 Mikel Etxebeste-Mitxeltorena
Mikel Etxebeste-Mitxeltorena Inés del Rincón-Loza
Inés del Rincón-Loza Beatriz Martín-Antonio
Beatriz Martín-Antonio
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 August 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.717850
This article is part of the Research Topic The Intricate Innate Immune-Cancer Cell Relationship in the Context of Tumor Angiogenesis, Immunity and Microbiota: the Angiogenic Switch in the Tumor Microenvironment as a Key Target for Immunotherapies View all 12 articles
Adoptive cellular immunotherapy using chimeric antigen receptor (CAR)-modified T cells and Natural Killer (NK) cells are common immune cell sources administered to treat cancer patients. In detail, whereas CAR-T cells induce outstanding responses in a subset of hematological malignancies, responses are much more deficient in solid tumors. Moreover, NK cells have not shown remarkable results up to date. In general, immune cells present high plasticity to change their activity and phenotype depending on the stimuli they receive from molecules secreted in the tumor microenvironment (TME). Consequently, immune cells will also secrete molecules that will shape the activities of other neighboring immune and tumor cells. Specifically, NK cells can polarize to activities as diverse as angiogenic ones instead of their killer activity. In addition, tumor cell phagocytosis by macrophages, which is required to remove dying tumor cells after the attack of NK cells or CAR-T cells, can be avoided in the TME. In addition, chemotherapy or radiotherapy treatments can induce senescence in tumor cells modifying their secretome to a known as “senescence-associated secretory phenotype” (SASP) that will also impact the immune response. Whereas the SASP initially attracts immune cells to eliminate senescent tumor cells, at high numbers of senescent cells, the SASP becomes detrimental, impacting negatively in the immune response. Last, CAR-T cells are an attractive option to overcome these events. Here, we review how molecules secreted in the TME by either tumor cells or even by immune cells impact the anti-tumor activity of surrounding immune cells.
Today, it is widely recognized that chronic inflammation is a driver of cancer (1), being estimated that 15-20% of cancers are inflammation-related (2). This association has been observed in different contexts, such as persistent Helicobacter pylori infection or autoimmune diseases like inflammatory bowel disease that increase the risk of developing gastric cancer (3) or colorectal cancer (4), respectively. Numerous studies have found associations of inflammatory markers with a higher risk of developing cancer. For instance, 15% of patients with cardiovascular disease, after a median follow-up of 8.3 years, developed different types of cancer whose incidence was associated with high C-reactive protein (CRP) levels (5). In addition, IL6 levels are also associated with an increased risk of developing different types of cancer (6). Moreover, IL1β inhibition reduced CRP and IL6 levels and the incidence of developing lung cancer in patients with atherosclerosis who had a myocardial infarction (7).
Both immune and tumor cells promote this pro-inflammatory microenvironment. Expressly, tumor cells release a secretome that displays an altered composition compared to the normal tissue from which they are derived (8). This secretome contains cytokines, chemokines, hormones, metabolites, and growth factors involved in cell-cell communication, angiogenesis, hypoxia, metastasis, extracellular matrix remodeling, and drug resistance (8, 9), where tumor cells employ it as a mechanism of immune evasion (10–12). On the other side, the different subsets of immune cells will also release immunosuppressive and inflammatory factors that will shape the tumor microenvironment (TME), promoting or inhibiting cancer progression (13).
The anti-tumor activity of immune cells infiltrating tumors led to the development of adoptive cellular immunotherapy administering natural killer (NK) cells, T cells, or genetically modified chimeric antigen receptor (CAR)-T cells in cancer patients (14–17). Clinical results administering different immune cells have been reviewed by others (Table 1). However, despite promising results in these studies for some malignancies (26), immune cells do not persist long for other malignancies, and patients end up relapsing (27). Once immune cells achieve the tumor, they will have to face tumor cells and their secretome that may polarize their anti-tumor activity to a pro-tumoral one, increasing angiogenesis and enhancing tumor growth (28). Moreover, after chemotherapy treatment, tumor cells can reach a senescent state, known as therapy-induced senescence (TIS), that shapes the tumor secretome to a variety of pro-inflammatory and angiogenic proteins known as “senescence-associated secretory phenotype” (SASP). The SASP may enhance the immune response at initial stages and contribute to a favorable environment for tumor growth at late stages (29). For example, senescent fibroblasts, much more than pre-senescent fibroblasts, secrete VEGF that causes premalignant and malignant epithelial cells to form tumors, suggesting that although cellular senescence suppresses tumorigenesis early in life, it may also promote cancer (30).

Table 1 Reviews indicating clinical results with different types of immune cells administered in immunotherapy studies in cancer patients.
Here, we review how the tumor secretome can shape the immune response achieving a state when immune cells no longer recognize tumor cells and instead, they secrete proteins that breed the TME. We will specifically focus on the impact on T cells, CAR-T cells, and NK cells, which are currently used in adoptive cellular immunotherapy (14–17, 31), and macrophages due to their relevant role in removing dying/senescent tumor cells after cancer treatment (32). The impact of these molecules is summarized in Table 2. Moreover, we will review the effect of the tumor secretome in the immune response when tumor cells become senescent due to chemotherapy treatments.
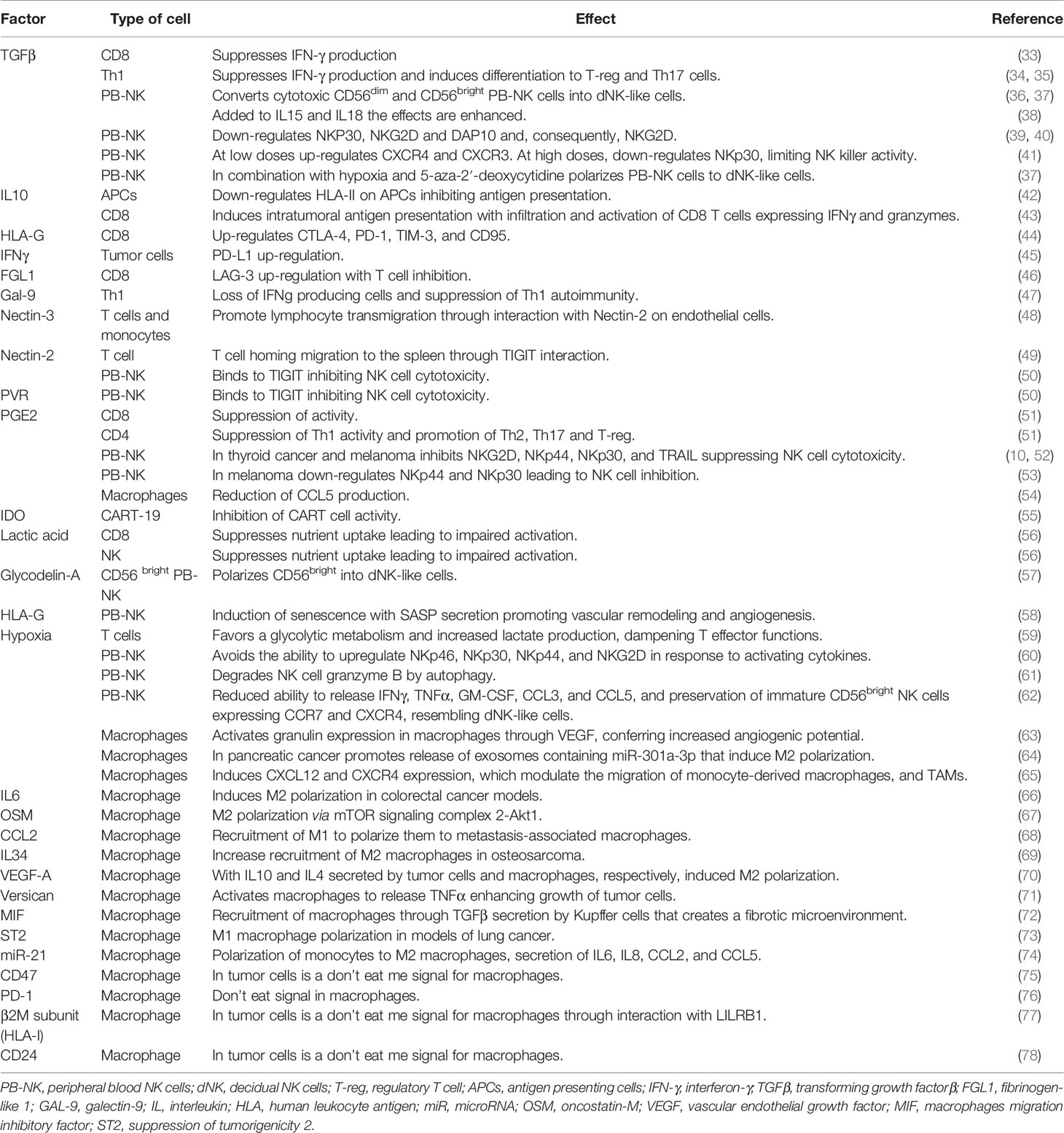
Table 2 Impact of secreted factors in the tumor microenvironment (TME) over the different immune cell populations and description of receptors acting as eat me or don’t eat me signals for phagocytic activity of macrophages.
Tumor cells with stromal cells, endothelial cells, fibroblasts, and immune cells create a suitable TME that favors tumor progression (79–81). The ability of T cells to infiltrate this TME has led to the development of adoptive cellular immunotherapy to treat cancer patients with tumor-infiltrating lymphocytes (TILs) or CAR-T cells (14, 15, 31). Interestingly, the TME can shape the anti-tumor activity of T cells depending on a variety of secreted molecules. We detail here the impact of some of these released factors.
TGF-β, a highly recognized immunosuppressive cytokine secreted by tumor cells (33), suppresses IFN-γ production by Th1 and effector CD8 T cells, inducing the differentiation of CD4 T cells to both regulatory (T-reg) cells and Th17 cells. T-reg cells that also release TFG-β and IL10 will further suppress the activation of CD8 T cells, promoting tumor cell growth (34, 35). IL10 production by tumor cells down-regulates HLA-I and HLA-II on tumor cells and HLA-II on antigen-presenting cells (APCs), inhibiting antigen presentation becoming an escape mechanism from immune surveillance (42, 82–84). On the other side, cancer models have shown that IL10 also induces intratumoral antigen presentation with infiltration and activation of tumor-specific cytotoxic CD8 T cells expressing IFNγ and granzymes (43) (Figure 1).
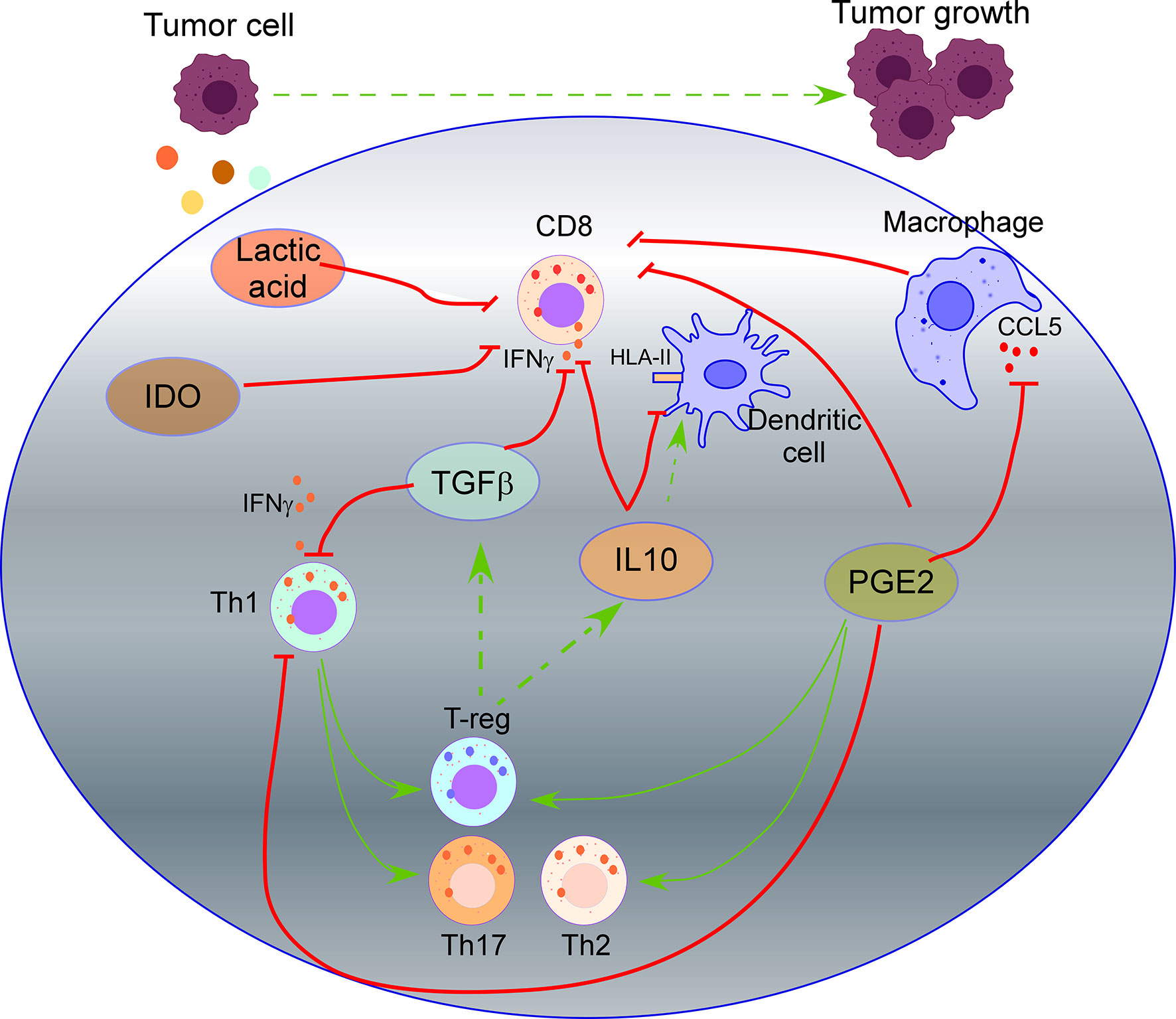
Figure 1 Impact of tumor secretome in T cell activity. TGF-β secreted by tumor cells suppresses IFN-γ production by Th1 and effector CD8 T cells, inducing the differentiation of CD4 T cells to regulatory (T-reg) cells and Th17 cells. T-reg cells also release TFG-β and IL10 that will suppress the activation of CD8 T cells. IL10 secreted by tumor cells down-regulates HLA-II on dendritic cells, inhibiting antigen presentation. Prostaglandin 2 (PGE2) secreted by tumor cells suppresses the functions of CD8 T cells and Th1 cells, and promotes Th2, Th17, and T-reg cell response. PGE2 reduces CCL5 production by macrophages, which is required for T cell proliferation. Secretion of Indoleamine 2,3 dioxygenase (IDO) by tumor cells produces metabolites that inhibit T cell activity. Lactic acid produced by tumor cells suppresses nutrient uptake by CD8 T cells.
