 Xinyu Lu
Xinyu Lu Qianhui Chen
Qianhui Chen Xiaoyong Zhang
Xiaoyong Zhang- State Key Laboratory of Organ Failure Research, Guangdong Provincial Key Laboratory of Viral Hepatitis Research, Department of Infectious Diseases, Nanfang Hospital, Southern Medical University, Guangzhou, China
The non-canonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway is an important component of NF-κB transcription complex. Activation of this pathway mediates the development and function of host immune system involved in inflammation and viral infection. During hepatitis B virus (HBV) infection, there is a complex interaction between infected hepatocytes and the immune cells, which can hinder antiviral immune responses and is associated with pathological changes in liver tissue. Consistently, the host immune system is closely related to the severity of liver damage and the level of viral replication. Previous studies indicated that the non-canonical NF-κB signaling pathway was affected by HBV and might play an important regulatory role in the antiviral immunity. Therefore, systematically elucidating the interplay between HBV and non-canonical NF-κB signaling will contribute the discovery of more potential therapeutic targets and novel drugs to treat HBV infection.
Introduction
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) encompasses an important family of transcription factors, can regulate the expression of multiple genes, and has been implicated in diverse biological processes including innate and adaptive immunity, inflammation, stress, immune cell development, and lymphatic organ formation (1, 2). Based on the components of the signaling cascade, NF-κB signaling pathways can be categorized as canonical or non-canonical (3). The canonical NF-κB pathway is rapid and transient, and mainly stimulated by proinflammatory cytokines such as IL-1β, tumor necrosis factor (TNF)-α, antigen ligands, and toll-like receptors (TLRs). Activation of the canonical NF-κB pathway relies on phosphorylation and ubiquitination of IκB kinase α/β (IKKα/β) causing the degradation of κB inhibitory factors (IκBs) protein, thereby countering inhibition of the nuclear transcription factor heterodimer RelA/p50 by IκBs (4, 5). The non-canonical NF-κB signaling pathway is slow and persistent, and generally activated by ligands of the TNF receptor superfamily, including lymphotoxin beta (LTB), CD40, OX40, RANK, TWEAK and B cell-activating factor (BAFF). These ligands stimulation induces NF-κB-inducing kinase (NIK, also called MAP3K14) stabilization and accumulation, results in IKKα phosphorylation. Activated IKKα subsequently phosphorylates p100, triggering K48 linked ubiquitination and degradation of p100, generating p52 (also known as NFKB2). The nuclear translocation of RelB-p52 heterodimer then initiates the expression of target genes (4, 5) (Figure 1). NIK is a key molecule for non-canonical NF-κB pathway activation, and this process requires both NIK expression and kinase activity (6, 7). Expression of human NIK-dominant inactivated mutant (NIKKA, KK429/430AA) results in the blockage of the non-canonical NF-κB pathway (8).
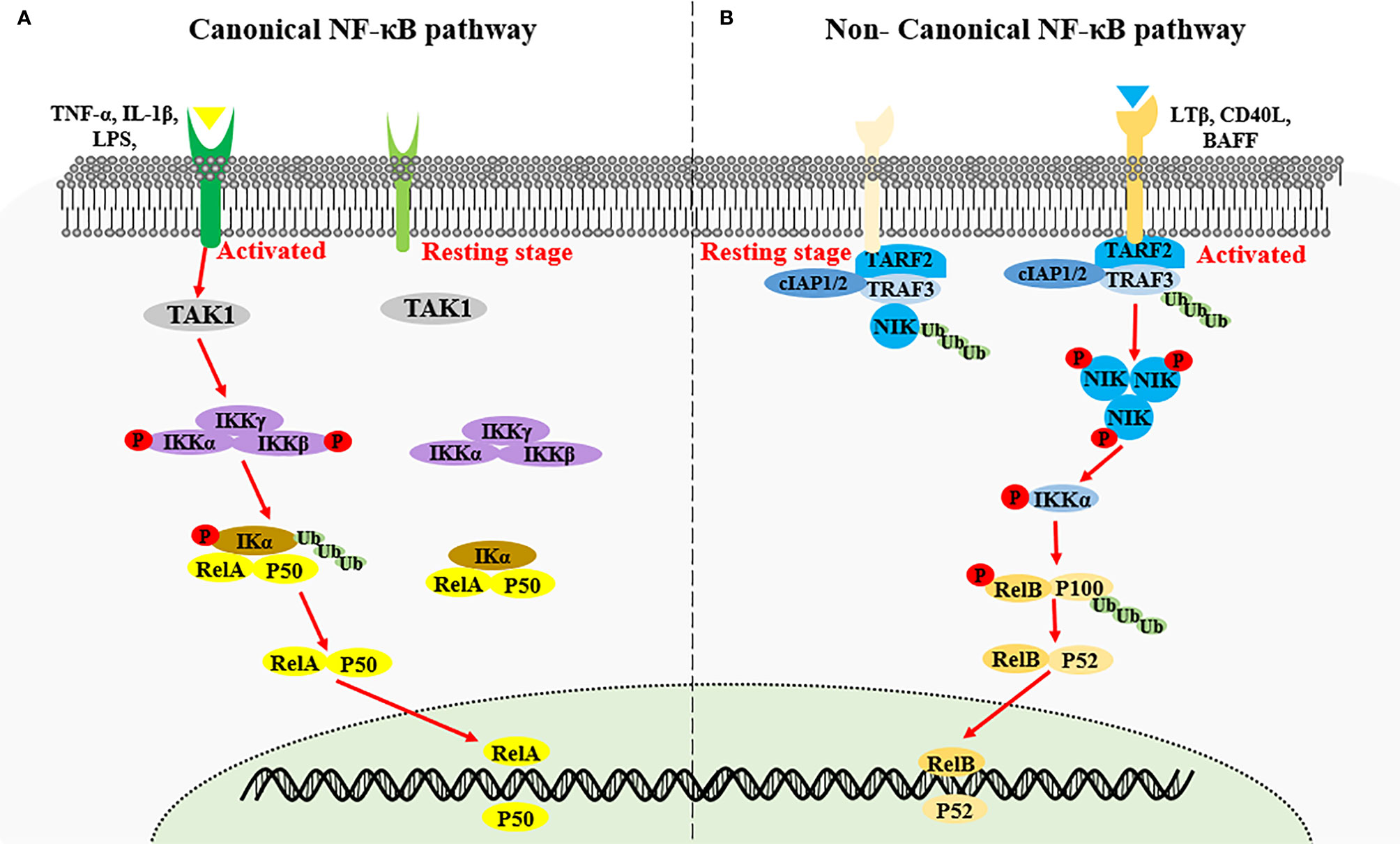
Figure 1 Activation of canonical NF-κB signaling and non-canonical NF-κB signaling. (A) Canonical NF-κB signaling is stimulated by proinflammatory cytokines such as IL-1β, TNF-α and LPS, inducing the activation of IKK complex by TAK1. The IKK complex then phosphorylates the IκB kinase α/β (IKKα/β), causing the ubiquitin-dependent degradation of κB inhibitory factors (IκBs) protein, thereby triggering the nuclear transcription factor heterodimer RelA/p50. (B) Non-canonical NF-κB signaling is activated by the specific TNFR superfamily. Receptor activation induce the recruitment of TRAF3-TRAF2-cIAP1/2 receptor complex, followed by the degradation of TRAF3 via ubiquitination, resulting in the stabilization and accumulation of NIK. NIK phosphorylates IKKα which in turn phosphorate p100, triggering the ubiquitination and degradation of p100 to generate p52 and the nuclear transduction of RelB-p52 heterodimer.
