 Phuong T. Le1
Phuong T. Le1 Ngoc Ha1†
Ngoc Ha1† Ngan K. Tran1†
Ngan K. Tran1† Andrew G. Newman2Katharine M. Esselen3
Andrew G. Newman2Katharine M. Esselen3 John L. Dalrymple3
John L. Dalrymple3 Eva M. Schmelz4
Eva M. Schmelz4 Avinash Bhandoola5‡
Avinash Bhandoola5‡ Hai-Hui Xue6‡
Hai-Hui Xue6‡ Prim B. Singh7‡
Prim B. Singh7‡ To-Ha Thai1,8*‡
To-Ha Thai1,8*‡- 1Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
- 2Institute of Cell and Neurobiology, Charité-Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin and Berlin Institute of Health, Berlin, Germany
- 3Division of Gynecologic Oncology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
- 4Department of Human Nutrition, Food, and Exercise, Virginia Tech, Blacksburg, VA, United States
- 5Center for Cancer Research, National Cancer Institute, Bethesda, MD, United States
- 6Center for Discovery and Innovation, Hackensack University Medical Center, Nutley, NJ, United States
- 7Nazarbayev University School of Medicine, Nur-Sultan, Kazakhstan
- 8Cancer Research Institute, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Immune checkpoint blockade (ICB) relieves CD8+ T-cell exhaustion in most mutated tumors, and TCF-1 is implicated in converting progenitor exhausted cells to functional effector cells. However, identifying mechanisms that can prevent functional senescence and potentiate CD8+ T-cell persistence for ICB non-responsive and resistant tumors remains elusive. We demonstrate that targeting Cbx3/HP1γ in CD8+ T cells augments transcription initiation and chromatin remodeling leading to increased transcriptional activity at Lef1 and Il21r. LEF-1 and IL-21R are necessary for Cbx3/HP1γ-deficient CD8+ effector T cells to persist and control ovarian cancer, melanoma, and neuroblastoma in preclinical models. The enhanced persistence of Cbx3/HP1γ-deficient CD8+ T cells facilitates remodeling of the tumor chemokine/receptor landscape ensuring their optimal invasion at the expense of CD4+ Tregs. Thus, CD8+ T cells heightened effector function consequent to Cbx3/HP1γ deficiency may be distinct from functional reactivation by ICB, implicating Cbx3/HP1γ as a viable cancer T-cell-based therapy target for ICB resistant, non-responsive solid tumors.
Introduction
Under persistent antigen exposure as in cancer or chronic infections, CD8+ effector T cells enter an altered differentiation program known as T-cell exhaustion (TEX) (1). In the tumor microenvironment (TME), CD8+ progenitor TEX cells express unique transcription factors as well as those shared with naïve and memory cells including the high mobility group (HMG) transcription factor, T-cell factor 1 (TCF-1, encoded by Tcf7). They have minimal effector capacity but can be reinvigorated by ICB to proliferate and differentiate into terminally exhausted cells having reduced proliferative capacity but efficient tumor killing ability mediated by perforin 1 (PRF1), granzyme B (GrB) and interferon γ (INF-γ) (2–4). TCF-1 is implicated in the conversion of CD8+ progenitor TEX to terminally differentiated TEX (5–9). However, given that only ~20% of all solid tumors and primarily those harboring high mutation loads respond to ICB, in addition to the emergence of ICB resistance (10), there is a need to identify novel targets. In humans and mice, TCF-1, and the related lymphoid enhancer-binding factor 1 (LEF-1) are functionally linked during T-cell development in the thymus and the generation of precursor as well as functional memory CD8+ and CD4+ T cells (11–19). Nevertheless, a role for LEF-1 in tumor response has not been documented despite Lef1 expression being detected in human and mouse stem-like tumor infiltrating lymphocyte (TIL) subsets that also express Tcf7 (7, 9, 20–22).
IL-21R is widely expressed on various innate and adaptive immune cell-lineages including activated CD8+ T, CD4+ TFH and NK cells. During a chronic viral infection or under IL-2-deprived conditions IL-21R signaling is critical for preventing CD8+ T-cell exhaustion (23, 24). In acute viral infections, IL-21R signaling is essential for the proliferation and survival of activated CD8+ T cells as well as the generation of long-lived memory cells (25–27). However, the function of IL-21R signaling in cancer is controversial and not completely understood (28–32).
Members of the mammalian heterochromatin protein 1 (HP1) family Cbx5/HP1α, Cbx1/HP1β and Cbx3/HP1γ are small modular proteins around 25kD consisting of an N-terminal chromodomain (CD) and a sequence related C-terminal sequence called the chromo-shadow domain (CSD) that are connected by an unstructured linker region (LR) (33). They are key epigenetic regulators that recognize and bind (“read”) the H3K9me2/3 determinant of the histone code (34). Cbx5/HP1α and Cbx1/HP1β are usually associated with constitutive heterochromatin while Cbx3/HP1γ has a more euchromatic distribution (35, 36). Outside of cytologically-visible heterochromatin, HP1 proteins and H3K9me2/3 assemble into heterochromatin-like domains/complexes having diverse chromatin template-dependent functions (37). HP1 proteins are involved in gene repression/activation, transcriptional elongation, RNA splicing and DNA repair (38–44). Cbx3/HP1γ likely controls gene activation through two intimately related processes: transcriptional elongation and co-transcriptional splicing of nascent RNA transcripts (39–41, 45). For example, in the immune system, Cbx3/HP1γ is found associated with the transcription elongation complex containing RNA polymerase II (Pol II) within the coding region of the actively transcribed IL-2 gene in stimulated primary T cells (40). By contrast, a role for gene repression is indicated during B-cell development where Cbx3/HP1γ is associated with the silenced κ allele implicating Cbx3/HP1γ in allelic exclusion (46).
Here, we demonstrate that targeting Cbx3/HP1γ promotes increased/sustained expression of LEF-1 and IL-21R in CD8+ effector T cells. Cbx3/HP1γ deficiency leads to augmented transcription initiation and chromatin remodeling that results in increased transcriptional activity at Lef1 and Il21r. Genetic ablation of Lef1 and Il21r in Cbx3/HP1γ-deficient mice causes a loss of tumor CD8+ effector T cells accompanied by the reduction of Ifng, Gzmb and Prf1 expression, which results in uncontrolled ovarian, melanoma and neuroblastoma growth. Our data establish that LEF-1 and IL-21R are necessary for Cbx3/HP1γ-deficient CD8+ T cells to maintain their effector capacity and persist in tumors. Our findings together with those of others (22, 47, 48) illustrate the complex mechanisms governing CD8+ T-cell effector differentiation and function within a given TME. They underscore the need for continuing exploration of novel targets that can reverse CD8+ T-cell dysfunction in ICB resistant and unresponsive solid tumors.
Methods
Mice
Cbx3/HP1γ floxed mice were generated and provided by Dr. Prim B. Singh (49). These mice were backcrossed to C57BL/6 for twelve generations; they bred and developed normally before and after Cre-mediated deletion. The Cbx3/HP1γ transgenic line was generated by inserting a 1 kb fragment of mouse Cbx3/HP1γ cDNA, derived from activated CD8+ T cells, into EcoR1/Sma1 cloning sites of the VA-hCD2 vector provided by Dr. Dimitris Kioussis (50). The purified Cbx3/HP1γ-VA-hCD2 DNA construct was injected into C57BL/6 pronuclei by Dr. Lina Du at the fee-based Dana-Farber/Harvard Cancer Center (DF/HCC) Transgenic Mouse Core. Two founder transgenic lines were produced. The Lef1 floxed mice were generated as described and provided by Dr. Hai-Hui Xue (12). The following mouse lines were purchased from The Jackson Laboratory: Il21r knock out (B6.129-Il21rtm1Kopf/J) (51), CD8α-Cre transgenic (C57BL/6-Tg(Cd8a-cre)1Itan/J) (52), ROSA26 (B6;129-Gt(ROSA)26Sortm2Sho/J) (53), Prf1 knock out (C57BL/6-Prf1tm1Sdz/J) (54), and Ifng knock out (B6.129S7-Ifngtm1Ts/J) (55). The B6.SJL line (B6.SJL-Ptprca/BoyAiTac) was purchased from Taconic.
Tumor Cell Lines
The mouse syngeneic ID8 ovarian tumor line was obtained from Dr. Katherine F. Roby under an MTA executed by the University of Kansas Medical Center (56). Dr. Eva M. Schmelz provided the mouse syngeneic MOSE-LTICv ovarian tumor line (57, 58). The NB9464 neuroblastoma cell line was a kind gift from Dr. Crystal L. MacKall (Stanford University) (59). The B16-F10 melanoma cell line was purchased from ATCC and tested negative for mycoplasma.
Antibodies
All flow cytometry fluorochrome-conjugated antibodies were purchased from BioLegend, eBio-sciences or BD Biosciences. The following Western blot and ChIP-tested antibodies were purchased from Cell Signaling: tri-methyl-histone H3 (Lys9) (D4W1U) rabbit mAb #13969, phospho-Rpb1 CTD (Ser2) (E1Z3G) rabbit mAb #13499, phospho-Rpb1 CTD (Ser5) (D9N5I) rabbit mAb #13523, LEF-1 (C12A5) rabbit mAb #2230, cleaved caspase-3 (Asp175) antibody #9661 and phospho-HP1γ (Ser83) Ab #2600. The Western blot and ChIP-tested mouse anti-HP1γ mAb (clone 42s2) was purchased from Millipore Sigma. The rabbit polyclonal to TBR2/Eomes (ab23345) was purchased from Abcam.
Tumor Induction
Ovarian tumor. Mice were injected intraperitoneally (IP) with ID8 (5 x 106/mouse in 200 µl PBS) or MOSE-LTICv (1 x 104/mouse in 200 µl PBS). On day 120 (ID8) or 36 (MOSE-LTICv), when abdominal distension was visible, mice were euthanized, ascites were collected, and volume measured using syringes fitted with 18-gauge needles.
Melanoma. Mice were implanted subcutaneously with B16 tumor cells (1 x 105/mouse in 100µL PBS). Using a digital caliber, B16 tumor size was measured and calculated on day 14 and at 2-day or 3-day intervals until day 20 depending on the size of the tumor, at which time mice were euthanized for analysis.
Neuroblastoma. Mice were implanted subcutaneously with NB-9464 tumor cells (1 x 106/mouse in 100µL PBS). Using a digital caliber, NB-9464 tumor volume was measured and calculated (W x L x 0.4) starting on day 22 and at 2-day intervals until day 30 depending on the size of the tumor, at which time mice were euthanized for analysis.
Flow Cytometry
Flow cytometry was performed on the Beckman Coulter CytoFLEX LX. Analysis was performed with FlowJo software version 10.8.0 (Tree Star, Inc.). Tumors and ascites were harvested, minced (NBL and melanoma), and cell-suspensions were filtered through 70μm cell-strainers (Fisherbrand). Cells were stained for appropriate surface markers as indicated in figures. Intracellular FOXP3 staining was performed according to manufacturer’s protocol (Biolegend). Briefly, tumor cells (1 x 106) were harvested and washed 2X with staining buffer [PBS, 2% fetal bovine serum (FBS)]. Standard surface staining for intratumoral CD4+CD25+ T cells was performed. Cells were washed and incubated in fixation/permeabilization buffer (BioLegend) for 30 minutes at room temperature (RT) or 18 hrs at 4°CC. Following incubation, cells were washed 2X with permeabilization buffer (BioLegend) and stained for FOXP3 using anti-mouse FOXP3-PE (BioLegend). Cells were incubated for 30 minutes at RT. Cells were washed 2X with permeabilization buffer and resuspended in FACS buffer for analysis. All antibodies for flow cytometry were purchased from BioLegend: Il-21R-PE/Cy5 (clone 4A9); B220-Pacific Blue (clone RA3-6B2); CD8-PB; CD8-APC/Cy7; CD8-PE/Cy7; CD8-APC (all CD8 antibodies were from clone 53-6.7); CD25-APC/Cy7; CD25-PE (clone PC61 and clone 3C7); NKG2D-PE (clone CX5); CD4-PE; CD4-APC; CD4-FITC (all CD4 antibodies were from clone GK1.5); CD62L-APC (clone MEL-14); CD44-PB; CD44-APC/Cy7 (all CD44 antibodies were from clone IM7); TIM3-APC (clone B8.2C12); Streptavidin-PE; NK1.1-APC (clone PK136); Gr1-APC/Cy7 (clone RB6-8C5); Mac-1-PE (clone M1/70); CXCR5-Biotin (L138D7); FOXP3-PE (MF-14); CD45.2-PE/Cy7 (clone 104).
In Vitro Activation/Differentiation of CD8+ T Cells
CD8+CD44– T cells were purified from spleen and peripheral lymph nodes using the MojoSort™ Mouse CD8 Naïve T-Cell Isolation Kit (BioLegend, #480044) followed by CD25-depletion using the mouse CD25 MicroBead Kit (Miltenyl Biotec, #130-091-072), all according to manufacturers’ protocols. Naïve CD8+ T cells (1 × 106/mL) were activated with plate-bound anti-CD3 (clone 145-2C11, 0.25 μg/ml, BioLegend) and anti-CD28 (clone 37.51, 0.5 μg/ml, BioLegend) in T-cell medium (high glucose DMEM, 10% FBS, penicillin/streptomycin, non-essential amino acids, HEPES, L-glutamate and sodium pyruvate) at 37°C in 10% CO2 for 2 days. Cells were then removed from CD3/CD28 activation and re-cultured (5 × 105/ml) in differentiation medium (T-cell medium supplemented with 10 IU/ml rhIL-2 from NCI) at 37°C in 10% CO2. Cells were then sub-cultured every day in differentiation medium at 5 × 105/ml each sub-culture. On day 5, cells were harvested and used for Western blots, ChIP-qPCR, qPCR or adoptive T-cell transfer.
Adoptive T-Cell Transfer
In vitro-generated donor CD8+ effector T cells (CD45.2+) were prepared as above. On day 0, B6.SJL (CD45.1+) mice were implanted subcutaneously with NB-9464 (1 x 106/mouse in 100μL PBS) or B16 (1 x 105/mouse in 100μL PBS) tumor cells. On day 10 (B16) or 14 (NB-9464) after tumor induction, mice were treated with donor CD8+ effector T cells (3 x 106/mouse in 100μL PBS) via tail vein intravenous injection. B16 tumor size and NB-9464 tumor volume were measured and calculated on indicated dates.
Co-Culturing Effector Cells With B16 Tumor Cells
Target B16 tumor cells were plated in 12-well plates at a density of 5 x 105 cells per well in 750μl of medium (high glucose DMEM, 10% FBS, penicillin/streptomycin, non-essential amino acids, HEPES, L-glutamate and sodium pyruvate). CD8+ or CD4+ effector T cells were added. For ratio of 1:1 (effector:target), 5 x 105 CD8+ or CD4+ T cells: 5 x 105 B16 cells were co-cultured in the same well; ratio 5:1, 2.5 x 106 CD8+ or CD4+ T cells: 5 x 105 B16 cells. Plates were incubated at 37°CC in 5% CO2 for 24 hours. Wells were washed to remove non-adherent T cells. Adherent B16 cells were collected and washed with 1 ml cold PBS. Pellets were resuspended in Radio-Immunoprecipitation Assay (RIPA) lysis buffer containing a protease inhibitor cocktail (Roche) and used immediately or stored at –80°CC for further analysis.
