 Olga Zimmermannova
Olga Zimmermannova Inês Caiado1,2,3,4
Inês Caiado1,2,3,4 Alexandra G. Ferreira
Alexandra G. Ferreira Carlos-Filipe Pereira
Carlos-Filipe Pereira- 1Cell Reprogramming in Hematopoiesis and Immunity Laboratory, Lund Stem Cell Center, Department of Molecular Medicine and Gene Therapy, Lund University, Lund, Sweden
- 2Wallenberg Center for Molecular Medicine, Lund University, Lund, Sweden
- 3Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
- 4Doctoral Programme in Experimental Biology and Biomedicine, University of Coimbra, Coimbra, Portugal
Advances in understanding how cancer cells interact with the immune system allowed the development of immunotherapeutic strategies, harnessing patients’ immune system to fight cancer. Dendritic cell-based vaccines are being explored to reactivate anti-tumor adaptive immunity. Immune checkpoint inhibitors and chimeric antigen receptor T-cells (CAR T) were however the main approaches that catapulted the therapeutic success of immunotherapy. Despite their success across a broad range of human cancers, many challenges remain for basic understanding and clinical progress as only a minority of patients benefit from immunotherapy. In addition, cellular immunotherapies face important limitations imposed by the availability and quality of immune cells isolated from donors. Cell fate reprogramming is offering interesting alternatives to meet these challenges. Induced pluripotent stem cell (iPSC) technology not only enables studying immune cell specification but also serves as a platform for the differentiation of a myriad of clinically useful immune cells including T-cells, NK cells, or monocytes at scale. Moreover, the utilization of iPSCs allows introduction of genetic modifications and generation of T/NK cells with enhanced anti-tumor properties. Immune cells, such as macrophages and dendritic cells, can also be generated by direct cellular reprogramming employing lineage-specific master regulators bypassing the pluripotent stage. Thus, the cellular reprogramming toolbox is now providing the means to address the potential of patient-tailored immune cell types for cancer immunotherapy. In parallel, development of viral vectors for gene delivery has opened the door for in vivo reprogramming in regenerative medicine, an elegant strategy circumventing the current limitations of in vitro cell manipulation. An analogous paradigm has been recently developed in cancer immunotherapy by the generation of CAR T-cells in vivo. These new ideas on endogenous reprogramming, cross-fertilized from the fields of regenerative medicine and gene therapy, are opening exciting avenues for direct modulation of immune or tumor cells in situ, widening our strategies to remove cancer immunotherapy roadblocks. Here, we review current strategies for cancer immunotherapy, summarize technologies for generation of immune cells by cell fate reprogramming as well as highlight the future potential of inducing these unique cell identities in vivo, providing new and exciting tools for the fast-paced field of cancer immunotherapy.
Introduction
Cancer progression entails close crosstalk between tumor cells and the immune system. The evolution of cancer is driven by oncogenic mutations and the deregulation of important signaling pathways. The unleashed proliferative capacity leads to the accumulation of additional mutations, endowing tumor cells with selective advantages such as improved fitness and survival. Yet, such cancer diversity resulting from high mutational burden comes with a cost. The more mutations cancer cells acquire, the more likely they are recognized and eliminated by the immune system due to the expression of various tumor-associated antigens (TAAs) (1–3). However, the pressure imposed by immune surveillance further shapes cancer development, and the evolving tumor heterogeneity provides opportunities for the selection of clones that have developed mechanisms to evade the host’s immune system (4). These mechanisms include the downregulation of Major Histocompatibility Complex (MHC)-I molecules, over-expression of inhibitory checkpoints that prevent or attenuate T-cell responses, editing of neoantigens, or hijacking immune cells by creating an immunosuppressive environment. Overall, the complex, adaptable, and heterogeneous nature of cancer is characterized by its ability to disengage a variety of immune cells controlling specific modules of the immune response. For example, the tolerogenic environment established in some tumors promotes the activity of regulatory T-cells (Treg) and compromises dendritic cell (DC) function, impacting the productive induction of anti-tumor immunity (1, 5).
The idea of harnessing the body’s immune system to elicit anti-cancer responses has now stepped into the spotlight of cancer research. The field was pioneered by William Coley and the development of vaccines that activated anti-tumor immunity via exposure to bacterial toxins (6). The intravesical administration of Bacillus Calmette Guerain (BCG) in bladder cancer (7) and the use of interleukin (IL)-2 in renal cell carcinoma (8) foreshadowed the era of modern immunotherapy. Due to immense research efforts and broadened understanding of tumor biology and its interactions with the immune system, the past decade witnessed rapid development of cancer immunotherapy, resulting in several U.S. Food and Drug Administration (FDA) approvals, and the highlight of seminal discoveries leading to immune checkpoint blockade with a Nobel Prize. Immunotherapy changed the paradigm of cancer treatment, and it is now established as a potent therapeutic modality with long-term effects for multiple cancers, adding to the clinical toolbox of more conventional approaches encompassing chemotherapy, radiotherapy and surgery. However, only a minority of patients respond to immunotherapy, and some cancer types are not yet open to these treatments, emphasizing the need for new strategies to deliver clinical benefits to a larger number of patients and indications. Harnessing the potential of the immune cell repertoire and their functional features represent the next frontier in immunotherapy.
Strategies for Cancer Immunotherapy
Cancer Vaccines
The cancer vaccine concept relies on the induction of polyclonal T-cell-specific immune responses against multiple TAAs, offering a potential mechanism to target cancer heterogeneity. Multiple strategies have been employed to generate cancer vaccines, encompassing administration of irradiated whole tumor cells, DCs preloaded with TAAs, or in vivo antigen delivery via DNA or RNA (9). Therefore, successful cancer vaccination requires 1) a supply of purified and functional DCs to direct adaptive and innate immune responses and 2) a selection of highly immunogenic and specific TAAs to engage a powerful T-cell response exclusively against tumor cells (10).
DCs are phagocytic cells that can be found in peripheral blood, lymphoid organs, and all tissues. Given their exceptional ability to capture, process, and present antigens, as well as to produce cytokines and migrate to lymph nodes to activate T-cells, DCs play a pivotal role in orchestrating adaptive and innate immune responses against pathogens and cancer (11). Human DCs are a heterogeneous group comprising four main subtypes with distinct ontology, phenotype, and function – conventional type 1 (cDC1), conventional type 2 (cDC2), plasmacytoid (pDC), and monocyte-derived (mo-DC) DCs (12, 13). Conventional DCs are defined by the expression of CD11c and HLA class II and include two main subsets. cDC1 are developmentally dependent on BATF3 and IRF8 transcription factors and can be identified by surface expression of XCR1, CLEC9A, and CD141. They produce pro-inflammatory cytokines including IL-12, IL-29, and CXCL10 and their hallmark function is antigen cross-presentation and the generation of T helper (Th)1 response (14–16). In contrast, cDC2 are characterized by the expression of CD1c and constitute a highly heterogeneous subset that relies partially on IRF4 and Notch signaling. Some cDC2 govern type 2 responses against parasites, some sense extracellular pathogens, whilst others mount type 3 response driven by the activation of Th2/17 and Innate Lymphoid Cells (ILCs) (17, 18). In contrast, CD123+CD11c- pDC cells, dependent on transcription factor E2-2 and high levels of IRF8, are endowed with exceptional ability to sense viral and bacterial pathogens and release type I interferon in response to acute infection (19). CD1c+CD14+ mo-DCs are usually rapidly recruited during acute inflammation and are also capable of T-cell activation. However, compared to cells primed with conventional DCs that tend to launch effector-prone differentiation, mo-DCs preferentially drive differentiation of activated CD8+ T-cells into memory cells (20). While crosstalk and synergy among DC subsets are important for the complexity and magnitude of anti-tumor immune response (21), experiments in mice have implicated the presence and uncompromised function of cDC1 as vital to elicit efficient cancer immunity. As shown by Hildner et al., selective loss of cDC1 in BATF3-/- mice leads to deficient cross-presentation and diminished CD8+ T-cell activation, resulting in deficient tumor control (22). In humans, genome-wide studies revealed that the presence of cDC1s within the tumor microenvironment correlates with a better outcome in patients (23). Given their role in orchestrating immunity, cDC1 activity is thus fundamental for effective response to immune checkpoint blockade or adoptive T-cell transfer (24–26).
Despite considerable efforts, the clinical translation of cancer vaccines into potent therapies has been challenging. Traditional DC vaccines are mostly based on DCs isolated from patients, or on DCs differentiated from autologous peripheral monocytes, which are further loaded with TAAs in tumor lysates. Spiluleucel-T is the only FDA-approved vaccine that targets the PAP antigen and increases overall survival in hormone-refractory prostate carcinoma by approximately 4 months (27) (Figure 1). However, mo-DCs have reduced capacity for efficient antigen presentation, compromising the efficiency of DC vaccines in most patients (20). An interesting approach may lie in cancer vaccines utilizing purified DC subsets. Importantly, vaccines derived from allogenic pDCs primed cytotoxic immune responses in advanced melanoma patients (28). Similarly, autologous cDC2 cells mounted CD8+ T-cell responses and delivered long-term progression-free responses in approximately 25% of melanoma patients (29). Combining DC subsets may be another interesting strategy to enhance the efficiency of DC vaccines as illustrated by pDC and cDC2 cooperation. pDCs produce high levels of CXCL3 that attract a variety of immune effectors, including CD8+ T-cells, CD56+ T-cells, and γδ T-cells, while cDC2 secure sufficient antigen presentation and T-cell priming (30). However, cancer vaccination strategies would potentially benefit if the preparation of vaccines could be done from pure, functional cDC1s, currently limited by their rarity. In this regard, identification of transcription factors imposing the identity of multiple DC subtypes by cellular reprogramming may open the door for the generation of pure populations of DCs (31).
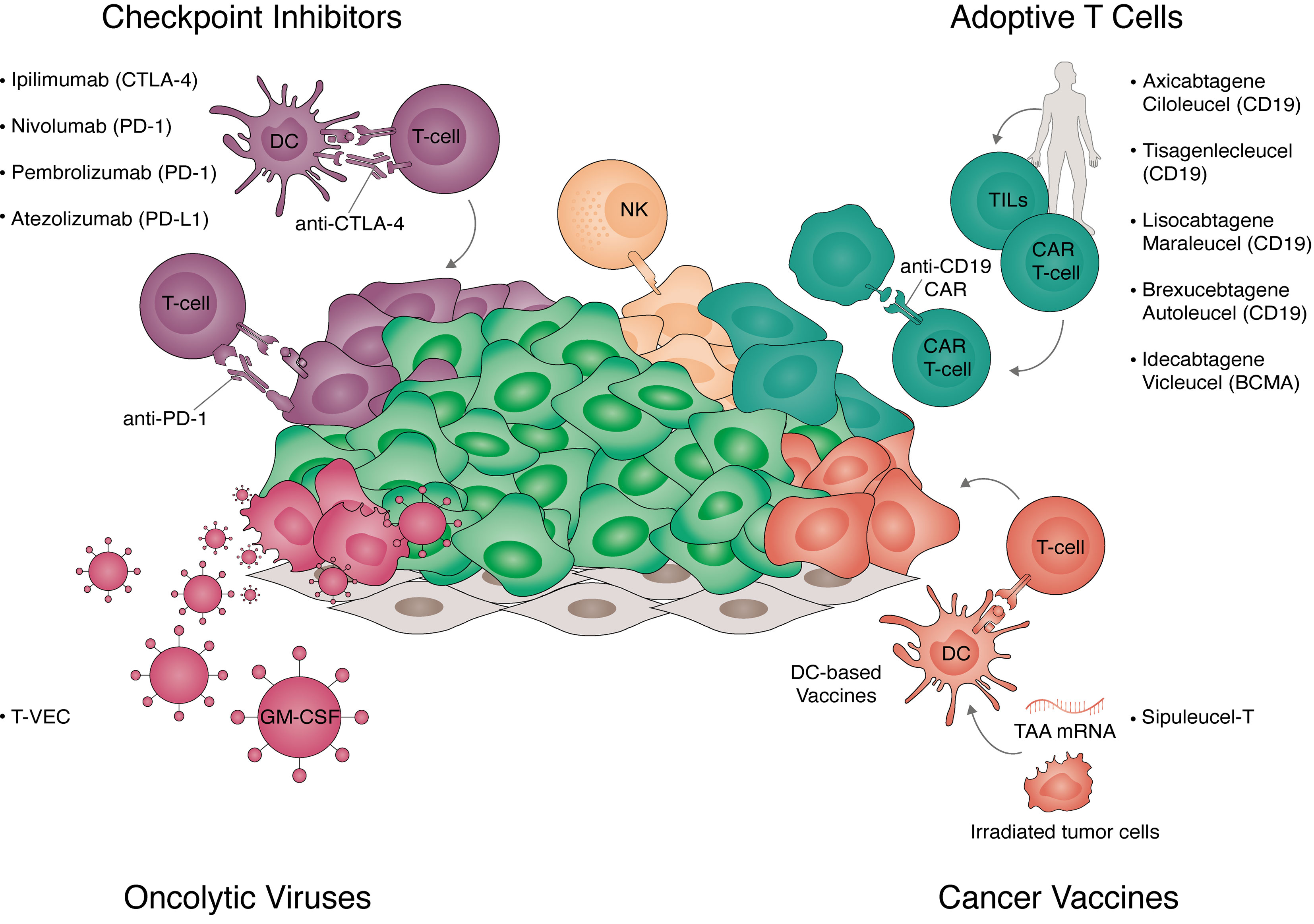
Figure 1 Strategies for cancer immunotherapy. Overview of the strategies for immunotherapy translated to clinical practice. Approved strategies include immune checkpoint inhibitors, adoptive T-cell therapy, cancer vaccines, and oncolytic viruses. Checkpoint inhibitors (top left panel) are antibodies targeting key negative regulators of T-cell activation. These include receptors expressed in T-cells targeted with anti-cytotoxic T-lymphocyte associated protein 4 (CTLA-4) (ipilimumab), anti-programmed cell death protein 1 (PD-1) (nivolumab, pembrolizumab) and receptors expressed in tumor cells targeted with anti-programmed death-ligand 1 (PD-L1) (atezolizumab). Adoptive T-cell transfer (top right panel) are autologous cellular therapies currently targeting hematological tumors. Approved Chimeric Antigen Receptor (CAR) T-cells target CD19 (axicabtagene ciloleucel, tisagenlecleucel, brexucabtagene autoleucel, and lisocabtagene maraleucel) or B-cell maturation antigen (BCMA) (idecabtagene vicleucel). Additionally, tumor infiltrated lymphocytes (TILs) expanded ex vivo and natural killer (NK) cells are currently in clinical trials. Cancer vaccines (bottom right panel) exploit the antigen presentation capacity of dendritic cells (DC) to trigger polyclonal T-cell responses to tumor-associated antigens (TAA). These include irradiated tumor cells expressing granulocyte-macrophage colony-stimulating factor (GM-CSF, GVAX), DCs loaded with tumor antigens (sipuleucel-T), or the delivery of mRNA in vivo encoding multiple TAAs. Oncolytic viruses (bottom left panel) target cancer cells own vulnerability to viral infections while inducing immunogenic cell death. Talimogene laherparepvec (T-VEC) is an approved intra-tumoral therapy consisting of a type-1 herpes virus expressing GM-CSF.
Despite the importance of using potent antigen-presenting cells, not all types of cancer vaccines include DCs directly. The development of GVAX is based on the use of irradiated tumor cells expressing GM-CSF which further modulate antigen presentation and support DC function (32).
Therapeutics based on nucleic acids have also emerged as an alternative to conventional vaccine approaches. mRNA vaccines recently attracted enormous attention as the basis of COVID-19 vaccination. In cancer immunotherapy, the mRNA approach has been explored for delivery of TAAs directly to DCs, offering an elegant solution for cancer vaccination. mRNA vaccines are delivered by lipid nanoparticles containing lipoplexed RNA molecules encoding selected neoantigens or TAAs (33). Promising results were recently reported by interim results of phase 1 clinical trial of FixVac, an RNA-LPX vaccine targeting four highly prevalent melanoma-associated antigens that are shared by most patients. When administered in a multiple-dose regimen, FixVac alone, or in combination therapy with checkpoint inhibitors, induced strong CD4+ and CD8+ immune responses and delivered durable responses in patients with unresectable melanoma (34). Nevertheless, a careful selection of suitable treatment combinations, as well identifications of biomarkers predicting response to cancer vaccines may help to maximize the clinical benefit of the vaccination strategy for a broader group of patients (35).
Oncolytic Viruses
Tumor cells frequently accumulate mutations in pathways securing cellular anti-viral defense, rendering themselves vulnerable to viral infections. This cancer vulnerability has been also exploited as a potential strategy to control tumor growth. Oncolytic viruses (OVs) are engineered to selectively infect and replicate in tumor cells and to induce immunogenic death. Hypothetically, the administration of OV therapy should be able to target natural cancer-immunity interactions at multiple levels: OV-mediated lysis of tumor cells promotes the release of TAAs and type I interferon, boosting anti-tumor immunity.
