 Anwen Ren1Wei Yin2Heather Miller3
Anwen Ren1Wei Yin2Heather Miller3 Lisa S. Westerberg4
Lisa S. Westerberg4 Fabio Candotti5Chan-Sik Park6Pamela Lee7Quan Gong8,9*Yan Chen10*
Fabio Candotti5Chan-Sik Park6Pamela Lee7Quan Gong8,9*Yan Chen10* Chaohong Liu1*
Chaohong Liu1*- 1Department of Pathogen Biology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3The Laboratory of Intracellular Parasites, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Hamilton, MT, United States
- 4Department of Microbiology Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
- 5Division of Immunology and Allergy, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- 6Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, South Korea
- 7Department of Paediatrics and Adolescent Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China
- 8Department of Immunology, School of Medicine, Yangtze University, Jingzhou, China
- 9Clinical Molecular Immunology Center, School of Medicine, Yangtze University, Jingzhou, China
- 10The Second Department of Pediatrics, Affiliated Hospital of Zunyi Medical University, Zunyi, China
With the expansion of our knowledge on inborn errors of immunity (IEI), it gradually becomes clear that immune dysregulation plays an important part. In some cases, autoimmunity, hyperinflammation and lymphoproliferation are far more serious than infections. Thus, immune dysregulation has become significant in disease monitoring and treatment. In recent years, the wide application of whole-exome sequencing/whole-genome sequencing has tremendously promoted the discovery and further studies of new IEI. The number of discovered IEI is growing rapidly, followed by numerous studies of their pathogenesis and therapy. In this review, we focus on novel discovered primary immune dysregulation diseases, including deficiency of SLC7A7, CD122, DEF6, FERMT1, TGFB1, RIPK1, CD137, TET2 and SOCS1. We discuss their genetic mutation, symptoms and current therapeutic methods, and point out the gaps in this field.
Introduction
The immune system is under regulation of several checkpoints during central and peripheral development. Disorder of regulation can cause abnormal activation and expansion of immune cells, leading to autoimmunity, hyperinflammation and even malignant proliferation. Inborn errors of immunity (IEI), used to widely known as primary immunodeficiency (PID), was historically defined by higher susceptibility to infections due to monogenic germline mutations. However, recent studies reveal that immune dysregulation accounts for a large proportion of manifestations in PID patients (1, 2). Additionally, it results in a worse prognosis in patients with immune dysregulation compared to those with only high infection susceptibility (3). International Union of Immunological Societies (IUIS) lists “Diseases of immune dysregulation” as an independent category of IEI (1), including familial hemophagocytic lymphohistiocytosis (FHL syndromes), FHL syndromes with hypopigmentation, regulatory T cell defects, autoimmunity with or without lymphoproliferation, immune dysregulation with colitis, autoimmune lymphoproliferative syndrome (ALPS, Canale-Smith syndrome) and susceptibility to EBV and lymphoproliferative conditions. Patients with these diseases suffer a combined manifestation of immune deficiency, autoimmunity, recurrent inflammation, lymphoproliferation and even predisposition to malignancy. Their immune cells like B cell, T cell and NK cell have abnormal amounts and functions. Due to the coexistence of autoimmunity and immunodeficiency in some cases, clinical treatment requires a delicate balance. Hematopoietic stem cell transplantation (HSCT) is a potential therapy, but improving survival rate still remains an issue (4). Considering its poor prognosis and difficult treatment, a deeper understanding of immune dysregulation in IEI is required for precise and timely diagnosis, disease monitoring and therapy.
Since the number of cases for any particular disease is usually few, a large-scale study of IEI can hardly be carried out. Thus, there is difficulty in studying and curing these diseases. With technical advancements in whole-exome sequencing/whole-genome sequencing (WES/WGS), tremendous progress has been made in the identification of mutations causing IEI, whose number has doubled in ten years (from 2009 to 2019) and continues to increase rapidly (1, 2). Here, we review novel discoveries of immune dysregulation in IEI, based on the genes listed in the Table 4 of IUIS 2019 IEI report (1) and its 2020 interim update (2), including SLC7A7 deficiency (FHL syndromes), CD122 deficiency (regulatory T cell defects), DEF6 (regulatory T cell defects), FERMT1 (regulatory T cell defects), SOCS1 (autoimmunity with or without lymphoproliferation), TGFB1 (immune dysregulation with colitis), RIPK1 (immune dysregulation with colitis), CD137 (susceptibility to EBV and lymphoproliferative conditions) and TET2 (susceptibility to EBV and lymphoproliferative conditions) (Table 1). We make a thorough review about their genetic mutations, clinical phenotypes and possible treatments.
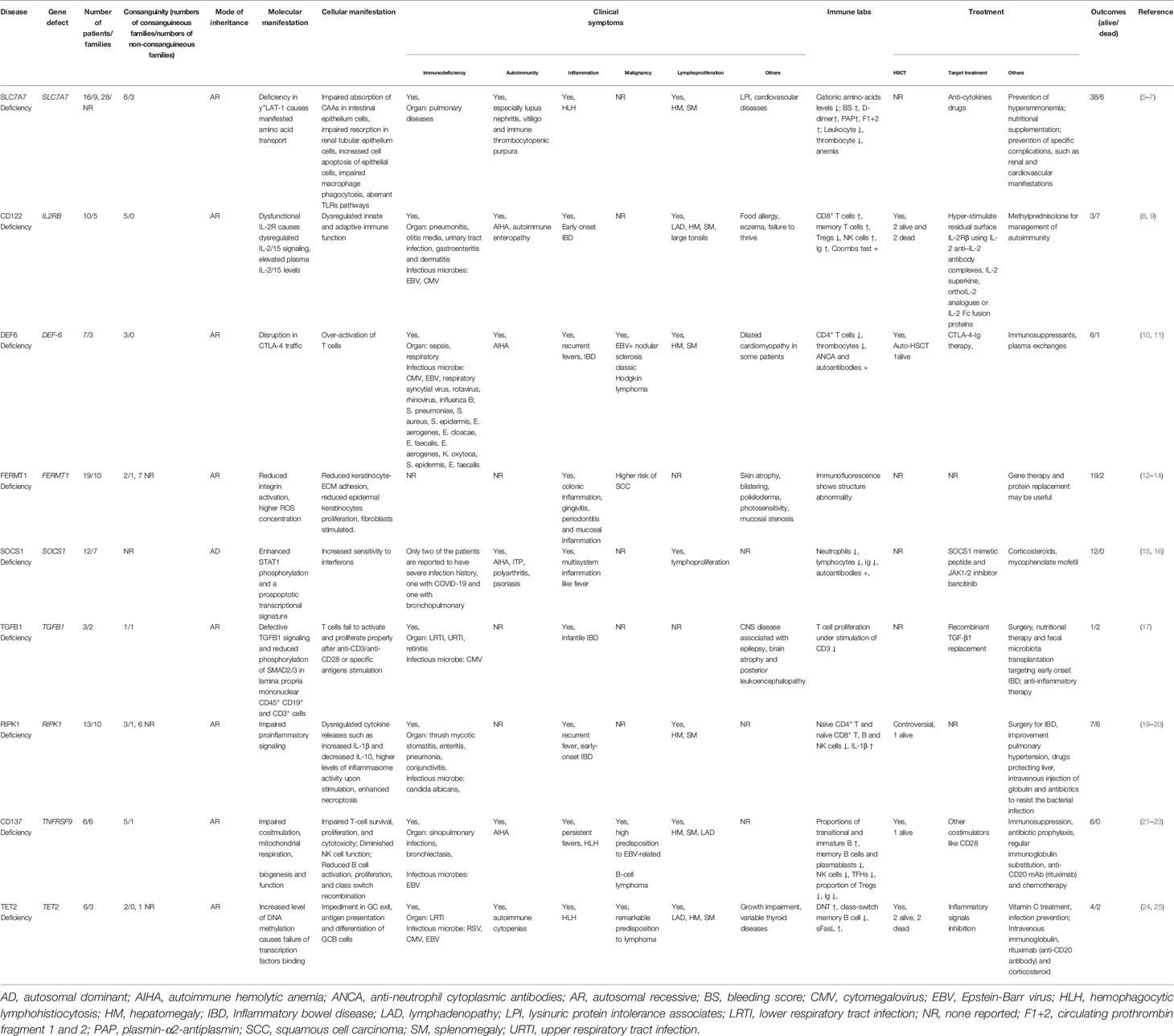
Table 1 Newly reported primary immune dysregulation diseases.
SLC7A7 Deficiency
Solute carrier family 7A member 7 (SLC7A7) encodes y+L amino acid transporter-1 (y+LAT-1). It is expressed mainly in monocyte‐derived macrophages, as well as intestinal and renal cells, while its homolog y+LAT-2, encoded by SLC7A6, is expressed ubiquitously, but with low levels in the cells mentioned above, explaining its inability to compensate for y+LAT-1 in LPI (26).
