 Zhuqing Jin
Zhuqing Jin En Zheng2
En Zheng2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 October 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.727861
This article is part of the Research TopicMolecular and Cellular Effectors in the Resolution of InflammationView all 33 articles
Inflammatory response is a host-protective mechanism against tissue injury or infections, but also has the potential to cause extensive immunopathology and tissue damage, as seen in many diseases, such as cardiovascular diseases, neurodegenerative diseases, metabolic syndrome and many other infectious diseases with public health concerns, such as Coronavirus Disease 2019 (COVID-19), if failure to resolve in a timely manner. Recent studies have uncovered a superfamily of endogenous chemical molecules that tend to resolve inflammatory responses and re-establish homeostasis without causing excessive damage to healthy cells and tissues. Among these, the monocyte chemoattractant protein-induced protein (MCPIP) family consisting of four members (MCPIP-1, -2, -3, and -4) has emerged as a group of evolutionarily conserved molecules participating in the resolution of inflammation. The focus of this review highlights the biological functions of MCPIP-1 (also known as Regnase-1), the best-studied member of this family, in the resolution of inflammatory response. As outlined in this review, MCPIP-1 acts on specific signaling pathways, in particular NFκB, to blunt production of inflammatory mediators, while also acts as an endonuclease controlling the stability of mRNA and microRNA (miRNA), leading to the resolution of inflammation, clearance of virus and dead cells, and promotion of tissue regeneration via its pleiotropic effects. Evidence from transgenic and knock-out mouse models revealed an involvement of MCPIP-1 expression in immune functions and in the physiology of the cardiovascular system, indicating that MCPIP-1 is a key endogenous molecule that governs normal resolution of acute inflammation and infection. In this review, we also discuss the current evidence underlying the roles of other members of the MCPIP family in the regulation of inflammatory processes. Further understanding of the proteins from this family will provide new insights into the identification of novel targets for both host effectors and microbial factors and will lead to new therapeutic treatments for infections and other inflammatory diseases.
Inflammatory response is an immunological defense mechanism of the host to infections and tissue damage (e.g., ischemic insults) or stress (1). Damaged or stressed cells are thought to release danger-associated molecular patterns (DAMPs) that can trigger the innate immune system in the same manner as microbial components, the so-called pathogen-associated molecular patterns (PAMPs), by binding to host pattern-recognition receptors (PRRs) presented on various cells, including immune cells (T-cells, B-cells and NK cells) and the tissue cells such as endothelial cells, cardiomyocytes, and even neurons (1). This recognition triggers a series of signaling cascades that initiate the activation of transcriptional factor nuclear factor-κB (NFκB) and NLRP3 inflammasome, leading to the release of inflammatory mediators, like tumor necrosis factor-alpha (TNFα), monocyte chemoattractant protein-1 (MCP-1), interleukin-1 (IL-1) and IL-6 (2, 3). The inflammatory response is beneficial to the host by eliminating the harmful agents (PAMPs or DAMPs) and is usually resolved in a timely manner; however, failure to resolve can cause excessive or persistent inflammation that is often disruptive and can cause marked tissue damage (4).
The resolution of inflammation is a programmed active process that involves the biosynthesis of a variety of active molecules that act on key events of inflammatory response to terminate the production of pro-inflammatory mediators and restore tissue homeostasis (5). There is an increasing body of evidence that many pro-inflammatory mediators produced during the inflammatory phase can simultaneously initiate a program for active resolution (6). For example, TNFα can effectively induce A20 (also known as tumor necrosis factor-α-induced protein 3 or TNFAIP3), an ubiquitin editing enzyme that negatively regulates the inflammatory response by interfering with NFκB signaling pathway, leading to the resolution of inflammation (7). The newly synthesized molecules not only act as signals for the termination of the inflammatory response, but also promote the clearance of dead cells to accelerate the resolution of inflammation (4, 8).
A major contribution of our group to the field was the discovery of the novel zinc-finger protein, named MCP-1–induced protein (MCPIP), which was originally detected in MCP-1 treated human peripheral blood monocytes (9, 10). Subsequent studies demonstrated that MCPIP belongs to a new Zc3h12 family consisting of four members (MCPIP-1, -2, -3, and -4) that are encoded by Zc3h12a, Zc3h12b, Zc3h12c, and Zc3h12d, respectively (11). MCPIP-1 is the most-studied protein that contains an N-terminal domain, a PilT N-terminus like (PIN) domain, a zinc finger domain, and a C-terminal domain (12). Emerging evidence indicates that MCPIP-1 plays an essential role in the regulation of inflammatory response, with additional roles in defense against viruses and various stresses, cellular differentiation, and apoptosis (13, 14), all of these are key cellular and molecular components that contribute to the successful resolution of inflammation (9, 10, 15–18). The focus of this review is to present evidence illustrating that the role played by MCPIP-1 is important in the resolution of inflammation initiated by virus infections or ischemic injuries and highlight recent advances on the actions of this protein and its potential clinical significance. We also discuss the available evidence regarding the role of other members (MCPIP-2, -3, and -4) from this family in the regulation of inflammatory processes.
MCPIP-1 was originally identified in human peripheral blood monocytes stimulated with MCP-1 (9). Subsequent studies demonstrated that MCPIP-1 is produced in many other cell types, either constitutively or after induction by a wide range of stimuli, such as inflammatory cytokines (e.g., IL-1β, IL-17 and TNFα) and oxidative stress (13, 19, 20). Further studies demonstreated that the expression of MCPIP-1 can be induced by ischemia in the heart and the brain (21, 22). Infections by virus such as hepatitis C virus (HCV), hepatitis B virus (HBV), influenza A virus (IAV), Japanese encephalitis virus (JEV) and Dengue virus as well as by bacteria and fungal increase the expression of MCPIP-1 (23–25). The extracellular high mobility group box 1 (HMGB1), a non-histone DNA-binding protein released from dying cells in response to tissue injuries, also increases microglium expression of MCPIP-1 that negatively regulates HMGB-1-mediated neuroinflammation and neuronal toxicity (26). Minocycline, a member of tetracycline antibiotics with anti-inflammatory properties, also induces MCPIP-1 expression in the heart and the brain (27, 28).
Although the molecular mechanisms responsible for MCPIP-1 expression are still poorly understood, MCP-1 binding to its cognate receptor CCR2 was thought to activate ERK or AKT pathways, leading to the expression of MCPIP-1 (9). Activation of NFκB signaling was suggested to induce the expresion of MCPIP-1 by inflammatory cytokines such as IL-1β (29). Transcription factors Elk-1 and SRF were also reported to mediate IL-1-dependent expression of MCPIP-1 by binding to the promoter region of MCPIP-1 (30). Activation of JAK/STAT3 signaling was also reported to mediate MCPIP-1 expression in epithelial cells (31). At the post-transcriptional level, MCPIP-1 mRNA was found to be downregulated by miR-9 in LPS-activated microglial cells (32). MCPIP-1 also cleaves its own transcript (33, 34). Moreover, the translated MCPIP-1 protein can be phosphorylated by IκB kinase (IKK) β and then undergoes ubiquitination and degradation (35) or cleaved by the paracaspase Malt-1 (33, 36). These data indicate that MCPIP-1expression is tightly controlled by an autoregulatory feedback mechanism, which ensures an appropriate level of MCPIP-1 aimed to minimize any disruption of immune homeostasis.
MCPIP-1 was first described as a transcriptional activator owing to the structural feature of a potential DNA binding zinc finger domain (9). Subsequent studies indicate that MCPIP is localized to both the cytoplasmic and nuclear compartments, depending on the distinct functions it plays in different cell types (11, 37, 38). Mutational analysis of MCPIP-1 has identified the two regions of the primary structure that are critical for its biological activity (12, 39–41). The first region consists of the ubiquitin-associated domain from 43-89, which is associated with the control of protein ubiquitination; whereas the second region consists of PIN domain from 133–270, which is associated with RNA-cleaving function and is why it was later renamed as Regnase-1 (regulatory RNase-1) (40, 42). In addition to these two regions, the C-terminal region of the NYN domain is crucial for suppressing microRNA (miRNA) biogenesis via cleavage of the terminal loops of precursor miRNAs (16, 40–44).
Ubiquitination has emerged as a crucial mechanism that regulates signal transduction in the inflammatory response (3, 45). The use of MCPIP-1-deficient mice has revealed the crucial role of MCPIP-1 in the regulation of inflammatory cytokine signaling pathways. Mice lacking MCPIP-1 are normal at birth but suffer growth retardation and die prematurely due to massive multi-organ inflammation, indicative of a key role for MCPIP in immune homeostasis of the host (41). Macrophages from MCPIP-1-deficient mice showed up-regulation of pro-inflammatory mediators together with a greatly increased ubiquitination of TRAFs (TNF receptor–associated factors) and the receptor-interacting protein (RIP) kinases, both of which play a central role in the LPS-, IL-1β- and TNF-induced activation of NFκB signaling pathway (46). The purified MCPIP-1 protein was shown to cleave K48- or K63-linked polyubiquitin chain, while this action of MCPIP-1 was inhibited by N-ethyl maleimide, a known inhibitor of cysteine proteinases (41). Consistent with this finding, treatment of high-molecular-weight K63-linked polyubiquitin with purified MCPIP-1 caused hydrolysis of the polyubiquitin, leading to the inhibition of phosphorylation of TAK1 (47), a critical factor for activation of the downstream kinase IKK, thereby mediating IκBα phosphorylation and NFκB activation (48). Accordingly, deletion of the ubiquitin association domain of MCPIP-1 resulted in the loss of inhibition of TNFα-induced NFκB activation (41). These results indicate that MCPIP-1 can act as a deubiquitinase to hydrolyze K63-, K48-linked polyubiquitin chains and inhibits NFκB transcriptional activity, thus contributing to suppression of the pro-inflammatory response (Figure 1). MCPIP-1 has also been reported to stabilize NFκB essential modulator by promoting deubiquitination, resulting in subsequent inhibition of NFκB activation induced by DNA damage (49). MCPIP-1 was also shown to stabilize the hypoxia-inducible factor 1alpha protein that is required for macrophage maturation under hypoxic conditions, in which deubiquitination plays a key regulatory role (50).
