 Eileen Haring
Eileen Haring Robert Zeiser
Robert Zeiser Petya Apostolova
Petya Apostolova
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 24 June 2021
Sec. Alloimmunity and Transplantation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.705342
This article is part of the Research TopicInterferons and Graft-versus-Host DiseaseView all 10 articles
The intestine can be the target of several immunologically mediated diseases, including graft-versus-host disease (GVHD) and inflammatory bowel disease (IBD). GVHD is a life-threatening complication that occurs after allogeneic hematopoietic stem cell transplantation. Involvement of the gastrointestinal tract is associated with a particularly high mortality. GVHD development starts with the recognition of allo-antigens in the recipient by the donor immune system, which elicits immune-mediated damage of otherwise healthy tissues. IBD describes a group of immunologically mediated chronic inflammatory diseases of the intestine. Several aspects, including genetic predisposition and immune dysregulation, are responsible for the development of IBD, with Crohn’s disease and ulcerative colitis being the two most common variants. GVHD and IBD share multiple key features of their onset and development, including intestinal tissue damage and loss of intestinal barrier function. A further common feature in the pathophysiology of both diseases is the involvement of cytokines such as type I and II interferons (IFNs), amongst others. IFNs are a family of protein mediators produced as a part of the inflammatory response, typically to pathogens or malignant cells. Diverse, and partially paradoxical, effects have been described for IFNs in GVHD and IBD. This review summarizes current knowledge on the role of type I, II and III IFNs, including basic concepts and controversies about their functions in the context of GVHD and IBD. In addition, therapeutic options, research developments and remaining open questions are addressed.
The intestine poses a unique environment for the immune system. Innate and adaptive immune cells cooperate at this physiological barrier surface to maintain homeostasis and prevent infection with pathogens that are ingested with the food. An interplay between intestinal microbiota and nutritional metabolites further shapes the microenvironment. Loss of homeostasis between these factors may result in local inflammation. Two disease groups that elicit immune-mediated intestinal tissue damage are graft-versus-host disease (GVHD) and inflammatory bowel disease (IBD). These diseases develop in distinct situations. IBD is the most prevalent autoimmune condition of the intestine, while the occurrence of GVHD is limited to the specific case of a patient who has received an allogeneic hematopoietic cell transplantation (allo-HCT). Nevertheless, both diseases share similar pathophysiological mechanisms. One of them is the involvement of interferons (IFNs) as soluble mediators shaping the microenvironment. Here, we review recent literature about the role of IFNs in intestinal GVHD and IBD. We first provide an introduction about the biology of both disease groups, followed by an overview of IFN production and signaling. In the second part, we discuss the function of different IFN subtypes in preclinical models and clinical studies of GVHD and IBD.
Allo-HCT is one essential curative therapy option for malignant diseases of the hematopoietic system such as leukemia or lymphoma. It is also used for the treatment of benign disorders, most predominantly immunodeficiency syndromes (1). The allograft recipient is conditioned for the graft transplantation by the administration of chemotherapy, and in some cases irradiation, followed by the intravenous infusion of allogeneic hematopoietic stem cells (HSCs). Along with HSCs, the allogeneic graft contains also pre-existing mature lymphocytes (2). These donor immune cells are able to attack residing tumor cells when the allo-HCT is performed to treat a malignant underlying disease. This process is termed graft-versus-leukemia or graft-versus-tumor effect and is essential for long-term malignancy control (3). On the other hand, the donor immune cells (especially T cells) can also harm healthy tissues in the recipient. This inflammatory process is known as GVHD and its high morbidity and mortality limit the therapeutic success of allo-HCT. Classically, GVHD presents itself in two different clinical manifestations: acute GVHD (aGVHD) and chronic GVHD (cGVHD). The main target organs in aGVHD are the liver, the skin and the gastrointestinal (GI) tract. Clinical symptoms may develop within a few weeks after allo-HCT and include a maculopapular rash, hyperbilirubinemia, cholestasis as well as voluminous diarrhea, abdominal pain and bleeding (4). In addition to the affected tissues in aGVHD, any other organ system such as oral, esophageal and ocular systems, but also hair, nails, genitalia, joint fascia and lungs can be involved in cGVHD, which occurs late (in most cases, up to one year) after allo-HCT (5). GVHD is a frequent complication of allo-HCT with 30-50% of all allo-HCT recipients being affected (4). Due to its high prevalence and the diversity of involved organs, GVHD poses a major challenge in the care of allo-HCT recipients together with the risk of infections and malignancy relapse.
