
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 September 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.729143
This article is part of the Research TopicNew Insights into B cell Subsets in Health and DiseaseView all 18 articles
 Arzoo M. Patel1
Arzoo M. Patel1 Yuxin S. Liu1,2
Yuxin S. Liu1,2 Scott P. Davies1Rachel M. Brown3Deirdre A. Kelly4
Scott P. Davies1Rachel M. Brown3Deirdre A. Kelly4 Dagmar Scheel-Toellner2
Dagmar Scheel-Toellner2 Gary M. Reynolds1,4†
Gary M. Reynolds1,4† Zania Stamataki1*†
Zania Stamataki1*†B lymphocytes are multitasking cells that direct the immune response by producing pro- or anti-inflammatory cytokines, by presenting processed antigen for T cell activation and co-stimulation, and by turning into antibody-secreting cells. These functions are important to control infection in the liver but can also exacerbate tissue damage and fibrosis as part of persistent inflammation that can lead to end stage disease requiring a transplant. In transplantation, immunosuppression increases the incidence of lymphoma and often this is of B cell origin. In this review we bring together information on liver B cell biology from different liver diseases, including alcohol-related and metabolic fatty liver disease, autoimmune hepatitis, primary biliary and primary sclerosing cholangitis, viral hepatitis and, in infants, biliary atresia. We also discuss the impact of B cell depletion therapy in the liver setting. Taken together, our analysis shows that B cells are important in the pathogenesis of liver diseases and that further research is necessary to fully characterise the human liver B cell compartment.
Liver disease is responsible for approximately 3.5% of deaths worldwide, with liver cirrhosis being the 11th most common cause of morbidity (1). As a consequence, there is high demand for donor livers for transplantation, the only effective current treatment. This makes the liver the second most frequent solid organ transplanted, with less than 10% of liver transplant needs being met (1); the discovery of alternative treatments is therefore essential in reducing the global demand for donor livers. In recent years, therapies which manipulate the immune system, an underlying factor in many disease settings, have reported efficacy in the liver (2). These approaches require an in-depth understanding of how cells of the adaptive immune response contribute to the progression of disease. B cells play a central role in the protection against pathogens, whilst also contributing to immune regulation and the maintenance of self-tolerance. B cells are also known to contribute to the pathogenesis of autoimmune disorders through the production of autoantibodies, antigen presentation and the secretion of pro-inflammatory cytokines (3, 4). The role of B cells in other chronic liver diseases is less clear. In this review, we will discuss descriptions of liver B cell subsets and how they may contribute to inflammation in the liver, with possibilities for therapeutic intervention.
B cells develop from haematopoietic stem cells (HSCs) in the bone marrow and progress from pro-B cell stages (expressing CD45 isoform B220) to pre-B cell stages (expressing CD19) (Figure 1) (8). The formation of the pre-B cell receptor (pre-BCR) involves the rearrangement and assembly of heavy and light immunoglobulins chains (8). B cells that possess a non-functional BCR are then deleted and those with an autoreactive pre-BCR either undergo apoptosis or receptor editing to produce a functional BCR (8, 9). These B cells further develop into immature B cells that express immunoglobulins (Ig)M and IgD. Immature B cells undergo another checkpoint where their BCR reactivity against autoantigens is monitored; B cells with high autoreactivity or low autoreactive BCRs are either deleted or undergo receptor editing to produce a functional BCR (10). Activation-induced cytidine deaminase (AID) is important in central B cell tolerance; Meyers et al., showed that there was an increase in the frequency of autoreactive clones, exiting the bone marrow, in AID-deficient patients (11). Developing B cells from humanized mice, deficient in AID expression failed to remove autoreactive clones displaying a vital role for AID expression in central B cell tolerance (12). Immature B cells with an autoreactive BCR, expressing recombination-activating gene 2 (RAG2) undergo secondary recombination to produce a non-autoreactive BCR (12). Those B cells with non-autoreactive BCRs then exit the bone marrow into the periphery and are termed transitional B cells (13, 14).
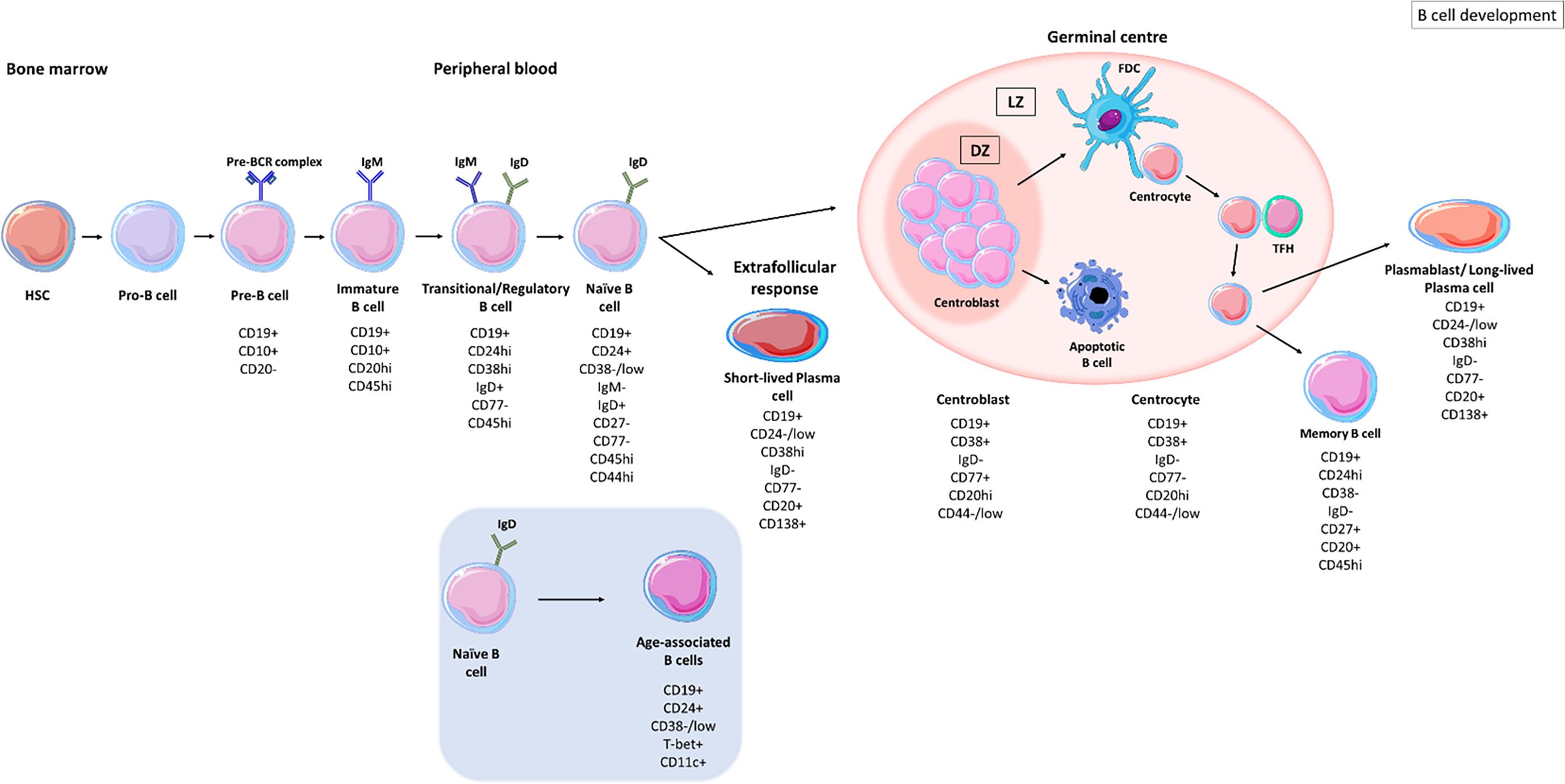
Figure 1 B cell development stages. B cells develop in the bone marrow from haematopoietic stem cells (HSCs), progressing from pro-B cell stages to pre-B cells before migrating into the circulation as transitional B cells. Upon antigen recognition, activated B cells migrate to secondary lymphoid organs and enter germinal centres where they undergo clonal expansion and somatic hypermutation (SHM) within the dark zones (DZ). B cells with disadvantageous mutations die by apoptosis whereas those B cells with improved receptor affinity interact with follicular dendritic cells (FDC) and T follicular helper cells (TFH), in the light zone (LZ). B cells undergo class switch recombination (CSR) and receive survival signals to differentiate into memory B cells and long-lived plasma cells (PCs) (5). Naïve B cells can differentiate into short-lived plasma cells through extrafollicular responses (6). Naïve B cells can also differentiate into age-associated B cells (ABCs) upon stimulation (7).
Transitional B cells are defined as CD19+ CD24hi CD38hi CD77- and express surface IgM (sIgM) and surface IgD (sIgD) (15). CD19+ CD24hi CD38hi populations also contain regulatory B cells (B-regs) which control the immune response through interleukin (IL)-10 and transforming growth factor β (TGF-β) secretion (16, 17). Transitional B cells migrate to secondary lymphoid organs (SLO) where they mature into naïve B cells, defined by CD19+ CD27- IgM+ IgD+ (CD24+ CD38-/low) waiting to encounter an antigen (5, 18, 19). If naïve B cells do not encounter their cognate antigen, they re-circulate back into the periphery and die within several days (5).
Upon antigen recognition, naïve B cells become activated and either differentiate to IgM-producing plasma cells as part of the extrafollicular response, where they form short-lived plasma cells (6) or enter secondary lymphoid tissues where they encounter T cells in the T cell zone. B cells that are co-stimulated by T cells enter B cell follicles where they differentiate into proliferating centroblasts forming a germinal centre (GC) (Figure 1) (20). Centroblasts rapidly proliferate in the dark zone of the germinal centre and somatic hypermutation (SHM) enters point mutations into the variable region genes. In the light zone, now differentiated to centrocytes (21), the B cells undergo selection based on affinity of their BCR. Centrocytes sample antigen from the surface of follicular dendritic cells and present it to follicular helper T cells (TFH) to undergo selection (22). Centrocytes may regain entry to the dark zone for further receptor editing or to undergo class-switch recombination (CSR) and leave the GC as memory B cells or as precursors to long-lived plasma cells (23).
Based on their IgD, CD27, CD38 and CD24 expression B cells can be separated into subpopulations of naïve and memory B cells. Common proteins that are used to identify B cell subsets are listed in Table 1. Unswitched memory B cells express IgM and CD27 on their surface, classical switched memory B cells are IgD- CD27+ and IgD- CD27- B cells, referred as double-negative (38, 57–60). This cell type is increased in inflammation caused by autoimmunity (58) or infection (61). Memory B cells that encounter antigen proliferate rapidly and mount a robust immune response (62). CD19+ CD24-/low CD38hi IgD- CD77- PBs are terminally differentiated B cells capable of secreting high affinity antibodies (15). PBs leave the GC and circulate in the blood to the bone marrow or to further target organs, where they further differentiate into long-lived plasma cells (PCs) (CD138+) that receive survival signals from their niche (62–65).

Table 1 Common proteins that are used to differentiate B cell subsets.
B cells with an exhausted memory-like phenotype are expanded in the peripheral blood of the elderly and are termed age-associated B cells (ABCs) (60). ABCs are characterised as CD19+ CD21lo CD11b+ CD11c+ and express the transcription factor, T-bet (60, 66–68). CD21 low B cell populations are likely to be heterogeneous and can show distinct stages of differentiation in different diseases. In SLE, they have been described as antibody secreting cells with germline-encoded Ig genes likely to belong to the extrafollicular response (69) while in other diseases, such as rheumatoid arthritis they have been described as memory B cells (70). This novel population of B cells has been found within the memory pool, contributing to inflammation associated with ageing, (‘inflammaging’) through the production of tumour necrosis factor-alpha (TNF-α) (60, 66). ABCs can be stimulated via BCR triggering or toll-like receptor (TLR) ligation to secrete pro-inflammatory cytokines (71, 72). Activation of ABCs also induces their differentiation into antibody secreting cells which may contribute to autoimmunity (72). Rubstov et al., showed that CD24- CD38- B cells are present at the onset of autoimmunity and that autoimmune mice depleted of CD24- CD38- B cells, had reduced number of autoantibodies, suggesting that this population plays a major role in the progression of autoimmunity (67, 73). A related population of B cells expressing the IgA receptor FcRL4 in the inflamed synovial tissue expresses RANKL and TNF in the inflamed synovium of patients with rheumatoid arthritis (50, 53, 74).
The liver is the largest internal organ with a remarkable ability to regenerate upon acute liver damage (75). Dual blood flow to the liver is supplied by the hepatic artery and portal vein, the latter accounting for over 80% of the liver’s blood supply that has passed through the spleen and gut (76). The liver is constantly exposed to gut-derived bacterial products, environmental toxins and food antigens and needs to maintain tolerance in order to prevent an over-active immune response resulting in hepatocyte damage (77–80). Frequent exposure to gut-derived toxins and antigens requires the liver to possess strong innate immune defences despite its constant state of immune tolerance (80–83). However, the liver can shift to a responsive state if an immune response is required (75, 77, 82, 84).
Acute hepatitis (liver inflammation) resolves upon the clearance of the pathogen or upon elimination of the cause of injury. Failure to clear the infection and resolve the inflammation results in the dysregulation of liver homeostasis and the progression to fibrosis (Figure 2) (76). Persistent liver insult can cause chronic inflammation and damage to hepatocytes, which can lead to cirrhosis, the major cause of mortality in chronic liver diseases (CLD) (85, 86). Patients with CLD are also at a higher risk of developing liver cancer (87).
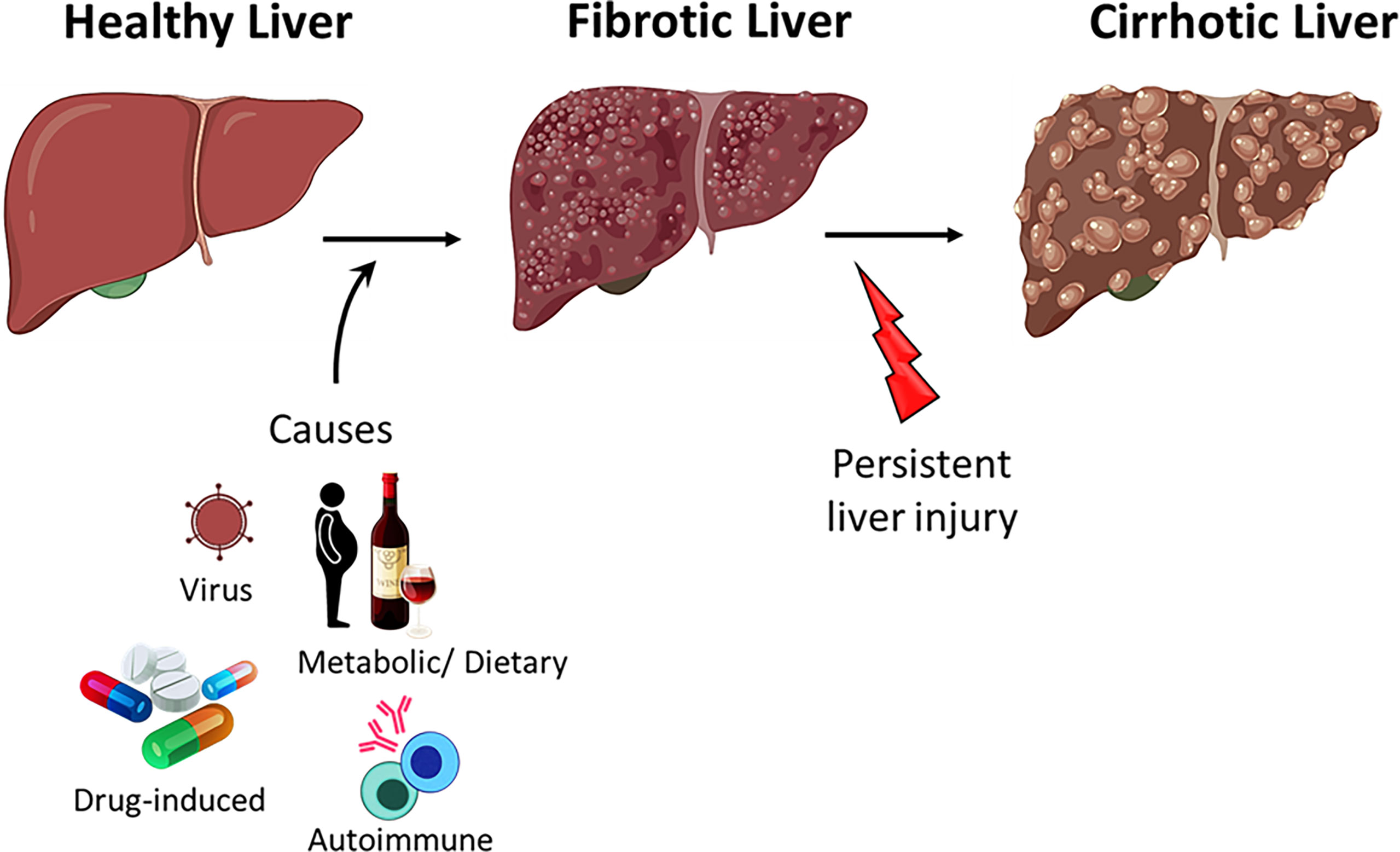
Figure 2 Progression of liver disease. Healthy liver can regenerate after acute injury however, persistent injury to the liver results in hepatocyte damage, inflammation and fibrosis. Persistent insult to the fibrotic liver may progress to cirrhosis.
B cells represent up to 50% of intrahepatic lymphocytes in mice with a higher expression of CD5 (88, 89). Novobrantseva et al., showed a role for B cells in fibrosis using carbon tetrachloride (CCL4) injections in mice deficient of B cells. B cell deficient mice showed a reduction in fibrotic deposition after 6 weeks of CCL4 injections when compared to wild-type mice, via accumulation of macrophages that contribute to fibroblast stimulation (88). B cells can contribute to collagen deposition by inducing the differentiation of hepatic stellate cells into myofibroblasts, via the production of IL-6 (90). In humans, B cells only account for 8% of the intrahepatic lymphocyte population (76).
