 Shuvasree SenGupta
Shuvasree SenGupta Lauren E. Hein
Lauren E. Hein Carole A. Parent
Carole A. Parent- 1Life Sciences Institute, University of Michigan, Ann Arbor, MI, United States
- 2Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI, United States
- 3Cancer Biology Graduate Program, University of Michigan Medical School, Ann Arbor, MI, United States
- 4Rogel Cancer Center, University of Michigan Medical School, Ann Arbor, MI, United States
- 5Department of Cell and Developmental Biology, University of Michigan Medical School, Ann Arbor, MI, United States
Neutrophils sense and migrate towards chemotactic factors released at sites of infection/inflammation and contain the affected area using a variety of effector mechanisms. Aside from these established immune defense functions, neutrophils are emerging as one of the key tumor-infiltrating immune cells that influence cancer progression and metastasis. Neutrophil recruitment to the tumor microenvironment (TME) is mediated by multiple mediators including cytokines, chemokines, lipids, and growth factors that are secreted from cancer cells and cancer-associated stromal cells. However, the molecular mechanisms that underlie the expression and secretion of the different mediators from cancer cells and how neutrophils integrate these signals to reach and invade tumors remain unclear. Here, we discuss the possible role of the epithelial to mesenchymal transition (EMT) program, which is a well-established promoter of malignant potential in cancer, in regulating the expression and secretion of these key mediators. We also summarize and review our current understanding of the machineries that potentially control the secretion of the mediators from cancer cells, including the exocytic trafficking pathways, secretory autophagy, and extracellular vesicle-mediated secretion. We further reflect on possible mechanisms by which different mediators collaborate by integrating their signaling network, and particularly focus on TGF-β, a cytokine that is highly expressed in invasive tumors, and CXCR2 ligands, which are crucial neutrophil recruiting chemokines. Finally, we highlight gaps in the field and the need to expand current knowledge of the secretory machineries and cross-talks among mediators to develop novel neutrophil targeting strategies as effective therapeutic options in the treatment of cancer.
Introduction
Neutrophils are the body’s first responders to injury or infection. They have an unparalleled ability to migrate toward gradients of chemoattractants, which are released at sites of inflammation, and to clear pathogens or cell debris by using a plethora of functions including phagocytosis, the release of cytotoxic enzymes or reactive oxygen species (ROS), and the release of neutrophil extracellular traps (NETs) (1). In addition, neutrophils have been reported in the tumor microenvironment (TME) (2), which has been described as a site of persistent inflammation similar to “wounds that do not heal” (3). The TME harbors a wide variety of diffusible mediators released from both tumor and stromal cells. These mediators induce neutrophil migration toward tumors and alter neutrophil function to promote or limit cancer progression. While studies have focused on understanding the tumor promoting or impairing properties of neutrophils, many questions remain unanswered about the identity of the mediators that control neutrophil recruitment to tumor sites and the function of tumor-associated neutrophils, referred to as TANs. In this perspective, we present an overview of the functions of TANs and the different classes of mediators that have been linked to neutrophil recruitment to tumors, discuss how cancer-associated changes such as the epithelial to mesenchymal transition (EMT) upregulate the expression of the mediators, and review the secretory mechanisms that potentially underlie the release of the mediators in the TME. Finally, we discuss our current understanding of the crosstalk between mediators, with a special focus on TGF-β and chemokines, to provide insights into the integrated mechanisms underlying neutrophil recruitment to the tumor niche and suggest gaps in knowledge that need to be filled for the development of anti-cancer therapeutic interventions.
Plasticity of Neutrophils in Cancer
To phenotypically classify TANs and their wide range of functions that impact the outcomes of tumors, several categories have emerged: high-density/low-density, immature/mature, and anti-tumor/pro-tumor/pro-metastatic. Evidence is now suggesting that neutrophils exhibit phenotypic plasticity and can exist on a spectrum within any of these overlapping categories (4–7). For example, neutrophils are often described as “N1” (anti-tumor) or “N2” (pro-tumor) (8), but Zilionis et al. recently described a range of five neutrophil subsets in human lung cancer based on transcriptome analysis, with gene expression ranging from canonical neutrophil genes (N1 subset) to genes that were tumor-specific (N5 subset) (4). These seemingly different phenotypes are mostly generated due to exposure to specific mediator(s), either systemically in the bone marrow/blood or locally at the tumor sites. Studies using murine tumor models report that the immunosuppressive cytokine TGF-β is responsible for promoting the generation of neutrophils with a pro-tumoral “N2” phenotype (8), while the type I interferon IFN-β gives rise to an anti-tumoral “N1” phenotype (9). Further, the enzyme protease cathepsin c, secreted by breast cancer cells, has been reported to initiate a signaling cascade in mouse models that recruits neutrophils to the lung metastatic niche and promotes a pro-metastatic phenotype, and its secretion is correlated with shortened metastasis-free survival in humans (10).
Several neutrophil effector functions that support tumor progression have been identified, including immunosuppression, remodeling of the extracellular matrix (ECM), and promoting angiogenesis (11). For example, it has been demonstrated in mouse models that immunosuppressive neutrophils promote metastasis by releasing high levels of inducible nitric oxide synthase (iNOS), which inhibits proliferation of cytotoxic T cells (12). Additionally, the release of NETs from neutrophils, which are mesh-like structures of DNA fibers studded with granule proteins, is associated with metastatic progression (13–16). In particular, granule proteins such as neutrophil elastase (NE) and matrix metalloproteinase 9 (MMP9) released with NETs have been shown to awaken dormant cancer cells by remodeling the ECM and inducing cancer cell proliferation in mouse models (17).
Although most studies have reported a pro-tumor effect of neutrophils, some contexts exist where neutrophils exhibit anti-tumor effects, seemingly maintaining their canonical role to protect the body from harm. These anti-tumor actions of neutrophils in mouse models include the direct killing of cancer cells through the release of cytotoxic ROS (including hydrogen peroxide) and the restriction of tumor growth by stimulating cancer cell detachment from the basement membrane by the release of MMP9 (18–20). Additionally, the neutrophil-dependent stimulation of T cell responses can indirectly contribute to anti-tumor activity, shown both in mouse models and in cells isolated from human tissue samples (11, 21). While it has been reported that specific mediators contribute to the pro- or anti-tumoral actions of neutrophils, the mechanisms underlying how the mediators are expressed and secreted by cancer cells and the interplay among the different mediators in regulating neutrophil function remain unknown.
Diffusible Mediators in the Tumor Niche
Cancer cells and stromal cells, including cancer-associated fibroblasts (CAFs), T cells, monocytes, tumor-associated macrophages, and TANs, can secrete diverse mediators that diffuse through tissues, potentially signaling to circulating or tissue-patrolling neutrophils to recruit them to the tumor niche. Table 1 provides a list of recently identified neutrophil mediators released in various mouse and human tissue or cell line models of cancer and their sources. These mediators primarily consist of chemokines, growth factors, and cytokines. The chemokines CXCL1, 2, 5, 6 and 8, which induce neutrophil chemotaxis through CXCR2/1 chemokine receptors, have been reported to be important for neutrophil recruitment in many cancer types (22, 25–28, 30–38). Chemokines are critical to recruit neutrophils not only to primary tumor sites but also to pre-metastatic niches and metastatic sites. For example, CXCL1, 2, and 5 released from tumor-associated mesenchymal stromal cells in a mouse model of breast cancer resulted in increased neutrophil recruitment to primary tumor sites (22). Additionally, CXCL5 and 7 released from tumor-activated platelets were reported to be crucial for neutrophil recruitment to the pre-metastatic niche and subsequent tumor cell seeding in the mouse lung (26). G-CSF and GM-CSF are important growth factors commonly upregulated in cancer, and their primary functions are to regulate the release of mature neutrophils from the bone marrow into the blood and to extend the survival of neutrophils (38, 39). In addition, cytokines, including interleukins (IL-17A, IL-6) (10, 12, 25, 40), interferons (IFN-β) (9), TNF-α (25), and TGF-β (5, 8, 24, 28), have been linked to neutrophil recruitment and extended neutrophil survival, as well as regulating neutrophil function. While a growing number of studies report the effect of individual mediators on neutrophil recruitment and function, it is likely that antagonistic, additive, or synergistic effects of different classes of mediators are crucial for neutrophil recruitment and function in the context of cancer.
