 Samaa T. Gobran
Samaa T. Gobran Petronela Ancuta
Petronela Ancuta Naglaa H. Shoukry
Naglaa H. Shoukry- 1Centre de Recherche du Centre hospitalier de l’Université de Montréal (CRCHUM), Montréal, QC, Canada
- 2Département de microbiologie, infectiologie et immunologie, Faculté de Médecine, Université de Montréal, Montréal, QC, Canada
- 3Department of Medical Microbiology and Immunology, Faculty of Medicine, Zagazig University, Zagazig, Egypt
- 4Département de médecine, Faculté de médecine, Université de Montréal, Montréal, QC, Canada
Nearly 2.3 million individuals worldwide are coinfected with human immunodeficiency virus (HIV) and hepatitis C virus (HCV). Odds of HCV infection are six times higher in people living with HIV (PLWH) compared to their HIV-negative counterparts, with the highest prevalence among people who inject drugs (PWID) and men who have sex with men (MSM). HIV coinfection has a detrimental impact on the natural history of HCV, including higher rates of HCV persistence following acute infection, higher viral loads, and accelerated progression of liver fibrosis and development of end-stage liver disease compared to HCV monoinfection. Similarly, it has been reported that HCV coinfection impacts HIV disease progression in PLWH receiving anti-retroviral therapies (ART) where HCV coinfection negatively affects the homeostasis of CD4+ T cell counts and facilitates HIV replication and viral reservoir persistence. While ART does not cure HIV, direct acting antivirals (DAA) can now achieve HCV cure in nearly 95% of coinfected individuals. However, little is known about how HCV cure and the subsequent resolution of liver inflammation influence systemic immune activation, immune reconstitution and the latent HIV reservoir. In this review, we will summarize the current knowledge regarding the pathogenesis of HIV/HCV coinfection, the effects of HCV coinfection on HIV disease progression in the context of ART, the impact of HIV on HCV-associated liver morbidity, and the consequences of DAA-mediated HCV cure on immune reconstitution and HIV reservoir persistence in coinfected patients.
Introduction
Hepatitis C virus (HCV) and human immunodeficiency virus type 1 (HIV-1) are two chronic viral infections that affect millions worldwide. They share similar routes of transmission through percutaneous exposure to infected blood, sexual activity and vertical transmission from infected mother-to-child. HCV is transmitted 10-times more efficiently than HIV through percutaneous exposure resulting in a high rate of HCV/HIV coinfection among people who inject drugs (PWID) (1). HIV infection on the other hand increases the risk of HCV acquisition through sexual contact, as demonstrated by recent outbreaks of HCV among men who have sex with men (MSM) (2, 3). Similarly, HIV infection increases the risk of vertical transmission of HCV from a mother to her infant (4, 5). Finally, people living in resource-limited countries with reduced access to diagnosis and medical care are at higher risk of being infected with both viruses (6). In absence of an effective vaccine against neither HCV nor HIV, coinfection is a seriously growing public health problem, with 2.3 million individuals affected, of whom 1.3 million are PWID (7).
The immunopathology of HCV/HIV coinfection is more deleterious than each infection separately. HIV accelerates the development of HCV-related liver disease including advanced liver fibrosis, cirrhosis, hepatocellular carcinoma (HCC) and death (8). Similarly, HCV coinfection is a major cause of non-AIDS-related morbidity and mortality in people living with HIV (PLWH) as it is associated with reduced or slower CD4+ T cell reconstitution after antiretroviral therapy (ART) (9). In addition, recent studies have reported that direct acting antiviral (DAA)-mediated HCV cure leads to an increase in cell-associated HIV-DNA in the blood of ART-treated PLWH who had low viral reservoirs at baseline, likely as a consequence of viral reservoir mobilization from tissues (10, 11). Whether DAA-mediated cure of HCV represents an obstacle to HIV reservoir elimination in coinfected subjects remains an important question to address.
In this review, we will discuss the interplay between HIV and HCV during coinfection. Specifically, how HIV influences HCV infectious outcome and liver disease progression and how HCV coinfection modulates the HIV latent reservoir and immune reconstitution following DAA-mediated cure and relevance to clinical management of coinfected patients.
HCV Monoinfection
Epidemiology
HCV is transmitted primarily through exposure to contaminated blood. Sexual transmission and vertical transmission from an infected mother to her infant are low, but both transmissions increase in the context of HIV coinfection (3–5). It is estimated that 71 million individuals are chronically infected with HCV globally (12). Injection drug use is the main source of HCV infection in high income countries, while unsafe medical injections/procedures are the main source in the developing world (13). The opioid crisis in the USA, has led to doubling the national incidence of HCV infection between 2010 and 2014 and the numbers continue to rise (14). Globally, drug use accounts for ~23% of new HCV infections (15).
Viral Replication Cycle
The HCV genome consists of an uncapped positive single stranded RNA of approximately 9.6 kb-pairs [reviewed in (16)]. The genome represents an uninterrupted open reading frame (17), encoding a polyprotein precursor of approximately 3,000 amino acids including three structural proteins (Core, envelope glycoproteins E1, and E2) and seven non-structural (NS) proteins (P7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (16). HCV replicates primarily in human hepatocytes. Although HCV uses multiple receptors for entry into host cells, it is dependent on four main receptors on the surface of hepatocytes: CD81, a cell membrane tetraspanin protein, the scavenger receptor class B type I (SR-BI), and the tight junction proteins claudin-1 (CLDN1) and occludin (OCLN) (16, 18). The viral envelope glycoproteins fuse with the cellular membrane by clathrin-mediated endocytosis then the viral genome is released into the cytosol (16). This is followed by translation of the open reading frame of the HCV genome generating a large polyprotein that is later processed into mature structural and non-structural proteins (16). Junctions between structural proteins are processed by host signal peptidases while the non-structural proteins are processed by the NS2/3 autoprotease and the NS3/4A serine protease. Replication of the HCV RNA takes place within endoplasmic reticulum derived structures known as the membranous web through a negative strand intermediate in a replication complex (16). This is mediated by the viral NS5B protein that acts as an RNA-dependent RNA polymerase and regulated by the NS5A protein that plays many pleiotropic functions during HCV replication and assembly (19). The progeny virion is assembled into a nucleocapsid built from the structural core proteins around the viral RNA. This is followed by virion release with a surrounding membrane derived from the human cell with embedded heterodimers of the envelope glycoproteins E1 and E2 (16).
Natural History, Pathogenesis, and Immune Responses
The acute phase of HCV is empirically defined as the first 6 months post infection. Acute HCV is asymptomatic in the majority of infected subjects. Approximately 20–30% of infected subjects are able to clear the virus spontaneously during the acute phase while 70–80% develop persistent infection (20). As the virus continues to replicate in hepatocytes, it elicits persistent inflammation with increased expression of pro-inflammatory cytokines and chemokines from hepatocytes, the liver resident macrophages known as Kupffer cells, and other immune cells such as monocyte derived macrophages, natural killer (NK) cells and dendritic cells (DCs) that are recruited to the liver. Virus replication in hepatocytes also triggers hepatocyte damage with production of reactive oxygen species (ROS), damage associated molecular patterns (DAMPs) and some apoptosis. Altogether these mediators trigger the fibrogenic process (21). Hepatic stellate cells (HSCs), the main mediators of fibrosis, are activated by these different inflammatory signals and the cytokine TGF-β and start to express alpha-smooth muscle actin (αSMA) and collagen type I. Persistent inflammation leads to continued deposition of matrix proteins and imbalance in their degradation through increased expression of tissue inhibitors of metalloproteases (TIMPs) leading to liver stiffness and gradual loss of function (22). This chronic liver damage can progress to different stages of fibrosis, cirrhosis and HCC over 5-30 years (20). Multiple host factors including age, male sex, alcohol consumption and/or coinfection with HIV can accelerate progression to end-stage liver disease (23, 24).
HCV infection of hepatocytes triggers innate immune responses that are reviewed extensively elsewhere (25). Briefly, the cytosolic RIG-I-like receptor (RLR), retinoic acid-inducible gene-I (RIG-I), recognizes 5′-triphosphate double-stranded RNA (dsRNA) replicative intermediate and/or polyuridine (poly(U)) motifs within the HCV RNA. RIG-I then translocates to the mitochondria where it interacts with the mitochondrial antiviral-signaling protein (MAVS) to activate the downstream transcription factors IRF-3 and NF-κB, resulting in the induction of type I and type III interferons (IFNs) (25). HCV dsRNA is also sensed by the toll-like receptor 3 (TLR3) resulting in innate immune signaling through the adaptor molecule TRIF. HCV is able to evade innate immune responses by virtue of its NS3/4A protease that cleaves MAVS and inactivates its downstream signaling effect(s) (25) and by blocking the TLR3-mediated interferon signaling via NS4B-induced TRIF degradation (26). Finally, infected hepatocytes also shed exosomes carrying HCV RNA that are taken up by liver infiltrating plasmacytoid DCs (pDCs) that are then activated to produce type I and type III IFNs in the liver microenvironment (27). This IFN response is reflected as increased levels of interferon-induced genes (ISG), chemokines and inflammatory mediators in the liver thus activating liver resident inflammatory cells (28, 29). This response is also detectable in the peripheral blood. One notable ISG is CXCL-10 or interferon-gamma-inducible protein-10 (IP-10) that is elevated in plasma during acute HCV where high levels can predict failure to spontaneously resolve HCV (30). Levels of ISG induction correlate with single nucleotide polymorphisms in the IFN-lambda 3 (IFNλ3)/IFNλ4 region that are also associated with acute infection outcome [reviewed in (31)]. Following spontaneous resolution, ISG and inflammatory mediators return to normal levels but remain elevated in those who develop chronic infection (32). Natural killer (NK) cells are also activated during acute infection irrespective of infectious outcome and exhibit reduced production of cytokines and enhanced cytotoxic functions in chronic infection [reviewed in (33)].
A broad, polyfunctional and sustained virus-specific CD4+ and CD8+ T cell response is essential for spontaneous viral clearance [reviewed in (34)] where CD127+ virus-specific memory T cells develop (35–38). The abrupt disappearance of HCV-specific CD4+ helper T cell responses compromises CD8+ T cell function(s) and facilitates emergence of viral escape mutants (39–42) resulting in chronic infection. In chronic HCV, CD8+ T cells become exhausted and express different levels of exhaustion markers like programmed death-1 (PD-1), T cell immunoglobulin and mucin domain-containing protein 3 (Tim-3), and others [reviewed in (34, 43)], and lose effector functions (34). This is associated with downregulation of the transcription factor T-bet and different levels of expression of the nuclear factors T cell factor 1 (TCF1), Eomesodermin (Eomes), and thymocyte selection-associated high mobility group box protein (TOX) that distinguish different subsets of exhausted CD8+ T cells (44–46). TCF1+CD127+PD1+T-betlo HCV-specific CD8+ T cells expressing both exhaustion and memory markers and of limited functionality were described in HCV chronically infected subjects and termed “memory-like” or “stem-like” T cells (47, 48). While PD-1hiEomeshiTOXhiCD127- were reported to be a terminally exhausted subset (49). Recent single cell RNA sequencing (scRNA-seq) analysis revealed that TCF1+CD127+PD1+ memory-like cells are likely the progenitors of the PD-1hiEomeshiTOXhiCD127- terminally exhausted cells (49). Finally, escape mutations that occur in epitopes targeted by CD8+ T cells influence their phenotype as they can no longer see their cognate antigen despite persistent viremia. CD8+ T cells targeting epitopes that have escaped, express fewer exhaustion markers and revert to a memory phenotype where they express the memory marker CD127 and acquire transcriptomic and functional signatures that partly resemble memory CD8+ T cells generated following spontaneous HCV clearance (50–52).
Increasing evidence suggest an important role for antibodies (Abs) against the HCV glycoproteins E1 and E2 in spontaneous clearance (53, 54). Development of neutralizing Abs (NAbs) is generally delayed during acute infection (55–57). Although Ab responses are short lived in resolvers (58), they appeared early during acute resolving infection(s) in some subjects following expansion of activated circulating T follicular helper cells (cTfh) expressing IL-21 that help expansion of HCV-specific B cells (59). Generation of NAbs correlated with spontaneous resolution in other studies (17, 60). Preincubation of virus inoculum with anti-HCV Abs or passive immunization resulted in reduced viral loads and/or sterilizing immunity in animal models (61–65). Broadly neutralizing Abs (bNAbs) neutralizing multiple HCV genotypes, blocked infection in humanized mice (66–68) and were isolated from spontaneous HCV resolvers (69) underscoring their protective role.
Spontaneous HCV clearance generates long-lived memory T cells with enhanced protective immunity upon reinfection (58, 70). Subsequent HCV reinfections are typically of lower viral loads, shorter viremia and higher clearance rate (80-50% vs 25%) (71–74). In contrast, correlates of long-term protective immunity upon repeated HCV exposures are not well understood. Depletion studies in chimpanzees underscored the essential and complementary protective roles of CD4+ and CD8+ memory T cells in preventing HCV persistence (42, 70). Protection in PWID was associated with increased magnitude, breadth and, polyfunctionality of HCV-specific memory T cells (71, 72) and NAbs (71). Altogether, these results suggest that long-term protective immunity against HCV is possible and effective and provide strong rationale for vaccine development.
Treatments for HCV
For nearly 30 years, interferon-based therapeutic regimens were the only option for treatment of chronic hepatitis C. These regimens were long (lasting nearly a year), had numerous side-effects, and were successful in only 50% of the treated individuals. Direct acting antivirals (DAA), targeting several key steps in the HCV replication cycle, have become increasingly available in recent years (75). Currently approved DAA primarily target the NS3/4A protease, the NS5B polymerase and NS5A and are usually given in different combinations (76). DAA has substantially changed the management of chronic HCV, as HCV cure is achieved in >95% of cases (76). Nevertheless, DAA regimens are costly and access to diagnosis and treatment remains limited especially among the highest risk groups like PWID and MSM. Furthermore, DAA-mediated cure does not protect against HCV reinfection (77). Hence, the development of an effective prophylactic anti-HCV vaccine remains a priority to achieve HCV elimination.
HIV Monoinfection
Epidemiology
HIV-1 is transmitted by sexual contact across mucosal surfaces, by vertical transmission through maternal-infant exposure, and by percutaneous exposure (78). It is estimated that 37.9 million people are infected with HIV worldwide (79). HIV incidence continues to rise among PWID through sharing HIV-contaminated materials. In addition, unprotected sex is another important HIV risk factor among PWID, female sex workers, and MSM (80). HIV-infected PWID transmit HIV sexually to non-injectors, and through vertical transmission (81). Whereas overall, 10% of HIV-infected persons are coinfected with HCV, among PWID, HCV coinfection rates range from 50% to >90% (81). Other factors are associated with higher rates of HIV-1 acquisition such as high number of recent sexual partners, anal sex, concurrent sexually transmitted diseases, as well as the viral load and the clinical stage of the transmitting partner (82, 83). Access of PLWH to ART increased from 2.98 million in 2006 to 21.8 million in 2017 and was accompanied by a 51% reduction in HIV-associated mortality from 1.95 million in 2006 to 0.95 million in 2017. However, the annual incidence of new HIV infections decreased only by 17%. The combination of decreased mortality associated with ART implementation and only a small decrease in the incidence of new HIV infection has led to an overall important increase in the number of PLWH world-wide from 8.74 million in 1990 to 36.82 million in 2017 (84).