The development of GVHD is a complex interplay between hematopoietic and non-hematopoietic cells, soluble mediators, metabolites and bacteria. The key cellular mediators of GVHD are the alloreactive T cells, which are contained in the donor graft and become activated by different signals during disease development. The conditioning regimen prior to allo-HCT damages tissues of the recipient resulting in the release of both danger- and pathogen-associated molecular patterns (DAMPs and PAMPs). Together with inflammatory cytokines such as TNF and IL-6, a local inflammatory environment is established (6–8). Antigen-presenting cells (APCs) get activated and present peptides from the recipient. This in turn leads to the activation and expansion of the alloreactive T cells, which recognize the host peptides as foreign based on differences in major and minor histocompatibility antigens between donor and recipient. Cellular mediators of tissue damage in the patient comprise cytotoxic CD8+ T cells, NK cells as well as macrophages (7). They act together with soluble inflammatory effectors to promote local tissue destruction and further enhance inflammation (Figure 1). Involvement of the GI tract is associated with a high morbidity and mortality (9, 10). Intestinal epithelial cell (IEC) numbers are markedly reduced in aGVHD, and their damage leads to a loss of intestinal barrier function associated with inferior survival (11). This in turn further elevates tissue damage accompanied by bacterial transmigration and therefore strengthens the local pro-inflammatory setting during disease pathogenesis (8, 12). Besides epithelial cells, intestinal stem cells (ISCs) and Paneth cells are a major target of GVHD. ISCs are located at the bottom of the intestinal crypts where they proliferate and differentiate to regenerate all intestinal cell types. Several studies could underline that damage of the ISC is a key event in disease pathogenesis and that supporting their regeneration improves GVHD outcome (13–16). Paneth cells are located in close proximity to the ISCs. They produce antimicrobial peptides, such as lysozyme and defensins. Paneth cell number reduction in GVHD has been associated with microbial dysregulation through the reduction of intestinal α-defensins (17, 18). In humans, low Paneth cell numbers at the onset of GVHD correlated with inferior survival (19). Besides Paneth cells, L cells were recently shown to be a target of aGVHD and their loss causes a lack of the enteroendocrine hormone Glucagon-like-peptide-2 (GLP-2) (16). Another major determinant of GVHD severity is the intestinal microbiome. Multiple studies observed a loss of general bacterial diversity with a shift between beneficial and detrimental bacterial species during GVHD (20–22). Fecal microbiota transplantation has shown efficacy in patients with steroid-resistant GVHD (23–26) pointing out to the significance of microbial regulation of inflammation. Due to this complex, multi-layer pathogenesis, GVHD has proven difficult to treat in a significant number of patients.
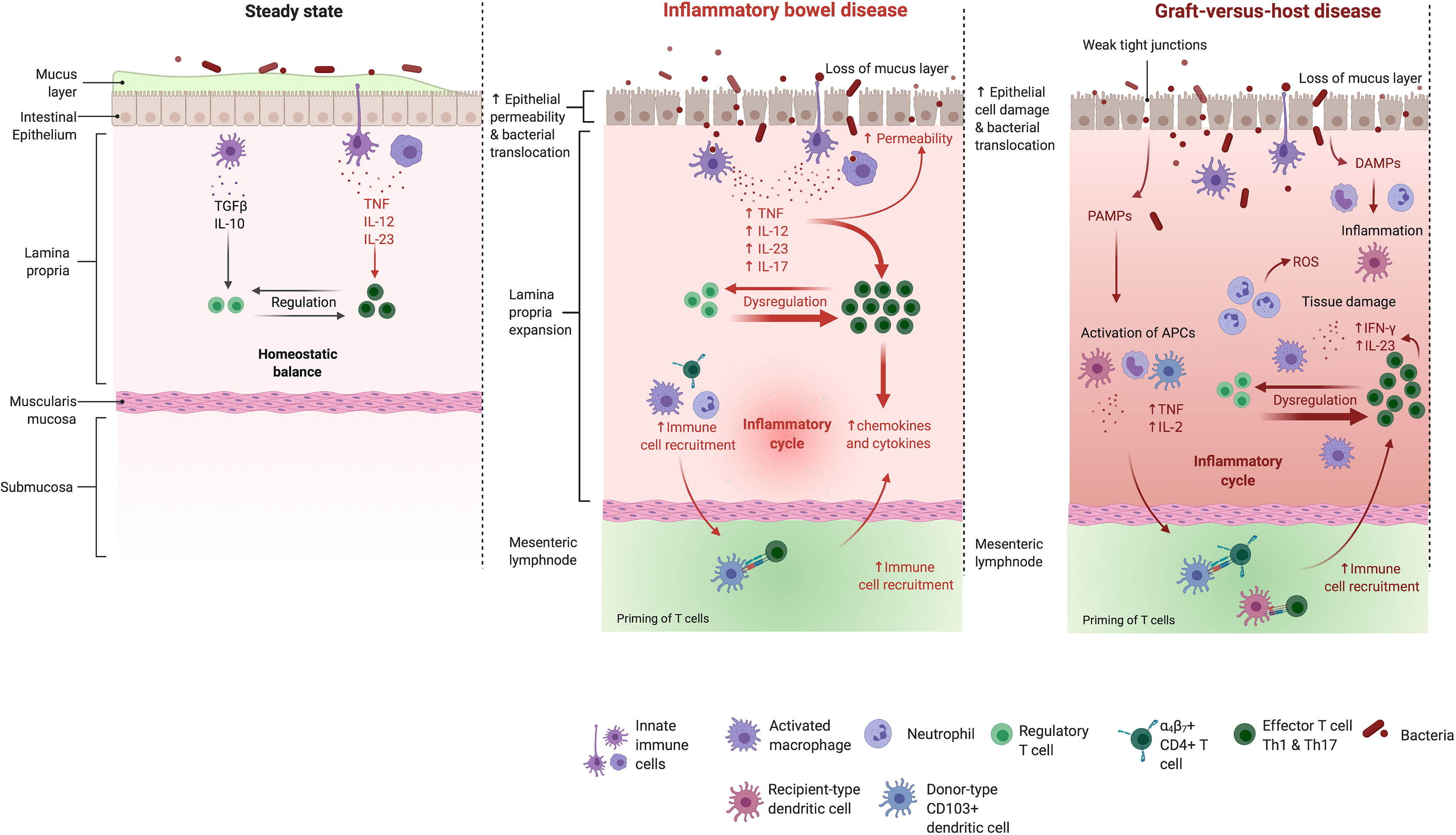
Figure 1 The intestinal mucosa in the healthy bowel, in IBD and GVHD. Mechanisms maintaining the healthy intestinal barrier (e.g. a thick mucus layer and tight junctions) are disrupted in the mucosa of IBD patients. The balance between effector- and regulatory T cells gets disturbed which leads to an activation of different effector T cell subtypes and their uncontrolled migration into the inflamed intestine. Also in GHVD pathogenesis, the intestinal barrier gets disrupted. Intestinal injury due to administered conditioning regiment leads to the translocation of bacteria, PAMPs and DAMPs. Neutrophils are recruited and promote tissue