Western Blots
Cells (1 x 106) were lysed with RIPA buffer (Boston BioProducts) containing a protease inhibitor cocktail on ice for 30 minutes. Lysates were centrifuged at 13,000 rpm for 10 minutes at 4°CC. Protein extracts were denatured at 95°CC for 10 minutes, separated by SDS-PAGE, and transferred to PVDF membranes (EMD Millipore). Membranes were probes with primary antibodies. Proteins of interest were detected with HRP-conjugated secondary antibodies and the Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific). Membranes were exposed with HyBlot CL Autoradiography films (Denville Scientific, Inc.), and developed with the Kodax X-OMAT 2000 Processor.
Chromatin Immunoprecipitation Followed by qPCR (ChIP-qPCR) and ChIP Followed by Deep Sequencing (ChIP-Seq)
ChIP was performed using the SimpleChIP Kit #9003 according to manufacturer’s protocol (Cell Signaling). Briefly, 2 x 107 CD8+ T cells were used for each ChIP. Cells were fixed in 1% formaldehyde to cross-link proteins to DNA, then lysed with 500μL of lysis buffer containing 1X ChIP buffer and protease inhibitor cocktail. Chromatin was sheared using a Sonic Dismembrator (Fisher Scientific Model 120) at 120W-20kHz power, at 15 seconds per cycle, 45 seconds break in between, for 3 cycles total. Chromatin was subjected to immunoprecipitation using specific antibodies at 4°C overnight with rotation. Following incubation, ChIP grade protein G magnetic beads were added to each ChIP and incubated for 2 hours at 4°C with rotation. Samples were placed in a magnetic separation rack for 2 minutes each time and washed 3X with high and low salt buffers. Chromatin was eluted from antibody/protein G magnetic beads using 1X ChIP elution buffer at 65°C for 30 minutes. Eluted chromatin was collected and subjected to cross-link reversal using 5M NaCl and 20 mg/ml Proteinase K, incubated for 2 hours at 65°C. After reversal of protein-DNA cross-link, the DNA was purified using DNA purification spin columns and eluted with DNA elution buffer provided in the kit. Purified ChIP DNA was immediately used for qPCR. BioRad hard-shell PCR Plates (BioRad) were used. In each well 2μL of purified ChIP DNA was added in triplicates along with 18μl primer mixture, which consisted of 1μL forward primer (5μM), 1μL reverse primer (5μM), 6μL nuclease-free water, and 10μL of 2X QuaniNova SYBR Green PCR Master Mix (Qiagen) or SimpleChIP Universal qPCR Master Mix (Cell Signaling). The plate was centrifuged at 300 RCF for 1 minute and read in the BioRad CFX384 Real-Time System (BioRad). The following qPCR settings were used: initial denaturation 95°C for 3:00 minutes, denatured at 95°C for 0:15 minutes, annealing and extension at 60°C for 1:00 minute, GOTO step denature and extension for a total of 40 cycles. Quantitative PCR result was analyzed, and the IP efficiency was calculated per formula provided in Simple ChIP Kit #9003 (Cell Signaling). Primers are listed in Table S2.
ChIP-Seq detailed experiments and analyses have been described previously (60).
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from tumors or activated/differentiated CD8+ T cells using TRIzol (Thermo Fisher Scientific). After DNAse I treatment and RNA cleanup with the RNeasy kit (Qiagen), 1 μg of total RNA was reversed transcribed into cDNA using the Invitrogen SuperScript II reverse transcriptase kit according to manufacturer’s recommendations (Thermo Fisher Scientific). The BioRad hard-shell PCR Plates (BioRad) were used. Typically, 5-10 ng of cDNA was added to each well in triplicates along with 7.5μL of the primer mixture, which consisted of 0.5μL forward primer (10μM), 0.5μL reverse primer (10μM), 1.7μL PCR grade water, and 5μL of Roches LightCycler 480 SYBR Green I Master (Roche). The plate was centrifuged at 300 RCF for 1 minute and read in the BioRad CFX384 Real-Time System (BioRad). The qPCR settings: 95°C for 10:00 minutes, 95°C for 0:10 minutes, 62°C for 0:20 minutes, 72°C for 0:20 minutes (Read Plate), GOTO step 2 for 39 times, 92°C for 0:05 minutes, melt curve 65°C to 97°C, at increment of 0.5°C for 1:00 minute (Read Plate), 40°C for 0:10 minutes. Primers are listed in Table S3.
Immunohistochemistry (IHC)
Tumor section processing and TUNEL staining were performed by HistoWiz (Brooklyn, NY). Briefly, all IHC staining is automated using BOND Rx. Slides were treated with Dewax, a solvent-based solution (Leica Biosystems). Tissue sections were fixed by adding 10% neutral buffered formalin for 15 minutes. Slides were treated with Proteinase K at 1:500 dilution. Tissue sections were re-fixed using 10% neutral buffered formalin. Tissue sections were then treated with the equilibration buffer and incubated for 12 minutes at room temperature (RT). Subsequently the TdT reaction mix was added and incubated for 60 minutes at 37°C. The TUNEL reaction was stopped by adding 2X SSC. Tissue slides were treated with Peroxide Block (3-4% Hydrogen Peroxide) followed by wash buffer. Streptavidin HRP was added and incubated for 30 minutes at RT. DAB was added onto the slides and incubated for 10 minutes. Counter-staining was done by adding hematoxylin. Slides were covered using the Sakura Tissue Tek strainer and cover slips then scanned at 40x using the Leica AT2 scanner.
Statistical Analysis
Statistical analysis of tumor growth was determined by the Graphpad Two-Way Anova, for survival the Log-rank (Mantel-Cox) test and for all others the Graphpad unpaired student t test (comparison between 2 groups) or One-Way Anova (multiple comparisons of group means) were performed. The number of mice used were determined using the GraphPad StatMate®, no animals were excluded from the analysis, and no randomization or blinding was applied.
Results
Cbx3/HP1γ Deficiency Modulates LEF-1 and IL-21R Expression in CD8+ Effector T Cells
Previously we showed that germline deletion of Cbx3/HP1γ impairs lymphoid-tissue germinal center (GC) reaction and high-affinity antibody response against a thymus (T)-dependent antigen (Ag) in a CD8+ T-cell-intrinsic manner (61). Cbx3/HP1γ deficiency releases the effector capacity of CD8+ T cells to control neuroblastoma (NBL) growth (60). However, it is not understood why Cbx3/HP1γ-deficient CD8+ effector T cells are not subjected to functional senescence and persist in NBL or if they can control tumors with varied mutation loads. To ensure Cbx3/HP1γ deletion was restricted to CD8+ T cells, all experiments were performed using the CD8α-Cre strain (52) crossed with our Cbx3/HP1γ-floxed mice (resulting animals were Cbx3/HP1γfl/+ and Cbx3/HP1γfl/fl; collectively designated as Cbx3/HP1γ-deficient). Cre recombinase from this CD8α-Cre strain was active in CD8+CD4– T cells not in CD4+CD8– or CD11b+ cells (Figure S1A). Ablation of Cbx3/HP1γ in CD8+ T cells resulted in a near or complete loss of protein expression and phosphorylation (Figure S1B). Expression of Bcl6 and Tbx21 (T-bet) (62) mRNAs was increased in Cbx3/HP1γ-deficient CD8+ effector T cells over control cells (Figure S1C). By contrast Eomes transcript and protein levels were reduced (Figures S1C, D). We observed an upregulation of Prf1, Gzmb and Ifng mRNA expression in Cbx3/HP1γ-deficient CD8+ effector T cells compared to control cells (Figure S1E). More cleaved caspase 3 (CC3) was detected in B16 melanoma tumor cells co-cultured with Cbx3/HP1γ-deficient CD8+ effector T cells compared to control co-cultures (Figure S1F). Cbx3/HP1γ deficiency in CD4+ T cells did not enhance tumor cells killing. Thus, after activation, Cbx3/HP1γ-deficient CD8+ T cells differentiate into effector-like cells armed with a heightened effector/killing capacity to induce tumor-cell apoptosis in vitro. To determine mechanisms conferring persistence on Cbx3/HP1γ-deficient CD8+ effector T cells, ChIP-Seq data was analyzed (60). In wild-type day 5 activated/differentiated CD8+ T cells, Cbx3/HP1γ was bound to the 5’ untranslated region (UTR) surrounding transcriptional start sites (TSSs) of Lef1 (Figures 1A, B) and Il21r (Figures 1D, E) (Table S1). Western immunoblots showed an induction of LEF-1 expression in activated/differentiated control CD8+ T cells; however, its expression was further increased when Cbx3/HP1γ was ablated (Figure 1C). LEF-1 levels in control and Cbx3/HP1γ-deficient thymocytes were similar (Figure S1G). The effects of IL-21 on IL-21R expression are controversial. In humans, IL-21 was shown to induce IL-21R expression on naïve CD8+ T cells (63). In mice, IL-21 exposure led to the downregulation of its receptor expression on all T-cell subsets (64). To mitigate IL-21 confounding effects, all experiments were done without added IL-21. In the absence of exogenous IL-21, activated/differentiated Cbx3/HP1γ-deficient CD8+ T cells expressed more IL-21R protein (measured as mean fluorescence intensity or MFI) and transcript, compared to control cells (Figures 1F–H). There was a steady expansion of Cbx3/HP1γ-deficient CD8+IL-21R+ T cells after activation/differentiation while the growth of control cells remained low throughout the activation/differentiation period (Figures 1F, I). IL-21R was not detected on thymocytes from control or Cbx3/HP1γ-deficient mice (Figure S1H). We establish that Cbx3/HP1γ deficiency induces the sustained increase of LEF-1 and IL-21R in CD8+ effector T cells as well as enhancing their effector capacity. IL-21R elevated expression likely provides signals mediating Cbx3/HP1γ-deficient CD8+ effector T-cell expansion in the absence of exogenous IL-21.
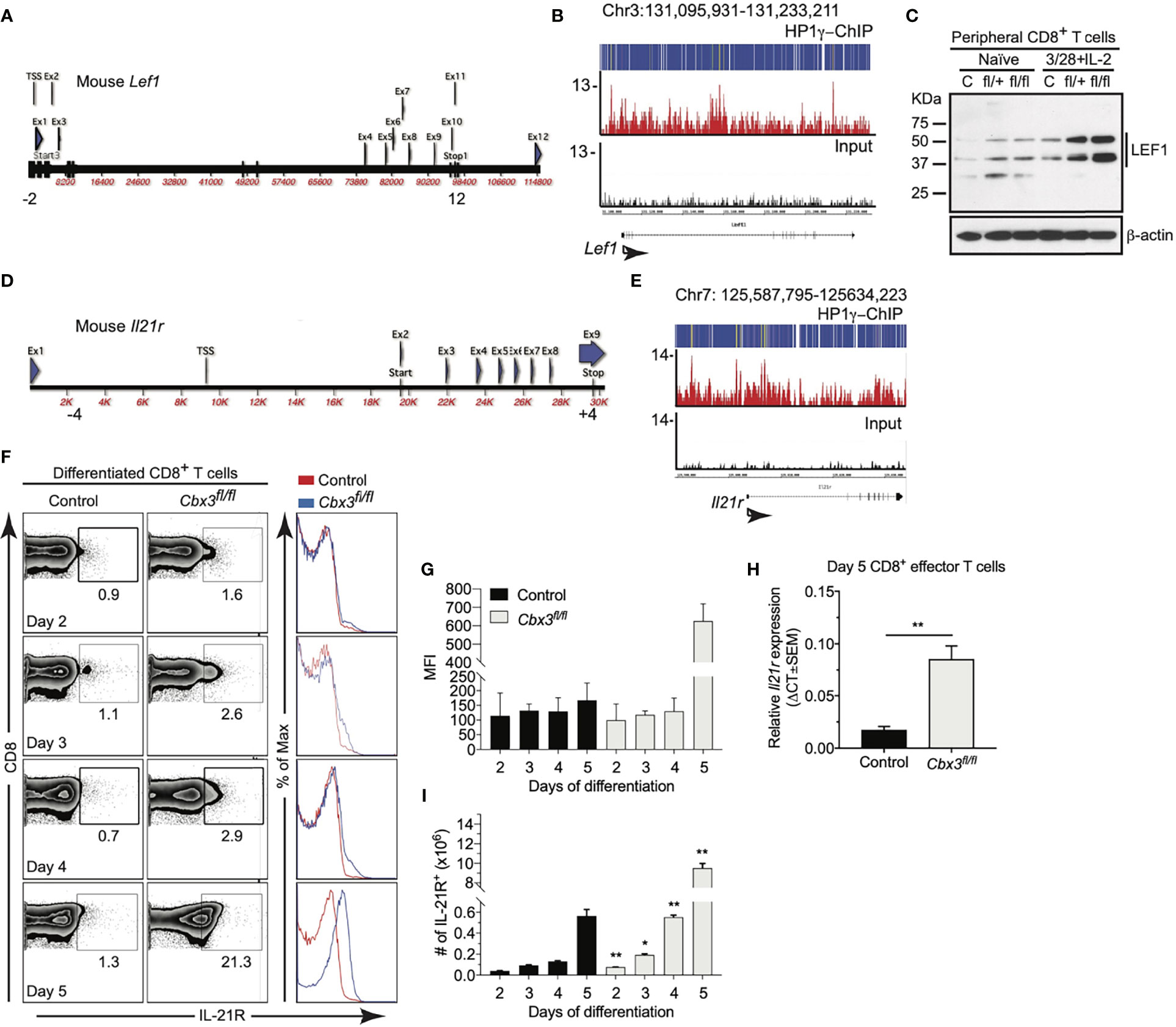
Figure 1 Cbx3/HP1γ deficiency modulates LEF-1 and IL-21R expression in CD8+ effector T cells. (A) Mouse Lef1 locus. -2 and 12: positions of first and last primer pairs, respectively. (B) Read-density tracks of Cbx3/HP1γ peaks across Lef1 identified by ChIP-Seq using chromatin from wt day 5 activated/differentiated CD8+ T cells (pooled from 3 mice); y axis: number of reads per million mapped per 25-bp window; representative of 2 independent ChIP-Seq runs. (C) LEF-1 expression was evaluated by Western immunoblot using total protein lysates from activated/differentiated control and Cbx3/HP1γ-deficient CD8+ T cells; 3/28+IL2: CD8+ T cells activated/differentiated for 5 days with plate-bound anti-CD3 and anti-CD28 plus IL-2; C: control (CD8α-Cre or wt); fl/+: Cbx3fl/+ and fl/fl: Cbx3fl/fl (CD8+ T-cell-restricted deletion of Cbx3/HP1γ using the CD8α-Cre mouse); n = 3; representative of 3 experiments. (D) Mouse Il21r locus. -4 and 4: positions of first and last pair of primers, respectively. (E) Read-density tracks of Cbx3/HP1γ peaks across Il21r were determined by ChIP-Seq as in (B). (F) IL-21R expression levels on activated/differentiated CD8+ T cells by flow; numbers: percent cells; representative of 3 experiments; n = 3. (G) IL-21R mean fluorescence intensity (MFI) was evaluated from (F). (H) Relative expression of Il21r in day 5 activated/differentiated CD8+ T cells was assessed by RT-qPCR and normalized to Gapdh; Graphpad unpaired student t-test: **p ≤ 0.01; representative of 3 experiments. (I) Number of CD8+IL-21R+ T cells in cultures was calculated from (F); Graphpad unpaired student t-test: *p ≤ 0.05, **p ≤ 0.01.