Viral Replication Cycle
HIV belongs to the genus Lentivirus within the Retroviridae family, with a virion containing two copies of positive single stranded RNA. The HIV genome of 9.2 Kb contains 9 genes encoding for multiple structural (gag, pol, env), regulatory (tat, rev), and accessory (nef, vpr, vif, vpu) proteins. The env gene encodes for the envelope glycoprotein gp160, that is further processed into gp120 and gp41. gp120 mediates HIV entry into target cells (e.g. CD4+ T cells and macrophages) through interaction with the CD4 molecule, which is the main receptor for the virus (85), as well as the chemokine receptors CCR5 and/or CXCR4, which are the main HIV coreceptors (86, 87). The molecular tropism of HIV, dictated by the capacity of specific viral strains to use CCR5 and/or CXCR4 for entry led to classification of HIV into CCR5-tropic (R5), CXCR4-tropic (X4) or dual-tropic (R5/X4) strains (87). Once gp120 binds to CD4, the envelope undergoes conformational changes, exposing the chemokine receptor binding domains of gp120 and allowing its interaction with the coreceptors (88). This leads to further conformational changes allowing gp41 to expose its fusion peptide, which penetrates the cell membrane to deliver the viral capsid inside the target cells (89). The HIV reverse transcriptase (RT) encoded by the pol gene initiates the process of reverse transcription by copying the positive-sense single-stranded RNA genome into a complementary DNA (cDNA) (90). The cDNA and its complement form a double-stranded viral DNA, which is then integrated into the host cell’s genome by the viral integrase (IN) encoded by the pol gene as well (91, 92). HIV integration is followed either by active viral transcription and production of new virion progeny or viral latency, with integrated proviral DNA persisting in many cells and tissues as latent HIV reservoirs (90, 93). The persistence of latent reservoirs despite viral-suppressive ART represents a major barrier to HIV cure (94, 95).
Natural History, Pathogenesis, and Immune Responses
The course of HIV infection includes three phases: acute, chronic, and Acquired Immunodeficiency Syndrome (AIDS) (96, 97). The acute HIV infection phase, between the time of HIV acquisition to seroconversion, can be further subdivided in four Fiebig stages (I-IV), based on the sequential detection of HIV RNA, proteins, and antibodies (98). The acute phase is associated with high levels of viremia and wide HIV dissemination into lymphoid organs (99), where the virus encounters its main targets: CD4+ T cells, macrophages and dendritic cells (100–102). The concurrent HIV-specific immunity initiated during the acute phase is associated with a dramatic decline in viremia (96, 97). However, this immunity is inadequate to suppress viral replication completely, with HIV replication persisting in lymph nodes even when plasma viremia is undetectable (96, 99). After the acute phase, most patients have a period of viral latency, in which the viral genome is stably integrated in the host cell genome (96, 99). While CD4 counts normalize in the peripheral blood at the end of the acute phase, alterations in CD4+ T cell homeostasis were documented in the gut-associated lymphoid tissue very early upon infection and persist during the chronic phase (96, 99, 103). These alterations are at the core of HIV pathogenesis, with a “leaky gut” being the source of microbial translocation, chronic immune activation, systemic inflammation and disease progression (104, 105). During the chronic phase, the state of viral latency is reversed upon activation of latently infected CD4+ T cells upon interaction with their cognate pathogens leading to productive infection (93) and massive depletion of central memory CD4+ T cells, which are the “self-renewing” source for tissue effector memory CD4+ T cells (97). The HIV-dependent depletion of CD4+ T cells is mediated through pyroptosis (106), apoptosis of uninfected bystander cells (107), and CD8+ cytotoxic T cell killing (108). When CD4+ T cell counts decline below the limit of 200 cells/μl of blood (109), cell-mediated immunity is severely compromised, resulting in AIDS, a fatal complication characterized by life-threatening opportunistic infections and cancers (97).
In the absence of ART, the duration of the chronic phase is highly variable and can last for several years. While the majority of PLWH progress rapidly to AIDS, a minority remain immune competent (long-term non-progressors, LTNP) and a subset of those efficiently control HIV replication (elite controllers, EC) (110). LTNP are often asymptomatic for 10–20 years with CD4+ T cell counts maintained within the normal range (>500 cells/μl) and plasma viral loads between 5,000 and 15,000 RNA copies/ml (111, 112). On the other hand, EC represent a small subset of the HIV-positive population (< 1%) who are able to control HIV viremia below the limit of detection in the absence of ART (113). Nevertheless, disease progression occurs as well in LTNP and EC, thus justifying the current recommendations of early ART initiation (114, 115). The natural control of HIV infection is achieved via multiple mechanisms including the expression of intrinsic restriction factors that limit HIV replication, the development of efficient cellular immune responses (116) in association with specific protective HLA class I alleles (117) and humoral immune responses. Furthermore, natural resistance to HIV acquisition was described in sex workers in Africa and represents a reference for studying correlates of HIV mucosal immunity (118). Finally, early treatment initiation renders a fraction of PLWH able to control HIV infection following treatment interruption (119), thus supporting the concept of “functional HIV cure” (120). However, about 10 to 40% of ART-treated individuals are immunological non-responders (INRs) (121, 122); these PLWH fail to reconstitute their CD4+ T cell frequencies and functions despite controlled viremia under ART (123). This is due to the incapacity of ART to clear HIV reservoirs and diminish inflammation (124). This sub-group is at higher risk for developing AIDS and non-AIDs related morbidity and mortality (124–127).
HIV-specific CD8+ T cells are also key determinants of the course of natural HIV infection. Almost all HIV infected individuals mount a strong virus-specific CD8+ T cell response during the acute phase and the polyfunctionality of these CD8+ T cells is key to long-term control of viremia and limiting disease progression [reviewed in (128)]. Many of the evasion mechanisms employed by HCV are also used by HIV including escape mutations in targeted epitopes and CD8+ T cell exhaustion and are detailed elsewhere (128). High-dimensional functional and phenotypic analysis revealed phenotypic heterogeneity among HIV-specific CD8+ T cells as well as common exhaustion signatures and pathways that are shared not only among chronic viral infections like HCV, HIV and LCMV but also in cancer (49, 129). These include increased expression of Eomes and TOX as key features of exhausted T cells associated with disease severity in HIV infection while CD8+ T cells expressing lower levels of inhibitory receptors like PD-1 and higher levels of CD127, TCF1 and/or CXCR5 were associated with higher functionality, proliferation and better control of viral replication (129–131). Moreover, CXCR5+CD8+ T cells express less inhibitory receptors levels with higher cytotoxicity (132), which could contribute to improved control of viral reservoirs in lymph nodes particularly since lymphoid tissue resident memory CD8+ T cells were shown to express higher levels of in CXCR5, compared with blood memory CD8+ T cells (133) and that higher frequencies of follicular CXCR5+ CD8+ T cells correlated with lower rates of peripheral viremia (134).
HIV Treatment
ART transformed HIV infection into a manageable chronic disease (78). Multiple antiviral drugs are now available that target different steps of the HIV replication cycle including, in order of discovery, reverse transcription, protease-mediated virion maturation, entry, fusion, and integration (78). However, ART does not eradicate HIV and viral rebound occurs rapidly upon treatment interruption due to the persistence of latent HIV reservoirs in long-lived memory CD4+ T cells (94). To date, HIV cure was achieved in only two people who received bone marrow transplantation to treat cancer using donor cells carrying the delta 32 mutation in the ccr5 gene (CCR5δ32/δ32) (135), a mutation known to confer resistance to CCR5-tropic HIV infection (86).
ART is also important as a preventative measure. Early initiation of ART reduces sexual transmission of HIV-1 (136), particularly in genetically linked HIV-1 infections among serodiscordant couples (137). Pre-exposure prophylaxis (PrEP) using daily oral doses of the combined nucleoside reverse transcriptase inhibitors, tenofovir and emtricitabine, conferred higher protection against HIV than placebo in controlled trials (138–140). Furthermore, an injectable, long-acting integrase inhibitor, cabotegravir, is safe and well-tolerated and two large efficacy trials are underway in uninfected high-risk individuals (140, 141). In the past ten years, broadly neutralizing antibodies (bNAbs) targeting different HIV-1 envelope epitopes isolated from PLWH with slow disease progression were engineered (142, 143) and are currently being tested for both efficacy and safety as a potential alternative or adjuvant for ART (144).
HCV/HIV Co-Infection
Epidemiology
Approximately 6.2% of PLWH are also co-infected with HCV. The odds of HCV infection are six times higher in PLWH than in the HIV-negative population, with the highest prevalence among PWID and MSM, consistent with shared routes of transmission (7). Although parenteral transmission remains the principal route of HCV transmission in PWID (7), sexual transmission of HCV occurs among HIV-positive MSM (3, 145, 146).
Natural History, Pathogenesis, and Immune Responses
Multiple factors shape the natural history of coinfection including genotype/subgroup of both viruses, host factors and socioeconomic factors including access to screening and treatment services (147). As reviewed above, virus-specific CD8+ T cell responses are essential for spontaneous resolution of acute HCV infection. The magnitude, breadth and polyfunctionality of the HCV-specific CD8+ T cells are key determinants of infectious outcome. These functions are dependent on CD4+ T cell help (34), and virus persistence during acute HCV is temporally associated with abrupt loss and/or dysfunction of virus-specific CD4+ helper T cells (148). As demonstrated in CD4+ T cell depletion studies in the chimpanzee model of HCV, loss of CD4+ T cells function was associated with decline in frequency of virus-specific CD8+ T cells and their cytokine production resulting in accumulation of escape mutations in the targeted CD8 epitopes with impairment of HCV-specific immune responses (42). CD4+ helper T cells are also essential for optimal B cell function and production of neutralizing antibodies. Specifically, antigen-specific B cell affinity maturation, isotype class switching and differentiation into antibody-secreting plasma cells in germinal centers are regulated by direct contact with CD4+ Tfh cells (59, 149).
Given the central role of CD4+ T cells in coordinating antiviral immune responses, it is not surprising that the immune response to HCV in the context of an HCV/HIV coinfection is compromised. HIV infection impairs the immune response to HCV, including in people who have cleared HCV infection (150). HIV-1-infected individuals with spontaneous control of HCV remain at significant risk for a second episode of HCV viremia (150). The breadth and magnitude of HCV-specific CD8+ T cells correlated with CD4+ T cell count (151). Similarly, coinfection and CD4+ T-cell counts <350/mm3 was associated with a global decline in the anti-HCV envelope antibody response, including binding antibody titers, NAb titers, and NAb breadth (152). These immunological factors are the likely cause of the observed lower rate of spontaneous clearance of acute HCV in HIV coinfected subjects (5-10%), as compared to HCV monoinfection (20-30%) (153), especially in patients with low CD4+ T cell counts (154, 155). Furthermore, the overall reduction in CD4+ T cells, microbial translocation, and systemic immune activation have deleterious effects on the liver with accelerated progression of liver fibrosis, early onset of cirrhosis and HCC, as compared to monoinfection (8)(discussed in the next section).
Liver Disease in HCV/HIV Coinfected Individuals
HCV/HIV co-infected individuals are at a higher risk of progressive liver disease, with accelerated progression to liver cirrhosis and higher rates of HCC, even when HIV is controlled by ART (24, 155, 156). HIV coinfection accelerates HCV-mediated fibrosis through both direct and indirect mechanisms that will be discussed in this section and are summarized in Figure 1.
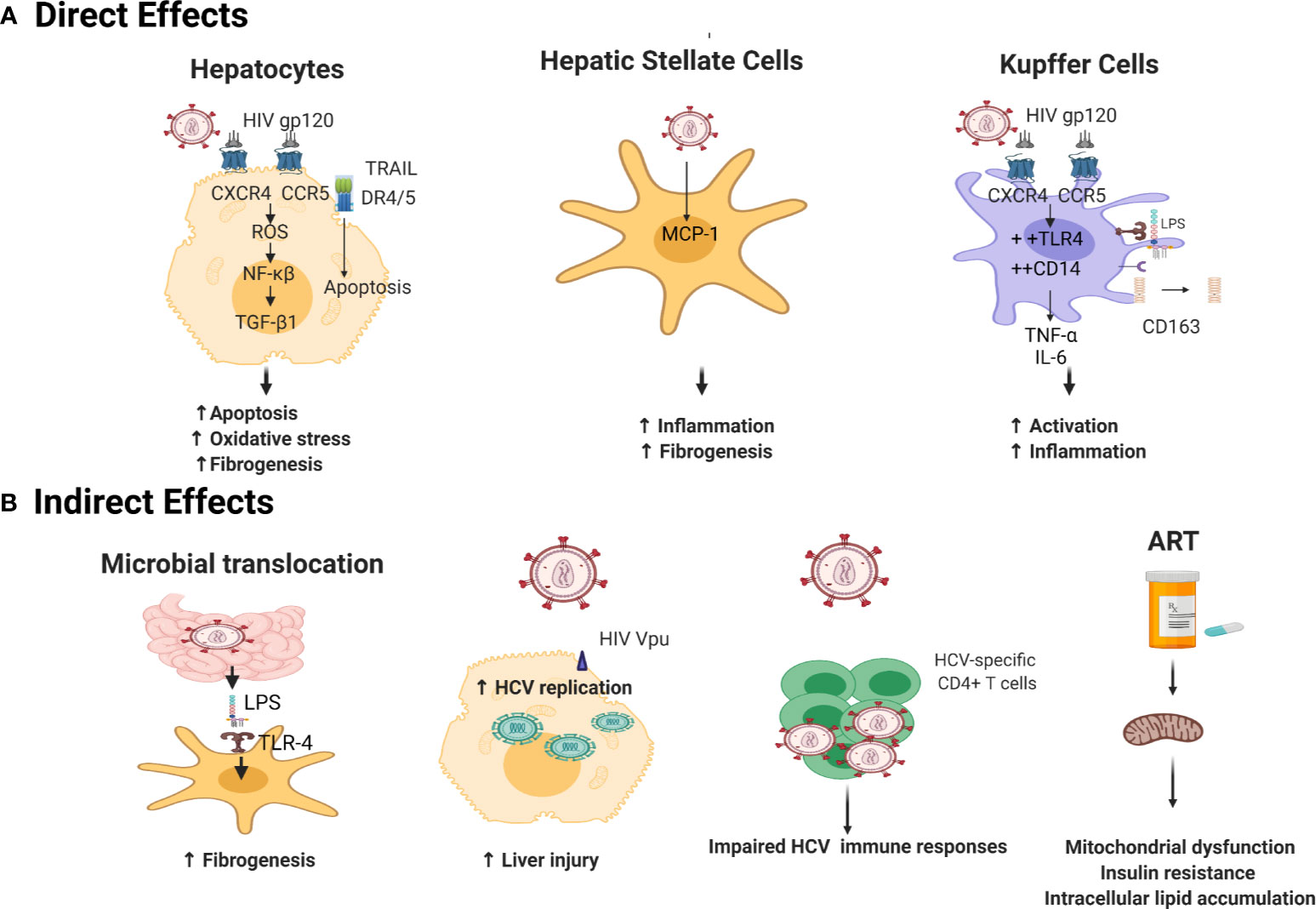
Figure 1 Mechanisms underlying acceleration of liver disease progression by HIV in HCV/HIV coinfection. (A) Direct Mechanisms: gp120 binds to HIV coreceptors (CXCR4 and CCR5) on hepatocytes surface (157) leading to accumulation of ROS which triggers an NF-κβ mediated oxidative stress (158, 159). HIV also activates TNF–related apoptosis-inducing ligand (TRAIL)-mediated apoptosis via upregulation of TRAIL receptor 1 (DR4), and 2 (DR5) (160), aggravating HCV fibrotic complications (161). HIV promotes hepatic stellate cells (HSC) collagen I expression and secretion of the proinflammatory cytokine monocyte chemoattractant protein-1 (MCP-1) inducing inflammation and fibrogenesis (162). HIV sensitizes Kupffer cells to lipopolysaccharide (LPS) via gp120 binding to CXCR4 and CCR5 (163) increasing cell surface expression of CD14 and TLR4, resulting in increased secretion of TNF-α and IL-6 (164). This is accompanied by shedding of CD163 in serum (165). (B) Indirect Mechanisms: HIV indirectly augments fibrosis mainly by microbial translocation: LPS translocate through impaired gut epithelium (105) and binds to Toll-like receptor 4 TLR4 on HSCs, leading to upregulated chemokine secretion and Kupffer cells chemotaxis (166). HIV accessory proteins Vpr (167) and Nef (168) enhance HCV RNA replication creating a state of inflammation and tissue damage. HIV causes functional alterations of HCV-specific immune responses with more HCV replication and hepatic inflammation (150, 169). ART elicits insulin resistance by altering mitochondrial function (170, 171) and increased intracellular lipid accumulation (172, 173) leading to enhanced development of liver fibrosis (174). Created with Biorender.com.
