 Paige M. Mortimer†
Paige M. Mortimer†- Centre for Inflammatory Disease, Department of Immunology & Inflammation, Imperial College, London, United Kingdom
Reactive oxygen species (ROS) derived from the phagocyte NADPH oxidase (NOX2) are essential for host defence and immunoregulation. Their levels must be tightly controlled. ROS are required to prevent infection and are used in signalling to regulate several processes that are essential for normal immunity. A lack of ROS then leads to immunodeficiency and autoinflammation. However, excess ROS are also deleterious, damaging tissues by causing oxidative stress. In this review, we focus on two particular aspects of ROS biology: (i) the emerging understanding that NOX2-derived ROS play a pivotal role in the development and maintenance of adaptive immunity and (ii) the effects of excess ROS in systemic disease and how limiting ROS might represent a therapeutic avenue in limiting excess inflammation.
1 Introduction
1.1 Reactive Oxygen Species
Reactive Oxygen Species (ROS) are small molecules that are derived from molecular oxygen. They can either be classed as radicals or non-radicals, depending on whether they have an unpaired electron (1). Superoxide (a radical) is typically restricted to the endosomal compartment, and can be converted into hydrogen peroxide (H2O2; a non-radical) at low pH. H2O2 can diffuse across membranes to oxidise specific targets, or can be converted to O2 and H2O (2).
H2O2 is a very useful signalling molecule because it can be rapidly generated and rapidly removed via specific enzymes such as catalase, superoxide dismutase and peroxiredoxin enzymes. It can also be quenched by non-enzymatic means such as glutathione (GSH) (3). As such, by predominantly facilitating cysteine and methionine oxidation, H2O2 is integral to regulating several crucial facets of the immune response.
ROS are produced during metabolic reactions within many cellular compartments, including the mitochondria, peroxisome and endoplasmic reticulum (4, 5). This review, however, will focus on ROS specifically produced in the phagosomes and at the cell membrane, by the phagocyte NADPH oxidase NOX2. ROS generation can occur from many sources in cells. These include mitochondria, peroxisomes and the P450 enzyme system. The NADPH oxidase is the first example of an enzyme where generating ROS is the primary function of the system, not a by-product of another process, e.g. the generation of ATP in mitochondria [discussed in (6)].
1.2 A Brief History of ROS Discovery
The physiological production of ROS was first described in 1908, by the German biochemist Otto Warburg, who identified that following the fertilisation of sea urchin eggs, H2O2 production succeeded a large and rapid increase in oxygen consumption (7). He suggested the existence of a respiratory enzyme that utilised oxygen to generate ROS, for which he won the Nobel Prize in Physiology and Medicine (8).
The ability of phagocytes to produce ROS was first noted by Baldridge and Gerrard in 1933 who described a marked increase in oxygen uptake by canine neutrophils following phagocytosis (9). Sbarra and Karnovsky extended these findings to show that “this burst of extra respiration” was accompanied by glucose consumption via the hexose monophosphate shunt and lactate production (10). Crucially, inhibitors of mitochondrial respiration have no effect on the oxygen consumption that accompanies phagocytosis. This is because the purpose of the oxygen consumption is independent from aerobic glycolysis, and is instead required to generate ROS.
Further key milestones followed, including (i) the finding that NADPH is the dominant physiological electron donor (although both NADH and NADPH can act in this capacity) that allows the production of ROS (11–13) and (ii) the seminal observation that the process starts with the generation of superoxide (14).
These findings show that neutrophils possess an enzyme that facilitates the donation of electrons to molecular oxygen. The identification of cytochrome b558, which we refer to as NOX2, as the relevant enzyme resulted from insightful biochemistry and the study of the monogenic immunodeficiency X-linked chronic granulomatous disease (X-CGD). This “fatal granulomatous disease of childhood” was first described in the 1950s. It described boys whose neutrophils were unable to kill certain bacteria and did not increase oxygen consumption or produce ROS (15).
In a landmark study for the field, Segal and colleagues showed that neutrophils from patients with CGD lacked both NADPH oxidase activity and a particular unusual b type cytochrome that localised to the plasma membrane (16, 17). The suspected causative genetic region was localised to Xp21 and cloned (18). The cDNA identified from such studies was used to make a translated protein and an anti-serum was raised to it. Elegant studies showed that the anti-sera stained a 91kDa protein found in “purified cytochrome b558” preparations. Crucially, it could not stain neutrophils from patients with X-CGD (19). Thus, the unusual cytochrome identified by Segal was indeed the product of the gene that was disrupted in X-CGD. However, it was clear that the story was not quite that simple. For instance (i) the 91kDa membrane-bound protein transcribed and translated from the X chromosome co-purified with a 22kDa protein (20, 21) and (ii) it transpired that there were autosomal recessive forms of CGD (AR-CGD) associated with a deficiency of other specific proteins (22, 23).
1.3 Chronic Granulomatous Disease
The phagocyte NADPH oxidase (NOX2) is a multi-subunit protein complex that, upon interaction, can form an active enzyme complex capable of producing superoxide. It is comprised of two integral membrane bound components; the 91kDa gp91phox and 22kDa p22phox, which together form cytochrome b558 (16, 21). p22phox binds to and stabilises gp91phox, preventing its degradation and its own in return. The cytosolic components comprise of p40phox (24), p47phox (22), p67phox (22) and Rac1 (25) or Rac2 (26). Following stimulation, p47phox becomes phosphorylated, allowing the complex to translocate to the membrane where it can associate with the gp91phox-p22phox heterodimer, forming the activated complex that transfers electrons from NADPH to molecular oxygen (Figure 1). This process is known as the respiratory burst, which is essential during the innate immune response (27–30). ROS can also be generated by the other NOX family members, NOX1, NOX3, NOX4, NOX5, DUOX1 and DUOX2 (28), however this review will focus on NOX2 derived ROS.
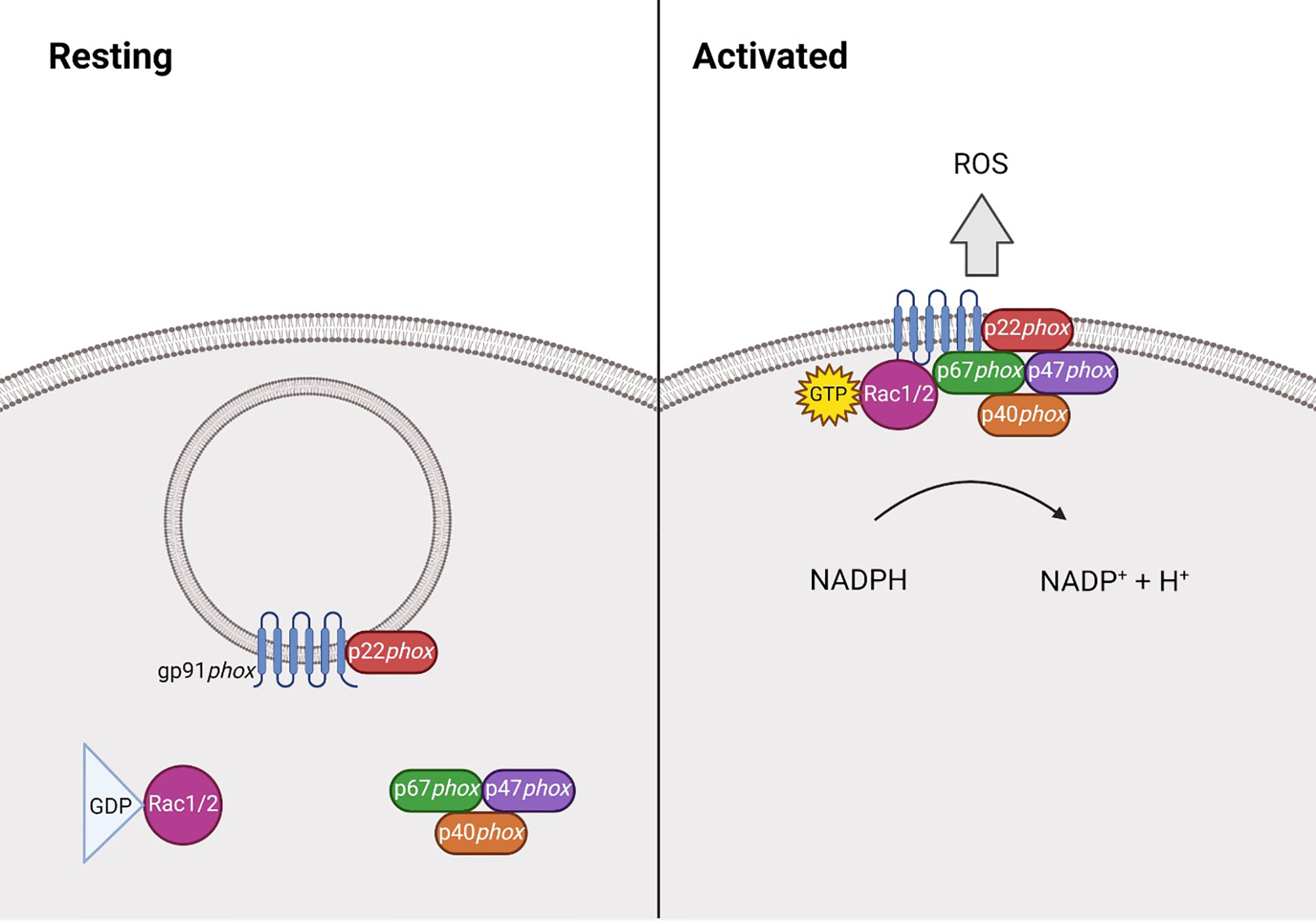
Figure 1 Structure of NADPH oxidase 2 during resting and activated states. At rest NADPH oxidase 2 (NOX2) is comprised of the membrane bound heterodimer gp91phox-p22phox, and the cytosolic component, comprised of p40phox (24), p47phox (22), p67phox (22) and the GTPase Rac1 (25) or Rac2 (26). Following stimulation, p47phox becomes phosphorylated and the Rac protein becomes GTP-bound, allowing the complex to translocate to the membrane where it can associate with the gp91phox-p22phox heterodimer. Together, this forms the activated complex that generates reactive oxygen species (ROS) by transferring electrons from NADPH.
