 Emma L. Lewis
Emma L. Lewis Natalia Tulina1
Natalia Tulina1 Lauren Anton
Lauren Anton Paige M. Porrett
Paige M. Porrett
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 04 October 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.741518
Intrauterine inflammation impacts prenatal neurodevelopment and is linked to adverse neurobehavioral outcomes ranging from cerebral palsy to autism spectrum disorder. However, the mechanism by which a prenatal exposure to intrauterine inflammation contributes to life-long neurobehavioral consequences is unknown. To address this gap in knowledge, this study investigates how inflammation transverses across multiple anatomic compartments from the maternal reproductive tract to the fetal brain and what specific cell types in the fetal brain may cause long-term neuronal injury. Utilizing a well-established mouse model, we found that mid-gestation intrauterine inflammation resulted in a lasting neutrophil influx to the decidua in the absence of maternal systemic inflammation. Fetal immunologic changes were observed at 72-hours post-intrauterine inflammation, including elevated neutrophils and macrophages in the fetal liver, and increased granulocytes and activated microglia in the fetal brain. Through unbiased clustering, a population of Gr-1+ γ/δ T cells was identified as the earliest immune cell shift in the fetal brain of fetuses exposed to intrauterine inflammation and determined to be producing high levels of IFNγ when compared to γ/δ T cells in other compartments. In a case-control study of term infants, IFNγ was found to be elevated in the cord blood of term infants exposed to intrauterine inflammation compared to those without this exposure. Collectively, these data identify a novel cellular immune mechanism for fetal brain injury in the setting of intrauterine inflammation.
Maternal inflammation is associated with fetal brain injury and long-term neurodevelopmental impairment. Intrauterine immune activation – whether from maternal autoinflammatory disease, infection, or microbial dysbiosis– contributes to offspring neurologic diseases from schizophrenia to autism spectrum disorder (ASD) (1–5). Overt symptoms of intrauterine inflammation, or clinical chorioamnionitis, occur in a small number of deliveries and likely do not identify the majority of infants at risk from exposure to intrauterine inflammation (6). In contrast, histological chorioamnionitis (HCA), postpartum pathologic evidence of intrauterine inflammation, occurs in 10-20% of term deliveries with an increasing prevalence observed with decreasing gestational age (6–8). However, histologic examination of postpartum tissue cannot explain how inflammation at the maternal-fetal interface initiates long-lasting injury in an entirely distinct anatomic compartment, the fetal brain.
Immune activation in the central nervous system is posited to be a necessary mechanistic link between a prenatal immune insult and select neurological disorders (1). Patients with schizophrenia have more activated microglia as measured by positron emission tomography (PET) scan uptake and more dense microglia in post-mortem studies (9, 10). Patients with ASD have greater T cell infiltration at the blood brain barrier in post-mortem studies (11). These two distant immunologic findings – in utero and post-mortem – outline the wide knowledge gap in the actual pathogenesis of these disorders. More mechanistic evidence exists for immune cell development and the “immune education” that occurs prenatally, demonstrating that immunologic memory begins in utero and influences immune cell responsiveness to allergens, pathogens, and other antigens later in life (12, 13). Neuroimmune “education” may also occur prenatally; for example, one mouse model of ASD is initiated by injections of IL-17A to the fetal brain (14). However, this model does not address how subclinical intrauterine inflammation could be transmitted across the maternal-fetal dyad and induce fetal brain injury.
Our laboratory has established a mouse model of low-dose intrauterine inflammation (IUI) that leads to fetal neuronal damage, including abnormal neuronal morphology and decreased dendritic counts in cortical culture (15–17). In this model, exposure to IUI induces transcriptional and metabolic alterations to both fetal and neonatal brains, and a reduction in early postnatal neurogenesis (17–19). This model of IUI also causes postnatal white matter damage and behavior abnormalities in adult offspring (20). These findings demonstrate the utility of this model for studying IUI as a cause of fetal brain injury with long-term consequences. However, the mechanisms by which IUI translate to neuronal damage have not been fully explored.
Therefore, the objective of this study is to identify the cellular immunologic changes that propagate across anatomic compartments from the uterus to the fetal brain. Immune cell populations and their responses are tissue-specific and must be analyzed as such. Using an established mouse model of IUI, immune cell composition and function were investigated by flow cytometry and ELISA over multiple prenatal time points in both maternal and fetal murine tissues, and human cord blood. We hypothesize that IUI will induce a cascade of cellular immune alterations, ultimately causing abnormal immune cell trafficking and activation within the fetal brain itself.
The goals of this study were to elucidate immune compositional and functional shifts associated with fetal brain injury in a mouse model and to correlate our findings to human cord blood samples. Flow cytometry and ELISA data are each compiled from a minimum of two experiments per time point with our mouse model and an N=4-6 mice/group/experiment. Human cord blood was obtained from an existing biorepository of cord blood serum collected for detection of biomarkers of fetal pathologies.
These studies utilize a well-established mouse model of intrauterine inflammation created by our lab and resulting in fetal and postnatal brain injury (16, 17). For these experiments, timed-pregnant CD-1 mice were purchased from Charles River Laboratories (Wilmington, MA) and arrived at our animal facility at embryonic day 11 (E11). On E15, pregnant mice underwent a mini-laparotomy, while under isoflurane anesthesia. Each mouse received an injection into their right uterine horn, between the first and second amniotic sacs proximal to the cervix, as previously described (21–23). Each injection was either 100µl sterile phosphate buffered saline (PBS; saline control group) or 50µg/100µl lipopolysaccharide (LPS)/sterile PBS (IUI-exposed group). LPS was from E. coli 055:B5 (Sigma, St. Louis, MO). Mice were then euthanized by CO2 either 48 or 72 hours post-IUI, with tissues collected only from mice that were still pregnant. All animal care and use procedures are approved by the University of Pennsylvania IACUC.
At either 48 or 72 hours post-IUI, mice were euthanized and tissues were collected into and washed with 10% charcoal-stripped fetal bovine serum (FBS; Gemini Bio, Sacramento, CA) in Hank’s balanced salt solution (HBSS) on ice. Maternal spleen was collected. Amniotic fluid was removed from each gestational sac with a 1ml syringe. The uterus was bisected and gestational sacs with fetal membranes and placentae were removed. The decidua was then scraped from uterus with a glass microscope slide into HBSS + 10% FBS on ice. Fetal tissues, placentae, fetal liver and fetal brains, were collected from the four fetuses proximal to the injection site and washed in HBSS + 10% FBS on ice. Tissues were either flash frozen in liquid nitrogen and stored at -80°C or immediately processed into a single cell suspension for flow cytometry.
Preparation of tissues for flow cytometry follows a protocol established with the placenta (24). Placentae were minced into 2mm pieces, suspended in 5ml of digest solution (HBSS + 10% FBS + 1mg/ml Collagenase IV, Gibco, Gaithersburg, MD), and incubated in a 37°C water bath for 30 minutes. Placentae (post-digest treatment), maternal spleens, decidua, fetal livers, and fetal brains were mechanically pressed through a 70µm cell strainer. The strainer was rinsed with 10ml of HBSS and strained cells were passed through a 40µm cell strainer into a 50ml conical tube. Cells were centrifuged at 1500rpm, 4°C for 10 minutes. Red blood cells were eliminated from placentae, maternal spleens, decidua, and fetal livers by suspension in 3ml of ACK lysing buffer (Gibco, Gaithersburg, MD) and incubation at room temperature for 10 minutes. ACK-treated cells and amniotic fluid samples were rinsed in 10ml of HBSS and centrifuged at 1500rpm, 4°C for 5 minutes. Single cell suspensions from maternal spleen, decidua, placentae, fetal livers, and amniotic fluid were suspended in 1ml of FACS buffer (PBS + 2% FBS + 20µM EDTA) and transferred into 5ml round-bottom tubes through 35µm filter caps. Cells were stored on ice until ready for staining.
Fetal brain single cell suspension was suspended in 5ml 35% Percoll and was slowly layered on top of 5ml 70% Percoll in a 15ml conical tube. Cells in Percoll gradient were centrifuged at 1000xg, 4°C, for 20 minutes without centrifuge break. Myelin and glial cells were removed from the top of the gradient and then 1ml at the interface was collected, containing mononuclear cells. Mononuclear cells were washed in 10ml HBSS and centrifuged at 1500rpm, 4°C for 10 minutes. Cells were resuspended in 1ml FACS buffer and transferred into 5ml round-bottom tubes through 35µm filter caps.
Cells were transferred to a 96-well round-bottom plate for staining, washed with 200µl of PBS, and the plate was centrifuged at 1500rpm, 4°C for 5 minutes. Cells were resuspended in 100µl of Live/Dead Fixable Aqua (Invitrogen, Waltham, MA) diluted 1:1000 in PBS and incubated for 30 minutes at 4°C in the dark. Cells were washed twice with 200µl of FACS buffer, centrifuging the plate between washes at 1500rpm, 4°C for 5 minutes. Cells were then resuspended in an extracellular antibody mix and incubated for 30 minutes at 4°C in the dark. Extracellular antibody mixes included: anti-CD3e-BV785, anti-CD4-BV605, anti-CD11c-PE.Cy7, anti-CD19-APC, anti-CD45-BV711, anti-CD49b-PacificBlue, anti-CD80-PacificBlue, anti-F4/80-FITC, anti-Ly6G-AF700, anti-TCRγ/δ-PerCP.Cy5.5 (BioLegend, San Diego, CA); anti-CD8a-APC.efluor780, and anti-Gr1-AF700 (eBioscience, San Diego, CA). The antibody clone and dilution used are listed in Table 1. Cells were washed twice with 200µl of FACS buffer, centrifuging between washes at 1500rpm, 4°C for 5 minutes. Cells were resuspended in 200µl fixation/permeabilization reagent (Transcription Factor Staining Buffer Set, eBioscience, San Diego, CA) and incubated for 30 minutes at 4°C in the dark. Cells were washed twice with 200µl of permeabilization buffer (Transcription Factor Staining Buffer Set, eBioscience, San Diego, CA), centrifuging at 2000rpm, 4°C for 5 minutes between washes. Cells were resuspended in anti-FoxP3-PE (eBioscience, San Diego, CA) diluted 1:300 in 100µl permeabilization buffer and incubated for 30 minutes at room temperature in the dark. Cells were washed twice with 200µl of permeabilization buffer, centrifuging at 2000rpm, 4°C for 5 minutes between washes. Cells were then resuspended in 200µl of FACS buffer and transferred into 5ml round-bottom tubes through 35µm filter caps and stored at 4°C in dark. Single-stained CompBeads (BD Biosciences, Franklin Lakes, NJ) were used as compensation controls to create a compensation matrix, and fluorescence minus one-stained splenocytes and placental cells (FMOs) were used as gating controls to draw gates. See gating scheme (Figure S1). All samples were analyzed by an LSR-II flow cytometer (BD Biosciences, Franklin Lakes, NJ).
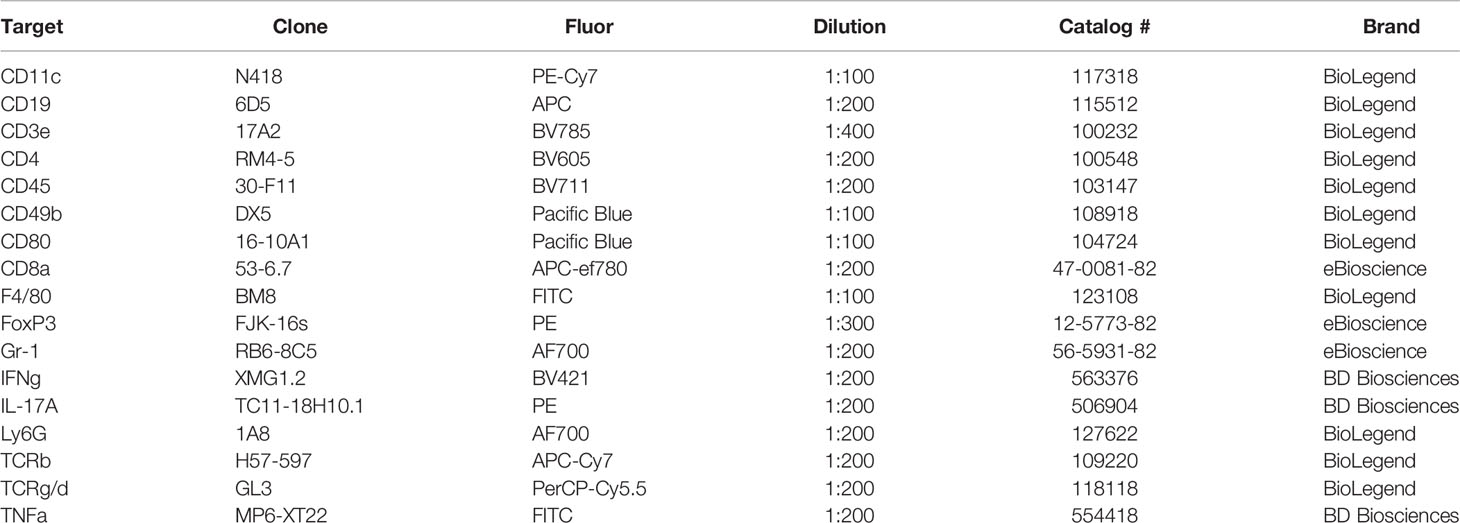
Table 1 Flow Cytometry Antibodies.
