 Stephan En Jie Chee
Stephan En Jie Chee Egle Solito
Egle Solito- 1Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, United Kingdom
- 2William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, United Kingdom
- 3Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Federico II University, Naples, Italy
Alzheimer’s Disease (AD) is a progressive neurodegenerative disease strongly associated with increasing age. Neuroinflammation and the accumulation of amyloid protein are amongst the hallmarks of this disease and most translational research to date has focused on targeting these two processes. However, the exact etiology of AD remains to be fully elucidated. When compared alongside, the immune response in AD closely resembles the central nervous system (CNS) immune changes seen in elderly individuals. It is possible that AD is a pathological consequence of an aged immune system secondary to chronic stimulation by a previous or ongoing insult. Pathological changes like amyloid accumulation and neuronal cell death may reflect this process of immunosenescence as the CNS immune system fails to maintain homeostasis in the CNS. It is likely that future treatments designed to modulate the aged immune system may prove beneficial in altering the disease course. The development of new tests for appropriate biomarkers would also be essential in screening for patients most likely to benefit from such treatments.
Introduction
Epidemiology
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized clinically by memory and cognitive impairments initially, and further neurocognitive deterioration with time (1). It is the most common cause of dementia, estimated to account for 60-70% of cases worldwide (2), and is strongly associated with increasing age, with about 10% of people aged 65 years and over and 32% of those over 85 years of age in the US having AD (2). Most cases are late onset AD (LOAD) and albeit being associated with various risk factors, most prominently age, family history in a first degree relative and specific genotypes, they are considered sporadic and idiopathic (1). Other types of AD include familial AD, early onset AD (EAOD) and AD associated with Down’s syndrome (DS), although these types are rare and constitute only a small proportion of cases (1).
Pathological Features of AD and the Aging CNS
Some of the classical hallmarks of AD pathology include amyloid β (Aβ) accumulation and neurofibrillary pathology. Aβ monomers are derived from the cleavage of amyloid precursor protein (APP), a membrane protein concentrated in neurons in the brain and whose function remains to be elucidated (3, 4). These monomers then aggregate to form Aβ oligomers which in turn form insoluble amyloid plaques extracellularly which are neurotoxic (5). It is worth noting that whilst much attention has traditionally been given to amyloid plaques, Aβ oligomers are now considered to be the most neurotoxic form (6).
Another hallmark of AD is the neurofibrillary pathology due to abnormal aggregation of tau protein. These proteins are normally modified physiologically to stabilize microtubules, coordinate axonal transport and maintain DNA integrity (5). As such, the pathological modification of tau in AD can lead to impaired neuronal health and loss (5).
Nine hallmarks of aging that characterizes the aging process have been described by Lopez-Otin et al. (7). Of particular note is the loss of proteostasis, whereby the various mechanisms that normally ensures the stabilization of correctly folded proteins and the degradation and removal of misfolded proteins are altered (7). For example, age-related LOAD has been demonstrated to have higher than normal expression of the chaperone heat-shock protein (HSP) 27 in degenerating areas of AD brains, and that this is thought to be a compensatory response to a proteotoxic CNS environment (8–11). Another important hallmark of note is altered intercellular communication, which includes changes in immunosurveillance and an inflammatory state resulting in “inflammaging” (7). For example, inflammaging can arise from various causes such as the accumulation of proinflammatory tissue damage and an increasingly dysfunctional immune system for maintaining homeostasis, resulting in the upregulation of pro-inflammatory pathways such as the NLRP3 inflammasome (12). As Aβ accumulation and neuroinflammation are amongst the key pathological features of AD, such age-related hallmarks suggest that AD pathogenesis might reflect a pathological consequence of the normal aging process.
CNS Immune Response in AD
Microglia
One immune cell type within the CNS that has been the subject of much research into neurodegenerative disease is the microglia. These are tissue-resident macrophages that are epigenetically and functionally different to peripheral monocytes and macrophages, and play an essential role in maintaining neuronal health and homeostasis. In the resting state, microglia extend various processes that allow it to monitor the neuronal environment, and alter their morphology and gene expression in response to different stimuli (13). Such stimuli include damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), ATP released by neuronal damage, cell debris and abnormal proteins such as Aβ amyloid (5, 13). The mechanisms employed by activated microglia to these stimuli are diverse, ranging from phagocytosis to the release of numerous cytokines and chemokines (14, 15). Given that microglia constitute part of the CNS immune system, AD pathogenesis has been suggested and traditionally believed to be due to inappropriate neuroinflammation. For example, microglia are commonly observed aggregating around Aβ plaques in response to chemotaxis from Aβ deposits (16). In addition, these microglia express increased levels of reactive oxygen species (ROS) production that inherently damage surrounding neurons. These activated microglia also express upregulated major histocompatibility complex (MHC) class II levels and increased secretion of pro-inflammatory cytokines interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF) and IL-8 (17, 18). Such findings support the theory that microglia may have a pro-inflammatory role in the perpetuation of AD.
However, as mentioned previously, microglia also play an important role in maintaining homeostasis in the neuronal environment and it has increasingly been suggested that AD pathogenesis occurs secondary to microglial dysfunction. For example, microglial dystrophy is a morphologically distinct phenotype to that of activated and resting microglia and includes features such as cytoplasmic fragmentation (19). This is characteristic of microglia which are abundant in areas of the AD brain showing high levels of neurofibrillary pathology (19). It is also worth noting that different microglial phenotypes exist in AD which are associated with and reflect an accelerated aging process and direct phagocytosis of Aβ plaques (20). This has led to the hypothesis that the AD microenvironment accelerates the transcriptional trajectory of aging microglia, but which can be altered upon phagocytosis of synaptic components around Aβ plaques – a process mediated by and reinforced by the Hif1a regulon (20).
In addition, the presence of these dystrophic microglia has been observed to precede that of the neuritic plaques (19). One manner which microglia may contribute to these plaques is through the inflammasome-mediated recruitment of the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) (21). For example, ASC was found to promote Aβ aggregation in AD patients and mice models, and to promote Aβ deposition in mice models of AD (21).
Brain regions showing elevated levels of dysmorphic microglia have also been observed to have increased cellular debris accompanied by an absence of brain macrophages, suggesting that microglial dystrophy might indicate an impairment of microglial phagocytic function and possibly microglial loss (22). It has also been shown that microglia play an essential role in synaptic pruning through various mechanisms like modifying the perisynaptic environment, remodeling and phagocytosing dendritic spines and axon terminals, and coordinates closely with astrocytes in synaptic pruning (23, 24). As such, microglial phagocytic dysfunction might also adversely affect synaptic pruning. It is also well-documented that neuronal synapse loss occurring early in AD is due to dysfunctional complement-mediated pruning of synapses by microglia and that this is strongly correlated with cognitive decline (25, 26).
Aβ deposits have also been observed to accumulate in the absence of microglia in transgenic mice, suggesting the role of microglia in removing Aβ deposits (27). As such, it has been postulated that microglial neurodegeneration results in the disturbed neuronal environment of tau and Aβ accumulation characteristic of AD pathophysiology (28).
Astrocytes
Another CNS immune cell type that has been implicated in AD is the astrocyte. Similar to microglia, astrocytes normally serve to maintain homeostasis within the neuronal environment. Some of the functions include maintaining the integrity of neuronal synapses via neurotrophic and metabolic support, maintaining the blood-brain barrier (BBB) with endothelial cells as part of the neurovascular unit (NVU), regulating cerebral blood flow, and removing neurotoxic waste products such as tau and amyloid via the glymphatic system (5, 18). Astrocytes are also attracted to and migrate towards Aβ deposits with evidence that there are attempts by these cells to remove them (5). The findings that Aβ has been found in astrocytes around Aβ deposits and that lower Aβ levels are associated with increased local astrocyte population suggest an active role played by astrocytes in the removal and degradation of Aβ deposits (29, 30). They have also been reported to adopt a pro-inflammatory phenotype in response to Aβ deposits, characterized by the release of various pro-inflammatory cytokines similar to those produced by activated microglia, a loss of their homeostatic function and an ability to induce apoptosis in neurons and oligodendrocytes (18, 31). As such, similarly to microglia, either an inappropriate neuroinflammatory response by astrocytes or astrocytic dysfunction may contribute to the pathogenesis of AD. It is also worth noting that astrocytes that accumulate Aβ can lyse to produce Aβ plaques, likely potentiating the pathological course of AD (32). It is also worth noting that IL-3 produced by astrocytes has recently been implicated as potentially having protective benefits in AD, with increased Aβ burden and worsening of memory impairment being recorded in knockout mice models of AD (33).
The role of astrocytes in maintaining the BBB and the glymphatic system has in recent years been particularly explored as a key contributor to AD pathogenesis. One of the features of AD is an impairment of the BBB, which is associated with reduced cerebral blood flow. This cerebrovascular dysfunction and pathology have also been associated with cognitive decline, neuronal loss, Aβ and tau pathology (34, 35). In addition, cerebral amyloid angiopathy (CAA) is considered one of the pathological hallmarks of AD and has also been observed to precede Aβ and tau pathology in a preclinical study (36, 37). The finding that the tight configuration of astrocytic end-feet that surround endothelial cells is altered with Aβ deposition in capillaries also suggest a possible general impairment of astrocytic capacity for regulating blood flow, in turn likely contributing to cognitive decline (34, 38). Furthermore, this impairment of the BBB with CAA also results in increased leakage across the BBB, including increased extravasation of peripheral immune cells into the CNS which is thought to contribute to AD pathogenesis (39). For example, reduced tight junction proteins and other intercellular structural proteins in endothelial cells have been observed with CAA (40). Aβ pathology of the BBB is also associated with overexpression of adhesion molecules like vascular adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1), facilitating the migration of peripheral leukocytes into the CNS (41).
In the same vein, astrocytic dysfunction has been implicated in the impairment of the recently described glymphatic system responsible for removing metabolic waste products including Aβ (42). The glymphatic system comprises a paravascular channel through which cerebrospinal fluid (CSF) from the subarachnoid space can flow and directly mix with interstitial fluid (ISF) in the brain parenchyma before returning as a CSF-ISF mixture to the subarachnoid space (42). As such, the glymphatic system plays an important role in removing metabolic waste products including Aβ (42). Central to this are astrocytes that regulate the flow of CSF between the paravascular spaces and the interstitium mainly via aquaporin 4 (AQP4) channels located mostly on astrocytic end-feet encapsulating the blood vessels (43). However, a loss of this polarization of AQP4 channels from the end-feet to the parenchymal processes instead has been observed in AD (44). It still yet remains to be elucidated if Aβ accumulation is a consequence of this polarization of AQP4 channels or if this polarization is secondary to Aβ pathology (43, 45, 46).
Adaptive Immune System
Research into the role of the CNS adaptive immune system in AD pathogenesis has been increasing in recent years, albeit slowly, and at present this topic remains poorly understood. Post-mortem brain tissue of AD patients has been reported to show increased numbers of T lymphocytes than those of healthy controls (47). In particular, most of these cells were found to be CD8+ T cells and were significantly correlated with tauopathy but not Aβ, suggesting a role for CD8+ T cells in neurofibrillary pathology (48). Increased CD8+ T cell numbers in the peripheral blood of AD patients has been reported, and of which are comprised mostly of clonal T cells expressing T cell receptors (TCR) with affinity to Ebstein-Barr Virus (EBV) antigens (49). With the majority of the CNS T cell population originating in the peripheral circulation and entering through the BBB, it has been posited that the CNS immune response in AD might be associated with a previous herpes virus infection or a reactivation of it (50, 51). However, a meta-analysis has recently shown that current evidence supporting either of these theories is inconsistent (52). It is also worth noting that T cell clonality and responses differ between compartments of the aging brain. For example, a population of clonally expanded T cells that are distinct from those in the peripheral blood have been reported in the aged brain and produce an IFNγ response which inhibits neural stem cell (NSC) proliferation, possibly contributing to the decline in NSCs during aging (53). Additionally, CNS-specific CD4+ effector memory-type T cells have been reported to reside in the choroid plexus (CP) epithelium which exhibit a Th2-like inflammatory response in the aged brain, with increased IL-4 production and decreased IFNγ production (54). The increased levels of IL-4 have been associated with increased production of the chemokine CCL11 that is in turn associated with age-related cognitive impairments (55). The raised IL-4 levels are also thought to impair brain plasticity by contributing to the dysregulation of brain derived growth factor (BDNF) and other neurotrophic factors (54).