Cbx3/HP1γ Regulates Lef1 and Il21r Transcription Initiation and Chromatin Remodeling
The rate of gene expression is governed in part by RNA Pol II initiation, elongation and/or chromatin remodeling. Pol II is phosphorylated at serine 5 (Pol II S5) during initiation of transcription while phosphorylation at serine 2 (Pol II S2) generally indicates chromatin remodeling concomitant with transcriptional elongation. To test whether alterations in transcriptional initiation, elongation and/or chromatin remodeling caused Lef1 and Il21r increased expression in Cbx3/HP1γ-deficient CD8+ effector T cells, ChIP-qPCR experiments were performed using ChIP-tested antibodies specific for Pol II S2 or S5, and H3K9me3. These results revealed augmented levels of Pol II S5 in or around TSSs of Lef1 and Il21r loci in Cbx3/HP1γ-deficient CD8+ effector T cells compared to control cells (Figures 2A, B). Similar levels of Pol II S2 density were observed in control and Cbx3/HP1γ-deficient CD8+ T cells (Figures 2C, D). The levels of H3K9me3 deposition at Lef1 locus was unaltered (Figure 2E). However, there was a general loss of H3K9me3 around the TSS of Il21r locus in Cbx3/HP1γ-deficient CD8+ effector T cells (Figure 2F). Thus, genetic deletion of Cbx3/HP1γ in CD8+ effector T cells results in enhanced transcription initiation at Lef1 and Il21r loci, with chromatin remodeling activity taking place in an extended region around the TSS of the Il21r locus. These changes likely underpinned the increased and sustained transcriptional activity at Lef1 and Il21r.
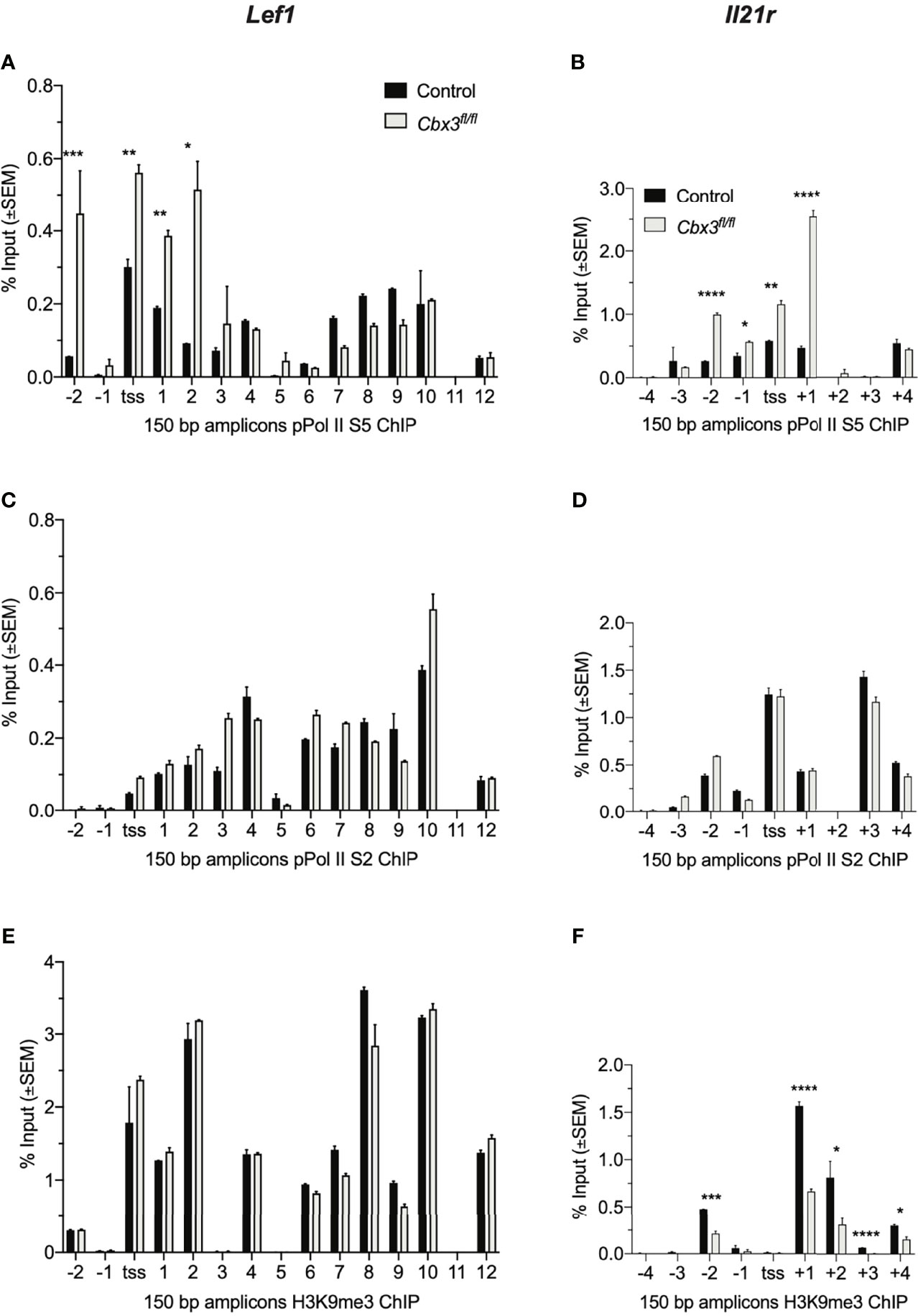
Figure 2 Cbx3/HP1γ regulates Lef1 and Il21r transcription initiation and chromatin remodeling. (A, B) Levels of Pol II S5 bound to Lef1 (A) and Il21r (B) were quantified by ChIP-qPCR using chromatin from day 5 activated/differentiated CD8+ T cells. TSS: transcription start site; numbers on X axis: positions of primers along the two loci; 150 bp products amplified by Lef1 or Il21r primers. (C, D) Levels of Pol II S2 bound to Lef1 (C) and Il21r (D) were quantified as in (A, B). (E, F) H3K9me3 deposition on Lef1 (E) and Il21r (F) was quantified as in (A, B). Graphpad unpaired student t-test: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; representative of 4 independent ChIPs using pooled chromatin from 3 mice.
Cbx3/HP1γ-Deficient CD8+ Effector T Cells Persist and Cause Tumor Rejection
The increased/sustained expression of LEF-1 and IL-21R together with the enhanced effector capacity exhibited by Cbx3/HP1γ-deficient CD8+ effector T cells suggest they can persist to control tumor development. Thus, the ability of Cbx3/HP1γ-deficient CD8+ T cells to eradicate solid tumors was evaluated using the mouse ID8 or MOSE-LTICv ovarian, B16 melanoma and NB-9464 NBL tumor models. Ovarian and NBL tumors have low mutation rates, no clear defining tumor-associated antigens (TAAs) and are minimally responsive to ICB (65–67). Accordingly, syngeneic ID8 or MOSE-LTICv ovarian tumor cells were injected intraperitoneally (IP) into control and Cbx3/HP1γ-deficient mice. Mice were monitored until abdominal distension was visible, which indicated increased ascites and mirrored metastasis observed in humans. Tumor growth and production of ascites were inhibited in Cbx3/HP1γ-deficient mice compared to controls or mice ectopically expressing Cbx3/HP1γ driven by the human T-cell-restricted Cd2 promoter (Cbx3/HP1γTg) (Figures 3A, B and S2A, B). As a result, Cbx3/HP1γ-deficient mice lived longer than controls (Figure 3C). Cbx3/HP1γ-deficient mice were equally effective in reducing B16 melanoma (Figure 3D) and NB-9464 NBL tumor burden (Figure 3F), leading to their increased survival compared to controls (Figures 3E, G). On day 120 after ID8 injection, there was an enrichment of CD8+NKG2D+ effector T cells and a decrease in CD4+CD25+FOXP3+ regulatory T cells (Tregs) in ascites from Cbx3/HP1γ-deficient mice compared to control or Cbx3/HP1γTg animals (Figures 3H, I and S2C, D). Similarly, enrichment of CD8+NKG2D+ effector T cells and decrease in CD4+CD25+FOXP3+ Tregs were observed in B16 melanoma as well as NBL tumors (Figures 3J–M and S2E–H). Comparable frequencies of NK1.1+NKG2D+ or CD4+NKG2D+ T cells were recovered from tumors of control, Cbx3/HP1γTg and Cbx3/HP1γ-deficient mice (Figure S3). Expression of Ifng, Gzmb and Prf1 was elevated in B16 melanoma and NBL tumors excised from Cbx3/HP1γ-deficient mice compared to controls or Cbx3/HP1γTg animals (Figures S4A–F). TUNEL staining revealed more apoptotic tumor cells (brown) in melanoma and NBL tumors from Cbx3/HP1γ-deficient mice compared to controls (Figures 3N, O). Genetic ablation of Prf1 in Cbx3/HP1γ-deficient animals led to uncontrolled B16 melanoma growth and decreased TUNEL positivity indicative of reduced tumor-cell apoptosis (Figures 4SG, H) while NBL tumor burden was incompletely inhibited and tumor-cell death was readily detected (Figures S4I, J). By contrast, Ifng deletion resulted in uncontrolled NBL tumor growth in Cbx3/HP1γ-deficient mice (Figure S4K). Thus, PRF1 and INF-γ likely mediate the killing of tumors in Cbx3/HP1γ-deficient mice. To show that Cbx3/HP1γ-deficient CD8+ effector T cells cause tumor rejection in vivo, CD8+ T cells (CD45.2+) were activated/differentiated in vitro (see Methods). On day 5 after activation/differentiation, CD8+ T cells were collected for adoptive T-cell therapy in tumor bearing congenic B6.SJL (CD45.1+) recipients (Figure S5A). Treatment with Cbx3/HP1γ-deficient CD8+ effector T cells alone resulted in a statistically significant decrease in B16 melanoma and NBL tumor burden compared to treatment with control cells (Figures S5B, C). Within B16 melanoma tumors, there was an enrichment of transferred Cbx3/HP1γ-deficient CD8+NKG2D+ effector T cells, not transferred control or endogenous CD8+ effector T cells (Figures S5D, E). Possible contaminating CD45.2+CD4+NKG2D+ and CD45.2+NK1.1+NKG2D+ T cells were not detected in tumors indicating that they were not the source of tumor killing (Figures S5F–I). These results suggested a role for NKG2D in tumor control by Cbx3/HP1γ-deficient CD8+ effector T cells. To test this hypothesis, B16 and NB-9464 tumor cells were injected subcutaneously into control and Cbx3/HP1γ-deficient mice. On day 2, a subset of tumor-bearing Cbx3/HP1γ-deficient mice were treated with the blocking/non-depleting anti-mouse NKG2D antibody HMG2D, and thereafter once per week. NKG2D blockade resulted in uncontrolled tumor growth and decreased survival of treated animals (Figures S6A–D), accompanied by the reduction of Cbx3/HP1γ-deficient CD8+NKG2D+ effector T cells in tumors, to a level similar to PBS-treated control mice (Figures S6E, F). NKG2D blockade did not affect CD4+NKG2D+ and NK1.1+NKG2D+ T-cell frequencies (Figures S6G, H). Together, our results demonstrate that Cbx3/HP1γ-deficient CD8+ effector T cells expressing NKG2D can persist and cause tumor rejection, irrespective of tumor mutation status. The engagement of NKG2D with its ligands expressed on tumor cells likely facilitates tumor killing by Cbx3/HP1γ-deficient CD8+ effector T cells through the enhanced production of PRF1, GrB and INF-γ. Additionally, B16 and NBL tumors have differential sensitivity to killing by PRF1 and IFN-γ, and surveying for their presence in the TME alone may not reveal the precise mechanism whereby tumor cells are eradicated. Furthermore, deletion of even one Cbx3/HP1γ allele is sufficient to control solid tumor growth suggesting that Cbx3/HP1γ is haploinsufficient.