Our understanding of CGD has improved as several large-scale cohort studies have been conducted. X-linked CGD is predominant in Europe (31–33), the United States (34) and Japan (35), accounting for approximately 60% of cases. p47phox deficiency accounts for around 30% of cases and p22phox and p67phox deficiency for the remaining 10%. AR-CGD is predominant in cohorts from countries such as Iran (36) or Turkey (37), where consanguineous marriage is more prevalent.
X-CGD patients have a more severe disease course than patients with the AR-CGD, presumably because they lack all oxidase activity (31). Residual oxidase activity in neutrophils is linked to reduced disease severity and modest production of ROS seems to confer a greater likelihood of long-term survival.
1.4 Anti-Microbial Action of NOX2-Derived ROS
The role of NOX2-derived ROS was first identified in killing microbes. This is well illustrated by the susceptibility of patients with CGD to an extensive, but nonetheless demarcated, range of pathogens. For example, neutrophils from patients with CGD can kill E. coli but not S. Aureus. The focus of this review is the extra-phagosomal role of ROS, but it is worthwhile describing briefly how neutrophil-mediated killing takes place. This can be both direct, where hydrogen peroxide is likely more microbicidal than superoxide, but also indirect through integration with other systems. Here reactive oxygen species collaborate with granule proteins such as myeloperoxidase (MPO). In the presence of H2O2, MPO catalyses the production of Hypochlorous acid (HOCL), a very potent anti-microbial agent. HOCL, in turn, can modify multiple proteins (both host and microbe) in the phagosome to generate chloramines and aldehydes.
While we do not cover these processes in detail, we would direct readers interested in this fascinating area to excellent reviews by Nauseef (38) and also, Winterbourn and Kettle (39). These expand more on a few key points:
(i) That there is synergy between ROS and other granule constituents, including HOCL and non-oxidative killing mechanisms such as cathelicidins, serine proteases and lactoferrin.
(ii) That microbes employ a variety of strategies to evade phagosomal killing.
(iii) That though the phagocyte NADPH oxidase is present in both neutrophils and macrophages, the intra-phagosomal environment in these cells differs greatly with respect to parameters such as pH and other anti-microbial components (such as MPO).
1.5 EROS Regulates the Expression of NOX2
An important recent addition to the biology of the phagocyte NADPH oxidase and CGD is the discovery of EROS (gene symbol CYBC1) (40). In 2017, we demonstrated that mice deficient in the previously uncharacterised open reading frame, bc017643, were exquisitely susceptible to infection with Salmonella Typhimurium and Listeria monocytogenes. It transpired that they failed to make ROS, owing to almost complete deficiency of gp91phox and p22phox. bc017643 encodes an ER-resident transmembrane protein that co-immunoprecipitates with gp91phox. We also observed that P2X7 receptor expression was downregulated in EROS deficient cells, which was later independently verified by another group (41), demonstrating that EROS mediates the expression of both gp91phox and P2X7. We characterised the protein encoded by bc017643 as EROS (Essential for Reactive Oxygen Species). Recent work in our laboratory suggests that EROS is a highly selective placeholder chaperone, binding to an “unsatisfied surface” on the gp91phox precursor and stabilising it until p22phox can bind (Randzavola, Mortimer et al., submitted). The lack of ROS and NOX2 expression in EROS-deficient mice, akin to that of gp91phox-/- mice, suggested that mutations in the human orthologue C17ORF62 might lead to CGD. In 2018, we and another group reported separate homozygous mutations in C17ORF62, leading to EROS-deficiency, as a novel cause of chronic granulomatous disease (CGD5) (42, 43). C17ORF62 has been re-named CYBC1 (CYtochrome B Chaperone 1).
Mutations in EROS represent the first type of CGD to arise as a result of altered stability or folding of gp91phox. Although they have not yet been implicated in CGD, several other proteins are capable of regulating gp91phox abundance. Negative Regulator of Reactive Oxygen Species (NRROS) was described in 2014 as an ER-resident protein that binds gp91phox and facilitates its degradation (44), its role appearing opposite to that of EROS. Similarly, the heat shock proteins, hsp90 and hsp70, also have opposing effects on gp91phox abundance, stabilising and degrading it respectively (45). The lamin beta receptor (LBR) has an intriguing role in regulating gp91phox and neutrophils that are LBR deficient show lower expression of gp91phox and generation of zymosan-induced ROS (46, 47).
Regulating gp91phox is one method of regulating ROS production. It is essential to tightly control the levels of ROS and subsequent sections of this review examine the tissue damage that can be caused by excess ROS production. However, too little ROS can also lead to inflammation. A key observation from studying CGD is that patients do not only experience opportunistic infections, but also present with autoinflammatory and autoimmune manifestations. These manifestations are characterised by sterile granulomatous inflammation, a hallmark of CGD (48). CGD patients often develop autoimmune diseases in which the pathogenesis is driven by autoantibody production, such as systemic lupus erythematosus and juvenile rheumatoid arthritis (31, 34). By producing H2O2, the phagocyte NAPDH oxidase regulates multiple pathways involved in innate anti-microbial defence, often serving to restrain inflammation in the process.
1.6 NOX2 Regulates Inflammation and Immune Signalling
The inflammatory manifestations that affect CGD patients arise as loss of ROS signalling impairs type 1 interferon signalling and autophagy (29). Patients with X-CGD are between 50-90% more likely to experience inflammatory episodes compared to patients with AR-CGD (49, 50), suggesting NOX2, particularly gp91phox, is essential for controlling the balance between a successful immune response and tissue damage.
2 NOX2 in Regulating Processes in Antigen Presentation
The regulation of type 1 interferon signalling, inflammasome activation and autophagy is well documented in the innate immune system and we have covered this in previous reviews (29, 30). The role of ROS in adaptive immunity starts with its pivotal role in antigen presentation.
2.1 MHC Class I Processing and Presentation
In normal circumstances, ROS generation occurs concomitantly with phagocytosis, regardless of cell type. Superoxide is a weak base and tends to alkalinise the phagosome, which influences proteolysis. A key study showed that the normal pH of dendritic cells (DCs) is neutral, tending towards slightly alkaline, but that NOX2-deficient DCs have acidic phagosomes which leads to enhanced antigen degradation and impaired cross presentation to CD8+ T cells via MHC Class I (51). The same group reported similar results in human DCs (52) and that Rac2 was key for NADPH oxidase assembly in CD8+ DCs (53). The small GTPase Rab27a was also necessary for NAPDH oxidase assembly (54). However another group, using slightly different conditions, found that while NOX2 did indeed reduce phagosomal proteolysis, this was not associated with significant changes in phagosomal pH. Rather, this group proposed that in DCs and macrophages, NOX2 affects proteolysis through reversible inhibition of the action of cysteine cathepsins via H2O2-driven oxidation of cysteine residues. Aspartic cathepsins are unaffected by the presence of NOX2 and thus the phagocyte NADPH oxidase was proposed to alter the activity of only a subset of proteases, skewing the peptide repertoire generated (55, 56).
A recent publication by Reis e Sousa and colleagues provided an intriguing new insight into the role of NOX2-derived ROS in antigen presentation (57). The DNGR1 receptor, expressed on the conventional DC1 (cDC1) subset of DCs, is essential for effective cross-presentation. DNGR1 binds F-actin on dead cell corpses, and has a short hemi-ITAM motif that can recruit and activate Syk. Mice that are deficient in DNGR1 or Syk expression in DCs have impaired cross presentation (58).
This group demonstrated that DNGR1 ligation facilitates Syk kinase activation and this, in turn, leads to NOX2 activation within phagosomes containing internalised antigen. The oxidative stress caused by the resulting free radicals damages the phagosome, causing membrane rupture, thus allowing leak of antigen into the cytosol and its translocation into the MHC class I presentation pathway. Cross presentation is markedly impaired in gp91phox-deficient DCs. How exactly Syk drives NOX2 activation in this context is unknown but the NOX2 activators Vav1 and Rac have previously been shown to be necessary for efficient cross-presentation and they are likely to be involved to some extent.
2.2 MHC Class II Processing and Presentation
There is also evidence that NOX2 can influence MHC class II peptide processing and presentation. Extending their previous work on cysteine cathepsins Yates and colleagues demonstrated that NOX2 not only affects the amount of proteolytic processing but affects it qualitatively too (59). They used the myelin oligodendrocyte glycoprotein (MOG) -induced model of experimental autoimmune encephalomyelitis (EAE) and showed that NOX2-derived macrophages (though interestingly not DCs) were defective in their ability to process and present the I-A(b)-immunodominant peptide of MOG. As such, p47phox or gp91phox deficient mice were partially protected from the central nervous system injury and inflammation that characterises EAE. MHC class II presentation is also impaired in human B cells deficient in p40phox (60). This was most marked for cytoplasmic and endogenous antigen but processing of membrane antigen was normal (see Altered Humoral Immune Response in NOX2 Deficiency below).