Th17 cells, like CD8+ T cells, have also been implicated in AD, with increased Th17 infiltration and upregulation of IL-17 and IL-22 in the hippocampus, blood and CSF being observed following the injection of Aβ into the hippocampi and induction of AD in rats (56). Additionally, IL-17 has been found to inhibit neurogenesis in the hippocampi of mice whilst genetic deletion of IL-17 promoted neurogenesis, possibly implicating Th17 cells in AD pathogenesis (57). T regulatory (Treg) cells have also been implicated in contributing to the pathogenesis of AD, albeit their exact role remains to be fully elucidated. For example, transient depletion of Treg cells have been associated with an IFNγ-dependent systemic immune response characterized by the peripheral recruitment of monocytes via the choroid plexus, and this process in turn was associated with increased Aβ plaque clearance and improvement in cognition in mice models of AD (58, 59). However, current studies showing conflicting evidence on the role of Treg cells as either beneficial or detrimental disallows any concrete conclusions on their contributions to AD pathogenesis (59–62).
Inflammasome
The role of inflammasomes in AD pathogenesis is currently an area of increasing research interest. Whilst various types of inflammasomes exist, NLRP3 in particular has been implicated in AD. The NLRP3 inflammasome is a protein complex comprised of the NLRP3 sensor molecule, the adaptor protein ASC and pro-caspase-1 and is found exclusively intracellularly in microglia (63). One of its main functions is the generation of active IL-1β and IL-18 from their precursors in response to DAMPs, chiefly Aβ in AD (63, 64). NLRP3 levels have been found to be increased in AD brains (65). Of particular interest, loss of NLRP3 inflammasome activation in murine models of AD is associated with decreased Aβ deposition and IL-1β levels, and increased phagocytic activity (65). Additionally, it has also been shown that IL-1β can inhibit Aβ clearance (63). Therefore, these findings suggest that NLRP3 play a significant role in the neuroinflammatory process in AD.
Meninges, Choroid Plexus, and Skull Bone Marrow
There has in recent years been an increasing interest and appreciation of various neuroimmune interfaces in AD pathology, namely the meninges, choroid plexus (CP) and skull bone marrow. The (re)discovery of the meningeal lymphatic system has led to the hypothesis that the dysfunction of these vessels contributes to AD pathology through impaired clearing of Aβ and consequently increased Aβ deposition (66–68). Supporting evidence for this includes the finding that macromolecules are drained into the cervical lymph nodes from the CNS via these lymphatic vessels, and that treatment of aged mice with vascular endothelial growth factor (VEGF) C improves drainage of macromolecules from the CSF and improves cognitive performance (68, 69). Conversely, the loss of these lymphatic vessels in transgenic mice led to increased Aβ deposition, and cognitive and behavioral symptoms (68, 69). In addition to these lymphatic vessels, the meninges serve as an important neuroimmune interface for modulating local immune responses. For example, the loss of C-C chemokine receptor (CCR) 7 in transgenic mice led to increased meningeal Treg responses, attenuated meningeal effector T cell responses, and cognitive deficits (59).
It has also recently been reported that a network of direct vascular channels linking the skull bone marrow and the meninges exists and serves as an important migratory route for the recruitment of myeloid cells into the brain (70, 71). Given the salient role of microglia in AD pathogenesis, these findings suggest that the skull bone marrow serves as an important source of peripherally-derived monocytes and microglia that contribute to the disease process (70). Additionally, it has recently been reported that the majority of myeloid cells in the meninges are derived from the skull bone marrow both in homeostatic and pathological states (72). Of note as well is the role of the skull bone marrow as a source of B cells to the meninges, with these developing B cells travelling via the direct vascular network to the meninges to complete their maturation (73). It is worth noting however that the role of B cells in AD pathogenesis still remains elusive. For example, in a recent study by (74), mice models lacking B cells and antibody responses were associated with the activation of endogenous retroviruses in the hippocampi which in turn was associated with memory impairments (74). Conversely, whilst B cells have been shown to produce immunoglobulins and cytokines that reduce Aβ plaques and ameliorate AD pathology, a recent study has reported an improvement in behavioral and memory symptoms with the genetic loss and transient depletion of B cells (75).
The CP is another neuroimmune interface that plays an important role in modulating the immune response in the CNS. For example, it has been shown that the CP integrates peripheral immune signals with those from the CNS, and is particularly important as a trafficking route for anti-inflammatory cells into the CNS, potentially influencing AD pathogenesis (76). It is likely that CP functioning is dependent upon a complex interplay between various cellular players including CP mesenchymal and epithelial cells, summarized by Dani et al. (77). In addition, it has recently been reported that various myeloid immune cell subset populations of microglia and dendritic cells reside in the CP and meninges and is summarized by Mrdjen et al. (78). However, much remains to be elucidated about the exact role of the CP in AD and the roles of these cells in health and disease (77, 78). Recently also the dural sinuses have been reported to be an important neuroimmune interface where CNS-derived antigens in the CSF are sampled by local antigen presenting cells (APCs) and presented to patrolling T cells (79). However as with the CP, much remains to be elucidated about its significance in various neurodegenerative pathologies including AD.
Age-Related Changes in the CNS Immune Response
Impaired Adaptive Immunity
Due to various age-related changes such as thymic involution, there is a decline in T cell numbers with age (80). However, more notably, there is a decrease in the number of naïve T cells and an increase in age-experienced cells, particularly the CD28-CD8+ T cell population (80, 81). These cells are only seen in the elderly compared to neonates and display an impaired functional phenotype, including a reduced proliferative response to TCR stimulation with a normal or augmented cytotoxic capacity and a resistance to apoptosis (82, 83). It is thought that these cells represent a population that has undergone terminal differentiation as a result of chronic stimulation by persistent infection with common pathogens such as cytomegalovirus (CMV) (80). This notion is supported by the finding that increased CD28-CD8+ T cell numbers are seen in patients with viral infections like CMV, and that naïve T cells are reduced in the elderly (80, 82). CD8+ T cells in the elderly generally also show significantly reduced interferon gamma (IFNγ) production and a reduced antigen-recognition repertoire by TCRs (80, 83). Therefore, the implication of this is two-fold: firstly, there is reduced adaptive immunity to previously encountered pathogens, and secondly that there is a reduced ability for the adaptive immune system to respond to novel pathogens (80). It is also possible that impaired IFN production in the elderly adversely affects the ability of the immune system to clear viral infections such as CMV and regulate neuroinflammation, resulting in a dysfunctional immune response that includes an impaired resolution of neuroinflammation by macrophages (84, 85). It is also worth noting that the clonal expansion of CD28-CD8+ T cells has been directly implicated in the increased infection rates and poor vaccine responses in the elderly (82).
CD4+ T cell function in the elderly is also altered but depends largely on when in the host’s lifetime naïve and memory CD4+ T cells are generated. Naïve CD4+ T cells from elderly humans and mice have been shown to display reduced responsiveness to TCR stimulation, and reduced IL-2 production and proliferation when presented with antigen compared to naïve CD4+ T cells from younger hosts (86). Naïve CD4+ T cells from elderly mice have also been shown to produce a poorer humoral immune response via the generation of germinal centers for B cells compared to those from younger hosts (87). Memory CD4+ T cells extracted from healthy elderly humans and mice have also been shown to respond normally to antigens and those generated early in life respond well to antigens over time (80). As such, it has been hypothesized that reduced memory CD4+ T cell function in old age might be due to their derivation from aged naïve CD4+ T cells that exhibit reduced clonal diversity and capacity for proliferation (80).
In addition to the impaired ability of CD4+ T cells to support the humoral immune response, other changes in humoral immunity are seen in the elderly. Similar to T cells, B cell production and numbers are reduced in the elderly along with reduced serum immunoglobulin levels (80, 88). The immunoglobulins produced also have altered specificities and affinities for antigens, with an increasing proportion of serum immunoglobulins in the elderly displaying specificities to autoantigens (89). These immunoglobulins are also low affinity IgM, reflecting an impaired capacity for class switching in B cells (80). This might also be a consequence of altered proportions of B cell subpopulations and an impaired ability for germinal center generation secondary to an impaired CD4+ T cell function (80).
Impaired Innate Immunity
One key characteristic of aging microglia is their propensity for pro-inflammatory cytokine production (90). Several mechanisms have been proposed for this phenotype. Firstly, age-related neuronal damage may result in a loss of inhibitory signals to microglia (90). Secondly, the accumulation of misfolded proteins like Aβ as a consequence of normal aging promotes a pro-inflammatory response as described earlier. Finally, chronic exposure to increased transforming growth factor β (TGFβ) might impair microglial capacity for anti-inflammatory cytokine production, possibly through the downregulation of transcription factors important for switching to an anti-inflammatory phenotype (90). Aging microglia have also been demonstrated to show reduced phagocytic activity for Aβ and neuronal waste products like myelin debris (91, 92). In addition, aging microglia show reduced chemotactic capacity in response to neuronal damage (93).
Neuroinflammation and Th1 Skewing of the Immune Response in AD
Given the age-related changes in the CNS immune system and the pathogenesis of AD described earlier, it seems that neuroinflammation and Th1 skewing of the immune response play a significant role in the pathogenesis of AD. Two dominant phenotypes of CD4+ T cells are the Th1 and Th2 types. Th1 cells are largely responsible for cytotoxic cell-mediated pro-inflammatory responses through coordinating other players such as CD8+ T cells and are essential for clearing intracellular infections (eg. viruses) and malignant cells. Th2 cells are largely responsible for humoral immunity through coordinating antibody generation by B cells (94). As described earlier, there is an increased number of CD8+ T cells, particularly CD8+ T CD28- cells, in the brains of AD patients (80). Memory CD8+ T cells can contribute to the control of CNS infections (95, 96). However, it remains unknown whether and how each subset contributes to CNS immunity. In addition, the presence of these cells positively correlates with the poor antibody response to the influenza vaccine in the elderly (95, 97). These cells also appear to produce high levels of IFNγ and low IL-5 levels, and promote a Th1 cytokine response by CD4+ T cells in response to auto antigenic stimulation (95). It is postulated that the chronic and ubiquitous exposure to autoantigens underpin the prevalence of these CD28-CD8+ clones and the high IFNγ levels in old age (95). Of note as well are the reduced rates of various cancers in AD patients, lending strength to the notion of Th1 skewing in AD pathology (98). In a mouse model of AD, the peripheral administration of antibodies against Aβ has also been shown to induce clearance of Aβ and reduce Aβ burden, suggesting that reduced antibody generation in the elderly might contribute to AD pathogenesis (99). This Th1 skewing of the immune response, namely the production of IFNγ and the reduced antibody production, could result in increased activation of microglia (94). IFNγ working in concert with TNF also triggers Aβ production in neuronal and extra neuronal cells and increased reactive oxygen species (ROS) production by microglia, likely perpetuating AD pathogenesis (80).
