 Jérôme Hadjadj
Jérôme Hadjadj Marie-Louise Frémond
Marie-Louise Frémond Bénédicte Neven
Bénédicte Neven
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 17 September 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.717388
This article is part of the Research TopicNovel Therapeutic Approaches to Primary ImmunodeficienciesView all 14 articles
Among inborn errors of immunity (IEIs), some conditions are characterized by inflammation and autoimmunity at the front line and are particularly challenging to treat. Monogenic diseases associated with gain-of-function mutations in genes critical for cytokine signaling through the JAK-STAT pathway belong to this group. These conditions represent good candidates for treatment with JAK inhibitors. Type I interferonopathies, a group of recently identified monogenic auto-inflammatory diseases characterized by excessive secretion of type I IFN, are also good candidates with growing experiences reported in the literature. However, many questions remain regarding the choice of the drug, the dose (in particular in children), the efficacy on the various manifestations, the monitoring of the treatment, and the management of potent side effects in particular in patients with infectious susceptibility. This review will summarize the current experiences reported and will highlight the unmet needs.
Use of next-generation sequencing (NGS) has permitted the identification of a growing number of inborn errors of immunity (IEIs), with more than 450 conditions following the latest classification from the International Union of Immunological Societies Committee on Inborn Errors of Immunity (1, 2). The clinical features are broad, ranging from increased susceptibility to infections to immune dysregulation, including multiple autoimmune manifestations, allergy, and lymphoproliferation. Increased susceptibility to malignancy can also occur. Several monogenic autoimmune and/or auto-inflammatory diseases have been identified, and the analysis of these genes has provided a wealth of information on the mechanisms of tolerance that control reactivity to self in humans. These last years, monogenic diseases affecting cytokine signaling and in particular the Janus kinase (JAK)/Signal transducer and activator of transcription (STAT) pathway were described (3–7) and responsible for overwhelming human immune dysregulation. These conditions, as well as the hematological diseases related to somatic gain-of-function mutations of JAK2 and STAT3 respectively in myeloproliferative diseases (8) (MPNs) and large granular leukemia (9) (LGL), illustrate the key role of this signaling pathway in the regulation of the immune response (Table 1). Furthermore, identification of these mutations may influence treatment options to offer targeted treatment. The goal of this review is to report the available data on drug inhibiting the JAK-STAT pathway, i.e., JAK inhibitor (JAKinib), in the context of IEIs.
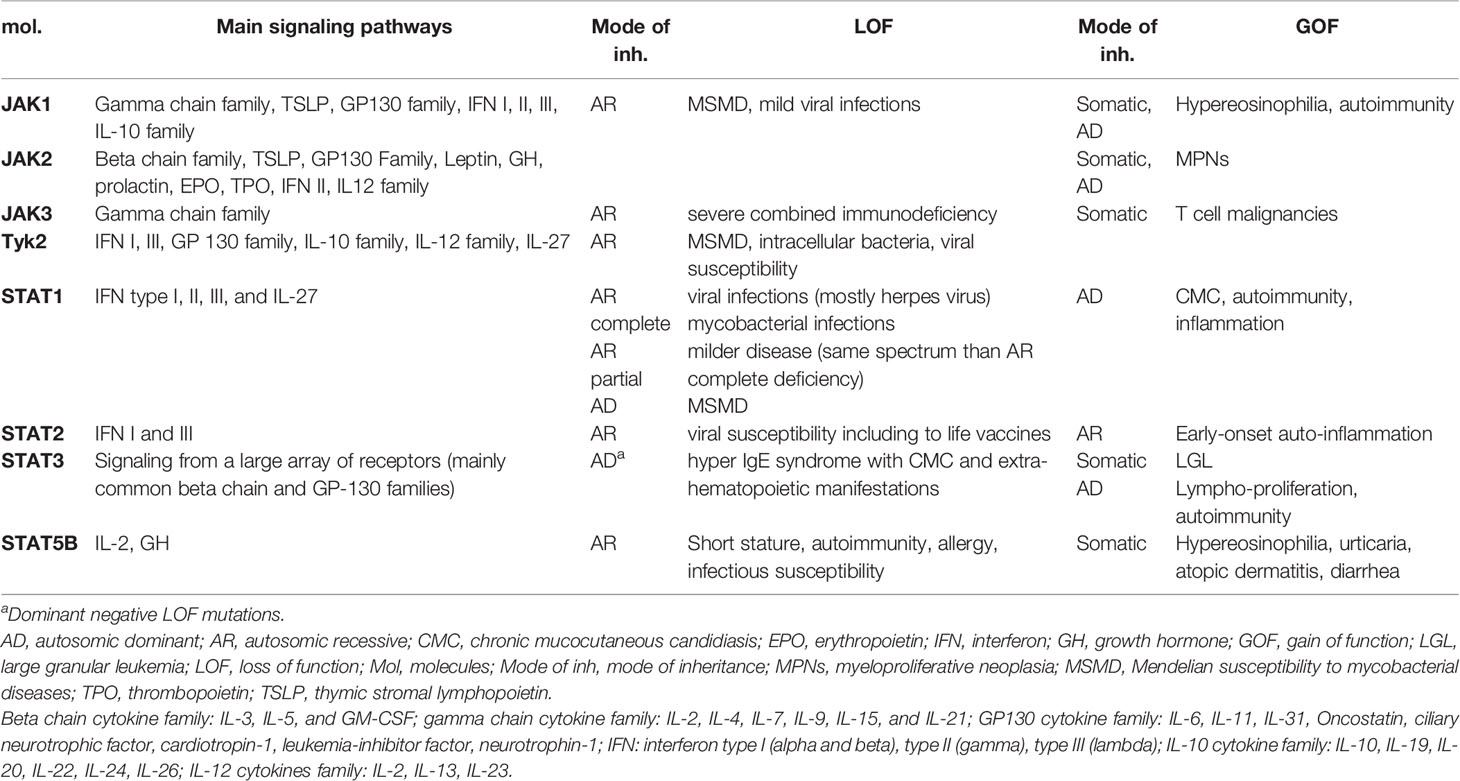
Table 1 Main IEIs and hematologic conditions in humans related to germline and somatic mutations in members of the JAK-STAT pathway.
The JAK-STAT signaling pathway is a direct, evolutionary conserved pathway allowing quick signaling from membrane to nucleus (10, 11). Numerous cytokine, interleukin, hormone, and growth factor (57 in total) signaling pathways rely on JAKs (11). These ligands bind to their cognate type I and II receptors, characterized by the lack of their own enzymatic activity that requires intracytoplasmic physically associated JAK tyrosine kinases. Each receptor uses a selective homo- or heterodimer composed of four JAK molecules (JAK1, JAK2, JAK3, TYK2) (10). Every JAK comprises four structural domains (the carboxy-terminal kinase domain, an adjacent pseudokinase domain, an Src homology 2 (SH2)-like domain, and the amino-terminal FERM domain which interacts with the cytosolic tail of the receptor) (10). Following engagement of the receptor, homo- or heterodimers of JAKs phosphorylate each other’s tyrosine residues as well as the intracellular tail of the receptor, creating a docking site that recruits downstream STAT DNA-binding proteins (10). The phosphorylation of STAT mediates dimerization, translocation, and accumulation into the nucleus and DNA binding to regulate gene expression. There are seven mammalian STATs: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6.
The implication of cytokines in many autoimmune diseases and the role of JAK2 GOF (gain of function) mutations in MPNs supported the rationale for development of molecules that block the kinase activity of JAKs, preventing phosphorylation of STAT (11).
Because JAKs are essential for signaling downstream from a wide range of substrates, the action of JAK inhibitors (JAKinibs) is large (12). The list of marketed drugs is shown in Table 2. Three JAKinibs of the first generation with poor specificity and a large range of cytokines signaling inhibition are approved in humans to treat autoimmune, inflammatory, and hematological conditions (13) such as rheumatoid arthritis (RA), psoriasis arthritis, ulcerative colitis (UC), MPNs, and acute graft versus host disease (GVHD) (11). Other sporadic autoimmune and hematological malignancies are good candidates for JAKinibs, and trials have been performed or are ongoing (in alopecia areata, atopic dermatitis, psoriasis, vitiligo, systemic sclerosis, spondyloarthritis, dermatomyositis, lupus, Crohn’s disease, primary biliary cholangitis, autoimmune hepatic disease, and type I diabetes).
