 Jiaqi Huang1,2†
Jiaqi Huang1,2† Jie Zhang2†
Jie Zhang2† Zhengyang Guo2
Zhengyang Guo2 Chen Li1,2Zhen Tan3Junjie Wang1
Chen Li1,2Zhen Tan3Junjie Wang1 Jianling Yang2*
Jianling Yang2* Lixiang Xue1,2*
Lixiang Xue1,2*- 1Department of Radiation Oncology, Peking University Third Hospital, Beijing, China
- 2Center of Basic Medical Research, Institute of Medical Innovation and Research, Peking University Third Hospital, Beijing, China
- 3Department of Bone and Joint Surgery, Peking University Shenzhen Hospital, Shenzhen, China
Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of polycomb repressive complex 2 (PRC2), which regulates downstream gene expression by trimethylation of lysine 27 in histone H3 (H3K27me3). EZH2 mutations or overexpressions are associated with many types of cancer. As inhibition of EZH2 activity could upregulate the expression of tumor suppressor genes, EZH2 has recently become an interesting therapeutic target for cancer therapy. Moreover, accumulating evidence has shown that EZH2 may contribute to the regulation of immune cells, especially T cells. EZH2 regulates T cell development, differentiation, and function, suggesting that EZH2 also regulates immune homeostasis in addition to tumor suppressor genes. Moreover, EZH2 can regulate T cell fate by targeting non-T cell factors such as metabolism, cytokines, and myeloid-derived suppressor cells. The role of EZH2 in this process has not been fully addressed. This review discusses up-to-date research on EZH2-mediated regulation of immunological function and the progress of immunological therapeutic strategies based on this regulation.
Introduction
Gene expression is epigenetically modified by changes in chromatin, which affects the accessibility of certain genomic loci to transcription enzymes and subsequent transcription. Histones can be methylated at lysine residues by histone methyltransferases that influence the chromatin condensation and thus regulate gene expression. Enhancer of zeste homolog 2 (EZH2) is the catalytic subunit of polycomb repressive complex 2 (PRC2), which can trimethylate H3K27 of the target gene, thereby silencing transcription and regulating cell development, oncogenesis, and stem cell plasticity (1–3). Emerging evidence has suggested that EZH2 also plays some unconventional roles in cancers, such as functioning as a non-histone protein transmethylase or a PRC2-independent transcriptional factor (4–6).
The functions of EZH2 in cancer cells have been extensively studied (7–9). Abnormal EZH2 expression contributes to increased aggressiveness and proliferation in various solid tumors (10–15). Furthermore, EZH2 may be involved in preserving the stemness of cancer stem cells (7–9). Even in non-cancerous cells, ectopic EZH2 expression promotes cell proliferation as well as neoplastic transformation (14, 16). However, missense mutations in EZH2 can also exert a cancer-promoting effect in hematological tumors (17, 18). EZH2 can also affect tumor progression by regulating metabolism (19). In general, EZH2 promotes stemness and proliferation by silencing the expression of transcription factors that determine cell fate.
In addition to focusing on the role and application of EZH2 in cancer cells, studies have also paid attention to the role of EZH2 in the development and function of the immune system. Considering its critical function in epigenetic regulation of target gene transcription, accumulating experimental evidence has shown that EZH2 contributes to the development, proliferation, and function of T cells and may act as an immunomodulatory factor. Abnormal EZH2 expression may be implicated in various immune-related diseases, including autoimmune diseases, infection, graft-versus-host disease, and cancer (20–22). In addition to its typical PRC2-dependent histone methyltransferase function, limited evidence has shown that EZH2 may also regulate T cell fate via unconventional manners. Further studies on the regulatory function of EZH2 in immunity are beneficial for identifying therapeutic strategies. In this review, we discuss the current knowledge of the functions of EZH2 in T cell development, differentiation, and function and its potential value in cancer treatment and immunotherapy.
EZH2 in T Cell Development
EZH2 is important in regulating T cell development and differentiation (Figure 1). In hematopoietic stem cells (HSCs), EZH2 plays an essential role in maintaining HSC stemness by negatively regulating cell cycle inhibitors such as Cdkn2 (23, 24). Deletion of EZH2 causes serious myelosuppression and anemia in fetal mice (24). However, hematopoietic phenotypes may not be significantly impacted by EZH2 deficiency because of the compensatory role of EZH1 in adult hematopoiesis (25). Double inactivation of EZH1 and EZH2 impairs the function of bone marrow HSCs (26). Deficiencies in other components of PRC2, such as SUZ12 or EED, can cause similar results (25, 27).
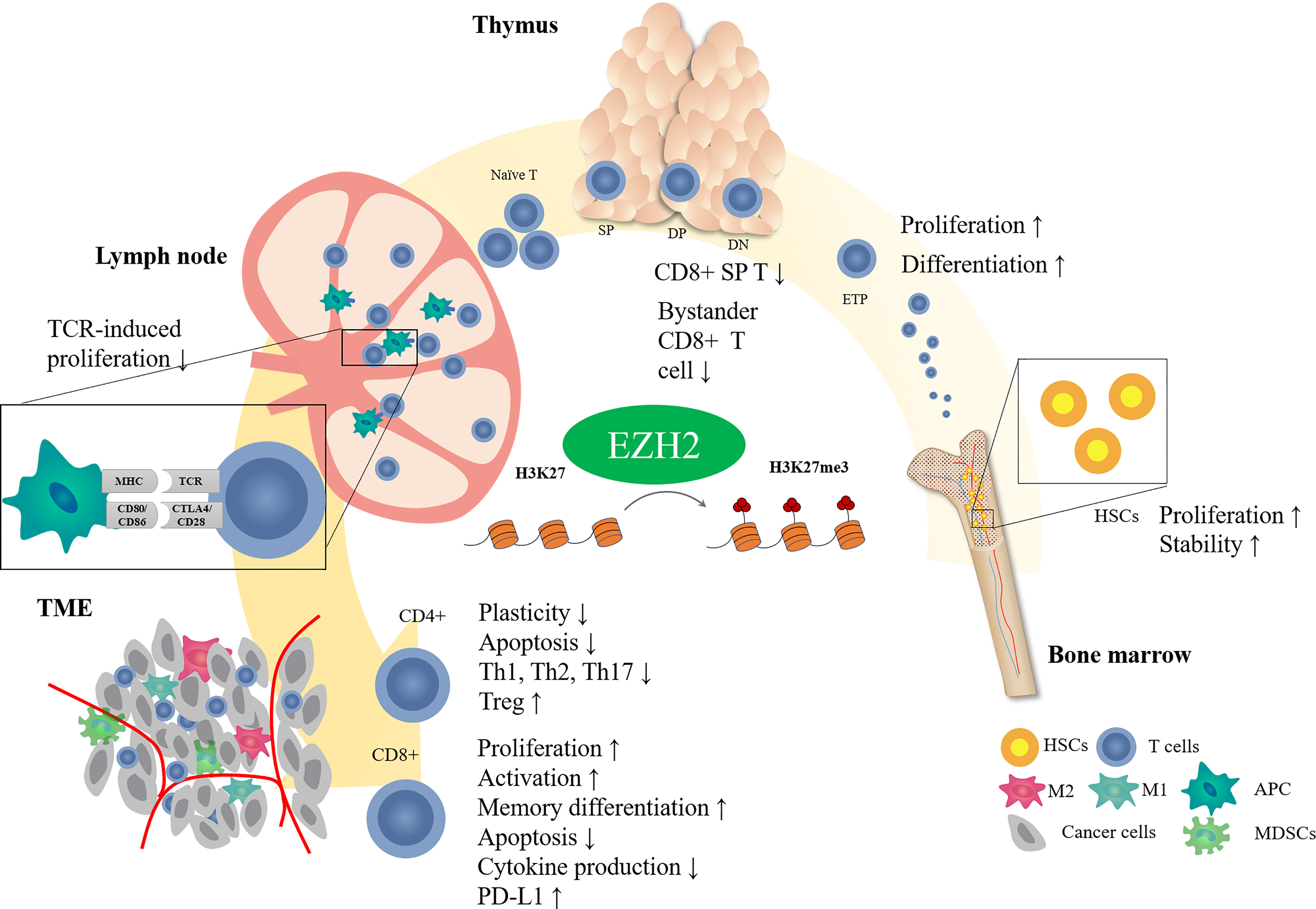
Figure 1 Effect of EZH2 on T cell development. T cells receive antigenic stimulation in lymph nodes after maturation in the thymus and are recruited into the tumor region to exert antitumor effects. EZH2 modifies different stages of T cell development through epigenetic regulatory mechanisms.
T cells in the thymus stage are also closely regulated by EZH2. EZH2 deficiency is associated with reduced H3K27me3 levels in thymic progenitors and upregulation of expression of the cell cycle inhibitor Cdkn2a. Inactivation of EZH2 in bone marrow cells causes increased arrest of thymocytes in the double negative (DN) phase (28), and the combined loss of EZH2 and Cdkn2a can partially rescue the cell number (28, 29). These results indicate that EZH2-induced methylation regulates thymocyte development by inhibiting cell cycle inhibitors.
