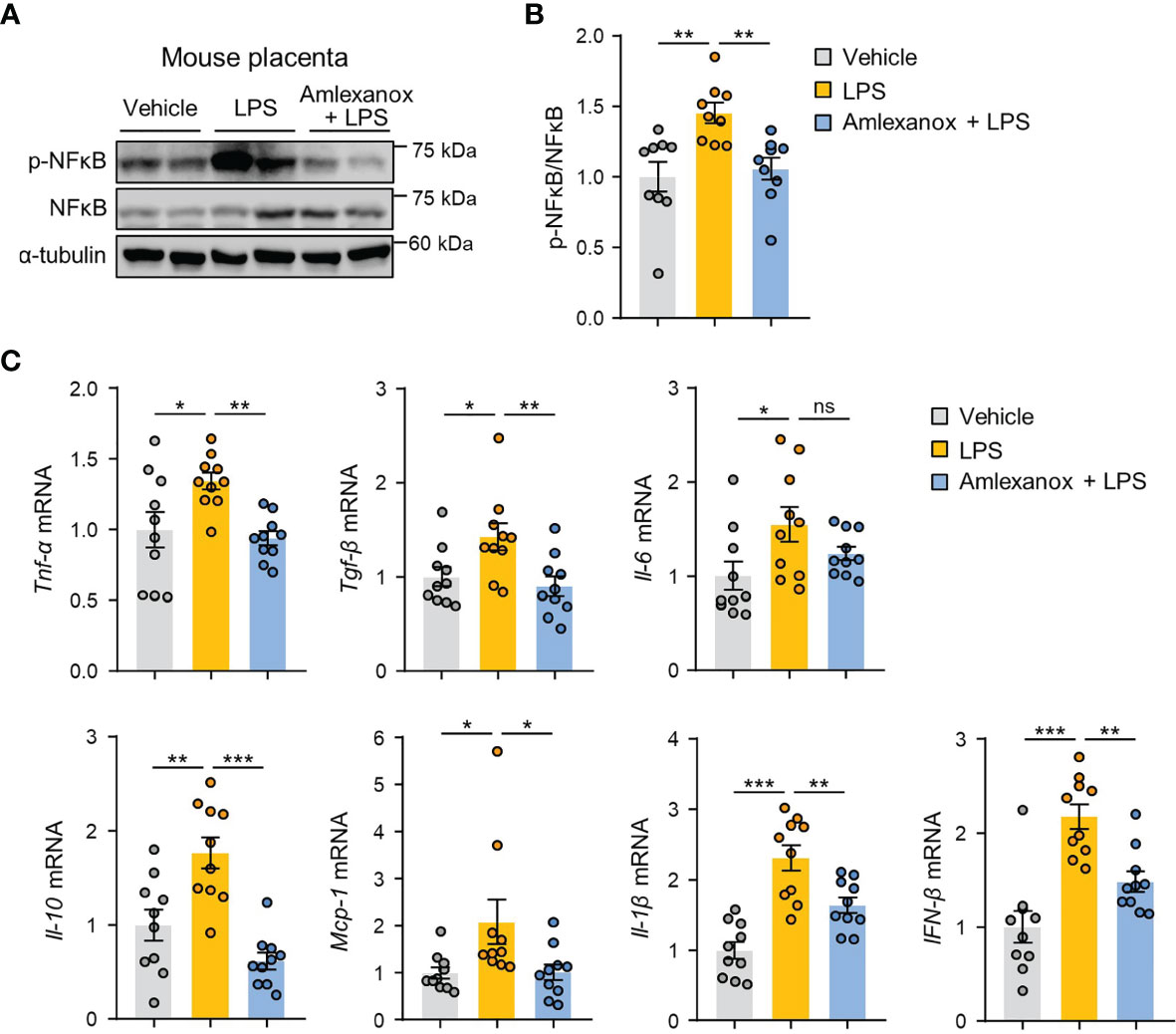
Figure 6 Amlexanox ameliorates LPS-induced inflammatory response in mouse placenta. (A–C) Pregnant mice were intraperitoneally injected with LPS (200 μg/kg) or amlexanox (2.5 mg/kg) on gestation day 17.5, as indicated in the figure. (A, B) Placental tissues were collected from mice and analyzed by immunoblotting with anti-p-p65 and anti-p65 antibodies (n = 9 per group). α-tubulin served as a loading control. Band intensities were quantified and normalized to control band intensities. (C) Placental tissues were collected from mice and analyzed by qRT-PCR for the expression of TNF-α, TGF-β, IL-6, IL-10, MCP-1, IL-1β, and IFN-β (n = 10 per group). Data are shown as means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant (One-way ANOVA, followed by Fisher’s LSD test).