SLC7A7 mutations contribute to lysinuric protein intolerance (LPI), whose symptoms compose of growth retardation, muscle hypotonia and hepatosplenomegaly (27). More related manifestations were revealed in later studies, such as pulmonary diseases, cardiovascular diseases, hemophagocytic-lymphohistiocytosis (HLH) and autoimmune diseases, especially lupus nephritis, vitiligo and immune thrombocytopenic purpura (5). It was initially thought that y+LAT-1 deficiency in polarized cells such as intestinal and renal tubular epithelium cells is the pathogenesis of LPI, since impaired absorption of cationic amino acids (CAAs) in the intestinal epithelium and impaired resorption in the kidneys cause an imbalance of amino acids, reduction of protein synthesis, hyperammonemia and growth disorders. However, later studies revealed that non-polarized cells like lymphocytes and macrophages are also involved, which contributes to renal, pulmonary, and immune disorders (28, 29). Immune dysregulation in LPI patients appears as a decrease in leukocyte phagocytic, cytotoxic, and natural killer (NK) cell activity and an increase in spontaneous proliferation of lymphocytes (28). Impairment of macrophage phagocytosis by amino acid transport leads to not only higher susceptibility to viral infection, but also abnormal inflammatory state and autoimmune diseases, since aberrant phagocytosis fails to remove apoptotic cells, which are related to inflammation and autoimmune responses (29). Furthermore, intracellular arginine of epithelial cells is accumulated since the influx of CAAs in LPI cells is intact, while efflux is abolished, possibly producing more nitric oxide (NO), which induces cell damage and apoptosis. Finally, cell responses to inflammatory and apoptosis increase (6). Together, increases in cell apoptosis and decreases in its clearance result in aberrant inflammation and autoimmunity. Apart from reduced phagocytosis, studies also demonstrate that there are other macrophage dysfunctions, such as aberrant toll-like receptor (TLR) pathways and a rise in serum inflammatory cytokine levels (30). These findings elucidate a central role of macrophages and the innate immune system in the pathogenesis of LPI. Recent studies reveal more functions other than arginine transposition of y+LAT-1. For example, the SLC7A7 mutation directly provokes production of proinflammatory cytokines, IL-1β and TNF-α, in macrophages and airway epithelial cells in an arginine independent way, partially explaining HLH and pulmonary diseases in y+LAT-1 deficient patients. This may ascribe to y+LAT-1’s ability to inhibit the nuclear factor kappa-B (NF-κB) pathway in a physiological scenario (31). Once this inhibition is lifted, cytokines are produced and released in large amounts. Besides, bleeding events were reported in some cases (7). Patients do not have spontaneous bleeding tendency but mucocutaneous bleeds can be triggered by invasive and surgical interventions or postpartum. Possible mechanisms include reduced NO production (32) and impaired hepatic clearance of F1 + 2, PAP, and D-dimer (7).
Difficulty in diagnosis lies in the heterogenetic phenotypes. Patients with the same point mutation (6) or even from the same family (5)may have different clinical manifestations and prognosis. Therefore, the SLC7A7 mutation is the only precise diagnostic method, but it still cannot predict the symptoms and disease development (5). Current therapy consists of three parts: ① prevention of hyperammonemia, meaning that hypoproteinemic regimen is required; ② nutritional supplementation, including L-citrulline, L-carnitine, vitamins and other nutritional supplementation; ③ prevention of specific complications, like renal and cardiovascular manifestations (5). With the development of our knowledge of LPI, new therapeutic strategies, like anti-cytokine drugs are proposed as well (31). Notably, pulmonary involvement is highly associated with death (5) and therapy targeting pulmonary is necessary.
Even though LPI was discovered more than fifty years ago, its pathogenesis still remains unclear. The relationship between SLC7A7 and LPI requires further study. The tamoxifen-induced ablation by UBC-Cre-ERT2 of Slc7a7 in the mouse (Slc7a7−/−) model reported by Bodoy et al. successfully mimics the phenotypes in human LPI (33). Viable animal models like this may accelerate the research of LPI pathogenesis, especially the complicated immune manifestations and possible available treatment.
IL-2Rβ (CD122) Deficiency
IL-2 is a critical immune regulation cytokine and IL-2Rs are expressed on the surface of T cells and NK cells, which are composed of IL-2Rα (CD25), IL-2Rβ (CD122) and IL-2Rγ (CD132). IL-2 is bi-functional in immune regulation. First of all, it boosts the immune response by promoting the proliferation, differentiation and function of effector T cells and NK cells (34, 35). Secondly, it participates in the maintenance and function of Tregs (regulatory T cells) which act as suppressive regulators (35, 36). In recent years, IL-2 and related biological production began to be used in the treatment of cancer and autoimmune diseases (37, 38). For the best application of these drugs, a full understanding of how IL-2 functions through IL-2Rs is imperative and the study of IL-2R deficiency can improve it vastly.
While the roles of IL-2Rα and IL-2Rγ deficiency in IEI have been known for a long time, IL-2Rβ deficiency in humans was just reported recently. Apart from IL-2R, IL-2Rβ also participates in the formation of IL-15R. Early studies revealed a connection between IL-2Rβ and autoimmune diseases like rheumatoid arthritis (RA) and type 1 diabetes (TID) (39, 40). Additionally, studies on IL-2Rβ deficient patients further confirm the connection and enrich the spectrum of primary diseases of immune regulation. Fernandez et al. reported a pair of siblings with homozygous IL2RB mutations which decrease IL-2Rβ expression and dysregulate IL-2 and IL-15 signaling. Plasma levels of IL-2 and IL-15 are increased, thus explaining the elevated CD8+ T cells and NK cells, while other proinflammatory cytokines are almost equal to healthy controls. Although the total number of NK cells in the patients is elevated, there is a block in the transition from immature ones to functional ones, contributing to higher susceptibility to cytomegalovirus (CMV). As for T cell subsets, there is a skew to memory T cells with reduced Tregs (8). Another group (9) reported patients with different mutations in IL2RB. Despite a slight difference in phenotypes and severity, all patients share common manifestations, namely immunodeficiency and autoimmune diseases including enteropathy, skin abnormalities, autoimmune hemolytic anemia, hypergammaglobulinemia and high susceptibility to infections, proving the pleiotropic functions of IL-2R signaling.
HSCT has a therapeutic effect on IL-2Rβ deficiency, but also causes a risk of complications of thrombotic microangiopathy, which can be lethal (8, 9). Another potential treatment is to hyper-stimulate residual surface IL-2Rβ, since expression of IL-2Rβ is decreased rather than abolished and downstream signaling pathways remain intact (9).
DEF6 Deficiency
Differentially expressed in FDCP6 homolog (DEF6), also known as IRF4 binding protein (IBP) or SWAP-70-like adaptor of T cells (SLAT), is a TCR downstream guanine nucleotide exchange factor (GEF).
Variations in phenotypes of DEF-6 knockout mice made its immune function mysterious (41–43). However, studying inborn DEF6 deficient patients clarified its role. Clinical manifestations of DEF6 deficient patients consist of T-cell lymphopenia, low class-switched B cells, hepatosplenomegaly, autoimmunity, bowel inflammation and susceptibility to EBV (10, 11). Cytotoxic T Lymphocyte antigen 4 (CTLA-4), expressed by activated T cells and Foxp3+ Tregs, is an antagonist of co-stimulator CD28 and competes with it in combination with CD80/CD86. Therefore, it has a negative role in co-stimulation of T cells (44). Membrane expressed CTLA-4 undergoes endocytosis constitutively and therefore CTLA-4 in healthy people is dominantly located in intracellular vesicles. Internalized CTLA-4 either goes back to the plasma membrane or is degraded (44). Previous studies have already shown that deficiency in lipopolysaccharide-responsive and beige-like anchor protein (LRBA), a protein involved in CTLA-4 traffic, causes increased CTLA-4 degradation and patients with LRBA deficiency have autoimmune and inflammatory symptoms just like those with DEF6 deficiency (45). Patients with DEF6 deficiency also show reduced availability of surface CTLA-4. DEF6 directly interacts with the small GTPase, RAB11, on recycling endosomes and therefore affects CTLA-4 shuttling. The attenuated availability of CTLA-4 accounts for defected CD80 uptake and autoimmune symptoms of the patients (10). In addition, it may also thwart Tregs maturation because Foxp3+CD25- regulatory T cells, likely being immature Tregs or precursors, are over-expanded in DEF6 deficient patients (11).
CTLA-4-Ig therapy helps to ameliorate these symptoms (10) and plasma exchanges and immune suppressors like corticosteroids, rituximab, azathioprine and bortezomib are also shown to be effective (11).
FERMT1 Deficiency
Mutations in FERMT1 (also known as KIND1), encoding the focal adhesion protein kindlin-1, cause Kindler syndrome (KS). KS patients are predominantly offspring of consanguineous couples (12) but there are exceptions (46). The major manifestation of KS is skin disorder such as atrophy, blistering, poikiloderma, photosensitivity (12, 13, 47). There is also increased risk of mucosal stenosis and muco-cutaneous cancer in KS patients. Extra-cutaneous manifestations mainly lie in inflammation, including colonic inflammation, gingivitis, periodontitis and mucosal inflammation (13). FERMT1 is critical to integrin activation. When FERMT1 is deficient, reduced β1 integrin activation causes attenuated keratinocyte-cell-extracellular matrix (ECM) adhesion, partially explaining the skin blistering in KS (47). Also, reduced β1 integrin activation is related to lower epidermal keratinocytes proliferation, accounting for skin atrophy in KS (47). While atrophy and blistering often occur at young patients, photosensitivity and squamous cell carcinoma (SCC) happen later. This is because with the patients aging, the effect of UV irradiation and chemical stressors accumulates, provoking reactive oxygen species (ROS). ROS leads to oxidative stress and molecular damage including DNA damage. Normal people can be protected from these damages by FERMT1 through activation of ERK pathway and inhibition of cyclin‐dependent kinase (CDK) activity while in KS patients, they accumulate and result in photosensitivity and high risk of SCC (48–50). Others hold the view that FERMT1 exerts a tumor-suppression role by balancing TGF-β–mediated growth-inhibitory signals and Wnt–β-catenin–mediated growth-promoting signals. Loss of balance when FERMT1 is deficient results in higher risk of muco-cutaneous cancer (51). However, in this animal experiment, tumors induced are basal cell carcinomas but not SCC. Inflammation of KS consists of increased cytokine secretion and macrophage infiltration but the precise mechanisms have not been revealed (52). Mucosal stenosis indicates the existence of fibrosis. Further studies reveal paracrine epithelial–mesenchymal signals accounting for the fibrosis. Keratinocytes lack FERMT1 over express and secret IL-20 and IL-24 under pressure, which stimulate fibroblasts and promote fibrosis (52). Genotype–phenotype correlation is not clear in KS, and possible influential factors consist of environmental, ethnic and geographical backgrounds (14). Studies on immune system in KS patients are rare up to now. However, integrin β1 is critical for CD4+ T cells migration (53), indicating that this process is possibly affected in KS patients.