damage through reactive oxygen species secretion. The costimulatory activity of host antigen presenting cells is enhanced. Donor T cells are primed, proliferate and differentiate in response to host stimulatory APCs. Th1 cytokines (IFN-γ, IL-2, and TNF) and chemokines are released in large quantities. A complex cascade including cellular mediators (e.g. cytotoxic T cells and macrophages) and soluble inflammatory effectors (e.g. TNF and IFN-γ) collectively promotes local tissue damage and further drives the inflammatory cycle. IL, interleukin; TGFβ, transforming growth factor β; TNF, tumor necrosis factor; IFN, interferon; ROS, reactive oxygen species; PAMPs, pathogen associated molecular patterns; DAMPs, danger associated molecular patterns. Adapted from “Immune response in IBD”, by Biorender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
Inflammatory bowel disease (IBD) is a group of chronic and recurrent nonspecific inflammatory autoimmune diseases of the intestinal tract. Several factors including genetic predisposition, environmental factors, the intestinal microbiome as well as immune dysregulation play a role for the development of IBD (27–29). The two main clinical presentations of IBD comprise Crohn’s disease (CD), characterized by inflammation in different parts of the intestine, and ulcerative colitis (UC), which leads to persistent inflammation and ulcers limited to the colon (30, 31). CD and UC are chronic, often progressive diseases. The major clinical symptoms are chronic diarrhea, abdominal pain and bleeding, weight loss, nausea, vomiting and fatigue (32). IBD can be accompanied by a wide range of serious complications such as abscesses, fistulas and inflammation-associated colon cancer. In particular in the case of CD, extra intestinal manifestations are frequent, with skin, eyes, bones and joints being affected (33, 34).
There has been strong evidence showing that - similarly to GVHD - a loss of intestinal barrier integrity contributes to the initiation of IBD (11, 35). The barrier disruption allows translocation of microbes and microbial products which results in the engagement of pattern-recognition-receptors (PRRs) present on IECs and various hematopoietic as well as non-hematopoietic cells within the mucosa. PRR stimulation ultimately leads to the induction of an immunologic response via inflammasome activation and the production and release of pro-inflammatory cytokines as well as chemokines (36, 37) (Figure 1). Previous studies could elucidate that an imbalance between pro-inflammatory Th17 cells and anti-inflammatory regulatory T cells (Tregs) was essential in the context of IBD initiation, progress and maintenance (38–40). Proinflammatory cytokines, including TNF and IFN-γ, were shown to be key players in driving the excessive and imbalanced immune response, accompanied by harmful leukocyte infiltration and intestinal mucosal damage (41, 42). Furthermore, it was demonstrated that the microbiome played a key role in IBD onset and pathogenesis as it was seen that the development of intestinal inflammation in mice was abolished under germ-free conditions in a variety of mouse models (43). In addition to similar intestinal clinical manifestations, both GVHD and IBD also share extra intestinal organ involvement such as bile duct damage, amongst others (37). Underlining the shared aspects of disease pathologies, corticosteroids and other immunosuppressive medication is utilized in both conditions (44, 45). Newer approaches in IBD therapy suggest that the earlier utilization of advanced therapies, including immunomodulatory drugs such as thiopurines and methotrexate effectively reduces disease progression and minimizes long-term complications for the patient (46, 47).