In response to LPS, TBK1 can directly activate mTORC1, but this function of TBK1 has been controversial (29, 30). Therefore, we checked the expression of downstream target proteins of mTORC1 in the placental tissues of control, vehicle-, and amlexanox-treated LPS mice. Immunoblot analysis indicated that mice treated with amlexanox had lower levels of phosphorylated p70 ribosomal protein S6 kinase (p70S6K) compared to that in LPS-treated mice (Figures 7A, B). Further, we evaluated the effect of amlexanox on LPS-induced mTORC1 activation in HTR-8/SVneo cells and found a decrease in the phosphorylation of p70S6K after pretreatment with amlexanox (Figures 7C, D). Consistent with the inhibitory effect of amlexanox on mTORC1 signaling, lentiviral knockdown of TBK1 blocked LPS-induced phosphorylation of p70S6K in HTR-8/SVneo cells (Figures 7E, F), indicating that TBK1 is involved in LPS-mediated mTORC1 activation. To investigate whether mTORC1 inhibition influences LPS-induced NLRP3 inflammasome activation, we pretreated HTR-8/SVneo cells with mTOR inhibitors, rapamycin and Torin 1. Immunoblot analysis revealed suppression of LPS-induced increase in NLRP3 and cleaved caspase-1 protein levels after pretreatment with rapamycin or Torin 1 (Figures 7G–J). Thus, these data suggest that TBK1 deficiency alleviates LPS-induced NLRP3 inflammasome activation in an mTORC1-dependent manner.
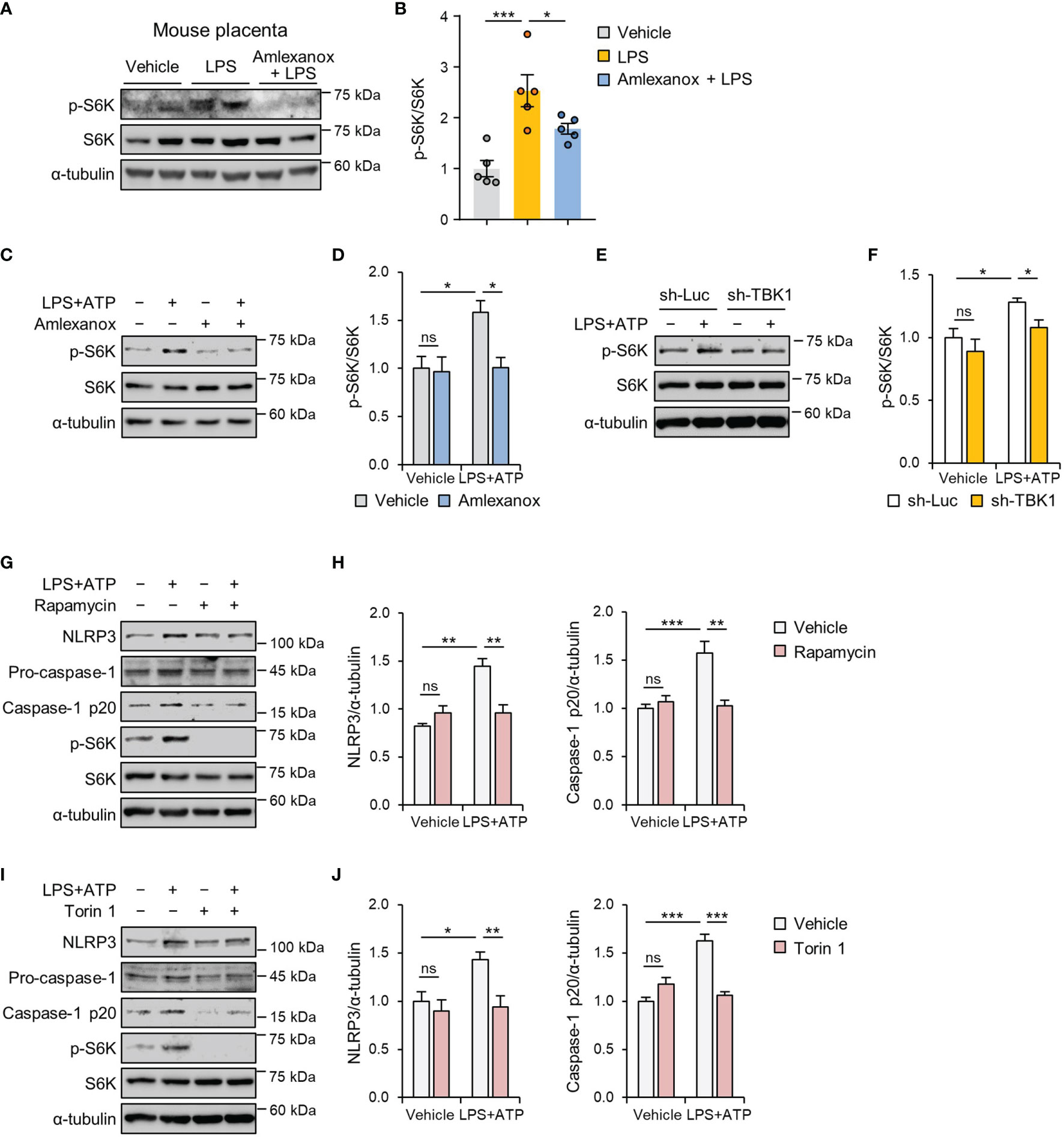
Figure 7 TBK1 deficiency suppresses LPS-induced NLRP3 inflammasome activation via an mTORC1-dependent mechanism. (A, B) Pregnant mice were intraperitoneally injected with LPS (200 μg/kg) or amlexanox (2.5 mg/kg) on gestation day 17.5, as indicated in the figure. Placental tissues were collected from mice and analyzed by immunoblotting with anti-p-S6K and anti-S6K antibodies (n = 5 per group). (C, D) HTR-8/SVneo cells were treated with 1 μg/mL LPS or 100 µM amlexanox for 24 h, as indicated in the figure. Cell lysates were analyzed by immunoblotting with anti-p-S6K and anti-S6K antibodies. α-tubulin served as a loading control. (E, F) HTR-8/SVneo cells were infected with lentiviruses expressing shRNAs targeting luciferase (sh-Luc) or TBK1 (sh-TBK1) and