It was reported that the alymphoplasia (aly/aly) mice with loss-of-function mutation of NIK exhibited aberrant development of spleen architecture and lymph nodes, impaired B cell proliferation and maturation, no formation of germinal center or isotype antibody switching in antigen-specific immune responses, and disordered thymus structure, and autoimmune inflammation in multiple organs (9, 10). Moreover, the phenotype of NFKB2 KO mice was similar to that of the aly/aly mice, with defects in splenic architecture, lymph nodes, and peripheral B cell maturation, as well as impaired antibody responses (11, 12). The severe immunodeficiencies observed in these mice strains indicates that the non-canonical NF-κB signaling pathway plays an essential role in multiple important biological processes, including the initiation and regulation of B cell and T cell immunity (13).
Host antiviral immunity is an important defense mechanism with respect to preventing virus invasion, and effectively clearing viral infection. Many RNA and DNA viruses, can regulate the activation of the non-canonical NF-κB pathway via different adaptor proteins, leading to the regulation of antiviral immunity (14). In contrast, some viruses can evade antiviral immunity by affecting the non-canonical NF-κB pathway. For example, the RNA virus EV71 upregulated NIK, the key protein of the non-canonical NF-κB pathway, to promote the secretion of inflammatory cytokines and apoptosis of EV71-infected cells (14, 15). Epstein-Barr virus, human T-lymphotropic virus 1, and Kaposi’s sarcoma associated herpesvirus could activate the non-canonical NF-κB pathway by encoding corresponding oncogenic proteins (16–18). Currently, increasing evidence suggests that the non-canonical NF-κB pathway also plays roles in host responses to hepatitis B virus (HBV) infection. In this review, we discussed the interplay between HBV and the non-canonical NF-κB pathway.
Regulation of Non-Canonical NF-κB Signaling Pathway by HBV Infection
HBV is a DNA virus that encodes multiple proteins that affect the activation of the non-canonical NF-κB pathway, thus enabling regulation of the host defense response. The type I interferon (IFN) system, an indispensable part of the innate immune response to HBV, is involved in an immediate antiviral response via the induction of numerous functional proteins against the viral life cycle, and activates the adaptive immune response (19). TBK1 can function as a positive and negative regulator of the non-canonical NF-κB pathway (20). HBV polymerase (Pol) can evidently inhibit the phosphorylation, dimerization, and nuclear translocation of IRF3, as well as IFN-β expression at the TBK1/IKKƐ level (19). Activating NIK promote phosphorylation of IRF3 to produce type I interferon (21, 22). Therefore, the mechanism by which HBV pol protein inhibits IFN-β production in human hepatocytes may also be associated with the level of non-canonical NF-κB pathway activation. Liu et al. (23) reported that Pol protein did not alter the level of NF-κB expression, but could prevent the activation of the non-canonical NF-κB pathway required for IFN-β activation by inhibiting the nuclear translocation of RelB/p52 and NF-κB subunits in hepatoma cells. These studies suggested that Pol protein could inhibit IFN-β production, which may be associated with the non-canonical NF-κB pathway in hepatocytes. However, additional studies showed some conflicting results. As observed in chronic HBV infection patients, the IFN-stimulated genes (ISGs) were not activated in liver tissues (24). In addition, results of the in vitro models of HBV-infected hepatocytes also found that HBV infection did not induce type I and III IFNs, and the downstream ISGs (25, 26), which further supported the inability of HBV to induce IFN response. Thus, the relationship between Pol protein, IFN signaling and non-canonical NF-κB pathway remains to be explored.
