 Matheus B. Carneiro
Matheus B. Carneiro Nathan C. Peters
Nathan C. Peters
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 September 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.728848
This article is part of the Research Topic Innate Immune Responses Against Leishmania Parasites View all 8 articles
Intracellular phagosomal pathogens represent a formidable challenge for innate immune cells, as, paradoxically, these phagocytic cells can act as both host cells that support pathogen replication and, when properly activated, are the critical cells that mediate pathogen elimination. Infection by parasites of the Leishmania genus provides an excellent model organism to investigate this complex host-pathogen interaction. In this review we focus on the dynamics of Leishmania amazonensis infection and the host innate immune response, including the impact of the adaptive immune response on phagocytic host cell recruitment and activation. L. amazonensis infection represents an important public health problem in South America where, distinct from other Leishmania parasites, it has been associated with all three clinical forms of leishmaniasis in humans: cutaneous, muco-cutaneous and visceral. Experimental observations demonstrate that most experimental mouse strains are susceptible to L. amazonensis infection, including the C57BL/6 mouse, which is resistant to other species such as Leishmania major, Leishmania braziliensis and Leishmania infantum. In general, the CD4+ T helper (Th)1/Th2 paradigm does not sufficiently explain the progressive chronic disease established by L. amazonensis, as strong cell-mediated Th1 immunity, or a lack of Th2 immunity, does not provide protection as would be predicted. Recent findings in which the balance between Th1/Th2 immunity was found to influence permissive host cell availability via recruitment of inflammatory monocytes has also added to the complexity of the Th1/Th2 paradigm. In this review we discuss the roles played by innate cells starting from parasite recognition through to priming of the adaptive immune response. We highlight the relative importance of neutrophils, monocytes, dendritic cells and resident macrophages for the establishment and progressive nature of disease following L. amazonensis infection.
Phagocytes and phagocytosis were first described by Elie Metchnikoff in 1883. At that time phagocytosis was primarily described in frogs and associated with homeostasis, nutrition and tissue reabsorption. Later, Metchnikoff described how this process could also act as a protective mechanism against pathogens (1). “Infection, a struggle between two organisms”, was the title Metchnikoff gave to the first of a series of lectures he delivered in 1891 (2), a title that accurately described the complex relationship, from an evolutionary perspective, between the host immune system and infectious pathogens. After 130 years, the challenge to understand how best to treat infectious diseases caused by pathogens that employ phagocytes as hosts for infection, replication and persistence via prophylactic vaccination, immunotherapy, or antibiotics remains. In the case of the intracellular parasite Leishmania, the infection poses significant challenges to treatment and prevention due to multiple mechanisms of immune evasion that allow the parasite to infect the very cells that the immune system employs to eliminate them (3, 4). Since the late 1970s Leishmania parasites have been described as phagosomal pathogens that reside and proliferate within phagocytic cells (5–8). We now understand that even a single Leishmania parasite transmitted by a sand fly bite is sufficient to establish infection (9, 10), corroborating data discussed throughout this review that innate mechanisms of immunity are not sufficient to provide protection and that the development of an adaptive immune response, mediated largely by Th1 CD4 cells, is required to activate phagocytic cells (11).
Leishmaniasis is a vector-borne disease associated with different clinical manifestations, determined by the intersection of the parasite species and the host immune response (12). In this review, we will discuss the paradoxical relationship between L. amazonensis (L.a.) parasites and phagocytic cells during cutaneous Leishmaniasis. Infection with L.a. is intriguing as it has been associated with a remarkably diverse clinical manifestations including localized cutaneous leishmaniasis (LCL), borderline disseminated cutaneous leishmaniasis (BDCL), anergic diffuse cutaneous leishmaniasis (ADCL) and, less frequently, with mucosal and visceral leishmaniasis (13, 14). We will focus on how living in a phagosome represents a challenge to L.a. and how this parasite has evolved to survive not only against innate mechanisms of immunity, but also to take advantage of Th1 immunity for the establishment and perpetuation of disease.
Phagocytosis is an important biological function mediated by monocytes, macrophages, neutrophils and dendritic cells (DCs), see Figure 1, that plays a role in both inflammation and homeostasis, where it can mediate both pathogen elimination and tissue healing (15). Phagocytosis is a complex and well-regulated process that has been reviewed in detail elsewhere (16). Briefly, phagocytosis is initiated by the binding of specific molecules expressed by pathogens, or the host, to surface receptors expressed on phagocytic cells and is therefore a contact-dependent process. The phagocyte receptors associated with Leishmania phagocytosis are either opsonic, when host-particles (opsonins) bind to the pathogen or non-opsonic, when they bind directly to pathogen-associated molecular patterns (PAMPs). Opsonic receptors are: FcγRs (CD64, CD32, CD16), complement receptors (CR1-4, Mac-1) and fibronectin receptors (a5B1), which bind respectively to antibodies (abs), complement and fibronectin. Examples of non-opsonic receptors are mannose receptor (MR or CD206), Dectin-1 and DC-SIGN (17–21). Specific receptors can also recognize apoptotic cells, such as TIM-1, TIM-4, which recognized phosphatidyl serine (PS), exposed on the membrane of dying cells, or MertK (tyrosine-protein kinase Mer), which binds to GAS6 (growth arrest-specific 6), a molecule that bridges PS to the membrane (22). In addition, in vitro, L.a. has been shown to use Toll-like receptor (TLR) 2 to infect neutrophils (23) and can also subvert endocytosis during cell plasma membrane repair to invade non-phagocytic cells, such as fibroblasts (24). As reviewed by Ueno and Wilson, the receptor involved in parasite entry may influence parasite fate, as it can influence phagolysosome maturation, reactive oxygen species (ROS) production and phagocytic cell activation (25). After binding to the phagocytic receptors, pseudopods surround Leishmania parasites and form the phagosome. During phagocytosis of L.a. metacyclic promastigotes, entrance is often mediated via the cell body rather than the flagellum, in a process that can take up to 10 minutes in vitro employing bone-marrow derived macrophages (BMDMs) (26). The engulfment of either promastigotes or amastigotes is associated with an arrangement of the cytoskeleton and a transient polymerization of F-actin around the parasites (26, 27).
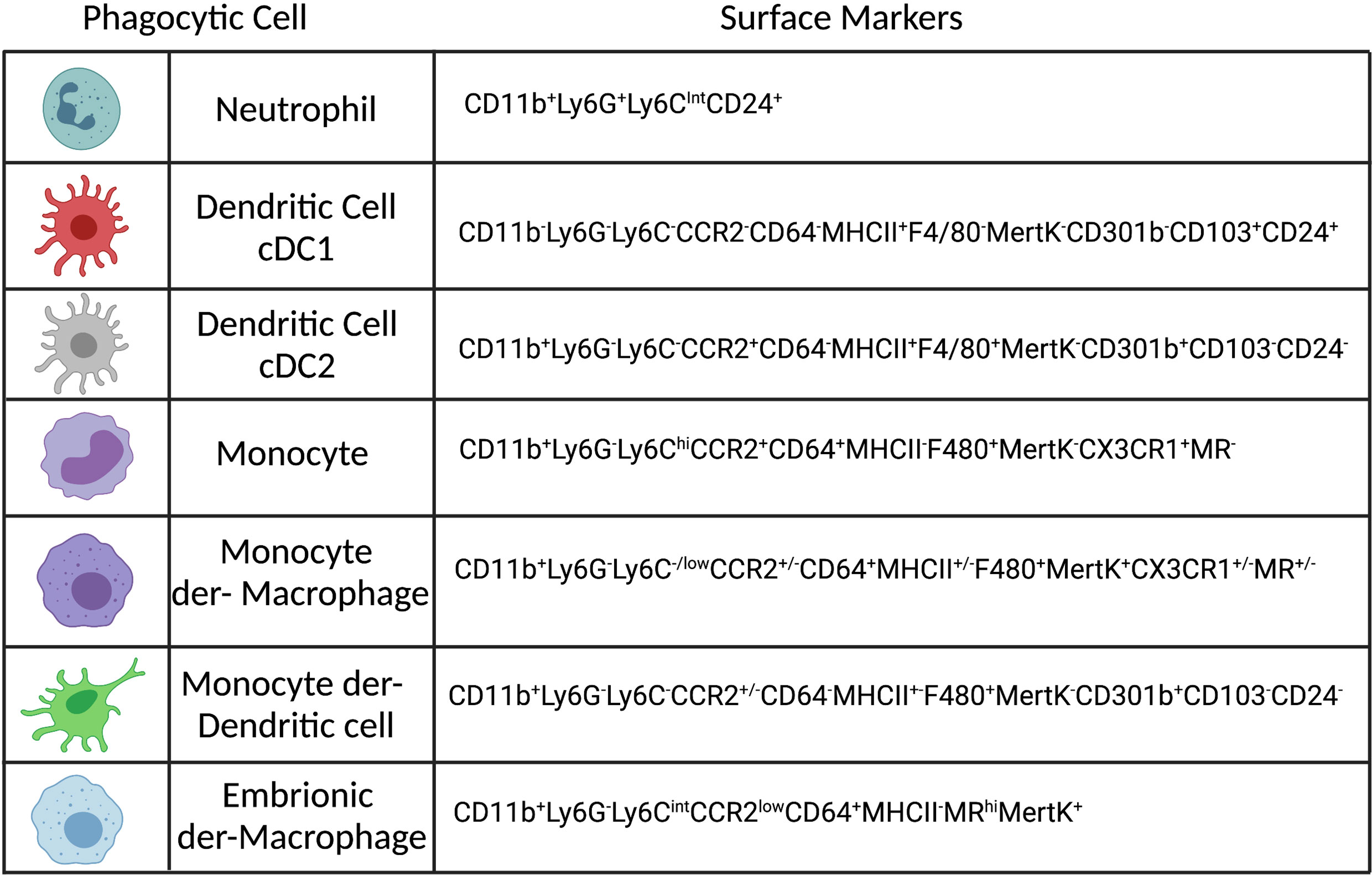
Figure 1 Phenotypes of the most abundant phagocytic cells in the dermis of Leishmania infected mice. Created with BioRender.com.
This initial phagosome will undergo a maturation process mediated by fusion with endosomes and lysosomes. After internalization, the early phagosome, characterized by moderate pH acidification, will fuse with early endosomes, a process that relies on the expression of Rab5, a GTPase (guanosine triphosphatase), that mediates membrane fusion events (16). The late phagosomes are formed when early phagosomes fuse with late endosomes. In this stage there is a shift from Rab5 to Rab7 at the phagosome membrane and the lumen becomes more acidic, due to an increase in the expression of V-ATPases (which translocate protons H+ from the cytosol to the phagosome at the cost of an ATP). The Rab7 mediated fusion of the late phagosome with lysosomes characterizing the formation of the phagolysosome or parasitophorous vacuole (PVs). The phagolysosome is characterized by membrane expression of lysosomal markers such as lysosomal associated membrane protein 1 and 2 (LAMP-1 and LAMP-2) and the presence of cathepsins, proteases and lysozymes in the lumen. In addition, L.a. PVs are considered hybrid compartments as the phagosome also presents molecules associated with the endoplasmic reticulum (ER) pathways such as calnexin (28).
Phagolysosomes are highly acidic, oxidative, contain antimicrobial peptides, proteases and restrict nutrient access, representing a highly hostile environment for an intracellular pathogen (16, 29). However, many intracellular pathogens have evolved different mechanisms to either avoid phagocytosis, escape from the phagolysosome or to resist phagolysosomal effector mechanisms (29). In contrast, Leishmania parasites are adapted to survive and replicate within phagolysosomes and different species of Leishmania have developed different strategies to do so. Survival primarily appears to be facilitated by allowing time for the parasite to differentiate into an amastigote before the hostile mature phagolysosome is formed. For instance, during in vitro infection of macrophages, promastigotes of L. donovani and L. major (L.m.) can delay phagolysosome maturation by preventing acidification (30), phagosome-endosome fusion (31) or NADPH (nicotinamide adenine dinucleotide phosphate) oxidase assembly (32). Curiously, L.a. parasites do not seem to interfere with phagolysosome maturation, an observation that is somewhat counterintuitive. Rather, several studies show that acidification, fusion between the phagosome and endosomes/lysosomes, lysosomal enzymes and protease activities and the expression of lysosome markers (Rab7, LAMP1, V-ATPase) are all present on L.a. containing phagolysosomes (26, 33–37). In fact, the formation of the L.a. phagolysosome is relatively rapid, occurring within 30 minutes post infection, when the phagolysosomes of most infected macrophages have already acquired lysosomal features in the lumen and membrane. This process occurs faster when the infection is initiated by the amastigote form of the parasite but is otherwise quite similar compared to promastigotes and it is not influenced by previous exposure to IFN-γ activation (26). Altogether, Leishmania amastigotes are extremely well-adapted to survive and proliferate inside mature phagolysosomes and L.a., in particular, does this without significantly interfering with this host defense mechanism compared with other species.
The structure of phagolysosomes differs when containing different Leishmania species. During L. major and L. donovani infection, the PVs are tight and harbor a single amastigote, while infections with species from the L. mexicana complex, such as L.a., generate large communal PVs that contain several amastigotes attached to the vacuole membrane (36). It has been suggested that the increased size of L.a.-containing vacuoles would dilute the effect of antimicrobial molecules, compromising parasite killing and favoring growth (38, 39), which could explain why killing L.a. amastigotes is harder to achieve then other species such as L.m (40, 41). The development of large PVs depends on the accumulation of CD36 at the sites where amastigotes attach to the phagosome membrane, suggesting that this process could occur either during or after amastigote differentiation. CD36-/- BMDMs infected with L.a. developed tight PVs that restricted L.a. growth but had no impact on L.m. growth (39). In vitro, the PV enlargement process is rapid and seen 8 h post-infection with promastigotes, reaching their maximum volume by 24 h (26, 39). CD36 likely induces PV enlargement by enhancing the fusion with late endocytic vesicles, more specifically with lysosomes (39). As fusion with ER vesicles has also been shown to participate discretely in the enlargement of L.a. PVs (42), it is possible that CD36 enhance this fusion pathway as well. Likely as a mechanism of defense, in vitro infected macrophages respond to L.a. infection by enhancing the lysosomal trafficking regulator (LYST)/Beige expression, a protein complex that limits the size of lysosomes, therefore reducing the size of the PVs and restricting parasite replication (38). However, it is unlikely that during in vivo infection the levels of LYST/Beige expression by host infected cells is enough to impact parasite growth, as chronic disease is associated with large PVs containing several amastigotes within mononuclear cells (43, 44). In addition, the engagement of CD36 driving PV enlargement is not associated with changes on LYST/Beige expression (39). Importantly, while the PV size is associated with L.a. growth restriction in tight PVs (39, 45), it did not impact parasite killing mediated by IFN-γ+ LPS on in vitro activated BMDM, arguing against the idea that large PVs would dilute microbicidal molecules, providing a shield to the parasites. However, direct in-vitro activation by IFN-γ + LPS might induce a stronger activation phenotype on host cells compared to in vivo infection and therefore it remains a possibility that the large PVs in L.a. infection provide a niche in host phagocytic cells that don’t get sufficiently activated.
Differences in the properties of phagolysosomes have also been reported between different phagocytic cells and this is relevant to Leishmania infection. Neutrophils are known for being able to produce up to 20 times more ROS than macrophages, which in combination with a lower expression of V-ATPase, due to the high activity of NADPH oxidase, characterize an early alkaline environment in the PVs (46–48). In addition, due to a lack of the endosomal pathway in neutrophils, the phagosome mostly fuses with the preformed granules found in these cells, which also contain NADPH oxidase in their membrane, contributing to higher ROS production (47). Fusion of phagosomes with azurophilic granules containing myeloperoxidase (MPO) has been described during infection of human neutrophils in vitro with L. major and L. donovani (49), however, little is known about the phagolysosome biology of neutrophils during Leishmania infection and an important question that remains to be elucidated is if the differences in phagolysosome maturation influences promastigote-amastigote differentiation in different phagocytic cells and how this might impact the development of disease.
The life experience of phagolysosome exposed amastigotes, including changes in temperature, pH, O2, and restricted access to nutrients, is significantly different compared to promastigotes, which are largely restricted to the sand fly vector midgut. In fact, some of these changes have been associated with the differentiation process from promastigotes to amastigotes, including the requirement for higher temperature and lower pH for the propagation of axenic amastigotes in-vitro (50). Interestingly, for L.a., ROS production and the acquisition of iron play critical roles in this process. The phagolysosome restricts the iron access to the parasites. Iron is a key cofactor to many enzymes and regulates important physiological pathways in different organisms. Iron deprivation induces an up-regulation of Leishmania iron transporter 1 (LIT1), that maintains internal iron content while reducing parasite proliferation. Parasites lacking LIT1 continue to proliferate until they show a robust cell death rate (51). In addition, Leishmania require iron as a co-factor for the activity of iron superoxide dismutase (FeSOD), a key enzyme that converts superoxide, mostly derived from parasite mitochondria, into hydrogen peroxide (H2O2), which drives amastigote differentiation (51, 52). In addition to iron, amastigotes need to uptake other nutrients from the phagolysosome lumen or cytoplasm including several essential amino acids, lipids, vitamins and purines, which are essential for parasite survival and growth (53). This uptake relies on phagosome-endosome fusion, on transporters present at the PVs membrane and on transporters expressed by the parasites themselves (53–56). A critical auxotrophic micronutrient for amastigotes is L-arginine. This amino acid is the only source for polyamines production, essential for parasite survival (57). L. amazonensis express their own arginase enzyme that hydrolyses L-arginine into L-ornithine and urea. Then ornithine decarboxylase (ODC), catalyzes the enzymatic decarboxylation of L-ornithine into polyamines (58). Amastigotes can also access polyamines generated by host cytoplasmatic arginase -1 (Arg1) activity (59). During infection, host cells increase the consumption of L-arginine as this amino acid is required for the activity of both arginase and inducible nitric oxide synthase (iNOS) enzymes. The up regulation of the receptor cationic amino acid transporter 2b (CAT-2B), modulated by IFN-γ, allows the necessary upregulation of L-arginine uptake. Interestingly, Leishmania parasites can sense the availability of L-arginine in their environment and change the expression of their own amino acid transporters (60–64).