treated with 1 μg/mL LPS for 24 h, as indicated in the figure. Cell lysates were analyzed by immunoblotting with anti-p-S6K and anti-S6K antibodies. (G–J) HTR-8/SVneo cells were treated with 1 μg/mL LPS, 100 nM rapamycin, or 250 nM Torin 1 for 24 h, as indicated in the figure. Cell lysates were analyzed by immunoblotting with anti-NLRP3, anti-caspase-1, anti-p-S6K, and anti-S6K antibodies. α-tubulin served as a loading control. Band intensities were quantified and normalized to control band intensities. Data are shown as means ± SEM. Results are representative of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant (One-way ANOVA, followed by Tukey’s test).
Excessive placental inflammation due to infectious and non-infectious stimuli is responsible for several adverse pregnancy outcomes, including preterm birth, intrauterine growth restriction, and preeclampsia. TBK1 is widely expressed in animals and is involved in the activation of the NF-κB pathway (31, 32). However, the regulation of TBK1 in the pathogenesis of pregnancy complications remains unclear. Here, we observed the phosphorylation of TBK1 during LPS-induced placental inflammation, as evident in mouse placental tissue and human trophoblasts. Elevated IL-1β production has been implicated in the pathogenesis of pregnancy syndromes associated with placental inflammation (33, 34). Growing evidence suggests that abnormal production of biologically active IL-1β is mediated by NLRP3 inflammasome activation in the maternal–placental interface trophoblasts (17, 26, 35, 36). Although experimental models have demonstrated that LPS stimulates the NLRP3 inflammasome in human trophoblasts, no studies have investigated the role of TBK1 in LPS-induced NLRP3 inflammasome activation. In this study, we demonstrated the molecular link between TBK1 phosphorylation and NLRP3 inflammasome activation in LPS-induced placental inflammation. Furthermore, TBK1 inhibition attenuates LPS-induced impairment of fetal and placental development, as assessed by fetal and placental weight and length (Figure 8), suggesting that TBK1 phosphorylation is involved in placental inflammation and NLRP3 inflammasome activation.