HBx protein plays a vital role in HBV replication, and is a potential inducer for hepatocellular carcinoma development (HCC) (27, 28). Previous studies indicate that HBx induced the phosphorylation and degradation of intracellular IkBα to activate the classical NF-κB pathway, accompanied with slight decrease in the level of p100 protein, suggesting that HBx may be involved in activation of the non-canonical NF-κB pathway (29, 30). Kim et al. (31) reported that HBx might disturb the activation of the non-canonical NF-κB pathway via a NIK-IKK-IkB-dependent mechanism in the liver, and result in the induction of inflammatory response or hepatic oncogenesis by stimulating hepatocyte proliferation. But whether the effect mediated by HBx is valid in the HBV infection need to be further studied.
HBV e antigen (HBeAg), a secreted protein and not required for viral replication, is thought to play an immunoregulatory role and promote viral persistence during viral infection. Researches by Wang et al. (32) has found that HBeAg can associate with NEMO, the regulatory subunit for IκB kinase (IKK) that controls the NF-κB signaling pathway, and thereby inhibited TRAF6-mediated K63-linked ubiquitination of NEMO induced by IL-1β, resulting in the downregulation of NF-κB activity and increased virus replication. HBeAg suppresses LPS-induced NLRP3 inflammasome activation, pro-IL-1β expression and IL-1β production in kuffer cells not only via inhibiting NF-κB phosphorylation but inhibiting ROS production (33). The roles of HBeAg in regulating the NF-κB pathway to affect host immune response are controversial during the infection of HBV. In addition, the HBeAg affected NEMO mainly functions in canonical NF-κB signaling pathway. Nevertheless, canonical and non-canonical NF-κB activation pathways are connected via many crosstalk mechanisms. Therefore, HBeAg affecting the host immune response also cannot exclude the role of non-canonical NF-κB pathway.
Regulation of HBV Infection by Non-Canonical NF-κB Signaling Pathway
Abnormal host immune function and continuous HBV replication are the main causes of disease progression in patients with chronic hepatitis B (CHB) (34). Concurrent continuous HBV replication and the activation of inflammatory pathways lead to chronic liver damage (35). Immune system is very important for the elimination of HBV, but CHB patients tend to exhibit defective innate and adaptive immune function, and cannot effectively remove viral products in the liver. Previous studies indicated that non-classical NF-κB pathway in the hepatocytes and immune cells played an important role in control of HBV replication in vitro and clearance of HBV in vivo (Figure 2).