Cirrhotic patients will eventually require a liver transplant. Orthotopic liver transplantation (OLT) requires chronic immunosuppressive therapy that can cause post-transplant lymphoproliferative disorders (PTLD) (91). Epstein-Barr virus (EBV) is associated with 60-70% of B cell PLD cases in patients on immunosuppressives (92). The suppressed immune system can no longer control the proliferation of EBV-transformed B cells (92).
Paediatric immune composition differs to that of adults (93). Dendritic cell and regulatory T cell numbers and functions are decreased in neonates (94). Neonates also have enhanced pro-inflammatory Th17 T cell responses and differences in the Th1/Th2 ratios, all of which could contribute to liver disease (94). The B cell compartment has not been widely characterised in neonatal livers. Duchamp et al., showed significant changes in B cell composition from birth to five years of age in peripheral blood (95). CD27+ IgM+ IgD+ memory B cells formed the largest compartment of B cells in the periphery of paediatric samples (95). B cell populations in paediatric livers remain to be thoroughly examined.
Neonates have incomplete development of the intrahepatic biliary tree and narrow bile ducts which affect the flow of bile and the production of mature bile acids (96). Infants may also have immature hepatocytes that are unable to detoxify and protect the liver from harmful substances (97). Stellate cells were shown to be increased in neonatal rats and they underwent myofibroblastic activation quicker than adult rat stellate cells (98). These may contribute to the rapid progression of liver disease in infants. Examining the B cell compartment in neonates may elucidate immune mechanisms that may contribute to liver disease progression.
We know that some liver diseases are specific to neonates (biliary atresia (97) and others that affect both neonates and adults (non-alcoholic fatty liver disease (NAFLD), autoimmune hepatitis (AIH), primary sclerosing cholangitis (PSC)) however, the differences between paediatric and adult hepatic immunity are poorly understood and not widely studied (96). Few studies have looked at differences in paediatric and adult NAFLD. These studies have found that neonatal NAFLD progressed more rapidly compared to adult NAFLD (99). Furthermore, paediatric NAFLD can be categorised into 2 phenotypes; adult-type (type 1 non-alcoholic steatohepatitis (NASH)) and paediatric-type (type 2 NASH) depending on histology (99, 100). Portal inflammation is mainly seen in children with NAFLD compared to lobular inflammation seen in adults (100). Adults have pericellular fibrosis whereas paediatric NAFLD show portal-periportal fibrosis (100). These discrepancies in histological features may contribute to the rapidly progressing NAFLD in children.
AIH in children presents with a more aggressive course compared to adults. Higher prevalence in females occurs in both paediatric and adult AIH (101). Infants and young children tend to present with type 2 AIH with IgA deficiency and raised levels of IgG (102). Those children with type 2 AIH that are positive for anti-liver kidney microsome type 1 (LKM1) have elevated bilirubin levels and can develop acute hepatic failure within 2-8 weeks of disease onset (102).
Alcohol related liver disease (ArLD) is associated with excessive consumption of alcohol causing hepatocyte damage and major shifts in metabolism leading to the retention of fat known as steatosis (86, 103, 104). Cessation of alcohol consumption at the point of early fibrosis and steatosis can reverse ArLD (105, 106). However, continued alcohol abuse can lead to the development of alcoholic steatohepatitis which progresses ultimately to cirrhosis (105–107). The toxic effects of acetaldehyde (the breakdown product of alcohol) cause enhanced lipogenesis resulting in the accumulation of fat molecules in the liver. Continued liver inflammation results in hepatic fibrosis and the formation of scar tissue which disrupts cellular formation (104).
ArLD patients have an altered B cell compartment; significant reductions in immature, memory and naïve B cells were reported in these patients, whilst the percentage of PBs were elevated (103). This increase in PBs may be responsible for high levels of IgA, IgG and IgM in ArLD. It can be hypothesised that a decline in regulatory B cells promotes the release of pro-inflammatory cytokines contributing to the exacerbation of inflammation, further activating immune cells and reducing the inhibitory function of regulatory B cell types (103, 108).
Programmed cell death ligand 1 (PD-L1), constitutively expressed on activated B cells, is the ligand for programmed cell death receptor 1 (PD-1) (109) and interaction between PD-1 and PD-L1 modulate immune responses (110). Kasztelan-Szczerbinska et al., showed a prevalence of PD-1/PD-L1 positive B cells in ALD females when compared to female controls. CD19+ PD-L1+ cells from female ALD patients correlated significantly with all conventional markers of inflammation (109). Sex hormones have been described to influence immune responses. There is evidence that oestrogen can regulate the immune response by modulating B cell function and impairing negative selection of high affinity auto-reactive B cells (111). Females with ArLD also present with elevated titres of circulating immunoglobulins and a variety of autoreactive antibodies (109). Steatohepatitis patients with more advanced disease have reduced numbers of sIgM+, soluble IgG+ (sIgG+) and soluble IgA+ (sIgA+)-reduced memory B cell numbers and increased sIgA+ class-switched memory B cells when compared to healthy controls (103, 108). In addition, alcoholic patients that show no sign of liver disease have a significant expansion of peripheral blood PBs and elevated sIgA+ memory cells (103).
Exposure to alcohol induces immune dysfunction and studies in human and animal models of ArLD show a decrease in B cell numbers (103, 107, 108). An impairment of B cell egress from the spleen to the blood, may account for the reduction in peripheral B cells (103). Despite this decline in B cells, ArLD is defined as an IgA-driven disorder with an increase in IgA complexes, and peripheral blood mononuclear cells (PBMCs) isolated from cirrhotic patients secrete significantly higher levels of IgA that correlate with serum IgA levels (103, 108). Deposition of IgA was observed in different organs and tissues in ArLD patients (103). Factors required for IgA class-switching, such as TGF-β were elevated in chronic ArLD patients together with a T-cell response from T-helper type 2 (Th2) cells (103).
A variety of toll-like receptors (TLRs) are expressed by B cells. TLR ligation activates B cells and is also required for B cell survival, antigen presentation and the production of cytokines and antibodies (112). In alcoholic cirrhosis, TLR-9 activated B cells were associated with a rise in IgA (80). However, Massonnet et al., noted a significant decrease in TLR-9 mRNA expression level in PBMCs from AC patients compared to healthy controls (108). Response to TLR stimulation was diminished in B cells isolated from alcoholic cirrhotic patients whereas, B cells from healthy controls produced IgA upon stimulation with CpG (103). However, B cells isolated from alcoholic cirrhotic patients exhibit an increase in IgA production when stimulated with CpG or R848, a TLR-7 agonist, compared to healthy controls (103). CpG-stimulated B cells, from cirrhotic ArLD patients, secreted more IgA, which may be due to the direct stimulation of B cells (108). B cells from ArLD patients, secreted a mean of 45 times more IgA in the absence of any stimulation compared to B cells from healthy controls (103). These studies show that TLR activation drives liver B cell responses in ArLD.
Alcohol has the ability to downregulate the expression of tight junction proteins permitting the transposition of bacterial constituents and causing a dysbiosis of gut flora, which may contribute to enhanced inflammation due to the presence of higher quantities of dangerous endotoxins (104). Altered intestinal permeability and bacterial translocation is often seen in ArLD patients (103, 107, 108). Impaired intestinal permeability results in the circulation of lipopolysaccharide (LPS), which was increased in the blood of ArLD patients (105, 106). LPS can activate immune cells via TLR-4 ligation resulting in further inflammation and damage to hepatocytes in ArLD (86, 105). Furthermore, alcoholic patients have elevated circulating levels of lipopolysaccharide binding protein (LBP) (103, 108). LBP elicits an immune response upon binding LPS, contributing to the inflammatory milieu and hepatocyte damage (105). LPS may trigger the migration of peripheral B cells towards gut-associated lymphoid tissue (GALT). Almeida et al., suggested that chronic alcoholic patients had increased numbers of GALT-derived sIgA+ B cells. This was supported by a significantly higher predominance of IgA+ memory B cells and IgA+ PBs in the peripheral blood of patients (103). In addition, they showed that peripheral blood sIgA+ memory B cells have GC-independent responses, similar to gut lamina propria IgA-producing cells, suggesting that this B cell population is the peripheral counterpart of gut lamina propria IgA-producing B cells (103). These results indicate that LPS, derived from the gut due to alcohol-induced intestinal permeability, could activate immune cells and initiate an inflammatory cascade, further exacerbating inflammation in ArLD.
Increased bacterial translocation results in chronic inflammation which coupled with alcohol abuse, damages hepatocytes (86, 107). Almedia et al., showed a reduction in circulating B cell numbers in ArLD patients; this may be due to alcohol-induced apoptosis of B cells (107). Hepatocyte and leukocyte damage was also mediated by reactive oxygen species (ROS) and acetaldehyde production (a product from the breakdown of alcohol), which destroys cell membranes (86). Bcl-2; a protein that regulates apoptosis, was strongly expressed on B cells in ArLD patients, correlating with the degree of portal and lobular inflammation (107). Significant volumes of cellular debris were produced due to Bcl-2-mediated B cell apoptosis and ROS-induced damage to hepatocytes and biliary epithelial cells (BECs). The release of cellular debris and intracellular proteins from cell debris may activate autoreactive B cells. ArLD patients had autoantibodies against modified liver, suggesting a dysregulated antibody response or impaired negative selection of B cells (105). This may be due to a breakdown in tolerance and a reduction in overall B-reg function.
25-60% of ArLD patients showed the presence of several self-recognising antibodies: mostly antiphospholipid, anti-nuclear, anti-dsDNA and anti-ssDNA (106). These autoantibodies arise due to alcohol-induced oxidative stress which damages cell structures and activates antigen presenting cells (APCs), which recognise haptens; a form of toxic metabolite (106). APCs induce the activation of T cells, which detect both self and non-self proteins, activating B cells to generate antibody secreting cells that release antibodies against proteins and haptens (106). TFH cell numbers were reduced in the blood as a result of excessive alcohol consumption (105). This may be due to the migration of TFH cells to local GC-like structures where they select the survival of B cells, allowing their differentiation into memory B cells and to high affinity antibody producing PCs, which are increased in ArLD patients.
To summarise, excessive alcohol consumption results in the breakdown of alcohol into acetaldehyde (Figure 3). This metabolite induces inflammation and damages cell membranes resulting in the exposure of cellular debris. Alcohol consumption also deregulates the gut barrier allowing bacterial translocation of LPS and other gut-derived pathogens, resulting in the secretion of inflammatory mediators which damage hepatocytes (113). Intracellular antigens from cell debris are engulfed by APCs and are presented to autoreactive T cells that become activated upon antigen recognition; B cells are activated as a consequence of T cell activation, migrate to the GC where they proliferate and differentiate into class-switched memory B cells and antibody secreting cells, with the aid of TFH cells. Increased immunoglobulin secretion ensues, forming immune complexes and further activating the immune response leading to liver injury.
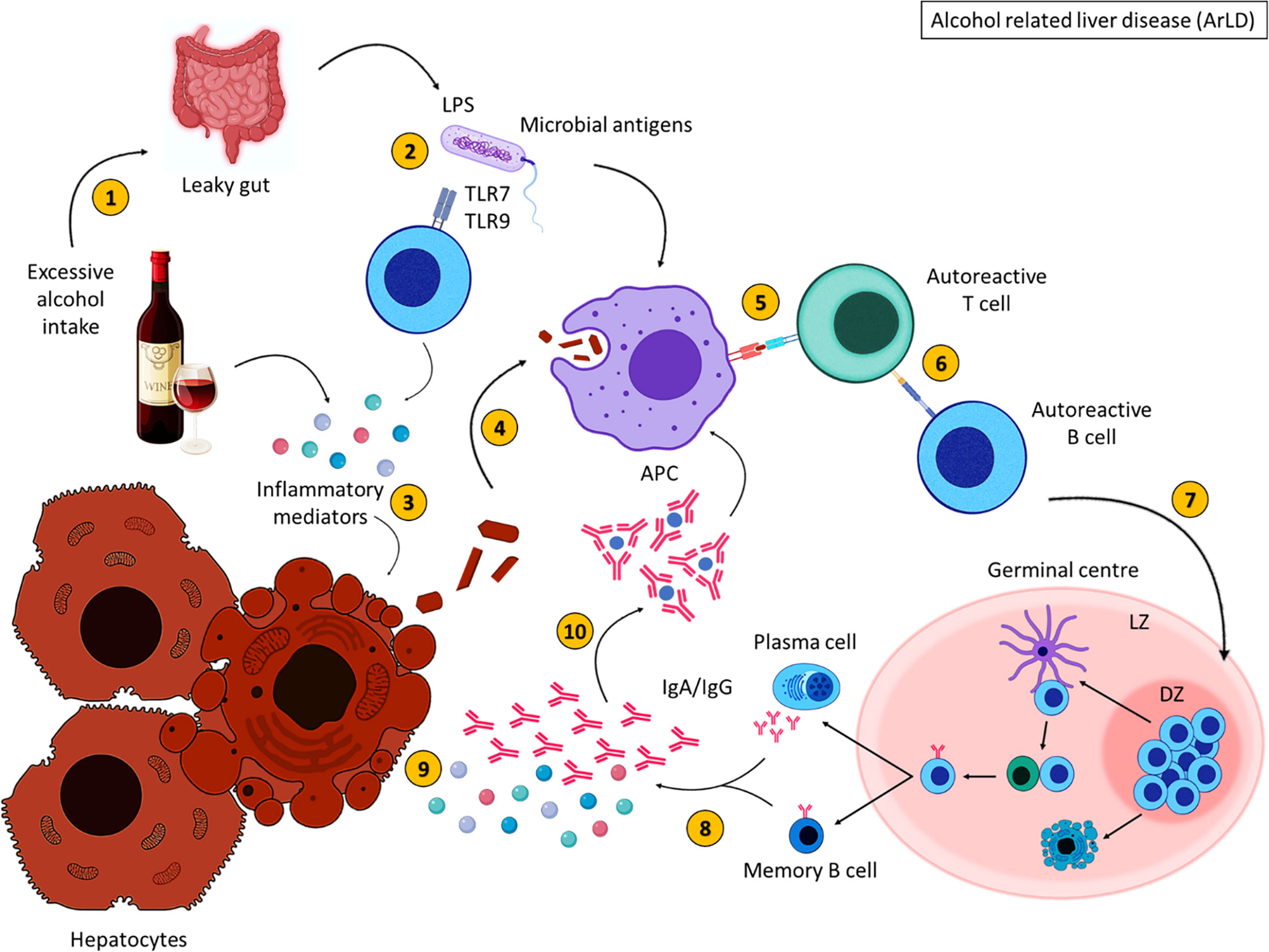
Figure 3 Alcohol related liver disease pathogenesis. Excessive alcohol consumption (1) induces inflammation and results in increased gut permeability (2), allowing bacterial translocation of LPS. Inflammatory mediators damage hepatocytes, resulting in the release of cellular debris (3). Self-antigens are engulfed by antigen presenting cells (4) and presented to autoreactive T cells (5), which stimulate autoreactive B cells (6). Activated B cells then migrate to secondary lymphoid tissues and undergo germinal centre reactions (7) where B cells with increased affinity receptors differentiate into memory B cells and PCs (8). The secretion of inflammatory mediators and autoantibodies from memory B cells and PCs further damage hepatocytes (9). The formation of immune complexes induces further inflammation (10). These immune complexes are engulfed by APCs. Created with BioRender.com.
Fat accumulation in the liver causes a range of conditions described as non-alcoholic fatty liver disease (NAFLD) (114). NAFLD can progress from the abnormal retention of lipids in the liver (steatosis) to non-alcoholic steatohepatitis (NASH), where lipid retention is accompanied with hepatic inflammation (114, 115). NASH patients have varying degrees of fibrosis, initiated due to the inflammatory damage of hepatocytes inducing their apoptosis (86). Fibrosis develops to cirrhosis with the eventual requirement of a liver transplant (114, 115). NAFLD patients frequently present with extrahepatic conditions such as obesity, type 2 diabetes, cardiovascular diseases and osteoporosis (115, 116). NAFLD/NASH patients have persistent injury to the hepatocytes due to ROS, lipotoxicity and the secretion of inflammatory mediators from immune cells (115).
The pathogenesis of NAFLD is considered to be a ‘two-hit’ theory; first-hit is the excessive lipid influx and/or a reduction in lipid clearance due to abnormal liver lipid metabolism and the second-hit is the inflammatory process (117), which leads to lobular and portal inflammation and infiltration of activated immune cells (115). Patients with NAFLD had altered hepatic lymphocyte compartments (114), and increased B cells (117) that were associated with disease severity (118). Ectopic lymphoid structures with B cell and T cell aggregates are seen in ~60% of patients with NASH, these aggregates correlate in size and prevalence with lobular inflammation (116). B cells may be involved in fibrosis through the production of inflammatory mediators that stimulate hepatic stellate cells, these cells support liver B cell survival and maturation into plasma cells (116). Isolated B cells from the visceral adipose tissue (VAT) of obese mice show elevated production of pro-inflammatory cytokines whilst a lack of B cells improves fat-induced inflammation (116), suggesting that B cells play an important role in the progression of NAFLD to NASH.
Obese people have altered distribution of adipose tissue. Obesity promotes B cell activation, an early event in the development of experimental NASH animal models, contributing to the progression of steatohepatitis (115). In mice, mesenteric adipose tissue (MAT), located between the gut and liver, affects the liver by secreting inflammatory cytokines, adipocytokines and releasing free fatty acids (FFA) that reach the liver via the portal vein (119). B cells from high fat diet (HFD)-fed mice produce IgG and promote epididymal adipose tissue (EAT) inflammation (119). The release of cytokines from inflamed adipose tissue combined with ROS production from dysregulated hepatocyte lipid metabolism, contribute to the progression from steatosis to NASH (86). Intestinal permeability was compromised in NAFLD allowing bacterial translocation and inducing the activation of hepatic inflammatory cells. Patients with NAFLD had elevated serum levels of endotoxin compared to healthy controls (118). Bacterial translocation and LPS promote hepatic inflammation, lipid accumulation and hepatocyte damage (86, 118). Furthermore, hepatic B cells encourage local inflammatory responses when stimulated with LPS (80).