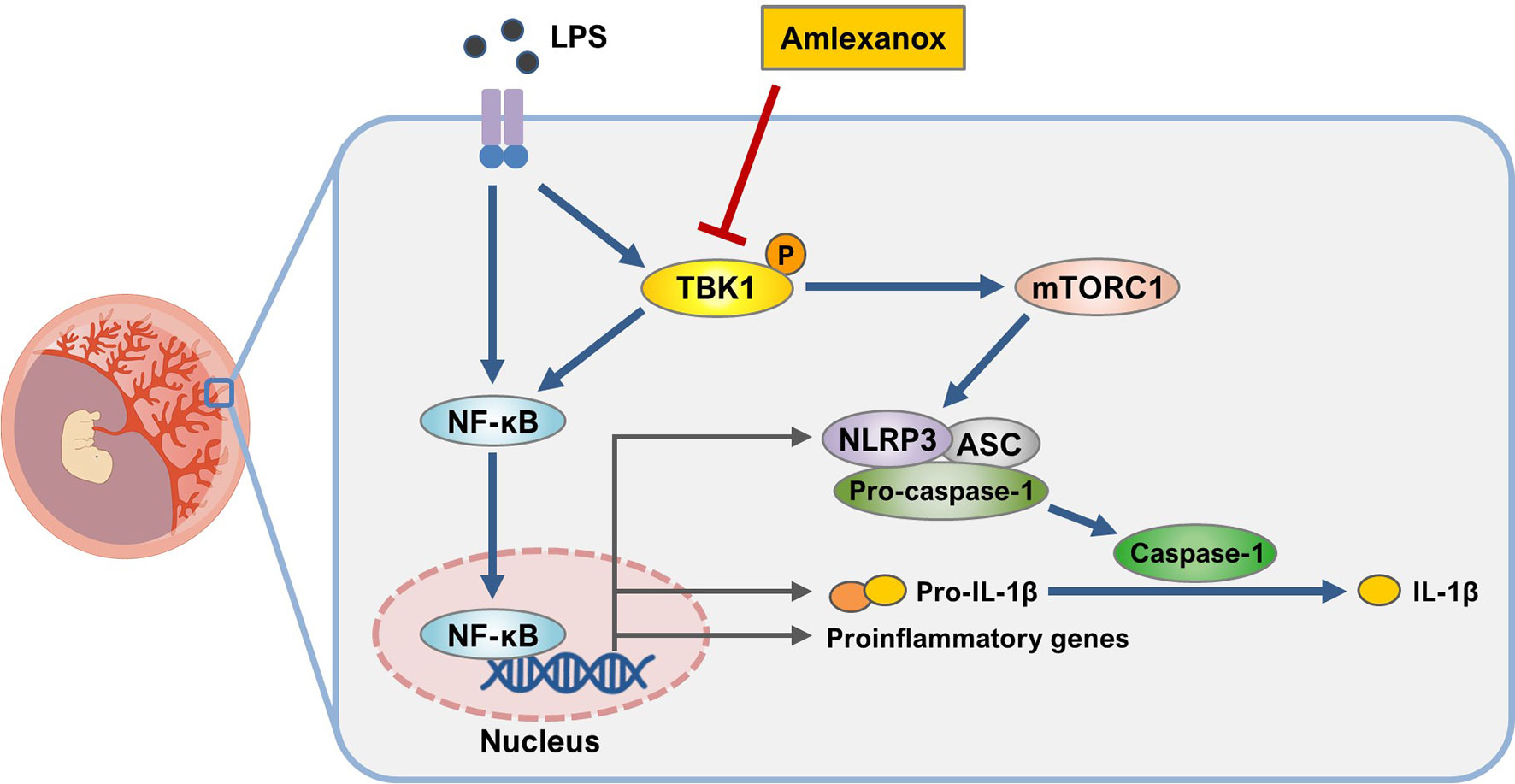
Figure 8 Schematic diagram of the role of TBK1 in trophoblasts upon exposure to LPS. Inhibition of TBK1 by amlexanox not only suppresses LPS-induced NLRP3 inflammasome activation but also attenuates inflammatory response by inhibiting mTORC1 signaling.