Pathogen engulfment triggers the assembly and activation of the enzyme complex NADPH oxidase in phagocytic cells. This enzyme reduces O2 into superoxide that can mediate damage to the parasite (47). Both in vitro infection of macrophages and neutrophils with either L.a. promastigotes or amastigotes induces this respiratory burst process (65, 66). However, the absence of a functional NADPH oxidase in mice lacking the membrane catalytic unit gp91 (Nox2) does not impact parasite survival significantly to have an impact on the establishment of disease or parasite loads over the course of infection (65, 67, 68). The production of nitric oxide (NO) requires the activity of iNOS, whose expression is induced by TNF, IFN-γ, IL-1β, and TLR ligands such as LPS (43, 69–71). L-arginine is the substrate for iNOS, therefore robust uptake of this amino acid is required when phagocytic cells are activated to ensure sustained and robust NO production (47, 72). iNOS expression can be found at the phagosome but its primary expressed on other vesicles in the cytosol (73, 74). As NO is a relatively stable uncharged molecule it can cross membranes such as the PV and the plasma membrane (75). NO mediates its cytotoxic effect by: a) reacting with heme-cytochrome C oxidase in pathogen mitochondria to initiate ROS production; b) compromising DNA synthesis by inhibition of ribonucleotide reductase activity; c) causing direct DNA damage (47, 76); d) reacting with ROS forming peroxynitrite anion (ONOO-), which is a stronger oxidant that promotes pathogen killing by compromising mitochondrial respiration and mediating tyrosine nitration that impacts proteins structure and function (77). Finally, NO can inhibit overall metabolism activity in Leishmania parasites restricting their growth (78). While ROS production does not provide protection against L.a, absence of NO is associated with enhanced parasite growth and pathology (44, 71, 79). L.a. can interfere with iNOS/NO production in different ways: by expression of ectonucleotidases, such as ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase), an enzyme that provides AMP for adenosine production, which then binds to adenosine A2B receptors on macrophages decreasing NO production and inhibiting TNF and IL-12 production (80); by activating NF-κB repressors (81, 82) or by inhibiting iNOS activity (83).
Innate cells detect pathogens, including Leishmania parasites, by recognizing PAMPs through the expression of pattern recognition receptors (PRRs). PRRs are expressed on the cell surface, in the cytoplasm, or are secreted into the blood stream or tissue fluids (84).
Leishmania metacyclic promastigotes are the infective stage of the parasite predominantly transmitted into the dermis of mammalian hosts by infected sandflies during acquisition of a blood meal (10, 85, 86). Metacyclic promastigotes are characterized by the presence of a thick glycocalyx on the cell surface where different proteins are expressed, representing a source for PAMPs. The most common components expressed on promastigotes are glycophosphatidylinositol lipids (GIPLs), lipophosphoglycan (LPG), proteophosphoglycan (PPG) and the GP63 zync-metalloprotease. Most of these molecules are attached to the parasite surface by glycosylphosphatidylinositol (GPIs) anchors (87). The amount of their expression can vary between the different parasite life-cycle stages and the structure of these components is different between different species of Leishmania, thereby explaining variations in the role played by these glycoconjugates following infection with different species (88).
Because of the pool feeding feature of sand flies, the first innate immune mechanism that challenges Leishmania survival is the exposure to complement components in the serum (89). L.a. metacyclic promastigotes are resistant to complement through at least two different mechanisms: cleavage of C3b into iC3b by GP63 or by interfering with correct C9 attachment to the parasite surface, in both cases avoiding the formation of the membrane attack complex (MAC), enabling parasite survival (90–92). After evading complement, L.a. must be phagocytosed by phagocytic cells to establish a niche for survival/replication. Interestingly, the presence of iC3b on the parasite surface is not only important to escape lysis mediated by the MAC, but also facilitates parasite phagocytosis by CR1/CD11b (93). It is possible that receptors such as CR3 bind simultaneously to both opsonin (iC3b) and the promastigote (LPG) in different binding sites enhancing phagocytosis (94). In addition, GP63 secretion can digest collagen type IV and fibronectin in an in vitro extracellular matrix, which might facilitate parasite-phagocyte encounters in the skin (95).
When phagocytic cells bind to and internalize Leishmania parasites host PRRs can be activated by PAMPs. The impact of TLR activation on leishmaniasis depends on parasite species, host genetic background, cell subset and has been linked to both anti and pro-inflammatory responses (96). Purified L.a.-LPG activates TLR2, triggering the production of type I interferon leading to activation of the IL27/IL-10 axis and inducing superoxide dismutase (SOD) expression, which impairs macrophage activation and favors parasite replication both in vitro and in murine infections (97–101). The production of type I interferon and IL-27/IL-10 axis is mediated by activation of ds-RNA dependent kinase (PKR), a PRR, which in this scenario is activated by type I interferon and IL-27 (99, 100). Importantly, in a model of co-infection of L.a. with an Amazonian Phlebovirus, which can infect sandflies, an exacerbation of lesion size and parasite load also correlated with an increase on IFN-β/IL-10 production associated with PKR activation (102). In addition, TLR2-deficient DCs infected in vitro showed higher IL-12p40 levels and induced stronger activation of naïve CD4 T cells as measured by higher replication rate and IFN-γ production compared to Wt DCs (103). Lastly, skin biopsies from patients infected with L.a. showed a higher expression of IFN-β/PKR for the more severe form of the disease, anergic diffuse cutaneous leishmaniasis (ADCL) compared to localized cutaneous leishmaniasis (LCL) (99). Thus, the TLR2/Interferon type I/PKR pathway plays a significant role in parasite evasion. Despite the pathogenic role played by TLR2 during L.a. infection, Myd88 deficiency is associated with higher susceptibility to disease, thus suggesting additional roles mediated by different TLRs that also signal via Myd88 (71, 104). In this context, TLR4, which can also be activated by LPG, is associated with both NO and TNF production following in-vitro macrophage infection (104, 105). The role played by TLR9 is controversial, L.a. amastigotes can release DNA on micro-vesicles, activating endosomal TLR9, which required both Myd88 and TRIF to induce CD200, which blocks iNOS expression, resulting in a transient higher resistance in TLR9-/- mice compared to Wt mice, in early but not chronic phases of the disease (106, 107). Lastly, the expression of TLR2, TLR4 and TLR9 is different during different clinical manifestations of L.a. infection in humans. Specifically, LCL showed a higher density of TLR2 expression compared to TLR4 and TLR9 by CD68+ macrophages from skin biopsies. On the other hand, for ADCL and BDCL forms, the density of TLR9 expression is higher comparing to TLR2 and TLR4. Importantly, no correlation between the expression pattern of TLRs and production of TNF, IL-10 or TGF-β by T cells were found (108).
Another important family of PRRs are the NOD-like receptors (NLRs), which work as a sensor for intracellular molecules. NLRs are important components in the inflammasome complex. In general, activation of the inflammasome multiprotein complex in the cytosol leads to activation of caspase-1 that cleaves the inactive proinflammatory cytokines (pro-IL-1β and pro-IL-18) into their respective active forms (109). The activation of the inflammasome through NRLP3 depends on ATP, released by dying cells at the site of infection, and ROS production by NADPH oxidase, released during parasite phagocytosis. In addition, L.a. LPG triggers a non-canonical pathway of inflammasome activation mediated by caspase-11, although it is not yet known how LPG from the parasites gains access to the host cell cytoplasm to activate the inflammasome complex (110–112). In general, IL-1β production during Leishmania infection has been associated with different disease outcomes: pathology in the case of L. major and L. braziliensis infection, dissemination following L. infantum infection, and a protective role against L.a (71, 113–115). BMDMs, previously primed with LPS, and infected by L.a. promastigotes induce IL-1β production, mediated by inflammasome activation. IL-1β was then shown to induce iNOS production, having a long-term impact on disease, as assessed by higher susceptibility of mice deficient for NLRP3, CASP1 and CASP11 (71, 110). IL-18 production, on the other hand, is associated with higher pathology and parasite loads during L.a. infection by an unknown mechanism (116). Interestingly, activation of the inflammasome seems to depend on the parasite stage. Different from promastigotes, infection by amastigote forms of L.a. inhibit several components of inflammasome activation including NRLP3, NLRP4 and AIM2 by modulating epigenetic changes at H3 (hypoacetylation) in macrophages (117). In addition, infection of DCs in vitro with L.a. amastigotes also inhibited inflammasome activation resulting in less IL-1β and IL-18 production (118). Considering the differences between how promastigotes and amastigotes modulate inflammasome activation it is not clear how inflammasome activation by promastigotes during the early stages of the infection would impact long-term disease by inducing iNOS expression, especially in the context of subsequent Th1-mediated iNOS production in response to IFN-γ. Currently, it is not known if inflammasome activation has an impact on infiltrating phagocytic cells during L.a. infection, thereby influencing permissive host cell availability. Another outstanding question is the impact of inflammasome activation on neutrophils, which represent important host cells infected by promastigote forms of the parasite, predominantly at acute stages of infection (10).
Neutrophils are generated in the bone marrow in adult mammals and represent the most abundant leukocyte population in the bloodstream, allowing for rapid recruitment into damaged tissue sites. Neutrophil recruitment to damaged tissues, following sterile or infection-associated injury, is a hallmark of early innate immunity (119). Neutrophil migration from bone-marrow to the circulation relies on the balance between retention/mobilizing signaling mediated by the expression of CXCR4/CXCR2, respectively (120). Blood neutrophils are short-lived (half-life of around 10 h), which can increase up to 24 h following tissue infiltration under homeostasis, or even up to 7 days in the context of inflammation (119). Recently, by employing both parabiosis and a tamoxifen-inducible Cre recombinase at the ly6g locus, allowing for tracking neutrophil fates in different tissues, it was observed that neutrophil half-life depends on the tissue, and, interestingly, the skin, amongst the tissues analyzed, is the organ where neutrophils have the longest half-life (around 18 h) (121). In addition, transcriptome analyses from skin neutrophil suggest that these cells might regulate epithelial growth in the skin under steady state (121). During systemic infections, a 10x increase in neutrophil production can be observed, a process called emergency granulopoiesis, which can be triggered by the increase in serum levels of the growth factors granulocyte and macrophage colony-stimulating factors (G-CSF/M-CSF, respectively) and cytokines (IL-6, type I interferons), which regulate hematopoietic stem cells (HSCs) in the bone-marrow (122). Neutrophil recruitment is also enhanced during inflammation. Neutrophil chemotaxis is mediated by different receptors: I) chemokine receptors such as CXCR2, CXCR4 and CXCR1 (123). The inflammatory chemokine receptors CCR1, CCR2, CCR3 and CCR5, can also be expressed by neutrophils but do not play a role on neutrophil recruitment during either homeostasis or inflammation, and instead, appear to be more relevant for monocyte and eosinophil recruitment (124); II) lipid mediator receptor, such as leukotriene B4 receptor (LTB4R), which binds to leukotriene B4 and III) complement receptors, as C5aR, CR3 and C3aR (123). The acute neutrophil recruitment to wounded skin can also be impaired in the absence of a microbiome (125).
The employment of multiphoton intravital microscopy to image leukocyte migration into different tissues has made a great impact on the understanding of neutrophil behavior after transmigration, for instance, into wounded skin. Tissue damage following sand fly bite or needle inoculation is enough to induce a robust neutrophil infiltrate to the dermis in the minutes to hours post-infection with L.m (10, 126, 127). Needle inoculation of L.a. parasites also induce early neutrophil recruitment to the skin (128, 129), see Figure 2. In addition to tissue damage, neutrophil recruitment is also associated with vector associated components, such as the sand fly gut microbiota, sand fly saliva, and promastigote secretory gel (PSG), rich in proteophosphoglycans and secreted by the parasites in the vector midgut, as well as factors released by the host immune response to the bite, including CXCL1, CXCL2 and C3 cleavage (114, 130–132). Importantly, while the presence of Leishmania is not required to induce neutrophil recruitment, the presence of parasites or sand fly derived factors is likely required for persistent neutrophil swarming and maintenance of the ‘neutrophil plug’ that forms where the sand fly proboscis has penetrated through the skin. In contrast, sterile wounds are associated with a mostly transient presence of neutrophils (10, 126, 133). All of these factors are likely to contribute to the intense neutrophil cluster formation at sites of sand fly bite and, up to 24 h post infection, neutrophils represent a majority of the inflammatory host cells for most Leishmania species following transmission by sand fly bite or needle inoculation (10, 43, 134). Recently, sand fly transmitted parasites of a non-healing L. major-RYN strain that were distributed to dermal sites distal to the site of vascular damage and neutrophil cluster formation resulting from sand fly probing were shown to be phagocytosed by a more diverse population of phagocytes, including resident dermal macrophages (135), suggesting the localization of parasite deposition can influence host cell phenotype. The biological relevance of neutrophil swarming and cluster formation during the early phase of Leishmania infection may be to restrict parasite dissemination, as neutrophils tend to concentrate at sites of tissue damage where Leishmania are also found and may limit their subsequent spread following phagocytosis even if they are unable to kill the parasite. Second, the co-localization between neutrophil clusters and parasites might restrict tissue pathology induced by neutrophil-derived inflammatory factors to the wound associated with sand fly bite versus a more generalized distribution in the skin. In fact, the robust acute neutrophil recruitment after either sand fly or needle injection does not correlate with the persistent tissue pathology associated with cutaneous disease, as clinically relevant lesions typically only appear after the induction of adaptive immunity (10, 43, 128, 134).
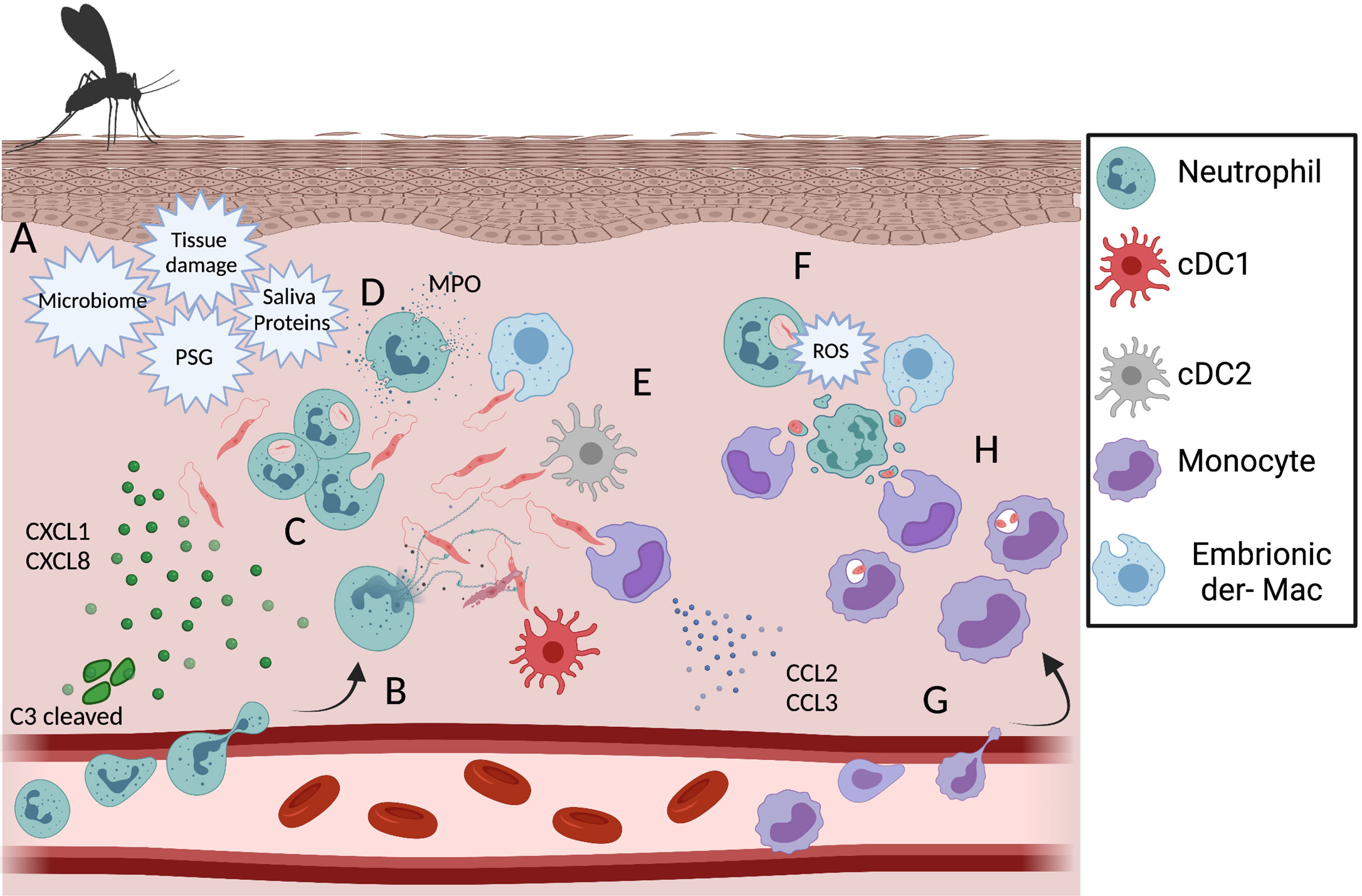
Figure 2 Dynamic of phagocytic cells during the acute phase of the immune response against L. amazonensis infection. (A) Recruitment of neutrophils is triggered by tissue damage, sand fly associated factors, chemokine production by keratinocytes and C3 cleavage. After recruitment to the dermis neutrophils can release NETs (B), phagocytose (C) and degranulate (D) in response to L.a. infection. Other infected phagocytic cells during early infection include embryonic dermal macrophages, resident DCs and monocytes (E). In response to metacyclic promastigote phagocytosis, neutrophils undergo respiratory burst and ROS mediated apoptosis (F). In response to changes in the innate chemokine production profile, a wave of inflammatory monocytes is recruited to the dermis skin (G) and become infected by phagocyting infected neutrophils, apoptotic bodies, a process known as the “Trojan Horse” model of infection (H) or parasites freed by apoptotic neutrophils. Created with BioRender.com.
In addition to sand fly bite associated factors, neutrophil cell death can also initiate or amplify neutrophil swarming and cluster formation (133). This observation is important in the context of Leishmania infection as parasite phagocytosis has been associated with the induction of neutrophil death. How fast Leishmania parasites induce neutrophil cell-death depends on parasite species and experimental settings (in vitro versus in vivo; and the source of neutrophils). L. major parasites can delay apoptosis during in vitro infected blood neutrophils or in vivo infected peritoneal neutrophils (136–138), but exposure of phosphatidylserine, often employed as a surrogate marker of apoptosis, has been reported within a few hours after infection of either bone-marrow derived (135) or dermal neutrophils (139), suggesting parasite-induced induction of apoptosis. After 18 h of infection by L. braziliensis peritoneal neutrophils also showed higher levels of PS exposure in the membrane (138). In addition, a few hours after the phagocytosis of L.a. neutrophils become annexin V positive, in a process that is driven by NADPH-dependent ROS production (67). It is important to highlight that neutrophil death may be induced by Leishmania phagocytosis, as shown by studies employing Leishmania-RFP expressing parasites, which allow for comparison of infected versus uninfected cells in the same environment, either in vitro or in vivo. This is relevant because variations in the infection rate can significantly impact the degree to which the total population will exhibit an infection related phenotype.