ROS and hepatocyte apoptosis result in the expulsion of hepatocyte cellular debris, inducing antibody production from B cells as a consequence. NAFLD/NASH patients had raised titres of IgG against oxidative stress-derived epitopes (OSE). Patients with increased anti-OSE IgG had a higher prevalence of fibrosis and/or cirrhosis with elevated serum levels of interferon gamma (IFN-γ) (115). PBs upregulate MHC class II as a result of B cell activation in NASH, suggesting that they have a role in presenting OSE to T cells that become activated and contribute to NASH progression (115). Aggregates of B and T cells were observed in 63% of NASH liver samples correlating with the severity of lobular infiltration and enhancement of fibrosis (115). These aggregates were also linked to an increase in anti-OSE IgG titres (115).
To summarise, NASH arises as a result of lipid accumulation within the liver which results in inflammation and fibrosis (Figure 4). Activation of various immune cells and the secretion of inflammatory mediators damages hepatocytes, further activating immune cells and initiating an inflammatory loop. B cells produce antibodies against OSE, contributing to increased cytokine production, activation of T cells and the production of ROS, all of which participate in damaging the liver. This vicious cycle of liver destruction results in fibrosis, progressing to cirrhosis making the liver unable to regenerate and heal.
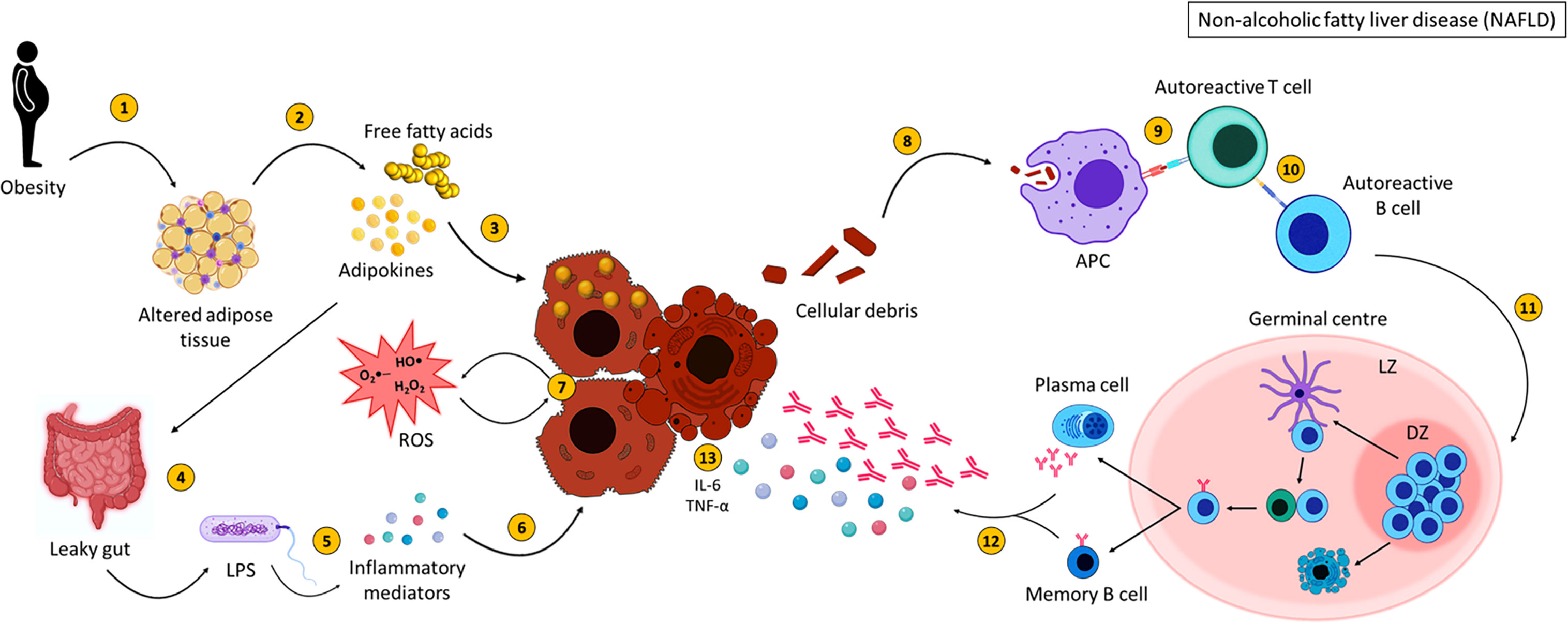
Figure 4 Non-alcoholic fatty liver disease pathogenesis. NASH pathogenesis is linked to obesity and altered adipose tissue distribution (1). Adipose tissue releases adipokines and free fatty acids (FFA) (2), which result in lipid accumulation within the liver (3) and affect intestinal permeability (4). This allows bacterial translocation of LPS and other gut-derived pathogens (5) resulting in the secretion of inflammatory mediators which could damage hepatocytes (6). FFA, adipokines, ROS and inflammatory mediators injure hepatocytes (7) resulting in the expulsion of cellular debris. Self-antigens are engulfed by antigen presenting cells (8) and presented to autoreactive T cells (9), which stimulate autoreactive B cells (10). Activated B cells then migrate to secondary lymphoid tissues and undergo germinal centre reactions (11), where B cells with increased affinity receptors differentiate into memory B cells and PCs (12). The secretion of pro-inflammatory mediators (interleukin-6 and tumour necrosis factor alpha) (120) and autoantibodies from memory B cells and PCs further damage hepatocytes (13). Created with BioRender.com.
There are five types of viral hepatitis, of which hepatitis B and hepatitis C viruses can cause chronic liver disease. Infection may lead to progressive inflammation and liver damage over decades that could lead to end-stage disease requiring a transplant, however these viruses are not directly cytopathic (75, 121).
Antibodies against both viruses are generated in infected individuals. In HBV, IgM antibodies against the core protein are used as a marker of early infection whereas, antibodies against the hepatitis surface (HBsAg) and e (HBeAg) antigens appear as the infection progresses (121, 122). HBsAg, a T cell-independent antigen, induces the activation of naïve B cells and mounts a robust antibody response (71, 123). Adults infected with HBV are able to resolve infection in the majority of cases, and there is an effective vaccine for HBV based on the HBsAg (124). Chronic HBV patients have circulating HBsAg-specific B cells, however these cells are unable to produce effective anti-HBs antibodies (71). The early humoral immune response in HCV is difficult to study as many patients are asymptomatic (125).
Intriguingly, antibodies to the envelope glycoproteins in HCV infection may emerge late and are abundant in chronic patients, while at the same time showing potent neutralising ability of heterologous viruses. Investigations in autologous virus neutralisation over years in a single patient showed that this RNA virus remained a step ahead of the antibody response by generating variants to escape neutralising antibodies (126). However, antibodies can prevent HCV infection in vivo and contribute to the eradication of the HCV infection (127). There is no vaccine against HCV infection, but immunisation of healthy volunteers with viral envelope glycoproteins resulted in the generation of neutralising antibodies (128), and antibodies were shown to be protective in a human liver chimeric mouse model (129). Immunisation of genetically humanised mice with soluble envelope glycoprotein 2 (sE2), derived from insect cells, produced high titres of broadly neutralising antibodies against diverse HCV envelopes and were protected from HCV infection, in vivo. Immunisation of non-primates with insect derived sE2 resulted in the induction of B and T cell immunity (127). The role of B cells and antibodies in the context of failure to control HCV infection was elegantly described by Dustin et al., (130).
An accumulation of circulating B cells within the liver is associated with severe liver damage (131) and elevated levels of activated B cells is seen in patients with HBV and HCV (121). However, these cells have a reduced proliferative capacity and express Fc receptor-like protein 4 (FcRL4), an inhibitory receptor overexpressed on exhausted memory B cells (121). This suggests that B cells are dysfunctional in infected livers as they are chronically activated and adopt an exhausted phenotype. Hepatic release of subviral particles (empty virions consisting of mostly HBsAg) (132) is an immune evasion mechanism in HBV which forms immune complexes by crosslinking neutralising antibodies targeting the virus. This leads to continual BCR triggering, promoting the expansion of exhausted memory B cells, also referred to as atypical memory B cells (71, 123, 133). Chronic hepatitis B patients had deposits of HBcAg-immune complexes in their liver (134). Fc receptor-like protein 5 (FcRL5) suppresses the activation of B cells by crosslinking to immune complexes and PD-1 inhibits B cell signalling; both these markers were enriched on the surface of atypical memory B cells; T-bet is also associated with the generation of atypical memory B cells (71, 135). This population of atypical memory B cells was found to be present in infected livers (71) Atypical memory B cells enriched in HBV were unable to escape apoptosis and differentiate into effective HBsAg-specific antibody secreting cells (71), impairing their ability to produce neutralising antibodies against the viruses (123, 133, 134).
IL-10 producing B-reg cells are another subset of regulatory B cells that are enriched in HBV and HCV patients, which may contribute to viral persistence (71, 121, 136–138). Eiza et al., showed an increased in IL-10 producing B-regs in chronic HBV patients, when compared to healthy controls and these cells were able to dampen down HBV-specific CD8+ T cell responses (138). A subset of B-reg cells that express high levels of CD5, CD1d and IgD are thought to be responsible for IL-10 production by B cells (136, 138). CD5+ B-regs produce IL-10 upon activation and correlate with poor virus elimination (138).
Beyond immune surveillance, we previously showed that B cells were vehicles for HCV transmission to hepatocytes (139). Stimulated B cells were able to bind viral particles using scavenger receptor B type 1 and C-type lectins DC-SIGN and L-SIGN and internalised the virus in compartments that prevented virus degradation. The intact virus was then recycled to the B cell surface within hours. B cell-transmitted virus was more infectious than cell-free virus, adding a pathogenic role for B cells in HCV infection. HCV RNA was detected in 83% (110/132) of patients with HCV genotype 1 (140). Inokuchi et al., reported that HCV RNA was detected more frequently in B cells compared to CD4+ and CD8+ T cells (141). The role of antibodies and adaptive immunity in HCV infection has been recently reviewed (142–144).
The most common B cell lymphoproliferative disease associated with HCV is mixed cryoglobulinemia (MC) (145, 146). MC presents with formations of cryoglobulins; abnormally precipitated immunoglobulins that can be coupled with rheumatoid factor (147), detected in the circulation of 40-60% of HCV-infected patients (148, 149). B cells contribute to the formation of cryoglobulins through uncontrolled autoantibody production and proliferation (149). These cryoglobulin-containing immune complexes deposit in small or medium vessels causing vasculitis (145, 150, 151). Whilst cryoglobulinemia is common in HCV, rare cases have been reported to exist in HBV infected patients (151). The clonal proliferation of B cells in MC (152), may cause the formation of ectopic lymphoid aggregates within the liver of HCV patients. Lauletta et al., has shown that cytokine (CXCL13) can cause B cell migration to intraportal lymphoid aggregates in the liver and create a microenvironment to sustain B cell aggregation (153).
Autoimmune hepatitis (AIH) is a chronic autoimmune disorder requiring life-long immunosuppressive therapy (75, 154–156). This disease affects all ages, races and sexes although it has a higher prevalence in females (75, 157–159). AIH is associated with other autoimmune diseases such as coeliac disease and can coexist with autoimmune family biliary liver diseases; primary biliary cholangitis (PBC) or primary sclerosing cholangitis (PSC) (159). This progressive, necro-inflammatory disease is linked to increased immune infiltration that destroys the hepatic parenchyma through immune-mediated hepatocyte damage (75, 107, 154, 155, 157, 158, 160, 161). Fibrosis and cirrhosis are ramifications of chronic inflammation and 40% of AIH patients present with cirrhosis at the time of diagnosis (154). Despite the use of corticosteroids and immunosuppressives, 10-20% of patients with AIH will progress to end-stage liver disease requiring liver transplantation (160).
AIH classification is dependent on antibody specificity. Patients with AIH can have numerous autoantibodies (162), including antinuclear antibodies (ANAs), smooth muscle antibodies (SMA) and antibodies directed against liver kidney microsome type 1 (LKM1) (107, 157, 159, 163). Type 1 AIH is characterised by the presence of ANA, SMA and perinuclear anti-neutrophil cytoplasmic antibodies (pANCA), the latter is present in 65-92% type 1 AIH patients (157, 159). Autoantibodies against liver cytosol type 1 (LC1) and/or anti-LKM1 antibodies are classified as type 2 AIH (159, 160); pANCA antibodies are not present in this type of AIH. CYP2D6 is the antigen for anti-LKM antibodies and anti-LC1 antibodies target a liver-specific metabolic enzyme, formiminotransferase cyclodeaminase (FTCD) (157, 160). CYPD26 autoantibodies are of the IgG isotype, supporting the role that T-dependent class-switching is essential to produce IgG+ PCs (158). Anti-LC1 antibody titres are associated with disease severity and are detected in 30-50% of patients with type 2 AIH (157). Type 3 AIH is proposed to be defined by the presence of anti-soluble liver antigen/liver pancreas antigen antibodies (anti-SLA/LP antibodies) which are present in 10-30% of AIH patients (157). 50-76% of AIH patients have antibodies against the asialoglycoprotein receptor (ASGPR) which is a component of the liver specific lipoprotein (LSP) expressed on hepatocyte surfaces (157, 163). Disease activity and poor outcome of AIH positively correlated with titres of anti-ASGPR in this group of patients (157).
Hepatic destruction in AIH is thought to be driven by T cells, however, the presence of several autoantibodies suggests a role for B cells in the pathogenesis of AIH (154). Elevated serum IgG levels are found in up to 85% of patients with AIH displaying ongoing inflammation within these patients (164). AIH liver biopsies showed mixed infiltration of T cells and B cells, including IgG+ B cells and PCs (157–159, 163, 165). AIH flare ups show an increased number of T and B cells present within the liver (159). B cells present self-antigens to autoreactive T cells which become activated, which then stimulate B cells to produce autoantibodies (163). Increased expression of CD86 is seen on B cells from new onset AIH patients, suggesting that these B cells are primed to co-stimulate T cells (166). Cytokines produced by Th2 cells also aid in B cell activation and differentiation via IL-4 production, which was elevated in AIH (157, 163).
IgG+ cells were significantly higher in AIH liver samples and were found distributed around the bile ducts and in portal tract areas, along with IgM+ cells (157, 158, 161, 165). Lymphocytes target IgG bound to hepatocytes in healthy individuals, mediating cellular injury and initiating inflammation (75).
Patients with systemic lupus erythematosus (SLE), an autoimmune disease driven by a dysregulated B cell response, frequently develop inflammation of the liver. Hepatic dysfunction in SLE patients can be caused independently of B cell responses for example by side effects of medication. However, two key examples for autoimmune liver conditions associated with SLE are lupus hepatitis (also known as SLE-associated hepatitis) and autoimmune hepatitis. Both involve extensive B cell activation and are often difficult to distinguish. They are both associated with hyperglobulinaemia but show differences in the profile of autoantibodies. Since their prognosis and therapeutic approach differs, it is an important goal to develop safe diagnostic criteria (167).
AIH is an autoimmune disease with many factors contributing to disease progression, however the trigger is unknown. The presence of autoantibodies, targeting many self-proteins, presents an important role for autoreactive B cells in the pathogenesis of AIH and suggests an impairment in central B cell tolerance. The survival and activation of autoreactive T and autoreactive B cells is a result of a breakdown in self-tolerance and a reduction in immune regulation.
Primary sclerosing cholangitis (PSC) is a cholestatic autoimmune disease in which fibrosis and chronic inflammation destroy the large bile ducts (157, 168–170). PSC is associated with inflammatory bowel disease (IBD); 87% of PSC patients present with ulcerative colitis (UC) and 13% have Crohn’s disease (CD) (83, 157, 168). Chronic destruction and scarring of the biliary tree leads to cirrhosis and many patients will eventually require liver transplants (169).
Anti-neutrophil cytoplasmic antibodies (ANCA) are detected in 88% of PSC patients however, these autoantibodies are not specific for PSC, but also seen in AIH and biliary atresia (BA) (157); PSC-specific autoantibodies have not been identified to date, but disease-relevant epitopes have been detected (171). PSC disease severity is associated with concentrations of anti-cardiolipin antibodies which were present in 2/3 of PSC patients (157).
Total numbers of B cells were significantly higher in PSC-derived PBMCs compared with healthy controls (64). Furthermore, 10% of PSC patients had elevated serum levels of IgG4 and a significant infiltration of IgG4 PCs (169, 172). IgG4+ PC aggregates were observed in PSC tissues and IgG4+ deposits were reported (169, 173). Fischer et al., showed that the intensity of IgG4+ immunostaining was linked to disease progression and infiltration of lymphocytes in PSC (169). B cells isolated from PSC liver explants produce a range of autoantibodies when cultured suggesting, that the targets in PSC are self-antigens or arise as a result of cross-reactivity of exogenous targets (168). Approximately 50% of explanted PSC liver specimens displayed evidence of IgG4+ cells and these tissue infiltrating IgG4+ cells were associated with a clinically aggressive disease course and a higher probability of liver transplantation (169). IgG4-related disease (IgG4RD) is an inflammatory disease associated with elevated numbers of IgG4-positive PCs which contribute to chronic damage and fibrosis (174, 175). IgG4RD-associated sclerosing cholangitis can be mistaken for PSC, which may explain the increase in IgG4+ cells seen by Fischer et al. (176).