There is a recent concept of microglial priming where previous and chronic disturbances in the CNS environment pushes microglia to a phenotype with more marked pro-inflammatory responses than the transient activated phenotype adopted during brief episodes of physiological insults (100). These insults include age-related oxidative stress and DNA damage, traumatic brain injuries (TBI) and CNS infections that ultimately induces a state of neuroinflammation required for microglial priming (100). With respect to microglial phenotypes, they are typically described in vivo as having either a pro-inflammatory activated phenotype associated with increased ROS and pro-inflammatory cytokine production and reduced neurotropic factor production, or a non-activated phenotype associated with increased anti-inflammatory cytokine production (eg. IL-10, TGFβ) and phagocytic activity (100, 101). In the case of primed microglia, these cells undergo morphological and functional changes when exposed to chronic stimulation to resemble a pro-inflammatory activated phenotype (102). However, they are characterized by a higher baseline production of inflammatory markers and mediators, a lower threshold for activations and switching to a pro-inflammatory state, and an exaggerated inflammatory response (102). The state of chronic neuroinflammation that follows can lead to the loss of neurons and the development and deposition of neurofibrillary tangles (NFT) and Aβ (100, 102). This as such might lead to a self-perpetuating cycle of Aβ deposition secondary to the accumulation of primed microglia which in turn stimulate a pro-inflammatory response by microglia. In addition, the reduced clearance of Aβ by mechanisms such as reduced microglial phagocytic capacity, increased IFNγ production and reduced antibody generation in the elderly may further exacerbate this neuroinflammation towards symptomatic AD (Figure 1). Comparisons between the immune system of a healthy individual and that of the elderly and AD patients are summarized in Figure 1.
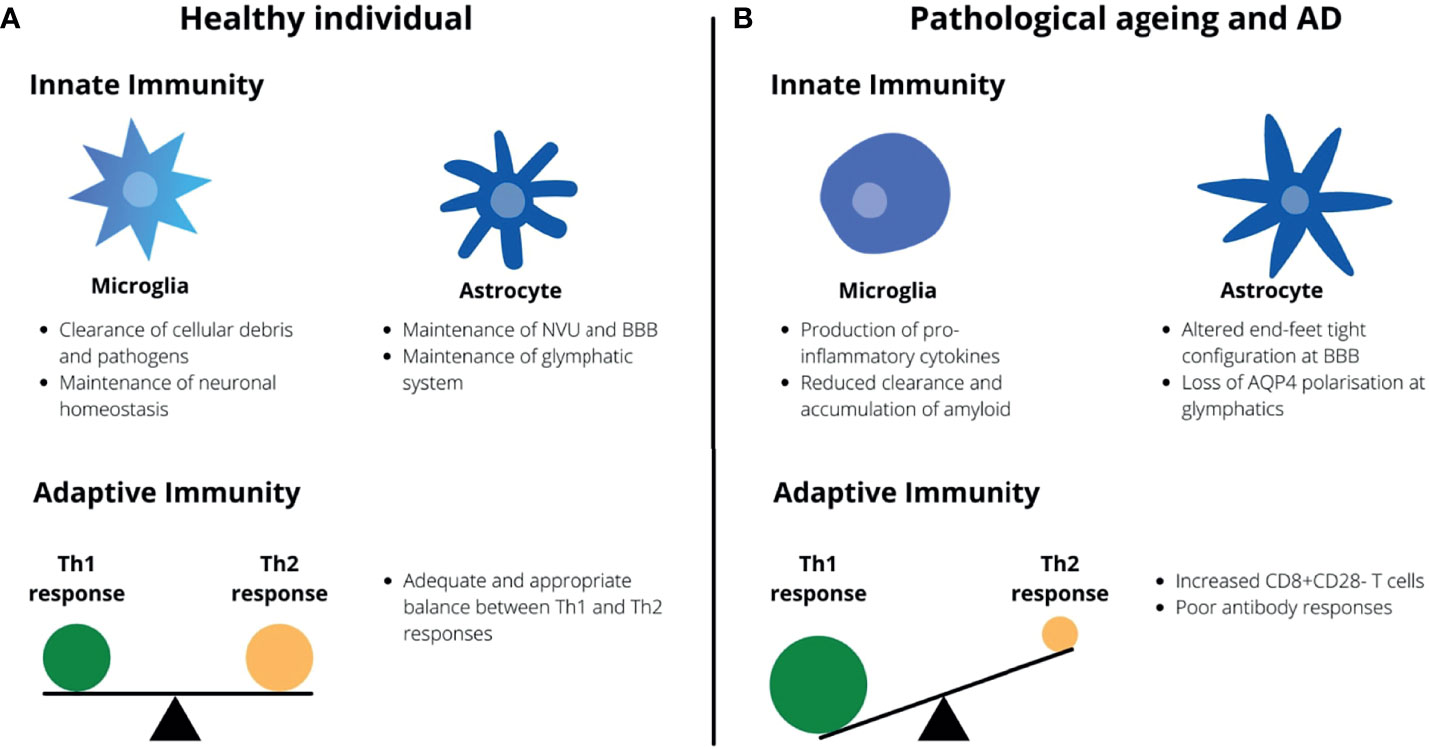
Figure 1 Healthy aging vs. pathological aging and AD. (A) In healthy individuals, microglia serve to provide neurotropic support and maintain neuronal homeostasis whilst astrocytes maintain the BBB and glymphatic system. There is also an appropriate balance between Th1 and Th2 responses by the adaptive immunity. (B) In AD and aging, microglia undergo morphological changes and adopt a pro-inflammatory phenotype whilst the capacity for astrocytes to maintain the BBB and glymphatic system is impaired. There is a Th1-skewing of the adaptive immune response with an accumulation of CD8+CD28- T cells.
There is evidence however that the neuroinflammatory process differs throughout the time course of AD pathogenesis. In a study by Vlad et. al., the risk of developing AD gradually reduced in proportion to the duration of NSAID therapy, reaching to levels almost half that of the general population after a few years (103). Subsequent studies have found similar results (104, 105). However, a Cochrane study has found no significant therapeutic benefit of anti-inflammatory drugs in the treatment of symptomatic AD (106). Supporting this notion of a changing neuroinflammatory process is the finding that oxidative damage is greatest early in AD and decreases with disease progression (80). It has been suggested as such that as AD worsens, Aβ deposition increases alongside compensatory changes to reduce oxidative damage, which might include dysregulated T cell responses (80).
It seems therefore that the pathogenesis of AD could potentially be divided into three broad stages. In the first instance, there is a period of marked neuroinflammation following physiological insults. This then progresses to the second stage characterized by a self-perpetuating state of chronic stimulation and neuroinflammation that potentially promotes the accumulation of primed microglia and a pro-inflammatory environment. This chronic neuroinflammation and stimulation, caused either by a transient and brief insult like a TBI or by chronic infection by viruses like CMV, pushes the CNS immune system towards immunosenescence, the third stage and final stage, where its ability to adequately maintain the homeostatic neuronal environment is impaired (Figure 2). Cellular senescence is characterized by the inability of cells to proliferate, this process being related to the erosion of telomere (107). Moreover, senescence is characterized by 3 main phases: the induction phase (characterized by telomere shortening, DNA damage, growth factor deprivation); the second phase – DNA damage response (characterized by DNA damage response); and phase 3 – growth arrest (signal transduction molecules p53, p21 trigger growth arrest). However not all primary cells senesce in the same way, for instance fibroblasts present a pre-senescence phase that may be reversible. In contrast T cell senescence does not seem related to shortening of telomere, and they die when they differentiate (107). Whatever the mechanism is, aging (p53 up regulation, mitochondrial dysfunction, DNA damage) and inflammation represent a two-way system in neurodegeneration.
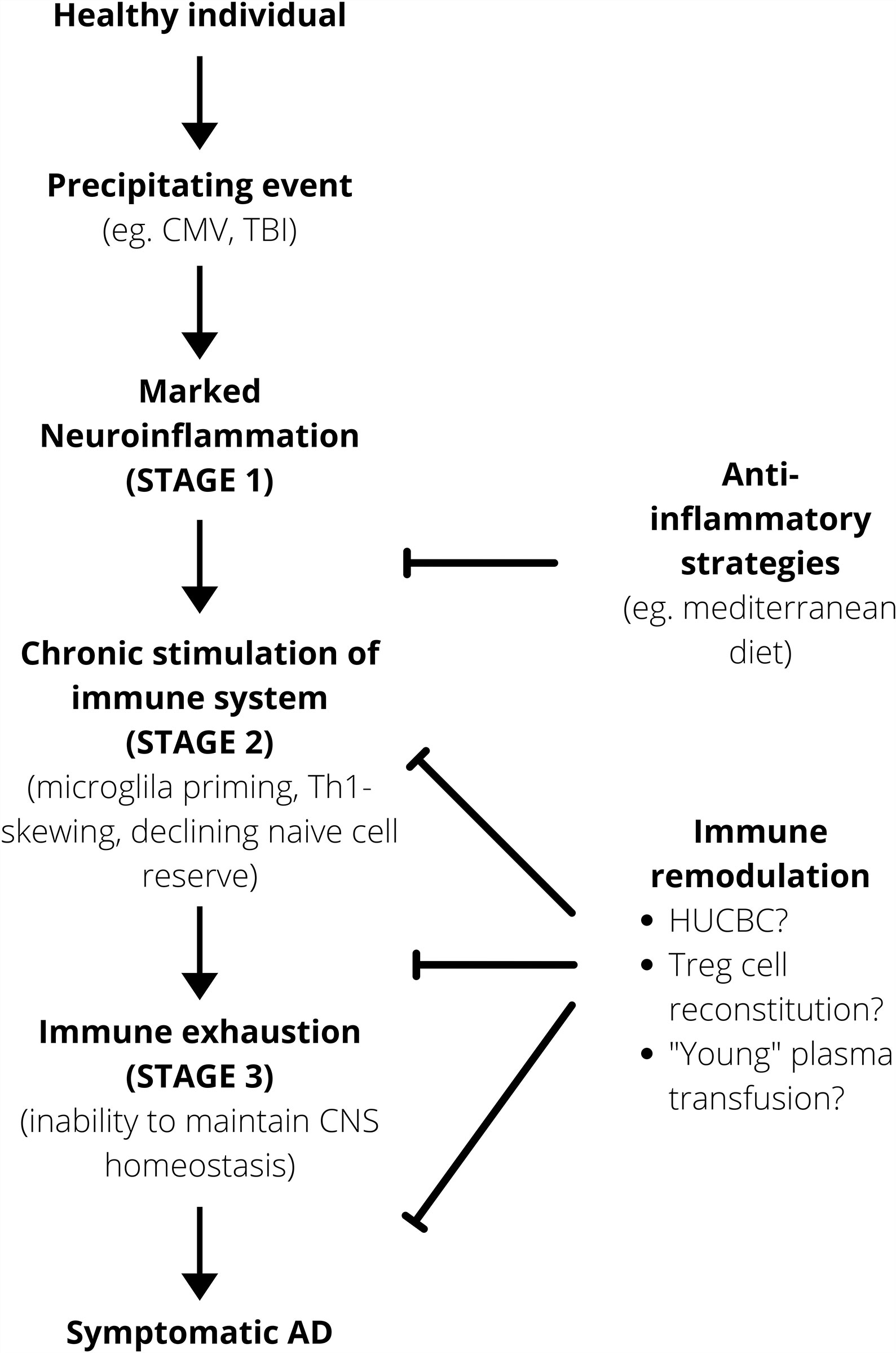
Figure 2 Working model for AD pathogenesis. Anti-inflammatory strategies are important prophylactically to minimize that risk of the immune system progressing onto a self-perpetuating path towards immunosenescence. After a certain point, immune remodulation is required to alter the disease course. HUCBC, Human Umbilical Cord Blood Cell.
Post-mortem brains of patients without a history of dementia have also been found to have sufficient Aβ and NFT amounts that would have otherwise prompted a diagnosis of AD, supporting the role of a dysfunctional CNS immune response in determining the onset of symptomatic AD (80, 108). One population that has been studied particularly with regards to health in old age are centenarians. Various anti-inflammatory mechanisms such as increased cortisol and IL-10 levels are well documented (80). This might enable centenarians to suppress neuroinflammation in the early stages before chronic neuroinflammation and significant CNS immune dysfunction develops. Interestingly, it has also been observed that administration of IFNβ-1, an antiviral agent, in patients with early AD for twenty-eight weeks resulted in significant improvements in some cognitive functions but not in inhibiting the disease progression (109). This raises the possibility of early antiviral treatment as a potential novel therapeutic option in early AD (109). It is also worth noting that whilst the role of Treg cells in AD pathogenesis remain elusive, centenarians have been noted to have increased numbers of Treg cells and that this positively correlated to improved survival outcomes (58, 110). It is possible that Treg and anti-inflammatory responses may differ significantly between mice models of AD and AD in humans in a manner that is currently poorly understood.
