 Theodore J. Cory1
Theodore J. Cory1 Kierstin L. Davis
Kierstin L. Davis Brandt D. Pence
Brandt D. Pence
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 11 November 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.733921
This article is part of the Research TopicImmunometabolic Mechanisms Underlying the Severity of COVID-19View all 25 articles
A hallmark of COVID-19 is a hyperinflammatory state associated with severity. Monocytes undergo metabolic reprogramming and produce inflammatory cytokines when stimulated with SARS-CoV-2. We hypothesized that binding by the viral spike protein mediates this effect, and that drugs which regulate immunometabolism could inhibit the inflammatory response. Monocytes stimulated with recombinant SARS-CoV-2 spike protein subunit 1 showed a dose-dependent increase in glycolytic metabolism associated with production of pro-inflammatory cytokines. This response was dependent on hypoxia-inducible factor-1α, as chetomin inhibited glycolysis and cytokine production. Inhibition of glycolytic metabolism by 2-deoxyglucose (2-DG) or glucose deprivation also inhibited the glycolytic response, and 2-DG strongly suppressed cytokine production. Glucose-deprived monocytes rescued cytokine production by upregulating oxidative phosphorylation, an effect which was not present in 2-DG-treated monocytes due to the known effect of 2-DG on suppressing mitochondrial metabolism. Finally, pre-treatment of monocytes with metformin strongly suppressed spike protein-mediated cytokine production and metabolic reprogramming. Likewise, metformin pre-treatment blocked cytokine induction by SARS-CoV-2 strain WA1/2020 in direct infection experiments. In summary, the SARS-CoV-2 spike protein induces a pro-inflammatory immunometabolic response in monocytes that can be suppressed by metformin, and metformin likewise suppresses inflammatory responses to live SARS-CoV-2. This has potential implications for the treatment of hyperinflammation during COVID-19.
The ongoing coronavirus disease 2019 (COVID-19) pandemic has presently claimed more than 3 million lives worldwide as of mid-April 2021 (1). COVID-19 is caused by a novel highly pathogenic coronavirus classified as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) (2). A hallmark of severe COVID-19 is hyperinflammation (3), although cytokine expression patterns in individuals are diverse, leading to controversy over classification of COVID-19 related inflammation as cytokine storm, macrophage activation syndrome, multisystem inflammatory syndrome, etc. Regardless, inflammatory cytokines appear to play a principal role in mediating COVID-19 symptoms, therefore therapies which target these responses are paramount to treating severe COVID-19. As such, a fuller understanding of the cellular and molecular mechanisms mediating hypercytokinemia during SARS-CoV-2 infection is necessary.
Mononuclear phagocytes such as monocytes and macrophages are key constituents of the innate immune system, and produce pro-inflammatory cytokines during viral infection (4–7). We have previously suggested a key role for these cells in mediating severity of COVID-19 (7–9). Monocyte and monocyte-derived macrophage infiltration into the lungs has been linked to severe COVID-19 in single cell RNA sequencing studies (10–13) and postmortem analyses (14–17) in human patients, as well as during experimental infections in animal models including mice (18, 19), hamsters (20), and various non-human primates (21–25). Monocytes in individuals infected with SARS-CoV-2 display phenotypic changes associated with hyperinflammation, including reduced HLA-DR expression (26–28), increased CD16 expression (27, 29–31), and increased cytokine production (32–35). Both monocytes (36–38) and monocyte-derived macrophages (39, 40) also produce pro-inflammatory cytokines under direct infection with SARS-CoV-2, although infection at least in macrophages appears to be abortive (39, 40).
The past decade has seen an explosion in scientific interest in the regulation of immune cell activation and function by metabolic reprogramming. Under pro-inflammatory conditions, immune cells – including myeloid cells – generally undergo a switch to aerobic glycolysis which provides ATP sufficient to support cellular functions which propagate pro-inflammatory and anti-pathogen host responses (41). Recently, Codo et al. demonstrated pro-inflammatory glycolytic reprogramming in monocytes infected with SARS-CoV-2 (36), and SARS-CoV-2 also appears to alter monocyte lipid metabolism to promote lipid droplet formation which is associated with pro-inflammatory cytokine production (37).
SARS-CoV-2 therefore appears to reprogram metabolism in monocytes, but the viral factors which mediate these responses are unclear. Research in the 2003 epidemic SARS-CoV-1 suggested that the viral spike protein could mediate pro-inflammatory activation in macrophages (42, 43), and recent evidence suggests the spike protein of SARS-CoV-2 also activates inflammatory responses in macrophages and monocytes both in vitro and in vivo (44, 45). Given this, we hypothesized that spike protein binding to monocytes mediates glycolytic reprogramming to promote pro-inflammatory responses of these cells to SARS-CoV-2. Our results herein support this hypothesis, and we additionally report outcomes from experiments aimed at evaluating the responsible cellular signaling mechanisms, as well as potential pharmaceutical strategies for inhibiting these responses.
Healthy 18–35-year-old subjects (N=14) were recruited without respect to sex or race. Participants reported to the laboratory approximately every two weeks for blood collection, and 8-24 ml blood was collected into EDTA-treated vacutainer tubes by venipuncture. Blood was immediately used for cell isolations as described below.
Assays were performed on purified human classical monocytes isolated using immunomagnetic negative sorting (EasySep Direct Human Monocyte Isolation Kit, StemCell Technologies, Cambridge, MA). As we have previously described (46), this procedure results in a highly pure (> 85%) population of classical monocytes, with depletion of intermediate and non-classical monocytes due to the presence of an anti-CD16 antibody in the cocktail. Isolation purity was verified at several points throughout the current study and averaged approximately 90% (not shown). Cells were counted at 10× dilution using a Scepter cell counter (Millipore Sigma, St. Louis, MO). Isolated monocytes were immediately utilized in downstream assays, and no cells were frozen for later use.
Unless otherwise specified, all assays were performed using Seahorse XF base DMEM medium (Agilent, Santa Clara, CA) supplemented with 10 mM glucose and 2 mM L-glutamine (Millipore Sigma, St. Louis, MO). Assays utilizing glucose deprivation omitted glucose from the media preparation. Media was not supplemented with fetal bovine serum or other additives. Recombinant spike protein subunit 1 (S1) was purchased from RayBiotech (Peachtree Corners, GA). 2-deoxyglucose, chetomin, compound C, and metformin were purchased from Millipore Sigma (St. Louis, MO). SARS-CoV-2 WA1/2020 strain was provided by Dr. Colleen Jonsson, Regional Biocontainment Laboratory, University of Tennessee Health Science Center.
Glycolysis and oxidative phosphorylation were respectively quantified via kinetic monitoring of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) on a Seahorse XFp analyzer (Agilent, Santa Clara, CA). For all assays, monocytes were plated at 1.5×105 cells per well, and wells A and H of the XFp plate were background wells with no cells. All analyses were run in duplicate. Plated cells were incubated at 37°C in a non-CO2 incubator for 1 hour prior to assays to stabilize pH. All wells were imaged at 10× magnification for cell counting in order to adjust raw measurements for cell number.
For quantification of dose response to S1, 5 basal measurements were made, followed by injection of media (wells B-C), 100 nM spike protein (wells D-E), or 300 nM spike protein (wells F-G). After injection into existing media in the well, spike protein concentrations were 10-fold lower than injection concentrations, thereby giving final spike protein concentrations of 0 nM, 10 nM, or 30 nM. Following injection, ECAR and OCR were monitored serially for 60 measurements. Following the assay, cell culture supernatants were removed, pooled by duplicate, and stored at -80°C. Cells were then lysed with 100 μl Trizol (Thermo Fisher Scientific, Waltham, MA), pooled by duplicate, and stored at -80°C as we have previously described (47).
For chetomin and metformin Seahorse assays, cells were incubated in media as above (wells B-E), or either 10 nM chetomin or 50 mM metformin during the 1-hour pre-incubation period (wells F-G). 5 basal ECAR/OCR measurements were performed, followed by injection of media (wells B-C) or 300 nM spike protein (wells D-G) for a final concentration of 0 nM (wells B-C) or 30 nM spike protein (wells D-G) as above. Following injection, ECAR and OCR were monitored serially for 30 measurements. Cell culture supernatants and Trizol lysates were processed as described above following the end of the assay.
For glycolysis inhibition assays, cells were incubated in media, 10 mM 2-deoxyglucose, or media without glucose (glucose deprivation) during the 1-hour pre-incubation period. 5 basal ECAR/OCR measurements were performed, followed by injection of 300 nM spike protein to all wells for a final concentration of 30 nM spike protein per well as above. Spike protein was prepared in non-glucose media for the glucose deprivation condition. Following injection, ECAR and OCR were monitored serially for 30 measurements. Cell culture supernatants and Trizol lysates were processed as described above following the end of the assay.
Isolated monocytes were incubated in RPMI-1640 media (Gibco, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Gibco), with or without 50 mM metformin, for 1-hour. Cells were then treated with media or infected with SARS-CoV-2 virus (WA1/2020 isolate) at 0.5 MOI and incubated for 24 hours. Cell culture supernatants were collected from untreated and infected cells and stored at -80°C until analysis. SARS-CoV-2 experiments were conducted under biosafety level 3 at the Regional Biocontainment Laboratory at the University of Tennessee Health Science Center.