IFNs are a group of cytokines which in humans can be divided into three categories: type I IFNs (comprising IFN-α, IFN-β, IFN-ϵ, IFN-κ, and IFN-ω), type II IFNs (IFN-γ) and type III IFNs (IFN-λ1, IFN-λ2, IFN-λ3, IFN-λ4), also known and described as IFN-like molecules. Type I IFNs bind to a common cell surface receptor named type I IFN receptor, which is composed of the two subunits IFNAR1 and IFNAR2 and is expressed on all nucleated cells (48, 49). The subunits are associated with the Janus activated kinases (JAKs) tyrosine kinase 2 (TYK2) and JAK1. Receptor engagement by type I IFN leads to tyrosine phosphorylation of signal transducer and activator of transcription 1 (STAT1) and STAT2. Together with interferon regulatory factor 9 (IRF9), both phosphorylated STAT proteins form a complex which is known as IFN-stimulated (IFN-stimulated gene (ISG) factor 3) ISGF3 complex (50, 51). This complex translocates into the nucleus and binds to IFN-stimulated response elements (ISREs) to initiate the transcription of different ISGs which mediate various biological processes (52). Aside from STAT1 and -2, type IFN I signaling can also induce STAT3-6, so that various homo- and heterodimer combinations can assemble (53). In contrast to the downstream signaling of the ISGF3 complex, which is comprised of STAT1, -2 and IRF9, the other complexes bind to another type of regulatory element: the IFN-γ-activated site (GAS) element. Various ISGs contain either only ISREs or GAS elements in their promoter regions, whereas some contain both. This shows that type I IFN signaling can induce a variety of functionally distinct target genes, although the exact mechanism behind the regulation of the various STAT engagements is not fully understood yet (51). IFN-γ, as the only type II IFN, binds to a different cell surface receptor: the type II receptor, composed of the two subunits IFNGR1 and IFNGR2, which are associated with JAK1 and JAK2, respectively (49, 54). Here, the STAT1 homodimer is the essential transcription factor, which gets activated via phosphorylation. Since the STAT1 homodimer does not bind to IRF9, it is not able to bind ISREs. Therefore, type II IFN signaling only induces transcription of genes, which possess GAS elements in their regulatory regions (55–57). Finally, all type III IFNs bind to a receptor complex composed of two subunits: CRF2-12 (also designated as IFN-λR1) and CRF2-4 (also known as IL-10R2), together named 65R1. Type III INFs are the “youngest” group of IFNs and were only discovered in 2003 (58, 59). Similar to type I IFNs, signaling via type III IFNs induces the trimerization of the heterodimer STAT1-STAT2 with IRF9 resulting in the assembly of the ISGF3 complex. Type III IFN signaling can therefore activate ISG with ISREs or GAS elements in their regulatory region (60) (Figure 2). In contrast to the wide receptor expression for type I and II interferons, expression of type III interferon receptor seems to be limited to certain tissues and cell types. Keratinocytes and epithelial cells of the lung and the GI tract have been shown to express significant amounts of IFNLR1. Interestingly, so far plasmacytoid dendritic cells (pDCs) seem to be the only hematopoietic cell type which is responsive to type III IFNs (61, 62). The various impacts and functions of ISGs were recently covered in a comprehensive review by Schoggins (63).
