 Yiming Ma
Yiming Ma Yingjiao Long*†
Yingjiao Long*† Yan Chen
Yan Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 21 July 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.720049
Cigarette smoke damages a wide range of immunological functions, including innate and adaptive immune responses. Emerging literature demonstrates that inflammasome constitutes an essential component in innate immune response. In this review, we focus on the cumulative mechanisms of inflammasome in cigarette smoke-related diseases and physiopathological disorders, and summarize potential therapeutic opportunities targeting inflammasome. This review suggests that inflammasomes (NLRP3, NLRP6, NLRP12 and AIM2) are involved in the pathogenesis of several cigarette smoke-related diseases (including COPD, ALI, atherosclerosis, kidney injury, bladder dysfunction, and oral leukoplakia) and physiopathological disorders (macrophage dysfunction, endothelial barrier dysfunction, podocyte injury, and ubiquitin-mediated proteasomal processing). MyD88/NF-κB, HMGB1, production of ROS, endoplasmic reticulum stress and mitochondrial dysfunction, and Ca2+ influx are potentially involved in cigarette smoke induced-inflammasome activation. Strategies targeting ROS/NLRP3 inflammasome axis are most widely investigated and show potential therapeutic effects.
Cigarette smoke remains to be a threat to the health of the world’s population, and a global survey has indicated that prevalence of daily tobacco smoking in the population older than 15 years is 31.1% for men and 6.2% for women (1). Previous studies have demonstrated that cigarette smoke is a major risk factor for occurrence and progress of diseases involving multiple systems throughout the body, such as respiratory system (2–4), cardiovascular system (4, 5), and nervous system (6, 7). Moreover, chronic cigarette smoke inhalation damages a wide range of immunological functions, including innate and adaptive immune responses (8).
Inflammasomes are multiprotein signaling platforms mediating inflammatory responses and coordinating antimicrobial host defenses (9–12). Assembly of an inflammasome complex requires cytosolic sensing of pathogen-associated molecular patterns or danger-associated molecular patterns by a nucleotide-binding domain and leucine-rich repeat receptor (NLR) or absent in melanoma (AIM) 2-like receptor (13). Inflammasomes could activate proinflammatory caspase-1, and caspase-11 (in mice)/caspase-4 and caspase-5 (in humans), which further leads to maturation of interleukins 1β and 18 (IL-1β and IL-18) through proteolytic cleavage of pro-IL-1β and pro-IL-18 (14–16); besides, activated caspase-1, and caspase-11 (in mice)/caspase-4 and caspase-5 (in humans) also cleave and activate Gasdermin D (GSDMD), which induces a type of cell death called pyroptosis (15, 17, 18). The NLRP3 inflammasome has been under intensive study due to its extensive connection with a variety of human diseases and a two-signal model has been proposed for NLRP3 inflammasome activation; in this model, the first signal (priming) is provided by microbial or endogenous molecules that induce NLRP3 and pro-IL-1β expression through activation of NF-κB; the second signal (activation) is triggered by K+ efflux, Ca2+ signaling, reactive oxygen species (ROS), mitochondrial dysfunction, and lysosomal rupture (19). In clinical practice, VX-765, an inhibitor that directly targets inflammasome downstream cytokines, is able to block the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients, which provides a novel alternative in treating immune-associated diseases (20). In this review, we focus on the cumulative mechanisms of inflammasome in cigarette smoke-related diseases and physiopathological disorders, and summarize potential therapeutic opportunities targeting inflammasome.
Roles of inflammasome in cigarette smoke-related diseases and physiopathological disorders are summarized in Table 1. The role of inflammasome in stable COPD has been widely investigated both in vitro and in vivo models, while the conclusion remains contradictory. In vitro, Mortaz et al. (38) found that cigarette smoke could increase the expression of caspase-1 and IL-1β in human alveolar epithelial cells, suggesting the potential role of inflammasome signaling in the pathogenesis of COPD. Thereafter, more studies further demonstrated cigarette smoke was able to activate NLRP3 inflammasome in human bronchial and alveolar epithelial cells, and the inflammasome activation was able to further increase the release of inflammatory cytokines (including IL-1β and IL-18) (21, 22, 26, 27). Particularly, Mahalanobish et al. (21) reported that the activation of NLRP3 inflammasome in alveolar epithelial might result from cigarette smoke induced endoplasmic reticulum (ER) stress and mitochondrial dysfunctions. Wang et al. (27) found that cigarette smoke-induced inflammasome activation was triggered by oxidative stress injury and Ca2+ influx in human bronchial and alveolar epithelial cells. Besides the activation of NLRP3 inflammasome, Singh et al. (31) observed that increase of NLRP10 and NLRP12 proteins in human alveolar type II epithelial cells challenged by cigarette smoke extract. Furthermore, Kaur et al. (32) proved that NLRP10 knockdown rescued cigarette smoke extract induced inflammatory responses in human alveolar type II epithelial cells, which might serve as an effective therapeutic target of COPD.

Table 1 Roles of inflammasome in cigarette smoke-related diseases and physiopathological disorders.