RNA isolation was performed using the Trizol procedure based on manufacturer’s instructions from cells lysed directly in the microplate or Seahorse plate wells as applicable. Isolated RNA (300-400 ng depending on experiment) was reverse-transcribed to cDNA using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA). Gene expression was analyzed using commercial pre-validated gene expression assays and Taqman reagents (Thermo Fisher Scientific, Waltham, MA). Relative gene expression was quantified using the 2-ΔΔCt method (48) against B2M or ACTB as housekeeping genes. Primer/probe IDs were: B2M Hs00187842_m1; ACTB Hs03023943_g1; IL1B Hs01555410_m1; IL6 Hs00174131_m1; CXCL8 Hs00174103_m1; TNF Hs00174128_m1; CPT1A Hs00912671_m1; G6PD Hs00166169_m1; HLA-DRA Hs00219575_m1; CDKN2A Hs00923894_m1; TLR4 Hs00152939_m1; IL10 Hs00961622_m1; TGFB1 Hs00998133_m1.
For protein quantification, cell culture supernatants harvested from microplates or Seahorse XFp plates were analyzed via ELISA. Commercial DuoSet matched-antibody reagent sets were purchased from R&D Systems (Minneapolis, MN) for quantifying human IL-6, human IL-1β, human IL-8, and human TNFα and were used according to manufacturer’s instructions. All samples were run in duplicate at 5× dilution (SARS-CoV-2 assays), or 50× dilution (Seahorse S1 dose response assays), or 10× dilution (all others) and assessed against a standard curve.
Protein concentration of angiotensin converting enzyme 2 (ACE2) and C-reactive protein (CRP) was performed by ELISA on plasma samples collected by venipuncture from subjects at the beginning of the study. Peripheral blood was collected by venipuncture into EDTA-coated vacutainer tubes, centrifuged at 1,500×g for 15 min, aliquoted, and stored at -80°C until analysis. Plasma samples were analyzed in duplicate at 10× (ACE2) or 10,000× (CRP) using commercial DuoSet matched-antibody reagent kits (R&D Systems) according to manufacturer’s instructions and assessed against a standard curve. ACE2 and CRP data are included in the FigShare repository (49), along with other clinical and anthropomorphic data for experimental subjects.
All data processing and statistical analyses were performed using R v. 3.6.2 (50). Isolated monocytes from each subject were given all treatments for each experiment, so data were paired and analyzed using within-subjects designs. Data were checked for normality by Shapiro-Wilk test and analyzed by one-way repeated measures ANOVA (RM-ANOVA, for data which met the normality assumption) or Friedman’s test (for data which did not meet the normality assumption). For analyses with significant main effects, post hoc mean separation was performed using pairwise paired T tests (for RM-ANOVA) or pairwise Wilcoxon signed-rank tests (for Friedman’s tests) with p-value adjustment using the Holm-Bonferroni method (51). Significance cutoff was p<0.05.
All raw data and analytical scripts (as R markdown files) are available in a dedicated FigShare repository (49). This manuscript was posted to the preprint server bioRxiv prior to submission for publication (52).
All human subjects activities were approved by the Institutional Review Board at the University of Memphis under protocol 4316, and subjects provided written informed consent prior to enrollment.
Recently it was demonstrated that SARS-CoV-2 promotes metabolic reprogramming in monocytes during infection (36, 37). Research in SARS-CoV-1 suggested that the viral spike protein induces inflammatory responses in macrophages (42, 43), and this has recently been replicated using spike protein from SARS-CoV-2 (44, 45). Likewise, spike protein binding to C-type lectins has recently been shown to mediate pro-inflammatory processes in myeloid cells (53, 54). Therefore, we hypothesized that the SARS-CoV-2 spike protein mediates a pro-inflammatory metabolic reprogramming in monocytes which could be a basis for hypercytokinemia. Stimulation of isolated human classical monocytes with recombinant spike protein subunit 1 (S1) from SARS-CoV-2 induced glycolytic activation (Figure 1A) and suppressed oxidative phosphorylation (OXPHOS, Figure 1C) in a dose-dependent manner. The effect of S1 dose was significant for both extracellular acidification rate (F2,14 = 72.44, p<0.0001, Figure 1B) and oxygen consumption rate (F2,14 = 5.785, p=0.0147, Figure 1D) as measured by quantification of area under the response curve.
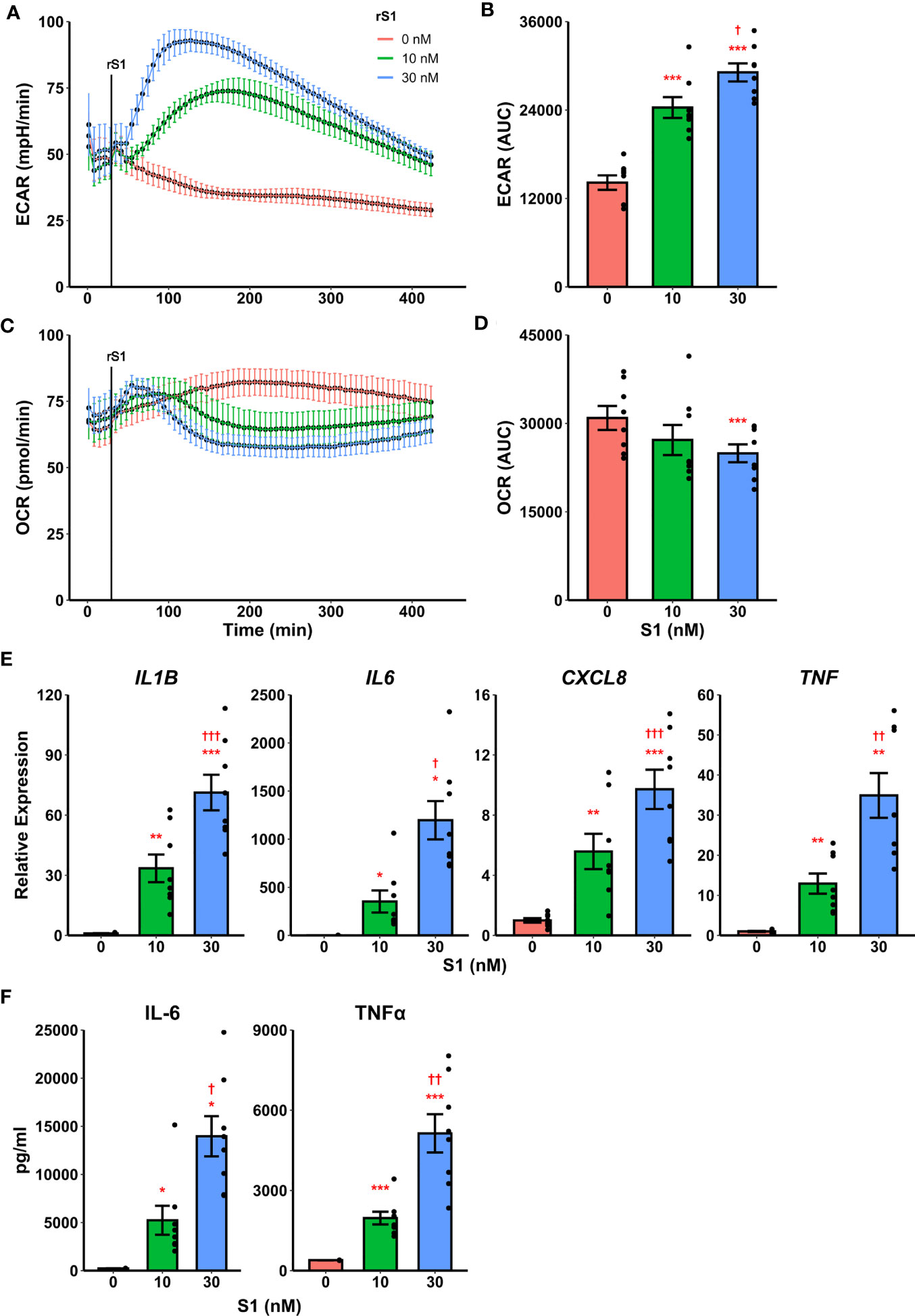
Figure 1 Recombinant SARS-CoV-2 spike protein subunit 1 (rS1) mediates immunometabolic activation of monocytes. (A) Monocytes increase extracellular acidification response rate (ECAR) in a dose-dependent manner when treated with rS1. (B) Quantification of ECAR by area under the curve (AUC). (C) rS1 treatment suppresses oxygen consumption rate (OCR) in monocytes in a dose-dependent fashion. (D) Quantification of OCR by AUC. (E) Gene expression analysis by qPCR reveals dose-dependent increases in responses of IL1B, IL6, CXCL8, and TNF to rS1 stimulation. (F) Protein expression analysis by ELISA reveals dose-dependent increases in responses of IL-6 and TNFα to rS1 stimulation. ECAR and OCR data in panels A-D are adjusted for values indexed to 1×105 cells/well. *, **, ***p < 0.05, p < 0.01, p < 0.001 vs. 0 nM rS1. †, ††, †††: p < 0.05, p < 0.01, p < 0.001 vs. 10 nM rS1. N = 8 biological replicates. Bars and error bars are mean ± SEM.
Additionally, recombinant S1 treatment caused a dose-dependent increase transcription of pro-inflammatory cytokines (Figure 1E) including IL1B (F2,14 = 50.98, p<0.001), IL6 (Friedman χ2 (df=2)=16, p<0.001), CXCL8 (F2,14 = 38.19, p<0.001), and TNF (F2,14 = 28.41, p<0.001) as measured by qPCR. These cytokines have been implicated in the pathogenesis of SARS-CoV-2 and in COVID-19-related hypercytokinemia in several studies (27, 55–60). To confirm that increased transcription resulted in increased protein expression, we evaluated protein concentrations of key cytokines in the supernatant of S1-stimulated monocytes by enzyme-linked immunosorbent assay (ELISA) (Figure 1F). S1 increased protein expression of interleukin (IL)-6 (Friedman χ2 (df=2)=16, p<0.001) and tumor necrosis factor (TNF)-α (F2,14 = 37.73, p<0.001) in a dose-dependent manner.