Since the first report of conditionally replicating Herpes Simplex type 1 (HSV-1) engineered virus which prolonged survival in nude mice with glioma (36), a myriad of viral backbones has been exploited, encompassing adenoviruses, measles, coxsackie, polio, reoviruses, or poxviruses. The selective tropism for tumor cells can be modulated by genetic modifications of the capsid, utilizing epitopes targeting receptors exclusively or highly expressed on tumor cells (37).
Furthermore, the engineering of OVs allows enforced expression of genes with therapeutic potential, including TAAs, costimulatory molecules, and cytokines (e.g. GM-CSF, Il-2, IL-12, or TNFα), which can further amplify immune responses and counteract tumor-induced immunosuppression. The only FDA-approved oncolytic viral-based therapy is talimogene laherparepvec (T-VEC), used in advanced melanoma patients (Figure 1). T-VEC is an attenuated HSV-1 virus harboring a deletion of neurovirulence genes, engineered to express GM-CSF to promote T-cell priming and boost immunogenicity. In advanced melanoma patients, T-VEC achieved durable responses and prolonged survival in nearly 20% of patients, benefiting particularly those at early stages of metastasized disease (38).
Given their unique characteristics, OVs are promising therapeutic agents, but their efficacy, when used as a single therapy, leaves room for improvement. In this way, the versatility of the oncolytic viral platform makes it suitable for combination with other immunotherapies and conventional treatments. The combination of T-VEC with immune checkpoint inhibitors showed improved efficacy in melanoma without an increase in adverse effects, delivering long-term response in up to 62% of patients (39–41). The effect is attributed to the ability of OVs to convert immunologically cold tumors into immune hotspots, overcoming systemic immunosuppression (42).
Immune Checkpoint Inhibitors
Immune checkpoints were originally discovered as a group of evolutionarily conserved molecules acting as negative regulators of T-cell activation, with cytotoxic T-lymphocyte Antigen 4 (CTLA-4) and PD-1 inhibitors being recently approved for clinical practice. CTLA-4 is primarily found on T-cells and controls early-stage T-cell activation by counteracting the activity of the co-stimulatory molecule CD28 (43–45). Unlike CTLA-4, PD-1 molecule is expressed following T-cell activation, and its major role is to tame T-cell activity in peripheral tissues, preventing tissue damage. As their natural role lies in the maintenance of self-tolerance, and modulation of the duration and magnitude of physiologic immune responses (46, 47), the engagement of immune checkpoints is one of the mechanisms which cancer cells deploy to hijack the host immune defense. Therefore, the expression of PD-1 ligand (PD-L1) was identified on many tumor cells including hematologic malignancies, glioblastoma, lung, kidney, prostate, breast, and ovarian carcinoma, and has a critical role in suppressing immune-surveillance mechanisms (48). Interestingly, within the tumor microenvironment, PD-L1 can be expressed by myeloid cells including macrophages and DCs. The deletion of PD-L1 in the DC compartment, but not in macrophages, strengthened cytotoxic CD8+ mediated responses and abrogated tumor growth (49).
Inhibition of immune checkpoint receptors employs synthesized antibodies as a therapeutic intervention disrupting the ability of some cancer cells to evade a patient´s immune response (45). Administration of anti-CTLA-4 (ipilimumab) prolongs survival in patients with metastatic melanoma where conventional treatment approaches failed (50). Notably, ipilimumab was the first FDA-approved checkpoint inhibitor, followed by anti-PD-1 inhibitors (nivolumab and pembrolizumab) for the treatment of squamous non-small cell lung carcinoma (NSCLC) as well as melanoma (51, 52) (Figure 1). Given their impact in reactivating anti-tumor immunity in humans, checkpoint inhibitors became a milestone achievement in cancer research of the past decade, as highlighted by the Nobel Prize awarded to James Allison and Tasuko Honjo in 2018.
Despite delivering unprecedented long-lasting anti-tumor responses (53), persisting for more than 10 years after discontinuation of therapy in some patients, important challenges remain: 1) only about 20% of patients benefit from immune checkpoint treatment (54) and, 2) cancer types that build a local strong immunosuppressive environment do not respond consistently to these therapies (55), suggesting that the future lies in combining interventions targeting mechanistically independent immune evasion mechanisms.
Following the therapeutic success in metastatic melanoma and NSCLC, checkpoint inhibitors became increasingly used as a treatment alternative in a range of solid tumors, particularly in cancers attributed to carcinogen exposure or infectious origin. In addition to NSCLC, smoking is associated with the development of bladder cancer, head and neck carcinoma, renal cell carcinoma, or stomach malignancies, conferring a high tumor mutational burden (56–60). Importantly, as high mutational load generally predicts a good response to checkpoint inhibition (61), patients with mismatch repair deficiency (MMR) are suitable candidates for checkpoint immunotherapy (62). Recent publications also reported therapeutic success of checkpoint inhibition in Hodgkin lymphoma (63), and pembrolizumab and anti PD-L1 (atezolizumab) are currently setting the way to improve the outcome in triple-negative breast carcinoma, as PD-L1 positive patients reached 25-month survival compared to 18 months in the control group (64). Overall, the efficacy of checkpoint inhibitors as an adjuvant therapy complementing surgery or chemotherapy is currently being evaluated in several clinical trials (65). On the other hand, the administration of checkpoint inhibitors is associated with excessive immune activation, with almost every third patient suffering from immune-related adverse effects, encompassing diarrhea, colitis, hepatitis, development of hypothyroidism, type 1 diabetes, or adrenocortical insufficiency (66). Notably, melanoma patients who received combined CTLA-4/PD-1 inhibition showed longer overall survival although at the cost of higher adverse effects (53, 67). Although the clinical benefit clearly outweighs toxicity, additional efforts should be made towards strategies to identify patients who can benefit the most from checkpoint therapy.
Adoptive Cellular Therapy
Harnessing T-cells for immunotherapy has long been of prime interest to cancer research. T-cells are unique immune cells: they are long-lived in resting conditions and proliferate extensively when encountering specific stimuli. T-cells elicit specific, robust, and enduring responses against tumor cells. As their action is systemic, they can reduce the tumor burden at the primary site and eradicate distant metastatic sites. On the downside, the manufacturing of lymphocytes for immunotherapy is challenging as it is so far tailored for each individual patient.
Tumor-Infiltrating Lymphocytes
First attempts to use T-cells in immunotherapy were based on tumor-infiltrating lymphocytes (TILs), a pool of T-lymphocytes pre-trained to recognize patient´s specific cancer cells. When expanded, pre-activated ex vivo, and reinfused into patients, TILs induce rapid tumor regression (68, 69). In the context of melanoma, around 20% of patients show durable responses to administration of ex vivo expanded TILs (5-year survival 93%) (70). Despite achieving outstanding responses in melanoma, the nature of the therapy makes it amenable only for highly mutated, immunologically hot tumors. Indeed, TIL-based therapies entered clinical trials also for the treatment of head and neck cancer, cervical carcinoma, ovarian carcinoma, or NSCLC.
CAR T-Cells
Chimeric antigen receptors (CAR) T-cells represent another breakthrough in immunotherapy. CAR T-cells are genetically engineered to express a synthetic T-cell receptor tailored to recognize surface antigens on cancer cells. The current generations of chimeric receptors usually consist of a single-chain variable fragment portion of the target receptor, along with the costimulatory intracellular domain of CD28, 4-1BB, or OX40 (71). CD19 targeting CAR T-cell-based immunotherapy encountered massive success in the treatment of B-cell hematologic malignancies, which translated into FDA approval of axicabtagene ciloleucel for relapsed or refractory large B cell lymphoma, as well as tisagenleucel in refractory pediatric B-cell acute lymphoblastic leukemia and B-cell lymphoma. More recently, B-cell maturation antigen (BCMA) CAR T-cells (idecabtagene vicleucel) were approved for therapy of advanced myeloma (72, 73), brexucabtagene autoleucel for mantle cell lymphoma, and lisocabtagene maraleucel for relapsed or refractory large B-cell lymphoma (Figure 1). However, the high efficacy of CAR T-cell treatments also comes with relatively high toxicity and particularly, with the risk of cytokine release syndrome (CRS) (74).
Despite achieving tremendous efficiency in the treatment of hematological malignancies, clinical success with CAR T-cells in the context of solid tumors is less encouraging, having hit multiple roadblocks from a limited number of targetable antigens to the inefficient trafficking of CAR T-cells into the tumor sites. Nevertheless, attempts are being made with CAR T-cells armed against ERBB2/HER2 receptor tyrosine kinase frequently over-expressed by tumor cells, as well as targeting TAAs including mesothelin, carcinoembryonic antigen (CEA), GD2 (75–79), or IL13Ra2 (80).
NK Cells
NK cells are innate cytotoxic lymphocytes critical for defense against abnormal cells, and, therefore, attracted plenty of attention in cancer immunotherapy. Unlike T-cells, their activation is induced via the direct interaction of native NK receptors with ligands on target cells. They are specially competent in recognizing cells that have lost MHC-I expression (81, 82), and lyse abnormal cells by granzyme B and perforin. NK cells also secrete a myriad of cytokines and chemokines, including IFN-γ, TNFα, IL-13, CCL3, CCL4, and GM-CSF, and, therefore, can modulate anti-tumor response by influencing trafficking other immune cells including cDC1s (83). However, NK cell function is severely compromised in patients, mostly due to the reduced expression of NK-activating ligands and suppressive effect of TGF-β within the tumor microenvironment (84–86), ultimately leading to their inability to lyse cancer cells and contributing to the progression of tumor development.
Adoptive NK cell therapy was originally successful in the treatment of hematologic malignancies, as administration of alloreactive NK cells elicited graft versus leukemia response and protected bone marrow transplanted acute myeloid leukemia (AML) patients from leukemia relapse (87), which largely prompted clinical endeavors in the application of NK transfer for solid tumors treatment.
In addition to AML, NK cell immunotherapy is delivering promising results also in children with neuroblastoma who were treated with anti-GD2 followed by allogenic NK transfer, eliciting a response in approximately 40% of patients (88). An exciting treatment opportunity further opens with the generation of NK cells expressing CARs – which are currently being tested, for example in glioblastoma (HER2-NK-92), pancreatic carcinoma (ROBO1-NK), colorectal carcinoma (Epcam-NK), or in patients with relapsed or refractory AML (CD33-NK-92) (89–91). As NK mediated cytotoxic happens independently on antigen processing, the use of NK cells may help overcome immunotherapy roadblocks imposed by lost antigen presentation or in tumors with a low mutational burden. NK therapy bypasses the need of the HLA-matched donors (92), and holds promise as off-the-shelf, bio-banked immunotherapy, which could be available on demand. Isolation of NK cells from donors at scale is problematic, but recent efforts have been made towards the manufacturing of NK cells via directed differentiation of induced Pluripotent Stem Cells (iPSCs), paving the way to harness cell reprogramming for immunotherapy (93, 94).
Immune Cell Programming and Reprogramming
The definition of individual immune cellular identities entails unique transcriptional, epigenetic, and functional signatures. Advances in next-generation sequencing and single-cell technologies have revealed additional layers of complexity in cell identity and cellular differentiation. For many years, cell differentiation was perceived as an irreversible, one-way road, metaphorically depicted as balls rolling down hills. Pioneering work with somatic cell nuclear transfer in frogs by John Gurdon in 1962 demonstrated that nuclei of terminally differentiated somatic cells retain the genetic information necessary to generate an entire individual, laying the foundation of cellular reprogramming (95). Decades later, Ian Wilmut cloned sheep from an adult mammary gland cell nucleus, providing evidence that somatic cell reprogramming can be achieved from adult cells in mammals (96). Discoveries with somatic cell nuclear transfer then prompted efforts to identify the factors that elicit reprogramming. The landmark discovery of Shinya Yamanaka in 2006, showing that enforced expression of the transcription factors OCT4, SOX2, KLF4, and MYC induced pluripotency in fibroblasts (97, 98) created a sea change in stem cell research.
The capacity to modify cell identity with a small set of transcription factors created exciting opportunities to investigate the fundamental principles of cell identity, provided a strategy to generate rare subtypes for drug screening and disease modeling, and opened avenues in regenerative medicine and cell replacement therapy.
Reprogramming to pluripotency was successfully shown from multiple somatic cell populations including fibroblasts, keratinocytes, neural progenitors, and peripheral blood cells (97–102), demonstrating the feasibility of iPSC generation from patient´s own biological material. iPSC reprogramming can generate target cell types on a large scale suitable for ex vivo production, as well as enable in vitro engineering to deliver desirable genetic modifications (e.g. CAR). However, clinical exploitation of iPSC technology has several major challenges.
iPSCs, just like embryonic stem cells (ESCs), are pluripotent and retain the capacity to generate all cell types of the embryo when provided with stage-specific signaling allowing differentiation to the cell type of interest. It is however a major challenge to fully recapitulate this process in vitro. In addition, the iPSC differentiation process is very dependent on line-to-line variability, and generates a heterogeneous population of cells, hampering clinical translation. Importantly, despite epigenetic resetting during induction of pluripotency, iPSCs may retain residual parental epigenetic memory which may bias their differentiation potential into certain cell types based on the source of donor cells used to establish iPSC (103). There is also the additional safety concern due to possibly contaminating iPSCs in the differentiated cell population as iPSCs are pluripotent and may generate teratomas in vivo. Therefore, to meet safety standards for clinical trials, more stringent protocols to purify differentiated cells are currently being developed. For example, a study exploiting iPSCs as a therapy of Parkinson disease is using a positive selection of differentiated dopaminergic neurons with anti-chorin antibody (104), while another clinical trial evaluating iPSCs in heart failure treatment uses the elimination of undifferentiated CD30+ cells by an antibody conjugated with an antimitotic agent (105).
iPSC technology holds the potential to provide a source of a wide range of blood cells. Differentiation of pluripotent stem cells recapitulates events known to occur during embryonic development. Given the complexity of hematopoiesis, which encompasses various anatomic locations and the formation of progenitors via endothelial-to-hematopoietic transition in several waves, in vitro differentiation of hematopoietic precursors then requires multiple steps involving the formation of embryonic bodies allowing mesodermal development. Furthermore, a range of differentiation cytokine cocktails, as well as co-culture with feeder layers promote cell growth and differentiation (Figure 2). Since the usage of multiple feeder cells is self-limiting for expansibility of target cell production, the development of feeder-free differentiation techniques is critical for wide-scale translation.