70% of PSC patients have IBD which is linked to defects in the intestinal barrier (177). The gut microbiota was altered in PSC patients when compared to UC and healthy controls (168). Gut-derived antigens may trigger the autoimmune response in PSC by allowing the translocation of bacterial and food antigens (78, 168). BECs propagate their own destruction when they are stimulated by LPS, which induces them to release chemokines and cytokines (83). These mediators activate various immune cells which damage the tissue leading to fibrosis and resulting in an inflammatory cascade (83). Other gut-derived bacterial motifs also stimulated BECs to drive their own destruction and analysis from PSC livers showed the presence of bacterial RNA (83).
The pathogenesis of PSC is reviewed by Lleo et al. (178). PSC may be initiated by a loss of self-tolerance due to bacterial antigens and the obliteration of BECs, resulting in the expulsion of self-antigens which activates autoreactive immune cells. Molecular mimicry may contribute to this initial loss in tolerance. Primed gut-derived T cells migrate to the liver where they may induce B cell proliferation and differentiation into IgG4+ secreting PCs (83). These immune cells will secrete many pro-inflammatory cytokines contributing to inflammation, the destruction of BECs and the progression of autoimmunity.
Primary biliary cholangitis (PBC) is a progressive autoimmune disease characterised by immune-mediated destruction of the intrahepatic small bile ducts (79, 107, 157, 179–182). This deregulated immune response results in liver inflammation and damage, causing fibrosis and eventually cirrhosis as an outcome of the accumulation of bile toxins (79, 183, 184).
There is a profound loss of B cell tolerance associated with PBC, which is supported by the presence of autoantibodies (83, 180, 185, 186); 90-95% of PBC patients have the presence of specific anti-mitochondrial antibodies (AMA), directed against the mitochondrial inner membrane member, 2-oxoacid dehydrogenase complexes (2-OADC) (79, 107, 157, 179, 181–183, 185–187). Autoantibodies targeting the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2) is a major autoantigen in PBC (157). 50% of PBC patients had antibodies targeting the nuclear pore complex members; gp210 and p62 (157). The anti-nuclear antibodies (ANAs) targeting gp210, correlate with disease severity (79, 157, 186). PBC patients had significantly higher PDC-E2 specific IgM, IgG, and IgA PB frequency (64). In addition, many PBC patients present with hyper-IgM expression in their serum (179, 186). Complement activation via agglutination by IgM plays a crucial role in innate immunity providing a link between innate and adaptive immunity as IgM enhances antigen-driven IgG responses (179).
GCs are essential for the production of class-switched immunoglobulins however, they also allow the differentiation of autoreactive B cells into autoreactive memory and autoantibody producing PCs in PBC (183, 185, 186, 188). TFH cells promote GC formation and allow B and T cell interaction promoting B cell activation, proliferation and differentiation into affinity matured, long-lived PCs (79, 183). TFH locate B cell follicles via the CXCL5 – CXCL13 chemokine axis and were present in vast numbers near damaged bile ducts, in lymphoid follicle-like structures (79). In healthy control livers, hepatic TFH cells were absent (183). PBC-derived TFH cells had a greater ability to induce B cell differentiation into class-switched memory B cells and mature PCs (183). Circulating TFH (cTFH) cell frequency was higher in PBC patients and in patients who do not respond to ursodeoxycholic acid (UDCA) treatment compared to UDCA responders (79). cTFH cells positively correlated with circulating PCs in PBC and secrete high levels of IL-21 inducing B cell proliferation, differentiation and secretion of autoantibodies suggesting that TFH cells contribute to PBC pathogenesis (79, 183, 185). Increased serum IL-21 levels positively correlated with concentrations of serum AMA and IgM (185). IL-21 is vital for the development of TFH cells and induces maturation of B cells in a paracrine manner whilst enhancing TFH function in an autocrine fashion (79, 183).
Tissue from the livers of PBC patients showed the presence of several bacterial products (83). TLR signalling pathway was activated in PBC patients and hyper IgM production which may be due to increased bacterial infections (83, 157). Studies have shown that the induction of PBC occurs due to molecular mimicry between PDC-E2 and bacterial proteins (79). Molecular mimicry may be the initial insult in the loss of self-tolerance, enabling the survival of autoreactive B cells that fail to enter apoptosis (186). Activation of TLRs induces the proliferation of B cells and the secretion of pro-inflammatory cytokines. TLR-9 expression was increased in B cells from PBC patients and CpG stimulation enhanced the secretion of IgM, cytokines and chemokines (83, 157, 179, 187). Kikuchi et al., showed a positive correlation between the intensity of TLR-9 expression and IgM+ memory B cells (83, 179). Bacterial motifs were required to increase TLR-9 expression on B cells and promote inflammation (83, 179). Furthermore, CpG stimulation of PBMCs derived from PBC patients resulted in vast production of AMAs compared to unstimulated controls (157). TLRs are also expressed by cholangiocytes which aid in immune activation and may contribute to PBC pathogenesis. TLR-4 and TLR-9 levels were highly expressed on cholangiocytes in PBC patients (75). Ma et al., showed increased TLR-4 expression on BECs in PBC and expression was seen in periportal and interlobular hepatocytes in patients with advanced disease (83).
BEC themselves may contribute to the initiation and progression of PBC rather than being the victims of the immune response. Damage to BEC is a hallmark of PBC and BEC obtained from PBC livers rapidly undergo apoptosis (186). BEC can engulf apoptotic BECs and translocate PDC-E2 into apoptotic bodies (186). The immunologically intact PDC-E2 is presented to autoreactive immune cells initiating their activation, secretion of pro-inflammatory mediators and AMA production (186).
Many factors contribute to the initiation and pathogenesis of PBC which is reviewed Carbone et al., (189). The initial insult in PBC is thought to be similar to that of PSC; molecular mimicry by bacterial motifs, subsequently activating the immune response and breaking down self-tolerance. The inflammatory milieu is further exacerbated by the destruction of BEC, which further activate autoreactive immune cells via antigen presentation of PDC-E2 in apoptotic bodies. The ongoing inflammatory cascade results in additional destruction of bile ducts, activation of autoreactive immune cells and the production of autoantibodies.
Biliary atresia (BA) affects 1 in 8,000-18,000 neonates and encompasses a host of potential aetiologies leading to progressive liver damage (190–192). Obliteration of the extrahepatic biliary tree and subsequent progressive destruction of the hepatic ducts leads to fibrosis and cirrhosis in BA infants (192, 193).
There are two forms of BA; acquired and congenital (194, 195). 80% of BA patients have the acquired form and 20% have the congenital form, both are characterised by destruction of bile ducts and fibrosis, with various degrees of inflammation (191, 195, 196). BA infants with the congenital form also present with other genetic abnormalities (195, 196). Kasai portoenterostomy (Kasai) is a surgical treatment performed at diagnosis in over 95% of BA infants (190). The Kasai procedure removes the damaged bile ducts and anastomoses the jejunum to patient intrahepatic bile ducts to allow bile flow from the liver to the gut; despite successful surgery, 80% of BA patients will require a liver transplant (191, 197). Medical management post-Kasai involves the use of antibiotics, vitamin supplementation, nutritional support and administration of UDCA to encourage bile flow (192). Kelly and Davenport show that having specialised centres for portoenterostomy surgery has improved survival to over 90% in the UK. This study also showed a reduced need for liver transplantation due to the centralisation of surgery (192).
BA livers showed increased immune infiltration and elevated lymphocyte activation in the portal tracts (191, 193). There was an increased presence of intrahepatic periductal B cells in BA patients at diagnosis and at the time of transplant (190). These activated B cells secrete IgM and IgG antibodies and Lu et al., found that IgG from the sera of BA patients reacted with the cholangiocyte cytosol (198, 199). Furthermore, 40% of BA infants had deposits of IgM and IgG along the basement membrane of the bile duct epithelia (190, 198, 200). Infants with BA show increased levels of high-affinity pathogenic IgG antibodies and a reduction in the level of natural IgM, which plays a protective role in immune function and the development of autoimmune disease (201). Anti-α-enolase and ANCA autoantibodies are observed in BA neonates and were detected in the sera of BA patients (190, 198). Anti-α-enolase IgM and IgG antibodies can be found in BA children who still have their own livers suggesting a role for B cells in BA pathogenesis (199).
There are various animal models of BA (195, 202) however the commonly used model is the rhesus group A-rotavirus (RRV)-induced mouse model of BA (193, 199, 203, 204). RRV-induced mice are able clear the virus by 2 weeks however, they show signs of extrahepatic bile duct obstruction and progressive inflammation, which leads to liver failure (199). Despite the evidence of viral insult in mouse models of BA, there are conflicting studies detecting the presence of rotavirus in BA patient samples. One study shows the presence of type C rotavirus RNA in 10 out of 20 BA liver samples (205) whereas Bobo et al., did not detect any rotavirus RNA from their BA liver cohort (n=10) (206).
To summarise the trigger for BA is unknown; viral, environmental, genetic and autoimmune factors are thought to contribute to BA pathogenesis (196, 198). A proposed theory for the pathogenesis of some types of BA is an initial infection with a cholangiotropic virus which may damage the bile duct epithelia directly, however this virus is still unidentified (195) (Figure 5). This initiates an immune response resulting in an exaggerated inflammatory response that further damages BEC (196). The injured bile ducts release altered self-antigens and may express self-antigens on their surface (194, 207). APCs recognise these self-antigens as foreign molecules subsequently, activating autoreactive T cells, mediating inflammatory destruction of the bile ducts (194). Activated autoreactive T cells also stimulate autoreactive B cells, augmenting the production of inflammatory mediators and initiating B cell differentiation. Despite the clearance of the virus, persistent inflammation contributes to the obliteration of the bile ducts leading to fibrosis and liver failure (199). It is important to stress, that inflammation is evident in a subsection of patients, and some children with BA show no inflammatory histological findings at Kasai or at end stage disease explant tissue. Histological characterisation of the immune compartment in BA may aid our understanding of disease pathogenesis.
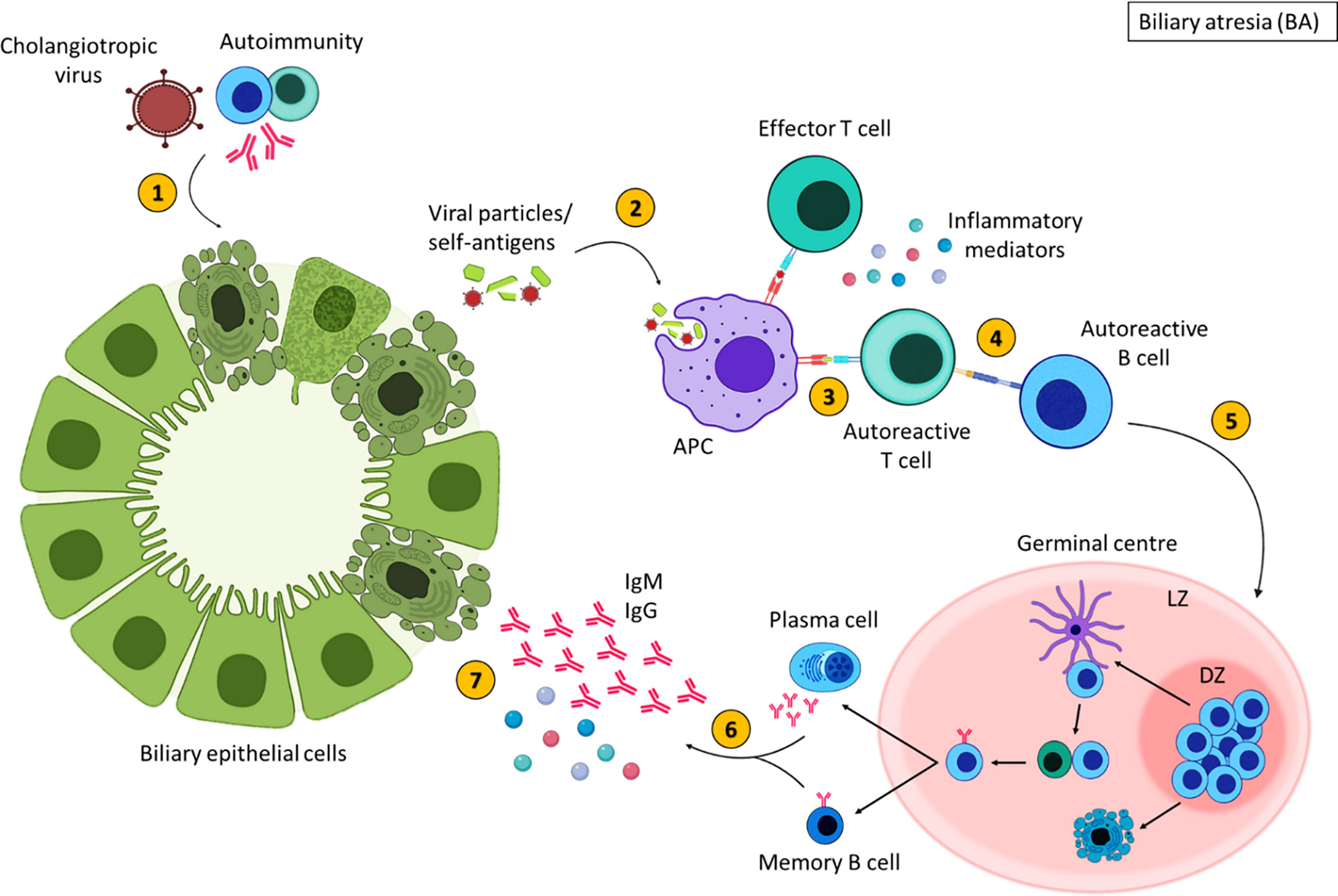
Figure 5 Inflammatory-mediated damage in biliary atresia. In some children with BA, damage to the extrahepatic bile ducts may occur due to cholangiotropic viruses or autoimmunity (1), resulting in the expulsion of viral or self-antigens. These antigens are engulfed by antigen presenting cells (2) and presented to T cells (3). Autoreactive T cells that recognise self-antigens stimulate autoreactive B cells (4). Activated B cells then migrate to secondary lymphoid tissues and undergo germinal centres reactions (5) where B cells with increased affinity receptors differentiate into memory B cells and PCs (6). The secretion of inflammatory mediators and autoantibodies from memory B cells and PCs further damage BECs (7). Created with BioRender.com.
Table 2 provides a brief summary of clinical features and immune involvement in liver diseases.

Table 2 Key clinical features and immune involvement in liver diseases.
Originally developed for the treatment of B cell lymphoma, rituximab is a human/murine chimeric monoclonal antibody that targets specifically the cell surface glycoprotein CD20 (208). CD20 is universally expressed by normal B cells through all stages of development from late pre-B cells in the bone marrow and right before terminal differentiation to plasma cells.
The true role of CD20 remains poorly understood; it has no known natural ligand, however its association with the BCR suggests a role in B cell signalling. CD20 is not immediately internalised upon antibody binding (209, 210), and thus monoclonal antibodies raised against it cannot be used to deliver cytotoxic moieties into the cell. As a result, the mode of action of anti-CD20 antibodies relies on the subsequent recruitment of the host immune response to opsonisation.
Multiple modes of actions have been proposed for rituximab mediated B cell depletion. Rituximab colocalises CD20 to lipid rafts (211), and through this induces B cell killing by NK cells through antibody-dependent cellular cytotoxicity (212). Efficacy of rituximab, however, differs greatly among different autoimmune diseases. Amongst these, Rituximab is approved for treatment of rheumatoid arthritis, granulomatosis with polyangiitis and microscopic polyangiitis and pemphigus vulgaris (213–215). In SLE, pilot trials and observational studies were initially promising but larger scale clinical trials did not show a clear benefit. Recent trials of a combination of Belimumab, which targets the cytokine BLyS with Rituximab, however, show promise in SLE (216). Direct cross-linking of CD20 on B cell tumour cell lines was shown to be sufficient for the induction of apoptosis through MAP kinase activation (212, 217). Rituximab may also induce complement dependent cytotoxicity (212, 217, 218). In a mouse model, Kupffer cells within the hepatic sinusoids have been shown to capture anti-CD20 antibody coated B cells (219).
During differentiation into mature antibody-secreting plasma cells, CD20 expression is lost (220). Due to this absence of CD20, rituximab treatment does not affect the production of long-lived PCs (221, 222), as rituximab does not deplete long-lived PCs. In multiple diseases where rituximab treatment has been trialled, 90-100% of peripheral B cells were depleted (223).
Efficacy of rituximab, however, differs greatly among different autoimmune diseases. Amongst others, rituximab has been shown to be an effective treatment for rheumatoid arthritis (224), systemic lupus erythematosus (225), thrombocytopenic purpura (226), and autoimmune haemolytic anaemia (227). B cells have now begun to be targeted in CLD (3).
Rituximab has been shown to be the most widely used treatment for HCV patients with cryoglobulinemia vasculitis (148, 228). One cycle of low-dose rituximab achieved a complete clinical response in 22 out of 31 (70.96%) of MC patients (229). Clinical manifestations of cryoglobulinemia such as skin ulcers, renal manifestations and sensitive-motor neuropathy have improved through the use of rituximab (228). Rituximab treatment reduces serum levels of cryoglobulins and rheumatoid factor through the clonal B cell depletion in the bone marrow (230).
Non-specific immunosuppression using prednisolone and azathioprine has improved symptoms and subsequently survival in patients with AIH (231). However, some patients either develop adverse side effects and as a consequence discontinue treatment or exhibit a suboptimal response to this standard therapy (232). As a result, more targeted immunotherapies for this disease are needed.
In a mouse model of AIH, administration of anti-CD20 antibodies resulted in a significant reduction in liver inflammation and ALT levels, but there was no reduction in the total IgG levels or autoantibody titres (233). The depletion of B cells resulted in a significant increase in naïve CD4+ and CD8+ T cells and a reduction in antigen-experienced T cells. In this model of AIH, B cells played an active role in disease pathogenesis through the antigen presentation process and modulated T cell functions (233).