3 NOX2 in Regulating Adaptive Immunity
Although most commonly associated with innate immunity, NOX2 also has a variety of signalling roles in T and B cell responses. Some of these include; modulating T helper differentiation, proliferation of B cells and inducing apoptosis.
3.1 NOX2 in CD4+ T Cells
3.1.1 NOX2 Signalling Influences CD4+T Helper Differentiation
There are many conflicting studies regarding the influence of NOX2 on the differentiation of T helper subsets, summarised in Table 1. Briefly, the first published study on T helper differentiation in NOX2 deficiency describes a preferential Th1 response in NOX2-/- CD4+ T cells (61). Secretion of IFNγ is typical of a Th1 response, IL-4 and IL-5 of a Th2 response, and IL-17 and TGFβ of a Th17 response. Jackson et al. (61) found an increase in IFNγ secretion and decrease in IL-4 and IL-5 secretion following stimulation with anti-CD3. However, Kwon et al. found an increase in IL-4 secretion following anti-CD3 and anti-CD28 stimulation, indicative of a Th2 response in NOX2 deficiency (62). In opposition to both, Tse et al. (63) describe that a Th17 response develops in NOX2-/- CD4+ T cells following anti-CD3 and anti-CD28 stimulation. They found decreased IFNγ and IL-4 secretion, but increased IL-17 and TGFβ secretion (63). Although, this group used the NOD strain of mice that are renowned for their autoimmune phenotype, whereas the studies mentioned previously used the C57BL/6 strain, which may account for the difference in findings. Most studies find a combined Th1/Th17 response in NOX2 deficiency, with increased levels of IFNγ, IL-17 and their associated transcription factors T-bet and RORγt (64–66). Interestingly, Lee et al. (60) found that under their specific polarising conditions, the differentiation of all T helper subsets was elevated when NOX2 was absent, demonstrating that ROS are required to balance the development of T cell responses. The importance of NOX2 in CD4+ T helper differentiation remains to be clarified, but it appears that proinflammatory Th1/Th17 skewing is favoured. There are currently no published studies on the effect of NOX2 deficiency on the differentiation of Th9 or T follicular helper cells (Tfh). It would be both interesting and important to understand the entire T cell phenotype in the context of NOX2 deficiency.
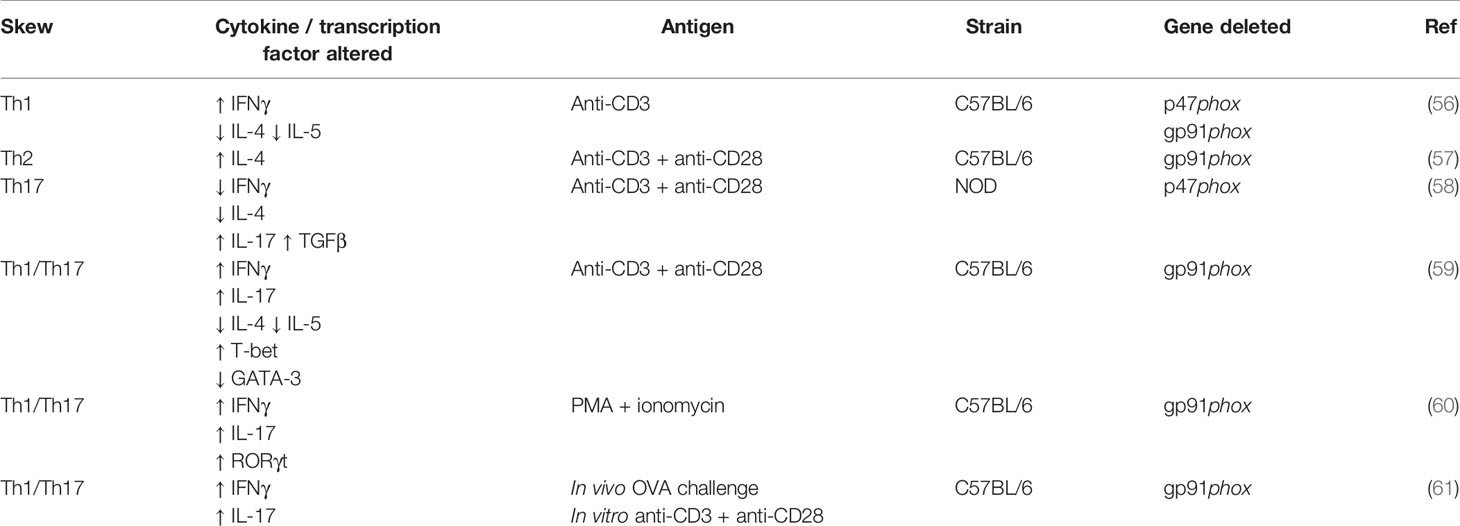
Table 1 Summary of studies describing T helper differentiation in NOX2 deficiency.
3.1.2 NOX2 Signalling Affects Treg Differentiation and Activity
NOX2 is involved in the differentiation of other T cell types, including T regulatory cells (Tregs). One study found fewer peripheral CD4+CD25+ Tregs and decreased FOXP3 expression in NOX2-/- mice, indicating that NOX2 derived ROS also play a role in controlling the development of Tregs (65). However, a recent study found no decrease in Treg number or function in CGD patients, except in those with X-linked gp91phox deficiency (67). The authors suggest this may coincide with the fact that gp91phox-/- CGD patients have more inflammatory symptoms than those with mutations in other NADPH oxidase subunits (49, 67).
NOX2 is required for restraining the expression of the immune suppressive molecules on Tregs. The expression of CTLA-4, GITR, CD39 and CD73 is significantly greater on gp91phox-/- Tregs. Additionally, gp91phox-/- Tregs have increased NF-kB activation and greater FOXP3 expression. Subsequently, Tregs deficient in gp91phox have greater suppressive activity than wildtype control Tregs (68). Interestingly, p47phox-/- Tregs have poorer suppressive capabilities compared to their wildtype counterparts (69). This may relate to the functions of p47phox independent of phagocyte NADPH oxidase (70, 71).
3.1.3 NOX2 Is Required for T Cell Apoptosis
NOX2 is required for inducing cell-intrinsic apoptosis in activated T cells during the resolution of an immune response (72). Apoptosis of excess T cells after antigen clearance is essential to prevent an over exuberant immune response when responding to new and repeated antigenic challenges. gp91phox-/- T cells display significantly improved survival in vivo following cytokine deprivation. Greater antigen-specific proliferative responses are also observed when compared to wildtype controls, due to the larger pool of T cells that remain after the initial antigen challenge (72). This increased T cell survival may account for the differences in cytokine secretion discussed in section NOX2 Signalling Influences CD4+T Helper Differentiation.
3.2 NOX2 in CD8+ T Cells
3.2.1 NOX2 Signalling Can Affect CD8+ T Cell Responses
CD8+ T cell responses are critical to eliminate intracellular pathogen infections. In the absence of NOX2, mice are highly susceptible to Trypanosoma cruzi infection. There are fewer CD8+ T cells present at baseline in p47phox-/- mice, and these fail to proliferate in response to T. cruzi infection (73). Conversely, p47phox-/- CD8+ T cells have improved survival and mice experience reduced viral titres in response to lymphocytic choriomenigitis virus (LCMV) infection. The authors state this improved CD8+ T cell viral response may be due to less immunopathology that occurs in the absence of p47phox (74). Similarly, gp91phox-/- mice have reduced inflammation and viral titres in response to influenza infection, however there was no difference in CD8+ T cell populations in vivo or influenza-specific CD8+ T cell responses in vitro (75). Therefore, the influence of NOX2 on CD8+ T cell responses may be dependent upon pathogen type.
3.2.2 NOX2 Is Critical for CD8+ Treg Driven Immunosupression
NOX2 is utilised by CD8+ Tregs to enable a novel Treg mediated suppression of CD4+ T cells (76). CD8+ Tregs are thought to release exosomes containing NOX2, which is taken up by CD4+ T cells located in nearby T cell zones of secondary lymphoid organs. NOX2 derived ROS inhibits the phosphorylation of the T cell receptor (TCR) signalling molecules ZAP70 and LAT, inhibiting TCR signal transduction. CD8+ Tregs treated with the flavoenzyme inhibitor diphenyleneiodonium (DPI), gp91ds-tat or short hairpin RNAs targeting NOX2 are unable to upregulate NOX2 and subsequently are unable to supress CD4+ T cell activation (76).
3.3 NOX2 in B Cells
3.3.1 NOX2 Elicits Bacterial Killing in B Cells
Similar to innate immune cells but unlike T cells, peritoneal B cells can utilise NOX2 derived ROS to kill intracellular bacteria. NOX2-/- B cells from NOX2 deficient mice have a reduced ability to produce the ROS required to kill engulfed pathogens, and therefore have greater survival of bacteria within phagosomes (77).
3.3.2 NOX2 Signalling Restrains Proliferation of B Cells
Following B cell receptor (BCR) stimulation, NOX2 is responsible for generating the rapid initial production of ROS, whereas the later stages of ROS production are not NOX2 dependant. NOX2-/- mice fail to produce ROS immediately after BCR stimulation, but BCR proximal signalling and subsequent downstream signalling pathways are normal (78, 79). However, NOX2-/- B cells have been found to undergo enhanced cell cycle entry following BCR stimulation (79, 80). This suggests NOX2 has a role in negatively modulating ROS-driven BCR induced proliferation in B cells.