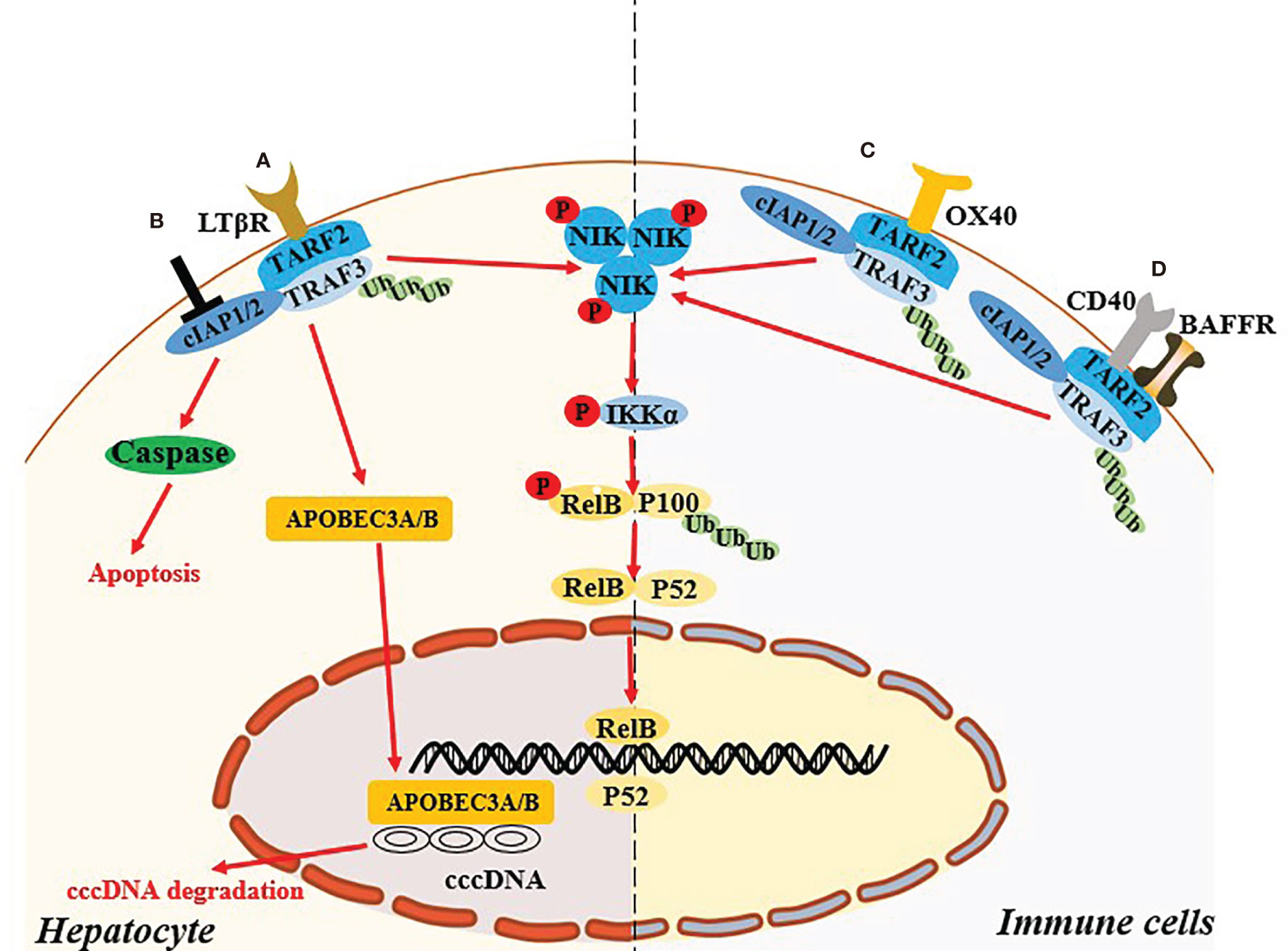
Figure 2 Non-canonical NF-κB signaling pathway regulates the HBV infection. (A) In hepatocyte, activation of LTB receptor in hepatocyte mediates the non-canonical NF-κB signaling pathway and induce the up-regulated expression of APOBCE3A/B protein, which degrade cccDNA. (B) Targeting cIAPs also mediate the activating the NIK-dependent non-canonical NF-κB signaling pathway and exert the antiviral effect in hepatocyte via TNF-mediated death of infected cells. (C, D) Specific ligation of OX40, BAFFR or CD40 in immune cells might recover the exhausted immune function during HBV infection through the activation of the NIK-dependent non-canonical NF-κB signaling pathway.
Hepatocytes-Intrinsic Non-Canonical NF-κB Signaling Pathway
HBV replication in hepatocytes may be related to NIK-dependent activation of the non-classical NF-κB pathway. Microarray analysis and western blotting validation suggested that mRNA and protein levels of TRAF2 and NIK in primary hepatocytes are upregulated during the early stage of HBV infection (36). Lymphotoxin beta receptor, a member of the TNF receptor superfamily, is able to activate the NIK-dependent non-canonical NF-κB pathway. Activation of this receptor could induce the expression of APOBEC3A/3B protein, which mediated deamination on the negative chain of cccDNA to degrade it (37). The above results suggested that the non-canonical NF-κB pathway activation in hepatocytes might exert a direct anti-HBV effect.
Cellular inhibitor of apoptosis proteins (cIAPs) are a family of highly conserved endogenous anti-apoptotic molecules (38). In the resting state, cIAP1/2 uses TRAF2 as an adaptor protein to connect with NIK binding protein TRAF3 to form a TRAF3-TRAF2-cIAP1/2 multi-subunit ubiquitin ligase complex, resulting in very low protein levels of cellular endogenous NIK (39). Degradation of cIAPs or loss of TRAF2 can continuously activate the NIK-dependent non-canonical NF-κB signaling pathway (39). Ebert et al. (40) reported that cIAPs can impair viral clearance by preventing TNF-mediated death of infected cells, and mice with hepatocyte-specific deficiency of cIAPs expression promoted the clearance chronic HBV infection. Moreover, birinapant, a chemical inhibitor of cIAPs, could reduce HBV DNA and hepatitis B surface antigen levels in serum, and induce hepatitis B core antigen-positive hepatocyte apoptosis in HBV persistence mouse model (41). Therefore, whether the antiviral effect mediated by targeting cIAPs is related to activation of the NIK-mediated non-canonical NF-κB signaling pathway need further investigation.