Rituximab has been trialed in both adult and paediatric patients with AIH which was unresponsive to prior treatments (234, 235). Rituximab was well tolerated, and complete remission was achieved and maintained. Serum IgG levels were also reduced, and ANA titres were decreased in 2 out of 6 subjects, becoming negative in one (234). More recently a multicentre retrospective study reported clinically meaningful reductions in liver enzyme values following the administration of rituximab in 22 patients with difficult to manage AIH (236). After treatment, 71% of patients were free from AIH flares (236).
Currently, therapy for PBC is limited to UDCA and, for patients with end-stage liver disease, liver transplantation. Although UDCA has demonstrated clinical benefits in liver biochemistries (237), up to 40% of patients have a suboptimal response to UDCA and 10% will go on to die or require liver transplantation (238).
At present, trials for the efficacy of rituximab in PBC have primarily enrolled patients who have demonstrated an unsatisfactory response to UDCA. Six patients with incomplete responses to UDCA were recruited in an open-label study (239). Patients were given 2 doses of rituximab separated by 2 weeks and followed for 52 weeks. This study showed a significant reduction in serum AMA titres and a reduction in ALP up to 36 weeks after treatment. A subsequent open-label study using the same method of treatment enrolled 14 patients with PBC refractory to UDCA (240). B cells were effectively depleted in 13 of the patients, and a reduction in serum AMA levels was observed at 6 months follow-up. However, the improvements in liver biochemistry were limited. Rituximab has also been used in a randomised trial of 57 PBC patients suffering with severe fatigue (241). Despite evidence to suggest that rituximab was effective for reduction of fatigue in a number of conditions including primary sjogrens syndrome (pSS) (242–245), a condition associated with PBC, this study showed no evidence of effectiveness for the treatment of fatigue in PBC.
Although these studies showed the limited efficacy of rituximab in PBC, they demonstrated that the drug is well tolerated by patients. This is in direct contrast to a study with a xenobiotic induced murine model of human PBC (246), where anti-CD20 treatment exacerbated liver pathology despite successful depletion of B cells and reduction in the production of AMAs (247). Conversely, in the genetic animal model of PBC, the dnTGF-βRII mouse (248), anti-CD20 treatment was effective at attenuating liver damage but exacerbates colitis (249). Moreover, this reduction in liver inflammation was only seen in young mice, as B cells depletion in old mice did not modify the course of liver disease (249). Interestingly, double transgenic mice with PBC and B cell depletion (lgμ-/-dnTGF-βRII mice) developed a more severe form of cholangitis (250), suggesting that during the initial inflammatory response in this model of PBC, B cells have a suppressive effect.
A lack of understanding of PSC pathogenesis has prevented the development of effective therapies. Transplantation was established as the only curative treatment option for PSC in 1983. A few years later, recurrence after liver transplantation was noted in some patients (251). It is estimated that recurrent PSC occurs in 20-25% of patients over a 10-year period after transplantation (252). A small study of 5 PSC patients who underwent ABO incompatible liver transplantation and were treated with rituximab, found that graft survival rate was 100% with no cases of recurrence over the median follow-up period of 7.2 years (253).
Despite advances in liver T cell biology, B cell biology and subset characterisation remains understudied in the context of chronic liver disease. Deep phenotyping approaches such as single cell RNA sequencing and spatial transcriptomics have yielded valuable information on liver immunity in the context of various liver cell types (254), and similar approaches are much needed for B cell biology. Mapping the B cell compartment in liver diseases will provide a better understanding of the roles of B cells in disease progression and offer new opportunities for therapeutic intervention.
AP, YL, DS-T, and ZS researched and composed the review. SD, RB, DK, and GR provided helpful critique of the manuscript. GMR and ZS are joint last authors. All authors contributed to the article and approved the submitted version.
This work is supported by a Birmingham Women’s and Children’s Hospital Charity BCHRF546 to AP, a NC3R trainee postdoctoral fellowship UKRI NC/R002061/1 to SD, a Medical Research Foundation intermediate career fellowship UKRI, MRF-169-0001-F-STAM-C0826 to ZS, an EU/EFPIA Innovative Medicines Initiative (IMI) 2 RTCure 777357 to DS-T and a Wellcome Trust funded PhD programme “MIDAS” 108871/B/15/Z to YL and DS-T.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Figures were created with BioRender.com.
1. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of Liver Diseases in the World. J Hepatol (2019) 70:151–71. doi: 10.1016/j.jhep.2018.09.014
2. Uhl P, Fricker G, Haberkorn U, Mier W. Current Status in the Therapy of Liver Diseases. Int J Mol Sci (2014) 15:7500–12. doi: 10.3390/ijms15057500
3. Cargill T, Culver EL. The Role of B Cells and B Cell Therapies in Immune-Mediated Liver Diseases. Front Immunol (2021) 12:661196. doi: 10.3389/fimmu.2021.661196
4. Hampe CS. B Cell in Autoimmune Diseases. Scientifica (Cairo) (2012) 2012:1–18. doi: 10.6064/2012/215308
5. Perez-Andres M, Paiva B, Nieto WG, Caraux A, Schmitz A, Almeida J, et al. Human Peripheral Blood B-Cell Compartments: A Crossroad in B-Cell Traffic. Cytometry B Clin Cytom (2010) 78:S47–60. doi: 10.1002/cyto.b.20547
6. Elsner RA, Shlomchik MJ. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity (2020) 53:1136–50. doi: 10.1016/j.immuni.2020.11.006
7. Ma K, Du W, Wang X, Yuan S, Cai X, Liu D, et al. Multiple Functions of B Cells in the Pathogenesis of Systemic Lupus Erythematosus. Int J Mol Sci (2019a) 20:1–19. doi: 10.3390/ijms20236021
8. Pieper K, Grimbacher B, Eibel H. B-Cell Biology and Development. J Allergy Clin Immunol (2013) 131:959–71. doi: 10.1016/j.jaci.2013.01.046
9. Meffre E, Casellas R, Nussenzweig MC. Antibody Regulation of B Cell Development. Nat Immunol (2000) 1:379–85. doi: 10.1038/80816
10. Matthias P, Rolink AG. Transcriptional Networks in Developing and Mature B Cells. Nat Rev Immunol (2005) 5:497–508. doi: 10.1038/nri1633
11. Meyers G, Ng YS, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, et al. Activation-Induced Cytidine Deaminase (AID) is Required for B-Cell Tolerance in Humans. Proc Natl Acad Sci USA (2011) 108:11554–9. doi: 10.1073/pnas.1102600108
12. Cantaert T, Schickel JN, Bannock JM, Ng YS, Massad C, Oe T, et al. Activation-Induced Cytidine Deaminase Expression in Human B Cell Precursors Is Essential for Central B Cell Tolerance. Immunity (2015) 43:884–95. doi: 10.1016/j.immuni.2015.10.002
13. Melchers F. Checkpoints That Control B Cell Development. J Clin Invest (2015) 125:2203–10. doi: 10.1172/JCI78083
14. Pelanda R, Torres RM. Central B-Cell Tolerance: Where Selection Begins. Cold Spring Harb Perspect Biol (2012) 4:a007146. doi: 10.1101/cshperspect.a007146
15. van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJ. Replication History of B Lymphocytes Reveals Homeostatic Proliferation and Extensive Antigen-Induced B Cell Expansion. J Exp Med (2007) 204:645–55. doi: 10.1084/jem.20060964
16. Liu Y, Cheng LS, Wu SD, Wang SQ, Li L, She WM, et al. IL-10-Producing Regulatory B-Cells Suppressed Effector T-Cells But Enhanced Regulatory T-Cells in Chronic HBV Infection. Clin Sci (Lond) (2016) 130:907–19. doi: 10.1042/CS20160069
17. Rosser EC, Mauri C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
18. Chung JB, Silverman M, Monroe JG. Transitional B Cells: Step by Step Towards Immune Competence. Trends Immunol (2003) 24:343–9. doi: 10.1016/S1471-4906(03)00119-4
19. Hobeika E, Maity PC, Jumaa H. Control of B Cell Responsiveness by Isotype and Structural Elements of the Antigen Receptor. Trends Immunol (2016) 37:310–20. doi: 10.1016/j.it.2016.03.004
20. MacLennan IC. Germinal Centers. Annu Rev Immunol (1994) 12:117–39. doi: 10.1146/annurev.iy.12.040194.001001
21. Carsetti R, Rosado MM, Wardmann H. Peripheral Development of B Cells in Mouse and Man. Immunol Rev (2004) 197:179–91. doi: 10.1111/j.0105-2896.2004.0109.x
22. Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. Regulation of the Germinal Center Response. Front Immunol (2018) 9:2469. doi: 10.3389/fimmu.2018.02469
23. Allen CD, Okada T, Cyster JG. Germinal-Center Organization and Cellular Dynamics. Immunity (2007) 27:190–202. doi: 10.1016/j.immuni.2007.07.009
24. Chaudhry MS, Karadimitris A. Role and Regulation of CD1d in Normal and Pathological B Cells. J Immunol (2014) 193:4761–8. doi: 10.4049/jimmunol.1401805
25. Gary-Gouy H, Harriague J, Bismuth G, Platzer C, Schmitt C, Dalloul AH. Human CD5 Promotes B-Cell Survival Through Stimulation of Autocrine IL-10 Production. Blood (2002) 100:4537–43. doi: 10.1182/blood-2002-05-1525
26. Mishra D, Singh S, Narayan G. Role of B Cell Development Marker CD10 in Cancer Progression and Prognosis. Mol Biol Int (2016) 2016:4328697. doi: 10.1155/2016/4328697
27. Vorup-Jensen T, Jensen RK. Structural Immunology of Complement Receptors 3 and 4. Front Immunol (2018) 9:2716. doi: 10.3389/fimmu.2018.02716
28. Karnell JL, Kumar V, Wang J, Wang S, Voynova E, Ettinger R. Role of CD11c(+) T-Bet(+) B Cells in Human Health and Disease. Cell Immunol (2017) 321:40–5. doi: 10.1016/j.cellimm.2017.05.008
29. Golinski ML, Demeules M, Derambure C, Riou G, Maho-Vaillant M, Boyer O, et al. CD11c(+) B Cells Are Mainly Memory Cells, Precursors of Antibody Secreting Cells in Healthy Donors. Front Immunol (2020) 11:32. doi: 10.3389/fimmu.2020.00032
30. Wang K, Wei G, Liu D. CD19: A Biomarker for B Cell Development, Lymphoma Diagnosis and Therapy. Exp Hematol Oncol (2012) 1:36. doi: 10.1186/2162-3619-1-36
31. Otero DC, Anzelon AN, Rickert RC. CD19 Function in Early and Late B Cell Development: I. Maintenance of Follicular and Marginal Zone B Cells Requires CD19-Dependent Survival Signals. J Immunol (2003) 170:73–83. doi: 10.4049/jimmunol.170.1.73
32. Lee KY, Jeon SY, Hong JW, Kim YH, Song KH, Kim KH. CD20 Positive T Cell Lymphoma Involvement of Skin. Ann Dermatol (2011) 23:529–35. doi: 10.5021/ad.2011.23.4.529
33. Pavlasova G, Mraz M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica (2020) 105:1494–506. doi: 10.3324/haematol.2019.243543
34. Cherukuri A, Cheng PC, Pierce SK. The Role of the CD19/CD21 Complex in B Cell Processing and Presentation of Complement-Tagged Antigens. J Immunol (2001) 167:163–72. doi: 10.4049/jimmunol.167.1.163
35. Fang X, Zheng P, Tang J, Liu Y. CD24: From A to Z. Cell Mol Immunol (2010) 7:100–3. doi: 10.1038/cmi.2009.119
36. Ayre DC, Pallegar NK, Fairbridge NA, Canuti M, Lang AS, Christian SL. Analysis of the Structure, Evolution, and Expression of CD24, an Important Regulator of Cell Fate. Gene (2016) 590:324–37. doi: 10.1016/j.gene.2016.05.038
37. Agematsu K, Hokibara S, Nagumo H, Komiyama A. CD27: A Memory B-Cell Marker. Immunol Today (2000) 21:204–6. doi: 10.1016/S0167-5699(00)01605-4
38. Wu YC, Kipling D, Dunn-Walters DK. The Relationship Between CD27 Negative and Positive B Cell Populations in Human Peripheral Blood. Front Immunol (2011) 2:81. doi: 10.3389/fimmu.2011.00081
39. Costa F, Dalla Palma B, Giuliani N. CD38 Expression by Myeloma Cells and Its Role in the Context of Bone Marrow Microenvironment: Modulation by Therapeutic Agents. Cells (2019) 8:1-12. doi: 10.3390/cells8121632
40. Glaria E, Valledor AF. Roles of CD38 in the Immune Response to Infection. Cells (2020) 9:1-16. doi: 10.3390/cells9010228
41. Kremmidiotis G, Zola H. Changes in CD44 Expression During B Cell Differentiation in the Human Tonsil. Cell Immunol (1995) 161:147–57. doi: 10.1006/cimm.1995.1021
42. Hogerkorp CM, Bilke S, Breslin T, Ingvarsson S, Borrebaeck CA. CD44-Stimulated Human B Cells Express Transcripts Specifically Involved in Immunomodulation and Inflammation as Analyzed by DNA Microarrays. Blood (2003) 101:2307–13. doi: 10.1182/blood-2002-06-1837
43. Giovannone N, Antonopoulos A, Liang J, Geddes Sweeney J, Kudelka MR, King SL, et al. Human B Cell Differentiation Is Characterized by Progressive Remodeling of O-Linked Glycans. Front Immunol (2018) 9:2857. doi: 10.3389/fimmu.2018.02857
44. Mangeney M, Richard Y, Coulaud D, Tursz T, Wiels J. CD77: An Antigen of Germinal Center B Cells Entering Apoptosis. Eur J Immunol (1991) 21:1131–40. doi: 10.1002/eji.1830210507
45. Mangeney M, Rousselet G, Taga S, Tursz T, Wiels J. The Fate of Human CD77+ Germinal Center B Lymphocytes After Rescue From Apoptosis. Mol Immunol (1995) 32:333–9. doi: 10.1016/0161-5890(95)00004-X
46. Hogerkorp CM, Borrebaeck CA. The Human CD77- B Cell Population Represents a Heterogeneous Subset of Cells Comprising Centroblasts, Centrocytes, and Plasmablasts, Prompting Phenotypical Revision. J Immunol (2006) 177:4341–9. doi: 10.4049/jimmunol.177.7.4341
47. Mongini PK, Tolani S, Fattah RJ, Inman JK. Antigen Receptor Triggered Upregulation of CD86 and CD80 in Human B Cells: Augmenting Role of the CD21/CD19 Co-Stimulatory Complex and IL-4. Cell Immunol (2002) 216:50–64. doi: 10.1016/S0008-8749(02)00512-9
48. Suvas S, Singh V, Sahdev S, Vohra H, Agrewala JN. Distinct Role of CD80 and CD86 in the Regulation of the Activation of B Cell and B Cell Lymphoma. J Biol Chem (2002) 277:7766–75. doi: 10.1074/jbc.M105902200
49. McCarron MJ, Park PW, Fooksman DR. CD138 Mediates Selection of Mature Plasma Cells by Regulating Their Survival. Blood (2017) 129:2749–59. doi: 10.1182/blood-2017-01-761643
50. Yeo L, Lom H, Juarez M, Snow M, Buckley CD, Filer A, et al. Expression of FcRL4 Defines a Pro-Inflammatory, RANKL-Producing B Cell Subset in Rheumatoid Arthritis. Ann Rheum Dis (2015) 74:928–35. doi: 10.1136/annrheumdis-2013-204116
51. Ehrhardt GR, Hsu JT, Gartland L, Leu CM, Zhang S, Davis RS, et al. Expression of the Immunoregulatory Molecule FcRH4 Defines a Distinctive Tissue-Based Population of Memory B Cells. J Exp Med (2005) 202:783–91. doi: 10.1084/jem.20050879
52. Li H, Dement-brown J, Liao PJ, Mazo I, Mills F, Kraus Z, et al. Fc Receptor-Like 4 and 5 Define Human Atypical Memory B Cells. Int Immunol (2020) 32:755–70. doi: 10.1093/intimm/dxaa053
53. Amara K, Clay E, Yeo L, Ramskold D, Spengler J, Sippl N, et al. B Cells Expressing the IgA Receptor FcRL4 Participate in the Autoimmune Response in Patients With Rheumatoid Arthritis. J Autoimmun (2017) 81:34–43. doi: 10.1016/j.jaut.2017.03.004
54. Franco A, Kraus Z, Li H, Seibert N, Dement-Brown J, Tolnay M. CD21 and FCRL5 Form a Receptor Complex With Robust B-Cell Activating Capacity. Int Immunol (2018) 30:569–78. doi: 10.1093/intimm/dxy052
55. Wang P, Wang Y, Xie L, Xiao M, Wu J, Xu L, et al. The Transcription Factor T-Bet Is Required for Optimal Type I Follicular Helper T Cell Maintenance During Acute Viral Infection. Front Immunol (2019) 10:606. doi: 10.3389/fimmu.2019.00606
56. Hansen IS, Baeten DLP, Den Dunnen J. The Inflammatory Function of Human IgA. Cell Mol Life Sci (2019) 76:1041–55. doi: 10.1007/s00018-018-2976-8
57. Fecteau JF, Cote G, Neron S. A New Memory CD27-IgG+ B Cell Population in Peripheral Blood Expressing VH Genes With Low Frequency of Somatic Mutation. J Immunol (2006) 177:3728–36. doi: 10.4049/jimmunol.177.6.3728
58. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A New Population of Cells Lacking Expression of CD27 Represents a Notable Component of the B Cell Memory Compartment in Systemic Lupus Erythematosus. J Immunol (2007) 178:6624–33. doi: 10.4049/jimmunol.178.10.6624