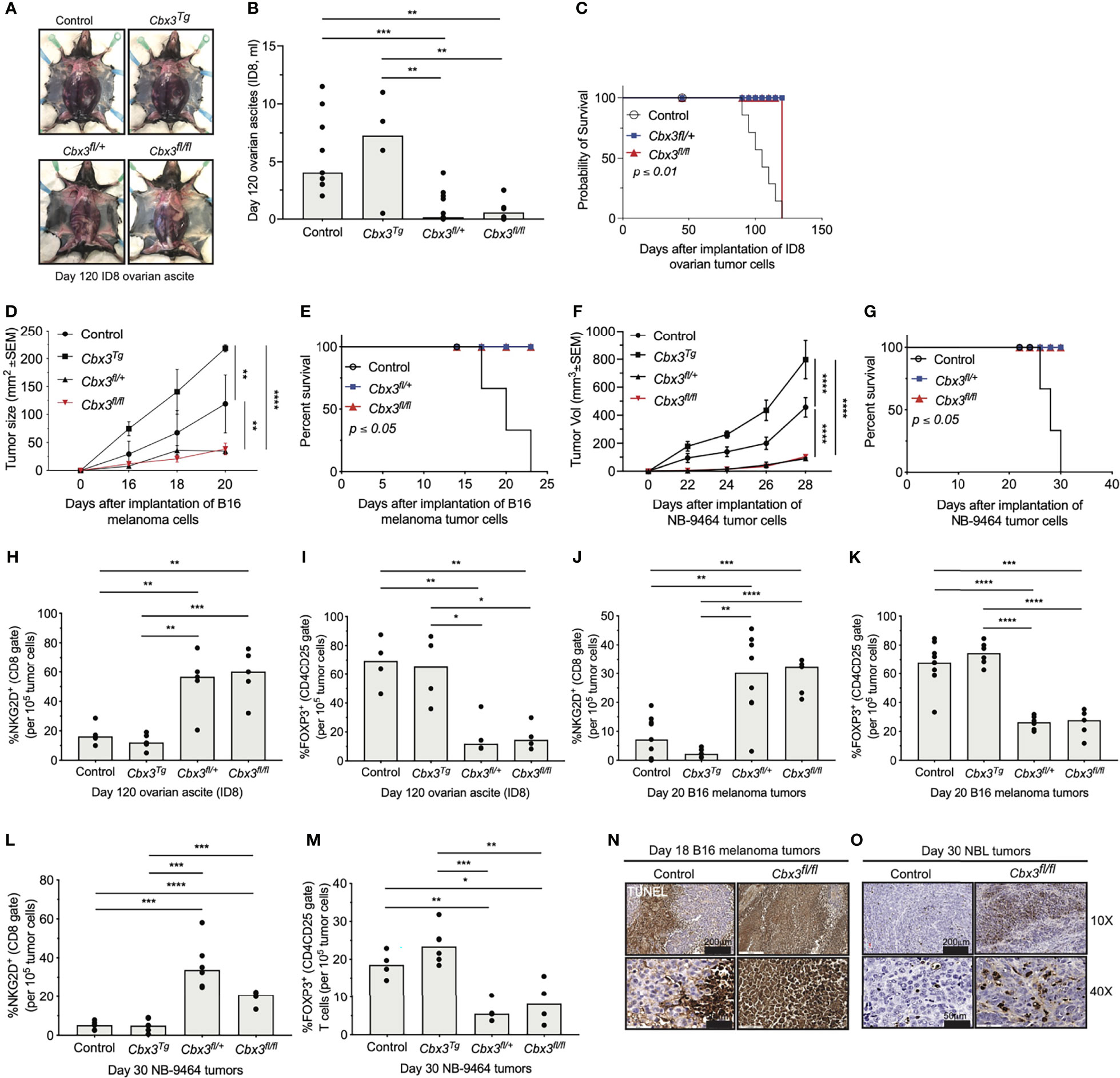
Figure 3 Cbx3/HP1γ-deficient CD8+ effector T cells can persist and cause tumor rejection. (A) Ascites from control (CD8α-Cre or wt), Cbx3Tg, Cbx3fl/+ and Cbx3fl/fl mice (X axis) were visualized on day 120 after IP injection of mouse ID8 ovarian tumor cells; Cbx3Tg: Cbx3/HP1γ T-cell-restricted expression driven by the human Cd2 promoter; Cbx3fl/+ and Cbx3fl/fl: CD8+ T-cell-restricted deletion of Cbx3/HP1γ using the CD8α-Cre mouse. (B) Ovarian ascites of mice in (A) were measured; bars: group median; Graphpad unpaired student t-test: **p≤0.01, ***p≤0.001; each symbol = one mouse; n = 5-8; representative of 2 experiments. (C) Survival curves of ovarian tumor-bearing mice were determined; Graphpad log-rank (Mantel-Cox) test; n = 5-8. (D) B16 tumor cells were injected subcutaneously (sc) and growth was assessed starting on day 14 after tumor-cell injection then every 2 days through day 20; Graphpad two-way ANOVA: **p ≤ 0.01, ****p ≤ 0.0001; n = 5; representative of 3 experiments. (E) Survival curves of B16 melanoma tumor-bearing mice; Graphpad log-rank (Mantel-Cox) test; n = 5. (F) NB-9464 tumor cells were injected sc, NBL tumor volume was determined every 2 days starting on day 22 through day 28; Graphpad two-way ANOVA: ****p ≤ 0.0001; n = 5; representative of 2 experiments. (G) Survival curves of NBL tumor-bearing mice; Graphpad log-rank (Mantel-Cox) test, n = 5. (H, J, L) Frequencies of CD8+NKG2D+ T cells in ovarian ascites, B16 and NBL tumors from control (CD8α-Cre or wt), Cbx3Tg, Cbx3fl/+ and Cbx3fl/fl mice; data were extracted from flow analysis (Figure S2C, E, G); bars: group median; Graphpad unpaired student t-test: **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; each symbol = one mouse; n = 5-8; representative of 2-3 experiments. (I, K, M) Frequencies of CD4+FOXP3+ Tregs in ovarian ascites, B16 and NBL tumors (Figure S2D, F, H); bars: group median; Graphpad unpaired student t-test: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; each symbol = one mouse; n = 5-8; representative of 2-3 experiments. (N, O) Apoptotic cells (brown) in B16 melanoma and NBL tumor sections were identified by TUNEL staining; representative of 4 tumors from each mouse strain.
Cbx3/HP1γ-Deficient CD8+ T Cells Remodel the Tumor Chemokine/Receptor Landscape
CD8+ effector T cells homing to tumors is mediated primarily by the interaction between CCL2/CCR2 and CXCL9/CXCL10/CXCR3 chemokine/receptor pairs while CD4+ Tregs trafficking to tumors is induced mostly by CCL28/CCR10 and CXCL12/CXCR4 (68–77). Chemokines are generally produced by tumor-associated myeloid cells including dendritic cells (DCs) and macrophages (MØs) whereas their cognate receptors are expressed on T cells as well as other immune cells. It has been proposed that changes in the tumor chemokine/receptor landscape influence anti-tumor responses. Whether CD8+ effector T cells residing in the TME participate in remodeling the tumor chemokine landscape is not clearly established. RT-qPCR assays demonstrated that Ccl2/Ccr2 and Cxcl9/Cxcl10/Cxcr3 levels were higher in B16 melanoma tumors from Cbx3/HP1γ-deficient mice than those from controls (Figures 4A, B). In NBL tumors from Cbx3/HP1γ-deficient mice, Ccl2/Ccr2 and Cxl10 remained similar to those of controls whereas Cxcl9/Cxcr3 levels were elevated (Figures 4C, D). These results indicate that homing of Cbx3/HP1γ-deficient CD8+ effector T cells to B16 melanoma tumors is likely mediated by CCL2/CCR2 and CXCL9/CXCL10/CXCR3 interactions. Cbx3/HP1γ-deficient CD8+ effector T cells likely depend on CXCL9/CXCR3 engagement to invade NBL tumors. Compared to controls, Ccr10 and Cxcl12/Cxcr4 levels were decreased in B16 melanoma tumors from Cbx3/HP1γ-deficient mice (Figures 4E, F). In NBL tumors from Cbx3/HP1γ-deficient mice, Ccl28/Ccr10 and Cxcl12 expression was reduced (Figures 4G, H). Thus, CCR10 and CXCL12/CXCR4 may regulate homing of CD4+ Tregs to B16 melanoma tumors while invasion of NBL tumors by CD4+ Tregs is likely governed by CCL28/CCR10 and CXCL12. Decreased levels of CD4+ Treg-attracting chemokine/receptor pairs may be causing the diminished presence of these cells in the respective tumors. In short, the presence of Cbx3/HP1γ-deficient CD8+ effector T cells in the TME induces remodeling of the chemokine/receptor landscape that favors their optimal trafficking into tumors at the expense of CD4+ Tregs.
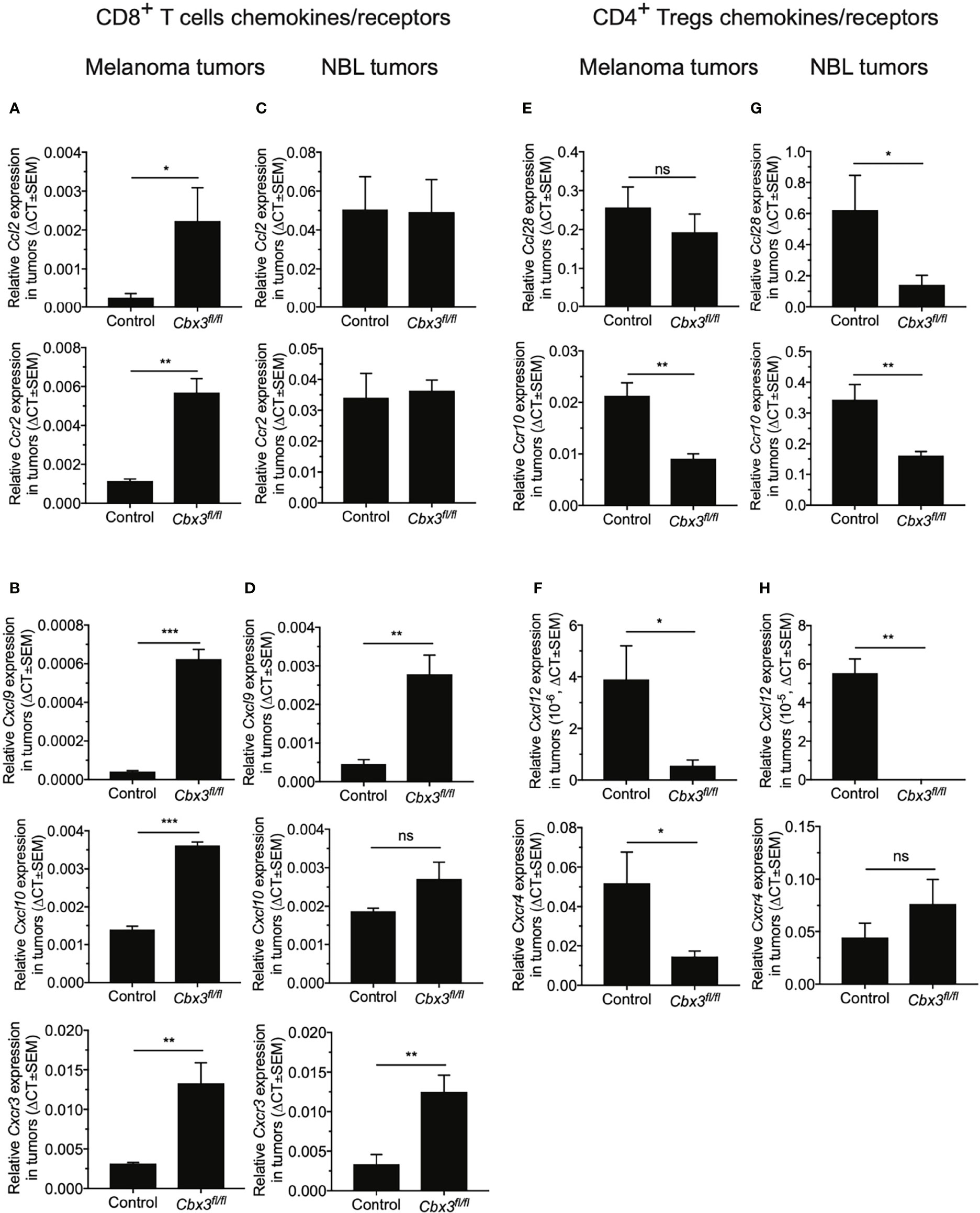
Figure 4 Cbx3/HP1γ-deficient CD8+ T cells remodel the tumor chemokine/receptor landscape. (A, B) Relative expression of chemokines and receptors mediating CD8+ T cells trafficking into B16 tumors was determined by RT-qPCR and normalized to Gapdh. (C, D) Relative expression of chemokines and receptors mediating CD8+ T cells trafficking into NBL tumors was evaluated by RT-qPCR and normalized to Gapdh. (E, F) Relative expression of chemokines and receptors inducing CD4+ Tregs homing into tumors was normalized to Gapdh. (G, H) Relative expression of chemokines and receptors inducing CD4+ Tregs homing into NBL tumors was assessed. X axis: mouse strains; Graphpad unpaired student t-test: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; ns, not significant; representative of 4 experiments; n = 4.
LEF-1 and IL-21R Are Indispensable for Halting Tumor Growth
To establish that LEF-1 and IL-21R contribute to the control of tumor growth by Cbx3/HP1γ-deficient CD8+ effector T cells, compound mutant mice were created (Figure S7A). In the Cbx3-Lef1-deficient mouse, Lef1 and Cbx3/HP1γ deletion was restricted to CD8+ T cells; in the Cbx3-Il21r-deficient mouse, Cbx3/HP1γ ablation was restricted to CD8+ T cells while Il21r was deleted in all tissues. LEF-1 and IL-21R expression was abolished in deficient CD8+ T cells compared to control cells (Figures 5A, B). Loss of one Lef1 or Il21r allele showed a decrease in protein levels that were almost completely abrogated upon deletion of both alleles. Progenitor T-cell development proceeded normally in the thymus of compound mutant animals (Figures S7B, C). Mesenteric lymph nodes (mLNs) of compound mutant mice displayed normal frequencies of mature CD8+ and CD4+ T cells (Figures S7D, E). Normal ratios of naïve (CD44–CD62L+), effector (CD44+CD62L–) and memory (CD44+CD62L+) T cells were observed in the mLNs of compound mutant animals (Figures S7F, G). In the mLNs, IL-21R expression was restricted to the CD8+ effector (CD44+CD62L–) T-cell population and was also dependent on gene dosage (Figure S7H). IL-21R was not detected on any CD4+ T-cell populations (Figure S7I). To determine LEF-1 contribution to tumor control, ID8 (ovarian), B16 (melanoma) or NB-9464 (NBL) tumor cells were injected into Lef1-deficient, Cbx3-Lef1-deficient and control mice. CD8+ T-cell-restricted Lef1 ablation resulted in uncontrolled ovarian, melanoma and NBL tumor growth (Figures 5C, D, F). Tumor burden in Lef1- and Cbx3-Lef1-deficient animals was higher than that observed for Cbx3/HP1γ-deficient and control mice. Because Il21r was deleted in all tissues, adoptive transfer of activated/differentiated CD8+ T cells from Il21r-/-, Cbx3-Il21r-deficient and control animals was performed. Melanoma and NBL growth proceeded unchecked in tumor-bearing B6.SJL congenic mice treated with control, Il21r-/- or Cbx3-Il21r-deficient CD8+ effector T cells compared to animals receiving Cbx3/HP1γ-deficient CD8+ effector T cells (Figures 5E, G). Our data establish that LEF-1 and IL-21R are necessary for the optimal control of ovarian, melanoma and NBL tumor growth. Moreover, LEF-1 expression in control CD8+ effector T cells is induced, yet they are less capable of persisting in tumors, suggesting there may be a threshold of expression level required for LEF-1 to function effectively. IL-21R expression is confined to the CD8+ T-cell effector population and its loss following deletion suggest that IL-21R function is essential to this population, which is critical for halting tumor development.