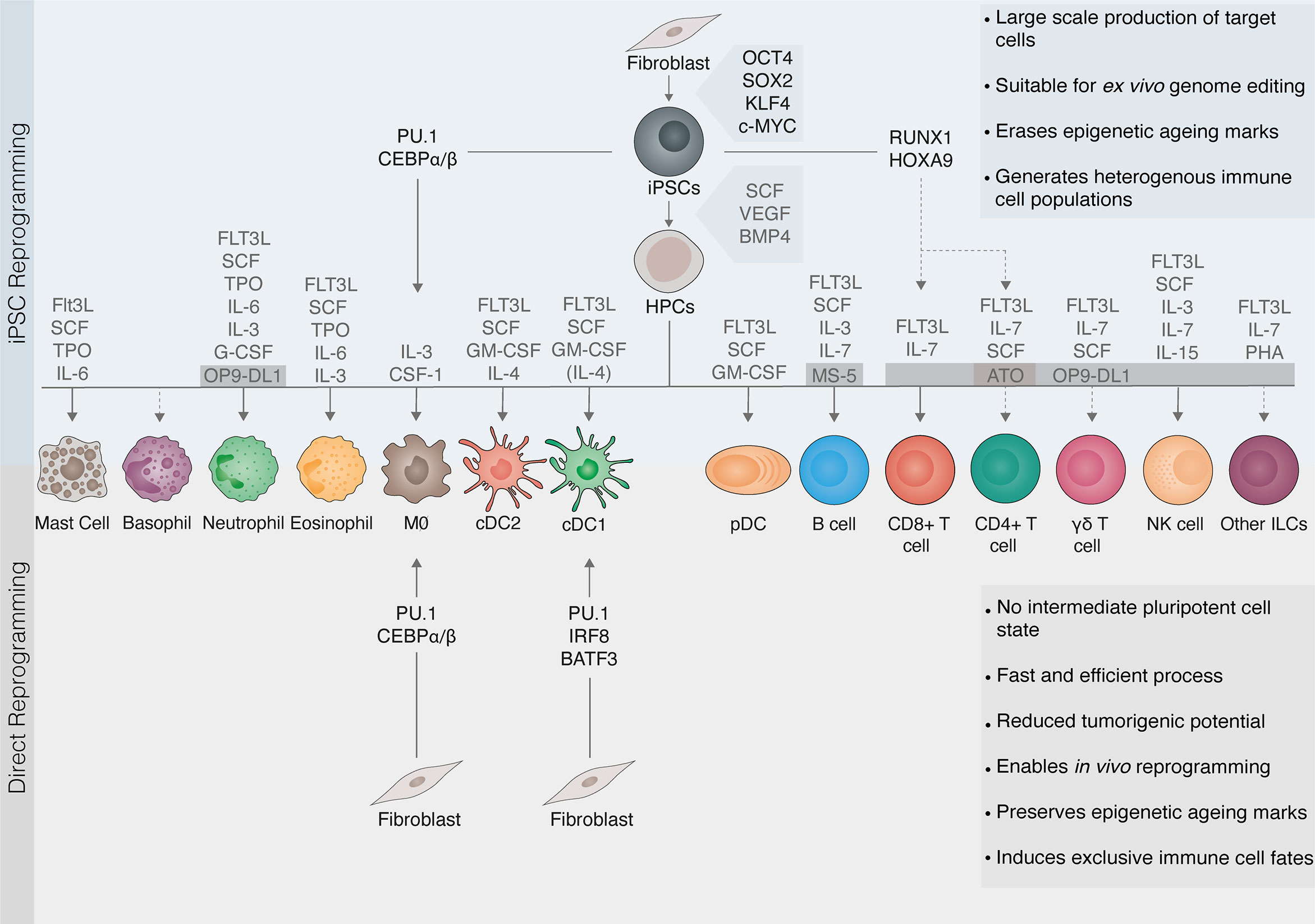
Figure 2 Cell reprogramming as a source of immune cells. Cellular reprogramming provides an opportunity to generate tailored immune cells for immunotherapy. Somatic cells can be reprogrammed to induced pluripotent stem cells (iPSCs) with the transcription factors OCT4, SOX2, KLF4, and c-MYC. iPSCs are then differentiated to hematopoietic progenitor cells (HPCs) with the cytokines stem cell factor (SCF), vascular endothelial growth factor (VEGF), and bone morphogenetic protein 4 (BMP4). Protocols for the differentiation of HPCs into myeloid and lymphoid lineages involve exposing cells to complex cytokine cocktails and/or feeder layers including MS-5, OP9-DL1, or artificial thymic organoids (ATO) supporting lymphopoiesis. Due to the self-renewal capacity of pluripotent stem cells, iPSC reprogramming (top panel) can generate immune cells on a large scale and is suitable for ex vivo genome editing. Cells undergo rejuvenation and erasure of epigenetic marks associated with aging or exhaustion. However, iPSC differentiation into mature immune cells is challenging (dashed lines) and leads to the generation of heterogeneous populations. Direct reprogramming (also known as transdifferentiation, bottom panel) refers to a change in cell fate mediated by lineage transcription factors that, unlike in iPSC reprogramming, does not involve pluripotent intermediates. The direct reprogramming of fibroblasts to macrophages (MØ) or conventional dendritic cells type 1 (cDC1) induce exclusively the target immune cell fate. Direct reprogramming is a faster and more efficient process. It can be elicited both in vitro and within the target tissue, thus having potential for in vivo reprogramming. It also reduces tumorigenic risks from contaminating stem cells. Moreover, direct reprogramming may retain epigenetic aging hallmarks, making it more suitable for modeling aging-related disease but more difficult to achieve complete epigenetic reprogramming. Transcription factors can also be applied to accelerate and direct the differentiation of iPSCs (also known as forward reprogramming, top panel). FLT3L, Fms-like tyrosine kinase 3 ligand; TPO, Thrombopoietin; IL, interleukin; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; cDC2, conventional dendritic cells type 2 DCs; pDCs, plasmacytoid DCs; NK, natural killer; ILCs, innate lymphoid cells.
The possibility of reverting somatic cells into the pluripotency encouraged investigation of combinatorial codes driving the conversion of somatic cells into another unrelated somatic cell type directly, without transiting through intermediate pluripotent or multipotent stages. Bypassing developmental stages, direct cellular reprogramming is specific, more efficient, faster, and easier from the manufacturing point of view, as well as suitable for in vivo applications as it does not impose the risk of malignant transformation associated with pluripotency. On the downside, cells generated via direct reprogramming may show premature phenotypes and higher infidelity to their physiologic counterparts caused mainly by the incomplete epigenetic memory loss during the reprogramming process. For instance, while both iPSC-derived and directly reprogrammed neurons launch gene regulatory networks specific for neurons, directly reprogrammed cells retain basal levels of the parental identity attributed to the incomplete erasure of DNA methylation and histone marks of the parental genome (106).
Nevertheless, the number of cell types generated via direct cellular reprogramming is rapidly increasing. The first example of direct reprogramming was in 1987, with the reprogramming of fibroblasts to myoblasts via forced expression of a single transcription factor, MyoD1 (107). The combinatorial approach has enabled the identification of master lineage transcription factors for the direct reprogramming into neurons, cardiomyocytes, blood progenitors, hepatocytes, neurons, Sertolli cells, adipocytes, chondrocytes, osteoblasts, or pancreatic insulin-producing β-cells (108–120).
Overall, a better understanding of cell fate plasticity opened new avenues for the potential application of cell reprogramming in regenerative medicine and tissue repair. Importantly, cell reprogramming is now providing the means to modulate tumor immunity either by iPSCs immunomodulatory properties or through the generation of discrete immune cell populations that can reinitiate the cancer-immunity cycle. Cell reprogramming may facilitate large-scale and standardized manufacturing of clinically relevant cell types which can be bio-banked and used as an off-the-shelf therapy. Such approach could meet limitations of current strategies for cancer immunotherapy and provide alternative sources of important immune players such as T-cells, NK-cells, or DCs, addressing current challenges of limited availability, and functional exhaustion.
iPSCs in Development of Cancer Vaccines
Given their proliferative potential and metabolic requirements, iPSCs share many features with cancer stem cells. Among these, iPSCs express TAAs which may be recognized by the immune system (121, 122). Interestingly, immune responses launched by iPSCs do not propagate the development of autoimmunity, indicating that fetal-like and adult settings are associated with a differential expression of antigens. To this end, their immunogenic properties render iPSCs as an interesting, patient-specific, source of cells for manufacturing prophylactic cancer vaccines, circumventing tumor mass collection. The efficacy of iPSCs-based vaccines has been addressed. Li et al. compared the efficiency of immunization with iPSCs and ESC lines in a mouse colorectal cancer model and demonstrated that while both vaccines activated IFN-γ and IL-4 producing T-cells, tumor growth was prevented only in the context of ESC-based vaccine (123). In contrast, transplantation of mouse fibroblast-derived iPSC into teratoma-bearing mice leads to T-cell infiltration and tumor shrinkage (124). More recently, iPSC-based vaccination halted growth of murine breast cancer, mesothelioma, and melanoma, as well as prevented melanoma relapse (122).
Reprogramming Into Macrophages
Macrophages are malleable immune cells that promptly respond to environmental cues and signals. Macrophages are essential for tissue homeostasis by clearing dying cells and invading pathogens, but their role is ambiguous and under certain conditions, macrophages actively contribute to the development of atherosclerosis or neurodegeneration. The ambiguity of macrophage functions is given by their polarization towards the promotion or suppression of immunity. In the context of cancer, pro-inflammatory macrophages (M1) can contribute to tumor clearance by the production of IL-12 and other pro-inflammatory cytokines, generation of reactive oxygen species, and presentation of antigens from engulfed dead tumor cells. However, cues produced by tumor cells can polarize macrophages into M2 anti-inflammatory phenotype, inducing an immunosuppressive microenvironment and supporting tumor development (125).
Several publications reported successful differentiation of macrophages from iPSCs upon culture with IL-3 and M-CSF (126, 127). iPSC-derived macrophages displayed a potent phagocytic capacity and rescued immunodeficient mice from Pseudomonas aeruginosa infection (126). Despite displaying functional features, differentiated macrophages showed low polarized phenotypes, expressing features of both pro-inflammatory M1 (CD195+, HLA-DR+) and suppressive M2 macrophages (CD163+, CD206+). Similarly, this ambiguity was mirrored by secretion of both pro-inflammatory cytokines, such as IL-6, CXCL8, CCL2, CXCL10, and M2-like cytokines, IL-10 and VEGF (128). A major challenge for the derivation of immune cells from iPSC resides in the specification of definitive hematopoietic stem cells (HSCs). Despite intense efforts in the last two decades, there is still no robust protocol for the generation of engraftable HSCs from iPSCs that would provide a renewable source of definite hematopoietic and immune cells (129). However, in contrast to other immune cells, microglia, the only lifelong resident immune cells of the central nervous system (CNS), have been suggested to be derived predominantly from yolk sac progenitors (130). This increases the feasibility of generating these highly specialized macrophages with fidelity from iPSCs that have recognized roles in neurodegenerative diseases (131–133).
Microglia can cross the blood-brain barrier, which makes them interesting vehicles for the delivery of therapeutic agents to the CNS. To this end, macrophages can be armed with a single chain variable fragments (scFv) specific to β-amyloid to help devour amyloid plaques, bringing new potential for the treatment of Alzheimer’s disease (134). A similar approach of macrophage modification can be also used to enhance the macrophage activity against certain tumors. As an example, macrophages generated from iPSCs engineered to express CD20-specific scFv eliminated B-lymphoblastic leukemic cells both in vitro and in a xenograft murine model (134). Macrophages modified with CARs to direct the engulfment of specific antigens have also been produced, showing an increased ability to phagocyte cancer cells (135).
Macrophages can also be generated by direct cellular reprogramming (Figure 2), via combined expression of the transcription factor CEBPα or CEBPβ, which favors myeloid development by the downregulation of B-cell commitment transcriptional factor PAX5, and PU.1 which is critical for instructing myeloid transcriptional program (136). By this approach, human fibroblasts were successfully reprogrammed into functional macrophage-like cells in less than a week, acquiring the ability to phagocyte bacteria and mount inflammatory responses (137). Transcription factors can also be applied to accelerate and direct the differentiation of iPSCs, a process also known as forward reprogramming (138, 139). Chen et al. have recently tested this strategy for the induction of immune cells. PU.1 and CEBPα were utilized to orchestrate rapid reprogramming of iPSCs into induced microglia endowed with several functional properties including LPS/IFN-γ-induced inflammatory responses, ADP/ATP-evoked migration, and phagocytic capacity (140). These findings highlight the advantage of employing direct reprogramming core regulatory circuits not only to define immune cellular identities but also to support their generation from somatic or pluripotent cells.
Reprogramming Into Dendritic Cells
DCs are key orchestrators of both innate and adaptive immunity. Given the low abundance of DCs in peripheral blood, cell reprogramming may offer an exciting opportunity to generate DC subsets at scale. The protocols to differentiate DC subsets from iPSCs are at their infancy, revealing heterogeneity of generated DC populations as one of the main challenges to overcome. DCs derived from iPSCs need multistep protocols, including differentiation with FLT3L, SCF, GM-CSF (that enrich in pDC-like cells), and IL-4 (enriching in cDC1 and cDC2-like cells), as well as the maturation signals TNFα and LPS (141).
Sachamitr et al. reported the directed differentiation of iPSCs into CD141+ cDC1 cells resembling naturally occurring cDC1s. Consistently with their capacity for antigen presentation, CD141+ iPSC-DCs acquired immunostimulatory CD40+ CD86+ phenotype upon TLR engagement and could elicit activation and proliferation of naïve T-cells. However, induced DCs produced only limited amounts of IL-12 cytokine and in a steady state displayed tolerogenic features characteristic for tissue-resident DCs (142). CD141+ DC1-like cells were differentiated also from iPSCs derived from T-cells and showed functional capacity to process antigens (143).
Additionally, iPSCs derived DCs displayed rather fetal, immature phenotypes, reflected by the low expression of HLA class II and costimulatory molecules, low secretion of the hallmark cytokine IL-12, and limited ability to activate antigen-specific cytotoxic T-cells via antigen cross-presentation (134). Nevertheless, immature fetal DC phenotype can be partially circumvented by differentiation of iPSCs generated from CD11c+ DCs, resulting in an overall improvement of DC-specific functions and a significant increase in DC-mediated T-cell activation (144). Interestingly, the utilization of the iPSC reprogramming platform also allows to introduce genetic modifications which can potentiate anti-tumor responses. For instance, vaccination with mo-DC-like DCs derived from modified, CEA expressing iPSCs restricted growth of CEA-positive murine colon carcinoma model (145).
We have recently reported the generation of cDC1-like cells by direct cellular reprogramming from mouse fibroblasts with the master regulators PU.1, IRF8, and BATF3 (Figure 2). Only nine days after reprogramming initiation, induced DCs established a cDC1 specific transcriptional program and morphology resembling their natural counterparts. Importantly, induced DCs were endowed with cDC1 hallmark functional properties including secretion of IL-12 and other pro-inflammatory cytokines, phagocytosis of dead cells, and cross-presentation (146). This study opens the way to employ direct cell reprogramming and forward reprogramming principles for the specification of homogeneous sub-populations of DCs.
Reprogramming Into Granulocytes
In contrast with numerous efforts towards reprogramming into macrophages and dendritic cells, differentiation towards granulocytic lineages was investigated less intensively. However, generation of autologous granulocytes holds promise to replenish immune competence in patients after HSC transplantation or chemotherapy to prevent the development of sepsis and severe bacterial infections. Using multistep protocols and growth factor cocktails, granulocytes have been successfully differentiated from iPSCs. iPSCs are first instructed towards hematopoietic precursors, followed by the induction of myeloid lineage and subsequent terminal differentiation towards granulocytic fate with FLT3L, SCF, TPO, IL-3, IL-6, G-CSF. This results in the generation of enriched populations of eosinophils, and upon OP9-DL1 co-culture, allows the specification of neutrophils (126, 147, 148). In accordance with their natural counterparts, iPSC-derived granulocytes acquired competence to phagocyte, produce reactive oxygen species, and, to a lesser extent, the ability to migrate in response to chemotactic stimuli (147). Alike many other immune cells, the instructor transcription factors that in combination impose granulocytic cell identity remain to be identified.
Reprogramming Into NK Cells
NK-based immunotherapy recently received plenty of interest. Many clinical and preclinical trials demonstrated significant efficacy of NKs particularly against AML and ovarian cancer (149). However, current NK-based immunotherapy products face hurdles imposed mainly by relatively high variability in the efficacy of NKs derived from different donors. Furthermore, allogenic NK products are usually prepared from donor blood by apheretic depletion of CD3 and CD19 cells, resulting in a monocyte/NK cell mixture containing approximately only 30-40% of NK cells in the final product (150). Therefore, immunotherapy approaches may leverage iPSCs to generate an off-the-shelf supply of NK cells with a known haplotype in a highly standardized manner. Indeed, iPSC-derived NK cells are currently being tested in the clinical setting, representing the first example of immune cells generated by cell fate reprogramming to reach patients.
NK cells can be differentiated from human iPSCs using a two-stage protocol without additional sorting or the use of xenogeneic feeder cells. In this way, iPSCs are first instructed into hematopoietic progenitors and then stimulated with IL-3, IL-7, IL-15, SCF, and FLT3L to further promote NK differentiation. At the end of a 30-day culture period, induced NK cells expressed inhibitory and activating receptors including KIR, CD16, NKG2D, or NKp46 (151). Recently, Dege et al. identified iPSC-derived erythro-myeloid progenitors-like NK cells that are more potent in terms of cytotoxicity than adult CD16+ NK cells. Despite using the same protocol to induce NK cells, hematopoietic progenitors that resemble yolk sack precursors gave rise to NK cells with higher degranulation response when stimulated with K562 cancer cells, suggesting that this new population has potential for cancer immunotherapy (152). In addition, protocols to expand NK cells by the co-culture with artificial antigen-presenting cells such as K562 expressing membrane-bound 4-1BBL, IL-15, or IL-21 have been developed (153, 154).
Anti-tumor activity of induced NK cells can be further potentiated with a non-cleavable CD16a activating receptor which confers increased antibody-dependent cellular toxicity (155). In terms of function, in an ovarian xenograft mouse model, iPSC-NK cells reduced tumor burden comparably to NK cells purified from peripheral blood. Furthermore, iPSC-derived NKs may serve as a platform allowing modification of NK cells with CARs to improve cytotoxicity against NK therapy-resistant tumors. In this regard, Ueda et al. developed a feeder-free differentiation protocol employing FLT3L, IL-7, and phytohemagglutinin (PHA) to yield CAR-expressing NK/ILC cells from an iPSC line transduced with anti-GPC3-CAR. Notably, CAR NK/ILC were capable to elicit cytotoxic response against GPC3 expressing tumor cells, and prolonged survival when injected into mice with ovarian cancer (156). While the transcriptional code for direct reprogramming of NK cells has not been unveiled, Li et al. have provided evidence for the transdifferentiation of mouse T-cells into NK cells by the deletion of Bcl11b, a transcription factor that acts as a guardian of T-cell development and identity maintenance, allowing a rapid switch to NK identity (157).