Tissues to be used for ELISA were flash frozen in liquid nitrogen at the time of tissue harvest and were stored at -80°C. Amniotic fluid samples to be used for ELISA were centrifuged at 3000rpm for 5 minutes and the supernatant was flash frozen for future use. Protein extracts were made from placentae or fetal liver tissue by submerging 50mg of frozen tissue in 1ml of RIPA buffer with cOmplete mini protease inhibitor cocktail (Roche, Basel, Switzerland) in a 2ml round bottom tube with a 5mm steel bead. Tissue was homogenized on Tissue Lyser II (Qiagen, Venlo, Netherlands) for 10 minutes at 30/second. Homogenate was rested on ice for 40 minutes and then centrifuged at 14000xg for 10 minutes. Total protein in supernatant was quantified by the BCA protein assay kit (Pierce, Rockford, IL) following the manufacturer’s protocol. Quantikine ELISA kits (R&D Systems, Minneapolis, MN) for IL-6, IL-10, IFNγ, CCL3, and CCL5 were used to measure cytokine levels, following the manufacturer’s protocol. Cytokine levels were normalized to total protein.
Fetal brains, decidua and maternal spleen were collected 72 hours post-intrauterine injection of LPS and processed into a single cell suspension as noted above. CD3+ cells were isolated from the single-cell suspension using mouse CD3e microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s protocol. Isolated CD3+ cells were resuspended at 106 cells/ml in T cell stimulation media: RPMI-1640 with L-glutamine (Corning Inc., Corning NY) supplemented with 50µM beta-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), 1X non-essential amino acids, 1X sodium pyruvate, 1X HEPES buffer (Gibco, Gaithersburg, MD), 1X Cell Stimulation Cocktail, 1X Protein Transport Inhibitor Cocktail (eBioscience, San Diego, CA), and 10% FBS. 100µl of each sample was plated on a 96-well round-bottom plate and incubated for 4 hours at 37°C. Cells were centrifuged at 400xg, 4°C, for 5 minutes, washed with PBS, resuspended in Live/Dead Fixable Aqua 1:2000 in 100µl of PBS, and incubated for 30 minutes at 4°C. Cells were then washed twice with FACS buffer; stained with extracellular markers, anti-CD45-BV711, anti-CD3ε-BV785, anti-TCRγ/δ -PerCP-Cy5.5, anti-TCRβ-APC-Cy7 (BioLegend, San Diego, CA); and incubated for 30 minutes at 4°C. Cells were washed twice with FACS buffer and resuspended in Fixation/Permeabilization reagent (eBioscience, San Diego, CA) for 30 minutes at 4°C. Cells were washed twice with Permeabilization Buffer and then incubated for 30 minutes at 4°C with intracellular cytokine staining antibodies: anti-TNFα-FITC (BioLegend, Sand Diego, CA), anti-IFNγ-BV421, and anti-IL-17A-PE (BD Biosciences, San Jose, CA). Cells were washed twice with Permeabilization Buffer and then resuspended in FACS buffer and stored at 4°C in dark until run on flow cytometer.
Cells were analyzed on an LSR-II flow cytometer (BD Biosciences, San Jose, CA) running FACSDiva software (BD Biosciences, San Jose, CA) in the University of Pennsylvania Flow Core. Flow cytometry data was then analyzed using FlowJo (BD Life Sciences, Ashland, OR) to identify immune cell subsets. High-dimensional, unbiased cell clustering and visualization tools were used to further analyze flow cytometry data. Specifically, t-SNE and FlowSOM algorithms were downloaded from the FlowJo Exchange and implemented using R/Bioconductor (25).
Cord blood was obtained from a previously published cohort of singleton pregnancies (N=723) (26, 27) of which 77 specimens were utilized in this study. Case specimens were selected as term neonates (>37 weeks gestational age at delivery) with documented HCA (N=41). Control specimens were also from term neonates (>37 weeks) that had placental histological examination but no HCA and were frequency matched for maternal race (N=36). All participants provided informed consent and the study was approved by the Institutional Review Board at the University of Pennsylvania (IRB #807678). Cord blood serum was analyzed by high sensitivity Milliplex assay (Millipore Sigma, Burlington, MA) for IFNγ and TNFα.
Statistical analyses were performed using GraphPad Prism. Data for every analysis is compiled from at least two experiments with 4-6 samples/group/experiment. Flow cytometry data was analyzed for differences between IUI and control samples collected at the same time – not for differences over time. All data was assessed for normality by Shapiro-Wilk test. Non-normally distributed data was analyzed by Mann-Whitney test. Normally distributed data was analyzed by unpaired student’s t-test, with Welch’s correction applied if the two groups differed in variance by F test. ELISA data and intracellular cytokine staining were analyzed by two-way ANOVA, followed by Tukey’s multiple comparison test if initial ANOVA was significant. Human cord blood data was non-normally distributed by Shapiro-Wilk test and analyzed for differences by Mann-Whitney test. Cord blood data was also divided by fetal sex and analyzed by two-way ANOVA to determine if fetal sex was a significant factor contributing to variance.
Our previous work utilizing our model of intrauterine inflammation (IUI) has demonstrated minimal maternal immune activation after IUI, specifically noted by a lack of maternal serum cytokine elevation (17). To confirm the hypothesis that the maternal systemic immune system is not impacted by IUI, we compared maternal splenic immune populations by flow cytometry from mice with IUI compared to saline-treated controls. From each spleen sample, 106 events were run on the flow cytometer, leading to the requisition of approximately 5x105 CD45+ cells (Figure S2A). At both 48- and 72-hours post-uterine injection, none of the following immune populations examined were significantly different between the two groups: macrophages, CD4+ T cells, CD8+ T cells, regulatory T cells, γ/δ T cells, B cells, NK cells, neutrophils, and dendritic cells (Figure S2B–J).
Decidua, the specialized uterine lining that develops during pregnancy, was collected from these same mice to examine immune changes local to the uterus at 48- and 72-hours post-uterine injection. The presence of IUI increased numbers of CD45+ cells in decidual tissue (Figures 1A, D), primarily due to an influx of decidual neutrophils (Figures 1B, C, E). This neutrophil influx was evident as both an increase in absolute number of decidual neutrophils (Figures 1B) and an increase in neutrophil frequency of decidual leukocytes (Figure 1C). The decidual neutrophil increase persisted in the mice with IUI at 72 hours post-uterine injection (Figures 1B, C) and these mice also had a slight elevation in decidual CD8+ T cells at 72 hours post-uterine injection (Figures 1F). The two most prevalent decidual immune cells, NK cells and macrophages (28, 29) did not differ between the IUI and control groups (Figures 1G, H).
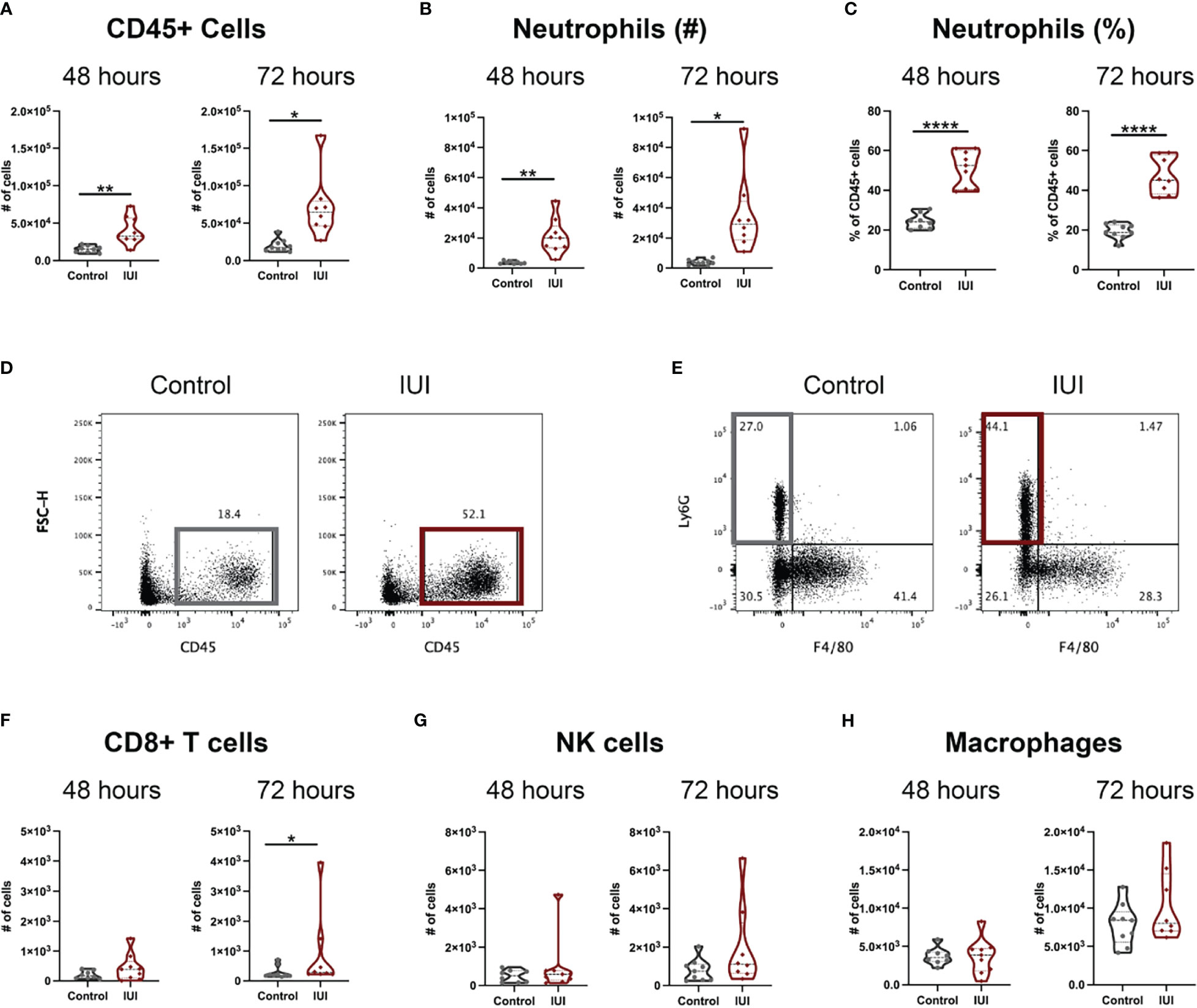
Figure 1 Intrauterine inflammation causes sustained influx of neutrophils to the decidua. Decidua was removed from the uterus at 48 and 72 hours post-uterine injection. All decidual cells were analyzed by flow cytometry to have full counts of (A) CD45+ cells. (D) Representative flow plots of total CD45+ cells are gated on non-debris, singlet, live cells. Immune cell subsets were reported both as cell counts and as a percent of total CD45+ cells, including (B, C) neutrophils, (F) CD8+ T cells, (G) NK cells, and (H) macrophages. (E) Representative flow plots of neutrophils were gated on non-debris, singlet, live, CD45+, CD19-, CD4-, CD8- cells. Significance was determined by unpaired t-test with Welch’s correction if positive F-test. *p<0.05, **p<0.01, ****p<0.0001.
To test the hypothesis that IUI would alter immune responses both within and across the placenta, flow cytometry was used to identify immune cells in the placentae and amniotic fluid of mice with IUI and controls. At 48 hours post-uterine injection, both the placentae and amniotic fluid of mice exposed to IUI had more total leukocytes than controls, but these levels normalized by 72 hours post-exposure (Figures 2A–D). Neutrophil increases in both the placenta and amniotic fluid accounted for most of the leukocyte increase in mice with IUI at 48 hours post-uterine injection and neutrophil counts in the placentae and amniotic fluid of mice with IUI also had normalized by 72 hours post-uterine injection (Figures 2E, F). Placentae, but not amniotic fluid, from mice with IUI had a small increase in macrophages (Figures 2G, H), but no other immune cell differences from control animals were observed by 72 hours post-uterine injection.
