 Ang Zhang1,2†
Ang Zhang1,2† Yao Sun3†
Yao Sun3† Jie Du4†
Jie Du4† Yansheng Dong4†Honggang Pang5†Lei Ma6Shaoyan Si1,7Zhong Zhang1,8Mingyi He1,2Yang Yue1,2Xiaoli Zhang1,2Weichao Zhao1,9
Yansheng Dong4†Honggang Pang5†Lei Ma6Shaoyan Si1,7Zhong Zhang1,8Mingyi He1,2Yang Yue1,2Xiaoli Zhang1,2Weichao Zhao1,9 Jianjun Pi1,9Mindong Chang10*Quanjun Wang11*Yikun Zhang1,2*
Jianjun Pi1,9Mindong Chang10*Quanjun Wang11*Yikun Zhang1,2*- 1Department of Hematology, Strategic Support Force Medical Center, Beijing, China
- 2The Department of Hematology, Beijing, China
- 3Department of Hematology, Fifth Medical Center of Chinese PLA General Hospital, Beijing, China
- 4SAFE Pharmaceutical Research Institute Co. Ltd, HeBei, China
- 5Department of Emergency, Affiliated Zhongshan Hospital, Dalian University, Dalian, China
- 6Academy of Military Medical Sciences, Academy of Military Sciences, Beijing, China
- 7Comprehensive Basic Experiment, Beijing, China
- 8The Department of Cardiovascular Medicine, Beijing, China
- 9The Department of Respiratory Medicine, Beijing, China
- 10Strategic Support Force Medical Center, The Department of Surgical Oncology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, China
- 11National Beijing Center for Drug Safety Evaluation and Research, State Key Laboratory of Medical Countermeasures and Toxicology, Institute of Pharmacology and Toxicology, Academy of Military Sciences, Beijing, China
Chimeric antigen receptor (CAR)-modified T cells targeting CD19 demonstrate unparalleled responses in B cell malignancies. However, high tumor burden limits clinical efficacy and increases the risk of cytokine release syndrome and neurotoxicity, which is associated with over-activation of the CAR-T cells. The hinge domain plays an important role in the function of CAR-T cells. We hypothesized that deletion of glycine, an amino acid with good flexibility, may reduce the flexibility of the hinge region, thereby mitigating CAR-T cell over-activation. This study involved generating a novel CAR by deletion of two consecutive glycine residues in the CD8 hinge domain of second-generation (2nd) CAR, thereafter named 2nd-GG CAR. The 2nd-GG CAR-T cells showed similar efficacy of CAR expression but lower hinge flexibility, and its protein affinity to CD19 protein was lower than that of 2nd CAR-T cells. Compared to the 2nd CAR-T cells, 2nd-GG CAR-T cells reduced proinflammatory cytokine secretion without diminishing the specific cytotoxicity toward tumor cells in vitro. Furthermore, 2nd-GG CAR-T cells prolonged overall survival in an immunodeficient mouse model bearing NALM-6 when tumor burden was high. This study demonstrated that a lower-flexibility of CD8α hinge improved survival under high tumor burden and reduced proinflammatory cytokines in preclinical studies. While there is potential for improved safety and efficacy, yet this needs validation with clinical trials.
Introduction
Chimeric antigen receptor T cell (CAR-T) therapy for hematological malignancies has demonstrated tremendous clinical outcomes (1, 2). Four CAR-T cell products have been approved globally, including Kite’s Yescarta and Tecartus, Novartis’s Kymriah, and BMS’s Breyanzi, all targeting CD19 antigen (2–4). However, a high tumor burden often indicates poor prognosis and significant adverse reactions after CAR-T therapy, which may be related to the over-activation of CAR-T cells (5–8). Therefore, patients with a high tumor burden have an unmet medical need for anti-CD19 CAR-T therapy.
Investigators are currently striving to improve the safety and efficacy of CAR-T cells by optimizing CAR designs to overcome their existing limitations (9). These include cytokine release syndrome (CRS) and immune-effector cell associated neurotoxicity syndrome (ICANS), both related to the excessive release of cytokines and limited persistence caused by activation-induced cell death (AICD) (10–15). The standard CAR design consists of four modular components: the antigen binding domain, hinge domain, transmembrane domain, and intracellular signaling domain, each of which has a specific function and thus the potential to be optimized (16). More attention has been paid to the improvement of signal regions, including antigen recognition and signaling argument regions such as the costimulatory domain and immunoreceptor tyrosine-based activation motif (ITAM) of CD3ξ (17, 18).
In recent years, a growing number of studies have demonstrated the significant function of non-signaling regions. The properties of the hinge and transmembrane domains also influence CAR-T cell cytokine production and AICD (19), which are related to the anti-tumor efficacy and the loss of CAR, respectively (20, 21). Ying et al. (22) constructed a new CAR design with longer extracellular and intracellular domains named CD19-BBz (86) CAR T cells, which produced a potent and durable anti-lymphoma response without causing neurotoxicity or severe CRS (greater than grade 1). The hinge provides sufficient flexibility to overcome steric hindrance, and length to facilitate access to the target antigen (23). It thus seems reasonable to down-regulate the activation of CAR-T cells by reducing the flexibility of the hinge region, thereby improving efficacy and safety. Glycine, the smallest amino acid is unique because unlike all others, it contains hydrogen as its side chain rather than a carbon (24), permitting much more conformational flexibility. (Gly4Ser)n is often used as a linker for different polypeptides because it is not prone to misfolding errors, and Gly plays an irreplaceable role in this structure (25).
Consequently, this study entailed designing a novel CAR by deleting two consecutive glycine residues in the CD8 hinge domain of traditional second-generation (2nd) CAR and named the FMC63-CD8(Gly2-deletion)-4-1BB-CD3ζ CAR as 2nd-GG CAR. Studies were then conducted to verify the flexibility and affinity of this new CAR, and compare the functions of 2nd and 2nd-GG CAR-T cells in vitro and in vivo.
Materials and Methods
Cell Lines and Cell Culture Conditions
Cell lines were cultured according to the manufacturers’ recommendations. NALM-6 is a pre-B cell acute lymphoblastic leukemia (ALL) cell line with high expression of CD19 (German DSMZ cell collection Cat#: ACC128). NALM-6-GFP-luciferase (luc) is a stable cell line engineered to express GFP-luciferase. K562 is a chronic myelogenous leukemia cell line (ATCC; Cat#: CCL-243). K562-CD19 and K562-CD19-GFP are stable cell lines engineered to express CD19 and/or GFP. 786o is a renal cell adenocarcinoma cell line (ATCC; Cat#: CRL-1932™). CD19 was transduced using a lentivirus system into 786o to produce 786o-CD19. The method of tumor cells culture refers to our previous study (26).
