 Qi Zhou
Qi Zhou Yunhe Xiong
Yunhe Xiong Bing Qu1†
Bing Qu1†- 1Reproductive Medical Center, Renmin Hospital of Wuhan University, Wuhan, China
- 2Urology Department, Renmin Hospital of Wuhan University, Wuhan, China
- 3Department of Clinical Laboratory, Renmin Hospital of Wuhan University, Wuhan, China
Recurrent pregnancy loss (RPL) is a common and severe pathological pregnancy, whose pathogenesis is not fully understood. With the development of epigenetics, the study of DNA methylation, provides a new perspective on the pathogenesis and therapy of RPL. The abnormal DNA methylation of imprinted genes, placenta-specific genes, immune-related genes and sperm DNA may, directly or indirectly, affect embryo implantation, growth and development, leading to the occurrence of RPL. In addition, the unique immune tolerogenic microenvironment formed at the maternal-fetal interface has an irreplaceable effect on the maintenance of pregnancy. In view of these, changes in the cellular components of the maternal-fetal immune microenvironment and the regulation of DNA methylation have attracted a lot of research interest. This review summarizes the research progress of DNA methylation involved in the occurrence of RPL and the regulation of the maternal-fetal immune microenvironment. The review provides insights into the personalized diagnosis and treatment of RPL.
Introduction
Recurrent pregnancy loss (RPL) refers to three or more consecutive spontaneous abortion with the same spouse before 28 weeks of gestation. This complication affects about 5% of women of childbearing age globally (1). The latest epidemiological data released by the American Reproductive Medicine showed a spontaneous abortion prevalence of 15-20%, of which the incidence of RPL comprised approximately 2-5% of the total pregnancy (2). The pathogenesis of RPL is a complex process comprising abnormal chromosome structure and function, immune dysfunction, endocrine disorder, abnormal reproductive structure, maternal factors, prethrombotic state, and the environment, among others (3, 4) (Figure 1). Although concentrated research has identified numerous causes for RPL, the etiology and pathogenesis of about 50% of RPL cases remain unclear. Such cases are referred to as, unexplained recurrent pregnancy loss (UPRL). Exploring the pathogenesis of URPL and providing early intervention are critical in ensuring a higher RPL live birth rate and a better pregnancy outcome.
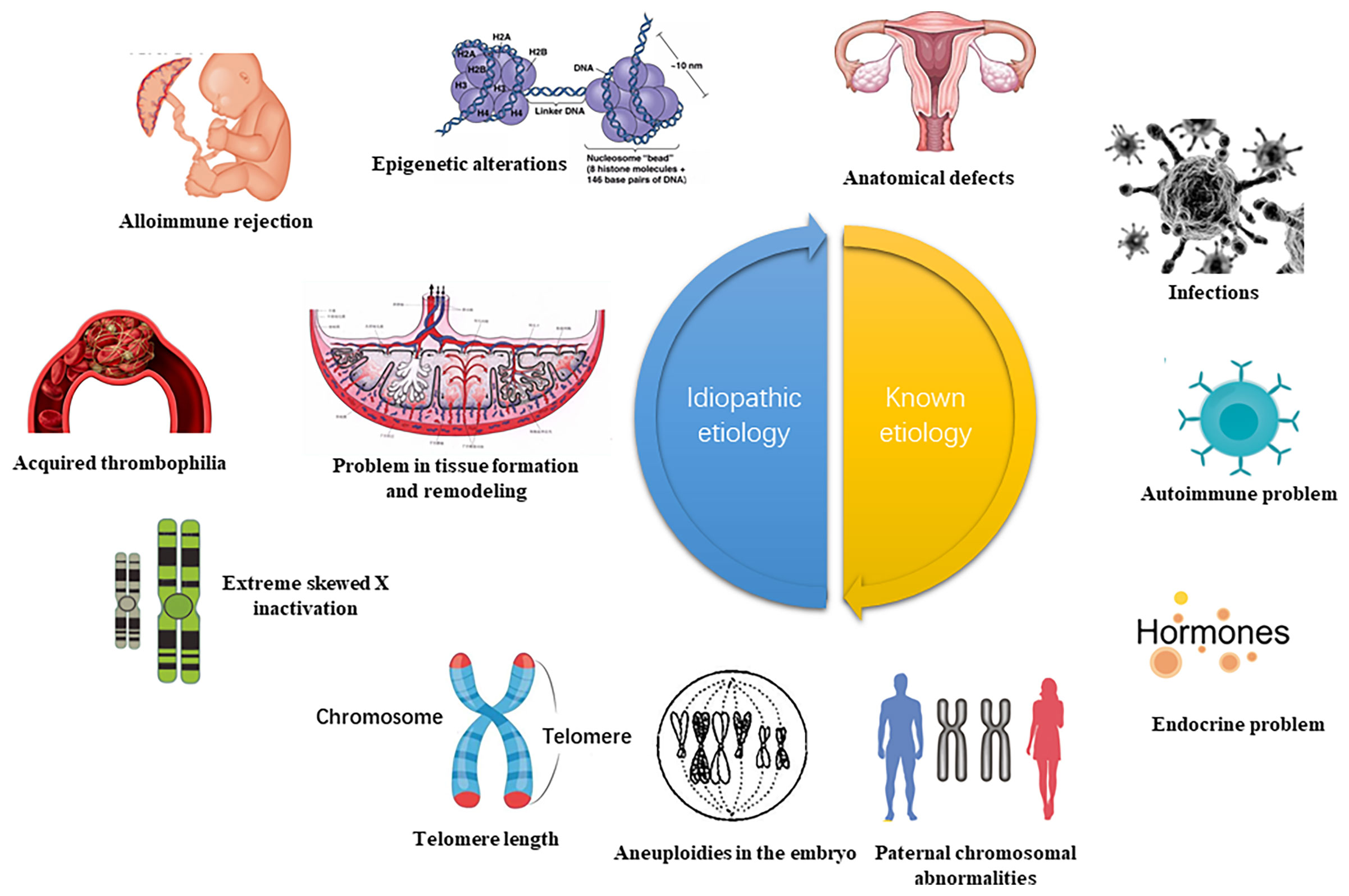
Figure 1 High risk of RPL (recurrent spontaneous abortion) (5). The etiology of RPL can be diagnosed by interrogation, clarifying the gestational age and characteristics of the miscarriage. Early RPL may be related to abnormal chromosome structure and function, immune dysfunction, endocrine disorder, abnormal reproductive system, prethrombotic state (PTS), poor environment and living habits. The function of the uterine cervical, the development of amniotic fluid, and the umbilical cord may also be linked to late RPL. This review focuses on the abnormal immune function of RPL and analyze the important role of DNA methylation.
The development of epigenetics provides a new perspective for the pathogenesis of RPL. Epigenetics refers that cells maintain or alter gene expression in stable or heritable manner without changing DNA sequence, including DNA or RNA methylation, histone or chromatin post-translational modification (PTM), and non-coding RNA (ncRNA) regulation et al. Epigenetics plays a vital role in the occurrence and development of RPL through regulating the expression of key genes determining cell differentiation, proliferation and apoptosis, which has been passed on to future generations by intergenerational and cross-generational inheritance and affected their health. These genes are expected to be the potential biomarkers for the diagnosis and new targets for the treatment of RPL. Numerous epigenetic studies have found that RPL is associated with defects in DNA methylation, methylation modification, and other related epigenetic modifications. Imprinting barriers, gene expression disorders, spermatozoa defect, and immune imbalance induced by abnormal DNA methylation may affect the process of embryo implantation, growth and development, and ultimately lead to the occurrence of RPL. To provide a new direction for the diagnosis and treatment of RPL, this review elaborates on the role of DNA methylation in the pathogenesis of RPL and its regulatory pathways.
DNA Methylation and Related Enzymes
DNA methylation is the earliest and most extensively studied epigenetic modification, which takes CpG Island as the central link. DNA methylation prevents the transcription factor (TF) from combining with DNA, or promotes the recruitment of methyl binding domain proteins for chromatin remodeling. It’s realized through generating 5-methylcytosine (m5C), which eventually leads to the silencing of gene expression (6). Extensive protein systems in cells write methylation patterns on DNA by de novo methylation (DNMT3a and DNMT3b) or demethylation (TET1, TET2 and TET3), and faithfully copy a series of factors (DNMT1 and UHRF1) of methylation patterns during DNA replication (Figure 2) (8). As an important epigenetic modification, the establishment, maintenance and clearance functions of DNA methylation play a key role in mammalian development and diseases (6). Previous studies have shown that DNA methylation is involved in the regulation of various biological processes. They include but are not limited to cell differentiation and development, gene transcription, chromatin structure remodeling, X chromosome inactivation, genome imprinting, and chromosome stability. At the same time, the pregnancy processes such as fertilization, embryo implantation, and placental development in human early embryo development are also closely related to DNA methylation (9, 10). The failure of cell invasion in early pregnancy, abnormal immune tolerance, and abnormal arterial remodeling of the decidua during placental development, which are controlled by a complex network of genetic-epigenetic modifications, may lead to the occurrence of URPL (9).
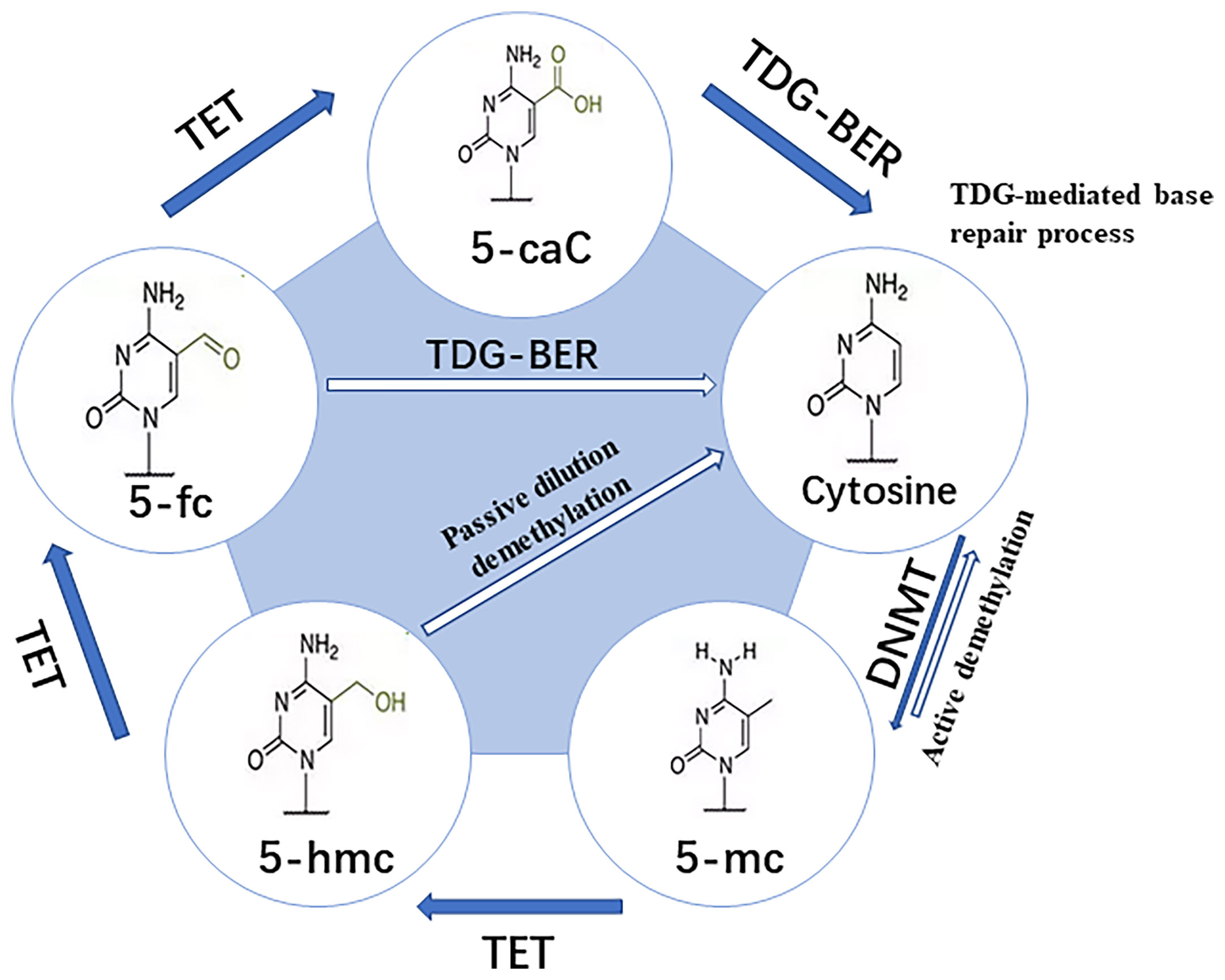
Figure 2 Pathway for dynamic modifications of cytosine. Methylation is achieved by DNMTs transferring the methyl to unmodified cytosine. It involves two steps: (A) Passive dilution, in which the methyl is directly removed to yield unmodified cytosine; and (B) Active demethylation, in which the methyl is catalyzed by TET into different oxidized forms and then combined with glycosylation, base repair, DNA replication, and other methods to yield unmodified cytosine (7).
DNA methylation is mainly established and maintained by DNA methyltransferases (DNMTs), that transfer a methyl group from S-adenyl methionine (SAM) to the fifth carbon of a cytosine residue to form 5Mc. The enzymes include DNMT1, DNMT3a, DNMT3b and DNMT3L (11). DNMT1 is responsible for maintaining the DNA methylation pattern during the S phase in cell division (12). DNMT3a and DNMT3b initiate de novo methylation in vivo and are required for establishing new DNA methylation patterns during embryonic development (13). Although it lacks the conserved catalytic domain common to DNA methyltransferases, DNMT3L acts as a necessary accessory protein for DNMT3a-mediated de novo methylation in the germline (14–16). DNMT3c, as a tandem copy of DNMT3b, is only found in rodents, and it’s responsible for the DNA methylation of repetitive elements in the male germline (17, 18). All the DNMTs are extensively involved in the development of an embryo. It’s obvious that the deposition of methylation marks relies on the catalytic activity of DNMTs, while the active removal relies on the activity of ten-eleven translocation enzymes (TET) and thymine DNA glycosylase (TDG) (19). Various biological processes, including preimplantation of the embryo and PGC development, ESC maintenance and differentiation, neurological function, and cancer, have been shown to involve DNA demethylation (7). Research has revealed that global DNA demethylation of the preimplantation embryo genome is mainly accomplished through passive dilution and TET3-mediated oxidation (20, 21). In female mice, TET1 deficiency leads to PGC meiosis defects, which has been linked to insufficient demethylation and failure of meiotic gene activation (22). TET1 deficiency, on the other hand, causes abnormal methylation patterns at imprinted genes in PGC and sperm cells in male mice (23). In addition, TET1 and TET2 are both expressed in mouse embryonic stem cells (mESC) (24, 25). Deletion of TET1 manipulates ESCs towards specific lineages, while deletion of TET2 can inhibit enhancer activity and delay transcriptional changes during differentiation (26). TET1/TET2/TET3 triple knock-out (TKO) of ESCs has basically normal morphology and expresses pluripotency markers, but their differentiation and developmental potential are impaired (27). In summary, abnormal DNA methylation may change the embryo development potential, jamming embryo implantation and placenta formation, which may lead to adverse pregnancy outcomes.