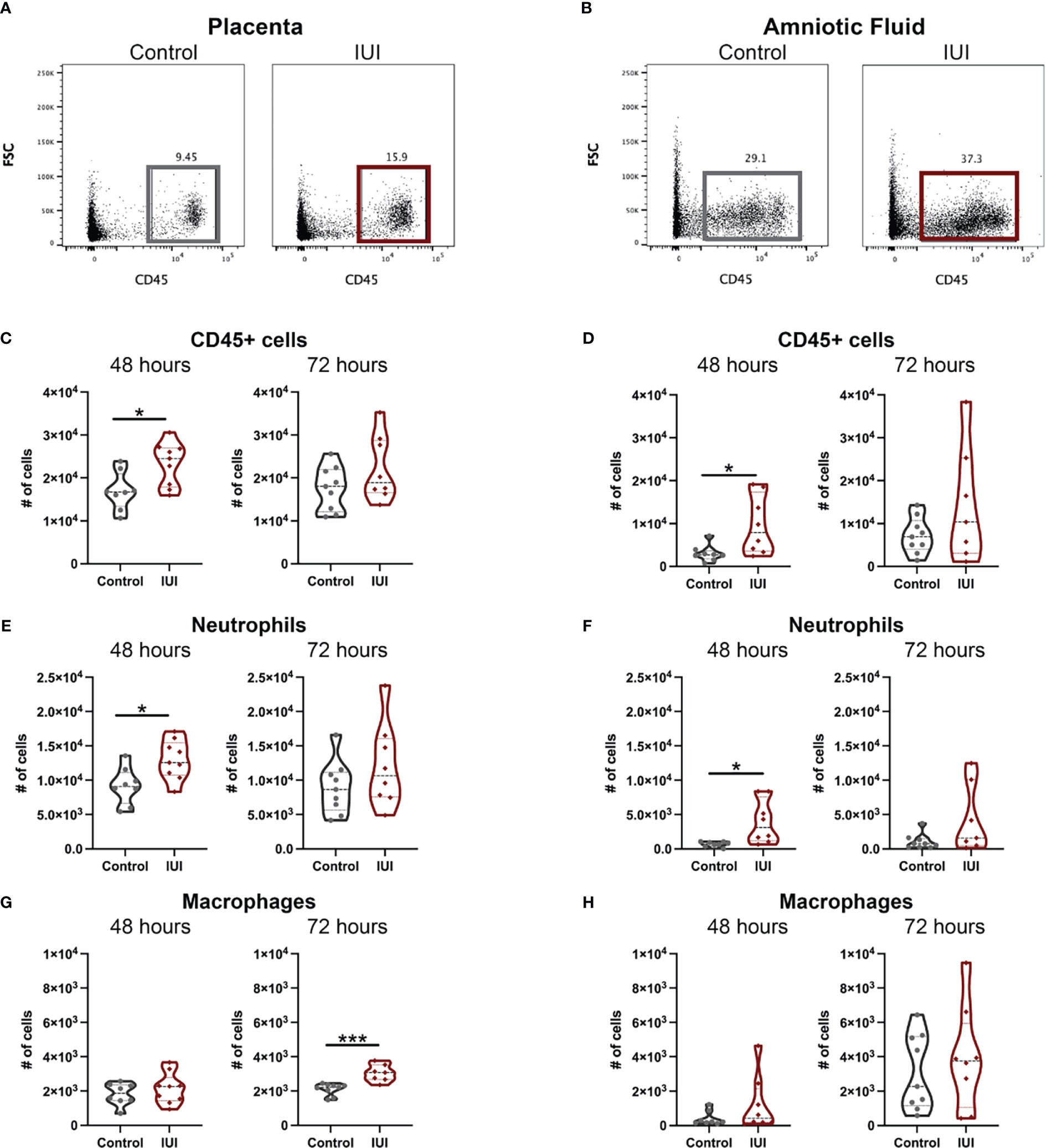
Figure 2 Intrauterine inflammation increases leukocytes found in the placenta and amniotic fluid. Placentae adjacent to the injection site and total amniotic fluid were collected at 48 and 72 hours post-uterine injection and immune cells were analyzed by flow cytometry. Representative flow plots of CD45+ cells in the (A) placenta and (B) amniotic fluid were gated on non-debris, singlet, live cells. (C, D) CD45+ cells, (E, F) neutrophils, and (G, H) macrophages were counted at each time point. Cell counts were tested for normal distribution by Shapiro-Wilk test. Normally distributed data was then analyzed by unpaired t-test and non-normally distributed data was analyzed by Mann-Whitney test. *p<0.05, ***p<0. 001.
Given the increase in CD45+ cells in the placentae and amniotic fluid of mice with IUI, we asked whether this was accompanied by an increase in chemokines that may recruit CD45+ cells to these tissues. Our previous work with this model of IUI had demonstrated significant elevations in CCL3, CCL5, and IL-6 in both the placenta and amniotic fluid at just 6 hours post-exposure (16, 17, 30), so we investigated whether these chemokines remained elevated at later time points. CCL5 was the most increased: 31-fold in the placenta and 8-fold in the amniotic fluid of mice with IUI at 48 hours post-uterine injection; but protein levels of this chemokine normalized to control levels by 72 hours post-uterine injection (Figures 3B, E). CCL3 was also elevated in the placenta and amniotic fluid in mice with IUI at 48 hours and normalized by 72 hours post-uterine injection (Figures 3A, D). IL-6 was elevated in the placenta but not the amniotic fluid of mice with IUI at 48 hours and was not significantly increased in either tissue at 72-hours post-injection (Figures 3C, F).
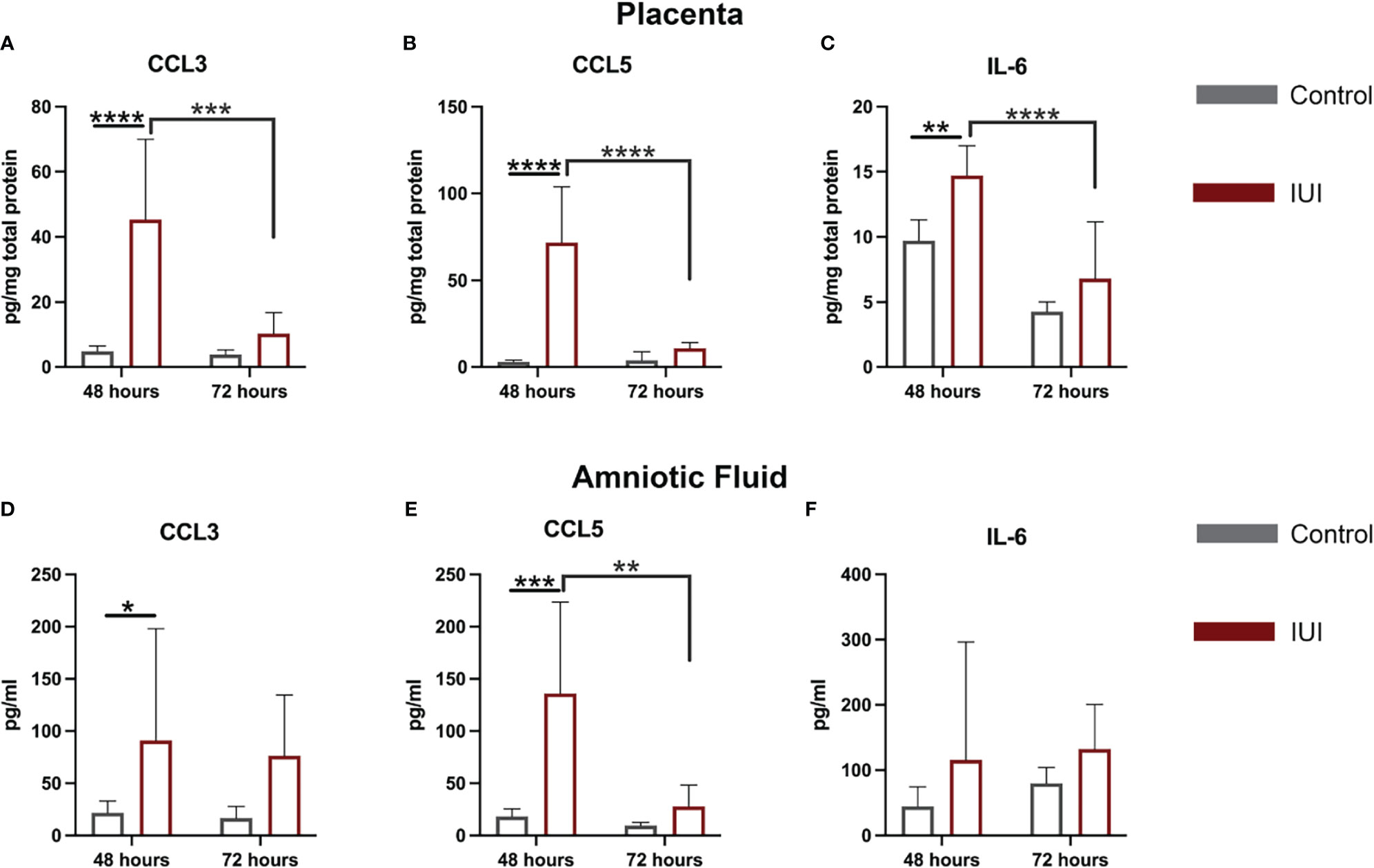
Figure 3 Elevated chemokines found in the placenta and amniotic fluid following intrauterine inflammation. Cytokines were measured in the placenta and amniotic fluid by ELISA, from samples collected at 48 and 72 hours post-uterine injection. (A–C) Placental ELISA values were normalized to total placental protein measured by BCA, while (D–F) amniotic fluid ELISA values were normalized to fluid volume. Significant differences were determined by two-way ANOVA followed by post-hoc Tukey’s multiple comparison test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Given that immune alterations were detected in the amniotic fluid of IUI-exposed mice, we hypothesized that the fetal immune system would be perturbed. In the fetal livers of mice exposed to IUI, CCL3 and IFNγ were elevated at 48 hours post-uterine injection and had normalized by 72 hours (Figures 4A, C). There was no significant difference in CCL5 levels between IUI-exposed and control fetuses (Figure 4B). No cellular immune changes were noted at 48 hours in the fetal liver. However, following the observed cytokine elevation at 48 hours, at 72 hours post-uterine injection, fetuses exposed to IUI had more CD45+ cells (Figures 4D, E), neutrophils (Figures 4F, G), and macrophages (Figures 4H, I) than control fetuses in their fetal livers.
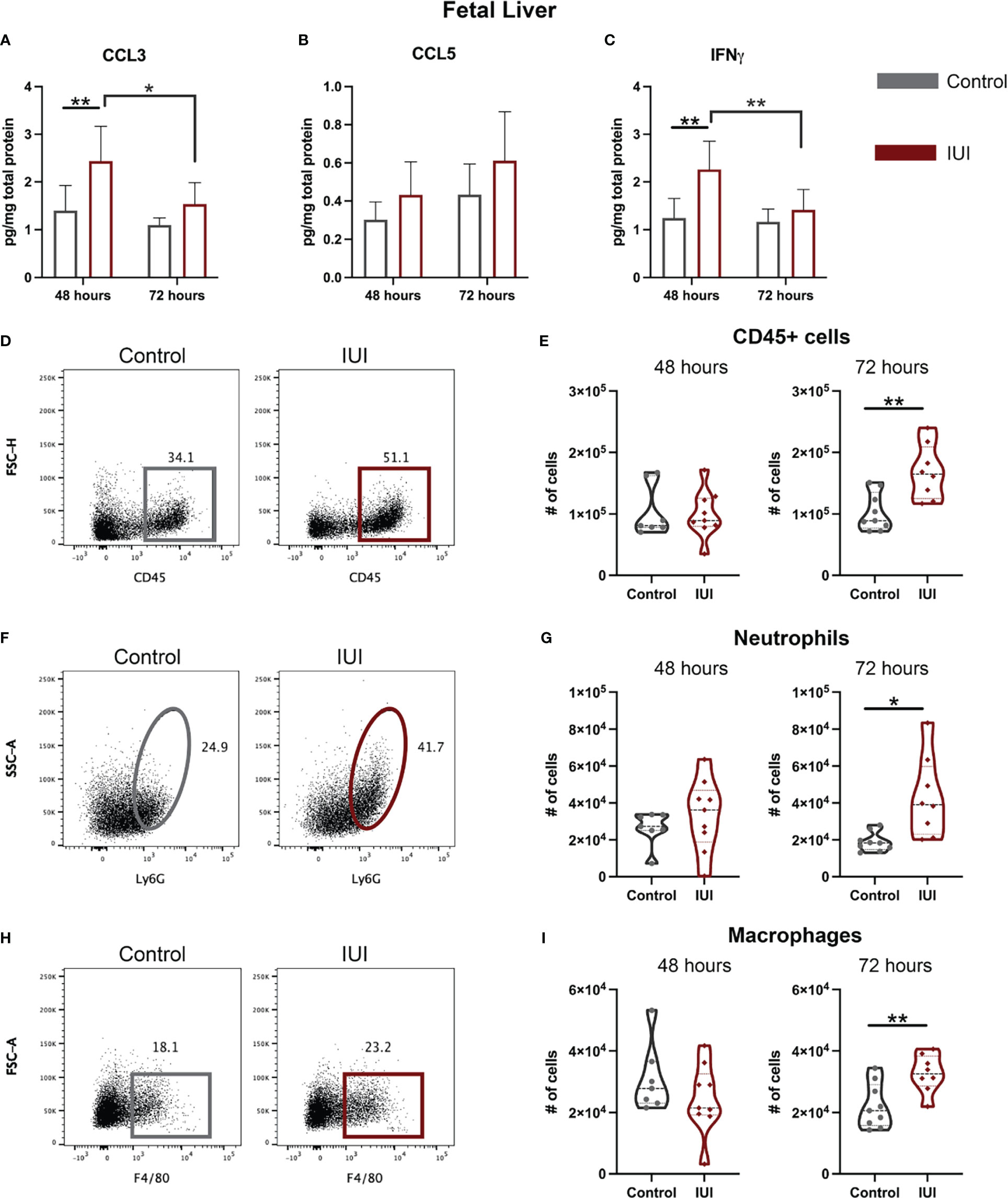
Figure 4 Neutrophil and cytokine elevation in the fetal liver demonstrate systemic fetal inflammation. Fetal livers were collected at 48 and 72 hours post-uterine injection and either frozen or processed for flow cytometry. (A–C) Frozen fetal livers were tested for cytokines by ELISA and normalized to total protein. Fresh fetal livers were analyzed by flow cytometry for immune cells. (D, E) Total CD45+ cells, (F, G) neutrophil, and (H, I) macrophage cell counts are shown. (D) Representative flow plots of CD45+ cells were gated on non-debris, singlet, live cells. (F) Representative flow plots of neutrophils were gated on non-debris, singlet, live, CD45+, CD3-, CD19- cells. (H) Representative flow plots of macrophages were gated on non-debris, singlet, live, CD45+, CD3-, CD19- cells. Cell count data was analyzed by Mann-Whitney test. Cytokine data were analyzed by two-way ANOVA with post-hoc Tukey’s multiple comparison test. *p<0.05, **p<0.01.