Generation of CAR Constructs
Generation of lentiviral constructs and production of lentiviral particles refer to our previous study (27). The conventional second-generation 2nd CAR was constructed by the fusion of CD19 scFv, CD8 hinge and transmembrane, 4-1BB, and CD3ζ. The structure of 2nd-GG is same to the 2nd CAR except for deletion of two consecutive glycine in the CD8 hinge. Nucleotide sequence of CD8 hinge in 2nd-CAR and 2nd-GG CAR are shown in Supplementary Figure 2.
Selection, Activation, and Lentivector Transduction of CD3+ T Cells
Blood samples from healthy volunteers were obtained using an approved protocol by the Ethics Committee of the Fifth Medical Center of Chinese PLA General Hospital (Ethical code: Ky-2018-5-37). These studies were conducted following the Declaration of Helsinki. All subjects provided written informed consent before participation in the present study. The methods of T cell isolation and culture and gene transfer refer to our previous study (26).
Binding Assay
Briefly, through the measurement of the fluorescence intensity of different CAR T cells to CD19 protein at various concentrations, their affinity for CD19 protein can be determined. Specifically, mock-T, 2nd CAR-T, and 2nd-GG CAR-T cells were washed twice by centrifugation with PBS (1% BSA). They were treated with CD19-Fc protein (11880- H02H) at final concentrations of 180 µg/mL, 72 µg/mL, 28.8 µg/mL, 11.52 µg/mL, 4.61 µg/mL, 1.84 µg/mL, 0.74 µg/mL, 0.29 µg/mL, 0.12 µg/mL, or 0.05 µg/mL, incubated at 4°C in darkness for 45 min, and washed twice with a PBS washing solution by centrifugation. Next, the cells were treated with 10 µL goat anti-human IgG (FC)/FITC, incubated at 4°C in darkness for 20 min, washed twice with a washing solution by centrifugation, and tested utilizing flow cytometry (NovoCyte D3010).
Cytotoxicity Assay
Briefly, CFSE-labeled targets were incubated at the indicated ratios with effector T cells for 12–16 h or 6–8 h. The cells were then harvested, and Annexin V and 7-AAD were added prior to flow cytometric analysis. The residual live target cells were CFSE+ Annexin V- 7-AAD-. E:T ratios designated the ratios of the absolute number of CAR T cells to target cells. The number of T cells was the same as that in the 2nd CAR group. All experiments were carried out in triplicate.
Cytokine Production
Effector cells (5 × 104) and target cells (5 × 104) were incubated at a 1:1 ratio in RPMI (10% FBS) media with 10% human serum for 24 h. Cytokine concentration in the culture supernatant and mouse serum was measured with enzyme-linked immunosorbent assay (ELISA) kits (MultiSciences Biotech Co., Ltd., China) for human IFN-γ, TNF-α, and IL-2. E:T ratio designated the ratio of the absolute number of CAR T cells to target cells. The number of T cells was the same as that in the 2nd CAR group.
Flow Cytometry
Anti-human antibodies were purchased from Becton Dickinson, BioLegend, and Miltenyi Biotec. The Accuri C6 (Becton Dickinson, USA), FACS Calibur (Becton Dickinson, USA), and BD FACSAriaTM II cell sorter were used for the analysis of various samples. Anti-human antibodies were purchased from BioLegend, eBioscience, Acrobiosystems, or BD. Cells were isolated from in vitro cultures or from animals, washed once with PBS supplemented with 2% FCS, and stained on ice after blocking Fc receptors. In all analyses, the population of interest was gated based on forward vs. side scatter characteristics followed by singlet gating.
Mouse Xenograft Tumor Model
Animal experiments were conducted at the National Beijing Center for Drug Safety Evaluation and Research and at the SAFE Pharmaceutical Research Institute Co.,Ltd (IACUC-2019-001). Female NSG mice (28) aged 6–8 weeks were used. For NALM-6-acute precursor B-ALL models, 106 tumor cells were intravenously injected with PBS, and tumors were measured by the total bioluminescent flux using a Xenogen Imaging System (PerkinElmer-IVIS Lumina III). Peripheral blood was collected via the submandibular vein.
Statistical Analysis
Statistical analyses were performed using Prism version 7.0 (GraphPad). For studies comparing two groups, we utilized a Students t-test. Log rank (Mantel Cox) test was used to analyze in vivo survival. Survival curves were constructed using Kaplan–Meier methodology.
Results
Deletion of Gly-Gly in CD8 Hinge Region of CAR Reduced the Flexibility of Hinge Without Affecting the CAR Expression Efficiency
The 2nd CAR-T cells, structured as FMC63-CD8-4-1BB-CD3ζ, have shown promising efficacy in clinical studies (1). To decrease the flexibility of the hinge region, deletion mutations were performed on two consecutive Glys in the wild-type CD8 hinge region of FMC63-CD8-4-1BB-CD3ζ CAR, and this novel CAR was named 2nd-GG CAR (Figure 1A). The specific nucleic acid sequences of the wild CD8 hinge region and the CD8 hinge region with deletion of 2 Gly are shown in Supplement Figure 1. The transduction efficiency of 2nd CAR and 2nd-GG CAR on human T cells was similar (approximately 70%) (Figures 1B, C). The S2 order parameters represent the restriction of movement of an atomic bond vector with respect to the molecular reference frame. The greater the value of S2, the less flexible the protein. Thus, the flexibility of the CD8-GG hinge region was less than that of the CD8 hinge region according to the index of S2 from DynaMine (29) (Figure 1D). Furthermore, when the two CAR-T cells were individually incubated with different concentrations of CD19 protein, the 2nd-GG CAR-T cells showed weaker binding ability to CD19 protein than 2nd CAR-T cells (Figure 1E).
