 Tharmarajan Ramprasath
Tharmarajan Ramprasath Young-Min Han
Young-Min Han Donghong Zhang
Donghong Zhang Chang-Jiang Yu
Chang-Jiang Yu Ming-Hui Zou
Ming-Hui Zou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 September 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.731701
This article is part of the Research Topic Multiple Implications of the Kynurenine Pathway in Inflammatory Diseases: Diagnostic and Therapeutic Applications View all 14 articles
Aortic diseases are the primary public health concern. As asymptomatic diseases, abdominal aortic aneurysm (AAA) and atherosclerosis are associated with high morbidity and mortality. The inflammatory process constitutes an essential part of a pathogenic cascade of aortic diseases, including atherosclerosis and aortic aneurysms. Inflammation on various vascular beds, including endothelium, smooth muscle cell proliferation and migration, and inflammatory cell infiltration (monocytes, macrophages, neutrophils, etc.), play critical roles in the initiation and progression of aortic diseases. The tryptophan (Trp) metabolism or kynurenine pathway (KP) is the primary way of degrading Trp in most mammalian cells, disturbed by cytokines under various stress. KP generates several bioactive catabolites, such as kynurenine (Kyn), kynurenic acid (KA), 3-hydroxykynurenine (3-HK), etc. Depends on the cell types, these metabolites can elicit both hyper- and anti-inflammatory effects. Accumulating evidence obtained from various animal disease models indicates that KP contributes to the inflammatory process during the development of vascular disease, notably atherosclerosis and aneurysm development. This review outlines current insights into how perturbed Trp metabolism instigates aortic inflammation and aortic disease phenotypes. We also briefly highlight how targeting Trp metabolic pathways should be considered for treating aortic diseases.
Aortic diseases are the primary public health concern caused by age, genetics, diabetes, obesity, sedentary lifestyle, infection and injury. A slow and gradual thickening of the arteries, otherwise called atherosclerosis, is the common cause of cardiovascular diseases. Further, human arteries become less flexible with increased ages, leading to aortic stiffness or a partially dilated artery called aneurysm. Being asymptomatic, abdominal aortic aneurysm (AAA) is a common and potentially life-threatening condition as it may lead to rupture. However, elective aortic surgery is also associated with risks; elective repair of the aneurysm is the only way to prevent rupture. Thus, this condition requires improved pharmacologic interventions, which lacks in this modern medical system.
Atherosclerosis and AAAs are multifactorial and polygenic diseases with known environmental and genetic risk factors contributing to disease development (1, 2). Atherosclerosis is a chronic progressive inflammatory disorder that presents with coronary artery disease (CAD) (3). CAD accounts for approximately 610,000 deaths annually (estimated 1 in 4 deaths) and is the leading cause of mortality in the United States (3). AAAs are majorly caused by aging, hypertension, nicotine usage and atherosclerosis (4). Traditionally plenty of evidence showed atherosclerosis as a common etiology for thoracic aortic aneurysms (TAAs) and AAAs. AAA is a focal progressive dilatation of the aorta with a diameter of at least 50% greater than the average proximal diameter due to irreversible structural aortic wall integrity loss. It is one of the significant causes of worldwide morbidity and mortality that affects>1 million people in the United States alone (5). According to CDC, AAAs were the cause of 9,923 deaths in 2018 in the United States (6), and the mortality rate associated with AAA rupture is 88%. Given the high mortality and morbidity related to ruptured AAAs, this disease has traditionally posed a heavy burden on healthcare systems (7), and there have been only modest improvements in mortality over the last three decades. The pathogenesis of AAA includes endothelial cell (EC) dysfunction and vascular smooth muscle cells (VSMC) apoptosis/senescence. Endothelial and VSMCs dysfunction both can contribute to atherogenesis, which is widely accepted (8, 9). A variety of anti-inflammatory, antioxidant, beta-blockers and hemodynamic modulator drugs and matrix metalloproteinase (MMP) inhibitors are being studied to slow aneurysm growth (9). However, there are no pharmacological treatments available to either prevent or reverse the development of AAA.
Our increasing knowledge suggests that inflammatory processes are involved in the pathogenesis of aortic diseases (10). An imbalance between the production and release of proinflammatory factors has been reported in AAA’s pathology (11) and atherosclerosis progression (12). The native and adaptive immune responses initiate and propagate the inflammatory response to AAA pathology (13). During the development of AAA, infiltration of many exogenous immune cells, including lymphocytes, macrophages, mast cells, neutrophils, and natural killer cells infiltrate gradually into the tissue from adventitia to the intima, elicit a continuous inflammatory response (10). The massive inflammatory cells infiltration was interpreted in human aortic aneurysm surgical samples. These infiltrations are usually absent among the healthy aortic specimens. In AAA tissues, the B lymphocytes, T lymphocytes and macrophages were majorly characterized cell populations. Whereas the mast cells and natural killer cells were characterized as minor cell populations in these AAA tissues (14). These infiltrated Th1 mononuclear cells secrete the cytokines such as IL-2, IFNγ, and TNFα, to stimulate proinflammatory osteopontin secretion from macrophages that can propagate the inflammatory response during the AAA development (15). Besides the immune cell infiltration, factors released from dysfunctional perivascular aortic tissue (PVAT), including several cytokines and adipokines, could also contribute to arterial remodeling via immune activation.
L-Tryptophan (Trp) is an essential amino acid that should be obtained from dietary intakes such as vegetal (potatoes, chickpeas, soybeans, cocoa beans, and nuts) and animal origin (dairy products, eggs, meat, and seafood) (16, 17). The tryptophan is so crucial for protein synthesis and thus it is required for normal cellular homeostasis. It also serves as an in vivo precursor for several bioactive compounds, including nicotinamide (vitamin B6), serotonin, melatonin, tryptamine, and kynurenines (18). Hepatic tryptophan-2,3-dioxygenase (TDO) is known to play a critical role in keeping the physiological concentrations of Trp and kynurenine (Kyn) at a controlled level via kynurenine pathway (KP). In humans’ serum concentrations of Trp are in the range of 70 ± 10 µmol/L for males and 65 ± 10 µmol/L for females and Kyn concentrations are around 1.8 ± 0.4 µM and do not differ between genders (19). Considering the KP metabolisms and their significant association to many biological activities, the perturbations in the KP have been linked to several diseases.
Two significant pathways that process Trp into other metabolites are serotonin and kynurenine pathways. Most dietary Trp (>95%) is fed into the KP, giving rise to several downstream metabolites (19, 20). The absolute and relative concentrations of kynurenines vary among different cell types due to different enzymatic repertoires (19). These Trp catabolites are activated in times of stress and inflammation (21). Three important rate-limiting enzymes indoleamine 2, 3-dioxygenase 1 and 2 (IDO1 and IDO2), and TDO utilize Trp as a substrate and generate N-formylkynurenine during the initial steps on Trp catabolism. This N-formylkynurenine is rapidly metabolized by formamidase into l-kynurenine (Kyn). Kyn is further catabolized into several potent metabolites such as 3-hydroxykynurenine (3-HK), 3-hydroxyanthranilic acid (3-HAA), kynurenic acid (KA), and xanthurenic acid (XA), quinolinic acid (QA), and produce the essential pyridine nucleotide end product, nicotinamide adenine dinucleotide+ (NAD+) (22). In brief, IDO produced Kyn further catabolized by kynureninase (Kynu) produces anthranilic acid (AA). Kynurenine-3-monooxygenase (KMO) also converts Kyn into 3-HK, which is further utilized by kynurenine aminotransferase (KAT) to produce XA or by the Kynu to form 3-HAA. Further, 3-HAA is converted into quinolinic acid (QA) or picolinic acid (PA) by a series of enzymatic conversions. In addition, KAT metabolizes Kyn into KA as well (Figure 1).