In vivo, COPD mouse model was established to explore the effect of cigarette smoke on inflammasome (21, 29, 32, 34, 37). In mouse lung tissue, increased expression of NLRP3 was observed after exposure to cigarette smoke (21, 29); moreover, it was also found that the release of downstream inflammation cytokines significantly increased in mouse bronchoalveolar lavage fluid (BALF) samples (21, 29, 34). Cao et al. (29) concluded that activation of NLRP3 inflammasome was related to cigarette smoke-induced ROS production. Nevertheless, an earlier study by Pauwels et al. (37) demonstrated that cigarette smoke-induced inflammation was independent on the activation of NLRP3/caspase-1/IL-1β axis. Moreover, NLRP10 activation might also be involved in the inflammation cytokine release induced by cigarette smoke (32).
Clinical samples from patients were also used to measure whether inflammasome played a role in the pathogenesis of stable COPD (33, 35). Inconsistent with results of studies in vitro and in vivo, Faner et al. (33) found that the NLRP3 inflammasome was primed, but not activated in the lung tissue of stable COPD patients; besides, both caspase-1 and ASC were mostly in inactive forms. Also, Di Stefano et al. (35) reported that the NLRP3 inflammasome is not activated in bronchial mucosa and BALF of stable COPD patients. However, gene expression analyses revealed that polymorphisms in NLRP1 rs12150220 were associated with COPD disease severity, which suggested the importance of NLRP1 inflammasome fine-tuning in maintaining lung tissue integrity and treating chronic airway inflammation (25). Significant heterogeneity of clinical presentation and disease progression exists in COPD patients, so phenotyping COPD patients is beneficial to identify patient subgroups with unique prognostic or therapeutic characteristics (52). The Spanish guidelines describe four clinical phenotypes for COPD, including chronic bronchitis phenotype, emphysema phenotype, asthma-COPD overlap syndrome phenotype, and non-exacerbator phenotype (53). Interestingly, chronic bronchitis phenotype of COPD is associated with worse respiratory symptoms and higher risk of exacerbations in contrast to other phenotypes (54). However, there have been no studies assessing activation levels of inflammasomes among different phenotypes of COPD patients. Importantly, it seems significant to evaluate whether a higher proportion of activated inflammasomes exists in chronic bronchitis phenotype of COPD patients compared with other phenotypes, as this may contribute a lot to the precision treatment of COPD.
Previous studies also proved that the activation of NLRP3 inflammasome was involved in the pathogenesis of AECOPD. Cigarette smoke could enhance the NLRP3 inflammasome activation, and further promote the downstream inflammatory cytokines’ release (including IL-1β and IL-18) in human bronchial epithelial cells (23, 24), and human alveolar epithelial cells (26). Of note, inducing factors of acute exacerbations varied in different studies, such as lipopolysaccharide (23, 26), lipoteichoic acid (23), extracellular heat shock protein 70 (23), and influenza A virus (24) etc. In addition, Rotta et al. (36) verified that there was an upregulation of the NLRP3 in murine macrophage cells and human alveolar macrophages from Nontypeable Haemophilus Influenzae infection (NTHi)-induced AECOPD disease model, which imitated the pathogenesis of bacterial exacerbations in COPD. In vivo, animal experiments showed that NLRP3 inflammasome activation existed in AECOPD rat lung tissue and BALF (24).
A growing number of studies demonstrated that there were significant increases of NLRP3 and IL-1β in human peripheral blood mononuclear cells from AECOPD patients (23, 30). Furthermore, potential NLRP3 inflammasome activation accompanied with inflammation cytokine release were also observed in human bronchial tissues (30, 36), serum (30), BALF (30), sputum (33), and plasma (33). Interestingly, Colarusso et al. (28) found that AIM2 inflammasome was also activated in human peripheral blood mononuclear cells derived from AECOPD patients, and could further lead to the release of IL-1α and TGF-β.
As for smoke inhalation-induced acute lung injury, Zhang et al. (39) reported that cigarette smoke was able to augment the formation of NLRP3 inflammasome, the activation of caspase-1 and IL-1β induced in mouse alveolar macrophages.
Recently, there have been studies investigating the role of inflammasome in cigarette smoke-induced atherosclerosis, and these studies mainly focused on NLRP3 inflammasome (40–42, 55). Mehta et al. (55) used human THP-1 monocytes, macrophages and foam cells to represent crucial stages of initiation, progression and development in cigarette smoke-induced atherogenesis, and it was found that cigarette smoke exposure could activate NLRP3 inflammasome in these cells with a stage-specific manner; furthermore, they proved that MyD88/NF-kappa B pathway was an upstream regulator of NLRP3 inflammasome (40). In rat vascular smooth muscle cells and rat aortic tissue, activation of reactive oxygen species (ROS)-NLRP3 inflammasome-C reactive protein (CRP) axis significantly enhanced after nicotine treatment (42). Moreover, nicotine induced ROS-NLRP3-mediated endothelial cell pyroptosis in human aortic endothelial cells (HAECs), which was evidenced by cleavage of caspase-1, production of downstream interleukin IL-1β and IL-18 (41). Importantly, one recent study found that transcriptional and translational expression of NLRP3 inflammasome markers (including caspase-1, pro-IL-1β, IL-1β, pro-IL-18 and IL-18) in mononuclear cells were significantly increased (2 to 7-fold) in smokers with coronary artery disease (CAD) in contrast to non-smokers with CAD (56).
