 Kai Kammers1
Kai Kammers1 Athena Chen2
Athena Chen2 Daniel R. Monaco3Sarah E. Hudelson4Wendy Grant-McAuley4Richard D. Moore5
Daniel R. Monaco3Sarah E. Hudelson4Wendy Grant-McAuley4Richard D. Moore5 Galit Alter6
Galit Alter6 Steven G. Deeks7
Steven G. Deeks7 Charles S. Morrison8Leigh A. Eller9,10
Charles S. Morrison8Leigh A. Eller9,10 Joel N. Blankson5Oliver Laeyendecker5,11Ingo Ruczinski2Susan H. Eshleman4*†
Joel N. Blankson5Oliver Laeyendecker5,11Ingo Ruczinski2Susan H. Eshleman4*† H. Benjamin Larman3*†
H. Benjamin Larman3*†- 1Division of Biostatistics and Bioinformatics, Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Department of Biostatistics, Johns Hopkins University Bloomberg School of Public Health, Baltimore, MD, United States
- 3Department of Pathology and the Institute for Cell Engineering, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 4Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 5Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 6Department of Medicine, Ragon Institute of Massachusetts General Hospital (MGH), Massachusetts Institute of Technology (MIT) and Harvard, Cambridge, MA, United States
- 7Department of Medicine, University of California, San Francisco (UCSF), San Francisco, CA, United States
- 8Behavioral, Epidemiologic and Clinical Sciences, Family Health International (FHI) 360, Durham, NC, United States
- 9U.S. Military HIV Research Program, Walter Reed Army Institute of Research, Silver Spring, MD, United States
- 10Henry M. Jackson Foundation for the Advancement of Military Medicine, Bethesda, MD, United States
- 11Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Baltimore, MD, United States
Introduction: Low HIV viral load is associated with delayed disease progression and reduced HIV transmission. HIV controllers suppress viral load to low levels in the absence of antiretroviral treatment (ART). We used an antibody profiling system, VirScan, to compare antibody reactivity and specificity in HIV controllers, non-controllers with treatment-induced viral suppression, and viremic non-controllers.
Methods: The VirScan library contains 3,384 phage-displayed peptides spanning the HIV proteome. Antibody reactivity to these peptides was measured in plasma from a Discovery Cohort that included 13 elite controllers, 27 viremic controllers, 12 viremic non-controllers, and 21 non-controllers who were virally suppressed on ART. Antibody reactivity to selected peptides was also assessed in an independent cohort of 29 elite controllers and 37 non-controllers who were virally suppressed on ART (Validation Cohort) and in a longitudinal cohort of non-controllers.
Results: In the Discovery Cohort, 62 peptides were preferentially targeted in HIV controllers compared to non-controllers who were virally suppressed on ART. These specificities were not significantly different when comparing controllers versus viremic non-controllers. Aggregate reactivity to these peptides was also high in elite controllers from the independent Validation Cohort. The 62 peptides formed seven clusters of homologous epitopes in env, gag, integrase, and vpu. Reactivity to one of these clusters located in gag p17 was inversely correlated with viral load set point in an independent cohort of non-controllers.
Conclusions: Antibody reactivity was low in non-controllers suppressed on ART, but remained high in viremic controllers despite viral suppression. Antibodies in controllers and viremic non-controllers were directed against epitopes in diverse HIV proteins; higher reactivity against p17 peptides was associated with lower viral load set point. Further studies are needed to determine if these antibodies play a role in regulation of HIV viral load.
Introduction
In the absence of antiretroviral treatment (ART), most persons living with HIV have on-going viral replication that leads to CD4+ T cell depletion and severe immunodeficiency. Effective ART suppresses viral replication to low levels which reduces morbidity, mortality and the risk HIV transmission (1). HIV controllers are individuals who control viral replication to low levels in the absence of ART (2). Elite controllers maintain viral loads of <50 copies/mL, while viremic controllers maintain viral loads of <2,000 copies/mL (3). These individuals usually maintain high CD4+ T cell counts for long periods before experiencing immune system decline (4, 5). In non-controllers, a viral load set point is usually established in the absence of treatment; higher viral load set points are associated with higher transmission rates and more rapid disease progression in the absence of treatment.
The mechanisms responsible for suppression of viral replication in HIV controllers are not well understood (4). Many elites and viremic controllers are infected with replication-competent virus (6–9), suggesting that host factors play a role in controlling viral replication. Class I major histocompatibility (MHC) alleles, such as HLA-B*57 and HLA-B*27, have been implicated in elite control through cohort studies (10) and genome-wide association studies (11, 12). This suggests a role for CD8+ T cells in HIV control. More effective CD8+ T cell responses have been observed in elite controllers compared to individuals with progressive HIV disease (13–16). Furthermore, in vivo depletion of CD8+ T cells in monkey elite controllers of simian immunodeficiency virus infection leads to loss of control of viral replication (17, 18). Other genetic studies suggest that certain natural killer cell receptors, such as the killer immunoglobulin-like receptors, may also be involved in elite control (19–21).
The role of humoral immunity in viremic control and control of viral load set point is not clear. Serologic assays used for cross-sectional HIV incidence estimation demonstrate that most elite controllers have lower antibody titers than viremic individuals (22). Broadly neutralizing antibodies are also more likely to be found in non-controllers with high viral loads (23). However, elite controllers do not have higher titers of neutralizing antibodies to autologous (24) or heterologous viruses (3, 25) compared to non-controllers. Some studies have suggested that some elite controllers may have higher levels of antibody-dependent cellular cytotoxicity (ADCC) than viremic persons (26), while other studies found that elite controllers do not have higher levels of either antibodies (27) or effector cells (28) that can mediate ADCC. A recent study suggested that while antibodies from elite controllers were not more effective for ADCC in any single assay, they were more likely to be polyfunctional than antibodies from non-controllers (29). However, it is not clear whether this polyfunctional antibody profile is a cause or consequence of elite control.
There are limited data comparing the fine specificity of anti-HIV antibodies in persons with suppressed viral load on ART and persons with low viral load set points. The VirScan assay is a highly multiplexed antibody profiling system that can be used to characterize the fine specificity of antibodies to viral peptides that lack highly conformational or glycosylated epitopes (30). We previously used VirScan to characterize the evolution of HIV antibodies in early- to late-stage HIV infection (31). In this report, we used VirScan to compare the antibody profiles in HIV controllers, non-controllers on ART, and viremic non-controllers, including persons with different viral load set points.
Materials and Methods
Samples Used for Analysis
Samples used to compare antibody reactivity in HIV controllers and non-controllers were grouped into two cohorts (Table 1). The Discovery Cohort included samples from elite controllers, viremic controllers, non-controllers suppressed on ART, and viremic non-controllers enrolled a clinic-based cohort [Study of the Consequences of the Protease Inhibitor Era (SCOPE) study], San Francisco, CA (32, 33). The Validation Cohort included samples from elite controllers enrolled in a clinic-based cohort [Johns Hopkins University (JHU) Elite Controller Cohort, Baltimore, MD (24)] and non-controllers from a clinic-based cohort who were virally suppressed on ART [JHU Moore Clinic (34), Baltimore, MD]. Samples used to analyze the association between viral load set point and antibody reactivity were obtained from a study of the natural history of HIV infection conducted in Uganda and Thailand [the Early Capture HIV Cohort Study (RV217 Cohort); this study recruited men and women at locations associated with transactional sex (35)]. Participants in the Discovery and Validation cohorts are from the United States, where most HIV strains are subtype B. The cohort used to evaluate antibody reactivity and viral load set point included samples from persons with diverse HIV subtypes and strains (Table 1).

Table 1 Samples used for analysis.
Informed Consent
The SCOPE, RV217 and JHU studies were approved by institutional review boards at the parent research institutions. Participants provided written informed consent for their samples to be used in HIV-related research.