Direct Effect of HIV on Liver Disease Progression
HIV gp120 interacts with CXCR4 and CCR5 on the surface of hepatocytes and HSCs, respectively (157, 175). This interaction activates HSCs and promotes ROS accumulation through an NF-κB-dependent mechanism, resulting in enhancement of the fibrogenic process (158, 159). Co-culture experiments in the presence of HCV, HIV or both HIV and HCV demonstrated an additive effect of HIV on the profibrogenic program in hepatocyte and HSC lines through ROS, NF-κB, and TGF-β1 up-regulation; resulting in increased expression of the extracellular matrix protein collagen I, and TIMP1 (176).
It was also reported that HIV can infect HSCs in a CD4/chemokine coreceptor-independent manner. HIV infection promoted HSC collagen I expression and secretion of the proinflammatory chemokine monocyte chemoattractant protein-1 (MCP-1) (162). HIV was also shown to infect Kupffer cells, the liver resident macrophages that orchestrates the intrahepatic inflammatory response. Kupffer cells express the HIV receptor CD4 and both coreceptors CCR5 and CXCR4 [reviewed in (163)]. In vitro infection of Kupffer cells by HIV led to their sensitization to lipopolysaccharide (LPS) treatment by increasing cell surface expression of CD14 and TLR4, resulting in increased secretion of TNF-α and IL-6. This effect was maintained even after suppression of HIV replication using antiretroviral drugs (164). Activation of macrophages/Kupffer cells is mirrored by increased levels of the soluble macrophage activation marker CD163 in serum of HIV/HCV coinfected subjects accompanied by periportal CD163+ macrophage accumulation during fibrosis progression but not in established cirrhosis suggesting that soluble CD163 is probably a marker of active fibrogenesis rather than accumulated fibrosis (165).
HIV also enhances apoptosis of hepatocytes, another accelerator of the fibrogenic process. This effect is mediated by TNF–related apoptosis-inducing ligand (TRAIL) signaling and upregulation of TRAIL receptor 1 (DR4), and 2 (DR5) (160). The TRAIL-dependent hepatocyte apoptosis in HCV infected individuals is aggravated by HIV coinfection (161). In addition, HIV gp120 and HCV-E2 protein, can collaboratively elicit hepatocyte apoptosis in vitro through upregulation of Fas ligand expression and dephosphorylation of the anti-apoptotic molecule AKT (161, 177).
Indirect Effect of HIV on Liver Disease Progression
The principal mechanism through which HIV indirectly accelerates liver disease progression is increased microbial translocation. HIV infection induces severe depletion of the gastrointestinal lymphoid tissue, impairing the gastrointenstinal tract epithelial integrity and allowing bacterial LPS to translocate from the gut to the systemic circulation and through the liver (105). LPS binds to TLR4 on the surface of quiescent HSCs, leading to upregulated chemokine secretion and chemotaxis of Kupffer cells. Concurrently, this interaction induces downregulation of the TGF-β pseudoreceptor Bambi to sensitize HSCs to TGF-β-induced signals from Kupffer cells (166). Activation of HSCs is further aggravated by HIV-mediated depletion of Kupffer cells that are responsible for LPS phagocytosis (163, 178).
Elevated levels of microbial translocation markers (e.g. LPS, LPS binding protein, soluble CD14) were reported in HCV-infected subjects upon HIV acquisition and compared to HIV uninfected subjects (179). In addition, decreased levels of endotoxin-core antibodies (EndoCAb IgM) and increased levels of IgG, specific for a heterophilic (alpha-galactose) epitope, which has previously been associated with stage of liver disease (180), were observed in HIV-infected compared to uninfected subjects (179). All these measures were strongly associated with HCV-related liver disease progression to cirrhosis (179).
Other indirect mechanisms include the enhancement of HCV RNA replication via the HIV accessory proteins Vpr (167) and Nef (168); the accumulation in the liver of HIV-specific CD8+ T cells creating a state of inflammation and tissue damage in co-infected patients (181, 182); and functional alterations of HCV-specific immune responses in the presence of HIV coinfection (150, 169). Also, the CD4/CD8 functional imbalance seen during HIV infection modifies the hepatic cytokine milieu, in favor of a profibrotic state in the liver (155, 183). Finally, HIV induces upregulation of the Triggering Receptor Expressed on Myeloid cells 1 (TREM1) on Kupffer cells and induces extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) cascade leading to increased macrophage inflammatory response (184).
The effect of ART on liver injury in co-infected patients is controversial. Early studies suggested that nucleoside reverse transcriptase inhibitors (NRTIs) and protease inhibitors may affect mitochondrial function leading to increased insulin resistance (170, 171), and development of liver fibrosis (174). This effect is mainly mediated by polymerase-gamma suppression, a DNA polymerase necessary for mitochondrial DNA replication (185). Another proposed mechanism for ART-induced mitochondrial insufficiency is the inhibition of mitochondrial adenylate kinase and adenosine nucleotide translocator (186, 187). Studies using hepatocyte cell lines have demonstrated that non-nucleoside reverse transcriptase inhibitors (NNRTIs) can interfere with mitochondrial function leading to increase intracellular lipid accumulation which can further enhance liver damage (172, 173). In contrast, other reports suggested that ART enhances HCV-specific T cell responses with a significant decline in HCV-RNA levels (188) and reduction in the rate of hepatic decompensation events and liver fibrosis (189, 190). Additional studies are needed to evaluate the effect new regimens of ART on liver functions.
Effect of HCV Infection on HIV Disease Progression
HCV infection impacts the natural history of HIV with increased risk of developing AIDS defining illness like HIV-related bacterial and mycotic infections in coinfected subjects (191). HCV likely contributes to increased immune activation in chronic HCV/HIV coinfection resulting in impaired immune responses and rapid progression to AIDS (192). The different effects of HCV on HIV disease progression are presented in this section and summarized in Table 1.

Table 1 Impact of HCV on HIV disease progression.
Impact of HCV on CD4+ and CD8+ T Cells
HCV seroconversion of HIV-infected subjects is associated with reduced CD4+ T cell counts during the first few years and their reduced recovery after ART (204, 205). This deficit could be attributed to the nature of the liver as a lymphoid organ essential for maintaining T cell homeostasis, where hepatic inflammation induced by HCV infection leads to altered T cell homeostasis (193). Similarly, persistent liver inflammation may contribute to enhanced immune activation and generalized exhaustion of CD8+ T cells with the upregulation of various exhaustion molecule like PD-1, Tim-3 and CD39 on total and virus-specific CD8+ T cells (195–197).
Modulation of HIV Replication by HCV
HCV proteins may modulate HIV replication through multiple mechanisms [reviewed in (206)]. The HCV non-structural protein NS3/4A can activate HIV transcription through its LTR by enhancing DNA binding of the transcription factor AP-1 (199). The NS3/4A effect on HIV transcription was facilitated by the HIV protein Vpu, which mediates ubiquitination and subsequent degradation of NS4A allowing the destabilization of the NS3/4A complex and the nuclear translocation of NS3 (200). Other studies demonstrated that both, HCV core protein and HIV Nef can bind directly to the tumor necrosis factor receptor-associated factor (TRAF) 2, TRAF5, and TRAF6 and initiate the NF-κB signaling cascade thus enhancing HIV replication in monocyte-derived macrophages (201). Another in vitro study demonstrated that HCV core potentiates HIV‐1 replication in macrophages via upregulation of TNF‐α and IL‐6 via TLR2, JNK, and MEK1/2‐dependent pathways. Furthermore, TNF‐α and IL‐6 secreted from HCV core‐treated macrophages reactivated pro-viral HIV-1 DNA in the latently infected monocytic U1 cell lines (207). In contrast, another study, using the HepG2 hepatocyte cell line, demonstrated that Tat-induced LTR activation was suppressed by HCV core protein restricting HIV-1 transcription and replication. This core mediated LTR inhibition was unchanged when NS3/4A protein was added to the culture. However, LTR activation and gene transcription were enhanced by infectious HCV virions suggesting the involvement of other viral or cellular proteins in this process (208).
HIV Reservoirs in HCV Coinfected Subjects
Few studies to date explored the effect of HCV coinfection on the HIV reservoir size (7). A recent study has demonstrated a larger HIV reservoir size in resting CD4+ T cells in ART-treated HCV/HIV coinfected individuals with chronic HCV or who have spontaneously resolved HCV as compared to HIV mono-infected subjects (209). This increased HIV reservoir size represents a major obstacle to HIV elimination in those co-infected subjects and can be explained by several theories. First, increased immune activation during HCV coinfection, where persistently activated HCV-specific and non-specific CD4+ T-cells provide new targets for HIV infection and latency, as previously observed with herpesviruses (202). This T cell activation may facilitate de novo infection, especially since the concentrations of antiretroviral drugs reaching lymphoid tissues are low (210). Another theory is the possibility that impaired HIV-specific cell-mediated immunity, responsible for clearance of HIV-infected cells, contributes to HIV reservoir persistence. This immune suppression may be a consequence of chronic HCV coinfection where higher frequencies of peripheral regulatory T cells (Tregs) and IL-10 producing cells were detected as compared to HIV monoinfected subjects (203). Finally, whether HCV-specific CD4+ T cells represent HIV infection targets and sites of HIV reservoir persistence remains to be investigated. Previous studies reveled that in addition to HIV-specific CD4+ T cells (211), increased HIV infection occurs in mycobacterium tuberculosis (MTb)-specific CD4+ T cells, while cytomegalovirus (CMV)-specific CD4+ T cells showed impaired permissiveness to HIV (212). Recently, several groups characterized the antigenic specificity of CD4+ T cells carrying HIV reservoirs, in an effort to therapeutically prevent HIV reservoir seeding in pathogen-specific cells or to target their specific elimination in HIV cure strategies (213, 214). We have recently demonstrated that integrative HIV infection in Staphylococcus aureus-reactive CD4+ T cells can be promoted by DCs in a retinoic acid-dependent manner (215) and the liver is an organ rich in retinoic acid (216). The liver is also an organ rich in Th7 cells (217), a subset of CD4+ T cells transcriptionally programmed to be HIV infection targets (218), with a considerable fraction of HCV-specific CD4+ T cells bearing the phenotypic and functional characteristics of Th17 cells (219). Therefore, it is tempting to speculate that HIV infection and viral reservoir persistence in HCV-specific CD4+ T cells may explain differences observed between HIV monoinfected and HIV/HCV coinfected individuals in terms of HIV reservoir size. The clonal expansion of HIV infected memory CD4+ T cells is a well-known mechanism, by which HIV latent reservoir is maintained and expanded (214, 220). A similar mechanism may be at play during concomitant HCV infection, potentially conferring preferential susceptibility of HCV-specific CD4+ T cells to HIV infection and reservoir expansion.
Effect of DAA-Mediated Cure of HCV in Coinfected Subjects
Before the DAA era, HCV treatment and cure in HCV/HIV coinfected subjects was a complex problem since long IFN-based therapeutic regimens had multiple side effects and the rate of sustained viral response (SVR) was much lower than in monoinfected subjects. IFN-free DAA regimens that are widely available now have been a major game changer given their relative short treatment duration, reduced side effects and a response rate of >95%. In the context of HIV/HCV coinfection however, early studies suggested a lower SVR rate to DAA (86.3%) as compared to monoinfection (94.9%) (221). Nevertheless, most recent studies reported response rates to DAA in HIV/HCV coinfection as comparable to those in HCV-monoinfected individuals (222). Predictors of failure to achieve SVR under DAA in coinfected individuals are not different from those observed in monoinfected people and include sex, immune status, HCV RNA load, severity of liver disease, and the use of suboptimal DAA-based regimens (129). Given these promising results, HCV/HIV coinfected subjects are no longer considered difficult to reach an HCV cure. Nevertheless, specific consideration should be given to negative predictors of SVR and barriers to treatment that may be more common in the coinfected population (223). Data from several cohorts have identified a clear benefit of HCV cure in limiting liver disease progression and reducing the risk of developing HCC in PLWH, especially if they are treated at an early fibrosis stage (224, 225). In this section, we will discuss the impact of DAA-mediated cure of HCV on liver disease progression, immune reconstitution and the HIV reservoir in coinfected subjects. These points are also summarized in Table 1.
Immune Reconstitution in HCV/HIV Coinfected Subjects Following DAA
DAA-mediated cure of HCV induces rapid decrease in serum levels of sCD163, a marker of inflammatory macrophage activity, and is associated with reduced histological inflammation in the liver (226). DAA therapy was also accompanied by a rapid decline in ISG expression in the liver and peripheral blood (227). Multianalyte profiling of 50 plasma proteins pre- and post DAA-mediated clearance in a cohort of 28 subjects with chronic HCV infection, including two HIV coinfected subjects, demonstrated that the elevated plasma cytokines and chemokines improve but do not completely normalize up to 8 months post cure (228). Interestingly, CXCL10/IP-10, an IFN induced chemokine, is usually elevated during chronic HCV infection and it rapidly decreased upon starting DAA therapy in line with reduced IFN-α signaling in the liver (228, 229). This was also associated with the normalization of NK cell phenotype and function (cytotoxicity and cytokine expression) (229)
The analysis of peripheral blood lymphocytes early during DAA therapy reported an increase in total CD4+ and CD8+ T cells but not NK cells or monocytes (230). At the same time, a reduction in activated (HLA-DR+ and CD38+) CD4+ and CD8+ T cells was observed in both monoinfected and coinfected subjects (230), with this effect being sustained up to one year post-SVR (231). Furthermore, there was an increase in the frequency of T cells expressing CXCR3, the CXCL10/IP-10 receptor (230), indicative of T cell redistribution in the periphery upon DAA treatment. Indeed, a reduction in liver inflammation may theoretically cause an efflux of lymphocytes from the liver and draining lymph nodes in the peripheral blood.
In HCV monoinfection, DAA treatment was associated with restoration of the proliferative capacity of HCV-specific CD8+ T cells (198). However, selective maintenance of TCF1+CD127+PD1+ memory like HCV-specific CD8+ T cells was observed following cessation of therapy (47), as well as a limited impact of DAA on the functional and mitochondrial impairment of HCV-specific CD8+ T cell responses (232). In contrast, Barili et al. reported partial improvement of the mitochondrial metabolic functions of HCV-specific CD8+ T cells following DAA treatment (233). However, they used in vitro stimulated PBMCs which may have affected mitochondrial function (233). Bulk RNA-seq and scRNA-seq analysis of HCV-specific CD8+ T cells identified an antigen-dependent core exhaustion signature, with memory-like CD8+ T cells targeting variant epitopes exhibiting a less pronounced exhaustion signature (49, 52). HCV-specific memory-like T cells harbored the same transcriptomic signatures before and after DAA treatment suggesting a permanent “exhaustion scar” that could not be reversed by DAA-mediated HCV cure (49). Only one study examined HCV-specific CD4+ T cells in subjects undergoing DAA treatment. In this study, the frequency of HCV-specific CD4+ T cells increased 2 weeks after starting DAA, with downregulation of the exhaustion and activation markers (CD38, CD39, ICOS, OX40 and PD-1) and upregulation of the memory-T cell markers (CD127 and CCR7) without complete normalization (234). As with total CD4+ T cells, this transient increase in HCV-specific CD4+ T cells early after starting DAA therapy is likely due to decreased liver inflammation and emigration of HCV-specific CD4+ T cells from the liver. It is noteworthy that HCV-specific CD4+ T cells with a Tfh phenotype and transcriptional signature were preferentially maintained following DAA-mediated HCV cure (234).
Conflicting results were reported in the setting of DAA therapy in HCV/HIV coinfection. One study reported a reduction in total activated (HLA-DR+ and CD38+) CD4+ and CD8+ T cells up to one year post-SVR (231). Another study did not observe any change in T cell activation at 12 weeks after DAA compared to baseline (235) but observed an increase in the frequency of total CD4+ and CD8+ T cells producing IFN-γ, IL-17, and IL-22 (235).