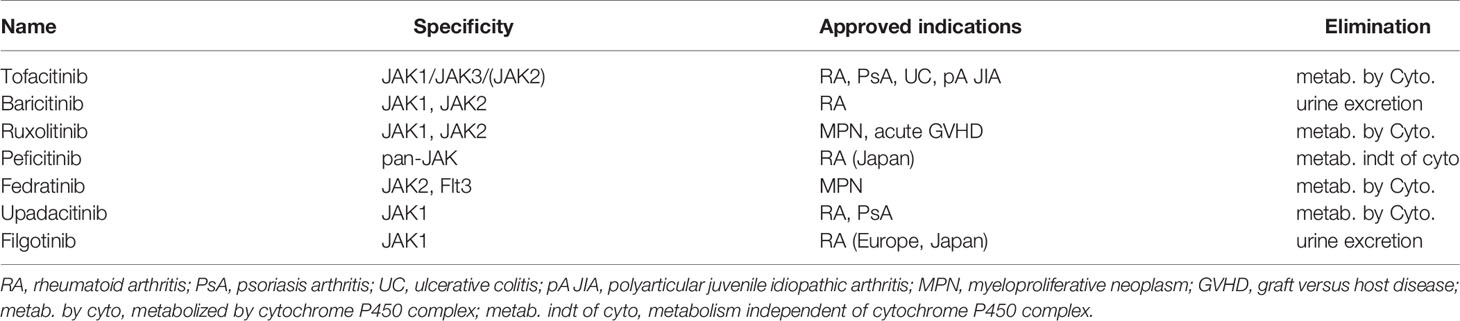
Table 2 Approved marketed JAKinibs.
Second-generation selective JAKinibs (11), with a more specific anti-JAK-1 (e.g., filgotinib and upadacitinib) or JAK2 (fedratinib) activity, narrow spectrum of action, and possibly improved safety, are now emerging. Topical and inhaled JAKinibs are also under investigation (11).
Most of the marketed JAKinibs are eliminated by metabolism via the cytochrome P450 enzymatic complex. This can lead to frequent drug–drug interactions (DDI) that need to be taken into consideration (14). In contrast to the others, baricitinib is mainly cleared by renal elimination through glomerular filtration and active secretion via transporters such as OAT3 (15). Thus far, inhibitors of this transporter such as probenecid can increase drug exposition of baricitinib. More broadly, marketed JAKinibs interact with drug transporters in several ways, which may trigger DDI, modify pharmacokinetics (PK), and increase risk of drug toxicities. These DDI are particularly relevant in the field of IEI where co-medications are frequent.
Pharmacokinetic (PK), pharmacodynamic (PD), and dose escalation studies have been performed mainly in adults and have defined the dose and treatment regimen for each JAKinib in approved conditions. FDA has approved (i) tofacitinib in children aged 2 years and older to treat patients with polyarticular juvenile idiopathic arthritis, (ii) baricitinib in children as young as 2 years for emergency use authorization for primary COVID-19 pneumonia in conjunction with remdesivir, and (iii) ruxolitinib down to age 12 for steroid refractory GVHD. PK studies allowed to define dosing regimen in this population for this condition (16). The ruxolitinib PK and preliminary PD phase I study has been performed in children with oncologic or hematologic malignancies with assessment of JAK2 and STAT5 phosphorylation as readout (17). Baricitinib PK and PD studies have been conducted in monogenic interferonopathies in a compassionate use program. Based on these data, a dose and scheme based on body weight and glomerular filtration rate have been proposed to optimize decrease of interferon (IFN) biomarkers (18).
Data from long-term extension studies and meta-analysis of tofacitinib and baricitinib in RA and ruxolitinib in MPNs provide important insight about the safe profile of these drugs, at least in adults (19–23) with infections being the most frequent followed by cytopenia and hyperlipidemia. Risk of side effects may vary depending on the underlying condition, the concomitant immunosuppression, the population (especially the age), and the dose of JAKinib required to control the disease (24).
The most frequent infectious complication regardless of the drug (tofacitinib, ruxolitinib, and baricitinib) and the underlying disease is reactivation of herpes zoster (23, 25). Other serious or opportunistic infections such as bacterial pneumonia, tuberculosis, BK virus nephropathy (24), and toxoplasmosis are also observed but with a lower incidence (26). One case of progressive multifocal leukoencephalopathy related to John Cunningham (JC) virus infection under ruxolitinib has been reported (27). The unexpected low incidence of serious infections may be due to the spared immune function relying on IL1, IL8, IL17, and TNFα with JAK-independent signaling. This may not be true when concomitant immunosuppressants are used in patients with IEIs.
Inhibition of JAK2 may be responsible for anemia and thrombocytopenia by interfering with erythropoietin and thrombopoietin signaling (10). Lymphopenia and a decreased number of NK cells can be observed with tofacitinib depending on the dose, likely due to the inhibition of JAK3-dependent T-cell functions.
A warning signal for increased risk of thromboembolic complications such as deep vein thrombosis and pulmonary embolism was raised by post-marketing safety studies of tofacitinib, ruxolitinib, and baricitinib, especially in patients carrying other risk factors for such complications (28, 29). These risks appear to be relatively low and might be disease-specific and dose-dependent. Increased low-density lipoprotein cholesterol and triglycerides are observed under JAKinib. Weight gain and increased body mass index (BMI) are reported with ruxolitinib (30) and tofacitinib (31). Such association with baricitinib is less clear. These side effects could be related to reduced postprandial leptine signaling due to JAK2 inhibition resulting in hyperphagia and contributing to weight gain, as demonstrated in mice (30).
JAKinibs might interfere with adaptive immune function in cancer immunosurveillance and with antineoplasic function of IFN. Thus far, data from a clinical trial in patients with RA or other immune-mediated diseases have not revealed an increased risk of hematological malignancies or solid tumors (32–34). A post-marketing surveillance study for tofacitinib conducted in >4,000 patients (50 years and older) with RA may suggest an increased risk of cancer (35). Additional long-term monitoring and caution is thus required. Long-term data in children are missing.
Independently to disease-specific recommendations, the following screening is advised before JAKinib initiation: complete blood count (CBC), liver function test, serum creatinine, fasting lipid panel, and screening for tuberculosis (36). BK viremia should also be excluded before treatment and monitored since BK nephropathy has been reported in patients treated for interferonopathies (24). Hepatitis B and C and HIV serology may be considered. Vaccination update prior to initiation of treatment is recommended, including varicella vaccine in case of negative serology and if the patient’s immune status allows for live vaccines. The latter will be contraindicated once treatment is initiated.
Ruxolitinib discontinuation syndrome was mainly reported in patients treated for MPNs (37–39). It is a life-threatening condition characterized by acute relapse of disease symptoms, sometimes mimicking septic shock that occurs 24 h to 3 weeks after drug cessation. To explain this withdrawal syndrome, it was shown that ruxolitinib blocked the dephosphorylation and ubiquitin degradation of JAK1 and JAK2, which accumulated and could lead to a notable activation of downstream signaling when ruxolitinib was removed (39). Similarly, we observed severe relapse of symptoms in patients with monogenic interferonopathies under ruxolitinib when treatment was temporarily stopped (references (40, 41) and unreported observations). Among baricitinib phase 3 trials in patients with RA, a brief interruption of baricitinib was associated with a minor increase of RA symptoms (42) while rebound phenomena were reported in animal models following abrupt withdrawal of JAK1 inhibitor oclacitinib (43) (a first-generation JAKinib prescribed in dogs). The risk of discontinuation syndrome may vary depending on the condition treated and the JAKinib prescribed, but it should not be overlooked. This also indicates the need for a careful tapering of the drug when JAKinib is interrupted.
Growth hormones signal through the JAK-STAT pathway via JAK2 (10). The JAK-STAT pathway is also involved in bone homeostasis in various ways since many cytokines with bone-protective and bone-degrading properties signal through JAK-STAT. Of note, Adam et al. showed in various models of mice at steady states and in inflammatory conditions that tofacitinib and baricitinib displayed a bone-sparing effect (44).
These last years, monogenic diseases associated with gain of function of cytokine signaling members and in particular involving the JAK-STAT pathway have been described to cause overwhelming immune dysregulation conditions (Table 1). These diseases represent good candidates for treatment with JAKinibs (Figure 1). IEIs associated with overproduction of cytokine signaling by the JAK-STAT pathway such as interferonopathies—a group of recently identified monogenic auto-inflammatory diseases characterized by excessive secretion of type I IFN—are also good candidates with growing experiences reported in the literature. All published patients are summarized in the Supplementary Table 1. In addition, JAKinibs are promising in primary hemophagocytic lymphohistiocytosis (HLH) (45), a family of diseases characterized by hyper-immune activation and massive release of inflammatory cytokines, mostly but not exclusively IFNγ. Because this topic will be covered in the framework of this series of mini-review, it will not be addressed here.