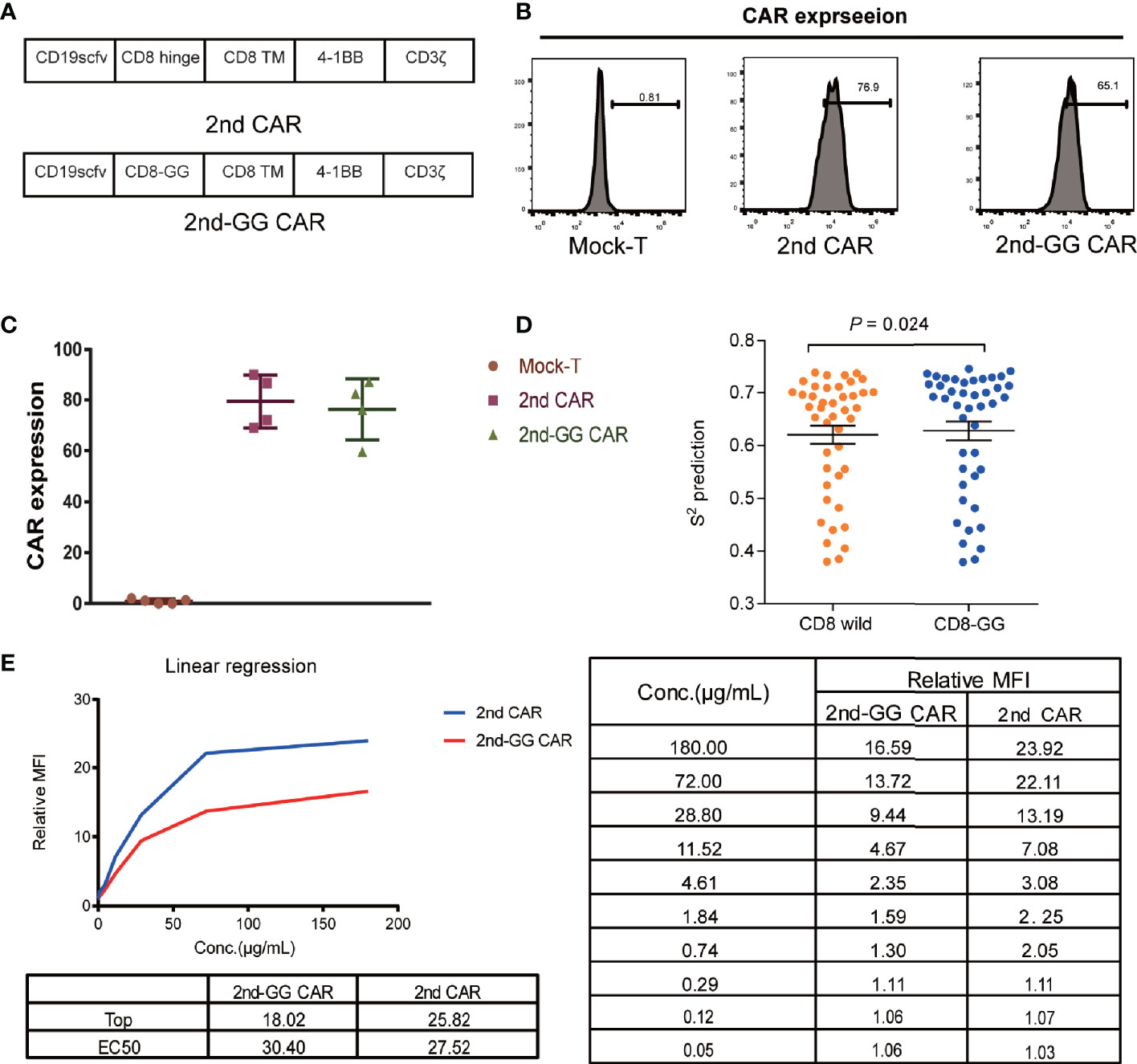
Figure 1 Schematic diagram and expression efficiency of 2nd and 2nd-GG CAR-T cells. (A) Diagrammatic model of 2nd and 2nd-GG CAR. Schematic of CAR containing scfv (FMC63), variations in the hinge, extra-membrane, and transmembrane domains. The hinge region of 2nd-GG deleted two Gly compared with that of the 2nd CAR, and the rest of the sequences were the same. (B) Typical flow cytometry detection of the expression efficiency of 2nd and 2nd-GG CAR on T cells. (C) Expression efficiency of 2nd and 2nd-GG on T cells 5-6 days after culture in vitro determined by flow cytometry (mean ± SD, n = 5). T cells are derived from at least three different healthy donors. (D). Comparison of the flexibility between the CD8 hinge and the CD8-GG hinge. S2 order parameter (S2 RCI) values were estimated from chemical shift values using the Random Coil Index (RCI) software. S2 is inversely proportional to the hinge region flexibility. (E). The affinity of CD19 protein to different CAR T cells: 2nd CAR-T cells > 2nd-GG CAR-T cells. The EC50 of 2nd and 2nd-GG CAR-T cells binding to CD19 protein was determined by flow cytometry. EC50, 50% maximal effective concentration. CAR, chimeric antigen receptor; FITC, fluorescein isothiocyanate.
2nd-GG CAR-T Cells Showed Similar Killing Efficiency but Secreted Less Proinflammatory Cytokines Compared to 2nd-GG CAR-T Cells In Vitro
To evaluate the effector function of the two different CAR-T cells, a killing (cytotoxicity) and cytokine secretion assays were conducted on different cell lines. These were: NALM-6, a precursor B-cell leukemia cell line that naturally expresses CD19, plus the 786o and K562 cell lines which are CD19 negative (Figure 2). The two CAR-T cells showed similar cytotoxic efficacy against the CD19-positive and negative cell lines, with no statistically significant differences.