The frequency of Foxp3+CD25+CD4+ Tregs is usually elevated in chronic HCV infection and contribute to the immune suppressive and anti-inflammatory immune response in the liver. However, DAA-mediated clearance of HCV does not completely normalize frequency of Tregs in neither monoinfected (236) nor coinfected subjects (237). Similarly, the frequency of HLA-DR-CD33+ CD11b+ myeloid-derived suppressor cells (MDSC) are expanded in HCV monoinfected (238) and coinfected subjects (237). MDSC play a crucial role in immune suppression via production of arginase‐1, inducible NOS, TGF-β and IL-10 that inhibit T cell functions (239). The frequency of MDSC remains elevated post-DAA therapy in HCV/HIV co-infected subjects and may contribute to generalized immune suppression (237).
In summary, studies examining DAA-mediated cure of HCV, mostly in monoinfected subjects, suggest only partial reconstitution of immune functions and persistence of immune suppressive cells like Tregs and MDSC. This immune suppressive environment may contribute to reduced immune response to vaccinations, limited immune surveillance against cancer, and increased reactivation of latent or occult infections of herpesviruses and hepatitis B virus, respectively (240, 241). Finally, this incomplete reconstitution of HCV-specific CD4+ and CD8+ T cells may increase the risk of HCV persistence upon re-exposure and reinfection in high-risk populations like PWID and MSM. Additional studies examining immune reconstitution post DAA-mediated cure in HCV/HIV coinfected subjects are warranted.
Liver Disease in HCV/HIV Coinfected Subjects Post DAA-Mediated Cure
As discussed above, DAA-mediated cure of HCV causes an improvement in liver fibrosis markers in both HCV monoinfected and HCV/HIV coinfected subjects (225, 242, 243). Furthermore, recent cohort studies suggest that HCV/HIV coinfected subjects with cirrhosis are no longer at higher risk for developing HCC or end-stage liver disease as compared to HCV monoinfection (225). However, the risk of developing HCC is not completely eliminated upon HCV cure and remains high in subjects with advanced fibrosis or cirrhosis pointing to an irreversible liver damage. Molecular studies using HCV-infected hepatocytes, as well as studies using liver biopsy samples from infected patients, have revealed that chronic HCV infection induces epigenetic and gene expression alterations associated with risk for HCC, alterations that persist after HCV cure (244, 245).
Other factors may contribute to continued risk of developing end-stage liver disease and HCC in HCV/HIV coinfected subjects. First, metabolic abnormalities including insulin resistance are quite common in HIV-infected individuals and result in hepatic steatosis with accumulation of triglycerides in hepatocytes, and can accelerate liver damage and increase risk of developing HCC irrespective of HCV status [reviewed in (246)]. Second, gut dysbiosis and intestinal damage are hallmarks of HIV infection that are not restored by ART [reviewed in (247)]. Considering the fact that DAA-mediated HCV clearance does not impact gut dysbiosis in cirrhotic HCV-monoinfected subjects (248), dysbiosis is highly likely to remain a prominent risk of liver damage in the context of HCV/HIV coinfection after DAA-mediated cure. Thus, new therapeutic interventions may be needed to normalize the metabolic status and restore intestinal health in DAA-treated HCV/HIV coinfected subjects.
Impact of DAA Treatment on HIV Reservoir
The HIV reservoir levels have been intensively studied in ART-treated HIV infected subjects (249, 250), but little is known about HIV persistence in the context of HIV/HCV coinfection, especially following DAA treatment. A study performed on PBMCs revealed that DAA-mediated HCV clearance did not induce a decrease in HIV reservoirs but rather an increase in integrated HIV-DNA levels, mainly in patients with low versus undetectable levels of HIV viremia before DAA (251). Similarly, Rozera et al. observed a significant rise in HIV-DNA levels in the peripheral blood of some successfully ART-treated HIV/HCV co-infected patients at the end of DAA treatment, which correlated with lower cellular HIV reservoir at baseline (10). Most recently, a study of 97 ART-treated subjects reported that HCV/HIV coinfected subjects with chronic HCV infection, as well as HCV/HIV coinfected subjects who have spontaneously cleared HCV, showed higher levels of HIV-DNA in resting CD4+ T cells (CD25-CD69-HLADR-) compared to HIV monoinfected individuals (209). This increase in HIV reservoir size following DAA therapy may be explained by the mobilization of cells carrying HIV reservoirs from lymph nodes, liver and other tissues into the peripheral blood in response to DAA-mediated reduction of HCV-elicited immune activation and hepatic inflammation (10, 252). This is mirrored by the rapid decline of CXCL10 levels in response to DAA therapy, similar to plasma HCV RNA levels (253). In contrast, a new study demonstrated that although all three forms of HIV DNA (total, integrated, and episomal) remained stable during DAA treatment, cell-associated unspliced HIV-RNA levels (giving rise to the progeny virions) were significantly increased 12 months after the end of DAA therapy, suggesting an increased HIV transcriptional activity in reservoir cells (11). This may be explained by a decrease in IFN-mediated antiviral immunity as reflected by the observed reduction in ISG signals (227) and normalization of the expression levels of IFN-β, IFI44, and CXCL10 in the peripheral blood following DAA therapy (254). In addition, as reported by Meissner et al, liver biopsies from patients who achieved SVR showed that HCV cure was accompanied by decreased expression of type II and III IFNs, but higher expression of type I IFNs compared to pre-treatment baseline (227). Recently, diverse and opposing effects of the type I IFNα on HIV latency were described. IFNα inhibits the establishment of latency. However, once latency is established, IFNα is able to reverse it through binding to its receptor on CD4+ T cells and triggering phosphorylation of STAT1, 3, and 5 proteins (255). It is well-established that STAT5 can activate HIV transcription via its binding to the HIV long terminal repeat (256).
Altogether, the limited data available so far suggest that the HIV reservoirs are higher in HCV/HIV coinfected compared to HIV monoinfected subjects and that DAA-mediated HCV cure does not reduce HIV persistence. However, several limitations prevent the generalization of these findings because some studies used purified memory CD4+ T cells while others used total PBMCs to quantify HIV DNA (10, 11). Another issue is the extensive use of PCR-based techniques to assess the integrated HIV-DNA. These techniques tend to overestimate the size of the reservoir due to the high prevalence of defective proviruses (257). Other confounding factors include the pre-ART HIV viral load, host genetic factors and liver condition. Additional studies with well-defined cohorts and extended follow-up after DAA-mediated cure of HCV are essential to accurately evaluate the long-term effects of DAA therapy on the HIV reservoir.
Conclusion
Since the discovery of HIV and HCV, tremendous advances were made in understanding the molecular steps of the viral replication cycle, natural infection, pathogenesis and immune responses. Studies also documented mechanisms by which HCV and HIV reciprocally influence their pathogenesis, thus leading to exacerbated alterations of immune competence as a consequence of impaired liver functions and altered intestinal barrier integrity. Despite the absence of efficient vaccines, DAA currently cures HCV infection, while ART controls viral replication at undetectable levels, thus improving the life quality of co-infected individuals. However, DAA treatment does not completely normalize immune and liver functions. Also, ART does not eradicate HIV reservoirs, which persist in long lived memory CD4+ T cells with various antigenic specificities, potentially including HCV-specific CD4+ T cells.
Future Directions
New longitudinal studies should address the effect of DAA on HIV reservoir persistence in ART-treated HCV/HIV coinfected individuals in relation to age, sex, metabolic status, liver damage, drug and alcohol use, and other comorbidities. Given the fact that the pool of HCV-specific CD4+ T cells contracts but still persists upon DAA, studies are also needed to evaluate whether these cells contribute to the pool of latent HIV reservoirs in ART-treated individuals and whether HCV re-exposure and/or reinfection in high-risk groups may promote HIV reservoir expansion. Finally, additional therapeutic interventions may be needed to restore immune competence and control residual HIV transcription in ART-treated individuals after DAA-mediated HCV cure.
Author Contributions
SG reviewed the literature and wrote the first draft. PA and NS reviewed the literature, revised the first draft, and modified the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Our research is funded through grants from the Canadian Institutes of Health Research CIHR (PJT-173467 (to NS), HOP-120239 and PJT-153052 (to PA)), the National Institutes of Health (NIH) (U01AI131313, R01AI136533 and U19AI159819 to NS), the Canadian HIV Cure Enterprise Team Grant (CanCURE 1.0) funded by CIHR in partnership with the Canadian Foundation for AIDS Research (CANFAR) and the International AIDS Society (IAS) (CanCURE 1.0; HIG-133050), and the CanCURE 2.0 Team Grant funded by CIHR (HB2-164064) to PA. SG is supported by a doctoral fellowship from the Canadian Network on Hepatitis C (CanHepC). CanHepC is funded by a joint initiative of the CIHR (NHC142832) and the Public Health Agency of Canada.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Coutinho RA. HIV and Hepatitis C Among Injecting Drug Users. BMJ (Clin Res Ed) (1998) 317:424–5. doi: 10.1136/bmj.317.7156.424
2. Hagan H, Jordan AE, Neurer J, Cleland CM. Incidence of Sexually Transmitted Hepatitis C Virus Infection in HIV-Positive Men Who Have Sex With Men. Aids (2015) 29:2335–45. doi: 10.1097/QAD.0000000000000834
3. Nijmeijer BM, Koopsen J, Schinkel J, Prins M, Geijtenbeek TB. Sexually Transmitted Hepatitis C Virus Infections: Current Trends, and Recent Advances in Understanding the Spread in Men Who Have Sex With Men. J Int AIDS Soc (2019) 22:e25348–8. doi: 10.1002/jia2.25348
4. Thomas DL, Villano SA, Riester KA, Hershow R, Mofenson LM, Landesman SH, et al. Perinatal Transmission of Hepatitis C Virus From Human Immunodeficiency Virus Type 1-Infected Mothers. Women Infants Transm Study J Infect Dis (1998) 177:1480–8. doi: 10.1086/515315
5. Ferrero S, Lungaro P, Bruzzone BM, Gotta C, Bentivoglio G, Ragni N. Prospective Study of Mother-to-Infant Transmission of Hepatitis C Virus: A 10-Year Survey (1990-2000). Acta Obstet Gynecol Scand (2003) 82:229–34. doi: 10.1034/j.1600-0412.2003.00107.x
6. Lemoine M, Nayagam S, Thursz M. Viral Hepatitis in Resource-Limited Countries and Access to Antiviral Therapies: Current and Future Challenges. Future Virol (2013) 8:371–80. doi: 10.2217/fvl.13.11
7. Platt L, Easterbrook P, Gower E, McDonald B, Sabin K, McGowan C, et al. Prevalence and Burden of HCV Co-Infection in People Living With HIV: A Global Systematic Review and Meta-Analysis. Lancet Infect Dis (2016) 16:797–808. doi: 10.1016/S1473-3099(15)00485-5
8. Chen JY, Feeney ER, Chung RT. HCV and HIV Co-Infection: Mechanisms and Management. Nat Rev Gastroenterol Hepatol (2014) 11:362–71. doi: 10.1038/nrgastro.2014.17
9. Chew KW, Bhattacharya D. Virologic and Immunologic Aspects of HIV-Hepatitis C Virus Coinfection. AIDS (Lond Engl) (2016) 30:2395–404. doi: 10.1097/QAD.0000000000001203
10. Rozera G, Fabbri G, Lorenzini P, Mastrorosa I, Timelli L, Zaccarelli M, et al. Peripheral Blood HIV-1 DNA Dynamics in Antiretroviral-Treated HIV/HCV Co-Infected Patients Receiving Directly-Acting Antivirals. PloS One (2017) 12:e0187095. doi: 10.1371/journal.pone.0187095
11. Ghiglione Y, Polo ML, Urioste A, Rhodes A, Czernikier A, Trifone C, et al. Hepatitis C Virus (HCV) Clearance After Treatment With Direct-Acting Antivirals in Human Immunodeficiency Virus (HIV)-HCV Coinfection Modulates Systemic Immune Activation and HIV Transcription on Antiretroviral Therapy. Open Forum Infect Dis (2020) 7:ofaa115. doi: 10.1093/ofid/ofaa115
12. World Health Organization (WHO). Hepatitis C - Fact Sheet. (2020). Available at: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (Accessed September 2020).
13. Trickey A, Fraser H, Lim AG, Peacock A, Colledge S, Walker JG, et al. The Contribution of Injection Drug Use to Hepatitis C Virus Transmission Globally, Regionally, and at Country Level: A Modelling Study. Lancet Gastroenterol Hepatol (2019) 4:435–44. doi: 10.1016/S2468-1253(19)30085-8
14. Suryaprasad AG, White JZ, Xu F, Eichler BA, Hamilton J, Patel A, et al. Emerging Epidemic of Hepatitis C Virus Infections Among Young Nonurban Persons Who Inject Drugs in the United States, 2006-2012. Clin Infect Dis (2014) 59:1411–9. doi: 10.1093/cid/ciu643
15. World Health Organization (WHO). WHO, Access to Hepatitis C Testing and Treatment for People Who Inject Drugs and People in Prisons - a Global Perspective. In: W.H.O. (WHO), editor. Geneva: World Health Organization (WHO (2019). p. Policy brief. Available at: https://apps.who.int/iris/bitstream/handle/10665/312116/WHO-CDS-HIV-19.6-eng.pdf?ua=1 (Accessed March 2020).
