
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 17 November 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.742834
 Anna Hogendorf1*
Anna Hogendorf1* Maciej Zieliński2
Maciej Zieliński2 Maria Constantinou3
Maria Constantinou3 Robert Śmigiel4Jolanta Wierzba5Krystyna Wyka6
Robert Śmigiel4Jolanta Wierzba5Krystyna Wyka6 Anna Wędrychowicz7
Anna Wędrychowicz7 Anna Jakubiuk-Tomaszuk8Edyta Budzynska3Malgorzata Piotrowicz9
Anna Jakubiuk-Tomaszuk8Edyta Budzynska3Malgorzata Piotrowicz9 Beata S. Lipska-Ziętkiewicz10Ewa Kaczorowska11Agata Cieślikowska12Anna Kutkowska-Kaźmierczak13Jolanta Fijak-Moskal14Monika Kugaudo15Małgorzata Kosińska-Urbańska16Agnieszka Szadkowska1
Beata S. Lipska-Ziętkiewicz10Ewa Kaczorowska11Agata Cieślikowska12Anna Kutkowska-Kaźmierczak13Jolanta Fijak-Moskal14Monika Kugaudo15Małgorzata Kosińska-Urbańska16Agnieszka Szadkowska1 Maciej Borowiec3Maciej Niedźwiecki17
Maciej Borowiec3Maciej Niedźwiecki17 Piotr Trzonkowski2
Piotr Trzonkowski2 Wojciech Młynarski6*
Wojciech Młynarski6*Introduction: Autoimmune disorders, IgA deficiency, and allergies seem to be common among individuals with 18q deletion syndrome [OMIM 601808]. We aimed to determine the prevalence, mechanism, and genetic background of autoimmunity, immune deficiency, and allergy in a cohort of patients with 18q deletions.
Material and Methods: Medical registries and social media were used to recruit the patients. Microarray oligonucleotide comparative genomic hybridization (aCGH) (Agilent, Santa Clara, CA, USA) was performed in all patients to identify size and location of chromosome 18 deletion. Clinical evaluation and medical record collection were performed in each of the study participants. The history of autoimmune disorders, severe and/or recurrent infections, and symptoms of allergy were noted. Total immunoglobulin IgG, IgA, IgM, IgE, and IgG1-4 serum levels were measured using nephelometry and ELISA methods. Lymphocyte T subset phenotyping was performed in 24 subjects from 18q del cohort. To predict the most promising candidate genes, we used the ENDEAVOUR—a free web resource for gene prioritization.
Results: 18q deletion was confirmed by means of array CGH analysis in 27 individuals, 15 (55.6%) females and 12 males, referred to the project by specialists in medical genetics, diabetology, or pediatric endocrinology between May 2015 and December 2019. The mean age at examination was 11.8 years (min–max: 4.0–33.5). Autoimmune disorders were present in 14/27 (51.8%) of the cohort. In eight of patients, symptoms of immune deficiency coexisted with autoimmunity. Allergy was reported in nine of 27 (33.4%) patients. Over 89% of patients presented with at list one type of immunoglobulin (IgA, IgM, IgG, IgE, and IgG1-4) deficiency and eight of 25 (32%) had abnormalities in at least two major immunoglobulin (IgG, IgA, IgM) measurements (CVID-like phenotype). Patients with 18q del exhibited a significantly decreased CD4, Treg FOXP3+, TregFOXP3+Helios+, and TemCD4 cell numbers in comparison with the control groups of 24 T1DM patients and 28 healthy controls.
Conclusions: Patients with 18q deletions frequently suffer from autoimmune disorders, recurrent infections, and allergy due to immune dysregulation presenting with variable antibody deficiencies and T-regulatory cell deficiency (CD4+CD25+CD127lowFOXP3+). The spectrum of speculations regarding which gene might be responsible for such phenotype ranges from single gene haploinsufficiency to deletion of a cluster of immunogenes located distally to 18q21.
Chromosome 18q deletion syndrome (de Grouchy syndrome, OMIM 601808) results from a loss of a fragment of the long (q) arm of 18 chromosomes. It was first described in 1964 (1) and occurs in about one in 40,000–55,000 live births (2). The syndrome is known to have a heterogeneous presentation that includes multiple congenital anomalies such as short stature, developmental delay, and intellectual disability, hypotonia, microcephaly, and other dysmorphic features such as hypoplasia of the midsection of the face with wide carp-like mouth, as well as skeletal deformities such as hand and foot deformities, genital anomalies, vision, and hearing impairment. Less common signs include cleft lip and palate, congenital heart disease, hypertelorism, clinodactyly, umbilical and inguinal hernia, convulsions, and feeding problems (3, 4). Most 18q deletions are terminal and localized in the distal half of the long arm (18q21.1-qter). However, smaller interstitial deletions, residing in the region between q12 and q21 and associated with a milder phenotype have also been described (5). About 94% of cases with 18q deletion syndrome appearance are de novo, and the remaining 6% are the inherited from a parent carrying a balanced chromosomal translocation.
We have previously reported on a girl with 18q del syndrome, type 1 diabetes mellitus, autoimmune thyroiditis, vitiligo, and recurrent infections due to immunoglobulin A, E, and G4 deficiency. She was also found to have CD3+CD4+FoxP3+, CD3+CD4+FoxP3+CD25+, and CD3+CD4+CD25+CD127+ regulatory T cell deficiency. Therefore, we hypothesized that the exceptional coincidence of the three autoimmune disorders occurring at an early age and immune deficiency is associated with the deletion of certain gene(s) located on deleted region at 18q21.32-q23 (chr18:58,660,699-78,012,870) (6).
Our hypothesis is strongly supported in literature by several case reports on the coexistence of autoimmune thyroiditis, pernicious anemia, vitiligo, and also immunoglobulin (Ig)A deficiency and CVID in patients with the syndrome (7–9).
This study was therefore undertaken to evaluate the prevalence of autoimmune disorders, recurrent/severe infections, and allergies among patients with 18q del syndrome and to check if the previously observed hypogammaglobulinemia and TregFOXP3+ deficiency are common features in these patients. Additional aim was to identify molecular background of the immune dysregulation by prioritization of deleted genes.
The study was approved by the Ethics Committee of the Medical University of Lodz. The information about the project was spread among stakeholders at the Departments of Medical Genetics and Pediatric Endocrinology countrywide, The Polish Registry of Congenital Defects, the Polish Association for Rare Diseases (patient society), Diabetes Poland (medical association) via internet and during Medical Conferences and Symposia. Social media, e.g., Facebook, Rare Connect were also helpful in the patients’ recruitment. Patients were recruited in collaboration with 12 medical genetics or endocrinology centers and cytogenetic laboratories from eight major Polish cities (Lodz, Warsaw, Krakow, Gdansk, Poznan, Wroclaw, Zielona Gora, and Bialystok). The deletions were previously diagnosed by standard cytogenetic methods. The written informed consent was obtained from each study subject and/or legal guardian in accordance with the Declaration of Helsinki.
The participants were evaluated on-site in the participating centers by our team of investigators; some of them several times. Data on medical history, especially concerning frequent and recurrent infections, autoimmune and nonautoimmune comorbidities and detailed family history, as well as laboratory results were obtained from the children’s parents and from all relevant medical records.
At the same time, peripheral venous blood samples were obtained, but only from those who were otherwise healthy.
For the purpose of the T-regulatory cells, assessment peripheral venous blood samples were also obtained from two age and sex-matched control groups—24 patients with autoimmune diabetes (T1D) and 28 healthy controls.
Peripheral venous blood drawn into 2.9 ml EDTA tubes was sent to the Department of Clinical Genetics, Medical University of Lodz for genetic evaluation (aCGH).
Serum samples (4 ml) were prepared by centrifugation at 3,000×g for 10 min, aliquoted, and sent to the “APC” laboratory until IgA, IgM, IgG, IgE, and IgG1-IgG4 level assessment and to study center of the Department of Pediatrics, Oncology and Hematology, Medical University of Lodz, where they were stored at −80°C for future analyses.
Peripheral venous blood (4–6 ml) for immunophenotyping was drawn into anticoagulanted EDTA tubes and sent overnight in styrofoam cooler boxes (not frozen) to the Department of Clinical Immunology and Transplantology, Medical University of Gdansk, Poland.
DNA was isolated from peripheral blood by DNA isolation kit (Qiagen, www.qiagen.com). Array comparative genome hybridization (aCGH) was performed by an Agilent Human Genome SurePrint G3 CGH ISCA v2 Microarray Kit, 8x60K (Agilent, www.agilent.com). A 60-mer oligonucleotyde-based microarray (60 k aCGH) with 18,851 probes in ISCA regions and 40,208 backbone probes was used, which allows for genome-wide survey and molecular aberration typing with resolution of approximately 180 kb. A 60-k array allows for detection of copy number variations (CNVs) larger than 500 kb. DNA—both the control one and that from the patient, was labeled using Klenov’s method, following the protocol of Agilent. A volume of 200 ng of purified patient’s DNA and DNA from a male control was double digested with restriction enzyme AluI and RSAI (Promega, www.promega.com) at 37°C for 2 h. Following purification on columns, 1 µg of digested patient’s DNA and of the male control’s DNA were labeled by Cy5-dUTP and Cy3-dUYP, respectively, using the random priming method (Invitrogen, www.invitrogen.com) at 37°C for 2 h. The labeled DNA was purified on the column, mixed, denaturated, and prehybridized with 50 µg of Cot-1 DNA at 37°C for 30 min, then applied onto the array and hybridized at 65°C in a hybridization stove with rotation for 40 h. After hybridization, the array was twice washed in aCGH wash buffers 1 and 2 and then analyzed by an INOPSYS 900 AL scanner and the Feature Extraction for CytoGenomics program. Genomic analysis and graphic presentation of the results was generated by the Agilent CytoGenomics software package (Edition 2.9.2.4).