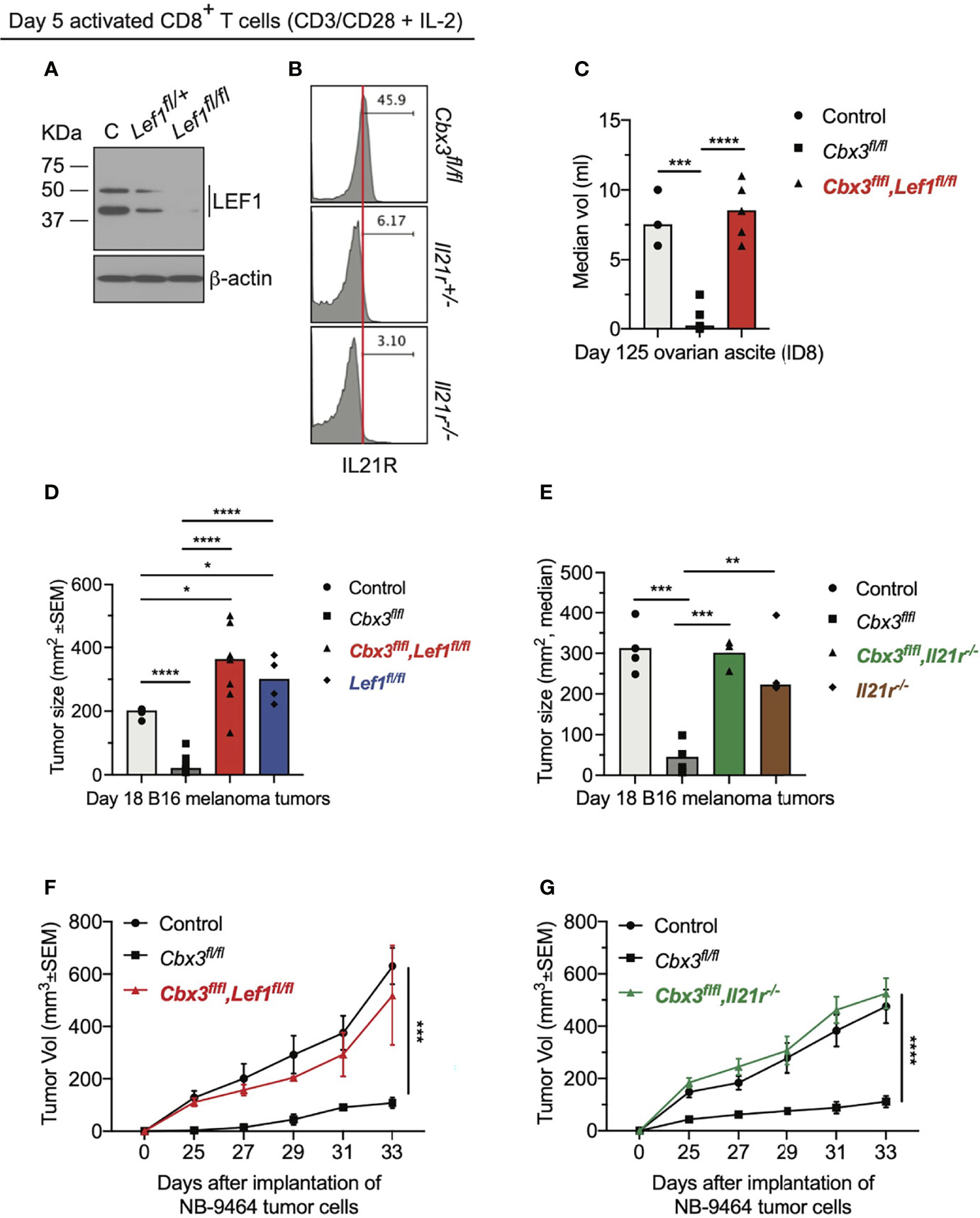
Figure 5 LEF-1 and IL-21R are indispensable for halting tumor growth. (A) Western blot of LEF-1 expression was done using total protein lysates of day 5 activated/differentiated CD8+ T cells; C: Cbx3/HP1γ-deficient; Lef1fl/+ and Lef1fl/fl: Cbx3-Lef1-deficient; representative of 4 experiments. (B) IL-21R expression was measured by flow; Cbx3fl/fl: Cbx3/HP1γ-deficient; Il21r+/- and Il21r-/-: Cbx3-Il21r-deficient; representative of 4 experiments. (C) Ovarian ascites were measured on day 125 after tumor injection; bars: group median; Graphpad unpaired student t-test: ***p ≤ 0.001, ****p ≤ 0.0001; each symbol = one mouse; n = 3-5; Cbx3fl/flLef1fl/fl: Cbx3-Lef1-deficient; representative of 2 experiments. (D) B16 melanoma tumor burden was determined on day 18 after tumor injection; bars: group median; Graphpad unpaired student t-test: *p ≤ 0.05, ****p ≤ 0.0001; each symbol = mouse; for each genotype n = 4-7; Lef1fl/fl: Lef1 ablation alone using CD8α-Cre mice; representative of 3 experiments. (E) On day 10 after injection of tumor cells, tumor bearing B6SJ/L mice (CD45.1+) were treated with in vitro-activated/differentiated CD8+ T cells (CD45.2+) from control, Cbx3fl/fl, Cbx3fl/fl,Il21r-/- (Cbx3-Il21r-deficient) or Il-21r-/- (germline deleted) mice; **p ≤ 0.01, ***p ≤ 0.001; n = 3-4 recipients; representative of 2 experiments. (F) NBL tumor burden was determined starting on day 25 after injection of tumor cells then every 2 days until day 33; Graphpad two-way ANOVA: ***p≤0.001; n = 2-5; representative of 2 experiments. (G) On day 14 after NBL injection, tumor bearing B6SJ/L mice were treated with in vitro-activated/differentiated CD8+ T cells from control, Cbx3fl/fl or Cbx3fl/fl,Il21r-/- (Cbx3-Il21r-deficient) mice; ****p ≤ 0.0001, n = 3-4 recipients; representative of 2 experiments.
LEF-1 and IL-21R Are Required for CD8+ Effector T Cells to Persist in Tumors
The inability of Lef1-, Il21r-, Cbx3-Lef1- and Cbx3-Il21r-deficient CD8+ T cells to halt tumor development suggests that LEF-1 and IL-21R are necessary for their persistence in tumors. Analyses of tumors from Cbx3-Lef1-deficient mice revealed that the frequencies of CD8+NKG2D+ T cells were reduced to control levels in all three tumors (Figures 6A, B, D). CD8+NKG2D+ T cells were also decreased to control levels in B16 tumors from Lef1-deficient mice (Figure 6B). There was no enrichment of CD8+NKG2D+ T cells in B16 or NBL tumors from animals treated with Cbx3-Il21r-deficient CD8+ effector T cells (Figures 6C, E). Likewise, CD8+NKG2D+ T cells were not augmented in B16 tumors from mice receiving Il21r-/- CD8+ effector T cells (Figure 6C). CD8+TIM3+CXCR5+ (progenitor exhausted) or CD8+TIM3+CXCR5- (terminally exhausted) T-cell populations were not perturbed (Figure S8). The frequency of endogenous NK1.1+NKG2D+, CD4+NKG2D+ T and myeloid cells in all tumor types was similar to that of controls (Figure S9). These results demonstrate that Lef1 and/or Il21r genetic ablation results in the exclusive loss of CD8+ effector T cells in all three tumor types. Our data underscore the fact that LEF-1 and IL-21R are requisite for the persistence of Cbx3/HP1γ-deficient CD8+ effector T cells in tumors with varied mutation loads that are ICB responsive (B16 melanoma) or non-responsive (ovarian and NBL).
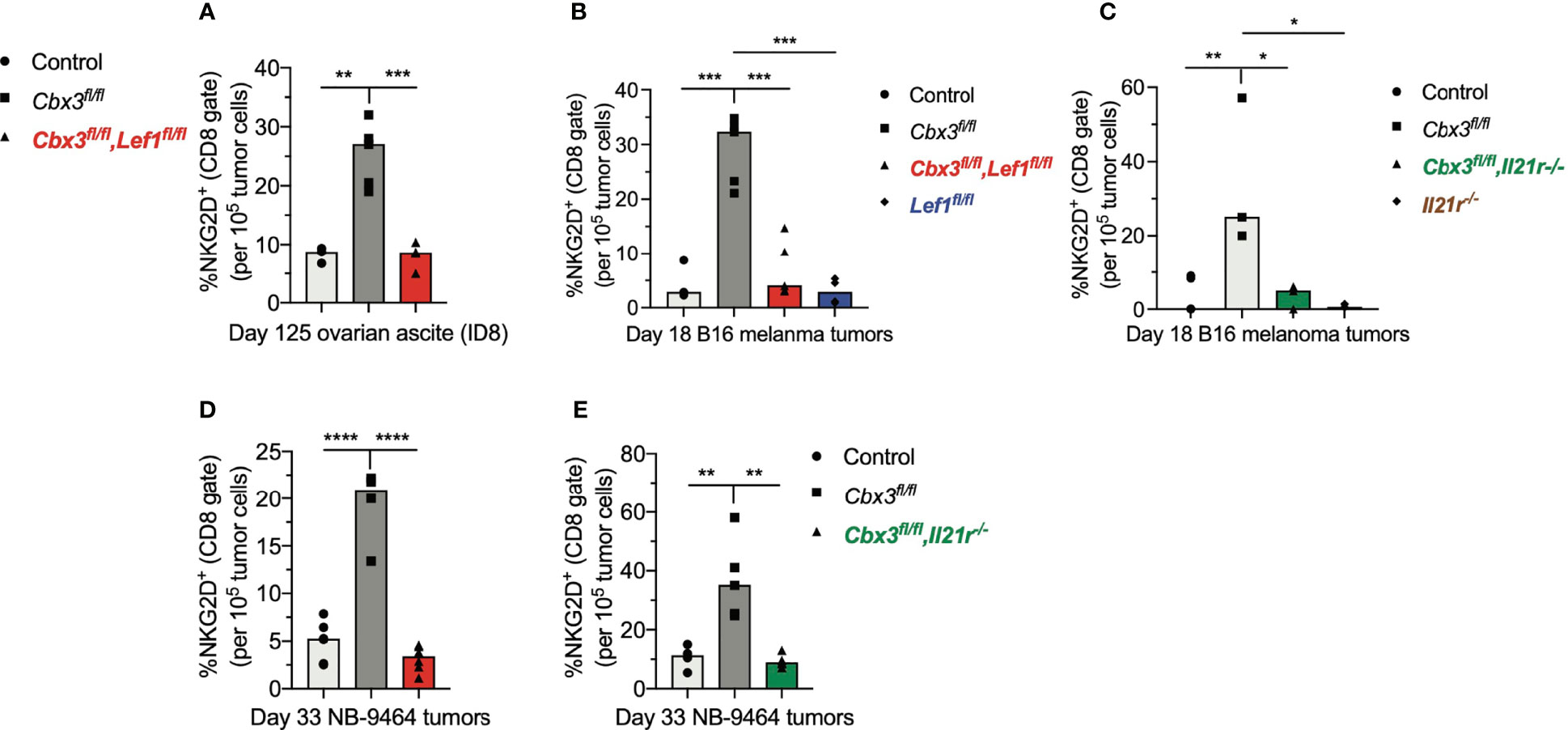
Figure 6 LEF-1 and IL-21R are required for CD8+ effector T cells to persist in tumors. (A) Frequencies of CD8+NKG2D+ T cells in ovarian ascites (from Figure 5) were extracted from flow analysis; bars: group median; Graphpad unpaired student t-test: **p ≤ 0.01, ***p ≤ 0.001; each symbol = one mouse. (B, C) Frequencies of CD8+NKG2D+ T cells in B16 melanoma tumors (from Figure 5) were extracted from flow analysis; bars: group median; Graphpad unpaired student t-test: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; each symbol = one mouse. (D, E) Frequencies of CD8+NKG2D+ T cells in NBL tumors (from Figure 5) were extracted from flow analysis; bars: group median; Graphpad unpaired student t-test: **p ≤ 0.01, ****p ≤ 0.0001; each symbol = one mouse.
LEF-1 and IL-21R Are Required to Maintain Effector Activity in Tumors
To show that uncontrolled tumor growth resulted primarily from the loss of CD8+ T-cell effector capacity, RT-qPCR assays were done using tumor RNA samples from compound mutant and control mice. Prf1 and Gzmb levels in melanoma tumors from control and compound mutant mice as well as those treated with Cbx3-Il21r- or Il21r-deficient CD8+ effector T cells were low compared to Cbx3/HP1γ-deficient mice (Figures 7A, B). Prf1, Gzmb and Ifng expression in NBL tumors from Cbx3-Lef1-deficient mice and those treated with Cbx3-Il21r-deficient CD8+ effector T cells was diminished compared to Cbx3/HP1γ-deficient animals (Figures 7C–E). Next, we asked whether the loss of effector capacity is inherent to Lef1 and/or Il21r deficiency or is caused by extrinsic factors in the TME. RT-qPCR analyses showed that Prf1, Gzmb and Ifng levels were reduced in in vitro-generated CD8+ effector T cells from compound mutant mice compared to those from Cbx3/HP1γ-deficient cells (Figures 7F–H). Therefore, our findings demonstrate that LEF-1 and IL-21R are indispensable for maintaining Prf1, Gzmb and Ifng in tumors. The loss of CD8+ effector T cells likely contributes to the observed blunted effector activity observed in tumors from Lef1- and Cbx3-Lef1-deficient mice as well as those treated with Il21r- or Cbx3-Il21r-deficient CD8+ T cells.
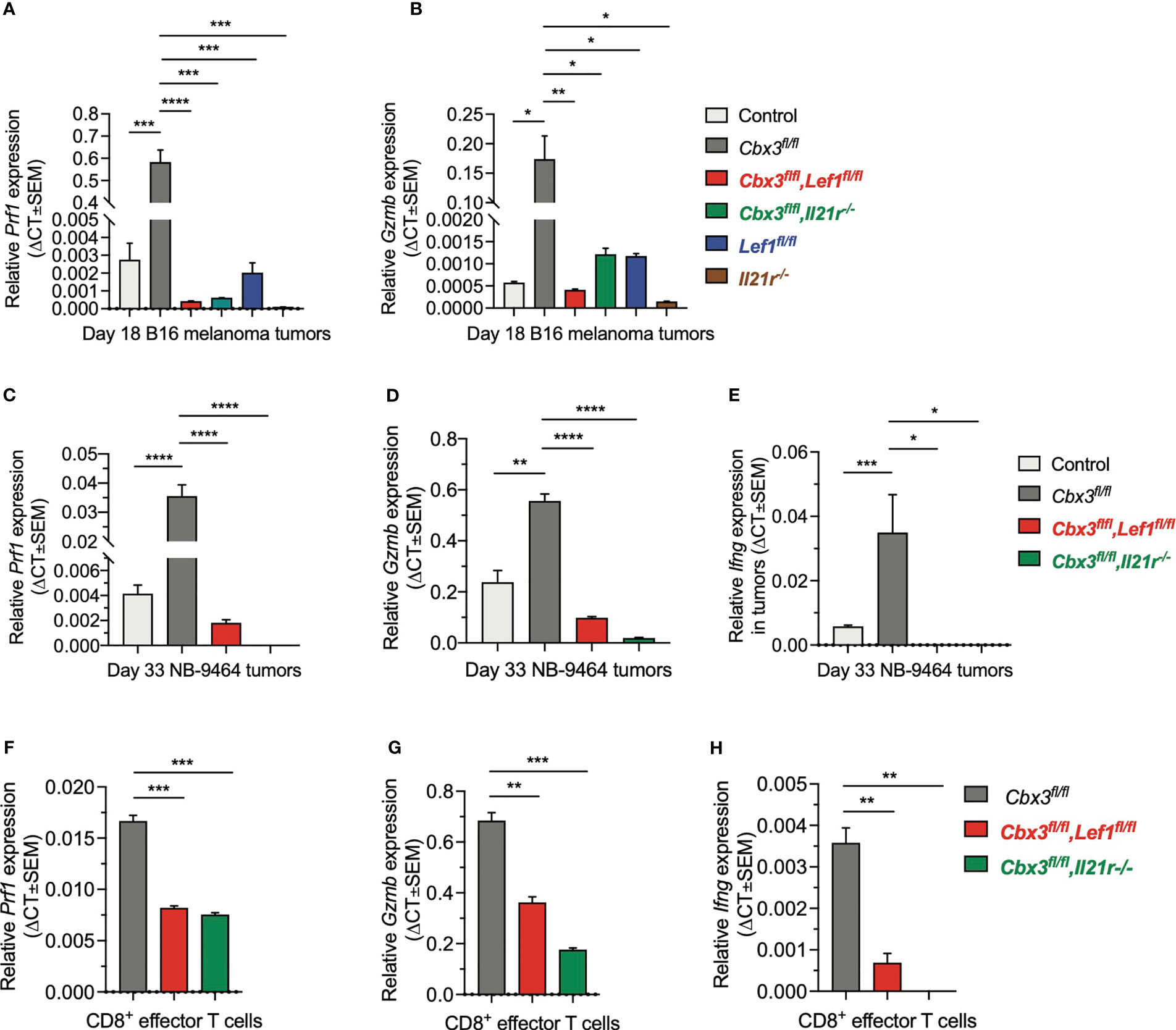
Figure 7 LEF-1 and IL-21R are required for CD8+ T cells to maintain their effector capacity. (A, B) Prf1 and Gzmb relative expression in B16 melanoma tumors was determined by RT-qPCR; Graphpad unpaired student t-test: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; n = 3 tumors; representative of 3 experiments. (C–E) Prf1, Gzmb and Ifng relative expression in NBL tumors was determined by RT-qPCR; Graphpad unpaired student t-test: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; n = 3 tumors; representative of 3 experiments. (F–H) Prf1, Gzmb and Ifng relative expression in day 5 in vitro-activated/differentiated CD8+ T cells from Cbx3/HP1γ-deficient and compound mutant mice was quantified by RT-qPCR; Graphpad unpaired student t-test: **p ≤ 0.01, ***p ≤ 0.001; representative of 3 experiments.