To address whether an inflammatory insult in the uterus could ultimately alter immune cell composition and function in the fetal brain, we isolated mononuclear cells from fetal brains and identified the immune cell types by flow cytometry (gating scheme in Figure 5A). At 48 hours post-uterine injection, the only detectable difference in the fetal brain immune cells was a 5-fold increase in granulocytes in the IUI-exposed group (Figure 5F). However, by 72 hours post-uterine injection, fetal brains from IUI-exposed dams had more overall CD45+ cells (Figure 5B) and specifically non-microglial CD45+ cells (Figure 5E), including 7-fold more granulocytes (Figure 5F). IUI-exposure increased the numbers of activated microglia and macrophages but did not affect the numbers of resting microglia (Figures 5C, D, G).
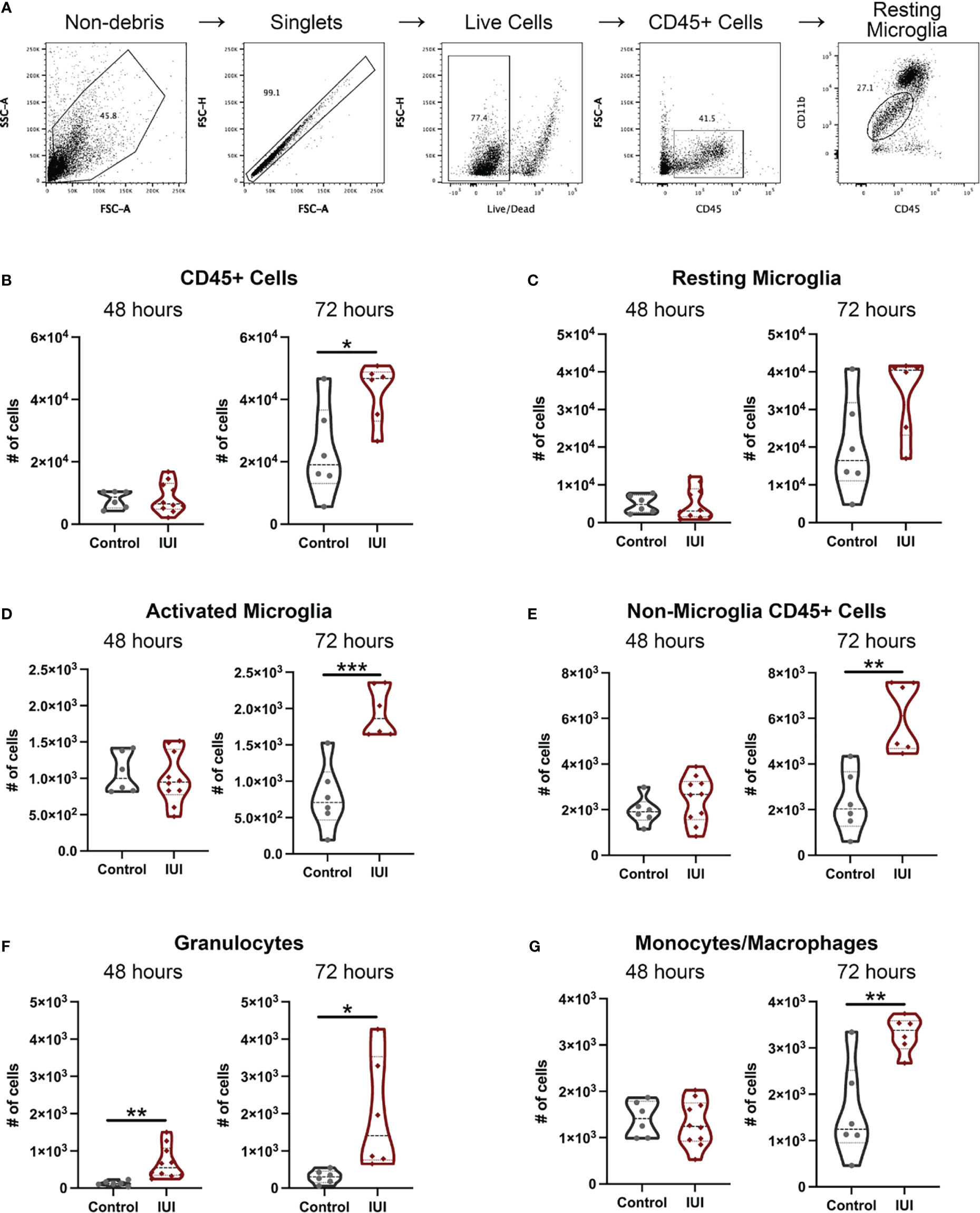
Figure 5 Intrauterine inflammation results in fetal microglial activation and an increase in non-microglial immune cells in the fetal brain. Fetal brains were collected at 48 and 72 hours post-uterine injection and processed for flow cytometry. (A) The gating scheme used to identify immune cells in the fetal brain is shown. (B) Total CD45+ cells and immune cell subtypes were counted. Subtypes include: (C) resting microglia, (D) activated microglia, (E) all non-microglia CD45+ cells, (F) granulocytes, and (G) monocytes/macrophages. All cell count data was analyzed by Shapiro-Wilk test to establish data distribution. Normally distributed data were analyzed by unpaired t-test and non-normally distributed data were analyzed by Mann-Whitney test. *p<0.05, **p<0.01, ***p<0.001.
Given limited studies on what immune cells can be found in the fetal brain, we relied on unbiased clustering algorithms to determine the immune population with the greatest difference between fetal brains from IUI-exposed and control dams. tSNE analysis creates a visual map of the different CD45+ cells found in the fetal brain, with a clear population enriched in the brains of fetuses exposed to IUI (black box, Figure 6A). The flowSOM algorithm generated ten different clusters of immune cells in the fetal brain (different colored clusters in Figure 6A). The absolute difference between the frequency of each flowSOM cluster in the fetal brains of IUI-exposed versus control mice was calculated and the population with the greatest difference was identified (purple population, Figures 6A, B). This population accounted for 7% of all CD45+ cells in the brains from IUI-exposed fetuses, but less than 1% of CD45+ cells in brains of control fetuses. This immune cell population was Gr-1+ CD11b+ γ/δ T cells (histograms in Figure 6B). Total γ/δ T cells were elevated in the fetal brains from IUI-exposed dams at both 48 and 72 hours post-uterine injection (Figures 6C, D). Total γ/δ T cells were also increased in the decidua of IUI-exposed dams at 72 hours post-uterine injection (Figure S3A), but were not changed in the spleen (Figure S2F), placenta, amniotic fluid or fetal liver (Figures S3B–D) by IUI.
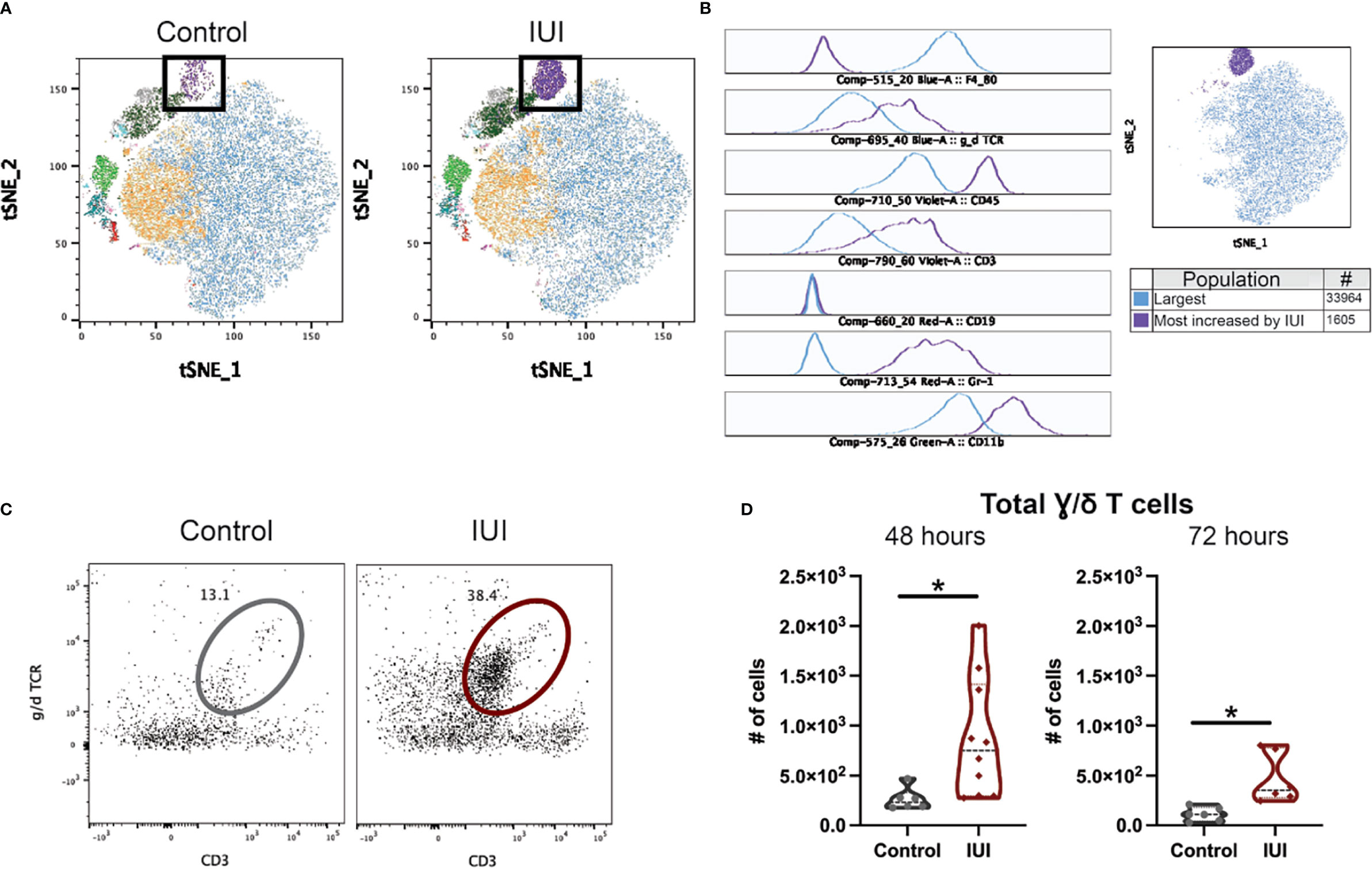
Figure 6 Intrauterine inflammation increases γ/δ T cells in the fetal brain. Fetal brains were collected at 48 and 72 hours post-uterine injection and processed for flow cytometry. CD45+ cells in the fetal brain at 72 hours were analyzed by FlowJo plug-ins tSNE and flowSOM to visualize and identify clusters, respectively. (A) Fetal brain CD45+ cells from saline-control and IUI-exposed dams were visualized by tSNE and colored by flowSOM populations. Black box indicates the most increased population by IUI. (B) Histograms of marker expression for two populations identified by flowSOM: the largest population (blue) and the population most increased by IUI-exposure (purple). (C) Representative flow plots of the γ/δ T cells in the fetal brain, gated on non-debris, singlet, live, CD45+, non-microglia, CD19- cells. (D) γ/δ T cells were counted in each fetal brain sample. All cell count data was analyzed by Shapiro-Wilk test to establish data distribution. Normally distributed data were analyzed by unpaired t-test and non-normally distributed data were analyzed by Mann-Whitney test. Normally distributed data with positive F-test for unequal variance were analyzed by unpaired t-test with Welch’s correction. *p<0.05.