Even though EZH2 inactivation impaired thymocyte development, it did not change the production of mature peripheral T cells (30). It suggested that there may be other factors, such as cytokines or transcription factors, restricting the EZH2-mediated regulation of thymocyte development except EZH2. In consistency, T-cell-specific knockout of both EZH1 and EZH2 did not affect the expression of characteristic T cell differentiation or activation markers but reduced T cell numbers slightly in secondary lymphoid organs (30). Interestingly, EZH2-deficient CD8+ thymocyte tended to express surface markers (CD24lo, CD44hi, and CD122hi) of memory-like T cells, also known as bystander T cells (31). However, Vasanthakumar et al. reported that neither T-cell-specific inactivation of EZH2, SUZ12, or EED does not alter the αβ or γδ T cell development in thyme (32). Based on the current understanding of EZH2 on T cell development, more studies are needed to uncover the effect of EZH2 on T cell development in thymus.
EZH2 in the Antitumor Immune Effects of T Cells
EZH2 in T Cell Activation
TCR recognition is essential for initiating an antitumor immune response. EZH2 plays an unconventional role in TCR stimulation. EZH2 forms a PRC2-like complex and is present in the cytoplasm of T cells. Cytoplasmic EZH2 is localized proximal to TCRβ and contributes to TCR-driven actin polymerization, which promotes interactions between T cells and antigen-presenting cells (30, 33). Immunoprecipitation demonstrated an interaction between EZH2 and CD3ϵ, which may form a complex with the TCR-signaling protein Vav1 and the Vav1-associated protein Nck (30). EZH2-deficient T cells maintained their proliferative ability in response to TCR stimulation, while EZH1/2 double-deficient T cells showed reduced IL-2 and IL-2RA expression and failed to proliferate in response to TCR triggering in vitro, which could not be rescued by exogenous IL-2 (30). This phenomenon may result from diminished TCR-mediated activation of Erk, which plays a key role in MAPK/Erk pathway-mediated regulation of IL-2 and IL-2RA expression and activation (34). Notably, EZH1/2 double deficiency in T cells showed no impact on Erk phosphorylation or T cell proliferation when stimulated by TCR-independent T cell activation with PMA in combination with ionomycin (30), suggesting the selective role of EZH1/2 in TCR-driven T cell activation. Although they exhibit impaired proliferation, EZH2-deficient CD4+ T cells retain their ability to initiate TCR-induced activation and proliferation in vitro (35, 36). Therefore, these results indicate that although EZH2 interacts with CD3, it may not regulate antigen recognition; rather, it may regulate TCR-induced proliferation together with EZH1. Additional studies are needed to confirm the effect of EZH2 on T cell activation in the tumor microenvironment.
EZH2 in CD8+ T Cells
CD8+ T cells play a key role in the defense against tumor cells. Antigen-specific CD8+ T cell responses display a remarkably reproducible pattern of activation, proliferation, differentiation, contraction, and memory cell formation (37). The characteristic epigenetic features of different CD8+ T cell stages have been validated (38). In line with its function in other immune cells, EZH2 regulates the activation, proliferation, and differentiation of CD8+ T cells via histone methylation modification (Figure 2).
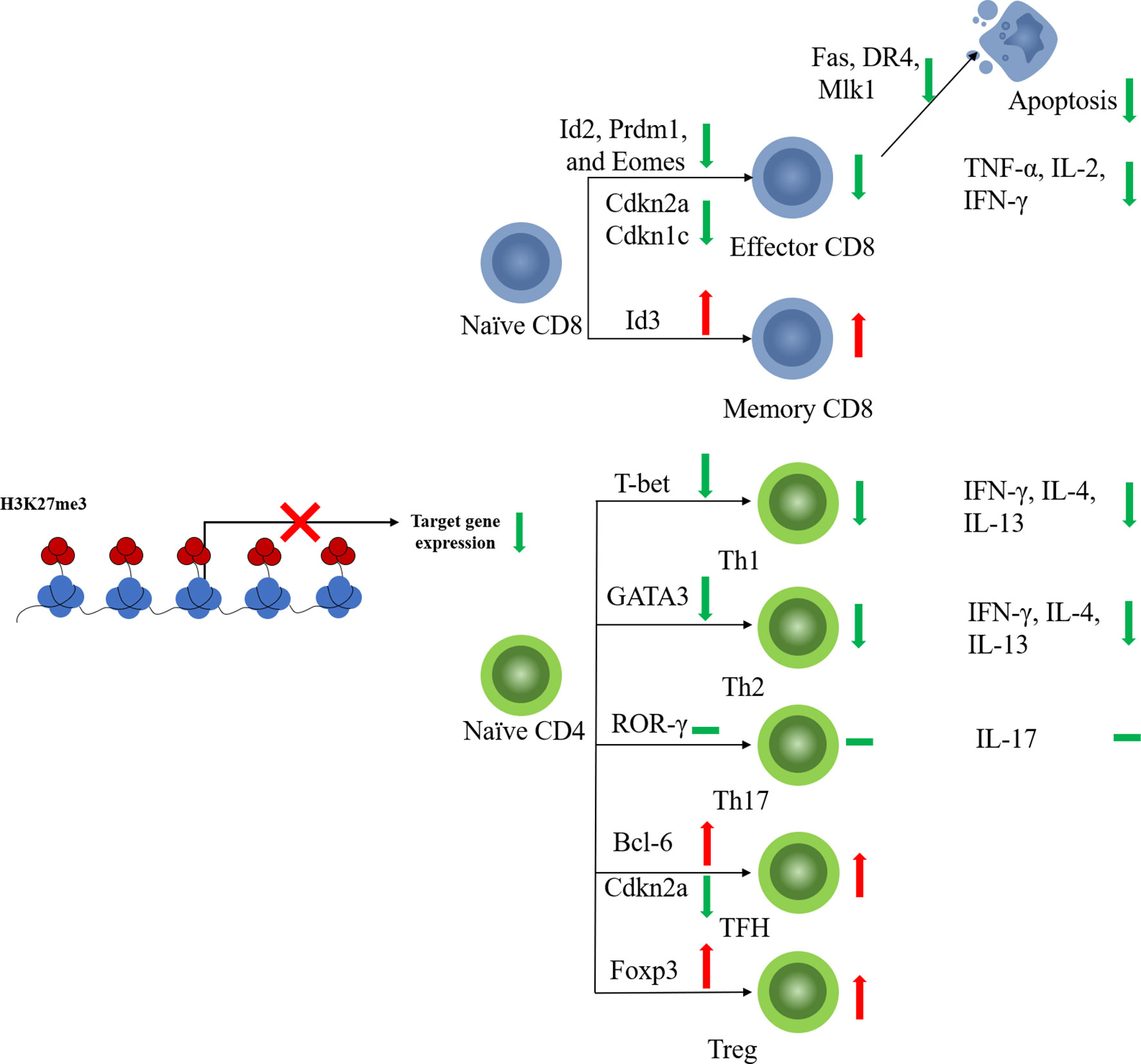
Figure 2 EZH2 function on T cell fates. EZH2 affects differentiation of CD4+ or CD8+ naïve T cells toward effector subtypes by regulating the expression of key transcription factors. EZH2 can also limit the apoptosis of mature effector T cells.
Naïve CD8+ T cells with EZH2 deficiency from thymocyte development (EZH2fl/flCD4Cre+) displayed impaired proliferation and increased apoptosis in response to antigen stimulation (39). EZH2 promotes CD8+ T cell proliferation by downregulating the cyclin-dependent kinase inhibitor. Deletion of EZH2 resulted in increased expression of Cdkn2a and Cdkn1c in activated naïve CD8+ T cells along with reduced levels of H3K27me3 at the loci of these two genes (39). Naïve CD8+T cells are activated and initiate proliferation after T cell receptor (TCR)-antigen stimulation. Consistent with the pro-proliferative role of EZH2, significantly elevated EZH2 protein levels were observed in activated T cells (40).
Besides, EZH2 also regulates naïve CD8+ T cell differentiation. EZH2-deficient CD8+ naïve T cells showed impaired memory cell formation after TCR stimulation (41). Conditional EZH2 deficiency in T cells causes upregulation of expression of pro-effector genes such as Id2, Prdm1, and Eomes upon TCR stimulation (41). Interestingly, Id3, a pro-memory transcription factor, was downregulated with EZH2 deficiency, suggesting that EZH2 activates rather than represses Id3 (41). It has been reported that transcription factors regulating T cell fate (e.g., Tbx21, Prdm1, Eomes, and Irf4) are modified by both repressive H3K27me3 and permissive H3K4me3 histone methylation (42, 43). Decreased H3K4me3 levels were observed at Id3 loci along with decreased H3K27me3 levels (41). EZH2 likely activates Id3 transcription in T cells by regulating the balance between H3K27me3 and H3K4me3, but the underlying mechanism remains unclear (41). These results suggest that EZH2 plays a pro-memory role in the selection process of T cell differentiation under different but shared congenerous epigenetic mechanisms. Moreover, the above function of EZH2 is also regulated by Akt-mediated EZH2 phosphorylation, which results in dysfunctional EZH2 and diminished T memory potential (41), suggesting that the regulatory function of EZH2 in CD8+ T cells is also affected by its posttranscriptional modification state.
CD8+ T cells kill malignant cells by secreting cytokines, such as TNF-α and IFN-γ. Increased evidence indicated that EZH2 may regulate cytokine production by CD8+T cells. EZH2-inactivated naïve CD8+ T cells showed increased production of IFN-γ, interleukin (IL-2), and GZMB after stimulation in vitro (39). Therefore, EZH2 appears to inhibit the production of cytokines, especially pro-inflammatory cytokines. However, other studies observed opposite results. Conditional EZH2 knockout in activated CD8+ T cells impaired TNF-α production but produced equivalent percentages of IFN-γ+ and IL-2+ cells compared to those of EZH2fl/fl cells 8 days after viral infection (41). In addition, both CD4+ and CD8+ T cells from the peripheral blood of healthy donors, which have high EZH2 expression levels, also produce multiple effector cytokines, suggesting that EZH2 plays pro-cytokine and polyfunctional roles in CD8+ T cells (44). Considering these contradictory results, the role of EZH2 in cytokine production by CD8+T cells need to be placed in each concrete cancer environment.