Immune Cells-Intrinsic Non-Canonical NF- κB Signaling Pathway
Abnormal expression and function of specific ligands and receptors involved in activation of the non-classical NF-κB pathway in the immune cells might be related to the chronicity of HBV infection. OX40 (also known as CD134) is an important costimulatory molecule in T cells, and its ligand OX40L (also known as CD252) is mainly expressed on antigen-presenting cells. OX40 ligation can promote T cell phenotypic transformation, maintain the activation state, and promote the proliferation of effector T cells and memory T cells, while also inhibiting the differentiation and activity of regulatory T cells (Tregs) (42, 43). Interestingly, studies suggest that the key to determining antiviral immunity is the expression of the costimulatory molecule OX40L on hepatic innate immune cells, and the expression of OX40L is positively correlated with age (44). OX40 agonists can increase the HBV clearance rate in a young mouse model of hepatitis B, as well as the strength of T cell responses in young mice and adult mice that were exposed to HBV when they were young and developed a CHB serological profile (44). Moreover, in adult humans HBV clearance is associated with increased OX40 expression in peripheral CD4+ T cells (44). In a recent study OX40 was highly expressed on CD4+ T cells in the immune microenvironment of HBV-related HCC (45). OX40 agonists combined with PD-1 blockers can enhance the function of HBV-specific CD4+ T cells (42). The above studies suggest that in innate immune cells and T cells the OX40-mediated non-canonical NF-κB pathway may participate in HBV clearance by affecting the function of immune cells. In a liver injury model in HBsAg transgenic mice mediated by natural killer (NK) cells induced with low or high doses of concanavalin A, the proportion of CD4+CD25+FoxP3+ Tregs in the liver increases, and the proportion of restored Tregs continued to increase with the development of liver damage (46). The mechanism of reducing liver injury in HBs transgenic mice is related to the direct inhibition of NK cell-mediated hepatotoxicity by Tregs via OX40/OX40L interaction in cell-cell contact (46). In patients with CHB the immune microenvironment is characterized by the exhaustion of virus-specific T cells and an increase in negatively regulated immune cells such as Tregs. Therefore, this may also be related to the non-classical NF-κB pathway regulated by OX40/OX40L.
BAFF is an essential cytokine for the activation of B lymphocytes, and BAFF receptor is expressed on B cells (47). Interaction between BAFF and BAFF receptor can also activate the non-canonical NF-κB signaling pathway, and its mediated activation plays an important role in regulating the survival and maturation of peripheral B cells (48). Studies indicate that in CHB patients’ serum BAFF is maintained at a high level, leading to excessive activation of B cells, thus changing T cell functions (upregulating PD-1, Tim-3, and Lag-3, i.e., exhaustion phenotypes) and reducing reactions to PEG-IFN (49, 50). Notably however, the mechanisms by which BAFF mediates the chronicity of HBV require further clarification. Studies suggest that in vitro administration of CD40L signals can partially restore the function of hepatitis B surface antigen (HBsAg) -specific B cells (51). CD40-CD40L interaction can induce activation of the non-canonical NF-κB pathway in mouse splenic B cells (52). The above results suggest that non-canonical NF-κB pathway dysregulation in immune cells of patients with CHB may be related to the failure of HBV clearance and disease progression.
NIK is a key adaptor protein associated with activation of the non-classical NF-κB pathway, and may also participate in the development of CHB disease by affecting the function of immune cells. In previous studies NIK knockout in dendritic cells affected antigen presentation to CD8+ T cells (53). T cell-specific NIK deficiency induces naive T cells to differentiate into effector T cells and memory T cells (54, 55). Moreover, in T cell-specific NIK-deficient mice the phosphorylation of Zap70, LAT, AKT, ERK1/2, and PLCγ is hindered, thereby blocking the T cell receptor signaling pathway (56). The continuous expression of viral antigens and the hepatic inflammatory microenvironment can downregulate the function and proportion of HBV-specific CD4+ cells, CD8+ T cells, dendritic cells, NK cells, and NK T cells in CHB patients. Therefore, the exhaustion of immune function in CHB patients may be related to the non-canonical NF-κB pathway dysregulation caused by downregulation of NIK in immune cells such as dendritic cells and T cells. Whether the inability of antiviral immunity in CHB patients is related to the activation of the non-canonical NF-κB pathway in these immune cells remains to be determined. In addition, IFN-γ and TNF-α produced by T cells can degrade the cccDNA through deamination, but not the cytolysis in hepatocyte, suggesting that the non-classical NF-κB pathway has a complex regulatory network in hosts with normal immunity (57).
Conclusion
To date studies indicate that the non-canonical NF-κB signaling pathway plays an important role in the development of the immune system and triggering inflammation, but research on the regulatory role of the HBV life cycle and host antiviral effects remains largely unknown. A variety of ligands and receptors, adaptor proteins, and regulatory factors involved in the formation of non-canonical NF-κB signaling pathway might play important roles in the establishment and development of HBV disease. Therefore, in-depth mechanistic studies investigating the interplay between HBV and non-canonical NF-κB signaling may provide potential strategies for HBV treatment.