The engulfment of infected apoptotic neutrophils or phagocytosis of parasites by other phagocytic cells that are concurrently involved in uptake of apoptotic blebs, a process referred to as efferocytosis, can enhance the chances of parasite survival. In the Leishmania literature, this process is commonly referred to as the “Trojan horse” model of infection (140). The neutrophil engulfment by dermal macrophages is mediated by the expression of tyrosinase kinases receptors, such as Axl and MertK, and has been recently shown during in vivo infection with L. major, where an up-regulation of wound-healing phenotype markers, such as resistin-like molecule alpha (Relmα) and Arg1 was reported (135). In vitro, the uptake of L.m. and L.a. infected apoptotic neutrophils by macrophages enhances transforming growth factor beta (TGF-β) and prostaglandin E2 (PGE2) production (140, 141). Meanwhile, when dermal DCs acquire L.m. via infected neutrophils, an impairment on antigen presentation and T cell priming is observed and is associated with decreased levels of MHC II, CD40 and CD86 expression by DCs (139). Therefore, it appears that Leishmania can take advantage of the anti-inflammatory bias that macrophages or DCs acquire after phagocyting apoptotic neutrophils at the site of infection (135, 139). The importance of neutrophil apoptosis in regulating tissue pathology during infection is highlighted in the studies of L.a. infection of ROS/NADPH-deficient gp91-/- mice mentioned above. In these studies, parasite loads are unaffected but NADPH-mediated progression of neutrophils to an apoptotic phenotype is abrogated, resulting in the accumulation of necrotic neutrophils and severe tissue pathology (67).
Interestingly, L.m can survive but do not appear to replicate within neutrophils during the acute innate phase of the disease (9, 10, 142). The subsequent host protective versus pathogenic roles played by neutrophils during Leishmania infection remains somewhat controversial. For instance, depletion of neutrophils in C57BL/6 mice just prior to exposure to the bites of L. major infected sand flies resulted in both a reduction in the number of productive transmission events and lower parasite loads (10). Similar observations were reported in neutrophil-deficient C57BL/6 mice infected with L. mexicana (143) and, more recently, enhanced neutrophil recruitment mediated by sand fly salivary proteins was also found to facilitate L.m. infection (132). On the other hand, depletion of neutrophils prior to a footpad infection in C57BL/6 mice with L. major was associated with an enhanced number of parasites in the draining lymph nodes (dLNs), suggesting a protective role for the neutrophils in this context, while the opposite was observed for BALB/c mice (144). In contrast, dermal needle inoculation of neutrophil-depleted BALB/c mice with L.a. resulted in increased lesions and parasite loads at the site of infection, while it did not affect the disease outcome in C57BL/6 mice (128, 131). It should be noted that interpretation of mouse studies can be complicated by the use of different Leishmania species and the site of infection employed, where intradermal inoculation in the ear is associated with robust neutrophil recruitment and a high proportion of infected cells being neutrophils, similar to sand fly transmission, versus sub-cutaneous inoculation in the footpad that is associated with limited neutrophil recruitment and infection (134, 145). In addition, some studies have employed the draining lymph node to determine parasite load without taking into account the impact of neutrophil depletion on dissemination from the skin or site-specific parasite loads.
Neutrophils have several antimicrobial mechanisms, such as: phagocytosis, degranulation, ROS production and release of neutrophil extracellular trap (NET), in a process known as NETosis (Figure 2) (146), and evidence is still being generated as to the relative importance of each of these mechanisms and the degree to which they can prevent Leishmania infection. Specifically, ROS is produced by murine neutrophils after uptake of promastigote or amastigote forms of L.a. (66). However, it is unlikely that ROS plays a major role mediating L.a. killing as genetic deficiency in the NADPH oxidase gp91 subunit, required for NOX2 ROS production, does not significantly impact parasite load during either the innate or adaptive phases of the disease (65, 67). Neutrophil degranulation during L.a. infection has also been reported in both human and murine settings (23, 97). Upon activation by skin extracellular matrix components (MEC), such as fibronectin, during transmigration, neutrophils degranulate releasing MPO, matrix metallopeptidase 9 (MMP9) and neutrophil elastase (NE), all of which can mediate parasite killing by infected macrophages in vitro (147). The degranulation of MPO and MMP9 by infected human neutrophils can mediate L.a. killing in vitro via an LTB4 dependent mechanism (23). In murine settings, increased MPO and NE has been associated with lower L.a. numbers in IFNAR-/- mice (97). NETs, primarily containing chromatin and coated by nuclear, cytosolic, and granular proteins as well as microRNAs are released into the extracellular milieu upon stimulation from either naïve or PMA-activated neutrophils (148–150). NETs can be found in cutaneous leishmaniasis biopsies, and their release can be induced by either promastigotes, amastigotes or L.a. purified LPG (150, 151). Other species such as L. infantum, L. major and L. mexicana can also induce NETs release by naïve neutrophils (143, 150). Histone dependent NETosis mediated L.a. killing within 10 minutes of parasite-neutrophil interactions, in a process that is depended on NE activity. Promastigotes induced higher amounts of NETs released and are also more susceptible to NETosis comparing to amastigotes forms of L.a (150–152). Lastly, NETs can impair, in vitro, monocyte maturation to a fully differentiated DC phenotype by downregulating IL-4R expression, instead inducing differentiation into macrophages (153). An intriguing open question in the field is how neutrophils “choose” between NETosis or phagocytosis, as currently the understanding is that the two processes are mechanistically mutually exclusive and irreversible. For NET release the enzymes NE and MPO must reach the nucleus to mediate chromatin decondensation, requiring their release from the granules into the cytosol. Meanwhile, during phagocytosis both enzymes are found in the vesicles associated with the endocytic pathway (154). Despite all the effector mechanisms triggered by neutrophils after L.a. infection, most parasites survive and are able to initiate the disease. L.a. parasites can evade NET mediate killing by LPG, gp63 and 3’nucleotidase/nuclease enzyme expression, which can cleave the NETs, releasing the promastigotes (151, 155). In addition, by expressing peroxidases, L.a. amastigotes are highly resistant against ROS production (66, 156).
While little is known about the role of neutrophils during chronic stages of Leishmaniasis, one study has shown that, at least in the case of L. mexicana, amastigotes can replicate inside neutrophil phagolysosomes, which expand in volume as described for macrophages (157). Neutrophils were also found to be the predominant infected population at dermal sites of early L. amazonensis infection in settings of monocyte deficiency, suggesting they can support a degree of ongoing infection (43). At sites of secondary challenge with L. major, where the majority of parasites are eliminated by IFN-γ activated inflammatory monocytes, a significant proportion of the parasites that are remaining, are found in neutrophils. Infection at secondary sites under conditions of monocyte deficiency is also associated with infection of neutrophils, suggesting that even in settings of robust pre-existing Th1 immunity, neutrophils remain a safe haven for infection (142). While these observations suggest neutrophils can maintain infection and even proliferation, studies implicating the mononuclear infiltrate as the host population that maintains parasite replication during the highly regulated late stages of the disease suggest neutrophils play a minor role in the ongoing maintenance of the parasite load under normal conditions (43, 134, 142, 157). In addition to directly providing a permissive host cell for parasites, neutrophils can also modulate the immune response by releasing several chemokines that promote monocyte, DCs, and T cells recruitment to sites of inflammation (158, 159).
Ultimately, any protective mechanism that does not achieve sterilizing immunity is, in fact, non-protective if infection can be initiated by a single parasite, as previously shown (9), and progress to the same peak parasite load and lesion size, albeit with different kinetics. Under this premise, the key biological events to take place in neutrophils, despite all the associated effector mechanisms, would be the differentiation from promastigotes to amastigotes, the stage adapted to survive within phagosomes, in addition to the modulation of cytokine/chemokine production and by potentially facilitating the silent entry of amastigotes into other phagocytic cells.
Dendritic cells were firstly described in the late 1970s (160) and are often referred to as the cells that “bridge” the innate and adaptive immune responses. DCs are a heterogenous population and different subsets have been described according primarily to their ontogeny and function. In the skin dermis conventional DCs (cDC1 and cDC2), minor populations of CD11b- DCs, such as plasmacytoid DCs (pDCs), and monocyte-derived DCs (mon-DCs) have been described (161). As professional antigen presenting cells (APCs), DCs can migrate from peripherical tissues to present antigen and activate naïve CD4 T cells in secondary lymphoid organs such as dLNs (Figure 3). Amongst the APCs active during infection with Leishmania, DCs represent the major source of IL-12 production and are critical for the development of Th1 immunity (162). After pathogen phagocytosis and during the migratory process, DCs undergo a process of further maturation, upregulating the expression of co-stimulatory molecules (CD86, CD80, CD40) and MHC II, which are essential for T cell priming (Figure 3B). The duration of this process presents an opportunity for pathogens to interfere with DC maturation and impair subsequent T cell priming (163). This is especially true for monocyte derived DCs that originate as CD11c-MHCII- cells in the blood and are permissive to L.a. replication (43). The specific role played by each DC subset during Leishmania infection is still not completely understood. Important questions such as if different DC subsets play a different role during (a) early versus late phases of the disease, (b) at the site of infection versus dLNs and (c) if and how they interact with each other remain to be elucidated.
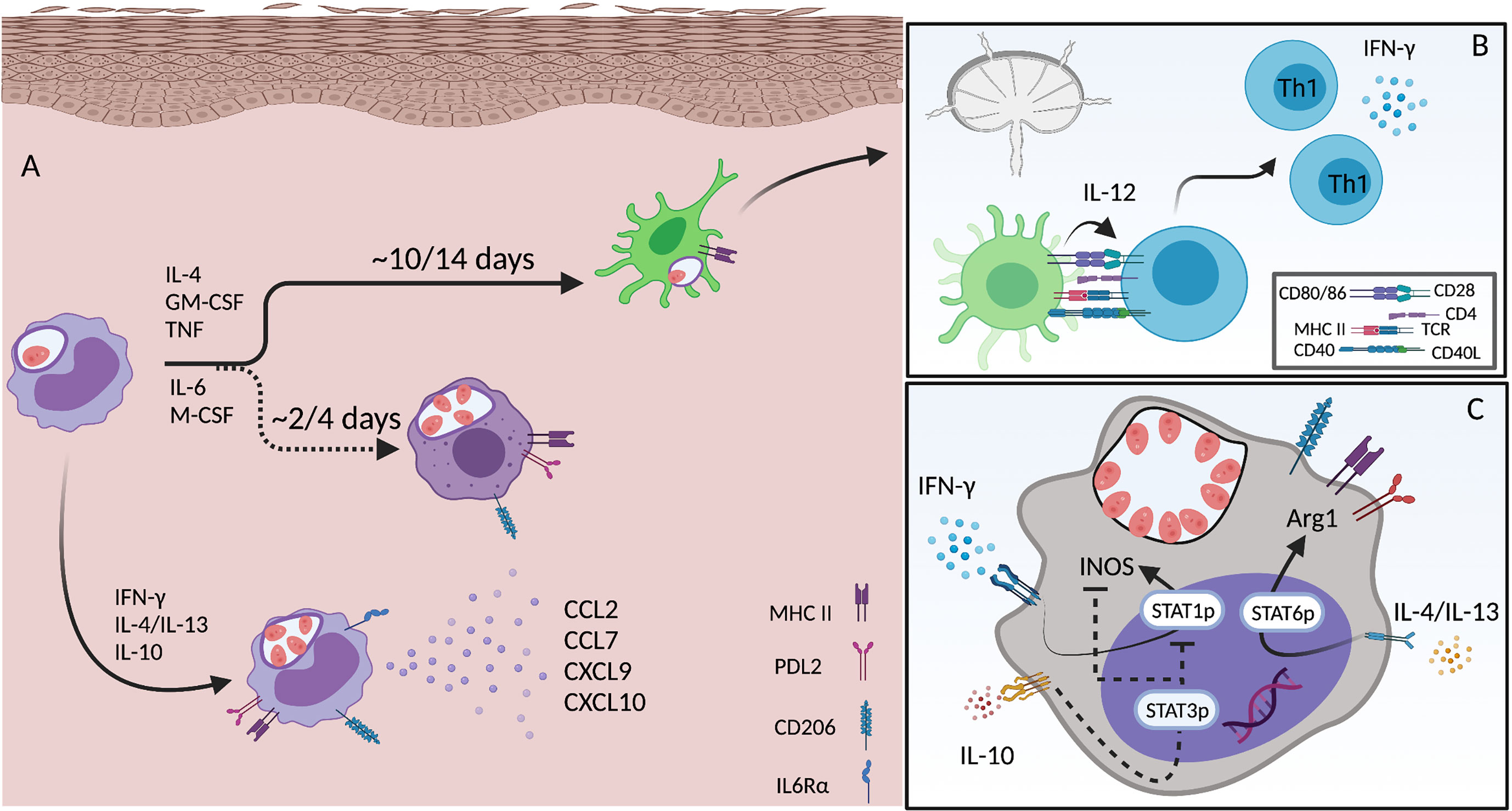
Figure 3 Monocyte differentiation and activation during L.a. infection. (A) Infected inflammatory monocytes acutely recruited to the site of infection become an important source of chemokines (CCL2, CCL7, CXCL9 and CXCL10) when activated by CD4 T cell derived IFN-γ. Infection triggers IL-6rα expression and signaling, which is important for the differentiation towards a macrophage phenotype. Mon-Macs are seen 2-4 days after monocyte recruitment to the site of infection. When activated by Th1 and Th2 cytokines both monocytes and mon-Macs upregulate the expression of MHC II, PDL2 and CD206. Inflammatory monocytes can also differentiate into DCs, a longer process that takes approximately 7-14 days. mon-DCs upregulate MHCII, but not PDL2 or CD206, and are critical for the priming and induction of Th1 immunity (B). The phenotype of both monocytes and mon-Macs exposed to different cytokines produced by different CD4 T cell subsets, leading to, in a single cell, activation of STAT1, STAT3 and STAT6, allowing the co-expression of both iNOS and Arg1 (C). Created with BioRender.com.
cDC1s are a minor population in the skin dermis of both humans and mice while cDC2s are the most abundant (161). cDC1s are usually associated with the cross-presentation of exogenous antigens and priming of CD8+ T cells and with the development of Th1 CD4 immunity. The role of cDC1s during Leishmania infection has been assessed using Baft3 (Basic leucine zipper transcriptional factor ATF-like 3) deficient mice, as cDC1 development is reliant on the expression of this transcriptional factor. During infection with L. major, Baft3-/- mice presented with a deficit in Th1 immunity and enhanced frequencies of Foxp3+ T regulatory cells in the skin that was associated with non-healing lesions and higher parasite loads versus Wt mice (164, 165). This occurred despite the fact that the priming of CD4+ or CD8+ T-cell responses was driven primarily by Baft3-independent DCs. Because Baft3 dependent DCs represent an important source of IL-12 in the skin, they likely impact Th1 CD4+ cells and CD8+ T cells activation at the site of infection despite their low numbers (164). We can also speculate that, as Baft3 dependent DCs represent a source of the chemokines CXCL9, CXCL10 and CXCL11 during bacterial infection, they may also participate in the recruitment and retention of CXCR3+ Th1 effector cells to the site of infection (166). Thus, it is not clear if the reduced numbers of Th1 cells in the dermis after L.m. infection is due to a deficit on T cell recruitment or/and in-situ activation mediated by IL-12 produced by cDC1s. Baft3 dependent DCs also play a protective role during L. infantum infection, as higher numbers of parasites were observed in the liver, but not spleen or bone-marrow, during the late stages of disease (7 weeks post-infection) in Baft3-/- versus Wt mice (167).
A few caveats associated with the use of Baft3 deficient mice to study the role of cDC1s or cross-presentation should be taken into account. cDC1 development may be restored in certain tissues under inflammatory conditions, probably due to the redundant role played by other BAFT proteins. In addition, while cDC1s are absent in the skin and spleen of Baft3-/- mice, these APCs or a very similar population are still present in lymph nodes (161, 168). In addition, other APCs, such as monocytes-derived DCs can cross-prime CD8+ T cells. In this case, monocytes rely on the expression of interferon regulatory factor 4 (IRF4), but not Baft3, to differentiate into DCs that mediate this process (169). It is important to mention that Baft3 deficient mice also show an impairment in monocyte maturation towards a DC phenotype. At early stages after L.m. infection (2-3 weeks), most Baft3 deficient monocytes remained Ly6C+MHCII-, indicating additional roles for Batf3 in different dermal APCs that could have significant secondary impacts during Leishmania infection (164). In regards to cross-presentation, it is known that L. major can impair the canonical endoplasmic reticulum transporter associated with antigen processing (TAP)/proteasome-dependent pathway, delaying CD8+ T cell priming (170). It is still not well described how peptides are processed for Leishmania cross-presentation but gp63 from L.m can cleave the SNARE VAMP8, present on late endosomes, preventing NADPH oxidase complex assembly, which impacts the phagosome pH and proteolytic activity, inhibiting cross-presentation (32). The role of cDC1s or cross-presentation specifically during L.a. infection is unknown, but macrophages can prime CD8+ T cells by presenting endogenous Leishmania antigen via the classical proteasome-dependent pathway (171).
cDC2s are usually associated with the development of a Th2 immune response, which requires IL-4 and alarmin (thymic stromal lymphopoietin (TSLP), IL-25 and IL-33) signaling (161). While cDC1s migrate towards the T cell zone of the lymph nodes, cDC2s localize at the subcapsular sinus space (172). Thus, in addition to expressing CCR7 to migrate into dLNs, CD301b+ cDC2 cells rely on CCR8 expression, during allergic immune response, to reach the LN parenchyma to trigger Th2 immunity (173). Thus, the anatomical location of different DCs subsets at dLNs play a significant role to determine the phenotype of adaptive immunity (174). To date, the exact role played by cDC2s on the establishment of the Th2 response during Leishmania, including L.a., infection is not clear.
Plasmacytoid DCs (pDCs) are found in the circulation in steady state but can be recruited to the skin during inflammation. These APCs are CD11b- CD11c+and represent an important source of type I IFNs (IFN-α/IFN-β), thus are critical for protective immunity during viral infections (161, 175). pDCS, when infected in vitro with L. infantum, can also induce type I IFNs, in a TLR9 dependent manner, in addition to IL-12 (176). During L. major infection pDCs can be found at skin dermis and dLNs, can be infected by the parasite and do not seem to produce significant amount of NO, which suggest these cells do not play a role in parasite killing (177). Compared to CD11b+ DCs, pDCs produce higher levels of IFN-α/β and lower levels of IL-12 in response to L. major infection (178). Type I IFNs have been associated with early iNOS expression at the site of infection during L. major infection (179) but a lack of type I IFN signaling does not impact the self-healing phenotype after L. major infection of C57BL/6 mice suggesting this function of pDCs might be dispensable (178), but more studies are still required.
While cDC1s might have an important role on eliciting Th1 effector cell activation in the skin, monocyte derived DCs seem to play the major role in priming the Th1 immune response in dLNs and mediate parasite elimination. Monocytes are recruited to the dermis after infection, differentiate into mon-DCs, and after migrating into dLNs prime Th1 immunity by producing IL-12 (180). In different experimental models, CCR2 deficient mice have been associated with a profound Th1 deficit and skewing towards Th2 immunity. Monocytes rely on CCR2 expression to leave the bone-marrow and reach the circulation, thus without this chemokine receptor monocytes are not available to enter peripherical tissues (181). The shift towards a Th2 immune response in CCR2-/- mice has been reported for infection by L. major (182) and L.a (43). During L. mexicana infection, the observed decrease in monocyte recruitment, versus L. major infected mice, may also be responsible for decreased Th1 priming (183). In addition, mon-DCs can express iNOS and represent an important cell that mediates parasite killing during L.m. infection (184). In contrast to Baft3-/- mice that have non-healing but stable lesions, monocyte-deficient ccr2-/- mice have highly significant deficiencies in iNOS+ cells and succumb to progressive disease at the late stages of infection in otherwise resistant C57BL/6 mice (164, 182, 184).