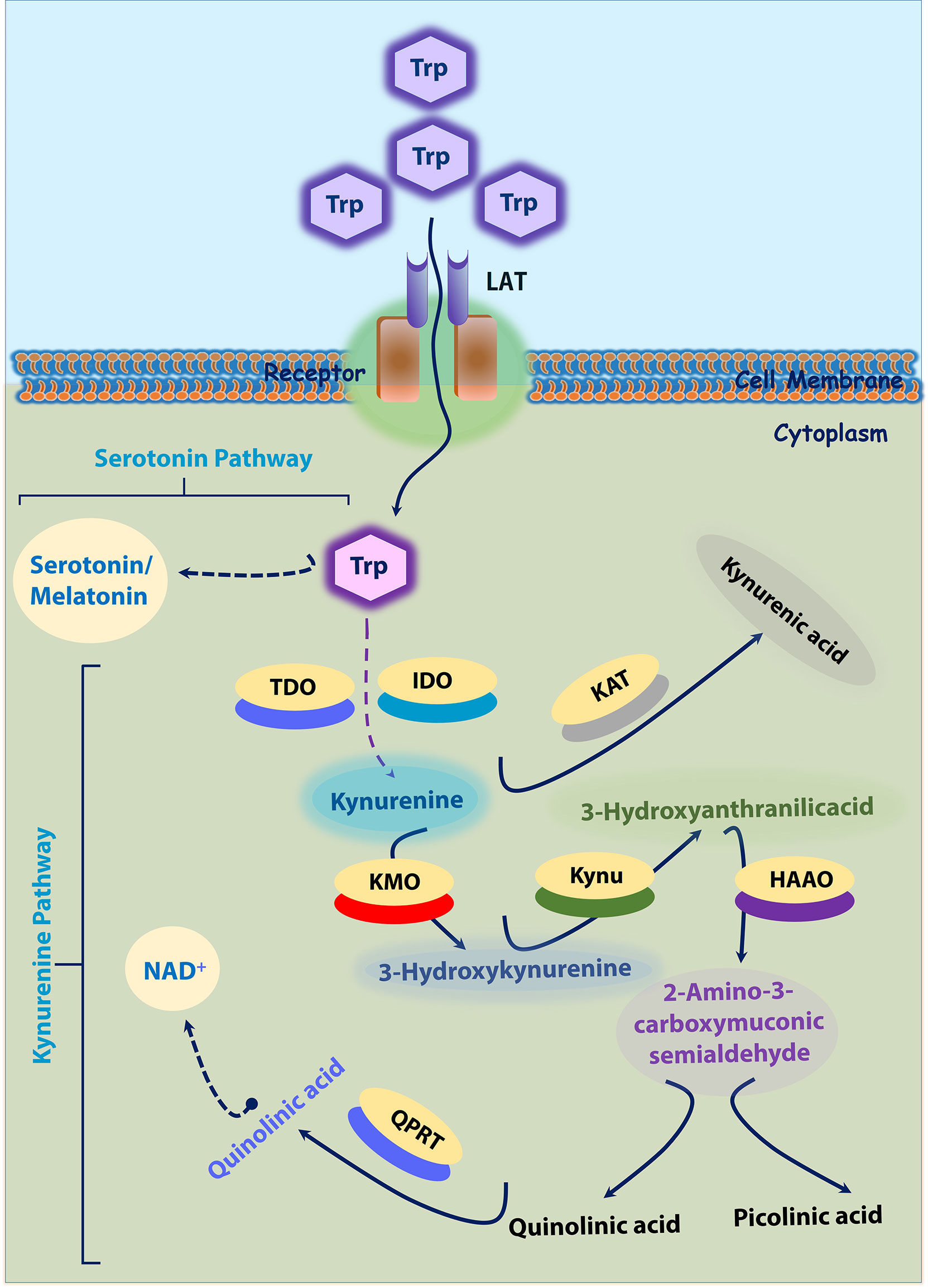
Figure 1 Tryptophan-Kynurenine and Serotonin Pathways. ~95% Trp is transported into cytoplasm by LAT. In cytoplasm, tryptophan is initially converted into kynurenine; Kynurenine into 3-Hydroxykynurenine by KMO; 3-Hydroxykynurenine into 3-Hydroxyanthranilicacid and further quinolinic acid or picolinic acid. In another axis, kynurenine is converted into kynurenic acid by KAT. In another hand, tryptophan (~5%) is converted into serotonin by serotonin pathway. 3-HK, 3-hydroxykynurenine; HAAO -3, hydroxyanthranilic acid dioxygenase; IDO, Indoleamine 2, 3-dioxygenase; KAT, kynurenine amino transferase; KAT, kynurenine amino transferase; KMO, kynurenine monooxygenase; Kynu, kynureninase; LAT, L-amino acid transporters; PA, picolinic acid; QA, quinolinic acid; QPRT, Quinolinate phosphoribosyl transferase; TDO, tryptophan-2,3-dioxygenase; Trp-tryptophan.
In hepatocytes TDO expression is stimulated by glucocorticoids. In contrast, the IDO1 present outside the liver is stimulated by proinflammatory cytokines (21). In the majority of cell types, IDO expression is induced by proinflammatory modulators, such as lipopolysaccharide (LPS), tumor necrosis factor-α (TNFα), interleukin 1 (IL-1), and IL-2 (23, 24). IDO isoforms, (IDO1 and IDO2) closely linked on chromosome 8 in humans, probably originating from an ancient gene duplication (23, 24). IDO1 is a heme-containing enzyme that catabolizes compounds containing indole rings, such as the essential amino acid Trp. The IDO1 isoform is expressed in various tissues, including dendritic cells, endothelial cells, macrophages, fibroblasts, and mesenchymal stromal cells, all are present in the arterial wall. This major isoform contributes to Trp degradation (25, 26). Transport of the amino acid l-tryptophan across the plasma membrane is known to occur through brush border L-amino acid transporters (LATs) (27). A well-known inducer interferon-gamma (IFN-γ) released from activated CD4+ T cells, robustly induces IDO1 expression contributing to Trp catabolism. IFN-γ coordinately induces LATs to maximize tryptophan depletion in IDO1-expressing cells and that the process involves a positive feedback mechanism via kynurenine-aryl hydrocarbon receptor (AhR) signaling (27). The IDO2 isoform is primarily expressed in the kidney, brain, colon, liver, and reproductive tract (25). Although the role of IDO1 is widely studied, the function of IDO2 is largely unexplored. Despite IDO1 and IDO2 exhibit critical functional differences, IDO2 was characterized as having a weaker catalytic activity than IDO1 in vitro (24). All these data strongly suggest the importance of the tissue-specific expression and localization of kynurenines producing proteins, which might regulate many signaling pathways and the body’s physiological status.
The altered amino acid metabolism and their metabolites were observed in the plasma of patients with AAA. Untargeted metabolic profiling of plasma showed statistically increased concentrations of amino acid metabolites in the plasma of people with large aneurysms when compared to the control population. Thus, beyond contributing to protein synthesis, amino acid metabolism plays a critical role in supporting various cell functions (28–30), which is positively as well as negatively correlated to vascular disease development. Among many amino acids, l-Arginine (Arg), l-homoarginine (hArg), and l-tryptophan (Trp) are important amino acids, and their metabolites have a putative role in determining cardiovascular diseases (31). For example, L-arginine, an essential amino acid, improves endothelial function and cardiovascular health (32). It is also known to alter inflammatory functions, and arginine supplementation has wound healing potential by reducing inflammation (33, 34).
Lines of evidence suggest that IDO1 and the Kyn pathway significantly contribute to cardiovascular diseases and thrombus formation. The incidence, development, and progression of vascular diseases are associated with body metabolism in general. Very importantly, accumulating evidence shows that Trp has a significant contribution to determine the AAA development. Trp metabolism, otherwise known as KP, is dysregulated during vascular inflammation and many cardiovascular diseases. A study conducted with the young Finns population showed IDO enzyme’s involvement in the immune regulation of early atherosclerosis (35). A significant positive correlation of IDO activity in serum was observed among the patients with more advanced atherosclerosis, which suggest that activated KP may play a crucial role in vascular diseases (36). We previously showed that angiotensin II (AngII) infusion activates IFNγ in immune cells, which induces the expression of IDO1 and Kynu and increases 3-HAA production in the plasma and aortas of (Apolipoprotein E) Apoe-/- mice, but not in Apoe-/- IDO-/- mice (37). Silencing of Kynu reduces the production of 3-HAA and further limits the production of matrix metallopeptidase-2 (MMP2) in SMCs, resulting in reduced AAA formation in Apoe-/- mice (37). We also showed that AngII triggers the conversion of Trp to the following product, 3-HK and activates the generation of NAD(P)H oxidase (NOX)–mediated superoxide anions in endothelial cells. The superoxide could accelerate the apoptotic process in endothelial cells, leading to endothelial dysfunction (38). Kyn also intensifies certain MMPs via the MEK-Erk1/2 signaling pathway (39), which is important in vascular disease development (40). Measurement of Trp degradation and the product/substrate ratio (Kyn or 3-HK or 3-HAA/Trp) will contribute to a better understanding of the interplay between inflammation and vascular diseases (22). As above discussed, the Trp metabolism is modulated by many risk factors during vascular disease developments, which will be discussed with more evidence in the following sections.
Cytokines are crucial mediators of inflammation and essential regulators of various immune and nonimmune cells in the aortic wall. The expression of IDO2 is basal, whereas that of IDO1, Kynu, etc, are induced by cytokines. A well-known cytokine is IFNγ, the most potent modulator of KP in vitro and in vivo models and humans (41). Activated IDO along with activated inflammatory parameters like IFNγ have a positive correlation with systemic chronic low-grade inflammation. However, LPS is not a strong inducer, it is also known to induce IDO. These findings reveal a direct link between the regulation of the KP and inflammation under aortic disease conditions. However, the functional contributions of secreted kynurenines by other cell types including neutrophils, monocyte/macrophages, mast cells, adipocytes, and platelets, remained to be determined. The following sections will provide some evidence on the role of these cells in aortic diseases and prove how kynurenine metabolism affects the inflammatory process (Table 1).

Table 1 Kynurenine metabolic members associated with aortic phenotype.
Kyn was shown to be a proinflammatory metabolite. The increased Kyn was accompanied by the Nod-like receptor protein 2 (NLPR2) inflammasome expression and activation (48). This was also evidenced by increased caspase-1 expression and IL-1β release. After Kyn treatment, nuclear factor kappa-B (NF-κB) could translocate into the nucleus and binds to the promoter of NLRP2, subsequently increased NLRP2 transcription in vitro (48). To examine the IDO1 associated transcription, a comparative transcriptome analysis was performed between Ido1-/- and Ido1+/+ rodent colon samples. Transcriptome analyses revealed that absence of IDO1 significantly down-regulated the pathways involving TLR and NF-kB signalings. Furthermore, dramatic changes in TLR and NF-kB signaling resulted in substantial changes in the expression of many inflammatory cytokines and chemokines (49). Similarly, 3-HK, and 3HAA both were reported to activate NF-kB signaling and mediate the EC apoptosis and SMC senescence respectively (37, 38, 50).
Many of the other kynurenines were also reported to modulate AhR, both at transcriptional as well at activity levels. AhR activation can influence inflammation and gene transcription through cross-regulation of many inflammatory signaling pathways. AhR activation was associated with activation of Toll-like receptor 2 (TLR2) and its downstream of NF-κB and the MAPKs, signaling pathways. Further, AhR activation also promotes phosphorylation of p65/NF-κB, JNK/MAPK, p38/MAPK, and ERK/MAPK pathways, which could further promote production of pro-inflammatory mediators including interleukin- 1β (IL-1β) and interleukin- 6 (IL-6) (51). Taken together these results demonstrate that the kynurenines including Kyn, 3-HK, and 3HAA (37, 38) are the molecular regulators of inflammation that can influence vascular inflammation.