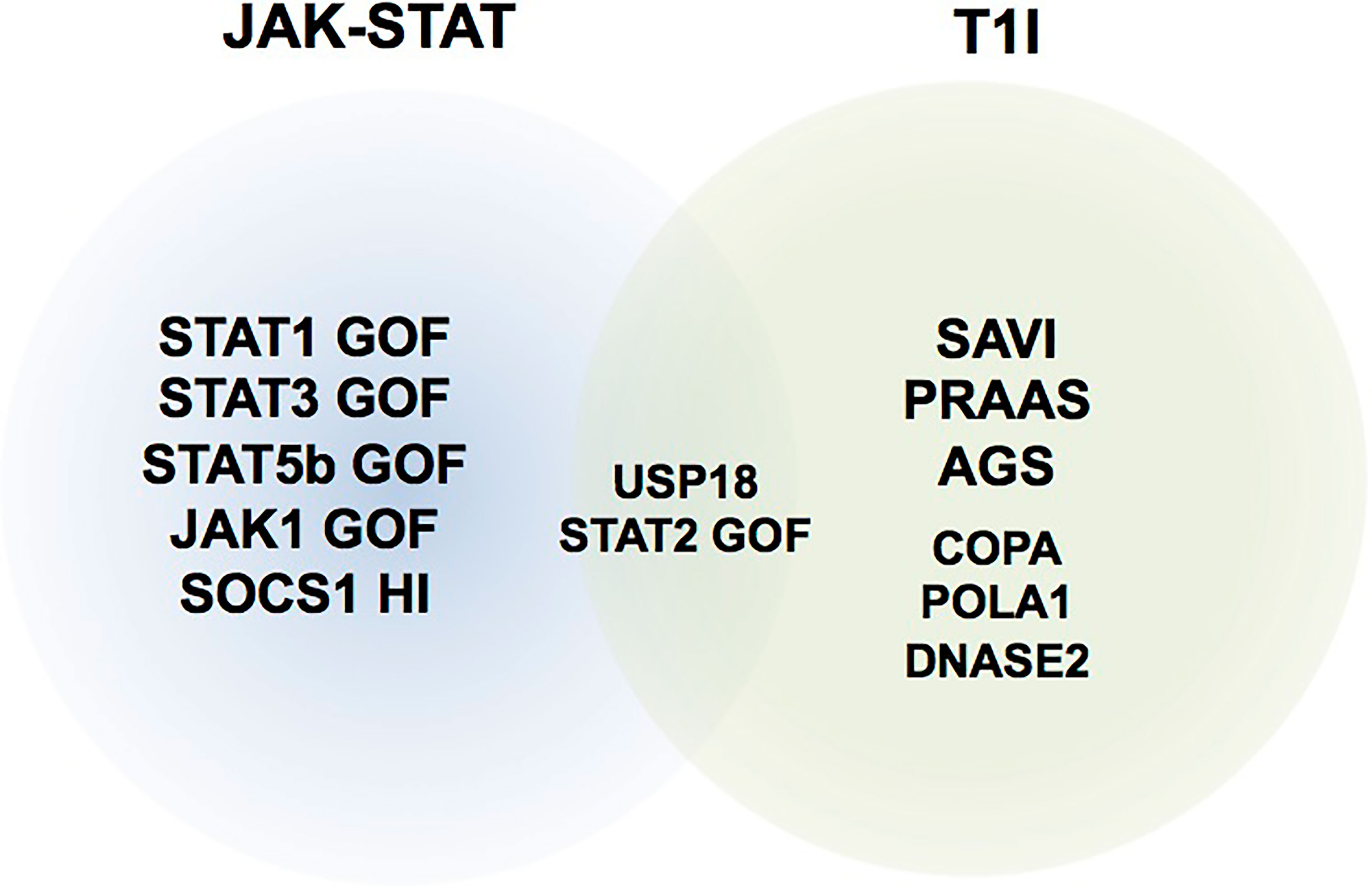
Figure 1 Representative schematic of inborn errors of immunity (IEI) where JAK inhibitors have been used. T1I, type I interferonopathies; JAK-STAT, IEIs related to mutations in components of the JAK-STAT pathway. HI, haploinsufficiency.
Heterozygous STAT1 mutations that confer hyper-responsiveness to stimulation are associated with a mixed phenotype characterized by infection, autoimmunity, and inflammation. Chronic mucocutaneous candidiasis (CMC) occurs in almost all patients, but a broader infectious susceptibility to invasive fungal infections and bacterial and viral infections is frequent (3, 46, 47). Various autoimmune and inflammatory manifestations are reported in one-third of the patients such as autoimmune endocrinopathies, hepatitis, cytopenia, skin involvement, and pseudo-IPEX enteropathy (48). Vascular aneurysms further enlarge the phenotype. The severity of the disease is variable with a broad spectrum. The increased risk of autoimmunity in these patients might be explained by an enhanced transcription of IFN-induced genes due to signal-induced increased levels of phosphorylated STAT1. CMC can be explained by a decreased proportion of circulating TH17 cells, but the molecular mechanism accounting for this decrease remains to be elucidated (49). Management of these patients included long-lasting antifungal and antibacterial prophylaxis and, when indicated, immunoglobulin replacement. Use of immunosuppression in case of autoimmunity needs to be balanced by the increased infectious susceptibility, and many treatments have failed. Experiences with hematopoietic stem cell transplantation (HSCT) are limited and associated with high morbidity and mortality (50, 51). Use of JAKinib was first reported in 2015 by Higgins et al. in an adult patient suffering from alopecia areata and CMC in whom ruxolitinib treatment allowed complete resolution of both (52). The latter relapsed after cessation of the drug. Since this first promising observation, 11 reports (see Supplementary Table 1) shared their experiences with JAKinibs in 18 patients suffering from STAT1 GOF (12 children from 1.1 to 17 years of age and 6 adults), mainly treated by ruxolitinib (n = 17) and baricitinib (n = 1). Forbes et al. reported the largest multicentric retrospective series of 11 patients (53). All patients had experienced CMC or other chronic fungal infections associated with recurrent or chronic viral and/or bacterial infections. Inflammation (mainly inflammatory bowel disease and chronic ulcers) and various autoimmune diseases (autoimmune cytopenia, aplastic anemia, hepatitis, type I diabetes, thyroiditis, etc.) were noticed in 9 and 10 patients respectively. Dysimmune manifestations required several lines of immunosuppressive drugs with limited benefits in most patients. Doses of ruxolitinib were variable, from 0.3 to 1 mg/kg (in two divided doses) in children and from 5 to 40 mg (in one or two divided doses) with a median follow-up of 5 months (range 0.5 to 14). Significant improvement of all main features of the disease was noticed. CMC resolved or improved while chronic severe dermatophytosis and disseminated coccidioidomycosis reported in one patient each got worse (54). Inflammatory and autoimmune manifestations dramatically improved including one teenage patient with reversible type 1 diabetes under ruxolitinib allowing long-lasting remission (55). The benefit of ruxolitinib on TH17 cell differentiation or IL-17 production remains controversial (49). Despite these very encouraging clinical results, many questions and uncertainties remain. The follow-up of the reported patients is short, and the optimal dose especially in children and the long-term effects in this particular IEI are unknown. The management of drug interactions of ruxolitinib with drugs metabolized, inhibiting or enhancing similar cytochrome metabolic pathways, in particular azoles and azithromycin, needs special consideration.
STAT3 hyperactivity due to germline heterozygous GOF mutations causes an early-onset immune dysregulation syndrome characterized by lymphoproliferation and a broad spectrum of autoimmune manifestations (4). Clinical phenotype is variable in terms of organ involved, age at onset, and severity. No clear genotype–phenotype correlation is described. In a systematic review describing clinical aspects of 42 patients from 18 publications, hematologic diseases was the most frequent finding (83% of cases), especially autoimmune cytopenia (56). Immunodeficiency with infection susceptibility and hypogammaglobulinemia were observed in 67% of cases. Other features included type I diabetes, enteropathy, interstitial lung disease (ILD) that can be life-threatening (56), arthritis, and growth failure.
Molecular consequences of STAT3 GOF mutations are variable (57). Autoimmunity might be caused by increased STAT3 activity, especially in response to IL-6 and IL-21, resulting in enhancement of TH17 cell fate determination (4, 58). It has also been suggested that reduction in regulatory T cell number and function caused by impaired cytokine-induced phosphorylation of STAT5 could lead to autoimmune manifestation (4).