Reprogramming Into T-Cells
There is wide interest in utilizing iPSC technology towards the generation of naïve T-cells, as patient-derived T-cells are frequently rare or functionally exhausted. Large-scale, standardized production of cytotoxic T-cells would have an immense impact on CAR T-cell manufacturing. These systems are still under development to recapitulate fully functional T-cells and require extensive cell culture steps with cytokines or artificial thymic organoids (ATO) (158). The iPSC differentiation process itself recapitulates the natural T-cell development, transiting through hematopoietic precursors, double-positive CD4+CD8+ thymocytes, and maturation of CD4+ or CD8+ cells upon culturing with FLT3L and IL-7 on OP9-DL1 feeder layers mimicking NOTCH-ligand rich thymic microenvironment. Although initial studies failed to produce T-cells with a high level of resemblance to their natural counterparts, CD8+ T-cells generated from iPSCs elicited specific cytotoxic responses, despite some phenotypic differences including the absence of CD28 (159, 160). However, the maturation into CD4+ T-cells is even more challenging and requires ATO-based T-cell differentiation supported by FLT3L, SCF, and IL-7 (161). In an effort to move T-cell reprogramming further towards clinical applications, Iriguchi et al. developed a clinically applicable T-cell differentiation technique based on SDF1α and p38 inhibitor, allowing differentiation without feeder layers (162). For the specification of T-cells, the somatic origin of iPSC significantly impacts differentiation. iPSCs derived from cells of hematopoietic origin are more efficient to produce CD4+CD8+ double-positive lymphoid precursors, probably due to partially retained epigenetic memory which alleviates differentiation blocks in T-cell development (160). Moreover, iPSC derived from T-cells confer parental genetically rearranged TCR loci which will remain unchanged during reprogramming and differentiation, allowing the generation of rejuvenated T-cells that maintain antigen specificity (159, 160). In this regard, taking advantage of pre-existing anti-tumor antigen specificity, tumor-specific T-cells clones were reprogrammed to iPSCs to produce rejuvenated and functionally competent T-cells recognizing common tumor-specific antigens including MART-1 (melanoma), WT1 (leukemia) or GPC3 (hepatic and ovarian carcinoma) (159, 163). In regards to additional T-cell populations, Themeli et al. differentiated CAR-modified T-iPSC with FLT3L, SCF, IL-7 into CD3+TCR+CD8α+ CAR T-cells resembling γδ T-cells in their transcriptomic profiles and functions, and demonstrated that despite expressing endogenous αβ TCR, the induced T-cells conferred functional characteristics of innate lymphocytes (164).
While direct reprogramming of T-cells has not been reported, Zhang et al. showed the in vivo conversion of pro-pre-B cells into functional early T-cell lineage progenitors by transient expression of HOXB5. Although the conversion occurred in the bone marrow, early T-cell lineage progenitors migrate to the thymus and mature into functional polyclonal T-cells (165). The forward reprogramming approach has also been attempted by Guo et al., reporting a generation of T-cells from iPSCs in vivo. In this protocol, hematopoietic progenitors with thymus-homing abilities were generated by the expression of RUNX1 and HOXA9 at the endothelial-to-hematopoietic transition stage. Upon engraftment, these induced progenitor cells gave rise to T-cells that restore immune surveillance in immune-deficient mice (166).
Reprogramming within the lymphocyte lineage has also been shown to occur in humans. Interestingly, mutations in FOXP3, a transcription factor tightly controlling Treg homeostasis, functionally reprogram Treg towards Th2-like phenotype, causing immune dysregulation, and driving autoimmune polyendocrinopathy (167).
An additional approach to T-cell reprogramming relies on de-differentiation. Several publications demonstrated functional reprogramming of T-cells into stem cell-like memory CD8+ T-cells by inducing metabolic changes or the introduction of transcription factors. Memory T-cells self-renew, live longer and exert stronger effector functions, and therefore can act as a reservoir for effector T-cells with a potent therapeutic potential. Lu et al. promoted reprogramming of terminally differentiated effector T-cells into a less differentiated state via the overexpression of early T-cell differentiation-specific transcription factors LEF1, KLF7, ID3, EOMES, BCL6, TCF7, FOXP1, and FOXO1 (168).
Another type of reprogramming that does not entail a change in cellular identity is metabolic reprogramming. While it is well described that cancer cells undergo metabolic reprogramming to enhance survival in the hypoxic tumor environment (169), there is a growing body of evidence that metabolic alterations also occur in immune cells altering their function. Changes in the metabolic profile impacts differentiation and the activity of T-cells and other immune cells, a topic recently discussed by others (170, 171). In lymphocytes, metabolic reprogramming following T-cell activation imprints different functional fates. Interestingly, the promotion of lipolysis along with the inhibition of glycolysis induced by PD-1/CTLA-4 inhibition hamper T-cell effector differentiation (172). Zhang et al. hinted that increasing fatty acid oxidation in CD8+ T-cells prevents T-cell functional exhaustion and works synergistically with checkpoint inhibitors to delay or prevent tumor growth in murine cancer models (173). Exhausted tumor-infiltrating CD8+ T-cells were recently reinvigorated by IL-10 which revived their proliferative capacity and cytotoxic functions against cancer cells (174). Moreover, Verma et al. demonstrated reprogramming of CD8+ T-cells into memory stem cells with enhanced functional capacities using a MEK1/2 inhibitor, enhancing respiratory capacity and fatty acid oxidation, metabolic features previously associated with memory cells. Functionally, MEK inhibition administered with vaccination against known antigens (gp100 in melanoma, HPV16E7 in cervical carcinoma model) in murine cancer models successfully increased numbers of circulating CD8+ T-cells, reduced tumor burden, and improved survival rates (175). Indeed, manipulations of T-cell metabolism may help to improve the efficacy of T-cell-based immunotherapies (176).
Reprogramming Into B-Cells
Generation of B-cells from iPSCs is equally challenging. Under specific conditions promoting lymphopoiesis, iPSCs can differentiate towards the B-cell lineage and form pre-B cells expressing CD45, CD19, and CD10. Reprogrammed B-cells upregulated the expression of pre-B specific genes including PAX5, IL7αR, and VpreB receptor. Interestingly, while they remained negative for surface IgM, induced cells displayed genomic D-J rearrangements, consistently with the adoption of pre-B identity (177). French et al. took the differentiation a step further and reported the generation of CD19+ CD10+ B lymphocytes via terminal differentiation with FLT3L, SCF, IL-3, and IL-7 on MS-5 stroma. iPSC-derived B-cells underwent full VDJ rearrangement and expressed surface IgM (178). While efforts in reprogramming and programming individual immune cell identities with fidelity will provide valuable cell sources for cancer immunotherapy, additional approaches take advantage of cellular reprogramming principles to modify cancer cell fate directly.
Modifying the Cell Fate of Cancer Cells
Tumor development is accompanied by both epigenetic and genetic changes of the genome. Unlike genetic changes, epigenetic changes do not alter the primary DNA sequence and are therefore reversible. So, reprogramming paradigms are also highly relevant to understand oncogenesis, from the initial cellular transformation to the hierarchical organization of established malignancies (179). Indeed, alike somatic cells, cancer cells may be endowed with similar epigenetic flexibility. During reprogramming, the whole epigenetic landscape undergoes massive remodeling, and changes in histone modification and DNA methylation patterns underlie dramatic changes in gene expression (179). It was quickly hypothesized that the fate of cancer cells could be modified by direct reprogramming leading to a potential loss of tumorigenicity. The concept of reversing tumorigenicity dates to 1969, when experiments performed by Henry Harris attenuated malignant phenotype by fusing mouse cancer cells with fibroblasts, providing first insights into the activity of tumor suppressor genes (180). Experiments with somatic cell nuclear transfer – where melanoma nuclei were fused to enucleated oocytes showed that it is possible to re-establish pluripotency in cancer cells leading to differentiation into multiple benign cell types. Therefore, these experiments demonstrated that genetic and epigenetic changes of a melanoma nucleus are compatible with broad developmental potential but predispose mice to melanomas (181).
Reprogramming the cancer cell phenotype with transcription factors draws two relevant implications. First, induced pluripotent cancer stem cells are offering a valuable model to study cancer heterogeneity, cancer stem cells, and their genetic drivers as iPSC clones derived from single cells retain mutational signatures (182). Second, the reorganization of cancer cell epigenome may allow cancer cells to acquire a benign phenotype (183).
Reprogramming of tumor cells into pluripotency was achieved in various types of malignant cells, however, given pre-existing reprogramming barriers, the efficiency remains low (184). Enforced expression of all or several of OSKM transcription factors generated iPSCs from multiple cancer cell lines including mouse melanoma (185), glioblastoma (186, 187), chronic myeloid leukemia (188), bladder cancer (189), breast carcinoma (190), and a range of gastrointestinal cancers (191) (Figure 3). Moreover, iPSC generation was achieved also from primary malignant leukemic blasts (192). In accordance with epigenetic rewiring, cancer reprogramming alleviated tumorigenicity in vitro and in vivo in most reprogrammed cells, even in those harboring constitutively active oncogenes. For instance, iPSC derived from BRAF mutated melanoma were further differentiated into non-tumorigenic neurons and fibroblasts displaying normal phenotype and functions (193). Furthermore, iPSC generated from primary BCR-ABL+ myeloid blasts differentiated into hematopoietic cells which did not retain leukemia potential. Upon transplantation, derived hematopoietic cells engrafted only temporarily and recipient animals did not develop chronic myeloid phenotype (192). Zhang et al. showed that sarcoma-derived iPSC generated primary connective tissue cells and erythroid cells. Moreover, when transplanted into mice, iPSC-derived adipocytes and osteoblasts did not form tumors, suggesting that reprogramming overrode tumorigenicity of parental sarcoma lines (183).
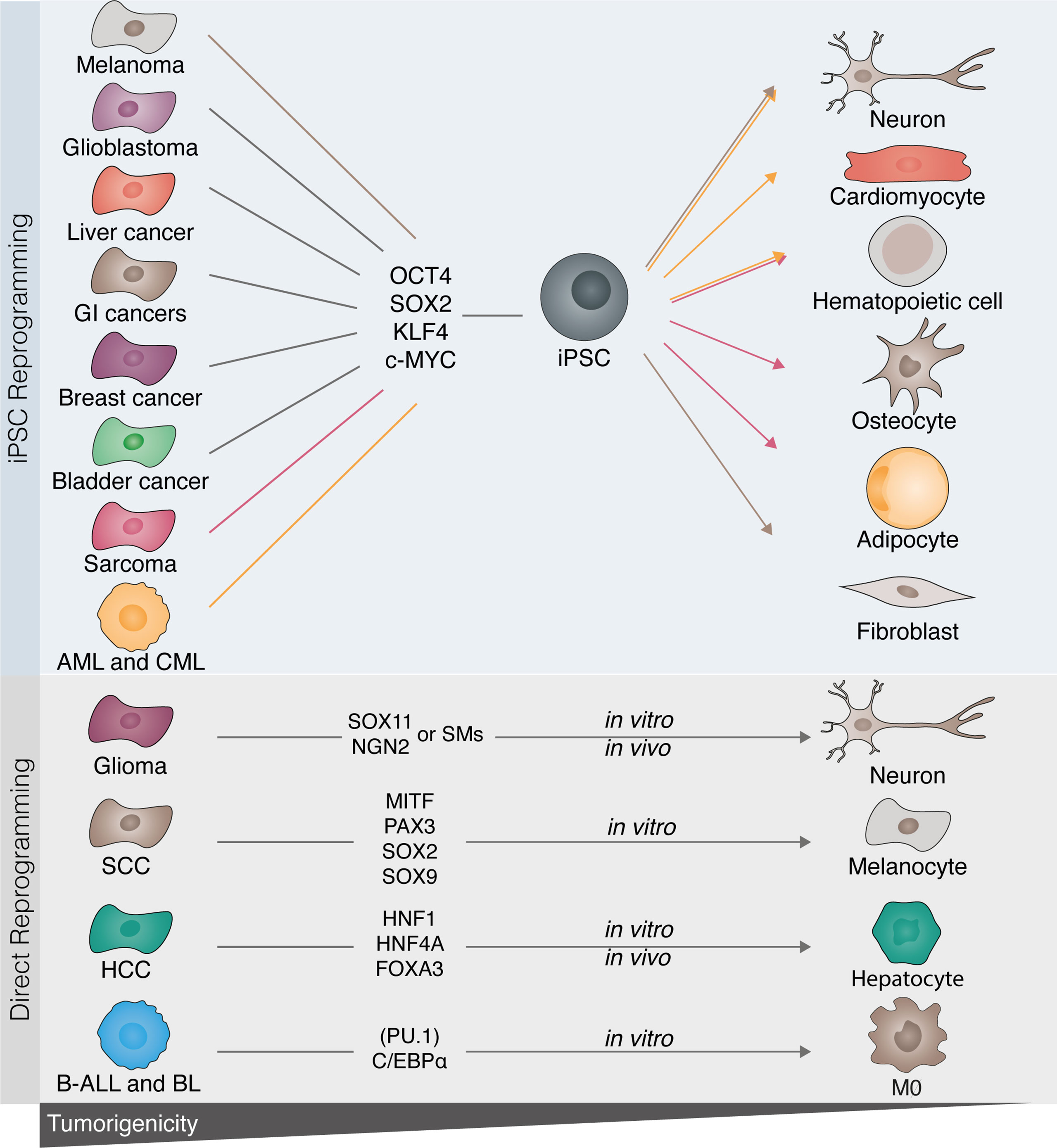
Figure 3 Disrupting cancer cell tumorigenicity with cellular reprogramming. Several tumor cell types have been described to be amenable to cell reprogramming towards induced pluripotent stem cells (iPSCs). Melanoma, glioblastoma, sarcoma, gastrointestinal (GI), as well as breast, bladder, and liver cancers, acute myeloid leukemia (AML), and chronic myelogenous leukemia (CML) have all been successfully reprogrammed towards iPSC with OCT4, SOX2, KLF4, and c-MYC. Following reprogramming to iPSCs, cancer cells can be differentiated into benignity by either differentiation to their original cell type or by converting them into a different lineage (highlighted by the colored lines). Similarly, in vitro direct reprogramming has been demonstrated in glioma, B-cell acute leukemia (B-ALL), Burkitt lymphoma (BL), squamous cell carcinoma (SCC), and hepatic cellular carcinoma (HCC). Lineage-specific factors rewrite the cancerous epigenetic and transcriptional program into mature terminally differentiated cells (glioma to neuron; SCC to melanocyte; HCC to hepatocyte), or by modifying cell fate and therefore disrupting the tumorigenic drive [B-leukemias to macrophage (MØ)].
Interestingly, iPSC derived from MLL-rearranged acute myeloid leukemia cells could be differentiated into non-tumorigenic cardiomyocytes and neurons exerting physiologic functions despite the presence of oncogenic drivers. However, hematopoietic differentiation of AML-iPSC resulted in retrieval of leukemia potential (194).
Direct Reprogramming of Cancer Cells Into Benignity
In contrast to the strategies forcing pluripotency in cancer cells, direct reprogramming allows the modification of somatic identity and may provide an undeviating way to disrupt the epigenetic tumorigenic drive. Considering the impact of CEBPα on reprogramming of B-cells into the myeloid lineage, Rapino et al. utilized its enforced expression to induce macrophage identity in human B-cell leukemia and lymphoma, generating quiescent phagocytic cells with transcriptomic profiles resembling macrophages (Figure 3). Importantly, activation of macrophage regulatory network, including endogenously expressed CEBPα, sustained the newly acquired macrophage identity (195). Similarly, McClellan et al. transdifferentiated primary acute leukemia blasts into macrophages either with overexpression of CEBPα and PU.1 or upon exposure to a myeloid cytokine cocktail comprising FLT3L, IL-7, IL-3, GM-CSF, and M-CSF, overcoming the differentiation block and reducing leukemogenicity (196).
Loss of tumorigenicity is also induced when cancer cells from the same lineage are induced into mature differentiated cells. Melanocytes induced from squamous cell carcinoma via overexpression of MITF, PAX3, SOX2, and SOX9 lose tumorigenicity, as shown by reduced proliferation, migration, and invasion (197). To explore enforced differentiation in the context of liver cancer, Cheng et al. deployed a combination of transcription factors HNF1A, HNF4A, and FOXA3 to reprogram hepatocellular carcinoma into hepatocytes. Importantly, reprogrammed hepatocytes retrieved benign functions including albumin and glycogen production, and actively contributed to liver regeneration in a murine model of genetic liver dysfunction (113). In glioblastoma, NGN2 and SOX11 were employed to force post-mitotic arrest and differentiation of glioma into neurons (198). In an attempt to develop transgene-free reprogramming methods, neuronal reprogramming in glioma was recently induced with ROCK and mTOR inhibitors (199). Alternatively, Yuan et al. induced a similar conversion with a small compound cocktail composed of forskolin, ISX9, DAPT, CHIR99021, and I-BET 151 (200).
Another solution that does not strictly reflect classical reprogramming was recently proposed by Ishay-Ronan et al. Exploiting the brief window of high cellular plasticity during epithelial-mesenchymal transition (EMT), breast carcinoma cells were diverted into post-mitotic functional adipocytes with a cocktail composed of TGF-β, BMP2, insulin, and dexametazone. Notably, rosiglitazone-induced EMT together with adipogenic differentiation therapy efficiently prevented cancer cell invasion and formation of metastasis in vivo (201). However, not all conversions of cellular identity entail the loss of tumorigenicity. Reprogramming of primary Pax+/-Ebf+/- pro-B leukemic cells into the T-cell lineage via constitutive induction of NOTCH1 forced lineage conversion into early-stage T-cells which displayed leukemic phenotype and potential (202).