Numerous studies demonstrated the crucial roles of inflammatory cells, including macrophages, dendritic cells and neutrophils for their contribution to the development of AAA (52). Aneurysm formation is associated with an accumulation of macrophages within the adventitia and the media. Monocytes/macrophages secrete TNFα, IFNγ and IL-6 inflammatory cytokines in the media and adventitia of aneurysmatic vessels (53). Under certain conditions, activated inflammatory macrophages express IDO and actively deplete their own Trp supply. In human macrophages and microglia cells, IFN-γ enhances the expression and activity of KMO (50, 54). A robust increase in KMO expression is associated with high levels of TNF-α and IL-6 following a systemic inflammatory challenge (55).
Moreover, accumulating evidence indicates that DCs can also induce tolerance, rather than immune activation, to the antigens they present. Similar to promoting immunity, promoting tolerance requires integrating information that DCs gather from the innate and adaptive immune systems. Plasmacytoid dendritic cells (pDCs) can produce type I interferons, such as IFNα and IFNβ, to promote proinflammatory responses by activating effector T cell, cytotoxic T cells, and NK cells and can further facilitate AAA development (52, 56, 57). Dendritic cells (DCs) respond actively to tolerogenic signals, such as transforming growth factor-β (TGF-β), which regulates Trp metabolism. Staining of the aneurysm aortas with a marker of activated DCs, CD83, showed rare CD83 cells located at the adventitial/medial border, whereas those cells were not found in control aortas (58). DCs have been shown to mediate immunoregulation contributed by Trp catabolism. A study conducted by Braidy et al. demonstrated the KP activation in human monocyte-derived DCs (MDDCs) compared to the human primary macrophages using mRNA expression assays, high-performance liquid chromatography, mass spectrometry, and immunocytochemistry. Following activation of the KP using IFNγ, MDDCs can mediate apoptosis of Th cells in vitro (59). KAT, kynurenine 3‐hydroxylase, and 3‐hydroxyanthranilic acid dioxygenase (HAAO) appeared to be constitutively expressed in murine macrophages. Whereas the kynurenine 3‐hydroxylase and Kynu activity alone need IFN‐γ stimulation for their expression (60). IDO1 has been suggested to play a protective role in atherosclerosis due to its potential immunomodulatory effect (61). IDO1 is expressed in human atherosclerosis where it co-localizes with macrophages (46). In the murine systems, the absence of IDO1 shown to protect against atherosclerosis. Thus, Metghalchi et al. addressed the direct role of IDO1 in the modulation of immuno-inflammatory responses and its potential impact on the development of atherosclerosis. They also showed IDO1 expression co-localized with macrophages and SMCs in the aortic sinus of low-density lipoprotein receptor knock out (Ldlr−/−) mice (62). On the other hand, high fat diet (HFD) dramatically increases IDO activity in macrophages and VSMC of aortic sinus and circulating levels of KA and QA in atherosclerosis-prone Ldlr−/− mice compared with the chow diet. A marginal increase of transcriptional expression of IDO1 and increased protein levels were observed in peritoneal macrophages after the LPS challenge (63). These results indicate that Kyn and 3-HAA produced by macrophages are independently associated with vascular inflammation, suggesting a connection between macrophage produced Kynu and arterial remodeling.
Peripheral blood of aortic dissection patients showed a significant reduction in total lymphocytes, T lymphocytes, and T helper fractions, with a substantial increase in neutrophils (64) that shows neutrophils must have a critical role in aortic pathogenesis. During the AAA progression period, initially neutrophils stimulate a network of immune cell types that together can direct a chronic pathological response (65). Activated neutrophils form neutrophil extracellular traps (NETs), propagating the inflammatory reactions and culminating in eventual AAA (56). Neutrophils are also known to secrete ECM-degrading collagenases such as MMP-8 and certain proteases (66). In angiotensin II-lysyl oxidase inhibitor (β-Aminopropionitrile monofumarate; BAPN)–preconditioned aortic dissection model mice, adventitial neutrophil recruitment and activation were detected. Furthermore, it was confirmed that neutrophil-derived IL-6 enhances the adventitial inflammation, leading to aortic rupture (67). Besides, kynurenines such as 3HAA and 3HK are toxic and can trigger apoptosis in certain cell types (37, 38). Thus, it could be possible that immune infiltrates are present in the aortas of patients with medial degeneration could contribute to the local expression of death-promoting mediators in the diseased aortas. Accumulating data also shows that these MMPs secretion in the AAA wall (68) are controlled by inflammatory kynurenines (37, 38). MMPs were thought to be secreted essentially by only mesenchymal and monocyte/macrophage lineages. However, the neutrophil has now been recognized as a significant cell type that secretes these enzymes (67). Neutrophils are thus the vital source of MMP-2 and MMP-9, two matrix-degrading enzymes known to be critical in the formation of AAA by regulating KP metabolism.
In many ways, endothelial and smooth muscle cell functions are linked to the health of the aorta (69). The widespread mechanisms link endothelial functions and aortic phenotypes are metalloproteinases and collagenase activation, collagen production and lysis, median and adventitial degradation, elastin lysis, and hypertension (70). In addition, endothelial cells respond to several stimulating factors, including smoking, hypertension and AT1 receptor stimulation and non-uniform distribution of the aortic wall (70). Besides, vascular smooth cells transformation and apoptosis also play a critical role in determining aortic health. The elaboration of cytokines, such as IL-2, IFNγ, and TNFα by a predominantly Th1 mononuclear response, stimulates proinflammatory osteopontin secretion from macrophages and vascular smooth muscle cells that further propagate the inflammatory response (15). Thus, the vascular phenotype majorly determined by the function and transformation of vascular cells. Despite the very first product of the kynurenine pathway, Kyn is a potential contributor to vessel relaxation; the other products are being studied for their involvement in vascular pathogenesis. Notable reports from our lab demonstrated the participation of kynurenine metabolism on endothelial dysfunction and aneurysm (37, 38) (Table 1).
Endothelial dysfunction and endothelial apoptosis are important factors in many aortic diseases’ pathogenesis including aortic aneurysm. Our group demonstrated the vasoactive peptide Ang II to induce vascular contractility, EC apoptosis, and dysfunction by mediating the activation of oxidative stress. We found that Trp catabolite, 3-HK mediates Ang II-induced EC apoptosis and subsequently endothelial dysfunction via the activation of NOX-derived superoxide anions in vivo. We further demonstrated that Ido1 silencing could block the effect of Ang II action on endothelium, resulting in normal endothelial function (38). Wang et al., showed that the metabolism of tryptophan to kynurenine by IDO expressed in endothelial cells contributes to arterial vessel relaxation and blood pressure control (45). Sepiapterin reductase (SPR), which is one of the crucial enzymes involves in the de novo synthesis of tetrahydrobiopterin (BH4) (42). This BH4 acts as a critical regulator of endothelial nitric oxide synthase (eNOS) function and suggests that BH4 is a rational therapeutic target in vascular disease states, particularly for hypertension (71). Several findings confirmed a causal link between eNOS uncoupling and BH4 deficiency in AAA formation (72–74). SPR activity was reported to be inhibited by XA one of the KP metabolites (43), which indicates that elevated XA arising out of upregulated KP could attenuate BH4 biosynthesis and consequently EC dysfunction. On the other hand, reduced bioavailability of the BH4 also leads to dysregulated eNOS that could increase superoxide production, which reacts with nitric oxide (NO) to generate peroxynitrite (75). Peroxynitrite consequently can nitrate IDO at Tyr15, Tyr345, and Tyr353, and inactivates IDO (76), which further leads to reduced production of kynurenine.
Some important cytokines like, IFNγ, usually show elevated level either in Ang II-treated mice or AAA patients (74). Studies from our group demonstrated a detrimental role of Kynu produced 3HAA in the pathogenesis of AAA in an AngII-Apoe-/- animal model. Intra-peritoneal injections of 3-HAA for 6 weeks increased the expression and activity of MMP2 in aortas without affecting metabolic parameters. The acute infusion of AngII markedly increased the incidence of AAA in Apoe−/− mice, but not in Apoe−/−IDO−/− mice, which presented decreased elastic lamina degradation and aortic expansion. Findings from another group also showed an enhanced survival of VSMC when Ido1 is silenced in murine model systems fed with HFD and either after infusion of AngII (dissecting AAA) or after topical peri-aortic elastase (non-dissecting AAA) (77). Mechanistically, 3HAA exposure in SMC mediates the NF-κB activation and further instigates the MMP2 upregulation (Figure 2). Hence, IDO1 deficiency can mitigate MMP2 upregulation in AAA model mice.