DNA Methylation Dynamics in Early Embryo Development
Methylation of DNA is a critical epigenetic regulatory mechanism in mammals during embryonic development. During gametogenesis and early embryo development stages in mammals, the whole genome undergoes a reprogramming event of demethylation-remethylation, realizing the continuation of the germline and the transmission of genetic material (28). Since 1987, it is believed that there are two demethylation-remethylation waves during development (Figure 3). The first waves of demethylation and remethylation occur during the migration of proliferating primordial germ cells and before the formation of mature gametes, respectively. During foetal life, the methylation marks on the imprinted genes are erased in the primordial germ cells. These marks are re-established during spermatogenesis up to the primary spermatocyte stage and faithfully maintained through the cell divisions by the DNA methyl-transferases. The second wave of demethylation occurs after fertilization, when the epigenetic memory of the gametes is erased on a large scale, and the lowest DNA methylation has been reached in the blastocyst (31). After embryo implantation, remethylation gradually occurs until it returns to the original high level (32). In other words, the methylation pattern of the early human embryo genome is reflected as follows: it’s at a high methylation level before fertilization, then the demethylation occurs during the cleavage stage, and the methylation level reaches a trough when the blastocyst is formed. Finally, it has been established a new methylation mode for interaction with the uterus after the embryo implantation to restore the original high methylation level. The periodic and orderly change of DNA methylation is the key to ensure the pluripotency of the developing embryo (33). Then, during the ovulation period, it is reflected through to the lowest level of methylation formed by the blastocyst, and finally the new methylation state in the mother-to-child interface. While researching on the reprogramming process of DNA methylation modification during preimplantation development of mouse cloned embryos, Gao Shaorong et al. revealed that abnormal DNA re-methylation is the key obstacle leading to abnormal development of cloned embryos after implantation (34). From this review and the existing literature, it can be suggested that the periodic and orderly changes in DNA methylation during early embryo development presents a novel research target of RPL.
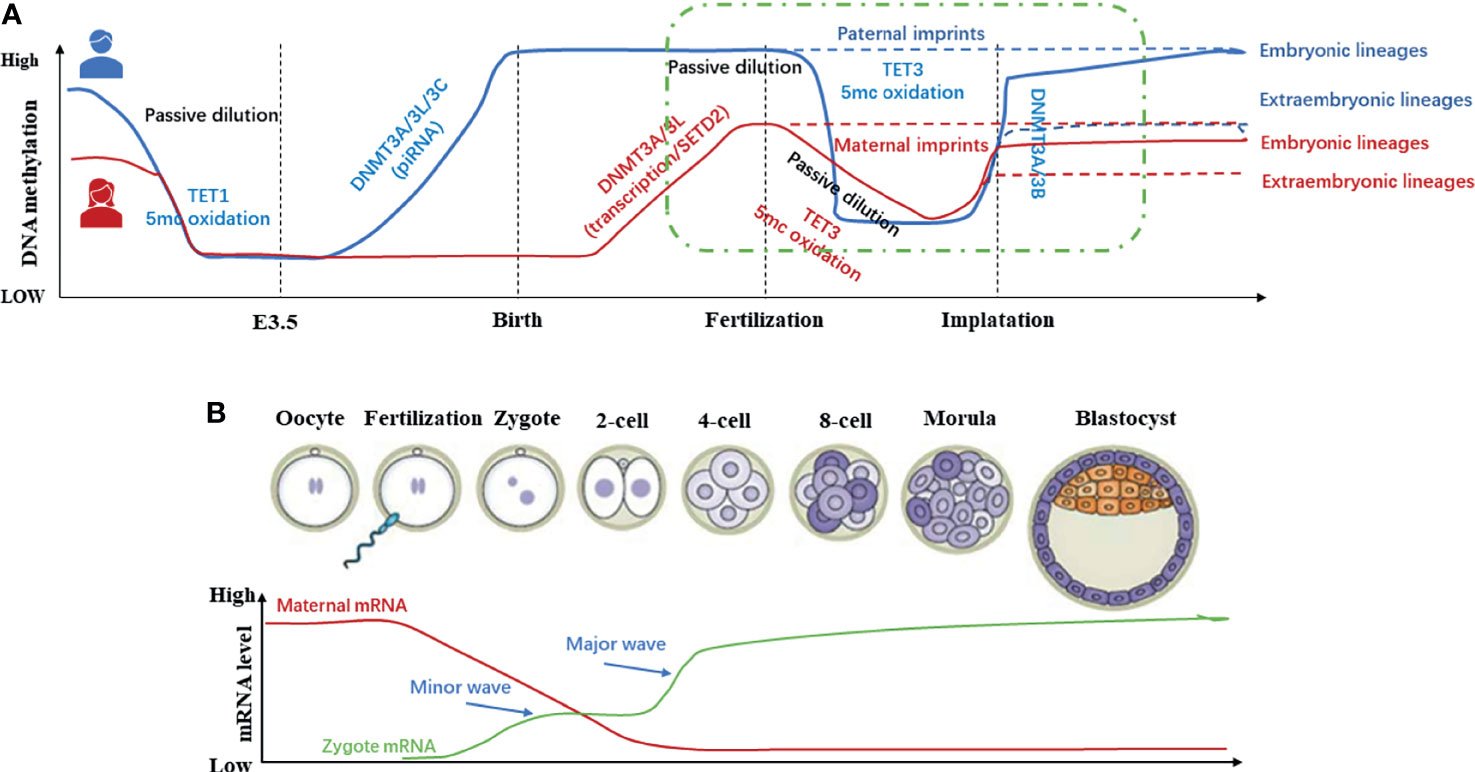
Figure 3 Methylation dynamics of gametes and early embryos during development (11, 29, 30). (A) In the germ line, DNA methylation in Primordial Germ Cells (PGCs) is almost completely eliminated to protect the PGCs from premature differentiation. Before mature gametes are formed, germ cells re-establish DNA methylation in a sex-specific manner. In early embryos, except imprinted genes and certain repetitive sequences, the genome undergoes global demethylation after fertilization and re-methylation after embryo implantation. (B) In general, global DNA methylation is mostly static throughout life. However, two waves of genome-wide DNA demethylation-remethylation occur in the germline and preimplantation embryos.
RPL and Abnormal or Defective DNA Methylation
DNA methylation runs through the entire reproductive process of gametogenesis, embryonic development, and maternal-fetal interface formation. Abnormal DNA methylation is a potential cause of early pregnancy loss (miscarriage or abortion within the sixth week of conception). Early embryo development (35), trophoblast cell invasion (36), maintenance of the uterine immune tolerance microenvironment, and uterine spiral artery remodeling are all controlled by a complex network of genetic-epigenetic modifications. Such modifications promote the occurrence of URPL. Globally, about 4% of spontaneous abortions have been linked to abnormal DNA methylation (37). Therefore, DNA methylation is an important molecular mechanism for the fertilization of the embryo and the normal development of the fetus. Vasconcelos et al. revealed for the first time, the disorder of epigenetic modifiers and imprinted genes in the placenta and fetal tissues of idiopathic spontaneous abortion. During the trimester, the author observed that TET2/3, IGF2, and CDKN1C were up-regulated; MEST was down-regulated; and methylation of MEST, KvDMR1, and H19 DMR was increased (38). DNA methylation may inhibit the expression of certain key genes in the embryo, which may interfere with the normal development and growth of the embryo, and promote the occurrence of RPL.
DNA methylation involves the selective addition of DNMTs and the selective deletion of TETs. Analysis of the chorionic villi and decidual tissues of URPL patients showed that they were hypomethylated and accompanied by down-regulation of DNA methyltransferases (DNMT1, DNMT3a, DNMT3b) and up-regulation of DNA demethylase (TET1/2/3) (39–42). The expression of DNMT1 and DNMT3a was significantly reduced in the receptive phase of the mouse endometrium. Clinical studies have revealed that the DNAMT3a (448A > G) gene polymorphism (43), and the DNMT3b gene polymorphism (rs1569686) may be a potential genetic marker of RPL susceptibility (44). Both DNMT1 and DNMT3a are found in the chorionic villus and decidua of normal early pregnancy. The interference of DNMT1 inhibitors may cause a decrease in the level of DNA methylation, impair embryo development, and inhibit the attachment between embryos and endometrial cells (45). Serman et al. found that after taking 5-Aza-CdR (DNA methylase inhibitor) in pregnant mice, the expression levels of DNMT1 and DNMT3a in the endometrial tissue decreased. Also, the proliferation of trophoblast cells was inhibited, and the embryo implantation rate was significantly decreased (46). Tanaka S et al. found that TET1 and TET2 regulate the expression of cyclin B1 through protein-protein interactions. As well, the enzyme regulate the integrity of trophoblast cells and the intracellular replication cycle, which affects the development of the fetus (47). In addition, demethylases (TET1/2/3) may induce the occurrence of URPL by interfering with the gamete meiosis, early embryo totipotency and differentiation of specific lineages (48, 49). We hope that through further experiments, we can find new means of preventing and treating URSA using methylation methyltransferase inhibitors.
Folic acid metabolism, as an important way to generate methyl donors (active methionine SAM), is inseparable from the occurrence of DNA methylation. the gene-specific methylation of 5,10-methykenetetrahydrofolate reductase (MTHFR) is a thrombophilic marker as well as it is positioned at the junction of the reactions producing methyl groups for the purpose of the DNA methylation, DNA repair and DNA synthesis (50). In RPL cases, MTHFR is significantly increased (p=0.002), which emphasizes the vital association between the methylation of MTHER and transformation (50). The clinical cases suggest that RPL’s spouse MTHFR (C677T/A1298C) polymorphism may alter sperm concentration and ratio of forward motility sperm (PR) by affecting DNA methylation, thereby increasing the risk of early spontaneous abortion (51, 52). In addition, the polymorphism of MTHFR (C677T) leads to low enzyme activity, changes the methylation cycle, and reduces the available free methyl. When these happen, they result in low global DNA methylation, which may be involved in the occurrence of preeclampsia (PE) and pregnancy loss (PL) (50). Rotondo et al. found that the promoter of MTHFR in the sperm of the RPL’s spouse showed a high degree of methylation. Based on the above researches, we suggested that abnormal methylation of MTHFR may affect the normal growth and development of the embryo, leading to the occurrence of RPL (53). We suspect whether simple supplementation of “folic acid” can compensate for the methylation defects in MTHFR-related RPL patients, thereby improving pregnancy outcomes? The feasibility of this measure requires further investigation of folate metabolism and other compensatory pathways in RPL patients.
DNA Methylation of Imprinted Genes and RPL
Genomic imprinting is a process in which parental alleles undergo modification as they are passed on to offspring through gametes. Alleles with parental imprinting acquire different expression characteristics. During development, the methylation and expression status of imprinted genes are tissue and stage specific during development (54). More than 50 percent of known imprinted genes are expressed in placenta, and they are crucial to cellular differentiation and embryonic development (55). Abnormal DNA methylation in the imprint control area (ICR) of imprinted genes can lead to abnormal silencing of active alleles or abnormal expression of inactive alleles. This causes imprinting disorders or deletions, fetal neurodevelopmental defects and metabolic disorders, which in turn affect embryo development and induce poor pregnancy outcomes. Previous studies have reported that imprinted gene defects lead to early pregnancy loss, fetal death, prolonged delivery, and the development of embryo tumors (56–59). Liang et al. found that the maintenance level of the imprinted gene PEGIO is very important for early embryo development, whose down-regulation leads to irreversible pregnancy loss (60). Zhang et al. verified that the changes in the expression of imprinted genes and the hypermethylation profile of the repetitive regions (PRE-1 and satellite DNA) in cloned pigs may be related to the developmental defects and early pregnancy loss of the cloned pigs (61). Clinical studies have supported that PHLDA2, a maternal gene, is up-regulated in the placental tissues of the RPL, and inactivation of Mash2/Peg10 can cause abnormal placental development, which may lead to early embryo death. Flisikowski et al. reported that a microdeletion of PEG3 (a maternal imprinted gene of cattle) enhanced inhibition of the paternal gene MIMT1, late pregnancy loss, and even stillbirth (62). In addition, the expression of IGF2/PHLDA2/PEG10/CDKN1C was increased in first/mid-pregnancy abortion, while PEG10 was lowered in late pregnancy abortion or stillbirth (63). A series of clinical trials have also verified the hypothesis that “improper methylation and expression patterns of imprinted genes may lead to RPL” (64). While evaluating the DNA methylation patterns of the maternal genetic imprinting gene GRB10 and the paternal inherited genes (IGF2 and PEG3) in the villus samples of RPL, Liu et al. reported that both showed significant high methylation (64). PEG3 plays a key role in p53-mediated cell death and offspring development. Deletion and mutation of PEG3 may lead to insufficient placental nutrient transport and retardation of offspring growth, which may trigger offspring death (65, 66). IGF2 participates in glycogen production and affects placental metabolism or fetal growth. The quality of the fetus and placenta in placenta-specific IGF2-knockout mice is significantly reduced (67). The transgenic mice with the proximal duplication of chromosome 11 where GRB10 is located, exhibited pre-/post-natal growth retardation (58). To summarize, the defects of imprinting genes related to abnormal DNA methylation can promote the disorders of fetal neurological, developmental, metabolic and jamming embryo development. The placenta is an important organ for the enriched expression of imprinted genes. The methylation of the imprinted gene (H19/PEG1/LIT1/SNRPN) in the placental villi of RPL was significantly higher than normal (68, 69). The abnormal increase of H19/ICR1 methylation in the villi indicates that the methylation changes at special sites in the placental villi may be closely related to the risk of RPL (42). DNMT3L acts as an important structural protein that coordinates DNMT3A/DNMT3b-mediated DNA methylation during embryo development. The DNMT3L-knockout oocytes were unable to establish maternal-specific methylation imprints, resulting in abnormal fetal neural-tube development, which manifested as SA in the second trimester. It’s also been proven that DNA methylation is the key to genomic imprinted markers (70). Sheng et al. obtained E7.5/E13.5/E19.5 placentas from pregnant C57/BL mice, and screened the imprinted LncRNA controlled by DNA methylation during placental development. The results showed that the methylation of the MEG8 promoter increased in RPL villi, and the abnormal up-regulation of lncRNA MEG8 (RIAN) in the trophoblast cell inhibited cell proliferation and invasion. The author linked this observation to the occurrence of URPL (71). All in all, genetic imprinting disorders or deletions related to abnormal DNA methylation may promote the occurrence of RPL by interfering with embryo development (neural-tube development and metabolism), placental nutrient transport, trophoblast proliferation and invasion (72). We believe that in cases of RPL especially (unexplained RPL), early finding of new prognostic marker would be of great help to obstetricians for early detection and management of RPL patients.