Recombinant S1 treatment also increased gene expression of the anti-inflammatory cytokine IL-10 (Wilcoxon Z=0, p=0.016, Figure 2A) and decreased gene expression of the anti-inflammatory cytokine TGF-β (Wilcoxon Z=28, p=0.016, Figure 2B). We additionally analyzed gene expression for a variety of polarization and reprogramming markers (Figure 2C), and found that rS1 treatment increased expression of the senescence-associated gene for p16 (CDKN2A, t(df=5)=-2.985, p=0.031) as well as HLA-DRA (t(df=5)=-2.887, p=0.034). Treatment with 30 nM rS1 also reduced expression of metabolism-related genes G6PD (Wilcoxon Z=21, p=0.031) and CPT1A (t(df=5)=15.373, p<0.001) and the LPS receptor TLR4 (t(df=5)=4.833, p=0.005). Therefore, monocytes appear to be activated by S1 and upregulate expression of both pro- and anti-inflammatory cytokines, and additionally show gene expression patterns reflecting altered metabolism.
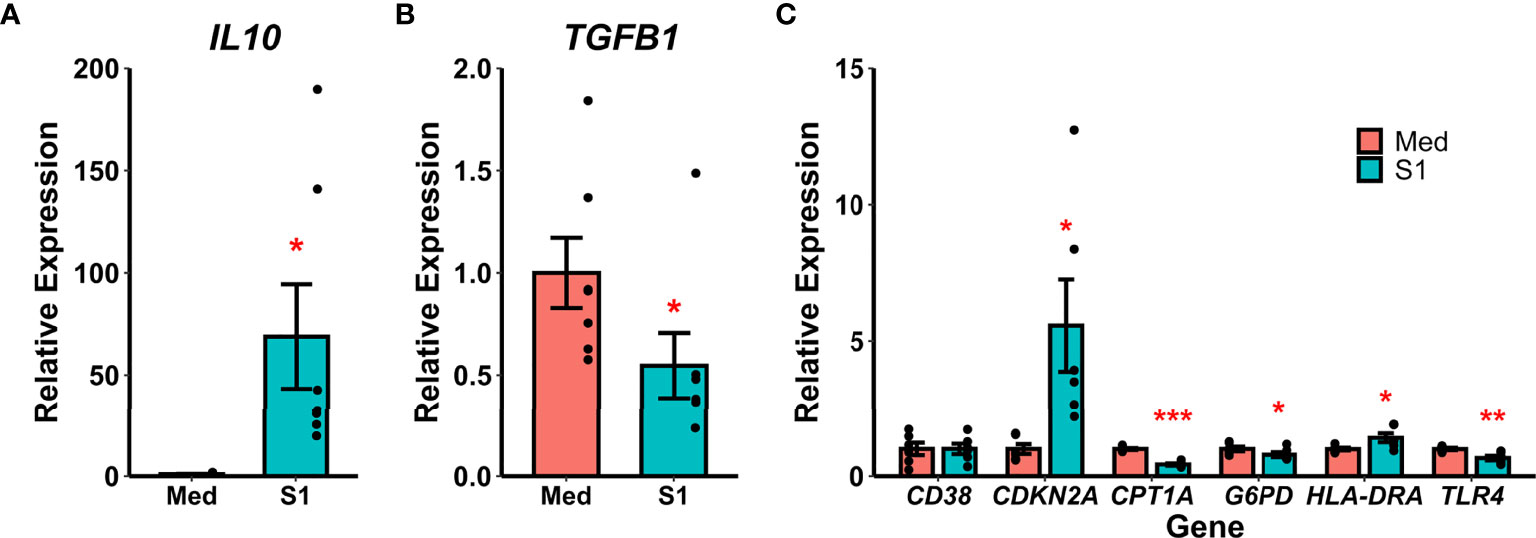
Figure 2 Recombinant SARS-CoV-2 spike protein subunit 1 (S1) alters expression of anti-inflammatory cytokine and polarization-related genes. (A) S1 treatment increased expression of IL10. (B) S1 treatment decreased expression of TGFB1. (C) S1 increased expression of CDKN2A and HLA-DRA, while decreasing expression of CPT1A, G6PD, and TLR4. *, *, ***p < 0.05, p < 0.01, p < 0.001 vs. media-treated cells. N = 7 biological replicates. Bars and error bars are mean ± SEM.
Hypoxia inducible factor (HIF)-1α was demonstrated nearly 20 years ago to mediate pro-inflammatory responses in myeloid cells (61), and has more recently been shown to regulate glycolytic activation in monocytes, macrophages, and other immune cells (62–64). SARS-CoV-2 activates HIF-1α-mediated glycolysis in monocytes (36), so we reasoned that this was a likely downstream mechanism by which the viral spike protein causes this similar glycolytic reprogramming in our experiments. As above, treatment of monocytes with S1 activated glycolysis, and this effect was abrogated by pre-treatment with chetomin (Figures 3A, B, F2,12 = 42.43, p<0.001), which disrupts the interaction between HIF-1α and p300 to block the effects of the former (65). Pre-treatment with chetomin also strongly suppressed the cytokine response due to S1 treatment (Figure 3C), including blunting transcription of IL1B (F2,12 = 27.35, p<0.001), IL6 (F2,12 = 16.11, p<0.001), CXCL8 (F2,12 = 25.54, p<0.001), and TNF (F2,12 = 29.04, p<0.001). Protein concentrations of these cytokines in culture supernatants showed similar patterns (Figure 3D). As such, HIF-1α appears to be a master regulator of both glycolytic reprogramming and inflammatory activation of monocytes under S1 stimulation.
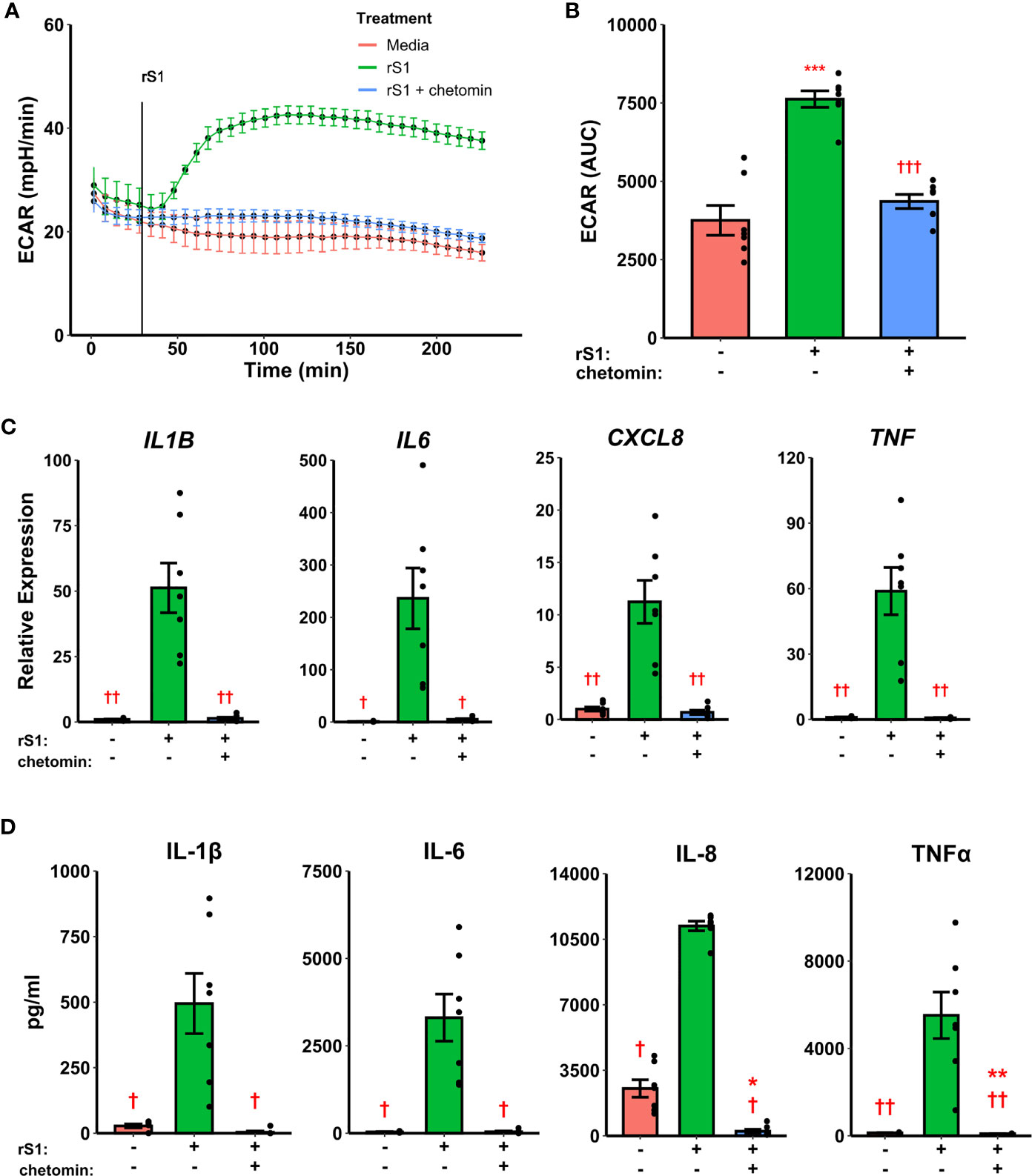
Figure 3 HIF-1α inhibition suppresses immunometabolic activation of monocytes due to recombinant spike protein (rS1). (A) Monocytes increase extracellular acidification response rate (ECAR) when treated with 30 nM rS1, but this is blocked by pre-treatment with chetomin. (B) Quantification of ECAR by area under the curve (AUC). (C) rS1 increase of expression of IL1B, IL6, CXCL8, and TNF is reversed by chetomin pre-treatment. (D) rS1 increase of IL-1β, IL-6, IL-8, and TNFα is reversed by chetomin pre-treatment. *, **, ***: p < 0.05, p < 0.01, p < 0.001 vs. untreated cells. †, ††, †††: p < 0.05, p < 0.01, p < 0.001 vs. rS1-treated cells. N = 7 biological replicates. Bars and error bars are mean ± SEM.