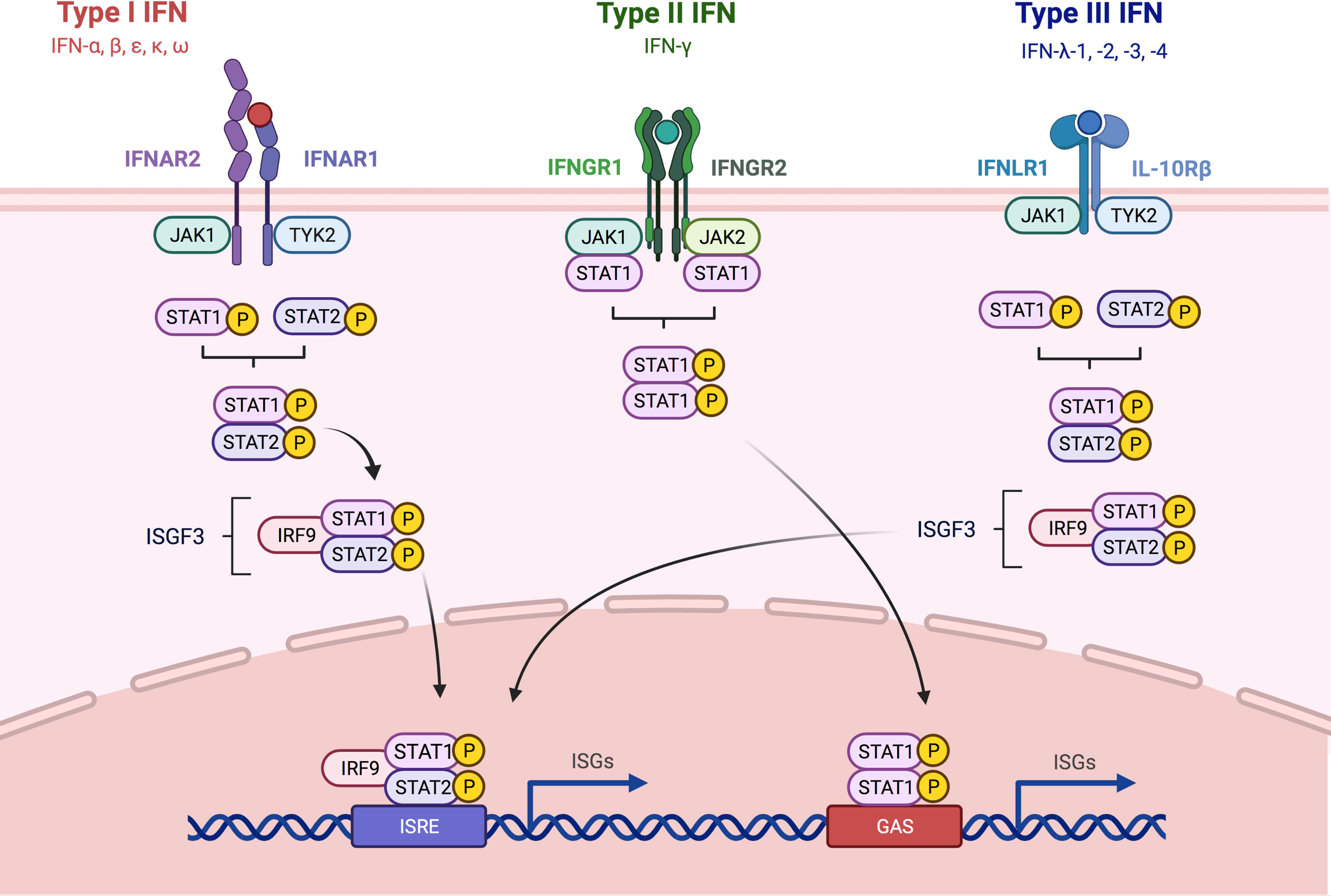
Figure 2 Overview about Type I, -II and -III IFN signaling pathways. The three different types of IFNs discussed in this review signal through distinct receptor complexes on the cell surface. Type I IFNs act through the type I IFN receptor which is composed of the two subunits IFNAR1 and IFNAR2; Type II IFNs act through heterodimers consisting of IFNGR1 and 2 IFNGR2 and type III IFNs signal via heterodimers consisting of IL-10R2 and IFNLR1. Binding of type I and type III IFN to their respective receptor complexes triggers phosphorylation of associated JAK1 and -2, leading to the recruitment and subsequent phosphorylation of STAT1 and -2. STAT 1 and -2 form together a complex, which in turn recruits IRF9 which results in the formation of ISGF3. Engagement of type II IFN to the IFNGR1/2 complex leads to phosphorylation JAK1 and -2, and subsequently STAT1 is recruited and phosphorylated. Both IRF9 and the homodimer consisting of phosphorylated STAT1 can then translocate into the nucleus and bind to ISRE and GAS elements in the promoter region of ISGs, leading to the induction of the expression of antiviral genes. IFN, interferon; STAT, signal transducer and activator of transcription; JAK, Janus kinase; TYK, tyrosine kinase; IL, interleukin; IFNAR, interferon alpha receptor; IFNGR, interferon gamma receptor; IFNLR, interferon lambda receptor; ISGF3, interferon-stimulated gene factor 3; IRF9, interferon regulatory factor 9; ISRE, interferon-stimulated response element; GAS, interferon gamma activated site; ISG, interferon-stimulated gene. Adapted from “Interferon pathay”, by Biorender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
Type I IFNs have a wide range of functions and are produced by various cell types in response to pathogenic - mostly viral but also bacterial - infections. The functions include anti-pathogen activity as well as anti-proliferative actions. During the last decades it became also clear, that type I IFN can exert immunomodulatory actions on cells of both the innate and the adaptive immune system (54, 64). Type I IFN production is triggered by various PRRs including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs) as well as NOD-like receptors (NLRs), that can be activated by sensing viral nucleic acids and other stimuli. PRR activation leads to the rapid induction of type I IFN during the early phases of viral infections before the adaptive immune response including antiviral CD8+ T cells is induced and established (65). As part of the innate immune system, plasmacytoid dendritic cells (pDCs) were implied as the most predominant IFN-α producing cells (66–68). Type I IFNs indirectly affect T cell activation by inducing the maturation, migration and antigen presentation capacity of DCs to facilitate their adaptive antiviral immune response (69–74).