Before the identification of STAT3 GOF mutations, patients were treated with years of various nonspecific immunosuppressive agents that were overall ineffective. Use of an anti-IL-6 receptor antibody (i.e., tocilizumab) was first reported (4), showing promising results, but the effect seems to plateau. Some patients received HSCT with high mortality (56). Experiences of JAKinibs were published in 13 patients in 6 reports suffering from STAT3 GOF (children from 1 month to 15 years of age) (53, 59–62), in combination with tocilizumab in 6 cases. Indications were mainly lymphoproliferation, autoimmune cytopenia, enteropathy, ILD, and arthritis not controlled with other therapies. Ten patients received ruxolitinib and three tofacitinib with various dosages. Ten patients had significant clinical improvement with a spectacular effect in some cases, i.e., cessation of oxygen therapy (including mechanical ventilation) and radiographic resolution of ILD, or independence of parenteral nutrition for enteropathy (53, 62). Three patients had no or minimal response. In the first case, respiratory failure progressed despite ruxolitinib leading to death. In the second, ruxolitinib was introduced during severe sepsis with multiorgan failure (53). In the last case, indication was pure red cell aplasia and ruxolitinib led to minor improvement (61). Tolerance was good in most cases. Adverse effects included thrombocytopenia, transiently increased transaminase and/or bilirubin levels, and influenza infection. In all cases, the follow-up of the reported patients is short (Supplementary Table 1).
Type I interferonopathies are a group of recently identified monogenic auto-inflammatory diseases characterized by constitutive signaling of type I IFN resulting from their excessive or dysregulated secretion. There are more than 25 monogenic diseases that are associated with increased production of type I IFNs (63) which in turn drive the expression of IFN-stimulated genes (ISG) (also called IFN signature) through the engagement of a common receptor that subsequently activates JAK1 and Tyk2. The concept of type I interferonopathies was raised in 2011 and supports the hypothesis that some if not all symptoms of these syndromes would be related to excessive or dysregulated type I IFN production warranting therapeutic intervention with drugs targeting this pathway (64).
STING-associated vasculopathy with onset in infancy (SAVI) was described in 2014 and is related to GOF mutation in STING1 (65, 66). Most patients harbor heterozygous mutations, but few patients with homozygous GOF mutation have been recently described (67, 68). STING is a central component in DNA sensing that leads to induction of type I IFNs, which in turn drives the expression of ISG. In a review from December 2020, 70 patients in 49 families are reported (69). The disease is variably characterized by early-onset systemic inflammation with fever, skin vasculopathy, and ILD leading to early-onset pulmonary fibrosis. Arthralgia or arthritis is also frequent, and infections are observed in patients with severe skin lesions and/or lung damage. Chronic elevated inflammatory markers and increased ISG are constant while T cell deficiency with lymphopenia and defect of T cell proliferation are also observed. SAVI is minimally responsive to conventional immunosuppressive therapies and thus is associated with a significant morbidity and increased mortality.
In vitro, the three JAKinibs tested on the patient’s lymphocytes (ruxolitinib, tofacitinib, and baricitinib) were able to block the constitutive phosphorylation of STAT1 (65). Based on the hypothesis that inhibiting JAK1 signaling would slow down IFN signaling and thus improve disease-related symptoms, JAKinibs have been proposed to patients suffering from SAVI. To date, 34 patients (14 female) (16 reports) received JAKinibs, i.e., ruxolitinib, baricitinib, and tofacitinib, in 19, 11, and 5 patients, respectively. Patients were less than 2 years in 7 cases and between 2 and 10 years in 13, while 8 were aged 11 to 18 years and 6 were adults. Skin involvement was reported in 22 patients including 9 with severe lesions (ulcers, ischemia of extremities). Lung disease was noticed in all but four patients; arthritis and failure to thrive were reported in eight patients each. All patients with appropriate clinical information provided presented systemic inflammation. Treatment failed on eight occasions, in four patients affected by severe lung involvement and respiratory failure at treatment initiation who died (n = 3) or required lung transplantation (n = 1), in two patients treated with a low dose of ruxolitinib (absence of response), and two patients worsened/deteriorated under tofacitinib (70, 71). Among the remaining patients, skin involvement improved in all cases with complete or partial remission, depending on the initial degree of severity. Lung involvement was also improved, and arthritis mostly resolved. General status and quality-of-life improvements were noticed by all authors. When reported, ISG were not normalized under treatment (24, 40, 41). Doses of the JAKinibs were extremely variable, from 0.2 to 1.5 mg/kg/day for ruxolitinib (10 to 35 mg/m2/day) in two divided doses, and from 2.5 to 5 mg bid for tofacitinib. Doses and scheme of administration of baricitinib were supported by PK studies associated with in vitro assessment of IFN biomarkers and more homogeneous (18). The main reported side effects were infectious (shingle, rhinovirus, and other viral respiratory infection, rotavirus enteritidis). Aspergilloma in lung cavities was also noticed (reference (41) and unpublished observations). Papillary edema and ruxolitinib discontinuation syndrome were also reported (40, 41).
Aicardi-Goutières syndrome (AGS) is the paradigm of the type I interferonopathies and is associated with high morbidity and mortality related to the prominent central nervous system involvement. AGS can be caused by any of the nine AGS-related genes (63, 72), with all the proteins encoded involved in either the processing or the sensing of nucleic acids. Given the marked efficacy of JAK1/2 inhibition in chilblain lupus due to TREX1 deficiency (73, 74) or STING GOF, the potential benefit of this treatment on the neurological component of AGS was also considered, despite uncertainty about the bioavailability of the drug in the central nervous system (75–78). Last year, the group of Adeline Vanderver published a large open-label study involving 35 AGS patients treated with baricitinib over a minimal period of 12 months (79). A clear efficacy was observed on skin vasculopathy, and neurological improvement was reported, although evaluation of neurological function is challenging especially in patients with differential onset and disease progression. Related to this, Neven et al. (78) reported an AGS child who presented first neurological symptoms at 14 months, despite that treatment with ruxolitinib started at age 5 months when the child was asymptomatic. This raises the question of (i) the drug penetration in the central nervous system [concentration of ruxolitinib in the cerebrospinal was measured at 10% of that in the blood (78)] and (ii) the role of additional triggers of the disease (e.g., infections and vaccinations). We note here that careful monitoring of pulmonary hypertension is recommended in patients treated with JAKinib considering the high risk of this AGS-related complication (79, 80) that can be exacerbated by JAK inhibition (81).
Heterozygous mutations in the gene encoding the coatomer subunit alpha (COPA) were described in 2015 to underlie an auto-inflammatory disorder associating mainly ILD and/or diffuse alveolar hemorrhage, joint involvement, and lupus-like nephritis (82). The disease is rare (less than 70 patients reported) and characterized by a high frequency of clinical non-penetrance (up to 25%) (69). COPA is part of a complex (COPI) involved in the intracellular trafficking of cargo proteins (83), and mutations in COPA were associated with enhanced endoplasmic reticulum (ER) stress and priming of a TH17 response (82). More recently, a positive IFN signature was recorded in the blood of several COPA patients (84). Studies from four different teams demonstrated that mutations in COPA led to STING-mediated IFN signaling and defined a role for wild-type COPA in STING retrieval from the Golgi back to the ER to prevent chronic immune activation (85–88). Considering these data, a few COPA patients (five in total) have been treated with JAKinib (85, 89–91) and follow-up on treatment has been reported for 3 (89–91). Complete or partial remission has been achieved for arthritis in two patients (89, 91) while a major improvement was observed in a COPA patient with a severe diffuse alveolar hemorrhage (90). However, she has subsequently experienced recurrences of diffuse alveolar hemorrhage associated with progression toward lung fibrosis on chest CT scan (authors’ personal observation).
In the large observational study of the use of baricitinib in type I interferonopathies published by Sanchez et al. (24), 10 patients with PRAAS (previously referred to as CANDLE-Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature), harboring monogenic mutations in PSMB8 (n = 6) and PSMB4 (n = 1), or digenic mutations in PSMB4/PSMB9 (n = 2) and PSMA3/PSMB8 (n = 1), were treated, with a major clinical improvement as compared to SAVI. Indeed, half of the patients reached durable remission with no disease symptoms, normalization of inflammatory markers, and discontinuation of steroid therapy (24). Interestingly, these five patients (who achieved remission) also normalized their IFN score, a unique observation to date in the literature in monogenic IFN-related diseases treated with JAKinib. Of note, one PRAAS patient discontinued the treatment due to acute renal injury related to BK virus infection and subsequently died after a relapse of his disease and a respiratory tract infection. Nevertheless, these very encouraging results in PRAAS were further confirmed by two single patient cases treated with another JAKinib (tofacitinib) (91, 92).