Sperm Abnormal DNA Methylation and RPL
RPL is also associated with certain male factors, including age, abnormal semen parameters, and sperm DNA damage. There is a growing body of evidence to support the hypothesis that the intact paternal epigenome is essential for normal embryo development (73). As the main epigenetic modification to maintain the normal development of sperm, DNA methylation can correctly compress the chromatin of the sperm head and permanently silence the gene promoters related to gene imprinting (22). Some studies have reported that the integrity and compactness of sperm chromatin in the RPL group are lacking (74–77). The abnormal DNA methylation of sperm interferes with spermatogenesis and embryonic development, leading to improper implantation and RPL (73, 78, 79). Sperm global methylation levels were found to be significantly reduced in the RPL group (80). The promoters of development-related genes in mature sperm often exhibit low methylation. The role of sperm DNA methylation in mammalian embryo develpment has been described by several authors. To identify the mechanism of how DNA methylation impacts sperm function and embryo development, several studies were performed in animal models. The earlier rodent studies have demonstrated that decreased global methylation levels in spermatozoa are associated with post-implantation embryo loss (81). DNA methyl-transferase family of proteins promote methylation and its maintenance (82). Studies in knockout mice and mutations in these proteins demonstrated an association with chromosomal deactivation, embryo growth interruption, and sub-sequent demise (12, 83, 84). In addition, studies that have used potent DNA methylation inhibitors in mice demonstrated an increased incidence of embryo mortality (85). It should be noted that embryo loss could be as a result of toxic effects of these inhibitors and not directly as a result of inhibition of DNA methylation (86). Furthermore, a recent study by Rogenhofer et al. demonstrated that P1 and P2 mRNA levels as well as the ratio of these two protamines were significantly different in male partners of women with unexplained recurrent pregnancy loss compared with healthy control men and subfertile couples undergoing in vitro fertilization/intracytoplasmic sperm injection (87). Based on the current body of evidence, we believe we can hypothesize that sperm epigenetics impact embryo development and alterations can be implicated in pregnancy loss. We await further studies on this idea.
Ankolkar et al. found that the methylation of imprinted gene H19 in the semen of the URPL spouse was significantly lower than normal, while there was no obvious difference in the methylation of PEG1/DLK1/GTL2/PLAGL1. Khambata et al. proposed several imprinted genes with abnormal DNA methylation in the sperm of URPL’s spouse that are related to RPL, such as IGF2-H19 DMR, intergenic differentially methylated region (IG-DMR), mesoderm specific transcript (MEST), zinc finger protein regulateing apoptosis and cell cycle arrest (ZAC, also called PLAGL1), DMR KCNQ1 intron 10 gene (KVDMR), paternally expressed gene 3 (PEG3) and paternally expressed gene 10 (PEG10), as well as decreased sperm global 5-methylcytosine (5mC) levels, are all related to RPL (80). IGF2-H19 DMR is an effective mitogen regulating the growth of embryos and placenta (88). The loss of DLK1 does not lead to embryo death, mainly manifested by defects in placenta formation and adipocyte differentiation (89). MEST, expressed in human villi and invasive trophoblasts during early pregnancy, contributes to angiogenesis during trophoblast invasion (90). PLAGL1 participates in the regulation of cell cycle, apoptosis and embryo development, whose lack may lead to embryo growth restriction and lethality (91). A meta-analysis showed that the hypomethylation and demethylation of H19 affect sperm concentration and motility (92, 93). The global 5mc level of sperm may be regarded as a diagnostic marker for RPL to detect “epigenetically abnormal” sperm, and the imprinted genes such as IGF-H19 will be the best candidate genes. In addition, MTHFR polymorphic variants are associated with decreased sperm count (53, 94, 95), leading to male oligospermia, infertility, and RA. Rotondo et al. believe that the hypermethylation of the MTHFR promoter in the sperm of URPL’s spouse may cause RA by abnormal embryo development and trophoblasts proliferation/apoptosis/differentiation (96). In conclusion, the paternal epigenome impacts sperm quality, early embryogenesis, and possible somatic health in future offspring (78). We need more transgenera-tional studies focused on environmental exposures in the F0 population, effects on sperm epigenetics, and any evidence of an impact on somatic health outcomes in offsprings. This will ultimately provide a path to preventative measures, diagnosis, and treatment.
DNA Methylation of Placenta/Decidual and RPL
The placenta is the first organ formed during pregnancy, which plays an important role in the intrauterine regulation of fetal growth. The placenta plays multiple functions, such as the exchange of nutrients, waste, and gas, maternal-fetal immune tolerance, and various kinds of metabolic and endocrine functions during fetal development (97). Placental insufficiency may cause preeclampsia (PE) or intrauterine growth restriction (IUGR). The inflammation or infection in the placenta can lead to chorioamnionitis (CA) or placental-related preterm birth (PTBs). Tumor formation in the placenta is also called choriocarcinoma (CA) or hydatidiform mole (HM) (54). Epigenetic modification is believed to play a vital role in placental function and development (4, 98). In early pregnancy, the human placenta has similar phenotypic characteristics and epigenetic patterns to tumors, such as extensive hypomethylation of the whole genome and local hypermethylation of CpG islands (99). The epigenetic regulation of imprinted and non-imprinted genes is important in the placenta, date from the preimplantation stage and maintaining it throughout the whole pregnancy (54). Furthermore, DNA methylation may be inherently more variable in the placenta, because of its need to be responsive to a variety of signals in its function as a mediator of exchange between the fetus and mother (100). Thus, the placenta acts as the most important organ for the expression of imprinted genes. Lim et al. used RRBS (bisulfite sequencing) and RNA-seq to perform DNA methylation and gene expression analysis on human placental tissue. The author suggested that significantly altered genes were enriched in pathways related to the cell cycle and immune response (101). It is worth noting that human chorionic gonadotropin (HCG), whose coding genes CGB5 and CGB8 carry imprinting sites of placental expression, is an important hormone for embryo development and pregnancy maintenance. When the CGB5 promoter is hemi-methylated in RPL, the secretion of HCG is reduced, and the abortion rate is significantly increased (22). In a study that involved use of BSP to detect the methylation of the villi in RPL, Hannah et al. reported that RPL is closely related to the DNA methylation of specific sites in the placenta (like the decreased defensin β1 and the elevated AXL) (102). Elsewhere, Du et al. compared and analyzed the genome-wide DNA methylation of placental villi in RPL. The authors proposed that the hypomethylation of the PRDM1 promoter induces the recruitment of GATA2-FOXA1complex, promotes the migration and apoptosis of trophoblast cells, and leads to the occurrence of RPL (70). PRDM1 is a transcriptional regulator of embryo cell fate, required for reprogramming of primordial germ cells and intestinal cells (103). Wu et al. analyzed the LncRNA expression profile by microarray, and proposed that the abnormal methylation of IGF2AS in the placental villi may affect the stability of early pregnancy (104). Previous studies have shown that the formation and localization of the placenta may be the result of a balance between matrix metalloproteinases (MMPs) and tissue inhibitors of matrix metalloproteinases (TIMPs) secreted by the extracellular matrix. That means the expression of MMPs/TIMPs and the changes in the methylation of the promoter lead to the imbalance of MMPs/TIMPs and the occurrence of URPL (39, 104). A clinical study suggested that changes in placental DNA methylation of genes involved in environmental adaptation, immune response, and imprinted genes may contribute to the etiology of RM (45). However, it is difficult to determine whether these changes are causal or a consequence of placental adaptation to an unhealthy embryo. Furthermore, studying DNA methylation in the placenta is complicated by the presence of different cell types which carry their own unique methylomes. The subsequent research should reinforce the need to focus DNA methylation sequencing experiments on individual cell populations rather than entire tissue extracts.
Decidua, the maternal part of the placenta, provides a delicate balance between immune tolerance and defense to maintain pregnancy (105). The establishment and maintenance of normal pregnancy is accompanied with the proliferation, migration and invasion of trophoblast cells and endometrial decidualization. The invasion disorders or excessive invasion of trophoblast cells and abnormal endometrial decidualization can cause RPL. The endometrial decidualization involves the endometrial stromal cells’ reprogramming, the production of various mediators such as cytokines and chemokines, and the selective recruitment of immune cells. As an important part of the maternal-fetal interface, decidua plays a crucial role in the establishment and maintenance of pregnancy. First, it limits the excessive invasion of trophoblasts cells to protect the maternal uterus (106). Second, decidual secrete lipid droplets, glycogen, and growth factors provide nutrition for embryo development (107). Third, decidual enhances the construction of the maternal-fetal immune tolerance microenvironment to maintain normal pregnancy (108). Abnormal DNA methylation interferes with the expression of decidual related genes, leading to a decrease in immune tolerance, cell invasion failure, and poor remodeling of the uterine spiral artery. When these happen, improper implantation, inflammation, and impaired placental development may occur (109). DNA methylation analysis of the decidual in RPL showed that overexpression of CREB5 (cAMP responsive element binding protein 5) and SGK3 (SGK is a serine-threonine protein kinase regulated by glucocorticoids, involved in epithelial ion transport and cell survival) (110) can promote the occurrence of RPL by interfering with trophoblasts migration, apoptosis and dysfunction (111). The GO analysis suggested that hypo-DMR near CREB5could up-regulate CREB5 gene expression by recruiting P53 and SP1, thus increasing cell migration and cell apoptosis, blocking cell cycle (22). In addition, some studies have shown the function of CREB5 in the immune response, including macrophage survival, regulation of T/B lymphocytes, and inducing the transcription of immune-related genes (such as IL-10) (112). That means the hypomethylation of CREB5 may promote the occurrence of RPL by regulating the immune response. Chen H, et al. constructed a miRNA-mRNA network in decidual and identified several small molecules involved in the occurrence and progression of URPL, such as FCGR1A/3A (important receptors for innate and adaptive immune responses), CXCL8 (also known as IL-8, an important inflammatory factor), HCK [expressed in bone marrow cells, B lymphocytes, and various cancer cells to enhance the expression of myelin growth factor and pro-inflammatory cytokines (113)], PLEK [an important substrate of protein kinase C, the phosphorylation of PLEK can promote the secretion of pro-inflammatory cytokines in phagocytes (113, 114)], IL10 [low IL-10 are associated with pregnancy complications, such as RA, premature delivery, fetal premature membrane rupture, PE, IUGR (115)], hsa-miR498 and hsa-miR-4530 (116). A retrospective observational case-control study that revealed the succinate dehydrogenase complex iron-sulfur subunit (SDHB) DNA methylation, SDHB expression increased, and succinate level decreased in the decidua of RPL. Low accumulation of chorionic succinic acid interferes with the invasion and proliferation of extravillous trophoblast cells through the PHD2-VHL-HIF-1α pathway, and affects embryo implantation (117). Xie et al. found that the DNA methylation of lncHZ08 promoter was decreased in the decidua of RPL, and the estrogen receptor (ER)-mediating the transcription of lncHZ08 was increased. The up-regulated lncHZ08 inhibits the PI3K/p-AKT/p-P21/CDK2 pathway by down-regulating PI3K, thereby reducing the proliferation, migration and invasion of trophoblast cells, and further inducing the occurrence of abortion (118). Fatima et al. analyzed the causal relationship between methyltransferase, methylation and cell apoptosis in abortion through structural equation modeling (119). Studies is the first to show that the methylation of p53 pathway (BAX/P53/CASPASE-6/BCL-2) achieved by methyltransferase (G9aMT/DNMT1) is closely related to the fate (maintenance or termination) of early pregnancy (119). It is worth noting that the abnormal response of human endometrial stromal cells (HESCs) to decidual signals is closely related to RPL. The MEDIP-Seq analysis of HESCs suggests that RPL is significantly associated with the decrease in methylation of CA-rich sequences, which are widely expressed throughout the genome and enriched in near telomeres. This study suggested that abnormal DNA methylation of HESCs in the uterine can cause them to lose their epigenetic stemness, promote stromal cell senescence, limit endometrium plasticity, prevent decidua, and further induce RPL (120). HESCs in vitro experiments have shown that the loss of DNMT3b in DSC can down-regulate the expression of decidual-specific IGFBP-1. This implies that inhibiting DNA methylation can eliminate decidualization before or after implantation, leading to pregnancy loss (121). The DNMT1 and DNMT3a in the endometrium of the mice injected with DNMT inhibitors (5-Aza-CR) at different pregnancy stages were down-regulated, and the subsequent endometrial decidualization and stromal cell proliferation defects showed the potential to directly reduce embryo implantation rate (116).
The Maternal-Fetal Immune Tolerance Microenvironment Is Regulated by DNA Methylation, As Is RPL
The endometrium is often regarded as an allograft that the embryo directly contacts. The main function of the endometrium is to create and maintain an optimal endocrine/paracrine, immune and molecular environment for proper attachment, implantation, invasion and development of the embryo. During pregnancy, the endometrial stromal cells undergo decidualization, a significant morphological and functional reprogramming of cells, cell reprogramming, tissue remodeling, gene expression, post-translational regulation, and change in the cell signal pathways. In the uterus, the activity of immune cells also undergoes major changes.
The maternal-fetal interface is an immune tolerance microenvironment composed of maternal decidua and fetal placenta, which are critical in ensuring the smooth progress of pregnancy. In this section, the composition of immune cells and related DNA methylation changes at the maternal-fetal interface of RPL is reviewed. Also, the effect of DNA methylation on the uterine immune microenvironment and the occurrence of RPL are explored.
