 Yiwen Zhang
Yiwen Zhang Wenlin Yang1
Wenlin Yang1 Yunjuan Zhao
Yunjuan Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 October 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.732933
This article is part of the Research TopicEmerging Insights in Controlling AutoimmunityView all 24 articles
Autoimmune diseases are a broad spectrum of human diseases that are characterized by the breakdown of immune tolerance and the production of autoantibodies. Recently, dysfunction of innate and adaptive immunity is considered to be a key step in the initiation and maintenance of autoimmune diseases. NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome is a multimeric protein complex, which can detect exogenous pathogen irritants and endogenous danger signals. The main function of NLRP3 inflammasome is to promote secretion of interleukin (IL)-1β and IL-18, and pyroptosis mediated by caspase-1. Served as a checkpoint in innate and adaptive immunity, aberrant activation and regulation of NLRP3 inflammasome plays an important role in the pathogenesis of autoimmune diseases. This paper reviewed the roles of NLRP3 inflammasome in autoimmune diseases, which shows NLRP3 inflammasome may be a potential target for autoimmune diseases deserved further study.
Autoimmune diseases are characterized by self-reactive cells and the overproduction of autoantibodies, which are led by the breakdown of immunological tolerance and aberrant autoreactive immune responses (1). Autoimmune diseases include organ-specific autoimmune diseases, such as type 1 diabetes, autoimmune thyroid diseases, and rheumatoid arthritis, and systemic autoimmune diseases, such as systemic lupus erythematosus and systemic sclerosis (2). Although the pathogenesis of autoimmune diseases is still unclear, numerous studies have shown that aberrant innate and adaptive immunity is involved in the pathogenesis of autoimmune diseases (3, 4). Recently, emerging appreciation showed that NLRP3 inflammasome plays an important role in recognizing innate immune signals and inducing autoreactive immune responses, which probably acts as a checkpoint in innate immunity to cause skewed adaptive immune responses (Figure 1).
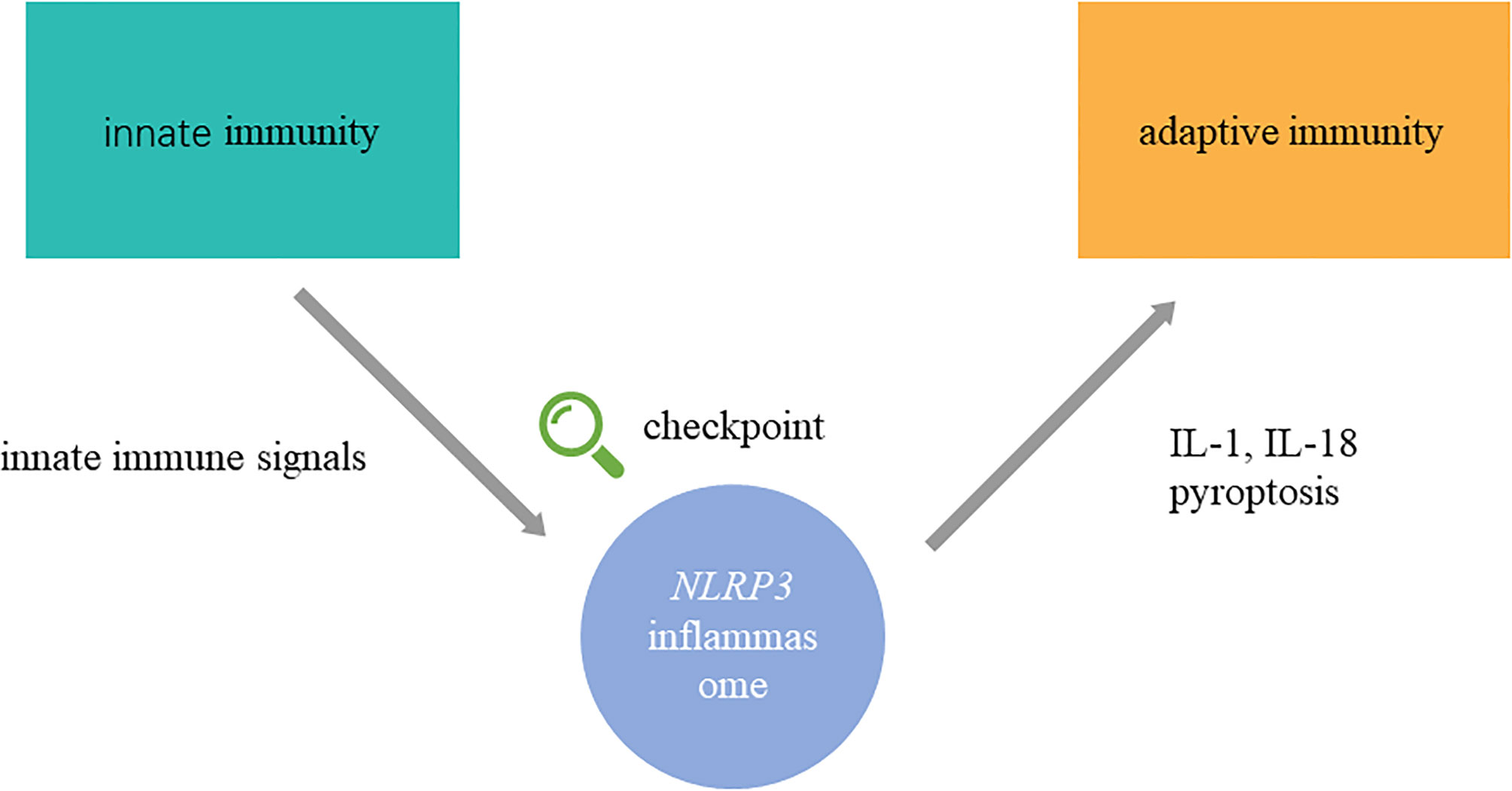
Figure 1 NLRP3 inflammasome connects innate and adaptive immunity in autoimmune diseases. NLRP3 inflammasome is activated abnormally in autoimmune diseases. In the upstream process, exogenous pathogen irritants and endogenous danger signals initiate assembly of NLRP3 inflammasome. In the upstream process, IL-1β, IL-18, and pyroptosis, which are modulated by NLRP3 inflammasome, regulate adaptive immune response. Namely, NLRP3 inflammasome might participate in the transition from innate immunity to adaptive immunity in the pathogenesis of autoimmune diseases as a checkpoint. The detailed process of activation, assembly, regulation, and effects on adaptive immunity are shown in Figure 2.
Inflammasomes are protein complexes composed of three parts: a sensor, an adaptor, and an effector. Tschopp et al. firstly proposed the concept of inflammasomes in 2002 (5), following which several inflammasome subtypes were discovered, including NLRP1 inflammasome, NLRP3 inflammasome, absent in melanoma 2 (AIM2) inflammasome, etc. In most of the inflammasome subtypes, the component of adaptor is usually apoptosis-associated speck-like protein (ASC), which contains a caspase activation and recruitment domain. The effector component is usually caspase-1. The differences among inflammasomes subtypes are the sensor component (Figure 2).
NOD-like receptor (NLR) is a typical type of pattern recognition receptors (PRRs), via which innate immune system recognizes pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). According to different subcellular localization, PRRs are classified into two categories: (a) PRRs located in the plasma membrane, which mainly play the role to recognize PAMPs and DAMPs, typically including Toll-like receptors (TLRs) and C-type lectin receptors (CLRs); (b) PRRs located in intracellular partitions, which mainly include NLRs, AIM2-like receptor (ALRs), and cytosolic sensor cyclic GMP-AMP (cGAMP) synthase (6, 7). NLRP3 inflammasome is one of the most widely studied inflammasomes. Effect of NLRP3 inflammasome to various physiological and pathogenic stimuli mainly includes caspase-1 activation, secretion of IL-1β and IL-18, and pyroptosis mediated by caspase-1. Under physiological conditions, inflammasomes play an important role in clearing pathogens and damaged cells, and serve as a critical composition of innate immune response. Whereas under pathological conditions, the overactivation of inflammasomes may trigger autoinflammatory and autoimmune responses and result in numerous diseases.
In this review, we summarized the references and presented that as the checkpoint, NLRP3 inflammasome connects innate and adaptive immunity in several autoimmune diseases, including inflammatory bowel disease, psoriasis, rheumatoid arthritis, systemic sclerosis, type 1 diabetes, systemic lupus erythematosus, and autoimmune thyroid diseases. Finally, we discussed the effect of new-onset inhibitors of NLRP3 inflammasome in autoimmune diseases, which implies their potential therapeutic value for clinical applications deserved further study.
NLRP3 inflammasome consists of three components including NLRP3 scaffold, a pyrin domain (PYD), and a caspase recruitment domain (CARD), known as ASC, and caspase-1 (8). NLRP3 belongs to NLR protein family which includes 22 members widely expressed in human histiocytes, such as dendritic cells, macrophages, and monocytes. NLRP3 contains three segments (Figure 2): (a) PYD in amino-terminal; (b) a NACHT domain in central part: executes the function of NLRP3 self-association via ATPase activity; (c) a leucine-rich repeat domain (LRR domain) in carboxy-terminal: depresses NLRP3 activation by inhibiting the ATPase activity of the NACHT domain. ASC includes PYD in amino-terminal and CARD in carboxy-terminal, which interact with each other to activate caspase-1. Caspase-1 consists of three parts from amino-terminal to carboxy-terminal: CARD, central large catalytic domain (p20), and small catalytic subunit domain (p10) (9, 10).
All of the substructures execute specific function in NLRP3 inflammasome assembly. Activated by upstream signals, the NACHT domains of NLRP3 interact with each other to induce NLRP3 oligomerization. And then the homotypic PYD-PYD interaction promotes ASC recruitments and formation of nucleates helical ASC filament. ASC recruits and activates caspase-1 via homotypic CARD-CARD interactions. At last, the clustered caspase-1 cleaves to a complex of p33 which comprises CARD and p20, a formation with proteolytical activation (11, Figure 2). NIMA-related kinase 7 (NEK7) is a new component in recent studies (Figure 2). Genetic research showed that NEK7 functioned as an indispensable component to NLRP3 inflammasome activation (12). NEK7 belongs to the family of mammalian NIMA-related kinases and is a serine or threonine kinase involved in mitosis. In the mediation of potassium efflux and mitochondrial reactive oxygen species (ROS), NEK7 binds to LRR domain of NLRP3, which is essential to NLRP3 inflammasome activation (13).
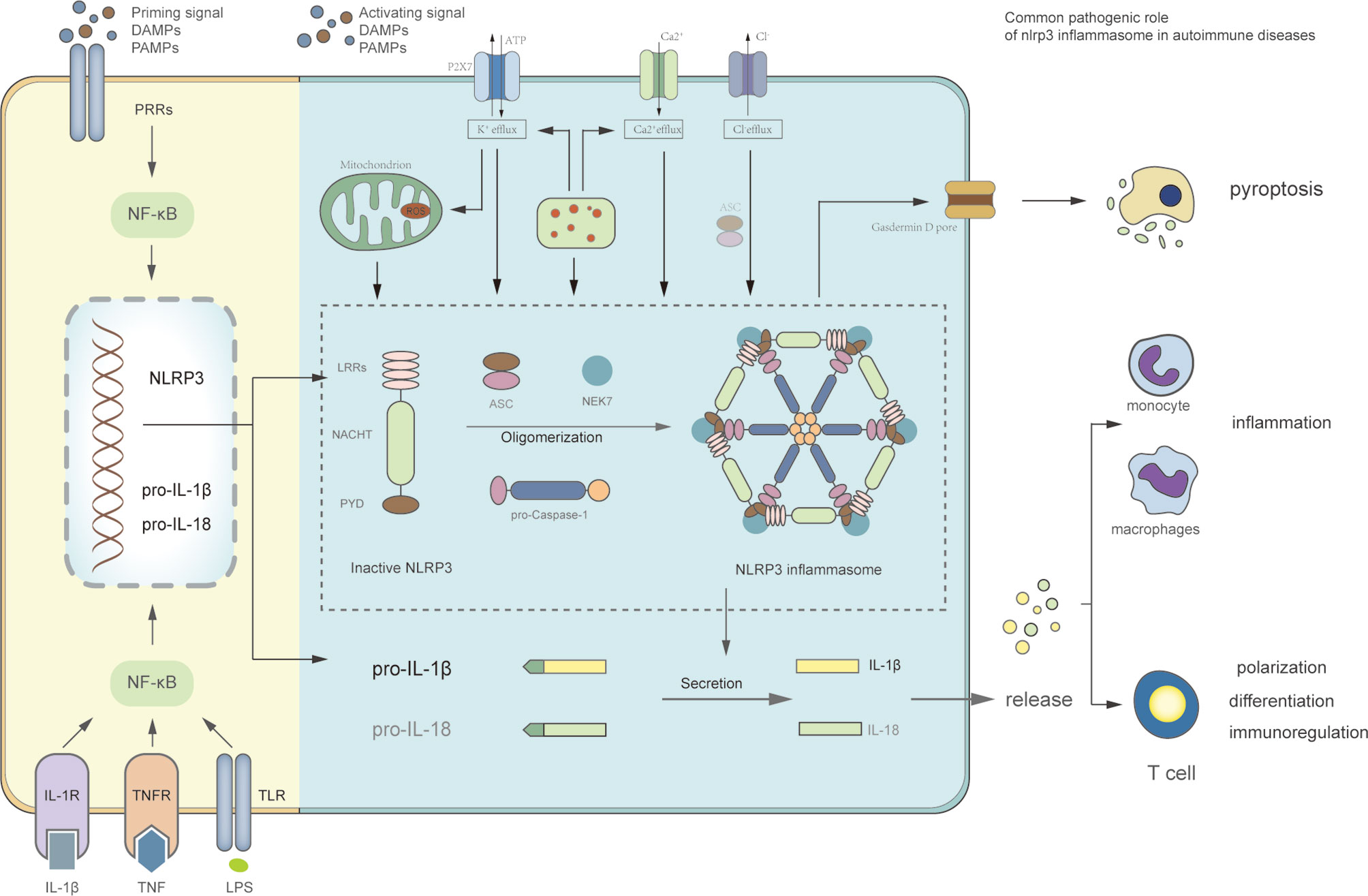
Figure 2 Signaling pathway of NLRP3 inflammasome and the role of NLRP3 inflammasome in autoimmune diseases. The priming stage: the binding of cytokines or PAMPs to its receptors can activate NF-κB signaling pathway, which upregulates transcription of NLRP3, pro-IL-1β, and pro-IL-18. The activation stage: this stage is stimulated by PAMPs and DAMPs. The activating signals, including K+ efflux, Ca2+flux, Cl− efflux, mitochondrial dysfunction, ROS production, lysosomal disruption, and NEK7 promote oligomerization of NLRP3, ASC, and pro-caspase-1 to format NLRP3 inflammasome complex. The active caspase-1 can cleave proinflammatory cytokines IL-1β and IL-18, and promote pyroptosis mediated by Gasdermin D. The role of NLRP3 inflammasome in autoimmune diseases: inflammation promoted by cytokines IL-1β and IL-18; adaptive immune dysfunction caused by cytokines, such as proliferation and differentiation of T cells; pyroptosis, which can cause histiocytic death such as insulin β-cells.