Author Contributions
XZ, XL, QC, and HL collected the data and drafted the manuscript. XZ made critical revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This review was partly supported by grants from the National Natural Science Foundation of China (No. 81871664 and No.81970539).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-Kappab: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
2. Hayden MS, Ghosh S. Shared Principles in NF-kappaB Signaling. Cell (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
3. Sun SC. The non-Canonical NF-kappaB Pathway in Immunity and Inflammation. Nat Rev Immunol (2017) 17:545–58. doi: 10.1038/nri.2017.52
4. Qu Z, Xiao G. Systematic Detection of Noncanonical NF-kappaB Activation. Methods Mol Biol (2015) 1280:121–54. doi: 10.1007/978-1-4939-2422-6_8
5. Sun SC. Non-Canonical NF-kappaB Signaling Pathway. Cell Res (2011) 21:71–85. doi: 10.1038/cr.2010.177
6. Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKalpha of a Second, Evolutionary Conserved, NF-Kappa B Signaling Pathway. Science (2001) 293:1495–9. doi: 10.1126/science.1062677
7. Thu YM, Richmond A. NF-kappaB Inducing Kinase: A Key Regulator in the Immune System and in Cancer. Cytokine Growth Factor Rev (2010) 21:213–26. doi: 10.1016/j.cytogfr.2010.06.002
8. Smith C, Andreakos E, Crawley JB, Brennan FM, Feldmann M, Foxwell BM. NF-kappaB-Inducing Kinase is Dispensable for Activation of NF-kappaB in Inflammatory Settings But Essential for Lymphotoxin Beta Receptor Activation of NF-kappaB in Primary Human Fibroblasts. J Immunol (2001) 167:5895–903. doi: 10.4049/jimmunol.167.10.5895
9. Miyawaki S, Nakamura Y, Suzuka H, Koba M, Yasumizu R, Ikehara S, et al. A New Mutation, Aly, That Induces a Generalized Lack of Lymph Nodes Accompanied by Immunodeficiency in Mice. Eur J Immunol (1994) 24:429–34. doi: 10.1002/eji.1830240224
10. Koike R, Nishimura T, Yasumizu R, Tanaka H, Hataba Y, Hataba Y, et al. The Splenic Marginal Zone is Absent in Alymphoplastic Aly Mutant Mice. Eur J Immunol (1996) 26:669–75. doi: 10.1002/eji.1830260324
11. Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, et al. Mice Deficient in Nuclear Factor (NF)-Kappa B/p52 Present With Defects in Humoral Responses, Germinal Center Reactions, and Splenic Microarchitecture. J Exp Med (1998) 187:147–59. doi: 10.1084/jem.187.2.147
12. Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, et al. Nuclear Factor (NF)-Kappa B2 (P100/P52) is Required for Normal Splenic Microarchitecture and B Cell-Mediated Immune Responses. J Exp Med (1998) 187:185–96. doi: 10.1084/jem.187.2.185
13. Shinkura R, Matsuda F, Sakiyama T, Tsubata T, Hiai H, Paumen M, et al. Defects of Somatic Hypermutation and Class Switching in Alymphoplasia (Aly) Mutant Mice. Int Immunol (1996) 8:1067–75. doi: 10.1093/intimm/8.7.1067
14. Struzik J, Szulc-Dabrowska L. Manipulation of Non-Canonical NF-kappaB Signaling by Non-Oncogenic Viruses. Arch Immunol Ther Exp (Warsz) (2019) 67:41–8. doi: 10.1007/s00005-018-0522-x
15. Khatiwada S, Delhon G, Nagendraprabhu P, Chaulagain S, Luo S, Diel DG, et al. A Parapoxviral Virion Protein Inhibits NF-kappaB Signaling Early in Infection. PloS Pathog (2017) 13:e1006561. doi: 10.1371/journal.ppat.1006561
16. Sun SC, Cesarman E. NF-kappaB as a Target for Oncogenic Viruses. Curr Top Microbiol Immunol (2011) 349:197–244. doi: 10.1007/82_2010_108
17. Bagneris C, Ageichik AV, Cronin N, Wallace B, Collins M, Boshoff C, et al. Crystal Structure of a Vflip-IKKgamma Complex: Insights Into Viral Activation of The IKK Signalosome. Mol Cell (2008) 30:620–31. doi: 10.1016/j.molcel.2008.04.029
18. Karrer U, Althage A, Odermatt B, Roberts CW, Korsmeyer SJ, Miyawaki S, et al. On the Key Role of Secondary Lymphoid Organs in Antiviral Immune Responses Studied in Alymphoplastic (Aly/Aly) and Spleenless (Hox11(-)/-) Mutant Mice. J Exp Med (1997) 185:2157–70. doi: 10.1084/jem.185.12.2157
19. Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z. Hepatitis B Virus Polymerase Inhibits RIG-I- and Toll-Like Receptor 3-Mediated Beta Interferon Induction in Human Hepatocytes Through Interference With Interferon Regulatory Factor 3 Activation and Dampening of the Interaction Between TBK1/IKKepsilon and DDX3. J Gen Virol (2010) 91:2080–90. doi: 10.1099/vir.0.020552-0
20. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-kappaB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct Target Ther (2020) 5:209. doi: 10.1038/s41392-020-00312-6
21. Chen B, Li C, Yao J, Shi L, Liu W, Wang F, et al. Zebrafish NIK Mediates IFN Induction by Regulating Activation of IRF3 and NF-Kappab. J Immunol (2020) 204:1881–91. doi: 10.4049/jimmunol.1900561
22. Parvatiyar K, Pindado J, Dev A, Aliyari SR, Zaver SA, Gerami H, et al. A TRAF3-NIK Module Differentially Regulates DNA vs RNA Pathways in Innate Immune Signaling. Nat Commun (2018) 9:2770. doi: 10.1038/s41467-018-05168-7
23. Liu D, Wu A, Cui L, Hao R, Wang Y, He J, et al. Hepatitis B Virus Polymerase Suppresses NF-kappaB Signaling by Inhibiting the Activity of IKKs via Interaction With Hsp90beta. PloS One (2014) 9:e91658. doi: 10.1371/journal.pone.0091658
24. Suslov A, Boldanova T, Wang X, Wieland S, Heim MH. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology (2018) 154:1778–90. doi: 10.1053/j.gastro.2018.01.034
25. Mutz P, Metz P, Lempp FA, Bender S, Qu B, Schoneweis K, et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology (2018) 154:1791–804. doi: 10.1053/j.gastro.2018.01.044
26. Cheng X, Xia Y, Serti E, Block PD, Chung M, Chayama K, et al. Hepatitis B Virus Evades Innate Immunity of Hepatocytes But Activates Cytokine Production by Macrophages. Hepatology (2017) 66:1779–93. doi: 10.1002/hep.29348
27. Chen HS, Kaneko S, Girones R, Anderson RW, Hornbuckle WE, Tennant BC, et al. The Woodchuck Hepatitis Virus X Gene is Important for Establishment of Virus Infection in Woodchucks. J Virol (1993) 67:1218–26. doi: 10.1128/jvi.67.3.1218-1226.1993
28. Benn J, Schneider RJ. Hepatitis B Virus HBx Protein Deregulates Cell Cycle Checkpoint Controls. Proc Natl Acad Sci USA (1995) 92:11215–9. doi: 10.1073/pnas.92.24.11215
29. Guo SP, Wang WL, Zhai YQ, Zhao YL. Expression of Nuclear Factor-Kappa B in Hepatocellular Carcinoma and its Relation With the X Protein of Hepatitis B Virus. World J Gastroenterol (2001) 7:340–4. doi: 10.3748/wjg.v7.i3.340
30. Su F, Schneider RJ. Hepatitis B Virus HBx Protein Activates Transcription Factor NF-kappaB by Acting On Multiple Cytoplasmic Inhibitors of Rel-Related Proteins. J Virol (1996) 70:4558–66. doi: 10.1128/jvi.70.7.4558-4566.1996
31. Kim WH, Hong F, Jaruga B, Hu Z, Fan S, Liang TJ, et al. Additive Activation of Hepatic NF-kappaB by Ethanol and Hepatitis B Protein X (HBX) or HCV Core Protein: Involvement of TNF-Alpha Receptor 1-Independent and -Dependent Mechanisms. FASEB J (2001) 15:2551–3. doi: 10.1096/fj.01-0217
32. Wang Y, Cui L, Yang G, Zhan J, Guo L, Chen Y, et al. Hepatitis B E Antigen Inhibits NF-kappaB Activity by Interrupting K63-Linked Ubiquitination of NEMO. J Virol (2019) 93:e00667-18. doi: 10.1128/JVI.00667-18
33. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV Inhibits LPS-Induced NLRP3 Inflammasome Activation and IL-1beta Production via Suppressing the NF-kappaB Pathway and ROS Production. J Hepatol (2017) 66:693–702. doi: 10.1016/j.jhep.2016.12.018
34. Chen CH, Chen CY, Wang JH, Lai HC, Hung CH, Lu SN, et al. Comparison of Incidence of Hepatocellular Carcinoma Between Chronic Hepatitis B Patients With Cirrhosis Treated With Entecavir or Tenofovir in Taiwan - a Retrospective Study. Am J Cancer Res (2020) 10:3882–95.
35. Bertoletti A, Ferrari C. Innate and Adaptive Immune Responses in Chronic Hepatitis B Virus Infections: Towards Restoration of Immune Control of Viral Infection. Gut (2012) 61:1754–64. doi: 10.1136/gutjnl-2011-301073
36. Ryu HM, Park SG, Yea SS, Jang WH, Yang YI, Jung G. Gene Expression Analysis of Primary Normal Human Hepatocytes Infected With Human Hepatitis B Virus. World J Gastroenterol (2006) 12:4986–95. doi: 10.3748/wjg.v12.i31.4986
37. Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science (2014) 343:1221–8. doi: 10.1126/science.1243462
38. Liu H, Hou J, Zhang X. Targeting cIAPs, a New Option for Functional Cure of Chronic Hepatitis B Infection? Virol Sin (2018) 33:459–61. doi: 10.1007/s12250-018-0062-x
39. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB Activation Requires Coordinated Assembly of a Regulatory Complex of the Adaptors Ciap1, Ciap2, TRAF2 and TRAF3 and the Kinase NIK. Nat Immunol (2008) 9:1371–8. doi: 10.1038/ni.1676
40. Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP Antagonists Target Ciap1 to Induce TNFalpha-Dependent Apoptosis. Cell (2007) 131:682–93. doi: 10.1016/j.cell.2007.10.037