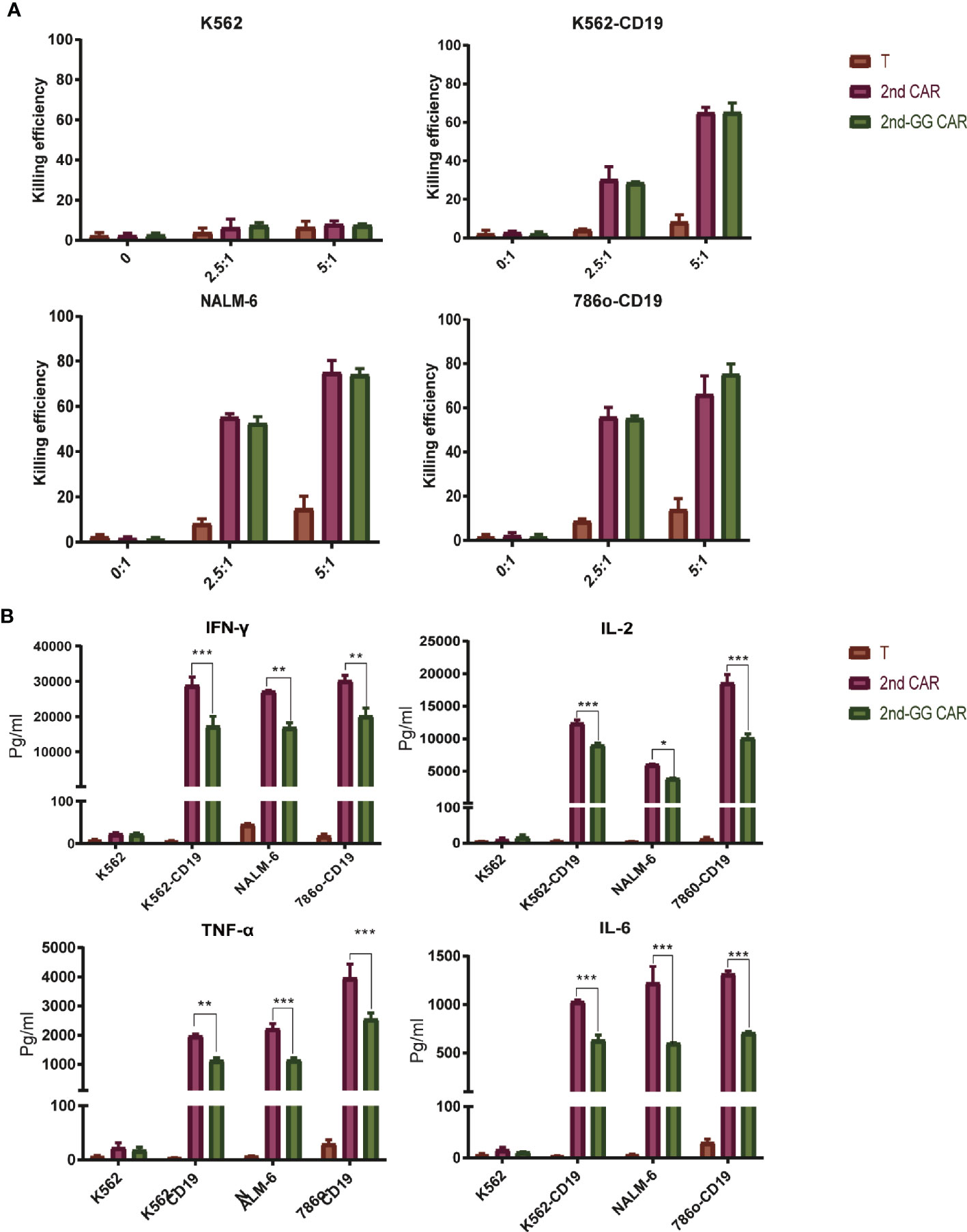
Figure 2 The killing efficiency and cytokine secretion of 2nd CAR-T and 2nd-GG CAR-T cells towards tumor cells. (A) Cytotoxic percentages of targeted cells by mock T, 2nd and 2nd-GG CAR-T cells after 8–10 h of co-culture in vitro. E: T (2.5:1 and 5:1) designate the ratios of the absolute number of CAR T cells to target cells, specifically K562, NALM-6, 786o-CD19, and K562-CD19. The number of mock T cells is the same as in the 2nd CAR-T cells group. Results are representative of at least three independent experiments with T cells from different healthy donors. (B) Human IFNγ, TNF-α, IL-2 and IL-6 production by mock T, 2nd and 2nd-GG CAR-T cells. Cytokine concentrations in the media were measured after 24 h of co-incubation with different target cells at E: T of 1:1. Values are mean ± SD of triplicate specimens obtained with T cells isolated from one healthy donor. *P < 0.05; **P < 0.01; ***P < 0.005.
It is well known that cytokines secreted from CAR-T cells trigger an overactivation of the immune system, ultimately leading to CRS (30). We therefore examined the pro-inflammatory factors released after the incubation of CAR T cells with different tumor cells. Following incubation with CD19+ target cells, the amount of proinflammatory cytokines secreted by 2nd-GG CAR-T cells was less than that of 2nd-GG CAR-T cells (P<0.01). None of the CAR-T cells produced specific killing effects or proinflammatory factors against K562, a CD19- tumor cell line, demonstrating the antigen-specificity towards CD19 by the 2nd-GG CAR-T cells.
2nd-GG CAR-T Cells Exhibited Similar Antitumor Efficacy but Less Proinflammatory Cytokines Release in Mouse Model With Moderate Tumor Burden
Although 2nd-GG CAR-T cells showed a similar specific immune response to CD19+ tumor cells in vitro compared with 2nd CAR-T cells, their antitumor efficacy in animal models needs to be further verified. The anti-tumor efficacy of CAR-T cells in NSG immunodeficient mice bearing NALM-6-GFP-luc(luciferase) was subsequently investigated, as detailed in Figure 3A. Both 2nd-GG and 2nd CAR-T cells exhibited improved overall survival (OS) and reduced tumor burden compared with the mock-T cells, demonstrating improved tumor control of both CAR-T cells (Figures 3B, D). Furthermore, compared to the 2nd CAR-T cell group, the OS in those administered 2nd-GG CAR-T cells was prolonged, although there was no statistical difference, as shown in Figure 3C. As expected, 2nd-GG CAR-T cells secreted less human proinflammatory cytokines, particularly IL-6 and IFN-γ, compared to the 2nd CAR-T cells in vivo (Figure 3E). In order to distinguish it from the following experiment with a higher tumor burden, this experiment was referred to as “with moderate tumor load”. The 2nd-GG CAR-T cells did not show sufficient advantage compared to the 2nd CAR-T cells in experiments with moderate tumor burden, owing to the relatively lower tumor load.