Defective negative regulation of the type IFN response, for example due to LOF (loss of function) mutations in USP18 and more recently GOF mutations in STAT2 (93, 94), supports the concept of type I interferonopathies. LOF mutations in USP18 have been described in six patients from three unrelated families (95, 96) to cause pseudo-TORCH syndrome, a severe condition mimicking the phenotype secondary to transplacental transmission of pathogens referred to as TORCH (97). USP18 is recruited by STAT2 to the type IFN receptor subunit IFNAR2 where it competes with JAK1 to enable negative-feedback control of type I IFN signaling (98). Ruxolitinib was trialed in one neonate with inherited USP18 deficiency and was associated with a promising complete recovery with 2 years of follow-up (96).
STAT2 homozygous GOF mutations were reported in three patients from two unrelated families characterized by a severe early-onset inflammation of type I interferonopathies (93, 94). This novel disease largely phenocopies USP18 deficiency in clinical presentation and molecular mechanism by an impaired regulation of late cellular response to type I IFN. Ruxolitinib was used in two cases and led to partial response, but these patients died despite this treatment (93).
The use of JAKinib in these rare inherited diseases with impaired IFN signaling regulation deserves additional reporting.
Few other case reports have described the use of JAKinib in the context of monogenic type I interferonopathies [i.e., due to DNASE2 (91, 99) and POLA1 (100) mutations, respectively]. These single observations together with a short follow-up do not allow any conclusion to be reached in these severe disorders, of which the pathophysiology is not yet completely understood.
JAK1 GOF mutations leading to activation of multiple STAT proteins were recently reported in four patients from two unrelated families (5, 6). These mutations give rise to a complex immune dysregulatory syndrome characterized by severe atopic dermatitis, profound eosinophilia with eosinophilic organ infiltration, failure to thrive, and autoimmune manifestations such as membranous nephropathy and thyroiditis. The description of this complex phenotype may reflect the scope of the various cytokine signaling pathways involved. JAKinib (ruxolitinib in two cases and tofacitinib in one case) resulted in remarkable improvement in clinical disease and biological abnormalities.
Germline LOF heterozygous SOCS1 mutations leading to haploinsufficiency were also recently described to be associated with a dominantly inherited predisposition to early-onset autoimmune disease including especially autoimmune cytopenias and systemic lupus, related to cytokine hypersensitivity (i.e., IFNγ, IL-2, and IL-4) in immune cells (7, 101). Given that these mutations were associated with uncontrolled JAK-STAT activation after cytokine stimulation, JAK1/2 inhibition was trialed and showed efficacy in vitro and ex-vivo. Until now, only one patient with SOCS1 deficiency and systemic lupus was treated with JAKinib (baricitinib), showing clinical remission, decrease in anti-DNA autoantibodies, and good tolerance (7). Therefore, JAK inhibitors may represent targeted therapies of value for SOCS1-insufficient patients.
Somatic STAT5B GOF mutations, while frequently described in T lymphocyte-derived neoplasms (102, 103), have been reported in three patients with IEI associating early-onset atopic disease, hyper-eosinophilia, urticaria, dermatitis, and diarrhea (104, 105). Use of ruxolitinib in two patients resulted in remarkable improvement of clinical symptoms and hypereosinophila (105).
The marketed JAKinib as well as the new more specific inhibitors in development find numerous indications in the field of IEIs, whether related to an intrinsic defect of the JAK-STAT pathway [mutations of one of its components or of a molecule involved in its regulation (i.e., SOCS1)] or extrinsic due to hypersecretion of one or more cytokines signaling through the JAK-STAT pathway. The clinical and biological data of JAKinibs in these conditions summarized above are very promising and open interesting perspectives, but remain preliminary, sporadic, and too heterogeneous to give firm and definitive therapeutic recommendations. Although this analysis does not raise red flags in terms of safety, particularly in terms of infection, caution remains in the context of IEIs. These data highlight the need for prospective, if possible multicenter, evaluation, which would provide answers to the most burning questions: i) what are the risk of infections in the short and long term? ii) what is the long-term safety of JAKinibs in children, particularly with regard to growth and bone metabolism? iii) are these molecules equivalent in terms of safety and efficacy? iv) how does this targeted treatment fit into the management of these conditions? Is it a long-term treatment or a bridge to transplant? v) what dosage and what administration scheme should be proposed according to the pathology and the age of the patient? Should the benefit of the treatment be monitored clinically and/or biologically, and what are the best readouts? The issue of blood–brain barrier crossing will also be important to consider in the context of AGS and HLH. Finally, the benefit of next-generation JAKinibs will need to be assessed. There are obviously more questions than answers, and everything must be done in the coming years to collectively provide answers to these matters.
All authors contributed to the article and approved the submitted version.
M-LF received a grant from the Institut National de la Santé et de la Recherche Médicale (reference: 000427993) and acknowledges La Fondation Square.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AW declared a past co-authorship with the author BN to the handling editor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.717388/full#supplementary-material
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
2. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J Clin Immunol (2021) 41:666–79. doi: 10.1007/s10875-021-00980-1
3. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 Gain-of-Function Mutations Underlie an Unexpectedly Broad Clinical Phenotype. Blood (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
4. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-Onset Lymphoproliferation and Autoimmunity Caused by Germline STAT3 Gain-of-Function Mutations. Blood (2015) 125:591–9. doi: 10.1182/blood-2014-09-602763
5. Del Bel KL, Ragotte RJ, Saferali A, Lee S, Vercauteren SM, Mostafavi SA, et al. JAK1 Gain-of-Function Causes an Autosomal Dominant Immune Dysregulatory and Hypereosinophilic Syndrome. J Allergy Clin Immunol (2017) 139:2016–20.e5. doi: 10.1016/j.jaci.2016.12.957
6. Gruber CN, Calis JJA, Buta S, Evrony G, Martin JC, Uhl SA, et al. Complex Autoinflammatory Syndrome Unveils Fundamental Principles of JAK1 Kinase Transcriptional and Biochemical Function. Immunity (2020) 53:672–84.e11. doi: 10.1016/j.immuni.2020.07.006
7. Hadjadj J, Castro CN, Tusseau M, Stolzenberg M-C, Mazerolles F, Aladjidi N, et al. Early-Onset Autoimmunity Associated With SOCS1 Haploinsufficiency. Nat Commun (2020) 11:5341. doi: 10.1038/s41467-020-18925-4
8. Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N Engl J Med (2005) 352:1779–90. doi: 10.1056/NEJMoa051113
9. Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 Mutations in Large Granular Lymphocytic Leukemia. N Engl J Med (2012) 366:1905–13. doi: 10.1056/NEJMoa1114885
10. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and Consequences of Jak-STAT Signaling in the Immune System. Nat Immunol (2017) 18:374–84. doi: 10.1038/ni.3691
11. Gadina M, Chisolm DA, Philips RL, McInness IB, Changelian PS, O’Shea JJ. Translating Jaks to Jakinibs. J Immunol (2020) 204:2011–20. doi: 10.4049/jimmunol.1901477
12. Gadina M, Le MT, Schwartz DM, Silvennoinen O, Nakayamada S, Yamaoka K, et al. Janus Kinases to Jakinibs: From Basic Insights to Clinical Practice. Rheumatol (Oxford) (2019) 58:i4–16. doi: 10.1093/rheumatology/key432
13. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK Inhibition as a Therapeutic Strategy for Immune and Inflammatory Diseases. Nat Rev Drug Discov (2017) 17:78. doi: 10.1038/nrd.2017.267
14. Veeravalli V, Dash RP, Thomas JA, Babu RJ, Madgula LMV, Srinivas NR. Critical Assessment of Pharmacokinetic Drug-Drug Interaction Potential of Tofacitinib, Baricitinib and Upadacitinib, the Three Approved Janus Kinase Inhibitors for Rheumatoid Arthritis Treatment. Drug Saf (2020) 43:711–25. doi: 10.1007/s40264-020-00938-z
15. Alim K, Bruyère A, Lescoat A, Jouan E, Lecureur V, Le Vée M, et al. Interactions of Janus Kinase Inhibitors With Drug Transporters and Consequences for Pharmacokinetics and Toxicity. Expert Opin Drug Metab Toxicol (2021) 17:259–71. doi: 10.1080/17425255.2021.1862084
16. Ruperto N, Brunner HI, Zuber Z, Tzaribachev N, Kingsbury DJ, Foeldvari I, et al. Pharmacokinetic and Safety Profile of Tofacitinib in Children With Polyarticular Course Juvenile Idiopathic Arthritis: Results of a Phase 1, Open-Label, Multicenter Study. Pediatr Rheumatol Online J (2017) 15:86. doi: 10.1186/s12969-017-0212-y
17. Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, et al. A Phase 1 Dosing Study of Ruxolitinib in Children With Relapsed or Refractory Solid Tumors, Leukemias, or Myeloproliferative Neoplasms: A Children’s Oncology Group Phase 1 Consortium Study (ADVL1011). Pediatr Blood Cancer (2015) 62:1717–24. doi: 10.1002/pbc.25575
18. Kim H, Brooks KM, Tang CC, Wakim P, Blake M, Brooks SR, et al. Pharmacokinetics, Pharmacodynamics, and Proposed Dosing of the Oral JAK1 and JAK2 Inhibitor Baricitinib in Pediatric and Young Adult CANDLE and SAVI Patients. Clin Pharmacol Ther (2018) 104:364–73. doi: 10.1002/cpt.936
19. Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, et al. Long-Term Safety of Tofacitinib Up to 9.5 Years: A Comprehensive Integrated Analysis of the Rheumatoid Arthritis Clinical Development Programme. RMD Open (2020) 6. doi: 10.1136/rmdopen-2020-001395
20. Burmester GR, Curtis JR, Yun H, FitzGerald O, Winthrop KL, Azevedo VF, et al. An Integrated Analysis of the Safety of Tofacitinib in Psoriatic Arthritis Across Phase III and Long-Term Extension Studies With Comparison to Real-World Observational Data. Drug Saf (2020) 43:379–92. doi: 10.1007/s40264-020-00904-9
21. Harrison C, Kiladjian J-J, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK Inhibition With Ruxolitinib Versus Best Available Therapy for Myelofibrosis. N Engl J Med (2012) 366:787–98. doi: 10.1056/NEJMoa1110556
22. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N Engl J Med (2012) 366:799–807. doi: 10.1056/NEJMoa1110557
23. Winthrop KL, Harigai M, Genovese MC, Lindsey S, Takeuchi T, Fleischmann R, et al. Infections in Baricitinib Clinical Trials for Patients With Active Rheumatoid Arthritis. Ann Rheum Dis (2020) 79:1290–7. doi: 10.1136/annrheumdis-2019-216852
24. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 Inhibition With Baricitinib in the Treatment of Autoinflammatory Interferonopathies. J Clin Invest (2018) 128:3041–52. doi: 10.1172/JCI98814
25. Bechman K, Subesinghe S, Norton S, Atzeni F, Galli M, Cope AP, et al. A Systematic Review and Meta-Analysis of Infection Risk With Small Molecule JAK Inhibitors in Rheumatoid Arthritis. Rheumatol (Oxford) (2019) 58:1755–66. doi: 10.1093/rheumatology/kez087
26. Wollenhaupt J, Lee E-B, Curtis JR, Silverfield J, Terry K, Soma K, et al. Safety and Efficacy of Tofacitinib for Up to 9.5 Years in the Treatment of Rheumatoid Arthritis: Final Results of a Global, Open-Label, Long-Term Extension Study. Arthritis Res Ther (2019) 21:89. doi: 10.1186/s13075-019-1866-2
27. Ballesta B, González H, Martín V, Ballesta JJ. Fatal Ruxolitinib-Related JC Virus Meningitis. J Neurovirol (2017) 23:783–5. doi: 10.1007/s13365-017-0558-4
28. Xie W, Huang Y, Xiao S, Sun X, Fan Y, Zhang Z. Impact of Janus Kinase Inhibitors on Risk of Cardiovascular Events in Patients With Rheumatoid Arthritis: Systematic Review and Meta-Analysis of Randomised Controlled Trials. Ann Rheum Dis (2019) 78:1048–54. doi: 10.1136/annrheumdis-2018-214846
29. Yates M, Mootoo A, Adas M, Bechman K, Rampes S, Patel V, et al. Venous Thromboembolism Risk With JAK Inhibitors: A Meta-Analysis. Arthritis Rheumatol (2021) 73:779–88. doi: 10.1002/art.41580
30. Mollé N, Krichevsky S, Kermani P, Silver RT, Ritchie E, Scandura JM. Ruxolitinib can Cause Weight Gain by Blocking Leptin Signaling in the Brain via JAK2/STAT3. Blood (2020) 135:1062–6. doi: 10.1182/blood.2019003050
31. Novikova DS, Udachkina HV, Markelova EI, Kirillova IG, Misiyuk AS, Demidova NV, et al. Dynamics of Body Mass Index and Visceral Adiposity Index in Patients With Rheumatoid Arthritis Treated With Tofacitinib. Rheumatol Int (2019) 39:1181–9. doi: 10.1007/s00296-019-04303-x
32. Maneiro JR, Souto A, Gomez-Reino JJ. Risks of Malignancies Related to Tofacitinib and Biological Drugs in Rheumatoid Arthritis: Systematic Review, Meta-Analysis, and Network Meta-Analysis. Semin Arthritis Rheum (2017) 47:149–56. doi: 10.1016/j.semarthrit.2017.02.007
33. Huss V, Bower H, Wadström H, Frisell T, Askling J, ARTIS group. Short- and Longer-Term Cancer Risks With Biologic and Targeted Synthetic Disease Modifying Antirheumatic Drugs as Used Against Rheumatoid Arthritis in Clinical Practice. Rheumatol (Oxford) (2021). doi: 10.1093/rheumatology/keab570
34. Olivera PA, Lasa JS, Bonovas S, Danese S, Peyrin-Biroulet L. Safety of Janus Kinase Inhibitors in Patients With Inflammatory Bowel Diseases or Other Immune-Mediated Diseases: A Systematic Review and Meta-Analysis. Gastroenterology (2020) 158:1554–73.e12. doi: 10.1053/j.gastro.2020.01.001
35. Pfizer Shares Co-Primary Endpoint Results From Post-Marketing Required Safety Study of XELJANZ® (Tofacitinib) in Subjects With Rheumatoid Arthritis (Ra) | pfpfizeruscom. Available at: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-shares-co-primary-endpoint-results-post-marketing.
36. Damsky W, Peterson D, Ramseier J, Al-Bawardy B, Chun H, Proctor D, et al. The Emerging Role of Janus Kinase Inhibitors in the Treatment of Autoimmune and Inflammatory Diseases. J Allergy Clin Immunol (2021) 147(3):814–26. doi: 10.1016/j.jaci.2020.10.022
37. Tefferi A, Pardanani A. Serious Adverse Events During Ruxolitinib Treatment Discontinuation in Patients With Myelofibrosis. Mayo Clin Proc (2011) 86:1188–91. doi: 10.4065/mcp.2011.0518
38. Beauverd Y, Samii K. Acute Respiratory Distress Syndrome in a Patient With Primary Myelofibrosis After Ruxolitinib Treatment Discontinuation. Int J Hematol (2014) 100:498–501. doi: 10.1007/s12185-014-1628-5
39. Tvorogov D, Thomas D, Liau NPD, Dottore M, Barry EF, Lathi M, et al. Accumulation of JAK Activation Loop Phosphorylation is Linked to Type I JAK Inhibitor Withdrawal Syndrome in Myelofibrosis. Sci Adv (2018) 4:eaat3834. doi: 10.1126/sciadv.aat3834
40. Frémond M-L, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, et al. Efficacy of the Janus Kinase 1/2 Inhibitor Ruxolitinib in the Treatment of Vasculopathy Associated With TMEM173-Activating Mutations in 3 Children. J Allergy Clin Immunol (2016) 138:1752–5. doi: 10.1016/j.jaci.2016.07.015