Total immunoglobulin IgG, IgA, IgM, and IgE serum levels were measured in all the patients in a reference “APC” laboratory (supported by Labquality and Cequal external quality assessment). IgG, IgA, IgM, and IgE concentrations were measured with nephelometry using a Binding Site, Birmingham, UK according to the manufacturer’s instructions. IgG subclasses IgG1, IgG2, IgG3, or IgG4 levels were determined in patients with IgGD and/or IgAD using PeliClass human IgG subclass ELISA kit M1551, Sanquin Reagents. Samples with an IgE concentration below the lower limit of detection of the nephelometric IgE assay were reevaluated by the total IgE low-range enzyme-linked fluorescent assay on a Mini Vidas instrument (bioMérieux S.A., Marcy l’Étoile, France) to obtain a precise determination.
As immunoglobulin levels in the pediatric population are age dependent, for the purpose of the study, age-adjusted reference values for IgA, IgM, IgG, and IgG1-4 levels, determined in healthy children in Mazovia district, were used (10).
Autoimmune diseases—diseases occurring when the immune system attacks self-molecules as a result of a breakdown of immunologic tolerance to autoreactive immune cells strongly associated with genetic, infectious, and/or environmental predisposing factors (e.g., autoimmune thyroiditis, type 1 diabetes, celiac disease, juvenile idiopathic arthritis, immune thrombocytopenic purpura, autoimmune neutropenia, ulcerative colitis, Crohn’s disease).
Severe infections—infections requiring hospitalization to administer specific treatment, not possible in-home care (intravenous antibiotic treatment, oxygen therapy, resuscitation).
Allergy—an unnecessary immune response to an innocuous substance (an allergen). Examples of common substances people are allergic to include pollens, dust mites, insect venom, and food such as nuts or shellfish. Allergic reactions can be grouped into two classes. The most common and best understood is mediated by a class of antibody called IgE. Other reactions are non-IgE mediated and typically cause symptoms to appear more slowly, sometimes several hours after exposure. In fact, non-IgE-mediated reactions do not necessarily involve antibodies but instead, cell reactions of the immune system. They are much less common and are generally less well-understood. The most common way to diagnose an IgE-mediated allergy is through a blood test to identify allergen-specific IgE or a skin prick test which results in a local inflammatory reaction after administration of the trigger allergen. Allergy may manifest as eczema, allergic rhinitis and conjunctivitis, food allergy, asthma, urticaria, angioedema, and anaphylactic shock.
Anaphylaxis is a serious, life-threatening, generalized hypersensitivity reaction that can occur via immunologic (either IgE-dependent or IgE-independent) or nonimmunologic mechanisms (11).
According to the diagnostic criteria for PID established by the European Society for Immunodeficiencies (ESID), selective IgAD deficiency was defined by serum levels of IgA less than 0.07 g/l in the presence of normal IgG and IgM in a patient older than 4 years, in whom other causes of hypogammaglobulinemia have been excluded (12). IgG and IgM deficiency was considered when it was below age-adjusted reference values (12). Selective IgE deficiency (IgED) was defined as a significant decrease in serum levels of IgE (<2 kIU/L) in the presence of normal IgG and IgM (13). Complex hypogammaglobulinemia or CVID-like phenotype—was defined by reduced total serum IgG and IgA and/or IgM levels. The definitions of immunoglobulin deficiencies are given in Table 1.

Table 1 Immunoglobulin deficiency categories considered in the studya.
Regulatory T cells were tested using EDTA whole blood samples. First, PBMC was isolated with a density gradient method, Ficoll Paque Plus (GE Healthcare, Chicago, IL, USA). Samples were checked for viability and 80% cutoff was applied, as tested with trypan blue method. Then 250,000 cells were stained with CD3 V500 (clone UCHT1), CD4 PerCP (SK3), CD25 PE (clone 2A3), CD127 FITC (clone HIL-7R-M21), CD45RA PE-Cy7 (clone L48) (all from BD Biosciences, San Jose, CA, USA), and CD62L APC eFluor 780 (clone DREG56) (Thermo Fisher Scientific, Waltham, MA, USA) monoclonal antibodies and permeabilized with Foxp3 Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, Waltham, MA, USA). Next, cells were stained with FoxP3 APC (clone), Helios eFluor 450 (clone 22F6) (all from Thermo Fisher Scientific, Waltham, MA, USA), and finally readout with FACS CANTOII equipped with FACS Diva software flow cytometer (BD Biosciences, San Jose, CA, USA). For data analysis, fluorescence minus one (FMO) approach was used to set gates adequately. First, doublets were excluded according to FSC A/H signal distribution, then CD3 high expressing lymphocytes were gated as CD4/CD3 double-positive T cells. Next, CD4+ T cells were gated and Tregs were identified as either CD127low/CD25+ or CD25/FoxP3 cells. Finally, Helios expression was checked in Tregs population and naïve/memory CD4 T lymphocytes were analyzed according to CD62L/CD45RA expression. For every sample, a minimum of 100,000 events were collected.
To predict the most promising candidate genes, we used the ENDEAVOUR—a free web resource for gene prioritization (https://endeavour.esat.kuleuven.be). Shortly, the strategy is based on how similar a candidate gene is to a profile derived from genes already known to be involved in the process of interest. The approach relies on the integration of multiple heterogeneous sources (e.g., coding sequence, gene expression, functional annotation, literature, regulatory information) that cover what we currently know about these genes (14). In the first step, we selected 20 genes which are widely known to be associated with autoimmunity and/or immunodeficiency (STAT1, STAT3, IL2RA, PIK3R1, CTLA4, FASLG, TYK2, CD19, FOXP3+, TNFRSF13B, PIK3CD, CD81, ICOS, CR2, ITCH, AICDA, AIRE, MS4A7, LRBA). These genes severed as a training set. In the second step, we selected the whole long arm of chromosome 18 with 284 identified genes for prioritization with the ENDEAVOUR approach. We performed four separate iterations, choosing sequence data, expression data, functional annotations (protein-protein interaction networks text mining, regulation information), and phenotypic information as the data source. Finally, the rankings (one per data source) were fused into a global ranking, using Order Statistics. We then selected top 10 genes that were ranked at the top in each iteration.
Having the top 10 candidates, we performed statistical analysis to study the association between the deletion of each candidate gene and the presence of disturbances within each CD4 T-cell compartment.
The Statistica 12.5 PL package (Statsoft, Tulsa, OK, USA) was used for the analysis. Continuous variables are presented as medians followed by interquartile ranges (IQR), while nominal variables are presented as numbers followed by percentages in brackets. The Shapiro-Wilk test was used to assess the normality of distribution. Continuous variables were compared using the Mann-Whitney U-test, Wilcoxon, and Kruskal-Wallis ANOVA in the case of a nonnormal distribution or t-test in case of normal distribution. p-values <0.05 were considered statistically significant.
Thirty-six patients were referred to the project by specialists in medical genetics, diabetology, or pediatric endocrinology between May 2015 and December 2019.
18q deletion was confirmed by array CGH analysis in 27 of them, 15 (55.6%) females and 12 males. The mean age at examination was 11.8 years (min–max: 4.0–33.5). Clinical characteristics are given in Table 2, and the particular phenotypic features are listed in Table 3.