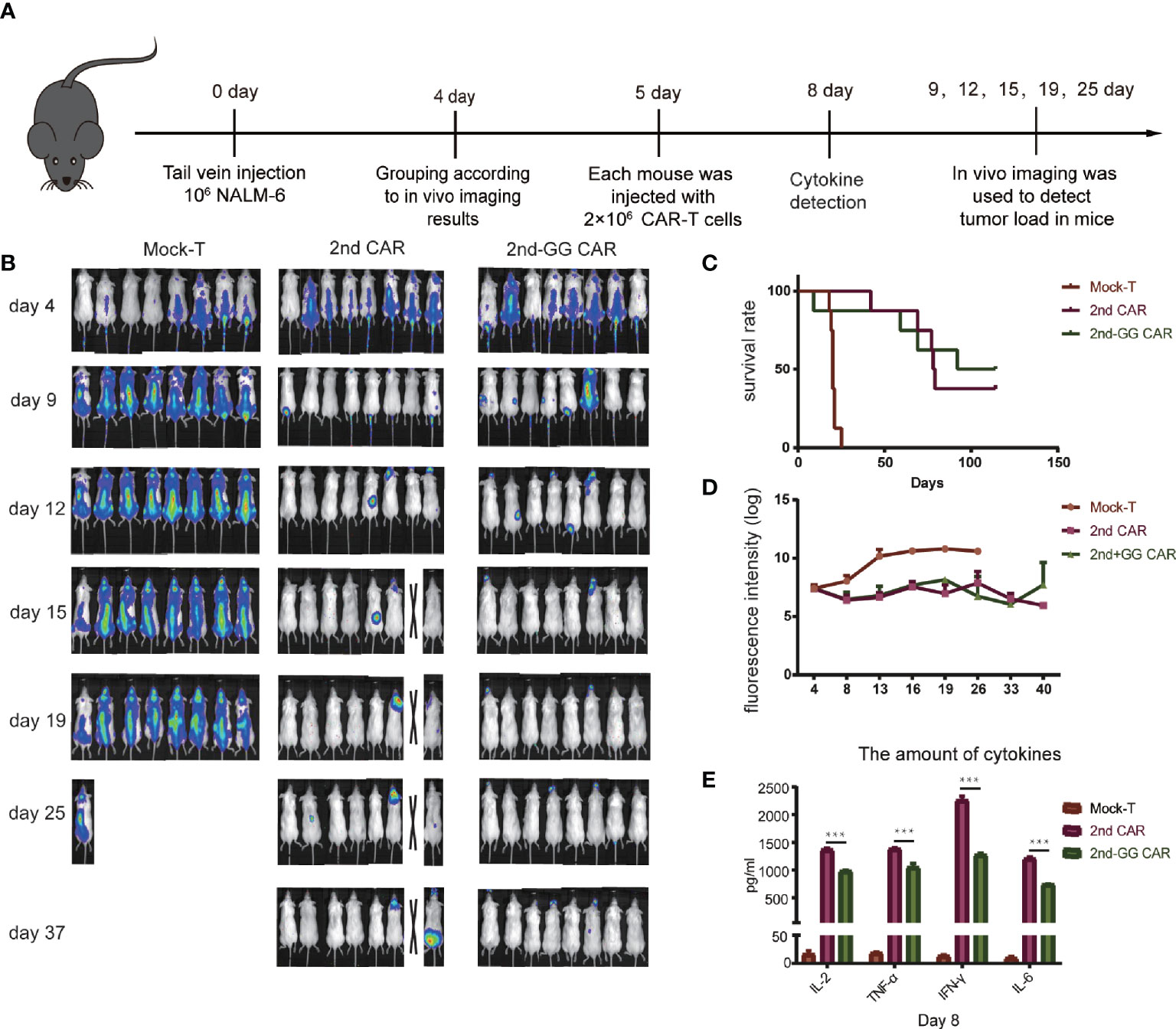
Figure 3 The antitumor efficacy and cytokines release of different CAR-T cells in moderate tumor load models. (A) Diagrammatic representations of the experimental procedures. (B) Representative bioluminescent images are shown. (C) Overall survival curves of NALM-6 -GFP-luc challenged mice (n = 8). (D) Tumor burden-total flux (log) for each mouse was quantified and averaged by group. (mean ± SEM) (E) On day 8, approximately 1,000 µL of blood were collected from the caudal vein of each mouse mixed to detect the concentration of human IL-2, TNF-α, IFN-γ, and IL-6 using an ELISA-kit. (mean ± SD, n = 2). ***P < 0.005.
2nd-GG CAR-T Cells Significantly Improved Antitumor Activity in Mouse Model With High Tumor Burden
A high tumor burden often indicates a poor prognosis and significant adverse reactions after CAR-T therapy (31). It is suggested that a high tumor burden might affect the efficacy of CAR-T cell therapy (32, 33). It was thus hypothesized that CAR T cells behave differently in mouse models with different tumor burdens. To mimic the clinical situation of a high tumor burden, NSG mice bearing NALM6-Luc tumors received delayed CAR-T cell infusion to increase the tumor load. The specific schedule is shown in Figure 4A. When NSG mice were challenged with high tumor burden, 2nd-GG CAR-T cells showed significantly improved overall survival compared with 2nd CAR-T cells, while the 2nd CAR-T cells showed no advantage over the mock-T cells (Figures 4B, D). The tumor load in group of 2nd-GG CAR-T was lower than that of 2nd CAR T (P>0.05) on day 15 and showed a downward trend (Figure 4C). The anergy of 2nd CAR-T cell in the mouse model with high tumor load is likely related to AICD. One mouse from each group was randomly selected on day 14, to evaluate the tumor load of peripheral blood (PB), bone marrow (BM), and spleen by flow cytometry. The results showed that the tumor burden of the 2nd-GG group was less than that of the other two groups after treatment (Figure 4E). Similarly, the amount of human proinflammatory cytokines secreted by 2nd-GG CAR T cells was lower than that of 2nd CAR T cells (Figure 4F).