When under persistent antigen stimulation, effector CD8+ T cells can develop into terminally differentiated (TE) T cells, which are also known as senescent CD8+ T cells. EZH2 suppresses the expression of pro-memory genes in KLRG1HiIL7RLo TE effector CD8+ T cells via deposition of H3K27me3 at these genes (40). FoxO1 is likely a key regulator that induces the functional change in EZH2. Decreased FoxO1 expression and binding to pro-memory genes were detected in TE CD8+ T cells compared to those in memory T cells. FoxO1 deficiency led to the accumulation of H3K27me3 in the pro-memory genes Tcf7 and Bach2 (40), suggesting that FoxO1 may promote and maintain the multipotency of memory T cells by shielding pro-memory genes from H3K27me3 deposition. Deficiencies in either EZH2 or FoxO1 lead to decreased TE cell formation (40). It can be concluded that EZH2 plays variable roles in different stages of CD8+ T cell development: in the early stage after TCR stimulation, EZH2 exerts a memory-promoting effect on CD8+ T cells, whereas in the late stage, EZH2 inhibits pro-memory gene expression and promotes TE cell formation.
The memory T cells are critical for T cell persistence and efficient tumor killing especially for cancer immunotherapy. Differentiated effector CD8+ cytotoxic T cells (CTLs) have a short life span and a tendency for apoptosis (45). EZH2 may play an important role in the regulation of apoptosis-related genes, such as Fas, DR4, and Mlk1, to avoid excessive attenuation of activated T cells (46). EZH2-deficient activated CD8+ T cells exhibit increased expression of apoptosis-related genes as well as elevated rates of apoptosis (39). As for memory CD8+ T cells, although EZH2 may not be required for memory CD8+ T cell formation, increased apoptosis-related gene expression was also observed in sorted EZH2-deficient central memory CD8+ T cells (41), which suggests that EZH2 is essential for memory T-cell persistence.
The studies mentioned above indicated the role of EZH2 in regulating CD8+ T cell function. These results should be considered for the therapeutic design of EZH2 inhibitors, especially in combination with immunotherapy.
EZH2 Regulates MDSCs Generation and Recruitment
After antigen stimulation, naïve CD4+T cells become non-polarizing Th0 cells and differentiate into distinct subsets of CD4+ T cells, such as Th1, Th2, Th17, regulatory T (Treg) cells, and follicular Th (TFH) cells. The lineage selection of Th0 cells is programmed by transcription factors and cytokines (47). The plasticity of CD4+ T cell differentiation is tightly regulated by the coordination of DNA methylation and histone modifications (48, 49), and the epigenetic mechanisms through which EZH2 regulates CD4+ T cell fate are highly complex (Figure 2). First, EZH2 inactivation leads to slight enhancement of CD4+ T cell polarization under short-term culture conditions. With T cell-specific EZH2 knockout, CD4+ T cells showed impaired H3K27me3 levels as well as increased expression of characteristic transcription factors, including T-bet and GATA-3, leading to improved cytokine production and enhanced differentiation of naïve CD4+ T cells into effector Th1 and Th2 cells (35). Consistent with this study, T-cell-specific knockout of EZH2 led to enhanced Th1 and Th2 differentiation but reduced Treg cell differentiation in vitro under the respective differentiation conditions for 3 days (50). Enhanced secretion of Th1 and Th2 cytokines, such as IFN-γ, IL-4, and IL-13, was also detected in EZH2-deficient Th1 and Th2 cells (36). Interestingly, EZH2 deficiency enhanced the plasticity of Th0 cells, enabling the transformation of differentiated Th1 cells into Th2 and vice versa (36). Second, in long-term culture conditions, this enhancement of effector helper T cells seems to be a kind of “false prosperity”. When stimulation was extended to 6 days, the proportion of EZH2-CKO CD4+ effector (Th0, Th1, Th2, Th17) T cells was decreased compared to that in the wild-type (WT) control (35). Similarly, EZH2-deficient Th1 cells showed an impaired ability to respond to IL-2 after long-term culture for 10 days (50). Consistent with the reduced cell number, upregulated levels of proteins of both intrinsic (tBid and Bax/Bak) and extrinsic (FasL, TNFR1, and TRAIL) apoptotic pathways were present in EZH2-CKO Th0 cells, but not in EZH2fl/fl cells. Inhibition of the apoptotic pathway provided significant but incomplete protection of the survival of EZH2-CKO Th0 cells compared with that of WT cells. These results suggest that EZH2 inactivation of T cells in long-term culture can promote cell death (35). Third, under the condition of chronic infection or tumor, EZH2 deficiency may not be conducive to a normal immune response. Decreases in the percentage and total number of CD4+ T cells were detected in both the spleen and peritoneum in an intraperitoneal parasite infection model, together with a reduced percentage of IFN-γ-producing CD4+ T cells and a poor survival (50). Similarly, mixed-bone marrow chimeric mice harboring WT and EZH2fl/fl/CD4-Cre cells showed a significantly reduced proportion of IFN-γ+ Th1 cells compared to that of the control group five days after infection with Listeria monocytogenes (35). A further reduction in the number of EZH2-null IFN-γ-producing CD4+ and CD8+ T cells was also observed in chimeric mice (35).
In addition to regulating CD4+ helper T cells, EZH2 can also mediate helper T cells recruitment by affecting chemokine expression. Both genetic and pharmacological inhibition of EZH2 in vitro enhanced IFN-γ-mediated induction of Th1-type chemokines, such as CXCL9 and CXCL10, in human ovarian cancer cells (51). Pharmacological EZH2 inhibition in vivo also led to increased levels of tumor-producing pro-Th1 chemokines and enhanced recruitment of effector T cells, resulting in reduced tumor volume and prolonged survival time in human ovarian cancers (51, 52). Similarly, in B cell lymphoma cell lines, pharmacological inhibition of EZH2 by EPZ6438 enhanced the expression of CCL17, a chemokine which attracts CCR4+ T cells and facilitates the T-cell response (53).
Few results have been reported regarding the regulatory effects of EZH2 on other helper T cell subsets, such as Th17 or TFH cells. Enhanced Th17 differentiation has also been observed in EZH2-deficient T cells (50). However, only a very slight increase in IL-17 production and RORγ expression was detected in EZH2-CKO CD4+ T cells cultured under Th17 cell-inducing conditions, with reduced EZH2 and H3K27me3 levels at Il17a and Rorc loci (36). This suggests that the epigenetic mechanisms of EZH2 may be insufficient to regulate Th17 differentiation; other means, such as cytokine production, are also involved in regulating Th17 cell differentiation. TFH cells are another specialized subgroup of CD4+ T cells that prompt B cells to undergo somatic hypermutation, affinity maturation, and transition into plasma cells and memory B cells (54). The promoter of Bcl6, the characteristic transcription factor of TFH cells (55), is associated with H3K27ac rather than H3K27me3, suggesting that EZH2 may not play the key function in this case. However, EZH2 is recruited by Tcf1 to directly activate Bcl6 transcription, and the function of Bcl6 requires EZH2 phosphorylation at Ser21, suggesting the unconventional role of EZH2 in regulating TFH cell fate (56). Furthermore, EZH2 represses Cdkn2a expression through H3K27me3, which regulates TFH cell proliferation and apoptosis (56). Therefore, EZH2 may regulate TFH differentiation, proliferation, and function in both PRC2-dependent and -independent manners.
Taken together, EZH2 inactivation enhanced the recruitment of T cells into the tumor region by upregulating chemokine expression and promoted naïve CD4+ T cells to differentiate into effector Th cells in the short term. However, EZH2 inactivation also impaired the long-term proliferative potential of T cells by activating multiple death pathways, including but not limited to apoptosis, and eventually impaired the antitumor immune response. Therefore, even though EZH2 impairs antitumor immune response by suppressing Th cell differentiation, inhibiting its plasticity and downregulating tumorous chemokine production, it also promotes the stability of effector T cells under tumor conditions.
EZH2 in Treg Cells
Treg cells are central inhibitory regulators in antitumor activity. Generally, Treg cells refer to a subgroup of CD4+ T cells that express the transcription factor Foxp3, which is a typical functional transcription factor of Treg cells. Foxp3 expression can be inhibited by STAT1 and STAT6, which are downstream activators of IFN-γ and IL-4, respectively (57). EZH2-deficient naïve T cells showed an impaired ability to differentiate into induced Treg (iTreg) cells under Treg-inducing conditions in vitro, which could be rescued by combining blockades of IFN-γ and IL-4 (50). Considering that T cell-specific inactivation of EZH2 enhances the production of proinflammatory cytokines, such as IFN-γ, in non-polarizing Th0 cells (35, 50), EZH2 may indirectly affect iTreg differentiation through aberrant production of cytokines, such as IFN-γ and IL-4, rather than through direct epigenetic regulation. However, whether inactivation of EZH2 increases the sensitivity of non-polarizing Th0 cells to cytokines remains unclear and warrants confirmation by further experiments.