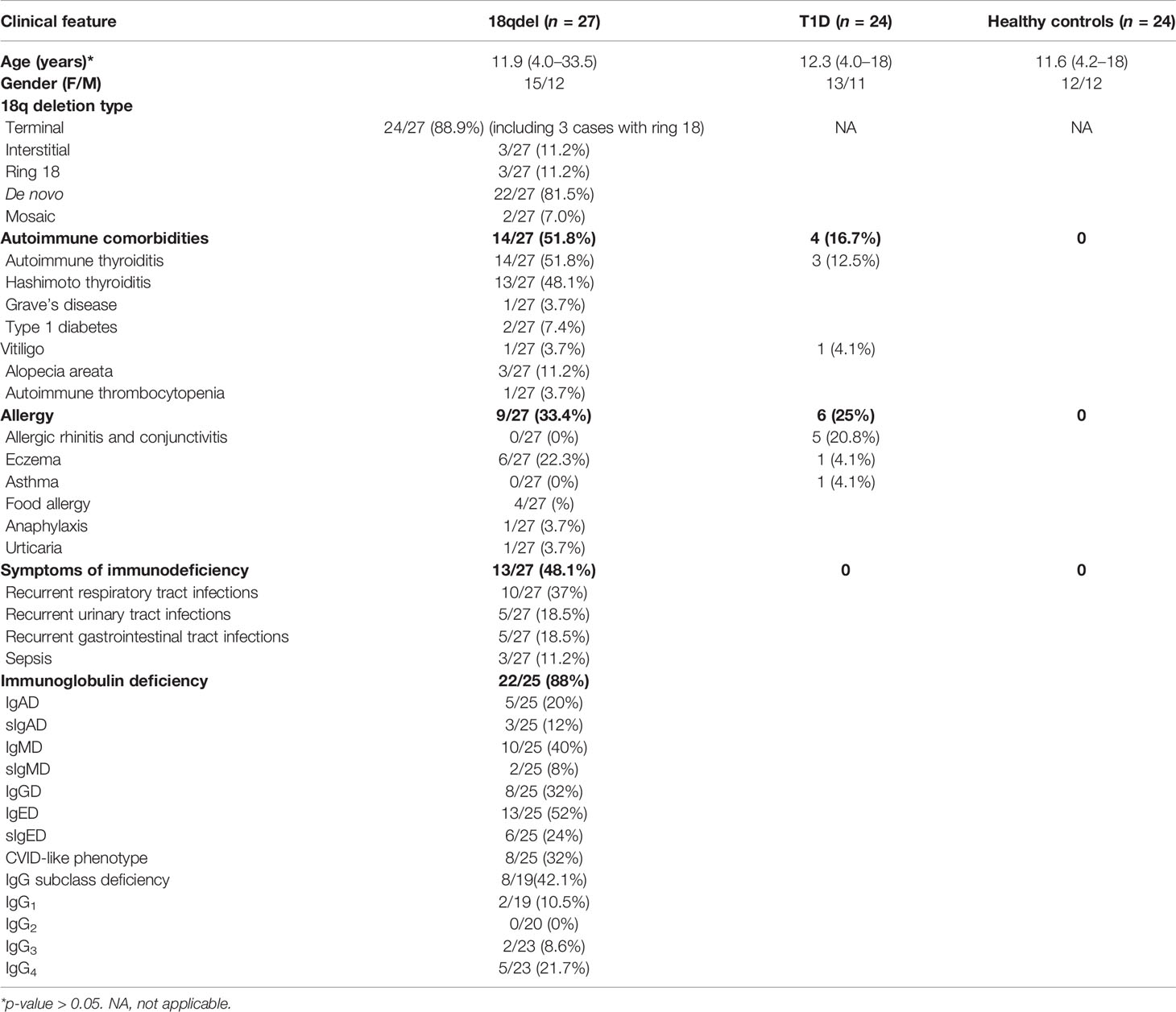
Table 2 Clinical characteristics of patients with 18q del syndrome.

Table 3 Phenotypic features of 27 patients with 18q deletion.
Twenty-four patients had “pure” 18q deletion and three of the patients had a ring chromosome 18 (deletion from both p and q arms). The majority of the patients 23/27 (85.2%) presented with the so-called distal 18q del syndrome due to a deletion distally to 18q21.1 (from 46,700,000 to the end of the chromosome at 78,077,248 bp *hg 19 nucleotide scale that includes 103 genes). One patient had proximal 18q del, one had both proximal and distal interstitial deletion, one proximal terminal deletion encompassing almost the whole arm of 18q (25% mosaic), and one proximal encompassing a little fragment distally to 18q21.1. The length of the q arm deletion varied from very small interstitial deletions encompassing 1,007 Mbp to almost the whole arm of 18q (55.00 Mbp). The deleted q fragments are depicted on the chromosome ideogram (Figure 1) and detailed microarray analysis results of each patient are given in the Supplementary Table S1. Two patients had a mosaic genotype. In 22/27(81.5%) of cases, aberration occurred de novo.
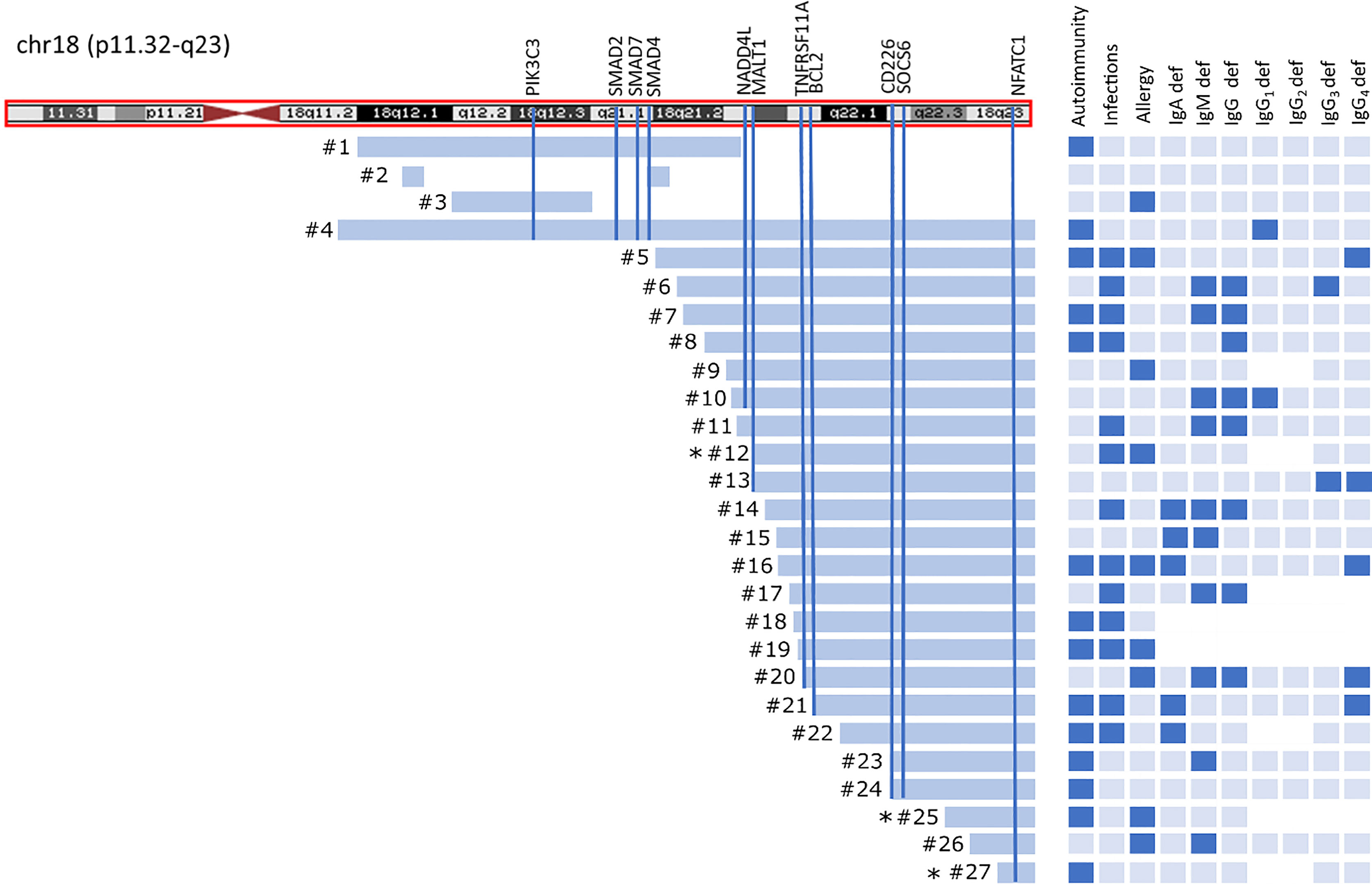
Figure 1 Chromosome 18 ideogram showing the mapping of breakpoints in 18qdel cohort. Light blue bars depict deleted fragments of q arm each of the 27 study subjects in relation to clinical and laboratory findings. Black asterisks indicates ring chromosome 18 with simultaneous deletion at 18p.
Autoimmune disorders were present in 14/27 (51.8%) of the cohort. Autoimmune thyroiditis was found in 14/27 (51.8%), alopecia areata in three of 27(11.2%), type 1 diabetes in two of 27 (7.4%), vitiligo in one of 27 (3.7%), and autoimmune thrombocytopenia in one of 27(3.7%).
Thirteen of 27 (48.1%) of the patients suffered from severe/recurrent infections. In eight of them, the symptoms of immune deficiency coexisted with autoimmunity. Recurrent respiratory tract infections at 10/27 (37%), followed by urinary tract infections and gastrointestinal tract infections (both in 5/27 (18.5%) were the most commonly reported. Sepsis was reported in three cases (11.2%). Only three patients had been initially diagnosed with immunodeficiency and had been referred to Clinical Immunology Outpatient Clinic. Allergy was reported in nine of 27 (33.4%) patients, including food allergy at four of 27 (14.8%), eczema at six of 27 (22.3%), urticaria at one of 27 (3.7%), and anaphylaxis at one of 27(3.7%). Autoimmunity, infections, and allergy coexisted in three of 27 cases, autoimmunity and allergy at one of 27, and autoimmunity and infections at five of 27 patients (Figure 1).
Immunoglobulins A, G, M, E, and IgG subclasses were measured in nearly all patients, with a few exceptions (Figure 1).
Seventeen of 19 (89.5%) patients presented with deficiency of at least one type of all measured immunoglobulins IgA, M, G, E, and IgG1-4.