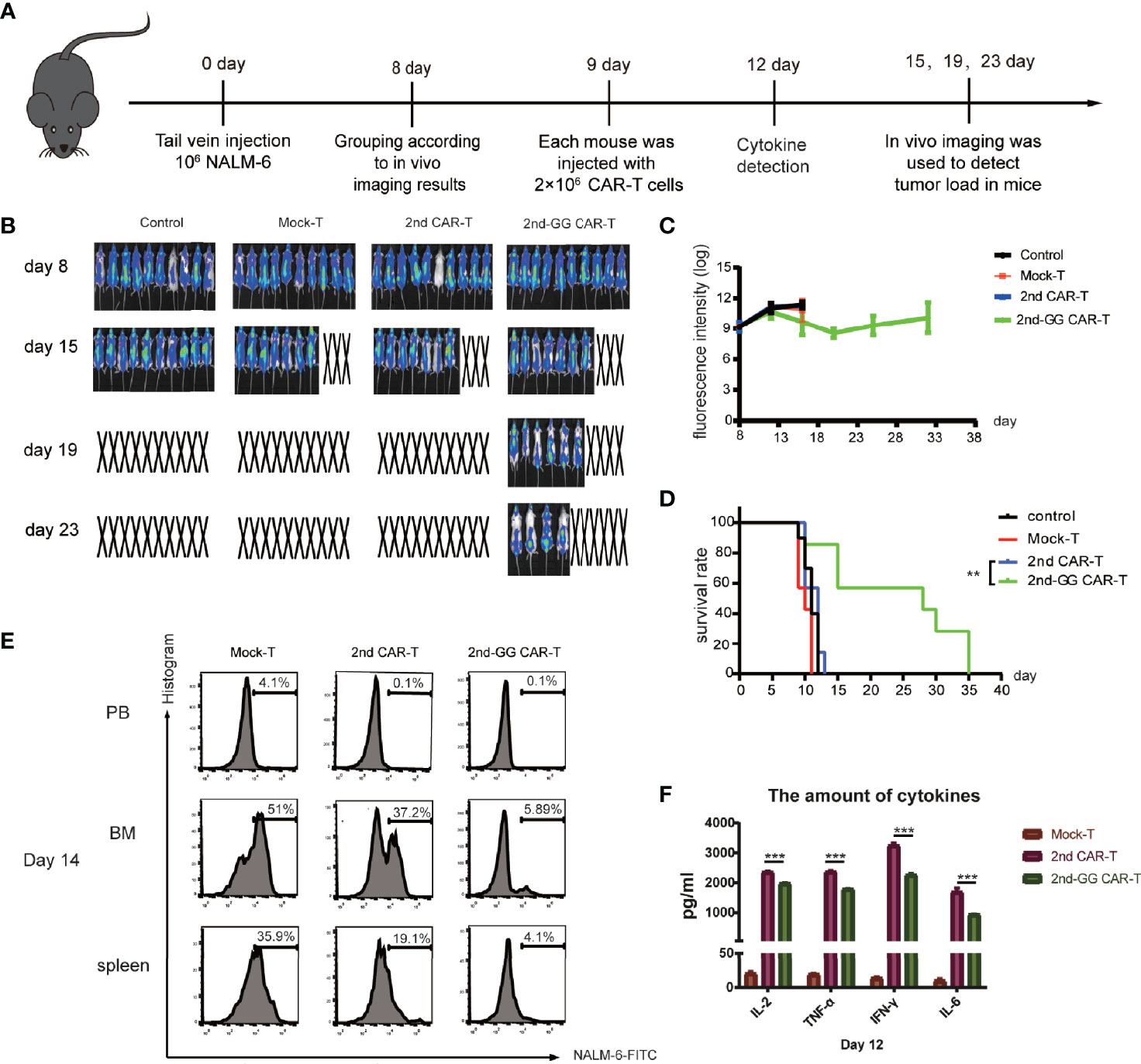
Figure 4 The antitumor efficacy and cytokines release of different CAR-T cells in high tumor load models. (A) Diagrammatic representations of the experimental procedures. (B) Representative bioluminescent images are shown. (C) Overall survival curves of NALM-6 -GFP-luc challenged mice (n = 8). (D) Tumor burden-total flux (log) for each mouse was quantified and averaged by group. (mean ± SEM) (E) On day 14, one mouse was randomly euthanized from the Mock-T, 2nd CAR-T and 2nd-GG CAR-T groups. Cell suspensions from peripheral blood, bone marrow and spleen were collected and ground for flow cytometry detection. Since the NALM-6 cells were engineered to express GFP, the tumor load was reflected by the expression percentage of GFP+ cells. (F) On day 12, approximately 1,000 µL of blood were collected from the caudal vein of each mouse to detect the concentration of human IL-2, TNF-α, IFN-γ, and IL-6 using an ELISA-kit. (mean ± SD, n = 2). **P < 0.01, ***P < 0.005.
Overall, 2nd-GG CAR-T cells exhibited stronger antitumor activity and lower cytokine release in the high tumor burden model than the 2nd CAR-T cells.
Discussion
This study demonstrated that 2nd-GG CAR exhibits lower flexibility and affinity for the CD19 antigen. The 2nd-GG CAR-T cells produced lower levels of cytokines, yet showed similar cytotoxicity to CD19+ tumor cells as 2nd CAR-T cells in vitro. However, 2nd-GG CAR-T cells show lower cytokine release in mouse models with moderate and high tumor burden, and prolong overall survival in animal models with high tumor burden.
Currently, the indication for anti-CD19 CAR T cells has been mainly for relapse and refractory B-cell malignancies, which are often insensitive to traditional radiotherapy and chemotherapy. Furthermore, an inevitable vein-to-vein time interval, typically 3-8 weeks, is required for patients preparing for CAR-T cell therapy. Pivotal trials of approved treatments have resulted in up to a third of the enrolled patients failing to receive the product. It has not been determined if bridging therapy is necessary during this gap, and which treatment regimen may be better (34). Although off-the-shelf cell therapy or Fast CAR-T cells may shorten the vein-to-vein time interval, it is still under clinical study (35). Therefore, the high tumor burden in patients before CAR-T cell therapy is an unavoidable problem. It has been reported that both the efficiency and the incidence of adverse reactions, such as CRS of the anti-CD19 second-generation CAR T cells, increased in patients with high tumor burden (36–38). Many studies have demonstrated that reduced activation of anti-CD19 CAR-T cells improves the safety and efficiency of CAR-T cells (22). This could be achieved through reducing anti-CD19 CAR T cell activation by diminishing scFv affinity (39), increasing the hinge and transmembrane region (22), replacing the co-stimulatory molecule from CD28 to 4-1BB (18), and mutation of the immunoreceptor tyrosine-based activation motif (ITAM) region of CD3ζ (40).
The hinge region has a significant impact on the function of CAR T cells, and its components are often derived from the IgG family or the co-receptor of T cells (CD4/CD8) (41), but the specific mechanism is still unclear (9). Studies have shown that the hinge region provides a spatial location for the recognition of scFv and antigens. When the epitope recognized by CAR is in a membrane proximal position, the hinge region is necessary for the recognition of CAR-T cells by antigens, such as when targeting NCAM or 5T4. Whereas if the epitope recognized by CAR is a membrane distal epitope, the hinge region is negligible for the recognition of CAR-T cells by antigens, such as when targeting CEA (42). In general, little is known about the role of the hinge domain, and strategies to optimize it need to be creatively explored.
The flexibility of the hinge region has been shown to affect the CAR T cell function. The addition of a flexible IgG hinge instead of a CD28 hinge alone (SD28ζ) led to more pro-cytokines produce and better recognition of the MUC1 epitope compared to S28ζ CAR-T cells (43). However, further verification is needed to determine whether reducing the flexibility of the hinge region can decrease CAR-T activity. We removed two consecutive glycine residues in the hinge region to reduce the flexibility of the hinge domain, thus resulting in better tumor control and lower release of inflammatory factors such as TNF-α and IL-6, which are the key molecules triggering the cytokine storm. This can be explained by the fact that reducing the flexibility of the hinge domain prevents overactivation of CAR-T cells, especially under high tumor load. However, the specific mechanism is unknown and warrants further investigation. Although studies have shown that the persistence of CAR-T cells is essential for immune surveillance of tumors, CAR gene copy numbers were unfortunately not measured (6). Studies have shown that the formation of immune synapses by CAR influences the function of CAR-T cells and changes the flexibility of the hinge region (44, 45). This may alter the formation of immune synapses in CAR, thus affecting the function of CAR-T cells, though it needs to be further explored.