In addition to regulating Treg differentiation, EZH2 is required for Treg activation, as evidenced by a strongly elevated expression of EZH2 in CD44hiCD62Llo-activated Treg cells compared to that in naïve or resting Treg cells (58). In tumor conditions, tumor-infiltrating lymphocytes have a higher proportion of total and activated Tregs than normal tissues (59). EZH2 expression is also upregulated in tumor-infiltrating Treg cells compared to that in effector T cells or Treg cells in lymph nodes (60). Increased H3K27me3 levels have been detected in activated Treg cells (61). Consistent with this finding, co-immunoprecipitation experiments showed that EZH2 forms complexes with Foxp3 in CD62Llo-activated Treg cells but not in resting Treg cells (58), suggesting that EZH2 plays a role in CD44hiCD62Llo-activated Treg cells by binding to Foxp3. Furthermore, similar differentially expressed genes were detected between EZH2- and Foxp3-deficient CD62Llo-activated Treg cells, but not in resting Treg cells (61), suggesting that the regulatory effect of activated Treg cells is mediated by the cooperation of EZH2 and Foxp3. These results also suggest that EZH2 may cooperate with Foxp3 under the non-classical regulatory mechanism.
Considering the important role of Treg cells in maintaining immune homeostasis, the loss of EZH2 may cause dysregulation of immune homeostasis (61). Foxp3-specific EZH2 deficiency increased T cell activation and infiltration in lymphoid organs, but it reduced the frequency in non-lymphoid tissues where effector Treg cells are resident (62). Conditional knockout of EZH2 in Treg cells causes impaired immune tolerance, including various symptoms of autoimmunity (such as reduced weight, hair loss, and scaly tails), extensive infiltration of mononuclear immune cells in non-lymphoid tissues (such as the lungs, liver, pancreas, and kidneys), and a reduced lifespan in mutant mice (61). Furthermore, Foxp3-specific EZH2 inactivation led to enhanced production of pro-inflammatory cytokines as well as reduced tumor size in murine bladder cancer (63). These results further confirmed that EZH2 is responsible for Treg cell activation, which is critical for the immunosuppressive function that maintains immune homeostasis.
EZH2 Compromises Antitumor Immune Effects of T Cells
In addition to its direct regulatory effect on T cells, EZH2 may also induce immune escape to some extent by affecting other factors in the TME, such as immune checkpoints, MDSCs, and cytokines.
EZH2 Regulates Immune Checkpoint Expression
Tumor cells in the TME may negatively influence the immune system via EZH2, thus impairing immune cell function and achieving immune escape. For example, tumor cells can express immune checkpoint ligands and bind to their receptors on T cells, thus suppressing antitumor immunity. Therefore, immune checkpoints may be potential therapeutic targets for the epigenetic regulation of EZH2. PD-1 and its ligand, PD-L1 (CD274), are the most well-known immune checkpoint targets (64). The PD-L1 expression in tumor-induced exhausted CD4+ T cells was found to be controlled by the interplay of chromatin remodeling SWI/SNF and PRC2 complexes, suggesting that EZH2 is involved in regulating PD-L1 expression in exhausted CD4+ T cells (65). However, contradictory results have been found regarding the regulatory effect of tumor EZH2 on PD-L1 expression (66, 67). A negative correlation between EZH2 and PD-L1 expression was observed in hepatocellular carcinoma and prostate cancer, which may result from the upregulation of H3K27me3 levels in the promoters of CD274 and IRF1 (66, 68). Therefore, inhibition of EZH2 function may enhance PD-L1 expression in tumor cells. Consistent with this hypothesis, both genetic and chemical inhibition of EZH2 in prostate cancer organoids repressed the H3K27me3 levels and upregulated the endogenous dsRNA levels. Accumulated endogenous dsRNA can activate the interferon-response pathway and upregulate tumor expression of PD-L1 in a STING-dependent manner (68). However, in another lung adenocarcinoma model, EZH2 expression was positively correlated with PD-L1 expression (67), but the mechanisms are uncertain. Although the exact regulatory effect of EZH2 on immune checkpoints remains unclear, EZH2 inhibitors in combination with immunotherapy seem to be a viable antitumor treatment strategy. Treatment with epigenetic modulators increases effector T cell infiltration, represses tumor progression, and promotes the therapeutic efficacy of PD-L1 checkpoint blockade in prostate or head and neck cancer (68, 69). Pharmacological inhibition of EZH2 also inhibits tumor growth and improves the efficacy of anti-CTLA-4 treatment in bladder cancer (63).
EZH2 Regulates MDSC Generation and Recruitment
MDSC is another cell population that exhibits an inhibitory regulator role in antitumor activity. Evidence has shown that MDSCs can be induced by EZH2-mediated epigenetic regulation. Upon induction of monocytic (M)-MDSCs in vitro, a gradual loss of EZH2 and H3K27me3 was observed along with a gradual expansion of the CD11b+Gr-1+ M-MDSC population (70, 71), suggesting that loss of EZH2 activity may be responsible for MDSC generation. Pharmacological inhibition of EZH2 enhanced MDSC generation in both tumor and non-tumor models. GSK343 and GSK126 both inhibit the histone methyltransferase activity of EZH2 without affecting its protein expression. In an inflammatory bowel disease model, GSK343 treatment caused the elevation of MDSCs in the colonic lamina propria and peripheral blood but did not alter the expression of MDSC-related chemokines, suggesting that EZH2 inhibition by GSK343 enhanced M-MDSC generation without affecting its chemotaxis (70). Similar results were observed in an MC38 mouse colon cancer tumor model. EZH2 inhibition by GSK126 was reported to promote M-MDSC formation, decrease T cell proliferation, and increase T cell apoptosis, leading to impaired antitumor immunity (71). Consistent with the results observed in the inflammatory bowel disease model, GSK126 treatment increased the frequencies and numbers of granulocyte/macrophage progenitors, which are also progenitors of MDSCs, within the hematopoietic progenitor cell (HPC) population, suggesting that GSK126-treated HPCs had an enhanced potential to differentiate into myeloid cells (71). Additionally, proliferation and apoptosis of MDSCs were unaffected by treatment with GSK126, suggesting that differentiated MDSCs are rarely regulated by EZH2, which is consistent with their low EZH2 and H3K27me3 levels (70, 71). Mechanistically, activation of the Jak-STAT and tumor necrosis factor (TNF) signaling pathways, which are known to regulate MDSC production (72, 73), was observed in GSK343-induced M-MDSCs in vitro, suggesting that GSK343 may enhance M-MDSC generation by activating Jak-STAT and TNF signaling (70). Additionally, EZH2 was reported to activate NF-κB as a PRC2-independent coactivator of transcriptional factors in ER-negative basal breast cancer (74). In hepatocellular carcinoma, cell cycle-related kinase (CCRK) enhanced IL-6 production via the abovementioned EHZ2/NF-κB signaling and induced increased accumulation of polymorphonuclear (PMN)-MDSCs (75), which suggests another mechanism by which EZH2 regulates MDSC generation. Although EZH2 inhibitors can enhance MDSC production via various pathways, not all EZH2 inhibition can induce the enhanced generation of MDSCs. Another EZH2 inhibitor, EPZ0011989, did not change the frequency of either M-MDSCs or PMN-MDSCs in mouse prostate tumor tissue (68). Therefore, further evidence is needed to exclude the influence of tissue origin specificity or different inhibitors.
Other Immune Subgroups
The regulatory effect of EZH2 on other immune cell subsets can also affect T cell function and antitumor immunity. Inhibition of EZH2 upregulated the expression of NKG2D, an activating receptor required for natural killer (NK) cells (76). Alternatively, EZH2 inhibition was also reported to induce the expression of NKG2D ligands in hepatocellular carcinoma cells (76). Taken together, EZH2 inhibition may promote NK-induced cytotoxicity by affecting both the tumor and NK cells. Tumor-associated macrophages are also an important part of the TME. EZH2 inhibition in glioblastoma induced an increase in M1 markers (iNOS and TNF-α) and reduction in M2 markers (tumor growth factor-β1, -β2, and stabilin-1) in both murine microglia and human peripheral blood mononuclear cell-derived macrophages (77), which is indicative of the pro-M2 function of EZH2. Furthermore, expanded M2 macrophages express increased levels of anti-inflammatory cytokines and play an immunosuppressive role in the TME (78).
Environmental Factors in the Regulation of T Cells by EZH2
Tumor development relies on the TME, such as hypoxia, nutritional restrictions, and metabolic reprogramming. These environmental factors are shaped jointly by multiple types of cells, including tumor cells, tumor vessels, immune cells, and stromal cells, and change dynamically with tumor progression. EZH2 expression is influenced by the TME, and conversely, it is involved in shaping the TME (Figure 3).