According to major immunoglobulin class division, IgE deficiency was the most common [13/25 (52%)], followed by IgM at 10/25 (40%), IgG at eight of 25 (32%), and IgA at five of 25 (20%). Eight of the 25 patients (32%) had abnormalities in at least two major immunoglobulin (IgG, IgA, IgM) measurements, suggesting CVID-like phenotype.
Six of the patients had selective IgED (24%), three had selective IgAD (12%), and two had selective IgMD (8%). With regard to IgG subclass deficiencies, the most prevalent was IgG4 deficiency at six of 24 (25%), followed by IgG1 at two of 20 (10%) and IgG3 at two of 24 (8.4%).
Patients with 18q del exhibited significantly decreased CD4, Treg FOXP3+, TregFOXP3+Helios+, TemCD4 cell numbers in comparison with the control groups. The control groups consisted of 24 T1DM patients and 28 healthy subjects.
Hypogammaglobulinemia was confirmed in 10/13 patients with the history of severe/recurrent infections. IgED was found in nine of 14 patients with autoimmunity; in five cases, it was selective. All patients with sIgAD had autoimmune phenomena.
Lymphocyte subset phenotyping was feasible in 24 subjects from the 18q del cohort. The controls comprised 24 patients with T1D (autoimmune) diabetes and 28 healthy subjects (Table 4).
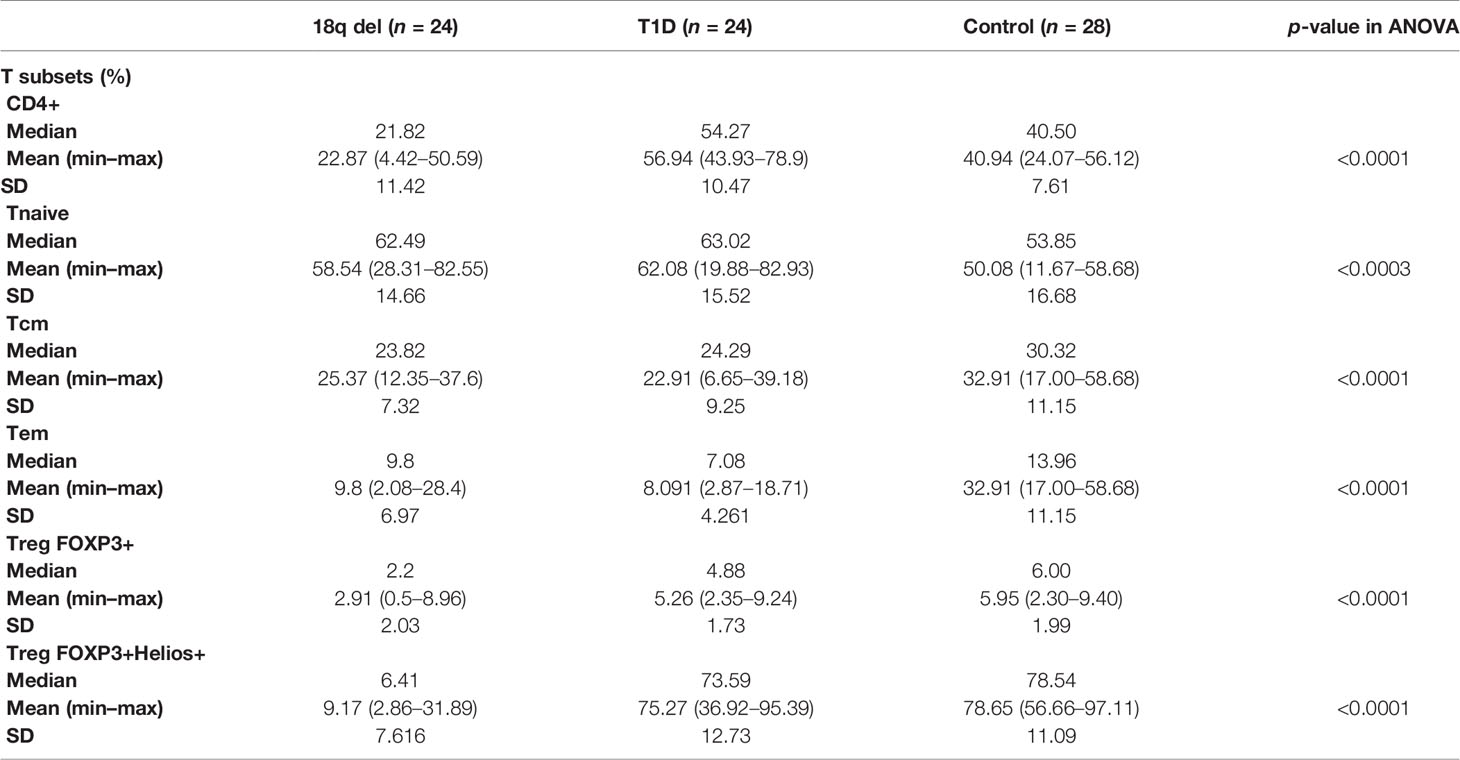
Table 4 The proportion (%) of CD4+ lymphocytes in patients with 18q del, patients with T1D (control group), and 28 healthy controls.
Total white blood cell (WBC) counts and lymphocyte counts as well as the CD4/CD8 ratio did not differ among the 18q del, T1DM subjects, and the healthy controls. Patients with 18q del exhibited significantly decreased percentage of CD4+CD25+ FOXP3+Treg and CD4+CD25+FOXP3+Helios+Treg (Helios+ cells originate from the thymus) cell subpopulations in comparison not only with healthy controls but also with T1DM groups (p < 0.00011 and p < 0.00027, respectively) (Figure 2A, B). This was also true for the percentage of total CD4+ lymphocytes (in both cases p < 0.00011). Scatter plots depicting the deficiency of CD4+CD25+FOXP3+ and CD4+CD25+FOXP3+ Helios+ lymphocyte subpopulations in a patient with 18q deletion are shown in Figure 3. There was no difference in the percentage of CD4-naïve lymphocytes, and CD4 Tem cells between 18q del and the heathy controls (p < 0.397 and p = 0.776, respectively). The percentage of CD4 Tcm lymphocytes was lower in both the 18q del and the T1DM group than in the healthy controls, p = 0.0289, and p = 0.0019, respectively, but there was no difference in the percentage of CD4 Tcm lymphocytes between the 18q del and the T1DM group, p = 0.548 (Figure 2E).
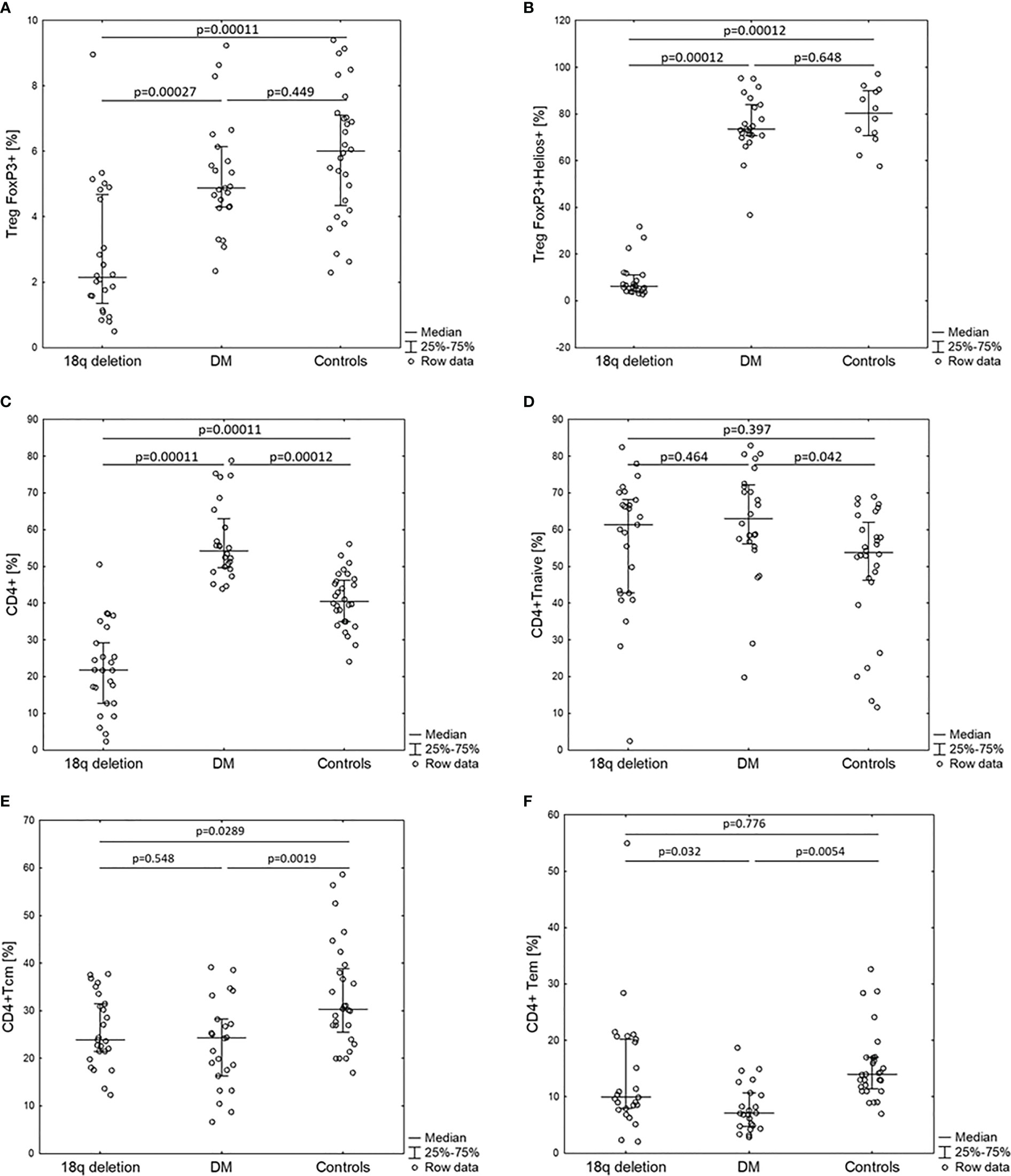
Figure 2 Percentage of CD4+ lymphocyte subpopulations of patients with 18q deletion syndrome, type 1 diabetes (DM), and healthy controls. (A) CD4+CD25+FOXP3+, (B) CD4+CD25+FOXP3+Helios+, (C) CD4+, (D) CD4+naïve, (E) CD4+T central memory, (F) CD4+T effective memory.