Mon-DCs are likely infected with amastigotes, rather than promastigotes. In-vitro infection of DCs by amastigotes has an important impact on activation and is associated with an overall anti-inflammatory response, associated with poor inflammasome activation, IL-12 production and CD4 T cell proliferation. How L.a. amastigotes compromise DC activation is not fully understood, but it does affect several important transcription factors signaling pathways, including: (a) reduces expression of IRF1, IRF8 and STAT1; (b) degrades STAT2 by proteases, (c) inhibits the NF-kB pathway by enhancing optineurin (OPTN) expression (118, 185). A caveat to the interpretation of these studies is that mon-DCs are likely to be infected when they are immature monocytes (43, 142, 180, 184), and therefore the relevant impact of infection may be the modulation of maturation towards a DC phenotype rather than the down-regulation of a cell that is already mature, such as is the case with bone-marrow derived DCs typically employed for these types of in-vitro experiments. These two processes are biologically distinct.
L.a. (both promastigotes and amastigotes) can infect and proliferate in both murine and human differentiated DCs through different receptors such as FcγR, complement, and proteoglycan receptors (186–189). Monocyte differentiation into DC is impacted by L.a. infection in vitro, as lower levels of CD80 and CD1a expression and IL-6 production were reported. However, the infection did not impact the expression of MHC II and increased CD86 levels, showing that this modulation does not occur equally across all maturation markers (189). How L.a. impacts DC activation varies between different studies because of differences on parasite stage, opsonization or not, genetic background and maturation status at the time of infection. In summary, parasite opsonization, either amastigotes or promastigotes, by antibodies, but not complement, plays a major role on DC activation. For DCs generated from BALB/c mice ab-opsonization of metacyclic promastigotes or amastigotes is associated with higher expression of CD40, CD86, CD54 and OX40L. In addition, ab-opsonization not only enhanced MHC II expression but it also accelerated its distribution on the cell surface, impacting antigen presentation (186). On the other hand, infection by non-opsonized metacyclics did not alter DC activation (186). Meanwhile, DCs infected with non-opsonized stationary promastigotes were able to weakly up-regulated MHC II, CD40, CD80 and CD86 expression (185, 187, 190). In DCs derived from C57BL/6 mice the use of non-opsonized metacyclic inhibited the expression of molecules such as MHC II, CD40, CD86 and IL-12p70. Particularly, the expression of CD40 is inhibited by the enhanced expression of DCs ectonucleotidases (CD39 and CD73) induced by L.a. infection, which is IL-4/IL-10 independent but ERK1/2 pathway dependent (191–194). These discrepant results reinforce the fact that the use of purified metacyclics has a big impact on infection, similar to opsonization. By inhibiting CD40 expression on DCs, L.a. might impair the development of a protective adaptive immune response as CD40 expression is relevant for IL-12 production and subsequent Th1 priming (195), including during L.a. infection (191, 196). In fact, L.a. parasites do impair both CD40 and IL-12 production on APCs to a higher degree then what is observed for self-healing parasite strains of L. major and L. braziliensis (190, 191). In summary, the infection of DCs by either metacyclic promastigotes or amastigotes have relevant impact on promoting anti-inflammatory response that may delay Th1 priming.
Other important questions related to DCs and still to be elucidated are: 1) if cDC1s represent an important source of IL-12 during early stages of the disease, before the recruitment of monocytes to the site of infection; 2) if continuous local activation of cDC1s, after the priming of Th1 immunity by mon-DCs in dLNs is required for ongoing protection; and 3) why the dermal resident cDC1s, which are already at the site of infection are not required for Th1 priming. This is a relevant point because cDC1s have higher migratory capacity and can induce stronger T cell activation, for both CD4+ and CD8+ T cells in vitro (197), compared to mon-DCs. However, as cDC1s might become immobile after, at least, the first hours post-infection, due to initial infection and inflammation at the dermis (198), we can speculate that, in addition to the fact that these are rare cells at the dermis, this immobilization might impact their priming capacity.
Embryonic-derived macrophages, Monocytes and Monocyte derived Macrophages (mon-Mac), are mononuclear phagocytic cells that play an essential role during homeostasis and inflammation in different organs, including a critical role mediating killing of intracellular pathogens and promoting wound healing [reviewed in (199, 200)]. During Leishmania infection these cells can play paradoxical roles, providing both a niche for parasite survival and replication or mediate parasite killing depending upon the activation state of the tissue environment they occupy (43, 142, 184, 201–203). It is important to highlight that for this review we are considering the current literature for macrophage ontogeny in the context of Leishmania infection in the skin. The reason why macrophage ontogeny matters is that in recent years, it has become clear that the ontogeny of macrophages has an impact on macrophage function, including differences on phagocytosis, cluster interaction with other immune cells and production of cytokines and chemokines. Differences on epigenetic and transcriptomic networks may explain these differences, but this is a field currently under intense investigation (197, 204, 205). It is well established that different tissues contain a heterogenous resident macrophage population, based on their origin, which can vary depending upon life history, including age and experience with inflammation. Resident tissue macrophages (RTMs) can originate from embryonic hematopoiesis and are self-maintained by replication or originate from the recruitment and differentiation of circulating CCR2hiLy6Chi monocytes after birth (197, 199). In vascularized tissue-barrier organs such the skin dermis, the replacement of embryonic derived macrophages by monocyte-derived cells is observed after birth and occurs more quickly compared to tissues such as the epidermis, brain and liver. After 36 months of age, around 80% of RTMs are monocyte-derived in the skin dermis (206, 207). It is relevant to mention that during cutaneous leishmaniasis two waves of monocyte recruitment can be observed. The first wave starts around 48 h post-infection and last up to around 72 h. Then, another monocyte wave is seen during adaptive immunity, starting around day 10 post-infection, and will persist for as long as it takes for parasite clearance (43, 139). The impact of this is that recently recruited immature monocytes, monocytes undergoing different stages of maturation toward a macrophage or DC phenotype, and fully differentiated monocyte-derived macrophages and DCs will co-exist at the site of infection. There are two main challenges when trying to define the role played by embryonic-derived macrophages, monocytes and mon-Macs: 1) studies relying on markers such as MHC II, F4/80 and CD11c to distinguish between these cells, and sometimes DCs, which we now know are not definitive, in addition to the fact that several studies do not take into account ontogeny when referring to macrophages (197). The most current markers to define innate cells at the skin can be seen in Figure 1, 2) much of what we know about macrophage function came from BMDMs, where monocytes are first fully differentiate towards a macrophage phenotype in vitro, and then infected by the parasite. The caveat here is that in most models of Leishmania infection, including L.a., recruited monocytes appear to be the cell type associated with ongoing parasite replication, though further work is required (43, 142, 208–211). Lastly, in addition to mon-Macs, a recently defined relevant macrophage population in a context of cutaneous leishmaniasis are the self-renewing embryonic-derived RTMs, which are maintained in an IL-4 dependent manner, which impacts the ability of these cells to get activated in a context of Th1 immunity (212). These cells in the dermis are characterized by high levels of MR expression and represent one of the first host cells for the L. major-RYN strain after sand fly transmission, which, at least in C57BL/6 mice, has a non-healing phenotype, and the L. major Seidman strain after needle inoculation, which exhibits a non-healing phenotype in both C57BL/6 mice and the patient from which it was derived (135, 201).
Finally, to best understand whether these cells provide a safe or a hostile environment for Leishmania parasites we cannot simply rely only on analyzing their activation status. Rather, we propose that to understand the big picture of the role of mononuclear phagocytic cells a balance between three equally important factors must be taken in consideration: 1) activation, 2) recruitment, and 3) spatial organization in the tissue.
Monocytes and Macrophages show high plasticity and heterogeneity in regard to their activation phenotype, which is determined by environmental cues. Depending on the cytokines, PAMPs and damage associated molecular patterns (DAMPs) in their environment, they can acquire different functions driven by both innate and adaptive immune responses. When exposed to cytokines such as IFN-γ and TNF these cells acquire a pro-inflammatory phenotype (or “M1-like”) and are able to produce high levels of iNOS, ROS, and cytokine and chemokine production such as TNF, CXCL9 and CXCL10. When activated by IL-4 and/or IL-13 they are associated with a wound-healing function (or “M2-like”). In this case these cells express molecules such as Relmα, Ym1, CCL24 and Arg1. If activated by immunocomplex or PGE2 these cells can play a suppressive function by producing high levels of anti-inflammatory cytokines such as IL-10 (200, 213, 214). Therefore, in the context of Th1 immunity these mononuclear cells, activated by IFN-γ to produce NO can control Leishmania growth (70, 78, 215, 216). Higher iNOS expression from skin biopsies from patients with American cutaneous disease has been associated with LCL and smaller number of parasites comparing to patients with DCL and higher parasite burden in the lesions (217). On the other hand, the expression of host cell Arg1 in highly Th2 polarized immune responses, such as infection of BALB/c mice, has been associated with parasite replication and susceptibility (59, 218, 219). Importantly, in a context of cutaneous leishmaniasis embryonic-derived macrophages, due to close cluster interaction with eosinophils, and discussed in more detail below, show a wound-healing phenotype despite the overall polarized Th1 immune response in the dermis (212).
In the context of Leishmania infection, monocytes, and both embryonic and monocyte-derived macrophages can also be suppressed by IL-10, which compromises the induction of effector mechanism, such as TNF, ROS and NO production, facilitating parasite growth or persistence (43, 201, 220–222). Most of the anti-inflammatory functions associated with IL-10 is associated with STAT3 signaling. STAT3 can induce the production of proteins that suppress the expression of several pro-inflammatory genes such as the suppressor of cytokine signaling 3 (SOCS3) and can inhibit NF-kB translocation by inducing IL-10 production (223), In addition, STAT3 can cross-regulate STAT1 signaling, the key transcription factor associated with IFN-γ activation. Depending on the experimental model, STAT3 can interfere with STAT1 tyrosine phosphorylation or can promote STAT1 sequestration, by binding to STAT1 forming STAT1-STAT3 heterodimers, which inhibits the DNA binding of STAT1 homodimers (224). The production of IL-10 by phagocytic cells during Leishmania infection is associated with ab-opsonized amastigotes, through binding to FcγR, inducing ERK1/2 activation, leading to IL-10 production (221, 225, 226). In addition, both Tregs (CD4+CD25+FoxP3+) as well as Th1 (CD4+CD25-FoxP3-T-bet+IFN-γ+) cells have been described as important sources of IL-10 during L. major and L. donovani infections (227, 228). Importantly, the lack of IL-10, however, is not enough to revert the susceptibility phenotype seen in C57BL/6 mice infected with L.a., despite the stronger Th1 immune response (43, 221, 229).
During in vivo infections by L.a. parasites, a mix of different CD4 T cell responses is observed (Th1, Th2), in addition to the presence of other cell types such as NK cells and ILC2, which can produce IFN-γ or IL-4 for instance (212, 230, 231). Thus, in addition to mononuclear phagocytic cells presenting highly differentiated M1-like or M2-like phenotypes in the skin lesions, the same cells, often simultaneously, express iNOS and Arg1, indicating that they are activated by diverse stimuli (Figure 3C) (43, 232). In fact, during L.a. infection most iNOS+ cells are also Arg1+ cells, even in a context of a Th1 prone environment (43), which could potentially prevent, in a single cell-manner, robust NO production by these cells, as previously described for L.m. infection (232). Our group also showed that infection of STAT6 deficient mice on the C57BL/6 background was associated with an almost complete abrogation of Arg1 expression by phagocytic cells, but this did not restrict parasite growth. Rather, by upregulating genes associated with L-arginine uptake and its own arginase versus WT infected mice, L.a. do not rely on host arginase to acquire polyamines, which is probably also true for other parasite species (43). For instance, in contrast to BALB/c infected mice, L. major growth in C57BL/6 mice also occurs independently of host Arg1 expression (233).
The idea of a weaker Th1 immune response during L.a. infection compared to L. major infection has been implicated as a cause of susceptibility (230, 234). However, this weaker Th1 immunity appears to be dose and/or route of infection dependent, as, following intradermal infection with lower doses, this phenotype is not readily apparent (43, 235). In addition, we and others have shown that the enhancement of Th1 immunity does not translate into a protective phenotype, despite the strong induction of iNOS expression by phagocytic cells (43, 79, 229, 236). There is no doubt that IFN-γ do mediate parasite killing during L.a. infection, however, the exponential parasite growth observed during early stages of the disease is not impacted by the absence of IFN-γ (43, 44, 236). We propose that, rather than eliciting a weak Th1 response or compromising the induction of this response, L.a. parasites have evolved to actually take advantage of some aspects of the otherwise protective type I immune response to establish a chronic and progressive disease (Figure 4). IFN-γ, the key cytokine produced by Th1 cells, modulates immunity through different ways, such as inducing effector mechanisms (NO/ROS production) in innate cells, but also by influencing immunometabolism, leukocyte trafficking, apoptosis and cell proliferation, to name a few (237). Two properties of IFN-γ function can favor L.a. growth: 1) it’s influence on cell metabolism and 2) leukocyte trafficking to skin lesions. When BMDMs are pre-activated with low doses of IFN-γ, and then infected with L.a. amastigotes in vitro, an enhancement in parasite replication, rather than their killing is observed. In this case, IFN-γ mediate CAT-2B up-regulation by BMDMs, increasing, therefore, the availability of this amino acid for Leishmania metabolism (60, 61). In addition, IFN-γ mediates the production of chemokines such as CCL2, CCL5, CXCL9 and CXCL-10 (44), significantly increasing leukocyte trafficking to sites of inflammation. In the context of L.a. infection, the influence of IFN-γ on monocyte recruitment has a large impact by providing host cells for parasite growth, as discussed below.

Figure 4 Dynamic of monocyte and monocyte derived cells during an ongoing adaptive immune response against L.a. infection: recruitment, activation and cell clusters. Infected monocytes (A) and CXCR3+ Th1 cells (B) produce CXCL9 and CXCL10 facilitating continuous recruitment of permissive immature inflammatory monocytes (C) that provides a niche for ongoing infection (D) and possibly favors the formation of monocyte-Th1 cell clusters. After infection, mon-Macs mediate some parasite killing when activated by IFN-γ (E); but when activated simultaneously by IFN-γ and Th2 cytokines (F) or by Th2 cytokines alone (G) support parasite replication. Infected monocytes can also be suppressed by IL-10, while receiving signals from IFN-γ and/or Th2 cytokines, which also favors parasite replication (H). Mature mon-DCs are more efficient at restricting parasite growth versus mon-Macs but this developmental program takes 7-14 days (I). Embryonic derived resident macrophages due to CCL24 production, cluster with eosinophils, which are an important source of IL-4 and IL-10 (J). Created with BioRender.com.
The observation that equivalent numbers of L.a. infected cells are seen in lesions of STAT6-/- mice compared to Wt mice, despite strong Th1 immunity, greater frequencies of iNOS+ phagocytic cells, and the absence of Arg1 expression is explained by an enhanced monocyte infiltration and IL-10 production. The opposite is found in ifnγ-deficient settings, where a lack of a Th1 response during the early phases of infection leads to a Th2 polarized environment but no enhancement of parasite numbers due to a lack of Th1-mediated monocyte recruitment (43). These data show how important the recruitment of permissive monocytes to the site of infection can be, which has been shown in different contexts for other Leishmania parasites as well (208, 211). In fact, emergency hematopoiesis, a process that facilitates the expansion of hematopoietic stem cells (HSCs) and myeloid progenitor cells (MPCs) is stimulated by systemic infection with L. donovani and is largely associated with the expansion of Ly6Chi monocytes rather than neutrophils, thereby providing a robust and ongoing supply of infiltrating monocytes that are permissive to infection (209). When in the tissue, monocytes differentiate into macrophages (mon-Macs) or DCs (mon-DCs), a process that takes time (Figure 3A). The different environmental cues, in different experimental models, associated with monocyte differentiation towards either mon-Macs or mon-DCs are still not completely understood. IL-4, TNF and GM-CSF has been associated with a mon-DC phenotype (238, 239) while IL-6 and M-CSF with a mon-Mac phenotype (240). If present during early stages of monocyte differentiation, IFN-γ, by modulating IL-6 expression and M-CSF expression and internalization, can shift human monocyte differentiation, in vitro, from a DC to a macrophage phenotype, even in the presence of IL-4 and GM-CSF (241). In addition, germ-free mice, under steady state, present lower numbers of mon-DCs in the dermis, but the same number of mon-Macs comparing to specific pathogen free mice, showing that the microbiota also influences monocyte fate in the skin (197).
Employing a tamoxifen inducible CCR2-Cre lineage tracing strategy we found that maturation from a circulating CCR2+Ly6C+ phenotype to a CCR2-Ly6C-CD11c+ DC phenotype requires approximately 2 weeks, during which the majority of maturing monocytes provided a safe niche for parasite replication, despite the Th1 immune response, likely as a result of L.a.-infection induced manipulation of cell activation by enhancing IL4R, MR and PDL2 expression on infected versus uninfected monocytes from the same dermal site of infection (43). While Mon-DCs represent the cell phenotype that is most efficient at restricting L.a. growth and adopts an MHC+PDL2- phenotype, even these cells can be found to harbor viable parasites, which has been shown for other pathogens (43, 184, 242). Importantly, permissive monocyte recruitment is mediate by IFN-γ produced by Th1 cells, as depletion of CD4+ T cells abrogated virtually all IFN-γ mRNA from the lesion site 21 days post-infection. Lastly, the monocytes and monocyte derived cells, recruited after the development of Th1 immunity, which coincide with parasite exponential growth, represent the host cells that harbor higher numbers of amastigotes, in contrast with the cells recruited during the innate phase of the disease and that are further along in their maturation (43). Altogether, these data suggest that L.a. parasites have evolved through evolution to subvert this property of Th1 immunity to facilitate infection. This represents one of the strongest examples of the paradox of a phagosomal lifestyle, where L.a. parasites evolved to depend upon monocyte recruitment mediated by IFN- γ to provide a permissive niche for replication.