Two recent researches concentrated on the change of inflammasome in cigarette smoke-related urinary diseases, including kidney injury and bladder dysfunction (43, 44). Zheng et al. (43) proposed that nicotine induced NLRP6 inflammasome activation via alpha7 nicotinic acetylcholine receptor in human kidney cells and mouse kidney tissue. Besides, Wu et al. (44) found that cigarette smoke induced the pyroptosis of urothelial cells through ROS/NLRP3/caspase-1 signaling pathway in bladder dysfunction models.
Inflammasome might also play an important role in some other physiopathological disorders induced by cigarette smoke, such as macrophage dysfunction (45), endothelial barrier dysfunction (47–49), podocyte injury (46), ubiquitin-mediated proteasomal processing (51), and oral leukoplakia (50) etc.
With regard to macrophage dysfunction, the study by Buscetta et al. (45) manifested that cigarette smoke restrained the expression of NLRP3, caspase-1, IL-1β, and IL-18 acting mainly at the transcriptional level in human monocyte-derived macrophages and THP-1 cells, and increased the caspase-1 activity via an NLRP3-independent and Toll-like receptor 4 (TLR4)-Toll/IL-1receptor domain containing adaptor inducing IFN-β (TRIF)-caspase-8-dependent pathway. On the contrary, Rumora et al. (22, 23) found that cigarette smoke was an activated factor of NLRP3 inflammasome in human monocyte-derived macrophages and THP-1 cells.
Endothelial barrier injury has been increasingly considered as an important pathophysiological process in COPD. Chen et al. (48) verified that cigarette smoke could increase the expression of caspase-1, NLRP3, and IL-1β in human umbilical vein endothelial cells. In mouse microvascular endothelial cells and mouse coronary arterial endothelium, nicotine was proved to increase high mobility group box 1(HMGB1) expression, cause NLRP3 inflammasome complex formation and enhance the inflammasome activity as demonstrated by increased cleavage of pro-caspase-1, and IL-1β production (47). In addition, Wang et al. (49) pointed out that cigarette smoke could activate ROS/NLRP3 axis in rat carotid artery tissue and human umbilical vein endothelial cells.
In addition, Singh et al. (46) found that nicotine instigated mouse podocyte cell injury via inducing ROS production and activating NLRP3 inflammasome. Han et al. (51) proved that cigarette could decrease NLRP3 protein abundance via increased ubiquitin-mediated proteasomal processing. Another interesting finding was that long-term cigarette smoking suppressed NLRP3 inflammasome activation in oral mucosal epithelium and attenuated host defense against Candida albicans in a rat model (50).
Inconsistent conclusions of cigarette smoke on NLRP3 expression among different studies may result from the following reasons. Firstly, Baroja-Mazo et al. (57) reported that the NLRP3 inflammasome particle was released from macrophages after inflammasome activation; thus, the decreased cellular level of NLRP3 protein may be due to the secretion of NLRP3 protein as extracellular oligomeric complexes. Secondly, the preparation method and treated concentration of cigarette smoke extract could not be homogenized among different experiments.
Table 2 illustrates therapeutic strategies to target inflammasome in cigarette smoke-induced diseases and physiopathological disorders. Scavenging ROS has become the most widely investigated strategy to inhibit NLRP3 inflammasome activation induced by cigarette smoke. N-Acetyl-L-cysteine (NAC) is an amino-acid derivative of cysteine and a precursor of the antioxidant enzyme glutathione (58). The application of NAC as an antioxidant to diminish ROS generation has been proved in previous studies (59, 60). In clinical practice, NAC has been used as a mucolytic to help clear mucus in patients with respiratory diseases (61). Several studies verified that NAC could decrease ROS generation and further inhibit NLRP3 inflammasome activation, and NAC-mediated disease/disorder involved atherosclerosis (41), bladder damage (44), and podocyte injury (46). Besides NAC, other therapies antagonizing ROS were also explored to modulate inflammasome activation in cigarette smoke-induced diseases. Melatonin (N-acetyl-5-methoxytryptamine) is a neuroendocrine hormone synthesized by tryptophan and serotonin metabolism, and secreted from the pineal gland (62, 63). Melatonin was found to play an important role in relieving oxidative stress injury (64, 65). One study by Wang et al. (49) illustrated that melatonin inhibited ROS production, NLRP3 inflammasome activation and pyroptosis in cigarette smoke-treated endothelial cells. Another effect of melatonin on NLRP3 inflammasome activation was to suppress endoplasmic reticulum stress and mitochondrial dysfunction in COPD (21). Inhibitors of transient receptor potential protein (TRP) ion channels (TRPA1 and TRPV1) might also attenuate NLRP3 inflammasome activation by reducing oxidative stress and blocking Ca2+ influx (27). Moreover, rosmarinic acid and lipoxin receptor agonist (BML-111) were also capable of mitigating ROS production and restraining NLRP3 inflammasome activation in cigarette smoke-induced diseases (29, 42).