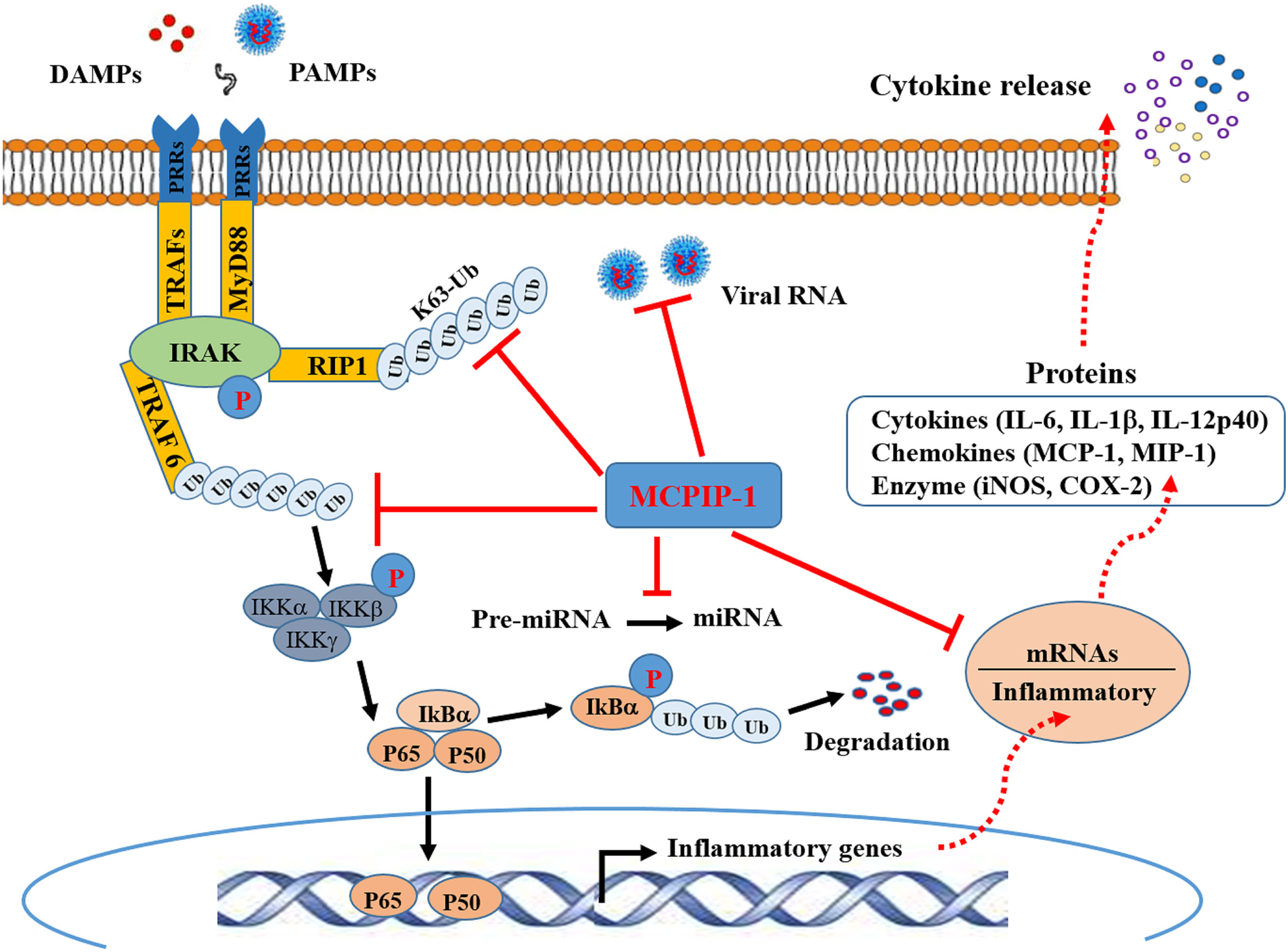
Figure 1 Schematic representation of anti-inflammatory activity of MCPIP-1. Binding of molecules (DAMPs) derived from tissue damage or pathogens (PAMPs) to PRRs triggers interactions between the cytoplasmic adaptor proteins and the kinases IRAK. This engages the ubiquitin ligase TRAF6 to make polyubiquitin chains that activate the IKK complex, leading to phosphorylation and subsequent ubiquitination IKBα. This releases P50/p65 dimer for entry into the nucleus to cause transcriptional activation of NFκB-dependent genes encoding inflammatory cytokines. Ubiquitylation of RIP1, and potentially other components of the complex, recruits IKKγ and TAK1 for NFκB and MAPK activation (not shown). MCPIP-1 hydrolyzes all of these K48- and K63-linked polyubiquitins to block NFκB activation. The RNase activity of MCPIP-1 also degrades viral RNA and some mRNAs encoding for inflammatory cytokines, leading to dampening of protein expression of the inflammatory cytokines. The anti-Dicer activity of MCPIP-1 can cleave the terminal loops of pre-miRNAs leading to destabilization of pre-miRNAs and suppression of the miRNA biogenesis.
MCPIP-1 can also act as an RNase to regulate mRNA stability (19, 33, 40, 51). MCPIP-1 can directly bind the 3′-untranslated region (UTR) of IL-6 mRNA and manifested RNase activity to degrade IL-6 transcripts (40). A similar finding was demonstrated in the expression of IL-1β mRNA that was degraded by the increased level of MCPIP-1 in HepG2 and U937 cells (19). Genome-wide association studies showed that the PIN domain of MCPIP-1 contains the RNase catalytic center that requires an interaction with the N-terminus for its full RNase activity and the zinc-finger domain is responsible for the recognition and direct binding of the mRNAs (12, 39, 51). With the cooperation of its domains, MCPIP-1 recognizes and degrades target mRNAs by recognizing stem-loop structures at the 3′-UTRs of these genes (40, 51). In T cells, MCPIP-1 downregulates a set of genes by cooperating with roquin, another CCCH type zinc finger protein (36, 51). However, a recent study in other type of cells indicates that, although roquin and MCPIP-1 control shared mRNAs, they do so in different mechanisms within different subcellular compartments (51). Thus, the co-operation between MCPIP-1 and roquin remains incompletely understood. In different cell types, MCPIP-1 recognizes and degrades target mRNAs may be cell-specific.
To date, MCPIP-1-dependent degradation of inflammatory mRNAs has been increasingly identified. In addition to IL-6 and IL-1β, mRNAs encoding for IL-2, IL-12 and IL-17 have been identified as direct targets of MCPIP-1 (51, 52). Both CXCL1 and CXCL2, two important chemokines contributing to early stage neutrophil recruitment during tissue inflammation (53), are also direct targets of MCPIP-1 (51). MCPIP-1 also degrades mRNAs encoding T-cell co-stimulatory receptors such as ICOS, TNFR2 and OX40 as well as T-cell activation marker CD44 (33), all of which play a key role in permitting T cell mature and activation. Importantly, MCPIP-1 degrades mRNA encoding for the anti-apoptotic immediate early response 3 (IER3) protein (54), resulting in apoptosis of macrophages that contributes to resolution of inflammation. Thus, MCPIP-1 controls inflammatory response not only by preventing the transcription of the inflammatory cytokines, but also by dampening of the protein expression of the inflammatory cytokines at the post-transcriptional level as well (Figure 1).
Emerging studies have shown that miRNAs modulate many aspects of the immune responses such as proliferation, differentiation, cell fate determination, immune cell function, and cytokine responses (55, 56). when miRNAs are aberrantly expressed they contributes to the pathogenesis of inflammatory and autoimmune diseases by regulating their cellular and molecular targets (55). Besides targeting mRNA, MCPIP-1 was shown to regulate miRNA biogenesis by counteracting Dicer, a central ribonuclease in miRNA biosynthesis (42, 57). MCPIP-1 can cleave the terminal loop of pre-miRNAs, thereby inhibiting their maturation (41). Studies have shown that miRNA-146a and miRNA-155 are specifically down-regulated by MCPIP-1 (42, 58). Both miR-146a and miR-155 have been proposed to regulate the macrophage activation by forming a combined negative and positive regulatory loop that alters NFκB activity (59). miR-155 is highly transcribed upon an inflammatory stimulus, which can amplify NFκB activity, while as an inflammatory response develops, miR-146a levels accumulate, which causes suppression of IRAK1 and TRAF6, leading to the inhibition of NFκB activation (59). However, a study by Mino et al. indicated that expression of both miR-155 and miR-146 was not altered in mouse embryonic fibroblasts from MCPIP-1-deficient mice (51). Therefore, MCPIP appears to utilize distinct mechanisms to keep the inflammatory signaling suppressed and to re-establish immune hemostasis (Figure 1).