The activation of NLRP3 inflammasome includes two steps: firstly, it should be primed, sequentially activated. Priming is a preparation stage for subsequent responses. On the one hand, upregulated expression of NLRP3, caspase-1, and pro-IL-1β is induced by gene transcription and activation of nuclear factor-κB (NF-κB). This process is initiated through three ways (Figure 2): (a) PRRs, such as NOD2 or TLRs, recognize PAMPs and DAMPs; (b) cytokines directly activate NF-κB pathway, including tumor necrosis factor (TNF) and IL-1β; (c) lipopolysaccharide (LPS) upregulates IL-1β transcription by shifting specific metabolism status (14–16). On the other hand, NLRP3 post-translational modifications (PTMs) are induced in the priming stage. NLRP3 is stabilized into a state in which the NLRP3 activity is auto-suppressed, but it still can respond to various signals (17). Importantly, NLRP3 PTMs occurs in the whole process of NLRP3 inflammasome activation, even in the unstimulated stage (18).
PAMPs and DAMPs stimuli varies in chemical properties and structures, and the direct binding of stimuli to NLRP3 are rarely detected. It is hypothesized that NLRP3 may sense common upstream signals which are induced by NLRP3 activators. This character of indirect activation is the core to understand molecular mechanism of NLRP3 activation. The comprehensive signal has not been determined, and relevant researches are contradictory. The common upstream signals identified include K+ efflux, Ca2+ flux, Cl− efflux, mitochondrial dysfunction, ROS production, lysosomal damage, trans-Golgi disassembly, metabolic changes, and so on. There are three typical consensus models of common upstream signals.
The ion fluxes are common triggers of NLRP3 inflammasome activation. They include K+ efflux, Ca2+ flux, and Cl− efflux, of which K+ efflux is an indispensable upstream event. It was first proposed that ATP-mediated P2X purinoceptor 7 (P2X7) promoted production of mature IL-1β through K+ efflux (19). And the high concentration of extracellular K+ can depress NLRP3 inflammasome activation (20, 21). It suggests that intracellular low potassium status may trigger NLRP3 inflammasome activation. Meanwhile, the activated P2X7-ATP-dependent pore recruits a pannexin-1 hemi-channel, through which extracellular agonists could enter the cytoplasm and interact with NLRP3 inflammasome complex to promote mature IL-1β secretion (22, 23). In addition, the silica, particulate stimuli alum and calcium pyrophosphate crystals are demonstrated to induce K+ efflux to trigger NLRP3 inflammasome activation (24). However, the mechanism of how intracellular decreased K+ levels trigger NLRP3 activation has not been precisely illuminated. One of the presuppositions suggested that the drop in intracellular K+ concentration may induce the conformational changes of NLRP3 to activate the following response (25, 26).
Particulate activation is a major factor to effect NLRP3 inflammasome. Self-derived particulate matter (such as uric acid, cholesterol crystals) and foreign-derived particulate matter (such as asbestos, alum, silica) could be endocytosed by lysosomal and cause subsequent lysosome membrane damages and release of the particulates and cathepsin B (27). NLRP3 inflammasome can be suppressed by broad-spectrum cathepsin inhibitors, such as CA-074-Me (a chemical cathepsin B inhibitor), which indicates that cathepsin is an important triggered signal of NLRP3 inflammasome (28, 29). Moreover, a variety of cathepsins exhibit function of promoting NLRP3 priming and activation, which is also blocked by CA-074-Me, and the NLRP3 activation process is rarely inhibited by the treatment of cathepsin B, X, L, or S gene deletion respectively (30). Hence, the redundancy among various cathepsin is vital for NLRP3 activation. In addition, lysosomal rupture can activate ion fluxes, including K+ efflux and Ca2+ influx. It indicates that ion fluxes may be a common-converged point in different NLRP3 activation models (25, Figure 2).
The activated NLRP3 inflammasome can eliminate dysfunctional mitochondria and reduce ROS, which are vital upstream events in NLRP3 inflammasome activation. NLRP3 inflammasome activation can be blocked by ROS scavenging agents or NAPDH oxidase inhibitors (31). NLRP3 inflammasome activation was also detected in mouse macrophages and human peripheral blood monocytes which are depleted of NAPDH oxidase activity (32). Further study is required to determine the comprehensive role of mitochondria in NLRP3 inflammasome activation. In addition, mitochondria can provide a docking site for assembly of NLRP3 inflammasome. Cardiolipin, mitochondrial antiviral signaling protein, and mitofusin 2 may serve as the connective point of NLRP3 to the mitochondria (33, 34).
The priming and activation stages result in a multimeric protein complex assembled by NLRP3, ASC and caspase-1. There are two major functions of activated caspase-1: (a) Promoting maturity and release of IL-1β and IL-18 by cleaving their precursors; (b) Initiating pyroptosis, a specific cell death between necrosis and apoptosis, by cleaving gasdermin D (11, 35). IL-1β and IL-18 are important members of the IL-1 family, which also include IL-1α, IL-33, IL 37, and so on. The IL-1 family play vital roles in inflammatory responses and immune regulation (36). IL-1β, as a typical pro-inflammatory cytokine, can induce autoinflammatory response and tissue destruction. It can improve macrophages’ functions, recruit leukocytes via upregulating adhesion molecules and chemokines, and promote leukocytes to produce proinflammatory mediators (36–38). IL-1β, as a kind of T cell co-stimulatory factor, can provide pro-survival and proliferation signals for T cells (36), which also induce differentiation and polarization of T cell. IL-1β can promote naive CD4+ T cells differentiation into Th17 cells and Th9 cells cooperating with other cytokines (39). IL-18, as a kind of IFN γ-inducing factor, can induce natural killer (NK) cells to product IFN-γ and IL-8 (40). IL-18 induces Th2 responses and promotes Th1 responses, synergizing with which induce IFN-γ production of T helper cells (36, 41). In epithelial cells, IL-18 regulates function of Th17 cell and Treg cell, which contributes to Th17/Treg imbalance (42).
By recognizing pathological infections and endogenous danger signals, NLRP3 inflammasome can trigger immune responses and inflammatory responses. The regulation of NLRP3 inflammasome activation is as critical for immune regulation in immune homeostasis, as for inflammose function itself. The aberrant regulation can induce NLRP3 inflammasome overactivation and excessive cytokine production, resulting in skewed inflammatory responses, which might induce adaptive immune dysfunction. The statue can be switched from physiological defense to pathogenic damage by aberrant regulation.
The strict regulation of NLRP3 inflammasome activation is essential to maintain immune system homeostasis. More importantly, accurate understanding of the regulatory mechanisms is crucial to identify triggers of autoimmune diseases involved in NLRP3 inflammasome, and treatments targeted on it may show great therapeutic potential. Various potential regulation mechanisms have been reported, such as cell surface associated mucin 1, MicroRNA (miRNA), small heterodimer partner (Table 1). Clarifying traits of regulations from the following aspects might contribute to a more comprehensive understanding: positive or negative regulation; the specific stage on which the regulator targets, including priming stage and activation stage; and specific subcellular location, such as mitochondria and Golgi apparatus (Table 1).
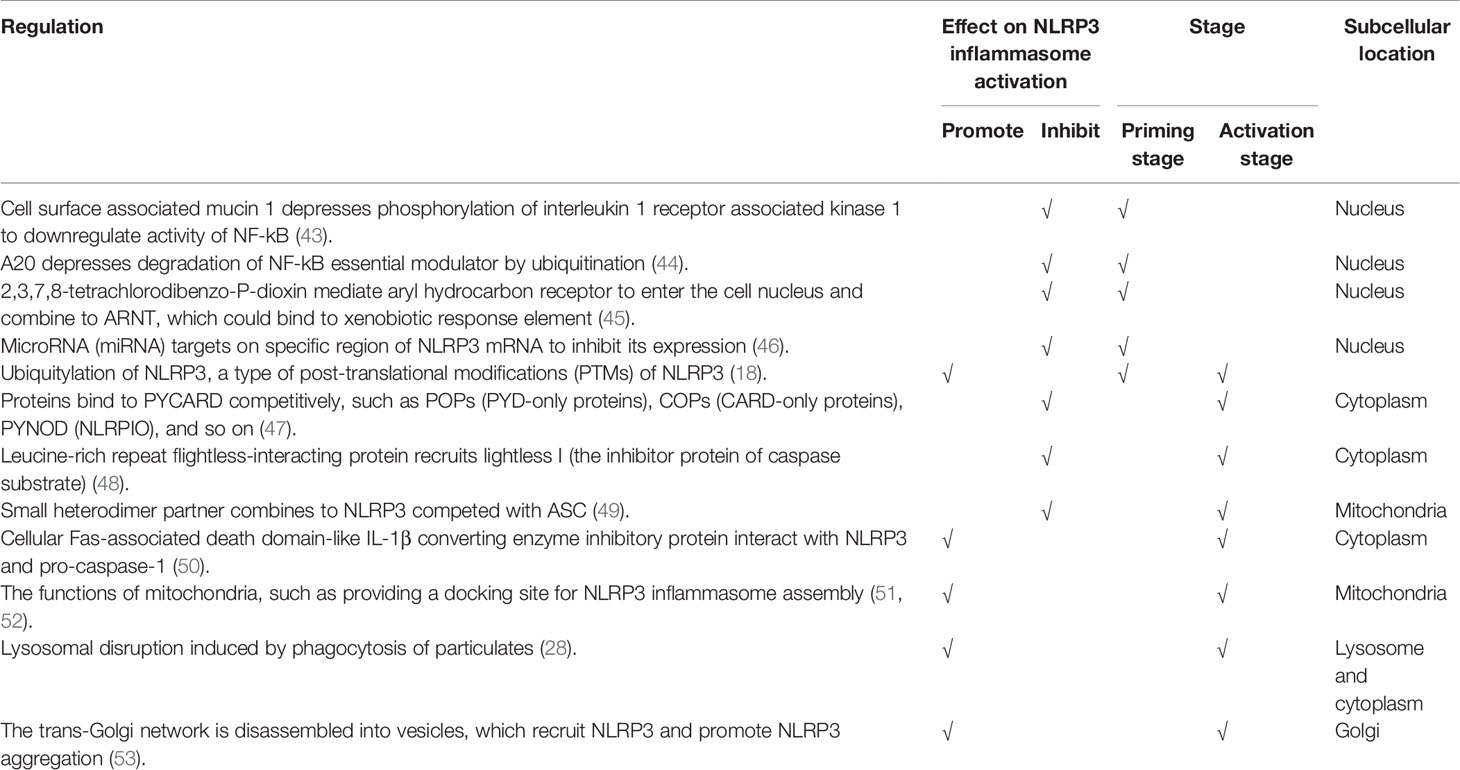
Table 1 Regulation of NLRP3 inflammasome.
PTMs of NLRP3 can participate in various phases of NLRP3 inflammasome activation and regulate innate immunity. Covalent additions including ubiquitylation, phosphorylation, and sumoylation have been reported in NLRP3 PTMs. For example, sumoylation by the protein E3 SUMO protein ligase MUL1 (also known as MAPL) depresses NLRP3 activation. However, both promoted and suppressed effect of phosphorylation on NLRP3 have been detected (54, 55). In addition, PTMs can interact with each other. Phosphorylation can promote deubiquitylation by interaction between BRCA1/BRCA2-containing complex subunit 3 and LRR domain of NLRP3 (54). It indicates that regulation of NLRP3 activation is a delicate and complex process.
Overreaction of innate immune responses may cause overactivity of cytokines and pyroptosis, which contribute to inflammatory and autoimmune diseases, the same as overreaction of skewed adaptive immune responses may cause hypersensitivity. Therefore, identifying critical triggers of the unbalanced immunoregulation would be an important direction in the future researches.
Autoimmune diseases are characterized by loss of immunological tolerance and inappropriately autoreactive immune responses against histocytes and organs. However, the exact pathogenesis of autoimmune diseases has not been identified, and existing treatments are unsatisfactory (56). Current studies have proposed the role of NLRP3 inflammasome in autoimmune diseases and its clinical therapeutic potential (Table 2).
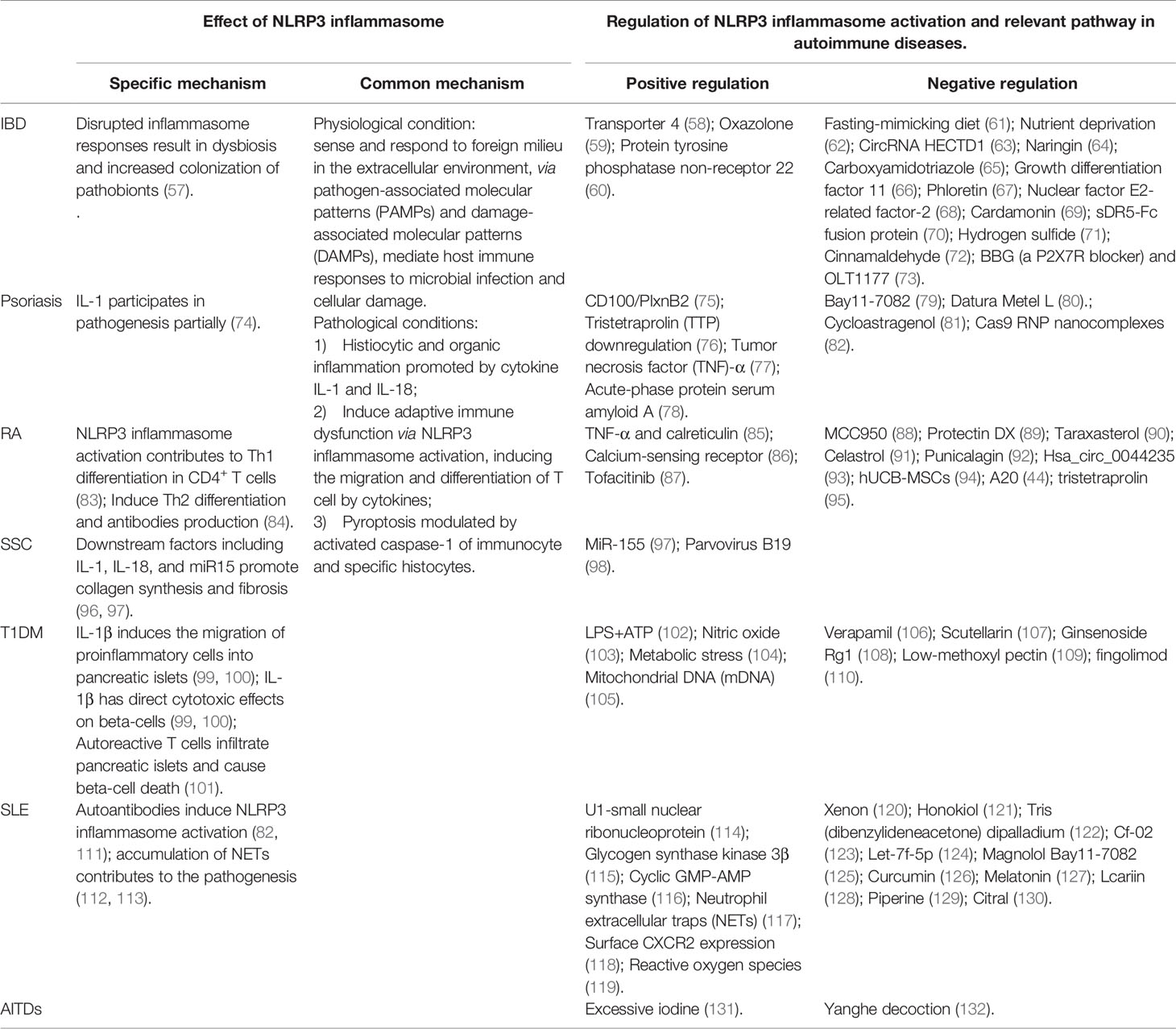
Table 2 Studies of the roles of NLRP3 inflammasome in autoimmune diseases.