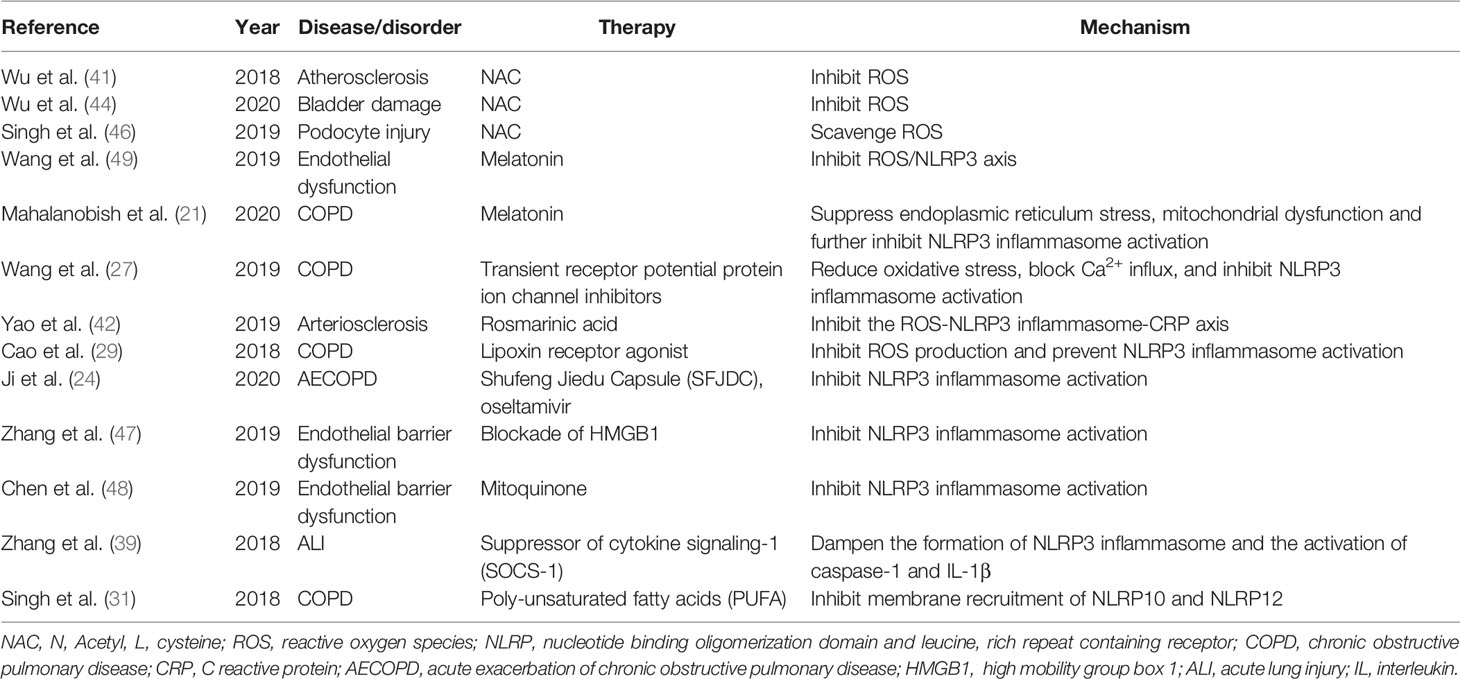
Table 2 Therapeutic strategies to target inflammasome in cigarette smoke-induced diseases and physiopathological disorders.
There were also some other strategies to target NLRP3 inflammasome in cigarette smoke-related diseases. Concerning endothelial barrier dysfunction induced by cigarette smoke, Zhang et al. (47) found that blockade of HMGB1 could inhibit NLRP3 inflammasome activation; similarly, mitoquinone was also able to diminish NLRP3 inflammasome activation in cigarette smoke-induced endothelial barrier dysfunction (48). With respect to cigarette smoke induced-ALI, suppressor of cytokine signaling-1 (SOCS-1) might dampen the formation of NLRP3 inflammasome and the activation of caspase-1 and IL-1β (39). Futhermore, Ji et al. (24) pointed out that Shufeng Jiedu Capsule (SFJDC) and oseltamivir significantly decreased NLRP3 inflammasome activation in influenza virus A-induced AECOPD disease models.
Targeting NLRP10 and NLRP12 inflammasome, poly-unsaturated fatty acids (PUFA) was capable of rescuing A549 cells from cigarette smoke extract (CSE)-mediated membrane recruitment of NLRP10 and NLRP12, and also from inflammatory responses (31). Finally, Supplemental Figure 1 summarized effects of cigarette smoke on NLRP3 inflammasome, and potential therapeutic strategies.