To determine whether metabolic reprogramming is responsible for altered cytokine responses to S1, we suppressed glycolytic responses during S1 treatment using 2-deoxyglucose (2-DG) pretreatment. Treatment of monocytes with 2-DG ablated monocyte glycolytic responses to S1 stimulation (Figure 4A) which was significant by comparison of area under the response curve (t6=-10.867, p<0.0001, Figure 4B). However, 2-DG also suppressed mitochondrial function in these cells (Figure 4C), though this was non-significant by area under the oxygen consumption (t6=-2.2284, p=0.0674, Figure 4D). This effect has been noted previously during responses to LPS (66). Anticipating this, we also included a condition where monocytes were cultured under glucose deprivation, as a second method of suppressing glycolytic activation. We noted a similar ablation of glycolytic responses to S1 using this strategy (Figure 4A) which was significant by area under the curve analysis (t6=-14.045, p<0.0001, Figure 4B). However, glucose deprivation caused an increase in oxygen consumption after S1 treatment (Figure 4C) which was significant compared to media- (t6 = 4.6618, p=0.0069) or 2-DG (t6=-15.607, p<0.001) pretreated monocytes (Figure 4D).
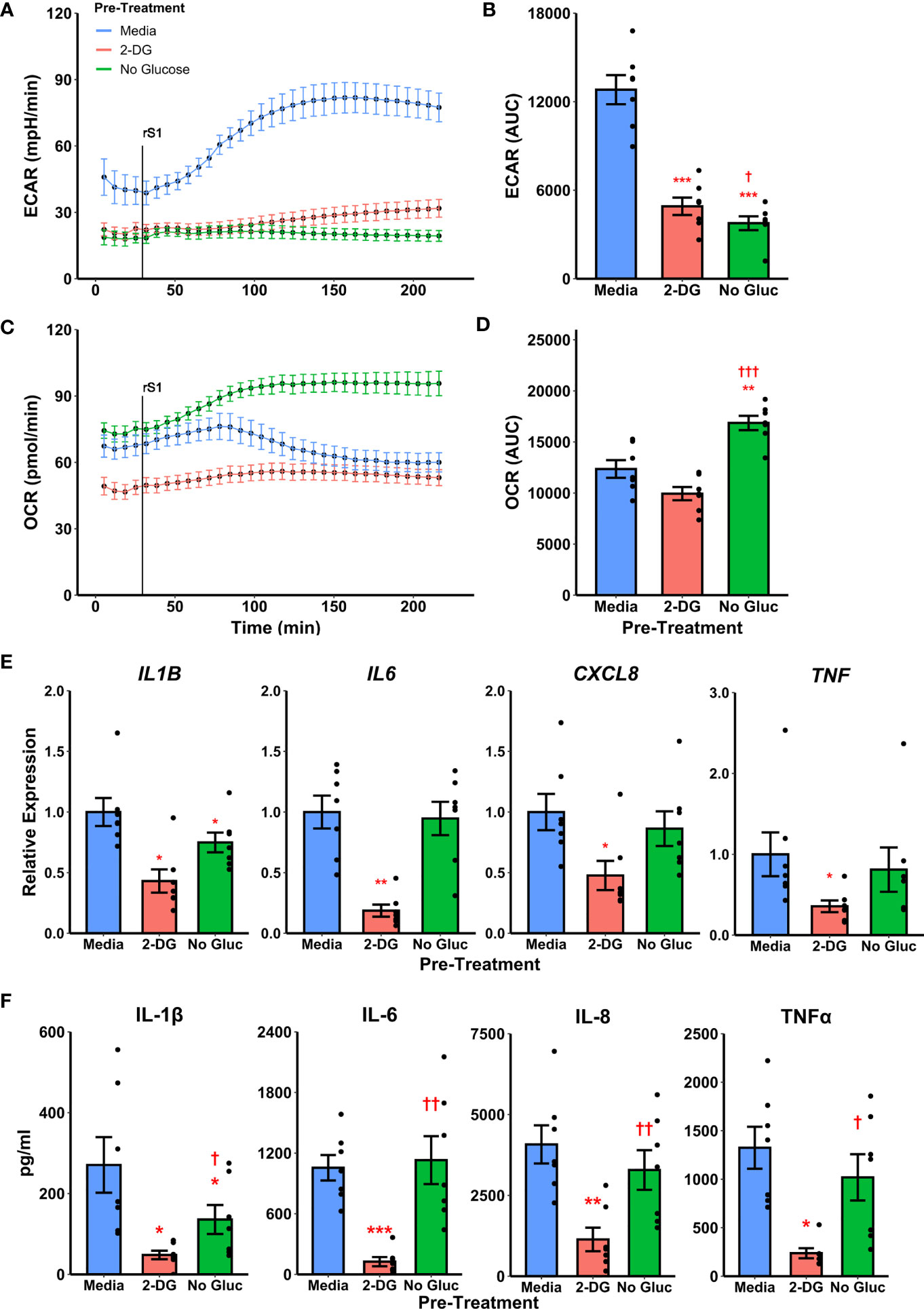
Figure 4 Targeting glycolysis has variable effects on recombinant spike protein (rS1) responses in monocytes. (A) Glucose deprivation or pre-treatment with 2-deoxyglucose (2-DG) block extracellular acidification rate (ECAR) increase due to rS1 treatment. (B) Quantification of ECAR by area under the curve (AUC). (C) 2-DG inhibits oxygen consumption rate in rS1-treated monocytes, but glucose-deprived monocytes upregulate OCR in response to rS1. (D) Quantification of OCR by AUC. (E) 2-DG blocks expression of IL1B, IL6, CXCL8, and TNF due to rS1 stimulation, but glucose deprivation has limited effects on cytokine expression. (F) 2-DG blocks protein production of pro-inflammatory cytokines, but glucose deprivation has limited effect. *, **, ***: p < 0.05, p < 0.01, p < 0.001 vs. rS1-treated cells. †, ††, †††: p < 0.05, p < 0.01, p < 0.001 vs. 2-DG-treated cells. N = 7 biological replicates. Bars and error bars are mean ± SEM.
Pre-treatment of monocytes with 2-DG also strongly inhibited cytokine expression compared to cells treated with S1 (Figure 4E), including transcription of IL1B (W=0, p=0.0313), IL6 (t6=-5.912, p=0.0021), CXCL8 (W=0, p=0.0313), and TNF (W=0, p=0.0313). However, glucose deprived monocytes generally maintained their ability to transcribe pro-inflammatory cytokines in response to S1, with only IL1B expression showing a modest 25.1% reduction in glucose deprived compared to S1-treated monocytes (Figure 4E, W=0, p=0.0313). Protein concentrations in cell culture supernatants followed gene expression patterns (Figure 4F). Monocytes appear to utilize fatty acid oxidation to compensate for loss of glycolysis during cytokine responses as has been previously demonstrated with LPS (66–69), and therefore the 2-DG-mediated suppression of S1-induced inflammation is likely due to its ability to suppress both glycolysis and mitochondrial metabolism in concert.
The small molecule compounds chetomin and 2-deoxyglucose inhibited immunometabolic activation in monocytes, suggesting a potential strategy for treating hypercytokinemia during COVID-19. However, chetomin is not approved for use in humans, although it has shown efficacy in vivo in animal models (65). Additionally, 2-DG has poor efficacy in humans due to rapid metabolism and limited bioavailability (70). Therefore, we investigated the ability of the common diabetes and geroprotector drug metformin to inhibit cytokine production in S1-stimulated monocytes. Metformin activates AMPK (71) and (independently of AMPK) opposes the action of HIF-1α (72, 73), and additionally inhibits mitochondrial metabolism through blocking complex I of the electron transport chain (74, 75), thus we hypothesized that it would have a qualitatively similar effect to 2-DG in inhibiting cytokine production through dual inhibition of glycolysis and OXPHOS.
Pre-treatment with metformin abrogated the glycolytic response to S1 in monocytes (Figures 5A, B, F2,12 = 60.05, p<0.001) and strongly inhibited cellular respiration (Figures 5C, D, Friedman χ2 (df=2)=12.286, p=0.0021) in Seahorse assays. Likewise, metformin pre-treatment suppressed cytokine responses to S1 treatment in monocytes (Figure 5E), including IL1B (Friedman χ2 (df=2)=12.286, p=0.0021), IL6 (Friedman χ2 (df=2)=10.571, p=0.0051), CXCL8 (F2,12 = 68.18, p<0.0001), and TNF (Friedman χ2 (df=2)=12.286, p=0.0021). As in previous experiments, protein concentrations (Figure 5F) followed gene expression patterns.