41. Frémond M-L, Hadchouel A, Berteloot L, Melki I, Bresson V, Barnabei L, et al. Overview of STING-Associated Vasculopathy With Onset in Infancy (SAVI) Among 21 Patients. J Allergy Clin Immunol Pract (2021) 9:803–18.e11. doi: 10.1016/j.jaip.2020.11.007
42. Emery P, Tanaka Y, Cardillo T, Schlichting D, Rooney T, Beattie S, et al. Temporary Interruption of Baricitinib: Characterization of Interruptions and Effect on Clinical Outcomes in Patients With Rheumatoid Arthritis. Arthritis Res Ther (2020) 22:115. doi: 10.1186/s13075-020-02199-8
43. Fukuyama T, Ganchingco JR, Bäumer W. Demonstration of Rebound Phenomenon Following Abrupt Withdrawal of the JAK1 Inhibitor Oclacitinib. Eur J Pharmacol (2017) 794:20–6. doi: 10.1016/j.ejphar.2016.11.020
44. Adam S, Simon N, Steffen U, Andes FT, Scholtysek C, Müller DIH, et al. JAK Inhibition Increases Bone Mass in Steady-State Conditions and Ameliorates Pathological Bone Loss by Stimulating Osteoblast Function. Sci Transl Med (2020) 12(530):eaay4447. doi: 10.1126/scitranslmed.aay4447
45. Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic Effect of JAK1/2 Blockade on the Manifestations of Hemophagocytic Lymphohistiocytosis in Mice. Blood (2016) 128:60–71. doi: 10.1182/blood-2016-02-700013
46. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LAB, Gilissen C, et al. STAT1 Mutations in Autosomal Dominant Chronic Mucocutaneous Candidiasis. N Engl J Med (2011) 365:54–61. doi: 10.1056/NEJMoa1100102
47. Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-Of-Function Human STAT1 Mutations Impair IL-17 Immunity and Underlie Chronic Mucocutaneous Candidiasis. J Exp Med (2011) 208:1635–48. doi: 10.1084/jem.20110958
48. Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant Gain-of-Function STAT1 Mutations in FOXP3 Wild-Type Immune Dysregulation-Polyendocrinopathy-Enteropathy-X-Linked-Like Syndrome. J Allergy Clin Immunol (2013) 131:1611–23. doi: 10.1016/j.jaci.2012.11.054
49. Okada S, Asano T, Moriya K, Boisson-Dupuis S, Kobayashi M, Casanova J-L, et al. Human STAT1 Gain-Of-Function Heterozygous Mutations: Chronic Mucocutaneous Candidiasis and Type I Interferonopathy. J Clin Immunol (2020) 40:1065–81. doi: 10.1007/s10875-020-00847-x
50. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic Stem Cell Transplantation in Patients With Gain-of-Function Signal Transducer and Activator of Transcription 1 Mutations. J Allergy Clin Immunol (2018) 141:704–17.e5. doi: 10.1016/j.jaci.2017.03.049
51. Kiykim A, Charbonnier LM, Akcay A, Karakoc-Aydiner E, Ozen A, Ozturk G, et al. Hematopoietic Stem Cell Transplantation in Patients With Heterozygous STAT1 Gain-of-Function Mutation. J Clin Immunol (2019) 39:37–44. doi: 10.1007/s10875-018-0575-y
52. Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, et al. Use of Ruxolitinib to Successfully Treat Chronic Mucocutaneous Candidiasis Caused by Gain-of-Function Signal Transducer and Activator of Transcription 1 (STAT1) Mutation. J Allergy Clin Immunol (2015) 135:551–3. doi: 10.1016/j.jaci.2014.12.1867
53. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the Treatment of Immune Dysregulation in Patients With Gain-of-Function Signal Transducer and Activator of Transcription 1 (STAT1) or STAT3 Mutations. J Allergy Clin Immunol (2018) 142:1665–9. doi: 10.1016/j.jaci.2018.07.020
54. Zimmerman O, Rösler B, Zerbe CS, Rosen LB, Hsu AP, Uzel G, et al. Risks of Ruxolitinib in STAT1 Gain-Of-Function-Associated Severe Fungal Disease. Open Forum Infect Dis (2017) 4:ofx202. doi: 10.1093/ofid/ofx202
55. Chaimowitz NS, Ebenezer SJ, Hanson IC, Anderson M, Forbes LR. STAT1 Gain of Function, Type 1 Diabetes, and Reversal With JAK Inhibition. N Engl J Med (2020) 383:1494–6. doi: 10.1056/NEJMc2022226
56. Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich P-S, et al. Clinical Aspects of STAT3 Gain-Of-Function Germline Mutations: A Systematic Review. J Allergy Clin Immunol Pract (2019) 7:1958–69.e9. doi: 10.1016/j.jaip.2019.02.018
57. Jägle S, Heeg M, Grün S, Rensing-Ehl A, Maccari ME, Klemann C, et al. Distinct Molecular Response Patterns of Activating STAT3 Mutations Associate With Penetrance of Lymphoproliferation and Autoimmunity. Clin Immunol (2020) 210:108316. doi: 10.1016/j.clim.2019.108316
58. Haapaniemi EM, Kaustio M, Rajala HLM, van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, Hypogammaglobulinemia, Lymphoproliferation, and Mycobacterial Disease in Patients With Activating Mutations in STAT3. Blood (2015) 125:639–48. doi: 10.1182/blood-2014-04-570101
59. Mauracher AA, Eekels JJM, Woytschak J, van Drogen A, Bosch A, Prader S, et al. Erythropoiesis Defect Observed in STAT3 GOF Patients With Severe Anemia. J Allergy Clin Immunol (2020) 145:1297–301. doi: 10.1016/j.jaci.2019.11.042
60. Parlato M, Charbit-Henrion F, Abi Nader E, Begue B, Guegan N, Bruneau J, et al. Efficacy of Ruxolitinib Therapy in a Patient With Severe Enterocolitis Associated With a STAT3 Gain-Of-Function Mutation. Gastroenterology (2019) 156:1206–10.e1. doi: 10.1053/j.gastro.2018.11.065
61. Wegehaupt O, Muckenhaupt T, Johnson MB, Schwab KO, Speckmann C. Ruxolitinib Controls Lymphoproliferation and Diabetes in a STAT3-GOF Patient. J Clin Immunol (2020) 40:1207–10. doi: 10.1007/s10875-020-00864-w
62. Sarfati E, Hadjadj J, Fusaro M, Klifa R, Grimaud M, Berteloot L, et al. Life-Saving, Dose-Adjusted, Targeted Therapy in a Patient With a STAT3 Gain-Of-Function Mutation. J Clin Immunol (2021) 41:807–10. doi: 10.1007/s10875-020-00914-3
63. Uggenti C, Lepelley A, Crow YJ. Self-Awareness: Nucleic Acid-Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol (2019) 37:247–67. doi: 10.1146/annurev-immunol-042718-041257
64. Crow YJ. Type I Interferonopathies: A Novel Set of Inborn Errors of Immunity. Ann N Y Acad Sci (2011) 1238:91–8. doi: 10.1111/j.1749-6632.2011.06220.x
65. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
66. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg M-C, et al. Inherited STING-Activating Mutation Underlies a Familial Inflammatory Syndrome With Lupus-Like Manifestations. J Clin Invest (2014) 124:5516–20. doi: 10.1172/JCI79100
67. Lin B, Berard R, Al Rasheed A, Aladba B, Kranzusch PJ, Henderlight M, et al. A Novel STING1 Variant Causes a Recessive Form of STING-Associated Vasculopathy With Onset in Infancy (SAVI). J Allergy Clin Immunol (2020) 146:1204–8.e6. doi: 10.1016/j.jaci.2020.06.032
68. Alghamdi MA, Mulla J, Saheb Sharif-Askari N, Guzmán-Vega FJ, Arold ST, Abd-Alwahed M, et al. A Novel Biallelic STING1 Gene Variant Causing SAVI in Two Siblings. Front Immunol (2020) 11:599564. doi: 10.3389/fimmu.2020.599564
69. Frémond M-L, Crow YJ. STING-Mediated Lung Inflammation and Beyond. J Clin Immunol (2021) 41(3):501–14. doi: 10.1007/s10875-021-00974-z
70. Balci S, Ekinci RMK, de Jesus AA, Goldbach-Mansky R, Yilmaz M. Baricitinib Experience on STING-Associated Vasculopathy With Onset in Infancy: A Representative Case From Turkey. Clin Immunol (2020) 212:108273. doi: 10.1016/j.clim.2019.108273
71. Tang X, Xu H, Zhou C, Peng Y, Liu H, Liu J, et al. STING-Associated Vasculopathy With Onset in Infancy in Three Children With New Clinical Aspect and Unsatisfactory Therapeutic Responses to Tofacitinib. J Clin Immunol (2020) 40:114–22. doi: 10.1007/s10875-019-00690-9
72. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher M-T, et al. cGAS-Mediated Induction of Type I Interferon Due to Inborn Errors of Histone pre-mRNA Processing. Nat Genet (2020) 52:1364–72. doi: 10.1038/s41588-020-00737-3