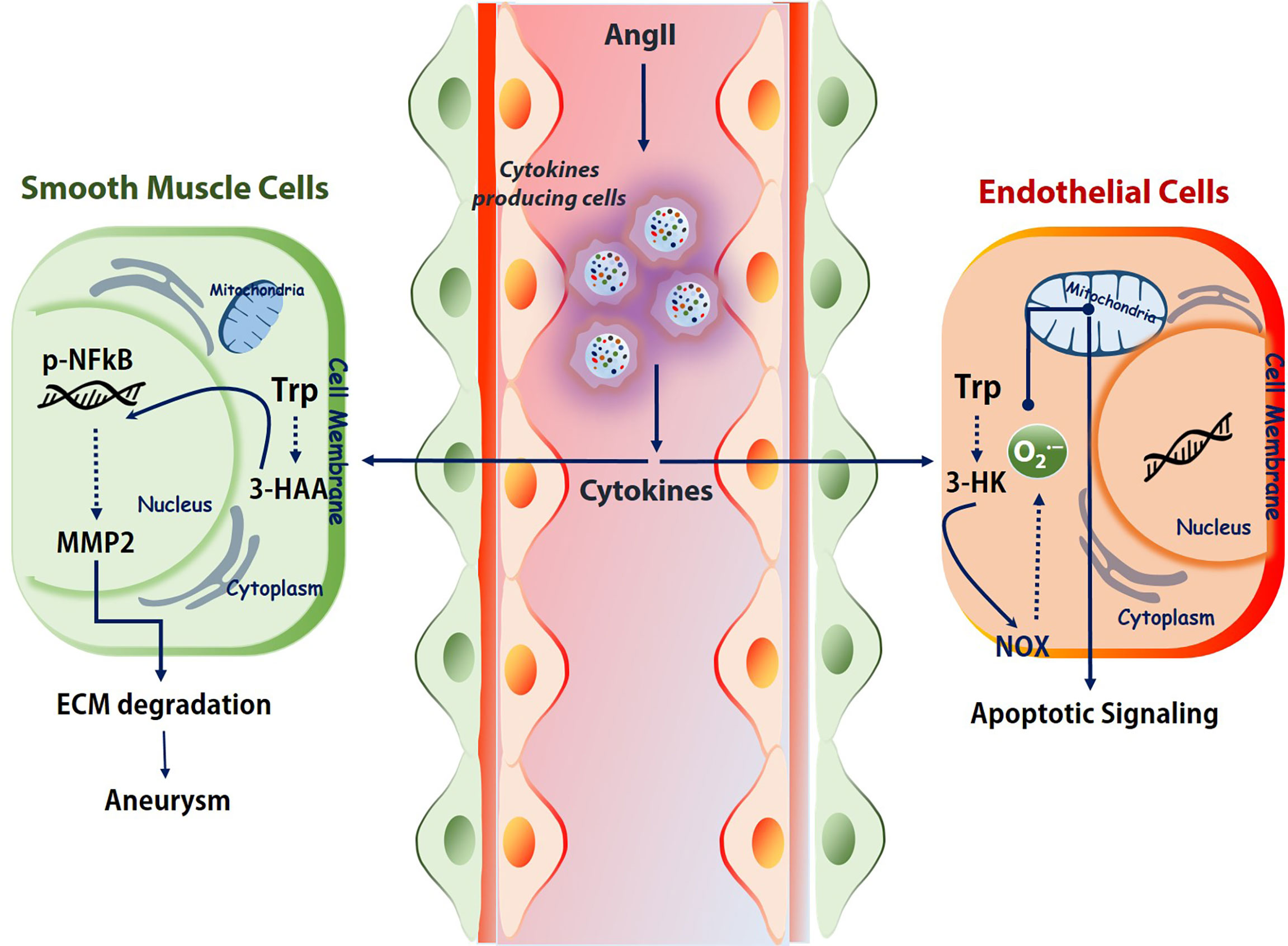
Figure 2 Kynurenines association to vascular diseases. Immune cells under challenged conditions release various cytokines (IFNγ, TGF-β, etc.) to regulate the activation and expression of the kynurenine pathway. Depends on the cell types and milieu, the activated KP effects differently. For example, in SMCs, 3-HAA triggers NF-κB and MMPs, which further degrades ECM. In endothelial cells, tryptophan catabolite 3-HK activates the NOX and produces superoxide. This superoxide further shoots up the apoptotic signaling. Trp, tryptophan; 3-HAA, 3-hydroxyanthranilic acid; AngII, angiotensin II; ECM, extracellular matrix; NOX, NADPH oxidase. 3-HK-3-hydroxykynurenine.
AAA is also an age-associated disease and the Trp pathway alters during aging (78, 79). Upregulation of KP in aging is due to IDO activation by age-related chronic inflammation (22). One key upstream mechanism that appears to target several pathways with age is kynurenine, a tryptophan metabolite and an endogenous aryl hydrocarbon receptor (AhR) agonist. The AhR signaling pathway has been reported to promote aging phenotypes across species and in different tissues (78, 80). Thus, mitigation of target receptors could prevent the kynurenine-induced increase in senescence-associated β-galactosidase and p21 levels and block aggregation of nuclear H3K9me3 (Histone 3 lysine 9 trimethylation) (79). Cellular senescence has historically been viewed as an irreversible cell-cycle arrest mechanism with complex biological processes such as development, tissue repair, ageing, and age-related disorders like aortic aneurysms. Thus, it is well understood that kynurenine metabolism involves triggering the senescence of vascular cells and targeting and controlling the activation of tryptophan metabolism may limit the development of AAA. Overall, casual relationships were well established between Kyn pathway and the development of AAA. Clinically, determining the Kyn or 3-HAA level at early stages in human patients could suggest that tryptophan-derived metabolites could be used as an early biomarker to identify AAA and atherosclerosis.
As reported above, many of these inflammatory signaling proteins are essential for cell cycle regulation. Hence, considerable enthusiasm remains for further investigations in this area, as well as it is yet to study using the KP pharmacological modulators for these KP enzyme proteins. Hence, it might be worth exploring the possible impact of modulating KP, which are regulated by cytokines for treating aortic diseases.
Despite, there is no clinical trial was carried out with KP inhibitors to target the vascular diseases, KP activation has been observed in inflammation-related vascular diseases, such as atherosclerosis, AAA, and endothelial dysfunction. Many of the available KP inhibitors are known to inhibit inflammation during in vivo experiments. For example, in vivo experiments using animals have demonstrated that targeting IDO1, KMO, KYNU, and KAT II KP enzymes can regress cardiovascular diseases by reducing inflammation (22). Hence, pharmacological manipulation of the KP enzymes employing the drugs based on structures becomes an attractive drug development area. Thus, we may expect the emergence of kynurenines enzyme based modulators in future. In the following sections, we briefly outline some KP modulators tested at pre-clinical and clinical levels.
A well-known IDO1 inhibitor used clinically is 1-MT (referred to as Indoximod), the first and widely used competitive inhibitor of IDO1. Other notable IDO1 inhibitors are INCB024360 and NLG919 (an imidazoleisoindole derivative). NLG919 a potent direct small molecule IDO1 inhibitor, was tested in clinical trials (81). In another study, navoximod (GDC-0919, NLG-919) intervention in patients with tumor showed transiently decreased plasma kynurenine from baseline levels with kinetics consistent with its half-life (82, 83). TDO is also actively being tested to use as a target for cancer (84). The indole structure (3-(2-(pyridyl)ethenyl)indoles) based TDO inhibitor had proven pharmacokinetic profile and was tested for preclinical evaluation in cancer patients (85). However, it should be taken into consideration that systemic TDO inhibition will result in increased levels of TRP metabolites such as KYN due to increased availability of TRP for IDO1 as observed in the TDO-deficient mice (84). Depends on the environment and cell types Ido1 deficiency as well as IDO1 inhibition, is known to enhance the atherogenesis. However, the IDO1 inhibitor epacadostat has contrasting effects on macrophages, which could reduce the tissue factor (TF). IDO1 expressed in coronary atherosclerotic plaques was reported to contribute to thrombus formation by upregulated expression of TF in activated macrophages. The IDO1 inhibition by epacadostat, significantly reduced the TF expression by reducing the Kyn/Trp ratio and activity, as well as NF-κB (p65) binding activity in activated macrophages. Further, epacadostat could inhibit the aryl hydrocarbon receptor (or binding of Kyn to AhR) and reduced Kyn-induced TF expression in activated macrophages (25). In another way, the inhibition of AHR by its specific inhibitor CH223191 also significantly inhibited Kyn-induced TF expression in activated macrophages (25) (Figure 3). Besides, recent research also indicates that 1-MT induces an increase of KYNA in ex vivo and in vivo. Consistently, IDO−/− mice also showed an increase of KYNA as Ido1 deficiency promotes a shift toward this branch of the kynurenine pathway (KP), which may be one potential mode of action by 1-MT and should be considered for further applications (86). These findings collectively suggest that the IDO1-mediated Kyn pathway plays a significant role in aortic diseases, and this area needs more study to decide pharmacological modulation of Ido1 and TDO enzyme activities to cure vascular inflammation associated diseases.