To elucidate the function of the γ/δ T cells in the IUI-exposed fetal brains, T cells were isolated from pregnant mice 72-hours after exposure to IUI from the maternal spleen, decidua, and fetal brain. T cells were stimulated in vitro for four hours and stained for intracellular cytokines. Cells were also stained for TCRβ and TCRγδ to differentiate between α/β and γ/δ T cells. Cytokine production by total T cells (Figures 7A–C) and specifically γ/δ T cells (Figures 7D–F) varied immensely by anatomic location. Approximately 10% of all splenic T cells produced at least one measurable cytokines, 33.7% of decidual T cells produced TNFα, and nearly 100% of T cells in the fetal brain produced at least one cytokine (Figures 7A, B). Of fetal brain γ/δ T cells, 9.1% produced IL-17A and 7.9% produced TNFα; while 89.4% of fetal brain γ/δ T cells produced IFNγ, with some γ/δ T cells producing multiple cytokines (Figures 7D, E). Whether examining total T cells or only γ/δ T cells, only those isolated from the fetal brain were making significant IFNγ (Figures 7C, F).
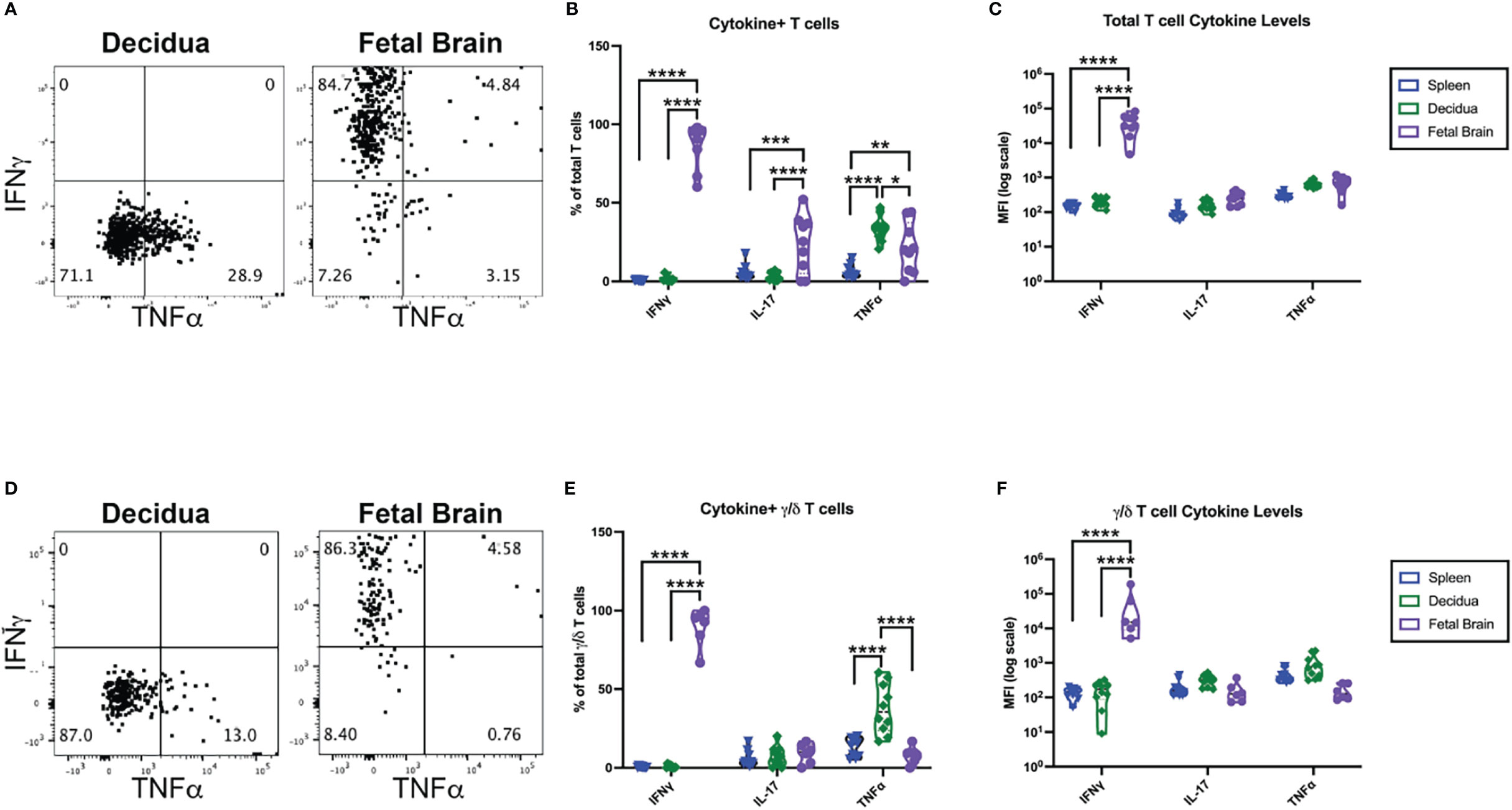
Figure 7 γ/δ T cells from the fetal brain, but not from the maternal spleen or decidua, are producing IFNγ;. CD3+ cells were isolated from tissues collected at 72 hours post-exposure to IUI. CD3+ cells were stimulated in vitro and stained for intracellular cytokines as well as T cell receptor subtype. (A) Representative flow plots from the decidua and fetal brain are gated on non-debris, singlet, live, CD45+, CD3+ cells. (D) Representative flow plots of cytokines in γ/δ T cells followed the same gating scheme, with the additional gates of TCRβ- and TCRγδ+. Cytokine expression was analyzed as (B) the % of T cells or (E) % of γ/δ T cells that were positive for each cytokine and as the MFI of either (C) total T cells or (F) γ/δ T cells. Cytokine data was analyzed by two-way ANOVA with post-hoc Tukey’s multiple comparison test comparing the expression of a single cytokine between tissues. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Cord blood was collected from term pregnancies (delivery at greater than 37 weeks) and placentae from these pregnancies were examined for histological chorioamnionitis (HCA) as part of clinical care. Term HCA was used as a human correlate to our mouse model of low dose intrauterine inflammation without preterm birth, as HCA is defined by placental neutrophil infiltrates (7). A case control study was performed. Cases were defined by the finding of HCA on placental pathology (N=36) compared to controls who did not have HCA (N=41). Cases and controls were frequency matched by self-reported race (Table 2). IFNγ and TNFα were measured in cord blood by a high-sensitivity Luminex assay. IFNγ levels were 1.8-fold higher in cord blood from neonates with HCA compared to controls (p<0.0001), but TNFα was equivalent between cases and controls (Figures 8A, B), indicating an IFNγ-specific inflammatory signature in neonates with HCA. Fetal sex did not contribute to cytokine level variance.
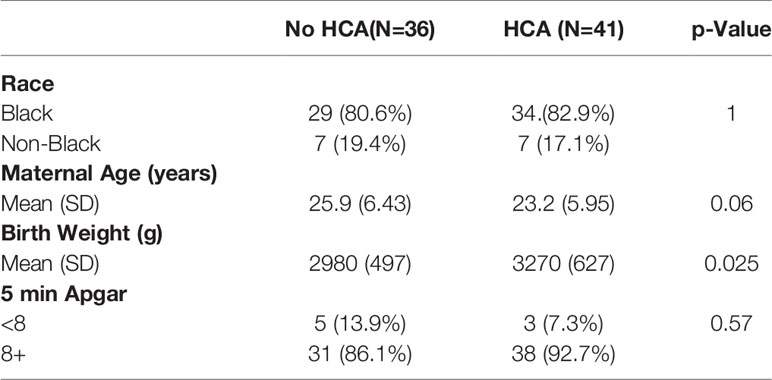
Table 2 Demographic table of study participants.
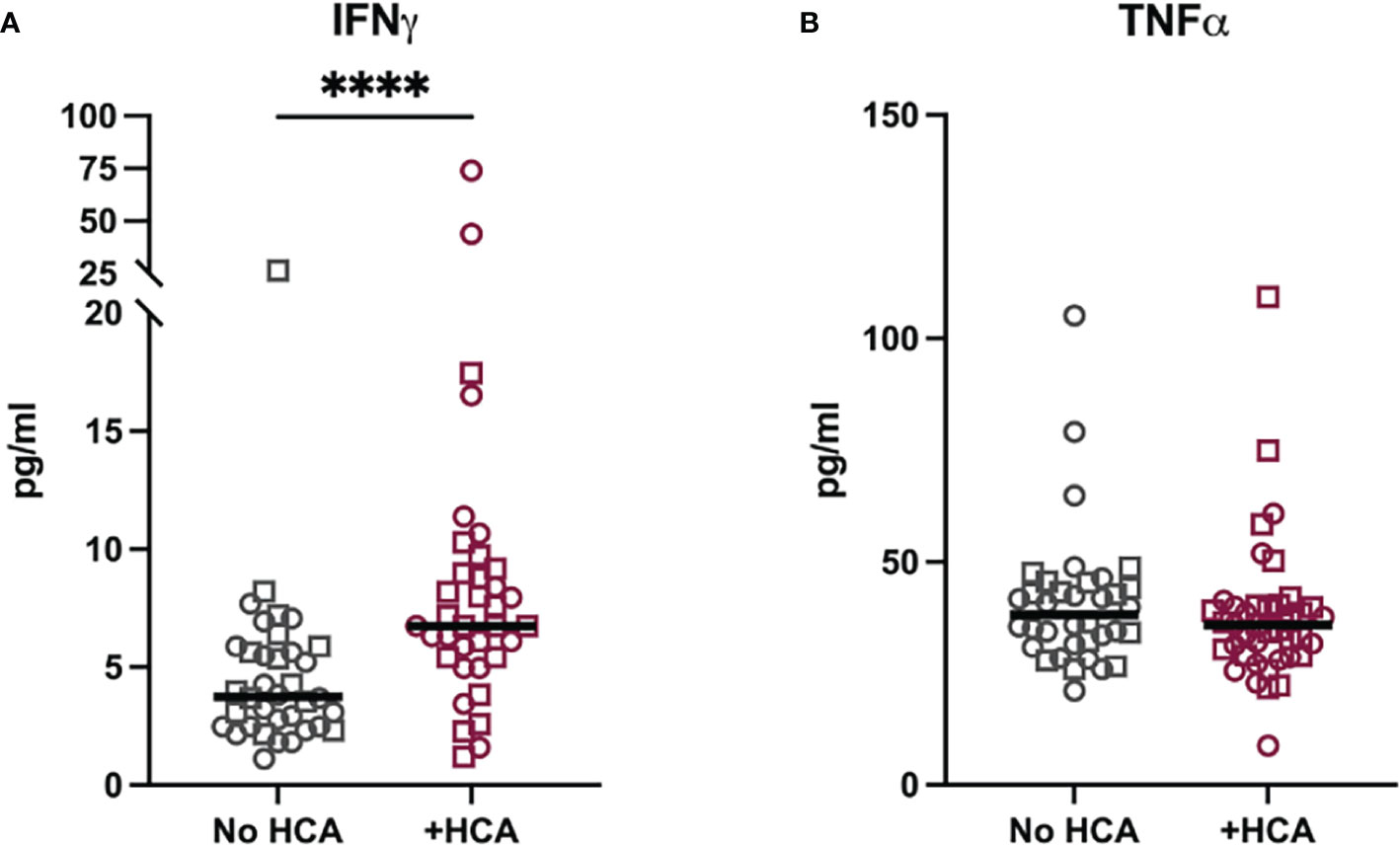
Figure 8 Cord blood from neonates with histological chorioamnionitis have elevated IFNγ. Cytokine levels were measured in a matched case-control study of cord blood plasma from term neonates (>37 weeks gestational age at delivery) with and without histological chorioamnionitis (HCA). (A) IFNγ and (B) TNFα were measured by Milliplex assay in 41 cases with HCA and 36 controls. Neonatal sex is noted by symbol shape: squares are male and circles are female. Sex did not impact variance in cytokine levels as measured by two-way ANOVA. Differences in cytokine levels were analyzed by Mann-Whitney test. ****p<0.0001.
The developing brain is exquisitely sensitive to intrauterine inflammation (IUI) such that the inflammatory insult may be asymptomatic in the mother but may cause long-lasting neuronal damage in the fetus. This study identifies a cascade of immunological events across the maternal-fetal dyad, demonstrating an active local immune response in the absence of systemic inflammation. The immune response in each compartment differed in cellular content, cytokine activation, and reaction time, emphasizing the importance of tissue-specific immunity and validating the necessity to examine each compartment across the maternal-fetal dyad. Activated IFNγ+ Gr-1+ γ/δ T cells in the fetal brain are a novel population that may be essential for fetal brain injury and IFNγ-driven inflammation may be critical for both identifying neonates at risk of brain injury and understanding the specific pathogenesis of neuroimmune diseases from exposure to prenatal inflammation.