Inflammatory bowel disease (IBD) is the most common intestinal tract disorders, characterized by dysfunction of innate immunity and aberrant inflammation in intestinal mucosa. It is comprised mainly of Crohn’s disease (CD) and ulcerative colitis (UC) (133). Though the specific pathogenesis of IBD has not been elucidated until now, several researches have revealed that NLRP3 inflammasome activation is upregulated in IBD. Serum concentration of NLRP3 is elevated, positively correlated with serum IL-1β level and severity of IBD patients (134). Furtherly, NLRP3 inflammasome is activated in early stage of CD patients, whereas in late stage of UC patients, which implies that there are differences between the development of UC and DC (135).
Downstream effects of NLRP3 inflammasome activation contribute to chronic inflammation, alterations in innate immune responses and disorders in mucosal immune response. These effects mainly include the consequent processes: pro-inflammatory cytokine release, macrophage hyperactivation with uncontrolled damage, and pyroptosis (136). In vitro, Monocarboxylate Transporter 4 contributed to intestinal enterocyte pyroptosis mediated by Caspase-1 through ERK1/2-NF-κB pathway, which can promote intestinal inflammation (58). Recent studies show that non-canonical endogenous irritants such as non-infectious stress conditions can also affect NLRP3 inflammasome activation. Fasting-mimicking diet reduced the expression of NLRP3 inflammasome and CD4+ T cells percentage in peripheral blood and spleen to alleviate intestinal inflammation, which indicated that calorie restriction might modulate immune response via NLRP3 inflammasome (61). Yun et al. presented that NLRP3 inflammasome was removed by ubiquitin-mediated degradation, which was promoted by autophagy under nutrient deprivation (62). The downstream proinflammatory cytokines IL-1and IL-17E/25 could be suppressed to strengthen intestinal barrier function (62). CircRNA HECTD1 (circHECTD1) can alleviate UC by inhibiting NLRP3 inflammasome. And in Caco-2 cells, circHECTD1 can induce human antigen R (HuR) via miR-182-5p, which contributes to NLRP3 inflammasome activation by autophagy (63). NLRP3 inflammasome may have a double effect on IBD: some reports showed that IL-1β and IL-18 play a protective role in gastrointestinal inflammation. Oxazolone can stimulate maturation of pro-caspase-1 and pro-IL-1β, and colitis induced by oxazolone can be ameliorated by exogenous IL-1β or IL-18 (59). Higher sensitivity to oxazolone treatment and decreased IL-1β or IL-18 production were detected in NLRP3(−/−) mice compared to wild-type mice, which indicates that IL-1β and IL-18 derived from NLRP3 inflammasome can protect against inflammation in gastrointestinal mucosa (59). In addition, IL-1β contributes to enhanced host defense against Clostridium difficile and Citrobacter rodentium during acute affection (137, 138). It induces ASC-dependent CXCL1 production to recruit neutrophils to the intestine, protects epithelial integrity, and reduces colonization (137, 138). There are several explanations for the protective effect. Marianne et al. presented that protein tyrosine phosphatase non-receptor 22 mediates dephosphorylation of NLRP3 inflammasome, resulting in its activation and release of mature IL-1β in mouse models (60). Under a physiological condition, this process contributes to effective host defense against harmful pathogens leading to gastrointestinal disorder and subsequent reconstitution (60). Meanwhile, another study had shown that NLRP3 inflammasome could promote neutrophil chemotaxis and antimicrobial secretions of the colon to sustain intestinal homeostasis (57). When epithelial barrier was destroyed, microbiota recruited immune cells to the lamina propria, and NLRP3 inflammasome activated in these immune cells played destructive effect (57). Therefore, NLRP3 inflammasome may have different effects in specific conditions, including physiological condition, infection by invading pathogenic bacteria and involving in innate immune disorder.
NLRP3 inflammasome inhibitors have been demonstrated to be effective in experimental models of IBD. Most inhibitors have a commonly downregulated effect on NLRP3, caspase-1, and pro-inflammatory cytokines. So far, therapeutic targets of NF-κB, Nuclear factor E2-related factor-2 (Nrf2), and ROS have been widely studied. Naringin, an activating ligand of peroxisome proliferator-activated receptor γ (PPARγ), can significantly decrease disease activity indexes (DAI), pathological damage of colon, and inflammation severity (64). Naringin alleviated UC via pressing NLRP3 inflammasome by activating PPARγ and degrading subsequent NF-κB activation (64). In a murine TNBS-induced colitis model, carboxyamidotriazolez downregulated NF-κB pathway by reduction of NF-κB p65 expression and phosphorylation of IκBα (65). Phloretin and Growth differentiation factor 11 play an anti-inflammatory effect targeting on the same pathway (66, 67). Nrf2 is a negative regulator for NLRP3 inflammasome by inhibiting priming of NLRP3 inflammasome (68). Cardamonin is a natural herbal extract of Alpinia katsumadai Hayata. Nrf2 and its target genes NQO1, Trx1, SOD2 and HO-1, especially NQO1, was elevated by cardamonin, which has the effect to depress NLRP3 inflammasome activation in a mouse model (69). And upregulation of Nrf2 has been detected after treatment of Resveratrol and Hydrogen sulfide (H2S) (70, 71). ROS is considered as a second messenger in pro-inflammatory responses. H2S can also reduce ROS production to prevent inflammation (71). Moreover, therapeutic strategies focused on miRNA has attracted more attention, Cinnamaldehyde can decrease MicroRNA-21 and miR-155 levels in colons and macrophages to ameliorate DSS-induced colitis (72). Recent studies showed that new mode of administration can also promote the efficacy of NLRP3 inflammasome inhibitors. The combined administration of BBG (a P2X7R blocker) and OLT1177 (a selective NLRP3 inhibitor) effectively alleviates UC by complementary effects (73). The effect of NLRP3 inflammasome inhibitors in IBD has not been comprehensively understood, which deserves further study.
Psoriasis is a chronic inflammatory skin disease mediated by immune responses. In psoriasis biopsy, the expression of NLRP3, caspase-1, and IL-1β was significantly upregulated compared to non-lesional psoriatic skin (139). The single nucleotide polymorphisms (SNPs) studies show that the genetic mutations in NLRP3 are associated with psoriasis susceptibility in Chinese Han population (140). And the genetic mutations in CARD8-C10X (rs2043211) are associated with Psoriatic arthritis (PsA) in northern Swedish population (141). All these findings indicate that NLRP3 inflammasome may be involved in the occurrence and development of psoriasis.
PlxnB2 and its ligand (such as CD100) participate in neuronal development and immune responses (75). The bound of soluble CD100 to PlxnB2 can upregulate NF-κB pathway, which induces NLRP3 inflammasome activation in keratinocytes of psoriasis patients (75). In fibroblasts deriving from patients with psoriasis, abnormal inflammasome activity can be induced by downregulation of tristetraprolin TTP (76). TTP can directly target the degradation of NLRP3 mRNA; therefore, TTP downregulation may contribute to pathogenesis of psoriasis via NLRP3 inflammasome activation (76). Verma et al. found that TNF-α could activate NLRP3 inflammasome, which is independent of priming signals, and caspase-1 reactivity, plasma IL-1β and IL-18 are reduced in psoriasis patients treated with anti-TNF therapy (77). In mouse model of psoriasis, NLRP3 inflammasome keratinocytes can be activated by TNF-α to induce inflammation, which was induced by inhibiting autophagy via PI3K/AKT/mTOR signaling pathway (142). Overexpression of lncRNA MEG3 could alleviate inflammation by promoting autophagy (142). Serum amyloid A (SAA) serves as an important trigger to promote expression of IL-1β by activating NF-κB pathway in psoriatic keratinocytes (78). And IL-1β plays a key role in the skin diseases mediated by T helper type 17 cells (78). In vitro, miR-155 silencing can depress NLRP3/caspase-1 signal pathway to alleviate inflammatory responses (79). In summary, these findings indicate that CD100/PlxnB2, TTP, TNF, miR-155, and SAA might be potential therapeutic targets for psoriasis.
Herbal extracts have been demonstrated to be effective treatment for psoriasis. In psoriasis mouse models induced by imiquimod, the expression of TLR7, TLR8, p-NF-κB, and NLRP3 were obviously suppressed by administration of Datura metel L (80). The production of key inflammatory cytokines including IL-1β, IL-17, and IL-23 was also inhibited, which implied that Datura metel L. plays a protective role by depressing the pathway of TLR7/8-MyD88-NF-κb-NLRP3 (80). Paeonia lactiflora Pallas extract and BAY11-7082 have an inhibitory effect against immune responses in keratinocytes probably by the similar pathway (79, 81). In addition, cycloastragenol specifically inhibits macrophages infiltration in dermis and pyroptosis mediated by NLRP3 inflammasome in mouse models (81). IL-1 inhibition might be another promising therapeutic strategy. Wan et al. reported a dissolvable microneedle, which consists of Cas9 RNP nanocomplexes and dexamethasone nanoparticles (82). The microneedle can be internalized by keratinocytes and immune cells, and disrupted NLRP3 inflammasome selectively to alleviate skin inflammations in a mouse model of psoriasis (82). Of note, new-onset plaque psoriasis has been shown to be a side effect in the RA patients treated with anti-IL-1 therapy (74). The role of NLRP3 inflammasome in psoriasis pathogenic mechanism is complicated, and the researches are comparatively limited, which deserves further studies.
Rheumatoid arthritis (RA) is a common chronic autoimmune disease characterized by persistent synovial inflammation, pannus formation, destruction of cartilage and small diarthrodial joints, which is mainly caused by autoantibody secretion and aberrant immune responses (143). Various NLRP3-releated SNPs have been shown to be associated with susceptibility of RA. NLRP3 SNPs are associated with susceptibility of RA and anti-TNF responses in Caucasian population (144). In addition, genetic mutations of NLRP3 inflammasome may increase the risk of stroke and TIA in Swedish population, but not of myocardial infarction and angina pectoris (145).
Several studies have shown the hyperactivity of NLRP3 Inflammasome and downstream factors in RA. The intracellular levels of NLRP3, active caspase-1, pro-IL-1β, and active IL-1β in whole blood cells are increased in active RA patients (146). This increased level can also be detected with the treatment of TLR4 or TLR3 agonist (146). Guo et al. found that MCC950, a selective NLRP3 inhibitor, is able to depress NLRP3 inflammasome activation in monocyte and macrophages to infiltrate into the synovia, resulting in lesser joint inflammation and bone destruction (88). It indicates that NLRP3 Inflammasome is involved in the pathogenesis of RA. NLRP3 inflammasome activation has not been detected in fibroblast-like synoviocytes (FLS), which suggests that FLS may not produce IL-1β mediated by NLRP3 inflammation (147). TLR1-9 have been demonstrated to play a modulatory role in joint inflammation in the pathogenesis of RA (148). Downregulation of NOD2 gene expression can decrease NF-κB and pro-inflammatory cytokines in FLS of RA patients, which indicates that NOD2 may promote NLRP3 Inflammasome activation by effecting the priming stage (149). Both in FLS and human umbilical vein endothelial cells of RA patients, TNF-α/CRT dual signaling promotes NLRP3 Inflammasome activation via enhancing the effect of caspase-1 (85). TNF-α may serve as an initiator for HuR translocating to mediate NLRP3 Inflammasome activation (85). The typical activated models of Ca2+ and P2X7 are also detected to initiate activation of NLRP3 Inflammasome in RA patients (86).
Zhao et al. demonstrated that NLRP3 Inflammasome promotes Th17 cell differentiation to enhance the adaptive immune dysfunction of RA (150). Th17 has been proven to have a vital role in the downstream cascade reactions of NLRP3 inflammasome in RA. Jin et al. firstly found that protectin DX downregulates Th17 cells and pro-inflammatory cytokines and upregulates Tregs and anti-inflammatory cytokines by inhibiting NLRP3 inflammasome activation via miR-20a (89). Tofacitinib effectively ameliorates the severity of RA by restoring Treg/Th17 cell balance via reducing NLRP3 inflammasome activation in arthritic joints and draining lymph nodes (87). NLRP3 inflammasome can also mediate secretion of Fas-associated death domain, which is a pivotal adaptor molecule in innate immunity and inflammation (151). However, the expression of NLRP3 and pro-caspase-1 was decreased in the peripheral neutrophils (152). The active caspase-1 expression was increased, which was positively correlated with serum level of IL-18, which suggested that IL-18 mediated by active caspase-1 plays a pro-inflammatory role in neutrophils of RA, independently of NLRP3 inflammasome (152). Thus, different cell types should be noted in the pathogenesis of RA.