Like many other IEI, mutation analysis is the most reliable diagnosis method for FERMT1 deficiency. Disrupted basement membrane found by indirect immunofluorescence (IIF) and transmission electron microscopy (TEM) also provides supportive evidence (54). It is same to other epidermolysis bullosa(EB) that there has not been a widely accepted, specific, safe and effective therapeutic method for KS, although gene therapy, protein replacement and HSCT have been reported (55). Further studies focusing on the exact pathogenesis of KS and precise functions of FERMT1 will shed light on this field.
SOCS1 Deficiency
Interferons (IFN) are critical for activating immune responses and providing antiviral protection. By binding to the receptor complex, they activate JAK, which then phosphorylates STAT. Activated STAT translocates to the nucleus and changes the expression of genes, contributing to phenotype alterations. However, their roles are not always beneficial to the host. Excessive IFN signaling causes severe toxicity and even lethal syndromes (56, 57). Consequently, a balance between activation and suppression of IFN is required. Suppressor of cytokine signaling (SOCS) 1 is an essential suppressor for type I and type II IFN signaling through inhibiting the JAK-STAT pathway and its expression can be induced by cytokines including type I and type II IFN, forming a negative feedback loop (58, 59). Homozygous deficiency of SOCS1 in mice is lethal with hypersensitivity to IFN-γ (57) and there are no homozygous SOCS1 deficient patients reported up to now, indicating its indispensable and uncompensated role in immune homeostasis. Mechanisms of SOCS1 in inhibiting type I and type II IFN signaling are quite different. As for type II IFN, both expression and sensitivity are increased in SOCS1 deficient mice, while only sensitivity is affected in the case of type I IFN (60). Among 8 family members of SOCS proteins, SOCS1 and SOCS3 are distinctive in inhibitory mechanisms. They not only inhibit downstream signaling by facilitating ubiquitination of signal intermediates, but they also have a kinase inhibitory region (KIR) that interacts with JAK directly. SOCS1 acts as a pseudo-substrate of JAK, blocking its interaction with real substrates, which mediate downstream signals. SOCS1 also binds to unphosphorylated JAK, which enhances its negative regulatory role (61). Also, accumulating studies show that SOCS1 can function in the nucleus in a different mode with JAK-STAT inhibition. It has a nuclear localization signal (NLS) that accounts for its directed location to the nucleus (62). Its roles in the nucleus have not been fully unraveled, but several functions have already been found, such as activating p53 (63) and limiting NF-κB signaling (64). Zimmer et al. adopted an elegant tool to study the nucleus-located SOCS1 by replacing its NLS. They found that mice lacking nuclear SOCS1, but not cytoplasmic SOCS1, do not have neonatal lethal symptoms, but have mild airway inflammation, indicating different roles of nucleus located and cytoplasm located SOCS1 (65).
SOCS1 is widely expressed in hematopoietic and stromal cells and therefore has multi-facet roles in immune regulation. In DCs, SOCS1 prevents aberrant overexpression of BAFF by breaking IFN induced “DC activation-IFNs release” positive loop, explaining autoimmune phenotypes, including overexpression of autoantibodies in SOCS1 deficient mice (66). In T cells, which are an important source of IFN-γ themselves, SOCS1 modulates their differentiation and terminates IFN-JAK-STAT signals to prevent overproduction of inflammatory cytokines (67, 68). SOCS1 is also found to sustain Foxp3 stability and Treg’s suppression function by preventing transformation to Th1- and Th17-like cells under inflammatory circumstances (69, 70). Deficiency of SOCS1 renders NKT cells abnormally active caused by loss of cross-talk inhibition of IFN-γ and IL-4 signaling, contributing to fulminant hepatitis (71).
Studies have shown a correlation between less SOCS1 serum levels and SLE. Abnormal activation of STAT1 in SLE patients contributes to over production of pro-inflammatory factors (72). Patients with heterozygous mutations in SOCS1 are reported to have immune cytopenia, autoimmune diseases, multisystem inflammation and lymphoproliferation (15, 16). Corresponding with the known function of SOCS1, it is observed that in patients with SOCS1 haploinsufficiency, phosphorylation of STAT1 after IFN-β and IFN-γ stimulation and basal expression of IFN-stimulated genes are increased compared to normal people, suggesting stronger type I and type II IFN signaling, which contributes to autoinflammation and cytopenia (15). Some SOCS1 mutation carriers are clinically asymptomatic, but still have an abnormal immune cell compartment, autoantibodies and higher IFN-γ-induced STAT1 phosphorylation (16).
A SOCS1 mimetic peptide alleviates SLE symptoms in MRL/lpr mice and significantly corrects their immune system by enhancing Foxp3 expression in Tregs and reducing abnormal T and B cell effects (73). Such peptides may also have therapeutic potential in SOCS1 deficient patients. Mycophenolate mofetil and the JAK1/2 inhibitor, baricitinib, are effective to mitigate manifestations in SOCS1 deficient patients (15, 16). Since SOCS1 expresses in both stromal and hematopoietic cells, the efficiency of HSCT remains unclear (15).
TGFB1 Deficiency
Transforming growth factor (TGF)-β1 is encoded by TGFB1 and is first translated into a precursor form containing an N-terminal signal peptide, a latency-associated peptide (LAP) and the C-terminal mature growth factor (TGF-β1). It is a strong immunosuppressive factor and functions through SMAD pathways (74). It has multifaceted roles in inflammation, oncogenic and fibrinogenic modulation. As for inflammatory regulation, TGF-β1 carried by extracellular vesicle mitigates inflammation in whole-blood cells by inhibiting IL1B transcription via upregulating SMAD7, as well as by amplifying the anti-inflammation role in endothelial cells via further upregulating TGFB1 transcription (75). Although TGF-β1 is expressed by many types of cells, Tregs are a non-redundant source of it, controlling allergic and autoimmune responses in a microbiota- and dose-dependent way in mice. Dose-dependent refers to different phenotypes between TGFB1 haploinsufficiency and biallelic deletion. TGFB1 haploinsufficiency in Tregs leads to food allergies while biallelic TGFB1 deletion results in autoimmunity, consisting of autoantibody release and dysregulations in DCs and effector T cells (76). Apart from Tregs, TGF-β1 also affects other subsets of T cells. It imposes constraints on activation-induced cell death (AICD) by downregulating Fas ligand via inhibiting c-Myc expressing, which is beneficial to the expansion of effector T cells and differentiation into memory T cells (77). It has a negative role in Th2 cell expansion, together with downregulating GATA-3 expression and IL-4-induced STAT6 activation (78). In cell populations other than T cells, TGF-β1 plays key roles as well. M2 macrophages not only bind to but also re-release TGF-β1, which is pivotal to their Treg induction role (79). In addition, TGF-β1 favors Langerhans cell (LC) differentiation in dendritic cells (DCs), while it shows a negative role in DC maturation, preventing DC activation in response to harmless environmental stimulation and therefore preparing them for response to dangerous signals (80). TGF-β inhibits maturation and activation of NK cells as well. It inhibits maturation from two facets, by preventing cell-cycle and by constraining transcription factors related to maturation (81). This inhibition has a double-sided nature. On one hand, it is useful for preventing harmful inflammation in early stages of development. And on the other hand, susceptibility to infection is extended since there is a lack of NK cells (81). A significant mediator of NK activation is mTOR induced by IL-2 or IL-15. By antagonizing the mTOR pathway, TGF-β negatively regulates activation of NK cells (82).
Typical manifestations of TGFB1 deficient patients include inflammatory bowel disease (IBD) and recurrent infections which can be lethal are reported as well (17). Numbers and distributions for T cells, B cells and NK cells can be normal or disturbed while proliferation of T cells stimulated with anti-CD3 is reduced. The immune dysregulation phenotypes of TGF-β1 deficient patients suggest an indispensable role of TGF-β1 in immune regulation. Besides, central nervous system (CNS) dysfunctions are also common in TGFB1 deficient patients. These dysfunctions include epilepsy, brain atrophy and posterior leukoencephalopathy (17). Former studies show reduced neuronal TGF-β signaling promotes Alzheimer’s disease (AD) and neurodegeneration (83). Mechanisms underlying neuronal roles of TGF-β has not been totally revealed. One possible explanation lies in its relationship with olfactory ensheathing cells (OECs). Clearance of degenerating or dying neurons and apoptotic neuron debris is critical to neuron regeneration, that is to say, it benefits the restoration of CNS injuries and neurodegeneration. TGF-β promotes this process by enhancing phagocytic activity of OECs through integrin/MFG-E8 signaling pathway and by shifting OECs shape to increase cellular surface area (84).