The mouse model used in this study recapitulates the clinical phenotype as exposed dams exhibit no sign of systemic illness or immunologic shifts. Yet, even in the absence of systemic immunological shifts, significant immune responses occur at the maternal-fetal interface and within the fetus. This model greatly differs from other models of maternal inflammation inducing fetal brain damage, in which polyI:C (3, 14, 31), LPS (32), IL-1β (33, 34), or pathogen-specific antigens (35) are injected systemically into the mother to mimic a viral-like illness (36, 37). Overt maternal illness occurs in only a minority of cases of IUI as indicated by histological chorioamnionitis (7, 8); as such, the model utilized in these studies more accurately recapitulates mechanism of disease in human pregnancy. Most cases of human IUI are thought to be caused by ascension of vaginal bacteria to the uterus (7). Maternal intestinal microbiota also influences the development of ASD in offspring, whereby the microbiota affects maternal immune and metabolic factors that reach the fetus and change fetal neurodevelopment (3, 5, 38, 39). Our model better mimics the subtle local inflammatory changes initiated by microbial products or microbial ascension to the uterus when compared to systemic inflammatory models.
Despite the different modes of inducing inflammation, our model demonstrates a neutrophil influx to the decidua, placenta, and amniotic fluid – which is consistent with other animal and human studies (37, 40–43). Neutrophil infiltrates to the placenta are also diagnostic of human histological chorioamnionitis (7). The local immune response at the maternal-fetal interface therefore appears similar whether the inflammatory insult is local or systemic. Importantly, this study demonstrates that a lack of maternal systemic immune activity does not reflect the immune activity of the maternal-fetal interface.
The duration of our study gives more context to the local immune cell infiltrates associated with IUI (Figure 9). Neutrophils are increased in the placenta and amniotic fluid at 48 hours post-uterine injection but have returned to control numbers by 72 hours. We find a similar trend with inflammatory cytokines in the placenta and amniotic fluid. Previous data from our laboratory has shown massive increases in IL-6 in the placenta and amniotic fluid in IUI-exposed pregnant mice at 6 hours post-uterine injection (17, 44). These current data show increases in CCL3, CCL5, and IL-6 in IUI-exposed pregnant mice at 48 hours, but by 72 hours post-uterine injection, these cytokines are returning to control levels. These data demonstrate a wave of inflammation through the placenta and amniotic fluid that is subsiding by 72-hours after initial insult.
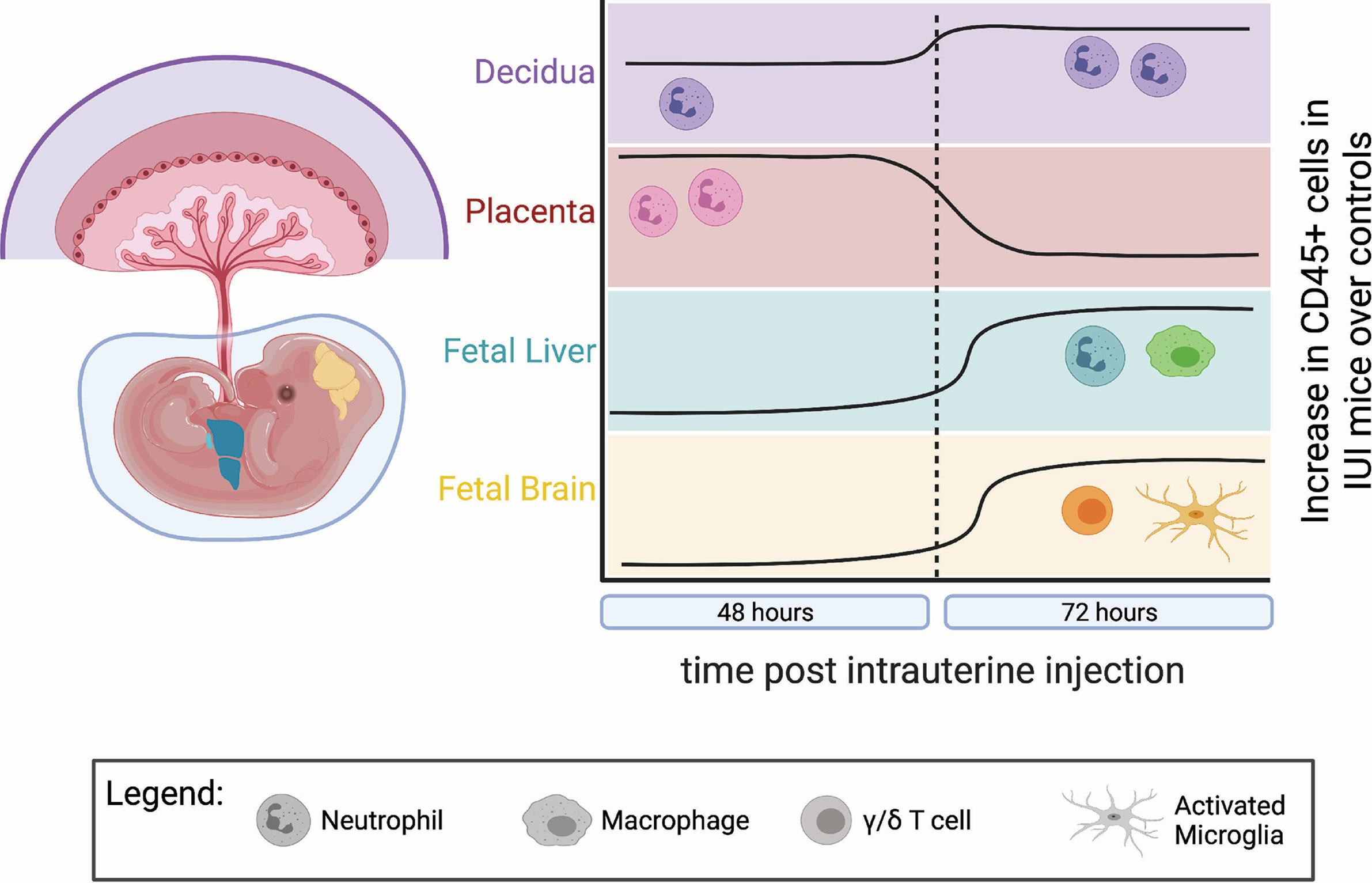
Figure 9 Temporal dynamics of immune infiltrates differed in each anatomic compartment. IUI-exposed mice had more CD45+ cells compared to controls in each examined tissue, but at different times post-uterine injection and with different immune subtypes. This figure demonstrates: the lasting neutrophil infiltrate in the decidua (purple) at 48 and 72 hours post-uterine injection; the neutrophil infiltrate to the placenta (pink) at 48 hours that normalizes to control levels by 72 hours; the increase in macrophages and neutrophils to the fetal liver (green) at 72 hours; and the increase in γ/δ T cells and activated microglia in the fetal brain (yellow) at 72 hours post-uterine injection. (Figure made using BioRender software.).
Even as inflammation in the placenta and amniotic fluid is lessening by 72-hours post-uterine injection, cellular changes to the fetal immune system in IUI-exposed fetuses are just becoming apparent (Figure 9). The increase in macrophages and neutrophils in the fetal livers of IUI-exposed fetuses is a novel finding that indicates IUI not only may affect neurodevelopment but also immune system development. These findings are consistent with the immune irregularities and increased autoimmunity that are present in patients with ASD (1, 45). Fetal immune activation in our mouse model may be a direct response to LPS, as LPS is able to cross the placenta into fetal tissue and fetal TLR4 activation is necessary for LPS-induced brain damage (30, 44). Prenatal exposures to microbial products impact allergic sensitivity and tolerance induction of fetal immune cells of both mice and humans (46, 47). Fetal blood brain barrier permeability is influenced by maternal microbiota and immune activity (31, 48). Fetal inflammation following maternal immune activation leads to an accumulation of immune cells at the embryonic choroid plexus and a weakening of tight junctions at the blood brain barrier (31). This weakened barrier likely allows for the increased infiltration of non-microglial CD45+ cells that we find in the brains of IUI-exposed fetuses (Figure 5E) and provides a model for our findings (Figure 10).
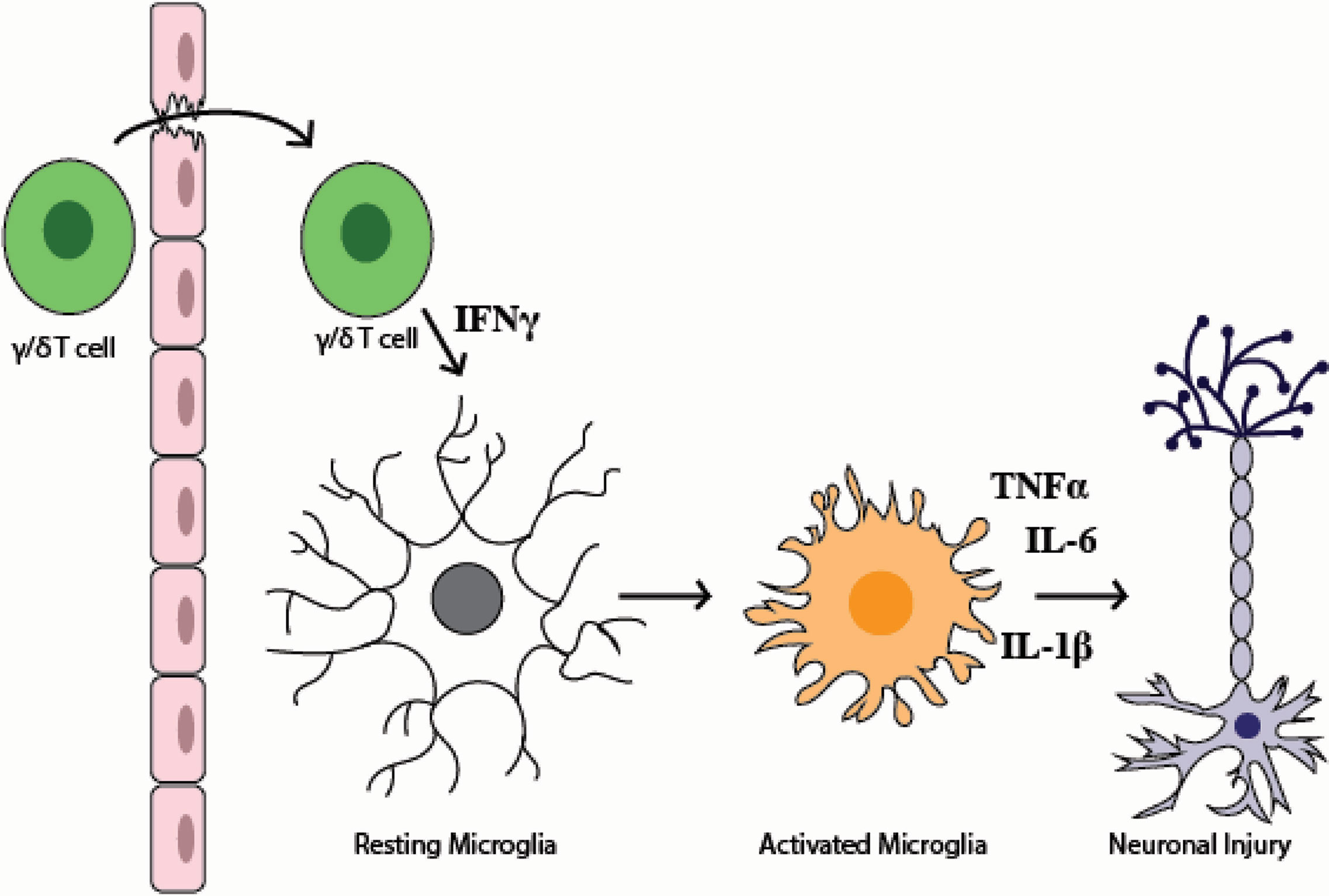
Figure 10 Model figure of the mechanism of fetal brain injury in the setting of intrauterine inflammation. This model demonstrates how γ/δ T cells in the fetal brain may lead to fetal brain injury. This model reflects both data in this paper and data from previously published literature that demonstrate that (1) the fetal blood brain barrier is weakened by maternal inflammation (2), during systemic inflammation γ/δ T cells may traffic to the brain (3), an increase in IFNγ+ γ/δ T cells are present in the fetal brain following IUI (4), IFNγ leads to microglial activation, and (5) prenatal microglial activation can cause neuronal injury.