Collectively, emerging evidence demonstrates that inflammasomes (NLRP3, NLRP6, NLRP12 and AIM2) are involved in the pathogenesis of several cigarette smoke-related diseases (including COPD, ALI, atherosclerosis, kidney injury, bladder dysfunction, and oral leukoplakia) and physiopathological disorders (macrophage dysfunction, endothelial barrier dysfunction, podocyte injury, and ubiquitin-mediated proteasomal processing). MyD88/NF-κB, HMGB1, production of ROS, endoplasmic reticulum stress and mitochondrial dysfunction, and Ca2+ influx are potentially involved in cigarette smoke induced-inflammasome activation. Strategies targeting ROS/NLRP3 inflammasome axis are most widely investigated and show potential therapeutic effects. Although this review reveals the potential relationship between cigarette smoke and inflammasome, more studies may be still needed to further confirm more detailed mechanisms so as to provide effective alternatives for treating cigarette smoke-related diseases.
All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (No. 81873410, No. 81800043, and No. 82070049), and National Natural Science Foundation of Hunan (No. 2020JJ5818).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.720049/full#supplementary-material
Supplementary Figure 1 | Effect of cigarette smoke on NLRP3 inflammasome, and potential therapeutic strategies. HMGB, high mobility group box 1; NAC, N-Acetyl-L-cysteine; ROS, reactive oxygen species; TRP, transient receptor potential protein; ER, endoplasmic reticulum; NLRP, Nucleotide binding oligomerization domain and leucine-rich repeat containing receptor.
1. Ng M, Freeman MK, Fleming TD, Robinson M, Dwyer-Lindgren L, Thomson B, et al. Smoking Prevalence and Cigarette Consumption in 187 Countries, 1980-2012. Jama-J Am Med Assoc (2014) 311(2):183–92. doi: 10.1001/jama.2013.284692
2. Ambrose JA, Barua RS. The Pathophysiology of Cigarette C-V Smoking and Cardiovascular Disease - An Update. J Am Coll Cardiol (2004) 43(10):1731–7. doi: 10.1016/j.jacc.2003.12.047
3. Yoshida T, Tuder RM. Pathobiology of Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. Physiol Rev (2007) 87(3):1047–82. doi: 10.1152/physrev.00048.2006
4. Pope CA III, Burnett RT, Turner MC, Cohen A, Krewski D, Jerrett M, et al. Lung Cancer and Cardiovascular Disease Mortality Associated With Ambient Air Pollution and Cigarette Smoke: Shape of the Exposure-Response Relationships. Environ Health Perspect (2011) 119(11):1616–21. doi: 10.1289/ehp.1103639
5. Huxley RR, Woodward M. Cigarette Smoking as a Risk Factor for Coronary Heart Disease in Women Compared With Men: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. Lancet (2011) 378(9799):1297–305. doi: 10.1016/s0140-6736(11)60781-2
6. Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A Meta-Analysis of Coffee Drinking, Cigarette Smoking, and the Risk of Parkinson’s Disease. Ann Neurol (2002) 52(3):276–84. doi: 10.1002/ana.10277
7. Heneka MT. Inflammasome Activation and Innate Immunity in Alzheimer’s Disease. Brain Pathol (2017) 27(2):220–2. doi: 10.1111/bpa.12483
8. Sopori M. Effects of Cigarette Smoke on the Immune System. Nat Rev Immunol (2002) 2(5):372–7. doi: 10.1038/nri803
9. Broz P, Dixit VM. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
10. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus Through Recognition of Viral RNA. Immunity (2009) 30(4):556–65. doi: 10.1016/j.immuni.2009.02.005
11. Man SM, Kanneganti T-D. Converging Roles of Caspases in Inflammasome Activation, Cell Death and Innate Immunity. Nat Rev Immunol (2016) 16(1):7–21. doi: 10.1038/nri.2015.7
12. Kanneganti T-D. The Inflammasome: Firing Up Innate Immunity. Immunol Rev (2015) 265(1):1–5. doi: 10.1111/imr.12297
13. Man SM, Kanneganti TD. Regulation of Inflammasome Activation. Immunol Rev (2015) 265(1):6–21. doi: 10.1111/imr.12296
14. Malik A, Kanneganti TD. Inflammasome Activation and Assembly at a Glance. J Cell Sci (2017) 130(23):3955–63. doi: 10.1242/jcs.207365
15. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for non-Canonical Inflammasome Signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
16. Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell (2014) 157(5):1013–22. doi: 10.1016/j.cell.2014.04.007