3.3.3 NOX2 Is Involved in B Cell Signalling
NOX2 derived ROS often acts as a second messenger during various signalling pathways. Tyrosine phosphorylation and IgM secretion is impaired following BCR or TLR4 stimulation in NOX2 deficient B cells. Accordingly, lentiviral induced expression of NOX2 components can restore signalling capabilities in NOX2 deficient cells following BCR stimulation (81). NOX2-/- B cells have increased expression of the Toll-like receptors (TLR) TLR7 and TLR9, and subsequently have greater responsiveness to TLR7/9 stimulation (82). These studies demonstrate that NOX2 can modulate BCR signalling in a number of ways.
3.3.4 NOX2 Regulates MHC Class II Antigen Presentation on B Cells
Presentation of exogenous antigens requires antigen uptake and processing in endosomal or lysosomal compartments to generate the peptides to be presented on MHC class II molecules [reviewed in (83)]. p40phox-/- B cells are less able to present exogenous antigen on their MHC class II. However, p40phox-/- B cells preferentially present self-membrane resident antigens, suggesting p40phox may skew epitope selection and have implications for CD4+ T cell activation (60).
3.3.5 Altered Humoral Immune Response in NOX2 Deficiency
NOX2 may have a role in the production of antibodies. NOX2-/- mice have greater antibody production following injection of collagen (84, 85) and challenge with UV-irradiated bacteria (86). Cachat et al. (88) found an increase in IgG1 and IgG2c production in NOX2-/- mice following ovalbumin injection. A later paper found NOX2-/- mice have increased production of IgA, IgG, IgG1, IgG2b and IgG3 levels following influenza A infection (88). The authors suggest that functional NOX2 activation during influenza A infection results in the suppression of antiviral cytokines, preventing the development of humoral immunity (88). Interestingly, there may be some differences between human and mouse. IgG1 levels are decreased whereas IgG2 levels are increased in CGD patient serum (87). CGD patients also have significantly increased levels of B cell activating factor (BAFF), a B cell survival factor, and subsequently have greater IgM levels compared to healthy controls (89). CGD patients have decreased numbers of influenza-specific peripheral memory B cells but increased numbers of nonconventional CD27- memory B cells compared to healthy controls (90, 91). Although, despite abnormal numbers of B memory cells, influenza specific memory B cell responses remain comparable to healthy controls (90). Therefore, NOX2 is involved in inducing and maintaining the humoral immune response, however the specific role of NOX2 in human B cell responses needs to be investigated further.
4 The Deleterious Role of ROS in Systemic Inflammation
In the sections above, we have seen that ROS are pivotal for both normal innate and adaptive immunity. We have also described how a lack of ROS in CGD can lead to autoinflammation and autoimmunity.
However, we have also seen how the generation of ROS must be tightly controlled and its generation can outstrip the capability of those systems that regulate it. Excess ROS can cause tissue damage in a variety of ways, causing protein and DNA damage and lipid peroxidation.
4.1 Oxidative Stress in Systemic Disease
Oxidative stress is well known to be a contributing factor in the development of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and Multiple Sclerosis. Dysregulation or overproduction of ROS leads to oxidative stress which is thought to disrupt immune homeostasis in the central nervous system (CNS) and promote prolonged neuroinflammation (92). NOX proteins are important generators of ROS in the CNS and NOX2 expression has been documented in the CNS in microglia, neurons and endothelial cells (93).
5 Reactive Oxygen Species in the CNS
5.1 Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by the progressive loss of motor neurons in the brain, brain stem and spinal cord. Disease progression is rapid, with a prognosis of only 2-5 years after diagnosis for most individuals (94).
Several studies have demonstrated that NOX2 contributes to disease progression in the SOD1G93A transgenic mouse, a common mouse model of ALS. NOX2 expression and activation was shown to be significantly upregulated in microglia in the spinal cord of SOD1G93A mice compared to controls (95). NOX2 expression was also found to be increased in the spinal cord of sporadic ALS patients. The authors demonstrated that NOX2 deletion in SOD1G93A transgenic mice prolonged survival and slowed disease progression, suggesting that NOX2 activity contributes to the degeneration of motor neurons and disease progression in ALS. Another study demonstrated that treatment of SOD1G93A mice with apocynin, a NOX inhibitor, also increased survival and slowed disease progression (96). The authors also demonstrated that SOD1 regulated RAC1/NOX2 dependent ROS generation in a redox-dependent manner. ALS-associated SOD1 mutants resulted in enhanced activation of RAC1/NOX2 and increased ROS production in both cell lines and the spinal cord of SOD1G93A mice.
However, in contrast to this, a recent study found that NOX2 inhibition did not extend survival in SOD1G93A mice (97). Deletion of NOX1 or NOX2 in SOD1G93A mice did not increase survival or influence microglia activation in this study. Treatment of SOD1G93A mice with the NOX inhibitors thioridazine and perphenazine did not significantly increase survival or prevent motor neuron degeneration. A study of the oxidative burst in granulocytes in the peripheral blood of ALS patients did not identify any significant difference in NOX2 activity between ALS patients and matched controls. However, patients with lower NOX2 activity were found to have a significant increase in survival (98). Therefore, whilst NOX2 has been shown to be upregulated in both mouse models and patients with ALS, the exact role of NOX2 in ALS pathogenesis remains unclear.
5.2 Multiple Sclerosis
Multiple sclerosis (MS) is a common neurodegenerative disorder characterised by inflammation and demyelination in the CNS. Microglial activation is hypothesised to play an important role in the pathogenesis of MS. NOX2 has also been shown to be upregulated in microglia in active lesions in MS patients (99).
The mouse experimental autoimmune encephalomyelitis (EAE) model is commonly used as a model to study MS. Recent evidence has demonstrated that disease severity is reduced in NOX2-/- mice after EAE induction. Lymphocyte and microglial infiltration in the CNS was also significantly decreased compared to heterozygous and wild-type controls. Interestingly, the authors showed that only immune infiltration in the CNS is reduced, as immune cell populations in peripheral tissues such as the spleen and cervical lymph node are similar in NOX2-/- and wild-type mice post EAE induction (100). Microglial activation was also decreased in NOX2-/- mice, with inflammatory cytokine and chemokine secretion levels in the CNS also decreased.
Another recent paper demonstrated that deletion of NOX2 in conventional DCs (cDCs) reduced disease severity and demyelination in an adoptive transfer model of EAE (101). Interestingly, the authors demonstrated that deletion of NOX2 in cDCs reduced accumulation and activation of autoimmune CD4+ T cells in the CNS in EAE mice, suggesting that NOX2 regulates CD4 infiltration. Deletion of NOX2 also abrogated LC3-associated phagocytosis and CD4+ T cell activation through reduced myelin antigen presentation. This study highlights an important role for NOX2 in promoting inflammation and demyelination in EAE mice. Therefore targeting NOX2-dependent ROS production may slow disease progression and provide therapeutic benefit for patients with MS.
5.3 Alzheimer’s Disease
Oxidative stress and damage have also been hypothesised to play a role in Alzheimer’s disease, although the role of NADPH oxidases remains unclear. As mentioned previously, microglia play a vital immune role in CNS homeostasis through clearance of dead cells and debris. However, dysregulation of microglia can lead to prolonged neuroinflammation and the development of neurodegenerative disorders. Microglia have been shown to associate with and promote clearance of amyloid-β (Aβ) deposits in the early stages of Alzheimer’s. However, in aging mice microglia appear to have a reduced ability to clear Aβ deposits and drive inflammation within the CNS (102).
NOX2 expression in microglia has also been hypothesised to play an important role in the pathogenesis of Alzheimer’s disease. A recent study demonstrated high NOX2 expression in microglia and increased microglial infiltration in aged wild-type brains, compared to young mice (103). Interestingly, NOX2-/- aged mice had significantly less Aβ deposition and plaque formation compared to aged wild-type controls. ROS production was also much lower in NOX2-/- mice than in wild-type mice, indicating that ROS production in the aged mice was NOX2-dependent. The authors also investigated ROS production in human brain tissue, and found older individuals had higher levels of ROS production when compared to young controls. Stimulation of the BV2 microglial cell line with Aβ42 peptide also resulted in significantly increased NOX2-depdendent ROS production, which could be inhibited using NOX2 inhibitors such as apocynin or Nox2tat. These results indicate that NOX2 may play an important role in the regulation of microglia and the microglial response to Aβ plaques and therefore it may be an important driver of the pathogenesis of Alzheimer’s disease.
Whilst it is clear that oxidative stress is involved in aging and the development of neurodegenerative diseases, the precise mechanisms defining how aberrant NOX2-depdendent ROS production drives neuroinflammation require further investigation. In addition, it remains to be investigated whether targeting of NOX2 through the use of inhibitors would provide therapeutic benefit in neurodegenerative disorders.
6 The Role of Reactive Oxygen Species in the Lung
ROS production by phagocytes plays a vital role in the innate immune response and the clearance of pathogens during infection. However, it is essential that the mechanisms which regulate ROS generation are tightly controlled. Failure to regulate the innate immune response results in excessive ROS production, or oxidative stress, which promotes inflammation. Oxidative stress and the resultant sustained inflammation can result in tissue damage, particularly in barrier sites such as the lung (104). Recent evidence has implicated excessive NOX2-derived ROS production in acute lung injury, particularly during influenza infection.