Although we observed a downward trend in tumor load in the 2nd-GG group, it is a limitation of our study that the lack of evidence for enhanced anti-tumor activity of 2nd-GG CAR-T in vivo. Mice in the group of Mock-T, which had very low level of cytokines, had the highest mortality at day 15. Therefore, the death of mice was not caused by excessive release of cytokines. Recent study demonstrated that patients with high tumor burden had higher immune dysregulation with increased serum inflammatory markers and tumor IFN signaling. IFN signaling is associated with the expression of multiple checkpoint ligands and inferior response to CAR-T therapy (46). Therefore, we considered the direct cause of death in high tumor burden model was the increased tumor load. We hypothesized that lower levels of inflammatory cytokine in vivo improved activity of 2nd-GG CAR-T through correct the immune dysregulation and reduce tumor IFN signaling, which requires further detection of phenotypes and exhaustion markers of T cells to confirm.
The present study demonstrated that a novel CD19 CAR with a less flexible hinge domain showed prolonged survival of mice under high tumor burden in preclinical studies. While there is potential for improved safety and efficacy, yet this needs validation with clinical trials.
Data Availability Statement
All data generated and analyzed for this study are included in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics committee of Fifth Medical Center of Chinese PLA General Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article The animal study was reviewed and approved by the Ethics committee of Fifth Medical Center of Chinese PLA General Hospital (ky-2018-5-37). Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
AZ designed the experiments. AZ and YS wrote the main body of the paper. AZ, YS, JD, YD, and HP performed the experiments and wrote the main body of the paper, with contributions from LM, SS, ZZ, MH, YY, XZ, WZ, and JP. YZ, QW, and MC supervise the experiments and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from 18-163-12-ZD-013-008-02, 2018ZX09711003-007 and 2018ZX09201017-003.
Conflict of Interest
Authors JD and YD were employed by SAFE Pharmaceutical Research Institute Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.724211/full#supplementary-material
References
1. Wei J, Guo Y, Wang Y, Wu Z, Bo J, Zhang B, et al. Clinical Development of CAR T Cell Therapy in China: 2020 Update. Cell Mol Immunol (2021) 18(4):792–804. doi: 10.1038/s41423-020-00555-x
2. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
3. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371(16):1507–17. doi: 10.1056/NEJMoa1407222
4. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene Maraleucel for Patients With Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet (2020) 396(10254):839–52. doi: 10.1016/s0140-6736(20)31366-0
5. Aldoss1 I, Bargou RC, Nagorsen D, Friberg GR, Baeuerle PA, Forman SJ, et al. Redirecting T Cells to Eradicate B-Cell Acute Lymphoblastic Leukemia: Bispecific T-Cell Engagers and Chimeric Antigen Receptors. Leukemia (2017). doi: 10.1038/leu.2016.391
6. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
7. Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in Refractory Diffuse Large B-Cell Lymphoma: Results From the International SCHOLAR-1 Study. Blood (2017) 130(16):1800–8. doi: 10.1182/blood-2017-03-769620
8. Singh N, Frey NV, Grupp SA, Maude SL. CAR T Cell Therapy in Acute Lymphoblastic Leukemia and Potential for Chronic Lymphocytic Leukemia. Curr Treat Options Oncol (2016) 17(6):28. doi: 10.1007/s11864-016-0406-4
9. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T Design: Elements and Their Synergistic Function. EBioMedicine (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
10. Han D, Xu Z, Zhuang Y, Ye Z, Qian Q. Current Progress in CAR-T Cell Therapy for Hematological Malignancies. J Cancer (2021) 12(2):326–34. doi: 10.7150/jca.48976
11. Cerrano M, Ruella M, Perales MA, Vitale C, Faraci DG, Giaccone L, et al. The Advent of CAR T-Cell Therapy for Lymphoproliferative Neoplasms: Integrating Research Into Clinical Practice. Front Immunol (2020) 11:888. doi: 10.3389/fimmu.2020.00888
12. Yanez L, Sanchez-Escamilla M, Perales MA. CAR T Cell Toxicity: Current Management and Future Directions. Hemasphere (2019) 3(2):e186. doi: 10.1097/HS9.0000000000000186
13. Neelapu SS. Managing the Toxicities of CAR T-Cell Therapy. Hematol Oncol (2019) 37(S1):48–52. doi: 10.1002/hon.2595
14. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated With Immune Effector Cells. Biol Blood marrow Transplant: J Am Soc Blood Marrow Transplant (2019) 25(4):625–38. doi: 10.1016/j.bbmt.2018.12.758
15. Fried S, Avigdor A, Bielorai B, Meir A, Besser MJ, Schachter J, et al. Early and Late Hematologic Toxicity Following CD19 CAR-T Cells. Bone Marrow Transplant (2019) 54(10):1643–50. doi: 10.1038/s41409-019-0487-3
16. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T Cells. biomark Res (2017) 5:22. doi: 10.1186/s40364-017-0102-y
17. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity (2016) 44(2):380–90. doi: 10.1016/j.immuni.2016.01.021
18. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
19. Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of Novel Anti-CD19 Chimeric Antigen Receptors With Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol Ther J Am Soc Gene Ther (2017) 25(11):2452–65. doi: 10.1016/j.ymthe.2017.07.013
20. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol Res (2015) 3(2):125–35. doi: 10.1158/2326-6066.CIR-14-0127
21. Almåsbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Kyte JA, et al. Inclusion of an IgG1-Fc Spacer Abrogates Efficacyof CD19 CAR T Cells in a Xenograft Mouse Model. Gene Ther (2015) 22(5):391–403. doi: 10.1038/gt.2015.4