Anti-inflammatories and Aβ Vaccines as Therapeutic Options for AD
Given the association between Aβ and AD, trials exploring the therapeutic potential Aβ vaccines have been conducted. The first attempt at developing an Aβ vaccine was by Schenk et al. who prepared a vaccine from Aβ1-42 peptide and Freud’s adjuvant (111). When tested on murine models of AD, the vaccine was found to confer both significant prophylactic and therapeutic benefits in the prevention of Aβ plaque formation and in inhibiting the progression and extent of Aβ deposition respectively (111). This vaccine was then developed for clinical trials but did not progress beyond phase II due to a proportion of patients developing aseptic meningoencephalitis (112). In a follow-up study on patients from this initial trial, post-mortem analysis of the brains of eight of the patients who had received the vaccine and had died with AD showed significant variation in Aβ burden. More notably however was the finding that in seven of the eight vaccinated patients including those with virtually complete Aβ plaque removal there was a severe end stage dementia prior to death (113). Although it is not possible to fully ascertain the therapeutic potential of therapies targeting Aβ from this study alone due to the small sample size analyzed post-mortem in the follow-up and the adverse events associated with the vaccine, it is possible however that future attempts at relieving Aβ burden may eventually reap limited rewards. This lack of association between Aβ burden and the clinical course of AD has been supported by other studies showing similar findings (108, 114). Attempts at using human intravenous immunoglobulins (IVIG) on the basis of preclinical findings showing Aβ clearance by IVIG have also failed to produce significant cognitive improvement in patients. Interestingly, however, was the finding that IVIG significantly reduced Aβ levels and slow cognitive decline in patients with the ApoE4 allele, although cognitive decline was only observed on neuropsychiatric examination with no corresponding improvement in activities of daily living (ADL) (115, 116). As such, it is possible that therapies targeting Aβ clearance may only prove efficacious for a small group of AD patients.
As described earlier, current evidence suggests a role for anti-inflammatory therapies in AD particularly as prophylaxis and treatment during the prodromal stage. At present, there is increasing interest in the inflammasome, particularly NLRP3, as a novel target for anti-inflammatory therapy research in AD. The NLRP3 inflammasome inhibitor dapansutrile (OLT1177) has been investigated for its therapeutic potential in other inflammatory conditions, having recently completed a phase II trial in treating osteoarthritis and acute gout flare (117). Given the association between NLRP3 and Aβ and pro-inflammatory responses in AD, dapansutrile has recently been investigated pre-clinically for the treatment of AD (118). In this study, dapansutrile was shown to reverse cognitive impairment, reduce microglial activation and cortical plaques, and normalize plasma metabolic markers of AD in murine models (118).
Since the success of the anti-TNF therapy infliximab in the treatment of Crohn’s disease, there has been a considerable interest in the following years in developing therapies targeting specific cytokines for various inflammatory conditions (119). One such drug currently of interest in the treatment of AD is etanercept. Similar to infliximab, etanercept is a TNF inhibitor. Various trials conducted have corroborated the positive therapeutic potential of etanercept in treating AD (120). However, in a recent phase II trial, although etanercept was found to be generally well tolerated by patients there was no statistically significant changes in cognition, behavior or global function (121). Interestingly, one mechanism by which etanercept might improve AD symptoms and neuropathology if any is that it modulates TNF regulatory function of synaptic transmission (120).
Naturally occurring anti-inflammatories and the role of certain diets have also been explored for their therapeutic potential in AD. One particular diet well-known for its anti-inflammatory properties is a Mediterranean diet (MD). This diet is characterized by large intake of fruit, vegetables, wholegrains, nuts and legumes; moderate consumption of fish, poultry and alcohol, and restricted intake of red and processed meats alongside olive oil as the main source of fat (122). Observational studies have found that greater adherence to the MD is associated with improved cognitive performance, slower cognitive decline and reduced risk of cognitive impairment and AD (122). The MD has also been associated with protective brain structures and function such as increased cortical thickness, greater brain volumes, reduced hippocampal atrophy rate and reduced Aβ accumulation (122). However, it seems that adherence to the MD for at least a few years is required to reap the cognitive benefits. In the PREDIMED study, adherence to the MD over 4-6 years was particularly associated with improved global cognition, memory and executive function (122). However, no cognitive benefits were seen in populations on the MD for up to one year, although improved global cognition and episodic memory were seen in those with greater adherence to the diet (123–125). These findings are not surprising as they reflect the findings of trials investigating the use of NSAIDs for treating AD as described earlier (103–106). It would be interesting however to explore in greater detail the role of a MD in managing symptomatic AD as it may be possible that there may be other beneficial compounds that may influence the disease course either through regulating neuroinflammation or by other pathways.
Rethinking the Role of Aβ in AD
The role of Aβ in AD pathogenesis was first proposed as part of the ‘amyloid cascade’ hypothesis following the observation that virtually all people with Down’s Syndrome (DS) will exhibit AD pathology by 40 years of age, and that there was concomitant overexpression of Aβ from the cloning of the APP gene with trisomy 21 (126). APP cloning and subsequent overexpression of Aβ also causes familial EOAD (127). However, as described earlier, post-mortem studies showing a lack of correlation between Aβ levels and clinical AD, and the current lack of therapeutic success in therapies targeting Aβ suggest that a dysfunctional immune response might play a more significant role in AD pathogenesis. In fact, people with DS typically have immune systems that are not dissimilar to that seen in AD and old age. For example, poor vaccine responses are frequently observed in DS patients alongside decreased immunoglobulin levels and B cell populations (128). Increased Th1/Th2 ratio with corresponding decreased risk of various cancers, decreased T cell proliferation and reduced naïve T cell populations are also seen (128, 129). Therefore, it is possible that a dysfunctional immune system might be the main player in the pathogenesis of AD, with Aβ likely exacerbating the process but not being a causative agent. For example, the observation that Aβ deposition is initially diffuse with gradual progression to neuritic plaques but accumulating at an exponential rate after 40 years of age in DS patients might be due to an earlier initiation and acceleration of the process driving the CNS immune system towards a state of immunosenescence resembling that seen in AD patients (126). Similarly, it is possible that overexpression of amyloid such as is seen with DS patients might be a key player driving the progression towards immunosenescence. Interestingly as well is the finding that DS individuals are more sensitive to IFN stimulation due to the overexpression of its receptor located on the extra chromosome 21 (130). It is possible that this might lead to increased activation of microglia and ROS production that in turn accelerates aging and depletion of immune reserves via chronic inflammation (131)
It has also recently been proposed that Aβ might be an innate effector molecule (132). For example, Aβ has been shown to have antimicrobial properties against viruses, bacteria and fungi and enact this by forming oligomers (133). In addition, overexpression of Aβ provides increased resistance to bacterial and viral infections (134). Aβ has also been proposed to functionally resemble that of cytokines and should be considered as such (132). This theory might account for the close spatial and temporal association between microglia and Aβ frequently observed in AD. It also strengthens the argument that AD might be brought on by CNS immunosenescence secondary to chronic stimulation like persistent CMV infection as the CNS immune system attempts to clear the infection (80).
Novel Strategies for Remodulating the CNS Immune Response
Human umbilical cord blood cells (HUCBC) have been garnering increasing interest as a potential strategy for remodulating the CNS immune response amid observations that HUCBC inhibits Th1 whilst promoting Th2 responses in murine models of stroke and neurodegenerative diseases (80, 135, 136). Hippocampal administration of HUCBC in murine models of AD have also been demonstrated to reduce neuropathological changes and improve cognitive impairment (137). Notably, there was a reduction in Aβ deposits and tau hyperphosphorylation, improved spatial learning and memory, a switching in cytokine profile exhibited by microglia from a pro-inflammatory phenotype to an anti-inflammatory one, and that these alternatively activated microglia were associated with Aβ (137). Such findings have led to the undertaking of a phase I clinical trial involving HUCBC mesenchymal stem cells (MSC) administration into the hippocampus of nine patients with mild to moderate AD (138). This method was found to be well tolerated and safe, although the efficacy was not able to be properly assessed in this study (138). Subsequent clinical trials have corroborated the safety profile of HUCBC-MSC administration into the brain. However, clinical outcomes have been less encouraging, with no significant improvement in cognition being reported (139). These clinical trials and their findings are summarized by Liu and colleagues (139). In addition to various technical issues such as the specific cell stage to be transplanted that remain to be resolved, it has been posited that the lack of clinical success with stem cell therapy research for AD could be due to inappropriate timing of stem cell transplantation (139). Although CSF biomarkers have strong predictive value in diagnosing or excluding AD in patients with mild dementia, individual clinical courses tend to be more variable which may reflect different stages and severity of neuropathology that may render subsequent stem cell transplantation ineffective (139).
Another strategy that might be useful to explore would be the reconstitution of Treg cells in AD patients. It has recently been shown that Treg anti-inflammatory function is impaired in patients with AD, but had increased suppressive activity on effector T cell proliferation and macrophage activation following ex vivo clonal expansion (140). This suggests that Treg function and immunophenotype are impaired in AD, but that administration of ex vivo clonally expanded of Tregs might be a viable therapeutic option (140). More recently, in vivo expansion of Tregs has been increasingly explored as a preferred method to ex vivo expansion, one main reason being that it bypasses the major obstacle of low Treg frequencies in lymphoid tissues that as such requires in vitro expansion prior to adoptive transfer (140). In vivo expansion of Tregs can be achieved via administration of low-dose IL-2 (141). IL-2 therapy has been shown to reduce Aβ load and improve neuroplasticity that was associated with recovery of memory deficits (60, 141). However, due to the short t1/2 of IL-2, one method that may help to overcome this is the co-administration of IL-2 with an IL-2 specific monoclonal antibody, such as JES6-1A12, which increases its t1/2 (141).
Findings such as the functional recovery of Tregs following ex vivo expansion in AD patients suggest that strategies tailored toward alteration of the AD microenvironment may help restore immune balance. One novel strategy could be in the form of transfusions of plasma or specific plasma constituents from young donors to elderly or AD recipients. Murine models of plasma transfusion including parabiosis studies have shown improvement in both neuropathological changes with reduction of Aβ and tau burden and improvement in cognition (142, 143). Various clinical trials exploring different types of therapeutic plasma exchange (TPE) in patients with established AD have generally yielded promising results, the findings of which are summarized by Imbimbo and colleagues (144). However, concrete conclusions are difficult to draw at this stage due to the inherent limitations of the study designs such as small sample sizes and strict exclusion criteria (144). It is also worth noting that one reason for the particular interest in TPE over other blood products such as whole blood might be due to previous associations found between blood transfusions and an increased risk of dementia or AD (145).
Whilst it is currently unclear how TPE with plasma from young donors might exert their therapeutic effects, several theories have been proposed. Vascular cell adhesion molecule 1 (VCAM1), an important molecule in mediating attachment of immune cells to the endothelial surface, has recently been found to be overexpressed in the brains and plasma of both aged mice and humans (146). In addition, systemic administration of anti-VCAM1 immunoglobulins and deletion of the Vcam1 gene in brain endothelial cells (BECs) was shown to reverse microglial reactivity and cognitive defects in mice (146). Therefore, it has been posited that increased inflammatory cytokine levels in old plasma increase VCAM1 expression on BECs, resulting in peripheral leukocyte recruitment and BEC inflammation, in turn activating microglia (146). Another theory for the mechanism of TPE in AD is that old and young plasma may contain youthful and pro-aging factors that influence that ultimately influence the CNS environment and immune response (142). Just as old plasma may contain higher levels of pro-aging pro-inflammatory cytokines, young plasma may contain factors that promote healthy aging of the CNS (142). For example, systemic administration of growth differentiation factor 11 (GDF11) was shown to increase neural stem cell numbers and vascular remodeling in the hippocampi of aged mice (142). However, more research is required to identify these factors in old and young plasma before specific supplementation with these factors can be considered as a viable therapeutic approach for AD.