Apoptosis is an evolutionarily conserved cell death program that is tightly regulated by the Bcl-2 family of proteins, which contains both pro-apoptotic and pro-survival members that balance the decision between cellular life and death (60). Microarray analysis revealed that MCPIP-1 upregulates the pro-apoptotic genes and downregulates the anti-apoptotic genes in the myocardium (9). Along this line, MCPIP-1 was found to mediate endothelial cell apoptosis and dysfunction upon MCP-1 treatment (61). The pro-apoptotic activity of MCPIP-1 was further documented in vitro assays in HEK 293 cells (9), H9c2 cardiomyoblasts (62), neonatal rat cardiomyocytes (63), macrophages (64, 65), neutrophils (66), T cells (67), and even cancer cells (68). Mechanistically, MCPIP-1 selectively binds and cleaves the mRNAs of anti-apoptotic genes, such as Bcl-2A1, Bcl-2L1, and RELB, leading to down-regulation of anti-apoptotic proteins and upregulation of pro-apoptotic proteins (66, 68). The inhibition of miRNA biogenesis by MCPIP-1 is also linked to its pro-apoptotic activity. MCPIP-1 has been shown to downregulate miR-3613-3p expression in neuroblastoma cells, which in turn upregulates apoptotic protease activating factor 1, causing apoptosis by caspase-9 proteolysis (69). The pro-apoptotic activity of MCPIP-1 was found to be associated with its influence on the formation of stress granules (SGs), one kind of non-membranous ribonucleoprotein complexes containing untranslated mRNA formed in response to stress exposure (70). MCPIP-1 can completely block SG formation and promote macrophage apoptosis (71).
It is well documented that the amount of reactive oxygen species (ROS) produced and the extent of oxidative stress in a cell determine the fate of the cell to die or survive (72). The pro-apoptotic activity of MCPIP-1 was strongly correlated with its ability to induce intracellular ROS that cause endoplasmic reticulum (ER) stress, resulting in autophagy and apoptosis in cardiomyocytes (62, 63), macrophages (64, 73), endothelial cells (74, 75), and in renal cell carcinoma (76). Deubiquitination of RIP1 by CYLD or A20 has been suggested to facilitate cell death (77, 78). As a new member of the deubiquitinase family, however, the role of the deubiquitinating activity of MCPIP-1 in cell death remains to be determined. While it has been reported that MG-132, a proteasome inhibitor, effectively upregulates MCPIP-1 expression, potently activating the apoptosis of cancer cells (79). Further research is needed to fully understand its significance in regulating cell death.
Resolution of inflammation is a coordinated and active process that involves the suppression of pro-inflammatory reaction, apoptosis and subsequent clearance of activated inflammatory cells, and repolarization of macrophages towards a resolving phenotype aimed at restoration of tissue integrity and function (5, 6). As mentioned above, the functions of MCPIP-1 support the notion that MCPIP-1 has critical roles in restricting inflammation (Figure 2). In the following we present the experimental findings to provide an overview of MCPIP-1 that drives these resolution processes.
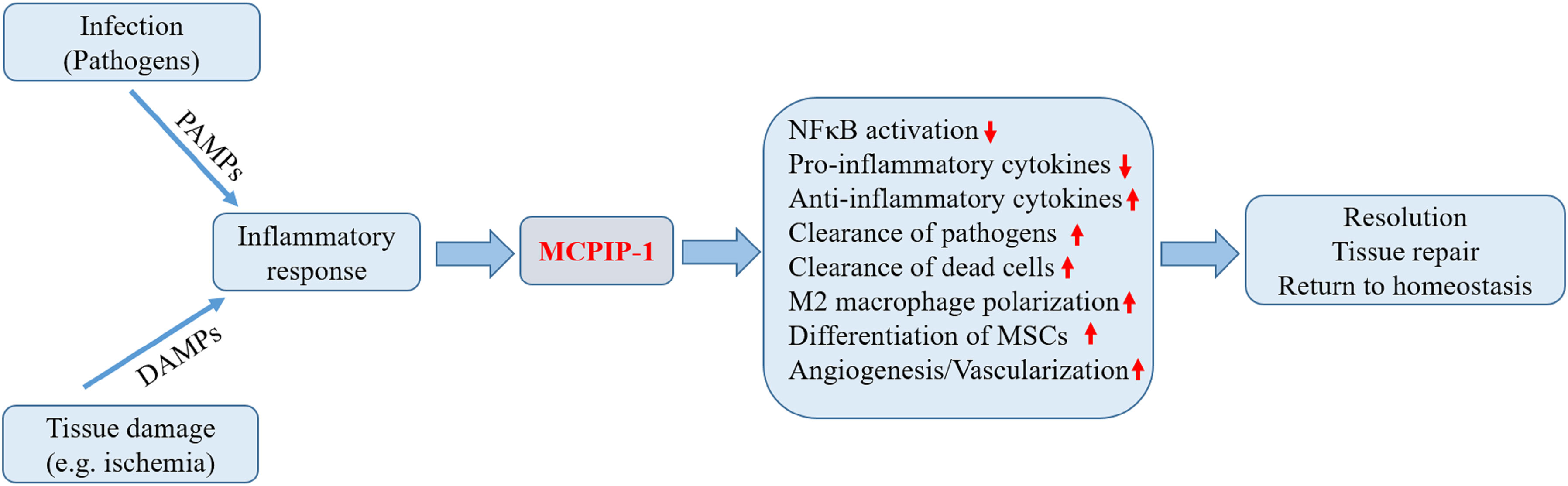
Figure 2 Biological roles of MCPIP-1 associated with resolution of inflammation. During the early phase of inflammation, inflammatory response initiated by infection or tissue injury activates the endogenous defense mechanisms aimed to bring about proper resolution. The expression of MCPIP-1 induced by the inflammatory response modulates a wide range of cellular and molecular events associated with the resolution of inflammation. The expression of MCPIP-1 results in the suppression of NFκB activation and reduced pro-inflammatory cytokines. MCPIP-1 is able to induce apoptosis of infected or damaged cells, leading to their clearance by macrophages. In addition, MCPIP-1 promotes M2 macrophage polarization and enhances angiogenic differentiation of mesenchymal stem cells (MSCs). These events will create a favorable environment contributing to the resolution and tissue homeostasis.
Acute inflammation occurs in response to pathogen infection. This process involves the activation of innate responses that enhance innate microbial killing and clearance to protect organ structure and function (80). MCPIP-1 has been evident in cells infected with various viruses and exerts antiviral activity (23–25, 81, 82). MCPIP-1 can distinguish mRNAs from the host genome and can selectively degrade foreign mRNAs through its RNase activity (83). Indeed, MCPIP-1 is activated upon viral infections and has been shown to restrict virus replication by directly binding and subsequently degrading viral RNAs, such as JEV, IAV, DEN, coxsackievirus B3, encephalomyocarditis virus, and HIV (23, 81, 84). The deubiquitinase activity has not been shown to play a role in the antiviral effect of MCPIP-1, even though ubiquitination has been implicated in virus replication (85, 86). MCPIP-1 also restricts HCV replication by directly degrading HCV RNA and inhibits HCV-mediated expression of pro-inflammatory response (23, 24). In patients with chronic hepatitis C, the expression of MCPIP-1 in the liver has been suggested to have a protective role in antiviral responses (87). Type I interferons (IFNs) are recognized as the first line of defense against viral infection (88). MCPIP-1 has been proven to be a positive feedback amplifier of IFN signaling and promotes innate antiviral immunity independently of its RNase and deubiquitinase activities (82). Therefore, MCPIP-1 seems to be a very promising target for antiviral infections by degrading viral genomes, suppressing virus-mediated expression of pro-inflammatory cytokines or through the induction of antiviral effector molecules, resulting in the resolution of inflammation. These findings also reveal an exciting possibility for MCPIP-1 protecting against the infection caused by the SARS-CoV-2, an RNA virus that may cause severe acute respiratory distress syndrome due to “cytokine storms” induced by a hyper-activation of inflammatory cytokine response (89, 90).
Inflammatory response represents one of the first immune processes following ischemic injury, which is usually self-limiting, followed by tissue repair and healing responses (91, 92). Ischemic preconditioning is a well-established phenomenon, in which brief episodes of sub-lethal ischemia and reperfusion elicit strong cellular protection against the subsequently sustained ischemia in the heart and the brain (93, 94). There is increasing evidence that ischemic preconditioning induces a powerful anti-inflammatory response (94–96), which has been well illustrated by ‘endotoxin tolerance’ and is thought to be an adaptive response conferring protection by suppression of the hyper-activation of the innate immune system through auto-regulatory network of cytokines (94, 97). Preconditioning stimuli with low doses of LPS, a primary ligand for TLR4, provides protection against subsequent challenges with injurious focal ischemia in the brain that is similar to ischemic preconditioning (98). Diminished activation of cellular inflammatory responses to ischemia by LPS preconditioning has been suggested to play an important role for protection against ischemic injury (98, 99). In a mouse model of middle cerebral artery (MCA) occlusion, LPS induces upregulation of MCPIP-1 in the brain and the neuroprotection offered by LPS preconditioning was diminished due to MCPIP-1 deficiency, suggesting MCPIP-1 expression initiated by LPS preconditioning may be an intrinsic cellular defense mechanism against the sustained ischemic injury (100). Consistently, mice lacking MCPIP-1 showed an enhanced leakage of blood–brain barrier, increased production of cytokines and a larger infarct volume after MCA occlusion (21, 101). This scenario was further demonstrated in the heart, where cardiac-specific expression of MCPIP-1 protected against LPS-induced myocardial inflammation and dysfunction (44). Consistently, mice with cardiac-specific expression of MCPIP-1 showed cardioprotective effects against myocardial infarction, as evidenced by the improved cardiac function, mitigated interstitial myocardial fibrosis, increased apoptosis of inflammatory cells, and decreased myocardial inflammation (22). In cardiomyocytes, NFκB activity was increased in response to LPS but suppressed by forced expression of MCPIP-1, thus linking MCPIP-1 to the suppression of myocardial inflammation in response to cardiac stress (22, 44). Consistent with this findings, NFκB activity initiated by myocardial infarction was inhibited by forced expression of MCPIP-1, suggesting that the preconditioning-like effects of MCPIP-1 probably involve its ability to inhibit NFκB activation (22, 44). The murine hearts expressing MCPIP-1 also displayed lower expression of inflammation-associated miR-126,-146,-155 and -199a when compared to those seen in the wild type mice (22). Preconditioning by minocycline is a pharmacological alternative to ischemic preconditioning, which enhances neuroprotection after ischemic stroke (102). Minocycline preconditioning inhibits the inflammatory cytokine response to ischemia through preferential induction of MCPIP-1, and the protective role of minocycline was diminished in the MCPIP-1-deficient mice subjected to focal cerebral ischemia/reperfusion (I/R) injury (27). Similar findings were observed in a mouse model of myocardial I/R injury, in which minocycline attenuated myocardial I/R injury via upregulating MCPIP-1 that subsequently inhibited NFκB activation and pro-inflammatory cytokine secretion (28). These findings are in agreement with the previous reports that MCPIP-1 deficiency results in massive multi-organ inflammation and premature death in mice (40, 41, 43).