Natural killer (NK) cells, natural killer T cells (NKT), CD4+ T helper type 1 (Th1) cells, CD8+ cytotoxic T cells as well as γδ T cells are the main IFN-γ-producing cell types (75). IFN-γ plays an essential role in MHC class I and II antigen presentation pathways. It induces the upregulation of MHC class I cell surface expression which is important for the immune response against intracellular pathogens and essential for the actions of cytotoxic effects of CD8+ T cells. All exact impacts of IFN-γ on genes which are associated with the MHC class I antigen presentation pathway have been reviewed by Schroder and colleagues (57). Notably, IFN-γ is the sole IFN, which is able to induce MHC class II expression on professional APCs such as DCs, macrophages and B cells. It thus plays an exclusive role in the activation of CD4+ T cells via specific MHC class II/peptide recognition (75). During the adaptive immune response, CD4+ Th1 cells as well as CD8+ cells are able to secrete IFN-γ after being activated and differentiated (63). Furthermore, IFN-γ can have both immune-stimulatory as well as -suppressive roles in all stages of the tumor immunoediting process (76–78).
Type III IFNs can promote an antiviral response, which is similar to the response to type I IFNs (79). A distinct feature of both IFN types lies in the production of the respective cytokine and the distribution of the corresponding receptors. Type III IFNs are especially important at epithelial barrier surfaces. Epithelial cells of the respiratory but also intestinal tract express high amounts of IFNLR1 demonstrating a predominant role of type III IFNs in the epithelial antiviral host defense (62, 80, 81).
IECs play a key role in balancing the intestinal immune homeostasis. They need to act tolerogenic to the vast amount of bacterial commensals but at the same time also be responsive to detrimental pathogens. In this context, there is increasing evidence that both type I and type III IFNs are important for the maintenance of the intestinal epithelial barrier integrity and the control of adaptive immune responses including antiviral responses (81, 82). In the intestine, type I IFNs are for example continuously produced by CD11c+ DCs of the lamina propria (83). In contrast to that, it was shown, that murine IECs preferentially expressed type III IFNs over type I IFNs upon infection with human reoviruses and that they expressed higher levels of IFNLR1 than IFNAR1 and -2 compared to the underlying lamina propria (84, 85). This differential distribution of IFN receptors demonstrated that type III IFN IFN-λ could be seen as the very first line of defense against intestinal pathogens and might represent a nonredundant part of the innate antiviral immune response (81). Proof for that concept was established by studies, which revealed that IFN-λ signaling in IECs was protective against intestinal virus infection using mice with a conditional knock-out of IFNRL1 in the intestine. Depleting IFN-λ signaling in IECs led to an increase in intestinal virus replication and fecal shedding (86). Additionally, it was demonstrated, that administration of IFN-λ could cure intestinal virus persistence of norovirus even independent of the adaptive immune system (87). Though IFNs type III were shown to have this very essential role for the antiviral response of IECs, type I IFNs are not expendable. The same studies underlined the hypothesis, that type I IFNs, rather than protecting the IECs directly, were in fact essential for the prevention of a systemic spread of the intestinal viral infection (85, 87). Broggi and colleagues concluded, that in the intestine, type I and III IFNs acted together in a compartmentalized system. In this synergy, the type III IFN IFN-λ had the primary role in protecting the epithelial barrier, and type I IFNs only came into action once this barrier has been penetrated by invading pathogens (88).
Nevertheless, IFN-α was found to prevent staurosporine-induced apoptosis murine model of the developing intestine via induction of the GTPase guanylate-binding protein-1 (GBP-1) expression, which was involved in regulating intestinal barrier function (89, 90). Using mice deficient for IFNAR1, it was demonstrated, that type I IFN signaling could determine Paneth- and goblet cell numbers in the intestine. Both epithelial cell hyper-proliferation and increased tumor burden were associated with the IFNAR1-deficient intestinal epithelium in a colitis-associated cancer model. Interestingly, intestinal cell hyper-proliferation as well as tumor promotion were reversed in the IFNAR1-deficient mice upon co-housing with WT littermates, underlining that IFNAR1 in IECs contributed to the regulation of the host-microbiome relationship which had consequences for intestinal cell regeneration as well as tumor formation (89). In the human setting it could also be demonstrated, that intestinal virus infection preferentially induced the upregulation of type III IFN to a higher extent compared to type I IFN, leading to a protective effect of type III IFN on the IECs expressing type III IFN receptors (91, 92). Recently it was discovered that, similar to the murine system, IFN-λ played an essential role in the context of epithelial cell protection during intestinal virus infection in humans. Human intestinal epithelial cells lacking IFNLR1, but not those lacking IFNAR1, showed diminished ability to control SARS-CoV-2 infection and replication in the intestine (93). Altogether, studies in both murine and human setting suggest a model, in which IECs favor type III IFN-mediated signaling over type I IFN signaling upon viral infection. This model allows an effective innate response to virus infection without triggering a systemic inflammatory process via type I IFN production and -signaling, thereby maintaining local intestinal gut homeostasis (91). In contrast to the protective role of type III IFNs on IECs, type II IFN IFN-γ was found to have negative effects on IECs and intestinal homeostasis (94–96). It was demonstrated, that IFN-γ produced by immune cells during mucosal immune response has destructive effects on Paneth cells (97, 98).