In 2006, Snelgrove et al. demonstrated that deletion of NOX2 in mice resulted in reduced viral load after infection with Influenza virus (105). NOX2-/- mice exhibited enhanced viral clearance, increased lung function and reduced lung damage when compared to wild-type mice. Increased macrophage and neutrophil infiltration into the airway epithelia was also observed. Another study by Vahlos et al. in 2011 also demonstrated that deletion of NOX2 resulted in reduced viral titres in mice infected with Influenza A virus (75). Apoptosis of lung alveolar epithelial cells was greatly reduced in lung tissue sections of NOX2-/- mice compared to wild-type mice. Interestingly, in contrast to the earlier findings by Snelgrove et al., the authors demonstrated that immune cell infiltration in the bronchoalveolar lavage fluid (BALF) was significantly decreased in NOX2-/- mice. However, the authors hypothesise that this could be due to sex differences between the mouse models used in the studies. Treatment of wild-type mice with the ROS inhibitor apocynin after Influenza A infection also significantly reduced macrophage and neutrophil infiltration and viral titres were reduced by 50%. These results indicate that NOX2 is driving inflammation and acute lung injury in response to Influenza A infection. Therefore, modulation of NOX2-dependent ROS production may provide therapeutic benefit and reduce lung damage in patients suffering from acute lung injury during infection.
A recent study identified that Influenza A infection drives the production of endosomal NOX2-dervied ROS in response to TLR7 stimulation by viral RNA (88). Endosomal ROS was also found to suppress cytokine secretion in a TLR7-dependent manner. Treatment with apocynin significantly increased IL-1β, TNF-α, IFN-β and IL-6 secretion in wild-type macrophages in response to imiquimod. Interestingly, the authors identified a single cysteine residue, Cys98, which is highly conserved and unique to TLR7, as a novel redox sensor. TLR7-/- macrophages transfected with a TLR7C98A mutant could not restore TLR7-dependent cytokine secretion. The authors hypothesise that ROS production by NOX2 may modify the Cys98 residue, resulting in reduced cytokine secretion and a dampened antiviral response (88). In Nox2 deficient mice infected with Influenza A virus, IL-1β and IFN-β secretion was significantly increased. Serum and BALF levels of IgG and IgA were also significantly increased compared to wild-type mice, indicating that NOX2-derived ROS can also suppress antibody production. These results indicate that ROS production can inhibit important antiviral responses, thereby reducing the host’s ability to efficiently clear viral pathogens.
Recent evidence has also demonstrated that NOX2 can modulate Type I Interferon (IFN) production in response to bacterial infections. NOX2-/- mice infected with Listeria monocytogenes exhibited an increased bacterial load, whereas Ifnar1-/- mice infected with L. monocytogenes. had a reduced bacterial load (106). Deletion of NOX2 in Ifnar1-/- mice also resulted in a reduced bacterial load, indicating that NOX2 regulation of Type I IFN controls L. monocytogenes infection. The number of infection foci was increased in NOX2-/- mice, however lymphocyte migration to infection foci was decreased in a Type I IFN-dependent manner. Interestingly, the authors also demonstrated that NOX2 deficiency upregulates IL-10 expression, which is known to play an anti-inflammatory role during infection. These results suggest a novel antimicrobial role for NOX2 in controlling L. monocytogenes infection through modulation of the Type I IFN response.
7 Conclusion
The balanced generation of ROS is essential to induce appropriate immune responses and avoid tissue damage by oxidative stress (Figure 2). ROS is required during the innate immune response to control invading pathogens and prevent infection. Too little ROS results in susceptibility to opportunistic pathogens, such as in CGD. However, increasing evidence has demonstrated that dysregulation of ROS production can result in sustained inflammation and tissue damage which can be fatal during severe infection. Recent evidence has also demonstrated that ROS can prevent antiviral immunity and reduce the body’s ability to clear viral infections. Therefore, inhibition of NOX2 during infection may help to promote antiviral responses and prevent excessive ROS generation, thereby reducing the occurrence of acute lung injury, for example in Influenza infection. It is important to further investigate the mechanisms underlying the regulation of ROS generation and how they become dysregulated during infection in order to understand how they may be targeted for clinical benefit in the future. Beyond influenza A, the utility of blocking NOX2 action should be examined in other settings such as acute lung injury and Acute Respiratory Distress Syndrome (ARDS). The question of blocking host inflammatory pathways in infection related lung pathology has been shown into sharp focus by the COVID-19 pandemic where treatment with dexamethasone (107) or IL-6 inhibition (108) can have a beneficial effect when used at the correct stage of the disease. It will be interesting to see if NOX2 inhibition has any beneficial effect in either in vitro or animal models of COVID-19 as it is likely that the virus will continue to mutate and become endemic, possibly escaping vaccine-mediated control in some instances.
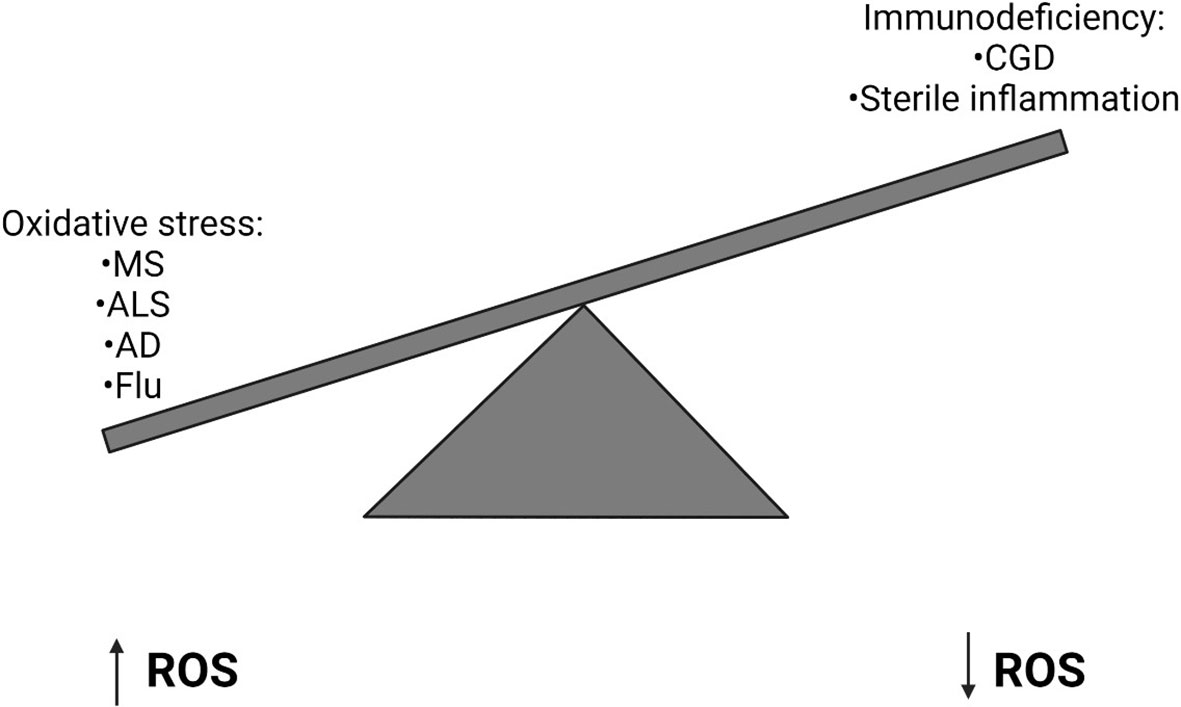
Figure 2 Imbalanced Reactive Oxygen Species generation can have implications for human health. High levels of Reactive Oxygen Species (ROS) can result in oxidative stress, which can lead to a number of diseases including Multiple Sclerosis (MS), Amyotrophic Lateral Sclerosis (ALS), Alzheimer’s Disease (AD) and Influenza. Alternatively in primary immunodeficiencies, such as Chronic Granulomatous Disease (CGD), where a genetic defect means ROS are not generated, sterile inflammation often develops.
Numerous NOX2 inhibitors have been developed (109) and it is likely that advances in in silico technology such as alpha-fold will also better inform our efforts to inhibit the action of this and other NADPH oxidases (110, 111).
Author Contributions
PM, SM, and DT: conceptualisation and writing. DT: funding acquisition and final revision. All authors contributed to the article and approved the submitted version.
Funding
DT is funded by a Wellcome-Beit Prize Trust Clinical Research Career Development Fellowship (grant code 098051) and Imperial College London. Research in DT lab is supported by a generous donation from the family of Sidharth Burman.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lushchak VI. Free Radicals, Reactive Oxygen Species, Oxidative Stress and its Classification. Chemico-Biological Interact (2014) 224:164–75. doi: 10.1016/j.cbi.2014.10.016
2. Winterbourn CC. Reconciling the Chemistry and Biology of Reactive Oxygen Species. Nat Chem Biol (2008) 4:278–86. doi: 10.1038/nchembio.85
3. Holmström KM, Finkel T. Cellular Mechanisms and Physiological Consequences of Redox-Dependent Signalling. Nat Rev Mol Cell Biol (2014) 15:411–21. doi: 10.1038/nrm3801
4. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
5. Peng H-Y, Lucav J, Ballard D, Das JK, Kumar A, Wang L, et al. Metabolic Reprogramming and Reactive Oxygen Species in T Cell Immunity. Front Immunol (2021) 0:900. doi: 10.3389/FIMMU.2021.652687
6. Bedard K, Krause KH. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol Rev (2007) 87:245–313. doi: 10.1152/physrev.00044.2005
7. Finkel T. Signal Transduction by Reactive Oxygen Species. J Cell Biol (2011) 194:7–15. doi: 10.1083/jcb.201102095
8. Brand RA. Biographical Sketch: Otto Heinrich Warburg, PhD, MD. Clin Orthop Relat Res (2010) 468:2831–2832. doi: 10.1007/s11999-010-1533-z
9. Baldridge CW, Gerard RW. The Extra Respiration of Phagocytosis. Am J Physiol Content (1932) 103:235–6. doi: 10.1152/ajplegacy.1932.103.1.235
10. Sbarra AJ, Karnovskyi ML. The Biochemical Basis of Phagocytosis I. Metabolic Changes During the Ingestion if Particles by Polymorphonuclear Leukocytes*. J Biol Chem (1959) 234(6):1355–62. doi: 10.1016/S0021-9258(18)70011-2
11. Rossi F, Zatti M. Changes in the Metabolic Pattern of Polymorpho-Nuclear Leucocytes. Br J Exp Pathol (1964) 45:548–59.