TBK1 is phosphorylated in adipose and liver tissues of obese mice (21, 37) and trophoblast cells in response to LPS stimulation (38), which agrees with our findings that LPS treatment induces TBK1 phosphorylation in mouse placental tissue and human trophoblasts. However, the relationship between LPS-associated placental inflammation and TBK1 activation had not been studied previously. A previous study from our group indicated that BX795, a TBK1 inhibitor, suppressed the expression of inflammatory cytokines, such as IL-6 and IL-10, in a nonalcoholic steatohepatitis mouse model; however, the relevant mechanism was not defined (21). Here, we found that maternal administration of amlexanox resulted in a decrease in the expression of inflammatory cytokine and chemokine genes, including IL-1β, IL-10, TNF-α, TGF-β1, MCP-1, and IFN-β in the placenta of LPS-treated mice. These effects are probably attributed to reduced TBK1 activity, which can regulate the expression of inflammatory mediators (39–41). IFN-β expression can be induced by pathogen-associated molecular patterns in trophoblast cells through TLR4/IRF3 signaling pathway (42–44). The role of IFN-β in pregnancy has been revealed recently. Kwon et al. reported that aberrant production of IFN-β and its associated IFN-stimulated gene activation result in fetal demise (38). Consistent with this finding, IFN-β gene expression was elevated in the placenta of LPS-treated mice as compared to that of control mice; this increase was largely attenuated in amlexanox-treated mice exposed to LPS. Although our current data clearly reveal a reduction in IL-1β expression following maternal administration of amlexanox in the placenta of LPS-treated mice, it is possible that blocking TBK1 activation might also affect IFN-β expression by activating IRF3. Given that TBK1 mediates phosphorylation and nuclear translocation of IRF3, which contributes to induction of type I IFNs (45, 46), our data suggest that the effect of TBK1 inhibition on the LPS-treated pregnant mice may be partially mediated through alteration of IFN-β production.
TBK1 contributes to the activation of the non-canonical NF-κB pathway (47–49). However, TBK1 may also inhibit the activation of the NF-κB pathway (50). Given that TBK1 is involved in cell- and stimulus-dependent NF-κB activation, we directly investigated the effect of TBK1 deficiency on the LPS-induced NF-κB pathway. Our results clearly demonstrated that the treatment of mice and human trophoblast cells with amlexanox reduced the LPS-induced NF-κB/p65 pathway. Thus, our data suggest that TBK1 inhibition is intimately involved in the negative regulation of the NF-κB/p65 pathway.
In conclusion, our results provide evidence for the potential role of TBK1 in LPS-induced activation of the placental NLRP3 inflammasome. LPS treatment upregulates the phosphorylation of TBK1, which mediates placental NLRP3 inflammasome activation and inflammation. Our study elucidates the TBK1-mTORC1 axis and its relevance to placental inflammation. Future investigation will be needed to explore the upstream signaling intermediates regulating the TBK1‐mTORC1 axis during LPS-induced placental inflammation. Our in vitro and in vivo study reveals a protective effect of pharmacological inhibition of TBK1 on LPS-induced NLRP3 inflammasome activation and inflammation, providing an interesting opportunity for repositioning of TBK inhibitors for pregnancy-related complications.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Konyang University.
S-KL and H-WP: Conceptualization and experimental design. SL, JiS, JoS, and H-WP: Investigation and analysis. J-SK, JoS, and S-KL: Resources J-SK, JiS, and JoS: methodology. SL, JiS, and H-WP: Writing—Original draft preparation. S-KL and H-WP: Writing— review and editing. S-KL and H-WP: Verification and supervision. All authors contributed to the article and approved the submitted version.