The maternal-fetal immune interaction plays an important role in embryo implantation and pregnancy maintenance. Some scholars have proposed that URPL is a type of allogeneic immune disease related to the failure of maternal-fetal immune tolerance, which means the success of pregnancy depends on the immune status of the gravida and the immune regulation ability of the embryo (122). Du et al. performed a genome-wide DNA methylation analysis on the placental villi of RPL and found many significant DMRs near dysregulated genes (such as PRDM1) in RPL. These differentially expressed genes are enriched in the immune response pathway, which implies that abnormal immune regulation may promote the occurrence of RPL (70). The imbalances of the immune microenvironment at the maternal-fetal interface are usually reflected in two aspects (123). One is the lack of negative signal activation to maternal-fetal tolerance, which impairs embryo implantation or embryo formation. Excessive immune activation, on the other hand, increases the uterine inflammatory environment or causes trophoblast damage. Chuang G et al. carried out a study to identify the complete cell lineage of the decidual immune microenvironment of RPL through single-cell high-throughput sequencing. They documented several cells including but not limited to natural killer cells, macrophages, dendritic cells and T cells (124).
Endometrial Stromal Cells Regulate Local Immune Function
Previous experiments have shown that proper decidualization of endometrial stromal cells plays an important role in implantation and maintenance of early pregnancy. The profound morphological and functional reprogramming of the endometrial stromal cells that differentiate into highly specialized cells with secretory capabilities, have been found in this progress. The overall emerging picture is that decidualization of the endometrium is a process involving profound cell reprogramming, tissue remodeling, changes in gene expression and post-translation regulation, and alterations in cell signaling pathways (125). These studies indicate that the endometrium is an excellent biosensor for the quality of implanted embryos (126–128). Normal female endometrial cells become sensitive to embryo signals after decidualization, thus low-quality embryos inhibit the secretion of factors beneficial to embryo implantation. On the contrary, embryos with strong developmental potential will produce signals promoting implantation (129). However, the RPL is not sensitive to this signal, allowing low-quality embryos to be implanted, leading to poor pregnancy outcomes (120).
Macrophages
There are few studies on macrophages in the progression of RPL. The number of macrophages in the non-pregnant state is relatively small, only significantly increased in the luteal phase. Macrophages, rapidly increased in pregnancy, are located close to invading trophoblast cells and remodeled uterine spiral artery (120). They participate in embryo implantation, trophoblast invasion, remodeling of the uterine spiral artery, removing apoptotic cells and cell debris, and protecting the fetus from microorganisms or pathogens (130–132). A clinical study aimed at determining the DNA methylation group of macrophages on the maternal-fetal interface (133) reported as follows: First, there are significant differences in the DNA methylation patterns of macrophages derived from maternal or fetal cells. Second, differentially methylated genes related to immune response are highly methylated in fetal-derived macrophages. The imbalance between decidua M1/M2 macrophages is related to abortion (134). M1 macrophages increase the inflammatory advantage by producing inflammatory cytokines such as TNF-α, IL-β, IL-6 and IL-12, whereas M2 macrophages promote immune tolerance by producing anti-inflammatory cytokines such as IL-10 and TGF-β (135, 136). Contrary to normal pregnancy, we can’t detect the reduction of M1 decidual macrophages in URPL cannot be detected (134). Although the current research on DNA methylation of macrophages in the uterine microenvironment of RPL is still blank. However, a MeDIP-seq of preterm placenta demonstrated that abnormal DNA methylation was enriched in Fcy receptor-mediated macrophage phagocytosis (48), suggesting that abnormal DNA methylation in macrophages may be involved in adverse pregnancy outcomes.
Natural Killer Cells (NK Cells): Are NK Cells the “BAD BOYS” That Reject Embryos?
As a member of the innate immune system, NK cells were originally defined as a cell population capable of killing autologous and allogeneic target cells through spontaneous cytotoxic activity. Based on the phenotype and function, NK cells can be divided into various subsets, such as peripheral NK (pNK) cells, uterine NK (uNK) cells, tissue-resident NK (trNK) cells and innate lymphoid cells-1 (ILC1s). This review focuses on uNK cells, mainly CD56bright cells, with low cytotoxicity, but they can secrete rich cytokines to protect the alloantigen and regulate the process of pregnancy (137). The uNK cells reach a peak in early pregnancy and are adjacent to the invading trophoblast cells (138, 139). Studies have shown that activated uNK can produce angiogenic factors (VEGF and ANG2) and a large number of cytokines (GM-CSF, CSF-1, TNF-α, IFN-γ, TGF-β, LIF, IL2, CXCL10, CXL12) (140). During pregnancy, uNK cells play a unique role in regulating trophoblast cell invasion (141, 142) and remodeling the uterine spiral artery (123), which is the necessary step for placenta formation. In addition, the uNK plays an immune role at the maternal-fetal interface, and assumes the responsibilities of immune killing (such as killing microorganisms to avoid various inflammations) and immune tolerance (such as normal pregnancy) at the same time (143, 144). A series of clinical studies have demonstrated that compared with normal pregnant women, the number, quality and cytotoxicity of uNk cells of URPL are disparate (145, 146). The dysregulation of uNK cells in RPL may cause pregnancy damage through the following five potential ways: (1) NK cell cytotoxicity is maintained but impaired (147); (2) the ability of NK cells to correctly interact with the specific HLA expressed by trophoblasts is impaired (148, 149); (3) the ability of NK cells to effectively participate in the complete uterine spiral artery remodeling is impaired (150, 151); (4) the limitation of T cell cytotoxicity is impaired and (5) NK cells interferes with the mode of cytokine production (152–154). To date, the fact that NK cells are related to RPL is well known, but its related epigenetic regulation mechanism is still unclear. Whether the proportion, functional status and methylation level of NK cells are related to RPL remains to be further explored.
The Balance of Th1/Th2 or Th17/Treg and RPL
In 2010, Saito et al. proposed that the Th1/Th2/Th17 and Treg cell paradigms play a crucial role in maternal immune tolerance (155). Th1 cells contribute to cellular immunity by producing IL2 and INF- α, which are thought to be the basis for allograft rejection (156, 157). On the contrary, Th2 cells release IL4, IL5 and IL13 and participate in humoral immunity (158), as the key to inducing and maintaining allograft tolerance (159). Th1 cells secrete cytokines (IL2/TNF-/IFN-), participate in immune surveillance, and protect against excessive trophoblast invasion (160). A pro-inflammatory Th1 immune response is required to promote tissue remodeling and angiogenesis during embryo implantation (149). Comparatively, pregnant women having RPL have higher Th1 levels in their peripheral blood than normal pregnant women (161). Th1 and NK cells secrete a large amount of TNF-α, which causes apoptosis, inhibits trophoblast growth, and inhibits the uterine epithelium’s secretion of granulocyte macrophage colony stimulating factor, ultimately leading to pregnancy loss (162, 163). Once the implantation period is over, the immunodominance of Th1 cells gradually shifts to Th2, participating in maintaining the maternal tolerance to fetal antigens (164) until delivery. Th2 immunity represses the development of Th1 and Th17 immunities by releasing IL-4 and IL-13, respectively, and promotes allograft tolerance (165). Meanwhile, Th2 cytokine, IL-4, was reported to induce autoreactive B cell activation and thus promotes autoimmunity (166). The exacerbated Th2 immunity during pregnancy may induce autoimmune diseases (systemic lupus erythematosus) (167), while the tolerogenic may induce uncontrolled viral infections (ZIKA virus) (168). Timely and adequate Th2 immunity is important for the immunotolerance and protection of fetus from infection. Therefore, the timely transfer and adequate balance of Th1 and Th2 cells during pregnancy seem to be essential for a successful pregnancy. Conversely, an improper balance during pregnancy is related to pregnancy complications, such as RPL and PE. Reversing the Th1/Th2 imbalance in RPL patients and supplementing the lack of related cytokines can provide new ideas for the targeted therapy of RPL. In the LPS-induced abortion mouse model, Administration of IL-10, which has immunoregulatory properties, and TNF-α receptor blocker etanercept, prevented LPS-induced pregnancy losses (169). When shifted Th1 immunity was regulated with intravenous immunoglobulin G (IVIg), TNF blockers, or T cell activation inhibitors, such as etanercept, adalimumab, or tacrolimus, pregnancy outcome was significantly improved in women with RPL (170–174).
Th17 cells, producing effective pro-inflammatory IL17, play an important role in inducing inflammation and immune rejection (175). The interaction between Th17 and Th1 is related to the pathogenicity of allergies, autoimmune diseases, immune rejection and adverse pregnancy outcomes (49, 176–179). In addition, Th17 can induce the activation of dNK cells and impair the vascular reactivity of the uterine artery (160), leading to embryo resorption (49, 180). The excessive Th17 cells were detected in the decidua and peripheral blood of unavoidable abortion (177, 181–183). Treg cells play an important role in embryo implantation and early pregnancy as participants in mediating maternal-fetal tolerance (162, 163). The proliferation of Tregs is related to normal pregnancy, while the reduction promotes embryo immune rejection and induce pregnancy loss (163). In animal models, the consumption of Tregs significantly increases the abortion rate (184, 185). Conversely, the transfer of Tregs can prevent pregnancy loss in miscarriage-prone mice (186). Similarly, in clinical trials, compared with normal pregnant women, the Tregs of the peripheral blood (187, 188), endometrium and decidua (189, 190) in RPL have been reduced, accompanied by increased Th17 cells (190, 191). The possibility of Tregs involved in RPL is supported by the key transcription factor FOXP3. Wang et al. found that the hypermethylation of Foxp3 promoter down-regulates the expression of the FOXP3, reduces the number of Tregs, leads to the imbalance of Treg/Th17 in the maternal-fetal immune interface, and promotes the occurrence of RPL (192–195). At the same time, Treg cells can participate in the down-regulation of maternal-fetal excessive inflammatory response and embryo implantation, whose imbalance or dysfunction may lead to the occurrence of RPL (189). Finally, the highly malleable Tregs can differentiate into effector T cells under certain conditions. Unbalanced Tregs may directly participate in fetal rejection (196). Although there is still a lack of direct evidence to confirm the driving effect of abnormal DNA methylation of Th cells and Treg cells in RPL. However, the existing research suggests that abnormal DNA methylation may promote the occurrence of RPL through regulating the proliferation, differentiation and cell activity of Th cells and Treg cells. Detecting the proportion of Th cell population at the maternal-fetal interface and intervening in Th cell activation is beneficial to the prevention and treatment of RPL patients.
Imbalanced Cytokine-Chemokine Network in RPL
Cytokines, chemokines and their receptors can be produced by endometrial glandular epithelial cells and stromal cells, decidual cells, cellular components of innate and adaptive immunity, and trophoblast cells. This network creates a local immune balance microenvironment, which is essential for successful embryo implantation, the regulation of trophoblast migration, and a normal pregnancy. In RPL patients or mouse models, the dysregulation of cytokines associated with embryo implantation and early pregnancy is present in or recruited to immune or non-immune cells in the endometrium and decidua. In turn, the unfavorable cytokine environment will impair the tolerance of the maternal immune system to the trophoblast, leading to immune rejection. RPL is related to the decrease of TGF-β produced by decidual dendritic cells (197). The increased IFN-γ in the decidua of RPL can induce apoptosis and embryotoxic effects and promote the occurrence of RPL by triggering the excessive inflammatory response of the decidua (198, 199), IL-10, like TGF- and IDO, can induce DC differentiation into a tolerance phenotype and regulate Treg population expansion. The reduction of IL-10 in the decidua of RPL will hinder this immune protection (200–202). In view of the complexity, sensitivity and plasticity of the endometrial cytokine-chemokine network, it is still difficult to determine all the cytokines involved and their cell sources in RPL. The mechanism and specific signaling pathways of the abnormal DNA methylation-induced cytokine-chemokine network imbalance in the maternal-fetal interface promoting the RPL are still to be further explored. The in-depth research of epigenetic regulation in immune regulation and related signaling pathways of RPL is expected to provide opportunities for personalized diagnosis and treatment of RPL.
In the last three decades, enormous progress has been made in the comprehension of the mechanisms establishing and maintaining the maternal-fetal interface immune tolerance microenvironment. The involvement of the maternal immune system is not limited to a correct immunologic dialogue at the maternal–fetal interface, but is extended to the endometrial decidualization, vascular remodeling, and placentation. It’s obvious that the maternal-fetal immune tolerance microenvironment is intimately involved in the establishment, maintenance, development, and termination of the normal pregnancy. Pregnancy loss due to maternal immune dysfunction can be prevented by inducing an overall immunological tolerance at maternal-fetal surface and maintaining full immunological reactivity against all the other foreign antigens. In fact, the currently available immune treatments for RPL are rather limited, empiric in the majority of cases, and with low efficacy. On the basis of the existing researches, the future perspectives in the immune treatment of RPL could be aimed to correct abnormal decidualization as well as dysfunctions of the maternal-fetal immune tolerance microenvironment. Although there is still a lack of direct evidence and related mechanisms of DNA methylation regulating the maternal-fetal immune microenvironment. Considering the similarity between pregnancy and tumors, we expect that new anti-tumor therapy “epi-drugs” can also shine in RPL immunotherapy.
Conclusion
Recurrent pregnancy loss (RPL) is one of the most frustrating and difficult areas in maternal-fetal medicine. The RPL rates have continued to rise during the last few decades, yet the underlying mechanisms remain poorly understood. An emerging area of interest is the mediation of essential gene expression by epigenetic modification during early pregnancy. Epigenetic intergenerational or cross-generational inheritance support “Fetal Origins of Adult disease (FOAD)”. Looking at it from this point of view, it may be considered lucky as the abnormal epigenetic load from the maternal side gets lost and not transferred to the next generation. The number of studies about DNA methylation has increased linearly since its discovery in the 1980s, while much remains unknown. For example, why do abnormal DNA methylation embryos die at such an early stage of development? How does abnormal DNA methylation lead to RPL? We are now in an era of unprecedented genetic tools, sensitive and highly quantitative sequencing technologies, and the ability to alter DNA methylation. All problems seem to be solved, but challenges still exist.