16. Scheel TK, Rice CM. Understanding the Hepatitis C Virus Life Cycle Paves the Way for Highly Effective Therapies. Nat Med (2013) 19:837–49. doi: 10.1038/nm.3248
17. Pestka JM, Zeisel MB, Blaser E, Schurmann P, Bartosch B, Cosset FL, et al. Rapid Induction of Virus-Neutralizing Antibodies and Viral Clearance in a Single-Source Outbreak of Hepatitis C. Proc Natl Acad Sci USA (2007) 104:6025–30. doi: 10.1073/pnas.0607026104
18. Gerold G, Moeller R, Pietschmann T. Hepatitis C Virus Entry: Protein Interactions and Fusion Determinants Governing Productive Hepatocyte Invasion. Cold Spring Harb Perspect Med (2020) 10(2):a036830. doi: 10.1101/cshperspect.a036830
19. Pawlotsky JM. NS5A Inhibitors in the Treatment of Hepatitis C. J Hepatol (2013) 59:375–82. doi: 10.1016/j.jhep.2013.03.030
20. Hoofnagle JH. Course and Outcome of Hepatitis C. Hepatology (2002) 36:S21–9. doi: 10.1053/jhep.2002.36227
21. Khatun M, Ray RB. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells (2019) 8(10):1249. doi: 10.3390/cells8101249
22. Zhang DY, Friedman SL. Fibrosis-Dependent Mechanisms of Hepatocarcinogenesis. Hepatology (2012) 56:769–75. doi: 10.1002/hep.25670
23. Poynard T, Bedossa P, Opolon P. Natural History of Liver Fibrosis Progression in Patients With Chronic Hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC Groups. Lancet (1997) 349:825–32. doi: 10.1016/S0140-6736(96)07642-8
24. Zeremski M, Dimova RB, Pillardy J, de Jong YP, Jacobson IM, Talal AH. Fibrosis Progression in Patients With Chronic Hepatitis C Virus Infection. J Infect Dis (2016) 214:1164–70. doi: 10.1093/infdis/jiw332
25. Schwerk J, Negash A, Savan R, Gale M Jr. Innate Immunity in Hepatitis C Virus Infection. Cold Spring Harb Perspect Med (2021) 11(2):a036988. doi: 10.1101/cshperspect.a036988
26. Liang Y, Cao X, Ding Q, Zhao Y, He Z, Zhong J. Hepatitis C Virus NS4B Induces the Degradation of TRIF to Inhibit TLR3-Mediated Interferon Signaling Pathway. PloS Pathog (2018) 14:e1007075. doi: 10.1371/journal.ppat.1007075
27. Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, et al. Plasmacytoid Dendritic Cells Sense Hepatitis C Virus-Infected Cells, Produce Interferon, and Inhibit Infection. Proc Natl Acad Sci USA (2010) 107:7431–6. doi: 10.1073/pnas.1002301107
28. Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, Chung J, et al. Simultaneous Detection of Hepatitis C Virus and Interferon Stimulated Gene Expression in Infected Human Liver. Hepatology (2014) 59:2121–30. doi: 10.1002/hep.26770
29. Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, et al. Genomic Analysis of the Host Response to Hepatitis C Virus Infection. Proc Natl Acad Sci USA (2002) 99:15669–74. doi: 10.1073/pnas.202608199
30. Grebely J, Feld JJ, Applegate T, Matthews GV, Hellard M, Sherker A, et al. Plasma Interferon-Gamma-Inducible Protein-10 (IP-10) Levels During Acute Hepatitis C Virus Infection. Hepatology (2013) 57:2124–34. doi: 10.1002/hep.26263
31. Boisvert M, Shoukry NH. Type III Interferons in Hepatitis C Virus Infection. Front Immunol (2016) 7:628. doi: 10.3389/fimmu.2016.00628
32. Rosenberg BR, Depla M, Freije CA, Gaucher D, Mazouz S, Boisvert M, et al. Longitudinal Transcriptomic Characterization of the Immune Response to Acute Hepatitis C Virus Infection in Patients With Spontaneous Viral Clearance. PloS Pathog (2018) 14:e1007290. doi: 10.1371/journal.ppat.1007290
33. Rehermann B. Natural Killer Cells in Viral Hepatitis. Cell Mol Gastroenterol Hepatol (2015) 1:578–88. doi: 10.1016/j.jcmgh.2015.09.004
34. Abdel-Hakeem MS, Shoukry NH. Protective Immunity Against Hepatitis C: Many Shades of Gray. Front Immunol (2014) 5:274. doi: 10.3389/fimmu.2014.00274
35. Golden-Mason L, Burton JR Jr., Castelblanco N, Klarquist J, Benlloch S, Wang C, et al. Loss of IL-7 Receptor Alpha-Chain (CD127) Expression in Acute HCV Infection Associated With Viral Persistence. Hepatology (2006) 44:1098–109. doi: 10.1002/hep.21365
36. Bengsch B, Spangenberg HC, Kersting N, Neumann-Haefelin C, Panther E, von Weizsacker F, et al. Analysis of CD127 and KLRG1 Expression on Hepatitis C Virus-Specific CD8+ T Cells Reveals the Existence of Different Memory T-Cell Subsets in the Peripheral Blood and Liver. J Virol (2007) 81:945–53. doi: 10.1128/JVI.01354-06
37. Badr G, Bedard N, Abdel-Hakeem MS, Trautmann L, Willems B, Villeneuve JP, et al. Early Interferon Therapy for Hepatitis C Virus Infection Rescues Polyfunctional, Long-Lived CD8+ Memory T Cells. J Virol (2008) 82:10017–31. doi: 10.1128/JVI.01083-08
38. Shin EC, Park SH, Nascimbeni M, Major M, Caggiari L, de Re V, et al. The Frequency of CD127+ HCV-Specific T Cells But Not the Expression of Exhaustion Markers Predict the Outcome of Acute Hepatitis C Virus Infection. J Virol (2013) 87(8):4772–4777. doi: 10.1128/JVI.03122-12
39. Bowen DG, Walker CM. Mutational Escape From CD8+ T Cell Immunity: HCV Evolution, From Chimpanzees to Man. J Exp Med (2005) 201:1709–14. doi: 10.1084/jem.20050808
40. Semmo N, Klenerman P. CD4+ T Cell Responses in Hepatitis C Virus Infection. World J Gastroenterol (2007) 13:4831–8. doi: 10.3748/wjg.v13.i36.4831
41. Schulze Zur Wiesch J, Ciuffreda D, Lewis-Ximenez L, Kasprowicz V, Nolan BE, Streeck H, et al. Broadly Directed Virus-Specific CD4+ T Cell Responses are Primed During Acute Hepatitis C Infection, But Rapidly Disappear From Human Blood With Viral Persistence. J Exp Med (2012) 209:61–75. doi: 10.1084/jem.20100388
42. Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J, et al. HCV Persistence and Immune Evasion in the Absence of Memory T Cell Help. Science (2003) 302:659–62. doi: 10.1126/science.1088774
43. Shin EC, Sung PS, Park SH. Immune Responses and Immunopathology in Acute and Chronic Viral Hepatitis. Nat Rev Immunol (2016) 16:509–23. doi: 10.1038/nri.2016.69
44. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX Reinforces the Phenotype and Longevity of Exhausted T Cells in Chronic Viral Infection. Nature (2019) 571:265–9. doi: 10.1038/s41586-019-1326-9
45. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science (2012) 338:1220–5. doi: 10.1126/science.1229620
46. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell Factor 1-Expressing Memory-Like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity (2016) 45:415–27. doi: 10.1016/j.immuni.2016.07.021
47. Wieland D, Kemming J, Schuch A, Emmerich F, Knolle P, Neumann-Haefelin C, et al. TCF1(+) Hepatitis C Virus-Specific CD8(+) T Cells are Maintained After Cessation of Chronic Antigen Stimulation. Nat Commun (2017) 8:15050. doi: 10.1038/ncomms15050
48. Lugli E, Galletti G, Boi SK, Youngblood BA. Stem, Effector, and Hybrid States of Memory CD8(+) T Cells. Trends Immunol (2020) 41:17–28. doi: 10.1016/j.it.2019.11.004
49. Hensel N, Gu Z, Sagar, Wieland D, Jechow K, Kemming J, et al. Memory-Like HCV-Specific CD8(+) T Cells Retain a Molecular Scar After Cure of Chronic HCV Infection. Nat Immunol (2021) 22:229–39. doi: 10.1038/s41590-020-00817-w
50. Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD-1, 2b4, CD160 and KLRG1 on Exhausted HCV-Specific CD8+ T Cells is Linked to Antigen Recognition and T Cell Differentiation. PloS Pathog (2010) 6:e1000947. doi: 10.1371/journal.ppat.1000947
51. Kasprowicz V, Kang YH, Lucas M, Schulze zur Wiesch J, Kuntzen T, Fleming V, et al. Hepatitis C Virus (HCV) Sequence Variation Induces an HCV-Specific T-Cell Phenotype Analogous to Spontaneous Resolution. J Virol (2010) 84:1656–63. doi: 10.1128/JVI.01499-09
52. Wolski D, Foote PK, Chen DY, Lewis-Ximenez LL, Fauvelle C, Aneja J, et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8(+) T Cells in Chronic Versus Acute Infection. Immunity (2017) 47:648–663.e8. doi: 10.1016/j.immuni.2017.09.006
53. Kinchen VJ, Cox AL, Bailey JR. Can Broadly Neutralizing Monoclonal Antibodies Lead to a Hepatitis C Virus Vaccine? Trends Microbiol (2018) 26:854–64. doi: 10.1016/j.tim.2018.04.002
54. Shoukry NH, Vaccines HC. Antibodies, and T Cells. Front Immunol (2018) 9:1480. doi: 10.3389/fimmu.2018.01480
55. Farci P, London WT, Wong DC, Dawson GJ, Vallari DS, Engle R, et al. The Natural History of Infection With Hepatitis C Virus (HCV) in Chimpanzees: Comparison of Serologic Responses Measured With First- and Second-Generation Assays and Relationship to HCV Viremia. J Infect Dis (1992) 165:1006–11. doi: 10.1093/infdis/165.6.1006
56. Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, et al. Neutralizing Antibody Response During Acute and Chronic Hepatitis C Virus Infection. Proc Natl Acad Sci USA (2004) 101:10149–54. doi: 10.1073/pnas.0403519101
57. Netski DM, Mosbruger T, Depla E, Maertens G, Ray SC, Hamilton RG, et al. Humoral Immune Response in Acute Hepatitis C Virus Infection. Clin Infect Dis (2005) 41:667–75. doi: 10.1086/432478
58. Takaki A, Wiese M, Maertens G, Depla E, Seifert U, Liebetrau A, et al. Cellular Immune Responses Persist and Humoral Responses Decrease Two Decades After Recovery From a Single-Source Outbreak of Hepatitis C. Nat Med (2000) 6:578–82. doi: 10.1038/75063
59. Salinas E, Boisvert M, Upadhyay AA, Bédard N, Nelson SA, Bruneau J, et al. Early T Follicular Helper Cell Activity Accelerates Hepatitis C Virus-Specific B Cell Expansion. J Clin Invest (2021) 131(2):e140590. doi: 10.1172/JCI140590
60. Osburn WO, Snider AE, Wells BL, Latanich R, Bailey JR, Thomas DL, et al. Clearance of Hepatitis C Infection Is Associated With Early Appearance of Broad Neutralizing Antibody Responses. Hepatology (2014) 59(6):2140–51. doi: 10.1002/hep.27013
61. Vanwolleghem T, Bukh J, Meuleman P, Desombere I, Meunier JC, Alter H, et al. Polyclonal Immunoglobulins From a Chronic Hepatitis C Virus Patient Protect Human Liver-Chimeric Mice From Infection With a Homologous Hepatitis C Virus Strain. Hepatology (2008) 47:1846–55. doi: 10.1002/hep.22244
62. Farci P, Alter HJ, Wong DC, Miller RH, Govindarajan S, Engle R, et al. Prevention of Hepatitis C Virus Infection in Chimpanzees After Antibody-Mediated In Vitro Neutralization. Proc Natl Acad Sci USA (1994) 91:7792–6. doi: 10.1073/pnas.91.16.7792
63. Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, et al. Prevention of Hepatitis C Virus Infection in Chimpanzees by Hyperimmune Serum Against the Hypervariable Region 1 of the Envelope 2 Protein. Proc Natl Acad Sci USA (1996) 93:15394–9. doi: 10.1073/pnas.93.26.15394
64. Krawczynski K, Alter MJ, Tankersley DL, Beach M, Robertson BH, Lambert S, et al. Effect of Immune Globulin on the Prevention of Experimental Hepatitis C Virus Infection. J Infect Dis (1996) 173:822–8. doi: 10.1093/infdis/173.4.822
65. Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, et al. A Genetically Humanized Mouse Model for Hepatitis C Virus Infection. Nature (2011) 474:208–11. doi: 10.1038/nature10168
66. Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, et al. Broadly Neutralizing Antibodies Protect Against Hepatitis C Virus Quasispecies Challenge. Nat Med (2008) 14:25–7. doi: 10.1038/nm1698
67. Bailey JR, Flyak AI, Cohen VJ, Li H, Wasilewski LN, Snider AE, et al. Broadly Neutralizing Antibodies With Few Somatic Mutations and Hepatitis C Virus Clearance. JCI Insight (2017) 2(9):e9287. doi: 10.1172/jci.insight.92872
68. Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, et al. Human Broadly Neutralizing Antibodies to the Envelope Glycoprotein Complex of Hepatitis C Virus. Proc Natl Acad Sci USA (2012) 109:6205–10. doi: 10.1073/pnas.1114927109
69. Kinchen VJ, Zahid MN, Flyak AI, Soliman MG, Learn GH, Wang S, et al. Broadly Neutralizing Antibody Mediated Clearance of Human Hepatitis C Virus Infection. Cell Host Microbe (2018) 24:717–730.e5. doi: 10.1016/j.chom.2018.10.012
70. Shoukry NH, Grakoui A, Houghton M, Chien DY, Ghrayeb J, Reimann KA, et al. Memory CD8+ T Cells Are Required for Protection From Persistent Hepatitis C Virus Infection. J Exp Med (2003) 197:1645–55. doi: 10.1084/jem.20030239
71. Osburn WO, Fisher BE, Dowd KA, Urban G, Liu L, Ray SC, et al. Spontaneous Control of Primary Hepatitis C Virus Infection and Immunity Against Persistent Reinfection. Gastroenterology (2010) 138:315–24. doi: 10.1053/j.gastro.2009.09.017
72. Abdel-Hakeem MS, Bedard N, Murphy D, Bruneau J, Shoukry NH. Signatures of Protective Memory Immune Responses During Hepatitis C Virus Reinfection. Gastroenterology (2014) 147:870–81.e8. doi: 10.1053/j.gastro.2014.07.005
73. Grebely J, Prins M, Hellard M, Cox AL, Osburn WO, Lauer G, et al. Hepatitis C Virus Clearance, Reinfection, and Persistence, With Insights From Studies of Injecting Drug Users: Towards a Vaccine. Lancet Infect Dis (2012) 12:408–14. doi: 10.1016/S1473-3099(12)70010-5
74. Sacks-Davis R, Grebely J, Dore GJ, Osburn W, Cox AL, Rice TM, et al. Hepatitis C Virus Reinfection and Spontaneous Clearance of Reinfection—the InC3 Study. J Infect Dis (2015) 212:1407–19. doi: 10.1093/infdis/jiv220
75. Zoulim F, Liang TJ, Gerbes AL, Aghemo A, Deuffic-Burban S, Dusheiko G, et al. Hepatitis C Virus Treatment in the Real World: Optimising Treatment and Access to Therapies. Gut (2015) 64:1824–33. doi: 10.1136/gutjnl-2015-310421
76. Pawlotsky JM, Feld JJ, Zeuzem S, Hoofnagle JH. From Non-A, Non-B Hepatitis to Hepatitis C Virus Cure. J Hepatol (2015) 62:S87–99. doi: 10.1016/j.jhep.2015.02.006
77. Cunningham EB, Hajarizadeh B, Amin J, Hellard M, Bruneau J, Feld JJ, et al. Reinfection Following Successful Direct-Acting Antiviral Therapy for Hepatitis C Infection Among People Who Inject Drugs. Clin Infect Dis (2020) 72(8):1392–400. doi: 10.1093/cid/ciaa253
78. Barre-Sinoussi F, Ross AL, Delfraissy JF. Past, Present and Future: 30 Years of HIV Research. Nat Rev Microbiol (2013) 11:877–83. doi: 10.1038/nrmicro3132
79. WHO. WHO, HIV/AIDS Fact Sheet. (2019). Available at: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (Accessed March 2020).