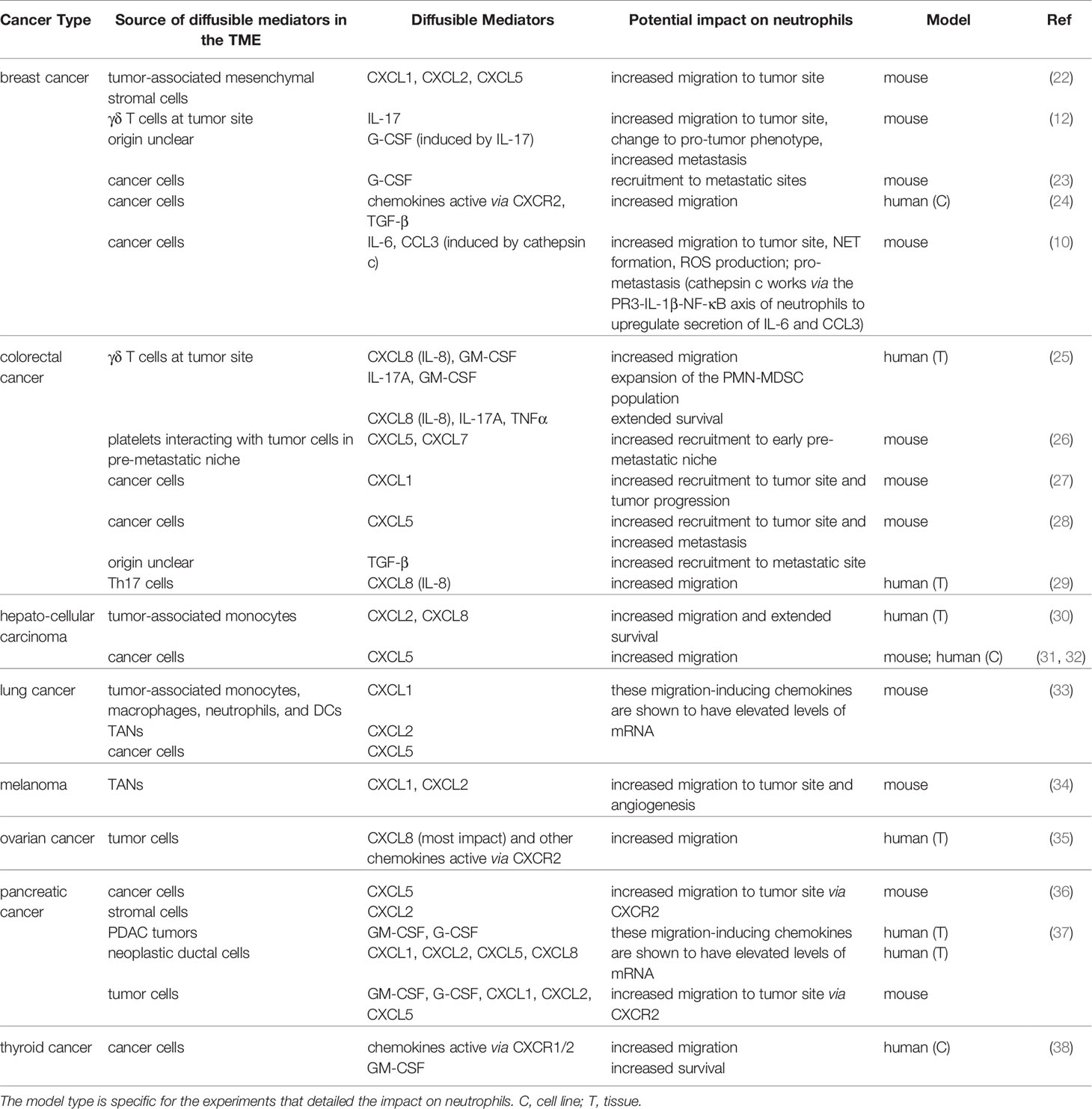
Table 1 List of diffusible mediators along with their cellular origin, potential impact on neutrophils and tumor progression, and the study models.
Mechanisms Regulating the Secretion of Diffusible Mediators From Cancer Cells
Cancer cells are known to upregulate the transcription of many diffusible mediators due to the constitutive activation or overexpression of oncoproteins (41). In many cases, the higher expression of the mediators correlates with poor clinical progression (42, 43). However, little is known about the secretory mechanisms that regulate the release of the mediators from cancer cells into the tumor niche and how the process of secretion may be altered due to cancer-associated changes compared to the mechanisms observed in non/early malignant cells. Here, we suggest that EMT induction is a key process that alters the tumor secretome and highlight the mechanisms and molecular players known to regulate the secretion of the mediators. We envision that similar mechanisms underlie the secretion of neutrophil recruiting mediators from cancer cells (Figure 1).
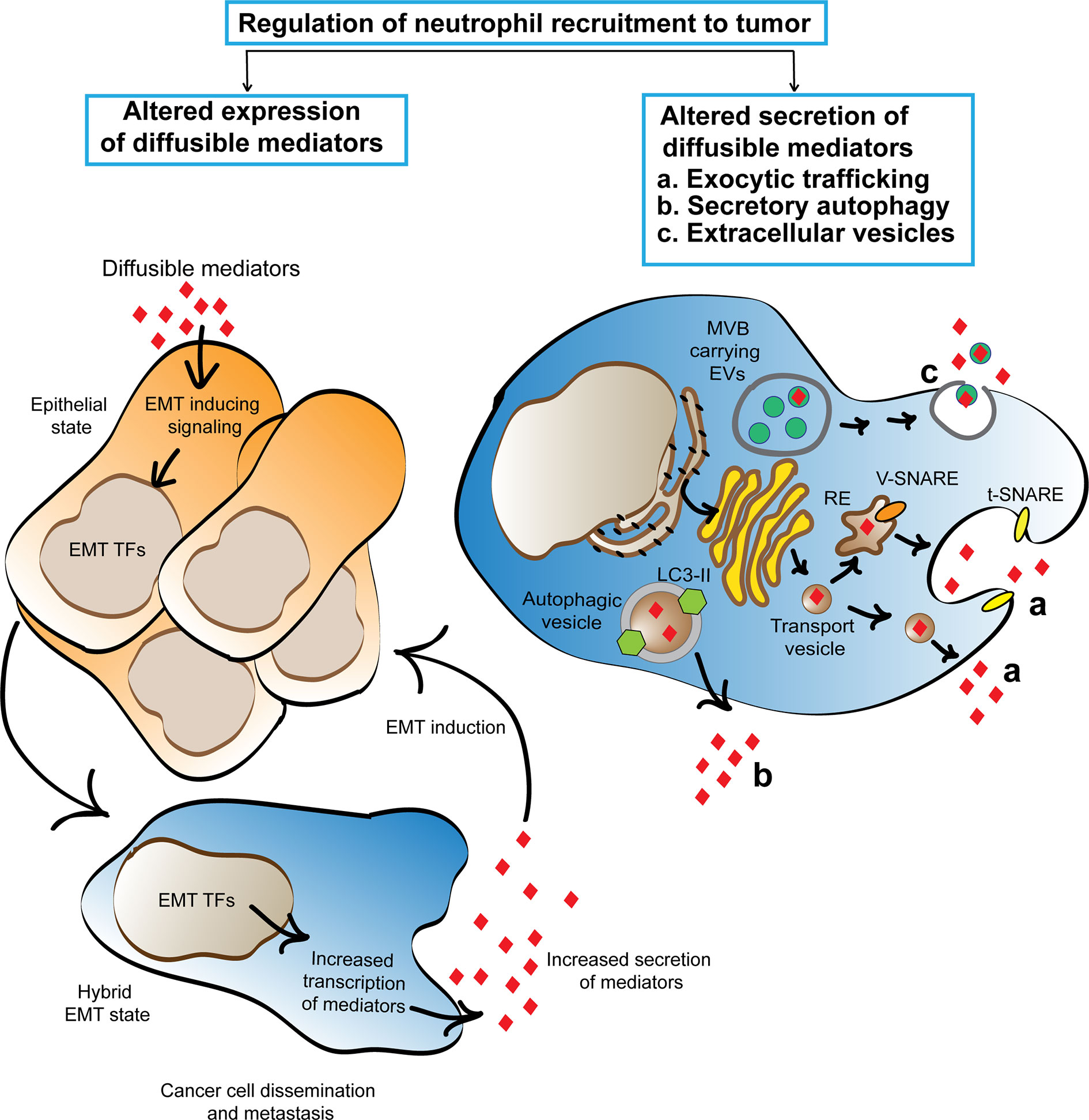
Figure 1 Cartoon depicting the proposed mechanisms that regulate the cancer secretome and favor neutrophil trafficking to tumors. Cancer associated EMT program activation alters cancer cell secretome by inducing the expression of neutrophil recruiting mediators. Three machineries, namely exocytic trafficking, secretory autophagy, and extracellular vesicles are proposed to enhance the release of neutrophil recruiting mediators from cancer cells. The secreted mediators promote EMT using a feed forward mechanism and initiate a chronic cycle of inflammation that supports cancer cell dissemination. TFs, transcription factors; RE, recycling endosome.