During infection with the non-healing Seidman or Ryan strains of L. major, embryonic derived RTMs, via close interactions with IL-4 producing eosinophils, maintain an M2-like, wound healing, activation state, despite highly polarized Th1 immunity at the site of infection, providing a perfect niche for replication (135, 212). In addition, monocytes and monocytes derived cells represent the majority of phagocytic cells responding to Th1/Th2 cytokines during infection with either the L.a. or L.m. (healing Fn) strain (43, 232). In CCR2 deficient mice, a huge deficit in the number of phagocytic cells with either a iNOS+ or Arg1+ phenotype strongly support the idea that monocytes and monocyte-derived cells are the cells either directly interacting with T cells or that are in close enough proximity to become activated by the T cell cytokine gradient (43). Specifically, Th1 CD4 T effector cells can mediate protection by interacting with only a fraction of infected cells, as a gradient of IFN-γ can reach and activate phagocytes that are not directly interacting with T cells to promote pathogen control. Furthermore, to reach an effective killing activity a certain threshold of phagocytic cells expressing iNOS might be necessary, where due to the diffusion capacity of NO through membranes, non-activated host cells are also able to kill the parasite (75, 243). In addition, dermal L.a. infected monocytes (43), as well as Th1 effectors cells, produce and are chemoattracted by the chemokines CXCL9 and CXCL10 (244, 245), which might favor cluster formation between these cell types (Figure 4). Lastly, the migratory capacity of Th1 and Th2 cells in the skin is different. Th2 cells, by expressing higher levels of the integrin αVβ3, can undertaking a broader “scanning” phenotype in the tissue and interact with more cells, compared to Th1 cells, which are more dependent on chemokine gradients to migrate (246).
We can speculate that ontogeny plays a role in the formation of phagocyte-T cell clusters, as embryonic derived RTMs are less mobile, while both monocytes and T cells migrate to the site of infection via the bloodstream, which may favor cluster formation between these cells. Clusters of embryonic derived RTMs associated with local skin nerves and clusters between T cells and monocytes during skin contact hypersensitivity has also been reported showing that the tissue spatial organization influences physiological and immunological events (247, 248). These relatively new data strongly support the idea that cell clusters among different cell subsets at the site of infection plays a major role on the outcome of disease, creating an opportunity for specific niches that facilitate pathogen growth or killing.
In a context of evolutionary host-parasite interactions the host must evolve and adapt to virulence factors associated with the pathogens, while the parasites have to evolve and adapt to the host-immune response. Phagosomal pathogens, such as Leishmania, have adapted to survive and replicate inside the innate cells that are recruited to eliminate them, representing a challenge for effective intervention. Innate effector mechanisms, for reasons discussed in this review, are not enough to eliminate Leishmania parasites and that creates an opportunity for the parasite to establish a niche for replication before the induction of Th1 adaptive immunity. During infection with the self-healing L. major strain, the development of Th1 immunity is enough to induce protection. However, in the context of L. amazonensis infection, Th1-dependent monocyte recruitment provides an opportunity for the parasite to replicate inside recently recruited immature monocytes and, while IFN-γ activation does ultimately provide some parasite killing, these infected cells are also responding to other environmental cues that are not associated with parasite elimination (Figure 4). The use of reporter parasites in-vivo has been instrumental in revealing the characteristics of infected monocytes and, while these cells eventually adopt characteristics of macrophages or DCs, to a certain degree they represent unique populations of monocyte-derived phagocytes that are more defined by their infection status rather than a classical macrophage or DC phenotype. Mechanistically reaching a subtle balance between monocyte recruitment by modulating the Th1/Th2 immune response to allow parasite killing while not providing a permissive host cell reservoir might represent one of the current big challenges in the field that could potentially lead to a vaccine or improvement of therapy.
MBC and NCP wrote the review. MBC made the figures and NCP did the revision of the figures. All authors contributed to the article and approved the submitted version.
This work was supported by Canadian Institutes of Health Research grant MOP-142302 and Canadian Foundation for Innovation grant RCP-16-027-SEG to NCP. A Snyder Institute for Chronic Diseases Beverley Phillips Rising Star Fellowship and Cumming School of Medicine Post-Doctoral Fellowship supported MBC. MBC received professional development training and travel funding from The University of Calgary Host-Parasite Interactions Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Merien F. A Journey With Elie Metchnikoff: From Innate Cell Mechanisms in Infectious Diseases to Quantum Biology. Front Public Health (2016) 4:125. doi: 10.3389/fpubh.2016.00125
2. Metchnikoff E. Lectures on the Comparative Pathology of Inflammation. London: Kegan Paul, Trench, Trubner & CO (1893).
3. Hohman LS, Peters NC. CD4(+) T Cell-Mediated Immunity Against the Phagosomal Pathogen Leishmania: Implications for Vaccination. Trends Parasitol (2019) 35(6):423–35. doi: 10.1016/j.pt.2019.04.002
4. Sacks D, Sher A. Evasion of Innate Immunity by Parasitic Protozoa. Nat Immunol (2002) 3(11):1041–7. doi: 10.1038/ni1102-1041
5. Chang KP, Dwyer DM. Multiplication of a Human Parasite (Leishmania Donovani) in Phagolysosomes of Hamster Macrophages In Vitro. Science (1976) 193(4254):678–80. doi: 10.1126/science.948742
6. Chang KP, Dwyer DM. Leishmania Donovani. Hamster Macrophage Interactions In Vitro: Cell Entry, Intracellular Survival, and Multiplication of Amastigotes. J Exp Med (1978) 147(2):515–30. doi: 10.1084/jem.147.2.515
7. Alexander J, Vickerman K. Fusion of Host Cell Secondary Lysosomes With the Parasitophorous Vacuoles of Leishmania Mexicana-Infected Macrophages. J Protozool (1975) 22(4):502–8. doi: 10.1111/j.1550-7408.1975.tb05219.x
8. Lewis DH, Peters W. The Resistance of Intracellular Leishmania Parasites to Digestion by Lysosomal Enzymes. Ann Trop Med Parasitol (1977) 71(3):295–312. doi: 10.1080/00034983.1977.11687192
9. Carneiro MB, Hohman LS, Egen JG, Peters NC. Use of Two-Photon Microscopy to Study Leishmania Major Infection of the Skin. Methods (2017) 127:45–52. doi: 10.1016/j.ymeth.2017.04.012
10. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In Vivo Imaging Reveals an Essential Role for Neutrophils in Leishmaniasis Transmitted by Sand Flies. Science (2008) 321(5891):970–4. doi: 10.1126/science.1159194
11. Scott P, Natovitz P, Coffman RL, Pearce E, Sher A. Immunoregulation of Cutaneous Leishmaniasis. T Cell Lines That Transfer Protective Immunity or Exacerbation Belong to Different T Helper Subsets and Respond to Distinct Parasite Antigens. J Exp Med (1988) 168(5):1675–84. doi: 10.1084/jem.168.5.1675
12. Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet (2018) 392(10151):951–70. doi: 10.1016/S0140-6736(18)31204-2
13. Silveira FT, Lainson R, Corbett CE. Clinical and Immunopathological Spectrum of American Cutaneous Leishmaniasis With Special Reference to the Disease in Amazonian Brazil: A Review. Mem Inst Oswaldo Cruz (2004) 99(3):239–51. doi: /S0074-02762004000300001 doi: 10.1590/S0074-02762004000300001
14. Almeida RP, Barral-Netto M, De Jesus AM, De Freitas LA, Carvalho EM, Barral A. Biological Behavior of Leishmania Amazonensis Isolated From Humans With Cutaneous, Mucosal, or Visceral Leishmaniasis in BALB/C Mice. Am J Trop Med Hyg (1996) 54(2):178–84. doi: 10.4269/ajtmh.1996.54.178
15. Rabinovitch M. Professional and Non-Professional Phagocytes: An Introduction. Trends Cell Biol (1995) 5(3):85–7. doi: 10.1016/s0962-8924(00)88955-2
16. Levin R, Grinstein S, Canton J. The Life Cycle of Phagosomes: Formation, Maturation, and Resolution. Immunol Rev (2016) 273(1):156–79. doi: 10.1111/imr.12439
17. Blackwell JM, Ezekowitz RA, Roberts MB, Channon JY, Sim RB, Gordon S. Macrophage Complement and Lectin-Like Receptors Bind Leishmania in the Absence of Serum. J Exp Med (1985) 162(1):324–31. doi: 10.1084/jem.162.1.324
18. Da Silva RP, Hall BF, Joiner KA, Sacks DL. CR1, the C3b Receptor, Mediates Binding of Infective Leishmania Major Metacyclic Promastigotes to Human Macrophages. J Immunol (1989) 143(2):617–22.
19. Guy RA, Belosevic M. Comparison of Receptors Required for Entry of Leishmania Major Amastigotes Into Macrophages. Infect Immun (1993) 61(4):1553–8. doi: 10.1128/IAI.61.4.1553-1558.1993
20. Mosser DM, Edelson PJ. The Mouse Macrophage Receptor for C3bi (CR3) Is a Major Mechanism in the Phagocytosis of Leishmania Promastigotes. J Immunol (1985) 135(4):2785–9.
21. Wyler DJ, Sypek JP, McDonald JA. In Vitro Parasite-Monocyte Interactions in Human Leishmaniasis: Possible Role of Fibronectin in Parasite Attachment. Infect Immun (1985) 49(2):305–11. doi: 10.1128/IAI.49.2.305-311.1985
22. Gordon S, Pluddemann A. Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front Immunol (2018) 9:127. doi: 10.3389/fimmu.2018.00127
23. Tavares NM, Araujo-Santos T, Afonso L, Nogueira PM, Lopes UG, Soares RP, et al. Understanding the Mechanisms Controlling Leishmania Amazonensis Infection In Vitro: The Role of LTB4 Derived From Human Neutrophils. J Infect Dis (2014) 210(4):656–66. doi: 10.1093/infdis/jiu158
24. Cavalcante-Costa VS, Costa-Reginaldo M, Queiroz-Oliveira T, Oliveira ACS, Couto NF, Dos Anjos DO, et al. Leishmania Amazonensis Hijacks Host Cell Lysosomes Involved in Plasma Membrane Repair to Induce Invasion in Fibroblasts. J Cell Sci (2019) 132(6). doi: 10.1242/jcs.226183
25. Ueno N, Wilson ME. Receptor-Mediated Phagocytosis of Leishmania: Implications for Intracellular Survival. Trends Parasitol (2012) 28(8):335–44. doi: 10.1016/j.pt.2012.05.002
26. Courret N, Frehel C, Gouhier N, Pouchelet M, Prina E, Roux P, et al. Biogenesis of Leishmania-Harbouring Parasitophorous Vacuoles Following Phagocytosis of the Metacyclic Promastigote or Amastigote Stages of the Parasites. J Cell Sci (2002) 115(Pt 11):2303–16. doi: 10.1242/jcs.115.11.2303
27. Love DC, Mentink Kane M, Mosser DM. Leishmania Amazonensis: The Phagocytosis of Amastigotes by Macrophages. Exp Parasitol (1998) 88(3):161–71. doi: 10.1006/expr.1998.4232
28. Ndjamen B, Kang BH, Hatsuzawa K, Kima PE. Leishmania Parasitophorous Vacuoles Interact Continuously With the Host Cell’s Endoplasmic Reticulum; Parasitophorous Vacuoles Are Hybrid Compartments. Cell Microbiol (2010) 12(10):1480–94. doi: 10.1111/j.1462-5822.2010.01483.x
29. Uribe-Querol E, Rosales C. Control of Phagocytosis by Microbial Pathogens. Front Immunol (2017) 8:1368. doi: 10.3389/fimmu.2017.01368
30. Vinet AF, Fukuda M, Turco SJ, Descoteaux A. The Leishmania Donovani Lipophosphoglycan Excludes the Vesicular Proton-ATPase From Phagosomes by Impairing the Recruitment of Synaptotagmin V. PloS Pathog (2009) 5(10):e1000628. doi: 10.1371/journal.ppat.1000628
31. Desjardins M, Descoteaux A. Inhibition of Phagolysosomal Biogenesis by the Leishmania Lipophosphoglycan. J Exp Med (1997) 185(12):2061–8. doi: 10.1084/jem.185.12.2061
32. Matheoud D, Moradin N, Bellemare-Pelletier A, Shio MT, Hong WJ, Olivier M, et al. Leishmania Evades Host Immunity by Inhibiting Antigen Cross-Presentation Through Direct Cleavage of the SNARE Vamp8. Cell Host Microbe (2013) 14(1):15–25. doi: 10.1016/j.chom.2013.06.003
33. Antoine JC, Prina E, Jouanne C, Bongrand P. Parasitophorous Vacuoles of Leishmania Amazonensis-Infected Macrophages Maintain an Acidic pH. Infect Immun (1990) 58(3):779–87. doi: 10.1128/IAI.58.3.779-787.1990
34. Prina E, Antoine JC, Wiederanders B, Kirschke H. Localization and Activity of Various Lysosomal Proteases in Leishmania Amazonensis-Infected Macrophages. Infect Immun (1990) 58(6):1730–7. doi: 10.1128/IAI.58.6.1730-1737.1990
35. Barbieri CL, Brown K, Rabinovitch M. Depletion of Secondary Lysosomes in Mouse Macrophages Infected With Leishmania Mexicana Amazonensis: A Cytochemical Study. Z Parasitenkd (1985) 71(2):159–68. doi: 10.1007/BF00926266
36. Real F, Mortara RA. The Diverse and Dynamic Nature of Leishmania Parasitophorous Vacuoles Studied by Multidimensional Imaging. PloS Negl Trop Dis (2012) 6(2):e1518. doi: 10.1371/journal.pntd.0001518
37. Antoine JC, Jouanne C, Ryter A, Zilberfarb V. Leishmania Mexicana: A Cytochemical and Quantitative Study of Lysosomal Enzymes in Infected Rat Bone Marrow-Derived Macrophages. Exp Parasitol (1987) 64(3):485–98. doi: 10.1016/0014-4894(87)90063-4
38. Wilson J, Huynh C, Kennedy KA, Ward DM, Kaplan J, Aderem A, et al. Control of Parasitophorous Vacuole Expansion by LYST/Beige Restricts the Intracellular Growth of Leishmania Amazonensis. PloS Pathog (2008) 4(10):e1000179. doi: 10.1371/journal.ppat.1000179
39. Okuda K, Tong M, Dempsey B, Moore KJ, Gazzinelli RT, Silverman N. Leishmania Amazonensis Engages CD36 to Drive Parasitophorous Vacuole Maturation. PloS Pathog (2016) 12(6):e1005669. doi: 10.1371/journal.ppat.1005669
40. Mukbel RM, Patten C Jr., Gibson K, Ghosh M, Petersen C, Jones DE. Macrophage Killing of Leishmania Amazonensis Amastigotes Requires Both Nitric Oxide and Superoxide. Am J Trop Med Hyg (2007) 76(4):669–75. doi: 10.4269/ajtmh.2007.76.669
41. Lemos de Souza V, Ascencao Souza J, Correia Silva TM, Sampaio Tavares Veras P, Rodrigues de-Freitas LA. Different Leishmania Species Determine Distinct Profiles of Immune and Histopathological Responses in CBA Mice. Microbes Infect (2000) 2(15):1807–15. doi: 10.1016/S1286-4579(00)01340-X
42. Canton J, Ndjamen B, Hatsuzawa K, Kima PE. Disruption of the Fusion of Leishmania Parasitophorous Vacuoles With ER Vesicles Results in the Control of the Infection. Cell Microbiol (2012) 14(6):937–48. doi: 10.1111/j.1462-5822.2012.01767.x
43. Carneiro MB, Lopes ME, Hohman LS, Romano A, David BA, Kratofil R, et al. Th1-Th2 Cross-Regulation Controls Early Leishmania Infection in the Skin by Modulating the Size of the Permissive Monocytic Host Cell Reservoir. Cell Host Microbe (2020) 27(5):752–68.e7. doi: 10.1016/j.chom.2020.03.011
44. Carneiro MB, Lopes ME, Vaz LG, Sousa LM, dos Santos LM, de Souza CC, et al. IFN-Gamma-Dependent Recruitment of CD4(+) T Cells and Macrophages Contributes to Pathogenesis During Leishmania Amazonensis Infection. J Interferon Cytokine Res (2015) 35(12):935–47. doi: 10.1089/jir.2015.0043
45. Pessoa CC, Reis LC, Ramos-Sanchez EM, Orikaza CM, Cortez C, de Castro Levatti EV, et al. ATP6V0d2 Controls Leishmania Parasitophorous Vacuole Biogenesis via Cholesterol Homeostasis. PloS Pathog (2019) 15(6):e1007834. doi: 10.1371/journal.ppat.1007834
46. Jankowski A, Scott CC, Grinstein S. Determinants of the Phagosomal pH in Neutrophils. J Biol Chem (2002) 277(8):6059–66. doi: 10.1074/jbc.M110059200
47. Piacenza L, Trujillo M, Radi R. Reactive Species and Pathogen Antioxidant Networks During Phagocytosis. J Exp Med (2019) 216(3):501–16. doi: 10.1084/jem.20181886
48. N’Diaye EN, Darzacq X, Astarie-Dequeker C, Daffe M, Calafat J, Maridonneau-Parini I. Fusion of Azurophil Granules With Phagosomes and Activation of the Tyrosine Kinase Hck Are Specifically Inhibited During Phagocytosis of Mycobacteria by Human Neutrophils. J Immunol (1998) 161(9):4983–91.