Antibody Profiling Using the VirScan Assay
Plasma samples were analyzed using the VirScan assay, as described previously (30, 31, 36). The VirScan library includes 3,384 HIV peptides spanning the viral genome, representing numerous HIV subtypes and strains (30). This includes 821 peptides in 21 proteins for subtype B; 203 peptides in 18 proteins in subtype A; and 224 peptides in 20 proteins in subtype D; the subtype/strain designation for other peptides is described previously (31). Peptides are expressed on bacteriophage from DNA tiles; the expressed peptides are 56 amino acids long with 28 amino acid overlaps. The number of peptides in each location varies across the HIV genome, reflecting the level of genetic diversity (31). The VirScan library also encodes peptides spanning the genomes of >200 other viruses that infect humans. Antibody reactivity to Ebola virus and rabies virus peptides was used to normalize HIV antibody binding data to account for differences in sequencing depth between samples (31). IgG concentrations in plasma samples were determined using an enzyme-linked immunosorbent assay (ELISA) for IgG, so that input to the VirScan assay was constant at 2 μg IgG per reaction (36). Data from mock immunoprecipitation reactions were used for normalization as described previously (31). After incubation with samples, antibody-bound phage was immunoprecipitated using magnetic beads coated with protein A and protein G. After bead washing, DNA tiles in the immunoprecipitated phage were amplified by polymerase chain reaction (PCR) using primers containing sample-specific barcodes. PCR products were sequenced using a NextSeq 500 instrument (Illumina, San Diego, CA) to determine the amino acid sequences of the antibody-bound peptides. The samples were tested via VirScan from May 2019 to August 2019.
VirScan Data Analysis
Antibody reactivity to each peptide was quantified using normalized read counts and relative fold change values, as described (31). Briefly, a raw read count was obtained for each peptide, reflecting the number of times the corresponding DNA sequence was detected in immunoprecipitated phage. Raw read counts were log10 transformed after adding one read count to the result for each peptide in each sample. The log-transformed values for each peptide and each sample were then normalized to the average read count obtained for all Ebola virus and rabies virus peptides for the same sample, in order to adjust for sample-to-sample differences in sequencing depth (none of the participants had these infections and antibody reactivity to these viruses most likely reflected non-specific antibody binding or spurious cross reactivity). Finally, a log10 relative fold change value for each peptide was calculated by subtracting the median of the normalized values for the same peptide observed in mock immunoprecipitation reactions run on the same plate from the log10 normalized value of the peptide. Six to eight mock immunoprecipitations were included on each 96 well immunoprecipitation plate.
Statistical Methods
For analysis of antibody reactivity in controllers versus non-controllers, statistical inference between groups was carried out by moderated t-tests (37). Multiple comparison corrected q-values (38) were calculated from the observed p-values. If a peptide had a q-value of 0.05, it was expected that 5% of the peptides with smaller p-values would be false positives. Peptides with a q-value <0.05 were considered to have significantly different levels of antibody reactivity in the two groups. To test for replication, permutation analysis was performed to compare antibody reactivity for a combined set of 62 pre-identified peptides in elite controllers vs. non-controllers. For this analysis, one-sided moderated t-tests were used to obtain a p-value for each of the 62 peptides. Fisher’s inverse chi-square test (39) was then used to calculate a test-statistic based on the full set of 62 p-values by adding the -log10 p-value for each peptide. Since the null distribution in Fisher’s test assumes independence of p-values, a permutation test with 10,000 permutations was used to assess statistical significance, accounting for dependence of test statistics (e.g., due to overlapping and homologous peptides). In each permutation, the group labels (29 elite controller labels, 37 non-controller labels) were randomly assigned to the 66 participants; a p-value for each of the 62 peptides was calculated using a one-sided moderated t-test statistic, and a test-statistic was obtained for the full set of peptides using Fisher’s method. Finally, a one-sided p-value of the permutation test was calculated as the proportion of sample permutations where the value of the test-statistic was greater than or equal to the observed test-statistic.
For analysis of viral load set point, the set point for each individual (log10 HIV RNA copies/mL) was calculated by taking the median pre-ART viral load for each person. Median antibody reactivity (log10 normalized fold change) was calculated for the same samples. Simple linear regression was conducted for each peptide with viral load set point as the dependent variable and median antibody reactivity as the independent variable. The estimate of the slope is reported as effect size; the statistical significance of the effect size (p-value) was derived by testing the hypothesis that the true slope was zero (i.e., no association between viral load set point and antibody reactivity). Multiple comparisons correction was carried out by controlling the family-wise error rate using the Bonferroni method. Due to the homology of peptides in the VirScan library and possible dependence of test statistics and p-values, a permutation test was also performed, jointly shuffling the viral load set point across all peptides, thereby maintaining the correlation structure.
The software environment R (version 4.0.1) was used for statistical computing. The software epitopefindr (GitHub: https://brandonsie.github.io/epitopefindr/) was used to identify amino acid motifs associated each peptide clusters.
Results
Comparison of Antibody Profiles in HIV Controllers and Non-Controllers
We first used VirScan to compare antibody reactivity among 40 HIV controllers and 33 non-controllers in the Discovery Cohort (Table 1). Antibody reactivity was quantified for each peptide in the VirScan assay. Sixty-two peptides had significantly higher levels of antibody reactivity in HIV controllers compared to non-controllers who were virally suppressed on ART (Figure 1A). Despite observing a similar trend for most of these peptides when comparing HIV controllers to viremic non-controllers none of the peptides displayed statistically significant differences in reactivity in these two groups after multiple testing correction (Figure 1B).
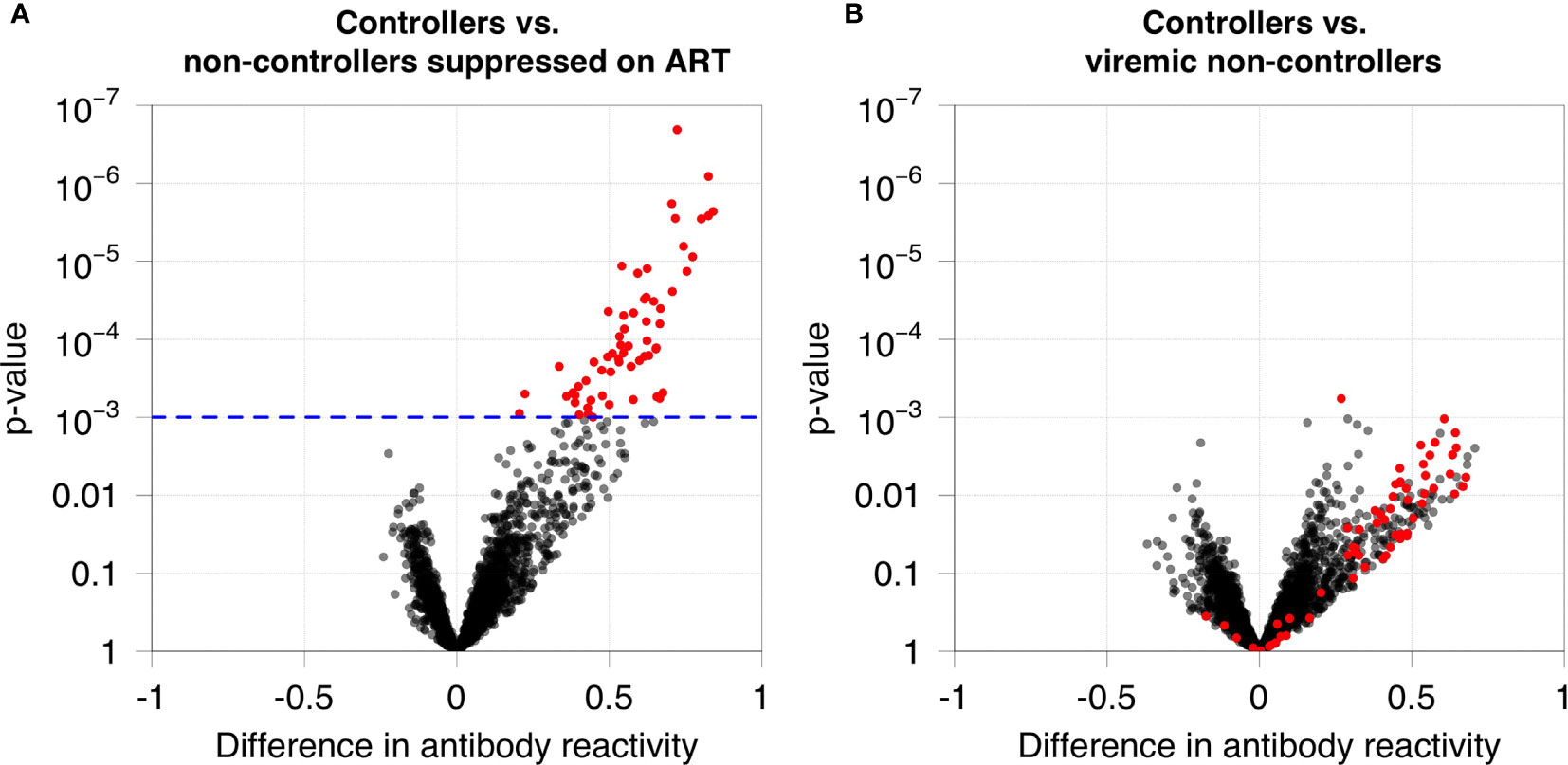
Figure 1 Antibody reactivities in HIV controllers compared to non-controllers. (A) Antibody reactivity was compared for 40 HIV controllers (13 elite controllers and 27 viremic controllers) and 21 non-controllers suppressed on ART. The volcano plot shows on the x-axis the difference in antibody reactivity in the two groups (estimated log10 fold change; positive numbers correspond to stronger reactivity in controllers) and the y-axis shows the -log10 p-value for each peptide based on moderated t-statistics. Red dots indicate the 62 peptides that are significantly more reactive in controllers at a false discovery rate of 5%. The blue dashed line indicates the highest q-value less than 5% (q=0.0485), which corresponds to a p-value of 0.001. (B) Antibody reactivity to each HIV peptide was compared for 40 HIV controllers and 12 viremic non-controllers. No significant differences in antibody reactivity were observed for these 62 peptides in these two groups.