Epithelial to Mesenchymal Transition
EMT is classified into three subtypes based on the biological context. Type I EMT is associated with embryonic development and multiple organ formation. Type 2 EMT is involved in wound healing through tissue repair and regeneration, which if unrestrained, could lead to tissue fibrosis, and organ damage. Type 3 EMT is exclusively associated with malignancy and metastatic spread, where cancer cells acquire the ability to invade locally and disseminate systemically (44). Epithelial cells undergoing all three types of EMT tend to lose their epithelial characteristics and acquire migratory mesenchymal cell-like properties. However, EMT is emerging as a dynamic process where cells adopt partial EMT or intermediate/hybrid states featuring a combination of phenotypes of both cell types (45–47). Type 3 EMT (subsequently referred to as EMT) triggers cytoskeletal remodeling, loss of cell-cell adhesion and cell polarity, and gain of migratory and invasive properties, which are proposed to be required for metastasis. A wide range of diffusible mediators released from transformed or non-transformed cells in the tumor niche are known to induce EMT, including growth factors (EGF), cytokines (TGF-β, TNF-α), chemokines (CXCL8, CXCL6), and lipid mediators (leukotriene B4 (LTB4)) (48, 49). Interestingly, EMT has been associated with altered secretory profiles of cancer cells (50–54). For instance, secreted factors from EMT-positive breast cancer cell lines have been reported to induce the in vivo recruitment of granulocytic myeloid derived suppressor cells (G-MDSC), which phenotypically resemble murine neutrophils and share immunosuppressive functions with “N2” neutrophils (50, 55). EMT-induced altered secretome of breast cancer cell lines also favors a tumor-permissive niche by activating tumor-associated macrophages, which further support EMT induction of cancer cells (54).
EMT inducing signals mediate their effects by stimulating a transcription program via the activation or enhanced expression of key EMT transcription factors: SNAIL, Twist and Zeb family proteins, and the T-box transcription factor Brachyury (56, 57). The role of these transcription factors in suppressing epithelial cell-cell adhesion proteins and inducing mesenchymal adhesion molecules has been widely studied (58, 59). Interestingly, the same transcription factors are also emerging as key regulators for the expression of mediators such as cytokines (TNF-α), chemokines (CCL2, CXCL6, GRO, CXCL8, CXCL11), and growth factors (GM-CSF) in cancer cells (50, 52, 53, 60–63). For example, chemokines, including CXCL6 and CXCL8, are one of the many secreted mediators that are upregulated in a Snail-dependent manner when EMT pathways are activated in cancer cells by EGF or TGF-β treatment (50). As many of the secreted mediators from EMT-activated tumors are established chemoattractants of neutrophils (GRO, CXCL8, GM-CSF) and monocytes (CCL2), the release of these mediators upon EMT induction is poised to regulate the immune landscape of the tumor niche.
Exocytic Trafficking Pathways
Conventional mechanisms that underlie the secretion of diffusible mediators, such as cytokines and chemokines, involve constitutive and regulated exocytosis pathways, depending on the cellular and inflammatory context (64–66). Much of our current knowledge comes from characterization in immune cells, particularly macrophages and dendritic cells (DCs) for constitutive secretion, and granulocytes for regulated secretion. In general, cytokines and chemokines carry a leader peptide sequence for secretion that facilitates their trafficking through the ER-Golgi network. Newly synthesized proteins are continuously exocytosed through trafficking from the Golgi network to the plasma membrane via small transport vesicles or tubules, which transport the cargo to the plasma membrane either directly or by merging with recycling endosomes (67). In contrast, pre-formed proteins after transiting through the Golgi network are stockpiled in vesicles or granules, which undergo regulated exocytosis in response to external inputs through receptor-ligand interactions (65). Key molecular players of the trafficking machinery include the evolutionary conserved membrane fusion proteins of the soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) protein family (67). Fusion of the vesicle and target membrane to form the core-SNARE complex is mediated by V (vesicle-associated)-SNARE and t (target membrane)-SNARE members for both constitutive and regulated exocytosis. For instance, the V-SNARE member vesicle-associated membrane protein 3 (VAMP3) localized in recycling endosomes mediates the membrane fusion of CXCL6- and TNF-α-carrying vesicles with the plasma membrane that leads to their constitutive secretion from macrophages and DCs (68–70). In granulocytes, such as eosinophils, however, CXCL6 is released through receptor mediated degranulation or regulated exocytosis, where the function of late endosomal V-SNARE members such as VAMP2 and VAMP7 have been implicated (71–74). VAMP7 is also reported to control the release of CXCL12 from VAMP7-positive late endosomal compartments in DCs, suggesting that the trafficking machinery varies greatly depending on cell types and mediators. Further, both V-SNAREs and t-SNAREs may play a rate-limiting role as their upregulation has been noted in stimulated macrophages and DCs with concomitant increase in mediator secretion (70, 75, 76). In addition, other molecular players such as Rho GTPases, including Rac1 and Cdc42, play important roles in TNF secretion in macrophage by delivering TNF-carrying recycling endosomes to the cell surface (77).
While cytokine trafficking in epithelial cells most likely utilizes the constitutive pathway (64), the mechanisms underlying mediator secretion are not well established in malignant cells even though cancer cells are known to abundantly secrete diverse mediators. An upregulation of signature genes associated with ER to Golgi trafficking pathways has been linked to the increased secretion of mediators, including CCL20, from murine breast cancer cell lines with high metastatic potential (78). Moreover, it has been shown that the secretion of CCL5 depends on the exocytosis of CCL5-carrying pre-made vesicles in the hormone receptor positive breast cancer cell line MCF-7 (79). Whether specific VAMP proteins mediate CCL5 trafficking and vesicle fusion with the plasma membrane and if/how the machinery differs in early- and late-stage malignant cells compared to non-malignant epithelial cells have yet to be determined. Of note, VAMP3 has been reported to be involved in CXCL6 and TNF-α release from the synovial sarcoma cell line SW982, indicating an active role of VAMP proteins in diffusible mediator secretion from cancer cells (80). Furthermore, studies have reported enhanced expression of V-SNARE and t-SNARE members in cancer cells along the course of tumor progression, which may further promote the exocytic release of the mediators in the TME (81).