59. Berkowska MA, Driessen GJ, Bikos V, Grosserichter-Wagener C, Stamatopoulos K, Cerutti A, et al. Human Memory B Cells Originate From Three Distinct Germinal Center-Dependent and -Independent Maturation Pathways. Blood (2011) 118:2150–8. doi: 10.1182/blood-2011-04-345579
60. Buffa S, Pellicano M, Bulati M, Martorana A, Goldeck D, Caruso C, et al. A Novel B Cell Population Revealed by a CD38/CD24 Gating Strategy: CD38(-)CD24 (-) B Cells in Centenarian Offspring and Elderly People. Age (Dordr) (2013) 35:2009–24. doi: 10.1007/s11357-012-9488-5
61. Bernard NJ. Double-Negative B Cells. Nat Rev Rheumatol (2018) 14:684. doi: 10.1038/s41584-018-0113-6
62. Bernasconi NL, Onai N, Lanzavecchia A. A Role for Toll-Like Receptors in Acquired Immunity: Up-Regulation of TLR9 by BCR Triggering in Naive B Cells and Constitutive Expression in Memory B Cells. Blood (2003) 101:4500–4. doi: 10.1182/blood-2002-11-3569
63. Caraux A, Klein B, Paiva B, Bret C, Schmitz A, Fuhler GM, et al. Circulating Human B and Plasma Cells. Age-Associated Changes in Counts and Detailed Characterization of Circulating Normal CD138- and CD138+ Plasma Cells. Haematologica (2010) 95:1016–20. doi: 10.3324/haematol.2009.018689
64. Zhang J, Zhang W, Leung PS, Bowlus CL, Dhaliwal S, Coppel RL, et al. Ongoing Activation of Autoantigen-Specific B Cells in Primary Biliary Cirrhosis. Hepatology (2014) 60:1708–16. doi: 10.1002/hep.27313
65. Arpin C, Dechanet J, Van Kooten C, Merville P, Grouard G, Briere F, et al. Generation of Memory B Cells and Plasma Cells In Vitro. Science (1995) 268:720–2. doi: 10.1126/science.7537388
66. Duggal NA, Upton J, Phillips AC, Sapey E, Lord JM. An Age-Related Numerical and Functional Deficit in CD19(+) CD24(hi) CD38(hi) B Cells is Associated With an Increase in Systemic Autoimmunity. Aging Cell (2013) 12:873–81. doi: 10.1111/acel.12114
67. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-Like Receptor 7 (TLR7)-Driven Accumulation of a Novel CD11c(+) B-Cell Population is Important for the Development of Autoimmunity. Blood (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
68. Trivedi N, Weisel F, Smita S, Joachim S, Kader M, Radhakrishnan A, et al. Liver Is a Generative Site for the B Cell Response to Ehrlichia Muris. Immunity (2019) 51:1088–101.e5. doi: 10.1016/j.immuni.2019.10.004
69. Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, Cellular Origin and Autoreactivity of Antibody-Secreting Cell Population Expansions in Acute Systemic Lupus Erythematosus. Nat Immunol (2015) 16:755–65. doi: 10.1038/ni.3175
70. Thorarinsdottir K, Camponeschi A, Cavallini N, Grimsholm O, Jacobsson L, Gjertsson I, et al. CD21(-/Low) B Cells in Human Blood are Memory Cells. Clin Exp Immunol (2016) 185:252–62. doi: 10.1111/cei.12795
71. Burton AR, Pallett LJ, Mccoy LE, Suveizdyte K, Amin OE, Swadling L, et al. Circulating and Intrahepatic Antiviral B Cells are Defective in Hepatitis B. J Clin Invest (2018) 128:4588–603. doi: 10.1172/JCI121960
72. Naradikian MS, Hao Y, Cancro MP. Age-Associated B Cells: Key Mediators of Both Protective and Autoreactive Humoral Responses. Immunol Rev (2016) 269:118–29. doi: 10.1111/imr.12380
73. Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. TLR7 Drives Accumulation of ABCs and Autoantibody Production in Autoimmune-Prone Mice. Immunol Res (2013) 55:210–6. doi: 10.1007/s12026-012-8365-8
74. Yeo L, Toellner KM, Salmon M, Filer A, Buckley CD, Raza K, et al. Cytokine mRNA Profiling Identifies B Cells as a Major Source of RANKL in Rheumatoid Arthritis. Ann Rheum Dis (2011) 70:2022–8. doi: 10.1136/ard.2011.153312
75. Bogdanos DP, Gao B, Gershwin ME. Liver Immunology. Compr Physiol (2013) 3:567–98. doi: 10.1002/cphy.c120011
76. Robinson MW, Harmon C, O’Farrelly C. Liver Immunology and its Role in Inflammation and Homeostasis. Cell Mol Immunol (2016) 13:267–76. doi: 10.1038/cmi.2016.3
78. Jeffery HC, van Wilgenburg B, Kurioka A, Parekh K, Stirling K, Roberts S, et al. Biliary Epithelium and Liver B Cells Exposed to Bacteria Activate Intrahepatic MAIT Cells Through MR1. J Hepatol (2016) 64:1118–27. doi: 10.1016/j.jhep.2015.12.017
79. Zhou ZQ, Tong DN, Guan J, Li MF, Feng QM, Zhou MJ, et al. Circulating Follicular Helper T Cells Presented Distinctively Different Responses Toward Bacterial Antigens in Primary Biliary Cholangitis. Int Immunopharmacol (2017) 51:76–81. doi: 10.1016/j.intimp.2017.08.004
80. Byun JS, Yi HS. Hepatic Immune Microenvironment in Alcoholic and Nonalcoholic Liver Disease. BioMed Res Int (2017) 2017:6862439. doi: 10.1155/2017/6862439
81. Kubes P, Jenne C. Immune Responses in the Liver. Annu Rev Immunol (2018) 36:247–77. doi: 10.1146/annurev-immunol-051116-052415
82. Nakashima M, Kinoshita M, Nakashima H, Habu Y, Miyazaki H, Shono S, et al. Pivotal Advance: Characterization of Mouse Liver Phagocytic B Cells in Innate Immunity. J Leukoc Biol (2012) 91:537–46. doi: 10.1189/jlb.0411214
83. Ma HD, Wang YH, Chang C, Gershwin ME, Lian ZX. The Intestinal Microbiota and Microenvironment in Liver. Autoimmun Rev (2015) 14:183–91. doi: 10.1016/j.autrev.2014.10.013
84. Zhang P, Lu Q. Genetic and Epigenetic Influences on the Loss of Tolerance in Autoimmunity. Cell Mol Immunol (2018) 15:575–85. doi: 10.1038/cmi.2017.137
85. Patten DA, Shetty S. Chronic Liver Disease: Scavenger Hunt for Novel Therapies. Lancet (2018) 391:104–5. doi: 10.1016/S0140-6736(17)32671-5
86. Seki E, Schwabe RF. Hepatic Inflammation and Fibrosis: Functional Links and Key Pathways. Hepatology (2015) 61:1066–79. doi: 10.1002/hep.27332
87. Pinter M, Trauner M, Peck-Radosavljevic M, Sieghart W. Cancer and Liver Cirrhosis: Implications on Prognosis and Management. ESMO Open (2016) 1:e000042. doi: 10.1136/esmoopen-2016-000042
88. Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, et al. Attenuated Liver Fibrosis in the Absence of B Cells. J Clin Invest (2005) 115:3072–82. doi: 10.1172/JCI24798
89. Holt AP, Stamataki Z, Adams DH. Attenuated Liver Fibrosis in the Absence of B Cells. Hepatology (2006) 43:868–71. doi: 10.1002/hep.21155
90. Xue H, McCauley RL, Zhang W. Elevated Interleukin-6 Expression in Keloid Fibroblasts. J Surg Res (2000) 89:74–7. doi: 10.1006/jsre.1999.5805
91. Moore CM, Lamzabi I, Bartels AK, Jakate S, VAN Thiel DH. Colonic Diffuse Large B-Cell Lymphoma in a Liver Transplant Patient With Historically Very Low Tacrolimus Levels. Case Rep Transplant (2012) 2012:952359. doi: 10.1155/2012/952359
92. Al-Mansour Z, Nelson BP, Evens AM. Post-Transplant Lymphoproliferative Disease (PTLD): Risk Factors, Diagnosis, and Current Treatment Strategies. Curr Hematol Malig Rep (2013) 8:173–83. doi: 10.1007/s11899-013-0162-5
93. Warner S, Richter A, Stamataki Z, Kelly D. Understanding COVID-19: Are Children the Key? BMJ Paediatr Open (2021) 5:e001063. doi: 10.1136/bmjpo-2021-001063
94. Mack CL. What Causes Biliary Atresia? Unique Aspects of the Neonatal Immune System Provide Clues to Disease Pathogenesis. Cell Mol Gastroenterol Hepatol (2015) 1:267–74. doi: 10.1016/j.jcmgh.2015.04.001
95. Duchamp M, Sterlin D, Diabate A, Uring-Lambert B, Guerin-el Khourouj V, le Mauff B, et al. B-Cell Subpopulations in Children: National Reference Values. Immun Inflammation Dis (2014) 2:131–40. doi: 10.1002/iid3.26
96. Wells RG. Hepatic Fibrosis in Children and Adults. Clin Liver Dis (Hoboken) (2017) 9:99–101. doi: 10.1002/cld.623
97. Harpavat S, Finegold MJ, Karpen SJ. Patients With Biliary Atresia Have Elevated Direct/Conjugated Bilirubin Levels Shortly After Birth. Pediatrics (2011) 128:e1428–33. doi: 10.1542/peds.2011-1869
98. Zeitlin L, Resnick MB, Konikoff F, Schuppan D, Bujanover Y, Lerner A, et al. Divergent Patterns of Extracellular Matrix Protein Expression in Neonatal Versus Adult Liver Fibrosis. Pediatr Pathol Mol Med (2003) 22:349–62. doi: 10.1080/pdp.22.4.349.362
99. Nobili V, Alisi A, Newton KP, Schwimmer JB. Comparison of the Phenotype and Approach to Pediatric vs Adult Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology (2016) 150:1798–810. doi: 10.1053/j.gastro.2016.03.009
100. Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, et al. Histopathology of Pediatric Nonalcoholic Fatty Liver Disease. Hepatology (2005) 42:641–9. doi: 10.1002/hep.20842
101. Mieli-Vergani G, Vergani D. Autoimmune Hepatitis in Children: What is Different From Adult AIH? Semin Liver Dis (2009) 29:297–306. doi: 10.1055/s-0029-1233529
102. Floreani A, Liberal R, Vergani D, Mieli-Vergani G. Autoimmune Hepatitis: Contrasts and Comparisons in Children and Adults - a Comprehensive Review. J Autoimmun (2013) 46:7–16. doi: 10.1016/j.jaut.2013.08.004
103. Almeida J, Polvorosa MA, Gonzalez-Quintela A, Madruga I, Marcos M, Perez-Nieto MA, et al. Altered Distribution of Peripheral Blood Maturation-Associated B-Cell Subsets in Chronic Alcoholism. Alcohol Clin Exp Res (2015) 39:1476–84. doi: 10.1111/acer.12783
104. Nowak AJ, Relja B. The Impact of Acute or Chronic Alcohol Intake on the NF-kappaB Signaling Pathway in Alcohol-Related Liver Disease. Int J Mol Sci (2020) 21:1-35. doi: 10.3390/ijms21249407
105. Hollister K, Kusumanchi P, Ross RA, Chandler K, Oshodi A, Heathers L, et al. Levels of Circulating Follicular Helper T Cells, T Helper 1 Cells, and the Prognostic Significance of Soluble Form of CD40 Ligand on Survival in Patients With Alcoholic Cirrhosis. Liver Res (2018) 2:52–9. doi: 10.1016/j.livres.2018.02.001
106. Matos LC, Batista P, Monteiro N, Ribeiro J, Cipriano MA, Henriques P, et al. Lymphocyte Subsets in Alcoholic Liver Disease. World J Hepatol (2013) 5:46–55. doi: 10.4254/wjh.v5.i2.46
107. Zwolak A, Surdacka A, Daniluk J. Bcl-2 and Fas Expression in Peripheral Blood Leukocytes of Patients With Alcoholic and Autoimmune Liver Disorders. Hum Exp Toxicol (2016) 35:799–807. doi: 10.1177/0960327115607078
108. Massonnet B, Delwail A, Ayrault JM, Chagneau-Derrode C, Lecron JC, Silvain C. Increased Immunoglobulin A in Alcoholic Liver Cirrhosis: Exploring the Response of B Cells to Toll-Like Receptor 9 Activation. Clin Exp Immunol (2009) 158:115–24. doi: 10.1111/j.1365-2249.2009.04004.x
109. Kasztelan-Szczerbinska B, Adamczyk K, Surdacka A, Rolinski J, Michalak A, Bojarska-Junak A, et al. Gender-Related Disparities in the Frequencies of PD-1 and PD-L1 Positive Peripheral Blood T and B Lymphocytes in Patients With Alcohol-Related Liver Disease: A Single Center Pilot Study. PeerJ (2021) 9:e10518. doi: 10.7717/peerj.10518
110. Capone I, Marchetti P, Ascierto PA, Malorni W, Gabriele L. Sexual Dimorphism of Immune Responses: A New Perspective in Cancer Immunotherapy. Front Immunol (2018) 9:552. doi: 10.3389/fimmu.2018.00552
111. Taneja V. Sex Hormones Determine Immune Response. Front Immunol (2018) 9:1931. doi: 10.3389/fimmu.2018.01931
112. Buchta CM, Bishop GA. Toll-Like Receptors and B Cells: Functions and Mechanisms. Immunol Res (2014) 59:12–22. doi: 10.1007/s12026-014-8523-2
113. Zhou Z, Zhong W. Targeting the Gut Barrier for the Treatment of Alcoholic Liver Disease. Liver Res (2017) 1:197–207. doi: 10.1016/j.livres.2017.12.004
114. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic Lipids Stored by Kupffer Cells Correlates With Their Pro-Inflammatory Phenotype at an Early Stage of Steatohepatitis. J Hepatol (2012) 57:141–9. doi: 10.1016/j.jhep.2012.02.028
115. Bruzzi S, Sutti S, Giudici G, Burlone ME, Ramavath NN, Toscani A, et al. B2-Lymphocyte Responses to Oxidative Stress-Derived Antigens Contribute to the Evolution of Nonalcoholic Fatty Liver Disease (NAFLD). Free Radic Biol Med (2018) 124:249–59. doi: 10.1016/j.freeradbiomed.2018.06.015
116. Sutti S, Albano E. Adaptive Immunity: An Emerging Player in the Progression of NAFLD. Nat Rev Gastroenterol Hepatol (2020) 17:81–92. doi: 10.1038/s41575-019-0210-2
117. Zeng F, Zhang Y, Han X, Zeng M, Gao Y, Weng J. Predicting Non-Alcoholic Fatty Liver Disease Progression and Immune Deregulations by Specific Gene Expression Patterns. Front Immunol (2020) 11:609900. doi: 10.3389/fimmu.2020.609900
118. Schwenger KJP, Chen L, Chelliah A, DA Silva HE, Teterina A, Comelli EM, et al. Markers of Activated Inflammatory Cells are Associated With Disease Severity and Intestinal Microbiota in Adults With Nonalcoholic Fatty Liver Disease. Int J Mol Med (2018) 42:2229–37. doi: 10.3892/ijmm.2018.3800
119. Wu Z, Xu J, Tan J, Song Y, Liu L, Zhang F, et al. Mesenteric Adipose Tissue B Lymphocytes Promote Local and Hepatic Inflammation in non-Alcoholic Fatty Liver Disease Mice. J Cell Mol Med (2019) 23:3375–85. doi: 10.1111/jcmm.14232
120. Zhang F, Jiang WW, Li X, Qiu XY, Wu Z, Chi YJ, et al. Role of Intrahepatic B Cells in non-Alcoholic Fatty Liver Disease by Secreting Pro-Inflammatory Cytokines and Regulating Intrahepatic T Cells. J Dig Dis (2016) 17:464–74. doi: 10.1111/1751-2980.12362
121. Loggi E, Gamal N, Bihl F, Bernardi M, Andreone P. Adaptive Response in Hepatitis B Virus Infection. J Viral Hepat (2014) 21:305–13. doi: 10.1111/jvh.12255
122. Tan A, Koh S, Bertoletti A. Immune Response in Hepatitis B Virus Infection. Cold Spring Harb Perspect Med (2015) 5:a021428. doi: 10.1101/cshperspect.a021428
123. Poonia B, Ayithan N, Nandi M, Masur H, Kottilil S. HBV Induces Inhibitory FcRL Receptor on B Cells and Dysregulates B Cell-T Follicular Helper Cell Axis. Sci Rep (2018) 8:15296. doi: 10.1038/s41598-018-33719-x
124. Hepatitis B vaccines: WHO position paper - July 2017. Wkly Epidemiol Rec (2017) 92:369–92. doi: 10.1016/j.vaccine.2017.07.046
125. Cashman SB, Marsden BD, Dustin LB. The Humoral Immune Response to HCV: Understanding is Key to Vaccine Development. Front Immunol (2014) 5:550. doi: 10.3389/fimmu.2014.00550
126. von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, et al. Hepatitis C Virus Continuously Escapes From Neutralizing Antibody and T-Cell Responses During Chronic Infection in vivo. Gastroenterology (2007) 132:667–78. doi: 10.1053/j.gastro.2006.12.008
127. Laidlaw SM, Dustin LB. An HCV Vaccine on the Fly. J Infect Dis (2020) 221:1216–8. doi: 10.1093/infdis/jiz231
128. Stamataki Z, Coates S, Abrignani S, Houghton M, Mckeating JA. Immunization of Human Volunteers With Hepatitis C Virus Envelope Glycoproteins Elicits Antibodies That Cross-Neutralize Heterologous Virus Strains. J Infect Dis (2011) 204:811–3. doi: 10.1093/infdis/jir399
129. Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, et al. Broadly Neutralizing Antibodies Protect Against Hepatitis C Virus Quasispecies Challenge. Nat Med (2008) 14:25–7. doi: 10.1038/nm1698
130. Dustin LB, Cashman SB, Laidlaw SM. Immune Control and Failure in HCV Infection–Tipping the Balance. J Leukoc Biol (2014) 96:535–48. doi: 10.1189/jlb.4RI0214-126R
131. Ni J, Hembrador E, di Bisceglie AM, Jacobson IM, Talal AH, Butera D, et al. Accumulation of B Lymphocytes With a Naive, Resting Phenotype in a Subset of Hepatitis C Patients. J Immunol (2003) 170:3429–39. doi: 10.4049/jimmunol.170.6.3429
132. Rydell GE, Prakash K, Norder H, Lindh M. Hepatitis B Surface Antigen on Subviral Particles Reduces the Neutralizing Effect of Anti-HBs Antibodies on Hepatitis B Viral Particles in vitro. Virology (2017) 509:67–70. doi: 10.1016/j.virol.2017.05.017
133. Hehle V, Beretta M, Bourgine M, Ait-Goughoulte M, Planchais C, Morisse S, et al. Potent Human Broadly Neutralizing Antibodies to Hepatitis B Virus From Natural Controllers. J Exp Med (2020) 217:1–17. doi: 10.1084/jem.20200840
134. Vanwolleghem T, Groothuismink ZMA, Kreefft K, Hung M, Novikov N, Boonstra A. Hepatitis B Core-Specific Memory B Cell Responses Associate With Clinical Parameters in Patients With Chronic HBV. J Hepatol (2020) 73:52–61. doi: 10.1016/j.jhep.2020.01.024
135. Ma Z, Zhang E, Gao S, Xiong Y, Lu M. Toward a Functional Cure for Hepatitis B: The Rationale and Challenges for Therapeutic Targeting of the B Cell Immune Response. Front Immunol (2019b) 10:2308. doi: 10.3389/fimmu.2019.02308
136. Fang Q, Deng Y, Liang R, Mei Y, Hu Z, Wang J, et al. CD19(+)CD24(hi)CD38(hi) Regulatory B Cells: A Potential Immune Predictive Marker of Severity and Therapeutic Responsiveness of Hepatitis C. Am J Transl Res (2020) 12:889–900.