The pathogenic factors of RPL are complex and diverse. DNA methylation plays a key role in early embryonic development, trophoblast cell invasion, uterine spiral artery remodeling and maintenance of the maternal-fetal interface by precisely regulating the inheritance and expression of genes. This review summarizes the relationship between abnormal or defective DNA methylation and RPL, and describes four aspects (imprinted genes, placental/decidual genes, sperm DNA, and immune-related genes) in detail. This review focuses on the composition of immune cells and related DNA methylation changes at the maternal-fetal interface of RPL. Earlier, the effect of DNA methylation on the uterine immune microenvironment and the occurrence of RPL were explored. This review fills a gap in the study of the important role of epigenetic regulatory networks in embryo development and the maternal-fetal interface. In this part, more basic experiments are needed to conduct in-depth research on the molecular mechanism and related signal pathways of epigenetic regulation in RPL. We also need to fully explore the complex association network between different regulation methods and their mechanism acting in early embryo development, embryo implantation, and the maternal-fetal interface. Our review opens up new avenues to screen women prior to pregnancy for the risk of miscarriage and point to the potential of immune therapies based on epigenetics in the prevention of RPL.
Author Contributions
QZ wrote the manuscript, made the figures, and critically reviewed the manuscript. YX and BQ wrote the manuscript and critically reviewed the manuscript. AB and YZ critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the following grants: National Key Research and Development Program of China (No. 2018YFC1004601), and the National Natural Science Foundation of China (No. 81801540, 81771662).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang J. Expert Consensus on the Diagnosis and Treatment of Recurrent Spontaneous Abortion. Fertil Steril(2016) 51:3–9.
2. Definitions of Infertility and Recurrent Pregnancy Loss: A Committee Opinion. Fertil Steril (2013) 99:63. doi: 10.1016/j.fertnstert.2012.09.023
3. El HH, Crepaux V, May-Panloup P, Descamps P, Legendre G, Bouet PE. Recurrent Pregnancy Loss: Current Perspectives. Int J Womens Health (2017) 9:331–45. doi: 10.2147/IJWH.S100817
4. Hocher B, Hocher CF. Epigenetics of Recurrent Pregnancy Loss. Ebiomedicine (2018) 35:18–9. doi: 10.1016/j.ebiom.2018.08.046
5. Arias-Sosa LA, Acosta ID, Lucena-Quevedo E, Moreno-Ortiz H, Esteban-Perez C, Forero-Castro M. Genetic and Epigenetic Variations Associated With Idiopathic Recurrent Pregnancy Loss. J Assist Reprod Genet (2018) 35:355–66. doi: 10.1007/s10815-017-1108-y
6. Michalak EM, Burr ML, Bannister AJ, Dawson MA. The Roles of DNA, RNA and Histone Methylation in Ageing and Cancer. Nat Rev Mol Cell Biol (2019) 20:573–89. doi: 10.1038/s41580-019-0143-1
7. Wu X, Zhang Y. TET-Mediated Active DNA Demethylation: Mechanism, Function and Beyond. Nat Rev Genet (2017) 18:517–34. doi: 10.1038/nrg.2017.33
8. Dor Y, Cedar H. Principles of DNA Methylation and Their Implications for Biology and Medicine. Lancet (2018) 392:777–86. doi: 10.1016/S0140-6736(18)31268-6
9. Law P, Holland ML. DNA Methylation at the Crossroads of Gene and Environment Interactions. Essays Biochem (2019) 63:717–26. doi: 10.1042/EBC20190031
10. Kazmi N, Sharp GC, Reese SE, Vehmeijer FO, Lahti J, Page CM, et al. Hypertensive Disorders of Pregnancy and DNA Methylation in Newborns. Hypertension (2019) 74:375–83. doi: 10.1161/HYPERTENSIONAHA.119.12634
11. Chen Z, Zhang Y. Role of Mammalian DNA Methyltransferases in Development. Annu Rev Biochem (2020) 89:135–58. doi: 10.1146/annurev-biochem-103019-102815
12. Li E, Bestor TH, Jaenisch R. Targeted Mutation of the DNA Methyltransferase Gene Results in Embryonic Lethality. Cell (1992) 69:915–26. doi: 10.1016/0092-8674(92)90611-F
13. Okano M, Bell DW. DNA Methyltransferases Dnmt3a and Dnmt3b are Essential for De Novo Methylation and Mammalian. Cell (1999) 99:247. doi: 10.1016/S0092-8674(00)81656-6
14. Bourc’His D, Bestor TH. Meiotic Catastrophe and Retrotransposon Reactivation in Male Germ Cells Lacking Dnmt3L. Nature (2004) 431:96–9. doi: 10.1038/nature02886
15. Bourc’His D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the Establishment of Maternal Genomic Imprints. Science (2001) 294:2536–9. doi: 10.1126/science.1065848
16. Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, et al. Essential Role for De Novo DNA Methyltransferase Dnmt3a in Paternal and Maternal Imprinting. Nature (2004) 429:900–3. doi: 10.1038/nature02633
17. Jain D, Meydan C, Lange J, Claeys BC, Lailler N, Mason CE, et al. Rahu is a Mutant Allele of Dnmt3c, Encoding a DNA Methyltransferase Homolog Required for Meiosis and Transposon Repression in the Mouse Male Germline. PloS Genet (2017) 13:e1006964. doi: 10.1371/journal.pgen.1006964
18. Barau J, Teissandier A, Zamudio N, Roy S, Nalesso V, Herault Y, et al. The DNA Methyltransferase DNMT3C Protects Male Germ Cells From Transposon Activity. Science (2016) 354:909–12. doi: 10.1126/science.aah5143
19. Parry A, Rulands S, Reik W. Active Turnover of DNA Methylation During Cell Fate Decisions. Nat Rev Genet (2021) 22:59–66. doi: 10.1038/s41576-020-00287-8
20. Saitou M, Kagiwada S, Kurimoto K. Epigenetic Reprogramming in Mouse Pre-Implantation Development and Primordial Germ Cells. Development (2012) 139:15–31. doi: 10.1242/dev.050849
21. Lee HJ, Hore TA, Reik W. Reprogramming the Methylome: Erasing Memory and Creating Diversity. Cell Stem Cell (2014) 14:710–9. doi: 10.1016/j.stem.2014.05.008
22. Yu M, Du G, Xu Q, Huang Z, Huang X, Qin Y, et al. Integrated Analysis of DNA Methylome and Transcriptome Identified CREB5 as a Novel Risk Gene Contributing to Recurrent Pregnancy Loss. Ebiomedicine (2018) 35:334–44. doi: 10.1016/j.ebiom.2018.07.042
23. Yamaguchi S, Hong K, Liu R, Shen L, Inoue A, Diep D, et al. Tet1 Controls Meiosis by Regulating Meiotic Gene Expression. Nature (2012) 492:443–7. doi: 10.1038/nature11709
24. Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, et al. Tet1 and Tet2 Regulate 5-Hydroxymethylcytosine Production and Cell Lineage Specification in Mouse Embryonic Stem Cells. Cell Stem Cell (2011) 8:200–13. doi: 10.1016/j.stem.2011.01.008
25. Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, et al. Distinct Roles of the Methylcytosine Oxidases Tet1 and Tet2 in Mouse Embryonic Stem Cells. Proc Natl Acad Sci USA (2014) 111:1361–6. doi: 10.1073/pnas.1322921111
26. Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mc Oxidation by Tet2 Modulates Enhancer Activity and Timing of Transcriptome Reprogramming During Differentiation. Mol Cell (2014) 56:286–97. doi: 10.1016/j.molcel.2014.08.026
27. Dawlaty MM, Breiling A, Le T, Barrasa MI, Raddatz G, Gao Q, et al. Loss of Tet Enzymes Compromises Proper Differentiation of Embryonic Stem Cells. Dev Cell (2014) 29:102–11. doi: 10.1016/j.devcel.2014.03.003
28. Stewart KR, Veselovska L, Kelsey G. Establishment and Functions of DNA Methylation in the Germline. Epigenomics-UK (2016) 8:1399–413. doi: 10.2217/epi-2016-0056
29. Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA Methylation and DNA Methyltransferases. Epigenet Chromatin (2017) 10:23. doi: 10.1186/s13072-017-0130-8
30. Li E, Zhang Y. DNA Methylation in Mammals. Cold Spring Harb Perspect Biol (2014) 6:a19133. doi: 10.1101/cshperspect.a019133
31. Smallwood SA, Kelsey G. De Novo DNA Methylation: A Germ Cell Perspective. Trends Genet (2012) 28:33–42. doi: 10.1016/j.tig.2011.09.004
32. Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, et al. The DNA Methylation Landscape of Human Early Embryos. Nature (2014) 511:606–10. doi: 10.1038/nature13544
33. Messerschmidt DM, Knowles BB, Solter D. DNA Methylation Dynamics During Epigenetic Reprogramming in the Germline and Preimplantation Embryos. Genes Dev (2014) 28:812–28. doi: 10.1101/gad.234294.113
34. Gao R, Wang C, Gao Y, Xiu W, Chen J, Kou X, et al. Inhibition of Aberrant DNA Re-Methylation Improves Post-Implantation Development of Somatic Cell Nuclear Transfer Embryos. Cell Stem Cell (2018) 23:426–35. doi: 10.1016/j.stem.2018.07.017
35. Novakovic B, Rakyan V, Ng HK, Manuelpillai U, Dewi C, Wong NC, et al. Specific Tumour-Associated Methylation in Normal Human Term Placenta and First-Trimester Cytotrophoblasts. Mol Hum Reprod (2008) 14:547–54. doi: 10.1093/molehr/gan046
36. Smith A, Witte E, McGee D, Knott J, Narang K, Racicot K. Cortisol Inhibits CSF2 and CSF3 via DNA Methylation and Inhibits Invasion in First-Trimester Trophoblast Cells. Am J Reprod Immunol (2017) 78(5):10. doi: 10.1111/aji.12741
37. Pliushch G, Schneider E, Weise D, El HN, Tresch A, Seidmann L, et al. Extreme Methylation Values of Imprinted Genes in Human Abortions and Stillbirths. Am J Pathol (2010) 176:1084–90. doi: 10.2353/ajpath.2010.090764
38. Vasconcelos S, Ramalho C, Marques CJ, Doria S. Altered Expression of Epigenetic Regulators and Imprinted Genes in Human Placenta and Fetal Tissues From Second Trimester Spontaneous Pregnancy Losses. Epigenetics-US (2019) 14:1234–44. doi: 10.1080/15592294.2019.1634988
39. Li J, Yu X, Wang L, Zhang Y. Expression and Methylation Level of MMPs/TIMPs in the Decidua Tissue of Patients With Unexplained Recurrent Spontaneous Abortion. Chin J of Clin Obstetrics Gynecol (2019) 20:514–8.
40. Li J, Yu X, Yang Y, Wang L, Zhang Y. DNA Hypomethylation in Villi of Patients With Unexplained Recurrent Spontaneous Abortion and Its Mechanism. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2019) 35:351–6.
41. Chao Y, Weng L, Zeng R. Correlation of Genomic DNA Methylation Level With Unexplained Early Spontaneous Abortion. Nan Fang Yi Ke Da Xue Xue Bao (2014) 34:1498–502.
42. Li J, Yu X, Yang Y, Wang L, Zhang Y. DNA Hypomethylation in Villi of Patients With Unexplained Recurrent Spontaneous Abortion and Its Mechanism. Chin J Cell Mol immunol (2019) 35:351–6.