80. Ochonye B, Folayan MO, Fatusi AO, Bello BM, Ajidagba B, Emmanuel G, et al. Sexual Practices, Sexual Behavior and HIV Risk Profile of Key Populations in Nigeria. BMC Public Health (2019) 19:1210. doi: 10.1186/s12889-019-7553-z
81. Taylor LE, Swan T, Matthews GV. Management of Hepatitis C Virus/HIV Coinfection Among People Who Use Drugs in the Era of Direct-Acting Antiviral-Based Therapy. Clin Infect Dis (2013) 57:S118–24. doi: 10.1093/cid/cit326
82. Dosekun O, Fox J. An Overview of the Relative Risks of Different Sexual Behaviours on HIV Transmission. Curr Opin HIV AIDS (2010) 5:291–7. doi: 10.1097/COH.0b013e32833a88a3
83. Shaw GM, Hunter E. HIV Transmission. Cold Spring Harb Perspect Med (2012) 2(11):a006965. doi: 10.1101/cshperspect.a006965
84. Pandey A, Galvani AP. The Global Burden of HIV and Prospects for Control. Lancet HIV (2019) 6:e809–11. doi: 10.1016/S2352-3018(19)30230-9
85. Klatzmann DR, McDougal JS, Maddon PJ. The CD4 Molecule and HIV Infection. Immunodefic Rev (1990) 2:43–66.
86. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a Major Co-Receptor for Primary Isolates of HIV-1. Nature (1996) 381:661–6. doi: 10.1038/381661a0
87. Berger EA, Doms RW, Fenyö EM, Korber BT, Littman DR, Moore JP, et al. A New Classification for HIV-1. Nature (1998) 391:240. doi: 10.1038/34571
88. Wyatt R, Sodroski J. The HIV-1 Envelope Glycoproteins: Fusogens, Antigens, and Immunogens. Science (1998) 280:1884–8. doi: 10.1126/science.280.5371.1884
89. Chan DC, Kim PS. HIV Entry and its Inhibition. Cell (1998) 93:681–4. doi: 10.1016/S0092-8674(00)81430-0
90. Zheng YH, Lovsin N, Peterlin BM. Newly Identified Host Factors Modulate HIV Replication. Immunol Lett (2005) 97:225–34. doi: 10.1016/j.imlet.2004.11.026
91. Chavez L, Calvanese V, Verdin E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PloS Pathog (2015) 11:e1004955. doi: 10.1371/journal.ppat.1004955
92. Gomez C, Hope TJ. The Ins and Outs of HIV Replication. Cell Microbiol (2005) 7:621–6. doi: 10.1111/j.1462-5822.2005.00516.x
93. Siliciano RF, Greene WC. HIV Latency. Cold Spring Harb Perspect Med (2011) 1:a007096. doi: 10.1101/cshperspect.a007096
94. Sengupta S, Siliciano RF. Targeting the Latent Reservoir for HIV-1. Immunity (2018) 48:872–95. doi: 10.1016/j.immuni.2018.04.030
95. Cohn LB, Chomont N, Deeks SG. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe (2020) 27:519–30. doi: 10.1016/j.chom.2020.03.014
96. Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV Infection: What the Virus Spares is as Important as What it Destroys. Nat Med (2006) 12:289–95. doi: 10.1038/nm1380
97. Douek DC, Roederer M, Koup RA. Emerging Concepts in the Immunopathogenesis of AIDS. Annu Rev Med (2009) 60:471–84. doi: 10.1146/annurev.med.60.041807.123549
98. Cohen MS, Gay CL, Busch MP, Hecht FM. The Detection of Acute HIV Infection. J Infect Dis (2010) 202:S270–7. doi: 10.1086/655651
99. Pantaleo G, Graziosi C, Fauci AS. The Immunopathogenesis of Human Immunodeficiency Virus Infection. N Engl J Med (1993) 328:327–35. doi: 10.1056/NEJM199302043280508
100. Feinberg MB, McCune JM, Miedema F, Moore JP, Schuitemaker H. HIV Tropism and CD4+ T-Cell Depletion. Nat Med (2002) 8:537. doi: 10.1038/nm0602-537a
101. Haase AT. Perils at Mucosal Front Lines for HIV and SIV and Their Hosts. Nat Rev Immunol (2005) 5:783–92. doi: 10.1038/nri1706
102. Orenstein JM, Fox C, Wahl SM. Macrophages as a Source of HIV During Opportunistic Infections. Science (1997) 276:1857–61. doi: 10.1126/science.276.5320.1857
103. Veazey RS, Lackner AA. Getting to the Guts of HIV Pathogenesis. J Exp Med (2004) 200:697–700. doi: 10.1084/jem.20041464
104. Brenchley JM, Price DA, Douek DC. HIV Disease: Fallout From a Mucosal Catastrophe? Nat Immunol (2006) 7:235–9. doi: 10.1038/ni1316
105. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial Translocation Is a Cause of Systemic Immune Activation in Chronic HIV Infection. Nat Med (2006) 12:1365–71. doi: 10.1038/nm1511
106. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell Death by Pyroptosis Drives CD4 T-Cell Depletion in HIV-1 Infection. Nature (2014) 505:509–14. doi: 10.1038/nature12940
107. Garg H, Mohl J, Joshi A. HIV-1 Induced Bystander Apoptosis. Viruses (2012) 4:3020–43. doi: 10.3390/v4113020
109. Loveday C, Hill A. Prediction of Progression to AIDS With Serum HIV-1 RNA and CD4 Count. Lancet (1995) 345:790–1. doi: 10.1016/S0140-6736(95)90668-1
110. Migueles SA, Connors M. Success and Failure of the Cellular Immune Response Against HIV-1. Nat Immunol (2015) 16:563–70. doi: 10.1038/ni.3161
111. Poropatich K, Sullivan DJ Jr. Human Immunodeficiency Virus Type 1 Long-Term non-Progressors: The Viral, Genetic and Immunological Basis for Disease non-Progression. J Gen Virol (2011) 92:247–68. doi: 10.1099/vir.0.027102-0
112. Mikhail M, Wang B, Saksena NK. Mechanisms Involved in non-Progressive HIV Disease. AIDS Rev (2003) 5:230–44.
113. Morley D, Lambert JS, Hogan LE, De Gascun C, Redmond N, Rutishauser RL, et al. Rapid Development of HIV Elite Control in a Patient With Acute Infection. BMC Infect Dis (2019) 19:815. doi: 10.1186/s12879-019-4485-2
114. Gebara NY, El Kamari V, Rizk N. HIV-1 Elite Controllers: An Immunovirological Review and Clinical Perspectives. J Virus Eradication (2019) 5:163–6. doi: 10.1016/S2055-6640(20)30046-7
115. Goulder P, Deeks SG. HIV Control: Is Getting There the Same as Staying There? PloS Pathog (2018) 14:e1007222. doi: 10.1371/journal.ppat.1007222
116. Walker BD, Yu XG. Unravelling the Mechanisms of Durable Control of HIV-1. Nat Rev Immunol (2013) 13:487–98. doi: 10.1038/nri3478
117. Carrington M, Walker BD. Immunogenetics of Spontaneous Control of HIV. Annu Rev Med (2012) 63:131–45. doi: 10.1146/annurev-med-062909-130018
118. Yao XD, Omange RW, Henrick BM, Lester RT, Kimani J, Ball TB, et al. Acting Locally: Innate Mucosal Immunity in Resistance to HIV-1 Infection in Kenyan Commercial Sex Workers. Mucosal Immunol (2014) 7:268–79. doi: 10.1038/mi.2013.44
119. Sáez-Cirión A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, et al. Post-Treatment HIV-1 Controllers With a Long-Term Virological Remission After the Interruption of Early Initiated Antiretroviral Therapy ANRS VISCONTI Study. PloS Pathog (2013) 9:e1003211. doi: 10.1371/journal.ppat.1003211
120. Hamimi C, Pancino G, Barré-Sinoussi F, Sáez-Cirión A. Will it be Possible to Live Without Antiretroviral Therapy? Curr Opin HIV AIDS (2013) 8:196–203. doi: 10.1097/COH.0b013e32835f94d5
121. Opportunistic Infections Project Team of the Collaboration of Observational, HIVERiEiE, Young J, Psichogiou M, Meyer L, Ayayi S, Grabar S, et al. CD4 Cell Count and the Risk of AIDS or Death in HIV-Infected Adults on Combination Antiretroviral Therapy With a Suppressed Viral Load: A Longitudinal Cohort Study From COHERE. PloS Med (2012) 9:e1001194–e1001194. doi: 10.1371/journal.pmed.1001194
122. Nakanjako D, Kiragga AN, Musick BS, Yiannoutsos CT, Wools-Kaloustian K, Diero L, et al. Frequency and Impact of Suboptimal Immune Recovery on First-Line Antiretroviral Therapy Within the International Epidemiologic Databases to Evaluate AIDS in East Africa. Aids (2016) 30:1913–22. doi: 10.1097/QAD.0000000000001085
123. Yang X, Su B, Zhang X, Liu Y, Wu H, Zhang T. Incomplete Immune Reconstitution in HIV/AIDS Patients on Antiretroviral Therapy: Challenges of Immunological non-Responders. J Leukoc Biol (2020) 107:597–612. doi: 10.1002/JLB.4MR1019-189R
124. Bruzzesi E, Sereti I. Residual Immune Activation and Latency. Curr Top Microbiol Immunol (2018) 417:157–80. doi: 10.1007/82_2018_118
125. Pacheco YM, Jarrin I, Rosado I, Campins AA, Berenguer J, Iribarren JA, et al. Increased Risk of Non-AIDS-Related Events in HIV Subjects With Persistent Low CD4 Counts Despite cART in the CoRIS Cohort. Antiviral Res (2015) 117:69–74. doi: 10.1016/j.antiviral.2015.03.002
126. Engsig FN, Zangerle R, Katsarou O, Dabis F, Reiss P, Gill J, et al. Long-Term Mortality in HIV-Positive Individuals Virally Suppressed for >3 Years With Incomplete CD4 Recovery. Clin Infect Dis (2014) 58:1312–21. doi:10.1093/cid/ciu038
127. Takuva S, Maskew M, Brennan AT, Long L, Sanne I, Fox MP. Poor CD4 Recovery and Risk of Subsequent Progression to AIDS or Death Despite Viral Suppression in a South African Cohort. J Int AIDS Soc (2014) 17:18651. doi: 10.7448/IAS.17.1.18651
128. Collins DR, Gaiha GD, Walker BD. CD8(+) T Cells in HIV Control, Cure and Prevention. Nat Rev Immunol (2020) 20:471–82. doi: 10.1038/s41577-020-0274-9
129. Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O’Brien S, et al. Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity (2018) 48:1029–1045.e5. doi: 10.1016/j.immuni.2018.04.026
130. Sekine T, Perez-Potti A, Nguyen S, Gorin JB, Wu VH, Gostick E, et al. TOX is Expressed by Exhausted and Polyfunctional Human Effector Memory CD8(+) T Cells. Sci Immunol (2020) 5(49):eaba7918. doi: 10.1126/sciimmunol.aba7918
131. Rutishauser RL, Deguit CDT, Hiatt J, Blaeschke F, Roth TL, Wang L, et al. TCF-1 Regulates HIV-Specific CD8+ T Cell Expansion Capacity. JCI Insight (2021) 6(3):e136648. doi: 10.1172/jci.insight.136648
132. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5- Expressing CD8(+) T Cells Curtail Chronic Viral Infection. Nature (2016) 537:412–28. doi: 10.1038/nature19317
133. Buggert M, Nguyen S, Salgado-Montes de Oca G, Bengsch B, Darko S, Ransier A, et al. Identification and Characterization of HIV-Specific Resident Memory CD8(+) T Cells in Human Lymphoid Tissue. Sci Immunol (2018) 3(24):eaar4526. doi: 10.1126/sciimmunol.aar4526
134. Reuter MA, Del Rio Estrada PM, Buggert M, Petrovas C, Ferrando-Martinez S, Nguyen S, et al. HIV-Specific CD8(+) T Cells Exhibit Reduced and Differentially Regulated Cytolytic Activity in Lymphoid Tissue. Cell Rep (2017) 21:3458–70. doi: 10.1016/j.celrep.2017.11.075
135. Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N Engl J Med (2009) 360:692–8. doi: 10.1056/NEJMoa0802905
136. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Prevention of HIV-1 Infection With Early Antiretroviral Therapy. N Engl J Med (2011) 365:493–505. doi: 10.1056/NEJMc1110588
137. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Antiretroviral Therapy for the Prevention of HIV-1 Transmission. N Engl J Med (2016) 375:830–9. doi: 10.1056/NEJMoa1600693
138. Anderson PL, Glidden DV, Liu A, Buchbinder S, Lama JR, Guanira JV, et al. Emtricitabine-Tenofovir Concentrations and Pre-Exposure Prophylaxis Efficacy in Men Who Have Sex With Men. Sci Transl Med (2012) 4:151ra125. doi: 10.1126/scitranslmed.3004006
139. McCormack S, Dunn DT, Desai M, Dolling DI, Gafos M, Gilson R, et al. Pre-Exposure Prophylaxis to Prevent the Acquisition of HIV-1 Infection (PROUD): Effectiveness Results From the Pilot Phase of a Pragmatic Open-Label Randomised Trial. Lancet (2016) 387:53–60. doi: 10.1016/S0140-6736(15)00056-2
140. Riddell JT, Amico KR, Mayer KH. HIV Preexposure Prophylaxis: A Review. Jama (2018) 319:1261–8. doi: 10.1001/jama.2018.1917
141. Markowitz M, Frank I, Grant RM, Mayer KH, Elion R, Goldstein D, et al. Safety and Tolerability of Long-Acting Cabotegravir Injections in HIV-Uninfected Men (ECLAIR): A Multicentre, Double-Blind, Randomised, Placebo-Controlled, Phase 2a Trial. Lancet HIV (2017) 4:e331–40. doi: 10.1016/S2352-3018(17)30068-1
142. Grobben M, Stuart RA, van Gils MJ. The Potential of Engineered Antibodies for HIV-1 Therapy and Cure. Curr Opin Virol (2019) 38:70–80. doi: 10.1016/j.coviro.2019.07.007
143. Caskey M, Klein F, Nussenzweig MC. Broadly Neutralizing Anti-HIV-1 Monoclonal Antibodies in the Clinic. Nat Med (2019) 25:547–53. doi: 10.1038/s41591-019-0412-8
144. Caskey M. Broadly Neutralizing Antibodies for the Treatment and Prevention of HIV Infection. Curr Opin HIV AIDS (2020) 15:49–55. doi: 10.1097/COH.0000000000000600
145. Jin F, Matthews GV, Grulich AE. Sexual Transmission of Hepatitis C Virus Among Gay and Bisexual Men: A Systematic Review. Sex Health (2017) 14:28–41. doi: 10.1071/SH16141
146. Nijmeijer BM, Sarrami-Forooshani R, Steba GS, Schreurs RR, Koekkoek SM, Molenkamp R, et al. HIV-1 Exposure and Immune Activation Enhance Sexual Transmission of Hepatitis C Virus by Primary Langerhans Cells. J Int AIDS Soc (2019) 22:e25268. doi: 10.1002/jia2.25268
147. Gupta P. Hepatitis C Virus and HIV Type 1 Co-Infection. Infect Dis Rep (2013) 5:e7. doi: 10.4081/idr.2013.s1.e7
148. Binder B, Thimme R. CD4+ T Cell Responses in Human Viral Infection: Lessons From Hepatitis C. J Clin Invest (2020) 130:595–7. doi: 10.1172/JCI133222
149. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
150. Kim AY, Schulze zur Wiesch J, Kuntzen T, Timm J, Kaufmann DE, Duncan JE, et al. Impaired Hepatitis C Virus-Specific T Cell Responses and Recurrent Hepatitis C Virus in HIV Coinfection. PloS Med (2006) 3:e492. doi: 10.1371/journal.pmed.0030492
151. Kim AY, Lauer GM, Ouchi K, Addo MM, Lucas M, Wiesch JS, et al. The Magnitude and Breadth of Hepatitis C Virus-Specific CD8+ T Cells Depend on Absolute CD4+ T-Cell Count in Individuals Coinfected With HIV-1. Blood (2005) 105:1170–8. doi: 10.1182/blood-2004-06-2336