The relative level of antibody reactivity to peptides in HIV controllers versus non-controllers who were virally suppressed on ART varied as a function of peptide location (Figures 2, S1). Sixty-one of the 62 peptides formed seven distinct clusters of homologous peptides (Figure 2B and Table S1). Twenty-two peptides were located in the gag region: 18 in the N-terminus of gag (p17, cluster 1) and four in the C-terminus of gag (p24, cluster 2). Four peptides were located in the C-terminus of integrase (cluster 3). Two peptides were located in the N-terminus of the vpu accessory protein (cluster 4). The remaining 34 peptides were located in the env region. Seven of these peptides spanned the V3 loop and CD4 binding loop of gp120 (six in cluster 5, one that was not in a cluster), 23 spanned the V5 loop of gp120 and the fusion peptide of gp41 (cluster 6), and four were in gp41, proximal to the membrane proximal external region (MPER, cluster 7). The peptides detected in each cluster are provided in Table 2.
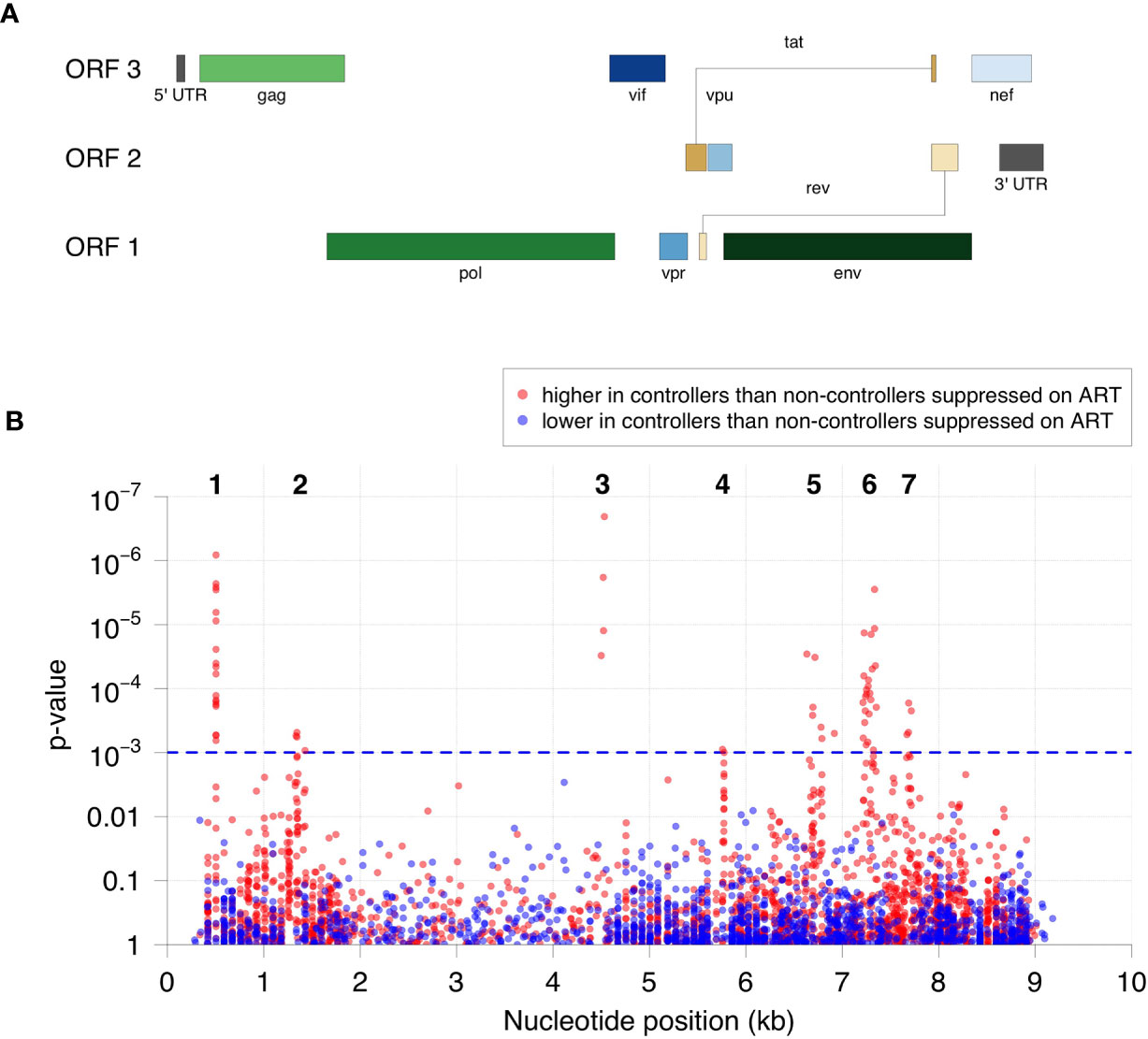
Figure 2 Specificity of antibodies targeted in HIV controllers compared to non-controllers who were virally suppressed on ART. (A) The positions and lengths of open reading frames (ORFs) in the HIV genome are plotted relative to genomic coordinates for HIV (HXB2, NCBI #NC_001802). (B) The significance for the difference in antibody reactivity in controllers versus non-controllers who were virally suppressed on ART is shown for each peptide. The x-axis shows the position of each peptide in the HIV genome. The y-axis shows the -log10 p-value based on moderated t-statistics for each peptide. Each dot shows the result for a single peptide. Red dots indicate peptides that had higher antibody reactivity in the HIV controller group. Blue dots indicate peptides that had lower antibody reactivity in the HIV controller group. The blue dashed line indicates the highest q-value less than 5% (q=0.0485), which corresponds to a p-value of 0.001. The 62 significant peptides with q-values <0.05 (above the blue dashed line) had significantly higher peptide reactivity in HIV controllers compared to non-controllers who were virally suppressed on ART. Cluster numbers (1-7) are noted above each group of clustered peptides (see Table 2).