41. Ebert G, Preston S, Allison C, Cooney J, Toe JG, Stutz MD, et al. Cellular Inhibitor of Apoptosis Proteins Prevent Clearance of Hepatitis B Virus. Proc Natl Acad Sci USA (2015) 112:5797–802. doi: 10.1073/pnas.1502390112
42. Shrimali RK, Ahmad S, Verma V, Zeng P, Ananth S, Gaur P, et al. Concurrent PD-1 Blockade Negates the Effects of OX40 Agonist Antibody in Combination Immunotherapy Through Inducing T-Cell Apoptosis. Cancer Immunol Res (2017) 5:755–66. doi: 10.1158/2326-6066.CIR-17-0292
43. Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, et al. OX40 Signaling Favors the Induction of T(H)9 Cells and Airway Inflammation. Nat Immunol (2012) 13:981–90. doi: 10.1038/ni.2390
44. Publicover J, Gaggar A, Jespersen JM, Halac U, Johnson AJ, Goodsell A, et al. An OX40/OX40L Interaction Directs Successful Immunity to Hepatitis B Virus. Sci Transl Med (2018) 10:eaah5766. doi: 10.1126/scitranslmed.aah5766
45. Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, et al. Multidimensional Analyses Reveal Distinct Immune Microenvironment in Hepatitis B Virus-Related Hepatocellular Carcinoma. Gut (2019) 68:916–27. doi: 10.1136/gutjnl-2018-316510
46. Chen Y, Sun R, Wu X, Cheng M, Wei H, Tian Z. CD4+CD25+ Regulatory T Cells Inhibit Natural Killer Cell Hepatocytotoxicity of Hepatitis B Virus Transgenic Mice via Membrane-Bound TGF-Beta and OX40. J Innate Immun (2016) 8:30–42. doi: 10.1159/000431150
47. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-Induced NEMO-Independent Processing of NF-Kappa B2 in Maturing B Cells. Nat Immunol (2002) 3:958–65. doi: 10.1038/ni842
48. Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, et al. An Essential Role for BAFF in the Normal Development of B Cells Through a BCMA-Independent Pathway. Science (2001) 293:2111–4. doi: 10.1126/science.1061964
49. Yang C, Li N, Wang Y, Zhang P, Zhu Q, Li F, et al. Serum Levels of B-Cell Activating Factor in Chronic Hepatitis B Virus Infection: Association With Clinical Diseases. J Interferon Cytokine Res (2014) 34:787–94. doi: 10.1089/jir.2014.0032
50. Khlaiphuengsin A, Chuaypen N, Hirankarn N, Avihingsanon A, Crane M, Lewin SR, et al. Circulating BAFF and CXCL10 Levels Predict Response to Pegylated Interferon in Patients With HBeAg-Positive Chronic Hepatitis B. Asian Pac J Allergy Immunol (2019) 39:129–35. doi: 10.12932/AP-050718-0365
51. Salimzadeh L, Le Bert N, Dutertre CA, Gill US, Newell EW, Frey C, et al. PD-1 Blockade Partially Recovers Dysfunctional Virus-Specific B Cells in Chronic Hepatitis B Infection. J Clin Invest (2018) 128:4573–87. doi: 10.1172/JCI121957
52. Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, et al. CD40 Regulates the Processing of NF-Kappab2 P100 to P52. EMBO J (2002) 21:5375–85. doi: 10.1093/emboj/cdf542
53. Lind EF, Ahonen CL, Wasiuk A, Kosaka Y, Becher B, Bennett KA, et al. Dendritic Cells Require the NF-Kappab2 Pathway for Cross-Presentation of Soluble Antigens. J Immunol (2008) 181:354–63. doi: 10.4049/jimmunol.181.1.354
54. Rowe AM, Murray SE, Raue HP, Koguchi Y, Slifka MK. Parker DC. A Cell-Intrinsic Requirement for NF-kappaB-Inducing Kinase in CD4 and CD8 T Cell Memory. J Immunol (2013) 191:3663–72. doi: 10.4049/jimmunol.1301328
55. Li Y, Wang H, Zhou X, Xie X, Chen X, Jie Z, et al. Cell Intrinsic Role of NF-kappaB-Inducing Kinase in Regulating T Cell-Mediated Immune and Autoimmune Responses. Sci Rep (2016) 6:22115. doi: 10.1038/srep22115
56. Lacher SM, Thurm C, Distler U, Mohebiany AN, Israel N, Kitic M, et al. NF-kappaB Inducing Kinase (NIK) is an Essential Post-Transcriptional Regulator of T-Cell Activation Affecting F-Actin Dynamics and TCR Signaling. J Autoimmun (2018) 94:110–21. doi: 10.1016/j.jaut.2018.07.017
Keywords: hepatitis B virus (HBV), non-canonical NF-κB signaling, antiviral immunity, NF-κB-inducing kinase (NIK), p100/p52 (NFKB2)
Citation: Lu X, Chen Q, Liu H and Zhang X (2021) Interplay Between Non-Canonical NF-κB Signaling and Hepatitis B Virus Infection. Front. Immunol. 12:730684. doi: 10.3389/fimmu.2021.730684
Received: 25 June 2021; Accepted: 13 September 2021;
Published: 29 September 2021.
Edited by:
Mengji Lu, Essen University Hospital, GermanyCopyright © 2021 Lu, Chen, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyong Zhang, eGlhb3l6aGFuZ0BzbXUuZWR1LmNu