Autophagy
An unconventional mechanism for diffusible mediator secretion is autophagy. Autophagy is traditionally known for intracellular degradation and recycling of cargos, including damaged organelles or protein aggregates, to maintain cellular homeostasis. The fusion of autophagic vacuole carrying cargos with lysosomes results in cargo degradation by acid hydrolysis and proteolysis (82). Our understanding of the machinery involved in autophagy-dependent secretion, also known as secretory autophagy (SA) (83), is however less clear. Under nutrient deprived conditions in yeast, SA is known to mediate the release of leader peptide-less proteins (84). However, there is emerging evidence that SA is involved in the secretion of mediators that are both leader-less, such as IL-1β and IL18, and leader-positive, such as CXCL6, CXCL8, and TGF-β (85, 86). Attempts to define the sequence of events leading to SA of IL-1β from macrophages in response to lysosomal damage identified several molecular players key to the process (87). For example, TRIM16 serves as the SA cargo receptor that together with VAMP member Sec22b sequesters IL-1β in LC3-II-positive vesicles, where LC3-II is a canonical autophagosome marker. Further, the fusion of cargo vesicles with the plasma membrane as well as the release of cargos are achieved by the coordinated action of dedicated SNARE proteins including syntaxin 3, syntaxin 4, SNAP-23, and SNAP-29.
SA in both cancer cells and CAFs has recently been implicated in shaping the tumor secretome and promoting cancer progression (88, 89). For instance, secretion of CXCL8, IL-1β, LIF (leukemia inhibitory factor), and Fam3 (family with sequence similarity 3 member C) were found elevated or impaired in a murine melanoma cell line when stimulated with an autophagy-inducing peptide or subjected to autophagy related gene knockdown approaches, respectively (90). More strikingly, a correlation between the elevated presence of the same mediators in the serum of patients with high-autophagy melanoma, compared to patients with low-autophagy melanoma, was reported in the same study. Autophagy is also recognized to contribute to oncogenic RAS driven cancer cell migration and invasion by inducing the secretion of the migration promoting CXCL6 chemokine and the transcription of pro-invasive molecules, including MMP2 and WNT5A (91). In addition, CXCL6 secretion was mediated by autophagy in the triple-negative breast cancer cell line MDA-MB-468, which depends on autophagy for survival (92). In contrast, autophagy inhibition promoted CXCL6 secretion from MCF-7 cells, which otherwise does not depend on autophagy for survival. The apparent contrasting effect of autophagy on CXCL6 secretion reflects context dependent regulation of cytokine secretion by autophagy and highlights the need to explore more comprehensively the role of autophagy in mediator secretion in different cancer types and subtypes.
Extracellular Vesicles
Extracellular vesicles (EVs) are heterogeneous in size (93). Exosomes are smaller EVs with a diameter less than 150 nm that originate as intraluminal vesicles (ILVs) by the inward budding of late endosomal vesicles that form multivesicular bodies (MVBs). Upon fusion of MVBs with the plasma membrane, ILVs are released as exosomes in the extracellular milieu. By contrast, other larger EVs (diameter up to 1000 nm) are generated through the outward budding of the plasma membrane (93) and come in different flavors, including microvesicles or ectosomes, migrasomes (secreted along retraction fibers of migrating cells), and oncosomes (secreted by cancer cells) (93–96). EVs are well established as vehicles for diverse cargos, including proteins, lipids, and nucleic acids that mediate intercellular communication. EVs released from cancer cells, CAFs, and immune cells have been shown to induce directional migration of the same or other cell types through autocrine and paracrine communication (93, 97). Interestingly, cancer cell-secreted exosomes were shown to mediate the systemic mobilization of neutrophils to the spleen in an in vivo model of breast cancer (98). However, the role of exosomes as the vehicle for tumor-secreted mediators that directly induce neutrophil migration remains to be determined. A diverse group of cytokines, chemokines and growth factors were found to be associated at the surface of EVs or encapsulated inside EVs that were isolated from cultured immune cells, tissue explants, and different types of biological fluids (99). The availability of the mediators in a free or EV-associated form was reported to depend on the activating stimuli and the cellular system studied. Furthermore, CCL chemokines were found to be enriched in exosomes when tumor cells were exposed to heat stress (100). The degree of exosomal chemokine release may therefore be tunable as tumor cells are exposed to changing physicochemical factors in the dynamic TME. EVs are also emerging as a vital means of tumor-stromal cell communication that further promote tumor progression and metastasis. For instance, osteosarcoma cells release EVs carrying membrane-associated TGF-β1, which was shown to educate mesenchymal stromal cells to release CXCL6, and promote further tumor growth and metastasis (101). In addition, osteosarcoma-derived EVs were shown to induce lung fibroblast differentiation in a TGF-β1 dependent manner, indicating a potential role of EV-associated immune mediators to endorse distant metastasis (102).
Interplay Between TGF-β and Chemokines to Maximize Neutrophil Recruitment
As mentioned, given the presence of diverse cell types in the tumor niche, the TME harbors multiple diffusible mediators. Neutrophil navigation to the tumor niche could therefore be orchestrated by the interplay of different mediators. We recently reported that CXCR2 ligands, potentially growth-related oncogene (GRO) members (CXCL1/2/3), and TGF-β1, which are abundantly secreted by triple-negative breast cancer cells, concertedly induce robust neutrophil migration (24). TGF-β ligands belong to the TGF-β subfamily, with three known mammalian isoforms: TGF- β1, TGF-β2 and TGF-β3, of which TGF-β1 is the most commonly expressed. Cells secrete all isoforms as a latent complex that is activated by the presence of integrins, ECM proteins, and proteolytic enzymes (103). Once released in an active form, all isoforms interact and activate the type II/type I TGF-β receptor complex and propagate signals through SMAD-dependent and -independent pathways (104, 105). TGF-β target genes are involved in regulating fundamental cellular functions such as proliferation, differentiation, migration, senescence, apoptosis, along with maintaining immune homeostasis. The signaling outcome of TGF-β is highly context dependent in cancer. During early stages of cancer, it can prevent tumorigenesis by inhibiting cell proliferation, regulating cell cycle progression, and promoting apoptosis. However, cancer-associated disruption of TGF-β receptor/signaling components and/or the activation of EMT inducing signaling of TGF-β may promote the dissemination of cancer cells (105, 106). The mechanistic basis for the complexity of TGF-β signaling outcome has achieved significant clarity over the years. However, the mechanism and outcome of the crosstalk of TGF-β with other diffusible mediators on tumor progression are only beginning to be understood.
Both cancer and immune cells express TGF-β receptors. Receptor expression on immune cells is further modulated by the mediators present in the tumor niche. For instance, mediators secreted from metastatic prostate cancer cells upregulate the gene expression of the type I TGF-β receptor (TGF-β RI) in neutrophils (107), suggesting that the effect of TGF-β on neutrophil function is tunable. In an in vivo murine model of lung cancer and mesothelioma, TGF-β has been reported to promote the tumor supporting functions of neutrophils and treatment with a systemic inhibitor of TGF-β RI led to increased neutrophil recruitment to tumors indicating a negative regulation of neutrophil migration by TGF-β signaling (8). Conversely, TGF-β signaling was reported to promote neutrophil recruitment to metastatic sites in a genetically engineered in vivo mouse model of metastatic colorectal cancer (28). Additionally, in vitro studies documented various ways by which TGF-β can foster or hinder other neutrophil responses, such as prolonging neutrophil survival, promoting phagocytosis and respiratory burst (108), and impairing granule exocytosis (109).
The role of TGF-β in directional migration of neutrophils is, surprisingly, not clear. Studies have reported strong to no direct effect of TGF-β on neutrophil chemotaxis (110, 111). Given its pleiotropic role, TGF-β may indirectly regulate neutrophil chemotaxis. Indeed, TGF-β1 was reported to promote chemotaxis of immature DCs to CC and CXC chemokines by upregulating chemokine receptor expression (112). Whether TGF-β1 uses a similar mechanism to regulate CXCR1/2 expression and mediate its effect on neutrophil chemotaxis remains unknown. TGF-β1 has also been reported to promote the secretion of CXCL5 from hepatocellular carcinoma cell lines, which in turn induces neutrophil migration (32). Furthermore, TGF-β1 is known to enhance the secretion of leukotrienes from monocyte-derived macrophages and DCs, of which LTB4 is a potent neutrophil recruiting lipid mediator (113, 114). Finally, chemokines may also synergize with TGF-β to optimize cellular responses by triggering the activation of downstream signaling components, such as SMAD3, which was reported to be phosphorylated by chemokines like CCL2 (115).