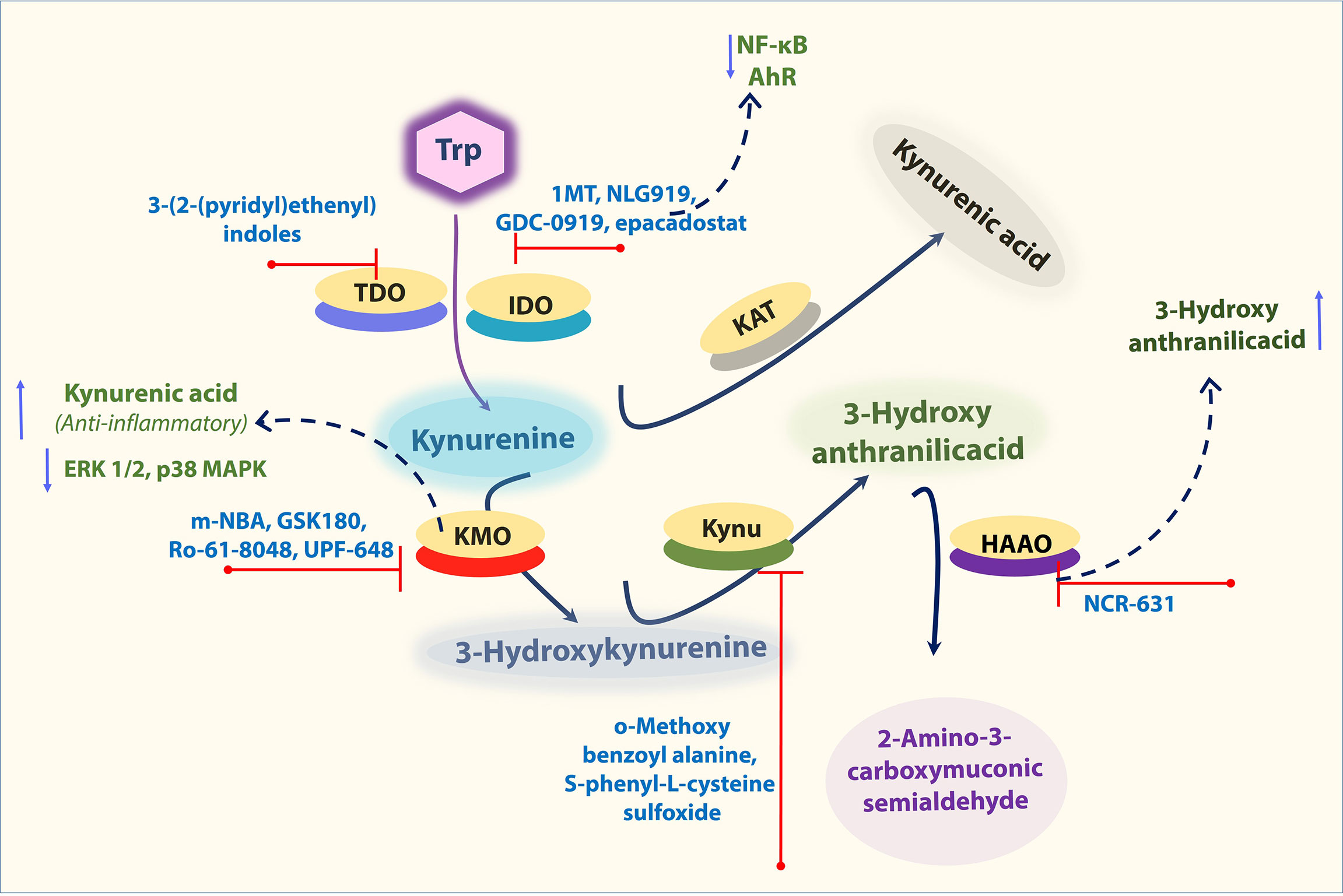
Figure 3 Important KP enzymes and their Inhibitors. Figure explains some KP modulators tested at pre-clinical and clinical levels. Refer to the text for the expanded form of abbreviations.
KMO expression was shown to be upregulated in response to challenging inflammatory conditions (55). Hence, KMO inhibition has been recognized as a potential therapeutic strategy to ameliorate inflammatory diseases in several animal models (87–89). From a therapeutic point of view, KMO is located at a critical branching point in the KP, its activity mediating opposite effects in the levels of 3-HK and QA versus KYNA.
KMO inhibitors based on the structure of the natural substrate KYN is m-Nitrobenzoylalanine (m-NBA), has an IC50 of 0.9 μM against KMO (90). Intraperitoneal administration (400 mg/kg) of this compound in rodents decreased the levels of 3-HK while increasing the levels of KYNA. Thus, inhibition of KMO should shunt the pathway away from the toxic metabolites 3-HK and QA and toward the formation of the protective metabolite KYNA (91). KYNA was assumed to have role as anti-inflammatory. KYNA decreased phosphorylation of extracellular signal-regulated kinases (ERK) 1/2, p38 MAPK, and Akt in colon epithelial cells. Further, it was also found to induce the accumulation of β-catenin. MAPK, PI3K/Akt and β-catenin pathways are well-known targets of GPR signaling (87, 88) (Figure 3). Therefore, it is possible that the observed inhibition of ERK and p38 and the induction of β-catenin accumulation after KYNA treatment are a consequence of GRP35 activation. Interestingly, all of these described effects of KYNA–GPR35 signaling might lead to the suppression or limitation of inflammation (92). Thus, inhibition of KMO can shunt the pathway away from the toxic metabolites 3-HK and QA and toward the formation of the protective metabolite KYNA. Similarly, many other KP enzyme inhibitors including, KMO inhibitors (GSK180, Ro-61-8048, UPF-648), KYNU inhibitors (o-Methoxybenzoyalanine, S-phenyl-L-cysteine sulfoxide), KAT inhibitors (PF-04859989, BFF 122) were also tested for their efficiency to reduce inflammatory diseases (22). In addition, Swainson et al. found that KMO inhibition using CHDI-340246 decreased acute simian immunodeficiency virus infection-induced increases in plasma levels of cytotoxic 3-HK and QA, and improved clinical outcomes as indicated by increased CD4+ T cell count and body weight (93). Despite this, the role of KMO inhibition in CVD is still unclear. Recently, Masanori Nishimura et al. found that both gene and protein expression levels of KMO were upregulated in macrophages in atherosclerotic aneurysmal samples obtained from patients (94). In another experiment, oxazolidinone GSK180 (3-(5,6-dichloro-2-oxobenzo[d]oxa-zol-3(2 H)-yl)propanoic acid; a KMO inhibitor), was shown to prevent extrapancreatic tissue injury to the many organs (88). However, further in vivo experiments using KMO inhibitor or knockout animals will help clarify the effects and potential mechanism of KMO in aortic aneurysms and related aortic diseases.
It was shown that 3-HAA, can modulate vascular inflammation and lipid metabolism. Supplementation of 3HAA reduced the athero formation (44) and blocking of HAAO (that catabolize 3HAA into other derivatives) by NCR-631 increased endogenous levels of 3-HAA, which reduced atherosclerosis (47) (Figure 3). Though many of these inhibitors were validated pre-clinically, their efficiency of inhibition of KP enzymes for vascular diseases and their application in the vascular field is largely unexplored. Understanding the outcome of multiple levels of KP inhibition would help us identify a potential target that would provide us with a choice to cure aortic diseases.
As per our current knowledge, in the United States, out of >200 000 new patients diagnosed for AAA, >40 000 patients are undergoing highly morbid aortic reconstructions. This approach is a catastrophic event associated with near-certain mortality, and no pharmaceutical currently exists to slow aneurysm growth (95). Evidence shows that targeting inflammation in vascular diseases could reduce secondary cardiovascular events (96).
Evidence-based studies confirmed the perturbed tryptophan metabolism and its association with many aortic diseases, particularly atherosclerosis and AAA. AAA most likely associated to the perturbed cytokine levels, which is linked to the disturbed tryptophan metabolism. Cytokines lead generation of kynurenines in immune or vascular cells, further triggers vascular inflammation. This metabolic behavior shows many commonalities to share with other vascular immune disorders. Generally, chronic inflammation drives initial aortic ectasia and dilation, and later, the tension on the aortic wall continues to expand. When wall tension drives sac expansion, no medical intervention will work except surgical aortic reconstruction (95). Hence, we may assume that limiting the inflammation-mediated downstream mechanisms, particularly the kynurenine pathway, emphasizes a further trend and application of these interventions.
Furthermore, as the product to substrate ratio of tryptophan metabolism is an indicator of vascular inflammation, controlling the patients’ Trp metabolic profile could be viable if anyone is diagnosed earlier. This will also allow early identification of patients at risk of vascular diseases that could be crucial to the success of nonsurgical treatment.
TR: Conceived idea and wrote the manuscript. Y-MH, DZ, and C-JY: Revised the manuscript. M-HZ: Conceived idea and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the AHA postdoctoral fellowship award: 19POST34380156 to TR. M-HZ is a recipient of the National Established Investigator Award of the American Heart Association.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Ping Song M.Sc., PhD, for his critical review and comments to improve the manuscript.
3-HAA, 3-hydroxyanthranilic acid; 3-HK, 3-hydroxykynurenine; AA, anthranilic acid; AAA, Abdominal aortic aneurysm; AhR, aryl hydrocarbon receptor; AngII, Angiotensin II; Apoe, Apolipoprotein E; BAPN, β-Aminopropionitrile monofumarate; BH4, tetrahydrobiopterin; CAD, coronary artery disease; EC, endothelial cells; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; H3K9me3, Histone 3 lysine 9 trimethylation; HAAO, 3‐hydroxyanthranilic acid dioxygenase; HFD, high fat diet; IDO, Indoleamine 2, 3-dioxygenase; IFNγ, interferon-gamma; IL-1, interleukin 1; IL-6, interleukin 6; KA, kynurenic acid; KAT, kynurenine aminotransferase; KMO, Kynurenine-3-monooxygenase; KP, Kynurenine pathway; Kyn, kynurenine; Kynu, kynureninase; Ldlr, low-density lipoprotein receptor; LKO, L-selectin-knockout mice; LPS, lipopolysaccharide; MDDCs, monocyte-derived DCs; MMP, Matrix metalloproteinase; m-NBA, Nitrobenzoylalanine; NAD+, Nicotinamide adenine dinucleotide+; NETs, neutrophil extracellular traps; NF-κB, nuclear factor kappa-B; NLPR2, Nod-like receptor protein 2; NO, nitric oxide; NOX, NADPH oxidase; , superoxide; O-MBA, ortho methoxy benzoyl alanine; pDC, Plasmacytoid dendritic cells; PMN, polymorphonuclear neutrophils; PVAT, perivascular aortic tissue; QA, quinolinic acid; QPRT, Quinolinate phosphoribosyl transferase; SPR, Sepiapterin reductase; TAA, Thoracic aortic aneurysm; TDO, tryptophan-2,3-dioxygenase; TGF-β, transforming growth factor-β; TLR2, Toll-like receptor 2; TNFα, tumor necrosis factor-alpha; Trp, Tryptophan; VSMC, vascular smooth muscle cells; XA, xanthurenic acid
1. Toghill BJ, Saratzis A, Bown MJ. Abdominal Aortic Aneurysm-an Independent Disease to Atherosclerosis? Cardiovasc Pathol (2017) 27:71–5. doi: 10.1016/j.carpath.2017.01.008
2. Ramprasath T, Senthil Murugan P, Prabakaran AD, Gomathi P, Rathinavel A, Selvam GS. Potential Risk Modifications of GSTT1, GSTM1 and GSTP1 (Glutathione-S-Transferases) Variants and Their Association to CAD in Patients With Type-2 Diabetes. Biochem Biophys Res Commun (2011) 407:49–53. doi: 10.1016/j.bbrc.2011.02.097
3. Li M, Qian M, Kyler K, Xu J. Endothelial-Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front Cardiovasc Med (2018) 5:151. doi: 10.3389/fcvm.2018.00151
4. Li ZZ, Dai QY. Pathogenesis of Abdominal Aortic Aneurysms: Role of Nicotine and Nicotinic Acetylcholine Receptors. Mediators Inflamm (2012) 2012:103120. doi: 10.1155/2012/103120
5. Abdulameer H, Al Taii H, Al-Kindi SG, Milner R. Epidemiology of Fatal Ruptured Aortic Aneurysms in the United States (1999-2016). J Vasc Surg (2019) 69:378–384 e2. doi: 10.1016/j.jvs.2018.03.435
6. Centers for Disease Control and Prevention. Facts About Aortic Aneurysm in the United States (2020). Available at: https://www.cdc.gov/heartdisease/aortic_aneurysm.htm.