Taraxasterol exerts anti-inflammatory effects by depressing NLRP3 inflammasome pathway via downregulating the expression of NF-κB in RA patients (90). Celastrol, a quinone-methylated triterpenoid extracted from Tripterygium wilfordii, alleviates RA inflammation by suppressing the ROS/NF-κB/NLRP3 pathway (91). In collagen-induced arthritis mouse models, Punicalagin (an active substance extracted from pomegranate peel) inhibits phenotype polarization and pyroptosis of M1 macrophages by downregulating NF-κB signaling (92). Hsa_circ_0044235 has been found to inhibit NLRP3-mediated pyroptosis in FLSs (93). Other potential therapeutic strategies focus on negative regulation of NLRP3 inflammasome. Human umbilical cord blood-derived MSCs suppress the expression of NLRP3 Inflammasome via a paracrine loop of IL-1β signaling in CIA mouse models and mononuclear cells from RA patients (94). The deficiency of A20 in macrophages can enhance the effect of NLRP3 inflammasome by promoting the biological activity of caspase-1, IL-1β secretion, and pyroptosis in mouse models (44). Activation of NLRP3 Inflammasome and secretion of IL-1β can be inhibited by TTP expression and activation of TTP in both in vivo and in vitro models (95). TTP can be activated by dephosphorylation mediated by protein phosphatase 2A (PP2A), and Arctigenin (PP2A agonist) reduces monosodium urate (MSU) crystal-induced inflammation (95). It is indicated that TTP might be a potential target for inflammation induced by MSU crystals (95). These studies indicate that treatments aiming to inhibit NLRP3 inflammasome and IL-1β might be potential therapy for RA.
Systemic sclerosis (SSc) is an idiopathic autoimmune disease targeting on connective tissue, characterized by autoimmune dysfunction, microvascular vessel alterations, and skin fibrosis. The fibrosis is triggered by inflammatory response, in which innate immunity serves as a key factor. However, the precise pathogenesis hasn’t been clarified, and few effective treatments have been available until now.
In skin of patients with SSc, NLRP3 is upregulated and correlates positively with skin thickness (153). A resistant feature to skin fibrosis can be detected in NLRP3(−/−) mice and ASC (−/−) mice, which implies that there is a correlativity between SNPs of NLRP3 and SSc (96). MiR-155 participates in innate immunity and adaptive immunity (154, 155). MiR-155 is involved in pulmonary fibrosis, hepatic fibrosis, as well as wound site fibrosis (156–158), and IL-1β can induce the expression of miR-155 (159). The expression of miR-155 was significantly increased in SSc lung fibroblasts, and fibrosis could be driven by inflammasome-dependent expression of miR-155 (97). Furthermore, there are positive feedbacks between NLRP3 inflammasome and miR-155, which explains sustaining fibrosis in SSc (97). MiR-155 might be a potential target for SSc, deserving future study.
Artlett et al. detected that caspase-1 inhibitor decreased secretion of IL-1β, IL-18, and collagens in dermal and lung fibroblasts of SSc, and reduced expression of α-smooth muscle actin in dermal fibroblasts (96). Shinohara presented that host receptors can identify pathogens to induce autoimmunity, which implies that infections and autoimmunity are closely connected (160). B19V infection induces caspase-1 mediated by NLRP3 inflammasome in monocytes of SSc patients (98). The expression of IL-1β and TNF-α can be induced by stimulus such as lipopolysaccharides (98). It indicates that viral components can enhance the sensitivity of NLRP3 inflammasome activation in monocytes (98). These findings may provide potential prevention strategy for SSc.
Type 1 diabetes (T1D) is an autoimmune disease targeting insulin-producing pancreatic β-cells specifically mediated by T lymphocyte (161). Both innate immunity and adaptive immunity participate in the development of T1D, and NLRP3 inflammasome acts as an important component of innate immunity to induce insulitis and β-cell death (162, 163). Polymorphisms of NLRP3 inflammasome-related gene correlate with T1D. Pontillo et al. found that SNPs in NLRP3 had a correlation specifically with T1D in Brazilian population (164). In Norwegian population and Chinese Han population, NLRP1 was associated strongly with T1DM (165). Wu et al. found that SNPs in NLRP3 correlated specifically with T1D, especially in Latin American population (166). In Slovenian population, the association of NLRP3 polymorphisms with T1D has not been observed (167). It is needed to confirm the specific gene polymorphisms of NLRP3 in different populations.
The important role of IL-1β in the pathogenesis of T1D has been determined in previous studies. IL-1β modulates pro-inflammatory cells to migrate into pancreatic islet, inducing direct cytotoxicity and β-cell apoptosis, which depends on the dose of IL-1β in T1D rat models (99, 100). Interestingly, NLRP3 inflammasome played a protective role in early stage of T1D in IRAK-M (−/−) NOD mice (168). In human islets, NLRP3 inflammasome can be activated, and secretion of IL-1β increases by the presence of LPS and ATP (102). As an upstream factor activating IL-1β, NLRP3 inflammasome may have a potential function in T1D. Resident peritoneal macrophages promote inflammation by inducing inflammatory cytokine/chemokine secretion such as IL-1β and upregulating the receptor expression of these proinflammatory cytokine/chemokine (103). Nitric oxide production is mediated by upregulated NLRP3/iNOS (nitric oxide synthase) pathway, which contributes to the proinflammatory state of resident PMs (103).
NLRP3 inflammasome has been shown to initiate and participate chronic complications of diabetes. Hyperglycemia can activate NLRP3 inflammasome, promote secretion of inflammatory cytokines, and trigger cascade of inflammatory response, finally inducing diabetic nephropathy, diabetic retinopathy, and so on (104, 169). Mitochondrial DNA activates NLRP3 inflammasome in endothelial cells via Ca2+ influx and mitochondrial ROS generation, which mediates endothelial dysfunction and vascular inflammation in diabetes complications (105). NLRP3 inflammasome activation can be detected in urothelial cells (170). It induces loss of in nerve density and Aδ-fibers, and leading to insensitivity of bladder fullness, which is a specific diabetic bladder dysfunction symptom (170). Verapamil, a calcium channel blocker, can diminish the release of IL-1β and TNF-α into the vitreous fluid and decrease retinal ganglion cell loss in diabetic retinopathy via inhibiting NLRP3 inflammasome mediated by TLR4 (106). Verapamil can also reduce pancreatic islets shrinkage and enhanced CD34 expression by interfering NLRP3 inflammasome assembly (106). In streptozotocin-induced mice with diabetic cardiomyopathy, the expression of NLRP3 inflammasome, IL-1β, and IL-18 in cardiac tissues was induced by nuclear NF-κB translocation, which can be inhibited by Scutellarin treatment (107). These researches imply that NLRP3 inhibitors could serve as a potential target for patients with diabetes.
MCC950, as an inhibitor of NLRP3 inflammasome, has been demonstrated to be a compelling treatment for diabetes in mouse models (171). Gao et al. found that Ginsenoside Rg1, a major active ingredient in ginseng, a traditional herb for diabetes in China, had a function to weaken NLRP3 activity in the liver and pancreas (108). In addition, low-methoxyl pectin can mediate decrease of NLRP3 inflammasome activation. And it can enhance cecal barrier function and shape intestinal homeostasis to ameliorate gut-pancreatic immune environment (109, 172). Low-methoxyl pectin may serve as a promising prevention drug of T1D. The TLR2/4, NF-κB, and NLRP3 inflammasome pathways are upregulated in intestinal tissues of NOD mice, which promotes the secretion of downstream signaling proteins such as IL-1β and IFN-γ (110). These downstream signaling proteins can induce activation and differentiation of T cells and the migration of these diabetogenic T cells to the pancreas (110). We propose that immunotherapy targeting on NLRP3 inflammasome is a promising approach to treat T1D.
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease characterized by production of autoantibodies against nuclear components, deposition of immune complex, and multiorgan damage resulting from aberrations of immune response. Gene polymorphisms of NLRP3 were significantly associated with susceptibility of SLE in Latin American individuals (173). However, in Chinese Han population, association between NLRP3 SNPs and SLE susceptibility has not been observed (174). It is warranted to study the relationship of gene polymorphisms of NLRP3 with SLE susceptibility in different populations. The expression of NLRP3 inflammasome, AIM2, and caspase-1 was increased in renal tissues, especially there was a positive relationship between the expression level of NLRP3 inflammasome and the activity index score in patients with SLE (175). Furthermore, the expression of NLRP3 mRNA was upregulated in macrophages, and the expression of AIM2 mRNA was decreased in female SLE patients (176). Meanwhile, in male SLE patients, the expression of AIM2 mRNA was increased, and SNP of CARD8 resulted in susceptibility of patients (176). It implied that there is a gender-dependent difference in mechanism of inflammasome activation in SLE patients.
Both innate and adaptive immunity are involved in the pathogenesis of SLE. Double-strand DNA and immune complex could activate NLRP3 inflammasome in histiocytes to promote inflammatory response mediated by IL-1β and IL-18 (49). Nucleic acid components including microbial nucleic acids, endogenous DNA, and endogenous RNA-containing U1-small nuclear ribonucleoprotein (U1-snRNP) can activate NLRP3 inflammasome in human monocytes in vitro (114). Shin MS et al. discovered that immune complexes could activate NLRP3 inflammasome by upregulating NF-kB pathway in animal models and patients with SLE (111). The caspase-1 activation and IL-1β production of bone marrow-derived macrophages was decreased after it was transfected with Glycogen synthase kinase 3β (GSK-3β) siRNA (115). The administration of GSK-3β inhibitor could alleviate severe proteinuria and nephritis, and anti-dsDNA antibody production, deposition of immune complex, and circulating cytokines were depressed (115). Zhao et al. suggested that GSK-3 promotes renal inflammation by activating NLRP3 inflammasome and mediating IL-1β release (115). Caspase-1 was activated especially in CD14-positive and CD16-positive monocytes from SLE patients, which were positively correlated with serum titers of anti-double-stranded DNA antibodies and negatively correlated with serum levels of complement component 3 and platelet count (116). NLRP3 inflammasome could be activated by cyclic GMP-AMP synthase stimulator of interferon genes pathway to promote caspase-1 activation and IL-1β secretion (116). Neutrophil extracellular traps (NETs) were also found to participate in the pathogenesis of SLE. NETs are a network consisting of chromatin fibers and granule-derived antimicrobial peptides (177). The impaired clearance of NETs and increased release of NETs are promoted by low-density granulocytes (LDGs), which contribute to accumulation of NETs in human monocytes in vitro (112, 113). NETs can activate caspase-1 with releasing of proinflammatory cytokines IL-1β and IL-18, and promote the formation of immune complex and type I interferon (117). Interestingly, IL-18 in turn induces perpetual NETosis in human neutrophils, which results in a feed-forward inflammatory loop (117). A recent study found that milk fat globule-EGF factor 8 (MFG-E8) could reduce neutrophil migration, accumulation, phagocytosis, and NETosis via reducing surface CXCR2 expression in pristane-induced lupus and patients with SLE (118).
NLRP3 inflammasome plays an important role in the development of SLE. Podocytes are highly differentiated epithelial cells and a critical component of glomerular basement membrane (GBM), which play a core role in maintaining the function of glomerular filtration (178). In the later stage of SLE, the expression of NLRP3 inflammasome and caspase-1 could be detected in podocytes in murine lupus models (115). The production of ROS promotes NLRP3 inflammasome activation in the podocytes line (119). Importantly, TLRs (such as TLR4) is expressed in glomerular podocytes of normal mice, which implies that podocytes also possess immune function in physiological condition (179). In addition, IL-18 release mediated by NLRP3 inflammasome is a potential pathogenic factor in cutaneous lupus lesions. Type I IFN is reported to be a main risk factor of cardiovascular disease (180). The feed-forward inflammatory loop mentioned above can also be detected in endothelial progenitor cells (181). Xenon can reduce NF-κB/NLRP3 inflammasome activation to ameliorating renal function in mouse models with spontaneous LN (120). Honokiol and dibenzylideneacetone (Tris) dipalladium both have potential therapeutic effect against accelerated and severe type of lupus nephritis by suppressing NLRP3 inflammasome activation (121, 122). The former interferes NLRP3 inflammasome via reducing NF-κB activation, suppressing reactive oxygen production and mitochondrial damage, and inducing sirtuin 1/autophagy axis activation (121). And the latter can reduce p38 MAPK signaling pathways and regulate the autophagy/NLRP3 inflammasome axis (122). Cf-02 can treat acute onset of severe lupus nephritis in mice by inhibiting the NF-κB/NLRP3 inflammasome axis and regulating T cell functions differentially (123).
Interestingly, NLRP3 inflammasome might have immunosuppressive effect on SLE (182–184). The expression of TGF-β target genes was depressed by deficiency of NLRP3 and ASC in mice models of spontaneous lupus-like autoimmunity (182). NLRP3 and ASC are demonstrated to downregulate TGF-β receptor signaling through SMAD2/3 phosphorylation, which contributes to the immunosuppressive effect (182). There are more and more new drugs targeting the NLRP3 inflammasome in the treatment for SLE, and some show to be effective (Table 2).
Autoimmune thyroid diseases (AITDs) are a series of thyroid diseases characterized by thyroid tissue damage and autoimmune disorders, including mainly Hashimoto’s thyroiditis (HT) and Graves’ disease (GD).
Guo et al. proposed firstly that multiple inflammasomes including NLRP3, NLRP1, NLRC4, and AIM2 participated in the development of AITDs (185). Excessive iodine promoted pyroptosis activity in thyroid follicular cells via the ROS-NF-κB-NLRP3 pathway which might be involved in the development of HT (186). However, Nagayama, Y., raised questions that Nthy-ori 3-1 cells aren’t capable of iodine uptake and the concentration of iodine (131). The role of NLRP3 inflammasome in Graves’ disease (GD) has not been reported so far.
It is reported that Yanghe decoction (a traditional Chinese herbal formulation) can alleviate autoimmune thyroiditis in rat models via downregulating NLRP3 inflammasome and adjusting the imbalance Th17/Treg (132). There are very few studies concerning the effect of NLRP3 inflammasome in the pathogenesis of AITDs, deserving further study.
NLRP3 inflammasome exists in various immune cells, including macrophages, granulocytes, antigen-presenting cells, B cells, and T cells. It has been found in tissues and cells such as platelet, podocyte, and keratinizing squamous epithelium of skin (187). As a vital component of innate immune system, NLRP3 inflammasome recognizes DAMPs and PAMPs, and initiates innate inflammatory responses via promoting proinflammatory cytokines secretion, which may also trigger adaptive immunity dysfunction (161). Besides its elementary physiological functions in immune responses, NLRP3 inflammasome also participates in pathogenesis process such as multiple autoimmune diseases. The common character of autoimmune diseases is histocyte and organ damage resulting from autoantibody overproduction, which leads to loss of immunological tolerance and aberrant autoreactive immune responses. The accurate role of NLRP3 inflammasome in autoimmune disease pathogenesis is complex and has not yet been completely illuminated.