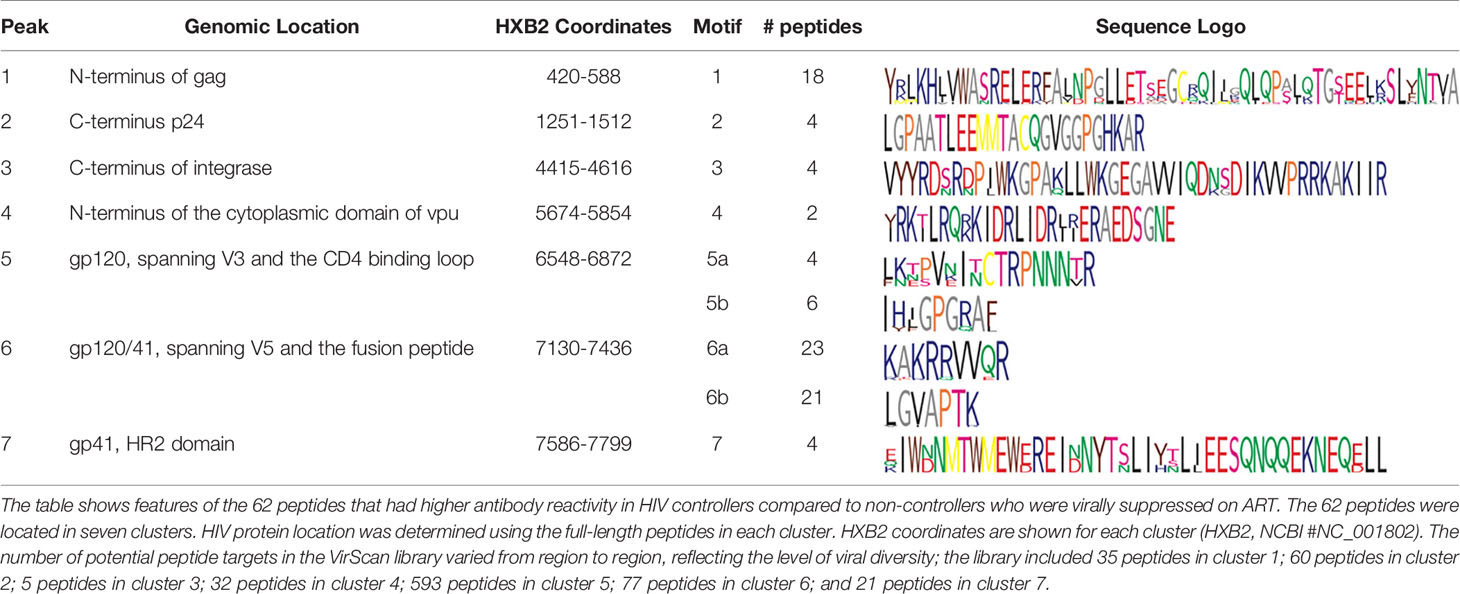
Table 2 Peptides with higher antibody reactivity in HIV controllers compared to non-controllers who were virally suppressed on antiretroviral therapy.
The program epitopefindr (40) was used to identify amino acid motifs that were common to peptides in each cluster. The number of peptides included in each motif varied from two to 23 (Table 2). Five clusters had one motif (clusters 1-4 and 7) and two had two motifs (clusters 5 and 6). Table 2 shows additional characteristics of the nine motifs. Note that one peptide near cluster 5 did not include either of the motifs associated with other peptides in that cluster. The pattern of antibody reactivity to peptides in each cluster varied among study participants (Figure 3). Most elite and viremic controllers had increased antibody reactivity to multiple peptides in multiple clusters. In contrast, antibody reactivity was minimal among non-controllers who were virally suppressed on ART and was predominantly observed for integrase peptides in viremic non-controllers (cluster 3).
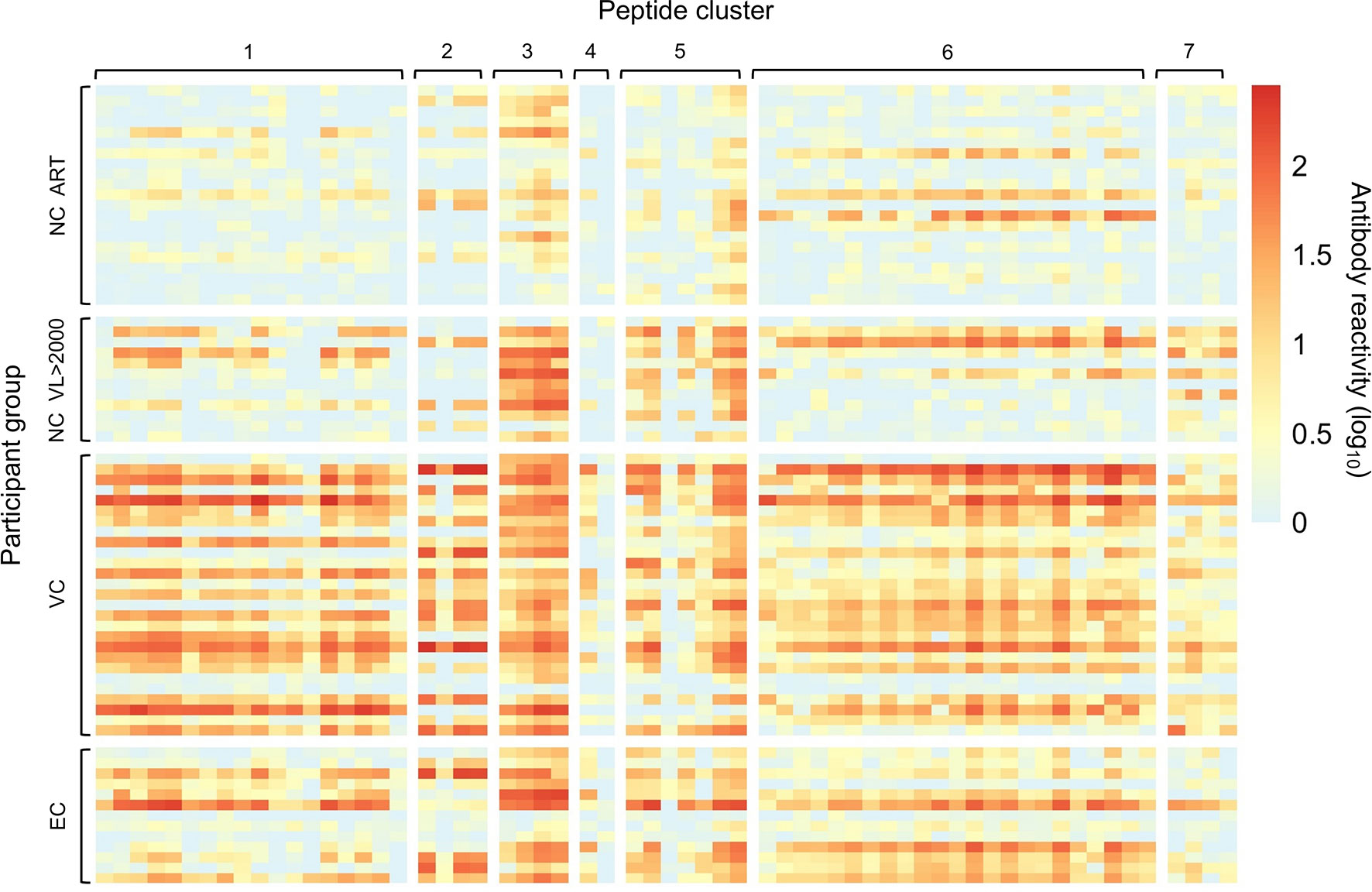
Figure 3 Individual patterns of antibody reactivity to peptides in peptide clusters. The patterns of antibody reactivity are shown for the 13 individual elite controllers (EC), 27 viremic controllers (VC), 12 non-controllers with viral loads >2,000 copies/mL (NC VL >2000), and non-controllers who were virally suppressed on ART (NC ART). Colors indicate the level of antibody reactivity (log10 fold change in VirScan read counts compared to mock immunoprecipitation reactions); values less than zero were assigned a value of zero. Each colored square represents data for a single peptide from a single individual. Darker colors indicate higher levels of antibody reactivity. Data are grouped by participant group and peptide cluster (see Table 2).
Analysis of Antibody Reactivity in Independent Cohorts
We next evaluated antibody reactivity to the 62 significant peptides using samples from an independent Validation Cohort that included samples from 29 elite controllers and 37 non-controllers who were virally-suppressed on ART (Table 1). Figure S2 shows the difference in antibody reactivity in these two groups. Reactivity in elite controllers was higher for 51 peptides and lower for 11 peptides. We used a permutation test to compare antibody reactivity for all 62 peptides combined in the two participant groups. The average antibody reactivity for the combined set of peptides was significantly higher for elite controllers than for virally suppressed non-controllers (p=0.024).
Association of HIV Peptide Reactivity With the Protective HLA-B*57 Allele
We then evaluated the association between antibody reactivity and the presence of the protective HLA-B*57 allele. This analysis was performed using samples from the 40 HIV controllers in the Discovery Cohort (21) (27 with the HLA-B*57 allele, 13 without the allele; Figure S3). None of the peptides in the VirScan library (including the 62 peptides described above) had antibody reactivity that was significantly different between these two groups.