22. Zhitao Y, Huang XF, Xiang XY, Liu Y, Kang X, Song Y, et al. A Safe and Potent Anti-CD19 CAR T Cell Therapy. Nat Med (2019) 25(6):947–53. doi: 10.1038/s41591-019-0421-7
23. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. Building Better Chimeric Antigen Receptors for Adoptive T Cell Therapy. Curr Gene Ther (2010) 10(2):77–90. doi: 10.2174/156652310791111001
24. Ishida A, Watanabe G, Oshikawa M, Ajioka I, Muraoka T. Glycine Substitution Effects on the Supramolecular Morphology and Rigidity of Cell-Adhesive Amphiphilic Peptides. Chemistry (2019) 25(59):13523–30. doi: 10.1002/chem.201902083
25. Spahr C, Shi SD, Lu HS. O-Glycosylation of Glycine-Serine Linkers in Recombinant Fc-Fusion Proteins: Attachment of Glycosaminoglycans and Other Intermediates With Phosphorylation at the Xylose Sugar Subunit. MAbs (2014) 6(4):904–14. doi: 10.4161/mabs.28763
26. Zhang A, Sun Y, Wang S, Du J, Gao X, Yuan Y, et al. Secretion of Human Soluble Programmed Cell Death Protein 1 by Chimeric Antigen Receptor-Modified T Cells Enhances Anti-Tumor Efficacy. Cytotherapy (2020) 22(12):734–43. doi: 10.1016/j.jcyt.2020.05.007
27. Kutner RH, Zhang XY, Reiser J. Production, Concentration and Titration of Pseudotyped HIV-1-Based Lentiviral Vectors. Nat Protoc (2009) 4(4):495–505. doi: 10.1038/nprot.2009.22
28. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized Mice for Immune System Investigation: Progress, Promise and Challenges. Nat Rev Immunol (2012) 12(11):786–98. doi: 10.1038/nri3311
29. Cilia E, Pancsa R, Tompa P, Lenaerts T, Vranken WF. From Protein Sequence to Dynamics and Disorder With DynaMine. Nature Communication. Nat Commun (2013) 4:2741. doi: 10.1038/ncomms3741
30. Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine Release Syndrome. J Immunother Cancer (2018) 6(1):56. doi: 10.1186/s40425-018-0343-9
31. Ataca P, Arslan O. Chimeric Antigen Receptor T Cell Therapy in Hematology. Turkish J Haematol (2015) 32(4):285–94. doi: 10.4274/tjh.2015.0049
32. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
33. Holzinger A, Abken H. CARs on the Highway: Chimeric Antigen Receptor Modified T Cells for the Adoptive Cell Therapy of Malignant Diseases. Immunother - Myths Reality Ideas Future (2017). doi: 10.5772/66496
34. Mohty M, Dulery R, Gauthier J, Malard F, Brissot E, Aljurf M, et al. CAR T-Cell Therapy for the Management of Refractory/Relapsed High-Grade B-Cell Lymphoma: A Practical Overview. Bone Marrow Transplant (2020) 55(8):1525–32. doi: 10.1038/s41409-020-0892-7
35. Zhang C, He J, Liu L, Wang J, Wang S, Liu L, et al. CD19-Directed Fast CART Therapy for Relapsed/Refractory Acute Lymphoblastic Leukemia: From Bench to Bedside. Blood (2019) 1340. doi: 10.1182/blood-2019-128006
36. Dean EA, Mhaskar RS, Lu H, Mousa MS, Krivenko GS, Lazaryan A, et al. High Metabolic Tumor Volume Is Associated With Decreased Efficacy of Axicabtagene Ciloleucel in Large B-Cell Lymphoma. Blood Adv (2020) 4(14):3268–76. doi: 10.1182/bloodadvances.2020001900
37. Jacobson CA, Hunter BD, Redd R, Rodig SJ, Chen P-H, Wright K, et al. Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J Clin Oncol (2020) 38(27):3095–106. doi: 10.1200/JCO.19.02103
38. Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Yi L, et al. Standard-Of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. Am Soc Clin Oncol (2020). doi: 10.1200/JCO.19.02104
39. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T Cell Expansion and Prolonged Persistence in Pediatric Patients With ALL Treated With a Low-Affinity CD19 CAR. Nat Med (2019) 25(9):1408–14. doi: 10.1038/s41591-019-0549-5
40. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR Activation Potential Directs Alternative T Cell Fates and Therapeutic Potency. Nat Med (2019) 25(1):82–8. doi: 10.1038/s41591-018-0290-5
41. Makita S, Yoshimura K, Tobinai K. Clinical Development of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Cancer Sci (2017) 108(6):1109–18. doi: 10.1111/cas.13239
42. Ryan D, Guest REH, Kirillova N, Cheadle EJ, et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors Evaluation of Four Different Scfvs and Antigens. J Immunother (2005) 28(3):203–11. doi: 10.1097/01.cji.0000161397.96582.59
43. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of Human T Cells to Tumor-Associated MUC1: The Evolution of a Chimeric Antigen Receptor. J Immunol (2008) 180(7):4901–9. doi: 10.4049/jimmunol.180.7.4901
44. Chuan T, Yajing Z, Yang L, Yao W, Zhiqiang W, Weidong H. Optimized Tandem CD19/CD20 CAR-Engineered T Cells in Refractory/Relapsed B-Cell Lymphoma. Blood (2020) 136(15):1632–44. doi: 10.1182/blood.2020005278
45. Xiong W, Chen Y, Kang X, Chen Z, Zheng P, Hsu YH, et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol Ther J Am Soc Gene Ther (2018). doi: 10.1016/j.ymthe.2018.01.020
Keywords: chimeric antigen receptor (CAR T), hinge region, cytokine release storm (CRS), structure optimization, cellular immunotherapy, gene modified T cell
Citation: Zhang A, Sun Y, Du J, Dong Y, Pang H, Ma L, Si S, Zhang Z, He M, Yue Y, Zhang X, Zhao W, Pi J, Chang M, Wang Q and Zhang Y (2021) Reducing Hinge Flexibility of CAR-T Cells Prolongs Survival In Vivo With Low Cytokines Release. Front. Immunol. 12:724211. doi: 10.3389/fimmu.2021.724211
Received: 12 June 2021; Accepted: 17 September 2021;
Published: 05 October 2021.
Edited by:
Ken Young, Duke University, United StatesReviewed by:
Sam Kung, University of Manitoba, CanadaNausheen Ahmed, University of Kansas, United States
Copyright © 2021 Zhang, Sun, Du, Dong, Pang, Ma, Si, Zhang, He, Yue, Zhang, Zhao, Pi, Chang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yikun Zhang, enlrMzA2QHNpbmEuY29t; Quanjun Wang, d2FuZ3F1YW5qdW5iZWlqaW5nQDE2My5jb20=; Mindong Chang, c2RtcXF3QDEyNi5jb20=
†These authors have contributed equally to this work