The detection of increased IFNγ in mouse fetal livers of IUI-exposed fetuses and in human cord blood supports a more systemic immune activation occurring in the fetus in response to IUI. This systemic fetal inflammation may specifically initiate γ/δ T cell trafficking to the brain, as has been reported to occur neonatal sepsis (49). Similarly, in experimental autoimmune encephalomyelitis, γ/δ T cells are thought to be primed in the periphery and then travel to the central nervous system (CNS), where they are able to augment CNS pathology (50). Consistent with reports that demonstrate peripheral γ/δ T cells traffic to the brain (49–51), in the fetus γ/δ T cells are found in the skin, intestines and meninges (52). Recent work has demonstrated that the presence of meningeal γ/δ T cells in neonates are critical for short-term memory and synaptic plasticity (53). Interestingly, these physiologic γ/δ T cells produce IL-17 (53), while the γ/δ T cells that we detected in the fetal brain produce IFNγ, suggesting a different, possibly pathologic, activation state.
As the bidirectional crossing of immune cells between mother and fetus remains a poorly studied area (54), one might argue that the γ/δ T cells may be maternal in origin. Based on prior data showing very few maternal-derived cells in the fetal liver (24), we suggest that the fetal brain γ/δ T cells are likely fetal in origin. Furthermore, the earliest fetal γ/δ T cell progenitors to exit the thymus at E13 express IFNγ (52), which is consistent with the phenotype of the γ/δ T cells in this study.
The expression of both Gr-1 and CD11b on the γ/δ T cells in this study is consistent with their IFNγ production. Gr-1 has been used as a marker of IFNγ-producing rather than IL-17-producing γ/δ T cells in the setting of bacterial pneumonia (55), and CD11b+ γ/δ T cells are reported to produce IFNγ and to present exogenous antigens (56). Together the phenotype of these γ/δ T cells suggests they are responding to and exacerbating fetal inflammation in this model. The production of IFNγ by γ/δ T cells in the fetal brain may is one plausible mechanism by which these γ/δ T cells cause brain injury, as microglial exposure to IFNγ initiates a microglial activation state that is pro-inflammatory and neurotoxic (57, 58). Microglia impacted by inflammation in the “pre-microglial” phase from E14 to one month of life are particularly sensitive to inflammation and rearrange their chromatin landscape to create lasting changes in gene expression patterns (59, 60). These early epigenetic changes to microglia may explain the long-term neurobehavioral outcomes from prenatal exposure to inflammation.
While the initial injury may be prenatal, neuroimmune pathologies may be treated postnatally. In multiple animal models of prenatal neuronal injury and inflammation, postnatal treatments have reversed neurologic, immunologic, and even behavior abnormalities in the exposed offspring (35, 61–63). The potential for treatment heightens the necessity for early diagnosis. Given the absence of a systemic maternal signal for the presence of intrauterine inflammation, other avenues to identify fetuses and neonates at risk is mandated. Assessing the immunological state of a neonate is feasible with testing of the cord blood immediately following delivery. Our finding of increased IFNγ in human cord blood from neonates with HCA demonstrates the plausibility of cord blood containing an immune signature of neuronal injury. The increased cord blood IFNγ indicates that inflammation from HCA is not isolated to the placenta and impacts the fetus itself. Additionally, the elevation of IFNγ but not TNFα suggests an adaptive over innate fetal response to HCA.
One limitation to this study is that T cell development in the mouse occurs later in gestation than in humans. In mice, γ/δ T cells do not develop until E13 and α/β T cells mostly mature postnatally, whereas human T cell development begins at week 11-12 of pregnancy (64, 65). Therefore, the fetal immune response observed in the mouse may not be wholly reflective of that in a human fetus. However, postmortem studies from human preterm infants have shown γ/δ T cells in neonatal brains with injury (specifically periventricular leukomalacia) but not in preterm infants without brain injury (66), indicating that γ/δ T cells may be critical to fetal brain injury regardless of the presence of α/β T cells. γ/δ T cells were also found in a sheep model of fetal asphyxia-induced brain injury (66), suggesting a conserved role for γ/δ T cell in prenatal neuropathology, regardless of the initial insult. In addition, in multiple mouse models of neonatal brain injury (sepsis, hypoxia-ischemia), γ/δ T cells were required to cause brain injury, and depletion of γ/δ T cells was protective (49, 66). γ/δ T cells are also implicated in multiple ‘adult’ neuroinflammatory diseases including encephalitis, multiple sclerosis, and ischemic stroke (50, 51, 67, 68). These findings suggest a specific role for γ/δ T cells, rather than α/β T cells, in neuroimmune disease.
Overall, our data support a model of fetal brain injury where local intrauterine inflammation progresses to systemic fetal inflammation, which then creates the conditions for activated γ/δ T cells to cross the blood brain barrier, make IFNγ, and activate fetal microglia (Figure 10). The lack of maternal systemic inflammation posits a challenge for prenatal diagnosis of neurodevelopmental disorders. However, detection of IFNγ in human cord blood provides the potential for early neonatal diagnosis, and thus treatment, of prenatal neuronal damage. These findings demonstrate a complex immune response in the maternal-fetal dyad that participate in fetal brain injury from exposure to prenatal inflammation. Importantly, this work identifies a potential target and/or marker of fetal brain inflammation through the identification of γ/δ T cells in our mouse model and elevated IFNγ in term HCA. Being able to identify, through cord blood biomarkers, neonates at greatest risk for neuroimmune activation, may provide a novel opportunity for pharmacologic interventions to prevent adverse neurobehavioral outcomes associated with prenatal inflammation.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Institutional Review Board, University of Pennsylvania (IRB #807678). Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements. The animal study was reviewed and approved by Institutional Animal Care & Use Committee, University of Pennsylvania.
EL and ME conceptualized this study. EL, NT, LA, and AB determined the methodology and conducted the experiments. EL and PP analyzed the data. ME secured funding for this study. EL wrote the initial draft of this manuscript with significant review and editing by ME and LA. All authors contributed to the article and approved the submitted version.
Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01-HD076032) and March of Dimes (21-FY08-539 – Elovitz).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge the University of Pennsylvania Human Immunology Core (P30-CA016520) for their help with the Luminex assays performed in this study and the University of Pennsylvania Maternal and Child Health Research Center research coordinators for their help in collecting cord blood samples, as well as the patients who consented to participate in perinatal research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.741518/full#supplementary-material
1. Anders S, Kinney DK. Abnormal Immune System Development and Function in Schizophrenia Helps Reconcile Diverse Findings and Suggests New Treatment and Prevention Strategies. Brain Res (2015) 1617:93–112. doi: 10.1016/j.brainres.2015.02.043
2. Easson A, Woodbury-Smith M. The Role of Prenatal Immune Activation in the Pathogenesis of Autism and Schizophrenia: A Literature Review. Res Autism Spect Dis (2014) 8:312–6. doi: 10.1016/j.rasd.2013.12.007
3. Lammert CR, Frost EL, Bolte AC, Paysour MJ, Shaw ME, Bellinger CE, et al. Cutting Edge: Critical Roles for Microbiota-Mediated Regulation of the Immune System in a Prenatal Immune Activation Model of Autism. J Immunol (2018) 201:845–50. doi: 10.4049/jimmunol.1701755
4. Madhusudan A, Vogel P, Knuesel I. Impact of Prenatal Immune System Disturbances on Brain Development. J Neuroimmune Pharmacol (2012) 8:79–86. doi: 10.1007/s11481-012-9374-z
5. Minakova E, Warner BB. Maternal Immune Activation, Central Nervous System Development and Behavioral Phenotypes. Birth Defects Res (2018) 110:1539–50. doi: 10.1002/bdr2.1416
6. Conti N, Torricelli M, Voltolini C, Vannuccini S, Clifton VL, Bloise E, et al. Term Histologic Chorioamnionitis: A Heterogeneous Condition. Eur J Obstetrics Gynecol Reprod Biol (2015) 188:34–8. doi: 10.1016/j.ejogrb.2015.02.034
7. Cappelletti M, Presicce P, Kallapur SG. Immunobiology of Acute Chorioamnionitis. Front Immunol (2020) 11:649. doi: 10.3389/fimmu.2020.00649
8. Lee SM, Lee KA, Kim SM, Park C-W, Yoon BH. The Risk of Intra-Amniotic Infection, Inflammation and Histologic Chorioamnionitis in Term Pregnant Women With Intact Membranes and Labor. Placenta (2011) 32:516–21. doi: 10.1016/j.placenta.2011.03.012
9. Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial Activity in People at Ultra High Risk of Psychosis and in Schizophrenia: An [(11)C]PBR28 PET Brain Imaging Study. Am J Psychiatry (2015) 173:44–52. doi: 10.1176/appi.ajp.2015.14101358
10. van Kesteren CFMG, Gremmels H, de Witte LD, Hol EM, Gool ARV, Falkai PG, et al. Immune Involvement in the Pathogenesis of Schizophrenia: A Meta-Analysis on Postmortem Brain Studies. Transl Psychiat (2017) 7:e1075–5. doi: 10.1038/tp.2017.4
11. DiStasio MM, Nagakura I, Nadler MJ, Anderson MP. T Lymphocytes and Cytotoxic Astrocyte Blebs Correlate Across Autism Brains. Ann Neurol (2019) 86:885–98. doi: 10.1002/ana.25610
12. Mandal M, Donnelly R, Elkabes S, Zhang P, Davini D, David BT, et al. Maternal Immune Stimulation During Pregnancy Shapes the Immunological Phenotype of Offspring. Brain Behav Immun (2013) 33:33–45. doi: 10.1016/j.bbi.2013.04.012
13. Renz H, Brandtzaeg P, Hornef M. The Impact of Perinatal Immune Development on Mucosal Homeostasis and Chronic Inflammation. Nat Rev Immunol (2011) 12:9–23. doi: 10.1038/nri3112
14. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The Maternal Interleukin-17a Pathway in Mice Promotes Autism-Like Phenotypes in Offspring. Science (2016) 351:933–9. doi: 10.1126/science.aad0314
15. Burd I, Bentz AI, Chai J, Gonzalez J, Monnerie H, Roux PDL, et al. Inflammation-Induced Preterm Birth Alters Neuronal Morphology in the Mouse Fetal Brain. J Neurosci Res (2010) 88:1872–81. doi: 10.1002/jnr.22368
16. Burd I, Brown A, Gonzalez JM, Chai J, Elovitz MA. A Mouse Model of Term Chorioamnionitis: Unraveling Causes of Adverse Neurological Outcomes. Reprod Sci (2011) 18:900–7. doi: 10.1177/1933719111398498
17. Elovitz MA, Brown AG, Breen K, Anton L, Maubert M, Burd I. Intrauterine Inflammation, Insufficient to Induce Parturition, Still Evokes Fetal and Neonatal Brain Injury. Int J Dev Neurosci (2011) 29:663–71. doi: 10.1016/j.ijdevneu.2011.02.011
18. Brown AG, Tulina NM, Barila GO, Hester MS, Elovitz MA. Exposure to Intrauterine Inflammation Alters Metabolomic Profiles in the Amniotic Fluid, Fetal and Neonatal Brain in the Mouse. PloS One (2017) 12:e0186656. doi: 10.1371/journal.pone.0186656
19. Hester MS, Tulina N, Brown A, Barila G, Elovitz MA. Intrauterine Inflammation Reduces Postnatal Neurogenesis in the Hippocampal Subgranular Zone and Leads to Accumulation of Hilar Ectopic Granule Cells. Brain Res (2018) 1685:51–9. doi: 10.1016/j.brainres.2018.02.005