17. Pang Y, Zhang PC, Lu RR, Li HL, Li JC, Fu HX, et al. Andrade-Oliveira Salvianolic Acid B Modulates Caspase-1-Mediated Pyroptosis in Renal Ischemia-Reperfusion Injury via Nrf2 Pathway. Front Pharmacol (2020) 11:541426. doi: 10.3389/fphar.2020.541426
18. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
19. He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
20. Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JC, et al. IL-Converting Enzyme/Caspase-1 Inhibitor VX-765 Blocks the Hypersensitive Response to an Inflammatory Stimulus in Monocytes From Familial Cold Autoinflammatory Syndrome Patients. J Immunol (2005) 175(4):2630–4. doi: 10.4049/jimmunol.175.4.2630
21. Mahalanobish S, Dutta S, Saha S, Sil PC. Melatonin Induced Suppression of ER Stress and Mitochondrial Dysfunction Inhibited NLRP3 Inflammasome Activation in COPD Mice. Food Chem Toxicol (2020) 144:111588. doi: 10.1016/j.fct.2020.111588
22. Rumora L, Somborac-Bačura A, Hlapčić I, Hulina-Tomašković A, Rajković MG. Cigarette Smoke and Extracellular Hsp70 Induce Secretion of ATP and Differential Activation of NLRP3 Inflammasome in Monocytic and Bronchial Epithelial Cells. Cytokine (2020) 135:155220. doi: 10.1016/j.cyto.2020.155220
23. Rumora L, Hlapčić I, Hulina-Tomašković A, Somborac-Bačura A, Bosnar M, Rajković MG. Pathogen-Associated Molecular Patterns and Extracellular Hsp70 Interplay in NLRP3 Inflammasome Activation in Monocytic and Bronchial Epithelial Cellular Models of COPD Exacerbations. APMIS: Acta Pathol Microbiol Immunol Scandinavica (2021) 129(2):80–90. doi: 10.1111/apm.13089
24. Ji S, Bai Q, Wu X, Zhang DW, Wang S, Shen JL, et al. Unique Synergistic Antiviral Effects of Shufeng Jiedu Capsule and Oseltamivir in Influenza A Viral-Induced Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Biomed Pharmacother Biomed Pharmacother (2020) 121:109652. doi: 10.1016/j.biopha.2019.109652
25. Ozretić P, da Silva Filho MI, Catalano C, Sokolović I, Vukić-Dugac A, Šutić M, et al. Association of NLRP1 Coding Polymorphism With Lung Function and Serum IL-1β Concentration in Patients Diagnosed With Chronic Obstructive Pulmonary Disease (COPD). Genes (2019) 10(10):783. doi: 10.3390/genes10100783
26. Nachmias N, Langier S, Brzezinski RY, Siterman M, Stark M, Etkin S, et al. NLRP3 Inflammasome Activity Is Upregulated in an in-Vitro Model of COPD Exacerbation. PloS One (2019) 14(5):e0214622. doi: 10.1371/journal.pone.0214622
27. Wang MY, Zhang YB, Xu MM, Zhang H, Chen YQ, Chung KF, et al. Roles of TRPA1 and TRPV1 in Cigarette Smoke-Induced Airway Epithelial Cell Injury Model. Free Radic Biol Med (2019) 134:229–38. doi: 10.1016/j.freeradbiomed.2019.01.004
28. Colarusso C, Terlizzi M, Molino A, Imitazione P, Somma P, Rega R, et al. AIM2 Inflammasome Activation Leads to IL-1α and TGF-β Release From Exacerbated Chronic Obstructive Pulmonary Disease-Derived Peripheral Blood Mononuclear Cells. Front Pharmacol (2019) 10:257. doi: 10.3389/fphar.2019.00257
29. Cao Y, Zhou X, Yin Z, Yu X, Yang Q, Guo Q, et al. The Anti-Inflammatory Effect of BML-111 on COPD may be Mediated by Regulating NLRP3 Inflammasome Activation and ROS Production. Prostaglandins Other Lipid Mediators (2018) 138:23–30. doi: 10.1016/j.prostaglandins.2018.08.001
30. Wang H, Lv C, Wang S, Ying H, Weng Y, Yu W. NLRP3 Inflammasome Involves in the Acute Exacerbation of Patients With Chronic Obstructive Pulmonary Disease. Inflammation (2018) 41(4):1321–33. doi: 10.1007/s10753-018-0780-0
31. Singh DP, Kaur G, Bagam P, Pinkston R, Batra S. Membrane Microdomains Regulate NLRP10- and NLRP12-Dependent Signalling in A549 Cells Challenged With Cigarette Smoke Extract. Arch Toxicol (2018) 92(5):1767–83. doi: 10.1007/s00204-018-2185-0
32. Kaur G, Bagam P, Pinkston R, Singh DP, Batra S. Cigarette Smoke-Induced Inflammation: NLRP10-Mediated Mechanisms. Toxicology (2018) 398-399:52–67. doi: 10.1016/j.tox.2018.02.010
33. Faner R, Sobradillo P, Noguera A, Gomez C, Cruz T, López-Giraldo A, et al. The Inflammasome Pathway in Stable COPD and Acute Exacerbations. ERJ Open Res (2016) 2(3):00002-2016. doi: 10.1183/23120541.00002-2016
34. Yang W, Ni H, Wang H, Gu H. NLRP3 Inflammasome is Essential for the Development of Chronic Obstructive Pulmonary Disease. Int J Clin Exp Pathol (2015) 8(10):13209–16.