Targeting Strategies/Perspective
Our knowledge of the multifaceted functions of neutrophils in cancer is rapidly expanding. Yet, a precise understanding of the diffusible mediators that are secreted in the TME and induce neutrophil trafficking to the tumor niche is lacking. Many cancer therapeutic strategies, such as chemotherapy, radiotherapy, and immune-checkpoint inhibitors, have the potential to affect the level of circulating neutrophils or modulate the recruitment or function of TANs, which may in turn impact patient prognosis (116). More effort should therefore be placed on directly targeting the diffusible mediators themselves or the pathways that underlie neutrophil recruitment to the TME. Integrating such neutrophil-focused approaches with routinely applied therapeutic strategies may lead to a synergistic protection against cancer progression. However, the fact that neutrophils are quintessential soldiers of the immune system requires careful consideration in developing neutrophil targeting strategies for cancer therapy. From a mechanistic standpoint, it is therefore crucial to address several questions in the context of neutrophil recruitment to specifically target the process without compromising the overall protective role of neutrophils. For example, (i) which trafficking molecules regulate the secretion of neutrophil recruiting mediators in cancer cells? (ii) Does EMT induction change the expression of these regulators and enhance the release of the mediators? (iii) Does the exocytic pathway/SA/EV-dependent release of mediators further promote EMT by triggering an autocrine-paracrine loop? and (iv) How do mediators from distinct classes such as chemokines and cytokines/growth factors/lipid mediators collaborate to optimize neutrophil recruitment to tumors and reprogram TAN function? Addressing these basic questions will provide a deeper understanding of the molecular players and signaling components that dictate neutrophil trafficking to tumors, which will assist in the design of effective therapeutic strategies.
Author Contributions
SS, LEH, and CAP equally contributed to conceptualizing, writing, and editing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the University of Michigan School of Medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
2. SenGupta S, Subramanian BC, Parent CA. Getting TANned: How the Tumor Microenvironment Drives Neutrophil Recruitment. J Leukocyte Biol (2019) 105(3):449–62. doi: 10.1002/JLB.3RI0718-282R
3. Dvorak HF. Tumors: Wounds That do Not Heal. Similarities Between Tumor Stroma Generation and Wound Healing. N Engl J Med (1986) 315(26):1650–9. doi: 10.1056/NEJM198612253152606
4. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations Across Individuals and Species. Immunity (2019) 50(5):1317–34.e10. doi: 10.1016/j.immuni.2019.03.009
5. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep (2015) 10(4):562–73. doi: 10.1016/j.celrep.2014.12.039
6. Hsu BE, Tabariès S, Johnson RM, Andrzejewski S, Senecal J, Lehuédé C, et al. Immature Low-Density Neutrophils Exhibit Metabolic Flexibility That Facilitates Breast Cancer Liver Metastasis. Cell Rep (2019) 27(13):3902–15.e6. doi: 10.1016/j.celrep.2019.05.091
7. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in Cancer: Neutral No More. Nat Rev Cancer (2016) 16(7):431–46. doi: 10.1038/nrc.2016.52
8. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: "N1" Versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
9. Andzinski L, Kasnitz N, Stahnke S, Wu CF, Gereke M, von Köckritz-Blickwede M, et al. Type I IFNs Induce Anti-Tumor Polarization of Tumor Associated Neutrophils in Mice and Human. Int J Cancer (2016) 138(8):1982–93. doi: 10.1002/ijc.29945
10. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, et al. Cathepsin C Promotes Breast Cancer Lung Metastasis by Modulating Neutrophil Infiltration and Neutrophil Extracellular Trap Formation. Cancer Cell (2021) 39(3):423–37.e7. doi: 10.1016/j.ccell.2020.12.012
11. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil Diversity and Plasticity in Tumour Progression and Therapy. Nat Rev Cancer (2020) 20(9):485–503. doi: 10.1038/s41568-020-0281-y
12. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-Producing γδ T Cells and Neutrophils Conspire to Promote Breast Cancer Metastasis. Nature (2015) 522(7556):345–8. doi: 10.1038/nature14282
13. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases After Surgical Stress. Cancer Res (2016) 76(6):1367–80. doi: 10.1158/0008-5472.CAN-15-1591
14. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer Cells Induce Metastasis-Supporting Neutrophil Extracellular DNA Traps. Sci Trans Med (2016) 8(361):361ra138. doi: 10.1126/scitranslmed.aag1711
15. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil Extracellular Traps Sequester Circulating Tumor Cells and Promote Metastasis. J Clin Invest (2013) 123(8):3446–58. doi: 10.1172/JCI67484
16. Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, et al. Primary Tumors Induce Neutrophil Extracellular Traps With Targetable Metastasis Promoting Effects. JCI Insight (2019) 5(16):e128008. doi: 10.1172/jci.insight.128008
17. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil Extracellular Traps Produced During Inflammation Awaken Dormant Cancer Cells in Mice. Sci (New York NY) (2018) 361(6409):eaao4227. doi: 10.1126/science.aao4227
18. Mahiddine K, Blaisdell A, Ma S, Créquer-Grandhomme A, Lowell CA, Erlebacher A. Relief of Tumor Hypoxia Unleashes the Tumoricidal Potential of Neutrophils. J Clin Invest (2020) 130(1):389–403. doi: 10.1172/JCI130952
19. Blaisdell A, Crequer A, Columbus D, Daikoku T, Mittal K, Dey SK, et al. Neutrophils Oppose Uterine Epithelial Carcinogenesis via Debridement of Hypoxic Tumor Cells. Cancer Cell (2015) 28(6):785–99. doi: 10.1016/j.ccell.2015.11.005
20. Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor Entrained Neutrophils Inhibit Seeding in the Premetastatic Lung. Cancer Cell (2011) 20(3):300–14. doi: 10.1016/j.ccr.2011.08.012
21. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-Associated Neutrophils Stimulate T Cell Responses in Early-Stage Human Lung Cancer. J Clin Invest (2014) 124(12):5466–80. doi: 10.1172/JCI77053
22. Yu PF, Huang Y, Han YY, Lin LY, Sun WH, Rabson AB, et al. Tnfα-Activated Mesenchymal Stromal Cells Promote Breast Cancer Metastasis by Recruiting CXCR2(+) Neutrophils. Oncogene (2017) 36(4):482–90. doi: 10.1038/onc.2016.217
23. Zhang L, Yao J, Wei Y, Zhou Z, Li P, Qu J, et al. Blocking Immunosuppressive Neutrophils Deters Py696-EZH2-Driven Brain Metastases. Sci Trans Med (2020) 12(545):eaaz5387. doi: 10.1126/scitranslmed.aaz5387