Two important regulators of IFN production are intestinal microbiota and their metabolites. Depletion of intestinal bacteria by antibiotic treatment reduced type I interferon responses in chicken after a challenge with influenza virus (99). In mice undergoing influenza A infection, decontamination of the gut by administration of antibiotic-containing water decreased ISG expression in stromal cells of the lung, indicating that changes of the intestinal microbiome have an impact on interferon signaling in the whole body. Interestingly, fecal transplantation was able to reverse the effects of antibiotic treatment and restored ISG expression (100). In a recent study, mice undergoing oral antibiotic treatment were also more susceptible to Chikungunya virus infection. The authors found by single-cell RNA sequencing that antibiotic treatment reduced type I IFN production by pDCs and subsequent expression of ISGs in infected monocytes. They further discovered that Clostridium scingens, by converting a primary bile acid into the secondary bile acid deoxycholic acid, was able to reconstitute IFN production by pDCs (101, 102). Other metabolites produced by intestinal microbiota, such as short-chain fatty acids (SCFAs), also play a significant role in colonic homeostasis and inflammation (103, 104). SCFAs can show modulatory effects on intestinal epithelial cells and neutrophils, as well as monocytes and macrophages (105). One of the most important SCFAs is butyrate, which is produced by Clostridia and Firmicutes, among others. Early on, butyrate enema therapy was found to be able to stimulate mucosal repair in experimental models of colitis in rats (106). Accordingly, several studies have been conducted highlighting the potential beneficial effect of butyrate on the course of UC in patients (107–109). In Crohn’s disease, butyrate was administered orally to patients in the form of tablets. Butyrate is able to antagonize colonic inflammation (110) and has been found to reduce the production of pro-inflammatory cytokines including IFN-γ. It does this by acting as a histone deacetylase inhibitor and interfering with transcription of IFN-γ by inhibiting IFN-γ-induced tyrosine and serine phosphorylation of STAT1 (111–113). In 2019, Chen and colleagues investigated whether butyrate treatment could regulate the differentiation of T cells into Th1 and Th17 cell fates. They found that, on the one hand, promotion of both T cell subtypes was induced and differentially regulated (including promotion of IFN-γ expression in Th1 cell development), but most interestingly, expression of anti-inflammatory IL-10 was induced in both cases. Rag1-deficient mice receiving these butyrate-treated T cells showed less severe colitis compared with animals receiving untreated T cells. These data provide important details about how butyrate might be used therapeutically in IBD (114). Another interesting study from the same year examined the relationship between the microbiome, their intestinal metabloites, and interferons. Zhai and colleagues tested the ability of strains of Akkermansia muciniphila, which may exert probiotic effects in obesity and diabetes, to decrease inflammation in chronic colitis in mice. Both strains used (namely 139 and ATCC) were able to improve colonic inflammation when introduced into mice suffering from DSS-induced colitis. In addition, the levels of proinflammatory TNF as well as IFN-γ were reduced in the colon of the mice. Most importantly, they found that strain ATCC was able to induce the production of beneficial SCFAs (115). Also beyond intestinal inflammation, butyrate production by Lachnospiraceae was found to inhibit STING-activated type I IFN production by DCs (116). Conversely, beneficial lactic acid bacteria were shown to induce interferon type I secretion (117). Oral administration of the SCFA acetate mediated an IFN-β response by increasing ISG expression (118). These data suggest that intestinal bacteria and their metabolites have the capability to modulate interferon production and thus impact the innate immune response.
One of the very first reports about IFNs in the context of GVHD was delivered in 1987, where Reyes and Klimpel measured the production of IFN-α/β/γ in sera of mice which were lethally irradiated and subjected to allo-BMT. They found that higher IFN activity positively correlated with GVHD occurrence. With these observations, they paved the way for following research work regarding the influence of IFNs on GVHD development (119).
There are controversial reports about the role of type I IFNs in GVHD. In early clinical studies from the 1990s, pre-transplant exogenous type I IFN administration in humans resulted in increased GVHD occurrence and transplant-related mortality (120, 121). In contrast, several experimental studies could demonstrate, that type I IFN signaling was able to positively modulate murine GVHD outcome (121–125). In 2011, Robb and colleagues were amongst the first researchers to investigate the role of type I IFNs in GVHD and GVL. Using IFNAR1-deficient mice as recipients or donors in a murine GVHD model as well as exogenous administration of IFN-α, they found that type I IFN signaling had pleiotropic effects. These included the suppression of CD4+ T cell-dependent GVHD and at the same time a paradoxical increase in CD8+ T cell-mediated GVHD (122). In 2017, Fischer and colleagues elegantly showed that mice deficient for mitochondrial antiviral-signaling protein (MAVS) or stimulator of interferon genes (STING), which are innate types of PRRs that induce the expression of type I IFNs, developed worse GVHD after allo-HCT. In line with that, they could ameliorate disease outcome triggering either the RIG-I/MAVS- or the STING pathway to induce protective type I IFN signaling and maintain intestinal epithelial barrier integrity (121, 123). Consistently, several studies could demonstrate, that the administration of type I IFN or type I IFN-inducing agonists was potent in protecting mice from GVHD in a MHC-mismatched model, when given before allo-HCT (124, 125). Interestingly, intestinal microbes that produce indole and indole derivatives, mitigate GVHD development, partly by induction of IFN type I-stimulated genes (124).