Association Between Viral Load Set Point and Antibody Reactivity
We next evaluated whether antibody reactivity to the 62 peptides was associated with viral load set point in non-controllers who were not on ART. This analysis was performed using samples from the RV217 Cohort, which included men and women from East Africa and Thailand with different risk factors for infection. Antibody reactivity to nine of the 62 peptides was significantly associated with viral load set point after using a permutation test for multiple testing correction (Figure 4). All nine peptides were located in gag protein, p17 (cluster 1; Table S2).
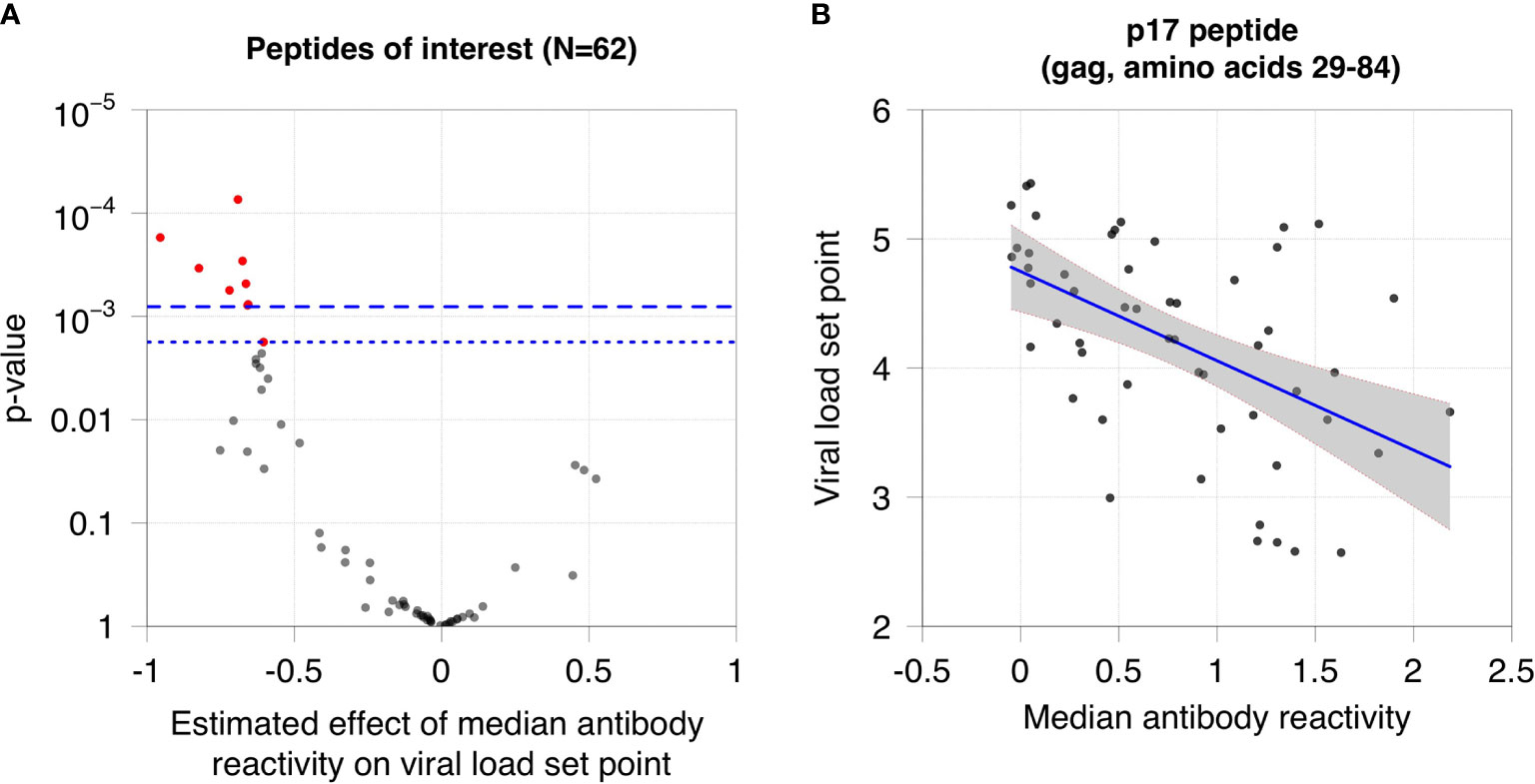
Figure 4 Association between viral load set point and antibody reactivity in the RV217 Cohort. (A) The plot shows the association between viral load set point and median antibody reactivity for the 62 peptides of interest. The x-axis shows the estimated effect of median antibody reactivity on viral load set point (effect size from the regression analysis); the y-axis shows the -log10 p-value of the association. Negative effect sizes indicate that antibody reactivity was higher for participants with lower viral load set points. The dashed line indicates the cutoff for significance using the Bonferroni correction (p=0.0008); the dotted line indicates the cutoff for significance using a permutation test (p=0.0018). Red dots indicate peptides that had a statistically significant association between viral load set point and median antibody reactivity. (B) The plot shows the median reactivity of the peptide (x-axis) that had the strongest observed association with viral load set point (y-axis). Each dot represents data from a single participant. The blue line indicates the least squares regression line. The grey shaded area represents the 95% confidence band for the mean viral load set point.
Discussion
Using the VirScan antibody profiling assay, we compared the antibody profiles in viremic non-controllers to persons with natural HIV control and those who were virally suppressed on ART. In a previous study, we demonstrated that most persons living with HIV express a diverse array of anti-HIV antibodies (31). This report was focused on characterization of antibodies in persons with natural and ART-induced viral suppression and persons with lower viral load set points. Previous studies show that overall HIV antibody expression is down-regulated in HIV controllers and individuals who are virally suppressed on ART (22, 41).
We identified groups of peptides that were preferentially targeted by antibodies in HIV controllers and persons with lower viral load set points. These peptides were grouped into homologous clusters in HIV env (gp120 and gp41), gag (p17 and p24), integrase, and vpu. Most of the HIV controllers in the Discovery Cohort had antibodies to peptides in all or most of the seven clusters. The level of reactivity to these peptides was similar in viremic non-controllers and HIV controllers, but was significantly lower in non-controllers who were suppressed on ART. Aggregate antibody reactivity to the same peptides was significantly higher in elite controllers compared to ART-suppressed non-controllers in an independent cohort. These findings demonstrate that elite and viremic HIV controllers produce many of the same antibodies as viremic non-controllers, and that these antibodies persist in HIV controllers despite long-term, natural suppression of viral replication. It is not clear whether the high levels of expression of these antibodies in HIV controllers reflects on-going viral replication (antigen stimulation) or other factors. It is also not clear whether these antibodies play a role in the natural control of HIV infection.
We also found that the pattern of antibody reactivity to peptides in the clusters varied among non-controllers, depending on HIV viral load. In the RV217 Cohort, antibody reactivity to peptides in the p17 cluster was significantly higher in non-controllers with lower viral load set points. These peptides are located in the p17 matrix protein. p17 is released from infected cells and plays a critical role in the viral replication cycle (42). This protein exerts cytokine-like activities that promote viral replication through interaction with target cell receptors (43). The functional epitope of p17 is located at the N-terminus of the protein (44), immediately upstream from the peptides identified in this report. Antibodies to p17 are associated with slower HIV disease progression (45). A synthetic peptide that includes the functional epitope of p17 was evaluated in a Phase 1 clinical trial as a therapeutic vaccine (46, 47). Further studies are needed to determine whether antibodies directed against p17 or other gag epitopes identified in this study play a functional role in natural control of HIV replication.
Three of the seven peptide clusters identified in this study were in HIV env. The capacity of env antibodies to modulate HIV replication is well-established. Broadly neutralizing monoclonal antibodies directed against HIV env are being evaluated in clinical trials as interventions for HIV prevention and treatment (48). These antibodies target epitopes on the surface of gp120 (e.g., in the CD4 binding site, the V3 loop, and V1/V2 region) or gp41 (in the MPER) (48). Most of the env peptides identified in this report were in gp120, spanning the V3 loop and CD4 binding loop or the V5 loop and fusion peptide; four peptides were located in the coiled coil domain of gp41, proximal to the MPER.
Antibodies to integrase peptides were also targeted by HIV controllers in this report. These antibodies were also observed in some viremic non-controllers; however, this reactivity was much lower when non-controllers were virally suppressed on ART. Antibody reactivity to non-surface proteins has been observed in other viral infections. For example, strong reactivity to nuclear antigens is observed in Epstein Barr virus infection (49), and reactivity to the nucleocapsid protein is often used to evaluate prior SARS-CoV-2 infection (50). In HIV-infected individuals, integrase antibodies often appear after antibodies to other HIV targets (51, 52). Intracellular antibody fragments directed against HIV integrase or its cellular target have been shown to inhibit viral replication (53, 54).