Collectively, the effect of NLRP3 inflammasome in autoimmune diseases involves the following two aspects (Table 2 and Figure 2): due to caspase-1 being the effector of inflammasome structure, IL-1β, IL-18, and pyroptosis modulated by activated caspase-1 play a major role in autoimmune diseases. Firstly, inflammation promoted by cytokines, especially IL-1, participates in the onset and development of most autoimmune diseases, such as RA and IBD (88, 188). Secondly, there is an emerging appreciation that NLRP3 inflammasome participates in adaptive immunity. Logically, NLRP3 inflammasome is involved in autoimmune diseases with adaptive immune dysfunction. For instance, IL-1 is a co-stimulatory factor and lymphocyte activating factor, which can provide signals of pro-survival and proliferation for T cells. It promotes autoreactive T cells to cause β-cell death (101). IL-1β induces migration of T cells into pancreatic islets by regulating chemotaxis (189). In RA, NLRP3 inflammasome of CD4+ T cells promote Th1 differentiation, which is induced by IL-1β in a caspase-1-dependent manner (83). In other autoimmune diseases, NLRP3 inflammasome can also induce differentiation and polarization of Th2, Th17, and dendritic cells (84). Thirdly, pyroptosis mediated by activated caspase-1 promotes development of autoimmune diseases. In IBD, NLRP3 inflammasome triggers pyroptosis in a caspace-1-dependent manner to cause death of histocytes such as macrophages and dendritic cells (190). The release of cellular debris reacts with immune cells, resulting in an enhanced circle of inflammation (190). Generally, IL-1β, IL-18, and pyroptosis play a function by inflammation, activating adaptive immunity and immunological regulation. Interestingly, the pathogenesis of IBD and psoriasis is mainly inflammation, and the pathogenesis of T1DM, SLE, and AITDs mainly involves adaptive immune response. Meanwhile, the pathogenesis of RA and SSC involves both above factors. So distinguishing emphasis of different autoimmune disease pathogenesis, which might involve a spectrum from auto-inflammatory to adaptive immune response, may contribute to understand the specific and accurate role of NLRP3 inflammasome.
On the other hand, besides effect of NLRP3 inflammasome pathway, NLRP3 inflammasome can promote autoimmune diseases in some other manners. In SSC, NLRP3 inflammasome activation upregulates miR-155, which participates in collagen synthesis of keratinocytes (156, 157). In summary, NLRP3 inflammasome participates in the initiation of autoimmune diseases, which serves as a checkpoint in innate immunity and adaptive immune dysfunction (Figure 1). Compared with clinical application focusing on downstream adaptive immune response, chemical inhibitor targeting on NLRP3 inflammasome pathway might be a more potential therapeutic strategy.
NLRP3 inflammasome is significantly increased in autoimmune diseases such as T1D, IBD, SLE, RA, SSC, psoriasis, and AITDs. However, the protective role and decrease of NLRP3 inflammasome have also been detected in T1D, IBD, SLE, and psoriasis. The controversial results may be explained by hypothesis as follows: (a) In different stages of the disease, NLRP3 inflammasome may conduct completely inverse function. For example, an experiment conducted with IRAK-M(−/−) NOD mice, which is characterized by early onset and rapid progression of T1D, shows that NLRP3 inflammasome is a protective factor in the initial stage of T1D (168). (b) Despite the effect of NLRP3 inflammasome signaling pathway, NLRP3 inflammasome may also affect other bioactive substances and signaling pathways, by which it plays an opposite role. In SLE, NLRP3 inflammasome modulates TGF-β and IFN-I to conduct immunosuppressive effect (182, 191). (c) In some special pathways, NLRP3 inflammasome plays a protective role. Gastrointestinal tract is characterized by communication with external environment indirectly and existence of intestinal microflora. NLRP3 inflammasome strengthens anti-inflammatory effect to maintain intestine homeostasis but promotes modulation of T cell in a physiological condition. Therefore, deficiency and overt activation of NLRP3 inflammasome can both promote the development of IBD. In summary, future researches to identify specific mechanism of NLRP3 inflammasome in different histiocytes, disease stages and conditions are warranted.
There are several common pathways in different autoimmune diseases, such as autophagy, mitochondrial DNA, TLR4, Nrf2 pathway, Bay11-7082, P2X7 receptor, microRNA, and so on (Table 2). Moreover, there are specific and subtle differences between these similarly pathogenic mechanisms. For instance, the main function of miR-155 is to promote inflammation in keratinocyte of psoriasis and in macrophage of IBD; however, it drives fibrosis in fibroblasts of SSC. All these contrasts and connect in various mechanism indicate the complexity of NLRP3 inflammasome in the pathogenesis of autoimmune diseases. So, further studies are needed to focus on the comparison of mechanisms in different active sites, such as intestine and pancreatic β cell. In addition, previous studies have shown that NLRP3 inflammasome plays a protective role in physiological condition, and both the upregulation and downregulation of NLRP3 inflammasome pathway stimulated by specific factor will initiate pathogenic progress. As the mechanism of NLRP3 inflammasome assembly and downstream cytokines effect are more studied, the initiating mechanism of regulation dysfunction is of great importance in future researches. Just as avoidance of allergen to hypersensitivity, therapy focusing on initiating mechanism of regulation dysfunction will be a simple but effective therapeutic application. The inhibition of NLRP3 inflammasome pathway, such as Ginsenoside Rg1, low methoxyl pectin, and Bay11-7082, may be a potential therapeutic strategy for autoimmune diseases (Table 2). The inhibition involves three levels: structural proteins of NLRP3 inflammasome, cytokines, and the signaling pathway. However, the effect of some inhibitions is limited and scant. Some inhibitions cause serious side effects, and some inhibitions even show paradoxical pro-inflammatory effect. Newly plaque psoriasis could be detected in RA patients with anti-IL-1 treatment (74). It indicates that the role of NLRP3 inflammasome inhibition is complicated and associates with multiple mechanisms. Consequently, further researches are needed to focus on the following points: inhibitions that target on junction point of pathway may show precise efficacy; in addition, focusing on upstream mechanism and finding inhibition that blocks the switch of the whole pathogenic progress may bring breakthrough to therapy of autoimmune diseases.
In conclusion, as a platform sensing dangerous stimuli from endogenous or exogenous environment, NLRP3 inflammasome participates in both innate and adaptive immunity via modulating secretion of cytokines and pyroptosis. Increasing experiments show that NLRP3 inflammasome may play different pathogenic roles in autoimmune diseases, which provides a promising therapeutic option for autoimmune diseases. Therefore, delineating a comprehensive molecular mechanism of the complex role of NLRP3 inflammasome in autoimmune diseases deserves further study.
YWZ, the main author, contributed in literature review and summary, manuscript drafting. WY assisted in reviewing literature. WL revised the manuscript. YJZ, the corresponding author, contributed in designing, reviewing, and editing the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (Grant No. 81700728), Science and Technology Projects of Guangzhou (Grant No. 201707010365), 2021 Medical Science Foundation of Guangdong Provincial Health Commission (Grant No. 202011179196403), 2021 Medical and Health Technology Projects of Guangzhou (Grant No. 20211A011083).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Davidson A, Diamond B. Autoimmune Diseases. N Engl J Med (2001) 345:340–50. doi: 10.1056/NEJM200108023450506
2. Papp G, Boros P, Nakken B, Szodoray P, Zeher M. Regulatory Immune Cells and Functions in Autoimmunity and Transplantation Immunology. Autoimmune Rev (2017) 16:435–44. doi: 10.1016/j.autrev.2017.03.011
3. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic Association Analyses Implicate Aberrant Regulation of Innate and Adaptive Immunity Genes in the Pathogenesis of Systemic Lupus Erythematosus. Nat Genet (2015) 47:1457–64. doi: 10.1038/ng.3434
4. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate Immunity and Intestinal Microbiota in the Development of Type 1 Diabetes. Nature (2008) 455:1109–13. doi: 10.1038/nature07336
5. Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol Cell (2002) 10:417–26. doi: 10.1016/s1097-2765(02)00599-3
6. Medzhitov R, Preston-Hurlburt P, Janeway CA. A Human Homologue of the Drosophila Toll Protein Signals Activation of Adaptive Immunity. Nature (1997) 388:394–7. doi: 10.1038/41131
7. Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung JU. TRIM56-Mediated Monoubiquitination of cGAS for Cytosolic DNA Sensing. Nat Commun (2018) 9:613. doi: 10.1038/s41467-018-02936-3
8. Franchi L, Warner N, Viani K, Nuñez G. Function of Nod-Like Receptors in Microbial Recognition and Host Defense. Immunol Rev (2009) 227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x
9. Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, et al. A Single Domain Antibody Fragment That Recognizes the Adaptor ASC Defines the Role of ASC Domains in Inflammasome Assembly. J Exp Med (2016) 213:771–90. doi: 10.1084/jem.20151790
10. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
11. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 Self-Cleavage Is an Intrinsic Mechanism to Terminate Inflammasome Activity. J Exp Med (2018) 215:827–40. doi: 10.1084/jem.20172222
12. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A Genome-Wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem (2016) 291:103–9. doi: 10.1074/jbc.C115.700492
13. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 Is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature (2016) 530:354–7. doi: 10.1038/nature16959
14. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting Edge: NF-κb Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J Immunol (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
15. Xing Y, Yao X, Li H, Xue G, Guo Q, Yang G, et al. Cutting Edge: TRAF6 Mediates TLR/IL-1r Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome. J Immunol (2017) 199:1561–6. doi: 10.4049/jimmunol.1700175
16. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate Is an Inflammatory Signal That Induces IL-1β Through HIF-1α. Nature (2013) 496:238–42. doi: 10.1038/nature11986
17. Liu J, Qian C, Cao X. Post-Translational Modification Control of Innate Immunity. Immunity (2016) 45:15–30. doi: 10.1016/j.immuni.2016.06.020
18. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-Transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation. J Biol Chem (2012) 287:36617–22. doi: 10.1074/jbc.M112.407130
19. Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-Inhibited Processing of IL-1 Beta in Human Monocytes. EMBO J (1995) 14:1607–14. doi: 10.1002/j.1460-2075.1995.tb07149.x
20. Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 Inflammasome Is Triggered by Low Intracellular Potassium Concentration. Cell Death Differ (2007) 14:1583–9. doi: 10.1038/sj.cdd.4402195
21. Franchi L, Kanneganti TD, Dubyak GR, Núñez G. Differential Requirement of P2X7 Receptor and Intracellular K+ for Caspase-1 Activation Induced by Intracellular and Extracellular Bacteria. J Biol Chem (2007) 282:18810–8. doi: 10.1074/jbc.M610762200
22. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 Mediates Non-Canonical Activation of the NLRP3 Inflammasome in Human Myeloid Cells. Eur J Immunol (2015) 45:2911–7. doi: 10.1002/eji.201545523
23. Ketelut-Carneiro N, Silva GK, Rocha FA, Milanezi CM, Cavalcanti-Neto FF, Zamboni DS, et al. IL-18 Triggered by the NLRP3 Inflammasome Induces Host Innate Resistance in a Pulmonary Model of Fungal Infection. J Immunol (2015) 194:4507–17. doi: 10.4049/jimmunol.1402321
24. Liu H, You L, Wu J, Zhao M, Guo R, Zhang H, et al. Berberine Suppresses Influenza Virus-Triggered NLRP3 Inflammasome Activation in Macrophages by Inducing Mitophagy and Decreasing Mitochondrial ROS. J Leukoc Biol (2020) 108:253–66. doi: 10.1002/JLB.3MA0320-358RR
25. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
26. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A Mutation in the Nlrp3 Gene Causing Inflammasome Hyperactivation Potentiates Th17 Cell-Dominant Immune Responses. Immunity (2009) 30:860–74. doi: 10.1016/j.immuni.2009.04.012
27. Chu J, Thomas LM, Watkins SC, Franchi L, Núñez G, Salter RD. Cholesterol-Dependent Cytolysins Induce Rapid Release of Mature IL-1beta From Murine Macrophages in a NLRP3 Inflammasome and Cathepsin B-Dependent Manner. J Leukoc Biol (2009) 86:1227–38. doi: 10.1189/jlb.0309164
28. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome Through Phagosomal Destabilization. Nat Immunol (2008) 9:847–56. doi: 10.1038/ni.1631
29. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-Beta. Nat Immunol (2008) 9:857–65. doi: 10.1038/ni.1636
30. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J Immunol (2015) 195:1685–97. doi: 10.4049/jimmunol.1500509
31. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate Immunee Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science (2008) 320:674–7. doi: 10.1126/science.1156995