152. Bailey JR, Dowd KA, Snider AE, Osburn WO, Mehta SH, Kirk GD, et al. CD4+ T-Cell-Dependent Reduction in Hepatitis C Virus-Specific Neutralizing Antibody Responses After Coinfection With Human Immunodeficiency Virus. J Infect Dis (2015) 212:914–23. doi: 10.1093/infdis/jiv139
153. Ganesan M, Poluektova LY, Kharbanda KK, Osna NA. Human Immunodeficiency Virus and Hepatotropic Viruses Co-Morbidities as the Inducers of Liver Injury Progression. World J Gastroenterol (2019) 25:398–410. doi: 10.3748/wjg.v25.i4.398
154. Sato H, Adachi E, Lim LA, Koga M, Koibuchi T, Tsutsumi T, et al. CD4/CD8 Ratio Predicts the Cellular Immune Response to Acute Hepatitis C in HIV-Coinfected Adults. J Infect Chemother (2019) 25:646–8. doi: 10.1016/j.jiac.2019.04.001
155. Hernandez MD, Sherman KE. HIV/hepatitis C Coinfection Natural History and Disease Progression. Curr Opin HIV AIDS (2011) 6:478–82. doi: 10.1097/COH.0b013e32834bd365
156. Thein HH, Yi Q, Dore GJ, Krahn MD. Natural History of Hepatitis C Virus Infection in HIV-Infected Individuals and the Impact of HIV in the Era of Highly Active Antiretroviral Therapy: A Meta-Analysis. Aids (2008) 22:1979–91. doi: 10.1097/QAD.0b013e32830e6d51
157. Xiao P, Usami O, Suzuki Y, Ling H, Shimizu N, Hoshino H, et al. Characterization of a CD4-Independent Clinical HIV-1 That can Efficiently Infect Human Hepatocytes Through Chemokine (C-X-C Motif) Receptor 4. Aids (2008) 22:1749–57. doi: 10.1097/QAD.0b013e328308937c
158. Lin W, Wu G, Li S, Weinberg EM, Kumthip K, Peng LF, et al. HIV and HCV Cooperatively Promote Hepatic Fibrogenesis via Induction of Reactive Oxygen Species and NFkappaB. J Biol Chem (2011) 286:2665–74. doi: 10.1074/jbc.M110.168286
159. Lin W, Weinberg EM, Chung RT. Pathogenesis of Accelerated Fibrosis in HIV/HCV Co-Infection. J Infect Dis (2013) 207:S13–8. doi: 10.1093/infdis/jis926
160. Jang JY, Shao RX, Lin W, Weinberg E, Chung WJ, Tsai WL, et al. HIV Infection Increases HCV-Induced Hepatocyte Apoptosis. J Hepatol (2011) 54:612–20. doi: 10.1016/j.jhep.2010.07.042
161. Zhou X, Jiang W, Liu Z, Liu S, Liang X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses (2017) 9(11):316. doi: 10.3390/v9110316
162. Tuyama AC, Hong F, Saiman Y, Wang C, Ozkok D, Mosoian A, et al. Human Immunodeficiency Virus (HIV)-1 Infects Human Hepatic Stellate Cells and Promotes Collagen I and Monocyte Chemoattractant Protein-1 Expression: Implications for the Pathogenesis of HIV/hepatitis C Virus-Induced Liver Fibrosis. Hepatology (2010) 52:612–22. doi: 10.1002/hep.23679
163. Zhang L, Bansal MB. Role of Kupffer Cells in Driving Hepatic Inflammation and Fibrosis in HIV Infection. Front Immunol (2020) 11:1086. doi: 10.3389/fimmu.2020.01086
164. Mosoian A, Zhang L, Hong F, Cunyat F, Rahman A, Bhalla R, et al. Frontline Science: HIV Infection of Kupffer Cells Results in an Amplified Proinflammatory Response to LPS. J Leukoc Biol (2017) 101:1083–90. doi: 10.1189/jlb.3HI0516-242R
165. Lidofsky A, Holmes JA, Feeney ER, Kruger AJ, Salloum S, Zheng H, et al. Macrophage Activation Marker Soluble CD163 Is a Dynamic Marker of Liver Fibrogenesis in Human Immunodeficiency Virus/Hepatitis C Virus Coinfection. J Infect Dis (2018) 218:1394–403. doi: 10.1093/infdis/jiy331
166. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 Enhances TGF-Beta Signaling and Hepatic Fibrosis. Nat Med (2007) 13:1324–32. doi: 10.1038/nm1663
167. Deng A, Chen C, Ishizaka Y, Chen X, Sun B, Yang R. Human Immunodeficiency Virus Type 1 Vpr Increases Hepatitis C Virus RNA Replication in Cell Culture. Virus Res (2014) 184:93–102. doi: 10.1016/j.virusres.2014.02.017
168. Park IW, Fan Y, Luo X, Ryou MG, Liu J, Green L, et al. HIV-1 Nef is Transferred From Expressing T Cells to Hepatocytic Cells Through Conduits and Enhances HCV Replication. PloS One (2014) 9:e99545. doi: 10.1371/journal.pone.0099545
169. Feuth T, Arends JE, Fransen JH, Nanlohy NM, van Erpecum KJ, Siersema PD, et al. Complementary Role of HCV and HIV in T-Cell Activation and Exhaustion in HIV/HCV Coinfection. PloS One (2013) 8:e59302. doi: 10.1371/journal.pone.0059302
170. van Vonderen MG, Blumer RM, Hassink EA, Sutinen J, Ackermans MT, van Agtmael MA, et al. Insulin Sensitivity in Multiple Pathways is Differently Affected During Zidovudine/Lamivudine-Containing Compared With NRTI-Sparing Combination Antiretroviral Therapy. J Acquir Immune Defic Syndr (2010) 53:186–93. doi: 10.1097/QAI.0b013e3181c190f4
171. Fleischman A, Johnsen S, Systrom DM, Hrovat M, Farrar CT, Frontera W, et al. Effects of a Nucleoside Reverse Transcriptase Inhibitor, Stavudine, on Glucose Disposal and Mitochondrial Function in Muscle of Healthy Adults. Am J Physiol Endocrinol Metab (2007) 292:E1666–73. doi: 10.1152/ajpendo.00550.2006
172. Blas-García A, Apostolova N, Ballesteros D, Monleón D, Morales JM, Rocha M, et al. Inhibition of Mitochondrial Function by Efavirenz Increases Lipid Content in Hepatic Cells. Hepatology (2010) 52:115–25. doi: 10.1002/hep.23647
173. Paemanee A, Sornjai W, Kittisenachai S, Sirinonthanawech N, Roytrakul S, Wongtrakul J, et al. Nevirapine Induced Mitochondrial Dysfunction in HepG2 Cells. Sci Rep (2017) 7:9194. doi: 10.1038/s41598-017-09321-y
174. Hull MW, Rollet K, Moodie EE, Walmsley S, Cox J, Potter M, et al. Insulin Resistance is Associated With Progression to Hepatic Fibrosis in a Cohort of HIV/hepatitis C Virus-Coinfected Patients. Aids (2012) 26:1789–94. doi: 10.1097/QAD.0b013e32835612ce
175. Bruno R, Galastri S, Sacchi P, Cima S, Caligiuri A, DeFranco R, et al. Gp120 Modulates the Biology of Human Hepatic Stellate Cells: A Link Between HIV Infection and Liver Fibrogenesis. Gut (2010) 59:513–20. doi: 10.1136/gut.2008.163287
176. Salloum S, Holmes JA, Jindal R, Bale SS, Brisac C, Alatrakchi N, et al. Exposure to Human Immunodeficiency Virus/Hepatitis C Virus in Hepatic and Stellate Cell Lines Reveals Cooperative Profibrotic Transcriptional Activation Between Viruses and Cell Types. Hepatology (2016) 64:1951–68. doi: 10.1002/hep.28766
177. Munshi N, Balasubramanian A, Koziel M, Ganju RK, Groopman JE, Hepatitis C. And Human Immunodeficiency Virus Envelope Proteins Cooperatively Induce Hepatocytic Apoptosis via an Innocent Bystander Mechanism. J Infect Dis (2003) 188:1192–204. doi: 10.1086/378643
178. Balagopal A, Ray SC, De Oca RM, Sutcliffe CG, Vivekanandan P, Higgins Y, et al. Kupffer Cells are Depleted With HIV Immunodeficiency and Partially Recovered With Antiretroviral Immune Reconstitution. Aids (2009) 23:2397–404. doi: 10.1097/QAD.0b013e3283324344
179. Balagopal A, Philp FH, Astemborski J, Block TM, Mehta A, Long R, et al. Human Immunodeficiency Virus-Related Microbial Translocation and Progression of Hepatitis C. Gastroenterology (2008) 135:226–33. doi: 10.1053/j.gastro.2008.03.022
180. Mehta AS, Long RE, Comunale MA, Wang M, Rodemich L, Krakover J, et al. Increased Levels of Galactose-Deficient Anti-Gal Immunoglobulin G in the Sera of Hepatitis C Virus-Infected Individuals With Fibrosis and Cirrhosis. J Virol (2008) 82:1259–70. doi: 10.1128/JVI.01600-07
181. Operskalski EA, Kovacs A. HIV/HCV Co-Infection: Pathogenesis, Clinical Complications, Treatment, and New Therapeutic Technologies. Curr HIV/AIDS Rep (2011) 8:12–22. doi: 10.1007/s11904-010-0071-3
182. Vali B, Yue FY, Jones RB, Sheth PM, Kaul R, Betts MR, et al. HIV-Specific T-Cells Accumulate in the Liver in HCV/HIV Co-Infection. PloS One (2008) 3:e3454. doi: 10.1371/journal.pone.0003454
183. Mastroianni CM, Lichtner M, Mascia C, Zuccala P, Vullo V. Molecular Mechanisms of Liver Fibrosis in HIV/HCV Coinfection. Int J Mol Sci (2014) 15:9184–208. doi: 10.3390/ijms15069184
184. Hyun J, McMahon RS, Lang AL, Edwards JS, Badilla AD, Greene ME, et al. HIV and HCV Augments Inflammatory Responses Through Increased TREM-1 Expression and Signaling in Kupffer and Myeloid Cells. PloS Pathog (2019) 15:e1007883. doi: 10.1371/journal.ppat.1007883
185. Pérez-Matute P, Pérez-Martínez L, Blanco JR, Oteo JA. Role of Mitochondria in HIV Infection and Associated Metabolic Disorders: Focus on Nonalcoholic Fatty Liver Disease and Lipodystrophy Syndrome. Oxid Med Cell Longevity (2013) 2013:493413. doi: 10.1155/2013/493413
186. Feeney ER, Mallon PW. Impact of Mitochondrial Toxicity of HIV-1 Antiretroviral Drugs on Lipodystrophy and Metabolic Dysregulation. Curr Pharm Des (2010) 16:3339–51. doi: 10.2174/138161210793563482
187. Apostolova N, Blas-García A, Esplugues JV. Mitochondrial Interference by Anti-HIV Drugs: Mechanisms Beyond Pol-γ Inhibition. Trends Pharmacol Sci (2011) 32:715–25. doi: 10.1016/j.tips.2011.07.007
188. Rohrbach J, Robinson N, Harcourt G, Hammond E, Gaudieri S, Gorgievski M, et al. Cellular Immune Responses to HCV Core Increase and HCV RNA Levels Decrease During Successful Antiretroviral Therapy. Gut (2010) 59:1252–8. doi: 10.1136/gut.2009.205971
189. Anderson JP, Tchetgen Tchetgen EJ, Lo Re V 3rd, Tate JP, Williams PL, Seage GR 3rd, et al. Antiretroviral Therapy Reduces the Rate of Hepatic Decompensation Among HIV- and Hepatitis C Virus-Coinfected Veterans. Clin Infect Dis (2014) 58:719–27. doi: 10.1093/cid/cit779
190. Price JC, Seaberg EC, Phair JP, Witt MD, Koletar SL, Thio CL. Brief Report: Highly Active Antiretroviral Therapy Mitigates Liver Disease in HIV Infection. J Acquir Immune Defic Syndr (2016) 72:319–23. doi: 10.1097/QAI.0000000000000981
191. d’Arminio Monforte A, Cozzi-Lepri A, Castagna A, Antinori A, De Luca A, Mussini C, et al. Risk of Developing Specific AIDS-Defining Illnesses in Patients Coinfected With HIV and Hepatitis C Virus With or Without Liver Cirrhosis. Clin Infect Dis (2009) 49:612–22. doi: 10.1086/603557
192. Boulougoura A, Sereti I. HIV Infection and Immune Activation: The Role of Coinfections. Curr Opin HIV AIDS (2016) 11:191–200. doi: 10.1097/COH.0000000000000241
193. Robinson MW, Harmon C, O’Farrelly C. Liver Immunology and its Role in Inflammation and Homeostasis. Cell Mol Immunol (2016) 13:267–76. doi: 10.1038/cmi.2016.3
194. Burchill MA, Golden-Mason L, Wind-Rotolo M, Rosen HR. Memory Re-Differentiation and Reduced Lymphocyte Activation in Chronic HCV-Infected Patients Receiving Direct-Acting Antivirals. J Viral Hepat (2015) 22:983–91. doi: 10.1111/jvh.12465
195. Rallon N, Garcia M, Garcia-Samaniego J, Rodriguez N, Cabello A, Restrepo C, et al. HCV Coinfection Contributes to HIV Pathogenesis by Increasing Immune Exhaustion in CD8 T-Cells. PloS One (2017) 12:e0173943. doi: 10.1371/journal.pone.0173943
196. Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PloS Pathog (2015) 11:e1005177. doi: 10.1371/journal.ppat.1005177
197. Barrett L, Trehanpati N, Poonia S, Daigh L, Sarin SK, Masur H, et al. Hepatic Compartmentalization of Exhausted and Regulatory Cells in HIV/HCV-Coinfected Patients. J Viral Hepat (2015) 22:281–8. doi: 10.1111/jvh.12291
198. Martin B, Hennecke N, Lohmann V, Kayser A, Neumann-Haefelin C, Kukolj G, et al. Restoration of HCV-Specific CD8+ T Cell Function by Interferon-Free Therapy. J Hepatol (2014) 61:538–43. doi: 10.1016/j.jhep.2014.05.043
199. Wu X, Ishaq M, Hu J, Guo D. HCV NS3/4A Protein Activates HIV-1 Transcription From its Long Terminal Repeat. Virus Res (2008) 135:155–60. doi: 10.1016/j.virusres.2008.03.006
200. Kang L, Luo Z, Li Y, Zhang W, Sun W, Li W, et al. Association of Vpu With Hepatitis C Virus NS3/4A Stimulates Transcription of Type 1 Human Immunodeficiency Virus. Virus Res (2012) 163:74–81. doi: 10.1016/j.virusres.2011.08.011
201. Khan KA, Abbas W, Varin A, Kumar A, Di Martino V, Dichamp I, et al. HIV-1 Nef Interacts With HCV Core, Recruits TRAF2, TRAF5 and TRAF6, and Stimulates HIV-1 Replication in Macrophages. J Innate Immun (2013) 5:639–56. doi: 10.1159/000350517
202. Gianella S, Anderson CM, Var SR, Oliveira MF, Lada SM, Vargas MV, et al. Replication of Human Herpesviruses Is Associated With Higher HIV DNA Levels During Antiretroviral Therapy Started at Early Phases of HIV Infection. J Virol (2016) 90:3944–52. doi: 10.1128/JVI.02638-15
203. Garcia-Broncano P, Medrano LM, Berenguer J, González-García J, Jiménez-Sousa M, Carrero A, et al. Dysregulation of the Immune System in HIV/HCV-Coinfected Patients According to Liver Stiffness Status. Cells (2018) 7(11): 196. doi: 10.3390/cells7110196
204. van Santen DK, van der Helm JJ, Touloumi G, Pantazis N, Muga R, Gunsenheimer-Bartmeyer B, et al. Effect of Incident Hepatitis C Infection on CD4+ Cell Count and HIV RNA Trajectories Based on a Multinational HIV Seroconversion Cohort. Aids (2019) 33:327–37. doi: 10.1097/QAD.0000000000002040
205. Potter M, Odueyungbo A, Yang H, Saeed S, Klein MB. Impact of Hepatitis C Viral Replication on CD4+ T-Lymphocyte Progression in HIV-HCV Coinfection Before and After Antiretroviral Therapy. Aids (2010) 24:1857–65. doi: 10.1097/QAD.0b013e32833adbb5
206. Liberto MC, Zicca E, Pavia G, Quirino A, Marascio N, Torti C, et al. Virological Mechanisms in the Coinfection Between HIV and HCV. Mediators Inflamm (2015) 2015:320532. doi: 10.1155/2015/320532