7. Png CYM, Wu J, Tang TY, Png IPL, Sheng TJ, Choke E. Decrease in Mortality From Abdominal Aortic Aneurysms (2001 to 2015): Is it Decreasing Even Faster? Eur J Vasc Endovasc Surg (2021) 61(6):900–7. doi: 10.1016/j.ejvs.2021.02.013
8. Summerhill VI, Sukhorukov VN, Eid AH, Nedosugova LV, Sobenin IA, Orekhov AN. Pathophysiological Aspects of the Development of Abdominal Aortic Aneurysm With a Special Focus on Mitochondrial Dysfunction and Genetic Associations. Biomol Concepts (2021) 12:55–67. doi: 10.1515/bmc-2021-0007
9. Sun J, Deng H, Zhou Z, Xiong X, Gao L. Endothelium as a Potential Target for Treatment of Abdominal Aortic Aneurysm. Oxid Med Cell Longev (2018) 2018:6306542. doi: 10.1155/2018/6306542
10. Li H, Bai S, Ao Q, Wang X, Tian X, Li X, et al. Modulation of Immune-Inflammatory Responses in Abdominal Aortic Aneurysm: Emerging Molecular Targets. J Immunol Res (2018) 2018:7213760. doi: 10.1155/2018/7213760
11. Villacorta L, Chang L. The Role of Perivascular Adipose Tissue in Vasoconstriction, Arterial Stiffness, and Aneurysm. Horm Mol Biol Clin Investig (2015) 21:137–47. doi: 10.1515/hmbci-2014-0048
12. Kasikara C, Doran AC, Cai B, Tabas I. The Role of non-Resolving Inflammation in Atherosclerosis. J Clin Invest (2018) 128:2713–23. doi: 10.1172/JCI97950
13. Nieuwland AJ, Kokje VB, Koning OH, Hamming JF, Szuhai K, Claas FH, et al. Activation of the Vitamin D Receptor Selectively Interferes With Calcineurin-Mediated Inflammation: A Clinical Evaluation in the Abdominal Aortic Aneurysm. Lab Invest (2016) 96:784–90. doi: 10.1038/labinvest.2016.55
14. Sagan A, Mikolajczyk TP, Mrowiecki W, MacRitchie N, Daly K, Meldrum A, et al. T Cells Are Dominant Population in Human Abdominal Aortic Aneurysms and Their Infiltration in the Perivascular Tissue Correlates With Disease Severity. Front Immunol (2019) 10:1979. doi: 10.3389/fimmu.2019.01979
15. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 Identifies Human and Mouse T Regulatory Type 1 Cells. Nat Med (2013) 19:739–46. doi: 10.1038/nm.3179
16. Palego L, Betti L, Rossi A, Giannaccini G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J Amino Acids (2016) 2016:8952520. doi: 10.1155/2016/8952520
17. Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nat Rev Drug Discov (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
18. Friedman M. Analysis, Nutrition, and Health Benefits of Tryptophan. Int J Tryptophan Res (2018) 11:1178646918802282. doi: 10.1177/1178646918802282
19. Gostner JM, Geisler S, Stonig M, Mair L, Sperner-Unterweger B, Fuchs D. Tryptophan Metabolism and Related Pathways in Psychoneuroimmunology: The Impact of Nutrition and Lifestyle. Neuropsychobiology (2020) 79:89–99. doi: 10.1159/000496293
20. Barik S. The Uniqueness of Tryptophan in Biology: Properties, Metabolism, Interactions and Localization in Proteins. Int J Mol Sci (2020) 21(22):8776. doi: 10.3390/ijms21228776
21. Evrensel A, Unsalver BO, Ceylan ME. Immune-Kynurenine Pathways and the Gut Microbiota-Brain Axis in Anxiety Disorders. Adv Exp Med Biol (2020) pp:155–67. doi: 10.1007/978-981-32-9705-0_10
22. Song P, Ramprasath T, Wang H, Zou MH. Abnormal Kynurenine Pathway of Tryptophan Catabolism in Cardiovascular Diseases. Cell Mol Life Sci (2017) 74:2899–916. doi: 10.1007/s00018-017-2504-2
23. Yuasa HJ, Ball HJ, Ho YF, Austin CJ, Whittington CM, Belov K, et al. Characterization and Evolution of Vertebrate Indoleamine 2, 3-Dioxygenases IDOs From Monotremes and Marsupials. Comp Biochem Physiol B Biochem Mol Biol (2009) 153:137–44. doi: 10.1016/j.cbpb.2009.02.002
24. Mandarano M, Bellezza G, Belladonna ML, Vannucci J, Gili A, Ferri I, et al. Indoleamine 2,3-Dioxygenase 2 Immunohistochemical Expression in Resected Human Non-Small Cell Lung Cancer: A Potential New Prognostic Tool. Front Immunol (2020) 11:839. doi: 10.3389/fimmu.2020.00839
25. Watanabe Y, Koyama S, Yamashita A, Matsuura Y, Nishihira K, Kitamura K, et al. Indoleamine 2,3-Dioxygenase 1 in Coronary Atherosclerotic Plaque Enhances Tissue Factor Expression in Activated Macrophages. Res Pract Thromb Haemost (2018) 2:726–35. doi: 10.1002/rth2.12128
26. Hubbard TD, Murray IA, Perdew GH. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab Dispos (2015) 43:1522–35. doi: 10.1124/dmd.115.064246
27. Bhutia YD, Babu E, Ganapathy V. Interferon-Gamma Induces a Tryptophan-Selective Amino Acid Transporter in Human Colonic Epithelial Cells and Mouse Dendritic Cells. Biochim Biophys Acta (2015) 1848:453–62. doi: 10.1016/j.bbamem.2014.10.021
28. Hou Y, Guo W, Fan T, Li B, Ge W, Gao R, et al. Advanced Research of Abdominal Aortic Aneurysms on Metabolism. Front Cardiovasc Med (2021) 8:630269. doi: 10.3389/fcvm.2021.630269
29. Kelly B, Pearce EL. Amino Assets: How Amino Acids Support Immunity. Cell Metab (2020) 32:154–75. doi: 10.1016/j.cmet.2020.06.010
30. Guo Y, Wan S, Han M, Zhao Y, Li C, Cai G, et al. Plasma Metabolomics Analysis Identifies Abnormal Energy, Lipid, and Amino Acid Metabolism in Abdominal Aortic Aneurysms. Med Sci Monit (2020) 26:e926766. doi: 10.12659/MSM.926766
31. Nitz K, Lacy M, Atzler D. Amino Acids and Their Metabolism in Atherosclerosis. Arterioscler Thromb Vasc Biol (2019) 39:319–30. doi: 10.1161/ATVBAHA.118.311572
32. Ramprasath T, Kumar PH, Puhari SS, Murugan PS, Vasudevan V, Selvam GS. L-Arginine Ameliorates Cardiac Left Ventricular Oxidative Stress by Upregulating eNOS and Nrf2 Target Genes in Alloxan-Induced Hyperglycemic Rats. Biochem Biophys Res Commun (2012) 428:389–94. doi: 10.1016/j.bbrc.2012.10.064
33. Tomita H, Egashira K, Ohara Y, Takemoto M, Koyanagi M, Katoh M, et al. Early Induction of Transforming Growth Factor-Beta via Angiotensin II Type 1 Receptors Contributes to Cardiac Fibrosis Induced by Long-Term Blockade of Nitric Oxide Synthesis in Rats. Hypertension (1998) 32:273–9. doi: 10.1161/01.HYP.32.2.273
34. Nijveldt RJ, Prins HA, Siroen MP, Rauwerda JA, Teerlink T, van Leeuwen PA. Low Arginine Plasma Levels in Patients After Thoracoabdominal Aortic Surgery. Eur J Clin Nutr (2000) 54:615–7. doi: 10.1038/sj.ejcn.1601062
35. Pertovaara M, Raitala A, Juonala M, Lehtimaki T, Huhtala H, Oja SS, et al. Indoleamine 2,3-Dioxygenase Enzyme Activity Correlates With Risk Factors for Atherosclerosis: The Cardiovascular Risk in Young Finns Study. Clin Exp Immunol (2007) 148:106–11. doi: 10.1111/j.1365-2249.2007.03325.x
36. Pawlak K, Mysliwiec M, Pawlak D. Kynurenine Pathway - a New Link Between Endothelial Dysfunction and Carotid Atherosclerosis in Chronic Kidney Disease Patients. Adv Med Sci (2010) 55:196–203. doi: 10.2478/v10039-010-0015-6
37. Wang Q, Ding Y, Song P, Zhu H, Okon I, Ding YN, et al. Tryptophan-Derived 3-Hydroxyanthranilic Acid Contributes to Angiotensin II-Induced Abdominal Aortic Aneurysm Formation in Mice In Vivo. Circulation (2017) 136:2271–83. doi: 10.1161/CIRCULATIONAHA.117.030972
38. Wang Q, Zhang M, Ding Y, Wang Q, Zhang W, Song P, et al. Activation of NAD(P)H Oxidase by Tryptophan-Derived 3-Hydroxykynurenine Accelerates Endothelial Apoptosis and Dysfunction In Vivo. Circ Res (2014) 114:480–92. doi: 10.1161/CIRCRESAHA.114.302113