43. Liu Y, Zheng H, Guo P, Feng S, Zhou X, Ye D, et al. DNA Methyltransferase 3A Promoter Polymorphism Is Associated With the Risk of Human Spontaneous Abortion After Assisted Reproduction Techniques and Natural Conception. J Assist Reprod Genet (2017) 34:245–52. doi: 10.1007/s10815-016-0837-7
44. Barisic A, Pereza N, Hodzic A, Ostojic S, Peterlin B. A Single Nucleotide Polymorphism of DNA Methyltransferase 3B Gene Is a Risk Factor for Recurrent Spontaneous Abortion. Am J Reprod Immunol (2017) 78:6. doi: 10.1111/aji.12765
45. Yin LJ, Zhang Y, Lv PP, He WH, Wu YT, Liu AX, et al. Insufficient Maintenance DNA Methylation Is Associated With Abnormal Embryonic Development. BMC Med (2012) 10:26. doi: 10.1186/1741-7015-10-26
46. Serman L, Vlahovic M, Sijan M, Bulic-Jakus F, Serman A, Sincic N, et al. The Impact of 5-Azacytidine on Placental Weight, Glycoprotein Pattern and Proliferating Cell Nuclear Antigen Expression in Rat Placenta. Placenta (2007) 28:803–11. doi: 10.1016/j.placenta.2007.04.001
47. Chrysanthou S, Senner CE, Woods L, Fineberg E, Okkenhaug H, Burge S, et al. A Critical Role of TET1/2 Proteins in Cell-Cycle Progression of Trophoblast Stem Cells. Stem Cell Rep (2018) 10:1355–68. doi: 10.1016/j.stemcr.2018.02.014
48. Schuster J, Uzun A, Stablia J, Schorl C, Mori M, Padbury JF. Effect of Prematurity on Genome Wide Methylation in the Placenta. BMC Med Genet (2019) 20:116. doi: 10.1186/s12881-019-0835-6
49. Crome SQ, Wang AY, Levings MK. Translational Mini-Review Series on Th17 Cells: Function and Regulation of Human T Helper 17 Cells in Health and Disease. Clin Exp Immunol (2010) 159:109–19. doi: 10.1111/j.1365-2249.2009.04037.x
50. Mishra J, Talwar S, Kaur L, Chandiok K, Yadav S, Puri M, et al. Differential Global and MTHFR Gene Specific Methylation Patterns in Preeclampsia and Recurrent Miscarriages: A Case-Control Study From North India. Gene (2019) 704:68–73. doi: 10.1016/j.gene.2019.04.036
51. Liu Y, Zhang F, Dai L. C677T Polymorphism Increases the Risk of Early Spontaneous Abortion. J Assist Reprod Genet (2019) 36:1737–41. doi: 10.1007/s10815-019-01500-2
52. Kim SY, Park SY, Choi JW, Kim DJ, Lee SY, Lim JH, et al. Association Between MTHFR 1298a>C Polymorphism and Spontaneous Abortion With Fetal Chromosomal Aneuploidy. Am J Reprod Immunol (2011) 66:252–8. doi: 10.1111/j.1600-0897.2011.00996.x
53. Rotondo JC, Bosi S, Bazzan E, Di Domenico M, De Mattei M, Selvatici R, et al. Methylenetetrahydrofolate Reductase Gene Promoter Hypermethylation in Semen Samples of Infertile Couples Correlates With Recurrent Spontaneous Abortion. Hum Reprod (2012) 27:3632–8. doi: 10.1093/humrep/des319
54. Deshpande SS, Balasinor NH. Placental Defects: An Epigenetic Perspective. Reprod Sci (2018) 25:1143–60. doi: 10.1177/1933719118766265
55. Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA Methylomes of Newborns and Centenarians. Proc Natl Acad Sci USA (2012) 109:10522–7. doi: 10.1073/pnas.1120658109
56. Xu P, Wu Z, Yang W, Wang L. Dysregulation of DNA Methylation and Expression of Imprinted Genes in Mouse Placentas of Fetal Growth Restriction Induced by Maternal Cadmium Exposure. Toxicology (2017) 390:109–16. doi: 10.1016/j.tox.2017.08.003
57. Janssen AB, Tunster SJ, Savory N, Holmes A, Beasley J, Parveen SA, et al. Placental Expression of Imprinted Genes Varies With Sampling Site and Mode of Delivery. Placenta (2015) 36:790–5. doi: 10.1016/j.placenta.2015.06.011
58. Shiura H, Nakamura K, Hikichi T, Hino T, Oda K, Suzuki-Migishima R, et al. Paternal Deletion of Meg1/Grb10 DMR Causes Maternalization of the Meg1/Grb10 Cluster in Mouse Proximal Chromosome 11 Leading to Severe Pre- and Postnatal Growth Retardation. Hum Mol Genet (2009) 18:1424–38. doi: 10.1093/hmg/ddp049
59. Anvar Z, Chakchouk I, Demond H, Sharif M, Kelsey G, Van den Veyver IB. DNA Methylation Dynamics in the Female Germline and Maternal-Effect Mutations That Disrupt Genomic Imprinting. Genes (Basel) (2021) 12(8):1214. doi: 10.3390/genes12081214
60. Shaker MM, Shalabi TA, Amr KS. Correlation of Methylation Status in MTHFR Promoter Region With Recurrent Pregnancy Loss. J Genet Eng Biotechnol (2021) 19:44. doi: 10.1186/s43141-021-00147-w
61. Zhang X, Wang D, Han Y, Duan F, Lv Q, Li Z. Altered Imprinted Gene Expression and Methylation Patterns in Mid-Gestation Aborted Cloned Porcine Fetuses and Placentas. J Assist Reprod Genet (2014) 31:1511–7. doi: 10.1007/s10815-014-0320-2
62. Flisikowski K, Venhoranta H, Bauersachs S, Hanninen R, Furst RW, Saalfrank A, et al. Truncation of MIMT1 Gene in the PEG3 Domain Leads to Major Changes in Placental Gene Expression and Stillbirth in Cattle. Biol Reprod (2012) 87:140. doi: 10.1095/biolreprod.112.104240
63. Doria S, Sousa M, Fernandes S, Ramalho C, Brandao O, Matias A, et al. Gene Expression Pattern of IGF2, PHLDA2, PEG10 and CDKN1C Imprinted Genes in Spontaneous Miscarriages or Fetal Deaths. Epigenetics-US (2010) 5:444–50. doi: 10.4161/epi.5.5.12118
64. Liu Y, Tang Y, Ye D, Ma W, Feng S, Li X, et al. Impact of Abnormal DNA Methylation of Imprinted Loci on Human Spontaneous Abortion. Reprod Sci (2018) 25:131–9. doi: 10.1177/1933719117704906
65. Angiolini E, Fowden A, Coan P, Sandovici I, Smith P, Dean W, et al. Regulation of Placental Efficiency for Nutrient Transport by Imprinted Genes. Placenta (2006) 27 Suppl A:S98–S102. doi: 10.1016/j.placenta.2005.12.008
66. Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA. Regulation of Maternal Behavior and Offspring Growth by Paternally Expressed Peg3. Science (1999) 284:330–3. doi: 10.1126/science.284.5412.330
67. Dilworth MR, Kusinski LC, Cowley E, Ward BS, Husain SM, Constancia M, et al. Placental-Specific Igf2 Knockout Mice Exhibit Hypocalcemia and Adaptive Changes in Placental Calcium Transport. Proc Natl Acad Sci USA (2010) 107:3894–9. doi: 10.1073/pnas.0911710107
68. Peng W, Chen Y, Luo X, Shan N, Lan X, Olson D, et al. DNA Methylation-Associated Repression of MEST/PEG1 Expression Contributes to the Invasion of Extravillous Trophoblast Cells. Placenta (2016) 46:92–101. doi: 10.1016/j.placenta.2016.08.093
69. Zheng HY, Tang Y, Niu J, Li P, Ye DS, Chen X, et al. Aberrant DNA Methylation of Imprinted Loci in Human Spontaneous Abortions After Assisted Reproduction Techniques and Natural Conception. Hum Reprod (2013) 28:265–73. doi: 10.1093/humrep/des358
70. Du G, Yu M, Xu Q, Huang Z, Huang X, Han L, et al. Hypomethylation of PRDM1 is Associated With Recurrent Pregnancy Loss. J Cell Mol Med (2020) 24:7072–7. doi: 10.1111/jcmm.15335
71. Sheng F, Sun N, Ji Y, Ma Y, Ding H, Zhang Q, et al. Aberrant Expression of Imprinted lncRNA MEG8 Causes Trophoblast Dysfunction and Abortion. J Cell Biochem (2019) 120:17378–90. doi: 10.1002/jcb.29002
72. Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC. Genomic Imprinting and Physiological Processes in Mammals. Cell (2019) 176:952–65. doi: 10.1016/j.cell.2019.01.043
73. Carrell DT, Salas-Huetos A, Hotaling J. Increasing Evidence of the Role of the Sperm Epigenome in Embryogenesis: Oligoasthenoteratozoospermia, Altered Embryo DNA Methylation, and Miscarriage. Fertil Steril (2018) 110:401–2. doi: 10.1016/j.fertnstert.2018.04.042
74. Zidi-Jrah I, Hajlaoui A, Mougou-Zerelli S, Kammoun M, Meniaoui I, Sallem A, et al. Relationship Between Sperm Aneuploidy, Sperm DNA Integrity, Chromatin Packaging, Traditional Semen Parameters, and Recurrent Pregnancy Loss. Fertil Steril (2016) 105:58–64. doi: 10.1016/j.fertnstert.2015.09.041
75. Zhang L, Wang L, Zhang X, Xu G, Zhang W, Wang K, et al. Sperm Chromatin Integrity may Predict Future Fertility for Unexplained Recurrent Spontaneous Abortion Patients. Int J Androl (2012) 35:752–7. doi: 10.1111/j.1365-2605.2012.01276.x
76. Ruixue W, Hongli Z, Zhihong Z, Rulin D, Dongfeng G, Ruizhi L. The Impact of Semen Quality, Occupational Exposure to Environmental Factors and Lifestyle on Recurrent Pregnancy Loss. J Assist Reprod Genet (2013) 30:1513–8. doi: 10.1007/s10815-013-0091-1
77. Kazerooni T, Asadi N, Jadid L, Kazerooni M, Ghanadi A, Ghaffarpasand F, et al. Evaluation of Sperm’s Chromatin Quality With Acridine Orange Test, Chromomycin A3 and Aniline Blue Staining in Couples With Unexplained Recurrent Abortion. J Assist Reprod Genet (2009) 26:591–6. doi: 10.1007/s10815-009-9361-3
78. Ibrahim Y, Hotaling J. Sperm Epigenetics and Its Impact on Male Fertility, Pregnancy Loss, and Somatic Health of Future Offsprings. Semin Reprod Med (2018) 36:233–9. doi: 10.1055/s-0039-1688453
79. Pan LJ, Ma JH, Zhang FL, Zhao D, Pan F, Zhang XY. Relationship of Abnormal Sperm DNA Methylation With Early Spontaneous Abortion. Zhonghua Nan Ke Xue (2016) 22:882–5.
80. Khambata K, Raut S, Deshpande S, Mohan S, Sonawane S, Gaonkar R, et al. DNA Methylation Defects in Spermatozoa of Male Partners From Couples Experiencing Recurrent Pregnancy Loss. Hum Reprod (2021) 36:48–60. doi: 10.1093/humrep/deaa278
81. Dumasia K, Kumar A, Deshpande S, Balasinor NH. Estrogen Signaling, Through Estrogen Receptor β, Regulates DNA Methylation and its Machinery in Male Germ Line in Adult Rats. Epigenetics-US (2017) 12:476–83. doi: 10.1080/15592294.2017.1309489
82. Eden S, Cedar H. Role of DNA Methylation in the Regulation of Transcription. Curr Opin Genet Dev (1994) 4:255–9. doi: 10.1016/S0959-437X(05)80052-8
83. Panning B, Jaenisch R. DNA Hypomethylation can Activate Xist Expression and Silence X-Linked Genes. Genes Dev (1996) 10:1991–2002. doi: 10.1101/gad.10.16.1991
84. Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP Endogenous Retroviruses Is Constrained by Cytosine Methylation. Nat Genet (1998) 20:116–7. doi: 10.1038/2413
85. Seifertova M, Vesely J, Cihak A. Enhanced Mortality in Offsprings of Male Mice Treated With 5-Azacytidine Prior to Mating. Morphological Changes in Testes. Neoplasma (1976) 23:53–60.
86. Oakes CC, Kelly TL, Robaire B, Trasler JM. Adverse Effects of 5-Aza-2’-Deoxycytidine on Spermatogenesis Include Reduced Sperm Function and Selective Inhibition of De Novo DNA Methylation. J Pharmacol Exp Ther (2007) 322:1171–80. doi: 10.1124/jpet.107.121699
87. Rogenhofer N, Ott J, Pilatz A, Wolf J, Thaler CJ, Windischbauer L, et al. Unexplained Recurrent Miscarriages are Associated With an Aberrant Sperm Protamine mRNA Content. Hum Reprod (2017) 32:1574–82. doi: 10.1093/humrep/dex224
88. DeChiara TM, Robertson EJ, Efstratiadis A. Parental Imprinting of the Mouse Insulin-Like Growth Factor II Gene. Cell (1991) 64:849–59. doi: 10.1016/0092-8674(91)90513-X
89. Moon YS, Smas CM, Lee K, Villena JA, Kim KH, Yun EJ, et al. Mice Lacking Paternally Expressed Pref-1/Dlk1 Display Growth Retardation and Accelerated Adiposity. Mol Cell Biol (2002) 22:5585–92. doi: 10.1128/MCB.22.15.5585-5592.2002
90. Mayer W, Hemberger M, Frank HG, Grummer R, Winterhager E, Kaufmann P, et al. Expression of the Imprinted Genes MEST/Mest in Human and Murine Placenta Suggests a Role in Angiogenesis. Dev Dyn (2000) 217:1–10. doi: 10.1002/(SICI)1097-0177(200001)217:1<1::AID-DVDY1>3.0.CO;2-4
91. Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, et al. Zac1 Regulates an Imprinted Gene Network Critically Involved in the Control of Embryonic Growth. Dev Cell (2006) 11:711–22. doi: 10.1016/j.devcel.2006.09.003
92. Zhang Y, Tycko B. Monoallelic Expression of the Human H19 Gene. Nat Genet (1992) 1:40–4. doi: 10.1038/ng0492-40
93. Rotondo JC, Selvatici R, Di Domenico M, Marci R, Vesce F, Tognon M, et al. Methylation Loss at H19 Imprinted Gene Correlates With Methylenetetrahydrofolate Reductase Gene Promoter Hypermethylation in Semen Samples From Infertile Males. Epigenetics-US (2013) 8:990–7. doi: 10.4161/epi.25798
94. Poorang S, Abdollahi S, Anvar Z, Tabei S, Jahromi BN, Moein-Vaziri N, et al. The Impact of Methylenetetrahydrofolate Reductase (MTHFR) Sperm Methylation and Variants on Semen Parameters and the Chance of Recurrent Pregnancy Loss in the Couple. Clin Lab (2018) 64:1121–8. doi: 10.7754/Clin.Lab.2018.171231
95. Gupta N, Gupta S, Dama M, David A, Khanna G, Khanna A, et al. Strong Association of 677 C>T Substitution in the MTHFR Gene With Male Infertility - A Study on an Indian Population and a Meta-Analysis. PloS One (2011) 6:e22277. doi: 10.1371/journal.pone.0022277
96. Wu W, Shen O, Qin Y, Niu X, Lu C, Xia Y, et al. Idiopathic Male Infertility is Strongly Associated With Aberrant Promoter Methylation of Methylenetetrahydrofolate Reductase (MTHFR). PloS One (2010) 5:e13884. doi: 10.1371/journal.pone.0013884
97. Uuskula L, Rull K, Nagirnaja L, Laan M. Methylation Allelic Polymorphism (MAP) in Chorionic Gonadotropin Beta5 (CGB5) and Its Association With Pregnancy Success. J Clin Endocrinol Metab (2011) 96:E199–207. doi: 10.1210/jc.2010-1647
98. Okae H, Toh H, Sato T, Hiura H, Takahashi S, Shirane K, et al. Derivation of Human Trophoblast Stem Cells. Cell Stem Cell (2018) 22:50–63. doi: 10.1016/j.stem.2017.11.004
99. Nordor AV, Nehar-Belaid D, Richon S, Klatzmann D, Bellet D, Dangles-Marie V, et al. The Early Pregnancy Placenta Foreshadows DNA Methylation Alterations of Solid Tumors. Epigenetics-US (2017) 12:793–803. doi: 10.1080/15592294.2017.1342912
100. Yuen RK, Robinson WP. Review: A High Capacity of the Human Placenta for Genetic and Epigenetic Variation: Implications for Assessing Pregnancy Outcome. Placenta (2011) 32(Suppl 2):S136–41. doi: 10.1016/j.placenta.2011.01.003
101. Lim YC, Li J, Ni Y, Liang Q, Zhang J, Yeo G, et al. A Complex Association Between DNA Methylation and Gene Expression in Human Placenta at First and Third Trimesters. PloS One (2017) 12:e181155. doi: 10.1371/journal.pone.0181155
102. Hanna CW, McFadden DE, Robinson WP. DNA Methylation Profiling of Placental Villi From Karyotypically Normal Miscarriage and Recurrent Miscarriage. Am J Pathol (2013) 182:2276–84. doi: 10.1016/j.ajpath.2013.02.021
103. Ohinata Y, Payer B, O’Carroll D, Ancelin K, Ono Y, Sano M, et al. Blimp1 is a Critical Determinant of the Germ Cell Lineage in Mice. Nature (2005) 436:207–13. doi: 10.1038/nature03813
104. Wu AH, Guo LY, Lu S, Chen XL, Wang AA, Wang XY, et al. Aberrant Methylation of IGF2-AS Promoter in Early Pregnancy Loss. Taiwan J Obstet Gynecol (2020) 59:109–14. doi: 10.1016/j.tjog.2019.11.017
105. Norwitz ER, Schust DJ, Fisher SJ. Implantation and the Survival of Early Pregnancy. N Engl J Med (2001) 345:1400–8. doi: 10.1056/NEJMra000763
106. Chen H, Cheng S, Liu C, Fu J, Huang W. Bioinformatics Analysis of Differentially Expressed Genes, Methylated Genes, and miRNAs in Unexplained Recurrent Spontaneous Abortion. J Comput Biol (2019) 26:1418–26. doi: 10.1089/cmb.2019.0158
107. Gao F, Ma X, Rusie A, Hemingway J, Ostmann AB, Chung D, et al. Epigenetic Changes Through DNA Methylation Contribute to Uterine Stromal Cell Decidualization. Endocrinology (2012) 153:6078–90. doi: 10.1210/en.2012-1457
108. Liu H, Huang X, Mor G, Liao A. Epigenetic Modifications Working in the Decidualization and Endometrial Receptivity. Cell Mol Life Sci (2020) 77:2091–101. doi: 10.1007/s00018-019-03395-9
109. Krieg SA, Fan X, Hong Y, Sang QX, Giaccia A, Westphal LM, et al. Global Alteration in Gene Expression Profiles of Deciduas From Women With Idiopathic Recurrent Pregnancy Loss. Mol Hum Reprod (2012) 18:442–50. doi: 10.1093/molehr/gas017
110. Feroze-Zaidi F, Fusi L, Takano M, Higham J, Salker MS, Goto T, et al. Role and Regulation of the Serum- and Glucocorticoid-Regulated Kinase 1 in Fertile and Infertile Human Endometrium. Endocrinology (2007) 148:5020–9. doi: 10.1210/en.2007-0659
111. Zhou L, Pu Y, Zhou Y, Wang B, Chen Y, Bai Y, et al. Genome Wide Methylation Analysis to Uncover Genes Related to Recurrent Pregnancy Loss. Genes Genomics (2021) 43:361–9. doi: 10.1007/s13258-020-01020-9
112. Lv XF, Sun LL, Cui CL, Han JS. NAc Shell Arc/Arg3.1 Protein Mediates Reconsolidation of Morphine CPP by Increased GluR1 Cell Surface Expression: Activation of ERK-Coupled CREB is Required. Int J Neuropsychopharmacol (2015) 18:9. doi: 10.1093/ijnp/pyv030
113. Poh AR, O’Donoghue RJ, Ernst M. Hematopoietic Cell Kinase (HCK) as a Therapeutic Target in Immune and Cancer Cells. Oncotarget (2015) 6:15752–71. doi: 10.18632/oncotarget.4199
114. Ding Y, Kantarci A, Badwey JA, Hasturk H, Malabanan A, Van Dyke TE. Phosphorylation of Pleckstrin Increases Proinflammatory Cytokine Secretion by Mononuclear Phagocytes in Diabetes Mellitus. J Immunol (2007) 179:647–54. doi: 10.4049/jimmunol.179.1.647