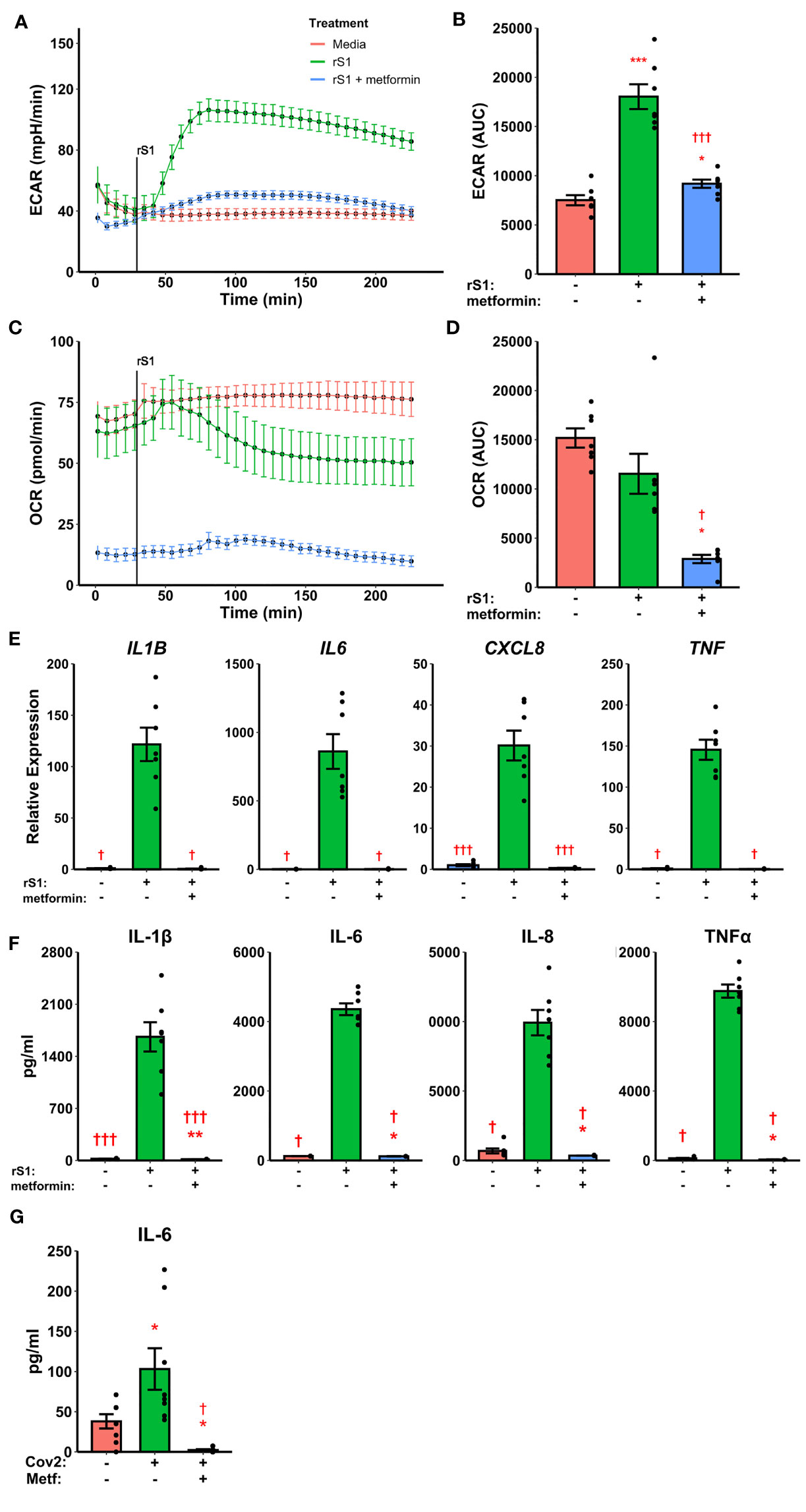
Figure 5 Metformin suppresses immunometabolic activation in monocytes treated with recombinant spike protein (rS1). (A) Metformin pre-treatment blocks the increase in extracellular acidification rate (ECAR) mediated by rS1. (B) Quantification of ECAR by area under the curve (AUC). (C) Metformin suppresses oxygen consumption rate (OCR). (D) Quantification of OCR by AUC. (E) Metformin suppresses cytokine responses, as demonstrated by gene expression of IL1B, IL6, CXCL8, and TNF, during rS1 stimulation in monocytes. (F) Metformin suppresses protein production of pro-inflammatory cytokines IL-1β, IL-6, IL-8, and TNFα during S1 stimulation in monocytes. (G) Metformin inhibits IL-6 production in monocytes infected with SARS-CoV-2 strain WA1/2020 (Cov2) at 0.5 MOI. *, **, ***: p < 0.05, p < 0.01, p < 0.001 vs. unstimulated cells. †, †††: p < 0.05, p < 0.001 vs. rS1-treated or Cov2-infected cells. N = 7-8 biological replicates. Bars and error bars are mean ± SEM.
Recent evidence suggests that myeloid cells recognize SARS-CoV-2 spike protein through C-type lectins (53, 54). However, the SARS-CoV-2 virion also contains additional immunoregulatory and pro-inflammatory proteins (76, 77), therefore we examined the ability of metformin to block cytokine responses to live SARS-CoV-2. Monocytes treated for 24 hr with SARS-CoV-2 increased expression of IL-6 protein, and this was suppressed by metformin pre-treatment (Figure 5G, F2,14 = 11.48, p=0.0011), suggesting that the anti-inflammatory effect of metformin is generalizable to SARS-CoV-2 infection of monocytes.
The present study resulted in several advances of major importance for the understanding of SARS-CoV-2 innate immune responses. First, we report here that monocytes treated with recombinant spike protein subunit 1 from the current pandemic SARS-CoV-2 undergo a dose-dependent increase in glycolysis which can be suppressed by a HIF-1α inhibitor and mediates the production of pro-inflammatory cytokines. These data suggest an initial signaling event which precipitates changes in glucose and lipid metabolism during SARS-CoV-2 infection in monocytes which have been previously reported to be linked to inflammatory activation (36, 37). Monocyte and monocyte-derived macrophages are substantially enriched in the lungs of SARS-CoV-2-infected individuals with severe COVID-19 (10, 11, 14–17) and respond to experimental viral infection by producing pro-inflammatory cytokines (36–40), therefore these results reflect a potential mechanism by which hypercytokinemia occurs during the early innate immune response to SARS-CoV-2.
Importantly, the available evidence suggests that infection of monocytes/macrophages by SARS-CoV-2 is abortive (39, 40, 78), thus recognition of SARS-CoV-2 structural proteins or genomic material is the likely mechanism by which direct infection precipitates inflammatory responses in this cell type. Our data suggest the spike protein is one such determinant, although we cannot conclude it is the only such mechanism given that recent reports have demonstrated inflammatory responses in macrophages treated with the SARS-CoV-2 envelope protein (76). It is also possible, however, that direct viral binding to monocytes is not the only way in which these cells can be exposed to the viral spike protein. Recent evidence suggests that vaccine antigens including S1 are released into the circulation following vaccination (albeit at very low levels) (79), and this represents a potential pro-inflammatory stimulus for monocytes. Monocyte/macrophage recognition of S1 may also contribute to the local (muscle) inflammatory response during vaccination. Additionally, the viral spike protein undergoes cleavage by furin during binding to ACE2 (80), and it has been suggested that this could lead to release of the S1 subunit during infection (81), although to date this is speculative.
ACE2 has limited expression on immune cells including monocytes and macrophages (82), which has called into question whether they can directly recognize SARS-CoV-2. In this study we did not identify the mechanism for monocyte recognition of S1, but several recent papers have shed light on this. Two reports recently demonstrated spike binding to C-type lectin receptors (53, 54) which mediates pro-inflammatory signaling in myeloid cells. Likewise, monocytes and macrophages express high levels of CD147 (82), and this receptor has been shown to recognize spike protein and contribute to activation of T cells (83). Monocytes therefore have multiple methods of recognizing S1, and the receptor(s) responsible for signaling to induce immunometabolic activation deserve further investigation.
The second major advance in this study is the identification of metformin as a potential immunometabolic regulator of inflammatory responses to SARS-CoV-2. Small molecule inhibitors of HIF-1α (chetomin) and glucose metabolism (2-deoxyglucose) blocked cytokine production in S1-treated monocytes, suggesting that interfering with downstream signaling pathways activated by spike protein binding is a potential therapeutic strategy to target inflammation during COVID-19. As these compounds are not approved for human use or have low efficacy in humans as described above, we evaluated the ability of metformin to suppress glycolytic reprogramming and cytokine production in S1-stimulated monocytes. Metformin reduced cytokine production and strongly inhibited both glycolysis and cellular respiration in culture, suggesting it as a potential treatment for hyperinflammation during COVID-19. Further, metformin blocked IL-6 production in monocytes infected with live SARS-CoV-2, suggesting this effect is not limited to artificial stimulation conditions with purified recombinant protein. However, we were unable to perform additional experiments using live SARS-CoV-2 to replicate further findings from this study, so a great deal of additional work is necessary to link our S1 data to our very preliminary SARS-CoV-2 observation.