We also identified vpu peptides that were preferentially targeted in HIV controllers compared to non-controllers who were suppressed on ART. Vpu is a transmembrane protein that enhances virion release from infected cells by antagonizing the host restriction factor, tetherin. Enhanced release of virions by vpu may reduce antibody binding to infected cells by limiting the amount of time that gp120 is expressed on the cell surface (55). In a previous study, antibodies directed against specific ADCC vpu epitopes were detected in long-term slow progressors (LTSP), but not in a control group of non-LTSP (56). Antibodies directed against a vpu peptide were also more likely to elicit responses in an ADCC assay in elite controllers compared to viremic individuals (57). The vpu peptides targeted by LTSP and elite controllers in those studies overlap with the vpu peptides that were preferentially targeted in HIV controllers in this report. The sequence of one peptide cluster from the LTSP study was contained within the vpu amino acid motif identified in this report; the sequence of another peptide cluster from the LTSP study overlapped with that motif (Table S3).
Many assays used to evaluate the specificity of HIV antibodies include antigen targets that are expressed as proteins rather than discrete peptides, or include a small number of protein and peptide targets (58–60). In contrast, the VirScan assay provides a comprehensive, unbiased platform for assessing the full range of linear, non-glycosylated HIV peptide targets. By aligning reactive peptides that share sequence homology, VirScan also provides information on the fine specificity of antibodies, including specific amino acid motifs that were preferentially targeted in HIV controllers. Further studies are needed to characterize antibody binding to non-linear and glycosylated epitopes.
Several factors may have limited our ability to identify the full range of antibody targets associated with viremic control of HIV infection. The sample sets used for peptide discovery were relatively small and were limited to samples from the United States that were likely to have subtype B HIV. These samples were collected from the general population and from a cohort comprised predominantly of men who have sex with men; other HIV subtypes and risk groups were not represented. Another limitation of this study is that the VirScan assay only detects antibody reactivity to linear epitopes in unglycosylated target peptides; the conformation of peptides expressed on bacteriophage may also differ from the natural conformation of peptides expressed in vivo. Antibodies to peptides in the HIV antisense protein 62 will also not be detected, since the current VirScan library does not include these peptides. This study also does not provide any information about cellular responses to HIV infection, which have been shown to play an important role in viremic control of HIV infection. However, T cell and B cell responses are coordinated and frequently target closely linked or overlapping epitopes. The antibody specificities reported here may therefore be associated with enhanced T cell recognition of linked epitopes presented on infected cells.
Conclusion
The studies cited above and findings in this report demonstrate that expression of antibodies against gp120, gp41, p17, integrase, and vpu persist in HIV controllers despite viral suppression. Additional research is needed to determine whether antibodies directed against peptides in these regions play a role in controlling HIV replication, and/or are indicators of enhanced T cell responses. This could be evaluated by comparing proviral and plasma sequences from HIV controllers to assess whether there is evidence of antibody-mediated selection against these peptides; this approach was used previously to identify cytotoxic T lymphocyte (CTL) escape mutations in HIV controllers reflecting selective pressure against HLA-B*57-restricted epitopes (61). It may also be possible to generate antibodies against epitopes identified in this report, or to isolate those antibodies from HIV controllers, and to test whether those antibodies can neutralize HIV or exert other functional effects on viral replication. Further characterization of antibodies in HIV controllers in other cohorts and populations may provide further insights into antibody-mediated control of HIV infection. This information may help inform development of therapeutic vaccines and monoclonal antibodies for HIV treatment and prevention. The VirScan assay also contains phage that express peptides spanning the full genomes of >200 other viruses that infect humans. The approach used in this report could also be used to identify peptides associated with control of viral replication for other clinically relevant viruses.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by IRB. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
HBL: Designed the study, analyzed data, and drafted the manuscript. SE: Designed the study, analyzed data, and drafted the manuscript. OL: Designed the study and provided input into data analysis. DM: Performed testing and analyzed data. AC: Contributed to study design and analyzed data. SH: Performed testing. WG-M: Assisted with literature review. GA: Provided samples and data from the SCOPE Study. RDM: Project lead for the JHU Moore Clinic Cohort. SD: Project lead for the SCOPE Study. LE: Investigator for the RV217 Study. CM: Project lead for the Genital Shedding Study. JB: Provided expertise on elite and viremic HIV controllers. IR: Provided statistical and bioinformatics expertise and analyzed data. KK: Provided statistical and bioinformatics expertise, analyzed data, and drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) through R01-AI095068 and the National Institute of General Medical Sciences (NIGMS) through R01-GM136724. Additional support was provided through the Laboratory Center of HIV Prevention Trials Network (HPTN) which is sponsored by the NIAID, National Institute on Drug Abuse, National Institute of Mental Health, and Office of AIDS Research, of the NIH, DHHS (UM1-AI068613), and through intramural funding from the Division of Intramural Research, NIAID, NIH. The Johns Hopkins HIV Cohort was supported by NIH grants U01-DA036935 and U01-AI069918. The Johns Hopkins Medicine Elite Controller Cohort was supported by NIH grant R01-AI140789. The RV217 study was supported by a cooperative agreement (W81XWH-18-2-0040) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., and the U.S. Department of Defense. This research was also funded, in part, by NIAID. The SCOPE cohort was supported by the UCSF/Gladstone Institute of Virology & Immunology CFAR (P30-AI027763).
Author Disclaimer
The views expressed in this report are those of the authors and should not be construed to represent the positions of the U.S. Army, the Department of Defense, or the Henry M. Jackson Foundation. The investigators in the RV217 Study adhered to the policies for protection of human subjects as prescribed in the Army Publishing Directorate AR 70–25.
Conflict of Interest
HBL is an inventor on a patent application (US20160320406A) filed by Brigham and Women’s Hospital that covers the use of the VirScan technology to identify pathogen antibodies and is a founder of ImmuneID, Portal Bioscience and Alchemab, and is an advisor to TScan Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the study teams and study participants who provided data and samples for this research. We are grateful to Stephen J. Elledge at Harvard Medical School and Brigham & Women’s Hospital for providing the VirScan phage display library, and to Mary Carrington at the National Cancer Institute for providing data for HLA typing. Thanks to Kwan Pui Chan for his critical reading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.740395/full#supplementary-material
References
1. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Antiretroviral Therapy for the Prevention of HIV-1 Transmission. N Engl J Med (2016) 375(9):830–9. doi: 10.1056/NEJMoa1600693
2. Deeks SG, Walker BD. Human Immunodeficiency Virus Controllers: Mechanisms of Durable Virus Control in the Absence of Antiretroviral Therapy. Immunity (2007) 27(3):406–16. doi: 10.1016/j.immuni.2007.08.010
3. Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, et al. Genetic and Immunologic Heterogeneity Among Persons Who Control HIV Infection in the Absence of Therapy. J Infect Dis (2008) 197(4):563–71. doi: 10.1086/526786
4. Gonzalo-Gil E, Ikediobi U, Sutton RE. Mechanisms of Virologic Control and Clinical Characteristics of HIV+ Elite/Viremic Controllers. Yale J Biol Med (2017) 90(2):245–59.