32. van Bruggen R, Köker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW, et al. Human NLRP3 Inflammasome Activation Is Nox1-4 Independent. Blood (2010) 115:5398–400. doi: 10.1182/blood-2009-10-250803
33. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The Mitochondrial Antiviral Protein MAVS Associates With NLRP3 and Regulates Its Inflammasome Activity. J Immunol (2013) 191:4358–66. doi: 10.4049/jimmunol.1301170
34. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial Protein Mitofusin 2 Is Required for NLRP3 Inflammasome Activation After RNA Virus Infection. Proc Natl Acad Sci USA (2013) 110:17963–8. doi: 10.1073/pnas.1312571110
35. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
36. Dinarello CA. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
37. Garlanda C, Dinarello CA, Mantovani A. The Interleukin-1 Family: Back to the Future. Immunity (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
38. Chan AH, Schroder K. Inflammasome Signaling and Regulation of Interleukin-1 Family Cytokines. J Exp Med (2020) 217:e20190314. doi: 10.1084/jem.20190314
39. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, Cytokine Profile and Function of Human Interleukin 17-Producing Helper T Cells. Nat Immunol (2007) 8:950–7. doi: 10.1038/ni1497
40. Poznanski SM, Lee AJ, Nham T, Lusty E, Larché MJ, Lee DA, et al. Combined Stimulation With Interleukin-18 and Interleukin-12 Potently Induces Interleukin-8 Production by Natural Killer Cells. J Innate Immun (2017) 9:511–25. doi: 10.1159/000477172
41. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 Is a Unique Cytokine That Stimulates Both Th1 and Th2 Responses Depending on Its Cytokine Milieu. Cytokine Growth Factor Rev (2001) 12:53–72. doi: 10.1016/s1359-6101(00)00015-0
42. Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE, et al. Epithelial-Derived IL-18 Regulates Th17 Cell Differentiation and Foxp3+ Treg Cell Function in the Intestine. Mucosal Immunol (2015) 8:1226–36. doi: 10.1038/mi.2015.13
43. Ng GZ, Menheniott TR, Every AL, Stent A, Judd LM, Chionh YT, et al. The MUC1 Mucin Protects Against Helicobacter Pylori Pathogenesis in Mice by Regulation of the NLRP3 Inflammasome. Gut (2016) 65:1087–99. doi: 10.1136/gutjnl-2014-307175
44. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative Regulation of the NLRP3 Inflammasome by A20 Protects Against Arthritis. Nature (2014) 512:69–73. doi: 10.1038/nature13322
45. Huai W, Zhao R, Song H, Zhao J, Zhang L, Zhang L, et al. Aryl Hydrocarbon Receptor Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting NLRP3 Transcription. Nat Commun (2014) 5:4738. doi: 10.1038/ncomms5738
46. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 Inflammasome Activity Is Negatively Controlled by miR-223. J Immunol (2012) 189:4175–81. doi: 10.4049/jimmunol.1201516
47. Le HT, Harton JA. Pyrin- and CARD-Only Proteins as Regulators of NLR Functions. Front Immunol (2013) 4:275. doi: 10.3389/fimmu.2013.00275
48. Jin J, Yu Q, Han C, Hu X, Xu S, Wang Q, et al. LRRFIP2 Negatively Regulates NLRP3 Inflammasome Activation in Macrophages by Promoting Flightless-I-Mediated Caspase-1 Inhibition. Nat Commun (2013) 4:2075. doi: 10.1038/ncomms3075
49. Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, et al. Small Heterodimer Partner Interacts With NLRP3 and Negatively Regulates Activation of the NLRP3 Inflammasome. Nat Commun (2015) 6:6115. doi: 10.1038/ncomms7115
50. Wu Y, Kuo W, Wu Y, Yang K, Chen S, Jiang S, et al. Participation of C-FLIP in NLRP3 and AIM2 Inflammasome Activation. Cell Death Differ (2014) 21:451–61. doi: 10.1038/cdd.2013.165
51. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
52. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell (2013) 153:348–61. doi: 10.1016/j.cell.2013.02.054
53. Chen J, Chen ZJ. PtdIns4P on Dispersed Trans-Golgi Network Mediates NLRP3 Inflammasome Activation. Nature (2018) 564:71–6. doi: 10.1038/s41586-018-0761-3
54. Song N, Liu Z, Xue W, Bai Z, Wang Q, Dai J, et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell (2017) 68:185–97. e6. doi: 10.1016/j.molcel.2017.08.017
55. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 Inflammasome Inhibition Is Disrupted in a Group of Auto-Inflammatory Disease CAPS Mutations. Nat Immunol (2016) 17:1176–86. doi: 10.1038/ni.3538
56. Chatenoud L. Immune Therapies of Autoimmune Diseases: Are We Approaching a Real Cure? Curr Opin Immunol (2006) 18:710–7. doi: 10.1016/j.coi.2006.09.004
57. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 Inflammasome Plays a Key Role in the Regulation of Intestinal Homeostasis. Inflammation Bowel Dis (2011) 17:1359–72. doi: 10.1002/ibd.21478
58. Wang Y, Zhou X, Zou K, Chen G, Huang L, Yang F, et al. Monocarboxylate Transporter 4 Triggered Cell Pyroptosis to Aggravate Intestinal Inflammation in Inflammatory Bowel Disease. Front Immunol (2021) 12:644862. doi: 10.3389/fimmu.2021.644862
59. Itani S, Watanabe T, Nadatani Y, Sugimura N, Shimada S, Takeda S, et al. NLRP3 Inflammasome has a Protective Effect Against Oxazolone-Induced Colitis: A Possible Role in Ulcerative Colitis. Sci Rep (2016) 6:39075. doi: 10.1038/srep39075
60. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, et al. NLRP3 Tyrosine Phosphorylation Is Controlled by Protein Tyrosine Phosphatase PTPN22. J Clin Invest (2016) 126:1783–800. doi: 10.1172/JCI83669
61. Song S, Bai M, Ling Z, Lin Y, Wang S, Chen Y. Intermittent Administration of a Fasting-Mimicking Diet Reduces Intestinal Inflammation and Promotes Repair to Ameliorate Inflammatory Bowel Disease in Mice. J Nutr Biochem (2021) 96:108785. doi: 10.1016/j.jnutbio.2021.108785
62. Yun Y, Baek A, Kim DE. Autophagy Down-Regulates NLRP3-Dependent Inflammatory Response of Intestinal Epithelial Cells Under Nutrient Deprivation. BMB Rep (2021) 54(5):260–5. doi: 10.5483/BMBRep.2021.54.5.211
63. Xu Y, Tian Y, Li F, Wang Y, Yang J, Gong H, et al. Circular RNA HECTD1 Mitigates Ulcerative Colitis by Promoting Enterocyte Autophagy Via miR-182-5p/HuR Axis. Inflamm Bowel Dis (2021) izab188:1–16. doi: 10.1093/ibd/izab188
64. Cao H, Liu J, Shen P, Cai J, Han Y, Zhu K, et al. Protective Effect of Naringin on DSS-Induced Ulcerative Colitis in Mice. J Agric Food Chem (2018) 66:13133–40. doi: 10.1021/acs.jafc.8b03942
65. Du X, Chen W, Wang Y, Chen C, Guo L, Ju R, et al. Therapeutic Efficacy of Carboxyamidotriazole on 2,4,6-Trinitrobenzene Sulfonic Acid-Induced Colitis Model Is Associated With the Inhibition of NLRP3 Inflammasome and NF-κb Activation. Int Immunopharmacol (2017) 45:16–25. doi: 10.1016/j.intimp.2017.01.015
66. Wang L, Wang Y, Wang Z, Qi Y, Zong B, Liu M, et al. Growth Differentiation Factor 11 Ameliorates Experimental Colitis by Inhibiting NLRP3 Inflammasome Activation. Am J Physiol Gastrointest Liver Physiol (2018) 315:G909–20. doi: 10.1152/ajpgi.00159.2018
67. Zhang Z, Li S, Cao H, Shen P, Liu J, Fu Y, et al. The Protective Role of Phloretin Against Dextran Sulfate Sodium-Induced Ulcerative Colitis in Mice. Food Funct (2019) 10:422–31. doi: 10.1039/c8fo01699b
68. Liu X, Zhang X, Ding Y, Zhou W, Tao L, Lu P, et al. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid Redox Signal (2017) 26:28–43. doi: 10.1089/ars.2015.6615
69. Wang K, Lv Q, Miao Y, Qiao S, Dai Y, Wei Z. Cardamonin, a Natural Flavone, Alleviates Inflammatory Bowel Disease by the Inhibition of NLRP3 Inflammasome Activation via an AhR/Nrf2/NQO1 Pathway. Biochem Pharmacol (2018) 155:494–509. doi: 10.1016/j.bcp.2018.07.039
70. Chen Y, Zheng Z, Li C, Pan Y, Tang X, Wang XJ. Synthetic Imine Resveratrol Analog 2-Methoxyl-3,6-Dihydroxyl-IRA Ameliorates Colitis by Activating Protective Nrf2 Pathway and Inhibiting NLRP3 Expression. Oxid Med Cell Longev (2019) 2019:7180284. doi: 10.1155/2019/7180284
71. Qin M, Long F, Wu W, Yang D, Huang M, Xiao C, et al. Hydrogen Sulfide Protects Against DSS-Induced Colitis by Inhibiting NLRP3 Inflammasome. Free Radic Biol Med (2019) 137:99–109. doi: 10.1016/j.freeradbiomed.2019.04.025
72. Qu S, Shen Y, Wang M, Wang X, Yang Y. Suppression of miR-21 and miR-155 of Macrophage by Cinnamaldehyde Ameliorates Ulcerative Colitis. Int Immunopharmacol (2019) 67:22–34. doi: 10.1016/j.intimp.2018.11.045
73. Sameh S, Mahmoud EY, Hossam S, Noha AA, Ruwyda ES, Farah HA, et al. BBG Enhances OLT1177-Induced NLRP3 Inflammasome Inactivation by Targeting P2X7R/NLRP3 and MyD88/NF-κb Signaling in DSS-Induced Colitis in Rats. Life Sci (2021) 270:119123. doi: 10.1016/j.lfs.2021.119123
74. González-López Ma, Martínez-Taboada Vm, González-Vela Mc, Fernández-Llaca H, Val-Bernal Jf. New-Onset Psoriasis Following Treatment With the Interleukin-1 Receptor Antagonist Anakinra. Br J Dermatol (2008) 158(5):1146–8. doi: 10.1111/j.1365-2133.2008.08470.x
75. Zhang C, Xiao C, Dang E, Cao J, Zhu Z, Fu M, et al. CD100-Plexin-B2 Promotes the Inflammation in Psoriasis by Activating NF-κb and the Inflammasome in Keratinocytes. J Invest Dermatol (2018) 138(2):375–83. doi: 10.1016/j.jid.2017.09.005
76. Matteo B, Sebastian F, Claudia A, Roberta L, Andrea M, Sandra P, et al. Promoter Methylation Leads to Decreased ZFP36 Expression and Deregulated NLRP3 Inflammasome Activation in Psoriatic Fibroblasts. Front Med (2021) 7:579383. doi: 10.3389/fmed.2020.579383
77. Deepti V, Shora ZF, Gunnthorunn S, Cecilia BE, Charlotta S, Charlotta E. Enhanced Inflammasome Activity in Patients With Psoriasis Promotes Systemic Inflammation. J Invest Dermatol (2021) 141(3):586–595.e5. doi: 10.1016/j.jid.2020.07.012
78. Yu N, Liu S, Yi X, Zhang S, Ding Y. Serum Amyloid A Induces Interleukin-1β Secretion From Keratinocytes via the NACHT, LRR and PYD Domains-Containing Protein 3 Inflammasome. Clin Exp Immunol (2015) 179(2):344–53. doi: 10.1111/cei.12458
79. Irrera N, Vaccaro M, Bitto A, Pallio G, Pizzino G, Lentini M, et al. BAY 11-7082 Inhibits the NF-κb and NLRP3 Inflammasome Pathways and Protects Against IMQ-Induced Psoriasis. Clin Sci (2017) 131(6):CS20160645. doi: 10.1042/CS20160645
80. Yang B, Cheng Y, Liu Y, Liu Y, Tan J, Guan W, et al. Datura Metel L. Ameliorates Imiquimod-Induced Psoriasis-Like Dermatitis and Inhibits Inflammatory Cytokines Production Through TLR7/8-MyD88-NF-κb-NLRP3 Inflammasome Pathway. Molecules (2019) 24(11):2157. doi: 10.3390/molecules24112157
81. Deng G, Chen W, Wang P, Zhan T, Zheng W, Gu Z, et al. Inhibition of NLRP3 Inflammasome-Mediated Pyroptosis in Macrophage by Cycloastragenol Contributes to Amelioration of Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice. Int Immunopharmacol (2019) 74:105682. doi: 10.1016/j.intimp.2019.105682
82. Wan T, Pan Q, Ping Y. Microneedle-Assisted Genome Editing: A Transdermal Strategy of Targeting NLRP3 by CRISPR-Cas9 for Synergistic Therapy of Inflammatory Skin Disorders. Sci Adv (2021) 7(11):eabe2888. doi: 10.1126/sciadv.abe2888
83. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T Helper 1 Immunity Requires Complement-Driven NLRP3 Inflammasome Activity in CD4+ T Cells. Science (2016) 352:aad1210. doi: 10.1126/science.aad1210
84. Xue G, Jin G, Fang J, Lu Y. IL-4 Together With IL-1β Induces Antitumor Th9 Cell Differentiation in the Absence of TGF-β Signaling. Nat Commun (2019) 10(1):1376. doi: 10.1038/s41467-019-09401-9
85. Liu Y, Wei W, Wang Y, Wan C, Bai Y, Sun X, et al. TNF-α/Calreticulin Dual Signaling Induced NLRP3 Inflammasome Activation Associated With HuR Nucleocytoplasmic Shuttling in Rheumatoid Arthritis. Inflammation Res (2019) 68:597–611. doi: 10.1007/s00011-019-01244-w
86. Jäger E, Murthy S, Schmidt C, Hahn M, Strobel S, Peters A, et al. Calcium-Sensing Receptor-Mediated NLRP3 Inflammasome Response to Calciprotein Particles Drives Inflammation in Rheumatoid Arthritis. Nat Commun (2020) 11:4243. doi: 10.1038/s41467-020-17749-6
87. Yang X, Zhan N, Jin Y, Ling H, Xiao C, Xie Z, et al. Tofacitinib Restores the Balance of γδtreg/γδt17 Cells in Rheumatoid Arthritis by Inhibiting the NLRP3 Inflammasome. Theranostics (2021) 11(3):1446–57. doi: 10.7150/thno.47860
88. Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, et al. NLRP3 Inflammasome Activation Contributes to the Pathogenesis of Rheumatoid Arthritis. Clin Exp Immunol (2018) 194:231–43. doi: 10.1111/cei.13167
89. Jin S, Sun S, Ling H, Ma J, Zhang X, Xie Z, et al. Protectin DX Restores Treg/T H 17 Cell Balance in Rheumatoid Arthritis by Inhibiting NLRP3 Inflammasome via miR-20a. Cell Death Dis (2021) 12(3):280. doi: 10.1038/s41419-021-03562-6
90. Chen J, Wu W, Zhang M, Chen C. Taraxasterol Suppresses Inflammation in IL-1β-Induced Rheumatoid Arthritis Fibroblast-Like Synoviocytes and Rheumatoid Arthritis Progression in Mice. Int Immunopharmacolog (2019) 70:274–83. doi: 10.1016/j.intimp.2019.02.029
91. Jing M, Yang J, Zhang L, Liu J, Xu S, Wang M, et al. Celastrol Inhibits Rheumatoid Arthritis Through the ROS-NF-κb-NLRP3 Inflammasome Axis. Int Immunopharmacol (2021) 98:107879. doi: 10.1016/j.intimp.2021.107879
92. Ge G, Bai J, Wang Q, Liang X, Tao H, Chen H, et al. Punicalagin Ameliorates Collagen-Induced Arthritis by Downregulating M1 Macrophage and Pyroptosis via NF-κb Signaling Pathway. Sci China Life Sci (2021) 64. doi: 10.1007/s11427-020-1939-1
93. Chen S, Luo Z, Chen X. Hsa_circ_0044235 Regulates the Pyroptosis of Rheumatoid Arthritis via MiR-135b-5p-SIRT1 Axis. Cell Cycle (Georgetown Tex) (2021) 20(12):1107–21. doi: 10.1080/15384101.2021.1916272
94. Shin TH, Kim HS, Kang TW, Lee BC, Lee HY, Kim YJ, et al. Human Umbilical Cord Blood-Stem Cells Direct Macrophage Polarization and Block Inflammasome Activation to Alleviate Rheumatoid Arthritis. Cell Death Dis (2016) 7:e2524. doi: 10.1038/cddis.2016.442