Discussion
We establish that genetic ablation of Cbx3/HP1γ induces the elevated, sustained expression of factors conferring both persistence and heightened effector/killing capacity on CD8+ T cells, which in turn enables them to control the growth of diverse tumor types. Once in the TME, Cbx3/HP1γ-deficient CD8+ T cells can remodel the chemokine/receptor landscape to ensure their optimal trafficking into tumors at the expense of CD4+ Tregs. Mechanistically, Cbx3/HP1γ deficiency allows for a higher rate of transcriptional initiation and chromatin remodeling at Lef1 and Il21r loci. Consequently, Cbx3/HP1γ-deficient CD8+ T cells express elevated levels of LEF-1 and IL-21R that can mitigate functional senescence and enable their persistence in tumors without the assistance of ICB.
To date, no essential functions for LEF-1 are identified in CD8+ effector T cells despite Lef1 expression being detected in human and mouse stem-like tumor infiltrating lymphocyte (TIL) subsets that also express Tcf7. Here, we provide compelling evidence that LEF-1 has a critical and non-redundant function in tumor rejection. The exclusive increase of Cbx3/HP1γ-deficient CD8+ effector T cells in tumors and their subsequent disappearance when Lef1 is ablated suggest that LEF-1 is requisite for their persistence in varied tumor types irrespective of mutational status. Lef1- and Cbx3-Lef1-deficient mice, in which Tcf7 is not deleted, fail to control tumor growth suggesting that Lef1 function is not redundant. Nevertheless, it is possible that in our models LEF-1+CD8+ and TCF-1+CD8+ T cells represent two separate populations with defined functions, and the former subset is endowed with an enhanced proliferative potential, or TCF-1 expression is downregulated in LEF-1+CD8+ T cells. Our data are distinct to those found with ICB where TCF-1 activity is implicated in the conversion of CD8+ TEX progenitor cells into terminally differentiated effector cells that can control the growth of tumors with high mutation rates. Instead, our findings are consistent with those recently published by Weber and colleagues (78). These authors demonstrate that CAR-T cells, when allowed to rest after receptor signaling is shut off, can recover from exhaustion, and regain their anti-tumor activity, in the absence of ICB or other pharmacological interventions. Lef1 expression is upregulated in rested CAR-T cells not in exhausted CAR-T cells, treated or not treated with anti-PD1. Although TCF-1 expression is similar in exhausted and rested CAR-T cells, the exhausted population cannot kill tumor cells while rested cells can. Moreover, Carr and colleagues report that LEF-1 plays a primary role in the post selection and effector fate differentiation of iNKT2 cells in the thymus (79). Similarly, Reya and colleagues show that LEF-1 is required for progenitor B-cell proliferation and survival through Wnt signaling (80). In the latter two models, the authors posit that LEF-1 exhibits a more dominant role while TCF-1 function is not sufficient for the differentiation and expansion of the immune-cell populations examined.
Our observation that IL-21R+CD8+ effector T cells increase in number after activation and are lost upon Il21r deletion suggest that IL-21R provides survival and expansion signals to these cells. This is in keeping with works showing that during a chronic viral infection or under IL-2-deprived conditions, IL-21R signaling is critical for preventing CD8+ T-cell exhaustion (23, 24). In acute viral infections, IL-21R signaling is essential for the proliferation and survival of activated CD8+ T cells as well as the generation of long-lived memory cells (25–27). In these models, IL-21R can activate the STAT1/STAT3 signaling pathways, which subsequently upregulate pro-survival factors BCL-2 and BCL-XL and downregulate TRAIL (26, 27). Furthermore, several lines of evidence indicate that IL-21R is involved in tumor immunity. First, high expression of IL-21R on CD8+ T cells in tumors correlates positively with overall survival and lack of tumor recurrence in hepatocellular carcinoma (HCC) patients (28). Second, in mice, IL-21R signaling reduces accumulation of myeloid derived suppressor cells (MDSCs) in the TME to control rapid HCC growth and maintains an immunological memory response to tumor re-challenge (28). Third, newly diagnosed HER2+ breast cancer patients with higher Il21r expression may have a reduced risk of distant relapse when treated with trastuzumab (anti-HER2/ErB2 mAb) in combination with chemotherapy; IL-21R expression on CD8+ effector T cells, not NK cells, is required for optimal anti-ErB2 mAb efficacy (29). Moreover, similar to HER2+ breast tumors, Il21 is not readily detected in B16 and NBL tumors from control or Cbx3/HP1γ-deficient mice. By contrast, IL-21 is visible in subcutaneous tumors (28). Differences in IL-21 expression are also observed in viral infection models. IL-21 is produced during both the acute and chronic phases; however, its levels decrease during the chronic phase (81, 82). This reduction is attributed to a contraction in the number of virus specific CD4+ T cells, which are the primary source of IL-21 production. Though reduced, IL-21 produced during the chronic phase can provide help to CD8+ T cells to resolve the infection. Thus, local endogenous IL-21 production in tumors may also be temporal. Because IL-21R expression is induced and sustained in Cbx3/HP1γ-deficient CD8+ effector T cells without exogenous IL-21, we posit that whether IL-21 production is high or low during tumor development, it can still positively affect Cbx3/HP1γ-deficient CD8+ T-cell effector functions.
Within tumors, activated tumor-associated-MØs and/or -DCs set up a milieu to be either activating or suppressing through the production of chemokines that attract CD8+ T cells or CD4+ Tregs (77). Our data demonstrate that Cbx3/HP1γ-deficient CD8+ effector T cells can remodel the chemokine landscape favoring their accumulation while preventing CD4+ Tregs from trafficking into tumors. It is tempting to speculate that the imbalance of chemokine production is caused by functional alterations and/or selective expansion of tumor-associated myeloid cells imposed by Cbx3/HP1γ-deficient CD8+ effector T cells.
Our results suggest a model whereby Cbx3/HP1γ normally restrains the effector and persistence potential of CD8+ T cells, which eventually succumb to functional inactivation and apoptosis thus incapable of controlling tumor development. Removal of Cbx3/HP1γ restraint allows CD8+ T cells to become effector-like cells armed with an intrinsic heightened killing and persistence capacity to control tumor growth; LEF-1 function and IL-21R signaling are necessary. Alternatively, Cbx3/HP1γ-deficient CD8+ effector T cells are preferentially recruited to tumors compared to control cells. Moreover, our model does not preclude the possibility that LEF-1 and IL-21R may also be implicated in CD8+ T-cell responses downstream of ICB. Since mouse and human Cbx3/HP1γ peptides are 100% identical, we suspect that human Cbx3/HP1γ will behave identically to the mouse ortholog: Cbx3/HP1γ deficiency would have the same effects on human CD8+ T cells. Ovarian and neuroblastoma are tumors with low mutation rates that respond minimally to ICB, yet their growth is effectively controlled by Cbx3/HP1γ-deficient CD8+ effector T cells. Thus, our findings provide a rationale for targeting Cbx3/HP1γ in human T cells to treat tumors harboring low mutation loads and do not respond to ICB.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/GSE183238.
Ethics Statement
The animal study was reviewed and approved by BIDMC Institutional Animal Care and Use Committee.
Author Contributions
THT, NH, and PL conceived the project and wrote the original draft with input and guidance from KE, JD, EMS, AB, H-HX, PS, THT, PL, NH, and NT designed and performed experiments. PL, NH, AB, H-HX, PS, and THT analyzed data and wrote the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants AI099012, CA198263, Friends for Life Neuroblastoma Research Program and The Mayer Family Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the Animal Technicians at the BIDMC Animal Research Facility for their dedication to and diligence in keeping our mouse colonies healthy. We are grateful to Drs. Crystal L. MacKall (Stanford University) and Katherine F. Roby (University of Kansas Medical Center) for the NB-9464 and ID8 tumor cell lines, respectively. We thank Dr. Dimitris Kioussis (MRC/National Institute for Medical Research, UK) for providing the VA-hCD2 vector. We are indebted to Dr. Lina Du at the Dana-Farber/Harvard Cancer Center (DF/HCC) Transgenic Mouse Core for her expertise in generating the Cbx3/HP1γTg mouse.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.738958/full#supplementary-material
Supplementary Figure 1 | Enhanced effector functions of Cbx3/HP1γ-deficient CD8+ T cells. (A) Cre activity (EGFP+) was restricted to CD8+CD4– spleen T cells from a cross between the reporter mouse ROSA-EGFP and the CD8α-Cre mouse. (B) Western immunoblot of total (tHP1γ) and phosphorylated (pHP1γ) Cbx3/HP1γ levels were evaluated in day 5 activated/differentiated pooled spleen and peripheral lymph node (pLN) CD8+ T cells using plate-bound anti-CD3/CD28 + IL-2 (10 IU/ml). Cbx3fl/+ and Cbx3fl/fl: CD8+ T-cell-restricted deletion of Cbx3/HP1γ in mice using the CD8α-Cre strain; collectively designated as Cbx3/HP1γ-deficient; representative of 3 experiments; n = 3 mice. (C) Relative expression of Bcl6, Eomes and Tbx21 was quantified by RT-qPCR using RNA samples of day 5 activated/differentiated CD8+ T cells. Unit 1 indicates no change; n = 3; representative of 3 experiments. (D) Western immunoblot showed EOMES expression in same cells as (B). (E) Relative expression of Gzmb, Prf1 and Ifng was etermined by RT-qPCR using same RNA samples as in (C). (F) Cleaved (activated) caspase 3 (CC3) in protein lysate of washed cells was detected 24 hours after coculture using an antibody specific to CC3. CD8:target = effector CD8+ T cells cocultured with target; control: CD8α-Cre; Cbx3fl/+: CD8-restricted deletion of Cbx3/HP1γ with the CD8α-Cre mouse. CD4:target = day 2 activated CD4+ T cells from control (CD4-Cre) or Cbx3fl/fl mice cocultured with target; Cbx3fl/fl: CD4-restricted deletion of Cbx3/HP1γ with the CD4-Cre mouse; target: B16 melanoma tumor cells. 1:1= 1 effector cell to 1 target cell; 5:1 = 5 effector cells to one target cell; Un: no added effector cells; representative of 3 experiments. (G) Western immunoblot showed LEF-1 expression in thymocytes from control and Cbx3/HP1γ-deficient mice; representative of 3 experiments. (H) Flow histograms showed lack of IL-21R expression on thymocytes from control and Cbx3/HP1γdeficient mice; representative of 3 experiments.
Supplementary Figure 2 | Reduction of ovarian ascites in Cbx3/HP1γ-deficient mice. (A) Images showed ovarian ascites in control and Cbx3/HP1γ-deficient mice injected (IP) with the syngeneic mouse ID8 cell line; control: CD8α-Cre; Cbx3fl/+ and Cbx3fl/fl: Cbx3/HP1γ-deficient mice; yellow outlines: tumor nodules (supporting data for Figures 3A, B). (B) Ovarian ascites in control and Cbx3/HP1γ-deficient mice injected (IP) with the syngeneic mouse MOSE-LTICv cell line were quantified and graphed as group median; control: CD8α-Cre; Cbx3fl/fl: Cbx3/HP1γ-deficient mice; Graphpad student unpaired t-test; *p≤0.05; n = 4-5.; representative of 2 experiments. (C, E, G) Flow analysis showed frequencies of CD8+NKG2D+ effector T cells in ovarian ascites, B16 and NBL tumors from control (CD8α-Cre or wt), Cbx3Tg, Cbx3fl/+ and Cbx3fl/fl mice; Cbx3Tg: T-cell-restricted expression of Cbx3/HP1γ driven by the human Cd2 promoter (supporting data for Figures 3H, J, L). (D, F, H) Flow analysis depicted the frequencies of CD4+FOXP3+ regulatory T cells (Tregs) in ovarian ascites, B16 and NBL tumors from control (CD8α-Cre or wt), Cbx3Tg, Cbx3fl/+ and Cbx3fl/fl mice (supporting data for Figures 3I, K, M).
Supplementary Figure 3 | No enrichment of CD4+NKG2D+ or NK1.1+NKG2D+ T cells in tumors (supporting data for Figure 3). (A, C, E) Frequencies of CD4+NKG2D+ T cells in ovarian ascites, B16 and NBL tumors were extracted from flow analysis; bars: group median; Graphpad unpaired student t-test: ns, not significant; each symbol = one mouse; n = 2-8. (B, D, F) Frequencies of NK1.1+NKG2D+ T cells in ovarian ascites, B16 and NBL tumors were extracted from flow analysis; bars: group median; each symbol = one mouse; n = 2-8.