115. Azizieh FY, Raghupathy R. IL-10 and Pregnancy Complications. Clin Exp Obstet Gynecol (2017) 44:252–8.
116. Logan PC, Ponnampalam AP, Rahnama F, Lobie PE, Mitchell MD. The Effect of DNA Methylation Inhibitor 5-Aza-2’-Deoxycytidine on Human Endometrial Stromal Cells. Hum Reprod (2010) 25:2859–69. doi: 10.1093/humrep/deq238
117. Wang XH, Xu S, Zhou XY, Zhao R, Lin Y, Cao J, et al. Low Chorionic Villous Succinate Accumulation Associates With Recurrent Spontaneous Abortion Risk. Nat Commun (2021) 12:3428. doi: 10.1038/s41467-021-23827-0
118. Xie J, Liang T, Zhao J, Xu Z, Tian P, Wang R, et al. Lnc-HZ08 Regulates BPDE-Induced Trophoblast Cell Dysfunctions by Promoting PI3K Ubiquitin Degradation and is Associated With Miscarriage. Cell Biol Toxicol (2021) 22. doi: 10.1007/s10565-021-09606-z
119. Fatima N, Ahmed SH, Chauhan SS, Mohammad O, Rehman S. Structural Equation Modelling Analysis Determining Causal Role Among Methyltransferases, Methylation, and Apoptosis During Human Pregnancy and Abortion. Sci Rep (2020) 10:12408. doi: 10.1038/s41598-020-68270-1
120. Lucas ES, Dyer NP, Murakami K, Lee YH, Chan YW, Grimaldi G, et al. Loss of Endometrial Plasticity in Recurrent Pregnancy Loss. Stem Cells (2016) 34:346–56. doi: 10.1002/stem.2222
121. Zhou M, Xu H, Zhang D, Si C, Zhou X, Zhao H, et al. Decreased PIBF1/IL6/p-STAT3 During the Mid-Secretory Phase Inhibits Human Endometrial Stromal Cell Proliferation and Decidualization. J Adv Res (2021) 30:15–25. doi: 10.1016/j.jare.2020.09.002
122. Mei S, Tan J, Chen H, Chen Y, Zhang J. Changes of CD4+CD25high Regulatory T Cells and FOXP3 Expression in Unexplained Recurrent Spontaneous Abortion Patients. Fertil Steril (2010) 94:2244–7. doi: 10.1016/j.fertnstert.2009.11.020
123. Alecsandru D, Klimczak AM, Garcia VJ, Pirtea P, Franasiak JM. Immunologic Causes and Thrombophilia in Recurrent Pregnancy Loss. Fertil Steril (2021) 115:561–6. doi: 10.1016/j.fertnstert.2021.01.017
124. Guo C, Cai P, Jin L, Sha Q, Yu Q, Zhang W, et al. Single-Cell Profiling of the Human Decidual Immune Microenvironment in Patients With Recurrent Pregnancy Loss. Cell Discovery (2021) 7:1. doi: 10.1038/s41421-020-00236-z
125. Ticconi C, Pietropolli A, Di Simone N, Piccione E, Fazleabas A. Endometrial Immune Dysfunction in Recurrent Pregnancy Loss. Int J Mol Sci (2019) 20(21):5332. doi: 10.3390/ijms20215332
126. Lucas ES, Dyer NP, Fishwick K, Ott S, Brosens JJ. Success After Failure: The Role of Endometrial Stem Cells in Recurrent Miscarriage. Reproduction (2016) 152:R159–66. doi: 10.1530/REP-16-0306
127. Macklon NS, Brosens JJ. The Human Endometrium as a Sensor of Embryo Quality. Biol Reprod (2014) 91:98. doi: 10.1095/biolreprod.114.122846
128. Quenby S, Brosens JJ. Human Implantation: A Tale of Mutual Maternal and Fetal Attraction. Biol Reprod (2013) 88:81. doi: 10.1095/biolreprod.113.108886
129. Brosens JJ, Salker MS, Teklenburg G, Nautiyal J, Salter S, Lucas ES, et al. Uterine Selection of Human Embryos at Implantation. Sci Rep (2014) 4:3894. doi: 10.1038/srep03894
130. Faas MM, de Vos P. Uterine NK Cells and Macrophages in Pregnancy. Placenta (2017) 56:44–52. doi: 10.1016/j.placenta.2017.03.001
131. Djurisic S, Hviid TV. HLA Class Ib Molecules and Immune Cells in Pregnancy and Preeclampsia. Front Immunol (2014) 5:652. doi: 10.3389/fimmu.2014.00652
132. Ning F, Liu H, Lash GE. The Role of Decidual Macrophages During Normal and Pathological Pregnancy. Am J Reprod Immunol (2016) 75:298–309. doi: 10.1111/aji.12477
133. Kim SY, Romero R, Tarca AL, Bhatti G, Kim CJ, Lee J, et al. Methylome of Fetal and Maternal Monocytes and Macrophages at the Feto-Maternal Interface. Am J Reprod Immunol (1989) (2012) 68:8–27. doi: 10.1111/j.1600-0897.2012.01108.x
134. Tsao FY, Wu MY, Chang YL, Wu CT, Ho HN. M1 Macrophages Decrease in the Deciduae From Normal Pregnancies But Not From Spontaneous Abortions or Unexplained Recurrent Spontaneous Abortions. J Formos Med Assoc (2018) 117:204–11. doi: 10.1016/j.jfma.2017.03.011
135. Lv R, Bao Q, Li Y. Regulation of M1type and M2type Macrophage Polarization in RAW264.7 Cells by Galectin9. Mol Med Rep (2017) 16:9111–9. doi: 10.3892/mmr.2017.7719
136. Edholm ES, Rhoo KH, Robert J. Evolutionary Aspects of Macrophages Polarization. Results Probl Cell Differ (2017) 62:3–22. doi: 10.1007/978-3-319-54090-0_1
137. Fu YY, Ren CE, Qiao PY, Meng YH. Uterine Natural Killer Cells and Recurrent Spontaneous Abortion. Am J Reprod Immunol (2021) 86:e13433. doi: 10.1111/aji.13433
138. Smith SD, Dunk CE, Aplin JD, Harris LK, Jones RL. Evidence for Immune Cell Involvement in Decidual Spiral Arteriole Remodeling in Early Human Pregnancy. Am J Pathol (2009) 174:1959–71. doi: 10.2353/ajpath.2009.080995
139. Helige C, Ahammer H, Moser G, Hammer A, Dohr G, Huppertz B, et al. Distribution of Decidual Natural Killer Cells and Macrophages in the Neighbourhood of the Trophoblast Invasion Front: A Quantitative Evaluation. Hum Reprod (2014) 29:8–17. doi: 10.1093/humrep/det353
140. Koopman LA, Kopcow HD, Rybalov B, Boyson JE, Orange JS, Schatz F, et al. Human Decidual Natural Killer Cells Are a Unique NK Cell Subset With Immunomodulatory Potential. J Exp Med (2003) 198:1201–12. doi: 10.1084/jem.20030305
141. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK Cells Regulate Key Developmental Processes at the Human Fetal-Maternal Interface. Nat Med (2006) 12:1065–74. doi: 10.1038/nm1452
142. Hiby SE, Walker JJ, O’Shaughnessy KM, Redman CW, Carrington M, Trowsdale J, et al. Combinations of Maternal KIR and Fetal HLA-C Genes Influence the Risk of Preeclampsia and Reproductive Success. J Exp Med (2004) 200:957–65. doi: 10.1084/jem.20041214
143. Wang W, Sung N, Gilman-Sachs A, Kwak-Kim J. T Helper (Th) Cell Profiles in Pregnancy and Recurrent Pregnancy Losses: Th1/Th2/Th9/Th17/Th22/Tfh Cells. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.02025
144. Bulmer JN, Williams PJ, Lash GE. Immune Cells in the Placental Bed. Int J Dev Biol (2010) 54:281–94. doi: 10.1387/ijdb.082763jb
145. Giuliani E, Parkin KL, Lessey BA, Young SL, Fazleabas AT. Characterization of Uterine NK Cells in Women With Infertility or Recurrent Pregnancy Loss and Associated Endometriosis. Am J Reprod Immunol (2014) 72:262–9. doi: 10.1111/aji.12259
146. Kuon RJ, Weber M, Heger J, Santillan I, Vomstein K, Bar C, et al. Uterine Natural Killer Cells in Patients With Idiopathic Recurrent Miscarriage. Am J Reprod Immunol (2017) 78:4. doi: 10.1111/aji.12721
147. PrabhuDas M, Bonney E, Caron K, Dey S, Erlebacher A, Fazleabas A, et al. Immune Mechanisms at the Maternal-Fetal Interface: Perspectives and Challenges. Nat Immunol (2015) 16:328–34. doi: 10.1038/ni.3131
148. Moffett A, Colucci F. Uterine NK Cells: Active Regulators at the Maternal-Fetal Interface. J Clin Invest (2014) 124:1872–9. doi: 10.1172/JCI68107
149. Wang S, Li YP, Ding B, Zhao YR, Chen ZJ, Xu CY, et al. Recurrent Miscarriage is Associated With a Decline of Decidual Natural Killer Cells Expressing Killer Cell Immunoglobulin-Like Receptors Specific for Human Leukocyte Antigen C. J obstet Gynaecol Res (2014) 40:1288–95. doi: 10.1111/jog.12329
150. Quenby S, Nik H, Innes B, Lash G, Turner M, Drury J, et al. Uterine Natural Killer Cells and Angiogenesis in Recurrent Reproductive Failure. Hum Reprod (2009) 24:45–54. doi: 10.1093/humrep/den348
151. El-Azzamy H, Dambaeva SV, Katukurundage D, Salazar GM, Skariah A, Hussein Y, et al. Dysregulated Uterine Natural Killer Cells and Vascular Remodeling in Women With Recurrent Pregnancy Losses. Am J Reprod Immunol (2018) 80:e13024. doi: 10.1111/aji.13024
152. Lee SK, Na BJ, Kim JY, Hur SE, Lee M, Gilman-Sachs A, et al. Determination of Clinical Cellular Immune Markers in Women With Recurrent Pregnancy Loss. Am J Reprod Immunol (2013) 70:398–411. doi: 10.1111/aji.12137
153. Fukui A, Ntrivalas E, Fukuhara R, Fujii S, Mizunuma H, Gilman-Sachs A, et al. Correlation Between Natural Cytotoxicity Receptors and Intracellular Cytokine Expression of Peripheral Blood NK Cells in Women With Recurrent Pregnancy Losses and Implantation Failures. Am J Reprod Immunol (2009) 62:371–80. doi: 10.1111/j.1600-0897.2009.00750.x
154. Fukui A, Kwak-Kim J, Ntrivalas E, Gilman-Sachs A, Lee SK, Beaman K. Intracellular Cytokine Expression of Peripheral Blood Natural Killer Cell Subsets in Women With Recurrent Spontaneous Abortions and Implantation Failures. Fertil Steril (2008) 89:157–65. doi: 10.1016/j.fertnstert.2007.02.012
155. Saito S, Nakashima A, Shima T, Ito M. Th1/Th2/Th17 and Regulatory T-Cell Paradigm in Pregnancy. Am J Reprod Immunol (2010) 63:601–10. doi: 10.1111/j.1600-0897.2010.00852.x
156. Erdmann AA, Jung U, Foley JE, Toda Y, Fowler DH. Co-Stimulated/Tc2 Cells Abrogate Murine Marrow Graft Rejection. Biol Blood Marrow Transplant (2004) 10:604–13. doi: 10.1016/j.bbmt.2004.06.006
157. Burns WR, Wang Y, Tang PC, Ranjbaran H, Iakimov A, Kim J, et al. Recruitment of CXCR3+ and CCR5+ T Cells and Production of Interferon-Gamma-Inducible Chemokines in Rejecting Human Arteries. Am J Transplant (2005) 5:1226–36. doi: 10.1111/j.1600-6143.2005.00892.x
158. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. 1986. J Immunol (2005) 175:5–14. doi: 10.1007/s00031-008-9037-6
159. Li XC, Zand MS, Li Y, Zheng XX, Strom TB. On Histocompatibility Barriers, Th1 to Th2 Immune Deviation, and the Nature of the Allograft Responses. J Immunol (1998) 161:2241–7.