95. Lv L, Qin T, Huang Q, Jiang H, Chen F, Long F, et al. Targeting Tristetraprolin Expression or Functional Activity Regulates Inflammatory Response Induced by MSU Crystals. Front Immunol (2021) 12:675534. doi: 10.3389/fimmu.2021.675534
96. Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The Inflammasome Activating Caspase 1 Mediates Fibrosis and Myofibroblast Differentiation in Systemic Sclerosis. Arthritis Rheum (2011) 63:3563–74. doi: 10.1002/art.30568
97. Artlett CM, Sassi-Gaha S, Hope JL, Feghali-Bostwick CA, Katsikis PD. Mir-155 Is Overexpressed in Systemic Sclerosis Fibroblasts and Is Required for NLRP3 Inflammasome-Mediated Collagen Synthesis During Fibrosis. Arthritis Res Ther (2017) 19:144. doi: 10.1186/s13075-017-1331-z
98. Zakrzewska K, Arvia R, Torcia MG, Clemente AM, Tanturli M, Castronovo G, et al. Effects of Parvovirus B19 In Vitro Infection on Monocytes From Patients With Systemic Sclerosis: Enhanced Inflammatory Pathways by Caspase-1 Activation and Cytokine Production. J Invest Dermatol (2019) 139:2125–33. doi: 10.1016/j.jid.2019.03.1144
99. Ohara-Imaizumi M, Cardozo AK, Kikuta T, Eizirik DL, Nagamatsu S. The Cytokine Interleukin-1beta Reduces the Docking and Fusion of Insulin Granules in Pancreatic Beta-Cells, Preferentially Decreasing the First Phase of Exocytosis. J Biol Chem (2004) 279:41271–4. doi: 10.1074/jbc.C400360200
100. Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, Berney T, et al. Leptin Modulates Beta Cell Expression of IL-1 Receptor Antagonist and Release of IL-1beta in Human Islets. Proc Natl Acad Sci USA (2004) 101:8138–43. doi: 10.1073/pnas.0305683101
101. Zheng Y, Wang Z, Zhou Z. miRNAs: Novel Regulators of Autoimmunity-Mediated Pancreatic β-Cell Destruction in Type 1 Diabetes. Cell Mol Immunol (2017) 14(6):488–96. doi: 10.1038/cmi.2017.7
102. Lebreton F, Berishvili E, Parnaud G, Rouget C, Bosco D, Berney T, et al. NLRP3 Inflammasome Is Expressed and Regulated in Human Islets. Cell Death Dis (2018) 9:726. doi: 10.1038/s41419-018-0764-x
103. Davanso MR, Crisma AR, Braga TT, Masi LN, Amaral CL, Leal VNC, et al. Macrophage Inflammatory State in Type 1 Diabetes: Triggered by NLRP3/iNOS Pathway and Attenuated by Docosahexaenoic Acid. Clin Sci (London England: 1979) (2021) 135(1):19–34. doi: 10.1042/CS20201348
104. Koenen TB, Stienstra R, van Tits LJ, de Graaf J, Stalenhoef AFH, Joosten LAB, et al. Hyperglycemia Activates Caspase-1 and TXNIP-Mediated IL-1beta Transcription in Human Adipose Tissue. Diabetes (2011) 60:517–24. doi: 10.2337/db10-0266
105. Pereira CA, Carlos D, Ferreira NS, Silva JF, Zanotto CZ, Zamboni DS, et al. Mitochondrial DNA Promotes NLRP3 Inflammasome Activation and Contributes to Endothelial Dysfunction and Inflammation in Type 1 Diabetes. Front Physiol (2019) 10:1557. doi: 10.3389/fphys.2019.01557
106. Eissa LD, Ghobashy WA, El-Azab MF. Inhibition of Thioredoxin-Interacting Protein and Inflammasome Assembly Using Verapamil Mitigates Diabetic Retinopathy and Pancreatic Injury. Eur J Pharmacol (2021) 901:174061. doi: 10.1016/j.ejphar.2021.174061
107. Xu L, Chen R, Zhang X, Zhu Y, Ma X, Sun G, et al. Scutellarin Protects Against Diabetic Cardiomyopathy via Inhibiting Oxidative Stress and Inflammatory Response in Mice. Ann Palliative Med (2021) 10(3):2481–93. doi: 10.21037/apm-19-516
108. Gao Y, Li J, Chu S, Zhang Z, Chen N, Li L, et al. Ginsenoside Rg1 Protects Mice Against Streptozotocin-Induced Type 1 Diabetic by Modulating the NLRP3 and Keap1/Nrf2/HO-1 Pathways. Eur J Pharmacol (2020) 866:172801. doi: 10.1016/j.ejphar.2019.172801
109. Wu C, Pan L, Niu W, Fang X, Liang W, Li J, et al. Modulation of Gut Microbiota by Low Methoxyl Pectin Attenuates Type 1 Diabetes in Non-Obese Diabetic Mice. Front Immunol (2019) 10:1733. doi: 10.3389/fimmu.2019.01733
110. Jia Li, Zhang Mi, Liu H, Sun J, Pan L. Early-Life Fingolimod Treatment Improves Intestinal Homeostasis and Pancreatic Immune Tolerance in Non-Obese Diabetic Mice. Acta Pharmacologica Sin (2021) 42(10):1620–9. doi: 10.1038/s41401-020-00590-4
111. Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, et al. Self Double-Stranded (Ds)DNA Induces IL-1β Production From Human Monocytes by Activating NLRP3 Inflammasome in the Presence of anti-dsDNA Antibodies. J Immunol (2013) 190:1407–15. doi: 10.4049/jimmunol.1201195
112. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
113. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A Distinct Subset of Proinflammatory Neutrophils Isolated From Patients With Systemic Lupus Erythematosus Induces Vascular Damage and Synthesizes Type I IFNs. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
114. Shin MS, Kang Y, Lee N, Kim SH, Kang KS, Lazova R, et al. U1-Small Nuclear Ribonucleoprotein Activates the NLRP3 Inflammasome in Human Monocytes. J Immunol (2012) 188:4769–75. doi: 10.4049/jimmunol.1103355
115. Zhao J, Wang H, Huang Y, Zhang H, Wang S, Gaskin F, et al. Lupus Nephritis: Glycogen Synthase Kinase 3β Promotion of Renal Damage Through Activation of the NLRP3 Inflammasome in Lupus-Prone Mice. Arthritis Rheumatol (2015) 67:1036–44. doi: 10.1002/art.38993
116. Inokuchi S, Mitoma H, Kawano S, Ayano M, Kimoto Y, Akahoshi M, et al. Activation of Caspase-1 Is Mediated by Stimulation of Interferon Genes and NLR Family Pyrin Domain Containing 3 in Monocytes of Active Systemic Lupus Erythematosus. Clin Exp Rheumatol (2021).
117. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil Extracellular Trap-Associated Protein Activation of the NLRP3 Inflammasome Is Enhanced in Lupus Macrophages. J Immunol (2013) 190:1217–26. doi: 10.4049/jimmunol.1202388
118. Huang W, Wu J, Yang H, Xiong Y, Jiang R, Cui T, et al. Milk Fat Globule-EGF Factor 8 Suppresses the Aberrant Immune Response of Systemic Lupus Erythematosus-Derived Neutrophils and Associated Tissue Damage. Cell Death differentiation (2017) 24(2):263–75. doi: 10.1038/cdd.2016.115
119. Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, et al. Podocyte Activation of NLRP3 Inflammasomes Contributes to the Development of Proteinuria in Lupus Nephritis. Arthritis Rheumatol (2017) 69:1636–46. doi: 10.1002/art.40155
120. Yang S, Hua K, Chu LJ, Hwu YK, Yang S, Wu C, et al. Xenon Blunts NF-κb/NLRP3 Inflammasome Activation and Improves Acute Onset of Accelerated and Severe Lupus Nephritis in Mice. Kidney Int (2020) 98(2):378–90. doi: 10.1016/j.kint.2020.02.033
121. Yang S, Hsu W, Wu C, Shang H, Liu F, Chen A. Accelerated, Severe Lupus Nephritis Benefits From Treatment With Honokiol by Immunoregulation and Differentially Regulating NF-κb/NLRP3 Inflammasome and Sirtuin 1/Autophagy Axis. FASEB Journal: Off Publ Fed Am Societies Exp Biol (2020) 34(10):13284–99. doi: 10.1096/fj.202001326R
122. Wu C, Hua K, Chu C, Yang S, Arbiser JL, Yang S, et al. Tris DBA Ameliorates Accelerated and Severe Lupus Nephritis in Mice by Activating Regulatory T Cells and Autophagy and Inhibiting the NLRP3 Inflammasome. J Immunol (Baltimore Md: 1950) (2020) 204(6):1448–61. doi: 10.4049/jimmunol.1801610
123. Yang S, Hua K, Yang C, Chen A, Weng J, Tsai Y, et al. Cf-02, a Novel Benzamide-Linked Small Molecule, Blunts NF-κb Activation and NLRP3 Inflammasome Assembly and Improves Acute Onset of Accelerated and Severe Lupus Nephritis in Mice. FASEB J (2021) 35(8):e21785. doi: 10.1096/fj.202100047R
124. Tan W, Gu Z, Leng J, Zou X, Chen H, Min F, et al. Let-7f-5p Ameliorates Inflammation by Targeting NLRP3 in Bone Marrow-Derived Mesenchymal Stem Cells in Patients With Systemic Lupus Erythematosus. BioMed Pharmacother (2019) 118:109313. doi: 10.1016/j.biopha.2019.109313
125. Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, et al. Bay11-7082 Attenuates Murine Lupus Nephritis via Inhibiting NLRP3 Inflammasome and NF-κb Activation. Int Immunopharmacol (2013) 17:116–22. doi: 10.1016/j.intimp.2013.05.027
126. Zhao J, Wang J, Zhou M, Li M, Li M, Tan H. Curcumin Attenuates Murine Lupus via Inhibiting NLRP3 Inflammasome. Int Immunopharmacol (2019) 69:213–6. doi: 10.1016/j.intimp.2019.01.046
127. Bonomini F, Dos Santos M, Veronese FV, Rezzani R. NLRP3 Inflammasome Modulation by Melatonin Supplementation in Chronic Pristane-Induced Lupus Nephritis. Int J Mol Sci (2019) 20(14):3466. doi: 10.3390/ijms20143466
128. Su B, Ye H, You X, Ni H, Chen X, Li L. Icariin Alleviates Murine Lupus Nephritis via Inhibiting NF-κb Activation Pathway and NLRP3 Inflammasome. Life Sci (2018) 208:26–32. doi: 10.1016/j.lfs.2018.07.009
129. Peng X, Yang T, Liu G, Liu H, Peng Y, He L. Piperine Ameliorated Lupus Nephritis by Targeting AMPK-Mediated Activation of NLRP3 Inflammasome. Int Immunopharmacol (2018) 65:448–57. doi: 10.1016/j.intimp.2018.10.025
130. Ka S, Lin J, Lin T, Liu F, Chao L, Ho C, et al. Citral Alleviates an Accelerated and Severe Lupus Nephritis Model by Inhibiting the Activation Signal of NLRP3 Inflammasome and Enhancing Nrf2 Activation. Arthritis Res Ther (2015) 17:331. doi: 10.1186/s13075-015-0844-6
131. Nagayama Y. Commentary: Excessive Iodine Promotes Pyroptosis of Thyroid Follicular Epithelial Cells in Hashimoto’s Thyroiditis Through the ROS-NF-κb-NLRP3 Pathway. Front Endocrinol (2020) 11:581. doi: 10.3389/fendo.2020.00581
132. Ma B, Chen D, Liu Y, Zhao Z, Wang J, Zhou G, et al. Yanghe Decoction Suppresses the Experimental Autoimmune Thyroiditis in Rats by Improving NLRP3 Inflammasome and Immune Dysregulation. Front Pharmacol (2021) 12:645354. doi: 10.3389/fphar.2021.645354
133. Goyette P, Labbé C, Trinh TT, Xavier RJ, Rioux JD. Molecular Pathogenesis of Inflammatory Bowel Disease: Genotypes, Phenotypes and Personalized Medicine. Ann Med (2007) 39:177–99. doi: 10.1080/07853890701197615
134. Chen Y, Wu D, Sun L. Clinical Significance of High-Mobility Group Box 1 Protein (HMGB1) and Nod-Like Receptor Protein 3 (NLRP3) in Patients With Ulcerative Colitis. Med Sci Monit (2020) 26:e919530. doi: 10.12659/MSM.919530
135. Lazaridis LD, Pistiki A, Giamarellos-Bourboulis EJ, Georgitsi M, Damoraki G, Polymeros D, et al. Activation of NLRP3 Inflammasome in Inflammatory Bowel Disease: Differences Between Crohn’s Disease and Ulcerative Colitis. Dig Dis Sci (2017) 62:2348–56. doi: 10.1007/s10620-017-4609-8
136. Zhen Y, Zhang H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front Immunol (2019) 10:276. doi: 10.3389/fimmu.2019.00276
137. Hasegawa M, Kamada N, Jiao Y, Liu MZ, Núñez G, Inohara N. Protective Role of Commensals Against Clostridium Difficile Infection via an IL-1β-Mediated Positive-Feedback Loop. J Immunol (2012) 189:3085–91. doi: 10.4049/jimmunol.1200821
138. Alipour M, Lou Y, Zimmerman D, Bording-Jorgensen MW, Sergi C, Liu JJ, et al. A Balanced IL-1β Activity Is Required for Host Response to Citrobacter Rodentium Infection. PloS One (2013) 8:e80656. doi: 10.1371/journal.pone.0080656
139. Su F, Xia Y, Huang M, Zhang L, Chen L. Expression of NLPR3 in Psoriasis Is Associated With Enhancement of Interleukin-1β and Caspase-1. Med Sci Monitor : Int Med J Exp Clin Res (2018) 24:7909–13. doi: 10.12659/MSM.911347
140. Yu P, Hao S, Zheng H, Zhao X, Li Y. Association of NLRP1 and NLRP3 Polymorphisms With Psoriasis Vulgaris Risk in the Chinese Han Population. BioMed Res Int (2018) 2018:4714836. doi: 10.1155/2018/4714836
141. Juneblad K, Kastbom A, Johansson L, Rantapää-Dahlqvist S, Söderkvist P, Alenius G-M. Association Between Inflammasome-Related Polymorphisms and Psoriatic Arthritis. Scandinavian J Rheumatol (2021) 50(3):206–12. doi: 10.1080/03009742.2020.1834611
142. Tang Z, Zhang K, Lv S, Xu G, Zhang J, Jia H. LncRNA MEG3 Suppresses PI3K/AKT/mTOR Signalling Pathway to Enhance Autophagy and Inhibit Inflammation in TNF-α-Treated Keratinocytes and Psoriatic Mice. Cytokine (2021) 148:155657. doi: 10.1016/j.cyto.2021.155657
143. Chang X, He H, Zhu L, Gao J, Wei T, Ma Z, et al. Protective Effect of Apigenin on Freund’s Complete Adjuvant-Induced Arthritis in Rats via Inhibiting P2X7/NF-κb Pathway. Chem Biol Interact (2015) 236:41–6. doi: 10.1016/j.cbi.2015.04.021
144. Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Eyre S, et al. Evidence of NLRP3-Inflammasome Activation in Rheumatoid Arthritis (RA); Genetic Variants Within the NLRP3-Inflammasome Complex in Relation to Susceptibility to RA and Response to Anti-TNF Treatment. Ann Rheum Dis (2014) 73:1202–10. doi: 10.1136/annrheumdis-2013-203276
145. Kastbom A, Ärlestig L, Rantapää-Dahlqvist S. Genetic Variants of the NLRP3 Inflammasome Are Associated With Stroke in Patients With Rheumatoid Arthritis. J Rheumatol (2015) 42:1740–5. doi: 10.3899/jrheum.141529
146. Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, et al. Enhanced Activity of NLRP3 Inflammasome in Peripheral Blood Cells of Patients With Active Rheumatoid Arthritis. Arthritis Res Ther (2015) 17:257. doi: 10.1186/s13075-015-0775-2
147. Kolly L, Busso N, Palmer G, Talabot-Ayer D, Chobaz V, So A. Expression and Function of the NALP3 Inflammasome in Rheumatoid Synovium. Immunology (2010) 129:178–85. doi: 10.1111/j.1365-2567.2009.03174.x
148. Takakubo Y, Barreto G, Konttinen YT, Oki H, Takagi M. Role of Innate Immune Sensors, TLRs, and NALP3 in Rheumatoid Arthritis and Osteoarthritis. J Long Term Eff Med Implants (2014) 24:243–51. doi: 10.1615/jlongtermeffmedimplants.2014011295
149. Kim HW, Kwon YJ, Park BW, Song JJ, Park YB, Park MC. Differential Expressions of NOD-Like Receptors and Their Associations With Inflammatory Responses in Rheumatoid Arthritis. Clin Exp Rheumatol (2017) 35:630–7.