49. Mollinedo F, Janssen H, de la Iglesia-Vicente J, Villa-Pulgarin JA, Calafat J. Selective Fusion of Azurophilic Granules With Leishmania-Containing Phagosomes in Human Neutrophils. J Biol Chem (2010) 285(45):34528–36. doi: 10.1074/jbc.M110.125302
50. Bates PA, Robertson CD, Tetley L, Coombs GH. Axenic Cultivation and Characterization of Leishmania Mexicana Amastigote-Like Forms. Parasitology (1992) 105(Pt 2):193–202. doi: 10.1017/s0031182000074102
51. Mittra B, Cortez M, Haydock A, Ramasamy G, Myler PJ, Andrews NW. Iron Uptake Controls the Generation of Leishmania Infective Forms Through Regulation of ROS Levels. J Exp Med (2013) 210(2):401–16. doi: 10.1084/jem.20121368
52. Mittra B, Laranjeira-Silva MF, Perrone Bezerra de Menezes J, Jensen J, Michailowsky V, Andrews NW. A Trypanosomatid Iron Transporter That Regulates Mitochondrial Function Is Required for Leishmania Amazonensis Virulence. PloS Pathog (2016) 12(1):e1005340. doi: 10.1371/journal.ppat.1005340
53. McConville MJ, de Souza D, Saunders E, Likic VA, Naderer T. Living in a Phagolysosome; Metabolism of Leishmania Amastigotes. Trends Parasitol (2007) 23(8):368–75. doi: 10.1016/j.pt.2007.06.009
54. Rabinovitch M, Topper G, Cristello P, Rich A. Receptor-Mediated Entry of Peroxidases Into the Parasitophorous Vacuoles of Macrophages Infected With Leishmania Mexicana Amazonensis. J Leukoc Biol (1985) 37(3):247–61. doi: 10.1002/jlb.37.3.247
55. Shepherd VL, Stahl PD, Bernd P, Rabinovitch M. Receptor-Mediated Entry of Beta-Glucuronidase Into the Parasitophorous Vacuoles of Macrophages Infected With Leishmania Mexicana Amazonensis. J Exp Med (1983) 157(5):1471–82. doi: 10.1084/jem.157.5.1471
56. Russell DG, Xu S, Chakraborty P. Intracellular Trafficking and the Parasitophorous Vacuole of Leishmania Mexicana-Infected Macrophages. J Cell Sci (1992) 103(Pt 4):1193–210. doi: 10.1242/jcs.103.4.1193
57. Roberts SC, Tancer MJ, Polinsky MR, Gibson KM, Heby O, Ullman B. Arginase Plays a Pivotal Role in Polyamine Precursor Metabolism in Leishmania. Characterization of Gene Deletion Mutants. J Biol Chem (2004) 279(22):23668–78. doi: 10.1074/jbc.M402042200
58. da Silva ER, da Silva MF, Fischer H, Mortara RA, Mayer MG, Framesqui K, et al. Biochemical and Biophysical Properties of a Highly Active Recombinant Arginase From Leishmania (Leishmania) Amazonensis and Subcellular Localization of Native Enzyme. Mol Biochem Parasitol (2008) 159(2):104–11. doi: 10.1016/j.molbiopara.2008.02.011
59. Kropf P, Fuentes JM, Fahnrich E, Arpa L, Herath S, Weber V, et al. Arginase and Polyamine Synthesis Are Key Factors in the Regulation of Experimental Leishmaniasis In Vivo. FASEB J (2005) 19(8):1000–2. doi: 10.1096/fj.04-3416fje
60. Qi H, Ji J, Wanasen N, Soong L. Enhanced Replication of Leishmania Amazonensis Amastigotes in Gamma Interferon-Stimulated Murine Macrophages: Implications for the Pathogenesis of Cutaneous Leishmaniasis. Infect Immun (2004) 72(2):988–95. doi: 10.1128/IAI.72.2.988-995.2004
61. Wanasen N, MacLeod CL, Ellies LG, Soong L. L-Arginine and Cationic Amino Acid Transporter 2B Regulate Growth and Survival of Leishmania Amazonensis Amastigotes in Macrophages. Infect Immun (2007) 75(6):2802–10. doi: 10.1128/IAI.00026-07
62. Goldman-Pinkovich A, Balno C, Strasser R, Zeituni-Molad M, Bendelak K, Rentsch D, et al. An Arginine Deprivation Response Pathway Is Induced in Leishmania During Macrophage Invasion. PloS Pathog (2016) 12(4):e1005494. doi: 10.1371/journal.ppat.1005494
63. da Silva MF, Zampieri RA, Muxel SM, Beverley SM, Floeter-Winter LM. Leishmania Amazonensis Arginase Compartmentalization in the Glycosome Is Important for Parasite Infectivity. PloS One (2012) 7(3):e34022. doi: 10.1371/journal.pone.0034022
64. Aoki JI, Muxel SM, Zampieri RA, Acuna SM, Fernandes JCR, Vanderlinde RH, et al. L-Arginine Availability and Arginase Activity: Characterization of Amino Acid Permease 3 in Leishmania Amazonensis. PloS Negl Trop Dis (2017) 11(10):e0006025. doi: 10.1371/journal.pntd.0006025
65. Roma EH, Macedo JP, Goes GR, Goncalves JL, Castro W, Cisalpino D, et al. Impact of Reactive Oxygen Species (ROS) on the Control of Parasite Loads and Inflammation in Leishmania Amazonensis Infection. Parasit Vectors (2016) 9:193. doi: 10.1186/s13071-016-1472-y
66. Carlsen ED, Hay C, Henard CA, Popov V, Garg NJ, Soong L. Leishmania Amazonensis Amastigotes Trigger Neutrophil Activation But Resist Neutrophil Microbicidal Mechanisms. Infect Immun (2013) 81(11):3966–74. doi: 10.1128/IAI.00770-13
67. Carneiro MBH, Roma EH, Ranson AJ, Doria NA, Debrabant A, Sacks DL, et al. NOX2-Derived Reactive Oxygen Species Control Inflammation During Leishmania Amazonensis Infection by Mediating Infection-Induced Neutrophil Apoptosis. J Immunol (2018) 200(1):196–208. doi: 10.4049/jimmunol.1700899
68. Horta MF, Mendes BP, Roma EH, Noronha FS, Macedo JP, Oliveira LS, et al. Reactive Oxygen Species and Nitric Oxide in Cutaneous Leishmaniasis. J Parasitol Res (2012) 2012:203818. doi: 10.1155/2012/203818
69. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of Interferon-Gamma as the Lymphokine That Activates Human Macrophage Oxidative Metabolism and Antimicrobial Activity. J Exp Med (1983) 158(3):670–89. doi: 10.1084/jem.158.3.670
70. Green SJ, Crawford RM, Hockmeyer JT, Meltzer MS, Nacy CA. Leishmania Major Amastigotes Initiate the L-Arginine-Dependent Killing Mechanism in IFN-Gamma-Stimulated Macrophages by Induction of Tumor Necrosis Factor-Alpha. J Immunol (1990) 145(12):4290–7.
71. Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva AL, Mineo TW, et al. Inflammasome-Derived IL-1beta Production Induces Nitric Oxide-Mediated Resistance to Leishmania. Nat Med (2013) 19(7):909–15. doi: 10.1038/nm.3221
72. Wu G, Morris SM Jr. Arginine Metabolism: Nitric Oxide and Beyond. Biochem J (1998) 336(Pt 1):1–17. doi: 10.1042/bj3360001
73. Vodovotz Y, Russell D, Xie QW, Bogdan C, Nathan C. Vesicle Membrane Association of Nitric Oxide Synthase in Primary Mouse Macrophages. J Immunol (1995) 154(6):2914–25.
74. Miller BH, Fratti RA, Poschet JF, Timmins GS, Master SS, Burgos M, et al. Mycobacteria Inhibit Nitric Oxide Synthase Recruitment to Phagosomes During Macrophage Infection. Infect Immun (2004) 72(5):2872–8. doi: 10.1128/iai.72.5.2872-2878.2004
75. Olekhnovitch R, Ryffel B, Muller AJ, Bousso P. Collective Nitric Oxide Production Provides Tissue-Wide Immunity During Leishmania Infection. J Clin Invest (2014) 124(4):1711–22. doi: 10.1172/JCI72058
76. Holzmuller P, Sereno D, Cavaleyra M, Mangot I, Daulouede S, Vincendeau P, et al. Nitric Oxide-Mediated Proteasome-Dependent Oligonucleosomal DNA Fragmentation in Leishmania Amazonensis Amastigotes. Infect Immun (2002) 70(7):3727–35. doi: 10.1128/iai.70.7.3727-3735.2002
77. Ferrer-Sueta G, Campolo N, Trujillo M, Bartesaghi S, Carballal S, Romero N, et al. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem Rev (2018) 118(3):1338–408. doi: 10.1021/acs.chemrev.7b00568
78. Muller AJ, Aeschlimann S, Olekhnovitch R, Dacher M, Spath GF, Bousso P. Photoconvertible Pathogen Labeling Reveals Nitric Oxide Control of Leishmania Major Infection In Vivo via Dampening of Parasite Metabolism. Cell Host Microbe (2013) 14(4):460–7. doi: 10.1016/j.chom.2013.09.008
79. Carneiro MB, de Andrade e Sousa LM, Vaz LG, Dos Santos LM, Vilela L, de Souza CC, et al. Short-Term Protection Conferred by Leishvacin(R) Against Experimental Leishmania Amazonensis Infection in C57BL/6 Mice. Parasitol Int (2014) 63(6):826–34. doi: 10.1016/j.parint.2014.07.010
80. Gomes RS, de Carvalho LC, de Souza Vasconcellos R, Fietto JL, Afonso LC. E-NTPDase (Ecto-Nucleoside Triphosphate Diphosphohydrolase) of Leishmania Amazonensis Inhibits Macrophage Activation. Microbes Infect (2015) 17(4):295–303. doi: 10.1016/j.micinf.2014.12.009
81. Calegari-Silva TC, Pereira RM, De-Melo LD, Saraiva EM, Soares DC, Bellio M, et al. NF-kappaB-Mediated Repression of iNOS Expression in Leishmania Amazonensis Macrophage Infection. Immunol Lett (2009) 127(1):19–26. doi: 10.1016/j.imlet.2009.08.009
82. Calegari-Silva TC, Vivarini AC, Pereira RMS, Dias-Teixeira KL, Rath CT, Pacheco ASS, et al. Leishmania Amazonensis Downregulates Macrophage iNOS Expression via Histone Deacetylase 1 (HDAC1): A Novel Parasite Evasion Mechanism. Eur J Immunol (2018) 48(7):1188–98. doi: 10.1002/eji.201747257
83. Balestieri FM, Queiroz AR, Scavone C, Costa VM, Barral-Netto M, Abrahamsohn Ide A. Leishmania (L.) Amazonensis-Induced Inhibition of Nitric Oxide Synthesis in Host Macrophages. Microbes Infect (2002) 4(1):23–9. doi: 10.1016/s1286-4579(01)01505-2
84. Janeway CA Jr., Medzhitov R. Innate Immune Recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
85. Sacks DL, Perkins PV. Identification of an Infective Stage of Leishmania Promastigotes. Science (1984) 223(4643):1417–9. doi: 10.1126/science.6701528
86. Giraud E, Martin O, Yakob L, Rogers M. Quantifying Leishmania Metacyclic Promastigotes From Individual Sandfly Bites Reveals the Efficiency of Vector Transmission. Commun Biol (2019) 2:84. doi: 10.1038/s42003-019-0323-8
87. Franco LH, Beverley SM, Zamboni DS. Innate Immune Activation and Subversion of Mammalian Functions by Leishmania Lipophosphoglycan. J Parasitol Res (2012) 2012:165126. doi: 10.1155/2012/165126
88. Turco SJ, Spath GF, Beverley SM. Is Lipophosphoglycan a Virulence Factor? A Surprising Diversity Between Leishmania Species. Trends Parasitol (2001) 17(5):223–6. doi: 10.1016/s1471-4922(01)01895-5
89. Dominguez M, Torano A. Immune Adherence-Mediated Opsonophagocytosis: The Mechanism of Leishmania Infection. J Exp Med (1999) 189(1):25–35. doi: 10.1084/jem.189.1.25
90. Brittingham A, Mosser DM. Exploitation of the Complement System by Leishmania Promastigotes. Parasitol Today (1996) 12(11):444–7. doi: 10.1016/0169-4758(96)10067-3
91. Nunes AC, Almeida-Campos FR, Horta MF, Ramalho-Pinto FJ. Leishmania Amazonensis Promastigotes Evade Complement Killing by Interfering With the Late Steps of the Cascade. Parasitology (1997) 115(Pt 6):601–9. doi: 10.1017/s0031182097001704
92. Laurenti MD, Orn A, Sinhorini IL, Corbett CE. The Role of Complement in the Early Phase of Leishmania (Leishmania) Amazonensis Infection in BALB/c Mice. Braz J Med Biol Res (2004) 37(3):427–34. doi: 10.1590/s0100-879x2004000300021
93. Brittingham A, Morrison CJ, McMaster WR, McGwire BS, Chang KP, Mosser DM. Role of the Leishmania Surface Protease Gp63 in Complement Fixation, Cell Adhesion, and Resistance to Complement-Mediated Lysis. J Immunol (1995) 155(6):3102–11. doi: 10.1016/0169-4758(95)80054-9
94. Talamas-Rohana P, Wright SD, Lennartz MR, Russell DG. Lipophosphoglycan From Leishmania Mexicana Promastigotes Binds to Members of the CR3, P150,95 and LFA-1 Family of Leukocyte Integrins. J Immunol (1990) 144(12):4817–24.
95. McGwire BS, Chang KP, Engman DM. Migration Through the Extracellular Matrix by the Parasitic Protozoan Leishmania Is Enhanced by Surface Metalloprotease Gp63. Infect Immun (2003) 71(2):1008–10. doi: 10.1128/iai.71.2.1008-1010.2003
96. Chauhan P, Shukla D, Chattopadhyay D, Saha B. Redundant and Regulatory Roles for Toll-Like Receptors in Leishmania Infection. Clin Exp Immunol (2017) 190(2):167–86. doi: 10.1111/cei.13014
97. Xin L, Vargas-Inchaustegui DA, Raimer SS, Kelly BC, Hu J, Zhu L, et al. Type I IFN Receptor Regulates Neutrophil Functions and Innate Immunity to Leishmania Parasites. J Immunol (2010) 184(12):7047–56. doi: 10.4049/jimmunol.0903273
98. Khouri R, Bafica A, Silva Mda P, Noronha A, Kolb JP, Wietzerbin J, et al. IFN-Beta Impairs Superoxide-Dependent Parasite Killing in Human Macrophages: Evidence for a Deleterious Role of SOD1 in Cutaneous Leishmaniasis. J Immunol (2009) 182(4):2525–31. doi: 10.4049/jimmunol.0802860
99. Vivarini Ade C, Pereira Rde M, Teixeira KL, Calegari-Silva TC, Bellio M, Laurenti MD, et al. Human Cutaneous Leishmaniasis: Interferon-Dependent Expression of Double-Stranded RNA-Dependent Protein Kinase (PKR) via TLR2. FASEB J (2011) 25(12):4162–73. doi: 10.1096/fj.11-185165
100. Barreto-de-Souza V, Ferreira PL, Vivarini Ade C, Calegari-Silva T, Soares DC, Regis EG, et al. IL-27 Enhances Leishmania Amazonensis Infection via ds-RNA Dependent Kinase (PKR) and IL-10 Signaling. Immunobiology (2015) 220(4):437–44. doi: 10.1016/j.imbio.2014.11.006
101. Guerra CS, Silva RM, Carvalho LO, Calabrese KS, Bozza PT, Corte-Real S. Histopathological Analysis of Initial Cellular Response in TLR-2 Deficient Mice Experimentally Infected by Leishmania (L.) Amazonensis. Int J Exp Pathol (2010) 91(5):451–9. doi: 10.1111/j.1365-2613.2010.00717.x
102. Rath CT, Schnellrath LC, Damaso CR, de Arruda LB, Vasconcelos P, Gomes C, et al. Amazonian Phlebovirus (Bunyaviridae) Potentiates the Infection of Leishmania (Leishmania) Amazonensis: Role of the PKR/IFN1/IL-10 Axis. PloS Negl Trop Dis (2019) 13(6):e0007500. doi: 10.1371/journal.pntd.0007500
103. Vargas-Inchaustegui DA, Tai W, Xin L, Hogg AE, Corry DB, Soong L. Distinct Roles for MyD88 and Toll-Like Receptor 2 During Leishmania Braziliensis Infection in Mice. Infect Immun (2009) 77(7):2948–56. doi: 10.1128/IAI.00154-09
104. Muxel SM, Acuna SM, Aoki JI, Zampieri RA, Floeter-Winter LM. Toll-Like Receptor and miRNA-Let-7e Expression Alter the Inflammatory Response in Leishmania Amazonensis-Infected Macrophages. Front Immunol (2018) 9:2792. doi: 10.3389/fimmu.2018.02792
105. Nogueira PM, Assis RR, Torrecilhas AC, Saraiva EM, Pessoa NL, Campos MA, et al. Lipophosphoglycans From Leishmania Amazonensis Strains Display Immunomodulatory Properties via TLR4 and Do Not Affect Sand Fly Infection. PloS Negl Trop Dis (2016) 10(8):e0004848. doi: 10.1371/journal.pntd.0004848
106. Sauter IP, Madrid KG, de Assis JB, Sa-Nunes A, Torrecilhas AC, Staquicini DI, et al. TLR9/MyD88/TRIF Signaling Activates Host Immune Inhibitory CD200 in Leishmania Infection. JCI Insight (2019) 4(10):e126207. doi: 10.1172/jci.insight.126207
107. Pratti JES, da Fonseca Martins AM, da Silva JP, Ramos TD, Pereira JC, Firmino-Cruz L, et al. The Role of TLR9 on Leishmania Amazonensis Infection and its Influence on Intranasal LaAg Vaccine Efficacy. PloS Negl Trop Dis (2019) 13(2):e0007146. doi: 10.1371/journal.pntd.0007146
108. Campos MB, Lima L, de Lima ACS, Vasconcelos Dos Santos T, Ramos PKS, Gomes CMC, et al. Toll-Like Receptors 2, 4, and 9 Expressions Over the Entire Clinical and Immunopathological Spectrum of American Cutaneous Leishmaniasis Due to Leishmania(V.) Braziliensis and Leishmania (L.) Amazonensis. PloS One (2018) 13(3):e0194383. doi: 10.1371/journal.pone.0194383
109. Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