Inducing Antigen Presentation in Cancer Cells with Cellular Reprogramming
A large body of evidence indicates that inhibition of antigen presentation is one of the major mechanisms underlying cancer immune evasion, allowing tumor cells to become invisible to immune effector cells. Therefore, restoration or enforcement of antigen presentation machinery in cancer cells holds enormous potential to reactivate cytotoxic anti-tumor responses. While all cell types can present their own antigens through HLA class I molecules, only professional antigen-presenting cells, such as DCs or macrophages, can prime productive T-cell responses by providing costimulatory signals besides HLA class I/II and TCR interactions. Interestingly, expression of class I and class II molecules in tumor cells can be, at least to a certain extent, induced by radiotherapy, several chemotherapeutic agents, a range of small compounds including histone deacetylase (HDAC) inhibitors, tyrosine kinase inhibitors (203), CDK4/6 inhibitors (204), or IFN-γ (205). Unfortunately, instead of boosting immune surveillance, these compounds may further escalate immunosuppression through induction of T-cell anergy triggered by TCR engagement in the absence of proper co-stimulation. In this context, reprogramming of cancer cells towards professional antigen-presenting cells may represent an unexplored avenue to elicit anti-tumor polyclonal T-cell responses. The establishment of dendritic-like cell identity in tumor cells could aid immune surveillance reactivation in a more complex way, removing two critical barriers of efficient anti-tumor immunity – deficient antigen presentation of tumor antigens and insufficient T-cell activation. Moreover, as DC-reprogrammed tumor cells could promote the recruitment of T-cells to the tumor site by the secretion of CXCL9 and CXCL10, such strategy could also potentiate the effect of other T-cell-based cancer immunotherapies including immune checkpoint blockade and adoptive cell transfer.
Reprogramming In Vivo
Inducing Cell Fate Reprogramming In Vivo
iPSC differentiation and direct reprogramming are now starting to provide the means to address the potential for cancer immunotherapy of unique patient-tailored immune cell types. However, long-term survival and functional integration within the target tissue remain long-term challenges for transplantation of in vitro generated cells. In parallel with ex vivo based therapy, the development of viral vectors for gene delivery has opened opportunities for in vivo reprogramming in regenerative medicine, an elegant strategy circumventing current limitations of in vitro cell manipulation. Therefore, the possibility of switching cell identity within the living organism gained attention for its immense therapeutic potential. The main advantages of inducing immune cell reprogramming in vivo would be the use of internal cell sources, limited immune rejection, limited risk for tumor formation and increased reprogramming efficiency and fidelity (206). In an in vivo setting, cells receive a full set of external cues that are difficult to recapitulate in vitro, maximizing the efficiency of reprogramming. However, efficient, safe, cell-specific delivery of transcription factors required for eliciting reprogramming represents the major bottleneck (Figure 4). Safety concerns accelerated the development of non-integrative viral vectors, non-viral delivery systems, as well as prompt efforts, and the design of cell type-specific promoters for more precise targeting (207–211).
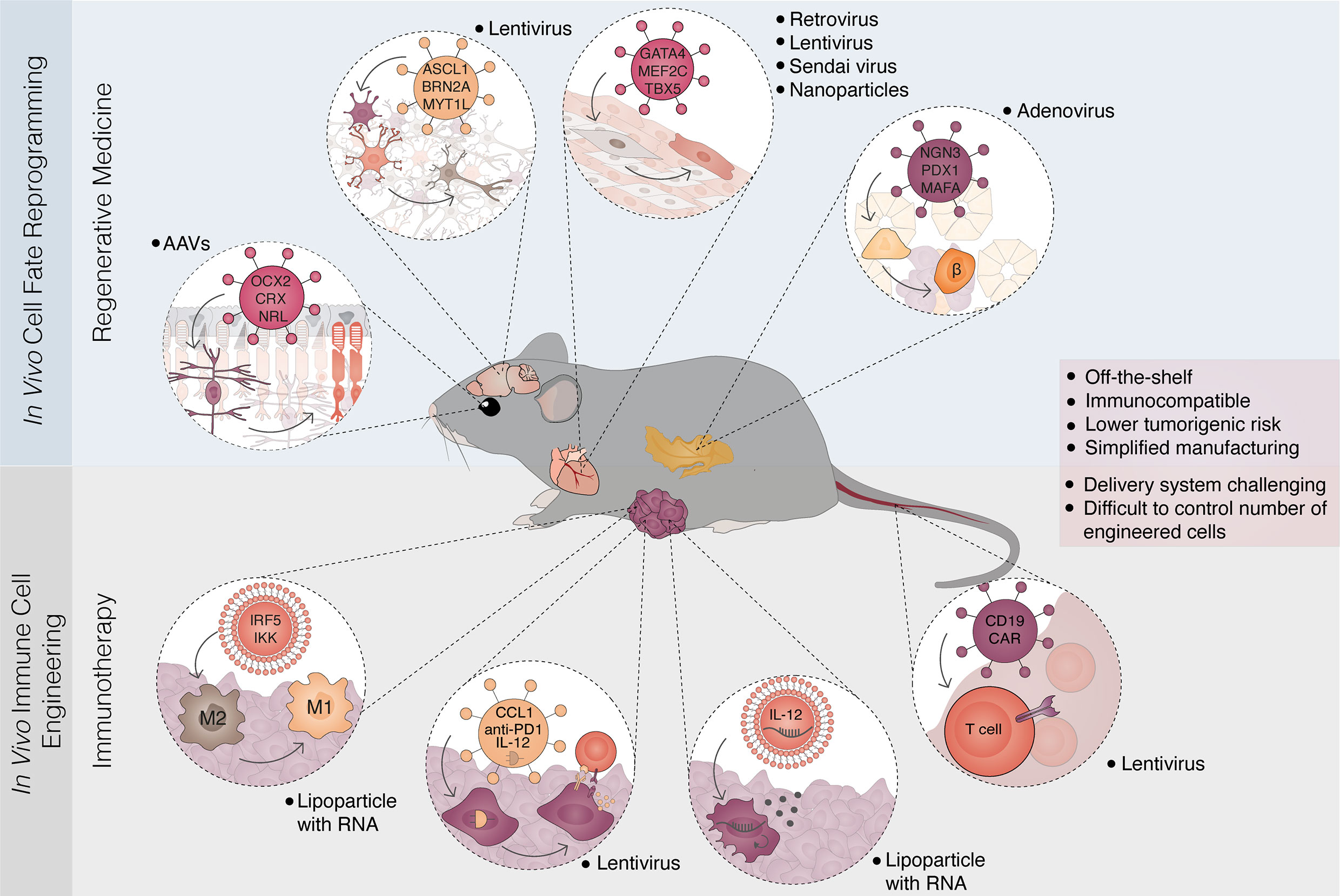
Figure 4 In vivo reprogramming strategies for regenerative medicine and immunotherapy. Overview of parallel in vivo approaches for cell fate reprogramming and immune cell engineering. In vivo cell fate reprogramming offers an approach to restore damaged cell types and tissue function (top panel). The concept has been demonstrated in multiple tissues and cellular conversions. Examples include the reprogramming of Müller glia cells to photoreceptors leading to restoration of vision in congenitally blind mice; reprogramming of astrocytes and glial cells to neurons; the conversion of fibrotic tissue after myocardial infarction to functional cardiomyocytes and the reprogramming of pancreatic acinar/exocrine cells into insulin-producing β-cells. These strategies mainly rely on the delivery of lineage-specific factors through viral vectors. In parallel, in vivo engineering of immune cells has been developed allowing the generation of genetically modified immune cells, often designated immune reprogramming (bottom panel). Recent examples in cancer immunotherapy include the conversion from macrophage type 2 (M2) to macrophage type 1 (M1), manipulation of cancer cells to express immunogenic circuitry and cytokines, and the in vivo delivery of chimeric antigen receptors (CAR) to circulating T-cells. Delivery strategies are diverse including lipid nanoparticles coupled with DNA or replicative RNA, and viral vectors (lentivirus, AAV, retrovirus, adenovirus, Sendai virus). In vivo approaches in regenerative medicine and immunotherapy converge towards the creation of off-the-shelf, immunocompatible therapies that harbor low tumorigenic risks. Simplified clinical translation and manufacturing are balanced by challenges in delivery and the control of cell-type specificity and numbers. AAV, adeno-associated virus.
In vivo reprogramming has been demonstrated with the expression of OCT4, SOX2, KLF4, and c-MYC and the induction of pluripotency. These studies demonstrated the feasibility of inducing transcription factor-mediated reprogramming in vivo but also highlighted the role of the tissue microenvironment. As the microenvironment and the presence of internal cues have a profound effect on cellular plasticity, local factors significantly impact the efficiency of the reprogramming process. Two independent studies highlighted the favorable effect of senescence, which in vitro significantly hampers reprogramming. Aging or cell damage triggers senescence and induces secretion of inflammatory cytokines, enhancing the natural plasticity of neighboring cells, and making them more amenable to undergo lineage conversion. Notably, lung epithelial cells could have been generated in vivo only after bleomycin-induced damage, accentuating the impact of injury regenerative response on intrinsic cell plasticity (212, 213).
Direct cell reprogramming between unrelated somatic cells holds more attractive therapeutic prospects. Injury response may lead to fibrotic tissue damage and loss of function, as seen for instance after myocardial infarction or brain injury. In this context, in vivo direct reprogramming offers an interesting alternative to restore damaged cell types and tissue function (Figure 4). In mice, resident cardiac fibroblasts were reprogrammed into functional cardiomyocytes with GATA4, MEF2C, and TBX5, counteracting adverse ventricular remodeling and improving function after a heart attack (112, 214). Similarly, overexpression of neural transcription factors in astrocytes and glial cells triggered reprogramming into neurons that were able to integrate within the existing neuronal circuits (215–217). Furthermore, encouraging results were delivered by conversion of pancreatic acinar/exocrine cells into insulin-producing β-cells (120, 218) or by induction of functional hepatocytes in the murine fibrotic liver, leading to an improvement in liver performance (219, 220). Notably, restoration of liver function was achieved also with hepatocytes reprogrammed in vivo from hepatocellular carcinoma cells (113). More recently, Kurita et al. turned wound resident mesenchymal cells into epithelial skin cells with DNP63A, GRHL2, TFAP2A, and MYC (221), while Yao et al. utilized in vivo reprogramming to restore vision in congenitally blind mice, with the overexpression of OCX2, CRX, and NRL. This transcription factor combination promoted photoreceptor identity in proliferating Müller glia cells preconditioned with β-catenin (222). Despite the numerous examples and vast range of applications in regenerative medicine, in vivo reprogramming paradigms have not yet been explored for immunotherapy.
In Vivo Immune Cell Engineering
An analogous approach has been recently developed in cancer immunotherapy by the generation of immune cells engineered in vivo. Thus, there is a conceptual parallel between in vivo cell fate reprogramming and in vivo immune genetic engineering (often named immune reprogramming), highlighting a possible convergence of approaches towards cancer immunotherapy (Figure 4). The engineering of immune cells may help to overcome cancer therapy limitations, including lack of targetable antigens or tumor-mediated immune suppression. Although immune-stimulatory factors such as cytokines and chemokines could be directly used to attempt to overcome these challenges, systemic administration often results in strong off-target activity and severe adverse effects, underscoring the need for in situ solutions.
The maintenance of tolerogenic tumor niche is attributed to tumor-associated macrophages (TAM). Their targeted elimination via CSF1R blockade emerged as a potential solution to reactivate localized immune responses (223). However, although CSF1R inhibition limited survival of TAMs and promoted favorable TAM polarization and elimination of glioblastoma in mice (224), also impacted tissue-resident macrophages and brought systemic toxicity. Interestingly, the mission to prevent pro-tumorigenic activities of TAMs may be also accomplished with the reprogramming of M2-polarized TAMs towards M1 pro-inflammatory phenotype. Exploiting the intrinsic phagocytic ability of macrophages, Zhang et al. reprogrammed TAMs with nanoparticles delivering M1 polarizing factors IRF5 and IKK and showed, that repeated delivery of therapeutic lipoparticles abrogated tumor growth and significantly prolonged survival in mice with ovarian carcinoma, melanoma, and glioblastoma. Notably, surviving mice did not form tumors when re-challenged with cancer cells, indicating that reprogramming-based macrophage polarization induced durable systemic immunity which may prevent the formation of metastasis (225).
The immunogenicity of cancer cells is another fundamental determinant of the immune response. The strategy to increase tumor immunogenicity reported by Wang et al. consisted of intratumoral delivery of activating CRISPRa components to augment endogenous expression of TAAs. In mouse melanoma and pancreatic adenocarcinoma models, CRISPRa-mediated enhanced expression of TAAs increased the proportion of antigen-presenting cells and T-lymphocytes within the tumor microenvironment and elicited systemic anti-tumor immunity. Notably, the developed platform can be modified to direct simultaneous activation of expression of multiple TAAs as well as other immunomodulatory molecules, chemokines, or cytokines to further support cancer immune responses (226). A strategy based on RNA was developed by Li et al. with an oncolytic nanoparticle-based system delivering self-replicating RNA encoding IL-12. The combination of immunogenic cell death with high levels of IL-12 elicited a systemic immune response and eradicated melanoma and colon carcinoma in mice (227). Similarly, functional engineering of tumors enhancing immunogenicity was recently accomplished by Tzeng et al. who used nanoparticles to transfect and overexpress 4-1BBL and IL-12 in cancer cells. Considering that most cancer cells retain at least low levels of HLA class I molecules displaying tumor antigens, additional expression of the costimulatory molecule and IL-12 may endow cancer cells with the ability to prime T-cells and trigger NK cell activation, mimicking actions of true professional antigen presenting cells. Notably, functionally reprogrammed cells elicited systemic cell mediated anti-tumor responses and in combination with checkpoint blockade significantly abrogated tumor growth in mice with melanoma and colorectal carcinoma, inducing systemic cancer immunity (228).
A riveting solution to boost tumor immunogenicity and to fuel local anti-cancer immune responses was recently suggested by Nissim et al. who engineered lentiviral-delivered synthetic gene circuits driving expression of immune-modulating components including a surface T-cell engager, the chemokine ligand CCL21, IL-12, and anti-PD1 antibody directly in tumor cells. To prevent unwanted toxicity and the risk of autoimmunity due to the off-target delivery into neighboring non-malignant cells, the expression of immune-modulating agents was linked to the activation of cancer-specific promoters, ensuring that the immune response is directed exclusively against cancer cells. Interestingly, the proposed immune-modulating therapy elicited a robust T-cell mediated response and resulted in the rejection of ovarian cancer (229).
In Vivo Generation of CAR T-Cells
Ex vivo produced CAR T-cells recognizing tumor antigens has proved as a highly effective treatment for patients with refractory B-cell malignancies. The manufacturing process which requires extensive and time-consuming procedures is the limiting step. To simplify the process, Pfeiffer et al. recently developed lentiviral vectors enabling highly selective targeting of CD8+ cells which is used for in vivo generation of CAR T-cells. Despite the relatively low transduction efficiency (not exceeding 5%), in vivo engineered CD19-CAR T-cells demonstrated the ability to eliminate CD19+ cells in mice (230). Importantly, a single injection of CD8 targeted lentivirus was sufficient to generate CD19-CAR T-cells and efficiently eradicate CD19+ leukemic blasts in mice within two weeks (231). Elimination of malignant B-cells was achieved also with in vivo modified CD4+ T-cells expressing CD19-CAR within 2-3 weeks. Interestingly, in tumor-bearing mice, CD4+ CD19-CAR T-cells exhibited higher anti-CD19 activity and outperformed CD8+ CAR T-cells especially in the context of higher tumor burden, suggesting that CD4+ CAR T-cells are less prone to exhaustion (232). In the clinical context, in vivo CAR generation may have major implications on the manufacturing process and facilitate the development of off-the-shelf CAR T-cell therapy, making it accessible for a larger group of patients.
Cancer immunotherapy has revolutionized cancer treatment. The limits of the current strategies both individually or in combination will be elucidated in the coming years. Harnessing cell reprogramming concepts towards immunotherapy represent an entirely new set of approaches with the potential to take advantage of the full immune repertoire, their identity, and the individual immunity modules under their control. Possibilities range from novel sources of allogeneic immune cells, rejuvenating functionally exhausted effector cells, disrupting vicious regulatory networks of cancer cells, or modifying the immunosuppressive tumor microenvironment by inducing immune cell identities in vivo. Overall, the redefinition of immune cell identity and their regulatory circuits by cell fate reprogramming is now providing new and exciting tools for the fast-paced field of cancer immunotherapy.