The mechanisms underlying the preconditioning-like cellular protection by MCPIP-1 need further investigation. MCPIP-1 is likely to alter the immune responses to the ischemic insults by limiting pro-inflammatory cytokine transcription, dampening of protein expression of pro-inflammatory cytokines, regulating synthesis of pro-inflammatory miRNAs or enhancing clearance of the infiltrated inflammatory cells (44, 100). The combination of these mechanisms may result in an effective resolution of inflammation, pointing to MCPIP-1 as a promising target for the development of new therapeutic strategies to treat post-ischemic inflammation, and therefore warrant future studies on the molecular mode of action of MCPIP-1 in inflammatory diseases.
Resolution of inflammation is a coordinated process that requires a tight interplay between macrophages, stem and progenitor cells, together with stromal cells to restoration of tissue integrity and function (5, 6). Beyond its anti-inflammatory activity, MCPIP-1 has also shown some beneficial pleiotropic effects, contributing to the resolution of inflammation and the restoration of tissue homeostasis.
MCPIP-1 was shown to induce angiogenesis by promoting the migration and apoptosis of human umbilical vein endothelial cells (HUVECs) and the expression of angiogenesis-related gene CDH12 and CDH19 (103). Moreover, MCPIP-1 inhibits the production of anti-angiogenetic miR-20b and miR-34a, which repress the translation of HIF-1α and SIRT-1 respectively, leading to promoting angiogenesis in the HUVECs (50). These findings agree with the animal data showing that forced expression of MCPIP-1 induces angiogenesis of bone marrow monocytic cells and accelerates post-ischemic neovascularization (104). Mesenchymal stem cells (MSCs) are candidates for cellular therapies aimed at promoting tissue repair or immunoregulation (105). MCPIP-1 was shown to increase angiogenic and cardiac differentiation capacity of bone marrow-derived MSCs, contributing to repair and regeneration of ischemic myocardium (106). Vascular endothelial and smooth muscle cells play critical roles in the stability and tonic regulation of vascular homeostasis. MCPIP-1 was shown to regulate the phenotypic switching of both endothelial and smooth muscle cells via suppression of synthesis of miRNAs, such as miR-126, 145, -146a, and -223 (43, 107). However, Marona et al. reported that MCPIP-1 reduces tumor vascularity in clear cell renal cell carcinoma by inhibiting the recruitment of bone marrow-derived endothelial progenitor cells (EPCs) and phosphorylation of VE-cadherin via the degradation of mRNAs encoding for IL8, VEGF and CXCL12 (108).
A key event required for resolution of inflammation is efferocytosis of apoptotic and necrotic cells, mostly by macrophages acquiring an alternative M2 phenotype (109). We reported the ability of MCPIP-1 to control macrophage reprogramming toward a M2 phenotype, resulting in reduced production of pro-inflammatory cytokines and increased release of anti-inflammatory and reparative mediators (16). With MCPIP-1 mutants that have only one of the two catalytic activities, both the deubiquitinase and RNase activities of MCPIP-1 were shown to play a critical role in M2 macrophage polarization (16, 110). MCPIP-1was also reported to suppress the synthesis of miR155 and upregulate miR-223 and miR-146 expression, contributing to M2 polarization (111). By its RNase activity, MCPIP-1 is capable of suppressing the expression of a group of mRNAs encoding factors involved in Th1 differentiation (36, 112). Similar effects were observed in Th17 differentiation, MCPIP-1 works cooperatively with roquin to suppress the differentiation of pro-inflammatory Th17 cells (36). IL-17, a cytokine produced by Th17 cells, has been indicated in the pathogenesis of chronic inflammatory and autoimmune diseases such as psoriasis (113). MCPIP-1 is induced by IL-17A via the phosphorylation of STAT3 (31) and negatively regulate IL-17-dependent inflammation through the degradation of IL-17A-induced target gene transcripts and IL-17RA mRNA (112). Ablation of MCPIP-1 in keratinocytes resulted in the upregulated expression of transcripts encoding factors related to inflammation and keratinocyte differentiation (114). Similar to the results obtained with Th17 cells, MCPIP-1 was shown to play a role in Th2 cell differentiation by dampening of Gata3 expression through the degradation of Gata3 mRNA (115). Mice lacking MCPIP-1 suffered severe airway inflammation, with increased numbers of airway Th2 cells and elevated level of IL-5 (115). These findings suggest MCPIP-1 has pleiotropic effects that contributes to maintaining homeostasis under inflammatory conditions.
The involvement of MCPIP-1 in adipogenesis was also reported, in which MCPIP-1 was thought to induce p47phox, a critical component of NADPH oxidase that contributes to the increase of ROS, which initiates the sequential differentiation process (116, 117). Other studies, however, reported an opposite effect of MCPIP-1 on adipogenesis (118), which shows that forced expression of MCPIP-1 decreases mRNA levels of the C/EBPβ and PPARγ, two key transcription factors controlling adipogenesis, leading to the impairment of adipogenesis (118). Although the evidence described above suggests that MCPIP-1 may exhibit diverse actions according to normal or pathological conditions and the types of cells, it should be noted that most of the evidence regarding the pleiotropic effects of MCPIP-1 were observed in vitro. In addition, most of the studies dissecting the effects of MCPIP-1 were associative and further investigations are warranted to address the pleiotropic effects of MCPIP-1.
Compared to MCPIP-1, the biological roles played by other three members of the Zc3h12 family are less characterized, although it appears that these members are also involved in inflammatory processes. Similar to MCPIP-1, MCPIP-2 has been shown to regulate the course of inflammation. MCPIP-2 participates in the degradation of IL-6 mRNA, resulting in reduced production of IL-6 protein upon stimulation with IL-1β (119). MCPIP-2 also interacts with other known substrates of MCPIP-1 and MCPIP-4, such as the 3’UTR of IER3 mRNA, leading to the degradation of the target mRNAs (119). In a separate study by Huang et al. (120) indicated that IL-6 mRNA is not a direct target of MCPIP-2, which could not exclusively attributable to the different cell line used. In addition, Suzuki et al. (42) showed that MCPIP-2 lacks the miRNA silencing activity, which is attributed to the lack of the proline-rich domain important for this activity. The biological roles of MCPIP-2 remain completely unknown, and further investigations are warranted to address this issue.
MCPIP-3 also contains an RNase domain at the N-terminus before the CCCH-zinc finger domain. Liu et al. (121) showed that MCPIP-3 is able to inhibit the endothelial cell inflammatory response in vitro by suppressing NFκB activation in human endothelial cells. Mice with MCPIP-3 deficiency developed hypertrophic lymph nodes and a higher proportion of immature B cells and innate immune cells, particularly macrophages, by regulating IFN signaling (122). Like MCPIP-1, MCPIP-3 is an RNase essential for immune homeostasis, which binds, degrades and regulates mRNAs, such as MCPIP-1 and IL-6, as observed by reduction in luciferase activity (123). Further comparative structural analysis of MCPIP-3 suggests that the RNA substrate is cooperatively recognized by the PIN and Zinc finger domains of MCPIP-3 (123). Unlike MCPIP-1, MCPIP-3 is specifically expressed in macrophages and is transcriptionally controlled by IFN signaling (122). In humans, MCPIP-3 has been linked with chronic immune disorders like psoriasis via regulating TNFα and Th1 activation (124, 125). Recently, Liu et al. reported that MCPIP-3 expression is positively associated with psoriasisform lesions, and highly expressed in macrophages and plasmacytoid dendritic cells (126). In the same study, the authors demonstrated that MCPIP-3 may promote TNFα/IL-12 via the degradation of MCPIP-1 and IL-6 via direct mRNA degradation, contributing to psoriatic skin inflammation. Consistently, mice with MCPIP-3 deficiency are protected from imiquimod-induced psoriasiform lesions. These data suggest that MCPIP-3 could be a potential inhibitory target to treat psoriasis and other autoimmune diseases (126).
MCPIP-4 was originally reported as a putative tumor suppressor that is deregulated in transformed follicular lymphoma in human (127). A single nucleotide polymorphism analysis indicated that MCPIP-4 is associated with the suppression of tumor cell growth both in vitro and in vivo (128). Similar to MCPIP-1, the expression of MCPIP-4 was markedly induced by TLR ligands through the activation of JNK and NFκB signal pathways, while forced expression of MCPIP-4 inhibited the activation of JNK, ERK, and NFκB signaling in macrophages (129). The latter is achieved by the inhibition of global protein ubiquitination, a key event in the regulation of NFκB activation, suggesting MCPIP-4 is a novel negative feedback regulator of TLR signaling and macrophage activation (129). MCPIP-4 also participates in the degradation of pro-inflammatory mRNAs, such as the mRNAs of IL-2, IL-6, IL-10, TNFα, IER3, and MCPIP-1 (130, 131). Mechanistically, it was demonstrated that MCPIP-4 interacts with MCPIP-1 to form a protein complex, but acts independently in the regulation of IL-6 mRNA degradation (120). To test the in vivo effect of MCPIP-4 in determining host immunity, Minagawa et al. generated a model with MCPIP-4 deletion in mice that displayed normal phenotypes under normal condition, but exhibited more activated lymphocytes, particularly Th17 cells, upon inflammatory stimulation (132). In experimental autoimmune encephalitis induced in the MCPIP-4-deficient mice, a higher proportion of Th17 cells with increased IL-17A mRNA levels were observed in the brain of MCPIP-4 deficient mice than did those in MCPIP-4 wild-type mice, suggesting MCPIP-4 may suppress excessive inflammation in the brain by inhibiting the infiltration and activation of Th17 cells in the experimental autoimmune encephalitis (132).