5. Leon A, Perez I, Ruiz-Mateos E, Benito JM, Leal M, Lopez-Galindez C, et al. Rate and Predictors of Progression in Elite and Viremic HIV-1 Controllers. AIDS (2016) 30(8):1209–20. doi: 10.1097/QAD.0000000000001050
6. Blankson JN, Bailey JR, Thayil S, Yang HC, Lassen K, Lai J, et al. Isolation and Characterization of Replication-Competent Human Immunodeficiency Virus Type 1 From a Subset of Elite Suppressors. J Virol (2007) 81(5):2508–18. doi: 10.1128/JVI.02165-06
7. Lamine A, Caumont-Sarcos A, Chaix ML, Saez-Cirion A, Rouzioux C, Delfraissy JF, et al. Replication-Competent HIV Strains Infect HIV Controllers Despite Undetectable Viremia (ANRS EP36 Study). AIDS (2007) 21(8):1043–5. doi: 10.1097/QAD.0b013e3280d5a7ac
8. Salgado M, Swanson MD, Pohlmeyer CW, Buckheit RW 3rd, Wu J, Archin NM, et al. HLA-B*57 Elite Suppressor and Chronic Progressor HIV-1 Isolates Replicate Vigorously and Cause CD4+ T Cell Depletion in Humanized BLT Mice. J Virol (2014) 88(6):3340–52. doi: 10.1128/JVI.03380-13
9. Veenhuis RT, Kwaa AK, Garliss CC, Latanich R, Salgado M, Pohlmeyer CW, et al. Long-Term Remission Despite Clonal Expansion of Replication-Competent HIV-1 Isolates. JCI Insight (2018) 3(18):1–12. doi: 10.1172/jci.insight.122795
10. Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, et al. HLA B*5701 Is Highly Associated With Restriction of Virus Replication in a Subgroup of HIV-Infected Long Term Nonprogressors. Proc Natl Acad Sci USA (2000) 97(6):2709–14. doi: 10.1073/pnas.050567397
11. Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A Whole-Genome Association Study of Major Determinants for Host Control of HIV-1. Science (2007) 317(5840):944–7. doi: 10.1126/science.1143767
12. International HIVCS, Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, et al. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science (2010) 330(6010):1551–7. doi: 10.1126/science.1195271
13. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV Nonprogressors Preferentially Maintain Highly Functional HIV-Specific CD8+ T Cells. Blood (2006) 107(12):4781–9. doi: 10.1182/blood-2005-12-4818
14. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, et al. HIV-Specific CD8+ T Cell Proliferation Is Coupled to Perforin Expression and Is Maintained in Nonprogressors. Nat Immunol (2002) 3(11):1061–8. doi: 10.1038/ni845
15. Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, et al. Lytic Granule Loading of CD8+ T Cells Is Required for HIV-Infected Cell Elimination Associated With Immune Control. Immunity (2008) 29(6):1009–21. doi: 10.1016/j.immuni.2008.10.010
16. Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, et al. HIV Controllers Exhibit Potent CD8 T Cell Capacity to Suppress HIV Infection Ex Vivo and Peculiar Cytotoxic T Lymphocyte Activation Phenotype. Proc Natl Acad Sci USA (2007) 104(16):6776–81. doi: 10.1073/pnas.0611244104
17. Friedrich TC, Valentine LE, Yant LJ, Rakasz EG, Piaskowski SM, Furlott JR, et al. Subdominant CD8+ T-Cell Responses Are Involved in Durable Control of AIDS Virus Replication. J Virol (2007) 81(7):3465–76. doi: 10.1128/JVI.02392-06
18. Pandrea I, Gaufin T, Gautam R, Kristoff J, Mandell D, Montefiori D, et al. Functional Cure of SIVagm Infection in Rhesus Macaques Results in Complete Recovery of CD4+ T Cells and Is Reverted by CD8+ Cell Depletion. PloS Pathog (2011) 7(8):e1002170. doi: 10.1371/journal.ppat.1002170
19. Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic Interaction Between KIR3DS1 and HLA-B Delays the Progression to AIDS. Nat Genet (2002) 31(4):429–34. doi: 10.1038/ng934
20. Martin MP, Naranbhai V, Shea PR, Qi Y, Ramsuran V, Vince N, et al. Killer Cell Immunoglobulin-Like Receptor 3DL1 Variation Modifies HLA-B*57 Protection Against HIV-1. J Clin Invest (2018) 128(5):1903–12. doi: 10.1172/JCI98463
21. Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, et al. Innate Partnership of HLA-B and KIR3DL1 Subtypes Against HIV-1. Nat Genet (2007) 39(6):733–40. doi: 10.1038/ng2035
22. Laeyendecker O, Rothman RE, Henson C, Horne BJ, Ketlogetswe KS, Kraus CK, et al. The Effect of Viral Suppression on Cross-Sectional Incidence Testing in the Johns Hopkins Hospital Emergency Department. J Acquir Immune Defic Syndr (2008) 48(2):211–5. doi: 10.1097/QAI.0b013e3181743980
23. Margolis DM, Koup RA, Ferrari G. HIV Antibodies for Treatment of HIV Infection. Immunol Rev (2017) 275(1):313–23. doi: 10.1111/imr.12506
24. Bailey JR, Lassen KG, Yang HC, Quinn TC, Ray SC, Blankson JN, et al. Neutralizing Antibodies Do Not Mediate Suppression of Human Immunodeficiency Virus Type 1 in Elite Suppressors or Selection of Plasma Virus Variants in Patients on Highly Active Antiretroviral Therapy. J Virol (2006) 80(10):4758–70. doi: 10.1128/JVI.80.10.4758-4770.2006
25. Lambotte O, Ferrari G, Moog C, Yates NL, Liao HX, Parks RJ, et al. Heterogeneous Neutralizing Antibody and Antibody-Dependent Cell Cytotoxicity Responses in HIV-1 Elite Controllers. AIDS (2009) 23(8):897–906. doi: 10.1097/QAD.0b013e328329f97d
26. Lambotte O, Pollara J, Boufassa F, Moog C, Venet A, Haynes BF, et al. High Antibody-Dependent Cellular Cytotoxicity Responses Are Correlated With Strong CD8 T Cell Viral Suppressive Activity But Not With B57 Status in HIV-1 Elite Controllers. PloS One (2013) 8(9):e74855. doi: 10.1371/journal.pone.0074855
27. Smalls-Mantey A, Doria-Rose N, Klein R, Patamawenu A, Migueles SA, Ko SY, et al. Antibody-Dependent Cellular Cytotoxicity Against Primary HIV-Infected CD4+ T Cells Is Directly Associated With the Magnitude of Surface IgG Binding. J Virol (2012) 86(16):8672–80. doi: 10.1128/JVI.00287-12
28. Isitman G, Lisovsky I, Tremblay-McLean A, Kovacs C, Harris M, Routy JP, et al. Antibody-Dependent Cellular Cytotoxicity Activity of Effector Cells From HIV-Infected Elite and Viral Controllers. AIDS Res Hum Retroviruses (2016) 32(10-11):1079–88. doi: 10.1089/aid.2016.0157
29. Ackerman ME, Mikhailova A, Brown EP, Dowell KG, Walker BD, Bailey-Kellogg C, et al. Polyfunctional HIV-Specific Antibody Responses Are Associated With Spontaneous HIV Control. PloS Pathog (2016) 12(1):e1005315. doi: 10.1371/journal.ppat.1005315
30. Xu GJ, Kula T, Xu Q, Li MZ, Vernon SD, Ndung’u T, et al. Viral Immunology. Comprehensive Serological Profiling of Human Populations Using a Synthetic Human Virome. Science (2015) 348(6239):aaa0698. doi: 10.1126/science.aaa0698
31. Eshleman SH, Laeyendecker O, Kammers K, Chen A, Sivay MV, Kottapalli S, et al. Comprehensive Profiling of HIV Antibody Evolution. Cell Rep (2019) 27(5):1422–33.e4. doi: 10.1016/j.celrep.2019.03.097
32. Asher AK, Santos GM, Evans J, Dokubo EK, Lee TH, Martin JN, et al. Human Leukocyte Antigen B*57 Does Not Fully Explain Hepatitis C Clearance in HIV Controllers. AIDS (2013) 27(17):2691–6. doi: 10.1097/01.aids.0000433242.86362.21
33. Hunt PW, Hatano H, Sinclair E, Lee TH, Busch MP, Martin JN, et al. HIV-Specific CD4+ T Cells May Contribute to Viral Persistence in HIV Controllers. Clin Infect Dis (2011) 52(5):681–7. doi: 10.1093/cid/ciq202
34. Moore RD. Understanding the Clinical and Economic Outcomes of HIV Therapy: The Johns Hopkins HIV Clinical Practice Cohort. J Acquir Immune Defic Syndr Hum Retrovirol (1998) 17(Suppl 1):S38–41. doi: 10.1097/00042560-199801001-00011
35. Robb ML, Eller LA, Kibuuka H, Rono K, Maganga L, Nitayaphan S, et al. Prospective Study of Acute HIV-1 Infection in Adults in East Africa and Thailand. N Engl J Med (2016) 374(22):2120–30. doi: 10.1056/NEJMoa1508952
36. Mohan D, Wansley DL, Sie BM, Noon MS, Baer AN, Laserson U, et al. Publisher Correction: PhIP-Seq Characterization of Serum Antibodies Using Oligonucleotide-Encoded Peptidomes. Nat Protoc (2019) 14(8):2596. doi: 10.1038/s41596-018-0088-4
37. Smyth GK. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat Appl Genet Mol Biol (2004) 3:Article3. doi: 10.2202/1544-6115.1027
38. Storey JD, Tibshirani R. Statistical Significance for Genomewide Studies. Proc Natl Acad Sci USA (2003) 100(16):9440–5. doi: 10.1073/pnas.1530509100
41. Wendel SK, Mullis CE, Eshleman SH, Blankson JN, Moore RD, Keruly JC, et al. Effect of Natural and ARV-Induced Viral Suppression and Viral Breakthrough on Anti-HIV Antibody Proportion and Avidity in Patients With HIV-1 Subtype B Infection. PloS One (2013) 8(2):e55525. doi: 10.1371/journal.pone.0055525
42. Fiorentini S, Marini E, Caracciolo S, Caruso A. Functions of the HIV-1 Matrix Protein P17. New Microbiol (2006) 29(1):1–10.