Author Contributions
OZ, IC, AGF, and C-FP conceptualized this manuscript. IC prepared the figures. AGF wrote figure legends. OZ and C-FP wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project was co-funded by Cancerfonden (20 0939 PjF), the Swedish research council (2020-00615), European Research Council (866448-TrojanDC), Novo Nordisk Foundation (0056527) and FCT (PTDC/MED-IMU/4520/2020). The Knut and Alice Wallenberg foundation, the Medical Faculty at Lund University and Region Skåne are acknowledged for generous financial support. OZ is supported by a Cancerfonden postdoctoral fellowship 190008. IC and AGF are supported by FCT doctoral fellowships (PD/BD/146424/2019 and SFRH/BD/133233/2017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
2. Chen DS, Mellman I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
3. Galon J, Bruni D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity (2020) 52(1):55–81. doi: 10.1016/j.immuni.2019.12.018
4. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell (2017) 168(4):613–28. doi: 10.1016/j.cell.2017.01.018
5. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
6. Coley WB. The Treatment of Malignant Tumors by Repeated Inoculations of Erysipelas. With a Report of Ten Original Cases. Clin Orthop Relat Res (1893) 1991(262):3–11. doi: 10.1097/00003086-199101000-00002
7. Morales A, Nickel JC. Immunotherapy of Superficial Bladder Cancer With BCG. World J Urol (1986) 3(4):209–14. doi: 10.1007/BF00632180
8. Rosenberg SA. Interleukin 2 for Patients With Renal Cancer. Nat Clin Pract Oncol (2007) 4(9):497. doi: 10.1038/ncponc0926
9. Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic Cancer Vaccines. Nat Rev Cancer (2021) 21:360–8. doi: 10.1038/s41568-021-00346-0
10. Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A. The Hallmarks of Successful Anticancer Immunotherapy. Sci Transl Med (2018) 10(459):eaat7807. doi: 10.1126/scitranslmed.aat7807
11. Merad M, Sathe P, Helft J, Miller J, Mortha A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu Rev Immunol (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
12. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic Cells, Monocytes and Macrophages: A Unified Nomenclature Based on Ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi: 10.1038/nri3712
13. Mildner A, Jung S. Development and Function of Dendritic Cell Subsets. Immunity (2014) 40(5):642–56. doi: 10.1016/j.immuni.2014.04.016
14. Idoyaga J, Lubkin A, Fiorese C, Lahoud MH, Caminschi I, Huang Y, et al. Comparable T Helper 1 (Th1) and CD8 T-Cell Immunity by Targeting HIV Gag P24 to CD8 Dendritic Cells Within Antibodies to Langerin, DEC205, and Clec9A. Proc Natl Acad Sci USA (2011) 108(6):2384–9. doi: 10.1073/pnas.1019547108
15. Segura E, Amigorena S. Cross-Presentation in Mouse and Human Dendritic Cells. Adv Immunol (2015) 127:1–31. doi: 10.1016/bs.ai.2015.03.002
16. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science (2017) 356(6335):eaah4573. doi: 10.1126/science.aah4573
17. Penna G, Vulcano M, Roncari A, Facchetti F, Sozzani S, Adorini L. Cutting Edge: Differential Chemokine Production by Myeloid and Plasmacytoid Dendritic Cells. J Immunol (2002) 169(12):6673–6. doi: 10.4049/jimmunol.169.12.6673
18. Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. Upon Viral Exposure, Myeloid and Plasmacytoid Dendritic Cells Produce 3 Waves of Distinct Chemokines to Recruit Immune Effectors. Blood (2006) 107(7):2613–8. doi: 10.1182/blood-2005-07-2965
19. Reizis B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity (2019) 50(1):37–50. doi: 10.1016/j.immuni.2018.12.027
20. Shinde P, Fernandes S, Melinkeri S, Kale V, Limaye L. Compromised Functionality of Monocyte-Derived Dendritic Cells in Multiple Myeloma Patients may Limit Their Use in Cancer Immunotherapy. Sci Rep (2018) 8(1):5705. doi: 10.1038/s41598-018-23943-w
21. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell (2019) 177(3):556–71.e16. doi: 10.1016/j.cell.2019.02.005
22. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 Deficiency Reveals a Critical Role for CD8alpha+ Dendritic Cells in Cytotoxic T Cell Immunity. Science (2008) 322(5904):1097–100. doi: 10.1126/science.1164206
23. Chou WC, Hsiung CN, Chen WT, Tseng LM, Wang HC, Chu HW, et al. A Functional Variant Near XCL1 Gene Improves Breast Cancer Survival via Promoting Cancer Immunity. Int J Cancer (2020) 146(8):2182–93. doi: 10.1002/ijc.32855
24. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell (2017) 31(5):711–23.e4. doi: 10.1016/j.ccell.2017.04.003
25. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity (2016) 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012
26. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell (2014) 26(6):938. doi: 10.1016/j.ccell.2014.11.010
27. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N Engl J Med (2010) 363(5):411–22. doi: 10.1056/NEJMoa1001294
28. Charles J, Chaperot L, Hannani D, Bruder Costa J, Templier I, Trabelsi S, et al. An Innovative Plasmacytoid Dendritic Cell Line-Based Cancer Vaccine Primes and Expands Antitumor T-Cells in Melanoma Patients in a First-in-Human Trial. Oncoimmunology (2020) 9(1):1738812. doi: 10.1080/2162402X.2020.1738812
29. Schreibelt G, Bol KF, Westdorp H, Wimmers F, Aarntzen EH, Duiveman-de Boer T, et al. Effective Clinical Responses in Metastatic Melanoma Patients After Vaccination With Primary Myeloid Dendritic Cells. Clin Cancer Res (2016) 22(9):2155–66. doi: 10.1158/1078-0432.CCR-15-2205
30. van Beek JJP, Florez-Grau G, Gorris MAJ, Mathan TSM, Schreibelt G, Bol KF, et al. Human pDCs Are Superior to Cdc2s in Attracting Cytolytic Lymphocytes in Melanoma Patients Receiving DC Vaccination. Cell Rep (2020) 30(4):1027–38.e4. doi: 10.1016/j.celrep.2019.12.096
31. Pires CF, Rosa FF, Kurochkin I, Pereira CF. Understanding and Modulating Immunity With Cell Reprogramming. Front Immunol (2019) 10:2809. doi: 10.3389/fimmu.2019.02809
32. Sabado RL, Balan S, Bhardwaj N. Dendritic Cell-Based Immunotherapy. Cell Res (2017) 27(1):74–95. doi: 10.1038/cr.2016.157
33. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature (2016) 534(7607):396–401. doi: 10.1038/nature18300
34. Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA Vaccine Drives Immunity in Checkpoint-Inhibitor-Treated Melanoma. Nature (2020) 585(7823):107–12. doi: 10.1038/s41586-020-2537-9
35. Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, et al. PD-1 Blockade in Subprimed CD8 Cells Induces Dysfunctional PD-1(+)CD38(hi) Cells and Anti-PD-1 Resistance. Nat Immunol (2019) 20(9):1231–43. doi: 10.1038/s41590-019-0441-y
36. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science (1991) 252(5007):854–6. doi: 10.1126/science.1851332
37. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic Viruses for Cancer Immunotherapy. J Hematol Oncol (2020) 13(1):84. doi: 10.1186/s13045-020-00922-1
38. Andtbacka RHI, Collichio F, Harrington KJ, Middleton MR, Downey G, hrling K, et al. Final Analyses of OPTiM: A Randomized Phase III Trial of Talimogene Laherparepvec Versus Granulocyte-Macrophage Colony-Stimulating Factor in Unresectable Stage III-IV Melanoma. J Immunother Cancer (2019) 7(1):145. doi: 10.1186/s40425-019-0623-z
39. Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J Clin Oncol (2018) 36(17):1658–67. doi: 10.1200/JCO.2017.73.7379
40. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol (2016) 34(22):2619–26. doi: 10.1200/JCO.2016.67.1529
41. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell (2017) 170(6):1109–19.e10. doi: 10.1016/j.cell.2017.08.027
42. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Sci Transl Med (2014) 6(226):226ra32. doi: 10.1126/scitranslmed.3008095
43. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed Death-1 Ligand 1 Interacts Specifically With the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity (2007) 27(1):111–22. doi: 10.1016/j.immuni.2007.05.016
44. Matzinger P. Friendly and Dangerous Signals: Is the Tissue in Control? Nat Immunol (2007) 8(1):11–3. doi: 10.1038/ni0107-11
45. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170(6):1120–33.e17. doi: 10.1016/j.cell.2017.07.024
46. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu Rev Immunol (2001) 19:565–94. doi: 10.1146/annurev.immunol.19.1.565
47. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
48. Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med (2016) 375(18):1767–78. doi: 10.1056/NEJMra1514296
49. Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, et al. PD-L1 Expression by Dendritic Cells Is a Key Regulator of T-Cell Immunity in Cancer. Nat Cancer (2020) 1(7):681–91. doi: 10.1038/s43018-020-0075-x
50. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab Plus Dacarbazine for Previously Untreated Metastatic Melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
51. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J Clin Oncol (2010) 28(19):3167–75. doi: 10.1200/JCO.2009.26.7609
52. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
53. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2017) 377(14):1345–56. doi: 10.1056/NEJMoa1709684
54. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
55. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
56. Lopez-Beltran A, Cimadamore A, Blanca A, Massari F, Vau N, Scarpelli M, et al. Immune Checkpoint Inhibitors for the Treatment of Bladder Cancer. Cancers (Basel) (2021) 13(1):131. doi: 10.3390/cancers13010131
57. Massari F, Di Nunno V, Cubelli M, Santoni M, Fiorentino M, Montironi R, et al. Immune Checkpoint Inhibitors for Metastatic Bladder Cancer. Cancer Treat Rev (2018) 64:11–20. doi: 10.1016/j.ctrv.2017.12.007
58. Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
59. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab Versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med (2015) 373(19):1803–13. doi: 10.1056/NEJMoa1510665
60. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-And-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol (2017) 18(8):1009–21. doi: 10.1016/S1470-2045(17)30516-8
61. Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
62. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
63. Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, Ouyang J, et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J Clin Oncol (2018) 36(10):942–50. doi: 10.1200/JCO.2017.77.3994
64. Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab Plus Nab-Paclitaxel as First-Line Treatment for Unresectable, Locally Advanced or Metastatic Triple-Negative Breast Cancer (IMpassion130): Updated Efficacy Results From a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2020) 21(1):44–59. doi: 10.1016/S1470-2045(19)30689-8
65. Rouanne M, Bajorin DF, Hannan R, Galsky MD, Williams SB, Necchi A, et al. Rationale and Outcomes for Neoadjuvant Immunotherapy in Urothelial Carcinoma of the Bladder. Eur Urol Oncol (2020) 3(6):728–38. doi: 10.1016/j.euo.2020.06.009
66. Postow MA, Hellmann MD. Adverse Events Associated With Immune Checkpoint Blockade. N Engl J Med (2018) 378(12):1165. doi: 10.1056/NEJMc1801663
67. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
68. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation With Antitumor Lymphocytes. Science (2002) 298(5594):850–4. doi: 10.1126/science.1076514
69. Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, et al. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. J Natl Cancer Inst (1994) 86(15):1159–66. doi: 10.1093/jnci/86.15.1159
70. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable Complete Responses in Heavily Pretreated Patients With Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi: 10.1158/1078-0432.CCR-11-0116
71. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T Cell Immunotherapy for Human Cancer. Science (2018) 359(6382):1361–5. doi: 10.1126/science.aar6711
72. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric Antigen Receptor T Cells Against CD19 for Multiple Myeloma. N Engl J Med (2015) 373(11):1040–7. doi: 10.1056/NEJMoa1504542
73. Wang QJ, Yu Z, Hanada KI, Patel K, Kleiner D, Restifo NP, et al. Preclinical Evaluation of Chimeric Antigen Receptors Targeting CD70-Expressing Cancers. Clin Cancer Res (2017) 23(9):2267–76. doi: 10.1158/1078-0432.CCR-16-1421
74. Singh AK, McGuirk JP. CAR T Cells: Continuation in a Revolution of Immunotherapy. Lancet Oncol (2020) 21(3):e168–78. doi: 10.1016/S1470-2045(19)30823-X
75. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi: 10.1200/JCO.2014.58.0225
76. Klampatsa A, Dimou V, Albelda SM. Mesothelin-Targeted CAR-T Cell Therapy for Solid Tumors. Expert Opin Biol Ther (2021) 21(4):473–86. doi: 10.1080/14712598.2021.1843628
77. Straathof K, Flutter B, Wallace R, Jain N, Loka T, Depani S, et al. Antitumor Activity Without On-Target Off-Tumor Toxicity of GD2-Chimeric Antigen Receptor T Cells in Patients With Neuroblastoma. Sci Transl Med (2020) 12(571):eabd6169. doi: 10.1126/scitranslmed.abd6169
78. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent Antitumor Efficacy of Anti-GD2 CAR T Cells in H3-K27M(+) Diffuse Midline Gliomas. Nat Med (2018) 24(5):572–9. doi: 10.1038/s41591-018-0006-x
79. Cha SE, Kujawski M PJY, Brown C, Shively JE. Tumor Regression and Immunity in Combination Therapy With Anti-CEA Chimeric Antigen Receptor T Cells and Anti-CEA-IL2 Immunocytokine. Oncoimmunology (2021) 10(1):1899469. doi: 10.1080/2162402X.2021.1899469
80. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
81. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
82. Myers JA, Miller JS. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat Rev Clin Oncol (2021) 18(2):85–100. doi: 10.1038/s41571-020-0426-7
83. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of Cdc1 Into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172(5):1022–37.e14. doi: 10.1016/j.cell.2018.01.004
84. Gao L, Wang L, Dai T, Jin K, Zhang Z, Wang S, et al. Tumor-Derived Exosomes Antagonize Innate Antiviral Immunity. Nat Immunol (2018) 19(3):233–45. doi: 10.1038/s41590-017-0043-5
85. Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-Beta Inhibits the Activation and Functions of NK Cells by Repressing the mTOR Pathway. Sci Signal (2016) 9(415):ra19. doi: 10.1126/scisignal.aad1884
86. Shimasaki N, Jain A, Campana D. NK Cells for Cancer Immunotherapy. Nat Rev Drug Discov (2020) 19(3):200–18. doi: 10.1038/s41573-019-0052-1
87. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science (2002) 295(5562):2097–100. doi: 10.1126/science.1068440
88. Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, et al. A Pilot Trial of Humanized Anti-GD2 Monoclonal Antibody (Hu14.18K322A) With Chemotherapy and Natural Killer Cells in Children With Recurrent/Refractory Neuroblastoma. Clin Cancer Res (2017) 23(21):6441–9. doi: 10.1158/1078-0432.CCR-17-0379
89. Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, et al. CD19-CAR Engineered NK-92 Cells Are Sufficient to Overcome NK Cell Resistance in B-Cell Malignancies. J Cell Mol Med (2016) 20(7):1287–94. doi: 10.1111/jcmm.12810
90. Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination Therapy With EpCAM-CAR-NK-92 Cells and Regorafenib Against Human Colorectal Cancer Models. J Immunol Res (2018) 2018:4263520. doi: 10.1155/2018/4263520
91. Genssler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual Targeting of Glioblastoma With Chimeric Antigen Receptor-Engineered Natural Killer Cells Overcomes Heterogeneity of Target Antigen Expression and Enhances Antitumor Activity and Survival. Oncoimmunology (2016) 5(4):e1119354. doi: 10.1080/2162402X.2015.1119354