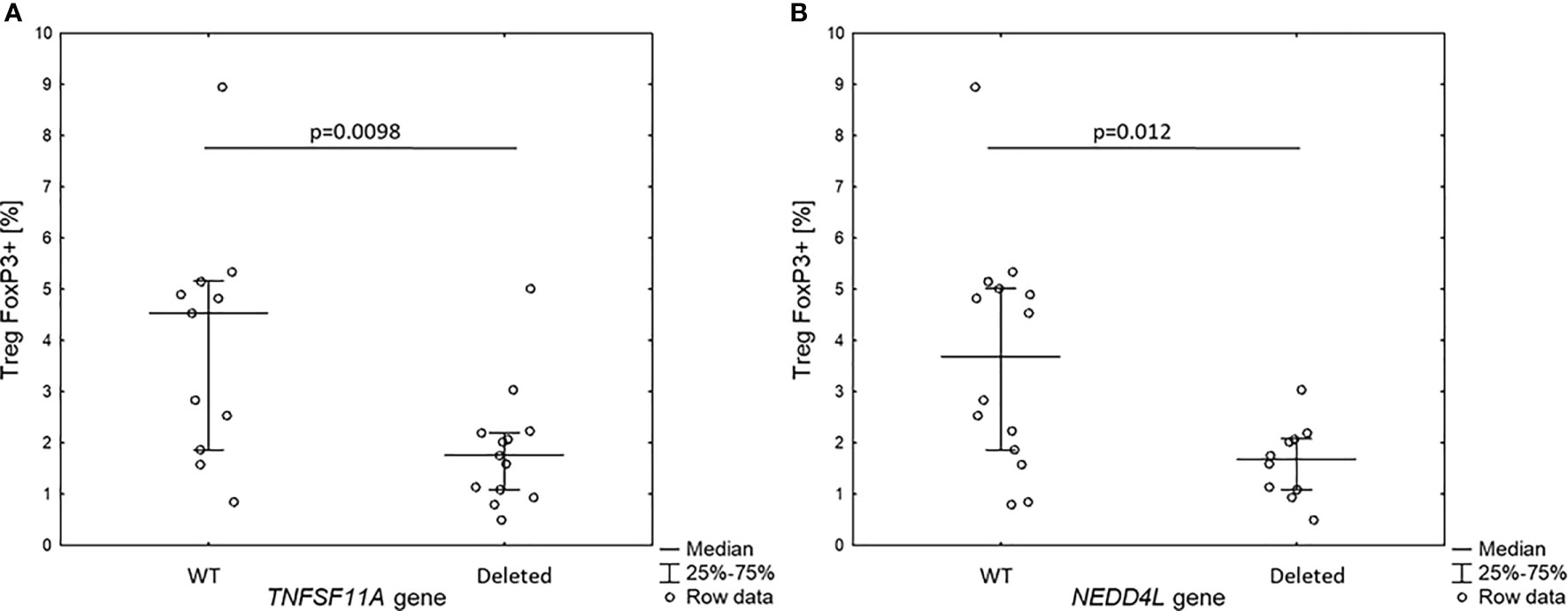
Figure 3 Percentage of Treg FOXP3+ cells in 18q del patients without and with haploinsufficiency of TNFSF11A (A) and NEDDL4 genes (B).
The T1DM group had significantly more CD4+ and CD4-naïve lymphocytes than the healthy controls, p < 0.00012 and p < 0.042, respectively (Figure 2C, D). On the other hand, the percentage of CD4+ Tcm lymphocytes was lower in the T1DM group than in the healthy controls, p = 0.0019. The percentage of CD4 Tem lymphocytes was statistically lower than in the controls but also lower than in patients with 18q del (Figure 2F)
After four consecutive iterations, 10 top candidate genes were selected, all of them having similar bioinformatic behavior to the learning set of genes associated with primary immunodeficiency and autoimmunity features (Supplementary Table S2).
This included the following genes: SOCS6, BCL2, MALT1, SMAD4, PIK3C3, NEDD4L, TNFRSF11A, NFATC1, SMAD2, and SMAD7. Short functional description of each of these genes is included in Table 5.

Table 5 Known functions of top 10 candidate immunogenes located on 18q.
Since the patients with 18q deletion syndrome had Treg FOXP3+ deficiency, we analyzed the level of Treg FOXP3+ in the context of gene dose for all 10 genes identified in prioritization approach. Interestingly, we found that patients with haploinsufficiency of TNFRSF11A and NEDD4L gene had low level of Treg FOXP3+ (1.9 ± 1.0% vs. 3.9 ± 12.3%, p = 0.0098 and 1.6 ± 0.7% vs. 3.8 ± 2.2%, p = 0.012, respectively) (Figure 4).
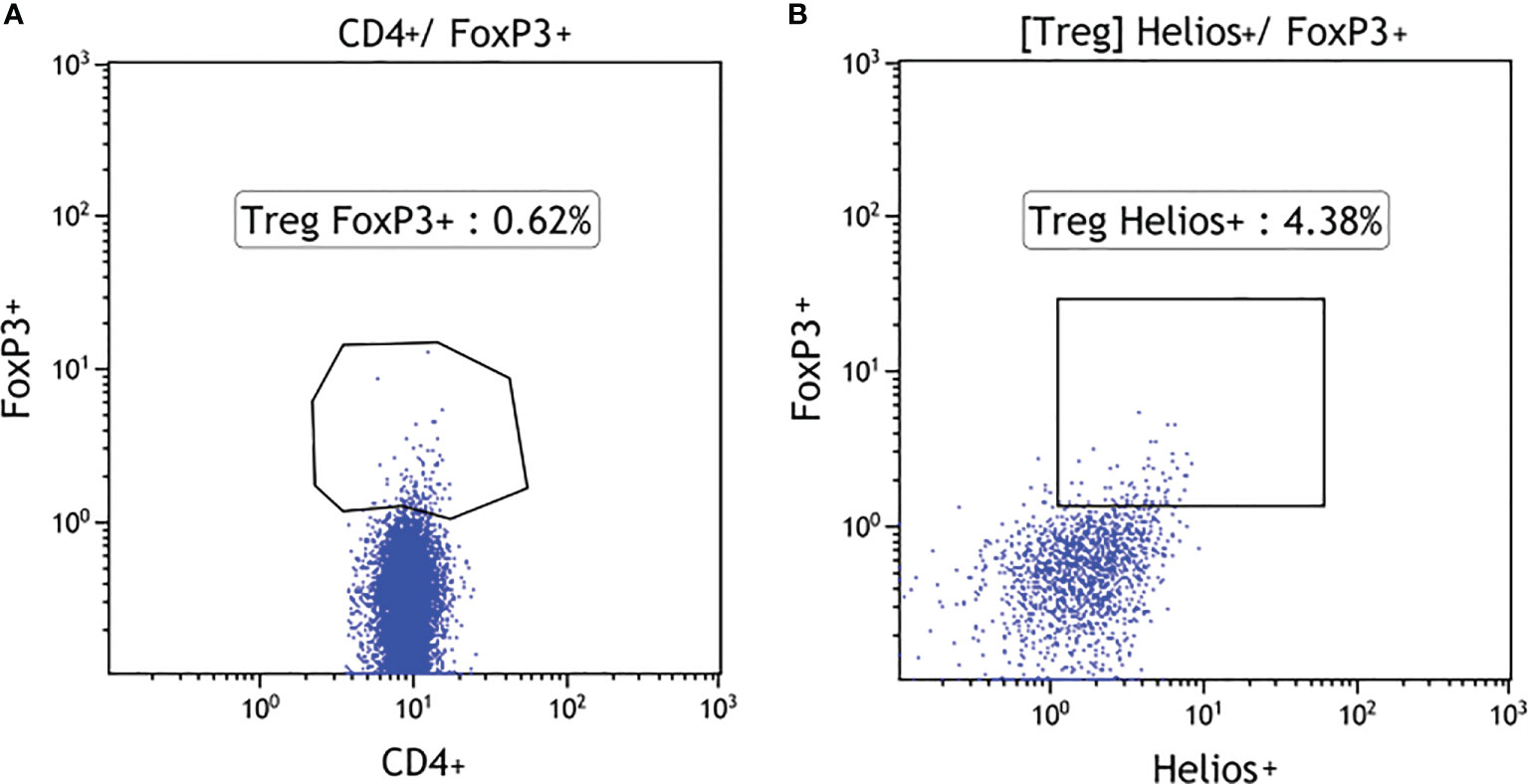
Figure 4 Percentage of Treg FOXP3+ cells in 18q del patients without and with haploin sufficiency of TNFSF11A (A) and NEDDL4 genes (B).