This research was supported by grants from the National Research Foundation of Korea funded by the Ministry of Science and ICT (No. 2015R1A5A1009701) and the Ministry of Education (No. NRF-2017R1A6A1A03015713). This research was also supported by a grant from the Korean Health Technology R&D Project funded by the Ministry of Health and Welfare, Republic of Korea (No. HI17C1238).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the H-WP lab members for their valuable contributions to this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.743700/full#supplementary-material
1. Riley JK, Nelson DM. Toll-Like Receptors in Pregnancy Disorders and Placental Dysfunction. Clin Rev Allergy Immunol (2010) 39:185–93. doi: 10.1007/s12016-009-8178-2
2. Mor G, Cardenas I, Abrahams V, Guller S. Inflammation and Pregnancy: The Role of the Immune System at the Implantation Site. Ann N Y Acad Sci (2011) 1221:80–7. doi: 10.1111/j.1749-6632.2010.05938.x
3. Benyo DF, Smarason A, Redman CW, Sims C, Conrad KP. Expression of Inflammatory Cytokines in Placentas From Women With Preeclampsia. J Clin Endocrinol Metab (2001) 86:2505–12. doi: 10.1210/jcem.86.6.7585
4. Boutsikou T, Mastorakos G, Kyriakakou M, Margeli A, Hassiakos D, Papassotiriou I, et al. Circulating Levels of Inflammatory Markers in Intrauterine Growth Restriction. Mediators Inflamm (2010) 2010:790605. doi: 10.1155/2010/790605
5. Sharma A, Satyam A, Sharma JB. Leptin, IL-10 and Inflammatory Markers (TNF-Alpha, IL-6 and IL-8) in Pre-Eclamptic, Normotensive Pregnant and Healthy non-Pregnant Women. Am J Reprod Immunol (2007) 58:21–30. doi: 10.1111/j.1600-0897.2007.00486.x
6. Abrahams VM, Mor G. Toll-Like Receptors and Their Role in the Trophoblast. Placenta (2005) 26:540–7. doi: 10.1016/j.placenta.2004.08.010
7. Patni S, Wynen LP, Seager AL, Morgan G, White JO, Thornton CA. Expression and Activity of Toll-Like Receptors 1-9 in the Human Term Placenta and Changes Associated With Labor at Term. Biol Reproduction (2009) 80:243–8. doi: 10.1095/biolreprod.108.069252
8. Tangeras LH, Stodle GS, Olsen GD, Leknes AH, Gundersen AS, Skei B, et al. Functional Toll-Like Receptors in Primary First-Trimester Trophoblasts. J Reprod Immunol (2014) 106:89–99. doi: 10.1016/j.jri.2014.04.004
9. Koga K, Izumi G, Mor G, Fujii T, Osuga Y. Toll-Like Receptors at the Maternal-Fetal Interface in Normal Pregnancy and Pregnancy Complications. Am J Reprod Immunol (2014) 72:192–205. doi: 10.1111/aji.12258
10. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of Interferon-Gamma Inducing Factor Mediated by Interleukin-1beta Converting Enzyme. Science (1997) 275:206–9. doi: 10.1126/science.275.5297.206
11. Van De Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome Activation and IL-1beta and IL-18 Processing During Infection. Trends Immunol (2011) 32:110–6. doi: 10.1016/j.it.2011.01.003
12. Heneka MT, Mcmanus RM, Latz E. Inflammasome Signalling in Brain Function and Neurodegenerative Disease. Nat Rev Neurosci (2018) 19:610–21. doi: 10.1038/s41583-018-0055-7
13. Latz E. The Inflammasomes: Mechanisms of Activation and Function. Curr Opin Immunol (2010) 22:28–33. doi: 10.1016/j.coi.2009.12.004
14. Gomez-Lopez N, Romero R, Garcia-Flores V, Leng Y, Miller D, Hassan SS, et al. Inhibition of the NLRP3 Inflammasome can Prevent Sterile Intra-Amniotic Inflammation, Preterm Labor/Birth, and Adverse Neonatal Outcomesdagger. Biol Reproduction (2019) 100:1306–18. doi: 10.1093/biolre/ioy264
15. Brien ME, Duval C, Palacios J, Boufaied I, Hudon-Thibeault AA, Nadeau-Vallee M, et al. Uric Acid Crystals Induce Placental Inflammation and Alter Trophoblast Function via an IL-1-Dependent Pathway: Implications for Fetal Growth Restriction. J Immunol (2017) 198:443–51. doi: 10.4049/jimmunol.1601179
16. I CW, Romao-Veiga M, Matias ML, Fioratti EG, Peracoli JC, Borges VT, et al. Increased Expression of NLRP3 Inflammasome in Placentas From Pregnant Women With Severe Preeclampsia. J Reprod Immunol (2017) 123:40–7. doi: 10.1016/j.jri.2017.09.002