A wide field of research in cancer immunotherapy consists of inhibiting immune-checkpoint receptors on immune cells and their ligands in tumor cells. The interaction of these receptors/ligands modulates the activity of immune cells to limit the development of auto-immunity and create immunotolerant T cells. Therefore, the inhibition of these interactions with monoclonal antibodies increases their anti-tumor activity. The most common immune checkpoints include cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed death 1 (PD-1), T cell immunoglobulin and mucin-3 (TIM-3), B and T lymphocyte attenuator (BTLA), lymphocyte activation gene 3 (LAG3), adenosine 2A receptor (A2AR) and T cell immunoglobulin and ITIM domain (TIGIT) (85–91). Secreted molecules by tumor cells impact the expression of immune-checkpoint receptors on immune cells. For instance, release of soluble HLA-G by tumor cells up-regulates CTLA-4, PD-1, TIM-3, and CD95 on CD8 T cells impacting their anti-tumor activity (44). On the other hand, cytokines released by activated immune cells can up-regulate ligands of immune-checkpoints in tumor cells. Thus, IFNγ release by activated T cells induces PD-L1 up-regulation in tumor cells (45) (Figure 2).
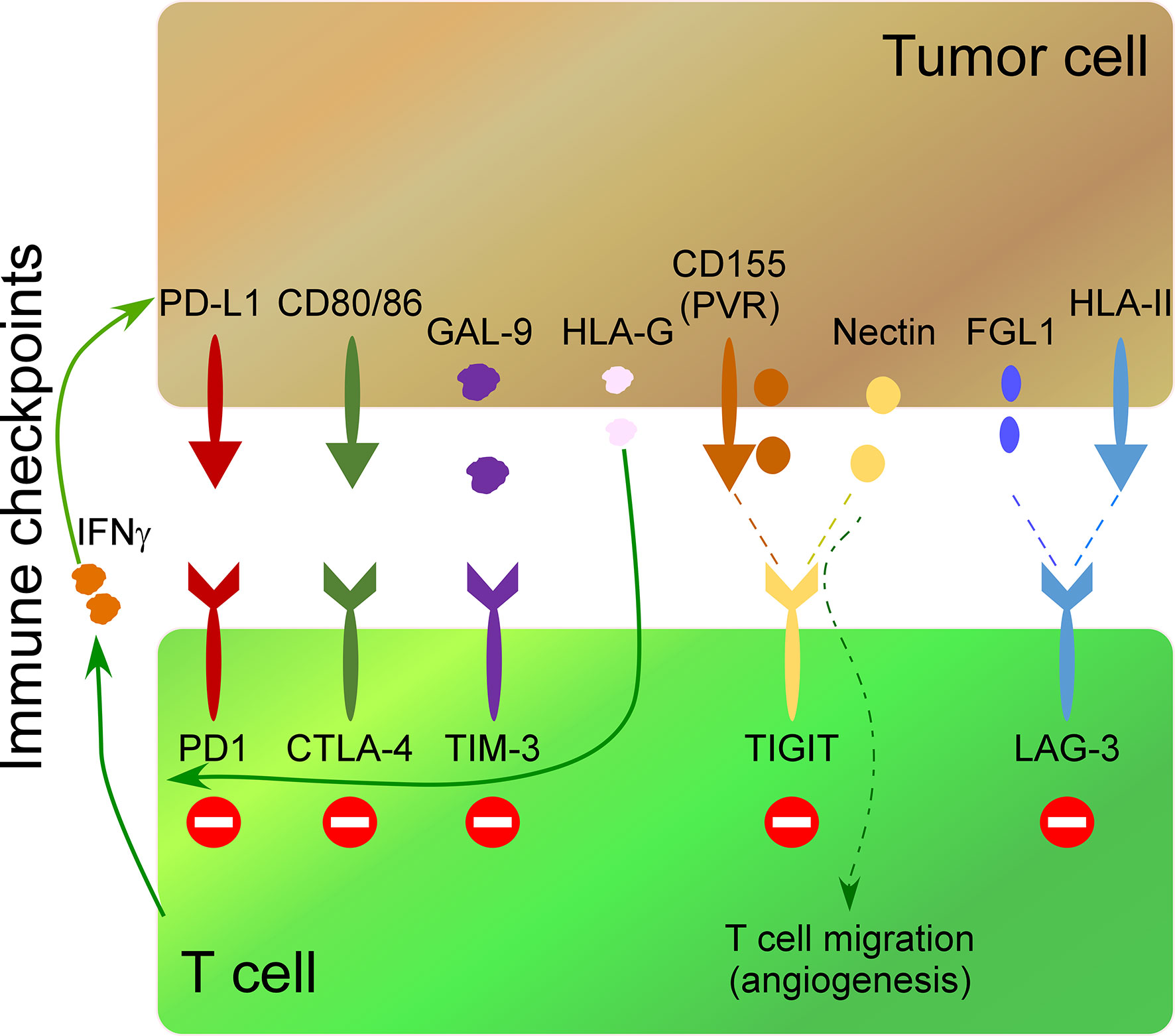
Figure 2 Impact of some secreted molecules in the TME on the expression of immunocheckpoints in T cells. The most common immune checkpoints on T cells include programmed death 1 (PD-1), cytotoxic T lymphocyte antigen 4 (CTLA-4), T cell immunoglobulin and mucin-3 (TIM-3), T cell immunoglobulin and ITIM domain (TIGIT) and lymphocyte activation gene 3 (LAG3), which interact with their ligands on tumor cells. IFNγ release by activated T cells induces PD-L1 up-regulation in tumor cells. TIM-3 interaction on Th1 cells with Galectin-9 (Gal-9) on tumor cells inhibits Th1 cell responses. Soluble HLA-G released by tumor cells up-regulates PD-1, CTLA-4, and TIM-3, on T cells. CD155 (PVR), and the Nectin family are ligands of TIGIT. Soluble PVR is released by tumor cells. Soluble Nectins released by cancer cells mediate transendothelial migration of immune cells promoting angiogenesis. HLA-II over-expression by tumor cells and fibrinogen-like 1 (FGL1) secreted by tumor cells impact the expression of LAG-3 in T cells.
HLA-II over-expression by tumor cells (92) and fibrinogen-like 1 (FGL1), a protein secreted by liver cells and tumor cells (46), are ligands of LAG-3, and their secretion impact the expression of LAG-3 in T cells, promoting an immunosuppressive function. TIM-3 is expressed on Th1 cells, and its interaction with its ligand Galectin-9 (Gal-9) on tumor cells inhibits Th1 cell responses (47) (Figure 2). Both overexpression of Gal-9 on gastric cancer cells and expression of TIM-3 on immune cells correlates negatively with poor outcomes in cancer patients (93) and lead to an increase in granulocytic myeloid-derived suppressor cells that inhibit immune responses impacting tumor growth (94).
TIGIT ligands include CD155 (PVR), and the Nectin family (95, 96) (Figure 2), which are over-expressed in many human malignancies (97). Specifically, soluble PVR is a valuable biomarker for cancer development, where higher soluble PVR levels are detected in lung, gastrointestinal, breast, and gynecologic cancers compared to healthy donors, being even higher at advanced stages of the disease (98). Of interest, Nectins promote the transendothelial migration of cells and associate with poor prognosis and advanced disease stages in different types of cancer (99). Soluble Nectin-4 released by cancer cells interacts with integrin-β4 on endothelial cells, promoting angiogenesis (100). Of interest, Nectins also mediate transendothelial migration of immune cells (48). For instance, Nectin-2 promotes endothelial cell migration, endothelial tube formation, and T cell homing migration to the spleen, promoting an angiogenic function (49); Nectin-3 expressed by T cells and monocytes binds to endothelial cells through Nectin-2 promoting the transmigration of immune cells (48). This angiogenic function of soluble Nectins released by tumor cells suggests an essential role of the tumor secretome polarizing the cytotoxic activity of T cells to an angiogenic one.
Prostaglandin 2 (PGE2) is a crucial mediator of immunopathology in chronic infections and cancer. PGE2 secreted by tumor cells suppresses the effector functions of CD8 T cells and Th1 cells, promotes Th2, Th17, and T-reg cell response, and inhibits the attraction of immune cells (51). Moreover, PGE2 reduces CCL5 production by macrophages (54), which is required for IL2, IFN-γ production, and T cell proliferation (101). Recent studies revealed that COX2/mPGES1/PGE2 pathway in tumor cells up-regulates PD-L1 in tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), which is followed by T cell elimination (102).
In addition, the tumor secretome impacts the metabolic activity of T cells through the competitive removal of essential nutrients for T lymphocytes. In this sense, secretion of “Indoleamine 2,3 dioxygenase” (IDO), which catalyzes tryptophan degradation, produces metabolites that inhibit T cell activity. In a murine lymphoma model with CAR-T cells targeting CD19, over-expression of IDO depleted the anti-tumor activity of CAR-T cells and inhibited the cytokine-dependent expansion of CAR-T cells, cytokine secretion, and increased their apoptosis (55) (Figure 1). Production of lactic acid by tumor cells also inhibits the activity of CD8 T cells and NK cells. In detail, most tumors rely on glycolytic metabolism to sustain rapid cell growth through the enzyme lactate dehydrogenase-A that produces lactic acid. CD8 T cells and NK cells undergo a similar metabolic switch activating a glycolytic metabolism when they evolve from a naive to an activated state. However, highly glycolytic tumor cells are superior competitors for glucose and amino acids than CD8 T cells and NK cells. In addition, lactic acid production further suppresses nutrient uptake by CD8 T cells and NK cells, dampening their metabolic programs, leading to impaired activation of CD8 T cells and NK cells with the subsequent overcoming of immune surveillance by tumor cells (56).
Adoptive cellular immunotherapy administering CAR-T cells has achieved outstanding and permanent responses in pediatric-B cell hematological malignancies with persistence of CAR-T cells over the years (26). However, in other hematological malignancies (15, 27) and solid tumors, results have been more inferior due to a short persistence of CAR-T cells and the barriers that CAR-T cells have to face in the TME, such as the impact of the tumor secretome. Fourth-generation CAR-T cells, termed armored or TRUCK CARs, are equipped with different features that can remodel the TME to enhance the activity of CAR-T cells.
Thus, a variety of armored CAR-T cells that secrete different cytokines have been developed. For instance, CART-19 cells that secrete IL12 show increased cytotoxicity and resistance to T-reg cell-mediated inhibition, better engraftment, and enhanced anti-tumor activity in models of B-cell malignancies (103) and ovarian cancer (104). Of note, severe adverse events were observed in a clinical trial with TILs secreting IL12 (105). Therefore, decreasing the amount of cytokines released by CART cells, in this case, IL12, could be modulated via different gene-expression cassettes, such as promoters in the CAR with inducible nuclear factor of activated T cells (NFAT) binding motifs (106). IL15 enhances the differentiation, homeostasis, and survival of T cells and NK cells. CART-19 cells secreting IL15 demonstrated increased expansion and efficacy, with decreased apoptosis and PD-1 expression, in models of Burkitt lymphoma (107). CAR-T cells secreting IL18 have caused increased M1-polarization in macrophages of the TME, depletion of M2-macrophages and T-reg cells (108), and recruitment of endogenous T cells (109). Nevertheless, as IL18 is pro-inflammatory, it has pathogenic roles in autoimmune diseases (110) and might also promote tumor progression, angiogenesis, immune escape, and metastasis (111). CAR-T cells secreting IL7 and CCL19 have also improved cell infiltration of dendritic cells (DCs) and survival of CAR-T cells (112). In addition, inhibition of TGFβ is achieved by co-expression in the CAR of a dominant-negative receptor for TGFβ that blocks TGFβ signaling, increasing proliferation and persistence of CAR-T cells in models of prostate cancer (113).
Armored CAR-T cells also avoid the negative impact of immune checkpoints. Thus, in lymphoma, the TME is marked by exacerbated lymphoid stroma activation and increased recruitment of follicular helper T cells, resulting from the disruption of the inhibitory checkpoint HVEM/BTLA. Secretion of HVEM by CAR-T cells binds BTLA avoiding this event (114). In addition, CAR-T cells that secrete anti-PD-L1 antibodies prevent T cell exhaustion and recruit NK cells to the tumors (115).
Furthermore, hypoxia is found in the TME and contributes to the rapid growth of tumor cells. Under hypoxia, glucose is fermented to lactate. The hypoxic TME also favors a glycolytic metabolism and increased lactate production, dampening T and NK cell effector functions and survival (59). Thus, armored CAR-T cells that secrete catalase (CAT-CAR) overcome hypoxia and reactive oxygen species (ROS) present in the TME (116). Another option to overcome these obstacles is to modify the CAR to express anti-oxidant factors such as N-acetylcysteine (NAC) that reduces DNA damage in CAR-T cells lowering activation induced-cell death in CAR-T cells (117).
The well-recognized anti-tumor activity of NK cells has led to many clinical studies administering either NK cells or CAR-modified NK cells, although results to date have shown mainly safety but not a high efficacy (18). These findings suggest the need to optimize NK cell anti-tumor efficacy. Here, we present studies that indicate that when NK cells arrive at the TME, events might happen that modify their killer activity.
In this regard, there are two main populations of NK cells in peripheral blood, the mature and cytotoxic NK with CD56lowCD16high expression, which constitutes 90% of NK cells, and the immature and immunoregulatory NK cells characterized by CD56highCD16low/negCD25+ expression, which comprise approximately 10% of peripheral blood (PB)-NK (18, 19). A third transient population, known as decidual NK (dNK) cells, present at the fetal-maternal interface during the first months of pregnancy, representing 70% of immune cells in the decidua. dNK cells are also known as uterine NK (uNK) cells, as classically, uNK cells were detected by Dolichos biflorus agglutinin (DBA) lectin staining, where DBA+ cells were defined as dNK cells. Decidualization is triggered during blastocyst implantation and the menstrual cycle, characterized by a marked increase in dNK cells. dNK or uNK cells are a dynamic population, and their origin is not clear. A recent model proposed that there is a first wave of proliferation of tissue-resident NK cells in the pregnant uterus at the onset of the decidualization process. Then, a second wave involves the recruitment of conventional PB-NK cells during the placentation process (118, 119).
dNK cells are immune-tolerant and characterized by CD56brightCD16−CD9+CD49a+ and Eomes+ expression (120, 121). They are angiogenic, regulate trophoblast invasion and vascular growth during the placental developmental process and cooperate with other cells to serve as constructive elements during early pregnancy. dNK cells produce large amounts of proangiogenic factors, including VEGF, PlGF, CXCL8, IL-10, and angiogenin, critical for decidual vascularization and spiral artery formation (122). dNK cells also express chemokine receptors, including CXCR3, CXCR4, CCR1, CCR9, and the integrin ITGA3 (120), and through the interaction of HLA-G on fetal trophoblast cells with ILT2 and KIR2DL4, they secrete other growth-promoting factors, including pleiotrophin and osteoglycin (121). Moreover, interaction of soluble HLA-G with KIR2DL4 induces a pro-inflammatory response in dNK cells, activating their senescence with SASP secretion that promotes vascular remodeling and angiogenesis in early pregnancy (58).
This “nurturing” role of dNK cells during early pregnancy presents many homologies to NK cells infiltrated in different types of tumors. Thus, a subset of NK cells in non-small cell lung cancer, squamous cell carcinoma, or colorectal cancer turns into dNK-like cells inducing human umbilical vein endothelial cell migration and formation of capillary-like structures (36, 123–125). Various studies have tried to determine different factors during early pregnancy that might be responsible for this polarization of PB-NK cells into dNK-like cells. Results suggest that this polarization seems more specific for CD56bright than for CD56dim NK cells. Of interest, NK cells administered in immunotherapy treatments undergo an in vitro expansion that turns them into CD56bright NK cells (17). Many of the factors responsible for this NK polarization are present in both the decidua and the TME, suggesting that these events occurring in the TME might impact the growth of tumor cells. In the next section, we detail the effect of secreted factors in the TME over the phenotype and polarization of NK cells.