35. Di Stefano A, Caramori G, Barczyk A, Vicari C, Brun P, Zanini A, et al. Innate Immunity But Not NLRP3 Inflammasome Activation Correlates With Severity of Stable COPD. Thorax (2014) 69(6):516–24. doi: 10.1136/thoraxjnl-2012-203062
36. Rotta Detto Loria J, Rohmann K, Droemann D, Kujath P, Rupp J, Goldmann T, et al. Nontypeable Haemophilus Influenzae Infection Upregulates the NLRP3 Inflammasome and Leads to Caspase-1-Dependent Secretion of Interleukin-1β - A Possible Pathway of Exacerbations in COPD. PloS One (2013) 8(6):e66818. doi: 10.1371/journal.pone.0066818
37. Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S, Vanden Berghe T, et al. Role of IL-1α and the Nlrp3/caspase-1/IL-1β Axis in Cigarette Smoke-Induced Pulmonary Inflammation and COPD. Eur Respir J (2011) 38(5):1019–28. doi: 10.1183/09031936.00158110
38. Mortaz E, Henricks PA, Kraneveld AD, Givi ME, Garssen J, Folkerts G. Cigarette Smoke Induces the Release of CXCL-8 From Human Bronchial Epithelial Cells via TLRs and Induction of the Inflammasome. Biochim Biophys Acta (2011) 1812(9):1104–10. doi: 10.1016/j.bbadis.2011.06.002
39. Zhang L, Xu C, Chen X, Shi Q, Su W, Zhao H. SOCS-1 Suppresses Inflammation Through Inhibition of NALP3 Inflammasome Formation in Smoke Inhalation-Induced Acute Lung Injury. Inflammation (2018) 41(4):1557–67. doi: 10.1007/s10753-018-0802-y
40. Mehta S, Dhawan V. Molecular Insights of Cigarette Smoke Condensate-Activated NLRP3 Inflammasome in THP-1 Cells in a Stage-Specific Atherogenesis. Int Immunopharmacol (2020) 88:107013. doi: 10.1016/j.intimp.2020.107013
41. Wu XX, Zhang HY, Qi W, Zhang Y, Li JM, Li ZG, et al. Nicotine Promotes Atherosclerosis via ROS-NLRP3-Mediated Endothelial Cell Pyroptosis. Cell Death Dis (2018) 9:12. doi: 10.1038/s41419-017-0257-3
42. Yao Y, Mao J, Xu S, Zhao L, Long L, Chen L, et al. Rosmarinic Acid Inhibits Nicotine-Induced C-Reactive Protein Generation by Inhibiting NLRP3 Inflammasome Activation in Smooth Muscle Cells. J Cell Physiol (2019) 234(2):1758–67. doi: 10.1002/jcp.27046
43. Zheng CM, Lee YH, Chiu IJ, Chiu YJ, Sung LC, Hsu YH, et al. Nicotine Causes Nephrotoxicity Through the Induction of NLRP6 Inflammasome and Alpha7 Nicotinic Acetylcholine Receptor. Toxics (2020) 8(4):92. doi: 10.3390/toxics8040092
44. Wu Z, Liu Q, Zhu K, Liu Y, Chen L, Guo H, et al. Cigarette Smoke Induces the Pyroptosis of Urothelial Cells Through ROS/NLRP3/caspase-1 Signaling Pathway. Neurourol Urodynamics (2020) 39(2):613–24. doi: 10.1002/nau.24271
45. Buscetta M, Di Vincenzo S, Miele M, Badami E, Pace E, Cipollina C. Cigarette Smoke Inhibits the NLRP3 Inflammasome and Leads to Caspase-1 Activation via the TLR4-TRIF-Caspase-8 Axis in Human Macrophages. FASEB J: Off Publ Fed Am Soc Exp Biol (2020) 34(1):1819–32. doi: 10.1096/fj.201901239R
46. Singh GB, Kshirasagar N, Patibandla S, Puchchakayala G, Koka S, Boini KM. Nicotine Instigates Podocyte Injury via NLRP3 Inflammasomes Activation. Aging (2019) 11(24):12810–21. doi: 10.18632/aging.102611
47. Zhang Y, Chen Y, Zhang Y, Li PL, Li X. Contribution of Cathepsin B-Dependent Nlrp3 Inflammasome Activation to Nicotine-Induced Endothelial Barrier Dysfunction. Eur J Pharmacol (2019) 865:172795. doi: 10.1016/j.ejphar.2019.172795
48. Chen S, Wang Y, Zhang H, Chen R, Lv F, Li Z, et al. The Antioxidant MitoQ Protects Against CSE-Induced Endothelial Barrier Injury and Inflammation by Inhibiting ROS and Autophagy in Human Umbilical Vein Endothelial Cells. Int J Biol Sci (2019) 15(7):1440–51. doi: 10.7150/ijbs.30193
49. Wang X, Bian Y, Zhang R, Liu X, Ni L, Ma B, et al. Melatonin Alleviates Cigarette Smoke-Induced Endothelial Cell Pyroptosis Through Inhibiting ROS/NLRP3 Axis. Biochem Biophys Res Commun (2019) 519(2):402–8. doi: 10.1016/j.bbrc.2019.09.005
50. Ye P, Wang X, Ge S, Chen W, Wang W, Han X. Long-Term Cigarette Smoking Suppresses NLRP3 Inflammasome Activation in Oral Mucosal Epithelium and Attenuates Host Defense Against Candida Albicans in a Rat Model. Biomed Pharmacother Biomed Pharmacother (2019) 113:108597. doi: 10.1016/j.biopha.2019.01.058