12. Rossi F, Zatti M. Biochemical Aspects of Phagocytosis in Poly-Morphonuclear Leucocytes. NADH and NADPH Oxidation by the Granules of Resting and Phagocytizing Cells. Experientia (1964) 20:21–3. doi: 10.1007/BF02146019
13. Babior BM, Kipnes RS. Superoxide-Forming Enzyme From Human Neutrophils: Evidence for a Flavin Requirement. Blood (1977) 50:517–24. doi: 10.1182/blood.V50.3.517.517
14. Babior BM, Kipnes RS, Curnutte JT. Biological Defense Mechanisms. The Production by Leukocytes of Superoxide, a Potential Bactericidal Agent. J Clin Invest (1973) 52:741–4. doi: 10.1172/JCI107236
15. Bridges RA, Berendes H, Good RA. A Fatal Granulomatous Disease of Childhood: The Clinical, Pathological, and Laboratory Features of a New Syndrome. AMA J Dis Child (1959) 97:387–408. doi: 10.1001/archpedi.1959.02070010389004
16. Segal AW, Jones OTG. Novel Cytochrome B System in Phagocytic Vacuoles of Human Granulocytes [25]. Nature (1978) 276:515–7. doi: 10.1038/276515a0
17. Segal AW, Jones OTG, Webster D, Allison AC. Absence of a Newly Described Cytochrome B From Neutrophils of Patients With Chronic Granulomatous Disease. Lancet (1978) 312:446–9. doi: 10.1016/S0140-6736(78)91445-9
18. Royer-Pokora B, Kunkel LM, Monaco AP, Goff SC, Newburger PE, Baehner RL, et al. Cloning the Gene for an Inherited Human Disorder - Chronic Granulomatous Disease - On the Basis of its Chromosomal Location. Nature (1986) 322:32–8. doi: 10.1038/322032a0
19. Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, Parkos CA. The Glycoprotein Encoded by the X-Linked Chronic Granulomatous Disease Locus Is a Component of the Neutrophil Cytochrome B Complex. Nature (1987) 327:717–20. doi: 10.1038/327717a0
20. Parkos CA, Allen RA, Cochrane CG, Jesaitis AJ. Purified Cytochrome B From Human Granulocyte Plasma Membrane Is Comprised of Two Polypeptides With Relative Molecular Weights of 91,000 and 22,000. J Clin Invest (1987) 80:732–42. doi: 10.1172/JCI113128
21. Segal AW. Absence of Both Cytochrome B-245 Subunits From Neutrophils in X-Linked Chronic Granulomatous Disease. Nature (1987) 326:88–91. doi: 10.1038/326088a0
22. Segal AW, Heyworth PG, Cockcroft S, Barrowman MM. Stimulated Neutrophils From Patients With Autosomal Recessive Chronic Granulomatous Disease Fail to Phosphorylate a Mr-44,000 Protein. Nature (1985) 316:547–9. doi: 10.1038/316547a0
23. Volpp BD, Nauseef WM, Clark RA. Two Cytosolic Neutrophil Oxidase Components Absent in Autosomal Chronic Granulomatous Disease. Science (1988) 242:1295–7. doi: 10.1126/science.2848318
24. Wientjes FB, Hsuan JJ, Totty NF, Segal AW. P40(Phox), a Third Cytosolic Component of the Activation Complex of the NADPH Oxidase to Contain Src Homology 3 Domains. Biochem J (1993) 296:557–61. doi: 10.1042/bj2960557
25. Abo A, Pick E. (PDF) Purification and Characterization of a Third Cytosolic Component of the Superoxide-Generating NADPH Oxidase of Macrophages. J Biol Chem (1992) 23577–85.
26. Roberts AW, Chaekyun K, Zhen L, Lowe JB, Kapur R, Petryniak B, et al. Deficiency of the Hematopoietic Cell-Specific Rho Family GTPase Rac2 Is Characterized by Abnormalities in Neutrophil Function and Host Defense. Immunity (1999) 10:183–96. doi: 10.1016/S1074-7613(00)80019-9
27. Lam GY, Huang J, Brumell JH. The Many Roles of NOX2 NADPH Oxidase-Derived ROS in Immunity. Semin Immunopathol (2010) 32:415–30. doi: 10.1007/s00281-010-0221-0
28. Panday A, Sahoo MK, Osorio D, Batra S. NADPH Oxidases: An Overview From Structure to Innate Immunity-Associated Pathologies. Cell Mol Immunol (2015) 12:5–23. doi: 10.1038/cmi.2014.89
29. Thomas DC. How the Phagocyte NADPH Oxidase Regulates Innate Immunity. Free Radical Biol Med (2018) 125:44–52. doi: 10.1016/j.freeradbiomed.2018.06.011
30. Thomas DC. The Phagocyte Respiratory Burst: Historical Perspectives and Recent Advances. Immunol Lett (2017) 192:88–96. doi: 10.1016/j.imlet.2017.08.016
31. van den Berg JM, van Koppen E, Åhlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic Granulomatous Disease: The European Experience. PloS One (2009) 4. doi: 10.1371/journal.pone.0005234
32. Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical Features, Long-Term Follow-Up and Outcome of a Large Cohort of Patients With Chronic Granulomatous Disease: An Italian Multicenter Study. Clin Immunol (2008) 126:155–64. doi: 10.1016/j.clim.2007.09.008
33. Jones LBKR, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special Article: Chronic Granulomatous Disease in the United Kingdom and Ireland: A Comprehensive National Patient-Based Registry. Clin Exp Immunol (2008) 152:211–8. doi: 10.1111/j.1365-2249.2008.03644.x
34. Winkelstein JA, Marino MC, Johnston RB, Boyle J, Curnutte J, Gallin JI, et al. Chronic Granulomatous Disease: Report on a National Registry of 368 Patients. Med (Baltimore) (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
35. Kobayashi S, Murayama S, Takanashi S, Takahashi K, Miyatsuka S, Fujita T, et al. Clinical Features and Prognoses of 23 Patients With Chronic Granulomatous Disease Followed for 21 Years by a Single Hospital in Japan. Eur J Pediatr (2008) 167:1389–94. doi: 10.1007/s00431-008-0680-7
36. Köker MY, Camcioglu Y, Van Leeuwen K, Kiliç SŞ, Barlan I, Yilmaz M, et al. Clinical, Functional, and Genetic Characterization of Chronic Granulomatous Disease in 89 Turkish Patients. J Allergy Clin Immunol (2013) 132. doi: 10.1016/j.jaci.2013.05.039
37. Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Fazlollahi SD, et al. Inheritance Pattern and Clinical Aspects of 93 Iranian Patients With Chronic Granulomatous Disease. J Clin Immunol (2011) 31:792–801. doi: 10.1007/s10875-011-9567-x
38. Nauseef WF. The Phagocyte NOX2 NADPH Oxidase in Microbial Killing and Cell Signaling. Curr Opin Immunol (2019) 60:130–40. doi: 10.1016/j.coi.2019.05.006
39. Winterbourn CC, Kettle AJ. Redox Reactions and Microbial Killing in the Neutrophil Phagosome. Antioxid Redox Signal (2013) 18:642–60. doi: 10.1089/ars.2012.4827
40. Thomas DC, Clare S, Sowerby JM, Pardo M, Juss JK, Goulding DA, et al. Eros Is a Novel Transmembrane Protein That Controls the Phagocyte Respiratory Burst and Is Essential for Innate Immunity. J Exp Med (2017) 214:1111–28. doi: 10.1084/jem.20161382
41. Ryoden Y, Fujii T, Segawa K, Nagata S. Functional Expression of the P2X7 ATP Receptor Requires Eros. J Immunol (2020) 204:559–68. doi: 10.4049/jimmunol.1900448
42. Thomas DC, Charbonnier L-M, Schejtman A, Aldhekri H, Coomber E, Dufficy ER, et al. EROS Is Required for Phagocyte NADPH Oxidase Function in Humans and its Deficiency Causes Chronic Granulomatous Disease. bioRxiv (2018) 331454. doi: 10.1101/331454
43. Arnadottir GA, Norddahl GL, Gudmundsdottir S, Agustsdottir AB, Sigurdsson S, Jensson BO, et al. A Homozygous Loss-of-Function Mutation Leading to CYBC1 Deficiency Causes Chronic Granulomatous Disease. Nat Commun (2018) 9. doi: 10.1038/s41467-018-06964-x
44. Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA, et al. NRROS Negatively Regulates Reactive Oxygen Species During Host Defence and Autoimmunity. Nature (2014) 509:235–9. doi: 10.1038/nature13152
45. Chen F, Yu Y, Qian J, Wang Y, Cheng B, Dimitropoulou C, et al. Opposing Actions of Heat Shock Protein 90 and 70 Regulate Nicotinamide Adenine Dinucleotide Phosphate Oxidase Stability and Reactive Oxygen Species Production. Arterioscler Thromb Vasc Biol (2012) 32:2989–99. doi: 10.1161/ATVBAHA.112.300361
46. Gaines P, Tien CW, Olins AL, Olins DE, Shultz LD, Carney L, et al. Mouse Neutrophils Lacking Lamin B-Receptor Expression Exhibit Aberrant Development and Lack Critical Functional Responses. Exp Hematol (2008) 36:965–76. doi: 10.1016/j.exphem.2008.04.006