Metformin is extremely inexpensive compared to many pharmaceuticals, with an estimated manufacturing cost under 10 USD per kg for the active ingredient (84) and a monthly wholesale cost as low as 25 USD (85). Metformin has been previously noted as a treatment for non-COVID acute respiratory distress syndrome (86) and is a potent suppressor of immune activation of monocytes and macrophages by other molecules including LPS (87–89). Additionally, several epidemiological studies have noted decreased mortality (90–94) and inflammation (systemic C-reactive protein) (95, 96) in COVID-19 patients who were taking metformin prior to diagnosis. These effects varied between studies, with mortality reductions of 20% - 80% across reports, but a meta-analysis found an overall reduction of 46% in mortality across 5 qualifying studies (94). Therefore, given these observations and its low cost, excellent safety profile, wide availability, and efficacy in inhibiting inflammatory responses to S1 in vitro, metformin is a promising candidate for further exploration as a COVID-19 therapeutic. Our study is limited to a single in vitro measure of metformin as a therapeutic for COVID-19, so a great deal of further study is necessary in order to establish this drug as a viable treatment. To this end, one recent study demonstrated metformin as efficacious in preventing acute respiratory distress syndrome in a preclinical animal model of COVID-19 (97). Therefore, although metformin is most likely to be useful as an adjuvant rather than front-line therapy for severe acute COVID-19, it remains an attractive option for targeting hyperinflammation in this disease to limit severity and mortality.
Limitations of this study include the small sample size, although the large effect sizes in most experiments are sufficient to convincingly demonstrate the interrogated mechanisms. More significantly, this study relies on healthy younger subjects and so does not address how comorbidities such as aging or obesity would affect the measured outcomes or the efficacy of the therapies we tested. We determined this to be appropriate, given our focus on mechanistic investigation in this study. However, further research is needed to determine if other populations have different responses to S1 and/or metformin. We additionally did not recruit with respect to race or sex and therefore have a relatively diverse sample which could increase variability. However, the magnitude of the responses did not show patterns consistent with race- or sex-based differences in our outcome measures. While it is conceivable that such differences exist, the effects are likely to be small enough that large studies would be necessary to detect small between-population differences. Finally, none of our subjects were previously taking metformin, and the ability of in vivo metformin to modulate monocyte responses to S1 requires further study.
In summary, we demonstrate here that the spike protein subunit 1 from SARS-CoV-2 causes activation of HIF-1α dependent glycolysis and inflammatory cytokine production in monocytes which can be suppressed by treatment with the diabetes drug metformin. These experiments detail a mechanism by which SARS-CoV-2 mediates metabolic reprogramming previously described in human monocytes, and additionally provides a potential mechanism for the observation that metformin is protective against mortality in COVID-19 patients. Continued research in this area has the potential to define therapeutic strategies and additional molecular targets for the treatment of COVID-19-associated hyperinflammation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://figshare.com/, DOI: 10.6084/m9.figshare.c.5463525.
The studies involving human participants were reviewed and approved by the University of Memphis Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
BP conceived the study. TC and BP designed experiments. TC, RE, JY, KD, and BP collected data. BP analyzed data and prepared the first manuscript draft. TC, RE, JY, KD, and BP edited the manuscript draft. All authors read and approved the final manuscript.
The study was primarily supported by a University of Memphis/University of Tennessee Health Science Center Collaborative Research Network (CORNET) award to BP and TC, with additional support from American Heart Association grants 18AIREA33961089 and 19TPA34910232 to BP, and a University of Memphis College of Health Sciences faculty research grant to BP. RE was supported by a postdoctoral fellowship funded by the University of Memphis Division of Research and Innovation through the Carnegie R1 Postdoc Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge the participants in this study. The authors thank Jyothi Parvathareddy and Colleen Jonsson from the Regional Biocontainment Laboratory at the University of Tennessee Health Science Center for providing SARS-CoV-2 virus stocks.
1. Worldometers. Covid-19 Coronavirus Pandemic (2020). Available at: https://www.worldometers.info/coronavirus/.
2. Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-nCoV and Naming it SARS-CoV-2. Nat Microbiol (2020) 5(4):536–44. doi: 10.1038/s41564-020-0695-z
3. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J Clin Invest (2020) 130(5):2620–9. doi: 10.1172/JCI137244
4. Vangeti S, Yu M, Smed-Sörensen A. Respiratory Mononuclear Phagocytes in Human Influenza A Virus Infection: Their Role in Immune Protection and As Targets of the Virus. Front Immunol (2018) 9:1521. doi: 10.3389/fimmu.2018.01521
5. Kimpen JLL. Respiratory Syncytial Virus and Asthma: The Role of Monocytes. Am J Respir Crit Care Med (2001) 163(3 II):S7–9. doi: 10.1164/ajrccm.163.supplement_1.2011110
6. Nikitina E, Larionova I, Choinzonov E, Kzhyshkowska J. Monocytes and Macrophages as Viral Targets and Reservoirs. Int J Mol Sci (2018) 19(9):2821. doi: 10.3390/ijms19092821
7. Pence BD. Severe COVID-19 and Aging: Are Monocytes the Key? GeroScience (2020) 42(4):1051–61. doi: 10.1007/s11357-020-00213-0
8. Pence BD. Aging and Monocyte Immunometabolism in COVID-19. Aging (Albany NY) (2021) 13(7):9154–5. doi: 10.18632/aging.202918
9. Pence BD. Atypical Monocytes in COVID-19: Lighting the Fire of Cytokine Storm? J Leukoc Biol (2021) 109(1):7–8. doi: 10.1002/JLB.5CE0920-613R
10. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
11. Xu G, Qi F, Li H, Yang Q, Wang H, Wang X, et al. The Differential Immune Responses to COVID-19 in Peripheral and Lung Revealed by Single-Cell RNA Sequencing. Cell Discov (2020) 6(1):73. doi: 10.1038/s41421-020-00225-2
12. Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe COVID-19. Cells (2020) 9(11):2374. doi: 10.3390/cells9112374
13. Ren X, Wen W, Fan X, Hou W, Su B, Cai P, et al. COVID-19 Immune Features Revealed by a Large-Scale Single-Cell Transcriptome Atlas. Cell (2021) 184(7):1895–913.e19. doi: 10.1016/j.cell.2021.01.053
14. Song JW, Zhang C, Fan X, Meng FP, Xu Z, Xia P, et al. Immunological and Inflammatory Profiles in Mild and Severe Cases of COVID-19. Nat Commun (2020) 11(1):3410. doi: 10.1038/s41467-020-17240-2
15. Nienhold R, Ciani Y, Koelzer VH, Tzankov A, Haslbauer JD, Menter T, et al. Two Distinct Immunopathological Profiles in Autopsy Lungs of COVID-19. Nat Commun (2020) 11(1):5086. doi: 10.1038/s41467-020-18854-2
16. Beigmohammadi MT, Jahanbin B, Safaei M, Amoozadeh L, Khoshavi M, Mehrtash V, et al. Pathological Findings of Postmortem Biopsies From Lung, Heart, and Liver of 7 Deceased COVID-19 Patients. Int J Surg Pathol (2021) 29(2):135–45. doi: 10.1177/1066896920935195
17. Li S, Jiang L, Li X, Lin F, Wang Y, Li B, et al. Clinical and Pathological Investigation of Patients With Severe COVID-19. JCI Insight (2020) 5(12):e138070. doi: 10.1172/jci.insight.138070
18. Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, et al. SARS-CoV-2 Infection of Human ACE2-Transgenic Mice Causes Severe Lung Inflammation and Impaired Function. Nat Immunol (2020) 21(11):1327–35. doi: 10.1038/s41590-020-0778-2
19. Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, et al. The Pathogenicity of SARS-CoV-2 in Hace2 Transgenic Mice. Nature (2020) 583(7818):830–3. doi: 10.1038/s41586-020-2312-y
20. Sia SF, Yan LM, Chin AWH, Fung K, Choy KT, Wong AYL, et al. Pathogenesis and Transmission of SARS-CoV-2 in Golden Hamsters. Nature (2020) 583(7818):834–8. doi: 10.1038/s41586-020-2342-5
21. Song TZ, Zheng HY, Han JB, Jin L, Yang X, Liu FL, et al. Delayed Severe Cytokine Storm and Immune Cell Infiltration in SARS-CoV-2-Infected Aged Chinese Rhesus Macaques. Zool Res (2020) 41(5):503–16. doi: 10.24272/j.issn.2095-8137.2020.202