51. Han S, Jerome JA, Gregory AD, Mallampalli RK. Cigarette Smoke Destabilizes NLRP3 Protein by Promoting its Ubiquitination. Respir Res (2017) 18(1):2. doi: 10.1186/s12931-016-0485-6
52. Barnes PJ. Inflammatory Mechanisms in Patients With Chronic Obstructive Pulmonary Disease. J Allergy Clin Immunol (2016) 138(1):16–27. doi: 10.1016/j.jaci.2016.05.011
53. Rubio MC, Casamor R, Miravitlles M, Grp FS. Identification and Distribution of COPD Phenotypes in Clinical Practice According to Spanish COPD Guidelines: The FENEPOC Study. Int J Chronic Obstructive Pulmonary Dis (2017) 12:2373–83. doi: 10.2147/copd.s137872
54. Kim V, Han MK, Vance GB, Make BJ, Newell JD, Hokanson JE, et al. The Chronic Bronchitic Phenotype of COPD: An Analysis of the COPDGene Study. Chest (2011) 140(3):626–33. doi: 10.1378/chest.10-2948
55. Mehta S, Dhawan V. Exposure of Cigarette Smoke Condensate Activates NLRP3 Inflammasome in THP-1 Cells in a Stage-Specific Manner: An Underlying Role of Innate Immunity in Atherosclerosis. Cell Signalling (2020) 72:109645. doi: 10.1016/j.cellsig.2020.109645
56. Mehta S, Vijayvergiya R, Dhawan V. Activation of NLRP3 Inflammasome Assembly is Associated With Smoking Status of Patients With Coronary Artery Disease. Int Immunopharmacol (2020) 87:106820. doi: 10.1016/j.intimp.2020.106820
57. Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 Inflammasome is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat Immunol (2014) 15(8):738–48. doi: 10.1038/ni.2919
58. Bretti C, Cardiano P, Irto A, Lando G, Milea D, Sammartano S. Interaction of N-Acetyl-L-Cysteine With Na(+), Ca(2+), Mg(2+) and Zn(2+). Thermodynamic Aspects, Chemical Speciation and Sequestering Ability in Natural Fluids. J Mol Liquids (2020) 319:114164. doi: 10.1016/j.molliq.2020.114164
59. Gupta A, Vijayaraghavan R, Gautam A. Combination Therapy of N-Acetyl-L-Cysteine and S-2(2-Aminoethylamino) Ethylphenyl Sulfide for Sulfur Mustard Induced Oxidative Stress in Mice. Toxicol Rep (2021) 8:599–606. doi: 10.1016/j.toxrep.2021.03.011
60. Lee CY, Su CH, Tsai PK, Yang ML, Ho YC, Lee SS, et al. Cadmium Nitrate-Induced Neuronal Apoptosis is Protected by N-Acetyl-L-Cysteine via Reducing Reactive Oxygen Species Generation and Mitochondria Dysfunction. Biomed Pharmacother Biomed Pharmacother (2018) 108:448–56. doi: 10.1016/j.biopha.2018.09.054
61. Holdiness MR. Clinical Pharmacokinetics of N-Acetylcysteine. Clin Pharmacokinetics (1991) 20(2):123–34. doi: 10.2165/00003088-199120020-00004
62. Shao A, Gao S, Wu H, Xu W, Pan Y, Fang Y, et al. Melatonin Ameliorates Hemorrhagic Transformation via Suppression of ROS-Induced NLRP3 Activation After Cerebral Ischemia in Hyperglycemic Rats. Oxid Med Cell Longevity (2021) 2021:6659282. doi: 10.1155/2021/6659282
63. Zhou R, Ma Y, Tao Z, Qiu S, Gong Z, Tao L, et al. Melatonin Inhibits Glucose-Induced Apoptosis in Osteoblastic Cell Line Through PERK-Eif2α-ATF4 Pathway. Front Pharmacol (2020) 11:602307. doi: 10.3389/fphar.2020.602307
64. Zhu HL, Shi XT, Xu XF, Zhou GX, Xiong YW, Yi SJ, et al. Melatonin Protects Against Environmental Stress-Induced Fetal Growth Restriction via Suppressing ROS-Mediated GCN2/ATF4/BNIP3-Dependent Mitophagy in Placental Trophoblasts. Redox Biol (2021) 40:101854. doi: 10.1016/j.redox.2021.101854
Keywords: inflammasome, cigarette smoke, NLRP3, ROS, innate immune
Citation: Ma Y, Long Y and Chen Y (2021) Roles of Inflammasome in Cigarette Smoke-Related Diseases and Physiopathological Disorders: Mechanisms and Therapeutic Opportunities. Front. Immunol. 12:720049. doi: 10.3389/fimmu.2021.720049
Received: 03 June 2021; Accepted: 08 July 2021;
Published: 21 July 2021.
Edited by:
Robson Coutinho-Silva, Federal University of Rio de Janeiro, BrazilReviewed by:
Cassio Almeida-da-Silva, University of the Pacific, United StatesCopyright © 2021 Ma, Long and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Chen, Y2hlbnlhbjk5NzI3QGNzdS5lZHUuY24=; Yingjiao Long, bG9uZ3lpbmdqaWFvQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.