Given the qualitatively and quantitatively poor production of immune cells seen in the elderly and in AD patients, bone marrow transplantation might be a potential therapy for AD. It has been found, for example, that transplantation of a young bone marrow into old recipient mice preserved cognitive functions, reduced microglial activation in the hippocampus and preserved synaptic connections (147). However, even if bone marrow transplantation was demonstrated to be highly effective in treating AD, the practical limitations in implementation are likely to exclude it as a treatment offered by healthcare systems. These may include availability of compatible bone marrows for transplantation and ethical considerations over which AD patients should receive it and if they should receive them at all in preference over other patient groups like childhood hematological cancers to name a few. Perhaps a more feasible approach might be strategies aimed at preserving and restoring thymic function. Various strategies to rejuvenate thymic function currently being explored are broad and diverse, and are discussed in detail in a review by Thomas and colleagues (148). It should also be noted, however, that in addition to these novel therapies simple strategies such as regular physical exercise have also been shown to have prophylactic and therapeutic benefits in reducing the risk of AD and improving cognitive decline in AD patients (149). The means through which they exert their beneficial effects include the slowing of age-related deterioration in thymic output and decreasing pro-inflammatory cytokine levels associated with neurodegeneration or “inflammaging” (150).
Biomarkers for AD
Identifying suitable biomarkers for monitoring different stages of the AD pathogenic course would be important for future research into therapeutic options for AD. Various biomarkers directly relating to the changing immune response in the AD continuum have been explored, one example being the blood compound neopterin. Neopterin is produced by IFNγ-stimulated macrophages and its association with cell-mediated responses has made it a common biomarker for monitoring such processes (80, 151–153). Notably, neopterin levels are increased in AD patients compared to age-matched controls and have been suggested to reflect serum immunity to CMV (80). In addition, neopterin levels were significantly associated with cognitive decline in AD patients (151). However, whilst neopterin levels were significantly higher in AD relative to MCI patients, there has been no significant difference in neopterin levels between MCI patients and cognitively normal individuals (154). Significantly increased neopterin levels were also only observed in patients with moderate dementia as assessed by their mini mental state score (MMSE) (153). This suggests that the usefulness of neopterin as a biomarker for AD is restricted to patients with established AD and is unlikely to be useful for this purpose during the preclinical phase for screening.
It is well acknowledged that immune changes are seen in the peripheral blood of AD patients. There is growing evidence showing peripheral immune changes in AD such as reduced CD4+ T cells and regulatory T cells (155). However, although T cell and B cell numbers and function have been shown to be altered in AD, there is a great amount of discordance between the results which are summarized in the review by Martorana et al. (155). The cytokine profile in the peripheral blood has also been documented reflecting the pro-inflammatory state associated with AD and are discussed in a review by Park and colleagues (156). Whilst attempts to accurately profile peripheral immune changes in AD may eventually lead to the discovery of new biomarkers, current understanding of these changes does not support their usefulness as biomarkers for monitoring the pre-symptomatic stage of AD. This is mainly due to virtually all current research into this being focused on patients with established AD. In addition, as discussed earlier, whilst a general state of neuroinflammation might predominate earlier in the AD continuum, the skewing of the immune response might be occurring much later in the course. It has also been found that the time spent in each phase of the continuum varies between individuals influenced by various factors (157). Essentially, what then remains to be more adequately understood is what the threshold is for pushing the immune system onto the self-perpetuating chronic inflammatory state towards immunosensence and what are the events that can precipitate this process. As discussed previously, events like CMV infection can lead to a state of chronic immune stimulation that depletes the limited immune reserves. This process of inflammaging is thought to play a key role in determining healthy and pathological aging (158). Better understanding and recognition of this threshold could help to identify patients for early intervention and produce better outcomes in old age.
It should be noted however that one main obstacle to research into peripheral immune changes in individuals with AD is the likely unwillingness of AD patients and their relatives to participate in the study given the substantial challenges already being experienced by them from the disease. This might be circumvented to some extent by gaining consent for post-mortem analysis of immune changes in the tissues, which might also have an added advantage of any findings directly reflecting the pathogenic process compared to peripheral blood immune changes which may differ. However, delays in acquiring and analyzing the samples and processing of the samples may alter the samples and potentially produce misleading findings. Alternatively, it might prove useful to further investigate the skull bone marrow especially for its therapeutic potential. The skull bone marrow provides a rapid source of immune cells to the brain parenchyma via direct vascular networks, which have been found in both mice and humans. Additionally, given the lack of a BBB in the dura mater, immune processes and cells may be more easily targeted by therapeutics and findings in mice models may be more easily translatable to humans (159).
Current attempts at producing a dementia risk assessment tool have begun making its way into clinical trials. For example, the Cardiovascular Risk Factors, Aging and Incidence of Dementia (CAIDE) score that takes into account risk factors like age, hypertension, hypercholesterolaemia, physical inactivity, obesity and educational attainment has demonstrated cognitive benefits in at-risk individuals identified by their CAIDE score following a multimodal intervention in the FINGER trial (160, 161). However, it may be possible that such risk scores may be limited in their effectiveness given the significant proportion of the population who may present with such a constellation of risk factors (162). This might make the large-scale implementation of multimodal interventions such as that used in the FINGER trial impracticable. It might prove more useful in future to design a risk assessment tool that factors in known precipitants and biomarkers of inflammaging such as CMV seropositivity and previous TBI to better target interventions to individuals that may already be progressing towards immunosenescence and AD.
Conclusion
There is good evidence to suggest that AD might be a consequence of CNS immunosenescence secondary to chronic inflammaging. The lack of therapeutic success of Aβ-directed and anti-inflammatory therapies alongside the recent discovery of novel properties of Aβ also prompts for a reconsideration of the role and function of Aβ, from being an etiological factor in AD pathogenesis to an innate immune response mediator. It is also important to note that whilst animal models have aided in our understanding of AD, there is still an unmet need for more humanized ex vivo models of the disease. This is due to the limitations intrinsic to current models such as 5XFADs that present an accelerated form of aging that does not occur in humans, in addition to differing brain areas and gene signatures between humans and the animal model. It is possible that single RNA sequencing may help in defining better understanding the disease. Additionally, characteristic AD features such as tau pathology may prove more useful as biomarkers rather than as potential therapeutic targets. However, future research into identifying new biomarkers especially in the preclinical stage of AD may aid development of early therapeutic interventions that may alter its clinical course.
Author Contributions
SEJC performed the scientific literature search, design the figures and wrote the review. ES supervised, completed and finalized the review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Alzheimer’s Research UK (Pilot Grant ARUKPPG2013B-2 to ES), Fondazione Italiana Sclerosi Multipla-FISM to ES (2014/R/21). And research contract grant number TMTL1DR to ES.
Conflict of Interest
The authors declare the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. DeTure MA, Dickson DW. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol Neurodegeneration (2019) 14(32). doi: 10.1186/s13024-019-0333-5
2. Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dementia (2018) 14(3):367–429. doi: 10.1016/j.jalz.2018.02.001
3. Thinakaran G, Koo EH. Amyloid Precursor Protein Trafficking, Processing, and Function. J Biol Chem (2008) 283(44):29615–9. doi: 10.1074/jbc.R800019200
4. Zheng H, Koo EH. The Amyloid Precursor Protein: Beyond Amyloid. Mol Neurodegeneration (2006) 1(5). doi: 10.1186/1750-1326-1-5
5. Leng F, Edison P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go From Here? Nat Rev Neurol (2021) 17:157–72. doi: 10.1038/s41582-020-00435-y
6. Cline EN, Bicca MA, Viola KL, Klein WL. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J Alzheimer’s Dis (2018) 64(Suppl 1):S567–610. doi: 10.3233/JAD-179941
7. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The Hallmarks of Aging. Cell (2013) 153(6):p1194–217. doi: 10.1016/j.cell.2013.05.039
8. Renkawek K, Bosman GJ, Gaestel M. Increased Expression of Heat-Shock Protein 27 kDa in Alzheimer Disease: A Preliminary Study. Neuroreport (1993) 5(1):14–6. doi: 10.1097/00001756-199310000-00003
9. Wilhelmus MMM, Boelens WC, Otte-Höller I, Kamps B, de Waal RMW, Verbeek MM. Small Heat Shock Proteins Inhibit Amyloid-β Protein Aggregation and Cerebrovascular Amyloid-β Protein Toxicity. Brain Res (2006) 1089(1):67–78. doi: 10.1016/j.brainres.2006.03.058
10. Koga H, Kaushik S, Cuervo AM. Protein Homeostasis and Aging: The Importance of Exquisite Quality Control. Aging Res Rev (2011) 10(2):205–15. doi: 10.1016/j.arr.2010.02.001
11. Franklin TB, Krueger-Naug AM, Clarke DB, Arrigo AP, Currie RW. The Role of Heat Shock Proteins Hsp70 and Hsp27 in Cellular Protection of the Central Nervous System. Int J Hyperthermia (2005) 21(5):379–92. doi: 10.1080/02656730500069955
12. Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: Disturbed Interplay Between Autophagy and Inflammasomes. Aging (2012) 4(3):166–75. doi: 10.18632/aging.100444
13. Wesselingh R, Butzkueven H, Buzzard K, Tarlinton D, O’Brien TJ, Monif M. Innate Immunity in the Central Nervous System: A Missing Piece of the Autoimmune Encephalitis Puzzle? Front Immunol (2019) 10:2066(2066). doi: 10.3389/fimmu.2019.02066
14. Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of Chemokines and Their Receptors in the Central Nervous System: Physiopathological Implications. J Neurochem (2002) 82:1311–29. doi: 10.1046/j.1471-4159.2002.01091.x
15. Owens T, Khorooshi R, Wlodarczyk A, Asgari N. Interferons in the Central Nervous System: A Few Instruments Play Many Tunes. Glia (2014) 62:339–55. doi: 10.1002/glia.22608
16. Rogers J, Lue LF. Microglial Chemotaxis, Activation, and Phagocytosis of Amyloid β-Peptide as Linked Phenomena in Alzheimer’s Disease. Neurochem Int (2001) 39(5-6):333–40. doi: 10.1016/S0197-0186(01)00040-7
17. Cuello AC. Early and Late CNS Inflammation in Alzheimer’s Disease: Two Extremes of a Continuum? Trends Pharmacol Sci (2017) 38(11):956–66. doi: 10.1016/j.tips.2017.07.005
18. Tuppo EE, Arias HR. The Role of Inflammation in Alzheimer’s Disease. Int J Biochem Cell Biol (2005) 37(2):289–305. doi: 10.1016/j.biocel.2004.07.009
19. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (Senescent) Rather Than Activated Microglial Cells are Associated With Tau Pathology and Likely Precede Neurodegeneration in Alzheimer’s Disease. Acta Neuropathol (2009) 118(4):475–85. doi: 10.1007/s00401-009-0556-6
20. Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, et al. Transcriptional Signature in Microglia Associated With Aβ Plaque Phagocytosis. Europe PMC (2021) 12(1):3015. doi: 10.1038/s41467-021-23111-1
21. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-Derived ASC Specks Cross-Seed Amyloid-β in Alzheimer’s Disease. Nature (2017) 552:355–61. doi: 10.1038/nature25158
22. Streit WJ. Microglial Senescence: Does the Brain’s Immune System Have an Expiration Date? Trends Neurosci (2006) 29(9):506–10. doi: 10.1016/j.tins.2006.07.001
23. Treblay ME. Majewska AK. A Role for Microglia in Synaptic Plasticity? Communicative Integr Biol (2011) 4(2):220–2. doi: 10.4161/cib.4.2.14506
24. Damisah EC, Hill RA, Rai A, Chen F, Rothlin CV, Ghosh S, et al. Astrocytes and Microglia Play Orchestrated Roles and Respect Phagocytic Territories During Neuronal Corpse Removal In Vivo. Sci Adv (2020) 6(26):eaba3239. doi: 10.1126/sciadv.aba3239
26. Galloway DA, Phillips AEM, Owen DRJ, Moore CS. Phagocytosis in the Brain: Homeostasis and Disease. Front Immunol (2019) 10:790. doi: 10.3389/fimmu.2019.00790
27. Neumann H, Kotter MR, Franklin JM. Debris Clearance by Microglia: An Essential Link Between Degeneration and Regeneration. BRAIN (2009) 132(2):288–95. doi: 10.1093/brain/awn109
28. Streit WJ, Xue QS. Human CNS Immune Senescence and Neurodegeneration. Curr Opin Immunol (2014) 29:93–6. doi: 10.1016/j.coi.2014.05.005
29. Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, et al. Astrocytes Containing Amyloid Beta-Protein (Abeta)-Positive Granules Are Associated With Abeta40-Positive Diffuse Plaques in the Aged Human Brain. Am J Pathol (1998) 152(4):983–92.
30. Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult Mouse Astrocytes Degrade Amyloid-β In Vitro and In Situ. Nat Med (2003) 9:452–7. doi: 10.1038/nm838
31. Liddelow SA, Guttenplan KA, Clarke LE, Bennet FC, Bohlen CJ, Schirmer L, et al. Neurotoxic Reactive Astrocytes are Induced by Activated Microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
32. Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes Accumulate A Beta 42 and Give Rise to Astrocytic Amyloid Plaques in Alzheimer Disease Brains. Brain Res (2003) 971(2):197–209. doi: 10.1016/S0006-8993(03)02361-8
33. Wood H. Astrocytic IL-3 Could Help Microglia Protect Against Alzheimer Disease. Nat Rev Neurol (2021) 17:525. doi: 10.1038/s41582-021-00546-0
34. Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral Blood Flow Regulation and Neurovascular Dysfunction in Alzheimer’s Disease. Nat Rev Neurosci (2017) 18(7):419–34. doi: 10.1038/nrn.2017.48
35. Sweeney MD, Sagare AP, Zlokovic BV. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat Rev Neurol (2018) 14:133–50. doi: 10.1038/nrneurol.2017.188
36. Hardy J, Allsop D. Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol Sci (1991) 12:383–8. doi: 10.1016/0165-6147(91)90609-V
37. Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC. The Alzheimer’s Disease Neuroimaging Initiative. Early Role of Vascular Dysregulation on Late-Onset Alzheimer’s Disease Based on Multifactorial Data-Driven Analysis. Nat Commun (2016) 7:11934. doi: 10.1038/ncomms11934
38. Zago W, Schroeter S, Guido T, Khan K, Seubert P, Yednock T, et al. Vascular Alterations in PDAPP Mice After Anti-Aβ Immunotherapy: Implications for Amyloid-Related Imaging Abnormalities. Alzheimer’s Dementia (2013) 9(55):S105–15. doi: 10.1016/j.jalz.2012.11.010
39. Zenaro E, Piacentino G, Constantin G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiol Dis (2017) 107:41–56. doi: 10.1016/j.nbd.2016.07.007
40. Yamazaki Y, Kanekiyo T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int J Mol Sci (2017) 18(9):1965. doi: 10.3390/ijms18091965
41. Zenaro E, Pietronigro E, Bianca VD, Piacentino G, Marongiu L, Budui S, et al. Neutrophils Promote Alzheimer’s Disease-Like Pathology and Cognitive Decline via LFA-1 Integrin. Nat Med (2015) 21:880–6. doi: 10.1038/nm.3913
42. Rustenhoven J, Kipnis J. Bypassing the Blood-Brain Barrier. Neuroimmunology (2019) 366(6472):1448–9. doi: 10.1126/science.aay0479
43. Silva I, Silva J, Ferreira R, Trigo D. Glymphatic System, AQP4, and Their Implications in Alzheimer’s Disease. Neurol Res Pract (2021) 3:5. doi: 10.1186/s42466-021-00102-7
44. Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld BS, et al. Impairment of Paravascular Clearance Pathways in the Aging Brain. Ann Neurol (2014) 76(6):845–61. doi: 10.1002/ana.24271
45. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci Trans Med (2012) 4(147):147ra111. doi: 10.1126/scitranslmed.3003748
46. Hoshi A, Yamamoto T, Shimizu K, Ugawa Y, Nishizawa M, Takahashi H, et al. Characteristics of Aquaporin Expression Surrounding Senile Plaques and Cerebral Amyloid Angiopathy in Alzheimer Disease. J Neuropathol Exp Neurol (2012) 71(8):750–9. doi: 10.1097/NEN.0b013e3182632566
47. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, et al. Occurrence of T Cells in the Brain of Alzheimer’s Disease and Other Neurological Diseases. J Neuroimmunol (2002) 124(1-2):83–92. doi: 10.1016/S0165-5728(01)00496-9
48. Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate With Tau But Not With Amyloid Pathology: An Immunohistochemical Study. Neurodegenerative Dis (2018) 18:49–56. doi: 10.1159/000486200
49. Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally Expanded CD8 T Cells Patrol the Cerebrospinal Fluid in Alzheimer’s Disease. Nature (2020) 577:399–404. doi: 10.1038/s41586-019-1895-7
50. Ousman SS, Kubes P. Immune Surveillance in the Central Nervous System. Nat Neurosci (2012) 15:1096–101. doi: 10.1038/nn.3161
51. Mayne K, White JA, McMurran CE, Rivera FJ, de la Fuente AG. Aging and Neurodegenerative Disease: Is the Adaptive Immune System a Friend or Foe? Front Aging Neurosci (2020) 12:572090. doi: 10.3389/fnagi.2020.572090
52. Warren-Gash C, Forbes HJ, Williamson E, Breuer J, Hayward AC, Mavrodaris A, et al. Human Herpesvirus Infections and Dementia or Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. Sci Rep (2019) 9:4743. doi: 10.1038/s41598-019-41218-w
53. Dulken BW, Buckley MT, Negredo PN, Saligrama N, Cayrol R, Leeman DS, et al. Single-Cell Analysis Reveals T Cell Infiltration in Old Neurogenic Niches. Nature (2019) 571:205–10. doi: 10.1038/s41586-019-1362-5
54. Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-Specific Immunity at the Choroid Plexus Shifts Toward Destructive Th2 Inflammation in Brain Aging. PNAS (2013) 110(6):2264–9. doi: 10.1073/pnas.1211270110
55. Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The Aging Systemic Milieu Negatively Regulates Neurogenesis and Cognitive Function. Nature (2011) 477(7362):90–4. doi: 10.1038/nature10357
56. Zhang J, Ke KF, Liu Z, Qiu YH, Peng YP. Th17 Cell-Mediated Neuroinflammation Is Involved in Neurodegeneration of Aβ1-42-Induced Alzheimer’s Disease Model Rats. PloS One (2013) 8(10):e75786. doi: 10.1371/journal.pone.0075786
57. Liu Q, Xin W, He P, Turner D, Yin J, Gan Y, et al. Interleukin-17 Inhibits Adult Hippocampal Neurogenesis. Sci Rep (2014) 4:7554. doi: 10.1038/srep07554
58. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking Immune Tolerance by Targeting Foxp3+ Regulatory T Cells Mitigates Alzheimer’s Disease Pathology. Nat Commun (2015) 6:7967. doi: 10.1038/ncomms8967
59. Mesquita SD, Herz J, Wall M, Dykstra T, de Lima KA, Norris GT, et al. Aging-Associated Deficit in CCR7 is Linked to Worsened Glymphatic Function, Cognition, Neuroinflammation, and β-Amyloid Pathology. Sci Adv (2021) 7(21):eabe4601. doi: 10.1126/sciadv.abe4601
60. Dansokho C, Ahmed DA, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T Cells Delay Disease Progression in Alzheimer-Like Pathology. Brain (2016) 139(4):1237–51. doi: 10.1093/brain/awv408
61. Baek H, Ye M, Kang GH, Lee C, Lee G, Choi DB, et al. Neuroprotective Effects of CD4+CD25+Foxp3+ Regulatory T Cells in a 3xtg-AD Alzheimer’s Disease Model. Oncotarget (2016) 7(43):69347–57. doi: 10.18632/oncotarget.12469
62. Alves S, Churlaud G, Audrain M, Michaelsen-Preusse K, Fol R, Souchet B, et al. Interleukin-2 Improves Amyloid Pathology, Synaptic Failure and Memory in Alzheimer’s Disease Mice. Brain (2016) 140(3):826–42. doi: 10.1093/brain/aww330
63. Venegas C, Heneka MT. Inflammasome-Mediated Innate Immunity in Alzheimer’s Disease. FASEB J (2019) 33(12):13075–84. doi: 10.1096/fj.201900439
64. Zhang Y, Dong Z, Song W. NLRP3 Inflammasome as a Novel Therapeutic Target for Alzheimer’s Disease. Nature (2020) 5(37). doi: 10.1038/s41392-020-0145-7
65. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature (2012) 493:674–8. doi: 10.1038/nature11729
66. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A Dural Lymphatic Vascular System That Drains Brain Interstitial Fluid and Macromolecules. J Exp Med (2015) 212(7):991–9. doi: 10.1084/jem.20142290
67. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and Functional Features of Central Nervous System Lymphatics. Nature (2015) 523(7560):337–41. doi: 10.1038/nature14432
68. Mesquita SD, Louveau A, Vaccari A, Smirnov I, Cornelison C, Kingsmore KM, et al. Functional Aspects of Meningeal Lymphatics in Aging and Alzheimer’s Disease. Nature (2018) 560:185–91. doi: 10.1038/s41586-018-0368-8
69. Mesquita SD, Papadopoulos Z, Dykstra T, Brase L, Farias FG, Wall M, et al. Meningeal Lymphatics Affect Microglia Responses and Anti-Aβ Immunotherapy. Nature (2021) 593:255–60. doi: 10.1038/s41586-021-03489-0
70. Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct Vascular Channels Connect Skull Bone Marrow and the Brain Surface Enabling Myeloid Cell Migration. Nat Neurosci (2018) 21:1209–17. doi: 10.1038/s41593-018-0213-2
71. Cai R, Pan C, Ghasemigharagoz A, Todorov MI, Forstera B, Zhao S, et al. Panoptic Imaging of Transparent Mice Reveals Whole-Body Neuronal Projections and Skull-Meninges Connections. Nat Neurosci (2019) 22:317–27. doi: 10.1038/s41593-018-0301-3
72. Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ, et al. Skull and Vertebral Bone Marrow are Myeloid Cell Reservoirs for the Meninges and CNS Parenchyma. Science (2021) 373(6553):eabf7844. doi: 10.1126/science.abf7844
73. Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, et al. Heterogeneity of Meningeal B Cells Reveals a Lymphopoietic Niche at the CNS Borders. Science (2021) 373(6553):eabf9277. doi: 10.1126/science.abf9277
74. Sankowski R, Strohl JJ, Huerta TS, Nasiri E, Mazzarello AN, D’Abramo C, et al. Endogenous Retroviruses are Associated With Hippocampus-Based Memory Impairment. PNAS (2019) 116(51):25982–90. doi: 10.1073/pnas.1822164116
75. Kim K, Wang X, Ragonnaud E, Bodogai M, Illouz T, DeLuca M, et al. Therapeutic B-Cell Depletion Reverses Progression of Alzheimer’s Disease. Nat Commun (2021) 12:2185. doi: 10.1038/s41467-021-22479-4
76. Schwartz M, Baruch K. The Resolution of Neuroinflammation in Neurodegeneration: Leukocyte Recruitment via the Choroid Plexus. EMBO J (2013) 33:7–22. doi: 10.1002/embj.201386609
77. Dani N, Herbst RH, McCabe C, Green GS, Kaiser K, Head JP, et al. A Cellular and Spatial Map of the Choroid Plexus Across Brain Ventricles and Ages. Cell (2021) 184(11):3056–3074.e21. doi: 10.1016/j.cell.2021.04.003
78. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity (2018) 48(2):p380–395.e6. doi: 10.1016/j.immuni.2018.01.011
79. Rustenhoven J, Drieu A, Mamladze T, de Lima KA, Dykstra T, Wall M, et al. Functional Characterization of the Dural Sinuses as a Neuroimmune Interface. Cell (2021) 184(4):1000–1016.e27. doi: 10.1016/j.cell.2020.12.040
80. Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, et al. Inflammaging as a Prodrome to Alzheimer’s Disease. J Neuroinflamm (2008) 5(51). doi: 10.1186/1742-2094-5-51
81. Azuma M, Phillips JH, Lanier LL. CD28-T Lymphocytes. Antigenic and Functional Properties. J Immunol (1993) 150(4):1147–59.
82. Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Würzner R, et al. Long-Term Cytomegalovirus Infection Leads to Significant Changes in the Composition of the CD8+ T-Cell Repertoire, Which May be the Basis for an Imbalance in the Cytokine Production Profile in Elderly Persons. J Virol (2005) 79(6):3675–83. doi: 10.1128/JVI.79.6.3675-3683.2005
83. Goronzy JJ, Weyand CM. T Cell Development and Receptor Diversity During Aging. Curr Opin Immunol (2005) 17(5):468–75. doi: 10.1016/j.coi.2005.07.020
84. Mudo G, Frinchi M, Nuzzo D, Scaduto P, Plescia F, Massenti MF, et al. Anti-Inflammatory and Cognitive Effects of Interferon-β1a (Ifnβ1a) in a Rat Model of Alzheimer’s Disease. J Neuroinflamm (2019) 16:44. doi: 10.1186/s12974-019-1417-4
85. Feng E, Balint E, Poznanski SM, Ashkar AA, Loeb M. Aging and Interferons: Impact on Inflammation and Viral Disease Outcomes. Europe PMC (2021) 10(3):708. doi: 10.3390/cells10030708
86. Swain S, Clise-Dwyer K, Haynes L. Homeostasis and the Age-Associated Defect of CD4 T Cells. Semin Immunol (2005) 17(5):370–7. doi: 10.1016/j.smim.2005.05.007
87. Eaton SM, Burns EM, Kusser K, Randall TD, Haynes L. Age-Related Defects in CD4 T Cell Cognate Helper Function Lead to Reductions in Humoral Responses. J Exp Med (2004) 200(12):1613–22. doi: 10.1084/jem.20041395
88. Ghia P, Melchers F, Rolink AG. Age-Dependent Changes in B Lymphocyte Development in Man and Mouse. Exp Gerontol (2000) 35(2):159–65. doi: 10.1016/S0531-5565(99)00095-9
89. Weksler ME. Changes in the B-Cell Repertoire With Age. Vaccine (2000) 18(16):1624–8. doi: 10.1016/S0264-410X(99)00497-1
90. Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW. Immunosenescence of Microglia and Macrophages: Impact on the Aging Central Nervous System. Brain (2016) 139(3):654–61. doi: 10.1093/brain/awv395
91. Njie EG, Boelen E, Stassen FR, Steinbusch HWM, Borchelt DR, Streit WJ. Ex Vivo Cultures of Microglia From Young and Aged Rodent Brain Reveal Age-Related Changes in Microglial Function. Neurobiol Aging (2012) 33(1):195. doi: 10.1016/j.neurobiolaging.2010.05.008
92. Natrajan MS, de la Fuente AG, Crawford AH, Linehan E, Nuñez V, Johnson KR, et al. Retinoid X Receptor Activation Reverses Age-Related Deficiencies in Myelin Debris Phagocytosis and Remyelination. Brain (2015) 138(12):3581–97. doi: 10.1093/brain/awv289
93. Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-Related Alterations in the Dynamic Behavior of Microglia. Aging Cell (2011) 10(2):263–76. doi: 10.1111/j.1474-9726.2010.00660.x
94. Marshall JS, Warrington R, Watson W, Kim HL. An Introduction to Immunology and Immunopathology. Allergy Asthma Clin Immunol (2018) 14(49). doi: 10.1186/s13223-018-0278-1
95. Saurwein-Teissl M, Lung TL, Marx F, Gschösser C, Asch E, Blasko I, et al. Lack of Antibody Production Following Immunization in Old Age: Association With CD8+CD28- T Cell Clonal Expansions and an Imbalance in the Production of Th1 and Th2 Cytokines. J Immunol (2021) 168:5893–9. doi: 10.4049/jimmunol.168.11.5893
96. Urban SL, Jensen IJ, Shan Q, Pewe LL, Xue HH, Badovinac VP, et al. Peripherally Induced Brain Tissue-Resident Memory CD8 + T Cells Mediate Protection Against CNS Infection. Nat Immunol (2020) 21:938–49. doi: 10.1038/s41590-020-0711-8
97. Gross PA, Hermogenes AW, Sacks HS, Lau J, Levandowski RA. The Efficacy of Influenza Vaccine in Elderly Persons. A Meta-Analysis and Review of the Literature. Ann Internal Med (1995) 123(7):518–27. doi: 10.7326/0003-4819-123-7-199510010-00008
98. Lee JE, Kim DW, Lee JH. Association Between Alzheimer’s Disease and Cancer Risk in South Korea: An 11-Year Nationwide Population-Based Study. Dementia Neurocognitive Disord (2018) 17(4):137–47. doi: 10.12779/dnd.2018.17.4.137
99. Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally Administered Antibodies Against Amyloid β-Peptide Enter the Central Nervous System and Reduce Pathology in a Mouse Model of Alzheimer Disease. Nat Med (2000) 6:916–9. doi: 10.1038/78682
100. Li JW, Zong Y, Cao XP, Tan L, Tan L. Microglial Priming in Alzheimer’s Disease. Ann Trans Med (2018) 6(10):176. doi: 10.21037/atm.2018.04.22
101. McPherson CA, Kraft AD, Harry GJ. Injury-Induced Neurogenesis: Consideration of Resident Microglia as Supportive of Neural Progenitor Cells. Neurotoxicity Res (2011) 19(2):341–52. doi: 10.1007/s12640-010-9199-6
102. Norden DM, Muccigrosso MM, Godbout JP. Microglial Priming and Enhanced Reactivity to Secondary Insult in Aging, and Traumatic CNS Injury, and Neurodegenerative Disease. Neuropharmacology (2015) 96(Pt A):29–41. doi: 10.1016/j.neuropharm.2014.10.028
103. Vlad SC, Miller DR, Kowall NW, Felson ST. Protective Effects of NSAIDs on the Development of Alzheimer Disease. Neurology (2008) 70(19):1672–7. doi: 10.1212/01.wnl.0000311269.57716.63
104. Etminan M, Gill S, Samii A. Effect of Non-Steroidal Anti-Inflammatory Drugs on Risk of Alzheimer’s Disease: Systematic Review and Meta-Analysis of Observational Studies. BMJ (2003) 327(7407):128. doi: 10.1136/bmj.327.7407.128
105. in ‘t Veld BA, LJ L, Breteler MMB, Hofman A, Stricker BHC. Pharmacologic Agents Associated With a Preventive Effect on Alzheimer’s Disease: A Review of the Epidemiologic Evidence. Epidemiol Rev (2002) 24(2):248–68. doi: 10.1093/epirev/mxf001
106. Jaturapaporn D, Isaac MGEKN, McCleery J, Tabet N. Aspirin, Steroidal and Non-Steroidal Anti-Inflammatory Drugs for the Treatment of Alzheimer’s Disease. Cochrane Database Systematic Rev (2012) 2):CD006378. doi: 10.1002/14651858.CD006378.pub2
107. Akbar AN, Henson SM. Are Senescence and Exhaustion Intertwined or Unrelated Processes That Compromise Immunity? Nat Rev Immunol (2011) 11:289–95. doi: 10.1038/nri2959
108. Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, et al. Heme Oxygenase-1 is Associated With the Neurofibrillary Pathology of Alzheimer’s Disease. Am J Pathol (1994) 145(1):42–7.