150. Zhao C, Gu Y, Zeng X, Wang J. NLRP3 Inflammasome Regulates Th17 Differentiation in Rheumatoid Arthritis. Clin Immunol (2018) 197:154–60. doi: 10.1016/j.clim.2018.09.007
151. Mouasni S, Gonzalez V, Schmitt A, Bennana E, Guillonneau F, Mistou S, et al. The Classical NLRP3 Inflammasome Controls FADD Unconventional Secretion Through Microvesicle Shedding. Cell Death Dis (2019) 10:190. doi: 10.1038/s41419-019-1412-9
152. Yang Z, Cao J, Yu C, Yang Q, Zhang Y, Han L. Caspase-1 Mediated Interleukin-18 Activation in Neutrophils Promotes the Activity of Rheumatoid Arthritis in a NLRP3 Inflammasome Independent Manner. Joint Bone Spine (2016) 83:282–9. doi: 10.1016/j.jbspin.2015.07.006
153. Martínez-Godínez MA, Cruz-Domínguez MP, Jara LJ, Domínguez-López A, Jarillo-Luna RA, Vera-Lastra O, et al. Expression of NLRP3 Inflammasome, Cytokines and Vascular Mediators in the Skin of Systemic Sclerosis Patients. Israel Med Assoc Journal: IMAJ (2015) 17(1):5–10.
154. O’Connell RM, Kahn D, Gibson WSJ, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 Promotes Autoimmune Inflammation by Enhancing Inflammatory T Cell Development. Immunity (2010) 33:607–19. doi: 10.1016/j.immuni.2010.09.009
155. Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, et al. The microRNA miR-155 Controls CD8(+) T Cell Responses by Regulating Interferon Signaling. Nat Immunol (2013) 14:593–602. doi: 10.1038/ni.2576
156. Bala S, Csak T, Saha B, Zatsiorsky J, Kodys K, Catalano D, et al. The Pro-Inflammatory Effects of miR-155 Promote Liver Fibrosis and Alcohol-Induced Steatohepatitis. J Hepatol (2016) 64(6):1378–87. doi: 10.1016/j.jhep.2016.01.035
157. Zheng L, Xu C, Chen W, Shen W, Ruan C, Zhu L, et al. MicroRNA-155 Regulates Angiotensin II Type 1 Receptor Expression and Phenotypic Differentiation in Vascular Adventitial Fibroblasts. Biochem Biophys Res Commun (2010) 400(4):483–8. doi: 10.1016/j.bbrc.2010.08.067
158. Yang L, Liu J, Bai X, Fan L, Han F, Jia W, et al. Acute Downregulation of miR-155 at Wound Sites Leads to a Reduced Fibrosis Through Attenuating Inflammatory Response. Biochem Biophys Res Commun (2014) 453:153–9. doi: 10.1016/j.bbrc.2014.09.077
159. Pottier N, Maurin T, Chevalier B, Puisségur MP, Lebrigand K, Robbe-Sermesant K, et al. Identification of Keratinocyte Growth Factor as a Target of microRNA-155 in Lung Fibroblasts: Implication in Epithelial-Mesenchymal Interactions. PloS One (2009) 4(8):e6718. doi: 10.1371/journal.pone.0006718
160. Shinohara ML. Yes, Research Matters. PloS Pathog (2017) 13:e1006420. doi: 10.1371/journal.ppat.1006420
161. Patin EC, Thompson A, Orr SJ. Pattern Recognition Receptors in Fungal Immunity. Semin Cell Dev Biol (2019) 89:24–33. doi: 10.1016/j.semcdb.2018.03.003
162. Esposito S, Toni G, Tascini G, Santi E, Berioli MG, Principi N. Environmental Factors Associated With Type 1 Diabetes. Front Endocrinol (Lausanne) (2019) 10:592. doi: 10.3389/fendo.2019.00592
163. Xia Y, Xie Z, Huang G, Zhou Z. Incidence and Trend of Type 1 Diabetes and the Underlying Environmental Determinants. Diabetes Metab Res Rev (2019) 35:e3075. doi: 10.1002/dmrr.3075
164. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 Gene Are Involved in the Predisposition to Type-1 Diabetes and Celiac Disease in a Pediatric Population From Northeast Brazil. Autoimmunity (2010) 43:583–9. doi: 10.3109/08916930903540432
165. Sun X, Xia Y, Liu Y, Wang Y, Luo S, Lin J, et al. Polymorphisms in NLRP1 Gene Are Associated With Type 1 Diabetes. J Diabetes Res (2019) 2019:7405120. doi: 10.1155/2019/7405120
166. Wu Z, Wu S, Liang T. Association of NLRP3 Rs35829419 and Rs10754558 Polymorphisms With Risks of Autoimmune Diseases: A Systematic Review and Meta-Analysis. Front Genet (2021) 12:690860. doi: 10.3389/fgene.2021.690860
167. Smigoc Schweiger D, Goricar K, Hovnik T, Mendez A, Bratina N, Brecelj J, et al. Dual Role of PTPN22 But Not NLRP3 Inflammasome Polymorphisms in Type 1 Diabetes and Celiac Disease in Children. Front Pediatr (2019) 7:63. doi: 10.3389/fped.2019.00063
168. Liu H, Xu R, Kong Q, Liu J, Yu Z, Zhao C. Downregulated NLRP3 and NLRP1 Inflammasomes Signaling Pathways in the Development and Progression of Type 1 Diabetes Mellitus. Biomedicine Pharmacotherapy (2017) 94:619–26. doi: 10.1016/j.biopha.2017.07.102
169. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
170. Hughes FM, Hirshman NA, Inouye BM, Jin H, Stanton EW, Yun CE, et al. NLRP3 Promotes Diabetic Bladder Dysfunction and Changes in Symptom-Specific Bladder Innervation. Diabetes (2019) 68:430–40. doi: 10.2337/db18-0845
171. Zhai Y, Meng X, Ye T, Xie W, Sun G, Sun X. Inhibiting the NLRP3 Inflammasome Activation With MCC950 Ameliorates Diabetic Encephalopathy in Db/Db Mice. Molecules (2018) 23(3):522. doi: 10.3390/molecules23030522
172. Wu C, Pan L, Luo Y, Niu W, Fang X, Liang W, et al. Low Methoxyl Pectin Protects Against Autoimmune Diabetes and Associated Caecal Dysfunction. Mol Nutr Food Res (2019) 63:e1900307. doi: 10.1002/mnfr.201900307
173. Lee YH, Bae SC. Association Between Functional NLRP3 Polymorphisms and Susceptibility to Autoimmune and Inflammatory Diseases: A Meta-Analysis. Lupus (2016) 25:1558–66. doi: 10.1177/0961203316644336
174. Su Z, Niu Q, Huang Z, Yang B, Zhang J. Association of Nucleotide-Binding Oligomerization Domain-Like Receptor Family Pyrin Domain-Containing Protein 3 Polymorphisms With Systemic Lupus Erythematosus Disease Activity and Biomarker Levels: A Case-Control Study in Chinese Population. Med (Baltimore) (2020) 99:e21888. doi: 10.1097/MD.0000000000021888
175. Huang T, Yin H, Ning W, Wang X, Chen C, Lin W, et al. Expression of Inflammasomes NLRP1, NLRP3 and AIM2 in Different Pathologic Classification of Lupus Nephritis. Clin Exp Rheumatol (2020) 38:680–90.
176. Yang C, Huang S, Chiang B. Sex-Dependent Differential Activation of NLRP3 and AIM2 Inflammasomes in SLE Macrophages. Rheumatol (Oxford) (2015) 54:324–31. doi: 10.1093/rheumatology/keu318
177. Brinkmann V, Zychlinsky A. Beneficial Suicide: Why Neutrophils Die to Make NETs. Nat Rev Microbiol (2007) 5:577–82. doi: 10.1038/nrmicro1710
178. Pavenstädt H, Kriz W, Kretzler M. Cell Biology of the Glomerular Podocyte. Physiol Rev (2003) 83:253–307. doi: 10.1152/physrev.00020.2002
179. Banas MC, Banas B, Hudkins KL, Wietecha TA, Iyoda M, Bock E, et al. TLR4 Links Podocytes With the Innate Immune System to Mediate Glomerular Injury. J Am Soc Nephrol (2008) 19:704–13. doi: 10.1681/ASN.2007040395
180. Denny MF, Thacker S, Mehta H, Somers EC, Dodick T, Barrat FJ, et al. Interferon-Alpha Promotes Abnormal Vasculogenesis in Lupus: A Potential Pathway for Premature Atherosclerosis. Blood (2007) 110:2907–15. doi: 10.1182/blood-2007-05-089086
181. Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome Activation of IL-18 Results in Endothelial Progenitor Cell Dysfunction in Systemic Lupus Erythematosus. J Immunol (2011) 187:6143–56. doi: 10.4049/jimmunol.1101284
182. Lech M, Lorenz G, Kulkarni OP, Grosser MOO, Stigrot N, Darisipudi MN, et al. NLRP3 and ASC Suppress Lupus-Like Autoimmunity by Driving the Immunosuppressive Effects of TGF-β Receptor Signalling. Ann Rheum Dis (2015) 74:2224–35. doi: 10.1136/annrheumdis-2014-205496
183. Thygesen SJ, Takizawa KE, Robertson AAB, Sester DP, Stacey KJ. Compromised NLRP3 and AIM2 Inflammasome Function in Autoimmune NZB/W F1 Mouse Macrophages. Immunol Cell Biol (2019) 97:17–28. doi: 10.1111/imcb.12193
184. Lu A, Li H, Niu J, Wu S, Xue G, Yao X, et al. Hyperactivation of the NLRP3 Inflammasome in Myeloid Cells Leads to Severe Organ Damage in Experimental Lupus. J Immunol (2017) 198:1119–29. doi: 10.4049/jimmunol.1600659
185. Guo Q, Wu Y, Hou Y, Liu Y, Liu T, Zhang H, et al. Cytokine Secretion and Pyroptosis of Thyroid Follicular Cells Mediated by Enhanced NLRP3, NLRP1, NLRC4, and AIM2 Inflammasomes Are Associated With Autoimmune Thyroiditis. Front Immunol (2018) 9:1197. doi: 10.3389/fimmu.2018.01197
186. Liu J, Mao C, Dong L, Kang P, Ding C, Zheng T, et al. Excessive Iodine Promotes Pyroptosis of Thyroid Follicular Epithelial Cells in Hashimoto’s Thyroiditis Through the ROS-NF-κb-NLRP3 Pathway. Front Endocrinol (2019) 10:778. doi: 10.3389/fendo.2019.00778
187. Liu G, Mateer S, Hsu A, Goggins B, Tay H, Mathe A, et al. Platelet Activating Factor Receptor Regulates Colitis-Induced Pulmonary Inflammation Through the NLRP3 Inflammasome. Mucosal Immunol (2019) 12(4):862–73. doi: 10.1038/s41385-019-0163-3
188. Ranson N, Veldhuis M, Mitchell B, Fanning S, Cook AL, Kunde D, et al. NLRP3-Dependent and -Independent Processing of Interleukin (IL)-1β in Active Ulcerative Colitis. Int J Mol Sci (2018) 20(1):57. doi: 10.3390/ijms20010057
189. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 Deficiency Protects From Type 1 Diabetes Through the Regulation of Chemotaxis Into the Pancreatic Islets. Proc Natl Acad Sci USA (2015) 112:11318–23. doi: 10.1073/pnas.1513509112
190. Lewis KL, Reizis B. Dendritic Cells: Arbiters of Immunity and Immunological Tolerance. Cold Spring Harb Perspect Biol (2012) 4(8):a007401. doi: 10.1101/cshperspect.a007401
Keywords: NLRP3, autoimmune diseases, inflammatory bowel disease, rheumatoid arthritis, type 1 diabetes, systemic lupus erythematosus
Citation: Zhang Y, Yang W, Li W and Zhao Y (2021) NLRP3 Inflammasome: Checkpoint Connecting Innate and Adaptive Immunity in Autoimmune Diseases. Front. Immunol. 12:732933. doi: 10.3389/fimmu.2021.732933
Received: 29 June 2021; Accepted: 20 September 2021;
Published: 11 October 2021.
Edited by:
Carlo Perricone, Sapienza University of Rome, ItalyReviewed by:
Ming-Kuei Shih, National Kaohsiung University of Hospitality and Tourism, TaiwanCopyright © 2021 Zhang, Yang, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunjuan Zhao, eW91Z3VrZWRyZWFtQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.