22. Shan C, Yao YF, Yang XL, Zhou YW, Gao G, Peng Y, et al. Infection With Novel Coronavirus (SARS-CoV-2) Causes Pneumonia in Rhesus Macaques. Cell Res (2020) 30(8):670–7. doi: 10.1038/s41422-020-0364-z
23. Chandrashekar A, Liu J, Martino AJ, McMahan K, Mercad NB, Peter L, et al. SARS-CoV-2 Infection Protects Against Rechallenge in Rhesus Macaques. Science (2020) 369(6505):812–7. doi: 10.1126/science.abc4776
24. Speranza E, Williamson BN, Feldmann F, Sturdevant GL, Pérez-Pérez L, Meade-White K, et al. Single-Cell RNA Sequencing Reveals SARS-CoV-2 Infection Dynamics in Lungs of African Green Monkeys. Sci Transl Med (2021) 13(578):eabe8146. doi: 10.1126/scitranslmed.abe8146
25. Fahlberg MD, Blair RV, Doyle-Meyers LA, Midkiff CC, Zenere G, Russell-Lodrigue KE, et al. Cellular Events of Acute, Resolving or Progressive COVID-19 in SARS-CoV-2 Infected Non-Human Primates. Nat Commun (2020) 11(1):6078. doi: 10.1101/2020.07.21.213777
26. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex Immune Dysregulation in COVID-19 Patients With Severe Respiratory Failure. Cell Host Microbe (2020) 27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009
27. Laing AG, Lorenc A, del Molino del Barrio I, Das A, Fish M, Monin L, et al. A Dynamic COVID-19 Immune Signature Includes Associations With Poor Prognosis. Nat Med (2020) 26(10):1623–35. doi: 10.1038/s41591-020-1038-6
28. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell (2020) 182(6):1419–40.e23. doi: 10.1016/j.cell.2020.08.001
29. Payen D, Cravat M, Maadadi H, Didelot C, Prosic L, Dupuis C, et al. A Longitudinal Study of Immune Cells in Severe COVID-19 Patients. Front Immunol (2020) 11:580250. doi: 10.3389/fimmu.2020.580250
30. Mann ER, Menon M, Knight SB, Konkel JE, Jagger C, Shaw TN, et al. Longitudinal Immune Profiling Reveals Key Myeloid Signatures Associated With COVID-19. Sci Immunol (2020) 5(51):eabd6197. doi: 10.1126/sciimmunol.abd6197
31. Kvedaraite E, Hertwig L, Sinha I, Ponzetta A, Myrberg IH, Lourda M, et al. Major Alterations in the Mononuclear Phagocyte Landscape Associated With COVID-19 Severity. Proc Natl Acad Sci U S A (2021) 118(6):e2018587118. doi: 10.1073/pnas.2018587118
32. Varchetta S, Mele D, Oliviero B, Mantovani S, Ludovisi S, Cerino A, et al. Unique Immunological Profile in Patients With COVID-19. Cell Mol Immunol (2021) 18(3):604–12. doi: 10.1038/s41423-020-00557-9
33. Su Y, Chen D, Yuan D, Lausted C, Choi J, Dai CL, et al. Multi-Omics Resolves a Sharp Disease-State Shift Between Mild and Moderate COVID-19. Cell (2020) 183(6):1479–95.e20. doi: 10.1016/j.cell.2020.10.037
34. Guo C, Li B, Ma H, Wang X, Cai P, Yu Q, et al. Single-Cell Analysis of Two Severe COVID-19 Patients Reveals a Monocyte-Associated and Tocilizumab-Responding Cytokine Storm. Nat Commun (2020) 11(1):3924. doi: 10.1038/s41467-020-17834-w
35. Kahn R, Schmidt T, Golestani K, Mossberg A, Gullstrand B, Bengtsson AA, et al. Mismatch Between Circulating Cytokines and Spontaneous Cytokine Production by Leukocytes in Hyperinflammatory COVID-19. J Leukoc Biol (2021) 109(1):115–20. doi: 10.1002/JLB.5COVBCR0720-310RR
36. Codo AC, Davanzo GG, Monteiro L de B, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response Through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab (2020) 32(3):437–446.e5. doi: 10.1016/j.cmet.2020.07.007
37. da Silva Gomes Dias S, Soares VC, Ferreira AC, Sacramento CQ, Fintelman-Rodrigues N, Temerozo JR, et al. Lipid Droplets Fuel SARS-CoV-2 Replication and Production of Inflammatory Mediators. PloS Pathog (2020) 16(12):e1009127. doi: 10.1101/2020.08.22.262733
38. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S da SG, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 Engages Inflammasome and Pyroptosis in Human Primary Monocytes. Cell Death Discov (2021) 7(1):43. doi: 10.1038/s41420-021-00428-w
39. Zheng J, Wang Y, Li K, Meyerholz DK, Allamargot C, Perlman S. Severe Acute Respiratory Syndrome Coronavirus 2-Induced Immune Activation and Death of Monocyte-Derived Human Macrophages and Dendritic Cells. J Infect Dis (2021) 223(5):785–95. doi: 10.1093/infdis/jiaa753
40. Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee ACY, et al. Attenuated Interferon and Proinflammatory Response in SARS-CoV-2-Infected Human Dendritic Cells is Associated With Viral Antagonism of STAT1 Phosphorylation. J Infect Dis (2020) 222(5):734–45. doi: 10.1093/infdis/jiaa356
41. O’Neill LAJ, Kishton RJ, Rathmell J. A Guide to Immunometabolism for Immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
42. Wang W, Ye L, Ye L, Li B, Gao B, Zeng Y, et al. Up-Regulation of IL-6 and TNF-α Induced by SARS-Coronavirus Spike Protein in Murine Macrophages via NF-κB Pathway. Virus Res (2007) 128(1–2):1–8. doi: 10.1016/j.virusres.2007.02.007
43. Dosch SF, Mahajan SD, Collins AR. SARS Coronavirus Spike Protein-Induced Innate Immune Response Occurs via Activation of the NF-κB Pathway in Human Monocyte Macrophages In Vitro. Virus Res (2009) 142(1–2):19–27. doi: 10.1016/j.virusres.2009.01.005
44. Ropa J, Cooper S, Capitano ML, Van’t Hof W, Broxmeyer HE. Human Hematopoietic Stem, Progenitor, and Immune Cells Respond Ex Vivo to SARS-CoV-2 Spike Protein. Stem Cell Rev Rep (2021) 17(1):253–65. doi: 10.1007/s12015-020-10056-z
45. Karwaciak I, Sałkowska A, Karaś K, Dastych J, Ratajewski M. Nucleocapsid and Spike Proteins of the Coronavirus Sars-Cov-2 Induce Il6 in Monocytes and Macrophages—Potential Implications for Cytokine Storm Syndrome. Vaccines (2021) 9(1):1–10. doi: 10.3390/vaccines9010054
46. Pence BD, Yarbro JR. Aging Impairs Mitochondrial Respiratory Capacity in Classical Monocytes. Exp Gerontol (2018) 108:112–7. doi: 10.1016/j.exger.2018.04.008
47. Pence BD, Yarbro JR. Classical Monocytes Maintain Ex Vivo Glycolytic Metabolism and Early But Not Later Inflammatory Responses in Older Adults. Immun Ageing (2019) 16:3. doi: 10.1186/s12979-019-0143-1
48. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods (2001) 25(4):402–8. doi: 10.1006/meth.2001.1262
49. Pence BD. Data for Metformin Suppresses Monocyte Immunometabolic Activation by SARS-CoV-2 and Spike Protein Subunit 1. (2021). doi: 10.6084/m9.figshare.c.5463525
50. Urbanek S, Bibiko H-J, Stefano ML. R: A Language and Environment for Statistical Computing. In: The R Foundation for Statistical Computing (2014). Available at: https://www.r-project.org.
51. Sture H. A Simple Sequentially Rejective Multiple Test Procedure. Scand J Stat (1979) 6(2):65–70.
52. Cory TJ, Emmons RS, Yarbro JR, Davis KL, Pence BD. Metformin Suppresses Immunometabolic Activation by SARS-CoV-2 and Spike Protein Subunit 1. bioRxiv (2021). doi: 10.1101/2021.05.27.445991
53. Thépaut M, Luczkowiak J, Vivès C, Labiod N, Bally I, Lasala F, et al. DC/L-SIGN Recognition of Spike Glycoprotein Promotes SARS-CoV-2 Trans-Infection and can be Inhibited by a Glycomimetic Antagonist. PLoS Pathog (2021) 17(5):e1009576. doi: 10.1371/journal.ppat.1009576
54. Lu Q, Liu J, Zhao S, Gomez Castro MF, Laurent-Rolle M, Dong J, et al. SARS-CoV-2 Exacerbates Proinflammatory Responses in Myeloid Cells Through C-Type Lectin Receptors and Tweety Family Member 2. Immunity (2021) 54(6):1304–19.e9. doi: 10.1016/j.immuni.2021.05.006
55. Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are Activated in Response to SARS-Cov-2 Infection and are Associated With COVID-19 Severity in Patients. J Exp Med (2020) 218(3):e20201707. doi: 10.1084/jem.20201707
56. Cauchois R, Koubi M, Delarbre D, Manet C, Carvelli J, Blasco VB, et al. Early IL-1 Receptor Blockade in Severe Inflammatory Respiratory Failure Complicating COVID-19. Proc Natl Acad Sci U S A (2020) 117(32):18951–3. doi: 10.1073/pnas.2009017117
57. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe COVID-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
58. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
59. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An Inflammatory Cytokine Signature Predicts COVID-19 Severity and Survival. Nat Med (2020) 26(10):1636–43. doi: 10.1038/s41591-020-1051-9
60. Chen L, Wang G, Tan J, Cao Y, Long X, Luo H, et al. Scoring Cytokine Storm by the Levels of MCP-3 and IL-8 Accurately Distinguished COVID-19 Patients With High Mortality. Signal Transduct Target Ther (2020) 5(1):292. doi: 10.1038/s41392-020-00433-y
61. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1α Is Essential for Myeloid Cell-Mediated Inflammation. Cell (2003) 112(5):645–57. doi: 10.1016/S0092-8674(03)00154-5
62. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate Is an Inflammatory Signal That Induces IL-1 Beta Through HIF-1 Alpha. Nature (2013) 496(7444):238–+. doi: 10.1038/nature11986
63. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-κB Links Innate Immunity to the Hypoxic Response Through Transcriptional Regulation of HIF-1α. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
64. Corcoran SE, O’Neill LAJ. HIF1α and Metabolic Reprogramming in Inflammation. J Clin Invest (2016) 126(10):3699–707. doi: 10.1172/JCI84431
65. Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, et al. Small Molecule Blockade of Transcriptional Coactivation of the Hypoxia-Inducible Factor Pathway. Cancer Cell (2004) 6(1):33–43. doi: 10.1016/j.ccr.2004.06.009
66. Raulien N, Friedrich K, Strobel S, Rubner S, Baumann S, von Bergen M, et al. Fatty Acid Oxidation Compensates for Lipopolysaccharide-Induced Warburg Effect in Glucose-Deprived Monocytes. Front Immunol (2017) 8:609. doi: 10.3389/fimmu.2017.00609
67. Yarbro JR, Pence BD. Classical Monocytes From Older Adults Maintain Capacity for Metabolic Compensation During Glucose Deprivation and Lipopolysaccharide Stimulation. Mech Ageing Dev (2019) 183:111146. doi: 10.1016/j.mad.2019.111146
68. Otto NA, Butler JM, Ramirez-Moral I, van Weeghel M, van Heijst JWJ, Scicluna BP, et al. Adherence Affects Monocyte Innate Immune Function and Metabolic Reprogramming After Lipopolysaccharide Stimulation In Vitro. J Immunol (2021) 206(4):827–38. doi: 10.4049/jimmunol.2000702
69. Woods PS, Kimmig LM, Meliton AY, Sun KA, Tian Y, O’Leary EM, et al. Tissue-Resident Alveolar Macrophages do Not Rely on Glycolysis for LPS-Induced Inflammation. Am J Respir Cell Mol Biol (2020) 62(2):243–55. doi: 10.1165/rcmb.2019-0244OC
70. Pajak B, Siwiak E, Sołtyka M, Priebe A, Zieliński R, Fokt I, et al. 2-Deoxy-D-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int J Mol Sci (2020) 21(1):234. doi: 10.3390/ijms21010234
71. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-Activated Protein Kinase in Mechanism of Metformin Action. J Clin Invest (2001) 108(8):1167–74. doi: 10.1172/JCI13505
72. Takiyama Y, Harumi T, Watanabe J, Fujita Y, Honjo J, Shimizu N, et al. Tubular Injury in a Rat Model of Type 2 Diabetes is Prevented by Metformin: A Possible Role of HIF-1α Expression and Oxygen Metabolism. Diabetes (2011) 60(3):981–92. doi: 10.2337/db10-0655
73. Zhou X, Chen J, Yi G, Deng M, Liu H, Liang M, et al. Metformin Suppresses Hypoxia-Induced Stabilization of HIF-1’ Through Reprogramming of Oxygen Metabolism in Hepatocellular Carcinoma. Oncotarget (2016) 7(1):873–84. doi: 10.18632/oncotarget.6418
74. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J Biol Chem (2000) 275(1):223–8. doi: 10.1074/jbc.275.1.223
75. Owen MR, Doran E, Halestrap AP. Evidence That Metformin Exerts its Anti-Diabetic Effects Through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochem J (2000) 348(3):607–14. doi: 10.1042/bj3480607
76. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 Senses the SARS-CoV-2 Envelope Protein to Produce Inflammatory Cytokines. Nat Immunol (2021) 22:829–38. doi: 10.1038/s41590-021-00937-x
77. Fu YZ, Wang SY, Zheng ZQ, Huang Y, Li WW, Xu ZS, et al. SARS-CoV-2 Membrane Glycoprotein M Antagonizes the MAVS-Mediated Innate Antiviral Response. Cell Mol Immunol (2021) 18(3):613–20. doi: 10.1038/s41423-020-00571-x
78. Boumaza A, Gay L, Mezouar S, Bestion E, Diallo AB, Michel M, et al. Monocytes and Macrophages, Targets of SARS-CoV-2: The Clue for Covid-19 Immunoparalysis. J Infect Dis (2021) 224(3):395–406. doi: 10.1101/2020.09.17.300996
79. Ogata AF, Cheng C, Desjardins M, Senussi Y, Sherman AC, Powell M, et al. Circulating SARS-CoV-2 Vaccine Antigen Detected in the Plasma of mRNA-1273 Vaccine Recipients. Clin Infect Dis (2021) ciab465. doi: 10.1093/cid/ciab465
80. Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, et al. Loss of Furin Cleavage Site Attenuates SARS-CoV-2 Pathogenesis. Nature (2021) 591(7849):293–9. doi: 10.1038/s41586-021-03237-4
81. Letarov AV, Babenko VV, Kulikov EE. Free SARS-CoV-2 Spike Protein S1 Particles May Play a Role in the Pathogenesis of COVID-19 Infection. Biochem (2021) 86(3):257–61. doi: 10.1134/S0006297921030032
82. Radzikowska U, Ding M, Tan G, Zhakparov D, Peng Y, Wawrzyniak P, et al. Distribution of ACE2, CD147, CD26, and Other SARS-CoV-2 Associated Molecules in Tissues and Immune Cells in Health and in Asthma, COPD, Obesity, Hypertension, and COVID-19 Risk Factors. Allergy (2020) 75(11):2829–45. doi: 10.1111/all.14429
83. Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, et al. CD147-Spike Protein is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduct Target Ther (2020) 5(1):283. doi: 10.1038/s41392-020-00426-x
84. Hill AM, Barber MJ, Gotham D. Estimated Costs of Production and Potential Prices for the WHO Essential Medicines List. BMJ Glob Health (2018) 3(1):e000571. doi: 10.1136/bmjgh-2017-000571
85. John M, Eisenberg Center for Clinical Decisions and Communications Science. Medicines for Rheumatoid Arthritis: A Review of the Research for Adults. In: Comparative Effectiveness Review Summary Guides for Consumers. Rockville, MD: Agency for Healthcare Research and Quality (2005). Available at: http://www.ncbi.nlm.nih.gov/pubmed/23285506.
86. Yu LL, Zhu M, Huang Y, Zhao YM, Wen JJ, Yang XJ, et al. Metformin Relieves Acute Respiratory Distress Syndrome by Reducing miR-138 Expression. Eur Rev Med Pharmacol Sci (2018) 22(16):5355–63. doi: 10.26355/eurrev_201808_15737
87. Kim J, Kwak HJ, Cha JY, Jeong YS, Rhee SD, Kim KR, et al. Metformin Suppresses Lipopolysaccharide (LPS)-Induced Inflammatory Response in Murine Macrophages via Activating Transcription Factor-3 (ATF-3) Induction. J Biol Chem (2014) 289(33):23246–55. doi: 10.1074/jbc.M114.577908
88. Kelly B, Tannahill GM, Murphy MP, O’Neill LAJ. Metformin Inhibits the Production of Reactive Oxygen Species From NADH: Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1β (IL-1β) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-Activated Macrophages. J Biol Chem (2015) 290(33):20348–59. doi: 10.1074/jbc.M115.662114
89. Arai M, Uchiba M, Komura H, Mizuochi Y, Harada N, Okajima K. Metformin, an Antidiabetic Agent, Suppresses the Production of Tumor Necrosis Factor and Tissue Factor by Inhibiting Early Growth Response Factor-1 Expression in Human Monocytes In Vitro. J Pharmacol Exp Ther (2010) 334(1):206–13. doi: 10.1124/jpet.109.164970
90. Crouse AB, Grimes T, Li P, Might M, Ovalle F, Shalev A. Metformin Use Is Associated With Reduced Mortality in a Diverse Population With COVID-19 and Diabetes. Front Endocrinol (Lausanne) (2021) 11:600439. doi: 10.3389/fendo.2020.600439
91. Bramante CT, Ingraham NE, Murray TA, Marmor S, Hovertsen S, Gronski J, et al. Metformin and Risk of Mortality in Patients Hospitalised With COVID-19: A Retrospective Cohort Analysis. Lancet Health Longev (2021) 2(1):e34–41. doi: 10.1016/S2666-7568(20)30033-7
92. Cariou B, Hadjadj S, Wargny M, Pichelin M, Al-Salameh A, Allix I, et al. Phenotypic Characteristics and Prognosis of Inpatients With COVID-19 and Diabetes: The CORONADO Study. Diabetologia (2020) 63(8):1500–15. doi: 10.1007/s00125-020-05180-x
93. Luo P, Qiu L, Liu Y, Liu XL, Zheng JL, Xue HY, et al. Metformin Treatment was Associated With Decreased Mortality in COVID-19 Patients With Diabetes in a Retrospective Analysis. Am J Trop Med Hyg (2020) 103(1):69–72. doi: 10.4269/ajtmh.20-0375
94. Hariyanto TI, Kurniawan A. Metformin Use is Associated With Reduced Mortality Rate From Coronavirus Disease 2019 (COVID-19) Infection. Obes Med (2020) 19:100290. doi: 10.1016/j.obmed.2020.100290
95. Chen Y, Yang D, Cheng B, Chen J, Peng A, Yang C, et al. Clinical Characteristics and Outcomes of Patients With Diabetes and COVID-19 in Association With Glucose-Lowering Medication. Diabetes Care (2020) 43(7):1399–407. doi: 10.2337/dc20-0660
96. Cheng X, Liu YM, Li H, Zhang X, Lei F, Qin JJ, et al. Metformin Is Associated With Higher Incidence of Acidosis, But Not Mortality, in Individuals With COVID-19 and Pre-Existing Type 2 Diabetes. Cell Metab (2020) 32(4):537–47.e3. doi: 10.1016/j.cmet.2020.08.013
Keywords: COVID-19, SARS-CoV-2, immunometabolism, inflammation, monocyte
Citation: Cory TJ, Emmons RS, Yarbro JR, Davis KL and Pence BD (2021) Metformin Suppresses Monocyte Immunometabolic Activation by SARS-CoV-2 Spike Protein Subunit 1. Front. Immunol. 12:733921. doi: 10.3389/fimmu.2021.733921
Received: 30 June 2021; Accepted: 26 October 2021;
Published: 11 November 2021.
Edited by:
Julia Kzhyshkowska, Heidelberg University, GermanyReviewed by:
Israel Torres Castro, U964 Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (INSERM), FranceCopyright © 2021 Cory, Emmons, Yarbro, Davis and Pence. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brandt D. Pence, YmRwZW5jZUBtZW1waGlzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.