137. Wang R, Xie R, Song Z. Circulating Regulatory TFH Cells are Enriched in Patients With Chronic Hepatitis B Infection and Induce the Differentiation of Regulatory B Cells. Exp Cell Res (2018) 365:171–6. doi: 10.1016/j.yexcr.2018.02.031
138. Eiza N, Zuckerman E, Carlebach M, Rainis T, Goldberg Y, Vadasz Z. Increased Killer B Cells in Chronic HCV Infection may Lead to Autoimmunity and Increased Viral Load. Clin Exp Immunol (2018) 193:183–93. doi: 10.1111/cei.13139
139. Stamataki Z, Shannon-Lowe C, Shaw J, Mutimer D, Rickinson AB, Gordon J, et al. Hepatitis C Virus Association With Peripheral Blood B Lymphocytes Potentiates Viral Infection of Liver-Derived Hepatoma Cells. Blood (2009) 113:585–93. doi: 10.1182/blood-2008-05-158824
140. Arai J, Ito T, Shimozuma Y, Uchikoshi M, Nakajima Y, Sakaki M, et al. Decreased Expression of Interferon-Stimulated Genes in B Cells of Patients With Chronic Hepatitis C During Interferon-Free Therapy Potentially Suggests the Eradication of Hepatitis C Virus in the B Cells: A Cohort Study. Health Sci Rep (2020) 3:e176. doi: 10.1002/hsr2.176
141. Inokuchi M, Ito T, Uchikoshi M, Shimozuma Y, Morikawa K, Nozawa H, et al. Infection of B Cells With Hepatitis C Virus for the Development of Lymphoproliferative Disorders in Patients With Chronic Hepatitis C. J Med Virol (2009) 81:619–27. doi: 10.1002/jmv.21388
142. Law M. Antibody Responses in Hepatitis C Infection. Cold Spring Harb Perspect Med (2020) 11(3):1-19. doi: 10.1101/cshperspect.a036962
143. Kemming J, Thimme R, Neumann-Haefelin C. Adaptive Immune Response Against Hepatitis C Virus. Int J Mol Sci (2020) 21:1–21. doi: 10.3390/ijms21165644
144. Stuart JD, Salinas E, Grakoui A. Immune System Control of Hepatitis C Virus Infection. Curr Opin Virol (2020) 46:36–44. doi: 10.1016/j.coviro.2020.10.002
145. Agnello V, Chung RT, Kaplan LM. A Role for Hepatitis C Virus Infection in Type II Cryoglobulinemia. N Engl J Med (1992) 327:1490–5. doi: 10.1056/NEJM199211193272104
146. Dammacco F, Sansonno D, Piccoli C, Racanelli V, D’Amore FP, Lauletta G. The Lymphoid System in Hepatitis C Virus Infection: Autoimmunity, Mixed Cryoglobulinemia, and Overt B-Cell Malignancy. Semin Liver Dis (2000) 20:143–57. doi: 10.1055/s-2000-9613
147. Charles ED, Orloff MI, Nishiuchi E, Marukian S, Rice CM, Dustin LB. Somatic Hypermutations Confer Rheumatoid Factor Activity in Hepatitis C Virus-Associated Mixed Cryoglobulinemia. Arthritis Rheum (2013) 65:2430–40. doi: 10.1002/art.38041
148. Tucci F, Kuppers R. Role of Hepatitis C Virus in B Cell Lymphoproliferations. Virol Sin (2014) 29:3–6. doi: 10.1007/s12250-014-3414-1
149. Boleto G, Vieira M, Saadoun D, Cacoub P. Hepatitis C Virus-Related Vasculitis. Clin Res Hepatol Gastroenterol (2020) 101575. doi: 10.1016/j.clinre.2020.11.005
150. Saadoun D, Landau DA, Calabrese LH, Cacoub PP. Hepatitis C-Associated Mixed Cryoglobulinaemia: A Crossroad Between Autoimmunity and Lymphoproliferation. Rheumatol (Oxford) (2007) 46:1234–42. doi: 10.1093/rheumatology/kem132
151. Alfraji N, Upadhyaya VD, Bekampis C, Kuzyshyn H. Mixed Cryoglobulinemia Syndrome (MCS) Due to Untreated Hepatitis B With Uncommon Presentation: Case Report and Literature Review. BMC Rheumatol (2020) 4:58. doi: 10.1186/s41927-020-00159-y
152. Dustin LB. Innate and Adaptive Immune Responses in Chronic HCV Infection. Curr Drug Targets (2017) 18:826–43. doi: 10.2174/1389450116666150825110532
153. Lauletta G, Russi S, Conteduca V, Sansonno L. Hepatitis C Virus Infection and Mixed Cryoglobulinemia. Clin Dev Immunol (2012) 2012:502156. doi: 10.1155/2012/502156
154. Dywicki J, Buitrago-Molina LE, Pietrek J, Lieber M, Broering R, Khera T, et al. Autoimmune Hepatitis Induction can Occur in the Liver. Liver Int (2020) 40:377–81. doi: 10.1111/liv.14296
155. Jeffery HC, Braitch MK, Bagnall C, Hodson J, Jeffery LE, Wawman RE, et al. Changes in Natural Killer Cells and Exhausted Memory Regulatory T Cells With Corticosteroid Therapy in Acute Autoimmune Hepatitis. Hepatol Commun (2018) 2:421–36. doi: 10.1002/hep4.1163
156. Taubert R, Hardtke-wolenski M, Noyan F, Wilms A, Baumann AK, Schlue J, et al. Intrahepatic Regulatory T Cells in Autoimmune Hepatitis are Associated With Treatment Response and Depleted With Current Therapies. J Hepatol (2014) 61:1106–14. doi: 10.1016/j.jhep.2014.05.034
157. Moritoki Y, Lian ZX, Ohsugi Y, Ueno Y, Gershwin ME. B Cells and Autoimmune Liver Diseases. Autoimmun Rev (2006) 5:449–57. doi: 10.1016/j.autrev.2006.02.006
158. Behairy BE, El-Araby HA, Abd el Kader HH, Ehsan NA, Salem ME, Zakaria HM, et al. Assessment of Intrahepatic Regulatory T Cells in Children With Autoimmune Hepatitis. Ann Hepatol (2016) 15:682–90. doi: 10.5604/16652681.1212319
159. Than NN, Jeffery HC, Oo YH. Autoimmune Hepatitis: Progress From Global Immunosuppression to Personalised Regulatory T Cell Therapy. Can J Gastroenterol Hepatol (2016) 2016:7181685. doi: 10.1155/2016/7181685
160. Lapierre P, Beland K, Yang R, Alvarez F. Adoptive Transfer of Ex Vivo Expanded Regulatory T Cells in an Autoimmune Hepatitis Murine Model Restores Peripheral Tolerance. Hepatology (2013) 57:217–27. doi: 10.1002/hep.26023
161. Ogawa S, Sakaguchi K, Takaki A, Shiraga K, Sawayama T, Mouri H, et al. Increase in CD95 (Fas/APO-1)-Positive CD4+ and CD8+ T Cells in Peripheral Blood Derived From Patients With Autoimmune Hepatitis or Chronic Hepatitis C With Autoimmune Phenomena. J Gastroenterol Hepatol (2000) 15:69–75. doi: 10.1046/j.1440-1746.2000.02044.x
162. Sebode M, Weiler-Normann C, Liwinski T, Schramm C. Autoantibodies in Autoimmune Liver Disease-Clinical and Diagnostic Relevance. Front Immunol (2018) 9:609. doi: 10.3389/fimmu.2018.00609
163. Schlaak JF, Lohr H, Gallati H, Meyer Zum Buschenfelde KH, Fleischer B. Analysis of the In Vitro Cytokine Production by Liver-Infiltrating T Cells of Patients With Autoimmune Hepatitis. Clin Exp Immunol (1993) 94:168–73. doi: 10.1111/j.1365-2249.1993.tb05996.x
164. Hartl J, Miquel R, Zachou K, Wong GW, Asghar A, Pape S, et al. Features and Outcome of AIH Patients Without Elevation of IgG. JHEP Rep (2020) 2:100094. doi: 10.1016/j.jhepr.2020.100094
165. Minaga K, Watanabe T, Chung H, Kudo M. Autoimmune Hepatitis and IgG4-Related Disease. World J Gastroenterol (2019) 25:2308–14. doi: 10.3748/wjg.v25.i19.2308
166. Ma L, Qin J, Ji H, Zhao P, Jiang Y. TFH and Plasma Cells are Correlated With Hypergammaglobulinaemia in Patients With Autoimmune Hepatitis. Liver Int (2014) 34:405–15. doi: 10.1111/liv.12245
167. Adiga A, Nugent K. Lupus Hepatitis and Autoimmune Hepatitis (Lupoid Hepatitis). Am J Med Sci (2017) 353:329–35. doi: 10.1016/j.amjms.2016.10.014
168. Chung BK, Henriksen EKK, Jorgensen KK, Karlsen TH, Hirschfield GM, Liaskou E. Gut and Liver B Cells of Common Clonal Origin in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease. Hepatol Commun (2018) 2:956–67. doi: 10.1002/hep4.1200
169. Fischer S, Trivedi PJ, Ward S, Greig PD, Therapondos G, Hirschfield GM. Frequency and Significance of IgG4 Immunohistochemical Staining in Liver Explants From Patients With Primary Sclerosing Cholangitis. Int J Exp Pathol (2014) 95:209–15. doi: 10.1111/iep.12076
170. Chapman MH, Thorburn D, Hirschfield GM, Webster GGJ, Rushbrook SM, Alexander G, et al. British Society of Gastroenterology and UK-PSC Guidelines for the Diagnosis and Management of Primary Sclerosing Cholangitis. Gut (2019) 68:1356–78. doi: 10.1136/gutjnl-2018-317993
171. Chung BK, Guevel BT, Reynolds GM, Gupta Udatha DB, Henriksen EK, Stamataki Z, et al. Phenotyping and Auto-Antibody Production by Liver-Infiltrating B Cells in Primary Sclerosing Cholangitis and Primary Biliary Cholangitis. J Autoimmun (2017) 77:45–54. doi: 10.1016/j.jaut.2016.10.003
172. Zen Y, Quaglia A, Portmann B. Immunoglobulin G4-Positive Plasma Cell Infiltration in Explanted Livers for Primary Sclerosing Cholangitis. Histopathology (2011) 58:414–22. doi: 10.1111/j.1365-2559.2011.03763.x
173. Zen Y, Britton D, Mitra V, Pike I, Heaton N, Quaglia A. A Global Proteomic Study Identifies Distinct Pathological Features of IgG4-Related and Primary Sclerosing Cholangitis. Histopathology (2016) 68:796–809. doi: 10.1111/his.12813
175. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus Statement on the Pathology of IgG4-Related Disease. Mod Pathol (2012) 25:1181–92. doi: 10.1038/modpathol.2012.72
176. Tanaka A. IgG4-Related Sclerosing Cholangitis and Primary Sclerosing Cholangitis. Gut Liver (2019) 13:300–7. doi: 10.5009/gnl18085
177. O’Hara SP, LaRusso NF. The Gut-Liver Axis in Primary Sclerosing Cholangitis: Are Pathobionts the Missing Link? Hepatology (2019) 70:1058–60. doi: 10.1002/hep.30673
178. Lleo A, Leung PSC, Hirschfield GM, Gershwin EM. The Pathogenesis of Primary Biliary Cholangitis: A Comprehensive Review. Semin Liver Dis (2020) 40:34–48. doi: 10.1055/s-0039-1697617
179. Kikuchi K, Lian ZX, Yang GX, Ansari AA, Ikehara S, Kaplan M, et al. Bacterial CpG Induces Hyper-IgM Production in CD27(+) Memory B Cells in Primary Biliary Cirrhosis. Gastroenterology (2005) 128:304–12. doi: 10.1053/j.gastro.2004.11.005
180. Zhang B, Hu M, Zhang P, Cao H, Wang Y, Wang Z, et al. BAFF Promotes Regulatory T-Cell Apoptosis and Blocks Cytokine Production by Activating B Cells in Primary Biliary Cirrhosis. Braz J Med Biol Res (2013) 46:433–9. doi: 10.1590/1414-431X20132665
181. Chen Q, Lai L, Chi X, Lu X, Wu H, Sun J, et al. CD19(+)CD24(hi)CD38(hi) B Cell Dysfunction in Primary Biliary Cholangitis. Mediators Inflamm (2020) 2020:3019378. doi: 10.1155/2020/3019378
182. Cichoz-Lach H, Grywalska E, Michalak A, Kowalik A, Mielnik M, Rolinski J. Deviations in Peripheral Blood Cell Populations are Associated With the Stage of Primary Biliary Cholangitis and Presence of Itching. Arch Immunol Ther Exp (Warsz) (2018) 66:443–52. doi: 10.1007/s00005-018-0515-9
183. Wang L, Sun Y, Zhang Z, Jia Y, Zou Z, Ding J, et al. CXCR5+ CD4+ T Follicular Helper Cells Participate in the Pathogenesis of Primary Biliary Cirrhosis. Hepatology (2015b) 61:627–38. doi: 10.1002/hep.27306
184. Hirschfield GM, Gershwin ME. The Immunobiology and Pathophysiology of Primary Biliary Cirrhosis. Annu Rev Pathol (2013) 8:303–30. doi: 10.1146/annurev-pathol-020712-164014
185. Wang L, Sun X, Qiu J, Cai Y, Ma L, Zhao P, et al. Increased Numbers of Circulating ICOS(+) Follicular Helper T and CD38(+) Plasma Cells in Patients With Newly Diagnosed Primary Biliary Cirrhosis. Dig Dis Sci (2015a) 60:405–13. doi: 10.1007/s10620-014-3372-3
186. Ma WT, Chen DK. Immunological Abnormalities in Patients With Primary Biliary Cholangitis. Clin Sci (Lond) (2019) 133:741–60. doi: 10.1042/CS20181123
187. Kikuchi K, Tsuneyama K, Yamada H, Kajiyama Y, Matsumoto K, Tsunashima H, et al. Splenic Lymph Follicles Generate Immunoglobulin M-Producing B Cells in Primary Biliary Cirrhosis. Hepatol Res (2014) 44:E253–6. doi: 10.1111/hepr.12231
188. Tan YG, Wang YQ, Zhang M, Han YX, Huang CY, Zhang HP, et al. Clonal Characteristics of Circulating B Lymphocyte Repertoire in Primary Biliary Cholangitis. J Immunol (2016) 197:1609–20. doi: 10.4049/jimmunol.1600096
189. Carbone M, Milani C, Gerussi A, Ronca V, Cristoferi L, Invernizzi P. Primary Biliary Cholangitis: A Multifaceted Pathogenesis With Potential Therapeutic Targets. J Hepatol (2020) 73:965–6. doi: 10.1016/j.jhep.2020.05.041
190. Taylor SA, Assis DN, Mack CL. The Contribution of B Cells in Autoimmune Liver Diseases. Semin Liver Dis (2019) 39:422–31. doi: 10.1055/s-0039-1688751
191. Mack CL, Sokol RJ. Unraveling the Pathogenesis and Etiology of Biliary Atresia. Pediatr Res (2005) 57:87R–94R. doi: 10.1203/01.PDR.0000159569.57354.47
192. Kelly DA, Davenport M. Current Management of Biliary Atresia. Arch Dis Child (2007) 92:1132–5. doi: 10.1136/adc.2006.101451
193. Lakshminarayanan B, Davenport M. Biliary Atresia: A Comprehensive Review. J Autoimmun (2016) 73:1–9. doi: 10.1016/j.jaut.2016.06.005
194. Brindley SM, Lanham AM, Karrer FM, Tucker RM, Fontenot AP, Mack CL. Cytomegalovirus-Specific T-Cell Reactivity in Biliary Atresia at the Time of Diagnosis is Associated With Deficits in Regulatory T Cells. Hepatology (2012) 55:1130–8. doi: 10.1002/hep.24807
195. Mack CL, Feldman AG, Sokol RJ. Clues to the Etiology of Bile Duct Injury in Biliary Atresia. Semin Liver Dis (2012) 32:307–16. doi: 10.1055/s-0032-1329899
196. Feldman AG, Mack CL. Biliary Atresia: Cellular Dynamics and Immune Dysregulation. Semin Pediatr Surg (2012) 21:192–200. doi: 10.1053/j.sempedsurg.2012.05.003
197. Mack CL, Tucker RM, Lu BR, Sokol RJ, Fontenot AP, Ueno Y, et al. Cellular and Humoral Autoimmunity Directed at Bile Duct Epithelia in Murine Biliary Atresia. Hepatology (2006) 44:1231–9. doi: 10.1002/hep.21366
198. Mack CL. The Pathogenesis of Biliary Atresia: Evidence for a Virus-Induced Autoimmune Disease. Semin Liver Dis (2007) 27:233–42. doi: 10.1055/s-2007-985068
199. Lu BR, Brindley SM, Tucker RM, Lambert CL, Mack CL. Alpha-Enolase Autoantibodies Cross-Reactive to Viral Proteins in a Mouse Model of Biliary Atresia. Gastroenterology (2010) 139:1753–61. doi: 10.1053/j.gastro.2010.07.042
200. Hadchouel M, Hugon RN, Odievre M. Immunoglobulin Deposits in the Biliary Remnants of Extrahepatic Biliary Atresia: A Study by Immunoperoxidase Staining in 128 Infants. Histopathology (1981) 5:217–21. doi: 10.1111/j.1365-2559.1981.tb01779.x
201. Wang J, Xu Y, Chen Z, Liang J, Lin Z, Liang H, et al. Liver Immune Profiling Reveals Pathogenesis and Therapeutics for Biliary Atresia. Cell (2020) 183:1867–83.e26. doi: 10.1016/j.cell.2020.10.048
202. Petersen C. Biliary Atresia: The Animal Models. Semin Pediatr Surg (2012) 21:185–91. doi: 10.1053/j.sempedsurg.2012.05.002
203. Mohanty SK, Donnelly B, Temple H, Tiao GM. A Rotavirus-Induced Mouse Model to Study Biliary Atresia and Neonatal Cholestasis. Methods Mol Biol (2019) 1981:259–71. doi: 10.1007/978-1-4939-9420-5_17
204. Feldman AG, Tucker RM, Fenner EK, Pelanda R, Mack CL. B Cell Deficient Mice are Protected From Biliary Obstruction in the Rotavirus-Induced Mouse Model of Biliary Atresia. PloS One (2013) 8:e73644. doi: 10.1371/journal.pone.0073644
205. Riepenhoff-Talty M, Gouvea V, Evans MJ, Svensson L, Hoffenberg E, Sokol RJ, et al. Detection of Group C Rotavirus in Infants With Extrahepatic Biliary Atresia. J Infect Dis (1996) 174:8–15. doi: 10.1093/infdis/174.1.8
206. Bobo L, Ojeh C, Chiu D, Machado A, Colombani P, Schwarz K. Lack of Evidence for Rotavirus by Polymerase Chain Reaction/Enzyme Immunoassay of Hepatobiliary Samples From Children With Biliary Atresia. Pediatr Res (1997) 41:229–34. doi: 10.1203/00006450-199702000-00013
207. Mack CL, Falta MT, Sullivan AK, Karrer F, Sokol RJ, Freed BM, et al. Oligoclonal Expansions of CD4+ and CD8+ T-Cells in the Target Organ of Patients With Biliary Atresia. Gastroenterology (2007) 133:278–87. doi: 10.1053/j.gastro.2007.04.032
208. Salles G, Barrett M, Foa R, Maurer J, O’Brien S, Valente N, et al. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv Ther (2017) 34:2232–73. doi: 10.1007/s12325-017-0612-x
209. Press OW, Appelbaum F, Ledbetter JA, Martin PJ, Zarling J, Kidd P, et al. Monoclonal Antibody 1F5 (Anti-CD20) Serotherapy of Human B Cell Lymphomas. Blood (1987) 69:584–91. doi: 10.1182/blood.V69.2.584.584
210. Johnson P, Glennie M. The Mechanisms of Action of Rituximab in the Elimination of Tumor Cells. Semin Oncol (2003) 30:3–8. doi: 10.1053/sonc.2003.50025
211. Cragg MS, Morgan SM, Chan HT, Morgan BP, Filatov AV, Johnson PW, et al. Complement-Mediated Lysis by Anti-CD20 mAb Correlates With Segregation Into Lipid Rafts. Blood (2003) 101:1045–52. doi: 10.1182/blood-2002-06-1761
212. Cardarelli PM, Quinn M, Buckman D, Fang Y, Colcher D, King DJ, et al. Binding to CD20 by Anti-B1 Antibody or F(Ab’)(2) is Sufficient for Induction of Apoptosis in B-Cell Lines. Cancer Immunol Immunother (2002) 51:15–24. doi: 10.1007/s00262-001-0247-1