Possible therapies include HSCT and recombinant TGF-β1 replacement, both of which have risks of severe comorbidities, and thus there is a need for safer and more efficient treatments (17). As for the symptoms of early onset IBD, surgery, nutritional therapy and some emerging new methods like complementary medicine and fecal microbiota transplantation may also be effective (85).
RIPK1 Deficiency
RIPK1 (receptor-interacting serine/threonine kinase 1) is a component of signal transduction complexes, mediating signals from surface receptors like Toll-like receptor 3 (TLR3), TLR4 and tumor necrosis factor receptor 1 (TNFR1) and controlling cell death and inflammation. RIPK1 can both mediate cell death and promote cell survival (86). Its role in cell death varies with cell types and contexts. There are two types of cell death inhibited by RIPK1 in hematopoietic cells, keratinocytes, epithelial cell and DCs, namely necroptosis via RIPK3 and pseudo-kinase mixed lineage kinase domain-like (MLKL), and apoptosis via caspase-8. These two types can switch from one to the other easily, when responding to conditional changes (86–88). Because necroptosis results in proinflammatory danger-associated molecular patterns (DAMP), RIPK1 can act as an inhibitor of inflammation in physiological situations (89–91). RIP homotypic interaction motif (RHIM) of RIPK1 is indispensable in competing with RIPK3 RHIM in ZBP1 (Z-DNA binding protein 1; also known as DAI or DLM1) binding and thus prevents RIPK3 autophosphorylation that is critical for necroptosis (92, 93). In the regulation of caspase-8, RIPK1 acts as both scaffold and kinase (88, 94, 95).
Manifestations in human patients include lymphopenia, susceptibility to infections, early-onset IBD and arthritis. Numbers of T, B and NK cells diminish to different extents (18–20). Cellular and molecular studies showed that patients with loss-of-function mutations in RIPK1 have dysregulation in cytokine release such as increased IL-1β and decreased IL-10, contributing to arthritis and IBD together (19). Although RIPK1 conditional deletion animal models show autoimmunity, patients with RIPK1 deficiency have not been reported to have autoimmune symptoms (90, 91).
A consensus has not been reached in therapeutic approaches for RIPK1 deficiency. Cuchet-Lourenço et al. proposed that HSCT is adequate since it greatly remitted clinical symptoms and dysregulated cytokine production in a patient (19), while Li et al. warranted that intrinsic intestinal phenotypes cannot be mitigated by HSCT (20).
In recent years, RIPK1 inhibitors are considered to be a curative therapy to diseases related to necroptosis. Studies with RIPK1 deficient patients further reveal the multi-faceted roles of RIPK1, so the use of RIPK1 inhibitors should be cautious and its safety requires deeper research (20). Besides, the lack of consensus on RIPK1 deficiency treatment also emphasizes the urgency of revealing the exact functions of RIPK1 in human.
CD137 Deficiency
CD137, also known as 4-1BB or tumor necrosis factor (TNF) receptor superfamily member 9 (TNFRSF9) is pivotal for immune homeostasis and tumor suppression. Ligation by CD137L leads to oligomerization of CD137 and galectin-9 (Gal-9) serves as a bridge. TRAFs are then recruited and activate NF-κB and PI3K-AKT pathways (96). Although it is expressed by various types of cells, its role in T cells as an inducible costimulator has been studied the most, which is significant for proper T cell survival, differentiation and cytokine secretion (97). The pro-survival role is mediated by upregulated anti-apoptosis protein Bcl-xL through the NF-κB pathway (97, 98). Generally, CD137 positively regulates the T cell response, but there are some exceptions. In the case of Tregs, it promotes clonal expansion, yet transiently neutralizes the suppressive activity of activated Tregs. Also, CD137 inhibits the expansion of Th17 in both IFN-γ dependent and independent ways, which is important for remission of autoimmunity (99). CD137 is also expressed on human B cells, but not murine B cells. Its functional mode in B cells resembles that in T cells in several facets. First, activation of BCRs/TCRs is a prerequisite for CD137 expression, which enhances survival, proliferation and cytokine production (97, 100). In invariant NKT (iNKT) cells, iNKT–monocyte interaction via CD137/CD137L promotes iNKT survival and proliferation and this interaction also affects monocytes survival (101). When CD137 is deficient, iNKT counts are reduced (102) due to at least partially the attenuated CD137/CD137L signal. Since iNKTs provide apoptosis signal to monocytes and prevent their over proliferation during inflammation, reduced iNKT number may account for the immune dysregulation of CD137 deficient patients. Additionally, like many TNF/TNFR superfamily members, CD137/CD137L evokes reverse signals to CD137L positive cells, making it hard to determine the respective functions of each one (103).
CD137 deficient mice have enhanced T cell proliferation, while the CTL response and IFN-γ expression are dampened (104). Patients having reduced or ablated expression of CD137 show hampered immune regulation, being susceptible to various pathogens including Epstein-Barr virus (EBV) and predisposed to EBV-related B-cell lymphoma as well as showing symptoms of autoimmunity (21, 22). Proliferation and function of both B cells and T cells are dysregulated, emphasizing the importance of CD137 in homeostasis of immune system. Notably, mitochondrial function is significant in T cell function and it is impaired in CD137 deficient patients (22). However, it is intriguing that some siblings harboring the same mutation as the patients do not show clinical manifestations, suggesting that the overt disease is affected by some other factors other than genes (21).
Possible therapeutic approaches lie in other costimulators like CD28, given that elevated CD28 signals ameliorated deficiency in T cell proliferation (21). Furthermore, the immunophenotype can be reversed by HSCT. As for patients with lymphoma, the less toxic and more specific therapies are recommended before HSCT, which can prevent the use of radiological treatment (23).
TET2 Deficiency
Ten-Eleven Translocation methylcytosine dioxygenase 2 (TET2) is a pivotal epigenetic regulatory factor in hematopoietic cells and it facilitates demethylation by oxidizing 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and other oxidation products (24). Accumulating evidence points out that its mutations are frequent in clonal hematopoiesis and myeloid malignancies (25). Therefore, it is reasonable to presume that TET2 has a crucial role in cell proliferation and differentiation. Homogeneous or heterogeneous loss of TET2 renders stem cell renewal more active in a cell intrinsic way, disturbs differentiation and increases the risk of myeloproliferation (105–108). CXCR4, a chemokine receptor critical for B cell development, is elevated in heterozygous TET2 mutant patients, suggesting a B cell regulatory role of TET2 (109). TET proteins are also essential for specific points of B cell development, for example the transition from pro-B to pre-B and the differentiation to plasma cells. For the former one, TET2/3 not only augment Igκ expression and rearrangement per se, but also assist the function of transcription factors (110). By demethylating CpG sites around Irf4, they allow high expression of IRF4, which is critical for the transition into plasma cells. However, it is not necessary for the initiation of Irf4 expression, but only the maintenance of it (111). In Tregs, TET is crucial for stable Foxp3 expression through regulation on the conserved non-coding DNA sequence-2 (CNS2) region and ‘upstream enhancer’ region. TET2/3 double-knockout Tregs show abnormal activity in proliferation and the counts of Th17 and Tfh-like cells are increased as well (112).
Autosomal homozygous TET2 missense or nonsense in humans results in immunodeficiency, growth impairment and autoimmune lymphoproliferative syndrome (ALPS) like phenotypes of raised proportion of double negative (CD4-CD8-) T-cells (DNTs), raised soluble Fas ligand level, lymphadenopathy, hepatosplenomegaly, autoimmunity and remarkable predisposition to lymphoma, which is reminiscent of phenotypes of TET2 deficient mice (24). Due to the loss-of-function mutation of TET2, levels of DNA methylation increase in hematologic cells, especially in the regions able to bind master transcription factors that have a strong regulatory effect on hematopoiesis, accounting for the skew to DNTs and failure of proper development in B cells (24, 109). Increased soluble Fas ligand accounts for reduced FasL-induced apoptosis and therefore the tendency to lymphoproliferation. Haploinsufficiency of TET2 is a wide spread mutation related to hematological neoplasia, although it is not able to induce cancer alone given that TET2 mutations also occur in healthy groups with clonal hematopoiesis (25, 109, 113, 114). Extrinsic factors may explain the phenotypes, but the underlying mechanisms have not been fully unraveled. Recent studies show that infection-induced inflammation is critical for the onset of malignance in TET2-/- mice, shedding light on the clinical prevention and treatment of malignancy related to TET2 deficiency (115, 116). In diffuse large B cell lymphomas (DLBCLs), gene alterations involved in TET2 mutation consist of both losing enhancer 5hmC and gaining promoter 5mC. Additionally, the chromatin accessibility and stability are also reduced. Activity of activation-induced cytidine deaminase (AID) and subsequent deamination are hampered in TET2-/- mice, which contributes to the disturbance of demethylation further. Together, these changes disrupt transcription of genes critical for GC (germinal center) exit, antigen presentation and differentiation of GC B cells, accounting for the occurrence of DLBCLs (113). In addition, mutations in TET2 are also a risk factor for neurodegenerative disorders such as early-onset Alzheimer’s disease and frontotemporal dementia (117).