110. de Carvalho RVH, Andrade WA, Lima-Junior DS, Dilucca M, de Oliveira CV, Wang K, et al. Leishmania Lipophosphoglycan Triggers Caspase-11 and the Non-Canonical Activation of the NLRP3 Inflammasome. Cell Rep (2019) 26(2):429–37 e5. doi: 10.1016/j.celrep.2018.12.047
111. Lima-Junior DS, Mineo TWP, Calich VLG, Zamboni DS. Dectin-1 Activation During Leishmania Amazonensis Phagocytosis Prompts Syk-Dependent Reactive Oxygen Species Production To Trigger Inflammasome Assembly and Restriction of Parasite Replication. J Immunol (2017) 199(6):2055–68. doi: 10.4049/jimmunol.1700258
112. Chaves MM, Sinflorio DA, Thorstenberg ML, Martins MDA, Moreira-Souza ACA, Rangel TP, et al. Non-Canonical NLRP3 Inflammasome Activation and IL-1beta Signaling Are Necessary to L. Amazonensis Control Mediated by P2X7 Receptor and Leukotriene B4. PloS Pathog (2019) 15(6):e1007887. doi: 10.1371/journal.ppat.1007887
113. Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N, et al. The Nlrp3 Inflammasome, IL-1beta, and Neutrophil Recruitment Are Required for Susceptibility to a Nonhealing Strain of Leishmania Major in C57BL/6 Mice. Eur J Immunol (2016) 46(4):897–911. doi: 10.1002/eji.201546015
114. Dey R, Joshi AB, Oliveira F, Pereira L, Guimaraes-Costa AB, Serafim TD, et al. Gut Microbes Egested During Bites of Infected Sand Flies Augment Severity of Leishmaniasis via Inflammasome-Derived IL-1beta. Cell Host Microbe (2018) 23(1):134–43.e6. doi: 10.1016/j.chom.2017.12.002
115. Novais FO, Carvalho AM, Clark ML, Carvalho LP, Beiting DP, Brodsky IE, et al. CD8+ T Cell Cytotoxicity Mediates Pathology in the Skin by Inflammasome Activation and IL-1beta Production. PloS Pathog (2017) 13(2):e1006196. doi: 10.1371/journal.ppat.1006196
116. Sousa LM, Carneiro MB, Dos Santos LM, Natale CC, Resende ME, Mosser DM, et al. IL-18 Contributes to Susceptibility to Leishmania Amazonensis Infection by Macrophage-Independent Mechanisms. Cytokine (2015) 74(2):327–30. doi: 10.1016/j.cyto.2015.01.021
117. Lecoeur H, Prina E, Rosazza T, Kokou K, N’Diaye P, Aulner N, et al. Targeting Macrophage Histone H3 Modification as a Leishmania Strategy to Dampen the NF-KappaB/NLRP3-Mediated Inflammatory Response. Cell Rep (2020) 30(6):1870–82.e4. doi: 10.1016/j.celrep.2020.01.030
118. Lecoeur H, Rosazza T, Kokou K, Varet H, Coppee JY, Lari A, et al. Leishmania Amazonensis Subverts the Transcription Factor Landscape in Dendritic Cells to Avoid Inflammasome Activation and Stall Maturation. Front Immunol (2020) 11:1098. doi: 10.3389/fimmu.2020.01098
119. Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, et al. Neutrophils: New Insights and Open Questions. Sci Immunol (2018) 3(30):eaat4579. doi: 10.1126/sciimmunol.aat4579
120. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 Antagonistically Regulate Neutrophil Trafficking From Murine Bone Marrow. J Clin Invest (2010) 120(7):2423–31. doi: 10.1172/JCI41649
121. Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernandez-Calvo G, et al. Co-Option of Neutrophil Fates by Tissue Environments. Cell (2020) 183(5):1282–97.e18. doi: 10.1016/j.cell.2020.10.003
122. Manz MG, Boettcher S. Emergency Granulopoiesis. Nat Rev Immunol (2014) 14(5):302–14. doi: 10.1038/nri3660
123. David BA, Kubes P. Exploring the Complex Role of Chemokines and Chemoattractants In Vivo on Leukocyte Dynamics. Immunol Rev (2019) 289(1):9–30. doi: 10.1111/imr.12757
124. Dyer DP, Medina-Ruiz L, Bartolini R, Schuette F, Hughes CE, Pallas K, et al. Chemokine Receptor Redundancy and Specificity Are Context Dependent. Immunity (2019) 50(2):378–89 e5. doi: 10.1016/j.immuni.2019.01.009
125. Canesso MC, Vieira AT, Castro TB, Schirmer BG, Cisalpino D, Martins FS, et al. Skin Wound Healing Is Accelerated and Scarless in the Absence of Commensal Microbiota. J Immunol (2014) 193(10):5171–80. doi: 10.4049/jimmunol.1400625
126. Peters NC, Kimblin N, Secundino N, Kamhawi S, Lawyer P, Sacks DL. Vector Transmission of Leishmania Abrogates Vaccine-Induced Protective Immunity. PloS Pathog (2009) 5(6):e1000484. doi: 10.1371/journal.ppat.1000484
127. Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, et al. Neutrophil Swarms Require LTB4 and Integrins at Sites of Cell Death In Vivo. Nature (2013) 498(7454):371–5. doi: 10.1038/nature12175
128. Sousa LM, Carneiro MB, Resende ME, Martins LS, Dos Santos LM, Vaz LG, et al. Neutrophils Have a Protective Role During Early Stages of Leishmania Amazonensis Infection in BALB/c Mice. Parasite Immunol (2014) 36(1):13–31. doi: 10.1111/pim.12078
129. Pompeu ML, Freitas LA, Santos ML, Khouri M, Barral-Netto M. Granulocytes in the Inflammatory Process of BALB/c Mice Infected by Leishmania Amazonensis. A Quantitative Approach. Acta Trop (1991) 48(3):185–93. doi: 10.1016/0001-706x(91)90046-m
130. Rogers M, Kropf P, Choi BS, Dillon R, Podinovskaia M, Bates P, et al. Proteophosophoglycans Regurgitated by Leishmania-Infected Sand Flies Target the L-Arginine Metabolism of Host Macrophages to Promote Parasite Survival. PloS Pathog (2009) 5(8):e1000555. doi: 10.1371/journal.ppat.1000555
131. Hurrell BP, Regli IB, Tacchini-Cottier F. Different Leishmania Species Drive Distinct Neutrophil Functions. Trends Parasitol (2016) 32(5):392–401. doi: 10.1016/j.pt.2016.02.003
132. Guimaraes-Costa AB, Shannon JP, Waclawiak I, Oliveira J, Meneses C, de Castro W, et al. A Sand Fly Salivary Protein Acts as a Neutrophil Chemoattractant. Nat Commun (2021) 12(1):3213. doi: 10.1038/s41467-021-23002-5
133. Kienle K, Lammermann T. Neutrophil Swarming: An Essential Process of the Neutrophil Tissue Response. Immunol Rev (2016) 273(1):76–93. doi: 10.1111/imr.12458
134. Ribeiro-Gomes FL, Roma EH, Carneiro MB, Doria NA, Sacks DL, Peters NC. Site-Dependent Recruitment of Inflammatory Cells Determines the Effective Dose of Leishmania Major. Infect Immun (2014) 82(7):2713–27. doi: 10.1128/IAI.01600-13
135. Chaves MM, Lee SH, Kamenyeva O, Ghosh K, Peters NC, Sacks D. The Role of Dermis Resident Macrophages and Their Interaction With Neutrophils in the Early Establishment of Leishmania Major Infection Transmitted by Sand Fly Bite. PloS Pathog (2020) 16(11):e1008674. doi: 10.1371/journal.ppat.1008674
136. Sarkar A, Aga E, Bussmeyer U, Bhattacharyya A, Moller S, Hellberg L, et al. Infection of Neutrophil Granulocytes With Leishmania Major Activates ERK 1/2 and Modulates Multiple Apoptotic Pathways to Inhibit Apoptosis. Med Microbiol Immunol (2013) 202(1):25–35. doi: 10.1007/s00430-012-0246-1
137. Charmoy M, Auderset F, Allenbach C, Tacchini-Cottier F. The Prominent Role of Neutrophils During the Initial Phase of Infection by Leishmania Parasites. J BioMed Biotechnol (2010) 2010:719361. doi: 10.1155/2010/719361
138. Falcao SA, Weinkopff T, Hurrell BP, Celes FS, Curvelo RP, Prates DB, et al. Exposure to Leishmania Braziliensis Triggers Neutrophil Activation and Apoptosis. PloS Negl Trop Dis (2015) 9(3):e0003601. doi: 10.1371/journal.pntd.0003601
139. Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL. Efficient Capture of Infected Neutrophils by Dendritic Cells in the Skin Inhibits the Early Anti-Leishmania Response. PloS Pathog (2012) 8(2):e1002536. doi: 10.1371/journal.ppat.1002536
140. van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, et al. Cutting Edge: Neutrophil Granulocyte Serves as a Vector for Leishmania Entry Into Macrophages. J Immunol (2004) 173(11):6521–5. doi: 10.4049/jimmunol.173.11.6521
141. Afonso L, Borges VM, Cruz H, Ribeiro-Gomes FL, DosReis GA, Dutra AN, et al. Interactions With Apoptotic But Not With Necrotic Neutrophils Increase Parasite Burden in Human Macrophages Infected With Leishmania Amazonensis. J Leukoc Biol (2008) 84(2):389–96. doi: 10.1189/jlb.0108018
142. Romano A, Carneiro MBH, Doria NA, Roma EH, Ribeiro-Gomes FL, Inbar E, et al. Divergent Roles for Ly6C+CCR2+CX3CR1+ Inflammatory Monocytes During Primary or Secondary Infection of the Skin With the Intra-Phagosomal Pathogen Leishmania Major. PloS Pathog (2017) 13(6):e1006479. doi: 10.1371/journal.ppat.1006479
143. Hurrell BP, Schuster S, Grun E, Coutaz M, Williams RA, Held W, et al. Rapid Sequestration of Leishmania Mexicana by Neutrophils Contributes to the Development of Chronic Lesion. PloS Pathog (2015) 11(5):e1004929. doi: 10.1371/journal.ppat.1004929
144. Ribeiro-Gomes FL, Otero AC, Gomes NA, Moniz-De-Souza MC, Cysne-Finkelstein L, Arnholdt AC, et al. Macrophage Interactions With Neutrophils Regulate Leishmania Major Infection. J Immunol (2004) 172(7):4454–62. doi: 10.4049/jimmunol.172.7.4454
145. Regli IB, Passelli K, Martinez-Salazar B, Amore J, Hurrell BP, Muller AJ, et al. TLR7 Sensing by Neutrophils Is Critical for the Control of Cutaneous Leishmaniasis. Cell Rep (2020) 31(10):107746. doi: 10.1016/j.celrep.2020.107746
146. Rosales C. Neutrophils at the Crossroads of Innate and Adaptive Immunity. J Leukoc Biol (2020) 108(1):377–96. doi: 10.1002/JLB.4MIR0220-574RR
147. Tavares N, Afonso L, Suarez M, Ampuero M, Prates DB, Araujo-Santos T, et al. Degranulating Neutrophils Promote Leukotriene B4 Production by Infected Macrophages To Kill Leishmania Amazonensis Parasites. J Immunol (2016) 196(4):1865–73. doi: 10.4049/jimmunol.1502224
148. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
149. Linhares-Lacerda L, Temerozo JR, Ribeiro-Alves M, Azevedo EP, Mojoli A, Nascimento MTC, et al. Neutrophil Extracellular Trap-Enriched Supernatants Carry microRNAs Able to Modulate TNF-Alpha Production by Macrophages. Sci Rep (2020) 10(1):2715. doi: 10.1038/s41598-020-59486-2
150. Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, et al. Leishmania Amazonensis Promastigotes Induce and Are Killed by Neutrophil Extracellular Traps. Proc Natl Acad Sci U S A (2009) 106(16):6748–53. doi: 10.1073/pnas.0900226106
151. Wang Y, Chen Y, Xin L, Beverley SM, Carlsen ED, Popov V, et al. Differential Microbicidal Effects of Human Histone Proteins H2A and H2B on Leishmania Promastigotes and Amastigotes. Infect Immun (2011) 79(3):1124–33. doi: 10.1128/IAI.00658-10
152. Rochael NC, Guimaraes-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF, et al. Classical ROS-Dependent and Early/Rapid ROS-Independent Release of Neutrophil Extracellular Traps Triggered by Leishmania Parasites. Sci Rep (2015) 5:18302. doi: 10.1038/srep18302
153. Guimaraes-Costa AB, Rochael NC, Oliveira F, Echevarria-Lima J, Saraiva EM. Neutrophil Extracellular Traps Reprogram IL-4/GM-CSF-Induced Monocyte Differentiation to Anti-Inflammatory Macrophages. Front Immunol (2017) 8:523. doi: 10.3389/fimmu.2017.00523
154. Manfredi AA, Ramirez GA, Rovere-Querini P, Maugeri N. The Neutrophil’s Choice: Phagocytose vs Make Neutrophil Extracellular Traps. Front Immunol (2018) 9:288. doi: 10.3389/fimmu.2018.00288
155. Guimaraes-Costa AB, DeSouza-Vieira TS, Paletta-Silva R, Freitas-Mesquita AL, Meyer-Fernandes JR, Saraiva EM. 3’-Nucleotidase/Nuclease Activity Allows Leishmania Parasites to Escape Killing by Neutrophil Extracellular Traps. Infect Immun (2014) 82(4):1732–40. doi: 10.1128/IAI.01232-13
156. Henard CA, Carlsen ED, Hay C, Kima PE, Soong L. Leishmania Amazonensis Amastigotes Highly Express a Tryparedoxin Peroxidase Isoform That Increases Parasite Resistance to Macrophage Antimicrobial Defenses and Fosters Parasite Virulence. PloS Negl Trop Dis (2014) 8(7):e3000. doi: 10.1371/journal.pntd.0003000
157. Hurrell BP, Beaumann M, Heyde S, Regli IB, Muller AJ, Tacchini-Cottier F. Frontline Science: Leishmania Mexicana Amastigotes can Replicate Within Neutrophils. J Leukoc Biol (2017) 102(5):1187–98. doi: 10.1189/jlb.4HI0417-158R
158. Charmoy M, Brunner-Agten S, Aebischer D, Auderset F, Launois P, Milon G, et al. Neutrophil-Derived CCL3 Is Essential for the Rapid Recruitment of Dendritic Cells to the Site of Leishmania Major Inoculation in Resistant Mice. PloS Pathog (2010) 6(2):e1000755. doi: 10.1371/journal.ppat.1000755
159. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the Activation and Regulation of Innate and Adaptive Immunity. Nat Rev Immunol (2011) 11(8):519–31. doi: 10.1038/nri3024
160. Steinman RM, Cohn ZA. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice. I. Morphology, Quantitation, Tissue Distribution. J Exp Med (1973) 137(5):1142–62. doi: 10.1084/jem.137.5.1142
161. Kashem SW, Haniffa M, Kaplan DH. Antigen-Presenting Cells in the Skin. Annu Rev Immunol (2017) 35:469–99. doi: 10.1146/annurev-immunol-051116-052215
162. von Stebut E, Belkaid Y, Jakob T, Sacks DL, Udey MC. Uptake of Leishmania Major Amastigotes Results in Activation and Interleukin 12 Release From Murine Skin-Derived Dendritic Cells: Implications for the Initiation of Anti-Leishmania Immunity. J Exp Med (1998) 188(8):1547–52. doi: 10.1084/jem.188.8.1547
163. Tiburcio R, Nunes S, Nunes I, Rosa Ampuero M, Silva IB, Lima R, et al. Molecular Aspects of Dendritic Cell Activation in Leishmaniasis: An Immunobiological View. Front Immunol (2019) 10:227. doi: 10.3389/fimmu.2019.00227
164. Martinez-Lopez M, Iborra S, Conde-Garrosa R, Sancho D. Batf3-Dependent CD103+ Dendritic Cells Are Major Producers of IL-12 That Drive Local Th1 Immunity Against Leishmania Major Infection in Mice. Eur J Immunol (2015) 45(1):119–29. doi: 10.1002/eji.201444651
165. Ashok D, Schuster S, Ronet C, Rosa M, Mack V, Lavanchy C, et al. Cross-Presenting Dendritic Cells Are Required for Control of Leishmania Major Infection. Eur J Immunol (2014) 44(5):1422–32. doi: 10.1002/eji.201344242
166. Arnold IC, Zhang X, Artola-Boran M, Fallegger A, Sander P, Johansen P, et al. BATF3-Dependent Dendritic Cells Drive Both Effector and Regulatory T-Cell Responses in Bacterially Infected Tissues. PloS Pathog (2019) 15(6):e1007866. doi: 10.1371/journal.ppat.1007866
167. Soto M, Ramirez L, Solana JC, Cook ECL, Hernandez-Garcia E, Charro-Zanca S, et al. Resistance to Experimental Visceral Leishmaniasis in Mice Infected With Leishmania Infantum Requires Batf3. Front Immunol (2020) 11:590934. doi: 10.3389/fimmu.2020.590934
168. Anderson DA 3rd, Dutertre CA, Ginhoux F, Murphy KM. Genetic Models of Human and Mouse Dendritic Cell Development and Function. Nat Rev Immunol (2021) 21(2):101–15. doi: 10.1038/s41577-020-00413-x
169. Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep (2016) 15(11):2462–74. doi: 10.1016/j.celrep.2016.05.025
170. Bertholet S, Goldszmid R, Morrot A, Debrabant A, Afrin F, Collazo-Custodio C, et al. Leishmania Antigens Are Presented to CD8+ T Cells by a Transporter Associated With Antigen Processing-Independent Pathway In Vitro and In Vivo. J Immunol (2006) 177(6):3525–33. doi: 10.4049/jimmunol.177.6.3525
171. Kima PE, Ruddle NH, McMahon-Pratt D. Presentation via the Class I Pathway by Leishmania Amazonensis-Infected Macrophages of an Endogenous Leishmanial Antigen to CD8+ T Cells. J Immunol (1997) 159(4):1828–34.
172. Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-Cytometry: A Method for Highly Multiplex Quantitative Tissue Imaging Analysis Applied to Dendritic Cell Subset Microanatomy in Lymph Nodes. Immunity (2012) 37(2):364–76. doi: 10.1016/j.immuni.2012.07.011
173. Sokol CL, Camire RB, Jones MC, Luster AD. The Chemokine Receptor CCR8 Promotes the Migration of Dendritic Cells Into the Lymph Node Parenchyma to Initiate the Allergic Immune Response. Immunity (2018) 49(3):449–63.e6. doi: 10.1016/j.immuni.2018.07.012
174. Leal JM, Huang JY, Kohli K, Stoltzfus C, Lyons-Cohen MR, Olin BE, et al. Innate Cell Microenvironments in Lymph Nodes Shape the Generation of T Cell Responses During Type I Inflammation. Sci Immunol (2021) 6(56):eabb9435. doi: 10.1126/sciimmunol.abb9435
175. Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, et al. Skin-Resident Murine Dendritic Cell Subsets Promote Distinct and Opposing Antigen-Specific T Helper Cell Responses. Immunity (2011) 35(2):260–72. doi: 10.1016/j.immuni.2011.06.005
176. Schleicher U, Liese J, Knippertz I, Kurzmann C, Hesse A, Heit A, et al. NK Cell Activation in Visceral Leishmaniasis Requires TLR9, Myeloid DCs, and IL-12, But is Independent of Plasmacytoid DCs. J Exp Med (2007) 204(4):893–906. doi: 10.1084/jem.20061293
177. Baldwin T, Henri S, Curtis J, O’Keeffe M, Vremec D, Shortman K, et al. Dendritic Cell Populations in Leishmania Major-Infected Skin and Draining Lymph Nodes. Infect Immun (2004) 72(4):1991–2001. doi: 10.1128/IAI.72.4.1991-2001.2004
178. Schleicher U, Liese J, Justies N, Mischke T, Haeberlein S, Sebald H, et al. Type I Interferon Signaling Is Required for CpG-Oligodesoxynucleotide-Induced Control of Leishmania Major, But Not for Spontaneous Cure of Subcutaneous Primary or Secondary L. Major Infection. Front Immunol (2018) 9:79. doi: 10.3389/fimmu.2018.00079
179. Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J, et al. Type 1 Interferon (IFNalpha/beta) and Type 2 Nitric Oxide Synthase Regulate the Innate Immune Response to a Protozoan Parasite. Immunity (1998) 8(1):77–87. doi: 10.1016/s1074-7613(00)80460-4
180. Leon B, Lopez-Bravo M, Ardavin C. Monocyte-Derived Dendritic Cells Formed at the Infection Site Control the Induction of Protective T Helper 1 Responses Against Leishmania. Immunity (2007) 26(4):519–31. doi: 10.1016/j.immuni.2007.01.017
181. Serbina NV, Pamer EG. Monocyte Emigration From Bone Marrow During Bacterial Infection Requires Signals Mediated by Chemokine Receptor CCR2. Nat Immunol (2006) 7(3):311–7. doi: 10.1038/ni1309
182. Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, et al. CC Chemokine Receptor (CCR)2 Is Required for Langerhans Cell Migration and Localization of T Helper Cell Type 1 (Th1)-Inducing Dendritic Cells. Absence of CCR2 Shifts the Leishmania Major-Resistant Phenotype to a Susceptible State Dominated by Th2 Cytokines, B Cell Outgrowth, and Sustained Neutrophilic Inflammation. J Exp Med (2000) 192(2):205–18. doi: 10.1084/jem.192.2.205
183. Petritus PM, Manzoni-de-Almeida D, Gimblet C, Gonzalez Lombana C, Scott P. Leishmania Mexicana Induces Limited Recruitment and Activation of Monocytes and Monocyte-Derived Dendritic Cells Early During Infection. PloS Negl Trop Dis (2012) 6(10):e1858. doi: 10.1371/journal.pntd.0001858
184. De Trez C, Magez S, Akira S, Ryffel B, Carlier Y, Muraille E. iNOS-Producing Inflammatory Dendritic Cells Constitute the Major Infected Cell Type During the Chronic Leishmania Major Infection Phase of C57BL/6 Resistant Mice. PloS Pathog (2009) 5(6):e1000494. doi: 10.1371/journal.ppat.1000494
185. Xin L, Li K, Soong L. Down-Regulation of Dendritic Cell Signaling Pathways by Leishmania Amazonensis Amastigotes. Mol Immunol (2008) 45(12):3371–82. doi: 10.1016/j.molimm.2008.04.018
186. Prina E, Abdi SZ, Lebastard M, Perret E, Winter N, Antoine JC. Dendritic Cells as Host Cells for the Promastigote and Amastigote Stages of Leishmania Amazonensis: The Role of Opsonins in Parasite Uptake and Dendritic Cell Maturation. J Cell Sci (2004) 117(Pt 2):315–25. doi: 10.1242/jcs.00860
187. Qi H, Popov V, Soong L. Leishmania Amazonensis-Dendritic Cell Interactions In Vitro and the Priming of Parasite-Specific CD4(+) T Cells In Vivo. J Immunol (2001) 167(8):4534–42. doi: 10.4049/jimmunol.167.8.4534
188. Bosetto MC, Giorgio S. Leishmania Amazonensis: Multiple Receptor-Ligand Interactions Are Involved in Amastigote Infection of Human Dendritic Cells. Exp Parasitol (2007) 116(3):306–10. doi: 10.1016/j.exppara.2007.01.003
189. Favali C, Tavares N, Clarencio J, Barral A, Barral-Netto M, Brodskyn C. Leishmania Amazonensis Infection Impairs Differentiation and Function of Human Dendritic Cells. J Leukoc Biol (2007) 82(6):1401–6. doi: 10.1189/jlb.0307187
190. Xin L, Li Y, Soong L. Role of Interleukin-1beta in Activating the CD11c(high) CD45RB- Dendritic Cell Subset and Priming Leishmania Amazonensis-Specific CD4+ T Cells In Vitro and In Vivo. Infect Immun (2007) 75(10):5018–26. doi: 10.1128/IAI.00499-07
191. Figueiredo AB, Serafim TD, Marques-da-Silva EA, Meyer-Fernandes JR, Afonso LC. Leishmania Amazonensis Impairs DC Function by Inhibiting CD40 Expression via A2B Adenosine Receptor Activation. Eur J Immunol (2012) 42(5):1203–15. doi: 10.1002/eji.201141926
192. Figueiredo AB, Souza-Testasicca MC, Mineo TWP, Afonso LCC. Leishmania Amazonensis-Induced cAMP Triggered by Adenosine A2B Receptor Is Important to Inhibit Dendritic Cell Activation and Evade Immune Response in Infected Mice. Front Immunol (2017) 8:849. doi: 10.3389/fimmu.2017.00849
193. Boggiatto PM, Jie F, Ghosh M, Gibson-Corley KN, Ramer-Tait AE, Jones DE, et al. Altered Dendritic Cell Phenotype in Response to Leishmania Amazonensis Amastigote Infection Is Mediated by MAP Kinase, ERK. Am J Pathol (2009) 174(5):1818–26. doi: 10.2353/ajpath.2009.080905
194. Jones DE, Buxbaum LU, Scott P. IL-4-Independent Inhibition of IL-12 Responsiveness During Leishmania Amazonensis Infection. J Immunol (2000) 165(1):364–72. doi: 10.4049/jimmunol.165.1.364
195. Campbell KA, Ovendale PJ, Kennedy MK, Fanslow WC, Reed SG, Maliszewski CR. CD40 Ligand Is Required for Protective Cell-Mediated Immunity to Leishmania Major. Immunity (1996) 4(3):283–9. doi: 10.1016/s1074-7613(00)80436-7
196. Soong L, Xu JC, Grewal IS, Kima P, Sun J, Longley BJ Jr., et al. Disruption of CD40-CD40 Ligand Interactions Results in an Enhanced Susceptibility to Leishmania Amazonensis Infection. Immunity (1996) 4(3):263–73. doi: 10.1016/S1074-7613(00)80434-3
197. Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, et al. Origins and Functional Specialization of Macrophages and of Conventional and Monocyte-Derived Dendritic Cells in Mouse Skin. Immunity (2013) 39(5):925–38. doi: 10.1016/j.immuni.2013.10.004
198. Ng LG, Hsu A, Mandell MA, Roediger B, Hoeller C, Mrass P, et al. Migratory Dermal Dendritic Cells Act as Rapid Sensors of Protozoan Parasites. PloS Pathog (2008) 4(11):e1000222. doi: 10.1371/journal.ppat.1000222
199. Guilliams M, Mildner A, Yona S. Developmental and Functional Heterogeneity of Monocytes. Immunity (2018) 49(4):595–613. doi: 10.1016/j.immuni.2018.10.005
200. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
201. Lee SH, Charmoy M, Romano A, Paun A, Chaves MM, Cope FO, et al. Mannose Receptor High, M2 Dermal Macrophages Mediate Nonhealing Leishmania Major Infection in a Th1 Immune Environment. J Exp Med (2018) 215(1):357–75. doi: 10.1084/jem.20171389
202. Glennie ND, Volk SW, Scott P. Skin-Resident CD4+ T Cells Protect Against Leishmania Major by Recruiting and Activating Inflammatory Monocytes. PloS Pathog (2017) 13(4):e1006349. doi: 10.1371/journal.ppat.1006349
203. Liew FY, Millott S, Parkinson C, Palmer RM, Moncada S. Macrophage Killing of Leishmania Parasite In Vivo Is Mediated by Nitric Oxide From L-Arginine. J Immunol (1990) 144(12):4794–7.
204. Gundra UM, Girgis NM, Ruckerl D, Jenkins S, Ward LN, Kurtz ZD, et al. Alternatively Activated Macrophages Derived From Monocytes and Tissue Macrophages Are Phenotypically and Functionally Distinct. Blood (2014) 123(20):e110–22. doi: 10.1182/blood-2013-08-520619
205. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
206. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant Replenishment From Circulating Monocytes Maintains the Macrophage Pool in the Intestine of Adult Mice. Nat Immunol (2014) 15(10):929–37. doi: 10.1038/ni.2967
207. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell (2019) 178(6):1509–25.e19. doi: 10.1016/j.cell.2019.08.009
208. Terrazas C, Varikuti S, Oghumu S, Steinkamp HM, Ardic N, Kimble J, et al. Ly6C(hi) Inflammatory Monocytes Promote Susceptibility to Leishmania Donovani Infection. Sci Rep (2017) 7(1):14693. doi: 10.1038/s41598-017-14935-3
209. Abidin BM, Hammami A, Stager S, Heinonen KM. Infection-Adapted Emergency Hematopoiesis Promotes Visceral Leishmaniasis. PloS Pathog (2017) 13(8):e1006422. doi: 10.1371/journal.ppat.1006422
210. Costa DL, Lima-Junior DS, Nascimento MS, Sacramento LA, Almeida RP, Carregaro V, et al. CCR2 Signaling Contributes to the Differentiation of Protective Inflammatory Dendritic Cells in Leishmania Braziliensis Infection. J Leukoc Biol (2016) 100(2):423–32. doi: 10.1189/jlb.4A0715-288R
211. Heyde S, Philipsen L, Formaglio P, Fu Y, Baars I, Hobbel G, et al. CD11c-Expressing Ly6C+CCR2+ Monocytes Constitute a Reservoir for Efficient Leishmania Proliferation and Cell-to-Cell Transmission. PloS Pathog (2018) 14(10):e1007374. doi: 10.1371/journal.ppat.1007374
212. Lee SH, Chaves MM, Kamenyeva O, Gazzinelli-Guimaraes PH, Kang B, Pessenda G, et al. M2-Like, Dermal Macrophages Are Maintained via IL-4/CCL24-Mediated Cooperative Interaction With Eosinophils in Cutaneous Leishmaniasis. Sci Immunol (2020) 5(46):eaaz4415. doi: 10.1126/sciimmunol.aaz4415
213. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
214. Carneiro MB, Vaz LG, Afonso LCC, Horta MF, Vieira LQ. Regulation of Macrophage Subsets and Cytokine Production in Leishmaniasis. Cytokine (2020) 147:155309. doi: 10.1016/j.cyto.2020.155309
215. Wakil AE, Wang ZE, Ryan JC, Fowell DJ, Locksley RM. Interferon Gamma Derived From CD4(+) T Cells Is Sufficient to Mediate T Helper Cell Type 1 Development. J Exp Med (1998) 188(9):1651–6. doi: 10.1084/jem.188.9.1651
216. Peters NC, Pagan AJ, Lawyer PG, Hand TW, Henrique Roma E, Stamper LW, et al. Chronic Parasitic Infection Maintains High Frequencies of Short-Lived Ly6C+CD4+ Effector T Cells That are Required for Protection Against Re-Infection. PloS Pathog (2014) 10(12):e1004538. doi: 10.1371/journal.ppat.1004538
217. Qadoumi M, Becker I, Donhauser N, Rollinghoff M, Bogdan C. Expression of Inducible Nitric Oxide Synthase in Skin Lesions of Patients With American Cutaneous Leishmaniasis. Infect Immun (2002) 70(8):4638–42. doi: 10.1128/iai.70.8.4638-4642.2002
218. Iniesta V, Carcelen J, Molano I, Peixoto PM, Redondo E, Parra P, et al. Arginase I Induction During Leishmania Major Infection Mediates the Development of Disease. Infect Immun (2005) 73(9):6085–90. doi: 10.1128/IAI.73.9.6085-6090.2005
219. Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-Regulated Expression of Arginase Isoforms in Murine Macrophages and Dendritic Cells. J Immunol (1999) 163(7):3771–7.
220. Bogdan C, Vodovotz Y, Nathan C. Macrophage Deactivation by Interleukin 10. J Exp Med (1991) 174(6):1549–55. doi: 10.1084/jem.174.6.1549
221. Kima PE, Constant SL, Hannum L, Colmenares M, Lee KS, Haberman AM, et al. Internalization of Leishmania Mexicana Complex Amastigotes via the Fc Receptor Is Required to Sustain Infection in Murine Cutaneous Leishmaniasis. J Exp Med (2000) 191(6):1063–8. doi: 10.1084/jem.191.6.1063
222. Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, et al. The Role of Interleukin (IL)-10 in the Persistence of Leishmania Major in the Skin After Healing and the Therapeutic Potential of Anti-IL-10 Receptor Antibody for Sterile Cure. J Exp Med (2001) 194(10):1497–506. doi: 10.1084/jem.194.10.1497
223. Mosser DM, Zhang X. Interleukin-10: New Perspectives on an Old Cytokine. Immunol Rev (2008) 226:205–18. doi: 10.1111/j.1600-065X.2008.00706.x
224. Hu X, Ivashkiv LB. Cross-Regulation of Signaling Pathways by Interferon-Gamma: Implications for Immune Responses and Autoimmune Diseases. Immunity (2009) 31(4):539–50. doi: 10.1016/j.immuni.2009.09.002
225. Kane MM, Mosser DM. The Role of IL-10 in Promoting Disease Progression in Leishmaniasis. J Immunol (2001) 166(2):1141–7. doi: 10.4049/jimmunol.166.2.1141
226. Yang Z, Mosser DM, Zhang X. Activation of the MAPK, ERK, Following Leishmania Amazonensis Infection of Macrophages. J Immunol (2007) 178(2):1077–85. doi: 10.4049/jimmunol.178.2.1077
227. Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(-)Foxp3(-) Th1 Cells Are the Source of IL-10-Mediated Immune Suppression in Chronic Cutaneous Leishmaniasis. J Exp Med (2007) 204(2):285–97. doi: 10.1084/jem.20061886
228. Owens BM, Beattie L, Moore JW, Brown N, Mann JL, Dalton JE, et al. IL-10-Producing Th1 Cells and Disease Progression Are Regulated by Distinct CD11c(+) Cell Populations During Visceral Leishmaniasis. PloS Pathog (2012) 8(7):e1002827. doi: 10.1371/journal.ppat.1002827
229. Jones DE, Ackermann MR, Wille U, Hunter CA, Scott P. Early Enhanced Th1 Response After Leishmania Amazonensis Infection of C57BL/6 Interleukin-10-Deficient Mice Does Not Lead to Resolution of Infection. Infect Immun (2002) 70(4):2151–8. doi: 10.1128/IAI.70.4.2151-2158.2002
230. Ji J, Sun J, Qi H, Soong L. Analysis of T Helper Cell Responses During Infection With Leishmania Amazonensis. Am J Trop Med Hyg (2002) 66(4):338–45. doi: 10.4269/ajtmh.2002.66.338
231. Scharton TM, Scott P. Natural Killer Cells Are a Source of Interferon Gamma That Drives Differentiation of CD4+ T Cell Subsets and Induces Early Resistance to Leishmania Major in Mice. J Exp Med (1993) 178(2):567–77. doi: 10.1084/jem.178.2.567
232. Schleicher U, Paduch K, Debus A, Obermeyer S, Konig T, Kling JC, et al. TNF-Mediated Restriction of Arginase 1 Expression in Myeloid Cells Triggers Type 2 NO Synthase Activity at the Site of Infection. Cell Rep (2016) 15(5):1062–75. doi: 10.1016/j.celrep.2016.04.001
233. Paduch K, Debus A, Rai B, Schleicher U, Bogdan C. Resolution of Cutaneous Leishmaniasis and Persistence of Leishmania Major in the Absence of Arginase 1. J Immunol (2019) 202(5):1453–64. doi: 10.4049/jimmunol.1801249
234. Afonso LC, Scott P. Immune Responses Associated With Susceptibility of C57BL/10 Mice to Leishmania Amazonensis. Infect Immun (1993) 61(7):2952–9. doi: 10.1128/iai.61.7.2952-2959.1993
235. Cortes DF, Carneiro MB, Santos LM, Souza TC, Maioli TU, Duz AL, et al. Low and High-Dose Intradermal Infection With Leishmania Major and Leishmania Amazonensis in C57BL/6 Mice. Mem Inst Oswaldo Cruz (2010) 105(6):736–45. doi: 10.1590/S0074-02762010000600002
236. Pinheiro RO, Rossi-Bergmann B. Interferon-Gamma Is Required for the Late But Not Early Control of Leishmania Amazonensis Infection in C57Bl/6 Mice. Mem Inst Oswaldo Cruz (2007) 102(1):79–82. doi: 10.1590/S0074-02762007000100013
237. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-Gamma: An Overview of Signals, Mechanisms and Functions. J Leukoc Biol (2004) 75(2):163–89. doi: 10.1189/jlb.0603252
238. Sallusto F, Lanzavecchia A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells is Maintained by Granulocyte/Macrophage Colony-Stimulating Factor Plus Interleukin 4 and Downregulated by Tumor Necrosis Factor Alpha. J Exp Med (1994) 179(4):1109–18. doi: 10.1084/jem.179.4.1109
239. Chomarat P, Dantin C, Bennett L, Banchereau J, Palucka AK. TNF Skews Monocyte Differentiation From Macrophages to Dendritic Cells. J Immunol (2003) 171(5):2262–9. doi: 10.4049/jimmunol.171.5.2262
240. Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 Switches the Differentiation of Monocytes From Dendritic Cells to Macrophages. Nat Immunol (2000) 1(6):510–4. doi: 10.1038/82763
241. Delneste Y, Charbonnier P, Herbault N, Magistrelli G, Caron G, Bonnefoy JY, et al. Interferon-Gamma Switches Monocyte Differentiation From Dendritic Cells to Macrophages. Blood (2003) 101(1):143–50. doi: 10.1182/blood-2002-04-1164
242. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-Producing Dendritic Cells Mediate Innate Immune Defense Against Bacterial Infection. Immunity (2003) 19(1):59–70. doi: 10.1016/S1074-7613(03)00171-7
243. Muller AJ, Filipe-Santos O, Eberl G, Aebischer T, Spath GF, Bousso P. CD4+ T Cells Rely on a Cytokine Gradient to Control Intracellular Pathogens Beyond Sites of Antigen Presentation. Immunity (2012) 37(1):147–57. doi: 10.1016/j.immuni.2012.05.015
244. Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, et al. Differential Expression of Chemokine Receptors and Chemotactic Responsiveness of Type 1 T Helper Cells (Th1s) and Th2s. J Exp Med (1998) 187(1):129–34. doi: 10.1084/jem.187.1.129
245. Vargas-Inchaustegui DA, Hogg AE, Tulliano G, Llanos-Cuentas A, Arevalo J, Endsley JJ, et al. CXCL10 Production by Human Monocytes in Response to Leishmania Braziliensis Infection. Infect Immun (2010) 78(1):301–8. doi: 10.1128/IAI.00959-09
246. Gaylo-Moynihan A, Prizant H, Popovic M, Fernandes NRJ, Anderson CS, Chiou KK, et al. Programming of Distinct Chemokine-Dependent and -Independent Search Strategies for Th1 and Th2 Cells Optimizes Function at Inflamed Sites. Immunity (2019) 51(2):298–309.e6. doi: 10.1016/j.immuni.2019.06.026
247. Liu Z, Yang F, Zheng H, Fan Z, Qiao S, Liu L, et al. Visualization of T Cell-Regulated Monocyte Clusters Mediating Keratinocyte Death in Acquired Cutaneous Immunity. J Invest Dermatol (2018) 138(6):1328–37. doi: 10.1016/j.jid.2018.01.018
Keywords: Leishmania, Leishmania amazonensis, neutrophils, monocytes, macrophages, dendritic cells, phagolysosome
Citation: Carneiro MB and Peters NC (2021) The Paradox of a Phagosomal Lifestyle: How Innate Host Cell-Leishmania amazonensis Interactions Lead to a Progressive Chronic Disease. Front. Immunol. 12:728848. doi: 10.3389/fimmu.2021.728848
Received: 22 June 2021; Accepted: 10 August 2021;
Published: 07 September 2021.
Edited by:
Esther Von Stebut, University of Cologne, GermanyReviewed by:
Salvador Iborra, Universidad Complutense de Madrid, SpainCopyright © 2021 Carneiro and Peters. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matheus B. Carneiro, bWF0aGV1cy5iYXRpc3RhaGVpdG9AdWNhbGdhcnkuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.