20. Makinson R, Lloyd K, Rayasam A, McKee S, Brown A, Barila G, et al. Intrauterine Inflammation Induces Sex-Specific Effects on Neuroinflammation, White Matter, and Behavior. Brain Behav Immun (2017) 66:277–88. doi: 10.1016/j.bbi.2017.07.016
21. Elovitz MA, Wang Z, Chien EK, Rychlik DF. Phillippe M. A New Model for Inflammation-Induced Preterm Birth The Role of Platelet-Activating Factor and Toll-Like Receptor-4. Am J Pathol (2003) 163:2103–11. doi: 10.1016/s0002-9440(10)63567-5
22. Elovitz MA, Mrinalini C, Sammel MD. Elucidating the Early Signal Transduction Pathways Leading to Fetal Brain Injury in Preterm Birth. Pediatr Res (2006) 59:50–5. doi: 10.1203/01.pdr.0000191141.21932.b6
23. Burd I, Chai J, Gonzalez J, Ofori E, Monnerie H, Roux PDL, et al. Beyond White Matter Damage: Fetal Neuronal Injury in a Mouse Model of Preterm Birth. Am J Obstet Gynecol (2009) 201:279.e1–8. doi: 10.1016/j.ajog.2009.06.013
24. Lewis EL, Sierra L-J, Barila GO, Brown AG, Porrett PM, Elovitz MA. Placental Immune State Shifts With Gestational Age. Am J Reprod Immunol (2018) 79:e12848. doi: 10.1111/aji.12848
25. Gassen SV, Callebaut B, Helden MJV, Lambrecht BN, Demeester P, Dhaene T, et al. FlowSOM: Using Self-Organizing Maps for Visualization and Interpretation of Cytometry Data. Cytom Part A (2015) 87:636–45. doi: 10.1002/cyto.a.22625
26. Bastek JA, Hirshberg A, Chandrasekaran S, Owen CM, Heiser LM, Araujo BA, et al. Biomarkers and Cervical Length to Predict Spontaneous Preterm Birth in Asymptomatic High-Risk Women. Obstetrics Gynecol (2013) 122:283–9. doi: 10.1097/aog.0b013e31829ab714
27. Elovitz MA, Anton L, Bastek J, Brown AG. Can microRNA Profiling in Maternal Blood Identify Women at Risk for Preterm Birth? Am J Obstet Gynecol (2015) 212:782.e1–5. doi: 10.1016/j.ajog.2015.01.023
28. Erlebacher A. Immunology of the Maternal-Fetal Interface. Annu Rev Immunol (2013) 31:387–411. doi: 10.1146/annurev-immunol-032712-100003
29. Yang F, Zheng Q, Jin L. Dynamic Function and Composition Changes of Immune Cells During Normal and Pathological Pregnancy at the Maternal-Fetal Interface. Front Immunol (2019) 10:2317. doi: 10.3389/fimmu.2019.02317
30. Tulina NM, Brown AG, Barila GO, Elovitz MA. The Absence of TLR4 Prevents Fetal Brain Injury in the Setting of Intrauterine Inflammation. Reprod Sci (2018) 26:1082–93. doi: 10.1177/1933719118805859
31. Cui J, Shipley FB, Shannon ML, Alturkistani O, Dani N, Webb MD, et al. Inflammation of the Embryonic Choroid Plexus Barrier Following Maternal Immune Activation. Dev Cell (2020) 55:617–628.e6. doi: 10.1016/j.devcel.2020.09.020
32. Prins JR, Zhang B, Schjenken JE, Guerin LR, Barry SC, Robertson SA. Unstable Foxp3+ Regulatory T Cells and Altered Dendritic Cells Are Associated With Lipopolysaccharide-Induced Fetal Loss in Pregnant Interleukin 10-Deficient Mice. Biol Reprod (2015) 93:95. doi: 10.1095/biolreprod.115.128694
33. Novak CM, Lee JY, Ozen M, Tsimis ME, Kucirka LM, McLane MW, et al. Increased Placental T Cell Trafficking Results in Adverse Neurobehavioral Outcomes in Offspring Exposed to Sub-Chronic Maternal Inflammation. Brain Behav Immun (2019) 75:129–36. doi: 10.1016/j.bbi.2018.09.025
34. Leitner K, Shammary MA, McLane M, Johnston MV, Elovitz MA, Burd I. IL-1 Receptor Blockade Prevents Fetal Cortical Brain Injury But Not Preterm Birth in a Mouse Model of Inflammation-Induced Preterm Birth and Perinatal Brain Injury. Am J Reprod Immunol (2014) 71:418–26. doi: 10.1111/aji.12216
35. Xu Z, Zhang X, Chang H, Kong Y, Ni Y, Liu R, et al. Rescue of Maternal Immune Activation-Induced Behavioral Abnormalities in Adult Mouse Offspring by Pathogen-Activated Maternal Treg Cells. Nat Neurosci (2021) 24:1–13. doi: 10.1038/s41593-021-00837-1
36. Smolders S, Notter T, Smolders SMT, Rigo J-M, Brône B. Controversies and Prospects About Microglia in Maternal Immune Activation Models for Neurodevelopmental Disorders. Brain Behav Immun (2018) 73:51–65. doi: 10.1016/j.bbi.2018.06.001
37. Lutz AB, Al-Nasiry S, Kramer BW, Mueller M. Understanding Host-Pathogen Interactions in Acute Chorioamnionitis Through the Use of Animal Models. Front Cell Infect Mi (2021) 11:709309. doi: 10.3389/fcimb.2021.709309
38. Jašarević E, Bale TL. Prenatal and Postnatal Contributions of the Maternal Microbiome on Offspring Programming. Front Neuroendocrin (2019) 55:100797. doi: 10.1016/j.yfrne.2019.100797
39. Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal Gut Bacteria Promote Neurodevelopmental Abnormalities in Mouse Offspring. Nature (2017) 549:528–32. doi: 10.1038/nature23910
40. Edey LF, O’Dea KP, Herbert BR, Hua R, Waddington SN, MacIntyre DA, et al. The Local and Systemic Immune Response to Intrauterine LPS in the Prepartum Mouse. Biol Reprod (2016) 95:125–5. doi: 10.1095/biolreprod.116.143289
41. Presicce P, Park C-W, Senthamaraikannan P, Bhattacharyya S, Jackson C, Kong F, et al. IL-1 Signaling Mediates Intrauterine Inflammation and Chorio-Decidua Neutrophil Recruitment and Activation. JCI Insight (2018) 3:e98306. doi: 10.1172/jci.insight.98306
42. Presicce P, Cappelletti M, Senthamaraikannan P, Ma F, Morselli M, Jackson CM, et al. TNF-Signaling Modulates Neutrophil-Mediated Immunity at the Feto-Maternal Interface During LPS-Induced Intrauterine Inflammation. Front Immunol (2020) 11:558. doi: 10.3389/fimmu.2020.00558
43. Gomez-Lopez N, Romero R, Xu Y, Miller D, Leng Y, Panaitescu B, et al. The Immunophenotype of Amniotic Fluid Leukocytes in Normal and Complicated Pregnancies. Am J Reprod Immunol (2018) 79:e12827. doi: 10.1111/aji.12827
44. Brown AG, Maubert ME, Anton L, Heiser LM, Elovitz MA. The Tracking of Lipopolysaccharide Through the Feto-Maternal Compartment and the Involvement of Maternal TLR4 in Inflammation-Induced Fetal Brain Injury. Am J Reprod Immunol (2019) 82:e13189. doi: 10.1111/aji.13189
45. Meltzer A, de Water JV. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacol (2016) 42:284–98. doi: 10.1038/npp.2016.158
46. Ege MJ, Herzum I, Büchele G, Krauss-Etschmann S, Lauener RP, Roponen M, et al. Prenatal Exposure to a Farm Environment Modifies Atopic Sensitization at Birth. J Allergy Clin Immun (2008) 122:407–412.e4. doi: 10.1016/j.jaci.2008.06.011
47. Mincham KT, Scott NM, Lauzon-Joset J-F, Leffler J, Larcombe AN, Stumbles PA, et al. Transplacental Immune Modulation With a Bacterial-Derived Agent Protects Against Allergic Airway Inflammation. J Clin Invest (2018) 128:4856–69. doi: 10.1172/jci122631
48. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The Gut Microbiota Influences Blood-Brain Barrier Permeability in Mice. Sci Transl Med (2014) 6:263ra158–263ra158. doi: 10.1126/scitranslmed.3009759
49. Zhang X, Rocha-Ferreira E, Li T, Vontell R, Jabin D, Hua S, et al. γδt Cells But Not αβt Cells Contribute to Sepsis-Induced White Matter Injury and Motor Abnormalities in Mice. J Neuroinflamm (2017) 14:255. doi: 10.1186/s12974-017-1029-9
50. Malik S, Want MY, Awasthi A. The Emerging Roles of Gamma-Delta T Cells in Tissue Inflammation in Experimental Autoimmune Encephalomyelitis. Front Immunol (2016) 7:14. doi: 10.3389/fimmu.2016.00014
51. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal Microbiota Affects Ischemic Stroke Outcome by Regulating Intestinal γδ T Cells. Nat Med (2016) 22:516–23. doi: 10.1038/nm.4068
52. Ribot JC, Lopes N, Silva-Santos B. γδ T Cells in Tissue Physiology and Surveillance. Nat Rev Immunol (2021) 21:221–32. doi: 10.1038/s41577-020-00452-4
53. Ribeiro M, Brigas HC, Temido-Ferreira M, Pousinha PA, Regen T, Santa C, et al. Meningeal γδ T Cell–Derived IL-17 Controls Synaptic Plasticity and Short-Term Memory. Sci Immunol (2019) 4:eaay5199. doi: 10.1126/sciimmunol.aay5199
54. Kinder JM, Stelzer IA, Arck PC, Way SS. Immunological Implications of Pregnancy-Induced Microchimerism. Nat Rev Immunol (2017) 17:483–94. doi: 10.1038/nri.2017.38
55. Wanke-Jellinek L, Keegan JW, Dolan JW, Lederer JA. Characterization of Lung Infection–Induced Tcrγδ T Cell Phenotypes by CyTOF Mass Cytometry. J Leukocyte Biol (2016) 99:483–93. doi: 10.1189/jlb.4a0315-115rr
56. Lebedev M, Faburay B, Richt JA, Young A. Myeloid-Like γδ T Cell Subset in the Immune Response to an Experimental Rift Valley Fever Vaccine in Sheep. Vet Immunol Immunop (2021) 233:110184. doi: 10.1016/j.vetimm.2021.110184
57. Papageorgiou IE, Lewen A, Galow LV, Cesetti T, Scheffel J, Regen T, et al. TLR4-Activated Microglia Require IFN-γ to Induce Severe Neuronal Dysfunction and Death in Situ. Proc Natl Acad Sci (2016) 113:212–7. doi: 10.1073/pnas.1513853113
58. Ta T-T, Dikmen HO, Schilling S, Chausse B, Lewen A, Hollnagel J-O, et al. Priming of Microglia With IFN-γ Slows Neuronal Gamma Oscillations in Situ. Proc Natl Acad Sci (2019) 116:201813562. doi: 10.1073/pnas.1813562116
59. Matcovitch-Natan O, Winter DR, Giladi A, Aguilar SV, Spinrad A, Sarrazin S, et al. Microglia Development Follows a Stepwise Program to Regulate Brain Homeostasis. Science (2016) 353:aad8670–aad8670. doi: 10.1126/science.aad8670
60. Cao M, Cortes M, Moore CS, Leong SY, Durosier LD, Burns P, et al. Fetal Microglial Phenotype In Vitro Carries Memory of Prior In Vivo Exposure to Inflammation. Front Cell Neurosci (2015) 9:294. doi: 10.3389/fncel.2015.00294
61. John CC, Black MM, Nelson CA. Neurodevelopment: The Impact of Nutrition and Inflammation During Early to Middle Childhood in Low-Resource Settings. Pediatrics (2017) 139:S59–71. doi: 10.1542/peds.2016-2828h
62. Wixey JA, Sukumar KR, Pretorius R, Lee KM, Colditz PB, Bjorkman ST, et al. Ibuprofen Treatment Reduces the Neuroinflammatory Response and Associated Neuronal and White Matter Impairment in the Growth Restricted Newborn. Front Physiol (2019) 10:541. doi: 10.3389/fphys.2019.00541
63. Yellowhair TR, Newville JC, Noor S, Maxwell JR, Milligan ED, Robinson S, et al. CXCR2 Blockade Mitigates Neural Cell Injury Following Preclinical Chorioamnionitis. Front Physiol (2019) 10:324. doi: 10.3389/fphys.2019.00324
64. Park J-E, Jardine L, Gottgens B, Teichmann SA, Haniffa M. Prenatal Development of Human Immunity. Science (2020) 368:600–3. doi: 10.1126/science.aaz9330
65. Khairallah C, Chu TH, Sheridan BS. Tissue Adaptations of Memory and Tissue-Resident Gamma Delta T Cells. Front Immunol (2018) 9:2636. doi: 10.3389/fimmu.2018.02636
66. Albertsson A-M, Zhang X, Vontell R, Bi D, Bronson RT, Supramaniam V, et al. γδ T Cells Contribute to Injury in the Developing Brain. Am J Pathol (2017) 188:757–67. doi: 10.1016/j.ajpath.2017.11.012
67. Mathern GW, Albayrak N. The Emerging Role of Gamma-Delta (γδ) T Cells in Neurological Diseases and Rasmussen Encephalitis. Int J Vaccines Vaccination (2016) 3:00068. doi: 10.15406/ijvv.2016.03.00068
Keywords: maternal-fetal interface, fetal brain injury, tissue-specific immunity, intrauterine inflammation, neuroimmune activation, γ/δ T cell, IFNγ
Citation: Lewis EL, Tulina N, Anton L, Brown AG, Porrett PM and Elovitz MA (2021) IFNγ-Producing γ/δ T Cells Accumulate in the Fetal Brain Following Intrauterine Inflammation. Front. Immunol. 12:741518. doi: 10.3389/fimmu.2021.741518
Received: 14 July 2021; Accepted: 01 September 2021;
Published: 04 October 2021.
Edited by:
Guillaume Dorothee, U938 Centre de Recherche Saint Antoine (CRSA) (INSERM), FranceReviewed by:
Antoine Louveau, Cleveland Clinic, United StatesCopyright © 2021 Lewis, Tulina, Anton, Brown, Porrett and Elovitz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michal A. Elovitz, ZWxvdml0ekBwZW5ubWVkaWNpbmUudXBlbm4uZWR1; Emma L. Lewis, ZW1tYS5sZXdpc0BwZW5ubWVkaWNpbmUudXBlbm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.