39. Li Y, Kilani RT, Rahmani-Neishaboor E, Jalili RB, Ghahary A. Kynurenine Increases Matrix Metalloproteinase-1 and -3 Expression in Cultured Dermal Fibroblasts and Improves Scarring In Vivo. J Invest Dermatol (2014) 134:643–50. doi: 10.1038/jid.2013.303
40. Zhang CF, Kang K, Li XM, Xie BD. MicroRNA-136 Promotes Vascular Muscle Cell Proliferation Through the ERK1/2 Pathway by Targeting PPP2R2A in Atherosclerosis. Curr Vasc Pharmacol (2015) 13:405–12. doi: 10.2174/1570161112666141118094612
41. Mangge H, Stelzer I, Reininghaus EZ, Weghuber D, Postolache TT, Fuchs D. Disturbed Tryptophan Metabolism in Cardiovascular Disease. Curr Med Chem (2014) 21:1931–7. doi: 10.2174/0929867321666140304105526
42. Wu Y, Ding Y, Ramprasath T, Zou MH, Stress O. GTPCH1, and Endothelial Nitric Oxide Synthase Uncoupling in Hypertension. Antioxid Redox Signal (2021) 34:750–64. doi: 10.1089/ars.2020.8112
43. Haruki H, Hovius R, Pedersen MG, Johnsson K. Tetrahydrobiopterin Biosynthesis as a Potential Target of the Kynurenine Pathway Metabolite Xanthurenic Acid. J Biol Chem (2016) 291:652–7. doi: 10.1074/jbc.C115.680488
44. Zhang L, Ovchinnikova O, Jonsson A, Lundberg AM, Berg M, Hansson GK, et al. The Tryptophan Metabolite 3-Hydroxyanthranilic Acid Lowers Plasma Lipids and Decreases Atherosclerosis in Hypercholesterolaemic Mice. Eur Heart J (2012) 33:2025–34. doi: 10.1093/eurheartj/ehs175
45. Wang Y, Liu H, McKenzie G, Witting PK, Stasch JP, Hahn M, et al. Kynurenine is an Endothelium-Derived Relaxing Factor Produced During Inflammation. Nat Med (2010) 16:279–85. doi: 10.1038/nm.2092
46. Niinisalo P, Oksala N, Levula M, Pelto-Huikko M, Jarvinen O, Salenius JP, et al. Activation of Indoleamine 2,3-Dioxygenase-Induced Tryptophan Degradation in Advanced Atherosclerotic Plaques: Tampere Vascular Study. Ann Med (2010) 42:55–63. doi: 10.3109/07853890903321559
47. Berg M, Polyzos KA, Agardh H, Baumgartner R, Forteza MJ, Kareinen I, et al. 3-Hydroxyanthralinic Acid Metabolism Controls the Hepatic SREBP/lipoprotein Axis, Inhibits Inflammasome Activation in Macrophages, and Decreases Atherosclerosis in Ldlr-/- Mice. Cardiovasc Res (2020) 116:1948–57. doi: 10.1093/cvr/cvz258
48. Zhang Q, Sun Y, He Z, Xu Y, Li X, Ding J, et al. Kynurenine Regulates NLRP2 Inflammasome in Astrocytes and its Implications in Depression. Brain Behav Immun (2020) 88:471–81. doi: 10.1016/j.bbi.2020.04.016
49. Shon WJ, Lee YK, Shin JH, Choi EY, Shin DM. Severity of DSS-Induced Colitis is Reduced in Ido1-Deficient Mice With Down-Regulation of TLR-MyD88-NF-kB Transcriptional Networks. Sci Rep (2015) 5:17305. doi: 10.1038/srep17305
50. Wang Q, Liu D, Song P, Zou MH. Tryptophan-Kynurenine Pathway is Dysregulated in Inflammation, and Immune Activation. Front Biosci (Landmark Ed) (2015) 20:1116–43. doi: 10.2741/4363
51. Wang L, Cheng B, Ju Q, Sun BK. AhR Regulates Peptidoglycan-Induced Inflammatory Gene Expression in Human Keratinocytes. J Innate Immun (2021) 1–11. doi: 10.1159/000517627
52. Yuan Z, Lu Y, Wei J, Wu J, Yang J, Cai Z. Abdominal Aortic Aneurysm: Roles of Inflammatory Cells. Front Immunol (2020) 11:609161. doi: 10.3389/fimmu.2020.609161
53. Cafueri G, Parodi F, Pistorio A, Bertolotto M, Ventura F, Gambini C, et al. Endothelial and Smooth Muscle Cells From Abdominal Aortic Aneurysm Have Increased Oxidative Stress and Telomere Attrition. PloS One (2012) 7:e35312. doi: 10.1371/journal.pone.0035312
54. Schroecksnadel K, Frick B, Winkler C, Fuchs D. Crucial Role of Interferon-Gamma and Stimulated Macrophages in Cardiovascular Disease. Curr Vasc Pharmacol (2006) 4:205–13. doi: 10.2174/157016106777698379
55. Connor TJ, Starr N, O’Sullivan JB, Harkin A. Induction of Indolamine 2,3-Dioxygenase and Kynurenine 3-Monooxygenase in Rat Brain Following a Systemic Inflammatory Challenge: A Role for IFN-Gamma? Neurosci Lett (2008) 441:29–34. doi: 10.1016/j.neulet.2008.06.007
56. Yan H, Zhou HF, Akk A, Hu Y, Springer LE, Ennis TL, et al. Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arterioscler Thromb Vasc Biol (2016) 36:1660–9. doi: 10.1161/ATVBAHA.116.307786
57. Schaheen B, Downs EA, Serbulea V, Almenara CC, Spinosa M, Su G, et al. B-Cell Depletion Promotes Aortic Infiltration of Immunosuppressive Cells and Is Protective of Experimental Aortic Aneurysm. Arterioscler Thromb Vasc Biol (2016) 36:2191–202. doi: 10.1161/ATVBAHA.116.307559
58. He R, Guo DC, Sun W, Papke CL, Duraisamy S, Estrera AL, et al. Characterization of the Inflammatory Cells in Ascending Thoracic Aortic Aneurysms in Patients With Marfan Syndrome, Familial Thoracic Aortic Aneurysms, and Sporadic Aneurysms. J Thorac Cardiovasc Surg (2008) 136:922–9.e1. doi: 10.1016/j.jtcvs.2007.12.063
59. Braidy N, Rossez H, Lim CK, Jugder BE, Brew BJ, Guillemin GJ. Characterization of the Kynurenine Pathway in CD8(+) Human Primary Monocyte-Derived Dendritic Cells. Neurotox Res (2016) 30:620–32. doi: 10.1007/s12640-016-9657-x
60. Alberati-Giani D, Ricciardi-Castagnoli P, Kohler C, Cesura AM. Regulation of the Kynurenine Metabolic Pathway by Interferon-Gamma in Murine Cloned Macrophages and Microglial Cells. J Neurochem (1996) 66:996–1004. doi: 10.1046/j.1471-4159.1996.66030996.x
61. Cuffy MC, Silverio AM, Qin L, Wang Y, Eid R, Brandacher G, et al. Induction of Indoleamine 2,3-Dioxygenase in Vascular Smooth Muscle Cells by Interferon-Gamma Contributes to Medial Immunoprivilege. J Immunol (2007) 179:5246–54. doi: 10.4049/jimmunol.179.8.5246
62. Metghalchi S, Ponnuswamy P, Simon T, Haddad Y, Laurans L, Clement M, et al. Indoleamine 2,3-Dioxygenase Fine-Tunes Immune Homeostasis in Atherosclerosis and Colitis Through Repression of Interleukin-10 Production. Cell Metab (2015) 22:460–71. doi: 10.1016/j.cmet.2015.07.004
63. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl Hydrocarbon Receptor Control of a Disease Tolerance Defence Pathway. Nature (2014) 511:184–90. doi: 10.1038/nature13323
64. del Porto F, Proietta M, Tritapepe L, Miraldi F, Koverech A, Cardelli P, et al. Inflammation and Immune Response in Acute Aortic Dissection. Ann Med (2010) 42:622–9. doi: 10.3109/07853890.2010.518156
65. Muller I, Munder M, Kropf P, Hansch GM. Polymorphonuclear Neutrophils and T Lymphocytes: Strange Bedfellows or Brothers in Arms? Trends Immunol (2009) 30:522–30. doi: 10.1016/j.it.2009.07.007
66. Nuche J, Palomino-Doza J, Ynsaurriaga FA, Delgado JF, Ibanez B, Oliver E, et al. Potential Molecular Pathways Related to Pulmonary Artery Aneurysm Development: Lessons to Learn From the Aorta. Int J Mol Sci (2020) 21(7):2509. doi: 10.3390/ijms21072509
67. Anzai A, Shimoda M, Endo J, Kohno T, Katsumata Y, Matsuhashi T, et al. Adventitial Cxcl1/G-CSF Expression in Response to Acute Aortic Dissection Triggers Local Neutrophil Recruitment and Activation Leading to Aortic Rupture. Circ Res (2015) 116:612–23. doi: 10.1161/CIRCRESAHA.116.304918
68. Reeps C, Pelisek J, Seidl S, Schuster T, Zimmermann A, Kuehnl A, et al. Inflammatory Infiltrates and Neovessels are Relevant Sources of MMPs in Abdominal Aortic Aneurysm Wall. Pathobiology (2009) 76:243–52. doi: 10.1159/000228900