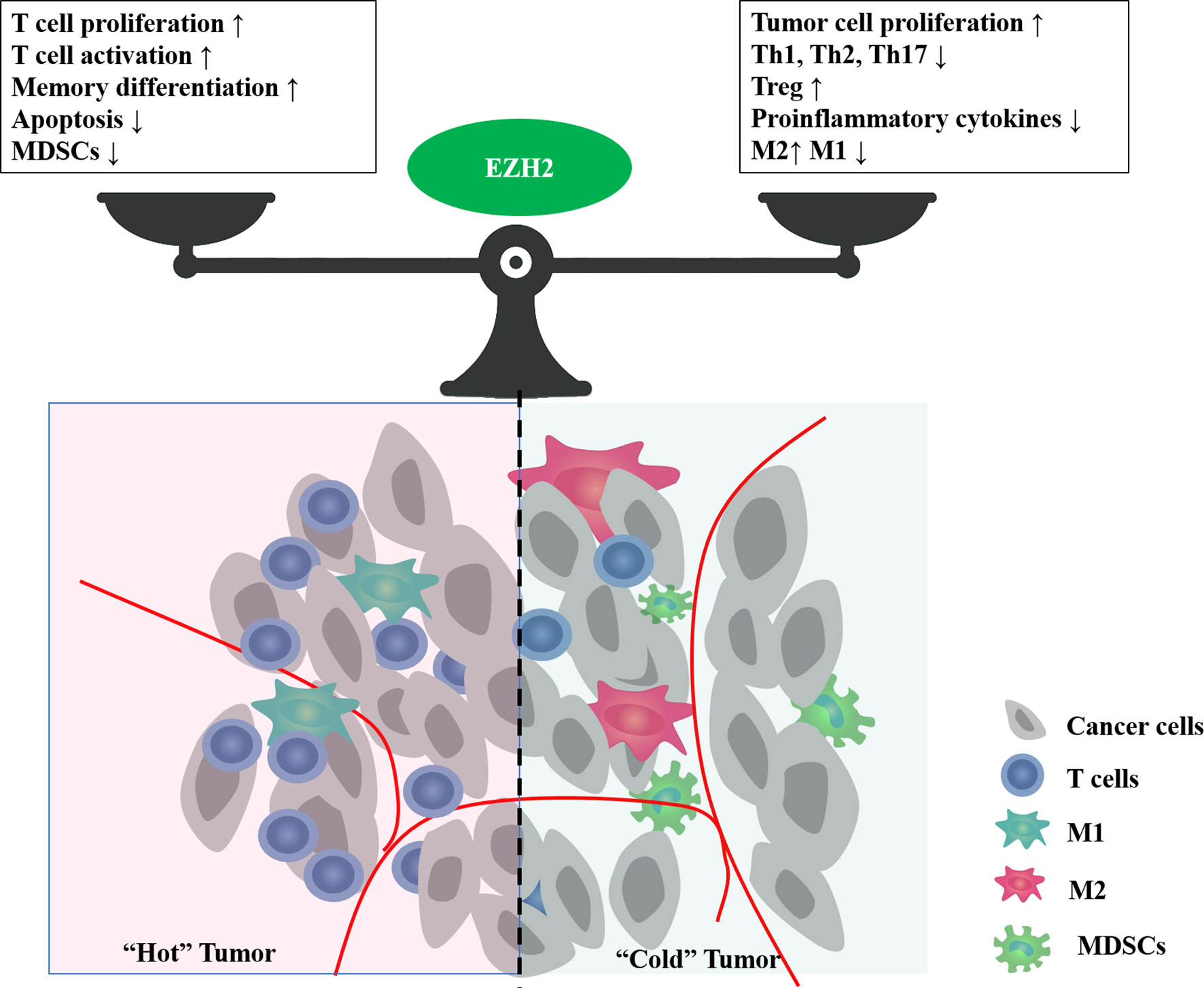
Figure 3 The bidirectional effect of EZH2 on tumor immune microenvironment. EZH2 can regulate multiple cellular components in the tumor microenvironment, such as cancer cells, T cells, macrophages, and myeloid-derived suppressor cells (MDSCs). Major phenotypes associated with EZH2 function in balancing the tumor immune microenvironment.
Hypoxia and Hypoxia-Inducible Factor (HIF)-1α
HIF-1α is a key downstream immunoregulator of EZH2. In cancer cells, under normoxia, EZH2 is recruited directly to the HIF-1α promoter and inhibits HIF-1α expression, whereas under hypoxia, this inhibitory effect is weakened, leading to unregulated HIF-1α expression (79). EZH2 knockdown further upregulates HIF-1α expression under hypoxic conditions (79). By contrast, studies have shown that HIF-1α can upregulate EZH2 expression in tumor models (80, 81). Activation of HIF-1α can recruit MDSCs to the primary tumor site by enhancing the expression of chemokine (C–C motif) ligand 26 in tumor cells (82), which can further inhibit antitumor T cell effector function. This may be one reason why EZH2 inhibition by GSK126 led to MDSC recruitment in the TME (71). The regulatory function of EZH2 on HIF-1α in immune cells has not been proven, but this effect, if present, may have a profound effect on the immune system. HIF-1α is a key regulator of T cell survival, proliferation, and effector function (83–85), which may be due to the hypoxic environment in the bone marrow and thymus. Therefore, upregulation of HIF-1α expression by EZH2 inhibition can protect T cells from hypoxia-induced apoptosis during early developmental stages. However, CD73, a HIF-1α-regulatory molecule that is mainly expressed on Treg cells or some tumor cells (86), regulates extracellular adenosine production. Adenosine plays an immunosuppressive role in the TME mainly through the adenosine 2A receptor on T cells (87–89). Thus, EZH2 inhibition in a hypoxic TME can further enhance extracellular adenosine production via the hypoxia/HIF-1α/adenosinergic pathway and impair antitumor immunity. Therefore, the bidirectional effect of HIF-1α at different stages of T cell development may be one reason for the complexity of the regulatory effect of EZH2 on T cells.
Metabolism
T cell fate is closely related to metabolism, which is highly regulated by epigenetics. Some EZH2-regulatory metabolic targets have been reported to have immunoregulatory effects. For instance, in lung cancer, EZH2 can suppress Lat1 and promote methionine uptake, which ultimately upregulate the synthesis of s-adenosine methionine (SAM), an important transporter of one carbon unit (90). SAM can induce T cell exhaustion in vitro, and interference with the synthesis of SAM via knockdown of MAT2A, a key SAM-producing enzyme, results in the inhibition of T-cell exhaustion in a mouse hepatocellular carcinoma model (91). Therefore, EZH2 inhibition can repress SAM synthesis and counteract its exhaustion-promoting effect. Additionally, SIRT6, a histone/protein deacetylase, regulates critical signaling pathways related to multiple metabolic processes, including PI3K/Akt/mTOR (92), HIF-1α (93), and NF-κB (94), which are also closely related to the immune response. SIRT6 is reported to be silenced by EZH2 (95). Thus, we can consider a SIRT6-participating pathway in the EZH2-regulatory function of the immune response. Furthermore, EZH2 functions in restricting glycolysis in T cells within the TME through the glycolytic pathway, which leads to reduced proinflammatory cytokine expression and poor survival of CD8+ T cells, thus ultimately increasing the tumor burden and metastatic potential of the melanoma model (44). Moreover, T cell functions in the TME are closely regulated by lipid metabolism (96). In an MC38 murine colon cancer model, high-fat diet (HFD)-induced obesity impaired CD8+ T cell function in the TME and led to poor outcomes, which resulted from distinct metabolic adaptations to obesity between the tumor and CD8+ T cells (97). Tumor cells increase fat uptake of the HFD and lead to altered fatty acid partitioning in HFD tumors, impairing CD8+ T cell infiltration and function (97). EZH2 is an important regulator of lipid metabolism. In glioma harboring telomerase reverse transcriptase (TERT) promoter mutations, TERT and EZH2 cooperate with fatty acid synthase (FASN) and induce fatty acid accumulation. Additionally, siRNA-mediated EZH2 knockdown reduced the abundance of intracellular fatty acids (98). EZH2 promotes fatty acid synthesis and lipid accumulation through the TERT-EZH2 network. However, under different experimental conditions, such as in hepatocyte cell lines (99) or breast cancer (100), inhibition of EZH2 induces lipid accumulation. This discrepancy may arise because of differences in tissue specificity. Although the effect of EZH2 on lipid metabolism in different cells or models varies, treatments targeting EZH2 have the potential to reshape lipid composition in the TME, which could further affect T cell function in the TME. More direct evidence is needed to verify the regulatory effect of EZH2 on T cells through metabolism.
Discussion
As the core catalytic subunit of PRC2, EZH2 plays a crucial role in the epigenetic control of chromatin accessibility for gene transcription via histone trimethylation. In addition to its known onco-promoting effects, EZH2 in the TME can influence the immune response and prognosis through its interaction with immune cells. This review focuses on T lymphocytes, which play an essential role in immune homeostasis. EZH2 regulates multiple aspects affecting the immune ability of lymphocytes, including maturation, differentiation, proliferation, function, and apoptosis. Abnormal EZH2 expression is closely associated with aberrant T-cell differentiation and function.
In clinical data, the roles of EZH2 in regulating T cell-mediated antitumor immunity can be complicated. EZH2 inactivation may activate T cell function in the short term, but it leads to increased apoptosis and impaired function in the long term, resulting in an impaired antitumor immune response. However, specific EZH2 deficiency in immunosuppressive cells, such as Treg cells, can lead to an enhanced immune response. A case study also confirmed the bidirectional complex effects of EZH2 inhibitors on the immune system (101). In a patient with metastatic, poorly differentiated chordoma, clinical use of EZH2 inhibitor EPZ6438 resulted in a significant increase in intratumoral and stromal infiltration by proliferative (high Ki-67) CD8+ T cells, Foxp3+ regulatory T cells, and immune cells expressing checkpoint regulators PD-1 and LAG-3 (101), which meant both immuno-activating and suppressive effects. In patients with ovarian cancer, high rate of EZH2+ CD8+ T cells positively affect cancer survival (44). However, systemic overexpression of EZH2 was significantly associated with impaired effector memory CD8+ T cell infiltration and poor survival in patients with hepatocellular carcinoma (102). These results suggest that EZH2 exerts opposite effects in tumor cells and T cells on antitumor immunity. Therefore, it is an urgent problem to explore how to amplify the immuno-activation effect of EZH2 inhibitors while avoiding their immuno-suppressive effect, to bring more clinical benefits. Some clinical trials are exploring the feasibility of EZH2 inhibitors in combination with immunotherapy for lymphoma and some solid tumors (NCT02220842, NCT03854474, NCT04407741, NCT03525795). However, most of them are phase I/II studies with insufficient data feedback. It is still not enough to analyze the effect of EZH2 inhibitors on immune cells, or to determine whether EZH2 inhibitors can be combined with immunotherapy to provide adequate clinical benefit.