In this review, we summarized the relevant literature about the role of MCPIP family proteins, in particular MCPIP-1, in the regulation of inflammatory response in different stress conditions. In vivo and in vitro studies revealed that MCPIP-1 expression by immune and non-immune cells contributes to the resolution of inflammation through distinct cellular and molecular programs. Overall, the expression of MCPIP-1 may be a promising target for the prevention and treatment of inflammatory disorders. Besides its crucial role the regulation of inflammation, MCPIP-1 appeared to play a significant role in diverse cellular functions in a variety of cell types, including macrophages, T cells, MSCs, endothelial progenitor cells and adipocytes, as we discussed above in this review. Macrophages are a major source of active inflammation associated with various chronic inflammatory diseases, such as cardiovascular disease, obesity, atherosclerosis, bone loss, and cancer. Understanding how MCPIP-1 regulates macrophage phenotype and modulates nflammatory response may offer promising opportunities for the development of novel therapeutic approach for these disorders. In addition, MCPIP-1 processes antiviral cellular response by degrading the genomic nucleic acids of both positive-sense and negative-sense RNA viruses and DNA viruses. There is a clear potential for MCPIP-1 to be considered as a therapeutic target to prevent the deleterious effects of cytokine storms caused by SARS−CoV−2 infection, although much remains to be investigated. On the other hand, other MCPIP members appear to be involved in the regulation of inflammatory processes. Additional studies are needed to elucidate the effects of other members on the regulation of inflammation, which would include the crosstalk of the proteins from this family and the mechanisms of their actions, especially those related to the resolution of inflammation. Finally, it should be assessed in the future whether the modulation of these proteins should contribute to the discovery of new pharmacological targets that allow us to design specific strategies to resolve inflammation, especially in the context of acute or chronic inflammatory diseases.
ZJ wrote the manuscript. EZ conducted literature collection and summary. JN prepared the final version of the manuscript. CS and PK critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported in part by the National Natural Science Foundation of China (Grant No. 81774010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We apologize to those investigators whose work was not cited due to space constraints.
1. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol (2018) 9:2379. doi: 10.3389/fimmu.2018.02379
2. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
3. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB Signaling in Inflammation. Signal Transduct Target Ther (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
4. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget (2018) 9(6):7204–18. doi: 10.18632/oncotarget.23208
5. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol (2016) 7:160. doi: 10.3389/fimmu.2016.00160
6. Serhan CN, Savill J. Resolution of Inflammation: The Beginning Programs the End. Nat Immunol (2005) 6(12):1191–7. doi: 10.1038/ni1276
7. Coornaert B, Carpentier I, Beyaert R. A20: Central Gatekeeper in Inflammation and Immunity. J Biol Chem (2009) 284(13):8217–21. doi: 10.1074/jbc.R800032200
8. Liu J, Cao X. Cellular and Molecular Regulation of Innate Inflammatory Responses. Cell Mol Immunol (2016) 13(6):711–21. doi: 10.1038/cmi.2016.58
9. Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, et al. Monocyte Chemoattractant Protein-1 Induces a Novel Transcription Factor That Causes Cardiac Myocyte Apoptosis and Ventricular Dysfunction. Circ Res (2006) 98(9):1177–85. doi: 10.1161/01.RES.0000220106.64661.71
10. Uehata T, Takeuchi O. Regnase-1 Is an Endoribonuclease Essential for the Maintenance of Immune Homeostasis. J Interferon Cytokine Res (2017) 37(5):220–9. doi: 10.1089/jir.2017.0001
11. Liang J, Wang J, Azfer A, Song W, Tromp G, Kolattukudy PE, et al. A Novel CCCH-Zinc Finger Protein Family Regulates Proinflammatory Activation of Macrophages. J Biol Chem (2008) 283(10):6337–46. doi: 10.1074/jbc.M707861200
12. Xu J, Peng W, Sun Y, Wang X, Xu Y, Li X, et al. Structural Study of MCPIP1 N-Terminal Conserved Domain Reveals a PIN-Like RNase. Nucleic Acids Res (2012) 40(14):6957–65. doi: 10.1093/nar/gks359
13. Mao R, Yang R, Chen X, Harhaj EW, Wang X, Fan Y. Regnase-1, a Rapid Response Ribonuclease Regulating Inflammation and Stress Responses. Cell Mol Immunol (2017) 14(5):412–22. doi: 10.1038/cmi.2016.70
14. Uehata T, Akira S. mRNA Degradation by the Endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim Biophys Acta (2013) 1829(6-7):708–13. doi: 10.1016/j.bbagrm.2013.03.001
15. Schett G, Neurath MF. Resolution of Chronic Inflammatory Disease: Universal and Tissue-Specific Concepts. Nat Commun (2018) 9(1):3261. doi: 10.1038/s41467-018-05800-6
16. Kapoor N, Niu J, Saad Y, Kumar S, Sirakova T, Becerra E, et al. Transcription Factors STAT6 and KLF4 Implement Macrophage Polarization via the Dual Catalytic Powers of MCPIP. J Immunol (2015) 194(12):6011–23. doi: 10.4049/jimmunol.1402797
17. Li Y, Huang X, Huang S, He H, Lei T, Saaoud F, et al. Central Role of Myeloid MCPIP1 in Protecting Against LPS-Induced Inflammation and Lung Injury. Signal Transduct Target Ther (2017) 2:17066. doi: 10.1038/sigtrans.2017.66
18. Cui X, Mino T, Yoshinaga M, Nakatsuka T, Hia F, Yamasoba D, et al. Regnase-1 and Roquin Nonredundantly Regulate Th1 Differentiation Causing Cardiac Inflammation and Fibrosis. J Immunol (2017) 199(12):4066–77. doi: 10.4049/jimmunol.1701211
19. Mizgalska D, Wegrzyn P, Murzyn K, Kasza A, Koj A, Jura J, et al. Interleukin-1-Inducible MCPIP Protein has Structural and Functional Properties of RNase and Participates in Degradation of IL-1beta mRNA. FEBS J (2009) 276(24):7386–99. doi: 10.1111/j.1742-4658.2009.07452.x
20. Dhamija S, Winzen R, Doerrie A, Behrens G, Kuehne N, Schauerte C, et al. Interleukin-17 (IL-17) and IL-1 Activate Translation of Overlapping Sets of mRNAs, Including That of the Negative Regulator of Inflammation, MCPIP1. J Biol Chem (2013) 288(26):19250–9. doi: 10.1074/jbc.M113.452649
21. Jin Z, Liang J, Wang J, Kolattukudy PE. Delayed Brain Ischemia Tolerance Induced by Electroacupuncture Pretreatment Is Mediated via MCP-Induced Protein 1. J Neuroinflamm (2013) 10:63. doi: 10.1186/1742-2094-10-63
22. Niu J, Jin Z, Kim H, Kolattukudy PE. MCP-1-Induced Protein Attenuates Post-Infarct Cardiac Remodeling and Dysfunction Through Mitigating NF-kappaB Activation and Suppressing Inflammation-Associated microRNA Expression. Basic Res Cardiol (2015) 110(3):26. doi: 10.1007/s00395-015-0483-8
23. Lin RJ, Chien HL, Lin SY, Chang BL, Yu HP, Tang WC, et al. MCPIP1 Ribonuclease Exhibits Broad-Spectrum Antiviral Effects Through Viral RNA Binding and Degradation. Nucleic Acids Res (2013) 41(5):3314–26. doi: 10.1093/nar/gkt019
24. Lin RJ, Chu JS, Chien HL, Tseng CH, Ko PC, Mei YY, et al. MCPIP1 Suppresses Hepatitis C Virus Replication and Negatively Regulates Virus-Induced Proinflammatory Cytokine Responses. J Immunol (2014) 193(8):4159–68. doi: 10.4049/jimmunol.1400337
25. Li M, Yang J, Zhao Y, Song Y, Yin S, Guo J, et al. MCPIP1 Inhibits Hepatitis B Virus Replication by Destabilizing Viral RNA and Negatively Regulates the Virus-Induced Innate Inflammatory Responses. Antiviral Res (2020) 174:104705. doi: 10.1016/j.antiviral.2020.104705
26. Liu XX, Wang C, Huang SF, Chen Q, Hu YF, Zhou L, et al. Regnase-1 in Microglia Negatively Regulates High Mobility Group Box 1-Mediated Inflammation and Neuronal Injury. Sci Rep (2016) 6:24073. doi: 10.1038/srep24073
27. Jin Z, Liang J, Wang J, Kolattukudy PE. MCP-Induced Protein 1 Mediates the Minocycline-Induced Neuroprotection Against Cerebral Ischemia/Reperfusion Injury In Vitro and In Vivo. J Neuroinflamm (2015) 12:39. doi: 10.1186/s12974-015-0264-1
28. Yi Q, Tan FH, Tan JA, Chen XH, Xiao Q, Liu YH, et al. Minocycline Protects Against Myocardial Ischemia/Reperfusion Injury in Rats by Upregulating MCPIP1 to Inhibit NF-kappaB Activation. Acta Pharmacol Sin (2019) 40(8):1019–28. doi: 10.1038/s41401-019-0214-z