69. Jia LX, Zhang WM, Li TT, Liu Y, Piao CM, Ma YC, et al. ER Stress Dependent Microparticles Derived From Smooth Muscle Cells Promote Endothelial Dysfunction During Thoracic Aortic Aneurysm and Dissection. Clin Sci (Lond) (2017) 131:1287–99. doi: 10.1042/CS20170252
70. Siasos G, Mourouzis K, Oikonomou E, Tsalamandris S, Tsigkou V, Vlasis K, et al. The Role of Endothelial Dysfunction in Aortic Aneurysms. Curr Pharm Des (2015) 21:4016–34. doi: 10.2174/1381612821666150826094156
71. Belik J, McIntyre BA, Enomoto M, Pan J, Grasemann H, Vasquez-Vivar J. Pulmonary Hypertension in the Newborn GTP Cyclohydrolase I-Deficient Mouse. Free Radic Biol Med (2011) 51:2227–33. doi: 10.1016/j.freeradbiomed.2011.09.012
72. Gao L, Siu KL, Chalupsky K, Nguyen A, Chen P, Weintraub NL, et al. Role of Uncoupled Endothelial Nitric Oxide Synthase in Abdominal Aortic Aneurysm Formation: Treatment With Folic Acid. Hypertension (2012) 59:158–66. doi: 10.1161/HYPERTENSIONAHA.111.181644
73. Li Q, Youn JY, Siu KL, Murugesan P, Zhang Y, Cai H. Knockout of Dihydrofolate Reductase in Mice Induces Hypertension and Abdominal Aortic Aneurysm via Mitochondrial Dysfunction. Redox Biol (2019) 24:101185. doi: 10.1016/j.redox.2019.101185
74. Silverstein MD, Pitts SR, Chaikof EL, Ballard DJ. Abdominal Aortic Aneurysm (AAA): Cost-Effectiveness of Screening, Surveillance of Intermediate-Sized AAA, and Management of Symptomatic AAA. Proc (Bayl Univ Med Cent) (2005) 18:345–67. doi: 10.1080/08998280.2005.11928095
75. Ramprasath T, Freddy AJ, Velmurugan G, Tomar D, Rekha B, Suvekbala V, et al. Context-Dependent Regulation of Nrf2/ARE Axis on Vascular Cell Function During Hyperglycemic Condition. Curr Diabetes Rev (2020) 16:797–806. doi: 10.2174/1573399816666200130094512
76. Fujigaki H, Saito K, Lin F, Fujigaki S, Takahashi K, Martin BM, et al. Nitration and Inactivation of IDO by Peroxynitrite. J Immunol (2006) 176:372–9. doi: 10.4049/jimmunol.176.1.372
77. Metghalchi S, Vandestienne M, Haddad Y, Esposito B, Dairou J, Tedgui A, et al. Indoleamine 2 3-Dioxygenase Knockout Limits Angiotensin II-Induced Aneurysm in Low Density Lipoprotein Receptor-Deficient Mice Fed With High Fat Diet. PloS One (2018) 13:e0193737. doi: 10.1371/journal.pone.0193737
78. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Grant R. Changes in Kynurenine Pathway Metabolism in the Brain, Liver and Kidney of Aged Female Wistar Rats. FEBS J (2011) 278:4425–34. doi: 10.1111/j.1742-4658.2011.08366.x
79. Kondrikov D, Elmansi A, Bragg RT, Mobley T, Barrett T, Eisa N, et al. Kynurenine Inhibits Autophagy and Promotes Senescence in Aged Bone Marrow Mesenchymal Stem Cells Through the Aryl Hydrocarbon Receptor Pathway. Exp Gerontol (2020) 130:110805. doi: 10.1016/j.exger.2019.110805
80. de Bie J, Guest J, Guillemin GJ, Grant R. Central Kynurenine Pathway Shift With Age in Women. J Neurochem (2016) 136:995–1003. doi: 10.1111/jnc.13496
81. Peng YH, Ueng SH, Tseng CT, Hung MS, Song JS, Wu JS, et al. Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives With IDO1. J Med Chem (2016) 59:282–93. doi: 10.1021/acs.jmedchem.5b01390
82. Indoleamine 2,3-Dioxygenase (IDO) Inhibitor in Advanced Solid Tumors (2017). Available at: https://www.clinicaltrials.gov/ct2/show/NCT02048709.
83. Nayak-Kapoor A, Hao Z, Sadek R, Dobbins R, Marshall L, Vahanian NN, et al. Phase Ia Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) in Patients With Recurrent Advanced Solid Tumors. J Immunother Cancer (2018) 6:61. doi: 10.1186/s40425-018-0351-9
84. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front Immunol (2014) 5:673. doi: 10.3389/fimmu.2014.00673
85. Dolusic E, Larrieu P, Moineaux L, Stroobant V, Pilotte L, Colau D, et al. Tryptophan 2,3-Dioxygenase (TDO) Inhibitors. 3-(2-(Pyridyl)Ethenyl)Indoles as Potential Anticancer Immunomodulators. J Med Chem (2011) 54:5320–34. doi: 10.1021/jm2006782
86. Wirthgen E, Leonard AK, Scharf C, Domanska G. The Immunomodulator 1-Methyltryptophan Drives Tryptophan Catabolism Toward the Kynurenic Acid Branch. Front Immunol (2020) 11:313. doi: 10.3389/fimmu.2020.00313
87. Bondulich MK, Fan Y, Song Y, Giorgini F, Bates GP. Ablation of Kynurenine 3-Monooxygenase Rescues Plasma Inflammatory Cytokine Levels in the R6/2 Mouse Model of Huntington’s Disease. Sci Rep (2021) 11:5484. doi: 10.1038/s41598-021-84858-7
88. Mole DJ, Webster SP, Uings I, Zheng X, Binnie M, Wilson K, et al. Kynurenine-3-Monooxygenase Inhibition Prevents Multiple Organ Failure in Rodent Models of Acute Pancreatitis. Nat Med (2016) 22:202–9. doi: 10.1038/nm.4020
89. Zhang S, Sakuma M, Deora GS, Levy CW, Klausing A, Breda C, et al. A Brain-Permeable Inhibitor of the Neurodegenerative Disease Target Kynurenine 3-Monooxygenase Prevents Accumulation of Neurotoxic Metabolites. Commun Biol (2019) 2:271. doi: 10.1038/s42003-019-0520-5
90. Chiarugi A, Carpenedo R, Moroni F. Kynurenine Disposition in Blood and Brain of Mice: Effects of Selective Inhibitors of Kynurenine Hydroxylase and of Kynureninase. J Neurochem (1996) 67:692–8. doi: 10.1046/j.1471-4159.1996.67020692.x
91. Toledo-Sherman LM, Prime ME, Mrzljak L, Beconi MG, Beresford A, Brookfield FA, et al. Development of a Series of Aryl Pyrimidine Kynurenine Monooxygenase Inhibitors as Potential Therapeutic Agents for the Treatment of Huntington’s Disease. J Med Chem (2015) 58:1159–83. doi: 10.1021/jm501350y
92. Wirthgen E, Hoeflich A, Rebl A, Gunther J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front Immunol (2017) 8:1957. doi: 10.3389/fimmu.2017.01957
93. Swainson LA, Ahn H, Pajanirassa P, Khetarpal V, Deleage C, Estes JD, et al. Kynurenine 3-Monooxygenase Inhibition During Acute Simian Immunodeficiency Virus Infection Lowers PD-1 Expression and Improves Post-Combination Antiretroviral Therapy CD4(+) T Cell Counts and Body Weight. J Immunol (2019) 203:899–910. doi: 10.4049/jimmunol.1801649
94. Nishimura M, Yamashita A, Matsuura Y, Okutsu J, Fukahori A, Hirata T, et al. Upregulated Kynurenine Pathway Enzymes in Aortic Atherosclerotic Aneurysm: Macrophage Kynureninase Downregulates Inflammation. J Atheroscler Thromb (2020). doi: 10.5551/jat.58248
95. Wang SK, Murphy MP. Immune Modulation as a Treatment for Abdominal Aortic Aneurysms. Circ Res (2018) 122:925–7. doi: 10.1161/CIRCRESAHA.118.312870
Keywords: aortic aneurysm, atherosclerosis, kynurenine pathway, tryptophan metabolism, vascular cells
Citation: Ramprasath T, Han Y-M, Zhang D, Yu C-J and Zou M-H (2021) Tryptophan Catabolism and Inflammation: A Novel Therapeutic Target For Aortic Diseases. Front. Immunol. 12:731701. doi: 10.3389/fimmu.2021.731701
Received: 28 June 2021; Accepted: 03 September 2021;
Published: 23 September 2021.
Edited by:
László Vécsei, University of Szeged, HungaryReviewed by:
Andrea Baragetti, University of Milan, ItalyCopyright © 2021 Ramprasath, Han, Zhang, Yu and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Hui Zou, bXpvdUBnc3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.