The results of the present study confirm our initial assumptions that the 18q deletion is responsible for the increased susceptibility to autoimmune disorders, infections, and allergy in the affected subjects. In this paper, we have also included some novel observations on the pathogenesis of immune dysregulation resulting in this phenotype of 18q deletion syndrome and ring 18.
Firstly, we have demonstrated that, despite relatively young age, more than half of our 18q del cohort suffered from autoimmune disorders. The most common of them were AITDs, followed by alopecia areata, T1DM, and vitiligo. All of these disorders, as well as juvenile rheumatoid arthritis, celiac disease, pernicious anemia, and autoimmune hepatitis, had been previously reported in association with 18q deletions (7, 9, 32–34). In some cases, they coexisted in autoimmune polyendocrine syndromes (6, 35), strongly suggesting impaired central immune tolerance development. Furthermore, we were able to confirm our initial observations (6), that in individuals with 18q del, autoimmunity often coexists with symptoms of immune deficiency and allergy. Nearly half of the cohort had history of severe/recurrent infections and over one-third of the patients reported allergy symptoms.
To the best of our knowledge, only two small studies on 14 and 16 patients aimed to assess the frequency of immune-mediated disorders in 18q del syndrome. Although their results correspond with ours, they should be considered with caution because autoimmune disorders have not been precisely defined and documented (36, 37). Linnankivi reported that three of 14(21.4%) had “autoimmune” disorders (hypothyroidism, iritis), eight of 14 (57%) suffered from frequent respiratory tract infections, and the same percentage had atopic disorders (eczema, food allergy, and asthma) (36).
Cody et al. selected 16 individuals (out of 290) with 18q hemizygosity who had terminal deletion between SERPINB8 and CDH7 that, despite unique breakpoints, encompassed the same set of genes and could be a reference group for distal 18q del. The disorders regarded by the authors as “autoimmune” (myalgia, arthritis, and hypothyroidism) occurred in 31% of subjects and only in females. Atopic diseases were present in 12 out of 16 (75%) of the participants (food allergy in 56%, allergic rhinitis in 50%, eczema in 44%, asthma in 21%) (37).
Secondly, we have shown that individuals with 18q deletion commonly presented with a variable immunoglobulin deficiency, rather than with IgAD alone, which has not been reported so far. Low or absent IgA levels have been regarded as phenotypic feature of 18q del syndrome since 1968 and have been recently estimated to be present in 24%–44% individuals (37–42). In our study, IgAD was present in 20% of our patients (in three of them as sIgAD), but the complex immunoglobulin deficiency mimicking CVID phenotype was far more common. Other types of antibody deficiency, such as selective IgM, IgG1-4 subclass deficiency, and IgE deficiency were also frequent (Figure 1).
Such alterations in immunoglobulin levels, as well as the progression to CVID (38) or CVID-like disease (immunoglobulin deficiency but good vaccine responses) (35), had been previously mentioned in a few case reports (39, 40). On the other hand, Smith et al. reported two individuals with interstitial deletions encompassing the region 18q21.2-21.3 with anaphylaxis, multiple food allergies, asthma, eczema, immunodeficiency, and extremely high levels of total IgE (43). Another case of persistently elevated IgE concerned a patient with atopic dermatitis and deletion encompassing 18q22.1-18.23 (41). In our study, however, over a half of the 18q del cohort had undetectable IgE levels. Six of them were found to have “selective IgED”—ultra-low IgE levels (<2 kIU/L), with normal levels of other immunoglobulins (13, 42), which was suggested to be a marker of immune dysregulation and autoimmunity (33). Moreover, all 18q del patients with selective IgED had coexisting autoimmune disorder. Unlike hyper-IgE syndrome, which is a well-known form of primary immune deficiency (44), low IgE level is currently not regarded as a clinically important immune deficit. However, it has been recently suggested to be a sensitive indicator for CVID. The variations in immunoglobulin levels point to profound defects in B-cell lineage and immunoglobulin class switching.
Thirdly, we have found that patients with 18q deletions, irrespective of their autoimmune or allergic status, had CD4+CD25+FOXP3+ Treg cell deficiency—the key players controlling reactivity to self-antigens and preventing autoimmunity. Thereby, this group share some striking similarities with the classic examples of the so-called Tregopathies like immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like monogenic syndromes caused by mutations in FOXP3 (43), and other genes (CTLA-4, CD25, LRBA, STAT1, STAT3, STAT5b, BACH2) mutations, respectively (45).
The profoundly low number of CD4+CD25+FOXP3+Helios+ Tregs (that originate from the thymus) (46), suggests intrinsic dysfunction of thymic maturation of nTregs in individuals with 18q deletions. The patients also exhibited CD4+ and TemCD4+ deficiency which, together with hypogammaglobulinemia, might explain their susceptibility to infections. We therefore suggest that 18q del syndrome should be regarded as another chromosomal syndrome, with compromised immune system, predisposing affected individuals to the development of autoimmune disorders and susceptibility to infections.
We have hypothesized that the observed immune dysregulation results from the deletion of certain genes located on 18q. To address this issue, we performed gene prioritization which indicated “top 10” genes that might be involved in immune system function and central tolerance development. However, only two of them had enough statistical significance for association with FOXP3+Treg deficiency. The first is a neural precursor cell-expressed developmentally downregulated four-like (NEDD4L), located on 18q21.31. It is an E3 ubiquitin protein ligase that regulates channel internalization and turnover. It is possibly involved in the termination of the transforming growth factor β (TGFβ)-induced signal transduction by ubiquitination of linker phosphorylated active Smad2/3 (24). Its role in Treg development and function is to be established.
The other gene is called tumor necrosis factor receptor superfamily member 11A (TNFRSF11A, RANK), located on 18q21.33. Interestingly, recent studies suggest that TNFRSF11A is essential for the correct differentiation of medullary thymic epithelial cells (mTECs) and serves as a surface marker of maturity of these cells in embryonic and adult thymus (25). mTECs play an important role in immune tolerance development by facilitating the clonal deletion of autoreactive T cells, while inducing the generation of thymic T-regulatory cells (47). After activation of transcription factor AIRE, they express the whole repertoire of self-antigens and present them to naïve T cells. Mutations in AIRE result in promiscuous gene expression in mTECs, leading to autoimmune poliendocrinopathy, candidiasis, ectodermal dystrophy (APECED)—another monogenic syndrome caused by central tolerance breakdown (48). It has been recently shown that TNFRSF11A gene product RANK is necessary for activation of AIRE (26). Therefore, it is possible that haploinsufficiency of TNFRSF11A could be also involved in both CD4+CD25+FOXP3+ cell deficiency.
It is worth mentioning that TNFRSF11A (RANK) might also be involved in immunoglobulin synthesis. Mutations of this gene were found in individuals with osteoclast-poor osteopetrosis associated with hypogammaglobulinemia (OMIM 612301), manifesting as bone fractures, hematologic abnormalities, recurrent infections, reduced immunoglobulin levels, and lack of antibody response to tetanus antigen (27). In addition, mutations in similar genes, like TNFRSF13 (TACI), TNFRSF13C (BAFF-R), TNFRSF12 (TWEAK), and TNFRSF7 (CD27) were found in individuals with selective IgA deficiency or CVID (49, 50). It is, therefore, possible that haploinsufficiency of this gene could explain immune dysregulation with immunoglobulin deficiency in 18q del syndrome.
Although not supported by statistical analysis, the loss of several other immune genes located in 18q might also be responsible for the observed dysregulation in 18q del syndrome (Table 5). The spectrum of speculations regarding which gene might be responsible for such phenotype ranges from single gene haploinsufficiency (like in IPEX syndrome) (51), to deletion of a cluster of genes, each gene having small effect, that disrupts gene-gene interactions. The latter scenario might have a greater effect than each of the genes taken separately.
Some practical implications can be seen in our study, such as careful clinical evaluation of patients and supervision since early childhood in terms of higher risk for the immune and endocrine manifestations. In some patients, immunoglobulin substitution may be needed to reduce the frequency of infections (35).