Additionally, the wide range of EZH2 targets also contributes to the complexity of its effect in the TME. Generally, chemical inhibition of EZH2 by an inhibitor will influence both tumor and immune cells in different aspects, including proliferation, apoptosis, metabolism, and cytokine as well as chemokine production. These nonspecific effects may be important barriers to the clinical application of EZH2 inhibitors (103). More research is needed to further reveal the effect, mechanism, and selectivity of different subgroups of EZH2 inhibitors on T cells. The following issues should be resolved (1): how EZH2 can be targeted in specific subgroups or differentiation stages of T lymphocytes (2); how the functional pattern of EZH2 on both tumor cells and T cells changes with tumor progression (3); how combination therapy strategies can be used to eliminate or weaken the adverse effects of EZH2 targeting; and (4) whether targeting EZH2 in combination with immunotherapy could be a new strategy for antitumor therapy. In terms of high tumor heterogeneity, existing studies have only revealed the tip of the iceberg. Further understanding of the mechanisms underlying the epigenetic effects of EZH2 on the development, differentiation, and immune function of T cells might provide new strategies for the combined or synergistic treatment of cancer as well as other immune-related diseases.
Author Contributions
JH and JZ contributed equally to this work. JY and LX conceived of the article. JH, JZ, ZG, LC,TZ and JY collected related studies and drafted the manuscript. LX, JY, JW and JH were responsible for the final review of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81972966 and No. 82072870) and the Youth Program of the Beijing Municipal Natural Science Foundation (7204328).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Prof. Yan Wang and Zhengyang Guo of the Peking University Health Science Center for their constructive discussions.
Abbreviations
EZH2, enhancer of zeste homolog 2; PRC2, polycomb repressive complex 2; HSCs, hematopoietic stem cells; WT, wild-type; CTLs, cytotoxic T cells; TME, tumor microenvironment; MDSCs, myeloid-derived suppressor cells; NK, natural killer; TCR, T cell receptor; HIF, hypoxia-inducible factor; Th, helper T; TFH, follicular helper T; Treg, regulatoty T; HPC, hematopoietic progenitor cell; TNF, tumor necrosis factor; CCRK, cell cycle-related kinase; PMN, polymorphonuclear; SAM, s-adenosine methionine.
References
1. Comet I, Riising EM, Leblanc B, Helin K. Maintaining Cell Identity: PRC2-Mediated Regulation of Transcription and Cancer. Nat Rev Cancer (2016) 16(12):803–10. doi: 10.1038/nrc.2016.83
2. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science (2002) 298(5595):1039–43. doi: 10.1126/science.1076997
3. Di Croce L, Helin K. Transcriptional Regulation by Polycomb Group Proteins. Nat Struct Mol Biol (2013) 20(10):1147–55. doi: 10.1038/nsmb.2669
4. He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, et al. PRC2 Directly Methylates GATA4 and Represses Its Transcriptional Activity. Genes Dev (2012) 26(1):37–42. doi: 10.1101/gad.173930.111
5. Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science (2012) 338(6113):1465–9. doi: 10.1126/science.1227604
6. Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, et al. PAF and EZH2 Induce Wnt/beta-Catenin Signaling Hyperactivation. Mol Cell (2013) 52(2):193–205. doi: 10.1016/j.molcel.2013.08.028
7. Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Control of Developmental Regulators by Polycomb in Human Embryonic Stem Cells. Cell (2006) 125(2):301–13. doi: 10.1016/j.cell.2006.02.043
8. Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb Complexes Repress Developmental Regulators in Murine Embryonic Stem Cells. Nature (2006) 441(7091):349–53. doi: 10.1038/nature04733
9. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 Promotes Expansion of Breast Tumor Initiating Cells Through Activation of RAF1-Beta-Catenin Signaling. Cancer Cell (2011) 19(1):86–100. doi: 10.1016/j.ccr.2010.10.035
10. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The Polycomb Group Protein EZH2 Is Involved in Progression of Prostate Cancer. Nature (2002) 419(6907):624–9. doi: 10.1038/nature01075
11. Li Z, Hou P, Fan D, Dong M, Ma M, Li H, et al. The Degradation of EZH2 Mediated by lncRNA ANCR Attenuated the Invasion and Metastasis of Breast Cancer. Cell Death Differ (2017) 24(1):59–71. doi: 10.1038/cdd.2016.95
12. Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, et al. CDK5/FBW7-Dependent Ubiquitination and Degradation of EZH2 Inhibits Pancreatic Cancer Cell Migration and Invasion. J Biol Chem (2017) 292(15):6269–80. doi: 10.1074/jbc.M116.764407
13. Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of Zeste Homolog 2 (EZH2) Induces Natural Killer Cell-Mediated Eradication of Hepatocellular Carcinoma Cells. Proc Natl Acad Sci USA (2018) 115(15):E3509–E18. doi: 10.1073/pnas.1802691115
14. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is Downstream of the pRB-E2F Pathway, Essential for Proliferation and Amplified in Cancer. EMBO J (2003) 22(20):5323–35. doi: 10.1093/emboj/cdg542
15. Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E, et al. Ink4a and Arf Differentially Affect Cell Proliferation and Neural Stem Cell Self-Renewal in Bmi1-Deficient Mice. Genes Dev (2005) 19(12):1438–43. doi: 10.1101/gad.1299305
16. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a Marker of Aggressive Breast Cancer and Promotes Neoplastic Transformation of Breast Epithelial Cells. Proc Natl Acad Sci USA (2003) 100(20):11606–11. doi: 10.1073/pnas.1933744100
17. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating Mutations of the Histone Methyltransferase Gene EZH2 in Myeloid Disorders. Nat Genet (2010) 42(8):722–6. doi: 10.1038/ng.621
18. Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, et al. Genetic Inactivation of the Polycomb Repressive Complex 2 in T Cell Acute Lymphoblastic Leukemia. Nat Med (2012) 18(2):298–301. doi: 10.1038/nm.2651
19. Zhang T, Gong Y, Meng H, Li C, Xue L. Symphony of Epigenetic and Metabolic Regulation-Interaction Between the Histone Methyltransferase EZH2 and Metabolism of Tumor. Clin Epigenet (2020) 12(1):72. doi: 10.1186/s13148-020-00862-0
20. Deb G, Thakur VS, Gupta S. Multifaceted Role of EZH2 in Breast and Prostate Tumorigenesis: Epigenetics and Beyond. Epigenetics (2013) 8(5):464–76. doi: 10.4161/epi.24532
21. Lund K, Adams PD, Copland M. EZH2 in Normal and Malignant Hematopoiesis. Leukemia (2014) 28(1):44–9. doi: 10.1038/leu.2013.288
22. He S, Xie F, Liu Y, Tong Q, Mochizuki K, Lapinski PE, et al. The Histone Methyltransferase Ezh2 Is a Crucial Epigenetic Regulator of Allogeneic T-Cell Responses Mediating Graft-Versus-Host Disease. Blood (2013) 122(25):4119–28. doi: 10.1182/blood-2013-05-505180
23. Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE, et al. Histone H3.3K27M Represses P16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol Cancer Res (2017) 15(9):1243–54. doi: 10.1158/1541-7786.MCR-16-0389
24. Mochizuki-Kashio M, Mishima Y, Miyagi S, Negishi M, Saraya A, Konuma T, et al. Dependency on the Polycomb Gene Ezh2 Distinguishes Fetal From Adult Hematopoietic Stem Cells. Blood (2011) 118(25):6553–61. doi: 10.1182/blood-2011-03-340554
25. Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, et al. Polycomb Repressive Complex 2 Regulates Normal Hematopoietic Stem Cell Function in a Developmental-Stage-Specific Manner. Cell Stem Cell (2014) 14(1):68–80. doi: 10.1016/j.stem.2013.10.001
26. Mochizuki-Kashio M, Aoyama K, Sashida G, Oshima M, Tomioka T, Muto T, et al. Ezh2 Loss in Hematopoietic Stem Cells Predisposes Mice to Develop Heterogeneous Malignancies in an Ezh1-Dependent Manner. Blood (2015) 126(10):1172–83. doi: 10.1182/blood-2015-03-634428
27. Lee SC, Miller S, Hyland C, Kauppi M, Lebois M, Di Rago L, et al. Polycomb Repressive Complex 2 Component Suz12 Is Required for Hematopoietic Stem Cell Function and Lymphopoiesis. Blood (2015) 126(2):167–75. doi: 10.1182/blood-2014-12-615898