Supplementary Figure 4 | Prf1, Gzmb and Ifng expression in tumors. (A–C) Relative expression of Ifng, Gzmb and Prf1 in B16 melanoma tumors were quantified by RT qPCR and normalized to Gapdh; Graphpad unpaired student t-test: *p≤0.05, **p≤0.01, n = 4 tumors from each mouse strain; representative of 4 experiments. (D–F) Relative expression of Ifng, Gzmb and Prf1 in NBL tumors was normalized to Gapdh; Graphpad unpaired student t-test: *p≤0.05, **p≤0.01, ***p≤0.001; n = 3 tumors from each mouse strain; representative of 3 experiments. (G) B16 melanoma tumor growth in Cbx3fl/fl,Prf1-/- (Cbx3/HP1γ-deficient mice lacking Prf1), Cbx3fl/fl (Cbx3/HP1γ-deficient) and control (CD8α-Cre and wt) mice was measured; Graphpad two-way ANOVA; ****p≤0.0001;n = 6; representative of 2 experiments. (H) Apoptotic cells (brown) in B16 melanoma tumors from mice in (A) were identified by TUNEL staining; representative of 2 experiments. (I) NBL tumor burden in mice implanted with NB-9464 tumor cells was measured; Cbx3fl/fl,Prf1-/-: Cbx3/HP1γ-deficient mice lacking Prf1; Cbx3fl/fl: Cbx3/HP1γ-deficient; control: CD8α-Cre and wt; Graphpad two-way ANOVA; ****p≤0.0001; n = 3-5; representative of 2 experiments. (J) Apoptotic cells (brown) in NBL tumors from mice in (C) were identified by TUNEL staining; representative of 2 experiments. (K) NBL tumor growth in Cbx3fl/fl,Ifng-/- (Cbx3/HP1γ-deficient mice lacking Ifng) was evaluated; Cbx3fl/fl (Cbx3/HP1γ-deficient) and control (CD8α-Cre and wt) mice; Graphpad two-way ANOVA; *p≤0.05, **p≤0.01; n = 3-4; representative of 2 experiments.
Supplementary Figure 5 | Direct cause of tumor rejection by Cbx3/HP1γ-deficient CD8+ effector T cells. (A) Schematic of ACT. (B) 10 days after implantation of B16 tumor cells, B6.SJL recipients (CD45.1+) were treated once with exogenous CD8+ effector T cells (CD45.2+); bars: group median; Graphpad unpaired student t-test: *p≤0.05, **p≤0.01; each symbol = one mouse; n = 4-5 recipients; representative of 2 experiments. (C) 14 days after implantation of N-9464 tumor cells, B6.SJL recipients were treated once with exogenous CD8+ effector T cells; Graphpad two-way ANOVA: ****p≤0.0001; n = 5 recipients; representative of 2 experiments. (D) Frequencies of CD8+ effector T cells in B16 melanoma tumors after ACT; A: B6.SJL mice treated with control CD8+ T cells, B: B6.SJL mice treated with Cbx3fl/+ CD8+ T cells, C: B6.SJL mice treated with Cbx3fl/fl CD8+ T cells; grey bars: transferred CD8+ effector T cells (CD45.2+); black bars: endogenous CD8+ effector T cells (CD45.1+); bars: group median; Graphpad unpaired student t-test: *p≤0.05, **p≤0.01, ns: not significant; each symbol = one mouse; n = 4-5 recipients from B. (E) Staining and gating strategies for panel D; gated on total live tumor cells obtained from tumors of B6.SJL recipients from B. (F, G) Frequencies of CD4+CD45.2+ T cells in B16 melanoma tumors (F) and spleens (G) from B6.SJL recipients treated with control (CD8α-Cre) or Cbx3/HP1γ-deficient CD8+ effector T cells; data were extracted from flow analysis; bars: group median; each symbol = one mouse; n = 4-5 recipients as in (D). (H, I) Frequencies of NK1.1+CD45.2+ T cells in B16 tumors (H) and spleens (I) from B6.SJL recipients treated with control or Cbx3/HP1γ-deficient CD8+ effector T cells; data were extracted from flow analysis; bars: group median; each symbol = one mouse; n = 4-5 recipients from B.
Supplementary Figure 6 | Effects of NKG2D blockade on tumor growth. (A) B16 melanoma tumor burden was assessed after NKG2D blockade using the anti-mouse NKG2D antibody HMG2D; Graphpad two-way ANOVA: ****p≤0.0001; Cbx3fl/fl-NKG2D TX: Cbx3fl/fl mice injected with HMG2D one day after injection of tumor cells; control and Cbx3fl/fl mice were injected with PBS; n = 5 received Ab, 10 received PBS; representative of 2 experiments. (B) Survival curves of B16 tumor-bearing mice after NKG2D blockade. (C) NBL tumor burden was evaluated after NKG2D blockade; Graphpad two-way ANOVA: **p≤0.01; n = 4 received Ab, 10 received PBS; representative of 2 experiments. (D) Survival curves of NBL tumor-bearing mice after NKG2D blockade. (E, F) Flow analysis showed frequencies of CD8+NKG2D+ T cells in B16 melanoma tumors; bars: group median; Graphpad student unpaired t-test: **p≤0.01; each symbol = one mouse; Cbx3fl/fl-NKG2D TX: Cbx3/HP1γ-deficient mice receiving the anti-mouse NKG2D antibody HMG2D; control and Cbx3/HP1γfl/fl: PBS treated. (G, H) Frequencies of CD4+NKG2D+ and NK1.1+NKG2D+ T cells in B16 melanoma tumors in (A).
Supplementary Figure 7 | Normal T-cell development in the thymus of compound mutant mice. (A) Generation of compound mutant mice. (B, C) T-cell development in the thymus of Cbx3-Lef1-deficient and Cbx3-Il21r-deficient mice was assessed; numbers: percent cells; DN1-DN4: double negative (CD4–CD8–) progenitor T-cell populations gated from bottom left quadrants of top flow plots. (D, E) CD4+ and CD8+ ratio was evaluated in mLNs from compound mutant mice; numbers: percent cells. (F, G) mLN naïve (CD44–CD62L+), effector (CD44+CD62L–) and memory (CD44+CD62L+) cells in CD4+ as well as CD8+ T-cell compartments of compound mutant mice were assessed; numbers: percent cells. (H, I) Flow analysis was done to evaluate IL21R expression on mLN CD8+ (H) and CD4+ (I) effector (CD44+CD62L–), naïve (CD44–CD62L+) and memory (CD44+CD62L+) T cells from Cbx3/HP1γ-deficient and Cbx3-Il21r-deficient mice; numbers: percent cells. For all panels, n = 3 for each genotype; representative of 3 experiments.
Supplementary Figure 8 | Analysis of CD8+ progenitor and terminally exhausted T cells. (A) Frequencies of progenitor exhausted (TIM3+CXCR5+) and terminally exhausted (TIM3+CXCR5–) CD8+ T cells in all tumors from Cbx3fl/flLef1fl/fl (Cbx3-Lef1-deficient), Cbx3fl/fl (Cbx3/HP1γ-deficient) and control (CD8α-Cre or wt) mice were assessed; n = 3-5; representative of 2 experiments. (B) Progenitor exhausted (TIM3+CXCR5+) and terminally exhausted (TIM3+CXCR5–) CD8+ T cells were identified in tumors from B6.SJL mice treated with Cbx3fl/flIl21r-/-, Cbx3fl/fl or control CD8+ effector T cells (CD45.2+); n = 3-5 recipients; representative of 2 experiments.
Supplementary Figure 9 | Analysis of tumor infiltrating immune populations. (A) Frequencies of NK1.1+NKG2D+, CD4+NKG2D+ T, Gr1+ and Mac1+ myeloid cells in tumors from Cbx3fl/flLef1fl/fl (Cbx3-Lef1-deficient) and control animals; representative of 2 experiments. (B) Frequencies of endogenous NK1.1+NKG2D+, CD4+NKG2D+ T, Gr1+ and Mac1+ myeloid cells in tumors from B6.SJL mice treated with Cbx3fl/flIl21r-/- and control CD8+ effector T cells; representative of 2 experiments.
References
1. Pauken KE, Wherry EJ. Overcoming T Cell Exhaustion in Infection and Cancer. Trends Immunol (2015) 36(4):265–76. doi: 10.1016/j.it.2015.02.008
2. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and Inflammation Induce Distinct Transcriptional Programs That Promote the Differentiation of Effector Cytolytic T Cells. Immunity (2010) 32(1):79–90. doi: 10.1016/j.immuni.2009.11.012
3. Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-Box Proteins Cooperate to Establish the Transcriptional Program of Effector CTLs. J Exp Med (2009) 206(1):51–9. doi: 10.1084/jem.20081242
4. Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, et al. The Transcription Factor Runx3 Guards Cytotoxic CD8(+) Effector T Cells Against Deviation Towards Follicular Helper T Cell Lineage. Nat Immunol (2017) 18(8):931–9. doi: 10.1038/ni.3773
5. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of Exhausted CD8+ T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
6. LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, et al. PTPN2 Regulates the Generation of Exhausted CD8+ T Cell Subpopulations and Restrains Tumor Immunity. Nat Immunol (2019) 20(10):1335–47. doi: 10.1038/s41590-019-0480-4
7. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1–CD8+ Tumor-Infiltrating T Cells. Immunity (2019) 50(1):181–94.e6. doi: 10.1016/j.immuni.2018.11.014
8. Im S, Hashimoto M, Gerner M, Lee J, Kissick H, Burger M, et al. Defining CD8+ T Cells That Provide the Proliferative Burst After PD-1 Therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330
9. Krishna S, Lowery FJ, Copeland AR, Bahadiroglu E, Mukherjee R, Jia L, et al. Stem-Like CD8 T Cells Mediate Response of Adoptive Cell Immunotherapy Against Human Cancer. Science (2020) 370(6522):1328–34. doi: 10.1126/science.abb9847
10. Liu D, Lin J-R, Robitschek EJ, Kasumova GG, Heyde A, Shi A, et al. Evolution of Delayed Resistance to Immunotherapy in a Melanoma Responder. Nat Med (2021) 27(6):985–92. doi: 10.1038/s41591-021-01331-8
11. Zhou X, Yu S, Zhao D-M, Harty J, Badovinac V, Xue H-H. Differentiation and Persistence of Memory CD8+ T Cells Depend on T Cell Factor 1. Immunity (2010) 33(2):229–40. doi: 10.1016/j.immuni.2010.08.002
12. Zhou X, Xue H-H. Cutting Edge: Generation of Memory Precursors and Functional Memory CD8+ T Cells Depends on T Cell Factor-1 and Lymphoid Enhancer-Binding Factor-1. J Immunol (2012) 189(6):2722–6. doi: 10.4049/jimmunol.1201150
13. Miron M, Kumar BV, Meng W, Granot T, Carpenter DJ, Senda T, et al. Human Lymph Nodes Maintain TCF-1hi Memory T Cells With High Functional Potential and Clonal Diversity Throughout Life. J Immunol (2018) 201(7):2132–40. doi: 10.4049/jimmunol.1800716
14. Jeannet G, Boudousquié C, Gardiol N, Kang J, Huelsken J, Held W. Essential Role of the Wnt Pathway Effector Tcf-1 for the Establishment of Functional CD8 T Cell Memory. Proc Natl Acad Sci (2010) 107(21):9777–82. doi: 10.1073/pnas.0914127107
15. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell Factor 1-Expressing Memory-Like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity (2016) 45(2):415–27. doi: 10.1016/j.immuni.2016.07.021
16. Ma G, Yasunaga J-i, Akari H, Matsuoka M. TCF1 and LEF1 Act as T-Cell Intrinsic HTLV-1 Antagonists by Targeting Tax. Proc Natl Acad Sci (2015) 112(7):2216–21. doi: 10.1073/pnas.1419198112
17. Okamura RM, Sigvardsson M, Galceran J, Verbeek S, Clevers H, Grosschedl R. Redundant Regulation of T Cell Differentiation and TCRalpha Gene Expression by the Transcription Factors LEF-1 and TCF-1. Immunity (1998) 8(1):11–20. doi: 10.1016/S1074-7613(00)80454-9
18. Weber BN, Chi AW-S, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, et al. A Critical Role for TCF-1 in T-Lineage Specification and Differentiation. Nature (2011) 476:63. doi: 10.1038/nature10279
19. Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, et al. Tcf1 and Lef1 Transcription Factors Establish CD8+ T Cell Identity Through Intrinsic HDAC Activity. Nat Immunol (2016) 17:695. doi: 10.1038/ni.3456
20. Acharya N, Madi A, Zhang H, Klapholz M, Escobar G, Dulberg S, et al. Endogenous Glucocorticoid Signaling Regulates CD8+ T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity (2020) 53(3):658–71.e6. doi: 10.1016/j.immuni.2020.08.005
21. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated With Response to Checkpoint Immunotherapy in Melanoma. Cell (2018) 175(4):998–1013.e20. doi: 10.1016/j.cell.2018.10.038
22. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-Jun Overexpression in CAR T Cells Induces Exhaustion Resistance. Nature (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
23. Khattar M, Miyahara Y, Schroder PM, Xie A, Chen W, Stepkowski SM. Interleukin-21 Is a Critical Regulator of CD4 and CD8 T Cell Survival During Priming Under Interleukin-2 Deprivation Conditions. PloS One (2014) 9(1):e85882. doi: 10.1371/journal.pone.0085882
24. Fröhlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, et al. IL-21R on T Cells Is Critical for Sustained Functionality and Control of Chronic Viral Infection. Science (2009) 324(5934):1576–80. doi: 10.1126/science.1172815
25. Noguchi N, Nakamura R, Hatano S, Yamada H, Sun X, Ohara N, et al. Interleukin-21 Induces Short-Lived Effector CD8(+) T Cells But Does Not Inhibit Their Exhaustion After Mycobacterium Bovis BCG Infection in Mice. Infect Immun (2018) 86(8):IAI.00147–18. doi: 10.1128/IAI.00147-18
26. Barker BR, Gladstone MN, Gillard GO, Panas MW, Letvin NL. Critical Role for IL-21 in Both Primary and Memory Anti-Viral CD8+ T-Cell Responses. Eur J Immunol (2010) 40(11):3085–96. doi: 10.1002/eji.200939939
27. Novy P, Huang X, Leonard WJ, Yang Y. Intrinsic IL-21 Signaling Is Critical for CD8 T Cell Survival and Memory Formation in Response to Vaccinia Viral Infection. J Immunol (2011) 186(5):2729–38. doi: 10.4049/jimmunol.1003009
28. Zheng X, Zhou Y, Yi X, Chen C, Wen C, Ye G, et al. IL-21 Receptor Signaling Is Essential for Control of Hepatocellular Carcinoma Growth and Immunological Memory for Tumor Challenge. OncoImmunology (2018) 7(12):e1500673. doi: 10.1080/2162402X.2018.1500673
29. Mittal D, Caramia F, Michiels S, Joensuu H, Kellokumpu-Lehtinen P-L, Sotiriou C, et al. Improved Treatment of Breast Cancer With Anti-HER2 Therapy Requires Interleukin-21 Signaling in CD8+ T Cells. Cancer Res (2016) 76(2):264–74. doi: 10.1158/0008-5472.CAN-15-1567
30. Wang LN, Cui YX, Ruge F, Jiang WG. Interleukin 21 and Its Receptor Play a Role in Proliferation, Migration and Invasion of Breast Cancer Cells. Cancer Genomics Proteomics (2015) 12(5):211–21.