Glycodelin-A is secreted in large amounts in the decidua and by tumor cells in malignancies, such as Non-Small Cell Lung Cancer (126), mesothelioma (127), ovarian cancer (128), and endometrial cancer (129). Glycodelin-A converts immunoregulatory CD56bright PB-NK cells into dNK-like cells, an effect that does not occur for mature CD56low PB-NK cells. This mechanism occurs through binding of Glycodelin-A to sialylated glycans on CD56bright NK cells and causes enhanced expression of CD9, CD49a, and production of VEGF and IGFBP-1 that regulate endothelial cell angiogenesis and trophoblast invasion (57).
Soluble HLA-G is associated with bad prognosis in different tumors (130–134). Of interest, soluble HLA-G mediates polarization of PB-NK cells to dNK-like cells, with a senescent phenotype, secretion of growth factors, and reduced killer activity (58), thus, emerging as an essential target that can polarize the activity of NK cells.
TGFβ secretion can be beneficial at early stages and detrimental at late-stage tumor development by remodeling the TME to favor tumor growth (130, 135). TGFβ converts both cytotoxic CD56dim and CD56bright PB-NK cells into dNK-like cells (36, 37) (Figure 3). Moreover, IL15 and IL18 added to TGFβ enhance the impact on the polarization of PB-NK cells toward a dNK cell phenotype with increased expression of CD9, CD49a, ITGA3, and CXCR4 (38). Of interest, as previously mentioned, IL15 and IL18 are beneficial for CAR-T cells (107–109), suggesting the negative role of these cytokines when TGFβ is added. Additional effects of TGFβ over NK cells include down-regulation of NKP30, NKG2D (39), and DAP10 and, consequently, NKG2D (40) inhibiting NK cell function (Figure 3). Of interest, this dual role of TGFβ in the TME is observed when at low doses facilitates NK cell recruitment to the tumor by up-regulating CXCR4 and CXCR3, markers of dNK; and at high doses, down-regulates NKp30, limiting NK killer activity (41).
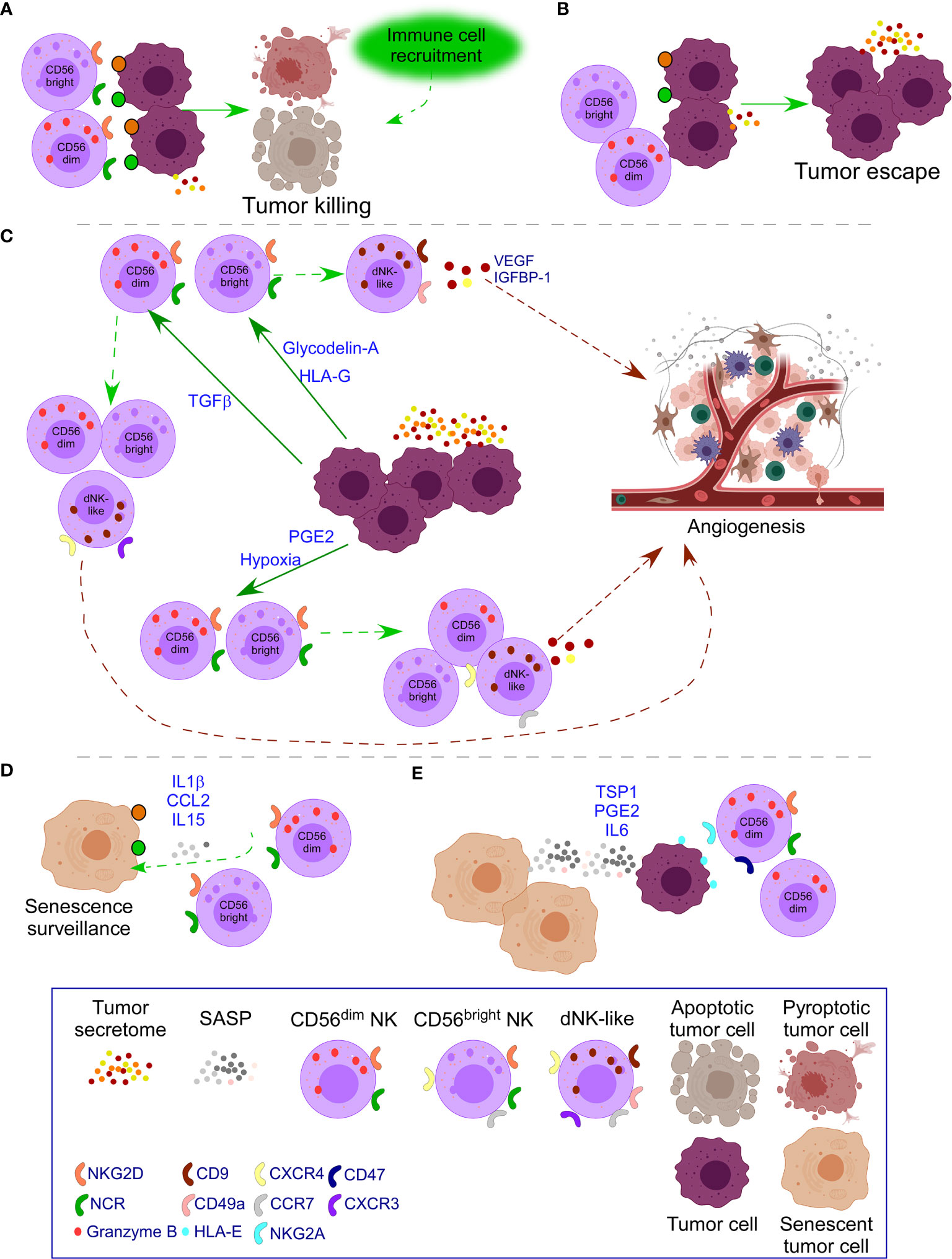
Figure 3 Impact of tumor secretome in NK cell activity. (A) In healthy conditions, NK cells recognize transformed cells through ligands of NKG2D and the family of NCR receptors (NKp30, NKp44, NKp46) which are over-expressed in transformed cells. Pro-inflammatory forms of cell death attract additional immune cells to cooperate in the killing. (B) In some cases, tumor cells down-regulate ligands for NK cell receptors or the tumor microenvironment (TME) causes down-regulation of activating NK cell receptors leading to tumor escape with additional secretion of tumor secretome. (C) When tumor escape occurs, increased tumor secretome leads to additional changes in NK cells. Specifically, release of Glycodelin-A and HLA-G converts immunoregulatory CD56bright PB-NK cells into dNK-like cells. TGFβ converts both cytotoxic CD56dim and CD56bright NK cells into dNK-like cells; and down-regulates NK cell activating receptors limiting NK killer activity. PGE2 and hypoxia inhibit the expression of NK cell activating receptors and their functional maturation leading to suppressed NK cell cytotoxicity. Moreover, hypoxia, preserves immature CD56bright NK cells with expression of receptors of dNK cells, resembling to dNK-like cells. In all cases, dNK-like cells will activate angiogenesis processes. (D) Emergence of senescent tumor cells leads to SASP secretion that attracts NK cells to mediate their clearance. (E) When the number of senescent cells increases, the SASP also does, leading to inhibition of NK cell activity, through mechanisms, such as the interaction of HLA-E with the inhibitory receptor NKG2A in NK cells and binding of TSP1 with CD47 that inhibit NK cell activity. PGE2 and IL6 in the SASP also down-regulate NK cell activating receptors. Moreover, therapy-induced senescence in established tumors down-regulates NK cell activating receptors on mature NK cells and their ligands on tumor cells.
PGE2 secretion in thyroid cancer and melanoma inhibits the expression of NKG2D, NKp44, NKp30, and TRAIL on PB-NK cells and their functional maturation leading to suppressed NK cell cytotoxicity (10, 52) (Figure 3). PGE2 release by cancer-associated fibroblasts in melanoma down-regulates NKp44 and NKp30 leading to NK cell inhibition (53). Soluble PVR and Nectin-2 released by tumor cells bind to TIGIT on NK cells inhibiting NK cell cytotoxicity (50).
Hypoxia is another factor present in both the decidua and the TME. Hypoxia in the TME avoids the ability of NK cells to upregulate NKp46, NKp30, NKp44, and NKG2D in response to activating cytokines (60) and degrades NK cell granzyme B by autophagy (61), impairing the ability to kill and promoting immune evasion (Figure 3). Moreover, exposure to a combination of hypoxia, TGFβ, and 5-aza-2′-deoxycytidine, results in the polarization of PB-NK cells to dNK-like cells. These changes are more pronounced when all the factors are together and lead to the expression of CD9, CD49a, chemokine receptors, and VEGF secretion that leads to dNK-like cells with capacity to promote invasion of trophoblast cell lines and reduced cytotoxicity. Significantly, these parameters are reversed after returning to normal conditions, indicating the plasticity of immune cells (37). Exposure of PB-NK cells to hypoxia also causes reduced NK cell ability to release IFNγ, TNFα, GM-CSF, CCL3, and CCL5, and preservation of immature CD56bright NK cells expressing CCR7 and CXCR4, resembling dNK-like cells (62).
The impact of these tumor secreted factors occur mainly on CD56bright PB-NK cells, and NK cells used in immunotherapy undergo an in vitro expansion that turn them into CD56bright NK cells (17). These events suggest that in cases that NK cells do not achieve complete removal of tumor cells they might have polarized into dNK-like cells. Therefore, monitoring these changes in immunotherapy NK cell studies will provide relevant information to improve the clinical outcome of patients.
Macrophages are innate immune cells with high plasticity which traditionally, have been classified as two extremes being either pro-inflammatory (M1: activated) or anti-inflammatory (M2: alternatively activated). M1 inhibits cell proliferation and causes tissue damage, while M2 promotes cell proliferation and tissue repair. M1 and M2 enable Th1, and Th2 responses, respectively, and consequently, Th1 and Th2 cytokines regulate their activity. Thus, M1 responds to IFN-γ, TNF-α, and TLR4 activation, and M2 to IL-4 and IL-13 (136). However, macrophages present high plasticity and convert to a wide variety of subpopulations depending on the stimuli they receive from the TME (63, 137). Macrophages represent the largest population of all infiltrating leukocytes in the tumor (138), where tumor-associated macrophages (TAMs), which present an M2-like phenotype, are considered highly responsible for tumor progression, and many studies have focused on trying to polarize M2-like macrophages to M1 (139). However, M2 are the macrophages with the highest phagocytic activity against apoptotic tumor cells (140), suggesting that removing this activity might also be detrimental. Therefore, efforts should be directed to preserve M1 macrophage activity while also enhancing the phagocytic activity of M2 macrophages. Here, we will pay special attention to the phagocytic function of M2 macrophages to remove tumor cells and how secreted molecules in the TME can polarize macrophages to an M2-like or M1 phenotype.
Phagocytosis of tumor cells by macrophages is performed after recognizing “eat me” or don’t eat me” signals that will or will not trigger phagocytosis. “Eat me” and “don’t eat me” signals act as ligands for phagocytic receptors that will or will not trigger the engulfment of the target. Different studies have shown the beneficial impact in tumor regression of inhibiting these “don’t eat me” signals. For instance, CD47 expression in small-cell lung cancer cells engages SIRPα on macrophages inhibiting their phagocytic activity, which is recovered with an anti-CD47 (75). Moreover, inhibition of CD47 in tumor cells promoted their phagocytosis and the anti-tumor activity of CD8 T cells while inhibiting T-reg cells (141). Blocking PD-1 expressed in TAMs or M2-like macrophages increases macrophage phagocytosis and reduces tumor growth (76) (Figure 4).
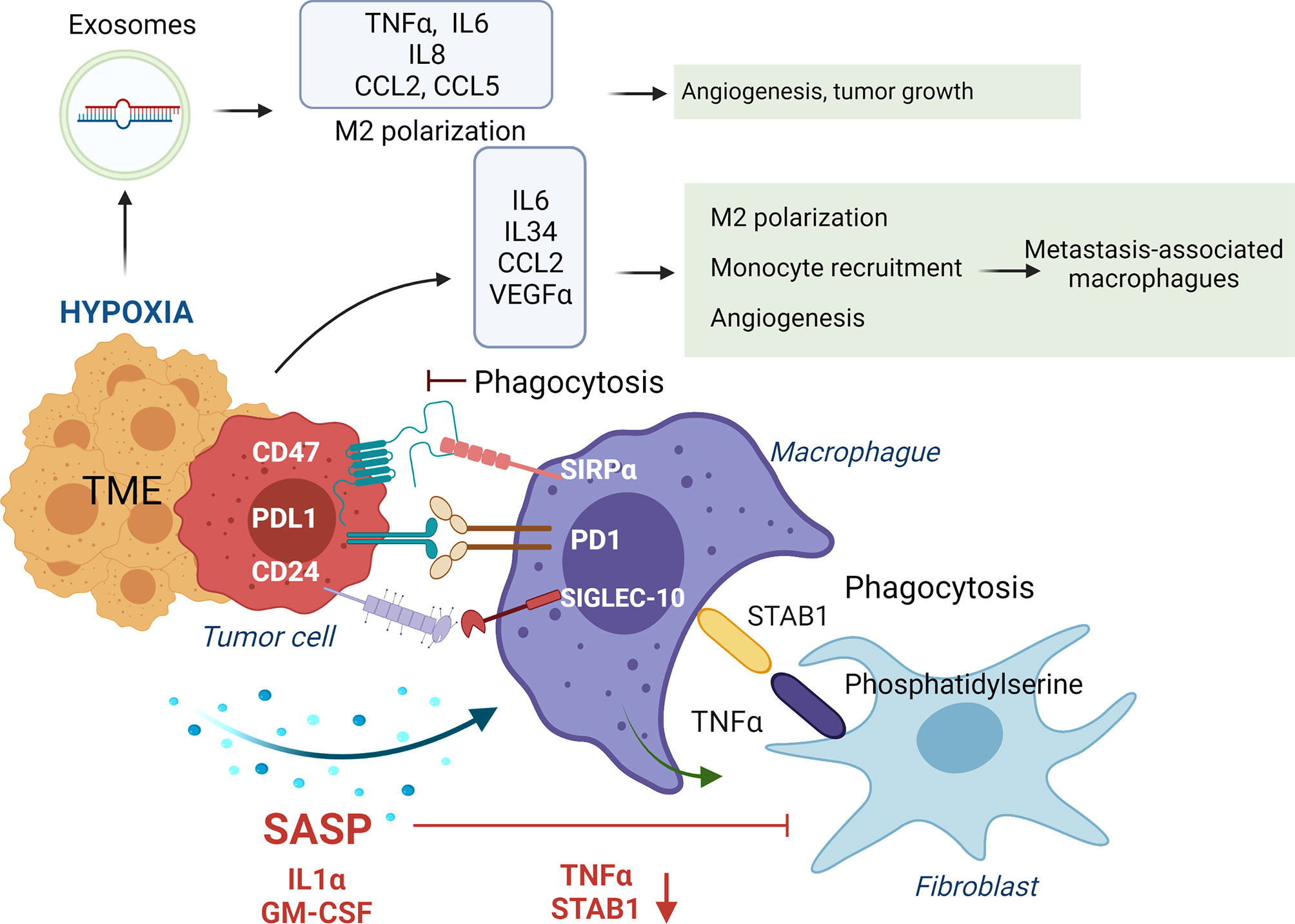
Figure 4 Impact of tumor secretome in the phagocytic activity of macrophages. In healthy conditions macrophages phagocyte transformed cells and senescent fibroblasts to maintain tissue homeostasis. Normally, macrophages, through release of TNFα, induce apoptosis in senescent fibroblasts, leading to expression of phosphatidylserine in their surface, which is recognized by STAB1 on macrophages to promote their phagocytosis. In advanced stages of senescence, phagocytic activity of macrophages is inhibited by over-expression of ligands of immune-checkpoints (CD47, PDL-1 and CD27) that interact with their receptors on macrophages (SIRPα, PD1 and SIGLEC-10). Moreover, SASP factors, including IL1α and GM-CSF, down-regulate STAB1 and TNFα expression, avoiding the phagocytosis of senescent fibroblasts by macrophages. In addition, IL6, IL34, CCL2 and VEGFa secretion in the TME, induce M2 macrophage polarization and recruitment of inflammatory monocytes that polarize to metastasis-associated macrophages that in summary promote tumor growth. Hypoxia in established tumors also promotes the release of exosomes containing the miRNAs miR-301a-3p and miR-21 that promote M2 polarization, and TNFα, IL6, IL8, CCL2 and CCL5 secretion impacting in higher angiogenesis, and tumor growth.