160. Torchinsky A, Shepshelovich J, Orenstein H, Zaslavsky Z, Savion S, Carp H, et al. TNF-Alpha Protects Embryos Exposed to Developmental Toxicants. Am J Reprod Immunol (2003) 49:159–68. doi: 10.1034/j.1600-0897.2003.01174.x
161. Williams Z. Inducing Tolerance to Pregnancy. N Engl J Med (2012) 367:1159–61. doi: 10.1056/NEJMcibr1207279
162. Keller CC, Eikmans M, van der Hoorn MP, Lashley L. Recurrent Miscarriages and the Association With Regulatory T Cells; A Systematic Review. J Reprod Immunol (2020) 139:103105. doi: 10.1016/j.jri.2020.103105
163. Ruocco MG, Chaouat G, Florez L, Bensussan A, Klatzmann D. Regulatory T-Cells in Pregnancy: Historical Perspective, State of the Art, and Burning Questions. Front Immunol (2014) 5:389. doi: 10.3389/fimmu.2014.00389
164. Piccinni MP, Lombardelli L, Logiodice F, Kullolli O, Romagnani S, Le Bouteiller P. T Helper Cell Mediated-Tolerance Towards Fetal Allograft in Successful Pregnancy. Clin Mol Allergy (2015) 13:9. doi: 10.1186/s12948-015-0015-y
165. Mitchell RE, Hassan M, Burton BR, Britton G, Hill EV, Verhagen J, et al. IL-4 Enhances IL-10 Production in Th1 Cells: Implications for Th1 and Th2 Regulation. Sci Rep (2017) 7:11315. doi: 10.1038/s41598-017-11803-y
166. Illera VA, Perandones CE, Stunz LL, Mower DJ, Ashman RF. Apoptosis in Splenic B Lymphocytes. Regulation by Protein Kinase C and IL-4. J Immunol (1993) 151:2965–73.
167. Jara LJ, Medina G, Cruz-Dominguez P, Navarro C, Vera-Lastra O, Saavedra MA. Risk Factors of Systemic Lupus Erythematosus Flares During Pregnancy. Immunol Res (2014) 60:184–92. doi: 10.1007/s12026-014-8577-1
168. Ashkar AA, Croy BA. Interferon-Gamma Contributes to the Normalcy of Murine Pregnancy. Biol Reprod (1999) 61:493–502. doi: 10.1095/biolreprod61.2.493
169. Renaud SJ, Cotechini T, Quirt JS, Macdonald-Goodfellow SK, Othman M, Graham CH. Spontaneous Pregnancy Loss Mediated by Abnormal Maternal Inflammation in Rats Is Linked to Deficient Uteroplacental Perfusion. J Immunol (2011) 186:1799–808. doi: 10.4049/jimmunol.1002679
170. Ahmadi M, Abdolmohammadi-Vahid S, Ghaebi M, Aghebati-Maleki L, Afkham A, Danaii S, et al. Effect of Intravenous Immunoglobulin on Th1 and Th2 Lymphocytes and Improvement of Pregnancy Outcome in Recurrent Pregnancy Loss (RPL). BioMed Pharmacother (2017) 92:1095–102. doi: 10.1016/j.biopha.2017.06.001
171. Muyayalo KP, Li ZH, Mor G, Liao AH. Modulatory Effect of Intravenous Immunoglobulin on Th17/Treg Cell Balance in Women With Unexplained Recurrent Spontaneous Abortion. Am J Reprod Immunol (2018) 80:e13018. doi: 10.1111/aji.13018
172. Nakagawa K, Kwak-Kim J, Kuroda K, Sugiyama R, Yamaguchi K. Immunosuppressive Treatment Using Tacrolimus Promotes Pregnancy Outcome in Infertile Women With Repeated Implantation Failures. Am J Reprod Immunol (2017) 78:3. doi: 10.1111/aji.12682
173. Fu J, Li L, Qi L, Zhao L. A Randomized Controlled Trial of Etanercept in the Treatment of Refractory Recurrent Spontaneous Abortion With Innate Immune Disorders. Taiwan J Obstet Gynecol (2019) 58:621–5. doi: 10.1016/j.tjog.2019.07.007
174. Winger EE, Reed JL. Treatment With Tumor Necrosis Factor Inhibitors and Intravenous Immunoglobulin Improves Live Birth Rates in Women With Recurrent Spontaneous Abortion. Am J Reprod Immunol (2008) 60:8–16. doi: 10.1111/j.1600-0897.2008.00585.x
175. Lash GE, Ernerudh J. Decidual Cytokines and Pregnancy Complications: Focus on Spontaneous Miscarriage. J Reprod Immunol (2015) 108:83–9. doi: 10.1016/j.jri.2015.02.003
176. Peck A, Mellins ED. Plasticity of T-Cell Phenotype and Function: The T Helper Type 17 Example. Immunology (2010) 129:147–53. doi: 10.1111/j.1365-2567.2009.03189.x
177. Lee SK, Kim JY, Lee M, Gilman-Sachs A, Kwak-Kim J. Th17 and Regulatory T Cells in Women With Recurrent Pregnancy Loss. Am J Reprod Immunol (2012) 67:311–8. doi: 10.1111/j.1600-0897.2012.01116.x
178. Steinborn A, Schmitt E, Kisielewicz A, Rechenberg S, Seissler N, Mahnke K, et al. Pregnancy-Associated Diseases are Characterized by the Composition of the Systemic Regulatory T Cell (Treg) Pool With Distinct Subsets of Tregs. Clin Exp Immunol (2012) 167:84–98. doi: 10.1111/j.1365-2249.2011.04493.x
179. Wang WJ, Hao CF, Qu QL, Wang X, Qiu LH, Lin QD. The Deregulation of Regulatory T Cells on Interleukin-17-Producing T Helper Cells in Patients With Unexplained Early Recurrent Miscarriage. Hum Reprod (2010) 25:2591–6. doi: 10.1093/humrep/deq198
180. Zhang P, Zhao M, Liang G, Yin G, Huang D, Su F, et al. Whole-Genome DNA Methylation in Skin Lesions From Patients With Psoriasis Vulgaris. J Autoimmun (2013) 41:17–24. doi: 10.1016/j.jaut.2013.01.001
181. Nakashima A, Ito M, Shima T, Bac ND, Hidaka T, Saito S. Accumulation of IL-17-Positive Cells in Decidua of Inevitable Abortion Cases. Am J Reprod Immunol (2010) 64:4–11. doi: 10.1111/j.1600-0897.2010.00812.x
182. Lee SK, Kim JY, Hur SE, Kim CJ, Na BJ, Lee M, et al. An Imbalance in Interleukin-17-Producing T and Foxp3+ Regulatory T Cells in Women With Idiopathic Recurrent Pregnancy Loss. Hum Reprod (2011) 26:2964–71. doi: 10.1093/humrep/der301
183. Wilczynski JR. Immunological Analogy Between Allograft Rejection, Recurrent Abortion and Pre-Eclampsia–the Same Basic Mechanism. (2006) 67(7):492–511. doi: 10.1016/j.humimm.2006.04.007
184. Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T Cells Mediate Maternal Tolerance to the Fetus. Nat Immunol (2004) 5:266–71. doi: 10.1038/ni1037
185. Darrasse-Jeze G, Klatzmann D, Charlotte F, Salomon BL, Cohen JL. CD4+CD25+ Regulatory/Suppressor T Cells Prevent Allogeneic Fetus Rejection in Mice. Immunol Lett (2006) 102:106–9. doi: 10.1016/j.imlet.2005.07.002
186. Wang WJ, Liu FJ, Xin-Liu, Hao CF, Bao HC, Qu QL, et al. Adoptive Transfer of Pregnancy-Induced CD4+CD25+ Regulatory T Cells Reverses the Increase in Abortion Rate Caused by Interleukin 17 in the CBA/JxBALB/c Mouse Model. Hum Reprod (2014) 29:946–52. doi: 10.1093/humrep/deu014
187. Zhang XX, Kang XM, Zhao AM. Regulation of CD4(+)FOXP3(+) T Cells by CCL20/CCR6 Axis in Early Unexplained Recurrent Miscarriage Patients. Genet Mol Res (2015) 14:9145–54. doi: 10.4238/2015.August.7.24
188. Wu M, Liu P, Cheng L. Galectin-1 Reduction and Changes in T Regulatory Cells may Play Crucial Roles in Patients With Unexplained Recurrent Spontaneous Abortion. Int J Clin Exp Patho (2015) 8:1973–8.
189. Inada K, Shima T, Ito M, Ushijima A, Saito S. Helios-Positive Functional Regulatory T Cells Are Decreased in Decidua of Miscarriage Cases With Normal Fetal Chromosomal Content. J Reprod Immunol (2015) 107:10–9. doi: 10.1016/j.jri.2014.09.053
190. Qian J, Zhang N, Lin J, Wang C, Pan X, Chen L, et al. Distinct Pattern of Th17/Treg Cells in Pregnant Women With a History of Unexplained Recurrent Spontaneous Abortion. Biosci Trends (2018) 12:157–67. doi: 10.5582/bst.2018.01012
191. Saifi B, Rezaee SA, Tajik N, Ahmadpour ME, Ashrafi M, Vakili R, et al. Th17 Cells and Related Cytokines in Unexplained Recurrent Spontaneous Miscarriage at the Implantation Window. Reprod BioMed Online (2014) 29:481–9. doi: 10.1016/j.rbmo.2014.06.008
192. Zhu L, Liu M, Zhang S, Ou Y, Chen Y, Wei J, et al. Foxp3 TSDR Hypermethylation Is Correlated With Decreased Tregs in Patients With Unexplained Recurrent Spontaneous Abortion. Reprod Sci (2021) 28:470–8. doi: 10.1007/s43032-020-00299-z
193. Hou W, Li Z, Li Y, Fang L, Li J, Huang J, et al. Correlation Between Protein Expression of FOXP3 and Level of FOXP3 Promoter Methylation in Recurrent Spontaneous Abortion. J Obstet Gynaecol Res (2016) 42:1439–44. doi: 10.1111/jog.13076
194. Tur-Torres MH, Garrido-Gimenez C, Alijotas-Reig J. Genetics of Recurrent Miscarriage and Fetal Loss. Best Pract Res Clin Obstet Gynaecol (2017) 42:11–25. doi: 10.1016/j.bpobgyn.2017.03.007
195. Wang YY, Wang Q, Sun XH, Liu RZ, Shu Y, Kanekura T, et al. DNA Hypermethylation of the Forkhead Box Protein 3 (FOXP3) Promoter in CD4+ T Cells of Patients With Systemic Sclerosis. Br J Dermatol (2014) 171:39–47. doi: 10.1111/bjd.12913
196. Sadlon T, Brown CY, Bandara V, Hope CM, Schjenken JE, Pederson SM, et al. Unravelling the Molecular Basis for Regulatory T-Cell Plasticity and Loss of Function in Disease. Clin Transl Immunol (2018) 7:e1011. doi: 10.1002/cti2.1011
197. Xu L, Qiu T, Wang Y, Chen Y, Cheng W. Expression of C-Type Lectin Receptors and Toll-Like Receptors in Decidua of Patients With Unexplained Recurrent Spontaneous Abortion. Reprod Fertil Dev (2017) 29:1613–24. doi: 10.1071/RD15489
198. Bao SH, Wang XP, De Lin Q, Wang WJ, Yin GJ, Qiu LH. Decidual CD4+CD25+CD127dim/- Regulatory T Cells in Patients With Unexplained Recurrent Spontaneous Miscarriage. Eur J Obstet Gynecol Reprod Biol (2011) 155:94–8. doi: 10.1016/j.ejogrb.2010.11.007
199. Yuan J, Li J, Huang SY, Sun X. Characterization of the Subsets of Human NKT-Like Cells and the Expression of Th1/Th2 Cytokines in Patients With Unexplained Recurrent Spontaneous Abortion. J Reprod Immunol (2015) 110:81–8. doi: 10.1016/j.jri.2015.05.001
200. Piccinni MP, Beloni L, Livi C, Maggi E, Scarselli G, Romagnani S. Defective Production of Both Leukemia Inhibitory Factor and Type 2 T-Helper Cytokines by Decidual T Cells in Unexplained Recurrent Abortions. Nat Med (1998) 4:1020–4. doi: 10.1038/2006
201. Comba C, Bastu E, Dural O, Yasa C, Keskin G, Ozsurmeli M, et al. Role of Inflammatory Mediators in Patients With Recurrent Pregnancy Loss. Fertil Steril (2015) 104:1467–74. doi: 10.1016/j.fertnstert.2015.08.011
Keywords: DNA methylation, recurrent pregnancy loss (RPL), maternal-fetal immune microenvironment, epigenetic, immune
Citation: Zhou Q, Xiong Y, Qu B, Bao A and Zhang Y (2021) DNA Methylation and Recurrent Pregnancy Loss: A Mysterious Compass? Front. Immunol. 12:738962. doi: 10.3389/fimmu.2021.738962
Received: 09 July 2021; Accepted: 04 October 2021;
Published: 21 October 2021.
Edited by:
Lei Huang, Newcastle University, United KingdomReviewed by:
Di Xie, Yale University, United StatesIonel Sandovici, University of Cambridge, United Kingdom
Copyright © 2021 Zhou, Xiong, Qu, Bao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anyu Bao, YmF5QHdodS5lZHUuY24=; Yan Zhang, cGVuZXl5YW5AbWFpbC51c3RjLmVkdS5jbg==
†These authors have contributed equally to this work