31. Linnebacher A, Mayer P, Marnet N, Bergmann F, Herpel E, Revia S, et al. Interleukin 21 Receptor/Ligand Interaction Is Linked to Disease Progression in Pancreatic Cancer. Cells (2019) 8(9):1104–21. doi: 10.3390/cells8091104
32. Davis MR, Zhu Z, Hansen DM, Bai Q, Fang Y. The Role of IL-21 in Immunity and Cancer. Cancer Lett (2015) 358(2):107–14. doi: 10.1016/j.canlet.2014.12.047
33. Jones DO, Cowell IG, Singh PB. Mammalian Chromodomain Proteins: Their Role in Genome Organisation and Expression. Bioessays (2000) 22(2):124–37. doi: 10.1002/(SICI)1521-1878(200002)22:2<124::AID-BIES4>3.0.CO;2-E
34. Jenuwein T, Allis CD. Translating the Histone Code. Science (2001) 293(5532):1074–80. doi: 10.1126/science.1063127
35. Minc E, Courvalin JC, Buendia B. HP1gamma Associates With Euchromatin and Heterochromatin in Mammalian Nuclei and Chromosomes. Cytogenet Cell Genet (2000) 90(3-4):279–84. doi: 10.1159/000056789
36. Minc E, Allory Y, Worman HJ, Courvalin JC, Buendia B. Localization and Phosphorylation of HP1 Proteins During the Cell Cycle in Mammalian Cells. Chromosoma (1999) 108(4):220–34. doi: 10.1007/s004120050372
37. Singh PB, Newman AG. On the Relations of Phase Separation and Hi-C Maps to Epigenetics. R Soc Open Sci (2020) 7(2):191976. doi: 10.1098/rsos.191976
38. Hediger F, Gasser SM. Heterochromatin Protein 1: Don’t Judge the Book by Its Cover! Curr Opin Genet Dev (2006) 16(2):143–50. doi: 10.1016/j.gde.2006.02.013
39. Smallwood A, Hon GC, Jin F, Henry RE, Espinosa JM, Ren B. CBX3 Regulates Efficient RNA Processing Genome-Wide. Genome Res (2012) 22(8):1426–36. doi: 10.1101/gr.124818.111
40. Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 Lysine 9 Methylation and HP1gamma Are Associated With Transcription Elongation Through Mammalian Chromatin. Mol Cell (2005) 19(3):381–91. doi: 10.1016/j.molcel.2005.06.011
41. Yearim A, Gelfman S, Shayevitch R, Melcer S, Glaich O, Mallm JP, et al. HP1 is Involved in Regulating the Global Impact of DNA Methylation on Alternative Splicing. Cell Rep (2015) 10(7):1122–34. doi: 10.1016/j.celrep.2015.01.038
42. Kwon SH, Workman JL. The Heterochromatin Protein 1 (HP1) Family: Put Away a Bias Toward HP1. Mol Cells (2008) 26(3):217–27.
43. Piacentini L, Fanti L, Berloco M, Perrini B, Pimpinelli S. Heterochromatin Protein 1 (HP1) Is Associated With Induced Gene Expression in Drosophila Euchromatin. J Cell Biol (2003) 161(4):707–14. doi: 10.1083/jcb.200303012
44. Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-Beta Mobilization Promotes Chromatin Changes That Initiate the DNA Damage Response. Nature (2008) 453(7195):682–6. doi: 10.1038/nature06875
45. Saint-Andre V, Batsche E, Rachez C, Muchardt C. Histone H3 Lysine 9 Trimethylation and HP1gamma Favor Inclusion of Alternative Exons. Nat Struct Mol Biol (2011) 18(3):337–44. doi: 10.1038/nsmb.1995
46. Goldmit M, Ji Y, Skok J, Roldan E, Jung S, Cedar H, et al. Epigenetic Ontogeny of the Igk Locus During B Cell Development. Nat Immunol (2005) 6(2):198–203. doi: 10.1038/ni1154
47. Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A Transcription Factors Limit CAR T Cell Function in Solid Tumours. Nature (2019) 567(7749):530–4. doi: 10.1038/s41586-019-0985-x
48. Yao C, Lou G, Sun H-W, Zhu Z, Sun Y, Chen Z, et al. BACH2 Enforces the Transcriptional and Epigenetic Programs of Stem-Like CD8+ T Cells. Nat Immunol (2021) 22(3):370–80. doi: 10.1038/s41590-021-00868-7
49. Oyama K, El-Nachef D, Fang C, Kajimoto H, Brown JP, Singh PB, et al. Deletion of HP1gamma in Cardiac Myocytes Affects H4K20me3 Levels But Does Not Impact Cardiac Growth. Epigenet Chromatin (2018) 11(1):18. doi: 10.1186/s13072-018-0187-z
50. Zhumabekov T, Corbella P, Tolaini M, Kioussis D. Improved Version of a Human CD2 Minigene Based Vector for T Cell-Specific Expression in Transgenic Mice. J Immunol Methods (1995) 185(1):133–40. doi: 10.1016/0022-1759(95)00124-S
51. Frohlich A, Marsland BJ, Sonderegger I, Kurrer M, Hodge MR, Harris NL, et al. IL-21 Receptor Signaling is Integral to the Development of Th2 Effector Responses In Vivo. Blood (2007) 109(5):2023–31. doi: 10.1182/blood-2006-05-021600
52. Maekawa Y, Minato Y, Ishifune C, Kurihara T, Kitamura A, Kojima H, et al. Notch2 Integrates Signaling by the Transcription Factors RBP-J and CREB1 to Promote T Cell Cytotoxicity. Nat Immunol (2008) 9(10):1140–7. doi: 10.1038/ni.1649
53. Mao X, Fujiwara Y, Chapdelaine A, Yang H, Orkin SH. Activation of EGFP Expression by Cre-Mediated Excision in a New ROSA26 Reporter Mouse Strain. Blood (2001) 97(1):324–6. doi: 10.1182/blood.V97.1.324
54. Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, et al. Cytotoxicity Mediated by T Cells and Natural Killer Cells Is Greatly Impaired in Perforin-Deficient Mice. Nature (1994) 369(6475):31–7. doi: 10.1038/369031a0
55. Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple Defects of Immune Cell Function in Mice With Disrupted Interferon-Gamma Genes. Science (1993) 259(5102):1739–42. doi: 10.1126/science.8456300
56. Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a Syngeneic Mouse Model for Events Related to Ovarian Cancer. Carcinogenesis (2000) 21(4):585–91. doi: 10.1093/carcin/21.4.585
57. Cohen CA, Shea AA, Heffron CL, Schmelz EM, Roberts PC. The Parity-Associated Microenvironmental Niche in the Omental Fat Band Is Refractory to Ovarian Cancer Metastasis. Cancer Prev Res (2013) 6(11):1182–93. doi: 10.1158/1940-6207.CAPR-13-0227
58. Roberts PC, Mottillo EP, Baxa AC, Heng HHQ, Doyon-Reale N, Gregoire L, et al. Sequential Molecular and Cellular Events During Neoplastic Progression: A Mouse Syngeneic Ovarian Cancer Model. Neoplasia (2005) 7(10):944–56. doi: 10.1593/neo.05358
59. Kroesen M, Nierkens S, Ansems M, Wassink M, Orentas RJ, Boon L, et al. A Transplantable TH-MYCN Transgenic Tumor Model in C57Bl/6 Mice for Preclinical Immunological Studies in Neuroblastoma. Int J Cancer J Int Cancer (2014) 134(6):1335–45. doi: 10.1002/ijc.28463
60. Sun M, Ha N, Pham DH, Frederick M, Sharma B, Naruse C, et al. Cbx3/HP1gamma Deficiency Confers Enhanced Tumor-Killing Capacity on CD8+ T Cells. Sci Rep (2017) 7:42888. doi: 10.1038/srep42888
61. Ha N, Pham DH, Shahsafaei A, Naruse C, Asano M, Thai TH. HP-1gamma Controls High-Affinity Antibody Response to T-Dependent Antigens. Front Immunol (2014) 5:271. doi: 10.3389/fimmu.2014.00271
62. Berrien-Elliott MM, Yuan J, Swier LE, Jackson SR, Chen CL, Donlin MJ, et al. Checkpoint Blockade Immunotherapy Relies on T-Bet But Not Eomes to Induce Effector Function in Tumor-Infiltrating CD8+ T Cells. Cancer Immunol Res (2015) 3(2):116–24. doi: 10.1158/2326-6066.CIR-14-0159
63. Alves NL, Arosa FA, van Lier RA. IL-21 Sustains CD28 Expression on IL-15-Activated Human Naive CD8+ T Cells. J Immunol (2005) 175(2):755–62. doi: 10.4049/jimmunol.175.2.755
64. Allard EL, Hardy MP, Leignadier J, Marquis M, Rooney J, Lehoux D, et al. Overexpression of IL-21 Promotes Massive CD8+ Memory T Cell Accumulation. Eur J Immunol (2007) 37(11):3069–77. doi: 10.1002/eji.200637017
65. Le TP, Thai TH. The State of Cellular Adoptive Immunotherapy for Neuroblastoma and Other Pediatric Solid Tumors. Front Immunol (2017) 8:1640. doi: 10.3389/fimmu.2017.01640
66. Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor Activity and Safety of Pembrolizumab in Patients With Advanced Recurrent Ovarian Cancer: Results From the Phase II KEYNOTE-100 Study. Ann Oncol (2019) 30(7):1080–7. doi: 10.1093/annonc/mdz135
67. Martin SD, Brown SD, Wick DA, Nielsen JS, Kroeger DR, Twumasi-Boateng K, et al. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PloS One (2016) 11(5):e0155189. doi: 10.1371/journal.pone.0155189
68. Bedognetti D, Spivey TL, Zhao Y, Uccellini L, Tomei S, Dudley ME, et al. CXCR3/CCR5 Pathways in Metastatic Melanoma Patients Treated With Adoptive Therapy and Interleukin-2. Br J Cancer (2013) 109:2412. doi: 10.1038/bjc.2013.557
69. Chen X, Wang Y, Nelson D, Tian S, Mulvey E, Patel B, et al. CCL2/CCR2 Regulates the Tumor Microenvironment in HER-2/Neu-Driven Mammary Carcinomas in Mice. PloS One (2016) 11(11):e0165595. doi: 10.1371/journal.pone.0165595
70. Chheda ZS, Sharma RK, Jala VR, Luster AD, Haribabu B. Chemoattractant Receptors BLT1 and CXCR3 Regulate Antitumor Immunity by Facilitating CD8(+) T Cell Migration Into Tumors. J Immunol (Baltimore Md: 1950) (2016) 197(5):2016–26. doi: 10.4049/jimmunol.1502376
71. Connolly KA, Belt BA, Figueroa NM, Murthy A, Patel A, Kim M, et al. Increasing the Efficacy of Radiotherapy by Modulating the CCR2/CCR5 Chemokine Axes. Oncotarget (2016) 7(52):86522–35. doi: 10.18632/oncotarget.13287
72. Liu J, Li F, Ping Y, Wang L, Chen X, Wang D, et al. Local Production of the Chemokines CCL5 and CXCL10 Attracts CD8(+) T Lymphocytes Into Esophageal Squamous Cell Carcinoma. Oncotarget (2015) 6(28):24978–89. doi: 10.18632/oncotarget.4617
73. Mikucki ME, Skitzki JJ, Frelinger JG, Odunsi K, Gajewski TF, Luster AD, et al. Unlocking Tumor Vascular Barriers With CXCR3: Implications for Cancer Immunotherapy. Oncoimmunology (2016) 5(5):e1116675. doi: 10.1080/2162402X.2015.1116675
74. Ramakrishnan R, Peña-Martínez P, Agarwal P, Rodriguez-Zabala M, Chapellier M, Högberg C, et al. CXCR4 Signaling Has a CXCL12-Independent Essential Role in Murine MLL-AF9-Driven Acute Myeloid Leukemia. Cell Rep (2020) 31(8):107684. doi: 10.1016/j.celrep.2020.107684
75. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour Hypoxia Promotes Tolerance and Angiogenesis via CCL28 and T(reg) Cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
76. Tubo NJ, McLachlan JB, Campbell JJ. Chemokine Receptor Requirements for Epidermal T-Cell Trafficking. Am J Pathol (2011) 178(6):2496–503. doi: 10.1016/j.ajpath.2011.02.031
77. Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular Profiling to Identify Relevant Immune Resistance Mechanisms in the Tumor Microenvironment. Curr Opin Immunol (2011) 23(2):286–92. doi: 10.1016/j.coi.2010.11.013
78. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient Rest Restores Functionality in Exhausted CAR-T Cells Through Epigenetic Remodeling. Science (2021) 372(6537):eaba1786. doi: 10.1126/science.aba1786
79. Carr T, Krishnamoorthy V, Yu S, Xue H-H, Kee BL, Verykokakis M. The Transcription Factor Lymphoid Enhancer Factor 1 Controls Invariant Natural Killer T Cell Expansion and Th2-Type Effector Differentiation. J Exp Med (2015) 212(5):793–807. doi: 10.1084/jem.20141849
80. Reya T, O’Riordan M, Okamura R, Devaney E, Willert K, Nusse R, et al. Wnt Signaling Regulates B Lymphocyte Proliferation Through a LEF-1 Dependent Mechanism. Immunity (2000) 13(1):15–24. doi: 10.1016/S1074-7613(00)00004-2
81. Elsaesser H, Sauer K, Brooks DG. IL-21 Is Required to Control Chronic Viral Infection. Science (2009) 324(5934):1569–72. doi: 10.1126/science.1174182
Keywords: Cbx3/HP1γ, LEF-1, IL-21 receptor, CD8+ T-cell persistence, ovarian cancer, melanoma
Citation: Le PT, Ha N, Tran NK, Newman AG, Esselen KM, Dalrymple JL, Schmelz EM, Bhandoola A, Xue H-H, Singh PB and Thai T-H (2021) Targeting Cbx3/HP1γ Induces LEF-1 and IL-21R to Promote Tumor-Infiltrating CD8 T-Cell Persistence. Front. Immunol. 12:738958. doi: 10.3389/fimmu.2021.738958
Received: 09 July 2021; Accepted: 08 September 2021;
Published: 06 October 2021.
Edited by:
William L. Redmond, Earle A. Chiles Research Institute, United StatesReviewed by:
Elena Jachetti, Istituto Nazionale dei Tumori (IRCCS), ItalyXin Yu, Amgen, United States
Copyright © 2021 Le, Ha, Tran, Newman, Esselen, Dalrymple, Schmelz, Bhandoola, Xue, Singh and Thai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: To-Ha Thai, dHRoYWlAYmlkbWMuaGFydmFyZC5lZHU=
†Present address: Ngoc Ha, Department of Physiology, Anatomy and Genetics, Oxford University, Oxford, Oxfordshire, United Kingdom Ngan K. Tran, Center for Drug Discovery, Department of Pharmaceutical Sciences, Northeastern University, Boston, MA, United States
‡These authors have contributed equally to this work