207. Swaminathan G, Pascual D, Rival G, Perales-Linares R, Martin-Garcia J, Navas-Martin S. Hepatitis C Virus Core Protein Enhances HIV-1 Replication in Human Macrophages Through TLR2, JNK, and MEK1/2-Dependent Upregulation of TNF-Alpha and IL-6. FEBS Lett (2014) 588:3501–10. doi: 10.1016/j.febslet.2014.08.009
208. Sengupta S, Powell E, Kong L, Blackard JT. Effects of HCV on Basal and Tat-Induced HIV LTR Activation. PloS One (2013) 8:e64956. doi: 10.1371/journal.pone.0064956
209. Lopez-Huertas MR, Palladino C, Garrido-Arquero M, Esteban-Cartelle B, Sanchez-Carrillo M, Martinez-Roman P, et al. HCV-Coinfection is Related to an Increased HIV-1 Reservoir Size in cART-Treated HIV Patients: A Cross-Sectional Study. Sci Rep (2019) 9:5606. doi: 10.1038/s41598-019-41788-9
210. Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Pond SLK, et al. Persistent HIV-1 Replication Maintains the Tissue Reservoir During Therapy. Nature (2016) 530:51–6. doi: 10.1038/nature16933
211. Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, et al. HIV Preferentially Infects HIV-Specific CD4+ T Cells. Nature (2002) 417:95–8. doi: 10.1038/417095a
212. Saharia KK, Koup RA. T Cell Susceptibility to HIV Influences Outcome of Opportunistic Infections. Cell (2013) 155:505–14. doi: 10.1016/j.cell.2013.09.045
213. Gantner P, Pagliuzza A, Pardons M, Ramgopal M, Routy JP, Fromentin R, et al. Single-Cell TCR Sequencing Reveals Phenotypically Diverse Clonally Expanded Cells Harboring Inducible HIV Proviruses During ART. Nat Commun (2020) 11:4089. doi: 10.1038/s41467-020-17898-8
214. Mendoza P, Jackson JR, Oliveira TY, Gaebler C, Ramos V, Caskey M, et al. Antigen-Responsive CD4+ T Cell Clones Contribute to the HIV-1 Latent Reservoir. J Exp Med (2020) 217(7):e20200051. doi: 10.1101/2020.01.10.902155
215. Cattin A, Wacleche VS, Fonseca Do Rosario N, Marchand LR, Dias J, Gosselin A, et al. RALDH Activity Induced by Bacterial/Fungal Pathogens in CD16(+) Monocyte-Derived Dendritic Cells Boosts HIV Infection and Outgrowth in CD4(+) T Cells. J Immunol (2021) 206:2638–51. doi: 10.4049/jimmunol.2001436
216. Zhong G, Kirkwood J, Won KJ, Tjota N, Jeong H, Isoherranen N. Characterization of Vitamin A Metabolome in Human Livers With and Without Nonalcoholic Fatty Liver Disease. J Pharmacol Exp Ther (2019) 370:92–103. doi: 10.1124/jpet.119.258517
217. Molina MF, Abdelnabi MN, Fabre T, Shoukry NH. Type 3 Cytokines in Liver Fibrosis and Liver Cancer. Cytokine (2019) 124:154497. doi: 10.1016/j.cyto.2018.07.028
218. Planas D, Routy JP, Ancuta P. New Th17-Specific Therapeutic Strategies for HIV Remission. Curr Opin HIV AIDS (2019) 14:85–92. doi: 10.1097/COH.0000000000000522
219. Kared H, Fabre T, Bédard N, Bruneau J, Shoukry NH. Galectin-9 and IL-21 Mediate Cross-Regulation Between Th17 and Treg Cells During Acute Hepatitis C. PloS Pathog (2013) 9:e1003422. doi: 10.1371/journal.ppat.1003422
220. Liu R, Simonetti FR, Ho YC. The Forces Driving Clonal Expansion of the HIV-1 Latent Reservoir. Virol J (2020) 17:4. doi: 10.1186/s12985-019-1276-8
221. Neukam K, Morano-Amado LE, Rivero-Juarez A, Mancebo M, Granados R, Tellez F, et al. HIV-Coinfected Patients Respond Worse to Direct-Acting Antiviral-Based Therapy Against Chronic Hepatitis C in Real Life Than HCV-Monoinfected Individuals: A Prospective Cohort Study. HIV Clin Trials (2017) 18:126–34. doi: 10.1080/15284336.2017.1330801
222. Sikavi C, Najarian L, Saab S. Similar Sustained Virologic Response in Real-World and Clinical Trial Studies of Hepatitis C/Human Immunodeficiency Virus Coinfection. Dig Dis Sci (2018) 63:2829–39. doi: 10.1007/s10620-018-5215-0
223. Sikavi C, Chen PH, Lee AD, Saab EG, Choi G, Saab S, et al. And Human Immunodeficiency Virus Coinfection in the Era of Direct-Acting Antiviral Agents: No Longer a Difficult-to-Treat Population. Hepatology (2018) 67:847–57. doi: 10.1002/hep.29642
224. Zahnd C, Salazar-Vizcaya L, Dufour JF, Müllhaupt B, Wandeler G, Kouyos R, et al. Modelling the Impact of Deferring HCV Treatment on Liver-Related Complications in HIV Coinfected Men Who Have Sex With Men. J Hepatol (2016) 65:26–32. doi: 10.1016/j.jhep.2016.02.030
225. Salmon-Ceron D, Nahon P, Layese R, Bourcier V, Sogni P, Bani-Sadr F, et al. Human Immunodeficiency Virus/Hepatitis C Virus (HCV) Co-Infected Patients With Cirrhosis Are No Longer at Higher Risk for Hepatocellular Carcinoma or End-Stage Liver Disease as Compared to HCV Mono-Infected Patients. Hepatology (2019) 70:939–54. doi: 10.1002/hep.30400
226. Lund Laursen T, Brøckner Siggard C, Kazankov K, Damgaard Sandahl T, Møller HJ, Ong A, et al. Rapid and Persistent Decline in Soluble CD163 With Successful Direct-Acting Antiviral Therapy and Associations With Chronic Hepatitis C Histology. Scand J Gastroenterol (2018) 53:986–93. doi: 10.1080/00365521.2018.1481996
227. Meissner EG, Wu D, Osinusi A, Bon D, Virtaneva K, Sturdevant D, et al. Endogenous Intrahepatic IFNs and Association With IFN-Free HCV Treatment Outcome. J Clin Invest (2014) 124:3352–63. doi: 10.1172/JCI75938
228. Hengst J, Falk CS, Schlaphoff V, Deterding K, Manns MP, Cornberg M, et al. Direct-Acting Antiviral-Induced Hepatitis C Virus Clearance Does Not Completely Restore the Altered Cytokine and Chemokine Milieu in Patients With Chronic Hepatitis C. J Infect Dis (2016) 214:1965–74. doi: 10.1093/infdis/jiw457
229. Serti E, Chepa-Lotrea X, Kim YJ, Keane M, Fryzek N, Liang TJ, et al. Successful Interferon-Free Therapy of Chronic Hepatitis C Virus Infection Normalizes Natural Killer Cell Function. Gastroenterology (2015) 149:190–200.e2. doi: 10.1053/j.gastro.2015.03.004
230. Meissner EG, Kohli A, Higgins J, Lee YJ, Prokunina O, Wu D, et al. Rapid Changes in Peripheral Lymphocyte Concentrations During Interferon-Free Treatment of Chronic Hepatitis C Virus Infection. Hepatol Commun (2017) 1:586–94. doi: 10.1002/hep4.1074
231. Emmanuel B, El-Kamary SS, Magder LS, Stafford KA, Charurat ME, Poonia B, et al. Immunological Recovery in T-Cell Activation After Sustained Virologic Response Among HIV Positive and HIV Negative Chronic Hepatitis C Patients. Hepatol Int (2019) 13:270–6. doi: 10.1007/s12072-019-09941-8
232. Aregay A, Owusu Sekyere S, Deterding K, Port K, Dietz J, Berkowski C, et al. Elimination of Hepatitis C Virus has Limited Impact on the Functional and Mitochondrial Impairment of HCV-Specific CD8+ T Cell Responses. J Hepatol (2019) 71:889–99. doi: 10.1016/j.jhep.2019.06.025
233. Barili V, Fisicaro P, Montanini B, Acerbi G, Filippi A, Forleo G, et al. Targeting P53 and Histone Methyltransferases Restores Exhausted CD8+ T Cells in HCV Infection. Nat Commun (2020) 11:604. doi: 10.1038/s41467-019-14137-7
234. Smits M, Zoldan K, Ishaque N, Gu Z, Jechow K, Wieland D, et al. Follicular T Helper Cells Shape the HCV-Specific CD4+ T Cell Repertoire After Virus Elimination. J Clin Invest (2020) 130:998–1009. doi: 10.1172/JCI129642
235. Najafi Fard S, Schietroma I, Corano Scheri G, Giustini N, Serafino S, Cavallari EN, et al. Direct-Acting Antiviral Therapy Enhances Total CD4+ and CD8+ T-Cells Responses, But Does Not Alter T-Cells Activation Among HCV Mono-Infected, and HCV/HIV-1 Co-Infected Patients. Clin Res Hepatol Gastroenterol (2018) 42:319–29. doi: 10.1016/j.clinre.2017.11.006
236. Langhans B, Nischalke HD, Kramer B, Hausen A, Dold L, van Heteren P, et al. Increased Peripheral CD4(+) Regulatory T Cells Persist After Successful Direct-Acting Antiviral Treatment of Chronic Hepatitis C. J Hepatol (2017) 66:888–96. doi: 10.1016/j.jhep.2016.12.019
237. Tumino N, Casetti R, Fabbri G, Cimini E, Romanelli A, Turchi F, et al. In HIV/HCV Co-Infected Patients T Regulatory and Myeloid-Derived Suppressor Cells Persist After Successful Treatment With Directly Acting Antivirals. J Hepatol (2017) 67:422–4. doi: 10.1016/j.jhep.2017.03.036
238. Cai W, Qin A, Guo P, Yan D, Hu F, Yang Q, et al. Clinical Significance and Functional Studies of Myeloid-Derived Suppressor Cells in Chronic Hepatitis C Patients. J Clin Immunol (2013) 33:798–808. doi: 10.1007/s10875-012-9861-2
239. Marvel D, Gabrilovich DI. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment: Expect the Unexpected. J Clin Invest (2015) 125:3356–64. doi: 10.1172/JCI80005
240. Macias J, Tellez F, Rivero-Juarez A, Palacios R, Morano LE, Merino D, et al. Early Emergence of Opportunistic Infections After Starting Direct-Acting Antiviral Drugs in HIV/HCV-Coinfected Patients. J Viral Hepat (2019) 26:48–54. doi: 10.1111/jvh.13003
241. Fabbri G, Mastrorosa I, Vergori A, Mazzotta V, Pinnetti C, Grisetti S, et al. Reactivation of Occult HBV Infection in an HIV/HCV Co-Infected Patient Successfully Treated With Sofosbuvir/Ledipasvir: A Case Report and Review of the Literature. BMC Infect Dis (2017) 17:182. doi: 10.1186/s12879-017-2287-y
242. Bachofner JA, Valli PV, Kröger A, Bergamin I, Künzler P, Baserga A, et al. Direct Antiviral Agent Treatment of Chronic Hepatitis C Results in Rapid Regression of Transient Elastography and Fibrosis Markers Fibrosis-4 Score and Aspartate Aminotransferase-Platelet Ratio Index. Liver Int (2017) 37:369–76. doi: 10.1111/liv.13256
243. Rial-Crestelo D, Sepúlveda MA, González-Gasca FJ, Geijo-Martínez P, Martínez-Alfaro E, Barberá JR, et al. Does Fibrosis Really Regress in HIV/hepatitis C Virus Co-Infected Patients After Treatment With Direct Antiviral Agents? Aids (2020) 34:427–32. doi: 10.1097/QAD.0000000000002433
244. Hamdane N, Jühling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology (2019) 156:2313–2329.e7. doi: 10.1053/j.gastro.2019.02.038
245. Perez S, Kaspi A, Domovitz T, Davidovich A, Lavi-Itzkovitz A, Meirson T, et al. Hepatitis C Virus Leaves an Epigenetic Signature Post Cure of Infection by Direct-Acting Antivirals. PloS Genet (2019) 15:e1008181. doi: 10.1371/journal.pgen.1008181
246. Jeyarajan AJ, Chung RT. Insights Into the Pathophysiology of Liver Disease in HCV/HIV: Does it End With HCV Cure? J Infect Dis (2020) 222:S802–s813. doi: 10.1093/infdis/jiaa279
247. Mudd JC, Brenchley JM. Gut Mucosal Barrier Dysfunction, Microbial Dysbiosis, and Their Role in HIV-1 Disease Progression. J Infect Dis (2016) 214:S58–66. doi: 10.1093/infdis/jiw258
248. Bajaj JS, Sterling RK, Betrapally NS, Nixon DE, Fuchs M, Daita K, et al. HCV Eradication Does Not Impact Gut Dysbiosis or Systemic Inflammation in Cirrhotic Patients. Aliment Pharmacol Ther (2016) 44:638–43. doi: 10.1111/apt.13732
249. Hong FF, Mellors JW. Changes in HIV Reservoirs During Long-Term Antiretroviral Therapy. Curr Opin HIV AIDS (2015) 10:43–8. doi: 10.1097/COH.0000000000000119
250. Pardons M, Baxter AE, Massanella M, Pagliuzza A, Fromentin R, Dufour C, et al. Single-Cell Characterization and Quantification of Translation-Competent Viral Reservoirs in Treated and Untreated HIV Infection. PloS Pathog (2019) 15:e1007619. doi: 10.1371/journal.ppat.1007619
251. Parisi SG, Andreis S, Basso M, Cavinato S, Scaggiante R, Franzetti M, et al. Time Course of Cellular HIV-DNA and Low-Level HIV Viremia in HIV-HCV Co-Infected Patients Whose HCV Infection had Been Successfully Treated With Directly Acting Antivirals. Med Microbiol Immunol (2017) 206:419–28. doi: 10.1007/s00430-017-0518-x
252. Schaefer CJ, Kossen K, Lim SR, Lin JH, Pan L, Bradford W, et al. Danoprevir Monotherapy Decreases Inflammatory Markers in Patients With Chronic Hepatitis C Virus Infection. Antimicrob Agents Chemother (2011) 55:3125–32. doi: 10.1128/AAC.00131-11
253. Balagopal A, Smeaton LM, Quinn J, Venuto CS, Morse GD, Vu V, et al. Intrahepatic Viral Kinetics During Direct-Acting Antivirals for Hepatitis C in HIV Co-Infection:The ACTG A5335S Substudy. J Infect Dis (2020) 222(4):601–10. doi: 10.1093/infdis/jiaa126
254. Sung PS, Lee EB, Park DJ, Lozada A, Jang JW, Bae SH, et al. Interferon-Free Treatment for Hepatitis C Virus Infection Induces Normalization of Extrahepatic Type I Interferon Signaling. Clin Mol Hepatol (2018) 24:302–10. doi: 10.3350/cmh.2017.0074
255. Van der Sluis RM, Zerbato JM, Rhodes JW, Pascoe RD, Solomon A, Kumar NA, et al. Diverse Effects of Interferon Alpha on the Establishment and Reversal of HIV Latency. PloS Pathog (2020) 16:e1008151. doi: 10.1371/journal.ppat.1008151
256. Selliah N, Zhang M, DeSimone D, Kim H, Brunner M, Ittenbach RF, et al. The Gammac-Cytokine Regulated Transcription Factor, STAT5, Increases HIV-1 Production in Primary CD4 T Cells. Virology (2006) 344:283–91. doi: 10.1016/j.virol.2005.09.063
Keywords: human immunodeficiency virus, hepatitis C, coinfection (HIV infection), direct acting antiviral, anti retro viral therapy, liver fibrosis, CD4 T cell
Citation: Gobran ST, Ancuta P and Shoukry NH (2021) A Tale of Two Viruses: Immunological Insights Into HCV/HIV Coinfection. Front. Immunol. 12:726419. doi: 10.3389/fimmu.2021.726419
Received: 16 June 2021; Accepted: 26 July 2021;
Published: 12 August 2021.
Edited by:
Vijayakumar Velu, Emory University, United StatesReviewed by:
Bertram Bengsch, University of Freiburg Medical Center, GermanyCarey Shive, Louis Stokes Cleveland VA Medical Center, United States
Copyright © 2021 Gobran, Ancuta and Shoukry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naglaa H. Shoukry, bmFnbGFhLnNob3VrcnlAdW1vbnRyZWFsLmNh