Recent studies have shown that vitamin C can mimic restoration of TET2, implicating a possible effect of high-dose vitamin C incorporation on TET2 deficient patients. Notably, the function of vitamin C requires a minimal existence of TET, meaning that the combined loss of TET2 and TET3 leads to poor response to vitamin C treatment (107). Besides, since mutations in TET2 alone are not sufficient for cancer onset, therapy targeting assistant factors, such as other genetic deficiencies and immunostimulation, are also effective treatments. As mentioned before, infection-induced inflammation, which can be corrected by antibiotics, shows positive correlation with myeloid expansion in TET2 deficient mice. Inhibition of bacterial inflammatory signals, such as inhibiting TNF-α, prevents tumor growth, providing a viable preventive method for TET2 deficient malignancy (116). With the high level of methylation being a positive factor for malignancy, it is not surprising that DNA methyltransferase inhibitors (DNMTi) are also effective (113).
Conclusion
In recent years, there has been a rapid growth of IEI discoveries and a deeper understanding of their mechanisms. It has come to our knowledge that IEI does not merely mean higher susceptibility to infections. Those patients with a combination of several types of autoimmune or inflammatory diseases should be considered for a scan of IEI. As for therapeutic methods, nonspecific immunosuppressive agents used to prevent autoimmunity sometimes have many side effects and they do not fit to the situation where autoimmune and susceptibility coexist. HSCT has similar shortcomings as well. Morbidity and mortality related to HSCT are still considerable problems (118). Besides, although it has a definitive effect in some cases, it acts less well when stromal cells, but not merely blood cells, are also affected (15). Thus, safer and more effective therapeutic methods are required. Pathogenesis studies of these immune dysregulations will shed light on this field. Clarifying the relationship between IEI and immune dysregulation helps not only to diagnose patients as early as possible, but also to improve the quality of the patients’ life and prolong their survival. Apart from the direct clinical use, studies on IEI also provide a wonderful way to understand the mechanism of immune regulation in our body and produce animal models for related research (10, 33).
There are several difficulties in identifying IEI: First, since every single inborn error has a relatively low prevalence rate, it is hard to figure out its clinical symptoms fully and quickly. Some patients harboring more than one mutation make it even more difficult to detect (11). Besides, it is intriguing and confusing that phenotypes of the same mutation vary from patient to patient, ranging from mild or even no obvious symptoms to life-threatening manifestations (21, 119). This complexity may come from both internal and external factors, such as incomplete penetrance and additional factors like infections and combination with other gene deficiencies (20, 21, 120).
The most crucial issue in IEI is to identify their pathogenesis and gene-phenotype relationship. It requires both clinical and laboratory efforts to address this problem. In clinical treatment, patients with immune dysregulations should be examined carefully and genetic identification should be executed if necessary and possible. By doing so, it can not only avoid misdiagnosis, but also find new mutations. Considering that the prevalence of IEI is very low and acquiring enough samples for studies is difficult, creating proper animal models is a useful alternative. However, it should be kept in mind that there are differences between humans and animals. Therefore, results from animal models only provide us with an indication and they should be examined on humans later to test the consistency.
Author Contributions
AR wrote the article. WY, HM, LW, FC, CP and PL reviewed and revised the draft. QG, YC and CL organized and revised the draft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (31970839), HUST Academic Frontier Youth Team (2018QYTD10) and Independent Innovation Research Fund of Huazhong University of Science and Technology (2020kfyXGYJ017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
2. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee (2021) 41:666–79. doi: 10.1007/s10875-021-00980-1.
3. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, Adoue D, et al. Autoimmune and Inflammatory Manifestations Occur Frequently in Patients With Primary Immunodeficiencies. J Allergy Clin Immunol (2017) 140:1388–93.e8. doi: 10.1016/j.jaci.2016.12.978
4. Chan AY, Leiding JW, Liu X, Logan BR, Burroughs LM, Allenspach EJ, et al. Hematopoietic Cell Transplantation in Patients With Primary Immune Regulatory Disorders (PIRD): A Primary Immune Deficiency Treatment Consortium (PIDTC) Survey. Front Immunol (2020) 11:239. doi: 10.3389/fimmu.2020.00239
5. Mauhin W, Habarou F, Gobin S, Servais A, Brassier A, Grisel C, et al. Update on Lysinuric Protein Intolerance, A Multi-Faceted Disease Retrospective Cohort Analysis From Birth to Adulthood. Orphanet J Rare Dis (2017) 12:3. doi: 10.1186/s13023-016-0550-8
6. Tringham M, Kurko J, Tanner L, Tuikkala J, Nevalainen OS, Niinikoski H, et al. Exploring the Transcriptomic Variation Caused by the Finnish Founder Mutation of Lysinuric Protein Intolerance (LPI). Mol Genet Metab (2012) 105:408–15. doi: 10.1016/j.ymgme.2011.12.007
7. Pitkänen HH, Kärki M, Niinikoski H, Tanner L, Näntö-Salonen K, Pikta M, et al. Abnormal Coagulation and Enhanced Fibrinolysis Due to Lysinuric Protein Intolerance Associates With Bleeds and Renal Impairment. Haemophilia (2018) 24:e312–21. doi: 10.1111/hae.13543
8. Fernandez IZ, Baxter RM, Garcia-Perez JE, Vendrame E, Ranganath T, Kong DS, et al. Correction: A Novel Human IL2RB Mutation Results in T and NK Cell–Driven Immune Dysregulation. J Exp Med (2019) 216:1465. doi: 10.1084/jem.2018201505102019c
9. Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, et al. Human Interleukin-2 Receptor β Mutations Associated With Defects in Immunity and Peripheral Tolerance. J Exp Med (2019) 216:1311–27. doi: 10.1084/jem.20182304
10. Serwas NK, Hoeger B, Ardy RC, Stulz SV, Sui Z, Memaran N, et al. Human DEF6 Deficiency Underlies an Immunodeficiency Syndrome With Systemic Autoimmunity and Aberrant CTLA-4 Homeostasis. Nat Commun (2019) 10:3106. doi: 10.1038/s41467-019-10812-x
11. Fournier B, Tusseau M, Villard M, Malcus C, Chopin E, Martin E, et al. DEF6 Deficiency, A Mendelian Susceptibility to EBV Infection, Lymphoma, and Autoimmunity. J Allergy Clin Immunol (2021) 147:740–3.e9. doi: 10.1016/j.jaci.2020.05.052
12. Fuchs-Telem D, Nousbeck J, Singer A, McGrath JA, Sarig O, Sprecher E. New Intragenic and Promoter Region Deletion Mutations in FERMT1 Underscore Genetic Homogeneity in Kindler Syndrome. Clin Exp Dermatol (2014) 39:361–7. doi: 10.1111/ced.12222
13. Techanukul T, Sethuraman G, Zlotogorski A, Horev L, Macarov M, Trainer A, et al. Novel and Recurrent FERMT1 Gene Mutations in Kindler Syndrome. Acta Derm Venereol (2011) 91:267–70. doi: 10.2340/00015555-1063
14. Kartal D, Borlu M, Has C, Fölster-Holst R. A Novel Mutation in the FERMT1 Gene in Turkish Siblings With Kindler Syndrome. J Eur Acad Dermatol Venereol (2016) 30:1233–5. doi: 10.1111/jdv.13163
15. Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune Dysregulation and Multisystem Inflammatory Syndrome in Children (MIS-C) in Individuals With Haploinsufficiency of SOCS1. J Allergy Clin Immunol (2020) 146:1194–200.e1. doi: 10.1016/j.jaci.2020.07.033
16. Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-Onset Autoimmunity Associated With SOCS1 Haploinsufficiency. Nat Commun (2020) 11:1–11. doi: 10.1038/s41467-020-18925-4