24. SenGupta S, Hein LE, Xu Y, Zhang J, Konwerski JR, Li Y, et al. Triple-Negative Breast Cancer Cells Recruit Neutrophils by Secreting TGF-β and CXCR2 Ligands. Front Immunol (2021) 12:659996. doi: 10.3389/fimmu.2021.659996
25. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. γδt17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity (2014) 40(5):785–800. doi: 10.1016/j.immuni.2014.03.013
26. Labelle M, Begum S, Hynes RO. Platelets Guide the Formation of Early Metastatic Niches. Proc Natl Acad Sci USA (2014) 111(30):E3053–61. doi: 10.1073/pnas.1411082111
27. Triner D, Xue X, Schwartz AJ, Jung I, Colacino JA, Shah YM. Epithelial Hypoxia-Inducible Factor 2α Facilitates the Progression of Colon Tumors Through Recruiting Neutrophils. Mol Cell Biol (2017) 37(5). doi: 10.1128/MCB.00481-16
28. Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, Lohuis JO, Ridgway RA, et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell (2019) 36(3):319–36.e7. doi: 10.1016/j.ccell.2019.08.003
29. Amicarella F, Muraro MG, Hirt C, Cremonesi E, Padovan E, Mele V, et al. Dual Role of Tumour-Infiltrating T Helper 17 Cells in Human Colorectal Cancer. Gut (2017) 66(4):692–704. doi: 10.1136/gutjnl-2015-310016
30. Peng ZP, Jiang ZZ, Guo HF, Zhou MM, Huang YF, Ning WR, et al. Glycolytic Activation of Monocytes Regulates the Accumulation and Function of Neutrophils in Human Hepatocellular Carcinoma. J Hepatol (2020) 73(4):906–17. doi: 10.1016/j.jhep.2020.05.004
31. Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. Overexpression of CXCL5 Mediates Neutrophil Infiltration and Indicates Poor Prognosis for Hepatocellular Carcinoma. Hepatol (Baltimore Md) (2012) 56(6):2242–54. doi: 10.1002/hep.25907
32. Haider C, Hnat J, Wagner R, Huber H, Timelthaler G, Grubinger M, et al. Transforming Growth Factor-β and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatol (Baltimore Md) (2019) 69(1):222–36. doi: 10.1002/hep.30166
33. Faget J, Groeneveld S, Boivin G, Sankar M, Zangger N, Garcia M, et al. Neutrophils and Snail Orchestrate the Establishment of a Pro-Tumor Microenvironment in Lung Cancer. Cell Rep (2017) 21(11):3190–204. doi: 10.1016/j.celrep.2017.11.052
34. Jablonska J, Wu CF, Andzinski L, Leschner S, Weiss S. CXCR2-Mediated Tumor-Associated Neutrophil Recruitment Is Regulated by IFN-β. Int J Cancer (2014) 134(6):1346–58. doi: 10.1002/ijc.28551
35. Yang M, Zhang G, Wang Y, He M, Xu Q, Lu J, et al. Tumour-Associated Neutrophils Orchestrate Intratumoural IL-8-Driven Immune Evasion Through Jagged2 Activation in Ovarian Cancer. Br J Cancer (2020) 123(9):1404–16. doi: 10.1038/s41416-020-1026-0
36. Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-Cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res (2016) 4(11):968–82. doi: 10.1158/2326-6066.CIR-16-0188
37. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting Both Tumour-Associated CXCR2(+) Neutrophils and CCR2(+) Macrophages Disrupts Myeloid Recruitment and Improves Chemotherapeutic Responses in Pancreatic Ductal Adenocarcinoma. Gut (2018) 67(6):1112–23. doi: 10.1136/gutjnl-2017-313738
38. Galdiero MR, Varricchi G, Loffredo S, Bellevicine C, Lansione T, Ferrara AL, et al. Potential Involvement of Neutrophils in Human Thyroid Cancer. PloS One (2018) 13(6):e0199740. doi: 10.1371/journal.pone.0199740
39. SenGupta S, Rane MJ, Uriarte SM, Woolley C, Mitchell TC. Human Neutrophils Depend on Extrinsic Factors Produced by Monocytes for Their Survival Response to TLR4 Stimulation. Innate Immun (2019) 25(8):473–86. doi: 10.1177/1753425919871994
40. Wu L, Awaji M, Saxena S, Varney ML, Sharma B, Singh RK. IL-17-CXC Chemokine Receptor 2 Axis Facilitates Breast Cancer Progression by Up-Regulating Neutrophil Recruitment. Am J Pathol (2020) 190(1):222–33. doi: 10.1016/j.ajpath.2019.09.016
41. Gorbachev AV, Fairchild RL. Regulation of Chemokine Expression in the Tumor Microenvironment. Crit Rev Immunol (2014) 34(2):103–20. doi: 10.1615/CritRevImmunol.2014010062
42. Khanna S, Graef S, Mussai F, Thomas A, Wali N, Yenidunya BG, et al. Tumor-Derived GM-CSF Promotes Granulocyte Immunosuppression in Mesothelioma Patients. Clin Cancer Res An Off J Am Assoc Cancer Res (2018) 24(12):2859–72. doi: 10.1158/1078-0432.CCR-17-3757
43. Yu S, Yi M, Xu L, Qin S, Li A, Wu K. CXCL1 as an Unfavorable Prognosis Factor Negatively Regulated by DACH1 in Non-Small Cell Lung Cancer. Front Oncol (2020) 9:1515–. doi: 10.3389/fonc.2019.01515
44. Kalluri R. EMT: When Epithelial Cells Decide to Become Mesenchymal-Like Cells. J Clin Invest (2009) 119(6):1417–9. doi: 10.1172/JCI39675
45. Aiello NM, Kang Y. Context-Dependent EMT Programs in Cancer Metastasis. J Exp Med (2019) 216(5):1016–26. doi: 10.1084/jem.20181827
46. Pastushenko I, Blanpain C. EMT Transition States During Tumor Progression and Metastasis. Trends Cell Biol (2019) 29(3):212–26. doi: 10.1016/j.tcb.2018.12.001
47. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell (2016) 166(1):21–45. doi: 10.1016/j.cell.2016.06.028
48. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and Inflammation: Inseparable Actors of Cancer Progression. Mol Oncol (2017) 11(7):805–23. doi: 10.1002/1878-0261.12095
49. Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, et al. Tumor-Derived CXCL5 Promotes Human Colorectal Cancer Metastasis Through Activation of the ERK/Elk-1/Snail and AKT/Gsk3β/β-Catenin Pathways. Mol Cancer (2017) 16(1):70. doi: 10.1186/s12943-017-0629-4
50. Suarez-Carmona M, Bourcy M, Lesage J, Leroi N, Syne L, Blacher S, et al. Soluble Factors Regulated by Epithelial-Mesenchymal Transition Mediate Tumour Angiogenesis and Myeloid Cell Recruitment. J Pathol (2015) 236(4):491–504. doi: 10.1002/path.4546
51. Dominguez C, David JM, Palena C. Epithelial-Mesenchymal Transition and Inflammation at the Site of the Primary Tumor. Semin Cancer Biol (2017) 47:177–84. doi: 10.1016/j.semcancer.2017.08.002
52. Koo YJ, Kim TJ, Min KJ, So KA, Jung US, Hong JH. CXCL11 Mediates TWIST1-Induced Angiogenesis in Epithelial Ovarian Cancer. Tumour Biol J Int Soc Oncodevelopmental Biol Med (2017) 39(5):1010428317706226. doi: 10.1177/1010428317706226
53. Brenot A, Knolhoff BL, DeNardo DG, Longmore GD. SNAIL1 Action in Tumor Cells Influences Macrophage Polarization and Metastasis in Breast Cancer Through Altered GM-CSF Secretion. Oncogenesis (2018) 7(3):32. doi: 10.1038/s41389-018-0042-x
54. Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A Positive Feedback Loop Between Mesenchymal-Like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell (2014) 25(5):605–20. doi: 10.1016/j.ccr.2014.03.021
55. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol (2018) 9:398. doi: 10.3389/fimmu.2018.00398