73. Briand C, Frémond M-L, Bessis D, Carbasse A, Rice GI, Bondet V, et al. Efficacy of JAK1/2 Inhibition in the Treatment of Chilblain Lupus Due to TREX1 Deficiency. Ann Rheum Dis (2019) 78:431–3. doi: 10.1136/annrheumdis-2018-214037
74. Zhang S, Song J, Yang Y, Miao H, Yang L, Liu Y, et al. Type I Interferonopathies With Novel Compound Heterozygous TREX1 Mutations in Two Siblings With Different Symptoms Responded to Tofacitinib. Pediatr Rheumatol Online J (2021) 19:1. doi: 10.1186/s12969-020-00490-1
75. McLellan KE, Martin N, Davidson JE, Cordeiro N, Oates BD, Neven B, et al. JAK 1/2 Blockade in MDA5 Gain-Of-Function. J Clin Immunol (2018) 38:844–6. doi: 10.1007/s10875-018-0563-2
76. Kothur K, Bandodkar S, Chu S, Wienholt L, Johnson A, Barclay P, et al. An Open-Label Trial of JAK 1/2 Blockade in Progressive IFIH1-Associated Neuroinflammation. Neurology (2018) 90:289–91. doi: 10.1212/WNL.0000000000004921
77. Zheng S, Lee PY, Wang J, Wang S, Huang Q, Huang Y, et al. Interstitial Lung Disease and Psoriasis in a Child With Aicardi-Goutières Syndrome. Front Immunol (2020) 11:985. doi: 10.3389/fimmu.2020.00985
78. Neven B, Al Adba B, Hully M, Desguerre I, Pressiat C, Boddaert N, et al. JAK Inhibition in the Aicardi-Goutières Syndrome. N Engl J Med (2020) 383:2190–1. doi: 10.1056/NEJMc2031081
79. Vanderver A, Adang L, Gavazzi F, McDonald K, Helman G, Frank DB, et al. Janus Kinase Inhibition in the Aicardi-Goutières Syndrome. N Engl J Med (2020) 383:986–9. doi: 10.1056/NEJMc2001362
80. Adang LA, Frank DB, Gilani A, Takanohashi A, Ulrick N, Collins A, et al. Aicardi Goutières Syndrome is Associated With Pulmonary Hypertension. Mol Genet Metab (2018) 125:351–8. doi: 10.1016/j.ymgme.2018.09.004
81. Low AT, Howard L, Harrison C, Tulloh RMR. Pulmonary Arterial Hypertension Exacerbated by Ruxolitinib. Haematologica (2015) 100:e244–5. doi: 10.3324/haematol.2014.120816
82. Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA Mutations Impair ER-Golgi Transport and Cause Hereditary Autoimmune-Mediated Lung Disease and Arthritis. Nat Genet (2015) 47:654–60. doi: 10.1038/ng.3279
83. Brandizzi F, Barlowe C. Organization of the ER-Golgi Interface for Membrane Traffic Control. Nat Rev Mol Cell Biol (2013) 14:382–92. doi: 10.1038/nrm3588
84. Volpi S, Tsui J, Mariani M, Pastorino C, Caorsi R, Sacco O, et al. Type I Interferon Pathway Activation in COPA Syndrome. Clin Immunol (2018) 187:33–6. doi: 10.1016/j.clim.2017.10.001
85. Lepelley A, Martin-Niclós MJ, Le Bihan M, Marsh JA, Uggenti C, Rice GI, et al. Mutations in COPA Lead to Abnormal Trafficking of STING to the Golgi and Interferon Signaling. J Exp Med (2020) 217(11):e20200600. doi: 10.1084/jem.20200600
86. Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A Defect in COPI-Mediated Transport of STING Causes Immune Dysregulation in COPA Syndrome. J Exp Med (2020) 217(11):e20201045. doi: 10.1084/jem.20201045
87. Mukai K, Ogawa E, Uematsu R, Kuchitsu Y, Kiku F, Uemura T, et al. Homeostatic Regulation of STING by Retrograde Membrane Traffic to the ER. Nat Commun (2021) 12:61. doi: 10.1038/s41467-020-20234-9
88. Steiner A, Hrovat Schaale K, Prigione I, De Nardo D, Dagley LF, Yu C-H, et al. Activation of STING Due to COPI-Deficiency. BioRxiv (2020). doi: 10.1101/2020.07.09.194399
89. Krutzke S, Rietschel C, Horneff G. Baricitinib in Therapy of COPA Syndrome in a 15-Year-Old Girl. Eur J Rheumatol (2019) 7(Suppl 1):1–4. doi: 10.5152/eurjrheum.2019.18177
90. Frémond M-L, Legendre M, Fayon M, Clement A, Filhol-Blin E, Richard N, et al. Use of Ruxolitinib in COPA Syndrome Manifesting as Life-Threatening Alveolar Haemorrhage. Thorax (2020) 75:92–5. doi: 10.1136/thoraxjnl-2019-213892
91. Pin A, Tesser A, Pastore S, Moressa V, Valencic E, Arbo A, et al. Biological and Clinical Changes in a Pediatric Series Treated With Off-Label JAK Inhibitors. Int J Mol Sci (2020) 21(20):7767. doi: 10.3390/ijms21207767
92. Patel PN, Hunt R, Pettigrew ZJ, Shirley JB, Vogel TP, de Guzman MM. Successful Treatment of Chronic Atypical Neutrophilic Dermatosis With Lipodystrophy and Elevated Temperature (CANDLE) Syndrome With Tofacitinib. Pediatr Dermatol (2021) 38(2):528–9. doi: 10.1111/pde.14517
93. Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe Type I Interferonopathy and Unrestrained Interferon Signaling Due to a Homozygous Germline Mutation in STAT2. Sci Immunol (2019) 4(42):eaav7501. doi: 10.1126/sciimmunol.aav7501
94. Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, et al. Homozygous STAT2 Gain-of-Function Mutation by Loss of USP18 Activity in a Patient With Type I Interferonopathy. J Exp Med (2020) 217(5):e20192319. doi: 10.1084/jem.20192319
95. Meuwissen MEC, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 Deficiency Underlies Type 1 Interferonopathy Leading to Severe Pseudo-TORCH Syndrome. J Exp Med (2016) 213:1163–74. doi: 10.1084/jem.20151529
96. Alsohime F, Martin-Fernandez M, Temsah M-H, Alabdulhafid M, Le Voyer T, Alghamdi M, et al. JAK Inhibitor Therapy in a Child With Inherited USP18 Deficiency. N Engl J Med (2020) 382:256–65. doi: 10.1056/NEJMoa1905633
97. Shin YH, Glass L, Evans HE. The "Torch Syndrome. Pediatr Ann (1976) 5:106–13. doi: 10.3928/0090-4481-19760201-11
98. Arimoto K-I, Löchte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, et al. STAT2 Is an Essential Adaptor in USP18-Mediated Suppression of Type I Interferon Signaling. Nat Struct Mol Biol (2017) 24:279–89. doi: 10.1038/nsmb.3378
99. Hong Y, Capitani M, Murphy C, Pandey S, Cavounidis A, Takeshita H, et al. Janus Kinase Inhibition for Autoinflammation in Patients With DNASE2 Deficiency. J Allergy Clin Immunol (2020) 145:701–5.e8. doi: 10.1016/j.jaci.2019.11.020
100. Légeret C, Meyer BJ, Rovina A, Deigendesch N, Berger CT, Daikeler T, et al. JAK Inhibition in a Patient With X-Linked Reticulate Pigmentary Disorder. J Clin Immunol (2021) 41(1):212–6. doi: 10.1007/s10875-020-00867-7
101. Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune Dysregulation and Multisystem Inflammatory Syndrome in Children (MIS-C) in Individuals With Haploinsufficiency of SOCS1. J Allergy Clin Immunol (2020) 146:1194–200.e1. doi: 10.1016/j.jaci.2020.07.033
102. Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, et al. Integrated Genomic Sequencing Reveals Mutational Landscape of T-Cell Prolymphocytic Leukemia. Blood (2014) 124:1460–72. doi: 10.1182/blood-2014-03-559542
103. Kontro M, Kuusanmäki H, Eldfors S, Burmeister T, Andersson EI, Bruserud O, et al. Novel Activating STAT5B Mutations as Putative Drivers of T-Cell Acute Lymphoblastic Leukemia. Leukemia (2014) 28:1738–42. doi: 10.1038/leu.2014.89
104. Ma CA, Xi L, Cauff B, DeZure A, Freeman AF, Hambleton S, et al. Somatic STAT5b Gain-of-Function Mutations in Early Onset Nonclonal Eosinophilia, Urticaria, Dermatitis, and Diarrhea. Blood (2017) 129:650–3. doi: 10.1182/blood-2016-09-737817
Keywords: Jakinib, inborn errors of immunity (IEIs), interferonopathies, STAT 1 and STAT3 gain of function, autoimmunity, novel therapies
Citation: Hadjadj J, Frémond M-L and Neven B (2021) Emerging Place of JAK Inhibitors in the Treatment of Inborn Errors of Immunity. Front. Immunol. 12:717388. doi: 10.3389/fimmu.2021.717388
Received: 30 May 2021; Accepted: 25 August 2021;
Published: 17 September 2021.
Edited by:
Claire Booth, University College London, United KingdomReviewed by:
Austen Worth, Great Ormond Street Hospital for Children NHS Foundation Trust, United KingdomCopyright © 2021 Hadjadj, Frémond and Neven. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bénédicte Neven, YmVuZWRpY3RlLm5ldmVuQGFwaHAuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.