17. Park S, Shin J, Bae J, Han D, Park SR, Shin J, et al. SIRT1 Alleviates LPS-Induced IL-1beta Production by Suppressing NLRP3 Inflammasome Activation and ROS Production in Trophoblasts. Cells (2020) 9:728. doi: 10.3390/cells9030728
18. Helgason E, Phung QT, Dueber EC. Recent Insights Into the Complexity of Tank-Binding Kinase 1 Signaling Networks: The Emerging Role of Cellular Localization in the Activation and Substrate Specificity of TBK1. FEBS Lett (2013) 587:1230–7. doi: 10.1016/j.febslet.2013.01.059
19. Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The Roles of Two IkappaB Kinase-Related Kinases in Lipopolysaccharide and Double Stranded RNA Signaling and Viral Infection. J Exp Med (2004) 199:1641–50. doi: 10.1084/jem.20040520
20. Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential Requirement for TANK-Binding Kinase-1 in Type I Interferon Responses to Toll-Like Receptor Activation and Viral Infection. J Exp Med (2004) 199:1651–8. doi: 10.1084/jem.20040528
21. Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, et al. Lipotoxicity Induces Hepatic Protein Inclusions Through TANK Binding Kinase 1-Mediated P62/Sequestosome 1 Phosphorylation. Hepatology (2018) 68:1331–46. doi: 10.1002/hep.29742
22. Fitzgerald KA, Mcwhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are Essential Components of the IRF3 Signaling Pathway. Nat Immunol (2003) 4:491–6. doi: 10.1038/ni921
23. Moser CV, Stephan H, Altenrath K, Kynast KL, Russe OQ, Olbrich K, et al. TANK-Binding Kinase 1 (TBK1) Modulates Inflammatory Hyperalgesia by Regulating MAP Kinases and NF-kappaB Dependent Genes. J Neuroinflamm (2015) 12:100. doi: 10.1186/s12974-015-0319-3
24. Niederberger E, Moser CV, Kynast KL, Geisslinger G. The non-Canonical IkappaB Kinases IKKepsilon and TBK1 as Potential Targets for the Development of Novel Therapeutic Drugs. Curr Mol Med (2013) 13:1089–97. doi: 10.2174/1566524011313070004
25. Lee S, Han D, Kang HG, Jeong SJ, Jo JE, Shin J, et al. Intravenous Sustained-Release Nifedipine Ameliorates Nonalcoholic Fatty Liver Disease by Restoring Autophagic Clearance. Biomaterials (2019) 197:1–11. doi: 10.1016/j.biomaterials.2019.01.008
26. Stodle GS, Silva GB, Tangeras LH, Gierman LM, Nervik I, Dahlberg UE, et al. Placental Inflammation in Pre-Eclampsia by Nod-Like Receptor Protein (NLRP)3 Inflammasome Activation in Trophoblasts. Clin Exp Immunol (2018) 193:84–94. doi: 10.1111/cei.13130
27. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB Signaling in Inflammation. Signal Transduct Target Ther (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
28. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 Inflammasome Activation. Ann N Y Acad Sci (2014) 1319:82–95. doi: 10.1111/nyas.12458
29. Antonia RJ, Castillo J, Herring LE, Serafin DS, Liu P, Graves LM, et al. TBK1 Limits Mtorc1 by Promoting Phosphorylation of Raptor Ser877. Sci Rep (2019) 9:13470. doi: 10.1038/s41598-019-49707-8
30. Bodur C, Kazyken D, Huang K, Ekim Ustunel B, Siroky KA, Tooley AS, et al. The IKK-Related Kinase TBK1 Activates Mtorc1 Directly in Response to Growth Factors and Innate Immune Agonists. EMBO J (2018) 37:19–38. doi: 10.15252/embj.201696164
31. Clement JF, Meloche S, Servant MJ. The IKK-Related Kinases: From Innate Immunity to Oncogenesis. Cell Res (2008) 18:889–99. doi: 10.1038/cr.2008.273
32. Yin N, Yang Y, Wang X, Yang C, Ma X, Shaukat A, et al. MiR-19a Mediates the Negative Regulation of the NF-kappaB Pathway in Lipopolysaccharide-Induced Endometritis by Targeting TBK1. Inflammation Res (2019) 68:231–40. doi: 10.1007/s00011-019-01213-3
33. Cierny JT, Unal ER, Flood P, Rhee KY, Praktish A, Olson TH, et al. Maternal Inflammatory Markers and Term Labor Performance. Am J Obstet Gynecol (2014) 210:447 e1–6. doi: 10.1016/j.ajog.2013.11.038
34. Gomez-Lopez N, Motomura K, Miller D, Garcia-Flores V, Galaz J, Romero R. Inflammasomes: Their Role in Normal and Complicated Pregnancies. J Immunol (2019) 203:2757–69. doi: 10.4049/jimmunol.1900901