Furthermore, anti-PD-L1 treatment reverses the immunosuppressive status of the TME and enhances specific T cell anti-tumor effects in murine models of cancer (142). Interaction of β2M subunit of HLA-I in tumor cells with LILRB1 on macrophages protects tumor cells from phagocytosis by TAMs, and disruption of this interaction potentiates phagocytosis of tumor cells (77). In ovarian cancer and triple-negative breast cancer, tumor cells evade clearance by macrophages through over-expression of CD24 that interacts with Siglec-10 in TAMs, and its blockade augments the phagocytosis of CD24-expressing tumors leading to a reduction of tumor growth (78). Dectin-2, a C-type lectin receptor in macrophages resident in the liver (Kupffer cells), promotes phagocytosis of cancer cells, avoiding liver metastasis (143) (Figure 4).
Phagocytosis requires an intimate contact of the macrophage and the target, where the glycocalyx, a layer that surrounds the plasma membrane containing glycolipids, glycoproteins, and surface-associated glycosaminoglycans, acts as a barrier for these contacts. The size and charge of this glycocalyx can be modified and modulated by enzymes or other molecules present on the TME to promote phagocytosis (144). Of interest, we described that NK cells release histones that bind to and degrade the syndecans on the glycocalyx of multiple myeloma cells (145), suggesting that by doing this, NK cells might also promote the phagocytosis of tumor cells by macrophages, an event observed during fungal infection (146).
Molecules secreted in the TME will also impact promoting an anti-inflammatory or pro-inflammatory environment that will polarize macrophages into TAMs/M2-like or M1 phenotypes. For instance, IL6 secretion in the TME induces M2 macrophage polarization in colorectal cancer models (66). The release of oncostatin M in the TME is involved in M2 polarization via mTOR signaling complex 2-Akt1 (67). At breast cancer, the release of CCL2 by tumor cells recruits inflammatory monocytes that polarize to metastasis-associated macrophages, which secrete CCL3, promoting lung metastasis (68). IL34 secretion by tumor cells binds to CSF1R in macrophages and polarizes them to M1 and M2 (147). Also, IL34 contributes to osteosarcoma growth by increasing the neo-angiogenesis and recruitment of M2 macrophages (69). In a skin carcinogenesis model, VEGF-A expression on tumor cells with IL10 and IL4 secreted by tumor cells and macrophages, respectively, induced M2 polarization that promoted tumor growth (70). Release of the proteglycan versican by lung carcinoma cells activates macrophages to release TNFα enhancing growth of tumor cells (71) (Figure 4).
Tumor hypoxia, a feature of the TME, promotes ID4 expression in cancer cells which, through VEGF, activates increased expression of granulin in macrophages, conferring increased angiogenic potential (63). In pancreatic cancer cells, the presence of hypoxia promotes the release of exosomes containing the miRNA miR-301a-3p that binds to TLR macrophages receptors, promoting M2 polarization, TNFα, and IL6 production, creating a pro-metastatic environment (64). Hypoxia induces CXCL12 and CXCR4 expression, which modulate the migration of monocytes, monocyte-derived macrophages, and TAMs (65) (Figure 4). Of interest, when hypoxia is absent in tumor cells, TAMs can enhance tumor hypoxia and glycolysis (148), being both features that promote tumor aggressiveness (149).
Exosomes released in liver tumors bind to macrophages through exosome integrins and prepare the pre-metastatic niche (150). In pancreatic ductal adenocarcinomas, tumor-derived exosomes with macrophage migration inhibitory factor are taken by Kupffer cells causing TGFβ secretion. Consequently, a fibrotic microenvironment emerges that recruits macrophages, creating a liver pre-metastatic niche (72). Of interest, the release of ST2 in Rab37 exosomes skewed M1 macrophage polarization leading to reduced tumor growth in models of lung cancer. Moreover, lung cancer patients with low Rab37, low soluble ST2, and low M1/M2 ratio presented worse overall survival (73). SNAIL, a transcription factor expressed during epithelial-mesenchymal transition, activates the production of tumor-derived exosomes containing miR-21 that will be phagocyted by monocytes leading to M2 macrophages, secretion of IL6, IL8, CCL2, and CCL5 impacting in higher angiogenesis, and tumor growth (74) (Figure 4).
Studies have demonstrated that chemotherapy treatment can lead to acquired resistance and the emergence of more aggressive tumor cells. In this regard, the tumor secretome is shaped by chemotherapy treatment that will impact the immune response and increase tumor aggressiveness. For instance, in breast cancer, IL6 release after treatment converts differentiated tumor cells to cancer stem cells through the IL6-JAK1-STAT3 pathway (151). In non-small cell lung cancer, cisplatin induces IL6 secretion that increases tumor progression and resistance to treatment through up-regulation of anti-apoptotic proteins and DNA repair associated genes (152). Paclitaxel enhances IRE1 RNase activity that leads to the production of IL6, IL8, CXCL1, GM-CSF, and TGFβ2 in breast cancer cells contributing to the expansion of tumor-initiating cells (153). Doxycycline treatment in squamous cell carcinoma leads to TGFβ secretion that activates the TGF-β/SMAD pathway increasing tumorigenic potential (154). Treatment with kinase inhibitors causes secretion of positive mediators of the AKT pathway, including IGF1, EGF, ANGPTL7, and PDGFD, accelerating the expansion and dissemination of drug-resistant clones (155). Docetaxel induces secretion of extracellular vesicle-encapsulated miRNAs, including miR-9-5p, miR-195-5p, and miR-203a-3p, which down-regulate the transcription factor ONECUT2, leading to up-regulation of stemness-associated genes, that stimulate cancer stem-like cells and resistance to therapy in breast cancer (156).
In addition, chemotherapy and radiotherapy treatments trigger a premature state of senescence in tumor cells termed “therapy-induced senescence” (TIS) that will shape the tumor secretome (29, 157). TIS can reactivate the cell cycle and bring on cancer daughter cells that survive therapy more transformed than the original population (158, 159). This secretome is unique because it is induced by senescence, being termed senescence-associated secretory phenotype (SASP). SASP includes various cytokines, chemokines, growth factors, and matrix metalloproteinases, such as IL1α, IL1β, IL6, IL8, CXCL1, CCL2, VEGF, and CXCR2 (29, 160, 161), that interfere with the paracrine activity of senescent cells. Of interest, SASP released by tumor cells after TIS induces transmission of senescence to non-senescent neighboring cells (162, 163). The SASP can foster an immunosuppressive environment favoring metastasis (160), and on the other side, attracts immune cells including macrophages, neutrophils, and NK cells to remove senescent cells, a process known as “senescence surveillance” (164–167).
Moreover, cancer is associated with aging. A physiological consequence of aging is the development of immunosenescence due to a functional degradation of the thymus, resulting in decreased functional naïve CD4 and CD8 T cells and a peripheral oligo-clonal expansion of memory T cells. These events provide a contracted T cell antigen receptor (TCR)-repertoire diversity with secretion of SASP (29, 168). Immunosenescence associated with aging also occurs due to exposure to virus infections or chronic inflammation (169); and additional factors such as nutrition, sex, genetics, previous diseases, or tumors (170, 171). Therefore, the immune cells of elderly cancer patients will probably be already senescent; and moreover, SASP secretion by senescent tumor cells after chemotherapy will accelerate this immunosenescence process (171). Here, we will mention some SASP factors released by tumor cells that impact the anti-tumor immune response.
Studies have shown a significant accumulation of senescent T cells in certain types of cancer patients (172), and that tumor SASP induces T cell senescence leading to suppression of responses of naïve/effector T cells (173), suggesting that this might be a strategy used by malignant cells to evade immune surveillance. Transformed senescent T cells are in cell cycle arrest and develop significant phenotypic alterations, such as down-regulation or loss of CD27 and CD28.Through SASP factors including pro-inflammatory cytokines or inhibitory molecules like IL10 or TGFβ, senescent T cells will amplify the immunosenescence process. Moreover, the development of exhaustion with high expression of immune checkpoints, such as TIM-3 and other co-inhibitory receptors as CD57 or KLRG-1, will promote replicative senescence of T cells (174).
TGFβ1 and TGFβ3 are early SASP factors that regulate thymic T cell homeostasis, inhibit cytotoxic T cell proliferation, and promote T-reg generation (175). Tumor senescent cells up-regulate NOTCH1 and drive a TGFβ-rich secretome that suppresses the release of a pro-inflammatory SASP and contributes to the transmission of senescence through cell-cell interaction via NOTCH-JAG1 pathway. Of interest, NOTCH1 inhibition recovers the secretion of pro-inflammatory cytokines, promoting lymphocyte recruitment and senescence surveillance (176). Senescent cells, after genotoxic stress, secrete IL6 and IL8 that promote epithelial-mesenchymal transition, increasing tumor cells’ invasiveness. Moreover, IL6 recruits myeloid cells that inhibit T cell responses (177).
MAPK signaling is a relevant pathway that controls T cell senescence (178) through activation of p53, p21, and p16 (179). Recent research demonstrated that tumor-derived T-reg cells exhibit an accelerated glucose uptake, competing with effector T cells for glucose through TLR8 signaling, leading to MAPK activation, which induces T cell senescence (180). Another study showed that T-reg cells, through p38, ERK1/2 signaling, p16, p21, and p53 induce senescence in responder naïve and effector T cells. This event is reverted by the block of TLR8 signaling and or by specific ERK1/2 and p38 inhibition (181). Moreover, the p53 isoforms Δ133p53 and p53β regulate proliferation and senescence in human T lymphocytes. Thus, decreased Δ133p53 and increased p53β expression in healthy individuals and lung cancer patients associated with age-dependent accumulation of senescent CD8 T cells (182).
The hypoxic TME leads to the accumulation of adenosine and tumor-derived cAMP. This cAMP is a SASP factor that induces T cell senescence in naïve/effector T cells. Of interest, activation of TLR8 signaling in tumor cells reverses this event resulting in enhanced anti-tumor immunity (183). Moreover, the accumulation of adenosine in the TME also inhibits the anti-tumor activity of T cells through the adenosine receptor A2AR, which in healthy conditions regulates immune cells protecting from inflammatory damage (184).
Whereas the immunosenescence process has been widely studied in T cells, there is a lack of information related to CAR-T cell senescence. It could be exciting to delve into the mechanisms of senescence of this type of cells to find pathways to inhibit senescence without impacting their anti-tumor activity. Specifically, CAR-T cells undergo a significant in vitro expansion (185) to obtain enough CAR-T cells to treat the patients. This expansion might impact the development of senescence due to continuous in vitro proliferation. Moreover, the transfer of senescence from tumor cells in the TME mediated by cell-cell contact or through factors present in the SASP will impact CAR-T cell activity. CAR-T cells can be engineered to avoid these events. Thus, recently, CAR-T cells have been used as senolytic agents in lung adenocarcinoma to remove chemically induced senescent cells by targeting the urokinase-type plasminogen activator receptor (186).
A tempting option that could be tested is to reverse early-stage senescent CAR-T cells by blocking critical mediators of this process, such as proteins involved in the DDR, p38, p53, p21, or ATM (187). However, these changes could also decrease T cell functionality by impacting other relevant functions. For instance, p38 is involved in the induction of senescence and IFNγ and TNFα secretion (188), and its inhibition have diminished these cytokines in different inflammation or virus infection models (189, 190). Moreover, blockage of DDR and p53 involves a risk of DNA damage on T cells that might induce malignancy (191).
NK cells have an essential role in the senescence surveillance of tumor cells. Senescence surveillance is initiated by the SASP that activates immune cells to clear senescent cells preventing tumor initiation (167), where both macrophages and NK cells have an important task (32, 192, 193). Proteins present in the SASP, such as CCL2, attract PB-NK cells to remove senescent cells through NKG2D (194). Of interest, this role of PB-NK cells removing senescent cells is also observed by decidual uterine NK cells to control embryo implantation. Specifically, dNK cells after being activated by IL15, present in the SASP, target and clear decidual cells that became senescent in an IL8 dependent manner. This mechanism of NK cells is mediated through granule exocytosis and involvement of NKG2D (195).
SASP secretion by senescent tumor cells up-regulates HLA-E, the ligand of the inhibitory NKG2A NK receptor (196), and cleave NKG2D ligands inhibiting NK cell activity (197).
Soluble Thrombospondin-1 (TSP1), released in the SASP, is involved in Ras-induced senescence (198). Moreover, TSP1 released by tumor cells binds CD47 on NK cells inhibiting its activity (199). CD47 is described as a relevant modulator of NK cell function in virus infection (200). Of interest, after TIS, binding of soluble TSP1 to CD47 causes emergence of tumor-resistant cells and metastasis in triple-negative breast cancer (201), and inhibits anti-melanoma NK cell activity with reduced granzyme B and IFNγ production (202) (Figure 3).
IL1β is another crucial molecule present in the SASP with a relevant pro-tumor activity (203). In detail, IL1 signaling controls the SASP production (204), and transmission of IL1β to neighboring cells induces cell senescence (205, 206). A dual role for IL1β is observed in NK cell activity. For example, IL1β is required by CD56bright NK cells to produce IFNγ (207) to activate pyroptosis, necessary for the anti-microbial (208) and anti-tumor (145) activity of NK cells. In addition, IL1β released by M1 macrophages increases NK cell cytotoxicity up-regulating NKp44 and NKG2D and triggering IFNγ production by NK cells. Of interest, these IL1β-primed NK cells can reverse M2 macrophage polarization (209). On the other side, a negative impact of IL1β has been described over NK activity. Thus, tumor-derived IL1β induces accumulation of MDSCs that impair NK cell development and functions (210). Moreover, a higher secretion of IL1β in endometrial cancer patients compared to healthy tissues correlates with infiltrating CD56bright NK cells in the tumor with exhausted phenotype, indicated by TIGIT and TIM3 expression (211).
IL6 and IL8, present in the SASP, favor the acquisition of migration/invasion and stem-like features, increasing tumor aggressiveness in breast cancer cells (212, 213). Moreover, IL6 also inhibits NK cytotoxic activity by down-regulating perforin and Granzyme B (214). In esophageal squamous cell carcinoma, tumor cells activate the STAT3 pathway on NK cells through IL6 and IL8, leading to down-regulation of NKp30 and NKG2D on NK cells and tumor progression (215) (Figure 3). In addition, increased levels of IL6 in the peritoneal fluid of endometriosis patients reduced the cytolytic activity of NK cells with down-regulation of granzyme B and perforin (216). IL8 activates and recruits immune cells (217) but also has tumor-promoting functions (218). IL8 is produced by CD56 bright NK cells (219), and stimulation with IL18 and IL12 induces higher IL8 production by NK cells (220).