28. Wang C, Oshima M, Sato D, Matsui H, Kubota S, Aoyama K, et al. Ezh2 Loss Propagates Hypermethylation at T Cell Differentiation-Regulating Genes to Promote Leukemic Transformation. J Clin Invest (2018) 128(9):3872–86. doi: 10.1172/JCI94645
29. Jacobsen JA, Woodard J, Mandal M, Clark MR, Bartom ET, Sigvardsson M, et al. EZH2 Regulates the Developmental Timing of Effectors of the Pre-Antigen Receptor Checkpoints. J Immunol (2017) 198(12):4682–91. doi: 10.4049/jimmunol.1700319
30. Dobenecker MW, Park JS, Marcello J, McCabe MT, Gregory R, Knight SD, et al. Signaling Function of PRC2 is Essential for TCR-Driven T Cell Responses. J Exp Med (2018) 215(4):1101–13. doi: 10.1084/jem.20170084
31. Dobenecker MW, Kim JK, Marcello J, Fang TC, Prinjha R, Bosselut R, et al. Coupling of T Cell Receptor Specificity to Natural Killer T Cell Development by Bivalent Histone H3 Methylation. J Exp Med (2015) 212(3):297–306. doi: 10.1084/jem.20141499
32. Vasanthakumar A, Xu D, Lun AT, Kueh AJ, van Gisbergen KP, Iannarella N, et al. A Non-Canonical Function of Ezh2 Preserves Immune Homeostasis. EMBO Rep (2017) 18(4):619–31. doi: 10.15252/embr.201643237
33. Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R, et al. Polycomb Group Protein Ezh2 Controls Actin Polymerization and Cell Signaling. Cell (2005) 121(3):425–36. doi: 10.1016/j.cell.2005.02.029
34. Gunawan M, Venkatesan N, Loh JT, Wong JF, Berger H, Neo WH, et al. The Methyltransferase Ezh2 Controls Cell Adhesion and Migration Through Direct Methylation of the Extranuclear Regulatory Protein Talin. Nat Immunol (2015) 16(5):505–16. doi: 10.1038/ni.3125
35. Zhang Y, Kinkel S, Maksimovic J, Bandala-Sanchez E, Tanzer MC, Naselli G, et al. The Polycomb Repressive Complex 2 Governs Life and Death of Peripheral T Cells. Blood (2014) 124(5):737–49. doi: 10.1182/blood-2013-12-544106
36. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The Polycomb Protein Ezh2 Regulates Differentiation and Plasticity of CD4(+) T Helper Type 1 and Type 2 Cells. Immunity (2013) 39(5):819–32. doi: 10.1016/j.immuni.2013.09.012
37. Shen CH, Talay O, Mahajan VS, Leskov IB, Eisen HN, Chen J. Antigen-Bearing Dendritic Cells Regulate the Diverse Pattern of Memory CD8 T-Cell Development in Different Tissues. Proc Natl Acad Sci USA (2010) 107(52):22587–92. doi: 10.1073/pnas.1016350108
38. Kaech SM, Cui W. Transcriptional Control of Effector and Memory CD8+ T Cell Differentiation. Nat Rev Immunol (2012) 12(11):749–61. doi: 10.1038/nri3307
39. Chen G, Subedi K, Chakraborty S, Sharov A, Lu J, Kim J, et al. Ezh2 Regulates Activation-Induced CD8(+) T Cell Cycle Progression via Repressing Cdkn2a and Cdkn1c Expression. Front Immunol (2018) 9:549. doi: 10.3389/fimmu.2018.00549
40. Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM. Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8(+) T Cell Terminal Differentiation and Loss of Multipotency. Immunity (2017) 46(4):596–608. doi: 10.1016/j.immuni.2017.03.012
41. He S, Liu Y, Meng L, Sun H, Wang Y, Ji Y, et al. Ezh2 Phosphorylation State Determines Its Capacity to Maintain CD8(+) T Memory Precursors for Antitumor Immunity. Nat Commun (2017) 8(1):2125. doi: 10.1038/s41467-017-02187-8
42. Russ BE, Olshanksy M, Smallwood HS, Li J, Denton AE, Prier JE, et al. Distinct Epigenetic Signatures Delineate Transcriptional Programs During Virus-Specific CD8(+) T Cell Differentiation. Immunity (2014) 41(5):853–65. doi: 10.1016/j.immuni.2014.11.001
43. Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. Active Genes Are Tri-Methylated at K4 of Histone H3. Nature (2002) 419(6905):407–11. doi: 10.1038/nature01080
44. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer Mediates Effector T Cell Dysfunction by Targeting microRNAs and EZH2 via Glycolysis Restriction. Nat Immunol (2016) 17(1):95–103. doi: 10.1038/ni.3313
45. Chang JT, Wherry EJ, Goldrath AW. Molecular Regulation of Effector and Memory T Cell Differentiation. Nat Immunol (2014) 15(12):1104–15. doi: 10.1038/ni.3031
46. Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, et al. Early Transcriptional and Epigenetic Regulation of CD8(+) T Cell Differentiation Revealed by Single-Cell RNA Sequencing. Nat Immunol (2017) 18(4):422–32. doi: 10.1038/ni.3688
47. Zhu J, Paul WE. CD4 T Cells: Fates, Functions, and Faults. Blood (2008) 112(5):1557–69. doi: 10.1182/blood-2008-05-078154
48. Liu H, Li P, Wei Z, Zhang C, Xia M, Du Q, et al. Regulation of T Cell Differentiation and Function by Epigenetic Modification Enzymes. Semin Immunopathol (2019) 41(3):315–26. doi: 10.1007/s00281-019-00731-w
49. Zhu J. T Helper Cell Differentiation, Heterogeneity, and Plasticity. Cold Spring Harb Perspect Biol (2018) 10(10). doi: 10.1101/cshperspect.a030338
50. Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, Sciume G, et al. EZH2 Is Crucial for Both Differentiation of Regulatory T Cells and T Effector Cell Expansion. Sci Rep (2015) 5:10643. doi: 10.1038/srep10643
51. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
52. Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res (2016) 76(2):275–82. doi: 10.1158/0008-5472.CAN-15-1938
53. Yuan H, Nishikori M, Otsuka Y, Arima H, Kitawaki T, Takaori-Kondo A. The EZH2 Inhibitor Tazemetostat Upregulates the Expression of CCL17/TARC in B-Cell Lymphoma and Enhances T-Cell Recruitment. Cancer Sci (2021). doi: 10.1111/cas.15122
54. Craft JE. Follicular Helper T Cells in Immunity and Systemic Autoimmunity. Nat Rev Rheumatol (2012) 8(6):337–47. doi: 10.1038/nrrheum.2012.58
55. Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, et al. Bcl6 Expressing Follicular Helper CD4 T Cells Are Fate Committed Early and Have the Capacity to Form Memory. J Immunol (2013) 190(8):4014–26. doi: 10.4049/jimmunol.1202963
56. Li F, Zeng Z, Xing S, Gullicksrud JA, Shan Q, Choi J, et al. Ezh2 Programs TFH Differentiation by Integrating Phosphorylation-Dependent Activation of Bcl6 and Polycomb-Dependent Repression of p19Arf. Nat Commun (2018) 9(1):5452. doi: 10.1038/s41467-018-07853-z
57. O'Malley JT, Sehra S, Thieu VT, Yu Q, Chang HC, Stritesky GL, et al. Signal Transducer and Activator of Transcription 4 Limits the Development of Adaptive Regulatory T Cells. Immunology (2009) 127(4):587–95. doi: 10.1111/j.1365-2567.2008.03037.x
58. Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-Induced Repression of Chromatin Bound by the Transcription Factor Foxp3 in Regulatory T Cells. Nat Immunol (2014) 15(6):580–7. doi: 10.1038/ni.2868
59. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T Cell Subpopulations Distinctly Control the Prognosis of Colorectal Cancers. Nat Med (2016) 22(6):679–84. doi: 10.1038/nm.4086
60. Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep (2018) 23(11):3262–74. doi: 10.1016/j.celrep.2018.05.050
61. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The Chromatin-Modifying Enzyme Ezh2 Is Critical for the Maintenance of Regulatory T Cell Identity After Activation. Immunity (2015) 42(2):227–38. doi: 10.1016/j.immuni.2015.01.007
62. Dias S, D'Amico A, Cretney E, Liao Y, Tellier J, Bruggeman C, et al. Effector Regulatory T Cell Differentiation and Immune Homeostasis Depend on the Transcription Factor Myb. Immunity (2017) 46(1):78–91. doi: 10.1016/j.immuni.2016.12.017
63. Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 Expression in T Cells Improves Efficacy of Anti-CTLA-4 Therapy. J Clin Invest (2018) 128(9):3813–8. doi: 10.1172/JCI99760
64. Ribas A. Tumor Immunotherapy Directed at PD-1. N Engl J Med (2012) 366(26):2517–9. doi: 10.1056/NEJMe1205943
65. Jancewicz I, Szarkowska J, Konopinski R, Stachowiak M, Swiatek M, Blachnio K, et al. PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4(+) T Cells. Cancers (Basel) (2021) 13(16):4148. doi: 10.3390/cancers13164148
66. Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 Negatively Regulates PD-L1 Expression in Hepatocellular Carcinoma. J Immunother Cancer (2019) 7(1):300. doi: 10.1186/s40425-019-0784-9
67. Toyokawa G, Takada K, Tagawa T, Hamamoto R, Yamada Y, Shimokawa M, et al. A Positive Correlation Between the EZH2 and PD-L1 Expression in Resected Lung Adenocarcinomas. Ann Thorac Surg (2019) 107(2):393–400. doi: 10.1016/j.athoracsur.2018.08.056
68. Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 Inhibition Activates a dsRNA-STING-Interferon Stress Axis That Potentiates Response to PD-1 Checkpoint Blockade in Prostate Cancer. Nat Cancer (2021) 2(4):444–56. doi: 10.1038/s43018-021-00185-w
69. Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin Cancer Res (2020) 26(1):290–300. doi: 10.1158/1078-0432.CCR-19-1351
70. Zhou J, Huang S, Wang Z, Huang J, Xu L, Tang X, et al. Targeting EZH2 Histone Methyltransferase Activity Alleviates Experimental Intestinal Inflammation. Nat Commun (2019) 10(1):2427. doi: 10.1038/s41467-019-10176-2
71. Huang S, Wang Z, Zhou J, Huang J, Zhou L, Luo J, et al. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res (2019) 79(8):2009–20. doi: 10.1158/0008-5472.CAN-18-2395
72. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
73. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor Necrosis Factor-Alpha Blocks Differentiation and Enhances Suppressive Activity of Immature Myeloid Cells During Chronic Inflammation. Immunity (2013) 38(3):541–54. doi: 10.1016/j.immuni.2013.02.007
74. Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, et al. Context-Specific Regulation of NF-kappaB Target Gene Expression by EZH2 in Breast Cancers. Mol Cell (2011) 43(5):798–810. doi: 10.1016/j.molcel.2011.08.011
75. Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma-Intrinsic CCRK Inhibition Diminishes Myeloid-Derived Suppressor Cell Immunosuppression and Enhances Immune-Checkpoint Blockade Efficacy. Gut (2018) 67(5):931–44. doi: 10.1136/gutjnl-2017-314032
76. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 Regulates Differentiation and Function of Natural Killer Cells Through Histone Methyltransferase Activity. Proc Natl Acad Sci USA (2015) 112(52):15988–93. doi: 10.1073/pnas.1521740112
77. Yin Y, Qiu S, Li X, Huang B, Xu Y, Peng Y. EZH2 Suppression in Glioblastoma Shifts Microglia Toward M1 Phenotype in Tumor Microenvironment. J Neuroinflamm (2017) 14(1):220. doi: 10.1186/s12974-017-0993-4
78. Yang L, Zhang Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J Hematol Oncol (2017) 10(1):58. doi: 10.1186/s13045-017-0430-2
79. Wang X, Wang Y, Li L, Xue X, Xie H, Shi H, et al. A lncRNA Coordinates With Ezh2 to Inhibit HIF-1alpha Transcription and Suppress Cancer Cell Adaption to Hypoxia. Oncogene (2020) 39(9):1860–74. doi: 10.1038/s41388-019-1123-9
80. Chen S, Chen JZ, Zhang JQ, Chen HX, Yan ML, Huang L, et al. Hypoxia Induces TWIST-Activated Epithelial-Mesenchymal Transition and Proliferation of Pancreatic Cancer Cells In Vitro and in Nude Mice. Cancer Lett (2016) 383(1):73–84. doi: 10.1016/j.canlet.2016.09.027
81. Huang Y, Tao Y, Hu K, Lin F, Li X, Feng T, et al. Hypoxia-Induced NIPP1 Activation Enhances Metastatic Potential and Predicts Poor Prognosis in Hepatocellular Carcinoma. Tumour Biol (2016) 37(11):14903–14. doi: 10.1007/s13277-016-5392-4
82. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia Induces Myeloid-Derived Suppressor Cell Recruitment to Hepatocellular Carcinoma Through Chemokine (C-C Motif) Ligand 26. Hepatology (2016) 64(3):797–813. doi: 10.1002/hep.28655
83. Palazon A, Tyrakis PA, Macias D, Velica P, Rundqvist H, Fitzpatrick S, et al. An HIF-1alpha/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell (2017) 32(5):669–83.e5. doi: 10.1016/j.ccell.2017.10.003
84. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-Hydroxyglutarate Regulates CD8(+) T-Lymphocyte Fate. Nature (2016) 540(7632):236–41. doi: 10.1038/nature20165
85. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic Control of Type 1 Regulatory T Cell Differentiation by AHR and HIF1-Alpha. Nat Med (2015) 21(6):638–46. doi: 10.1038/nm.3868
86. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-Deficient Mice Have Increased Antitumor Immunity and Are Resistant to Experimental Metastasis. Cancer Res (2011) 71(8):2892–900. doi: 10.1158/0008-5472.CAN-10-4246
87. Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, et al. A2AR Antagonism With CPI-444 Induces Antitumor Responses and Augments Efficacy to Anti-PD-(L)1 and Anti-CTLA-4 in Preclinical Models. Cancer Immunol Res (2018) 6(10):1136–49. doi: 10.1158/2326-6066.CIR-18-0056
88. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-Inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-Tumor Immune Responses. Cancer Cell (2016) 30(3):391–403. doi: 10.1016/j.ccell.2016.06.025
89. Sitkovsky MV. T Regulatory Cells: Hypoxia-Adenosinergic Suppression and Re-Direction of the Immune Response. Trends Immunol (2009) 30(3):102–8. doi: 10.1016/j.it.2008.12.002
90. Dann SG, Ryskin M, Barsotti AM, Golas J, Shi C, Miranda M, et al. Reciprocal Regulation of Amino Acid Import and Epigenetic State Through Lat1 and EZH2. EMBO J (2015) 34(13):1773–85. doi: 10.15252/embj.201488166
91. Hung MH, Lee JS, Ma C, Diggs LP, Heinrich S, Chang CW, et al. Tumor Methionine Metabolism Drives T-Cell Exhaustion in Hepatocellular Carcinoma. Nat Commun (2021) 12(1):1455. doi: 10.1038/s41467-021-21804-1
92. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, et al. The Sirtuin SIRT6 Blocks IGF-Akt Signaling and Development of Cardiac Hypertrophy by Targeting C-Jun. Nat Med (2012) 18(11):1643–50. doi: 10.1038/nm.2961
93. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1alpha. Cell (2010) 140(2):280–93. doi: 10.1016/j.cell.2009.12.041
94. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 Links Histone H3 Lysine 9 Deacetylation to NF-kappaB-Dependent Gene Expression and Organismal Life Span. Cell (2009) 136(1):62–74. doi: 10.1016/j.cell.2008.10.052
95. Dong Z, Li C, Yin C, Xu M, Liu S, Gao M. LncRNA PU.1 AS Regulates Arsenic-Induced Lipid Metabolism Through EZH2/Sirt6/SREBP-1c Pathway. J Environ Sci (China) (2019) 85:138–46. doi: 10.1016/j.jes.2019.05.019
96. Lochner M, Berod L, Sparwasser T. Fatty Acid Metabolism in the Regulation of T Cell Function. Trends Immunol (2015) 36(2):81–91. doi: 10.1016/j.it.2014.12.005
97. Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, Gassaway BM, et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell (2020) 183(7):1848–66 e26. doi: 10.1016/j.cell.2020.11.009
98. Ahmad F, Patrick S, Sheikh T, Sharma V, Pathak P, Malgulwar PB, et al. Telomerase Reverse Transcriptase (TERT) - Enhancer of Zeste Homolog 2 (EZH2) Network Regulates Lipid Metabolism and DNA Damage Responses in Glioblastoma. J Neurochem (2017) 143(6):671–83. doi: 10.1111/jnc.14152
99. Vella S, Gnani D, Crudele A, Ceccarelli S, De Stefanis C, Gaspari S, et al. EZH2 Down-Regulation Exacerbates Lipid Accumulation and Inflammation in In Vitro and In Vivo NAFLD. Int J Mol Sci (2013) 14(12):24154–68. doi: 10.3390/ijms141224154
100. Hayden A, Johnson PW, Packham G, Crabb SJ. S-Adenosylhomocysteine Hydrolase Inhibition by 3-Deazaneplanocin A Analogues Induces Anti-Cancer Effects in Breast Cancer Cell Lines and Synergy With Both Histone Deacetylase and HER2 Inhibition. Breast Cancer Res Treat (2011) 127(1):109–19. doi: 10.1007/s10549-010-0982-0
101. Gounder MM, Zhu G, Roshal L, Lis E, Daigle SR, Blakemore SJ, et al. Immunologic Correlates of the Abscopal Effect in a SMARCB1/INI1-Negative Poorly Differentiated Chordoma After EZH2 Inhibition and Radiotherapy. Clin Cancer Res (2019) 25(7):2064–71. doi: 10.1158/1078-0432.CCR-18-3133
102. Zeng F, Zhang Y, Han X, Zeng M, Gao Y, Weng J. Employing Hypoxia Characterization to Predict Tumour Immune Microenvironment, Treatment Sensitivity and Prognosis in Hepatocellular Carcinoma. Comput Struct Biotechnol J (2021) 19:2775–89. doi: 10.1016/j.csbj.2021.03.033
Keywords: EZH2, T cells, T cell differentiation, T cell activation, antitumor immunity
Citation: Huang J, Zhang J, Guo Z, Li C, Tan Z, Wang J, Yang J and Xue L (2021) Easy or Not—The Advances of EZH2 in Regulating T Cell Development, Differentiation, and Activation in Antitumor Immunity. Front. Immunol. 12:741302. doi: 10.3389/fimmu.2021.741302
Received: 14 July 2021; Accepted: 28 September 2021;
Published: 19 October 2021.
Edited by:
Yong Zhao, Institute of Zoology (CAS), ChinaCopyright © 2021 Huang, Zhang, Guo, Li, Tan, Wang, Yang and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lixiang Xue, bGl4aWFuZ3h1ZUBiam11LmVkdS5jbg==; Jianling Yang, amlhbmxpbmd5YW5nQGJqbXUuZWR1LmNu
†These authors have contributed equally to this work