35. Tamura K, Ishikawa G, Yoshie M, Ohneda W, Nakai A, Takeshita T, et al. Glibenclamide Inhibits NLRP3 Inflammasome-Mediated IL-1beta Secretion in Human Trophoblasts. J Pharmacol Sci (2017) 135:89–95. doi: 10.1016/j.jphs.2017.09.032
36. Abrahams VM, Tang Z, Mor G, Guller S. NLRP3 Inflammasome Function and Pyroptotic Cell Death in Human Placental Hofbauer Cells. J Reprod Immunol (2020) 142:103214. doi: 10.1016/j.jri.2020.103214
37. Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An Inhibitor of the Protein Kinases TBK1 and IKK-Varepsilon Improves Obesity-Related Metabolic Dysfunctions in Mice. Nat Med (2013) 19:313–21. doi: 10.1038/nm.3082
38. Kwon JY, Aldo P, You Y, Ding J, Racicot K, Dong X, et al. Relevance of Placental Type I Interferon Beta Regulation for Pregnancy Success. Cell Mol Immunol (2018) 15:1010–26. doi: 10.1038/s41423-018-0050-y
39. Yu T, Shim J, Yang Y, Byeon SE, Kim JH, Rho HS, et al. 3-(4-(Tert-Octyl)phenoxy)propane-1,2-Diol Suppresses Inflammatory Responses via Inhibition of Multiple Kinases. Biochem Pharmacol (2012) 83:1540–51. doi: 10.1016/j.bcp.2012.02.018
40. Xie XH, Zang N, Li SM, Wang LJ, Deng Y, He Y, et al. Resveratrol Inhibits Respiratory Syncytial Virus-Induced IL-6 Production, Decreases Viral Replication, and Downregulates TRIF Expression in Airway Epithelial Cells. Inflammation (2012) 35:1392–401. doi: 10.1007/s10753-012-9452-7
41. Yu T, Yi YS, Yang Y, Oh J, Jeong D, Cho JY. The Pivotal Role of TBK1 in Inflammatory Responses Mediated by Macrophages. Mediators Inflamm (2012) 2012:979105. doi: 10.1155/2012/979105
42. Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, Peri F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines (Basel) (2017) 5:34. doi: 10.3390/vaccines5040034
43. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 Signal Transduction Pathway. Cytokine (2008) 42:145–51. doi: 10.1016/j.cyto.2008.01.006
44. Firmal P, Shah VK, Chattopadhyay S. Insight Into TLR4-Mediated Immunomodulation in Normal Pregnancy and Related Disorders. Front Immunol (2020) 11:807. doi: 10.3389/fimmu.2020.00807
45. Rathinam VA, Fitzgerald KA. Cytosolic Surveillance and Antiviral Immunity. Curr Opin Virol (2011) 1:455–62. doi: 10.1016/j.coviro.2011.11.004
46. Yum S, Li M, Fang Y, Chen ZJ. TBK1 Recruitment to STING Activates Both IRF3 and NF-kappaB That Mediate Immune Defense Against Tumors and Viral Infections. Proc Natl Acad Sci U S A (2021) 118:e2100225118. doi: 10.1073/pnas.2100225118
47. Pomerantz JL, Baltimore D. NF-kappaB Activation by a Signaling Complex Containing TRAF2, TANK and TBK1, a Novel IKK-Related Kinase. EMBO J (1999) 18:6694–704. doi: 10.1093/emboj/18.23.6694
48. Kravchenko VV, Mathison JC, Schwamborn K, Mercurio F, Ulevitch RJ. IKKi/IKKepsilon Plays a Key Role in Integrating Signals Induced by Pro-Inflammatory Stimuli. J Biol Chem (2003) 278:26612–9. doi: 10.1074/jbc.M303001200
49. Lee SH, Kim KW, Min KM, Kim KW, Chang SI, Kim JC. Angiogenin Reduces Immune Inflammation via Inhibition of TANK-Binding Kinase 1 Expression in Human Corneal Fibroblast Cells. Mediators Inflamm (2014) 2014:861435. doi: 10.1155/2014/861435
Keywords: TBK1, NLRP3 inflammasome, maternal inflammation, trophoblast, placenta, mTORC1
Citation: Lee S, Shin J, Kim J-S, Shin J, Lee SK and Park H-W (2021) Targeting TBK1 Attenuates LPS-Induced NLRP3 Inflammasome Activation by Regulating of mTORC1 Pathways in Trophoblasts. Front. Immunol. 12:743700. doi: 10.3389/fimmu.2021.743700
Received: 19 July 2021; Accepted: 22 October 2021;
Published: 09 November 2021.
Edited by:
Markus H. Hoffmann, University of Erlangen Nuremberg, GermanyReviewed by:
Chien-Kuo Lee, National Taiwan University, TaiwanCopyright © 2021 Lee, Shin, Kim, Shin, Lee and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hwan-Woo Park, aHdhbndvb3BhcmtAa29ueWFuZy5hYy5rcg==; Sung Ki Lee, aHNrbGVlQGt5dWguYWMua3I=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.