17. Kotlarz D, Marquardt B, Barøy T, Lee WS, Konnikova L, Hollizeck S, et al. Human TGF-β1 Deficiency Causes Severe Inflammatory Bowel Disease and Encephalopathy. Nat Genet (2018) 50:344–8. doi: 10.1038/s41588-018-0063-6
18. Lin L, Wang Y, Liu L, Ying W, Wang W, Sun B, et al. Clinical Phenotype of a Chinese Patient With RIPK1 Deficiency Due to Novel Mutation. Genes Dis (2020) 7:122–7. doi: 10.1016/j.gendis.2019.10.008
19. Cuchet-Lourenço D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 Mutations in Humans Cause Severe Immunodeficiency, Arthritis, and Intestinal Inflammation. Science (80-) (2018) 361:810–3. doi: 10.1126/science.aar2641originally
20. Li Y, Führer M, Bahrami E, Socha P, Klaudel-Dreszler M, Bouzidi A, et al. Human RIPK1 Deficiency Causes Combined Immunodeficiency and Inflammatory Bowel Diseases. Proc Natl Acad Sci USA (2019) 116:970–5. doi: 10.1073/pnas.1813582116
21. Somekh I, Thian M, Medgyesi D, Gülez N, Magg T, Duque AG, et al. CD137 Deficiency Causes Immune Dysregulation With Predisposition to Lymphomagenesis. Blood (2019) 134:1510–6. doi: 10.1182/blood.2019000644
22. Alosaimi MF, Hoenig M, Jaber F, Platt CD, Jones J, Wallace J, et al. Immunodeficiency and EBV-Induced Lymphoproliferation Caused by 4-1BB Deficiency. J Allergy Clin Immunol (2019) 144:574–83.e5. doi: 10.1016/j.jaci.2019.03.002
23. Wildermann C, Alosaimi M, Liebenehm S, Jacobsen EM, Barth TFE, Möller P, et al. Successful Hematopoietic Stem Cell Transplantation in a 4-1BB Deficient Patient With EBV-Induced Lymphoproliferation. Clin Immunol (2021) 222:2020–2. doi: 10.1016/j.clim.2020.108639
24. Spegarova JS, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 Loss of Function Causes Childhood Immunodeficiency and Lymphoma. Blood (2020) 136:1055–66. doi: 10.1182/blood.2020005844
25. Duployez N, Goursaud L, Fenwarth L, Bories C, Marceau-Renaut A, Boyer T, et al. Familial Myeloid Malignancies With Germline TET2 Mutation. Leukemia (2020) 34:1450–3. doi: 10.1038/s41375-019-0675-6
26. Rotoli BM, Barilli A, Visigalli R, Ferrari F, Dall’Asta V. Y+LAT1 and Y+LAT2 Contribution to Arginine Uptake in Different Human Cell Models: Implications in the Pathophysiology of Lysinuric Protein Intolerance. J Cell Mol Med (2020) 24:921–9. doi: 10.1111/jcmm.14801
27. Torrents D, Mykkänen J, Pineda M, Feliubadaló L, Estévez R, De Rafael C, et al. Identification of SLC7A7, Encoding Y+LAT-1, as the Lysinuric Protein Intolerance Gene. Nat Genet (1999) 21:293–6. doi: 10.1038/6809
28. Noguchi A, Takahashi T. Overview of Symptoms and Treatment for Lysinuric Protein Intolerance. J Hum Genet (2019) 64:849–58. doi: 10.1038/s10038-019-0620-6
29. Barilli A, Rotoli BM, Visigalli R, Bussolati O, Gazzola GC, Gatti R, et al. Impaired Phagocytosis in Macrophages From Patients Affected by Lysinuric Protein Intolerance. Mol Genet Metab (2012) 105:585–9. doi: 10.1016/j.ymgme.2012.01.008
30. Kurko J, Vähä-Mäkilä M, Tringham M, Tanner L, Paavanen-Huhtala S, Saarinen M, et al. Dysfunction in Macrophage Toll-Like Receptor Signaling Caused by an Inborn Error of Cationic Amino Acid Transport. Mol Immunol (2015) 67:416–25. doi: 10.1016/j.molimm.2015.07.006
31. Rotoli BM, Barilli A, Visigalli R, Ingoglia F, Milioli M, Di Lascia M, et al. Downregulation of SLC7A7 Triggers an Inflammatory Phenotype in Human Macrophages and Airway Epithelial Cells. Front Immunol (2018) 9:508. doi: 10.3389/fimmu.2018.00508
32. Kayanoki Y, Kawata S, Yamasaki E, Kiso SI, Inoue S, Tamura S, et al. Reduced Nitric Oxide Production by L-Arginine Deficiency in Lysinuric Protein Intolerance Exacerbates Intravascular Coagulation. Metabolism (1999) 48:1136–40. doi: 10.1016/S0026-0495(99)90127-0
33. Bodoy S, Sotillo F, Espino-Guarch M, Sperandeo MP, Ormazabal A, Zorzano A, et al. Inducible Slc7a7 Knockout Mouse Model Recapitulates Lysinuric Protein Intolerance Disease. Int J Mol Sci (2019) 20:5294. doi: 10.3390/ijms20215294
34. Sharma R, Das A. IL-2 Mediates NK Cell Proliferation But Not Hyperactivity. Immunol Res (2018) 66:151–7. doi: 10.1007/s12026-017-8982-3
35. Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T Cell Activation and Autoimmunity in Mice Lacking Interleukin-2 Receptor β. Science (80-) (1995) 268:1472–6. doi: 10.1126/science.7770771
36. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, et al. Cutting Edge: Mechanisms of IL-2–Dependent Maintenance of Functional Regulatory T Cells. J Immunol (2010) 185:6426–30. doi: 10.4049/jimmunol.0903940
37. Ghelani A, Bates D, Conner K, Wu MZ, Lu J, Hu YL, et al. Defining the Threshold IL-2 Signal Required for Induction of Selective Treg Cell Responses Using Engineered IL-2 Muteins. Front Immunol (2020) 11:1106. doi: 10.3389/fimmu.2020.01106
38. Drerup JM, Deng Y, Pandeswara SL, Padrón ÁS, Reyes RM, Zhang X, et al. CD122-Selective IL2 Complexes Reduce Immunosuppression, Promote Treg Fragility, and Sensitize Tumor Response to PD-L1 Blockade. Cancer Res (2020) 80:5063–75. doi: 10.1158/0008-5472.CAN-20-0002
39. Yang Y, Yuan S, Che M, Jing H, Yuan L, Dong K, et al. Genetic Analysis of the Relation Between IL2RA/IL2RB and Rheumatoid Arthritis Risk. Mol Genet Genomic Med (2019) 7:1–11. doi: 10.1002/mgg3.754
40. Yuan X, Dong Y, Tsurushita N, Tso JY, Fu W. CD122 Blockade Restores Immunological Tolerance in Autoimmune Type 1 Diabetes via Multiple Mechanisms. JCI Insight (2018) 3:e96600. doi: 10.1172/jci.insight.96600
41. Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, et al. IRF-4-Binding Protein Inhibits Interleukin-17 and Interleukin-21 Production by Controlling the Activity of IRF-4 Transcription Factor. Immunity (2008) 29:899–911. doi: 10.1016/j.immuni.2008.10.011
42. Bécart S, Charvet C, Balancio AJC, De Trez C, Tanaka Y, Duan W, et al. SLAT Regulates Th1 and Th2 Inflammatory Responses by Controlling Ca 2+/NFAT Signaling. J Clin Invest (2007) 117:2164–75. doi: 10.1172/JCI31640
43. Canonigo-Balancio AJ, Fos C, Prod’homme T, Bécart S, Altman A. SLAT/Def6 Plays a Critical Role in the Development of Th17 Cell-Mediated Experimental Autoimmune Encephalomyelitis. J Immunol (2009) 183:7259–67. doi: 10.4049/jimmunol.0902573
44. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A Moving Target in Immunotherapy. Blood (2018) 131:58–67. doi: 10.1182/blood-2017-06-741033.CTLA-4
45. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients With LRBA Deficiency Show CTLA4 Loss and Immune Dysregulation Responsive to Abatacept Therapy. Science (80-) (2015) 349:436–40. doi: 10.1126/science.aaa1663
46. Meng L, Yang X, Wu Y, Zhao Z, Yang L, Li M, et al. A Novel Frameshift Mutation in the FERMT1 Gene in a Chinese Patient With Kindler Syndrome. Exp Ther Med (2020) 20:1–1. doi: 10.3892/etm.2020.9233
47. Lai-Cheong JE, Parsons M, Tanaka A, Ussar S, South AP, Gomathy S, et al. Loss-Of-Function FERMT1 Mutations in Kindler Syndrome Implicate a Role for Fermitin Family Homolog-1 in Integrin Activation. Am J Pathol (2009) 175:1431–41. doi: 10.2353/ajpath.2009.081154
48. Emmert H, Patel H, Brunton VG. Kindlin-1 Protects Cells From Oxidative Damage Through Activation of ERK Signalling. Free Radic Biol Med (2017) 108:896–903. doi: 10.1016/j.freeradbiomed.2017.05.013
49. Zhang X, Luo S, Wu J, Zhang L, Wang W, Degan S, et al. KIND1 Loss Sensitizes Keratinocytes to UV-Induced Inflammatory Response and DNA Damage. J Invest Dermatol (2017) 137:475–83. doi: 10.1016/j.jid.2016.09.023
50. Emmert H, Culley J, Brunton VG. Inhibition of Cyclin-Dependent Kinase Activity Exacerbates H2O2-Induced DNA Damage in Kindler Syndrome Keratinocytes. Exp Dermatol (2019) 28:1074–8. doi: 10.1111/exd.14000
51. Rognoni E, Widmaier M, Jakobson M, Ruppert R, Ussar S, Katsougkri D, et al. Kindlin-1 Controls Wnt and TGF-β Availability to Regulate Cutaneous Stem Cell Proliferation. Nat Med (2014) 20:350–9. doi: 10.1038/nm.3490