47. Subramanian G, Chaudhury P, Malu K, Fowler S, Manmode R, Gotur D, et al. Lamin B Receptor Regulates the Growth and Maturation of Myeloid Progenitors via its Sterol Reductase Domain: Implications for Cholesterol Biosynthesis in Regulating Myelopoiesis. J Immunol (2012) 188:85–102. doi: 10.4049/jimmunol.1003804
49. Magnani A, Brosselin P, Beauté J, de Vergnes N, Mouy R, Debré M, et al. Inflammatory Manifestations in a Single-Center Cohort of Patients With Chronic Granulomatous Disease. J Allergy Clin Immunol (2014) 134:655–62.e8. doi: 10.1016/j.jaci.2014.04.014
50. Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O’Brien S, et al. Gastrointestinal Involvement in Chronic Granulomatous Disease. Pediatrics (2004) 114:462–8. doi: 10.1542/peds.114.2.462
51. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 Controls Phagosomal pH to Regulate Antigen Processing During Crosspresentation by Dendritic Cells. Cell (2006) 126:205–18. doi: 10.1016/j.cell.2006.05.035
52. Mantegazza AR, Savina A, Vermeulen M, Pérez L, Geffner J, Hermine O, et al. NADPH Oxidase Controls Phagosomal pH and Antigen Cross-Presentation in Human Dendritic Cells. Blood (2008) 112:4712–22. doi: 10.1182/blood-2008-01-134791
53. Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, et al. The Small GTPase Rac2 Controls Phagosomal Alkalinization and Antigen Crosspresentation Selectively in CD8+ Dendritic Cells. Immunity (2009) 30:544–55. doi: 10.1016/j.immuni.2009.01.013
54. Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PMC, et al. Rab27a Regulates Phagosomal pH and NADPH Oxidase Recruitment to Dendritic Cell Phagosomes. Nat Cell Biol (2007) 9:367–78. doi: 10.1038/ncb1552
55. Rybicka JM, Balce DR, Khan MF, Krohn RM, Yates RM. NADPH Oxidase Activity Controls Phagosomal Proteolysis in Macrophages Through Modulation of the Lumenal Redox Environment of Phagosomes. Proc Natl Acad Sci USA (2010) 107:10496–501. doi: 10.1073/pnas.0914867107
56. Rybicka JM, Balce DR, Chaudhuri S, Allan ERO, Yates RM. Phagosomal Proteolysis in Dendritic Cells Is Modulated by NADPH Oxidase in a pH-Independent Manner. EMBO J (2012) 31:932–44. doi: 10.1038/emboj.2011.440
57. Canton J, Blees H, Henry CM, Buck MD, Schulz O, Rogers NC, et al. The Receptor DNGR-1 Signals for Phagosomal Rupture to Promote Cross-Presentation of Dead-Cell-Associated Antigens. Nat Immunol (2021) 22:140–53. doi: 10.1038/s41590-020-00824-x
58. Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, Hernanz-Falcón P, et al. Identification of a Dendritic Cell Receptor That Couples Sensing of Necrosis to Immunity. Nature (2009) 458:899–903. doi: 10.1038/nature07750
59. Allan ERO, Tailor P, Balce DR, Pirzadeh P, McKenna NT, Renaux B, et al. NADPH Oxidase Modifies Patterns of MHC Class II–Restricted Epitopic Repertoires Through Redox Control of Antigen Processing. J Immunol (2014) 192:4989–5001. doi: 10.4049/jimmunol.1302896
60. Crotzer VL, Matute JD, Arias AA, Zhao H, Quilliam LA, Dinauer MC, et al. Cutting Edge: NADPH Oxidase Modulates MHC Class II Antigen Presentation by B Cells. J Immunol (2012) 189:3800–4. doi: 10.4049/jimmunol.1103080
61. Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T Cells Express a Phagocyte-Type NADPH Oxidase That Is Activated After T Cell Receptor Stimulation. Nat Immunol (2004) 5:818–27. doi: 10.1038/ni1096
62. Kwon BI, Kim TW, Shin K, Kim YH, Yuk CM, Yuk JM, et al. Enhanced Th2 Cell Differentiation and Function in the Absence of Nox2. Allergy Eur J Allergy Clin Immunol (2017) 72:252–65. doi: 10.1111/all.12944
63. Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH Oxidase Deficiency Regulates Th Lineage Commitment and Modulates Autoimmunity. J Immunol (2010) 185:5247–58. doi: 10.4049/jimmunol.1001472
64. Shatynski KE, Chen H, Kwon J, Williams MS. Decreased STAT5 Phosphorylation and GATA-3 Expression in NOX2-Deficient T Cells: Role in T Helper Development. Eur J Immunol (2012) 42:3202–11. doi: 10.1002/eji.201242659
65. Lee K, Won HY, Bae MA, Hong JH, Hwang ES. Spontaneous and Aging-Dependent Development of Arthritis in NADPH Oxidase 2 Deficiency Through Altered Differentiation of CD11b+ and Th/Treg Cells. Proc Natl Acad Sci USA (2011) 108:9548–53. doi: 10.1073/pnas.1012645108
66. Won HY, Jang EJ, Min HJ, Hwang ES. Enhancement of Allergen-Induced Airway Inflammation by NOX2 Deficiency. Immune Netw (2011) 11:169. doi: 10.4110/in.2011.11.3.169
67. van de Geer A, Cuadrado E, Slot MC, van Bruggen R, Amsen D, Kuijpers TW. Regulatory T Cell Features in Chronic Granulomatous Disease. Clin Exp Immunol (2019) 197:222–9. doi: 10.1111/cei.13300
68. Emmerson A, Trevelin SC, Mongue-Din H, Becker PD, Ortiz C, Smyth LA, et al. Nox2 in Regulatory T Cells Promotes Angiotensin II–induced Cardiovascular Remodeling. J Clin Invest (2018) 128:3088–101. doi: 10.1172/JCI97490
69. Efimova O, Szankasi P, Kelley TW. Ncf1 (P47phox) Is Essential for Direct Regulatory T Cell Mediated Suppression of CD4+ Effector T Cells. PloS One (2011) 6:e16013. doi: 10.1371/journal.pone.0016013
70. Chose O, Sansilvestri-Morel P, Badier-Commander C, Bernhardt F, Fabiani J-N, Rupin A, et al. Distinct Role of Nox1, Nox2, and P47phox in Unstimulated Versus Angiotensin II-Induced NADPH Oxidase Activity in Human Venous Smooth Muscle Cells. J Cardiovasc Pharmacol (2008) 51:131–9. doi: 10.1097/FJC.0b013e31815d781d
71. Li JM, Wheatcroft S, Fan LM, Kearney MT, Shah AM. Opposing Roles of P47phox in Basal Versus Angiotensin II-Stimulated Alterations in Vascular O2- Production, Vascular Tone, and Mitogen-Activated Protein Kinase Activation. Circulation (2004) 109:1307–13. doi: 10.1161/01.CIR.0000118463.23388.B9
72. Purushothaman D, Sarin A. Cytokine-Dependent Regulation of NADPH Oxidase Activity and the Consequences for Activated T Cell Homeostasis. J Exp Med (2009) 206:1515–23. doi: 10.1084/jem.20082851
73. Dhiman M, Garg NJ. P47phox–/– Mice Are Compromised in Expansion and Activation of CD8+ T Cells and Susceptible to Trypanosoma Cruzi Infection. PloS Pathog (2014) 10(12):e1004516. doi: 10.1371/journal.ppat.1004516
74. Lang PA, Xu HC, Grusdat M, McIlwain DR, Pandyra AA, Harris IS, et al. Reactive Oxygen Species Delay Control of Lymphocytic Choriomeningitis Virus. Cell Death Differ (2013) 20:649–58. doi: 10.1038/cdd.2012.167
75. Vlahos R, Stambas J, Bozinovski S, Broughton BRS, Drummond GR, Selemidis S. Inhibition of Nox2 Oxidase Activity Ameliorates Influenza a Virus-Induced Lung Inflammation. PloS Pathog (2011) 7:1001271. doi: 10.1371/journal.ppat.1001271
76. Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, et al. NADPH Oxidase Deficiency Underlies Dysfunction of Aged CD8+ Tregs. J Clin Invest (2016) 126:1953–67. doi: 10.1172/JCI84181
77. Kovács I, Horváth M, Lányi Á., Petheő GL, Geiszt M. Reactive Oxygen Species-Mediated Bacterial Killing by B Lymphocytes. J Leukoc Biol (2015) 97:1133–7. doi: 10.1189/jlb.4AB1113-607RR
78. Wheeler ML, DeFranco AL. Prolonged Production of Reactive Oxygen Species in Response to B Cell Receptor Stimulation Promotes B Cell Activation and Proliferation. J Immunol (2012) 189:4405–16. doi: 10.4049/jimmunol.1201433
79. Richards SM, Clark EA. BCR-Induced Superoxide Negatively Regulates B-Cell Proliferation and T-Cell-Independent Type 2 Ab Responses. Eur J Immunol (2009) 39:3395–403. doi: 10.1002/eji.200939587