109. Grimaldi LME, Zappalà G, Iemolo F, Castellano AE, Ruggieri S, Bruno G, et al. A Pilot Study on the Use of Interferon Beta-1a in Early Alzheimer’s Disease Subjects. J Neuroinflamm (2014) 11:30. doi: 10.1186/1742-2094-11-30
110. Derhovanessian E, Chen S, Maier AB, Hähnel K, de Craen AJM, Roelofs H, et al. CCR4+ Regulatory T Cells Accumulate in the Very Elderly and Correlate With Superior 8-Year Survival. J Gerontol: Ser A (2015) 70(8):917–23. doi: 10.1093/gerona/glu128
111. Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization With Amyloid-β Attenuates Alzheimer-Disease-Like Pathology in the PDAPP Mouse. Nature (1999) 400:173–7. doi: 10.1038/22124
112. Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute Meningoencephalitis in a Subset of Patients With AD After Abeta42 Immunization. Neurology (2003) 61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8
113. Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-Term Effects of Aβ42 Immunisation in Alzheimer’s Disease: Follow-Up of a Randomised, Placebo-Controlled Phase I Trial. Lancet (2008) 372(9634):216–23. doi: 10.1016/S0140-6736(08)61075-2
114. Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, et al. The Adaptive Immune System Restrains Alzheimer’s Disease Pathogenesis by Modulating Microglial Function. PNAS (2016) 113(9):E1316–25. doi: 10.1073/pnas.1525466113
115. Knight EM, Gandy S. Immunomodulation and AD – Down But Not Out. J Clin Immunol (2014) 34:70–3. doi: 10.1007/s10875-014-0039-y
116. Relkin N. Clinical Trials of Intravenous Immunoglobulin for Alzheimer’s Disease. J Clin Immunol (2014) 34:74–9. doi: 10.1007/s10875-014-0041-4
117. Klück V, Jansen TLTA, Janssen M, Comarniceanu A, Efdé M, Tengesdal IW, et al. Dapansutrile, an Oral Selective NLRP3 Inflammasome Inhibitor, for Treatment of Gout Flares: An Open-Llabel, Dose-Adaptive, Proof-Of-Concept, Phase 2a Trial. Lancet Rheumatol (2020) 2(5):e270–80. doi: 10.1016/S2665-9913(20)30065-5
118. Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D’Alessandro A, et al. The NLRP3 Inflammasome Inhibitor OLT1177 Rescues Cognitive Impairment in a Mouse Model of Alzheimer’s Disease. PNAS (2020) 117(50):32145–54. doi: 10.1073/pnas.2009680117
119. Pogglioli G, Laureti S, Campieri M, Pierangeli F, Gionchetti P, Ugolini F, et al. Infliximab in the Treatment of Crohn’s Disease. Ther Clin Risk Manage (2007) 3(2):301–8. doi: 10.2147/tcrm.2007.3.2.301
120. Tobinick EL, Gross H. Rapid Cognitive Improvement in Alzheimer’s Disease Following Perispinal Etanercept Administration. J Neuroinflamm (2008) 5(2). doi: 10.1186/1742-2094-5-2
121. Butchart J, Brook L, Hopkins V, Teeling J, Püntener U, Culliford D, et al. Etanercept in Alzheimer Disease. Neurology (2015) 84(21):2161–8. doi: 10.1212/WNL.0000000000001617
122. McGrattan AM, McGuiness B, McKinley MC, Kee F, Passmore P, Woodside JV, et al. Diet and Inflammation in Cognitive Aging and Alzheimer’s Disease. Curr Nutr Rep (2019) 8:53–6. doi: 10.1007/s13668-019-0271-4
123. Valls-Pedret C, Sala-Vila A, Serra-Mir M, Corella D, de la Torre R, Martínez-González MA, et al. Mediterranean Diet and Age-Related Cognitive Decline: A Randomized Clinical Trial. JAMA Internal Med (2015) 175(7):1094–103. doi: 10.1001/jamainternmed.2015.1668
124. Marseglia A, Xu W, Fratiglioni L, Fabbri C, Berendsen AAM, Bialecka-Debek A, et al. Effect of the NU-AGE Diet on Cognitive Functioning in Older Adults: A Randomized Controlled Trial. Front Physiol (2018) 9:349. doi: 10.3389/fphys.2018.00349
125. Knight A, Bryan J, Wilson C, Hodgson JM, Davis CR, Murphy KJ. The Mediterranean Diet and Cognitive Function Among Healthy Older Adults in a 6-Month Randomised Controlled Trial: The MedLey Study. Nutrients (2016) 8(9):579. doi: 10.3390/nu8090579
126. Lott IT, Head E. Dementia in Down Syndrome: Unique Insights for Alzheimer Disease Research. Nat Rev Neurol (2019) 15:135–47. doi: 10.1038/s41582-018-0132-6
127. Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrièreet A, Vital A, et al. APP Locus Duplication Causes Autosomal Dominant Early-Onset Alzheimer Disease With Cerebral Amyloid Angiopathy. Nat Genet (2006) 38:24–6. doi: 10.1038/ng1718
128. Dieudonné Y, Uring-Lambert B, Jeljeli MM, Gies V, Alembik Y, Korganow AS, et al. Immune Defect in Adults With Down Syndrome: Insights Into a Complex Issue. Front Immunol (2020) 11:840. doi: 10.3389/fimmu.2020.00840
129. Satgé D, Seidel MG. The Pattern of Malignancies in Down Syndrome and Its Potential Context With the Immune System. Front Immunol (2018) 9:3058. doi: 10.3389/fimmu.2018.03058
130. Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, et al. Trisomy 21 Consistently Activates the Interferon Response. eLife (2016) 5:e16220. doi: 10.7554/eLife.16220
131. Ponnappan S, Ponnappan U. Aging and Immune Function: Molecular Mechanisms to Interventions. Antioxid Redox Signalling (2011) 14(8):1551–85. doi: 10.1089/ars.2010.3228
132. Weaver DF. Amyloid Beta is An Early Responder Cytokine and Immunopeptide of the Innate Immune System. Trans Res Clin Interventions (2020) 6(1):e12100. doi: 10.1002/trc2.12100
133. Moir RD, Lathe R, Tanzi RE. The Antimicrobial Protection Hypothesis of Alzheimer’s Disease. Alzheimer’s Dementia (2018) 14(12):1602–14. doi: 10.1016/j.jalz.2018.06.3040
134. Eimer WA, Kumar DKV, Shanmugam NKN, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect Against Brain Infection. Neuron (2018) 99(1):56–63.e3. doi: 10.1016/j.neuron.2018.06.030
135. Vendrame M, Cassady J, Newcomb J, Butler T, Pennypacker KR, Zigova T, et al. Infusion of Human Umbilical Cord Blood Cells in a Rat Model of Stroke Dose-Dependently Rescues Behavioral Deficits and Reduces Infarct Volume. Stroke (2004) 35(10):2390–5. doi: 10.1161/01.STR.0000141681.06735.9b
136. Xie Q, Liu R, Jiang J, Peng J, Yang C, Zhang W, et al. What is the Impact of Human Umbilical Cord Mesenchymal Stem Cell Transplantation on Clinical Treatment? Stem Cell Res Ther (2020) 11:519. doi: 10.1186/s13287-020-02011-z
137. Lee HJ, Lee JK, Lee H, Carter JE, Chang JW, Oh W, et al. Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells Improve Neuropathology and Cognitive Impairment in an Alzheimer’s Disease Mouse Model Through Modulation of Neuroinflammation. Neurobiol Aging (2012) 33(3):588–602. doi: 10.1016/j.neurobiolaging.2010.03.024
138. Kim HJ, Seo SW, Chang JW, Lee JI, Kim CH, Chin J, et al. Stereotactic Brain Injection of Human Umbilical Cord Blood Mesenchymal Stem Cells in Patients With Alzheimer’s Disease Dementia: A Phase 1 Clinical Trial. Alzheimer’s Dementia: Trans Res Clin Interventions (2015) 1(2):95–102. doi: 10.1016/j.trci.2015.06.007
139. Liu XY, Yang LP, Zhao L. Stem Cell Therapy for Alzheimer’s Disease. World J Stem Cells (2020) 12(8):787–802. doi: 10.4252/wjsc.v12.i8.787
140. Faridar A, Thome AD, Zhao W, Thonhoff JR, Beers DR, Pascual B, et al. Restoring Regulatory T-Cell Dysfunction in Alzheimer’s Disease Through Ex Vivo Expansion. Brain Commun (2020) 2(2):fcaa112. doi: 10.1093/braincomms/fcaa112
141. Tahvidari M, Dana R. Low-Dose IL-2 Therapy in Transplantation, Autoimmunity, and Inflammatory Diseases. J Immunol (2019) 203(11):2749–55. doi: 10.4049/jimmunol.1900733
142. Kang S, Moser VA, Svendsen CN, Goodridge HS. Rejuvenating the Blood and Bone Marrow to Slow Aging-Associated Cognitive Decline and Alzheimer’s Disease. Commun Biol (2020) 3:69. doi: 10.1038/s42003-020-0797-4
143. Zhao Y, Qian R, Zhang J, Liu F, Iqbal K, Dai CL, et al. Young Blood Plasma Reduces Alzheimer’s Disease-Like Brain Pathologies and Ameliorates Cognitive Impairment in 3×Tg-AD Mice. Alzheimer’s Res Ther (2020) 12:70. doi: 10.1186/s13195-020-00639-w
144. Imbimbo BP, Ippati S, Ceravolo F, Watling M. Perspective: Is Therapeutic Plasma Exchange a Viable Option for Treating Alzheimer’s Disease? Alzheimer’s Dementia: Trans Res Clin Interventions (2020) 6(1):e12004. doi: 10.1002/trc2.12004
145. Lin SY, Hsu WH, Lin CC, Lin CL, Yeh HC, Kao CH. Association of Transfusion With Risks of Dementia or Alzheimer’s Disease: A Population-Based Cohort Study. Front Psychiatry (2019) 10:571. doi: 10.3389/fpsyt.2019.00571
146. Yousef H, Czupalla CJ, Lee D, Chen MB, Burke AN, Zera KA, et al. Aged Blood Impairs Hippocampal Neural Precursor Activity and Activates Microglia Via Brain Endothelial Cell Vcam1. Nat Med (2019) 25:988–1000. doi: 10.1038/s41591-019-0440-4
147. Das MM, Godoy M, Chen S, Moser VA, Avalos P, Roxas KM, et al. Young Bone Marrow Transplantation Preserves Learning and Memory in Old Mice. Commun Biol (2019) 2:73. doi: 10.1038/s42003-019-0298-5
148. Thomas R, Wang W, Su DM. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun Aging (2020) 17(2). doi: 10.1186/s12979-020-0173-8
149. Meng Q, Lin MS, Tzeng IS. Relationship Between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front Neurosci (2020) 14:131. doi: 10.3389/fnins.2020.00131
150. Duggal NA, Pollock RD, Lazarus NR, Harridge S, Lord JM. Major Features of Immunesenescence, Including Reduced Thymic Output, are Ameliorated by High Levels of Physical Activity in Adulthood. Aging Cell (2018) 17(2):e12750. doi: 10.1111/acel.12750
151. Murr C, Hainz U, Asch E, Berger P, Jenewein B, Saurwein-Teissl M, et al. Association of Increased Neopterin Production With Decreased Humoral Immunity in the Elderly. Exp Gerontol (2003) 38(5):583–7. doi: 10.1016/S0531-5565(03)00062-7
152. Wirleitner B, Schroecksnadel K, Winkler C, Fuchs D. Neopterin in HIV-1 Infection. Mol Immunol (2005) 42(2):183–94. doi: 10.1016/j.molimm.2004.06.017
153. Blasko I, Knaus G, Weiss E, Kemmler G, Winkler C, Falkensammer G, et al. Cognitive Deterioration in Alzheimer’s Disease is Accompanied by Increase of Plasma Neopterin. J Psychiatr Res (2007) 41(8):694–701. doi: 10.1016/j.jpsychires.2006.02.001
154. Parker DC, Mielke MM, Yu Q, Rosenberg PB, Jain A, Lyketsos G, et al. Plasma Neopterin Level as a Marker of Peripheral Immune Activation in Amnestic Mild Cognitive Impairment and Alzheimer’s Disease. Int J Geriatric Psychiatry (2013) 28(2):149–54. doi: 10.1002/gps.3802
155. Martorana A, Bulati M, Buffa S, Pellicanò M, Caruso C, Candore G, et al. Immunosenescence, Inflammation and Alzheimer’s Disease. Longevity Healthspan (2012) 1:8. doi: 10.1186/2046-2395-1-8
156. Park JC, Han SH, Mook-Jung I. Peripheral Inflammatory Biomarkers in Alzheimer’s Disease: A Brief Review. BMB Rep (2020) 53(1):10–9. doi: 10.5483/BMBRep.2020.53.1.309
157. Vermunt L, Sikkes SAM, van der Hout A, Handels R, Bos I, van der Flier WM, et al. Duration of Preclinical, Prodromal and Dementia Alzheimer Disease Stages in Relation to Age, Sex, and APOE Genotype. Alzheimer’s Dementia (2019) 15(7):888–98. doi: 10.1016/j.jalz.2019.04.001
158. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol (2018) 8:1960. doi: 10.3389/fimmu.2017.01960
159. Engelhardt B. Private Immune Protection at the Border of the Central Nervous System. Nature (2021) 596:38–40. doi: 10.1038/d41586-021-01962-4
160. Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soninen H, Tuomilehto J. Risk Score for the Prediction of Dementia Risk in 20 Years Among Middle Aged People: A Longitudinal, Population-Based Study. Lancet Neurol (2006) 5(9):735–41. doi: 10.1016/S1474-4422(06)70537-3
161. Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 Year Multidomain Intervention of Diet, Exercise, Cognitive Training, and Vascular Risk Monitoring Versus Control to Prevent Cognitive Decline in At-Risk Elderly People (FINGER): A Randomised Controlled Trial. Lancet (2015) 385(9984):2255–63. doi: 10.1016/S0140-6736(15)60461-5
162. Yusuf S, Joseph P, Rangarajan S, Islam S, Mente A, Hystad P, et al. Modifiable Risk Factors, Cardiovascular Disease, and Mortality in 155 722 Individuals From 21 High-Income, Middle-Income, and Low-Income Countries (PURE): A Prospective Cohort Study. Lancet (2019) 395(10226):795–808. doi: 10.1016/S0140-6736(19)32008-2
Keywords: immunosenescence, Alzheimer, neuroinflammation, biomarker, screening, aging
Citation: Chee SEJ and Solito E (2021) The Impact of Ageing on the CNS Immune Response in Alzheimer’s Disease. Front. Immunol. 12:738511. doi: 10.3389/fimmu.2021.738511
Received: 09 July 2021; Accepted: 01 September 2021;
Published: 17 September 2021.
Edited by:
Guillaume Dorothee, U938 Centre de Recherche Saint Antoine (CRSA) (INSERM), FranceReviewed by:
Sandro Dá Mesquita, Mayo Clinic Florida, United StatesAleksandra Deczkowska, Institut Pasteur, France
Copyright © 2021 Chee and Solito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan En Jie Chee, cy5jaGVlQHNlMTQucW11bC5hYy51aw==; Egle Solito, ZS5zb2xpdG9AcW11bC5hYy51aw==