29. Skalniak L, Mizgalska D, Zarebski A, Wyrzykowska P, Koj A, Jura J. Regulatory Feedback Loop Between NF-kappaB and MCP-1-Induced Protein 1 RNase. FEBS J (2009) 276(20):5892–905. doi: 10.1111/j.1742-4658.2009.07273.x
30. Kasza A, Wyrzykowska P, Horwacik I, Tymoszuk P, Mizgalska D, Palmer K, et al. Transcription Factors Elk-1 and SRF Are Engaged in IL1-Dependent Regulation of ZC3H12A Expression. BMC Mol Biol (2010) 11:14. doi: 10.1186/1471-2199-11-14
31. Ruiz-Romeu E, Ferran M, Gimenez-Arnau A, Bugara B, Lipert B, Jura J, et al. MCPIP1 RNase Is Aberrantly Distributed in Psoriatic Epidermis and Rapidly Induced by IL-17a. J Invest Dermatol (2016) 136(8):1599–607. doi: 10.1016/j.jid.2016.04.030
32. Yao H, Ma R, Yang L, Hu G, Chen X, Duan M, et al. MiR-9 Promotes Microglial Activation by Targeting MCPIP1. Nat Commun (2014) 5:4386. doi: 10.1038/ncomms5386
33. Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, et al. Malt1-Induced Cleavage of Regnase-1 in CD4(+) Helper T Cells Regulates Immune Activation. Cell (2013) 153(5):1036–49. doi: 10.1016/j.cell.2013.04.034
34. Wawro M, Kochan J, Kasza A. The Perplexities of the ZC3H12A self-mRNA Regulation. Acta Biochim Pol (2016) 63(3):411–5. doi: 10.18388/abp.2016_1325
35. Iwasaki H, Takeuchi O, Teraguchi S, Matsushita K, Uehata T, Kuniyoshi K, et al. The IkappaB Kinase Complex Regulates the Stability of Cytokine-Encoding mRNA Induced by TLR-IL-1R by Controlling Degradation of Regnase-1. Nat Immunol (2011) 12(12):1167–75. doi: 10.1038/ni.2137
36. Jeltsch KM, Hu D, Brenner S, Zoller J, Heinz GA, Nagel D, et al. Cleavage of Roquin and Regnase-1 by the Paracaspase MALT1 Releases Their Cooperatively Repressed Targets to Promote T(H)17 Differentiation. Nat Immunol (2014) 15(11):1079–89. doi: 10.1038/ni.3008
37. Chan FK, Luz NF, Moriwaki K. Programmed Necrosis in the Cross Talk of Cell Death and Inflammation. Annu Rev Immunol (2015) 33:79–106. doi: 10.1146/annurev-immunol-032414-112248
38. Koenig U, Robenek H, Barresi C, Brandstetter M, Resch GP, Groger M, et al. Cell Death Induced Autophagy Contributes to Terminal Differentiation of Skin and Skin Appendages. Autophagy (2020) 16(5):932–45. doi: 10.1080/15548627.2019.1646552
39. Yokogawa M, Tsushima T, Noda NN, Kumeta H, Enokizono Y, Yamashita K, et al. Structural Basis for the Regulation of Enzymatic Activity of Regnase-1 by Domain-Domain Interactions. Sci Rep (2016) 6:22324. doi: 10.1038/srep22324
40. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Miyake T, Satoh T, et al. Zc3h12a Is an RNase Essential for Controlling Immune Responses by Regulating mRNA Decay. Nature (2009) 458(7242):1185–90. doi: 10.1038/nature07924
41. Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, et al. MCP-Induced Protein 1 Deubiquitinates TRAF Proteins and Negatively Regulates JNK and NF-kappaB Signaling. J Exp Med (2010) 207(13):2959–73. doi: 10.1084/jem.20092641
42. Suzuki HI, Arase M, Matsuyama H, Choi YL, Ueno T, Mano H, et al. MCPIP1 Ribonuclease Antagonizes Dicer and Terminates microRNA Biogenesis Through Precursor microRNA Degradation. Mol Cell (2011) 44(3):424–36. doi: 10.1016/j.molcel.2011.09.012
43. Jin Z, Niu J, Kapoor N, Liang J, Becerra E, Kolattukudy PE. Essential Role of Endothelial MCPIP in Vascular Integrity and Post-Ischemic Remodeling. Int J Mol Sci (2019) 20(1):172. doi: 10.3390/ijms20010172
44. Niu J, Wang K, Graham S, Azfer A, Kolattukudy PE. MCP-1-Induced Protein Attenuates Endotoxin-Induced Myocardial Dysfunction by Suppressing Cardiac NF-Small Ka, CyrillicB Activation via Inhibition of Ismall Ka, CyrillicB Kinase Activation. J Mol Cell Cardiol (2011) 51(2):177–86. doi: 10.1016/j.yjmcc.2011.04.018
45. Hu H, Sun SC. Ubiquitin Signaling in Immune Responses. Cell Res (2016) 26(4):457–83. doi: 10.1038/cr.2016.40
46. Wu Y, Kang J, Zhang L, Liang Z, Tang X, Yan Y, et al. Ubiquitination Regulation of Inflammatory Responses Through NF-kappaB Pathway. Am J Transl Res (2018) 10(3):881–91.
47. Kolattukudy PE, Niu J. Inflammation, Endoplasmic Reticulum Stress, Autophagy, and the Monocyte Chemoattractant Protein-1/CCR2 Pathway. Circ Res (2012) 110(1):174–89. doi: 10.1161/CIRCRESAHA.111.243212
48. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential Function for the Kinase TAK1 in Innate and Adaptive Immune Responses. Nat Immunol (2005) 6(11):1087–95. doi: 10.1038/ni1255
49. Niu J, Shi Y, Xue J, Miao R, Huang S, Wang T, et al. USP10 Inhibits Genotoxic NF-kappaB Activation by MCPIP1-Facilitated Deubiquitination of NEMO. EMBO J (2013) 32(24):3206–19. doi: 10.1038/emboj.2013.247
50. Roy A, Zhang M, Saad Y, Kolattukudy PE. Antidicer RNAse Activity of Monocyte Chemotactic Protein-Induced Protein-1 Is Critical for Inducing Angiogenesis. Am J Physiol Cell Physiol (2013) 305(10):C1021–32. doi: 10.1152/ajpcell.00203.2013
51. Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels H, Ori D, et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell (2015) 161(5):1058–73. doi: 10.1016/j.cell.2015.04.029
52. Li M, Cao W, Liu H, Zhang W, Liu X, Cai Z, et al. MCPIP1 Down-Regulates IL-2 Expression Through an ARE-Independent Pathway. PloS One (2012) 7(11):e49841. doi: 10.1371/journal.pone.0049841
53. De Filippo K, Dudeck A, Hasenberg M, Nye E, Rooijen NV, Hartmann K, et al. Mast Cell and Macrophage Chemokines CXCL1/CXCL2 Control the Early Stage of Neutrophil Recruitment During Tissue Inflammation. Blood (2013) 121(24):4930–7. doi: 10.1182/blood-2013-02-486217
54. Kochan J, Wawro M, Kasza A. IF-Combined smRNA FISH Reveals Interaction of MCPIP1 Protein With IER3 mRNA. Biol Open (2016) 5(7):889–98. doi: 10.1242/bio.018010
55. Singh RP, Massachi I, Manickavel S, Singh S, Rao NP, Hasan S, et al. The Role of miRNA in Inflammation and Autoimmunity. Autoimmun Rev (2013) 12(12):1160–5. doi: 10.1016/j.autrev.2013.07.003
56. Zhou X, Li X, Wu M. miRNAs Reshape Immunity and Inflammatory Responses in Bacterial Infection. Signal Transduct Target Ther (2018) 3:14. doi: 10.1038/s41392-018-0006-9
57. Song MS, Rossi JJ. Molecular Mechanisms of Dicer: Endonuclease and Enzymatic Activity. Biochem J (2017) 474(10):1603–18. doi: 10.1042/BCJ20160759
58. Qu B, Cao J, Zhang F, Cui H, Teng J, Li J, et al. Type I Interferon Inhibition of MicroRNA-146a Maturation Through Up-Regulation of Monocyte Chemotactic Protein-Induced Protein 1 in Systemic Lupus Erythematosus. Arthritis Rheumatol (2015) 67(12):3209–18. doi: 10.1002/art.39398
59. Mann M, Mehta A, Zhao JL, Lee K, Marinov GK, Garcia-Flores Y, et al. An NF-kappaB-microRNA Regulatory Network Tunes Macrophage Inflammatory Responses. Nat Commun (2017) 8(1):851. doi: 10.1038/s41467-017-00972-z
60. Singh R, Letai A, Sarosiek K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat Rev Mol Cell Biol (2019) 20(3):175–93. doi: 10.1038/s41580-018-0089-8
61. He M, Liang X, He L, Wen W, Zhao S, Wen L, et al. Endothelial Dysfunction in Rheumatoid Arthritis: The Role of Monocyte Chemotactic Protein-1-Induced Protein. Arterioscler Thromb Vasc Biol (2013) 33(6):1384–91. doi: 10.1161/ATVBAHA.113.301490
62. Younce CW, Kolattukudy PE. MCP-1 Causes Cardiomyoblast Death via Autophagy Resulting From ER Stress Caused by Oxidative Stress Generated by Inducing a Novel Zinc-Finger Protein, MCPIP. Biochem J (2010) 426(1):43–53. doi: 10.1042/BJ20090976
63. Younce CW, Wang K, Kolattukudy PE. Hyperglycaemia-Induced Cardiomyocyte Death Is Mediated via MCP-1 Production and Induction of a Novel Zinc-Finger Protein MCPIP. Cardiovasc Res (2010) 87(4):665–74. doi: 10.1093/cvr/cvq102
64. Wang X, Zhang Y, Zhang W, Liu H, Zhou Z, Dai X, et al. MCPIP1 Regulates Alveolar Macrophage Apoptosis and Pulmonary Fibroblast Activation After In Vitro Exposure to Silica. Toxicol Sci (2016) 151(1):126–38. doi: 10.1093/toxsci/kfw029
65. Shu S, Zhang Y, Li W, Wang L, Wu Y, Yuan Z, et al. The Role of Monocyte Chemotactic Protein-Induced Protein 1 (MCPIP1) in Angiotensin II-Induced Macrophage Apoptosis and Vulnerable Plaque Formation. Biochem Biophys Res Commun (2019) 515(2):378–85. doi: 10.1016/j.bbrc.2019.05.145
66. Dobosz E, Wadowska M, Kaminska M, Wilamowski M, Honarpisheh M, Bryzek D, et al. MCPIP-1 Restricts Inflammation via Promoting Apoptosis of Neutrophils. Front Immunol (2021) 12:627922. doi: 10.3389/fimmu.2021.627922
67. Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 Programs Long-Lived Effector T Cells for Cancer Therapy. Nature (2019) 576(7787):471–6. doi: 10.1038/s41586-019-1821-z
68. Lu W, Ning H, Gu L, Peng H, Wang Q, Hou R, et al. MCPIP1 Selectively Destabilizes Transcripts Associated With an Antiapoptotic Gene Expression Program in Breast Cancer Cells That Can Elicit Complete Tumor Regression. Cancer Res (2016) 76(6):1429–40. doi: 10.1158/0008-5472.CAN-15-1115
69. Boratyn E, Nowak I, Horwacik I, Durbas M, Mistarz A, Kukla M, et al. Monocyte Chemoattractant Protein-Induced Protein 1 Overexpression Modulates Transcriptome, Including MicroRNA, in Human Neuroblastoma Cells. J Cell Biochem (2016) 117(3):694–707. doi: 10.1002/jcb.25354
70. Kedersha N, Anderson P. Mammalian Stress Granules and Processing Bodies. Methods Enzymol (2007) 431:61–81. doi: 10.1016/S0076-6879(07)31005-7
71. Qi D, Huang S, Miao R, She ZG, Quinn T, Chang Y, et al. Monocyte Chemotactic Protein-Induced Protein 1 (MCPIP1) Suppresses Stress Granule Formation and Determines Apoptosis Under Stress. J Biol Chem (2011) 286(48):41692–700. doi: 10.1074/jbc.M111.276006
72. Circu ML, Aw TY. Reactive Oxygen Species, Cellular Redox Systems, and Apoptosis. Free Radic Biol Med (2010) 48(6):749–62. doi: 10.1016/j.freeradbiomed.2009.12.022
73. Liu H, Fang S, Wang W, Cheng Y, Zhang Y, Liao H, et al. Macrophage-Derived MCPIP1 Mediates Silica-Induced Pulmonary Fibrosis via Autophagy. Part Fibre Toxicol (2016) 13(1):55. doi: 10.1186/s12989-016-0167-z
74. Chao J, Wang X, Zhang Y, Zhu T, Zhang W, Zhou Z, et al. Role of MCPIP1 in the Endothelial-Mesenchymal Transition Induced by Silica. Cell Physiol Biochem (2016) 40(1-2):309–25. doi: 10.1159/000452547
75. Xie X, Zhu T, Chen L, Ding S, Chu H, Wang J, et al. MCPIP1-Induced Autophagy Mediates Ischemia/Reperfusion Injury in Endothelial Cells via HMGB1 and CaSR. Sci Rep (2018) 8(1):1735. doi: 10.1038/s41598-018-20195-6
76. Lee CH, Hung PF, Lu SC, Chung HL, Chiang SL, Wu CT, et al. MCP-1/MCPIP-1 Signaling Modulates the Effects of IL-1beta in Renal Cell Carcinoma Through ER Stress-Mediated Apoptosis. Int J Mol Sci (2019) 20(23):6101. doi: 10.3390/ijms20236101
77. Ganjam GK, Terpolilli NA, Diemert S, Eisenbach I, Hoffmann L, Reuther C, et al. Cylindromatosis Mediates Neuronal Cell Death In Vitro and In Vivo. Cell Death Differ (2018) 25(8):1394–407. doi: 10.1038/s41418-017-0046-7
78. Garcia-Carbonell R, Wong J, Kim JY, Close LA, Boland BS, Wong TL, et al. Elevated A20 Promotes TNF-Induced and RIPK1-Dependent Intestinal Epithelial Cell Death. Proc Natl Acad Sci U S A (2018) 115(39):E9192–200. doi: 10.1073/pnas.1810584115
79. Skalniak L, Koj A, Jura J. Proteasome Inhibitor MG-132 Induces MCPIP1 Expression. FEBS J (2013) 280(11):2665–74. doi: 10.1111/febs.12264
80. Serhan CN. Treating Inflammation and Infection in the 21st Century: New Hints From Decoding Resolution Mediators and Mechanisms. FASEB J (2017) 31(4):1273–88. doi: 10.1096/fj.201601222R
81. Liu S, Qiu C, Miao R, Zhou J, Lee A, Liu B, et al. MCPIP1 Restricts HIV Infection and Is Rapidly Degraded in Activated CD4+ T Cells. Proc Natl Acad Sci U S A (2013) 110(47):19083–8. doi: 10.1073/pnas.1316208110
82. Qian L, Zuo Y, Deng W, Miao Y, Liu J, Yuan Y, et al. MCPIP1 Is a Positive Regulator of Type I Interferons Antiviral Activity. Biochem Biophys Res Commun (2018) 498(4):891–7. doi: 10.1016/j.bbrc.2018.03.076
83. Qian Y, Li X, Miao R, Liu S, Xin HB, Huang X, et al. Selective Degradation of Plasmid-Derived mRNAs by MCPIP1 RNase. Biochem J (2019) 476(19):2927–38. doi: 10.1042/BCJ20190646
84. Li M, Yan K, Wei L, Yang Y, Qian Q, Xu W. MCPIP1 Inhibits Coxsackievirus B3 Replication by Targeting Viral RNA and Negatively Regulates Virus-Induced Inflammation. Med Microbiol Immunol (2018) 207(1):27–38. doi: 10.1007/s00430-017-0523-0
85. Liao TL, Wu CY, Su WC, Jeng KS, Lai MM. Ubiquitination and Deubiquitination of NP Protein Regulates Influenza A Virus RNA Replication. EMBO J (2010) 29(22):3879–90. doi: 10.1038/emboj.2010.250
86. Si X, Gao G, Wong J, Wang Y, Zhang J, Luo H. Ubiquitination Is Required for Effective Replication of Coxsackievirus B3. PloS One (2008) 3(7):e2585. doi: 10.1371/journal.pone.0002585
87. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic Variation in IL28B Predicts Hepatitis C Treatment-Induced Viral Clearance. Nature (2009) 461(7262):399–401. doi: 10.1038/nature08309
88. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
89. Hirano T, Murakami M. COVID-19: A New Virus, But a Familiar Receptor and Cytokine Release Syndrome. Immunity (2020) 52(5):731–3. doi: 10.1016/j.immuni.2020.04.003
90. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/S0140-6736(20)30566-3
91. Shichita T, Sakaguchi R, Suzuki M, Yoshimura A. Post-Ischemic Inflammation in the Brain. Front Immunol (2012) 3:132. doi: 10.3389/fimmu.2012.00132
92. Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol Biochem (2018) 46(4):1650–67. doi: 10.1159/000489241
93. Tomai F, Crea F, Chiariello L, Gioffre PA. Ischemic Preconditioning in Humans: Models, Mediators, and Clinical Relevance. Circulation (1999) 100(5):559–63. doi: 10.1161/01.CIR.100.5.559
94. Smith RM, Lecour S, Sack MN. Innate Immunity and Cardiac Preconditioning: A Putative Intrinsic Cardioprotective Program. Cardiovasc Res (2002) 55(3):474–82. doi: 10.1016/S0008-6363(02)00288-2
95. McDonough A, Weinstein JR. Neuroimmune Response in Ischemic Preconditioning. Neurotherapeutics (2016) 13(4):748–61. doi: 10.1007/s13311-016-0465-z
96. Obrenovitch TP. Molecular Physiology of Preconditioning-Induced Brain Tolerance to Ischemia. Physiol Rev (2008) 88(1):211–47. doi: 10.1152/physrev.00039.2006
97. Sullivan PJ, Sweeney KJ, Hirpara KM, Malone CB, Curtin W, Kerin MJ. Cyclical Ischaemic Preconditioning Modulates the Adaptive Immune Response in Human Limb Ischaemia-Reperfusion Injury. Br J Surg (2009) 96(4):381–90. doi: 10.1002/bjs.6554
98. Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin Preconditioning Prevents Cellular Inflammatory Response During Ischemic Neuroprotection in Mice. Stroke (2004) 35(11):2576–81. doi: 10.1161/01.STR.0000143450.04438.ae
99. Dong JW, Vallejo JG, Tzeng HP, Thomas JA, Mann DL. Innate Immunity Mediates Myocardial Preconditioning Through Toll-Like Receptor 2 and TIRAP-Dependent Signaling Pathways. Am J Physiol Heart Circ Physiol (2010) 298(3):H1079–1087. doi: 10.1152/ajpheart.00306.2009
100. Liang J, Wang J, Saad Y, Warble L, Becerra E. Kolattukudy PE. Participation of MCP-Induced Protein 1 in Lipopolysaccharide Preconditioning-Induced Ischemic Stroke Tolerance by Regulating the Expression of Proinflammatory Cytokines. J Neuroinflamm (2011) 8:182. doi: 10.1186/1742-2094-8-182
101. Jin Z, Liang J, Li J, Kolattukudy PE. Absence of MCP-Induced Protein 1 Enhances Blood-Brain Barrier Breakdown After Experimental Stroke in Mice. Int J Mol Sci (2019) 20(13):3214. doi: 10.3390/ijms20133214
102. Sakata H, Niizuma K, Yoshioka H, Kim GS, Jung JE, Katsu M, et al. Minocycline-Preconditioned Neural Stem Cells Enhance Neuroprotection After Ischemic Stroke in Rats. J Neurosci (2012) 32(10):3462–73. doi: 10.1523/JNEUROSCI.5686-11.2012
103. Niu J, Azfer A, Zhelyabovska O, Fatma S, Kolattukudy PE. Monocyte Chemotactic Protein (MCP)-1 Promotes Angiogenesis via a Novel Transcription Factor, MCP-1-Induced Protein (MCPIP). J Biol Chem (2008) 283(21):14542–51. doi: 10.1074/jbc.M802139200
104. Niu J, Wang K, Zhelyabovska O, Saad Y, Kolattukudy PE. MCP-1-Induced Protein Promotes Endothelial-Like and Angiogenic Properties in Human Bone Marrow Monocytic Cells. J Pharmacol Exp Ther (2013) 347(2):288–97. doi: 10.1124/jpet.113.207316
105. Uccelli A, Moretta L, Pistoia V. Mesenchymal Stem Cells in Health and Disease. Nat Rev Immunol (2008) 8(9):726–36. doi: 10.1038/nri2395
106. Labedz-Maslowska A, Lipert B, Berdecka D, Kedracka-Krok S, Jankowska U, Kamycka E, et al. Monocyte Chemoattractant Protein-Induced Protein 1 (MCPIP1) Enhances Angiogenic and Cardiomyogenic Potential of Murine Bone Marrow-Derived Mesenchymal Stem Cells. PloS One (2015) 10(7):e0133746. doi: 10.1371/journal.pone.0133746
107. Shu B, Zhuo M, Liu Z, Lu Z, Qian M. Cholesterol Induces Dedifferentiation of Vascular Smooth Muscle Cells by Regulating Monocyte Chemotactic Protein-1-Induced Protein 1. Int J Clin Exp Pathol (2019) 12(9):3258–67.