Despite several advantages, such as the highest to date number of involved patients, the novelty of results, as well as the clinical value, our study has some limitations. First of all, the prevalence of the autoimmune disorders, which typically more often appear in adults, might be influenced by the relatively young age of the subjects. Secondly, we have not been able at this point to perform B lymphocyte assessment, as it is done when CVID is suspected, including response to vaccines, which may not necessarily be impaired (35, 52). Also, it might be difficult to find gene(s) involved in immune dysregulation with the approach we used, because the appearance and/or severity of some phenotypic features of the syndrome (e.g., congenital heart defects or narrow ear canals) varies significantly even among family members carrying similar 18q deletion (5, 52). It is thus possible that haploinsufficiency of certain gene(s) predisposing to autoimmunity causes an abnormal phenotype only in the presence of an additional genetic or environmental factors. Since we have not been able to distinguish, which of the top-scoring genes is really associated with the phenotypic features of immunodeficiency in our cohort of patients with 18q deletion, the identification of more patients with different breakpoints within 18q would be helpful to find the responsible gene(s). Moreover, these top 10 genes could be good candidates for future genetic search for mutation among patients with primary immunodeficiency who are negative for mutation in the known PID-responsible genes.
For the purpose of the study, we focused only on some aspects of immunity and further research is needed to precisely describe all possible defects in the innate and adaptive immune system in 18q del syndrome. Taking into account our results and some case reports (35, 53), B-cell immunophenotyping and functional studies on B-cell compartment as well as immunoglobulin class-switching are necessary. Moreover, genomic, transcriptomic, and proteomic data are needed to illicit how genes located on 18q interact with each other and what pathways they create. Also, studies on the influence of knock out of certain 18q genes on the expression of FOXP3+, Helios, AIRE, etc. can provide critical information of Treg cell biology, the role of Treg cell-associated molecules, and the regulation of central and peripheral tolerance in humans.
Patients with 18q deletions frequently suffer from autoimmune disorders, recurrent infections, and allergy due to immune dysregulation presenting with variable antibody deficiencies and T-regulatory cell deficiency (CD4+CD25+CD127lowFOXP3+). The spectrum of speculations regarding which gene might be responsible for such phenotype ranges from single gene haploin sufficiency to deletion of a cluster of immunogenes located distally to 18q21.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: the Decipher database (DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources; (https://www.deciphergenomics.org/). The accession numbers of the deposited variants in Decipher are 438816–438849.
The studies involving human participants were reviewed and approved by Ethics Committee of Medical University of Lodz, Poland. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
AH and WM contributed to the study design, protocol writing, data collection, analysis, interpretation, and writing and reviewing of the manuscript. MZ, PT, and KW contributed to cell preparation, cell separation, data collection, data analysis and reviewed the report. All other authors contributed to data collection, interpretation and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a Diabetes Poland grant (AH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank 18q del patients and their families and control subjects for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.742834/full#supplementary-material
Supplementary Table 1 | Microarray (aCGH) analysis showing cytogenic location and genomic coordinates (GRCh38) of 18 chromosome gains (x3) and losses (x1) of genetic material of the 27 studied subjects.
Supplementary Table 2 | The rank of the top 10 candidate genes located on 18q by the ENDEAVOUR prioritization iterations for primary immunodeficiency and autoimmunity.
1. De Grouchy J, Royer P, Salmon C, Lamy M. Partial Deletion of the Long Arm of the Chromosome 18. Pathol Biol (1964) 12:579–82.
2. Andler W, Heüveldop A, Polichronidou T. Endocrinologic Disorders in Deletion of Chromosome 18. Monatsschr Kinderheilkd (1992) 140(5):303–6.
3. Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, et al. Congenital Anomalies and Anthropometry of 42 Individuals With Deletions of Chromosome 18q. Am J Med Genet (1999) 85(5):455–62. doi: 10.1002/(sici)1096-8628(19990827)85:5<455::aid-ajmg5>3.0.co;2-z
4. Cody JD, Heard PL, Crandall AC, Carter EM, Li J, Hardies LJ, et al. Narrowing Critical Regions and Determining Penetrance for Selected 18q- Phenotypes. Am J Med Genet A (2009) 149a(7):1421–30. doi: 10.1002/ajmg.a.32899
5. Cody JD, Sebold C, Heard P, Carter E, Soileau B, Hasi-Zogaj M, et al. Consequences of Chromsome18q Deletions. Am J Med Genet C Semin Med Genet (2015) 169(3):265–80. doi: 10.1002/ajmg.c.31446
6. Hogendorf A, Lipska-Zietkiewicz BS, Szadkowska A, Borowiec M, Koczkowska M, Trzonkowski P, et al. Chromosome 18q Deletion Syndrome With Autoimmune Diabetes Mellitus: Putative Genomic Loci for Autoimmunity and Immunodeficiency. Pediatr Diabetes (2016) 17(2):153–9. doi: 10.1111/pedi.12235
7. Lomenick JP, Smith WJ, Rose SR. Autoimmune Thyroiditis in 18q Deletion Syndrome. J Pediatr (2005) 147(4):541–3. doi: 10.1016/j.jpeds.2005.04.064
8. Stricker RB, Linker CA. Pernicious Anemia, 18q Deletion Syndrome, and IgA Deficiency. JAMA (1982) 248(11):1359–60. doi: 10.1001/jama.1982.03330110055029
9. Tutunculer F, Darendeliler F, Gunoz H, Karaman B, Kayserili H. 18q Deletion Syndrome Associated With Autoimmune Thyroid Disease Presenting as Hyperthyroidism. J Pediatr Endocrinol Metab (2005) 18(4):419–20. doi: 10.1515/jpem.2005.18.4.419
10. Piatosa B, Wolska-Kusnierz B, Bernatowska E, Pac M, Gregorek H, Zapasnik A, et al. Wartości Referencyjne Stężeń Immunoglobulin G, A, M I D W Surowicy Zdrowych Dzieci I Osób Dorosłych, Mieszkańców Województwa Mazowieckiego. Standardy Medyczne (2010) 7(3):524–32.
11. Simons FE, Ardusso L,R, Bilò M,B, El-Gamal Y,M, Ledford D,K, Ring J, et al. World Allergy Organization Guidelines for the Assessment and Management of Anaphylaxis. World Allergy Organ J (2011) 4(2):13–37. doi: 10.1097/WOX.0b013e318211496c
12. Seidel M,G, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract (2019) 7(6):1763–70. doi: 10.1016/j.jaip.2019.02.004
13. Magen E, Schlesinger M, David M, Ben-Zion I, Vardy D. Selective IgE Deficiency, Immune Dysregulation, and Autoimmunity. Allergy Asthma Proc (2014) 35(2):e27–33. doi: 10.2500/aap.2014.35.3734
14. Tranchevent LC, Ardeshirdavani A, ElShal S, Alcaide D, Aerts J, Auboeuf D, et al. Candidate Gene Prioritization With Endeavour. Nucleic Acids Res (2016) 44(W1):W117–21. doi: 10.1093/nar/gkw365
15. Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, Inflammation, and Autoimmunity. Front Immunol (2012) 3:58. doi: 10.3389/fimmu.2014.00058
16. Del Pino Molina L, Torres Canizales JM, Pernía O, Rodríguez Pena R, Ibanez de Caceres I, López Granados E. Defective Bcl-2 Expression in Memory B Cells From Common Variable Immunodeficiency Patients. Clin Exp Immunol (2021) 203(3):341–50. doi: 10.1111/cei.13522
17. Kamada S, Shimono A, Shinto Y, Tsujimura T, Takahashi T, Noda T, et al. Bcl-2 Deficiency in Mice Leads to Pleiotropic Abnormalities: Accelerated Lymphoid Cell Death in Thymus and Spleen, Polycystic Kidney, Hair Hypopigmentation, and Distorted Small Intestine. Cancer Res (1995) 55(2):354–9.
18. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-Deficient Mice Demonstrate Fulminant Lymphoid Apoptosis, Polycystic Kidneys, and Hypopigmented Hair. Cell (1993) 75(2):229–40. doi: 10.1016/0092-8674(93)80065-m
19. Brüstle A, Brenner D, Knobbe-Thomsen CB, Cox M, Lang PA, Lang KS, et al. MALT1 is an Intrinsic Regulator of Regulatory T Cells. Cell Death Differ (2017) 24(7):1214–23. doi: 10.1038/cdd.2015.104
20. Ruland J, Duncan GS, Wakeham A, Mak TW. Differential Requirement for Malt1 in T and B Cell Antigen Receptor Signaling. Immunity (2003) 19(5):749–58. doi: 10.1016/s1074-7613(03)00293-0
21. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A Homozygous Mucosa-Associated Lymphoid Tissue 1 (MALT1) Mutation in a Family With Combined Immunodeficiency. J Allergy Clin Immunol (2013) 132(1):151–8. doi: 10.1016/j.jaci.2013.04.047
22. Malhotra N, Kang J. SMAD Regulatory Networks Construct a Balanced Immune System. Immunology (2013) 139(1):1–10. doi: 10.1111/imm.12076
23. Yang G, Song W, Postoak JL, Chen J, Martinez J, Zhang J, et al. Autophagy-Related Protein PIK3C3/VPS34 Controls T Cell Metabolism and Function. Autophagy (2021) 17(5):1193–204. doi: 10.1080/15548627.2020.1752979
24. Gao S, Alarcón C, Sapkota G, Rahman S, Chen PY, Goerner N, et al. Ubiquitin Ligase Nedd4L Targets Activated Smad2/3 to Limit TGF-Beta Signaling. Mol Cell (2009) 36(3):457–68. doi: 10.1016/j.molcel.2009.09.043
25. Sobacchi C, Menale C, Villa A. The RANKL-RANK Axis: A Bone to Thymus Round Trip. Front Immunol (2019) 10:629. doi: 10.3389/fimmu.2019.00629
26. Rossi SW, Kim MY, Leibbrandt A, Parnell SM, Jenkinson WE, Glanville SH, et al. RANK Signals From CD4(+)3(-) Inducer Cells Regulate Development of Aire-Expressing Epithelial Cells in the Thymic Medulla. J Exp Med (2007) 204(6):1267–72. doi: 10.1084/jem.20062497
27. Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, et al. Human Osteoclast-Poor Osteopetrosis With Hypogammaglobulinemia Due to TNFRSF11A (RANK) Mutations. Am J Hum Genet (2008) 83(1):64–76. doi: 10.1016/j.ajhg.2008.06.015
28. Vaeth M, Schliesser U, Muller G, Reissig S, Satoh K, Tuettenberg A, et al. Dependence on Nuclear Factor of Activated T-Cells (NFAT) Levels Discriminates Conventional T Cells From Foxp3+ Regulatory T Cells. Proc Natl Acad Sci USA (2012) 109(40):16258–63. doi: 10.1073/pnas.1203870109
29. Giri PS, Dwivedi M, Begum R. Decreased Suppression of CD8(+) and CD4(+) T Cells by Peripheral Regulatory T Cells in Generalized Vitiligo Due to Reduced NFATC1 and FOXP3 Proteins. Exp Dermatol (2020) 29(8):759–75. doi: 10.1111/exd.14157
30. Shioda T, Lechleider RJ, Dunwoodie SL, Li H, Yahata T, de Caestecker MP, et al. Transcriptional Activating Activity of Smad4: Roles of SMAD Hetero-Oligomerization and Enhancement by an Associating Transactivator. Proc Natl Acad Sci USA (1998) 95(17):9785–90. doi: 10.1073/pnas.95.17.9785
31. Klein J, Ju W, Heyer J, Wittek B, Haneke T, Knaus P, et al. B Cell-Specific Deficiency for Smad2 In Vivo Leads to Defects in TGF-Beta-Directed IgA Switching and Changes in B Cell Fate. J Immunol (2006) 176(4):2389–96. doi: 10.4049/jimmunol.176.4.2389
32. Dostal A, Linnankivi T, Somer M, Kahkonen M, Litzman J, Tienari P. Mapping Susceptibility Gene Locus for IgA Deficiency at Del(18)(Q22.3-Q23); Report of Familial Cryptic Chromosome T(18q; 10p) Translocations. Int J Immunogenet (2007) 34(3):143–7. doi: 10.1111/j.1744-313X.2007.00652.x
33. Rosen P, Hopkin RJ, Glass DN, Graham TB. Another Patient With Chromosome 18 Deletion Syndrome and Juvenile Rheumatoid Arthritis. J Rheumatol (2004) 31(5):998–1000.
34. Sabouni MA, Benedict D, Alom MS, Petty S, Patel K. Atrial Septal Defect can be Easily Missed in Chromosome 18q Deletion Syndrome. Oxf Med Case Rep (2018) 10):omy076. doi: 10.1093/omcr/omy076
35. Calvo Campoverde K, Gean E, Piquer Gibert M, Martinez Valdez L, Deyà-Martínez A, Rojas Volquez M, et al. Humoral Deficiency in Three Paediatric Patients With Genetic Diseases. Allergol Immunopathol (Madr) (2016) 44(3):257–62. doi: 10.1016/j.aller.2015.07.007
36. Linnankivi T, Tienari P, Somer M, Kähkönen M, Lönnqvist T, Valanne L, et al. 18q Deletions: Clinical, Molecular, and Brain MRI Findings of 14 Individuals. Am J Med Genet A (2006) 140(4):331–9. doi: 10.1002/ajmg.a.31072
37. Cody JD, Hasi M, Soileau B, Heard P, Carter E, Sebold C, et al. Establishing a Reference Group for Distal 18q-: Clinical Description and Molecular Basis. Hum Genet (2014) 133(2):199–209. doi: 10.1007/s00439-013-1364-6
38. Slyper AH, Pietryga D. Conversion of Selective IgA Deficiency to Common Variable Immunodeficiency in an Adolescent Female With 18q Deletion Syndrome. Eur J Pediatr (1997) 156(2):155–6.
39. Lawrence MG, Palacios-Kibler TV, Workman LJ, Schuyler AJ, Steinke JW, Payne SC, et al. Low Serum IgE is a Sensitive and Specific Marker for Common Variable Immunodeficiency (CVID). J Clin Immunol (2018) 38(3):225–33. doi: 10.1007/s10875-018-0476-0
40. Ferastraoaru D, Bax HJ, Bergmann C, Capron M, Castells M, Dombrowicz D, et al. AllergoOncology: Ultra-Low IgE, a Potential Novel Biomarker in Cancer-a Position Paper of the European Academy of Allergy and Clinical Immunology (EAACI). Clin Transl Allergy (2020) 10:32. doi: 10.1186/s13601-020-00335-w
41. Yasharpour M, Agarwal S, Jerome D, Yel L. Is it Hyper IgE Syndrome or Something Else? MOJ Immunol (2014) 1(2):10. doi: 10.15406/moji.2014.01.00010
42. Pate MB, Smith JK, Chi DS, Krishnaswamy G. Regulation and Dysregulation of Immunoglobulin E: A Molecular and Clinical Perspective. Clin Mol Allergy (2010) 8:3. doi: 10.1186/1476-7961-8-3
43. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) is Caused by Mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
44. Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE Syndrome With Recurrent Infections–an Autosomal Dominant Multisystem Disorder. N Engl J Med (1999) 340(9):692–702. doi: 10.1056/NEJM199903043400904
45. Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG. Tregopathies: Monogenic Diseases Resulting in Regulatory T-Cell Deficiency. J Allergy Clin Immunol (2018) 142(6):1679–95. doi: 10.1016/j.jaci.2018.10.026
46. Baine I, Basu S, Ames R, Sellers RS, Macian F. Helios Induces Epigenetic Silencing of IL2 Gene Expression in Regulatory T Cells. J Immunol (2013) 190(3):1008–16. doi: 10.4049/jimmunol.1200792
47. Anderson G, Takahama Y. Thymic Epithelial Cells: Working Class Heroes for T Cell Development and Repertoire Selection. Trends Immunol (2012) 33(6):256–63. doi: 10.1016/j.it.2012.03.005
48. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional Cloning of the APECED Gene. Nat Genet (1997) 17(4):393–8. doi: 10.1038/ng1297-393
49. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is Mutant in Common Variable Immunodeficiency and IgA Deficiency. Nat Genet (2005) 37(8):829–34. doi: 10.1038/ng1601
50. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, González-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
51. Shamriz O, Patel K, Marsh RA, Bleesing J, Joshi AY, Lucas L, et al. Hypogammaglobulinemia With Decreased Class-Switched B-Cells and Dysregulated T-Follicular-Helper Cells in IPEX Syndrome. Clin Immunol (2018) 197:219–23. doi: 10.1016/j.clim.2018.10.005
52. Heard PL, Carter EM, Crandall AC, Sebold C, Hale DE, Cody JD. High Resolution Genomic Analysis of 18q- Using Oligo-Microarray Comparative Genomic Hybridization (aCGH). Am J Med Genet A (2009) 149a(7):1431–7. doi: 10.1002/ajmg.a.32900
Keywords: 18q deletion syndrome, immune deficiency, type 1 diabetes, autoimmune diseases, thyroiditis, T regulatory cells, antibody deficiency
Citation: Hogendorf A, Zieliński M, Constantinou M, Śmigiel R, Wierzba J, Wyka K, Wędrychowicz A, Jakubiuk-Tomaszuk A, Budzynska E, Piotrowicz M, Lipska-Ziętkiewicz BS, Kaczorowska E, Cieślikowska A, Kutkowska-Kaźmierczak A, Fijak-Moskal J, Kugaudo M, Kosińska-Urbańska M, Szadkowska A, Borowiec M, Niedźwiecki M, Trzonkowski P and Młynarski W (2021) Immune Dysregulation in Patients With Chromosome 18q Deletions—Searching for Putative Loci for Autoimmunity and Immunodeficiency. Front. Immunol. 12:742834. doi: 10.3389/fimmu.2021.742834
Received: 16 July 2021; Accepted: 22 October 2021;
Published: 17 November 2021.
Edited by:
Sinisa Savic, University of Leeds, United KingdomReviewed by:
Catharina Schuetz, University Hospital Carl Gustav Carus, GermanyCopyright © 2021 Hogendorf, Zieliński, Constantinou, Śmigiel, Wierzba, Wyka, Wędrychowicz, Jakubiuk-Tomaszuk, Budzynska, Piotrowicz, Lipska-Ziętkiewicz, Kaczorowska, Cieślikowska, Kutkowska-Kaźmierczak, Fijak-Moskal, Kugaudo, Kosińska-Urbańska, Szadkowska, Borowiec, Niedźwiecki, Trzonkowski and Młynarski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Hogendorf, YW5uYS5ob2dlbmRvcmZAZ21haWwuY29t; Wojciech Młynarski, d29qY2llY2gubWx5bmFyc2tpQHVtZWQubG9kei5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.