43. Caccuri F, Marsico S, Fiorentini S, Caruso A, Giagulli C. HIV-1 Matrix Protein P17 and Its Receptors. Curr Drug Targets (2016) 17(1):23–32. doi: 10.2174/1389450116666150825110840
44. De Francesco MA, Baronio M, Fiorentini S, Signorini C, Bonfanti C, Poiesi C, et al. HIV-1 Matrix Protein P17 Increases the Production of Proinflammatory Cytokines and Counteracts IL-4 Activity by Binding to a Cellular Receptor. Proc Natl Acad Sci USA (2002) 99(15):9972–7. doi: 10.1073/pnas.142274699
45. Fiorentini S, Giagulli C, Caccuri F, Magiera AK, Caruso A. HIV-1 Matrix Protein P17: A Candidate Antigen for Therapeutic Vaccines Against AIDS. Pharmacol Ther (2010) 128(3):433–44. doi: 10.1016/j.pharmthera.2010.08.005
46. Foca E, Iaria ML, Caccuri F, Fiorentini S, Motta D, Giagulli C, et al. Long-Lasting Humoral Immune Response Induced in HIV-1-Infected Patients by a Synthetic Peptide (AT20) Derived From the HIV-1 Matrix Protein P17 Functional Epitope. HIV Clin Trials (2015) 16(4):157–62. doi: 10.1179/1528433614Z.0000000018
47. Iaria ML, Fiorentini S, Foca E, Zicari S, Giagulli C, Caccuri F, et al. Synthetic HIV-1 Matrix Protein P17-Based AT20-KLH Therapeutic Immunization in HIV-1-Infected Patients Receiving Antiretroviral Treatment: A Phase I Safety and Immunogenicity Study. Vaccine (2014) 32(9):1072–8. doi: 10.1016/j.vaccine.2013.12.051
48. Gruell H, Klein F. Antibody-Mediated Prevention and Treatment of HIV-1 Infection. Retrovirology (2018) 15(1):73. doi: 10.1186/s12977-018-0455-9
49. Corrales I, Gimenez E, Navarro D. Evaluation of the Architect Epstein-Barr Virus (EBV) Viral Capsid Antigen (VCA) IgG, VCA IgM, and EBV Nuclear Antigen 1 IgG Chemiluminescent Immunoassays for Detection of EBV Antibodies and Categorization of EBV Infection Status Using Immunofluorescence Assays as the Reference Method. Clin Vaccine Immunol (2014) 21(5):684–8. doi: 10.1128/CVI.00104-14
50. Burbelo PD, Riedo FX, Morishima C, Rawlings S, Smith D, Das S, et al. Sensitivity in Detection of Antibodies to Nucleocapsid and Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus 2 in Patients With Coronavirus Disease 2019. J Infect Dis (2020) 222(2):206–13. doi: 10.1093/infdis/jiaa273
51. Fiebig EW, Wright DJ, Rawal BD, Garrett PE, Schumacher RT, Peddada L, et al. Dynamics of HIV Viremia and Antibody Seroconversion in Plasma Donors: Implications for Diagnosis and Staging of Primary HIV Infection. AIDS (2003) 17(13):1871–9. doi: 10.1097/00002030-200309050-00005
52. Rikhtegaran Tehrani Z, Azadmanesh K, Mostafavi E, Gharibzadeh S, Soori S, Azizi M, et al. High Avidity Anti-Integrase Antibodies Discriminate Recent and Non-Recent HIV Infection: Implications for HIV Incidence Assay. J Virol Methods (2018) 253:5–10. doi: 10.1016/j.jviromet.2017.12.003
53. Bao L, Hannon C, Cruz-Mignoni A, Ptchelkine D, Sun MY, Miller A, et al. Intracellular Immunization Against HIV Infection With an Intracellular Antibody That Mimics HIV Integrase Binding to the Cellular LEDGF Protein. Sci Rep (2017) 7(1):16869. doi: 10.1038/s41598-017-16742-2
54. Rondon IJ, Marasco WA. Intracellular Antibodies (Intrabodies) for Gene Therapy of Infectious Diseases. Annu Rev Microbiol (1997) 51:257–83. doi: 10.1146/annurev.micro.51.1.257
55. Kramski M, Stratov I, Kent SJ. The Role of HIV-Specific Antibody-Dependent Cellular Cytotoxicity in HIV Prevention and the Influence of the HIV-1 Vpu Protein. AIDS (2015) 29(2):137–44. doi: 10.1097/QAD.0000000000000523
56. Wren LH, Chung AW, Isitman G, Kelleher AD, Parsons MS, Amin J, et al. Specific Antibody-Dependent Cellular Cytotoxicity Responses Associated With Slow Progression of HIV Infection. Immunology (2013) 138(2):116–23. doi: 10.1111/imm.12016
57. Madhavi V, Wines BD, Amin J, Emery S, Group ES, Lopez E, et al. HIV-1 Env- and Vpu-Specific Antibody-Dependent Cellular Cytotoxicity Responses Associated With Elite Control of HIV. J Virol (2017) 91(18):e00700–17. doi: 10.1128/JVI.00700-17
58. Dotsey EY, Gorlani A, Ingale S, Achenbach CJ, Forthal DN, Felgner PL, et al. A High Throughput Protein Microarray Approach to Classify HIV Monoclonal Antibodies and Variant Antigens. PloS One (2015) 10(5):e0125581. doi: 10.1371/journal.pone.0125581
59. Curtis KA, Kennedy MS, Charurat M, Nasidi A, Delaney K, Spira TJ, et al. Development and Characterization of a Bead-Based, Multiplex Assay for Estimation of Recent HIV Type 1 Infection. AIDS Res Hum Retroviruses (2012) 28(2):188–97. doi: 10.1089/aid.2011.0037
60. Kong W, Li Y, Cheng S, Yan C, An S, Dong Z, et al. Luminex xMAP Combined With Western Blot Improves HIV Diagnostic Sensitivity. J Virol Methods (2016) 227:1–5. doi: 10.1016/j.jviromet.2015.10.007
Keywords: HIV control, viral load set point, antibody profiling, phage display, VirScan
Citation: Kammers K, Chen A, Monaco DR, Hudelson SE, Grant-McAuley W, Moore RD, Alter G, Deeks SG, Morrison CS, Eller LA, Blankson JN, Laeyendecker O, Ruczinski I, Eshleman SH and Larman HB (2021) HIV Antibody Profiles in HIV Controllers and Persons With Treatment-Induced Viral Suppression. Front. Immunol. 12:740395. doi: 10.3389/fimmu.2021.740395
Received: 13 July 2021; Accepted: 10 August 2021;
Published: 26 August 2021.
Edited by:
Nicholas Funderburg, The Ohio State University, United StatesReviewed by:
Rebecca M. Lynch, George Washington University, United StatesCarey Shive, Louis Stokes Cleveland VA Medical Center, United States
Copyright © 2021 Kammers, Chen, Monaco, Hudelson, Grant-McAuley, Moore, Alter, Deeks, Morrison, Eller, Blankson, Laeyendecker, Ruczinski, Eshleman and Larman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. Benjamin Larman, aGxhcm1hbjFAamhtaS5lZHU=; Susan H. Eshleman, c2VzaGxlbUBqaG1pLmVkdQ==
†These authors have contributed equally to this work