PGE2 secretion, present in the tumor secretome, inhibits NK cell activity (10, 52, 53). Moreover, PGE2 is also present in the SASP at early tumorigenesis stages, secreted by COX-2, a critical regulator of the SASP, and promotes senescence surveillance (221) (Figure 3).
Senescent cells show high ROS levels and lactate production that induce and maintain cell senescence (222, 223). ROS can present contradictory effects on the activity of NK cells. Specifically, lactate production by metastatic colorectal cancer cells induces mitochondrial stress, increased ROS, and apoptosis in NK cells (224). On the other side, ROS is required for the anti-tumor activity of NK cells (225). Moreover, TIS up-regulates NKG2D ligands (MICA, MICB, and PVR) in an oxidant-dependent manner, resulting in enhanced NK cell activity against myeloma cells (226). This up-regulation of NKG2D ligands upon oxidative stress was also observed in colon carcinoma cells, leading to improved NK cell killing (227). However, in established tumors, ROS down-regulates NKp46 and NKG2D on mature CD56dim NK cells inducing suppression of NK activity against melanoma (228) and acute myeloid leukemia cells (229). Of interest, we previously observed that cord blood-derived NK cells reduce ROS levels in multiple myeloma cells (230). This negative role of ROS in tumors has led to antioxidant treatments in cellular immunotherapy studies. For instance, as previously mentioned, in solid tumors, CAR-T cells modified to express the enzyme catalase presented an anti-oxidant capacity to protect bystander T cells and NK cells (116).
All these studies suggest the beneficial and detrimental role of the SASP at early and late stages of tumorigenesis, respectively. As high levels of SASP inhibit NK cell activity, a strategy to treat advanced cancer patients with cellular immunotherapy, could be to administer senescence inhibitors to decrease the number of senescent cells. Once reduced levels of SASP are achieved, immune cells could be administered, that would be attracted to remove the remaining senescent tumor cells.
Macrophages are attracted and stimulated by SASP factors including MCP-1, MIP-1α, and GM-CSF to remove senescent cells (231). Macrophages are also affected by age-related immunosenescence and the consequences of inflammaging, a chronic inflammation occurring with aging, leading to macrophage dysfunction. Increased levels of A20, a suppressor of the NFκB and MAPK signaling, mediated this dysfunction, leading to poor NFκB and MAPK activation following TLR stimulation (232).
There is a disparity in the impact of TIS and the SASP in macrophage polarization and their phagocytic activity. Thus, in a model of skin aging, macrophage activity is inhibited when there are a high number of senescent cells (233). Specifically, through TNFα release, macrophages induce apoptosis in senescent fibroblasts, leading to the expression of phosphatidylserine on their surface. Phosphatidylserine is recognized by the STAB1 receptor on macrophages to promote their phagocytosis. However, SASP factors, including IL1α and GM-CSF, down-regulate STAB1 and TNFα expression, avoiding the killing and phagocytosis of macrophages, with no impact observed in the macrophage polarization (233).
In a model of thyroid cancer, monocytes exposed to conditioned media from senescent thyrocytes and thyroid tumor cells, undergo M2-like polarization displaying tumor-promoting. These events were related to the production of PGE2 (234). In liver fibrosis and cirrhosis, hepatic stellate cells made senescent by carbon tetrachloride treatment produce cytokines that recruit M1 macrophages, promoting a tumor-suppressive environment. However, in the absence of p53, a promoter of senescence, the released secretome induces M2 polarization, enhancing premalignant cells’ proliferation (235). In a model of pancreatic cancer with oncogene-induced senescence, the SASP factor CXCL1 activates CXCR2 that leads to recruitment of M1 macrophages, inhibiting carcinogenesis. However, oncogene-induced senescence and SASP are bypassed at late stages, and M2 macrophages are recruited to enhance the proliferation of the transformed pancreatic cancer cells (236).
Finally, we call the reader’s attention to the type of cell death activated in tumor cells after the attack of immune cells in adoptive cellular immunotherapy. Inflammatory forms of cell death include pyroptosis, which activates the NLRP3 inflammasome, leading to IL1β production (237). As previously mentioned, IL1 signaling controls the SASP production (204). Of interest, CAR-T cells and NK cells used in adoptive cellular immunotherapy activate pyroptosis when they encounter the tumor cell (145, 238). These events suggest that the consequences of this IL1β release should be considered. Expressly, inflammasome activation and pyroptosis execution represent a double edge-sword in cancer immunotherapy, as on one side, pyroptosis executes cell death. On the other side, pyroptosis and IL1β production activate multiple signaling pathways and inflammatory mediators that promote tumor growth and metastasis in cancer models (239, 240), triggering TAMs to boost tumor angiogenesis (241). Moreover, the role of pyroptosis is highly relevant to attracting other immune cells through IL1β and IL18 secretion. These events are observed in microbial infections, where pyroptosis attract immune cells to kill the previously trapped pathogen and remove the infected cell (208, 242). In adoptive cellular immunotherapy, removing dead tumor cells after being killed by immune cells is required, suggesting an advantage of pyroptosis in this context.
To conclude, adoptive cellular immunotherapy has emerged as a promising treatment to treat cancer patients in the last years. However, results still need to be improved in a variety of malignancies. Immune cells present a high capacity of plasticity when they receive stimuli from secreted molecules in the TME. Thus, if immune cells do not remove tumor cells, tumor secretome could modify their killer activity to an angiogenic or immunosuppressive one. A highly relevant aspect that needs to be considered to avoid these events is an efficient removal by macrophages of dying/dead tumor cells after the attack of immune cells, such as NK cells or CAR-T cells. Of interest, NK cells present additional functions to their classic killer activity that might help in this tumor cell surveillance. Inflammatory forms of cell death activated by in vitro expanded immune cells might also impact these processes. In summary, to achieve complete and permanent responses in cancer patients treated with adoptive cellular immunotherapy, all these aspects together need to be considered and count on the activity of the whole immune response and not just one immune cell population.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This research was funded by Fondos Feder with a grant of the Institute of Health Carlos III, grant number PI20/00991.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Some sections of the Figures were made with Biorender.
1. Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
2. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-Related Inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
3. Kumar S, Metz DC, Ellenberg S, Kaplan DE, Goldberg DS. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter Pylori Infection: A Large Cohort Study. Gastroenterology (2020) 158:527–36.e7. doi: 10.1053/j.gastro.2019.10.019
4. Waldner MJ, Neurath MF. Colitis-Associated Cancer: The Role of T Cells in Tumor Development. Semin Immunopathol (2009) 31:249–56. doi: 10.1007/s00281-009-0161-8
5. van’t Klooster CC, Ridker PM, Hjortnaes J, van der Graaf Y, Asselbergs FW, Westerink J, et al. On Behalf of the UCC-SMART Study Group. The Relation Between Systemic Inflammation and Incident Cancer in Patients With Stable Cardiovascular Disease: A Cohort Study. Eur Heart J (2019) 40:3901–9. doi: 10.1093/eurheartj/ehz587
6. Heikkilä K, Harris R, Lowe G, Rumley A, Yarnell J, Gallacher J, et al. Associations of Circulating C-Reactive Protein and Interleukin-6 With Cancer Risk: Findings From Two Prospective Cohorts and a Meta-Analysis. Cancer Causes Control (2009) 20:15–26. doi: 10.1007/s10552-008-9212-z
7. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. CANTOS Trial Group. Effect of Interleukin-1β Inhibition With Canakinumab on Incident Lung Cancer in Patients With Atherosclerosis: Exploratory Results From a Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
8. da Cunha BR, Domingos C, Stefanini ACB, Henrique T, Polachini GM, Castelo-Branco P, et al. Cellular Interactions in the Tumor Microenvironment: The Role of Secretome. J Cancer (2019) 10:4574–87. doi: 10.7150/jca.21780
9. Mukherjee P, Mani S. Methodologies to Decipher the Cell Secretome. Biochim Biophys Acta (2013) 1834:2226–32. doi: 10.1016/j.bbapap.2013.01.022
10. Park A, Lee Y, Kim MS, Kang YJ, Park Y-J, Jung H, et al. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front Immunol (2018) 9:1859. doi: 10.3389/fimmu.2018.01859
11. Teng MWL, Galon J, Fridman W-H, Smyth MJ. From Mice to Humans: Developments in Cancer Immunoediting. J Clin Invest (2015) 125:3338–46. doi: 10.1172/JCI80004
12. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
13. Bui JD, Schreiber RD. Cancer Immunosurveillance, Immunoediting and Inflammation: Independent or Interdependent Processes? Curr Opin Immunol (2007) 19:203–8. doi: 10.1016/j.coi.2007.02.001
14. Perez-Amill L, Suñe G, Antoñana-Vildosola A, Castella M, Najjar A, Bonet J, et al. Preclinical Development of a Humanized Chimeric Antigen Receptor Against B Cell Maturation Antigen for Multiple Myeloma. Haematologica (2021) 106:173–84. doi: 10.3324/haematol.2019.228577
15. Zhao W-H, Liu J, Wang B-Y, Chen Y-X, Cao X-M, Yang Y, et al. A Phase 1, Open-Label Study of LCAR-B38M, a Chimeric Antigen Receptor T Cell Therapy Directed Against B Cell Maturation Antigen, in Patients With Relapsed or Refractory Multiple Myeloma. J Hematol Oncol (2018) 11:141. doi: 10.1186/s13045-018-0681-6
16. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
17. Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJN, et al. Antigen Presenting Cell-Mediated Expansion of Human Umbilical Cord Blood Yields Log-Scale Expansion of Natural Killer Cells With Anti-Myeloma Activity. PloS One (2013) 8:e76781. doi: 10.1371/journal.pone.0076781
18. Bachiller M, Battram AM, Perez-Amill L, Martín-Antonio B. Natural Killer Cells in Immunotherapy: Are We Nearly There? Cancers (Basel) (2020) 12(11):3139. doi: 10.3390/cancers12113139
19. Martín-Antonio B, Suñe G, Perez-Amill L, Castella M, Urbano-Ispizua A. Natural Killer Cells: Angels and Devils for Immunotherapy. Int J Mol Sci (2017) 18(9):1868. doi: 10.3390/ijms18091868
20. Perez-Amill L, Marzal B, Urbano-Ispizua A, Juan M, Martín-Antonio B. CAR-T Cell Therapy: A Door is Open to Find Innumerable Possibilities of Treatments for Cancer Patients. Turk J Haematol (2018) 35:217–28. doi: 10.4274/tjh.2018.0196
21. Marofi F, Rahman HS, Achmad MH, Sergeevna KN, Suksatan W, Abdelbasset WK, et al. A Deep Insight Into CAR-T Cell Therapy in non-Hodgkin Lymphoma: Application, Opportunities, and Future Directions. Front Immunol (2021) 12:681984. doi: 10.3389/fimmu.2021.681984
22. Martino M, Alati C, Canale FA, Musuraca G, Martinelli G. Cerchione C. A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia. Int J Mol Sci (2021) 22:2150. doi: 10.3390/ijms22042150
23. Dafni U, Michielin O, Lluesma SM, Tsourti Z, Polydoropoulou V, Karlis D, et al. Efficacy of Adoptive Therapy With Tumor-Infiltrating Lymphocytes and Recombinant Interleukin-2 in Advanced Cutaneous Melanoma: A Systematic Review and Meta-Analysis. Ann Oncol (2019) 30:1902–13. doi: 10.1093/annonc/mdz398
24. Moreno V, Hernandez T, de Miguel M, Doger B, Calvo E. Adoptive Cell Therapy for Solid Tumors: Chimeric Antigen Receptor T Cells and Beyond. Curr Opin Pharmacol (2021) 59:70–84. doi: 10.1016/j.coph.2021.05.004
25. Lin B, Du L, Li H, Zhu X, Cui L, Li X. Tumor-Infiltrating Lymphocytes: Warriors Fight Against Tumors Powerfully. Biomed Pharmacother (2020) 132:110873. doi: 10.1016/j.biopha.2020.110873
26. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
27. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
28. Bruno A, Pagani A, Pulze L, Albini A, Dallaglio K, Noonan DM, et al. Orchestration of Angiogenesis by Immune Cells. Front Oncol (2014) 4:131. doi: 10.3389/fonc.2014.00131
29. Battram AM, Bachiller M, Martín-Antonio B. Senescence in the Development and Response to Cancer With Immunotherapy: A Double-Edged Sword. Int J Mol Sci (2020) 21:4346. doi: 10.3390/ijms21124346
30. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent Fibroblasts Promote Epithelial Cell Growth and Tumorigenesis: A Link Between Cancer and Aging. Proc Natl Acad Sci USA (2001) 98:12072–7. doi: 10.1073/pnas.211053698
31. Wolf B, Zimmermann S, Arber C, Irving M, Trueb L, Coukos G. Safety and Tolerability of Adoptive Cell Therapy in Cancer. Drug Saf (2019) 42:315–34. doi: 10.1007/s40264-018-0779-3
32. Kale A, Sharma A, Stolzing A, Desprez P-Y, Campisi J. Role of Immune Cells in the Removal of Deleterious Senescent Cells. Immun Ageing (2020) 17:16. doi: 10.1186/s12979-020-00187-9
34. Sun X, Cui Y, Feng H, Liu H, Liu X. TGF-β Signaling Controls Foxp3 Methylation and T Reg Cell Differentiation by Modulating Uhrf1 Activity. J Exp Med (2019) 216:2819–37. doi: 10.1084/jem.20190550
35. Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
36. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The Proangiogenic Phenotype of Natural Killer Cells in Patients With non-Small Cell Lung Cancer. Neoplasia (2013) 15:133–42. doi: 10.1593/neo.121758
37. Cerdeira AS, Rajakumar A, Royle CM, Lo A, Husain Z, Thadhani RI, et al. Conversion of Peripheral Blood NK Cells to a Decidual NK-Like Phenotype by a Cocktail of Defined Factors. J Immunol (2013) 190:3939–48. doi: 10.4049/jimmunol.1202582
38. Siewiera J, Gouilly J, Hocine H-R, Cartron G, Levy C, Al-Daccak R, et al. Natural Cytotoxicity Receptor Splice Variants Orchestrate the Distinct Functions of Human Natural Killer Cell Subtypes. Nat Commun (2015) 6:10183. doi: 10.1038/ncomms10183
39. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming Growth Factor Beta 1 Inhibits Expression of Nkp30 and NKG2D Receptors: Consequences for the NK-Mediated Killing of Dendritic Cells. Proc Natl Acad Sci USA (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
40. Park YP, Choi S-C, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, et al. Complex Regulation of Human NKG2D-DAP10 Cell Surface Expression: Opposing Roles of the Γc Cytokines and TGF-β1. Blood (2011) 118:3019–27. doi: 10.1182/blood-2011-04-346825
41. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-Derived TGF-β1 Modulates the Chemokine Receptor Repertoire of Human Resting NK Cells. J Immunol (2013) 190:5321–8. doi: 10.4049/jimmunol.1202693
42. Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4(+) and CD8(+) Anergic T Cells Induced by Interleukin-10-Treated Human Dendritic Cells Display Antigen-Specific Suppressor Activity. Blood (2002) 99:2468–76. doi: 10.1182/blood.v99.7.2468
43. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 Elicits Ifnγ-Dependent Tumor Immune Surveillance. Cancer Cell (2011) 20:781–96. doi: 10.1016/j.ccr.2011.11.003
44. Schwich E, Hò G-GT, LeMaoult J, Bade-Döding C, Carosella ED, Horn PA, et al. Soluble HLA-G and HLA-G Bearing Extracellular Vesicles Affect ILT-2 Positive and ILT-2 Negative CD8 T Cells Complementary. Front Immunol (2020) 11:2046. doi: 10.3389/fimmu.2020.02046
45. Chen J, Feng Y, Lu L, Wang H, Dai L, Li Y, et al. Interferon-Γ-Induced PD-L1 Surface Expression on Human Oral Squamous Carcinoma via PKD2 Signal Pathway. Immunobiology (2012) 217:385–93. doi: 10.1016/j.imbio.2011.10.016
46. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-Like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell (2019) 176:334–47.e12. doi: 10.1016/j.cell.2018.11.010
47. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 Ligand Galectin-9 Negatively Regulates T Helper Type 1 Immunity. Nat Immunol (2005) 6:1245–52. doi: 10.1038/ni1271
48. Devilard E, Xerri L, Dubreuil P, Lopez M, Reymond N. Nectin-3 (CD113) Interacts With Nectin-2 (CD112) to Promote Lymphocyte Transendothelial Migration. PloS One (2013) 8:e77424. doi: 10.1371/journal.pone.0077424
49. Russo E, Runge P, Jahromi NH, Naboth H, Landtwing A, Montecchi R, et al. CD112 Regulates Angiogenesis and T Cell Entry Into the Spleen. Cells (2021) 10(1):169. doi: 10.3390/cells10010169
50. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The Interaction of TIGIT With PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc Natl Acad Sci USA (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
51. Kalinski P. Regulation of Immune Responses by Prostaglandin E2. J Immunol (2012) 188:21–8. doi: 10.4049/jimmunol.1101029
52. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma Cells Inhibit Natural Killer Cell Function by Modulating the Expression of Activating Receptors and Cytolytic Activity. Cancer Res (2012) 72:1407–15. doi: 10.1158/0008-5472.CAN-11-2544
53. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-Associated Fibroblasts Modulate NK Cell Phenotype and Antitumor Cytotoxicity. Proc Natl Acad Sci USA (2009) 106:20847–52. doi: 10.1073/pnas.0906481106
54. Qian X, Zhang J, Liu J. Tumor-Secreted PGE2 Inhibits CCL5 Production in Activated Macrophages Through Camp/PKA Signaling Pathway. J Biol Chem (2011) 286:2111–20. doi: 10.1074/jbc.M110.154971
55. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor Indoleamine 2,3-Dioxygenase (IDO) Inhibits CD19-CAR T Cells and Is Downregulated by Lymphodepleting Drugs. Blood (2015) 125:3905–16. doi: 10.1182/blood-2015-01-621474
56. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
57. Lee C-L, Vijayan M, Wang X, Lam KKW, Koistinen H, Seppala M, et al. Glycodelin-a Stimulates the Conversion of Human Peripheral Blood CD16-CD56bright NK Cell to a Decidual NK Cell-Like Phenotype. Hum Reprod (2019) 34:689–701. doi: 10.1093/humrep/dey378
58. Rajagopalan S, Long EO. Cellular Senescence Induced by CD158d Reprograms Natural Killer Cells to Promote Vascular Remodeling. Proc Natl Acad Sci USA (2012) 109:20596–601. doi: 10.1073/pnas.1208248109
59. Schurich A, Magalhaes I, Mattsson J. Metabolic Regulation of CAR T Cell Function by the Hypoxic Microenvironment in Solid Tumors. Immunotherapy (2019) 11:335–45. doi: 10.2217/imt-2018-0141
60. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia Downregulates the Expression of Activating Receptors Involved in NK-Cell-Mediated Target Cell Killing Without Affecting ADCC. Eur J Immunol (2013) 43:2756–64. doi: 10.1002/eji.201343448
61. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B Degradation by Autophagy Decreases Tumor Cell Susceptibility to Natural Killer-Mediated Lysis Under Hypoxia. Proc Natl Acad Sci USA (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
62. Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front Immunol (2018) 9:2358. doi: 10.3389/fimmu.2018.02358
63. Donzelli S, Milano E, Pruszko M, Sacconi A, Masciarelli S, Iosue I, et al. Expression of ID4 Protein in Breast Cancer Cells Induces Reprogramming of Tumour-Associated Macrophages. Breast Cancer Res (2018) 20:1–15. doi: 10.1186/s13058-018-0990-2
64. Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, et al. Hypoxic Tumor-Derived Exosomal MiR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res (2018) 78:4586–98. doi: 10.1158/0008-5472.CAN-17-3841
65. Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, et al. Regulation of the Chemokine Receptor CXCR4 by Hypoxia. J Exp Med (2003) 198:1391–402. doi: 10.1084/jem.20030267
66. Chen L, Wang S, Wang Y, Zhang W, Ma K, Hu C, et al. IL-6 Influences the Polarization of Macrophages and the Formation and Growth of Colorectal Tumor. Oncotarget (2018) 9:17443–54. doi: 10.18632/oncotarget.24734
67. Shrivastava R, Asif M, Singh V, Dubey P, Ahmad Malik S, Lone M-U-D, et al. M2 Polarization of Macrophages by Oncostatin M in Hypoxic Tumor Microenvironment is Mediated by Mtorc2 and Promotes Tumor Growth and Metastasis. Cytokine (2019) 118:130–43. doi: 10.1016/j.cyto.2018.03.032
68. Kitamura T, Qian B-Z, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-Induced Chemokine Cascade Promotes Breast Cancer Metastasis by Enhancing Retention of Metastasis-Associated Macrophages. J Exp Med (2015) 212:1043–59. doi: 10.1084/jem.20141836
69. Ségaliny AI, Mohamadi A, Dizier B, Lokajczyk A, Brion R, Lanel R, et al. Interleukin-34 Promotes Tumor Progression and Metastatic Process in Osteosarcoma Through Induction of Angiogenesis and Macrophage Recruitment. Int J Cancer (2015) 137:73–85. doi: 10.1002/ijc.29376
70. Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular Endothelial Growth Factor-Induced Skin Carcinogenesis Depends on Recruitment and Alternative Activation of Macrophages. J Pathol (2012) 227:17–28. doi: 10.1002/path.3989
71. Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-Produced Factors Activate Myeloid Cells Through TLR2 to Stimulate Metastasis. Nature (2009) 457:102–6. doi: 10.1038/nature07623
72. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic Cancer Exosomes Initiate Pre-Metastatic Niche Formation in the Liver. Nat Cell Biol (2015) 17:816–26. doi: 10.1038/ncb3169
73. Tzeng H-T, Su C-C, Chang C-P, Lai W-W, Su W-C, Wang Y-C. Rab37 in Lung Cancer Mediates Exocytosis of Soluble ST2 and Thus Skews Macrophages Toward Tumor-Suppressing Phenotype. Int J Cancer (2018) 143:1753–63. doi: 10.1002/ijc.31569
74. Hsieh C-H, Tai S-K, Yang M-H. Snail-Overexpressing Cancer Cells Promote M2-Like Polarization of Tumor-Associated Macrophages by Delivering Mir-21-Abundant Exosomes. Neoplasia (2018) 20:775–88. doi: 10.1016/j.neo.2018.06.004
75. Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, et al. CD47-Blocking Immunotherapies Stimulate Macrophage-Mediated Destruction of Small-Cell Lung Cancer. J Clin Invest (2016) 126:2610–20. doi: 10.1172/JCI81603
76. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
77. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC Class I by the Inhibitory Receptor LILRB1 Suppresses Macrophages and Is a Target of Cancer Immunotherapy. Nat Immunol (2018) 19:76–84. doi: 10.1038/s41590-017-0004-z
78. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 Signalling Through Macrophage Siglec-10 Is a Target for Cancer Immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
79. Paltridge JL, Belle L, Khew-Goodall Y. The Secretome in Cancer Progression. Biochim Biophys Acta (2013) 1834:2233–41. doi: 10.1016/j.bbapap.2013.03.014
80. Hanash S, Schliekelman M. Proteomic Profiling of the Tumor Microenvironment: Recent Insights and the Search for Biomarkers. Genome Med (2014) 6:12. doi: 10.1186/gm529
81. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
82. Mocellin S, Panelli MC, Wang E, Nagorsen D, Marincola FM. The Dual Role of IL-10. Trends Immunol (2003) 24:36–43. doi: 10.1016/S1471-4906(02)00009-1
83. Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the Tumor Microenvironment: A Target for Anticancer Immunotherapy. Immunol Res (2011) 51:170–82. doi: 10.1007/s12026-011-8262-6
84. Yue FY, Dummer R, Geertsen R, Hofbauer G, Laine E, Manolio S, et al. Interleukin-10 Is a Growth Factor for Human Melanoma Cells and Down-Regulates HLA Class-I, HLA Class-II and ICAM-1 Molecules. Int J Cancer (1997) 71:630–7. doi: 10.1002/(sici)1097-0215(19970516)71:4<630::aid-ijc20>3.0.co;2-e
85. Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
86. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol Cell Biol (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
87. Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 Inhibits T Helper Type 1-Mediated Auto- and Alloimmune Responses and Promotes Immunological Tolerance. Nat Immunol (2003) 4:1093–101. doi: 10.1038/ni987
88. Zhang H, Wang Z, Zhang J, Zhang X, Gui Z, Sun L, et al. The Synergism of B and T Lymphocyte Attenuator (BTLA) and Cytotoxic T Lymphocyte Associated Antigen-4 (CTLA-4) Attenuated Acute T-Cell Mediated Rejection and Prolonged Renal Graft Survival. Transl Androl Urol (2020) 9:1990–9. doi: 10.21037/tau-20-728
89. Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, et al. LAG-3 Regulates Plasmacytoid Dendritic Cell Homeostasis. J Immunol (2009) 182:1885–91. doi: 10.4049/jimmunol.0800185
90. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ (2002) 9:1069–77. doi: 10.1038/sj.cdd.4401073
91. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10:48–57. doi: 10.1038/ni.1674
92. Yu X, Huang X, Chen X, Liu J, Wu C, Pu Q, et al. Characterization of a Novel Anti-Human Lymphocyte Activation Gene 3 (LAG-3) Antibody for Cancer Immunotherapy. MAbs (2019) 11:1139–48. doi: 10.1080/19420862.2019.1629239
93. Wang Y, Zhao E, Zhang Z, Zhao G, Cao H. Association Between Tim−3 and Gal−9 Expression and Gastric Cancer Prognosis. Oncol Rep (2018) 40:2115–26. doi: 10.3892/or.2018.6627
94. Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, et al. Tim-3/Galectin-9 Pathway: Regulation of Th1 Immunity Through Promotion of CD11b+Ly-6G+ Myeloid Cells. J Immunol (2010) 185:1383–92. doi: 10.4049/jimmunol.0903275
95. Lozano E, Mena M-P, Díaz T, Martin-Antonio B, León S, Rodríguez-Lobato L-G, et al. Nectin-2 Expression on Malignant Plasma Cells Is Associated With Better Response to TIGIT Blockade in Multiple Myeloma. Clin Cancer Res (2020) 26:4688–98. doi: 10.1158/1078-0432.CCR-19-3673
96. Reches A, Ophir Y, Stein N, Kol I, Isaacson B, Charpak Amikam Y, et al. Nectin4 Is a Novel TIGIT Ligand Which Combines Checkpoint Inhibition and Tumor Specificity. J Immunother Cancer (2020) 8(1):e000266. doi: 10.1136/jitc-2019-000266
97. Harjunpää H, Guillerey C. TIGIT as an Emerging Immune Checkpoint. Clin Exp Immunol (2020) 200:108–19. doi: 10.1111/cei.13407
98. Iguchi-Manaka A, Okumura G, Kojima H, Cho Y, Hirochika R, Bando H, et al. Increased Soluble CD155 in the Serum of Cancer Patients. PloS One (2016) 11(4):e0152982. doi: 10.1371/journal.pone.0152982
99. Li M, Qiao D, Pu J, Wang W, Zhu W, Liu H. Elevated Nectin-2 Expression Is Involved in Esophageal Squamous Cell Carcinoma by Promoting Cell Migration and Invasion. Oncol Lett (2018) 15:4731–6. doi: 10.3892/ol.2018.7953
100. Siddharth S, Nayak A, Das S, Nayak D, Panda J, Wyatt MD, et al. The Soluble Nectin-4 Ecto-Domain Promotes Breast Cancer Induced Angiogenesis Via Endothelial Integrin-β4. Int J Biochem Cell Biol (2018) 102:151–60. doi: 10.1016/j.biocel.2018.07.011
101. Dorner BG, Scheffold A, Rolph MS, Huser MB, Kaufmann SHE, Radbruch A, et al. MIP-1alpha, MIP-1beta, RANTES, and ATAC/Lymphotactin Function Together With IFN-Gamma as Type 1 Cytokines. Proc Natl Acad Sci USA (2002) 99:6181–6. doi: 10.1073/pnas.092141999
102. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/Mpges1/PGE2 Pathway Regulates PD-L1 Expression in Tumor-Associated Macrophages and Myeloid-Derived Suppressor Cells. Proc Natl Acad Sci USA (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
103. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-Targeted T Cells Modified to Secrete IL-12 Eradicate Systemic Tumors Without Need for Prior Conditioning. Blood (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
104. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors. vivo. Oncoimmunol (2015) 4:e994446. doi: 10.4161/2162402X.2014.994446
105. Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-Infiltrating Lymphocytes Genetically Engineered With an Inducible Gene Encoding Interleukin-12 for the Immunotherapy of Metastatic Melanoma. Clin Cancer Res (2015) 21:2278–88. doi: 10.1158/1078-0432.CCR-14-2085
106. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving Adoptive T Cell Therapy by Targeting and Controlling IL-12 Expression to the Tumor Environment. Mol Ther (2011) 19:751–9. doi: 10.1038/mt.2010.313
107. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-Specific T Lymphocytes With Interleukin-15 and a Suicide Gene to Enhance Their Anti-Lymphoma/Leukemia Effects and Safety. Leukemia (2010) 24:1160–70. doi: 10.1038/leu.2010.75
108. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bethigh Foxo1low Effectors That Exhibit Augmented Activity Against Advanced Solid Tumors. Cell Rep (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
109. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and Through Activation of the Endogenous Immune System. Cell Rep (2018) 23:2130–41. doi: 10.1016/j.celrep.2018.04.051
110. Sedimbi SK, Hägglöf T, Karlsson MCI. IL-18 in Inflammatory and Autoimmune Disease. Cell Mol Life Sci (2013) 70:4795–808. doi: 10.1007/s00018-013-1425-y
111. Vidal-Vanaclocha F, Mendoza L, Telleria N, Salado C, Valcárcel M, Gallot N, et al. Clinical and Experimental Approaches to the Pathophysiology of Interleukin-18 in Cancer Progression. Cancer Metastasis Rev (2006) 25:417–34. doi: 10.1007/s10555-006-9013-3
112. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat Biotechnol (2018) 36:346–51. doi: 10.1038/nbt.4086
113. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation and Augments Prostate Cancer Eradication. Mol Ther (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
114. Boice M, Salloum D, Mourcin F, Sanghvi V, Amin R, Oricchio E, et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell (2016) 167:405–18.e13. doi: 10.1016/j.cell.2016.08.032
115. Suarez ER, Chang DK, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric Antigen Receptor T Cells Secreting Anti-PD-L1 Antibodies More Effectively Regress Renal Cell Carcinoma in a Humanized Mouse Model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
116. Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, et al. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as Well as Bystander Cells From Oxidative Stress-Induced Loss of Antitumor Activity. J Immunol (2016) 196:759–66. doi: 10.4049/jimmunol.1401710
117. Scheffel MJ, Scurti G, Simms P, Garrett-Mayer E, Mehrotra S, Nishimura MI, et al. Efficacy of Adoptive T-Cell Therapy is Improved by Treatment With the Antioxidant N-Acetyl Cysteine, Which Limits Activation-Induced T-Cell Death. Cancer Res (2016) 76:6006–16. doi: 10.1158/0008-5472.CAN-16-0587
118. Sojka DK, Yang L, Yokoyama WM. Uterine Natural Killer Cells. Front Immunol (2019) 10:960. doi: 10.3389/fimmu.2019.00960
119. Huhn O, Zhao X, Esposito L, Moffett A, Colucci F, Sharkey AM. How do Uterine Natural Killer and Innate Lymphoid Cells Contribute to Successful Pregnancy? Front Immunol (2021) 12:607669. doi: 10.3389/fimmu.2021.607669
120. Jabrane-Ferrat N. Features of Human Decidual NK Cells in Healthy Pregnancy and During Viral Infection. Front Immunol (2019) 10:1397. doi: 10.3389/fimmu.2019.01397
121. Fu B, Zhou Y, Ni X, Tong X, Xu X, Dong Z, et al. Natural Killer Cells Promote Fetal Development Through the Secretion of Growth-Promoting Factors. Immunity (2017) 47:1100–13.e6. doi: 10.1016/j.immuni.2017.11.018
122. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK Cells Regulate Key Developmental Processes at the Human Fetal-Maternal Interface. Nat Med (2006) 12:1065–74. doi: 10.1038/nm1452
123. Albini A, Noonan DM. Decidual-Like NK Cell Polarization: From Cancer Killing to Cancer Nurturing. Cancer Discov (2021) 11:28–33. doi: 10.1158/2159-8290.CD-20-0796
124. Bassani B, Baci D, Gallazzi M, Poggi A, Bruno A, Mortara L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities Into Potent Anti-Tumor Effects. Cancers (Basel) (2019) 11(4):461. doi: 10.3390/cancers11040461
125. Bruno A, Bassani B, D’Urso DG, Pitaku I, Cassinotti E, Pelosi G, et al. Angiogenin and the MMP9-TIMP2 Axis are Up-Regulated in Proangiogenic, Decidual NK-Like Cells From Patients With Colorectal Cancer. FASEB J (2018) 32:5365–77. doi: 10.1096/fj.201701103R
126. Schneider MA, Granzow M, Warth A, Schnabel PA, Thomas M, Herth FJF, et al. Glycodelin: A New Biomarker With Immunomodulatory Functions in non–Small Cell Lung Cancer. Clin Cancer Res (2015) 21:3529–40. doi: 10.1158/1078-0432.CCR-14-2464
127. Schneider MA, Muley T, Kahn NC, Warth A, Thomas M, Herth FJF, et al. Glycodelin Is a Potential Novel Follow-Up Biomarker for Malignant Pleural Mesothelioma. Oncotarget (2016) 7:71285–97. doi: 10.18632/oncotarget.12474
128. Scholz C, Heublein S, Lenhard M, Friese K, Mayr D, Jeschke U. Glycodelin a is a Prognostic Marker to Predict Poor Outcome in Advanced Stage Ovarian Cancer Patients. BMC Res Notes (2012) 5:551–1. doi: 10.1186/1756-0500-5-551
129. Lenhard M, Heublein S, Kunert-Keil C, Vrekoussis T, Lomba I, Ditsch N, et al. Immunosuppressive Glycodelin a Is an Independent Marker for Poor Prognosis in Endometrial Cancer. BMC Cancer (2013) 13:616. doi: 10.1186/1471-2407-13-616
130. Marletta S, Girolami I, Munari E, Pantanowitz L, Bernasconi R, Torresani E, et al. HLA-G Expression in Melanomas. Int Rev Immunol (2021) 40(5):330–43. doi: 10.1080/08830185.2020.1869732
131. Lázaro-Sánchez AD, Salces-Ortiz P, Velásquez LI, Orozco-Beltrán D, Díaz-Fernández N, Juárez-Marroquí A. HLA-G as a New Tumor Biomarker: Detection of Soluble Isoforms of HLA-G in the Serum and Saliva of Patients With Colorectal Cancer. Clin Transl Oncol (2020) 22:1166–71. doi: 10.1007/s12094-019-02244-2
132. Wlasiuk P, Stec A, Piechnik A, Kaminska W, Dmoszynska A, Ksiazek A, et al. Expression of Soluble HLA-G in Multiple Myeloma Patients and Patients With Renal Failure. Leuk Res (2012) 36:881–3. doi: 10.1016/j.leukres.2012.02.015
133. Caocci G, Greco M, Arras M, Cusano R, Orrù S, Martino B, et al. HLA-G Molecules and Clinical Outcome in Chronic Myeloid Leukemia. Leuk Res (2017) 61:1–5. doi: 10.1016/j.leukres.2017.08.005
134. Ullah M, Azazzen D, Kaci R, Benabbou N, Pujade Lauraine E, Pocard M, et al. High Expression of HLA-G in Ovarian Carcinomatosis: The Role of Interleukin-1β. Neoplasia (2019) 21:331–42. doi: 10.1016/j.neo.2019.01.001
136. Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol (2012) 32:463–88. doi: 10.1615/critrevimmunol.v32.i6.10
137. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
138. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells Across Human Cancers. Nat Med (2015) 21:938–45. doi: 10.1038/nm.3909
139. Genard G, Wera A-C, Huart C, Le Calve B, Penninckx S, Fattaccioli A, et al. Proton Irradiation Orchestrates Macrophage Reprogramming Through Nfκb Signaling. Cell Death Dis (2018) 9:728. doi: 10.1038/s41419-018-0757-9
140. Schulz D, Severin Y, Zanotelli VRT, Bodenmiller B. In-Depth Characterization of Monocyte-Derived Macrophages Using a Mass Cytometry-Based Phagocytosis Assay. Sci Rep (2019) 9:1925. doi: 10.1038/s41598-018-38127-9
141. Tseng D, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 Antibody-Mediated Phagocytosis of Cancer by Macrophages Primes an Effective Antitumor T-Cell Response. Proc Natl Acad Sci USA (2013) 110:11103–8. doi: 10.1073/pnas.1305569110
142. Sun N-Y, Chen Y-L, Wu W-Y, Lin H-W, Chiang Y-C, Chang C-F, et al. Blockade of PD-L1 Enhances Cancer Immunotherapy by Regulating Dendritic Cell Maturation and Macrophage Polarization. Cancers (Basel) (2019) 11(9):1400. doi: 10.3390/cancers11091400
143. Kimura Y, Inoue A, Hangai S, Saijo S, Negishi H, Nishio J, et al. The Innate Immune Receptor Dectin-2 Mediates the Phagocytosis of Cancer Cells by Kupffer Cells for the Suppression of Liver Metastasis. Proc Natl Acad Sci USA (2016) 113:14097–102. doi: 10.1073/pnas.1617903113
144. Imbert PRC, Saric A, Pedram K, Bertozzi CR, Grinstein S, Freeman SA. An Acquired and Endogenous Glycocalyx Forms a Bidirectional “Don’t Eat” and “Don’t Eat Me” Barrier to Phagocytosis. Curr Biol (2021) 31:77–89.e5. doi: 10.1016/j.cub.2020.09.082
145. Martín-Antonio B, Suñe G, Najjar A, Perez-Amill L, Antoñana-Vildosola A, Castella M, et al. Extracellular NK Histones Promote Immune Cell Anti-Tumor Activity by Inducing Cell Clusters Through Binding to CD138 Receptor. J Immunother Cancer (2019) 7:259. doi: 10.1186/s40425-019-0739-1
146. Gaforio JJ, Ortega E, Algarra I, Serrano MJ, Alvarez de Cienfuegos G. NK Cells Mediate Increase of Phagocytic Activity But Not of Proinflammatory Cytokine (Interleukin-6 [IL-6], Tumor Necrosis Factor Alpha, and IL-12) Production Elicited in Splenic Macrophages by Tilorone Treatment of Mice During Acute Systemic Candidiasis. Clin Diagn Lab Immunol (2002) 9:1282–94. doi: 10.1128/CDLI.9.6.1282-1294.2002
147. Boulakirba S, Pfeifer A, Mhaidly R, Obba S, Goulard M, Schmitt T, et al. IL-34 and CSF-1 Display an Equivalent Macrophage Differentiation Ability But a Different Polarization Potential. Sci Rep (2018) 8:256. doi: 10.1038/s41598-017-18433-4
148. Duan Z, Luo Y. Targeting Macrophages in Cancer Immunotherapy. Signal Transduction Targeted Ther (2021) 6:1–21. doi: 10.1038/s41392-021-00506-6
149. Jeong H, Kim S, Hong B-J, Lee C-J, Kim Y-E, Bok S, et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res (2019) 79:795–806. doi: 10.1158/0008-5472.CAN-18-2545
150. Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, et al. Macrophage Polarity in Cancer: A Review. J Cell Biochem (2019) 120:2756–65. doi: 10.1002/jcb.27646
151. Kim S-Y, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, et al. Role of the IL-6-JAK1-STAT3-Oct-4 Pathway in the Conversion of non-Stem Cancer Cells Into Cancer Stem-Like Cells. Cell Signal (2013) 25:961–9. doi: 10.1016/j.cellsig.2013.01.007
152. Duan S, Tsai Y, Keng P, Chen Y, Lee SO, Chen Y. IL-6 Signaling Contributes to Cisplatin Resistance in Non-Small Cell Lung Cancer via the Up-Regulation of Anti-Apoptotic and DNA Repair Associated Molecules. Oncotarget (2015) 6:27651–60. doi: 10.18632/oncotarget.4753
153. Logue SE, McGrath EP, Cleary P, Greene S, Mnich K, Almanza A, et al. Inhibition of IRE1 RNAse Activity Modulates the Tumor Cell Secretome and Enhances Response to Chemotherapy. Nat Commun (2018) 9:3267. doi: 10.1038/s41467-018-05763-8
154. Brown JA, Yonekubo Y, Hanson N, Sastre-Perona A, Basin A, Rytlewski JA, et al. TGF-β-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell (2017) 21:650–64.e8. doi: 10.1016/j.stem.2017.10.001
155. Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-Induced Tumour Secretomes Promote Resistance and Tumour Progression. Nature (2015) 520:368–72. doi: 10.1038/nature14336
156. Shen M, Dong C, Ruan X, Yan W, Cao M, Pizzo D, et al. Chemotherapy-Induced Extracellular Vesicle Mirnas Promote Breast Cancer Stemness by Targeting ONECUT2. Cancer Res (2019) 79:3608–21. doi: 10.1158/0008-5472.CAN-18-4055
157. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-Induced Senescence is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature (2006) 444:638–42. doi: 10.1038/nature05327
158. Pluquet O, Abbadie C, Coqueret O. Connecting Cancer Relapse With Senescence. Cancer Lett (2019) 463:50–8. doi: 10.1016/j.canlet.2019.08.004
159. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat Med (2015) 21:1424–35. doi: 10.1038/nm.4000
160. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PloS Biol (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
161. Kojima H, Inoue T, Kunimoto H, Nakajima K. IL-6-STAT3 Signaling and Premature Senescence. JAKSTAT (2013) 2(4):e25763. doi: 10.4161/jkst.25763
162. Le Duff M, Gouju J, Jonchère B, Guillon J, Toutain B, Boissard A, et al. Regulation of Senescence Escape by the Cdk4-EZH2-AP2M1 Pathway in Response to Chemotherapy. Cell Death Dis (2018) 9:199. doi: 10.1038/s41419-017-0209-y
163. Maybruck BT, Pfannenstiel LW, Diaz-Montero M, Gastman BR. Tumor-Derived Exosomes Induce CD8+ T Cell Suppressors. J Immunother Cancer (2017) 5:65. doi: 10.1186/s40425-017-0269-7
164. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and Tumour Clearance Is Triggered by P53 Restoration in Murine Liver Carcinomas. Nature (2007) 445:656–60. doi: 10.1038/nature05529
165. Yi F, Frazzette N, Cruz AC, Klebanoff CA, Siegel RM. Beyond Cell Death: New Functions for TNF Family Cytokines in Autoimmunity and Tumor Immunotherapy. Trends Mol Med (2018) 24:642–53. doi: 10.1016/j.molmed.2018.05.004
166. Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence Surveillance of Pre-Malignant Hepatocytes Limits Liver Cancer Development. Nature (2011) 479:547–51. doi: 10.1038/nature10599
167. d’Adda di Fagagna F. Living on a Break: Cellular Senescence as a DNA-Damage Response. Nat Rev Cancer (2008) 8:512–22. doi: 10.1038/nrc2440
168. Thomas R, Wang W, Su D-M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun Ageing (2020) 17:2. doi: 10.1186/s12979-020-0173-8
169. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: A Key Player in Cancer Development. J Hematol Oncol (2020) 13:151. doi: 10.1186/s13045-020-00986-z
170. Su D-M, Aw D, Palmer DB. Immunosenescence: A Product of the Environment? Curr Opin Immunol (2013) 25:498–503. doi: 10.1016/j.coi.2013.05.018
171. Fane M, Weeraratna AT. How the Ageing Microenvironment Influences Tumour Progression. Nat Rev Cancer (2020) 20:89–106. doi: 10.1038/s41568-019-0222-9
172. Tsukishiro T, Donnenberg AD, Whiteside TL. Rapid Turnover of the CD8(+)CD28(-) T-Cell Subset of Effector Cells in the Circulation of Patients With Head and Neck Cancer. Cancer Immunol Immunother (2003) 52:599–607. doi: 10.1007/s00262-003-0395-6
173. Ye J, Ma C, Hsueh EC, Eickhoff CS, Zhang Y, Varvares MA, et al. Tumor-Derived Γδ Regulatory T Cells Suppress Innate and Adaptive Immunity Through the Induction of Immunosenescence. J Immunol (2013) 190:2403–14. doi: 10.4049/jimmunol.1202369
174. Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, et al. Expression of CD57 Defines Replicative Senescence and Antigen-Induced Apoptotic Death of CD8+ T Cells. Blood (2003) 101:2711–20. doi: 10.1182/blood-2002-07-2103
175. Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol (2017) 9(6):a022236. doi: 10.1101/cshperspect.a022236
176. Hoare M, Ito Y, Kang T-W, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 Mediates a Switch Between Two Distinct Secretomes During Senescence. Nat Cell Biol (2016) 18:979–92. doi: 10.1038/ncb3397
177. Ruhland MK, Loza AJ, Capietto A-H, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal Senescence Establishes an Immunosuppressive Microenvironment That Drives Tumorigenesis. Nat Commun (2016) 7:11762. doi: 10.1038/ncomms11762
178. Iwasa H, Han J, Ishikawa F. Mitogen-Activated Protein Kinase P38 Defines the Common Senescence-Signalling Pathway. Genes Cells (2003) 8:131–44. doi: 10.1046/j.1365-2443.2003.00620.x