92. Rezvani K, Rouce R, Liu E, Shpall E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol Ther (2017) 25(8):1769–81. doi: 10.1016/j.ymthe.2017.06.012
93. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered With Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell (2018) 23(2):181–92.e5. doi: 10.1016/j.stem.2018.06.002
94. Cichocki F, Bjordahl R, Gaidarova S, Mahmood S, Abujarour R, Wang H, et al. iPSC-Derived NK Cells Maintain High Cytotoxicity and Enhance In Vivo Tumor Control in Concert With T Cells and Anti-PD-1 Therapy. Sci Transl Med (2020) 12(568):eaaz5618. doi: 10.1126/scitranslmed.aaz5618
95. Gurdon JB. The Developmental Capacity of Nuclei Taken From Intestinal Epithelium Cells of Feeding Tadpoles. J Embryol Exp Morphol (1962) 10:622–40. doi: 10.1242/dev.10.4.622
96. Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable Offspring Derived From Fetal and Adult Mammalian Cells. Nature (1997) 385(6619):810–3. doi: 10.1038/385810a0
97. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of Pluripotent Stem Cells From Adult Human Fibroblasts by Defined Factors. Cell (2007) 131(5):861–72. doi: 10.1016/j.cell.2007.11.019
98. Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells From Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell (2006) 126(4):663–76. doi: 10.1016/j.cell.2006.07.024
99. Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, et al. Reprogramming of Human Peripheral Blood Cells to Induced Pluripotent Stem Cells. Cell Stem Cell (2010) 7(1):20–4. doi: 10.1016/j.stem.2010.06.002
100. Loh YH, Hartung O, Li H, Guo C, Sahalie JM, Manos PD, et al. Reprogramming of T Cells From Human Peripheral Blood. Cell Stem Cell (2010) 7(1):15–9. doi: 10.1016/j.stem.2010.06.004
101. Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, et al. Efficient and Rapid Generation of Induced Pluripotent Stem Cells From Human Keratinocytes. Nat Biotechnol (2008) 26(11):1276–84. doi: 10.1038/nbt.1503
102. Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell (2009) 4(6):472–6. doi: 10.1016/j.stem.2009.05.005
103. Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, et al. Donor Cell Type can Influence the Epigenome and Differentiation Potential of Human Induced Pluripotent Stem Cells. Nat Biotechnol (2011) 29(12):1117–9. doi: 10.1038/nbt.2052
104. Kikuchi T, Morizane A, Doi D, Magotani H, Onoe H, Hayashi T, et al. Human iPS Cell-Derived Dopaminergic Neurons Function in a Primate Parkinson’s Disease Model. Nature (2017) 548(7669):592–6. doi: 10.1038/nature23664
105. Sougawa N, Miyagawa S, Fukushima S, Kawamura A, Yokoyama J, Ito E, et al. Immunologic Targeting of CD30 Eliminates Tumourigenic Human Pluripotent Stem Cells, Allowing Safer Clinical Application of hiPSC-Based Cell Therapy. Sci Rep (2018) 8(1):3726. doi: 10.1038/s41598-018-21923-8
106. Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, Collins JJ. CellNet: Network Biology Applied to Stem Cell Engineering. Cell (2014) 158(4):903–15. doi: 10.1016/j.cell.2014.07.020
107. Davis RL, Weintraub H, Lassar AB. Expression of a Single Transfected cDNA Converts Fibroblasts to Myoblasts. Cell (1987) 51(6):987–1000. doi: 10.1016/0092-8674(87)90585-X
108. Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, et al. Direct Conversion of Fibroblasts to Neurons by Reprogramming PTB-Regulated microRNA Circuits. Cell (2013) 152(1-2):82–96. doi: 10.1016/j.cell.2012.11.045
109. Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, et al. MicroRNA-Mediated Conversion of Human Fibroblasts to Neurons. Nature (2011) 476(7359):228–31. doi: 10.1038/nature10323
110. Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. Direct Reprogramming of Fibroblasts Into Functional Cardiomyocytes by Defined Factors. Cell (2010) 142(3):375–86. doi: 10.1016/j.cell.2010.07.002
111. Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, et al. Heart Repair by Reprogramming non-Myocytes With Cardiac Transcription Factors. Nature (2012) 485(7400):599–604. doi: 10.1038/nature11139
112. Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, et al. MicroRNA-Mediated In Vitro and In Vivo Direct Reprogramming of Cardiac Fibroblasts to Cardiomyocytes. Circ Res (2012) 110(11):1465–73. doi: 10.1161/CIRCRESAHA.112.269035
113. Cheng Z, He Z, Cai Y, Zhang C, Fu G, Li H, et al. Conversion of Hepatoma Cells to Hepatocyte-Like Cells by Defined Hepatocyte Nuclear Factors. Cell Res (2019) 29(2):124–35. doi: 10.1038/s41422-018-0111-x
114. Sekiya S, Suzuki A. Direct Conversion of Mouse Fibroblasts to Hepatocyte-Like Cells by Defined Factors. Nature (2011) 475(7356):390–3. doi: 10.1038/nature10263
115. Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, et al. Induction of Functional Hepatocyte-Like Cells From Mouse Fibroblasts by Defined Factors. Nature (2011) 475(7356):386–9. doi: 10.1038/nature10116
116. Liang J, Wang N, He J, Du J, Guo Y, Li L, et al. Induction of Sertoli-Like Cells From Human Fibroblasts by NR5A1 and GATA4. Elife (2019) 8:e48767. doi: 10.7554/eLife.48767
117. Kishida T, Ejima A, Yamamoto K, Tanaka S, Yamamoto T, Mazda O. Reprogrammed Functional Brown Adipocytes Ameliorate Insulin Resistance and Dyslipidemia in Diet-Induced Obesity and Type 2 Diabetes. Stem Cell Rep (2015) 5(4):569–81. doi: 10.1016/j.stemcr.2015.08.007
118. Hiramatsu K, Sasagawa S, Outani H, Nakagawa K, Yoshikawa H, Tsumaki N. Generation of Hyaline Cartilaginous Tissue From Mouse Adult Dermal Fibroblast Culture by Defined Factors. J Clin Invest (2011) 121(2):640–57. doi: 10.1172/JCI44605
119. Yamamoto K, Kishida T, Sato Y, Nishioka K, Ejima A, Fujiwara H, et al. Direct Conversion of Human Fibroblasts Into Functional Osteoblasts by Defined Factors. Proc Natl Acad Sci USA (2015) 112(19):6152–7. doi: 10.1073/pnas.1420713112
120. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In Vivo Reprogramming of Adult Pancreatic Exocrine Cells to Beta-Cells. Nature (2008) 455(7213):627–32. doi: 10.1038/nature07314
121. Ghosh Z, Huang M, Hu S, Wilson KD, Dey D, Wu JC. Dissecting the Oncogenic and Tumorigenic Potential of Differentiated Human Induced Pluripotent Stem Cells and Human Embryonic Stem Cells. Cancer Res (2011) 71(14):5030–9. doi: 10.1158/0008-5472.CAN-10-4402
122. Kooreman NG, Kim Y, de Almeida PE, Termglinchan V, Diecke S, Shao NY, et al. Autologous iPSC-Based Vaccines Elicit Anti-Tumor Responses In Vivo. Cell Stem Cell (2018) 22(4):501–13 e7. doi: 10.1016/j.stem.2018.01.016
123. Li Y, Zeng H, Xu RH, Liu B, Li Z. Vaccination With Human Pluripotent Stem Cells Generates a Broad Spectrum of Immunological and Clinical Responses Against Colon Cancer. Stem Cells (2009) 27(12):3103–11. doi: 10.1002/stem.234
124. Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of Induced Pluripotent Stem Cells. Nature (2011) 474(7350):212–5. doi: 10.1038/nature10135
125. Mehla K, Singh PK. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer (2019) 5(12):822–34. doi: 10.1016/j.trecan.2019.10.007
126. Lachmann N, Ackermann M, Frenzel E, Liebhaber S, Brennig S, Happle C, et al. Large-Scale Hematopoietic Differentiation of Human Induced Pluripotent Stem Cells Provides Granulocytes or Macrophages for Cell Replacement Therapies. Stem Cell Rep (2015) 4(2):282–96. doi: 10.1016/j.stemcr.2015.01.005
127. Karlsson KR, Cowley S, Martinez FO, Shaw M, Minger SL, James W. Homogeneous Monocytes and Macrophages From Human Embryonic Stem Cells Following Coculture-Free Differentiation in M-CSF and IL-3. Exp Hematol (2008) 36(9):1167–75. doi: 10.1016/j.exphem.2008.04.009
128. Nenasheva T, Gerasimova T, Serdyuk Y, Grigor’eva E, Kosmiadi G, Nikolaev A, et al. Macrophages Derived From Human Induced Pluripotent Stem Cells Are Low-Activated “Naive-Like” Cells Capable of Restricting Mycobacteria Growth. Front Immunol (2020) 11:1016. doi: 10.3389/fimmu.2020.01016
129. Daniel MG, Pereira CF, Lemischka IR, Moore KA. Making a Hematopoietic Stem Cell. Trends Cell Biol (2016) 26(3):202–14. doi: 10.1016/j.tcb.2015.10.002
130. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity (2015) 42(4):665–78. doi: 10.1016/j.immuni.2015.03.011
131. Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, et al. Efficient Derivation of Microglia-Like Cells From Human Pluripotent Stem Cells. Nat Med (2016) 22(11):1358–67. doi: 10.1038/nm.4189
132. Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, et al. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Rep (2017) 8(6):1516–24. doi: 10.1016/j.stemcr.2017.04.023
133. Svoboda DS, Barrasa MI, Shu J, Rietjens R, Zhang S, Mitalipova M, et al. Human iPSC-Derived Microglia Assume a Primary Microglia-Like State After Transplantation Into the Neonatal Mouse Brain. Proc Natl Acad Sci USA (2019) 116(50):25293–303. doi: 10.1073/pnas.1913541116
134. Senju S, Haruta M, Matsumura K, Matsunaga Y, Fukushima S, Ikeda T, et al. Generation of Dendritic Cells and Macrophages From Human Induced Pluripotent Stem Cells Aiming at Cell Therapy. Gene Ther (2011) 18(9):874–83. doi: 10.1038/gt.2011.22
135. Morrissey MA, Williamson AP, Steinbach AM, Roberts EW, Kern N, Headley MB, et al. Chimeric Antigen Receptors That Trigger Phagocytosis. Elife (2018) 7:e36688. doi: 10.7554/eLife.36688
136. Xie H, Ye M, Feng R, Graf T. Stepwise Reprogramming of B Cells Into Macrophages. Cell (2004) 117(5):663–76. doi: 10.1016/S0092-8674(04)00419-2
137. Feng R, Desbordes SC, Xie H, Tillo ES, Pixley F, Stanley ER, et al. PU.1 and C/EBPalpha/beta Convert Fibroblasts Into Macrophage-Like Cells. Proc Natl Acad Sci USA (2008) 105(16):6057–62. doi: 10.1073/pnas.0711961105
138. Lange L, Hoffmann D, Schwarzer A, Ha TC, Philipp F, Lenz D, et al. Inducible Forward Programming of Human Pluripotent Stem Cells to Hemato-Endothelial Progenitor Cells With Hematopoietic Progenitor Potential. Stem Cell Rep (2020) 14(1):122–37. doi: 10.1016/j.stemcr.2019.11.005
139. Pawlowski M, Ortmann D, Bertero A, Tavares JM, Pedersen RA, Vallier L, et al. Inducible and Deterministic Forward Programming of Human Pluripotent Stem Cells Into Neurons, Skeletal Myocytes, and Oligodendrocytes. Stem Cell Rep (2017) 8(4):803–12. doi: 10.1016/j.stemcr.2017.02.016
140. Chen SW, Hung YS, Fuh JL, Chen NJ, Chu YS, Chen SC, et al. Efficient Conversion of Human Induced Pluripotent Stem Cells Into Microglia by Defined Transcription Factors. Stem Cell Rep (2021) 16(5):1363–80. doi: 10.1016/j.stemcr.2021.03.010
141. Sontag S, Forster M, Qin J, Wanek P, Mitzka S, Schuler HM, et al. Modelling IRF8 Deficient Human Hematopoiesis and Dendritic Cell Development With Engineered iPS Cells. Stem Cells (2017) 35(4):898–908. doi: 10.1002/stem.2565
142. Sachamitr P, Leishman AJ, Davies TJ, Fairchild PJ. Directed Differentiation of Human Induced Pluripotent Stem Cells Into Dendritic Cells Displaying Tolerogenic Properties and Resembling the CD141(+) Subset. Front Immunol (2017) 8:1935. doi: 10.3389/fimmu.2017.01935
143. Iizuka-Koga M, Asashima H, Ando M, Lai CY, Mochizuki S, Nakanishi M, et al. Functional Analysis of Dendritic Cells Generated From T-iPSCs From CD4+ T Cell Clones of Sjogren’s Syndrome. Stem Cell Rep (2017) 8(5):1155–63. doi: 10.1016/j.stemcr.2017.04.010
144. Horton C, Davies TJ, Lahiri P, Sachamitr P, Fairchild PJ. Induced Pluripotent Stem Cells Reprogrammed From Primary Dendritic Cells Provide an Abundant Source of Immunostimulatory Dendritic Cells for Use in Immunotherapy. Stem Cells (2020) 38(1):67–79. doi: 10.1002/stem.3095
145. Kitadani J, Ojima T, Iwamoto H, Tabata H, Nakamori M, Nakamura M, et al. Cancer Vaccine Therapy Using Carcinoembryonic Antigen - Expressing Dendritic Cells Generated From Induced Pluripotent Stem Cells. Sci Rep (2018) 8(1):4569. doi: 10.1038/s41598-018-23120-z
146. Rosa FF, Pires CF, Kurochkin I, Ferreira AG, Gomes AM, Palma LG, et al. Direct Reprogramming of Fibroblasts Into Antigen-Presenting Dendritic Cells. Sci Immunol (2018) 3(30):eaau4292. doi: 10.1126/sciimmunol.aau4292
147. Sweeney CL, Teng R, Wang H, Merling RK, Lee J, Choi U, et al. Molecular Analysis of Neutrophil Differentiation From Human Induced Pluripotent Stem Cells Delineates the Kinetics of Key Regulators of Hematopoiesis. Stem Cells (2016) 34(6):1513–26. doi: 10.1002/stem.2332
148. Hansen M, Varga E, Aarts C, Wust T, Kuijpers T, von Lindern M, et al. Efficient Production of Erythroid, Megakaryocytic and Myeloid Cells, Using Single Cell-Derived iPSC Colony Differentiation. Stem Cell Res (2018) 29:232–44. doi: 10.1016/j.scr.2018.04.016
149. Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front Immunol (2019) 10:1205. doi: 10.3389/fimmu.2019.01205
150. Williams SM, Sumstad D, Kadidlo D, Curtsinger J, Luo X, Miller JS, et al. Clinical-Scale Production of cGMP Compliant CD3/CD19 Cell-Depleted NK Cells in the Evolution of NK Cell Immunotherapy at a Single Institution. Transfusion (2018) 58(6):1458–67. doi: 10.1111/trf.14564
151. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Transl Med (2013) 2(4):274–83. doi: 10.5966/sctm.2012-0084
152. Dege C, Fegan KH, Creamer JP, Berrien-Elliott MM, Luff SA, Kim D, et al. Potently Cytotoxic Natural Killer Cells Initially Emerge From Erythro-Myeloid Progenitors During Mammalian Development. Dev Cell (2020) 53(2):229–39.e7. doi: 10.1016/j.devcel.2020.02.016
153. Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 Clinical Trial Using Mbil21 Ex Vivo-Expanded Donor-Derived NK Cells After Haploidentical Transplantation. Blood (2017) 130(16):1857–68. doi: 10.1182/blood-2017-05-785659
154. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous Growth and Increased Cytotoxicity of Natural Killer Cells Expressing Membrane-Bound Interleukin-15. Blood (2014) 124(7):1081–8. doi: 10.1182/blood-2014-02-556837
155. Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, et al. Identification of an ADAM17 Cleavage Region in Human CD16 (FcgammaRIII) and the Engineering of a non-Cleavable Version of the Receptor in NK Cells. PloS One (2015) 10(3):e0121788. doi: 10.1371/journal.pone.0121788
156. Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non-Clinical Efficacy, Safety and Stable Clinical Cell Processing of Induced Pluripotent Stem Cell-Derived Anti-Glypican-3 Chimeric Antigen Receptor-Expressing Natural Killer/Innate Lymphoid Cells. Cancer Sci (2020) 111(5):1478–90. doi: 10.1111/cas.14374
157. Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, et al. Reprogramming of T Cells to Natural Killer-Like Cells Upon Bcl11b Deletion. Science (2010) 329(5987):85–9. doi: 10.1126/science.1188063
158. Nianias A, Themeli M. Induced Pluripotent Stem Cell (iPSC)-Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr Hematol Malig Rep (2019) 14(4):261–8. doi: 10.1007/s11899-019-00528-6
159. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of Rejuvenated Antigen-Specific T Cells by Reprogramming to Pluripotency and Redifferentiation. Cell Stem Cell (2013) 12(1):114–26. doi: 10.1016/j.stem.2012.11.002
160. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S, et al. Regeneration of Human Tumor Antigen-Specific T Cells From iPSCs Derived From Mature CD8(+) T Cells. Cell Stem Cell (2013) 12(1):31–6. doi: 10.1016/j.stem.2012.12.006
161. Montel-Hagen A, Seet CS, Li S, Chick B, Zhu Y, Chang P, et al. Organoid-Induced Differentiation of Conventional T Cells From Human Pluripotent Stem Cells. Cell Stem Cell (2019) 24(3):376–89.e8. doi: 10.1016/j.stem.2018.12.011
162. Iriguchi S, Yasui Y, Kawai Y, Arima S, Kunitomo M, Sato T, et al. A Clinically Applicable and Scalable Method to Regenerate T-Cells From iPSCs for Off-the-Shelf T-Cell Immunotherapy. Nat Commun (2021) 12(1):430. doi: 10.1038/s41467-020-20658-3