80. Feng Y-Y, Tang M, Suzuki M, Gunasekara C, Anbe Y, Hiraoka Y, et al. Essential Role of NADPH Oxidase–Dependent Production of Reactive Oxygen Species in Maintenance of Sustained B Cell Receptor Signaling and B Cell Proliferation. J Immunol (2019) 202:2546–57. doi: 10.4049/jimmunol.1800443
81. Bertolotti M, Farinelli G, Galli M, Aiuti A, Sitia R. AQP8 Transports NOX2-Generated H 2 O 2 Across the Plasma Membrane to Promote Signaling in B Cells. J Leukoc Biol (2016) 100:1071–9. doi: 10.1189/jlb.2AB0116-045R
82. Mcletchie S, Volpp BD, Dinauer MC, Blum JS. Hyper-Responsive Toll-Like Receptor 7 and 9 Activation in NADPH Oxidase-Deficient B Lymphoblasts. Immunology (2015) 146:595–606. doi: 10.1111/imm.12530
83. Roche PA, Furuta K. The Ins and Outs of MHC Class II-Mediated Antigen Processing and Presentation. Nat Rev Immunol (2015) 15:203–16. doi: 10.1038/nri3818
84. Hultqvist M, Olofsson P, Holmberg J, Bäckström BT, Tordsson J, Holmdahl R. Enhanced Autoimmunity, Arthritis, and Encephalomyelitis in Mice With a Reduced Oxidative Burst Due to a Mutation in the Ncf1 Gene. Proc Natl Acad Sci USA (2004) 101:12646. doi: 10.1073/pnas.0403831101
85. Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, et al. Macrophages Suppress T Cell Responses and Arthritis Development in Mice by Producing Reactive Oxygen Species. J Clin Invest (2007) 117:3020. doi: 10.1172/JCI31935
86. Vasilevsky S, Liu Q, Koontz SM, Kastenmayer R, Shea K, Jackson SH. Role of P47phox in Antigen-Presenting Cell-Mediated Regulation of Humoral Immunity in Mice. Am J Pathol (2011) 178:2774. doi: 10.1016/j.ajpath.2011.02.038
87. Cachat J, Deffert C, Alessandrini M, Roux-Lombard P, Gouellec A Le, Stasia MJ, et al. Altered Humoral Immune Responses and IgG Subtypes in NOX2-Deficient Mice and Patients: A Key Role for NOX2 in Antigen-Presenting Cells. Front Immunol (2018) 9:11. doi: 10.3389/fimmu.2018.01555
88. To EE, Vlahos R, Luong R, Halls ML, Reading PC, King PT, et al. Endosomal NOX2 Oxidase Exacerbates Virus Pathogenicity and Is a Target for Antiviral Therapy. Nat Commun (2017) 8. doi: 10.1038/s41467-017-00057-x
89. Matharu K, Zarember KA, Marciano BE, Kuhns DB, Spalding C, Garofalo M, et al. B-Cell Activating Factor (BAFF) Is Elevated in Chronic Granulomatous Disease. Clin Immunol (2013) 148:258–64. doi: 10.1016/j.clim.2013.05.007
90. Moir S, De Ravin SS, Santich BH, Kim JY, Posada IG, Ho J, et al. Humans With Chronic Granulomatous Disease Maintain Humoral Immunologic Memory Despite Low Frequencies of Circulating Memory B Cells. Blood (2012) 120:4850–8. doi: 10.1182/blood-2012-05-430959
91. Bleesing JJ, Souto-Carneiro MM, Savage WJ, Brown MR, Martinez C, Yavuz S, et al. Patients With Chronic Granulomatous Disease Have a Reduced Peripheral Blood Memory B Cell Compartment. J Immunol (2006) 176:7096–103. doi: 10.4049/jimmunol.176.11.7096
92. Simpson DSA, Oliver PL. Ros Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (2020) 9:1–27. doi: 10.3390/antiox9080743
93. Fan LM, Geng L, Cahill-Smith S, Liu F, Douglas G, Mckenzie CA, et al. Nox2 Contributes to Age-Related Oxidative Damage to Neurons and the Cerebral Vasculature. J Clin Invest (2019) 129:3374–86. doi: 10.1172/JCI125173
94. Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J, et al. Prevalence of Amyotrophic Lateral Sclerosis — United States, 2015. MMWR Morb Mortal Wkly Rep (2018) 67:1285–9. doi: 10.15585/mmwr.mm6746a1
95. Wu DC, Ré DB, Nagai M, Ischiropoulos H, Przedborski S. The Inflammatory NADPH Oxidase Enzyme Modulates Motor Neuron Degradation in Amyotrophic Lateral Sclerosis Mice. Proc Natl Acad Sci USA (2006) 103:12132–7. doi: 10.1073/pnas.0603670103
96. Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, et al. SOD1 Mutations Disrupt Redox-Sensitive Rac Regulation of NADPH Oxidase in a Familial ALS Model. J Clin Invest (2008) 118:659–70. doi: 10.1172/JCI34060
97. Seredenina T, Nayernia Z, Sorce S, Maghzal GJ, Filippova A, Ling SC, et al. Evaluation of NADPH Oxidases as Drug Targets in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Free Radic Biol Med (2016) 97:95–108. doi: 10.1016/j.freeradbiomed.2016.05.016
98. Marrali G, Casale F, Salamone P, Fuda G, Caorsi C, Amoroso A, et al. NADPH Oxidase (NOX2) Activity Is a Modifier of Survival in ALS. J Neurol (2014) 261:2178–83. doi: 10.1007/s00415-014-7470-0
99. Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, et al. NADPH Oxidase Expression in Active Multiple Sclerosis Lesions in Relation to Oxidative Tissue Damage and Mitochondrial Injury. Brain (2012) 135:886–99. doi: 10.1093/brain/aws012
100. Hu CF, Wu SP, Lin GJ, Shieh CC, Hsu CS, Chen JW, et al. Microglial Nox2 Plays a Key Role in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Front Immunol (2021) 12:991. doi: 10.3389/fimmu.2021.638381
101. Keller CW, Kotur MB, Mundt S, Dokalis N, Ligeon LA, Shah AM, et al. CYBB/NOX2 in Conventional DCs Controls T Cell Encephalitogenicity During Neuroinflammation. Autophagy (2021) 17:1244–58. doi: 10.1080/15548627.2020.1756678
102. Hickman SE, Allison EK, El Khoury J. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J Neurosci (2008) 28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
103. Geng L, Fan LM, Liu F, Smith C, Li JM. Nox2 Dependent Redox-Regulation of Microglial Response to Amyloid-β Stimulation and Microgliosis in Aging. Sci Rep (2020) 10:1–11. doi: 10.1038/s41598-020-58422-8
104. Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, Black SM, et al. “ROS Signaling in the Pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS)”. In: Advances in Experimental Medicine and Biology, vol. 967. Springer New York LLC (2017). p. 105–137.
105. Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An Absence of Reactive Oxygen Species Improves the Resolution of Lung Influenza Infection. Eur J Immunol (2006) 36:1364–73. doi: 10.1002/eji.200635977
106. Rojas Márquez JD, Li T, McCluggage ARR, Tan JMJ, Muise A, Higgins DE, et al. Cutting Edge: NOX2 NADPH Oxidase Controls Infection by an Intracellular Bacterial Pathogen Through Limiting the Type 1 IFN Response. J Immunol (2021) 206:323–8. doi: 10.4049/jimmunol.2000694
107. Group, T. R. C. Dexamethasone in Hospitalized Patients With Covid-19 — Preliminary Report. N Engl J Med (2021) 384:693–704. doi: 10.1056/NEJMoa2021436
108. Gordon AC, Mouncey PR, Al Beidh F, Rowan KM, Nichol AD, Arabi YM, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients With Covid-19. N Engl J Med (2021) 384:1491–502. doi: 10.1056/NEJMoa2100433
109. Reis J, Massari M, Marchese S, Ceccon M, Aalbers F, Corana F, et al. A Closer Look Into NADPH Oxidase Inhibitors: Validation and Insight Into Their Mechanism of Action. Redox Biol (2020) 32. doi: 10.1016/j.redox.2020.101466
110. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly Accurate Protein Structure Prediction With AlphaFold. Nature (2021). doi: 10.1038/S41586-021-03819-2
Keywords: NOX2, ROS, CGD, oxidative stress, systemic inflammation
Citation: Mortimer PM, Mc Intyre SA and Thomas DC (2021) Beyond the Extra Respiration of Phagocytosis: NADPH Oxidase 2 in Adaptive Immunity and Inflammation. Front. Immunol. 12:733918. doi: 10.3389/fimmu.2021.733918
Received: 30 June 2021; Accepted: 16 August 2021;
Published: 01 September 2021.
Edited by:
Michael Walch, Université de Fribourg, SwitzerlandReviewed by:
Kalyani Pyaram, Kansas State University, United StatesBalazs Rada, University of Georgia, United States
Copyright © 2021 Mortimer, Mc Intyre and Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David C. Thomas, david.thomas1@imperial.ac.uk
†These authors have contributed equally to this work and share first authorship