179. Herbig U, Jobling WA, Chen BPC, Chen DJ, Sedivy JM. Telomere Shortening Triggers Senescence of Human Cells Through a Pathway Involving ATM, P53, and P21(CIP1), But Not P16(INK4a). Mol Cell (2004) 14:501–13. doi: 10.1016/s1097-2765(04)00256-4
180. Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab (2019) 29:103–23.e5. doi: 10.1016/j.cmet.2018.09.020
181. Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y, et al. Human Regulatory T Cells Induce T-Lymphocyte Senescence. Blood (2012) 120:2021–31. doi: 10.1182/blood-2012-03-416040
182. Mondal AM, Horikawa I, Pine SR, Fujita K, Morgan KM, Vera E, et al. P53 Isoforms Regulate Aging- and Tumor-Associated Replicative Senescence in T Lymphocytes. J Clin Invest (2013) 123:5247–57. doi: 10.1172/JCI70355
183. Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S, et al. TLR8 Signaling Enhances Tumor Immunity by Preventing Tumor-Induced T-Cell Senescence. EMBO Mol Med (2014) 6:1294–311. doi: 10.15252/emmm.201403918
184. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A Adenosine Receptor Protects Tumors From Antitumor T Cells. Proc Natl Acad Sci USA (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
185. Castella M, Caballero-Baños M, Ortiz-Maldonado V, González-Navarro EA, Suñé G, Antoñana-Vidósola A, et al. Point-of-Care CAR T-Cell Production (ARI-0001) Using a Closed Semi-Automatic Bioreactor: Experience From an Academic Phase I Clinical Trial. Front Immunol (2020) 11:482. doi: 10.3389/fimmu.2020.00482
186. Amor C, Feucht J, Leibold J, Ho Y-J, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T Cells Reverse Senescence-Associated Pathologies. Nature (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
187. Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of Human Cellular Senescence: Roles of the P53 and P16 Pathways. EMBO J (2003) 22:4212–22. doi: 10.1093/emboj/cdg417
188. Akbar AN, Henson SM. Are Senescence and Exhaustion Intertwined or Unrelated Processes That Compromise Immunity? Nat Rev Immunol (2011) 11:289–95. doi: 10.1038/nri2959
189. Lee M, Kim DW, Khalmuratova R, Shin S-H, Kim Y-M, Han DH, et al. The IFN-Γ-P38, ERK Kinase Axis Exacerbates Neutrophilic Chronic Rhinosinusitis by Inducing the Epithelial-to-Mesenchymal Transition. Mucosal Immunol (2019) 12:601–11. doi: 10.1038/s41385-019-0149-1
190. Nayak TK, Mamidi P, Sahoo SS, Kumar PS, Mahish C, Chatterjee S, et al. P38 and JNK Mitogen-Activated Protein Kinases Interact With Chikungunya Virus Non-Structural Protein-2 and Regulate TNF Induction During Viral Infection in Macrophages. Front Immunol (2019) 10:786. doi: 10.3389/fimmu.2019.00786
191. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML Regulates P53 Acetylation and Premature Senescence Induced by Oncogenic Ras. Nature (2000) 406:207–10. doi: 10.1038/35018127
192. Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V. Granule Exocytosis Mediates Immune Surveillance of Senescent Cells. Oncogene (2013) 32:1971–7. doi: 10.1038/onc.2012.206
193. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
194. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. P53-Dependent Chemokine Production by Senescent Tumor Cells Supports NKG2D-Dependent Tumor Elimination by Natural Killer Cells. J Exp Med (2013) 210:2057–69. doi: 10.1084/jem.20130783
195. Brighton PJ, Maruyama Y, Fishwick K, Vrljicak P, Tewary S, Fujihara R, et al. Clearance of Senescent Decidual Cells by Uterine Natural Killer Cells in Cycling Human Endometrium. Elife (2017) 6:e31274. doi: 10.7554/eLife.31274
196. Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, et al. Senescent Cells Evade Immune Clearance via HLA-E-Mediated NK and CD8+ T Cell Inhibition. Nat Commun (2019) 10:2387. doi: 10.1038/s41467-019-10335-5
197. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-Associated MICA Is Shed by ADAM Proteases. Cancer Res (2008) 68:6368–76. doi: 10.1158/0008-5472.CAN-07-6768
198. Baek K-H, Bhang D, Zaslavsky A, Wang L-C, Vachani A, Kim CF, et al. Thrombospondin-1 Mediates Oncogenic Ras-Induced Senescence in Premalignant Lung Tumors. J Clin Invest (2013) 123:4375–89. doi: 10.1172/JCI67465
199. Kaur S, Bronson SM, Pal-Nath D, Miller TW, Soto-Pantoja DR, Roberts DD. Functions of Thrombospondin-1 in the Tumor Microenvironment. Int J Mol Sci (2021) 22(9):4570. doi: 10.3390/ijms22094570
200. Nath PR, Gangaplara A, Pal-Nath D, Mandal A, Maric D, Sipes JM, et al. CD47 Expression in Natural Killer Cells Regulates Homeostasis and Modulates Immune Response to Lymphocytic Choriomeningitis Virus. Front Immunol (2018) 9:2985. doi: 10.3389/fimmu.2018.02985
201. Guillon J, Petit C, Moreau M, Toutain B, Henry C, Roché H, et al. Regulation of Senescence Escape by TSP1 and CD47 Following Chemotherapy Treatment. Cell Death Dis (2019) 10(3):199. doi: 10.1038/s41419-019-1406-7
202. Nath PR, Pal-Nath D, Mandal A, Cam MC, Schwartz AL, Roberts DD. Natural Killer Cell Recruitment and Activation are Regulated by CD47 Expression in the Tumor Microenvironment. Cancer Immunol Res (2019) 7:1547–61. doi: 10.1158/2326-6066.CIR-18-0367
203. Baker KJ, Houston A, Brint E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front Immunol (2019) 10:1197. doi: 10.3389/fimmu.2019.01197
204. Man SM, Karki R, Kanneganti T-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol Rev (2017) 277:61–75. doi: 10.1111/imr.12534
205. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat Cell Biol (2013) 15:978–90. doi: 10.1038/ncb2784
206. Shang D, Hong Y, Xie W, Tu Z, Xu J. Interleukin-1β Drives Cellular Senescence of Rat Astrocytes Induced by Oligomerized Amyloid β Peptide and Oxidative Stress. Front Neurol (2020) 11:929. doi: 10.3389/fneur.2020.00929
207. Cooper MA, Fehniger TA, Ponnappan A, Mehta V, Wewers MD, Caligiuri MA. Interleukin-1beta Costimulates Interferon-Gamma Production by Human Natural Killer Cells. Eur J Immunol (2001) 31:792–801. doi: 10.1002/1521-4141(200103)31:3<792::aid-immu792>3.0.co;2-u
208. Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM, et al. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity (2015) 43:987–97. doi: 10.1016/j.immuni.2015.10.010
209. Pesant M, Mavilio D. Priming of Human Resting NK Cells by Autologous M1 Macrophages Via the Engagement of IL-1β, IFN-β, and IL-15 Pathways. J Immunol (2015) 195:2818–28. doi: 10.4049/jimmunol.1500325
210. Elkabets M, Ribeiro VSG, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, et al. IL-1β Regulates a Novel Myeloid-Derived Suppressor Cell Subset That Impairs NK Cell Development and Function. Eur J Immunol (2010) 40:3347–57. doi: 10.1002/eji.201041037
211. Degos C, Heinemann M, Barrou J, Boucherit N, Lambaudie E, Savina A, et al. Endometrial Tumor Microenvironment Alters Human NK Cell Recruitment, and Resident NK Cell Phenotype and Function. Front Immunol (2019) 10:877. doi: 10.3389/fimmu.2019.00877
212. Ortiz-Montero P, Londoño-Vallejo A, Vernot J-P. Senescence-Associated IL-6 and IL-8 Cytokines Induce a Self- and Cross-Reinforced Senescence/Inflammatory Milieu Strengthening Tumorigenic Capabilities in the MCF-7 Breast Cancer Cell Line. Cell Commun Signal (2017) 15:17. doi: 10.1186/s12964-017-0172-3
213. Barajas-Gómez BA, Rosas-Carrasco O, Morales-Rosales SL, Pedraza Vázquez G, González-Puertos VY, Juárez-Cedillo T, et al. Relationship of Inflammatory Profile of Elderly Patients Serum and Senescence-Associated Secretory Phenotype With Human Breast Cancer Cells Proliferation: Role of IL6/IL8 Ratio. Cytokine (2017) 91:13–29. doi: 10.1016/j.cyto.2016.12.001
214. Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of Natural Killer Cell Cytotoxicity by Interleukin-6: Implications for the Pathogenesis of Macrophage Activation Syndrome. Arthritis Rheumatol (2015) 67:3037–46. doi: 10.1002/art.39295
215. Wu J, Gao F, Wang C, Qin M, Han F, Xu T, et al. IL-6 and IL-8 Secreted by Tumour Cells Impair the Function of NK Cells Via the STAT3 Pathway in Oesophageal Squamous Cell Carcinoma. J Exp Clin Cancer Res (2019) 38:321. doi: 10.1186/s13046-019-1310-0
216. Kang Y-J, Jeung IC, Park A, Park Y-J, Jung H, Kim T-D, et al. An Increased Level of IL-6 Suppresses NK Cell Activity in Peritoneal Fluid of Patients With Endometriosis Via Regulation of SHP-2 Expression. Hum Reprod (2014) 29:2176–89. doi: 10.1093/humrep/deu172
217. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat Commun (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
218. Campbell LM, Maxwell PJ, Waugh DJJ. Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer. Pharmaceuticals (Basel) (2013) 6:929–59. doi: 10.3390/ph6080929
219. Sabry M, Zubiak A, Hood SP, Simmonds P, Arellano-Ballestero H, Cournoyer E, et al. Tumor- and Cytokine-Primed Human Natural Killer Cells Exhibit Distinct Phenotypic and Transcriptional Signatures. PloS One (2019) 14:e0218674. doi: 10.1371/journal.pone.0218674
220. Poznanski SM, Lee AJ, Nham T, Lusty E, Larché MJ, Lee DA, et al. Combined Stimulation With Interleukin-18 and Interleukin-12 Potently Induces Interleukin-8 Production by Natural Killer Cells. JIN (2017) 9:511–25. doi: 10.1159/000477172
221. Gonçalves S, Yin K, Ito Y, Chan A, Olan I, Gough S, et al. COX2 Regulates Senescence Secretome Composition and Senescence Surveillance Through PGE2. Cell Rep (2021) 34:108860. doi: 10.1016/j.celrep.2021.108860
222. Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca DROS, Senescence C. And Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid Med Cell Longevity (2016) 2016:e3565127. doi: 10.1155/2016/3565127
223. Sabbatinelli J, Prattichizzo F, Olivieri F, Procopio AD, Rippo MR, Giuliani A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front Physiol (2019) 10:1523. doi: 10.3389/fphys.2019.01523
224. Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol Res (2019) 7:335–46. doi: 10.1158/2326-6066.CIR-18-0481
225. Duwe AK, Werkmeister J, Roder JC, Lauzon R, Payne U. Natural Killer Cell-Mediated Lysis Involves an Hydroxyl Radical-Dependent Step. J Immunol (1985) 134:2637–44.
226. Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, et al. Reactive Oxygen Species- and DNA Damage Response-Dependent NK Cell Activating Ligand Upregulation Occurs at Transcriptional Levels and Requires the Transcriptional Factor E2F1. J Immunol (2014) 193:950–60. doi: 10.4049/jimmunol.1400271
227. Yamamoto K, Fujiyama Y, Andoh A, Bamba T, Okabe H. Oxidative Stress Increases MICA and MICB Gene Expression in the Human Colon Carcinoma Cell Line (CaCo-2). Biochim Biophys Acta (2001) 1526:10–2. doi: 10.1016/s0304-4165(01)00099-x
228. Aydin E, Johansson J, Nazir FH, Hellstrand K, Martner A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol Res (2017) 5:804–11. doi: 10.1158/2326-6066.CIR-16-0382
229. Romero AI, Thorén FB, Brune M, Hellstrand K. Nkp46 and NKG2D Receptor Expression in NK Cells With CD56dim and CD56bright Phenotype: Regulation by Histamine and Reactive Oxygen Species. Br J Haematol (2006) 132:91–8. doi: 10.1111/j.1365-2141.2005.05842.x
230. Martin-Antonio B, Najjar A, Robinson SN, Chew C, Li S, Yvon E, et al. Transmissible Cytotoxicity of Multiple Myeloma Cells by Cord Blood-Derived NK Cells is Mediated by Vesicle Trafficking. Cell Death Differ (2015) 22:96–107. doi: 10.1038/cdd.2014.120
231. Lgpl P, Ig O TT, Jl K. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin Immunol (2018) 40:101275. doi: 10.1016/j.smim.2019.04.003
232. Hinojosa CA, Akula Suresh Babu R, Rahman MM, Fernandes G, Boyd AR, Orihuela CJ. Elevated A20 Contributes to Age-Dependent Macrophage Dysfunction in the Lungs. Exp Gerontol (2014) 54:58–66. doi: 10.1016/j.exger.2014.01.007
233. Ogata Y, Yamada T, Hasegawa S, Sanada A, Iwata Y, Arima M, et al. SASP-Induced Macrophage Dysfunction may Contribute to Accelerated Senescent Fibroblast Accumulation in the Dermis. Exp Dermatol (2021) 30:84–91. doi: 10.1111/exd.14205
234. Mazzoni M, Mauro G, Erreni M, Romeo P, Minna E, Vizioli MG, et al. Senescent Thyrocytes and Thyroid Tumor Cells Induce M2-Like Macrophage Polarization of Human Monocytes via a PGE2-Dependent Mechanism. J Exp Clin Cancer Res (2019) 38:208. doi: 10.1186/s13046-019-1198-8
235. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, et al. Non-Cell-Autonomous Tumor Suppression by P53. Cell (2013) 153:449–60. doi: 10.1016/j.cell.2013.03.020
236. Lesina M, Wörmann SM, Morton J, Diakopoulos KN, Korneeva O, Wimmer M, et al. Rela Regulates CXCL1/CXCR2-Dependent Oncogene-Induced Senescence in Murine Kras-Driven Pancreatic Carcinogenesis. J Clin Invest (2016) 126:2919–32. doi: 10.1172/JCI86477
237. Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, et al. N-GSDMD Trafficking to Neutrophil Organelles Facilitates IL-1β Release Independently of Plasma Membrane Pores and Pyroptosis. Nat Commun (2020) 11:2212. doi: 10.1038/s41467-020-16043-9
238. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-Mediated Target Cell Pyroptosis by CAR T Cells Triggers Cytokine Release Syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
239. Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, et al. The Role of Pyroptosis in Cancer: Pro-Cancer or Pro-“Host”? Cell Death Dis (2019) 10:1–13. doi: 10.1038/s41419-019-1883-8
240. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting Inflammasome/IL-1 Pathways for Cancer Immunotherapy. Sci Rep (2016) 6:36107. doi: 10.1038/srep36107
241. Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on Tumor-Associated Macrophages Promotes Lymphangiogenesis and Metastasis via NLRP3/IL-1β. J Exp Med (2017) 214:2695–713. doi: 10.1084/jem.20160392
Keywords: tumor secretome, SASP, senescence, immunotherapy, macrophages, CAR-T cells, NK cells, T cells
Citation: Etxebeste-Mitxeltorena M, del Rincón-Loza I and Martín-Antonio B (2021) Tumor Secretome to Adoptive Cellular Immunotherapy: Reduce Me Before I Make You My Partner. Front. Immunol. 12:717850. doi: 10.3389/fimmu.2021.717850
Received: 31 May 2021; Accepted: 26 July 2021;
Published: 10 August 2021.
Edited by:
Lorenzo Mortara, University of Insubria, ItalyReviewed by:
Cristina Eguizabal, Biocruces Bizkaia Health Research Institute, SpainCopyright © 2021 Etxebeste-Mitxeltorena, del Rincón-Loza and Martín-Antonio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz Martín-Antonio, YmVhdHJpei5hbnRvbmlvQHF1aXJvbnNhbHVkLmVz
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.