108. Marona P, Gorka J, Mazurek Z, Wilk W, Rys J, Majka M, et al. MCPIP1 Downregulation in Clear Cell Renal Cell Carcinoma Promotes Vascularization and Metastatic Progression. Cancer Res (2017) 77(18):4905–20. doi: 10.1158/0008-5472.CAN-16-3190
109. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The Role of Macrophages in the Resolution of Inflammation. J Clin Invest (2019) 129(7):2619–28. doi: 10.1172/JCI124615
110. Xu R, Li Y, Yan H, Zhang E, Huang X, Chen Q, et al. CCL2 Promotes Macrophages-Associated Chemoresistance via MCPIP1 Dual Catalytic Activities in Multiple Myeloma. Cell Death Dis (2019) 10(10):781. doi: 10.1038/s41419-019-2012-4
111. Miao R, Huang S, Zhou Z, Quinn T, Van Treeck B, Nayyar T, et al. Targeted Disruption of MCPIP1/Zc3h12a Results in Fatal Inflammatory Disease. Immunol Cell Biol (2013) 91(5):368–76. doi: 10.1038/icb.2013.11
112. Garg AV, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, et al. MCPIP1 Endoribonuclease Activity Negatively Regulates Interleukin-17-Mediated Signaling and Inflammation. Immunity (2015) 43(3):475–87. doi: 10.1016/j.immuni.2015.07.021
113. Beringer A, Noack M, Miossec P. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol Med (2016) 22(3):230–41. doi: 10.1016/j.molmed.2016.01.001
114. Konieczny P, Lichawska-Cieslar A, Kwiecinska P, Cichy J, Pietrzycka R, Szukala W, et al. Keratinocyte-Specific Ablation of Mcpip1 Impairs Skin Integrity and Promotes Local and Systemic Inflammation. J Mol Med (Berl) (2019) 97(12):1669–84. doi: 10.1007/s00109-019-01853-2
115. Peng H, Ning H, Wang Q, Lu W, Chang Y, Wang TT, et al. Monocyte Chemotactic Protein-Induced Protein 1 Controls Allergic Airway Inflammation by Suppressing IL-5-Producing TH2 Cells Through the Notch/Gata3 Pathway. J Allergy Clin Immunol (2018) 142(2):582–94. doi: 10.1016/j.jaci.2017.09.031
116. Younce CW, Azfer A, Kolattukudy PE. MCP-1 (Monocyte Chemotactic Protein-1)-Induced Protein, a Recently Identified Zinc Finger Protein, Induces Adipogenesis in 3T3-L1 Pre-Adipocytes Without Peroxisome Proliferator-Activated Receptor Gamma. J Biol Chem (2009) 284(40):27620–8. doi: 10.1074/jbc.M109.025320
117. Younce C, Kolattukudy P. MCP-1 Induced Protein Promotes Adipogenesis via Oxidative Stress, Endoplasmic Reticulum Stress and Autophagy. Cell Physiol Biochem (2012) 30(2):307–20. doi: 10.1159/000339066
118. Lipert B, Wegrzyn P, Sell H, Eckel J, Winiarski M, Budzynski A, et al. Monocyte Chemoattractant Protein-Induced Protein 1 Impairs Adipogenesis in 3T3-L1 Cells. Biochim Biophys Acta (2014) 1843(4):780–8. doi: 10.1016/j.bbamcr.2014.01.001
119. Wawro M, Wawro K, Kochan J, Solecka A, Sowinska W, Lichawska-Cieslar A, et al. ZC3H12B/MCPIP2, a New Active Member of the ZC3H12 Family. RNA (2019) 25(7):840–56. doi: 10.1261/rna.071381.119
120. Huang S, Liu S, Fu JJ, et al. Monocyte Chemotactic Protein-Induced Protein 1 and 4 Form a Complex But Act Independently in Regulation of Interleukin-6 mRNA Degradation. J Biol Chem (2015) 290(34):20782–92. doi: 10.1074/jbc.M114.635870
121. Liu L, Zhou Z, Huang S, Tony Wang T, Yao X, Kumar A, et al. Zc3h12c Inhibits Vascular Inflammation by Repressing NF-kappaB Activation and Pro-Inflammatory Gene Expression in Endothelial Cells. Biochem J (2013) 451(1):55–60. doi: 10.1042/BJ20130019
122. von Gamm M, Schaub A, Jones AN, Wolf C, Behrens G, Lichti J, et al. Immune Homeostasis and Regulation of the Interferon Pathway Require Myeloid-Derived Regnase-3. J Exp Med (2019) 216(7):1700–23. doi: 10.1084/jem.20181762
123. Garg A, Roske Y, Yamada S, Uehata T, Takeuchi O, Heinemann U. PIN and CCCH Zn-Finger Domains Coordinate RNA Targeting in ZC3H12 Family Endoribonucleases. Nucleic Acids Res (2021) 49(9):5369–81. doi: 10.1093/nar/gkab316
124. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 New Psoriasis Susceptibility Loci Highlights the Role of Innate Immunity. Nat Genet (2012) 44(12):1341–8. doi: 10.1038/ng.2467
125. Munir S, ber Rahman S, Rehman S, Saba N, Ahmad W, Nilsson S, et al. Association Analysis of GWAS and Candidate Gene Loci in a Pakistani Population With Psoriasis. Mol Immunol (2015) 64(1):190–4. doi: 10.1016/j.molimm.2014.11.015
126. Liu B, Huang J, Ashraf A, Rahaman O, Lou J, Wang L, et al. The RNase MCPIP3 Promotes Skin Inflammation by Orchestrating Myeloid Cytokine Response. Nat Commun (2021) 12(1):4105. doi: 10.1038/s41467-021-24352-w
127. Minagawa K, Yamamoto K, Nishikawa S, Ito M, Sada A, Yakushijin K, et al. Deregulation of a Possible Tumour Suppressor Gene, ZC3H12D, by Translocation of IGK@ in Transformed Follicular Lymphoma With T(2;6)(P12;Q25). Br J Haematol (2007) 139(1):161–3. doi: 10.1111/j.1365-2141.2007.06752.x
128. Wang M, Vikis HG, Wang Y, Jia D, Wang D, Bierut LJ, et al. Identification of a Novel Tumor Suppressor Gene P34 on Human Chromosome 6q25.1. Cancer Res (2007) 67(1):93–9. doi: 10.1158/0008-5472.CAN-06-2723
129. Huang S, Qi D, Liang J, Miao R, Minagawa K, Quinn T, et al. The Putative Tumor Suppressor Zc3h12d Modulates Toll-Like Receptor Signaling in Macrophages. Cell Signal (2012) 24(2):569–76. doi: 10.1016/j.cellsig.2011.10.011
130. Zhang H, Wang WC, Chen JK, Zhou L, Wang M, Wang ZD, et al. ZC3H12D Attenuated Inflammation Responses by Reducing mRNA Stability of Proinflammatory Genes. Mol Immunol (2015) 67(2 Pt B):206–12. doi: 10.1016/j.molimm.2015.05.018
131. Wawro M, Kochan J, Krzanik S, Jura J, Kasza A. Intact NYN/PIN-Like Domain Is Crucial for the Degradation of Inflammation-Related Transcripts by ZC3H12D. J Cell Biochem (2017) 118(3):487–98. doi: 10.1002/jcb.25665
Keywords: inflammation, viral infection, ischemic inflammation, resolution, MCPIP-1
Citation: Jin Z, Zheng E, Sareli C, Kolattukudy PE and Niu J (2021) Monocyte Chemotactic Protein-Induced Protein 1 (MCPIP-1): A Key Player of Host Defense and Immune Regulation. Front. Immunol. 12:727861. doi: 10.3389/fimmu.2021.727861
Received: 19 June 2021; Accepted: 08 September 2021;
Published: 01 October 2021.
Edited by:
Sylvaine You, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Dong Li, Jilin University, ChinaCopyright © 2021 Jin, Zheng, Sareli, Kolattukudy and Niu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianli Niu, am5pdUBtaHMubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.