163. Minagawa A, Yoshikawa T, Yasukawa M, Hotta A, Kunitomo M, Iriguchi S, et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell (2018) 23(6):850–8 e4. doi: 10.1016/j.stem.2018.10.005
164. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of Tumor-Targeted Human T Lymphocytes From Induced Pluripotent Stem Cells for Cancer Therapy. Nat Biotechnol (2013) 31(10):928–33. doi: 10.1038/nbt.2678
165. Zhang M, Dong Y, Hu F, Yang D, Zhao Q, Lv C, et al. Transcription Factor Hoxb5 Reprograms B Cells Into Functional T Lymphocytes. Nat Immunol (2018) 19(3):279–90. doi: 10.1038/s41590-018-0046-x
166. Guo R, Hu F, Weng Q, Lv C, Wu H, Liu L, et al. Guiding T Lymphopoiesis From Pluripotent Stem Cells by Defined Transcription Factors. Cell Res (2020) 30(1):21–33. doi: 10.1038/s41422-019-0251-7
167. Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, et al. A Mutation in the Transcription Factor Foxp3 Drives T Helper 2 Effector Function in Regulatory T Cells. Immunity (2019) 50(2):362–77 e6. doi: 10.1016/j.immuni.2018.12.016
168. Lu H, Wang H, Yan L, Shao H, Zhang W, Shen H, et al. Overexpression of Early T Cell Differentiation-Specific Transcription Factors Transforms the Terminally Differentiated Effector T Cells Into Less Differentiated State. Cell Immunol (2020) 353:104118. doi: 10.1016/j.cellimm.2020.104118
169. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The Cancer Metabolic Reprogramming and Immune Response. Mol Cancer (2021) 20(1):28. doi: 10.1186/s12943-021-01316-8
170. Lim AR, Rathmell WK, Rathmell JC. The Tumor Microenvironment as a Metabolic Barrier to Effector T Cells and Immunotherapy. Elife (2020) 9:e55185. doi: 10.7554/eLife.55185
171. Madden MZ, Rathmell JC. The Complex Integration of T-Cell Metabolism and Immunotherapy. Cancer Discov (2021) 11:1636–43. doi: 10.1158/2159-8290.CD-20-0569
172. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
173. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism Within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell (2017) 32(3):377–91.e9. doi: 10.1016/j.ccell.2017.08.004
174. Guo H, Wang Q, Ghneim K, Wang L, Rampanelli E, Holley-Guthrie E, et al. Multi-Omics Analyses Reveal That HIV-1 Alters CD4(+) T Cell Immunometabolism to Fuel Virus Replication. Nat Immunol (2021) 22(4):423–33. doi: 10.1038/s41590-021-00898-1
175. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. MEK Inhibition Reprograms CD8(+) T Lymphocytes Into Memory Stem Cells With Potent Antitumor Effects. Nat Immunol (2021) 22(1):53–66. doi: 10.1038/s41590-020-00818-9
176. Pellegrino M, Del Bufalo F, De Angelis B, Quintarelli C, Caruana I, de Billy E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells (2020) 10(1):14. doi: 10.3390/cells10010014
177. Carpenter L, Malladi R, Yang CT, French A, Pilkington KJ, Forsey RW, et al. Human Induced Pluripotent Stem Cells Are Capable of B-Cell Lymphopoiesis. Blood (2011) 117(15):4008–11. doi: 10.1182/blood-2010-08-299941
178. French A, Yang CT, Taylor S, Watt SM, Carpenter L. Human Induced Pluripotent Stem Cell-Derived B Lymphocytes Express Sigm and can be Generated via a Hemogenic Endothelium Intermediate. Stem Cells Dev (2015) 24(9):1082–95. doi: 10.1089/scd.2014.0318
179. Suva ML, Riggi N, Bernstein BE. Epigenetic Reprogramming in Cancer. Science (2013) 339(6127):1567–70. doi: 10.1126/science.1230184
180. Harris H, Miller OJ, Klein G, Worst P, Tachibana T. Suppression of Malignancy by Cell Fusion. Nature (1969) 223(5204):363–8. doi: 10.1038/223363a0
181. Hochedlinger K, Blelloch R, Brennan C, Yamada Y, Kim M, Chin L, et al. Reprogramming of a Melanoma Genome by Nuclear Transplantation. Genes Dev (2004) 18(15):1875–85. doi: 10.1101/gad.1213504
182. Wesely J, Kotini AG, Izzo F, Luo H, Yuan H, Sun J, et al. Acute Myeloid Leukemia iPSCs Reveal a Role for RUNX1 in the Maintenance of Human Leukemia Stem Cells. Cell Rep (2020) 31(9):107688. doi: 10.1016/j.celrep.2020.107688
183. Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I. Terminal Differentiation and Loss of Tumorigenicity of Human Cancers via Pluripotency-Based Reprogramming. Oncogene (2013) 32(18):2249–60, 60 e1-21. doi: 10.1038/onc.2012.237
184. Gong L, Yan Q, Zhang Y, Fang X, Liu B, Guan X. Cancer Cell Reprogramming: A Promising Therapy Converting Malignancy to Benignity. Cancer Commun (2019) 39(1):48. doi: 10.1186/s40880-019-0393-5
185. Utikal J, Maherali N, Kulalert W, Hochedlinger K. Sox2 Is Dispensable for the Reprogramming of Melanocytes and Melanoma Cells Into Induced Pluripotent Stem Cells. J Cell Sci (2009) 122(Pt 19):3502–10. doi: 10.1242/jcs.054783
186. Stricker SH, Feber A, Engstrom PG, Caren H, Kurian KM, Takashima Y, et al. Widespread Resetting of DNA Methylation in Glioblastoma-Initiating Cells Suppresses Malignant Cellular Behavior in a Lineage-Dependent Manner. Genes Dev (2013) 27(6):654–69. doi: 10.1101/gad.212662.112
187. Liu Z, Che P, Mercado JJ, Hackney JR, Friedman GK, Zhang C, et al. Characterization of iPSCs Derived From Low Grade Gliomas Revealed Early Regional Chromosomal Amplifications During Gliomagenesis. J Neurooncol (2019) 141(2):289–301. doi: 10.1007/s11060-018-03047-1
188. Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, Camargo FD, et al. Generation of iPSCs From Cultured Human Malignant Cells. Blood (2010) 115(20):4039–42. doi: 10.1182/blood-2009-07-231845
189. Iskender B, Izgi K, Canatan H. Reprogramming Bladder Cancer Cells for Studying Cancer Initiation and Progression. Tumour Biol (2016) 37(10):13237–45. doi: 10.1007/s13277-016-5226-4
190. Corominas-Faja B, Cufi S, Oliveras-Ferraros C, Cuyas E, Lopez-Bonet E, Lupu R, et al. Nuclear Reprogramming of Luminal-Like Breast Cancer Cells Generates Sox2-Overexpressing Cancer Stem-Like Cellular States Harboring Transcriptional Activation of the mTOR Pathway. Cell Cycle (2013) 12(18):3109–24. doi: 10.4161/cc.26173
191. Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, Tanaka F, et al. Defined Factors Induce Reprogramming of Gastrointestinal Cancer Cells. Proc Natl Acad Sci USA (2010) 107(1):40–5. doi: 10.1073/pnas.0912407107
192. Kumano K, Arai S, Hosoi M, Taoka K, Takayama N, Otsu M, et al. Generation of Induced Pluripotent Stem Cells From Primary Chronic Myelogenous Leukemia Patient Samples. Blood (2012) 119(26):6234–42. doi: 10.1182/blood-2011-07-367441
193. Bernhardt M, Novak D, Assenov Y, Orouji E, Knappe N, Weina K, et al. Melanoma-Derived iPCCs Show Differential Tumorigenicity and Therapy Response. Stem Cell Rep (2017) 8(5):1379–91. doi: 10.1016/j.stemcr.2017.03.007
194. Chao MP, Gentles AJ, Chatterjee S, Lan F, Reinisch A, Corces MR, et al. Human AML-iPSCs Reacquire Leukemic Properties After Differentiation and Model Clonal Variation of Disease. Cell Stem Cell (2017) 20(3):329–44. e7. doi: 10.1016/j.stem.2016.11.018
195. Rapino F, Robles EF, Richter-Larrea JA, Kallin EM, Martinez-Climent JA, Graf T. C/EBPalpha Induces Highly Efficient Macrophage Transdifferentiation of B Lymphoma and Leukemia Cell Lines and Impairs Their Tumorigenicity. Cell Rep (2013) 3(4):1153–63. doi: 10.1016/j.celrep.2013.03.003
196. McClellan JS, Dove C, Gentles AJ, Ryan CE, Majeti R. Reprogramming of Primary Human Philadelphia Chromosome-Positive B Cell Acute Lymphoblastic Leukemia Cells Into Nonleukemic Macrophages. Proc Natl Acad Sci USA (2015) 112(13):4074–9. doi: 10.1073/pnas.1413383112
197. Fehrenbach S, Novak D, Bernhardt M, Larribere L, Boukamp P, Umansky V, et al. Loss of Tumorigenic Potential Upon Transdifferentiation From Keratinocytic Into Melanocytic Lineage. Sci Rep (2016) 6:28891. doi: 10.1038/srep28891
198. Su Z, Zang T, Liu ML, Wang LL, Niu W, Zhang CL. Reprogramming the Fate of Human Glioma Cells to Impede Brain Tumor Development. Cell Death Dis (2014) 5:e1463. doi: 10.1038/cddis.2014.425
199. Yuan J, Zhang F, Hallahan D, Zhang Z, He L, Wu LG, et al. Reprogramming Glioblastoma Multiforme Cells Into Neurons by Protein Kinase Inhibitors. J Exp Clin Cancer Res (2018) 37(1):181. doi: 10.1186/s13046-018-0857-5
200. Yuan ZD, Zhu WN, Liu KZ, Huang ZP, Han YC. Small Molecule Epigenetic Modulators in Pure Chemical Cell Fate Conversion. Stem Cells Int (2020) 2020:8890917. doi: 10.1155/2020/8890917
201. Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, et al. Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells Into Adipocytes Inhibits Cancer Metastasis. Cancer Cell (2019) 35(1):17–32 e6. doi: 10.1016/j.ccell.2018.12.002
202. Somasundaram R, Ahsberg J, Okuyama K, Ungerback J, Lilljebjorn H, Fioretos T, et al. Clonal Conversion of B Lymphoid Leukemia Reveals Cross-Lineage Transfer of Malignant States. Genes Dev (2016) 30(22):2486–99. doi: 10.1101/gad.285536.116
203. de Charette M, Marabelle A, Houot R. Turning Tumour Cells Into Antigen Presenting Cells: The Next Step to Improve Cancer Immunotherapy? Eur J Cancer (2016) 68:134–47. doi: 10.1016/j.ejca.2016.09.010
204. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature (2017) 548(7668):471–5. doi: 10.1038/nature23465
205. Zhang S, Kohli K, Black RG, Yao L, Spadinger SM, He Q, et al. Systemic Interferon-Gamma Increases MHC Class I Expression and T-Cell Infiltration in Cold Tumors: Results of a Phase 0 Clinical Trial. Cancer Immunol Res (2019) 7(8):1237–43. doi: 10.1158/2326-6066.CIR-18-0940
206. Li H, Chen G. In Vivo Reprogramming for CNS Repair: Regenerating Neurons From Endogenous Glial Cells. Neuron (2016) 91(4):728–38. doi: 10.1016/j.neuron.2016.08.004
207. Athanasopoulos T, Munye MM, Yanez-Munoz RJ. Nonintegrating Gene Therapy Vectors. Hematol Oncol Clin North Am (2017) 31(5):753–70. doi: 10.1016/j.hoc.2017.06.007
208. Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral Vector Platforms Within the Gene Therapy Landscape. Signal Transduct Target Ther (2021) 6(1):53. doi: 10.1038/s41392-021-00487-6
209. Goswami R, Subramanian G, Silayeva L, Newkirk I, Doctor D, Chawla K, et al. Gene Therapy Leaves a Vicious Cycle. Front Oncol (2019) 9:297. doi: 10.3389/fonc.2019.00297
210. Juttner J, Szabo A, Gross-Scherf B, Morikawa RK, Rompani SB, Hantz P, et al. Targeting Neuronal and Glial Cell Types With Synthetic Promoter AAVs in Mice, non-Human Primates and Humans. Nat Neurosci (2019) 22(8):1345–56. doi: 10.1038/s41593-019-0431-2
211. Montano-Samaniego M, Bravo-Estupinan DM, Mendez-Guerrero O, Alarcon-Hernandez E, Ibanez-Hernandez M. Strategies for Targeting Gene Therapy in Cancer Cells With Tumor-Specific Promoters. Front Oncol (2020) 10:605380. doi: 10.3389/fonc.2020.605380
212. Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, Rovira M, et al. Tissue Damage and Senescence Provide Critical Signals for Cellular Reprogramming In Vivo. Science (2016) 354(6315):aaf4445. doi: 10.1126/science.aaf4445
213. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev (2017) 31(2):172–83. doi: 10.1101/gad.290635.116
214. Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, et al. In Vivo Reprogramming of Murine Cardiac Fibroblasts Into Induced Cardiomyocytes. Nature (2012) 485(7400):593–8. doi: 10.1038/nature11044
215. Wang LL, Su Z, Tai W, Zou Y, Xu XM, Zhang CL. The P53 Pathway Controls SOX2-Mediated Reprogramming in the Adult Mouse Spinal Cord. Cell Rep (2016) 17(3):891–903. doi: 10.1016/j.celrep.2016.09.038
216. Guo Z, Zhang L, Wu Z, Chen Y, Wang F, Chen G. In Vivo Direct Reprogramming of Reactive Glial Cells Into Functional Neurons After Brain Injury and in an Alzheimer’s Disease Model. Cell Stem Cell (2014) 14(2):188–202. doi: 10.1016/j.stem.2013.12.001
217. Torper O, Pfisterer U, Wolf DA, Pereira M, Lau S, Jakobsson J, et al. Generation of Induced Neurons via Direct Conversion In Vivo. Proc Natl Acad Sci USA (2013) 110(17):7038–43. doi: 10.1073/pnas.1303829110
218. Li W, Nakanishi M, Zumsteg A, Shear M, Wright C, Melton DA, et al. In Vivo Reprogramming of Pancreatic Acinar Cells to Three Islet Endocrine Subtypes. Elife (2014) 3:e01846. doi: 10.7554/eLife.01846
219. Song G, Pacher M, Balakrishnan A, Yuan Q, Tsay HC, Yang D, et al. Direct Reprogramming of Hepatic Myofibroblasts Into Hepatocytes In Vivo Attenuates Liver Fibrosis. Cell Stem Cell (2016) 18(6):797–808. doi: 10.1016/j.stem.2016.01.010
220. Rezvani M, Espanol-Suner R, Malato Y, Dumont L, Grimm AA, Kienle E, et al. In Vivo Hepatic Reprogramming of Myofibroblasts With AAV Vectors as a Therapeutic Strategy for Liver Fibrosis. Cell Stem Cell (2016) 18(6):809–16. doi: 10.1016/j.stem.2016.05.005
221. Kurita M, Araoka T, Hishida T, O’Keefe DD, Takahashi Y, Sakamoto A, et al. In Vivo Reprogramming of Wound-Resident Cells Generates Skin Epithelial Tissue. Nature (2018) 561(7722):243–7. doi: 10.1038/s41586-018-0477-4
222. Yao K, Qiu S, Wang YV, Park SJH, Mohns EJ, Mehta B, et al. Restoration of Vision After De Novo Genesis of Rod Photoreceptors in Mammalian Retinas. Nature (2018) 560(7719):484–8. doi: 10.1038/s41586-018-0425-3
223. Stanley ER, Chitu V. CSF-1 Receptor Signaling in Myeloid Cells. Cold Spring Harb Perspect Biol (2014) 6(6):a021857. doi: 10.1101/cshperspect.a021857
224. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
225. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, et al. Genetic Programming of Macrophages to Perform Anti-Tumor Functions Using Targeted mRNA Nanocarriers. Nat Commun (2019) 10(1):3974. doi: 10.1038/s41467-019-11911-5
226. Wang G, Chow RD, Bai Z, Zhu L, Errami Y, Dai X, et al. Multiplexed Activation of Endogenous Genes by CRISPRa Elicits Potent Antitumor Immunity. Nat Immunol (2019) 20(11):1494–505. doi: 10.1038/s41590-019-0500-4
227. Li Y, Su Z, Zhao W, Zhang X, Momin N, Zhang C, et al. Multifunctional Oncolytic Nanoparticles Deliver Self-Replicating IL-12 RNA to Eliminate Established Tumors and Prime Systemic Immunity. Nat Cancer (2020) 1(9):882–93. doi: 10.1038/s43018-020-0095-6
228. Tzeng SY, Patel KK, Wilson DR, Meyer RA, Rhodes KR, Green JJ. In Situ Genetic Engineering of Tumors for Long-Lasting and Systemic Immunotherapy. Proc Natl Acad Sci USA (2020) 117(8):4043–52. doi: 10.1073/pnas.1916039117
229. Nissim L, Wu MR, Pery E, Binder-Nissim A, Suzuki HI, Stupp D, et al. Synthetic RNA-Based Immunomodulatory Gene Circuits for Cancer Immunotherapy. Cell (2017) 171(5):1138–50.e15. doi: 10.1016/j.cell.2017.09.049
230. Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, et al. In Vivo Generation of Human CD19-CAR T Cells Results in B-Cell Depletion and Signs of Cytokine Release Syndrome. EMBO Mol Med (2018) 10(11):e9158. doi: 10.15252/emmm.201809158
231. Agarwal S, Weidner T, Thalheimer FB, Buchholz CJ. In Vivo Generated Human CAR T Cells Eradicate Tumor Cells. Oncoimmunology (2019) 8(12):e1671761. doi: 10.1080/2162402X.2019.1671761
Keywords: cancer immunotherapy, cellular reprogramming, tumor immunology, CAR-T, transcription factor, dendritic cell, antigen presentation, cancer vaccine
Citation: Zimmermannova O, Caiado I, Ferreira AG and Pereira C-F (2021) Cell Fate Reprogramming in the Era of Cancer Immunotherapy. Front. Immunol. 12:714822. doi: 10.3389/fimmu.2021.714822
Received: 25 May 2021; Accepted: 06 July 2021;
Published: 21 July 2021.
Edited by:
Laura Lynne Eggink, Susavion Biosciences, Inc., United StatesReviewed by:
Georgina Xanthou, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceGiulio Fracasso, University of Verona, Italy
Copyright © 2021 Zimmermannova, Caiado, Ferreira and Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos-Filipe Pereira, ZmlsaXBlLnBlcmVpcmFAbWVkLmx1LnNl