213. NICE. RITUXIMAB [Online] (2021). Available at: https://bnf.nice.org.uk/drug/rituximab.html.
214. European Medicines Agency. MabThera [Online]. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/mabthera.
215. Favas C, Isenberg DA. B-Cell-Depletion Therapy in SLE–what are the Current Prospects for its Acceptance? Nat Rev Rheumatol (2009) 5:711–6. doi: 10.1038/nrrheum.2009.218
216. Shipa M, Embleton-Thirsk A, Parvaz M, Santos Ribeiro L, Muller P, Chowdhury K, et al. OP0129 Belimumab After Rituximab Significantly Reduced IGG Anti-DSDNA Antibody Levels and Prolonged Time to Severe Flare in Patients With Systemic Lupus Erythematosus. Ann Rheumatic Dis (2021) 80:74–4. doi: 10.1136/annrheumdis-2021-eular.553
217. Pedersen IM, Buhl AM, Klausen P, Geisler CH, Jurlander J. The Chimeric Anti-CD20 Antibody Rituximab Induces Apoptosis in B-Cell Chronic Lymphocytic Leukemia Cells Through a P38 Mitogen Activated Protein-Kinase-Dependent Mechanism. Blood (2002) 99:1314–9. doi: 10.1182/blood.V99.4.1314
218. Golay J, Zaffaroni L, Vaccari T, Lazzari M, Borleri GM, Bernasconi S, et al. Biologic Response of B Lymphoma Cells to Anti-CD20 Monoclonal Antibody Rituximab In Vitro: CD55 and CD59 Regulate Complement-Mediated Cell Lysis. Blood (2000) 95:3900–8. doi: 10.1182/blood.V95.12.3900.012k14_3900_3908
219. Montalvao F, Garcia Z, Celli S, Breart B, Deguine J, van Rooijen N, et al. The Mechanism of Anti-CD20-Mediated B Cell Depletion Revealed by Intravital Imaging. J Clin Invest (2013) 123:5098–103. doi: 10.1172/JCI70972
220. Withers DR, Fiorini C, Fischer RT, Ettinger R, Lipsky PE, Grammer AC. T Cell-Dependent Survival of CD20+ and CD20- Plasma Cells in Human Secondary Lymphoid Tissue. Blood (2007) 109:4856–64. doi: 10.1182/blood-2006-08-043414
221. Ferraro AJ, Drayson MT, Savage CO, Maclennan IC. Levels of Autoantibodies, Unlike Antibodies to All Extrinsic Antigen Groups, Fall Following B Cell Depletion With Rituximab. Eur J Immunol (2008) 38:292–8. doi: 10.1002/eji.200737557
222. Teng YK, Wheater G, Hogan VE, Stocks P, Levarht EW, Huizinga TW, et al. Induction of Long-Term B-Cell Depletion in Refractory Rheumatoid Arthritis Patients Preferentially Affects Autoreactive More Than Protective Humoral Immunity. Arthritis Res Ther (2012) 14:R57. doi: 10.1186/ar3770
223. Hofmann K, Clauder AK, Manz RA. Targeting B Cells and Plasma Cells in Autoimmune Diseases. Front Immunol (2018) 9:835. doi: 10.3389/fimmu.2018.00835
224. Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for Rheumatoid Arthritis Refractory to Anti-Tumor Necrosis Factor Therapy: Results of a Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase III Trial Evaluating Primary Efficacy and Safety at Twenty-Four Weeks. Arthritis Rheum (2006) 54:2793–806. doi: 10.1002/art.22025
225. Sanz I. Connective Tissue Diseases: The Conundrum of B Cell Depletion in SLE. Nat Rev Rheumatol (2009) 5:304–5. doi: 10.1038/nrrheum.2009.100
226. Stasi R, del Poeta G, Stipa E, Evangelista ML, Trawinska MM, Cooper N, et al. Response to B-Cell Depleting Therapy With Rituximab Reverts the Abnormalities of T-Cell Subsets in Patients With Idiopathic Thrombocytopenic Purpura. Blood (2007) 110:2924–30. doi: 10.1182/blood-2007-02-068999
227. Quartier P, Brethon B, Philippet P, Landman-Parker J, le Deist F, Fischer A. Treatment of Childhood Autoimmune Haemolytic Anaemia With Rituximab. Lancet (2001) 358:1511–3. doi: 10.1016/S0140-6736(01)06573-4
228. Roccatello D, Sciascia S, Rossi D, Solfietti L, Fenoglio R, Menegatti E, et al. The Challenge of Treating Hepatitis C Virus-Associated Cryoglobulinemic Vasculitis in the Era of Anti-CD20 Monoclonal Antibodies and Direct Antiviral Agents. Oncotarget (2017) 8:41764–77. doi: 10.18632/oncotarget.16986
229. Basile U, Gulli F, Napodano C, Pocino K, Basile V, Marrapodi R, et al. Biomarkers of Minimal Residual Disease in Rituximab-Treated Patients With Mixed Cryoglobulinemia. Biotechnol Appl Biochem (2020) 68:319–29. doi: 10.1002/bab.1929
230. Roccatello D, Baldovino S, Rossi D, Mansouri M, Naretto C, Gennaro M, et al. Long-Term Effects of Anti-CD20 Monoclonal Antibody Treatment of Cryoglobulinaemic Glomerulonephritis. Nephrol Dial Transplant (2004) 19:3054–61. doi: 10.1093/ndt/gfh469
231. Czaja AJ, Freese DK, American Association for the Study of Liver, D. Diagnosis and Treatment of Autoimmune Hepatitis. Hepatology (2002) 36:479–97. doi: 10.1053/jhep.2002.34944
232. Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vergani D, et al. Diagnosis and Management of Autoimmune Hepatitis. Hepatology (2010) 51:2193–213. doi: 10.1002/hep.23584
233. Beland K, Marceau G, Labardy A, Bourbonnais S, Alvarez F. Depletion of B Cells Induces Remission of Autoimmune Hepatitis in Mice Through Reduced Antigen Presentation and Help to T Cells. Hepatology (2015) 62:1511–23. doi: 10.1002/hep.27991
234. Burak KW, Swain MG, Santodomingo-Garzon T, Lee SS, Urbanski SJ, Aspinall AI, et al. Rituximab for the Treatment of Patients With Autoimmune Hepatitis Who are Refractory or Intolerant to Standard Therapy. Can J Gastroenterol (2013) 27:273–80. doi: 10.1155/2013/512624
235. D’Agostino D, Costaguta A, Alvarez F. Successful Treatment of Refractory Autoimmune Hepatitis With Rituximab. Pediatrics (2013) 132:e526–30. doi: 10.1542/peds.2011-1900
236. Than NN, Hodson J, Schmidt-Martin D, Taubert R, Wawman RE, Botter M, et al. Efficacy of Rituximab in Difficult-to-Manage Autoimmune Hepatitis: Results From the International Autoimmune Hepatitis Group. JHEP Rep (2019) 1:437–45. doi: 10.1016/j.jhepr.2019.10.005
237. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, et al. Primary Biliary Cirrhosis. Hepatology (2009) 50:291–308. doi: 10.1002/hep.22906
238. Pares A, Caballeria L, Rodes J. Excellent Long-Term Survival in Patients With Primary Biliary Cirrhosis and Biochemical Response to Ursodeoxycholic Acid. Gastroenterology (2006) 130:715–20. doi: 10.1053/j.gastro.2005.12.029
239. Tsuda M, Moritoki Y, Lian ZX, Zhang W, Yoshida K, Wakabayashi K, et al. Biochemical and Immunologic Effects of Rituximab in Patients With Primary Biliary Cirrhosis and an Incomplete Response to Ursodeoxycholic Acid. Hepatology (2012) 55:512–21. doi: 10.1002/hep.24748
240. Myers RP, Swain MG, Lee SS, Shaheen AA, Burak KW. B-Cell Depletion With Rituximab in Patients With Primary Biliary Cirrhosis Refractory to Ursodeoxycholic Acid. Am J Gastroenterol (2013) 108:933–41. doi: 10.1038/ajg.2013.51
241. Khanna A, Jopson L, Howel D, Bryant A, Blamire A, Newton JL, et al. Rituximab Is Ineffective for Treatment of Fatigue in Primary Biliary Cholangitis: A Phase 2 Randomized Controlled Trial. Hepatology (2019) 70:1646–57. doi: 10.1002/hep.30099
242. Meijer JM, Meiners PM, Vissink A, Spijkervet FK, Abdulahad W, Kamminga N, et al. Effectiveness of Rituximab Treatment in Primary Sjogren’s Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheum (2010) 62:960–8. doi: 10.1002/art.27314
243. Devauchelle-Pensec V, Morvan J, Rat AC, Jousse-Joulin S, Pennec Y, Pers JO, et al. Effects of Rituximab Therapy on Quality of Life in Patients With Primary Sjogren’s Syndrome. Clin Exp Rheumatol (2011) 29:6–12.
244. Fluge O, Bruland O, Risa K, Storstein A, Kristoffersen EK, Sapkota D, et al. Benefit From B-Lymphocyte Depletion Using the Anti-CD20 Antibody Rituximab in Chronic Fatigue Syndrome. A Double-Blind and Placebo-Controlled Study. PloS One (2011) 6:e26358. doi: 10.1371/journal.pone.0026358
245. Rigby W, Ferraccioli G, Greenwald M, Zazueta-Montiel B, Fleischmann R, Wassenberg S, et al. Effect of Rituximab on Physical Function and Quality of Life in Patients With Rheumatoid Arthritis Previously Untreated With Methotrexate. Arthritis Care Res (Hoboken) (2011) 63:711–20. doi: 10.1002/acr.20419
246. Wakabayashi K, Lian ZX, Leung PS, Moritoki Y, Tsuneyama K, Kurth MJ, et al. Loss of Tolerance in C57BL/6 Mice to the Autoantigen E2 Subunit of Pyruvate Dehydrogenase by a Xenobiotic With Ensuing Biliary Ductular Disease. Hepatology (2008) 48:531–40. doi: 10.1002/hep.22390
247. Dhirapong A, Lleo A, Yang GX, Tsuneyama K, Dunn R, Kehry M, et al. B Cell Depletion Therapy Exacerbates Murine Primary Biliary Cirrhosis. Hepatology (2011) 53:527–35. doi: 10.1002/hep.24044
248. Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, et al. Anti-Mitochondrial Antibodies and Primary Biliary Cirrhosis in TGF-Beta Receptor II Dominant-Negative Mice. J Immunol (2006) 177:1655–60. doi: 10.4049/jimmunol.177.3.1655
249. Moritoki Y, Lian ZX, Lindor K, Tuscano J, Tsuneyama K, Zhang W, et al. B-Cell Depletion With Anti-CD20 Ameliorates Autoimmune Cholangitis But Exacerbates Colitis in Transforming Growth Factor-Beta Receptor II Dominant Negative Mice. Hepatology (2009a) 50:1893–903. doi: 10.1002/hep.23238
250. Moritoki Y, Zhang W, Tsuneyama K, Yoshida K, Wakabayashi K, Yang GX, et al. B Cells Suppress the Inflammatory Response in a Mouse Model of Primary Biliary Cirrhosis. Gastroenterology (2009b) 136:1037–47. doi: 10.1053/j.gastro.2008.11.035
251. Lerut J, Demetris AJ, Stieber AC, Marsh JW, Gordon RD, Esquivel CO, et al. Intrahepatic Bile Duct Strictures After Human Orthotopic Liver Transplantation. Recurrence of Primary Sclerosing Cholangitis or Unusual Presentation of Allograft Rejection? Transpl Int (1988) 1:127–30. doi: 10.1007/BF00348833
252. Graziadei IW, Wiesner RH, Marotta PJ, Porayko MK, Hay JE, Charlton MR, et al. Long-Term Results of Patients Undergoing Liver Transplantation for Primary Sclerosing Cholangitis. Hepatology (1999) 30:1121–7. doi: 10.1002/hep.510300501
253. Yamada Y, Hoshino K, Fuchimoto Y, Matsubara K, Hibi T, Yagi H, et al. Rituximab Induction to Prevent the Recurrence of PSC After Liver Transplantation-The Lessons Learned From ABO-Incompatible Living Donor Liver Transplantation. Transplant Direct (2018) 4:e342. doi: 10.1097/TXD.0000000000000760
Keywords: B cell, liver, liver fibrosis, biliary atresia, paediatric liver disease, liver diseases
Citation: Patel AM, Liu YS, Davies SP, Brown RM, Kelly DA, Scheel-Toellner D, Reynolds GM and Stamataki Z (2021) The Role of B Cells in Adult and Paediatric Liver Injury. Front. Immunol. 12:729143. doi: 10.3389/fimmu.2021.729143
Received: 22 June 2021; Accepted: 16 August 2021;
Published: 23 September 2021.
Edited by:
Mirjam van der Burg, Leiden University Medical Center, NetherlandsReviewed by:
Xiaohui Wang, The University of Hong Kong, Hong Kong, SAR ChinaCopyright © 2021 Patel, Liu, Davies, Brown, Kelly, Scheel-Toellner, Reynolds and Stamataki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zania Stamataki, ei5zdGFtYXRha2lAYmhhbS5hYy51aw==
†These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.