52. Heinemann A, He Y, Zimina E, Boerries M, Busch H, Chmel N, et al. Induction of Phenotype Modifying Cytokines by FERMT1 Mutations. Hum Mutat (2011) 32:397–406. doi: 10.1002/humu.21449
53. Ali N, Rosenblum MD. Regulatory T Cells in Skin. Immunology (2017) 152:372–81. doi: 10.1111/imm.12791
54. Has C, Castiglia D, del Rio M, Garcia Diez M, Piccinni E, Kiritsi D, et al. Kindler Syndrome: Extension of FERMT1 Mutational Spectrum and Natural History. Hum Mutat (2011) 32:1204–12. doi: 10.1002/humu.21576
55. Gonzalez ME. Evaluation and Treatment of the Newborn With Epidermolysis Bullosa. Semin Perinatol (2013) 37:32–9. doi: 10.1053/j.semperi.2012.11.004
56. Gutterman JU. Cytokine Therapeutics: Lessons From Interferon α. Proc Natl Acad Sci USA (1994) 91:1198–205. doi: 10.1073/pnas.91.4.1198
57. Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a Critical Inhibitor of Interferon γ Signaling and Prevents the Potentially Fatal Neonatal Actions of This Cytokine. Cell (1999) 98:597–608. doi: 10.1016/S0092-8674(00)80047-1
58. Qin H, Wilson CA, Lee SJ, Benveniste EN, Qin H, Wilson CA, et al. IFN-β-Induced SOCS-1 Negatively Regulates CD40 Gene Expression in Macrophages and Microglia. FASEB J (2006) 20:985–7. doi: 10.1096/fj.05-5493fje
59. Sato T, Saito R, Jinushi T, Tsuji T, Matsuzaki J, Koda T, et al. IFN-γ-Induced SOCS-1 Regulates STAT6-Dependent Eotaxin Production Triggered by IL-4 and TNF-α. Biochem Biophys Res Commun (2004) 314:468–75. doi: 10.1016/j.bbrc.2003.12.124
60. Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, et al. Suppressor of Cytokine Signaling 1 Regulates the Immune Response to Infection by a Unique Inhibition of Type I Interferon Activity. Nat Immunol (2006) 7:33–9. doi: 10.1038/ni1287
61. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat Commun (2018) 9:1–14. doi: 10.1038/s41467-018-04013-1
62. Baetz A, Koelsche C, Strebovsky J, Heeg K, Dalpke AH. Identification of a Nuclear Localization Signal in Suppressor of Cytokine Signaling 1. FASEB J (2008) 22:4296–305. doi: 10.1096/fj.08-116079
63. Calabrese V, Mallette FA, Deschênes-Simard X, Ramanathan S, Gagnon J, Moores A, et al. SOCS1 Links Cytokine Signaling to P53 and Senescence. Mol Cell (2009) 36:754–67. doi: 10.1016/j.molcel.2009.09.044
64. Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of Cytokine Signaling 1 (SOCS1) Limits Nfκb Signaling by Decreasing P65 Stability Within the Cell Nucleus. FASEB J (2011) 25:863–74. doi: 10.1096/fj.10-170597
65. Zimmer J, Weitnauer M, Boutin S, Küblbeck G, Thiele S, Walker P, et al. Nuclear Localization of Suppressor of Cytokine Signaling-1 Regulates Local Immunity in the Lung. Front Immunol (2016) 7:514. doi: 10.3389/fimmu.2016.00514
66. Hanada T, Yoshida H, Kato S, Tanaka K, Masutani K, Tsukada J, et al. Suppressor of Cytokine Signaling-1 is Essential for Suppressing Dendritic Cell Activation and Systemic Autoimmunity. Immunity (2003) 19:437–50. doi: 10.1016/S1074-7613(03)00240-1
67. Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, et al. SOCS1 Deficiency Causes a Lymphocyte-Dependent Perinatal Lethality. Cell (1999) 98:609–16. doi: 10.1016/S0092-8674(00)80048-3
68. Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, et al. Loss of Suppressor of Cytokine Signaling 1 in Helper T Cells Leads to Defective Th17 Differentiation by Enhancing Antagonistic Effects of IFN-γ on STAT3 and Smads. J Immunol (2008) 180:3746–56. doi: 10.4049/jimmunol.180.6.3746
69. Takahashi R, Nishimoto S, Muto G, Sekiya T, Tamiya T, Kimura A, et al. SOCS1 is Essential for Regulatory T Cell Functions by Preventing Loss of Foxp3 Expression as Well as IFN-γ and IL-17A Production. J Exp Med (2011) 208:2055–67. doi: 10.1084/jem.20110428
70. Takahashi R, Nakatsukasa H, Shiozawa S, Yoshimura A. SOCS1 Is a Key Molecule That Prevents Regulatory T Cell Plasticity Under Inflammatory Conditions. J Immunol (2017) 199:149–58. doi: 10.4049/jimmunol.1600441
71. Naka T, Tsutsui H, Fujimoto M, Kawazoe Y, Kohzaki H, Morita Y, et al. SOCS-1/SSI-1-Deficient NKT Cells Participate in Severe Hepatitis Through Dysregulated Cross-Talk Inhibition of IFN-γ and IL-4 Signaling In Vivo. Immunity (2001) 14:535–45. doi: 10.1016/S1074-7613(01)00132-7
72. Sukka-Ganesh B, Larkin J. Therapeutic Potential for Targeting the Suppressor of Cytokine Signalling-1 Pathway for the Treatment of SLE. Scand J Immunol (2016) 84:299–309. doi: 10.1111/sji.12475
73. Sharma J, Collins TD, Roach T, Mishra S, Lam BK, Mohamed ZS, et al. Suppressor of Cytokine Signaling − 1 Mimetic Peptides Attenuate Lymphocyte Activation in the MRL/lpr Mouse Autoimmune Model. Sci Rep (2021) 11:6354. doi: 10.1038/s41598-021-86017-4
74. Pang N, Zhang F, Ma X, Zhu Y, Zhao H, Xin Y, et al. TGF-β/Smad Signaling Pathway Regulates Th17/Treg Balance During Echinococcus Multilocularis Infection. Int Immunopharmacol (2014) 20:248–57. doi: 10.1016/j.intimp.2014.02.038
75. Halder LD, Jo EAH, Hasan MZ, Ferreira-Gomes M, Krüger T, Westermann M, et al. Immune Modulation by Complement Receptor 3-Dependent Human Monocyte TGF-β1-Transporting Vesicles. Nat Commun (2020) 11:2331. doi: 10.1038/s41467-020-16241-5
76. Turner JA, Stephen-Victor E, Wang S, Rivas MN, Abdel-Gadir A, Harb H, et al. Regulatory T Cell-Derived TGF-β1 Controls Multiple Checkpoints Governing Allergy and Autoimmunity. Immunity (2020) 53:1202–14.e6. doi: 10.1016/j.immuni.2020.10.002
77. Genestier BL, Kasibhatla S, Brunner T, Green DR. Transforming Growth Factor β1 Inhibits Fas Ligand Expression and Subsequent Activation-Induced Cell Death in T Cells via Downregulation of C-Myc. J Exp Med (1999) 189:231–9. doi: 10.1084/jem.189.2.231
78. Heath VL, Murphy EE, Crain C, Tomlinson MG, O’Garra A. TGF-β1 Down-Regulates Th2 Development and Results in Decreased IL-4-Induced STAT6 Activation and GATA-3 Expression. Eur J Immunol (2000) 30:2639–49. doi: 10.1002/1521-4141(200009)30:9<2639::AID-IMMU2639>3.0.CO;2-7
79. Schmidt A, Zhang XM, Joshi RN, Iqbal S, Wahlund C, Gabrielsson S, et al. Human Macrophages Induce CD4 + Foxp3 + Regulatory T Cells via Binding and Re-Release of TGF-β. Immunol Cell Biol (2016) 94:747–62. doi: 10.1038/icb.2016.34
80. Ohtani T, Mizuashi M, Nakagawa S, Sasaki Y, Fujimura T, Okuyama R, et al. TGF-β1 Dampens the Susceptibility of Dendritic Cells to Environmental Stimulation, Leading to the Requirement for Danger Signals for Activation. Immunology (2009) 126:485–99. doi: 10.1111/j.1365-2567.2008.02919.x
81. Marcoe JP, Lim JR, Schaubert KL, Fodil-Cornu N, Matka M, McCubbrey AL, et al. TGF-β is Responsible for NK Cell Immaturity During Ontogeny and Increased Susceptibility to Infection During Mouse Infancy. Nat Immunol (2012) 13:843–50. doi: 10.1038/ni.2388
82. Viel S, Marçais A, Guimaraes FSF, Loftus R, Rabilloud J, Grau M, et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the mTOR Pathway. Sci Signal (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
83. Tesseur I, Zou K, Esposito L, Bard F, Berber E, Van Can J, et al. Deficiency in Neuronal TGF-β Signaling Promotes Neurodegeneration and Alzheimer’s Pathology. J Clin Invest (2006) 116:3060–9. doi: 10.1172/JCI27341
84. Li Y, Zou T, Xue L, Yin ZQ, Huo S, Xu H. TGF-β1 Enhances Phagocytic Removal of Neuron Debris and Neuronal Survival by Olfactory Ensheathing Cells via Integrin/MFG-E8 Signaling Pathway. Mol Cell Neurosci (2017) 85:45–56. doi: 10.1016/j.mcn.2017.08.006
85. Ouahed J, Spencer E, Kotlarz D, Shouval DS, Kowalik M, Peng K, et al. Very Early Onset Inflammatory Bowel Disease: A Clinical Approach With a Focus on the Role of Genetics and Underlying Immune Deficiencies. Inflammation Bowel Dis (2020) 26:820–42. doi: 10.1093/ibd/izz259
86. Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, et al. RIPK1 Maintains Epithelial Homeostasis by Inhibiting Apoptosis and Necroptosis. Nature (2014) 513:90–4. doi: 10.1038/nature13608
87. Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 Blocks Early Postnatal Lethality Mediated by Caspase-8 and RIPK3. Cell (2014) 157:1189–202. doi: 10.1016/j.cell.2014.04.018