56. Roselli M, Fernando RI, Guadagni F, Spila A, Alessandroni J, Palmirotta R, et al. Brachyury, a Driver of the Epithelial-Mesenchymal Transition, Is Overexpressed in Human Lung Tumors: An Opportunity for Novel Interventions Against Lung Cancer. Clin Cancer Res An Off J Am Assoc Cancer Res (2012) 18(14):3868–79. doi: 10.1158/1078-0432.CCR-11-3211
57. Tsai JH, Yang J. Epithelial-Mesenchymal Plasticity in Carcinoma Metastasis. Genes Dev (2013) 27(20):2192–206. doi: 10.1101/gad.225334.113
58. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell (2004) 117(7):927–39. doi: 10.1016/j.cell.2004.06.006
59. Chen M, Wu Y, Zhang H, Li S, Zhou J, Shen J. The Roles of Embryonic Transcription Factor BRACHYURY in Tumorigenesis and Progression. Front Oncol (2020) 10:961. doi: 10.3389/fonc.2020.00961
60. Low-Marchelli JM, Ardi VC, Vizcarra EA, van Rooijen N, Quigley JP, Yang J. Twist1 Induces CCL2 and Recruits Macrophages to Promote Angiogenesis. Cancer Res (2013) 73(2):662–71. doi: 10.1158/0008-5472.CAN-12-0653
61. Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, et al. SNAIL Regulates Interleukin-8 Expression, Stem Cell-Like Activity, and Tumorigenicity of Human Colorectal Carcinoma Cells. Gastroenterology (2011) 141(1):279–91, 91.e1-5. doi: 10.1053/j.gastro.2011.04.008
62. Hsu DS, Wang HJ, Tai SK, Chou CH, Hsieh CH, Chiu PH, et al. Acetylation of Snail Modulates the Cytokinome of Cancer Cells to Enhance the Recruitment of Macrophages. Cancer Cell (2014) 26(4):534–48. doi: 10.1016/j.ccell.2014.09.002
63. Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. IL-8 Signaling Plays a Critical Role in the Epithelial-Mesenchymal Transition of Human Carcinoma Cells. Cancer Res (2011) 71(15):5296–306. doi: 10.1158/0008-5472.CAN-11-0156
64. Stanley AC, Lacy P. Pathways for Cytokine Secretion. Physiol (Bethesda Md) (2010) 25(4):218–29. doi: 10.1152/physiol.00017.2010
65. Stow JL, Murray RZ. Intracellular Trafficking and Secretion of Inflammatory Cytokines. Cytokine Growth Factor Rev (2013) 24(3):227–39. doi: 10.1016/j.cytogfr.2013.04.001
66. Lacy P, Stow JL. Cytokine Release From Innate Immune Cells: Association With Diverse Membrane Trafficking Pathways. Blood (2011) 118(1):9–18. doi: 10.1182/blood-2010-08-265892
67. Murray RZ, Stow JL. Cytokine Secretion in Macrophages: SNAREs, Rabs, and Membrane Trafficking. Front Immunol (2014) 5:538. doi: 10.3389/fimmu.2014.00538
68. Manderson AP, Kay JG, Hammond LA, Brown DL, Stow JL. Subcompartments of the Macrophage Recycling Endosome Direct the Differential Secretion of IL-6 and TNFalpha. J Cell Biol (2007) 178(1):57–69. doi: 10.1083/jcb.200612131
69. Verboogen DRJ, Revelo NH, Ter Beest M, van den Bogaart G. Interleukin-6 Secretion Is Limited by Self-Signaling in Endosomes. J Mol Cell Biol (2019) 11(2):144–57. doi: 10.1093/jmcb/mjy038
70. Murray RZ, Kay JG, Sangermani DG, Stow JL. A Role for the Phagosome in Cytokine Secretion. Sci (New York NY) (2005) 310(5753):1492–5. doi: 10.1126/science.1120225
71. Lacy P, Logan MR, Bablitz B, Moqbel R. Fusion Protein Vesicle-Associated Membrane Protein 2 Is Implicated in IFN-Gamma-Induced Piecemeal Degranulation in Human Eosinophils From Atopic Individuals. J Allergy Clin Immunol (2001) 107(4):671–8. doi: 10.1067/mai.2001.113562
72. Logan MR, Lacy P, Bablitz B, Moqbel R. Expression of Eosinophil Target SNAREs as Potential Cognate Receptors for Vesicle-Associated Membrane Protein-2 in Exocytosis. J Allergy Clin Immunol (2002) 109(2):299–306. doi: 10.1067/mai.2002.121453
73. Willetts L, Felix LC, Jacobsen EA, Puttagunta L, Condjella RM, Zellner KR, et al. Vesicle-Associated Membrane Protein 7-Mediated Eosinophil Degranulation Promotes Allergic Airway Inflammation in Mice. Commun Biol (2018) 1:83. doi: 10.1038/s42003-018-0081-z
74. Davoine F, Lacy P. Eosinophil Cytokines, Chemokines, and Growth Factors: Emerging Roles in Immunity. Front Immunol (2014) 5:570–. doi: 10.3389/fimmu.2014.00570
75. Chiaruttini G, Piperno GM, Jouve M, De Nardi F, Larghi P, Peden AA, et al. The SNARE VAMP7 Regulates Exocytic Trafficking of Interleukin-12 in Dendritic Cells. Cell Rep (2016) 14(11):2624–36. doi: 10.1016/j.celrep.2016.02.055
76. Pagan JK, Wylie FG, Joseph S, Widberg C, Bryant NJ, James DE, et al. The T-SNARE Syntaxin 4 Is Regulated During Macrophage Activation to Function in Membrane Traffic and Cytokine Secretion. Curr Biol CB (2003) 13(2):156–60. doi: 10.1016/S0960-9822(03)00006-X
77. Stanley AC, Wong CX, Micaroni M, Venturato J, Khromykh T, Stow JL, et al. The Rho GTPase Rac1 Is Required for Recycling Endosome-Mediated Secretion of TNF in Macrophages. Immunol Cell Biol (2014) 92(3):275–86. doi: 10.1038/icb.2013.90
78. Howley BV, Link LA, Grelet S, El-Sabban M, Howe PH. A CREB3-Regulated ER-Golgi Trafficking Signature Promotes Metastatic Progression in Breast Cancer. Oncogene (2018) 37(10):1308–25. doi: 10.1038/s41388-017-0023-0
79. Soria G, Lebel-Haziv Y, Ehrlich M, Meshel T, Suez A, Avezov E, et al. Mechanisms Regulating the Secretion of the Promalignancy Chemokine CCL5 by Breast Tumor Cells: CCL5's 40s Loop and Intracellular Glycosaminoglycans. Neoplasia (2012) 14(1):1–IN3. doi: 10.1593/neo.111122
80. Boddul SV, Meng J, Dolly JO, Wang J. SNAP-23 and VAMP-3 Contribute to the Release of IL-6 and Tnfα From a Human Synovial Sarcoma Cell Line. FEBS J (2014) 281(3):750–65. doi: 10.1111/febs.12620
81. Raja SA, Abbas S, Shah STA, Tariq A, Bibi N, Yousuf A, et al. Increased Expression Levels of Syntaxin 1A and Synaptobrevin 2/Vesicle-Associated Membrane Protein-2 Are Associated With the Progression of Bladder Cancer. Genet Mol Biol (2019) 42(1):40–7. doi: 10.1590/1678-4685-gmb-2017-0339
82. Dikic I, Elazar Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat Rev Mol Cell Biol (2018) 19(6):349–64. doi: 10.1038/s41580-018-0003-4
83. Gonzalez CD, Resnik R, Vaccaro MI. Secretory Autophagy and Its Relevance in Metabolic and Degenerative Disease. Front Endocrinol (Lausanne) (2020) 11:266. doi: 10.3389/fendo.2020.00266
84. Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional Secretion of Pichia Pastoris Acb1 Is Dependent on GRASP Protein, Peroxisomal Functions, and Autophagosome Formation. J Cell Biol (2010) 188(4):537–46. doi: 10.1083/jcb.200911149
85. New J, Thomas SM. Autophagy-Dependent Secretion: Mechanism, Factors Secreted, and Disease Implications. Autophagy (2019) 15(10):1682–93. doi: 10.1080/15548627.2019.1596479
86. Nüchel J, Ghatak S, Zuk AV, Illerhaus A, Mörgelin M, Schönborn K, et al. TGFB1 Is Secreted Through an Unconventional Pathway Dependent on the Autophagic Machinery and Cytoskeletal Regulators. Autophagy (2018) 14(3):465–86. doi: 10.1080/15548627.2017.1422850
87. Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, et al. Dedicated SNAREs and Specialized TRIM Cargo Receptors Mediate Secretory Autophagy. EMBO J (2017) 36(1):42–60. doi: 10.15252/embj.201695081
88. Bustos SO, Antunes F, Rangel MC, Chammas R. Emerging Autophagy Functions Shape the Tumor Microenvironment and Play a Role in Cancer Progression - Implications for Cancer Therapy. Front Oncol (2020) 10:606436. doi: 10.3389/fonc.2020.606436
89. New J, Arnold L, Ananth M, Alvi S, Thornton M, Werner L, et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res (2017) 77(23):6679–91. doi: 10.1158/0008-5472.CAN-17-1077
90. Kraya AA, Piao S, Xu X, Zhang G, Herlyn M, Gimotty P, et al. Identification of Secreted Proteins That Reflect Autophagy Dynamics Within Tumor Cells. Autophagy (2015) 11(1):60–74. doi: 10.4161/15548627.2014.984273
91. Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-Dependent Production of Secreted Factors Facilitates Oncogenic RAS-Driven Invasion. Cancer Discovery (2014) 4(4):466–79. doi: 10.1158/2159-8290.CD-13-0841
92. Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol Cancer Res MCR (2015) 13(4):651–8. doi: 10.1158/1541-7786.MCR-14-0487
93. Sung BH, Parent CA, Weaver AM. Extracellular Vesicles: Critical Players During Cell Migration. Dev Cell (2021) 56(13):1861–74. doi: 10.1016/j.devcel.2021.03.020
94. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular Transfer of the Oncogenic Receptor EGFRvIII by Microvesicles Derived From Tumour Cells. Nat Cell Biol (2008) 10(5):619–24. doi: 10.1038/ncb1725
95. Cocucci E, Meldolesi J. Ectosomes and Exosomes: Shedding the Confusion Between Extracellular Vesicles. Trends Cell Biol (2015) 25(6):364–72. doi: 10.1016/j.tcb.2015.01.004
96. Ma L, Li Y, Peng J, Wu D, Zhao X, Cui Y, et al. Discovery of the Migrasome, an Organelle Mediating Release of Cytoplasmic Contents During Cell Migration. Cell Res (2015) 25(1):24–38. doi: 10.1038/cr.2014.135
97. Harris DA, Patel SH, Gucek M, Hendrix A, Westbroek W, Taraska JW. Exosomes Released From Breast Cancer Carcinomas Stimulate Cell Movement. PloS One (2015) 10(3):e0117495. doi: 10.1371/journal.pone.0117495
98. Bobrie A, Krumeich S, Reyal F, Recchi C, Moita LF, Seabra MC, et al. Rab27a Supports Exosome-Dependent and -Independent Mechanisms That Modify the Tumor Microenvironment and can Promote Tumor Progression. Cancer Res (2012) 72(19):4920–30. doi: 10.1158/0008-5472.CAN-12-0925
99. Fitzgerald W, Freeman ML, Lederman MM, Vasilieva E, Romero R, Margolis L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci Rep (2018) 8(1):8973. doi: 10.1038/s41598-018-27190-x
100. Chen T, Guo J, Yang M, Zhu X, Cao X. Chemokine-Containing Exosomes Are Released From Heat-Stressed Tumor Cells via Lipid Raft-Dependent Pathway and Act as Efficient Tumor Vaccine. J Immunol (Baltimore Md 1950) (2011) 186(4):2219–28. doi: 10.4049/jimmunol.1002991
101. Baglio SR, Lagerweij T, Pérez-Lanzón M, Ho XD, Léveillé N, Melo SA, et al. Blocking Tumor-Educated MSC Paracrine Activity Halts Osteosarcoma Progression. Clin Cancer Res An Off J Am Assoc Cancer Res (2017) 23(14):3721–33. doi: 10.1158/1078-0432.CCR-16-2726
102. Mazumdar A, Urdinez J, Boro A, Migliavacca J, Arlt MJE, Muff R, et al. Osteosarcoma-Derived Extracell Vesicles Induce Lung Fibroblast Reprogramming. Int J Mol Sci (2020) 21(15):5451. doi: 10.3390/ijms21155451
103. Munger JS, Sheppard D. Cross Talk Among TGF-β Signaling Pathways, Integrins, and the Extracellular Matrix. Cold Spring Harbor Perspect Biol (2011) 3(11):a005017. doi: 10.1101/cshperspect.a005017
104. Bierie B, Moses HL. Tumour Microenvironment: TGFbeta: The Molecular Jekyll and Hyde of Cancer. Nat Rev Cancer (2006) 6(7):506–20. doi: 10.1038/nrc1926
106. Gu S, Feng X-H. TGF-β Signaling in Cancer. Acta Biochim Biophys Sin (2018) 50(10):941–9. doi: 10.1093/abbs/gmy092
107. Costanzo-Garvey DL, Keeley T, Case AJ, Watson GF, Alsamraae M, Yu Y, et al. Neutrophils Are Mediators of Metastatic Prostate Cancer Progression in Bone. Cancer Immunol Immunother CII (2020) 69(6):1113–30. doi: 10.1007/s00262-020-02527-6
108. Lagraoui M, Gagnon L. Enhancement of Human Neutrophil Survival and Activation by TGF-Beta 1. Cell Mol Biol (Noisy-le-Grand France) (1997) 43(3):313–8.
109. Shen L, Smith JM, Shen Z, Eriksson M, Sentman C, Wira CR. Inhibition of Human Neutrophil Degranulation by Transforming Growth Factor-Beta1. Clin Exp Immunol (2007) 149(1):155–61. doi: 10.1111/j.1365-2249.2007.03376.x
110. Brandes ME, Mai UE, Ohura K, Wahl SM. Type I Transforming Growth Factor-Beta Receptors on Neutrophils Mediate Chemotaxis to Transforming Growth Factor-Beta. J Immunol (Baltimore Md 1950) (1991) 147(5):1600–6.
111. Parekh T, Saxena B, Reibman J, Cronstein BN, Gold LI. Neutrophil Chemotaxis in Response to TGF-Beta Isoforms (TGF-Beta 1, TGF-Beta 2, TGF-Beta 3) Is Mediated by Fibronectin. J Immunol (Baltimore Md 1950) (1994) 152(5):2456–66.
112. Sato K, Kawasaki H, Nagayama H, Enomoto M, Morimoto C, Tadokoro K, et al. TGF-Beta 1 Reciprocally Controls Chemotaxis of Human Peripheral Blood Monocyte-Derived Dendritic Cells via Chemokine Receptors. J Immunol (Baltimore Md 1950) (2000) 164(5):2285–95. doi: 10.4049/jimmunol.164.5.2285
113. Esser J, Gehrmann U, D'Alexandri FL, Hidalgo-Estévez AM, Wheelock CE, Scheynius A, et al. Exosomes From Human Macrophages and Dendritic Cells Contain Enzymes for Leukotriene Biosynthesis and Promote Granulocyte Migration. J Allergy Clin Immunol (2010) 126(5):1032–40, 40.e1-4. doi: 10.1016/j.jaci.2010.06.039
114. Subramanian BC, Majumdar R, Parent CA. The Role of the LTB(4)-BLT1 Axis in Chemotactic Gradient Sensing and Directed Leukocyte Migration. Semin Immunol (2017) 33:16–29. doi: 10.1016/j.smim.2017.07.002
115. Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N. CCL2/CCR2 Chemokine Signaling Coordinates Survival and Motility of Breast Cancer Cells Through Smad3 Protein- and P42/44 Mitogen-Activated Protein Kinase (MAPK)-Dependent Mechanisms. J Biol Chem (2012) 287(43):36593–608. doi: 10.1074/jbc.M112.365999
Keywords: neutrophils, tumor-associated neutrophils (TANs), chemokines, TGF-β, EMT, secretory pathways, secretory autophagy, EVs
Citation: SenGupta S, Hein LE and Parent CA (2021) The Recruitment of Neutrophils to the Tumor Microenvironment Is Regulated by Multiple Mediators. Front. Immunol. 12:734188. doi: 10.3389/fimmu.2021.734188
Received: 30 June 2021; Accepted: 26 August 2021;
Published: 10 September 2021.
Edited by:
Jörg Renkawitz, Ludwig Maximilian University of Munich, GermanyReviewed by:
Leo Marc Carlin, University of Glasgow, United KingdomGeert Van Den Bogaart, University of Groningen, Netherlands
Copyright © 2021 SenGupta, Hein and Parent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuvasree SenGupta, c3JlZXNnQHVtaWNoLmVkdQ==