88. Takahashi N, Vereecke L, Bertrand MJM, Duprez L, Berger SB, Divert T, et al. RIPK1 Ensures Intestinal Homeostasis by Protecting the Epithelium Against Apoptosis. Nature (2014) 513:95–9. doi: 10.1038/nature13706
89. Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 Regulates RIPK3-MLKL-Driven Systemic Inflammation and Emergency Hematopoiesis. Cell (2014) 157:1175–88. doi: 10.1016/j.cell.2014.04.019
90. Roderick JE, Hermane N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, et al. Hematopoietic RIPK1 Deficiency Results in Bone Marrow Failure Caused by Apoptosis and RIPK3-Mediated Necroptosis. Proc Natl Acad Sci USA (2014) 111:14436–41. doi: 10.1073/pnas.1409389111
91. O’Donnell JA, Lehman J, Roderick JE, Martinez-Marin D, Zelic M, Doran C, et al. Dendritic Cell RIPK1 Maintains Immune Homeostasis by Preventing Inflammation and Autoimmunity. J Immunol (2018) 200:737–48. doi: 10.4049/jimmunol.1701229
92. Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 Inhibits ZBP1-Driven Necroptosis During Development. Nature (2016) 540:129–33. doi: 10.1038/nature20559
93. Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, et al. RIPK1 Counteracts ZBP1-Mediated Necroptosis to Inhibit Inflammation. Nature (2016) 540:124–8. doi: 10.1038/nature20558
94. Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 Contributes to TNFR1-Mediated RIPK1 Kinase-Dependent Apoptosis in Conditions of Ciap1/2 Depletion or TAK1 Kinase Inhibition. Cell Death Differ (2013) 20:1381–92. doi: 10.1038/cdd.2013.94
95. Newton K, Dugger DL, Wickliffe KE, Kapoor N, De Almagro MC, Vucic D, et al. Activity of Protein Kinase RIPK3 Determines Whether Cells Die by Necroptosis or Apoptosis. Science (80-) (2014) 343:1357–60. doi: 10.1126/science.1249361
96. Bitra A, Doukov T, Wang J, Picarda G, Benedict CA, Croft M, et al. Crystal Structure of Murine 4-1BB and Its Interaction With 4-1BBL Support a Role for Galectin-9 in 4-1BB Signaling. J Biol Chem (2018) 293:1317–29. doi: 10.1074/jbc.M117.814905
97. Laderach D, Movassagh M, Johnson A, Mittler RS, Galy A. 4-1BB Co-Stimulation Enhances Human CD8+ T Cell Priming by Augmenting the Proliferation and Survival of Effector CD8+ T Cells. Int Immunol (2002) 14:1155–67. doi: 10.1093/intimm/dxf080
98. Lee H-W, Park S-J, Choi BK, Kim HH, Nam K-O, Kwon BS. 4-1bb Promotes the Survival of CD8 + T Lymphocytes by Increasing Expression of Bcl-X L and Bfl-1. J Immunol (2002) 169:4882–8. doi: 10.4049/jimmunol.169.9.4882
99. Kim YH, Choi BK, Shin SM, Kim CH, Oh HS, Park SH, et al. 4-1bb Triggering Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating the Balance Between Th17 and Regulatory T Cells. J Immunol (2011) 187:1120–8. doi: 10.4049/jimmunol.1002681
100. Zhang X, Voskens CJ, Sallin M, Maniar A, Montes CL, Zhang Y, et al. CD137 Promotes Proliferation and Survival of Human B Cells. J Immunol (2010) 184:787–95. doi: 10.4049/jimmunol.0901619
101. Cole SL, Benam KH, McMichael AJ, Ho L-P. Involvement of the 4-1BB/4-1BBL Pathway in Control of Monocyte Numbers by Invariant NKT Cells. J Immunol (2014) 192:3898–907. doi: 10.4049/jimmunol.1302385
102. Vinay DS, Choi BK, Bae JS, Kim WY, Gebhardt BM, Kwon BS. CD137-Deficient Mice Have Reduced NK/NKT Cell Numbers and Function, Are Resistant to Lipopolysaccharide-Induced Shock Syndromes, and Have Lower IL-4 Responses. J Immunol (2004) 173:4218–29. doi: 10.4049/jimmunol.173.6.4218
103. Choi BK, Lee HW. The Murine CD137/CD137 Ligand Signalosome: A Signal Platform Generating Signal Complexity. Front Immunol (2020) 11:553715. doi: 10.3389/fimmu.2020.553715
104. Kwon BS, Hurtado JC, Lee ZH, Kwack KB, Seo SK, Choi BK, et al. Immune Responses in 4-1BB (CD137)-Deficient Mice. J Immunol (2002) 168:5483–90. doi: 10.4049/jimmunol.168.11.5483
105. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell (2011) 20:11–24. doi: 10.1016/j.ccr.2011.06.001
106. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) Negatively Regulates Homeostasis and Differentiation of Hematopoietic Stem Cells in Mice. Proc Natl Acad Sci USA (2011) 108:14566–71. doi: 10.1073/pnas.1112317108
107. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell (2017) 170:1079–95.e20. doi: 10.1016/j.cell.2017.07.032
108. Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood (2011) 118:4509–18. doi: 10.1182/blood-2010-12-325241
109. Kaasinen E, Kuismin O, Rajamäki K, Ristolainen H, Aavikko M, Kondelin J, et al. Impact of Constitutional TET2 Haploinsufficiency on Molecular and Clinical Phenotype in Humans. Nat Commun (2019) 10:1252. doi: 10.1038/s41467-019-09198-7
110. Lio CW, Zhang J, González-Avalos E, Hogan PG, Chang X, Rao A. Tet2 and Tet3 Cooperate With B-Lineage Transcription Factors to Regulate DNA Modification and Chromatin Accessibility. Elife (2016) 5:1–26. doi: 10.7554/eLife.18290
111. Fujii K, Tanaka S, Hasegawa T, Narazaki M, Kumanogoh A, Koseki H, et al. Tet DNA Demethylase Is Required for Plasma Cell Differentiation by Controlling Expression Levels of IRF4. Int Immunol (2020) 32:683–90. doi: 10.1093/intimm/dxaa042
112. Nakatsukasa H, Oda M, Yin J, Chikuma S, Ito M, Koga-Iizuka M, et al. Loss of TET Proteins in Regulatory T Cells Promotes Abnormal Proliferation, Foxp3 Destabilization and IL-17 Expression. Int Immunol (2019) 31:335–47. doi: 10.1093/intimm/dxz008
113. Rosikiewicz W, Chen X, Dominguez PM, Ghamlouch H, Aoufouchi S, Bernard OA, et al. TET2 Deficiency Reprograms the Germinal Center B Cell Epigenome and Silences Genes Linked to Lymphomagenesis. Sci Adv (2020) 6:1–16. doi: 10.1126/sciadv.aay5872
114. Muto H, Sakata-Yanagimoto M, Nagae G, Shiozawa Y, Miyake Y, Yoshida K, et al. Reduced TET2 Function Leads to T-Cell Lymphoma With Follicular Helper T-Cell-Like Features in Mice. Blood Cancer J (2014) 4:e264. doi: 10.1038/bcj.2014.83
115. Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial Signals Drive Pre-Leukaemic Myeloproliferation in a Tet2-Deficient Host. Nature (2018) 557:580. doi: 10.1038/s41586-018-0125-z
116. Zeng H, He H, Guo L, Li J, Lee M, Han W, et al. Antibiotic Treatment Ameliorates Ten-Eleven Translocation 2 (TET2) Loss-of-Function Associated Hematological Malignancies. Cancer Lett (2019) 467:1–8. doi: 10.1016/j.canlet.2019.09.013
117. Cochran JN, Geier EG, Bonham LW, Newberry JS, Amaral MD, Thompson ML, et al. Non-Coding and Loss-Of-Function Coding Variants in TET2 Are Associated With Multiple Neurodegenerative Diseases. Am J Hum Genet (2020) 106:632–45. doi: 10.1016/j.ajhg.2020.03.010
118. Spierings J, van Laar JM. Is There a Place for Hematopoietic Stem Cell Transplantation in Rheumatology? Rheum Dis Clin North Am (2019) 45:399–416. doi: 10.1016/j.rdc.2019.04.003
119. Salzer E, Zoghi S, Kiss MG, Kage F, Rashkova C, Stahnke S, et al. The Cytoskeletal Regulator HEM1 Governs B Cell Development and Prevents Autoimmunity. Sci Immunol (2020) 5:eabc3979. doi: 10.1126/sciimmunol.abc3979
Keywords: inborn errors of immunity, immune dysregulation, primary immune dysregulation disease, autoimmunity, hyperinflammation, lymphoproliferation
Citation: Ren A, Yin W, Miller H, Westerberg LS, Candotti F, Park CS, Lee P, Gong Q, Chen Y and Liu C (2021) Novel Discoveries in Immune Dysregulation in Inborn Errors of Immunity. Front. Immunol. 12:725587. doi: 10.3389/fimmu.2021.725587
Received: 15 June 2021; Accepted: 27 July 2021;
Published: 27 August 2021.
Edited by:
Markus G. Seidel, Medical University of Graz, AustriaReviewed by:
Hassan Abolhassani, Karolinska University Hospital, SwedenSvetlana O. Sharapova, Belarusian Research Center for Pediatric Oncology and Hematology, Belarus
Copyright © 2021 Ren, Yin, Miller, Westerberg, Candotti, Park, Lee, Gong, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaohong Liu, Y2hhb2hvbmdsaXU4MEAxMjYuY29t; Quan Gong, cXVhbmdvbmcxOTk4QDE2My5jb20=; Yan Chen, Y3l6NjAwQDE2My5jb20=