Another study investigated the synergy between IL-22, known to be a key player in promoting aGVHD development, and type I IFN (126). For this, the authors used IFNAR- as well as IL-22-deficient mice as recipients of allogeneic wild-type BM cells in combination with allogeneic T cells from either IFNAR- or IL-22-deficient donors. They observed lower GVHD severity in IFNAR-deficient recipient animals when IL-22-deficient donor T cells were transferred in a major MHC mismatch model. Therefore, interference with IL-22 and type I IFN signaling could be a novel treatment approach. Additionally, the authors could connect the increased GVHD severity to elevated STAT1 activation and CXCL10 expression. It was speculated, that the synergy between donor-derived IL-22 and recipient type I IFN signaling could favor the loss of intestinal barrier integrity in aGVHD pathogenesis (126). Also in a model of systemic sclerosis (Ssc) -like cutaneous GVHD, protection was achieved by blocking type I IFN signaling via usage of a neutralizing Ab against IFNAR1. Notably, the central question in this study was to elucidate the role of type I IFN blocking in SSc, and the cutaneous model of GVHD was only used to mimic this disease. The authors investigated fibrogenesis, but important features such as survival rate after GVHD induction and histopathological score of the intestine were not obtained (127). Altogether, these data show that type I IFNs signaling has complex and partly opposite effects on GVHD development, depending on the preclinical model used.
Over the last decades it became clear, that IFN-γ has pleiotropic effects in GVHD pathogenesis as well, depending on the examined cell type. It is well established, that intestinal damage during GVHD results in large parts from the increased release of IFN-γ and IL-12 from alloreactive Th1 T cells (128). IFN-γ induced intestinal cell apoptosis and, together with LPS originating from transmigrated bacteria, it stimulated the secretion of proinflammatory cytokines such as TNF, further supporting the inflammatory setting (129, 130). In mouse intestinal organoids, activated T cells induced tissue damage and reduction in Paneth cell and ISC numbers via IFN-γ signaling (131). Organoids deficient for the IFN-γ receptor remained unaffected by T cells, and in vivo IFN-γ administration elicited enteric inflammation (131). These data were supported by murine in vivo studies, where IFN-γ was described as the major mediator of ISC reduction in the colonic crypts (132). When GVHD was induced by T cells lacking IFN-γ or in mice deficient for the IFN-γ receptor in ISC, the stem cell compartment was protected (132). Collectively, these data indicate that IFN-γ has detrimental effects on the intestinal epithelium. In line with this hypothesis, already in 1989, Mowat described positive effects of the administration of an anti-IFN-γ antibody in two murine GVHD models (133).
Contrarily, a number of studies have also reported protective roles of the type II IFN in the context of GVHD. In a murine model of fully MHC-mismatched allo-BMT, IFN-γ-deficient donor CD8+ T cells, but not WT donor cells, were able to induce lethal GVHD (134, 135). GVHD protection appeared to be mediated by effects of IFN-γ on T cells, either through a direct mechanism or via modulation of IL-12 signaling. IL-12 is essential in promoting the differentiation of naïve T cells into Th1 cells (136). IL-12 is produced by APCs and stimulates IFN-γ production by T cells as well as NK cells (137). In lethally irradiated mice, one single injection of recombinant murine IL-12 simultaneously with the BMT led to the protection of mice against aGVHD in both in fully MHC- as well as minor antigen-mismatched strain combinations (138–140). In another study, the authors pinpointed that dose as well as timing of recombinant IL-12 administration determined whether this cytokine had protective or rather detrimental effects. They found that administration of IL-12 1-12h prior to BMT resulted in protective actions of IL-12 whereas administration more than 36h after BMT completely abrogated these positive effects (141). Interestingly, in the study of Yang and colleagues from 1999, protection against GVHD was completely lost upon treatment with the neutralizing anti-IFN-γ monoclonal antibody (mAb) R4-6A2 (141). Altogether, this led to the assumption, that IFN-γ is required for the protective effects of IL-12, but is not per se responsible for GVHD induction (142). To decipher, whether recipient or donor IFN-γ was responsible for the protective effects via IL-12, Dey and colleagues transplanted C57/BL6 mice with allogeneic HSCs from IFN-γ KO BALB/c mice and could not achieve prolonged survival rates via treatment with IL-12. This data supported the hypothesis that the IFN-γ which was mediating the protective effects of IL-12 was donor-derived. Mechanistically, the authors could show that Fas-mediated donor CD4+ T cell apoptosis was one of the underlying mechanisms involved in the protective effects of IL-12 on GHVD pathogenesis (139). Apart from regulation of IL-12 signaling, a direct protective role of IFN-γ was also observed using IFN-γ KO mice. In one study, the authors could show that the dosing of conditioning regimen plays a pivotal role considering disease outcome: IFN-γ KO animals were used as donors in lethal and sublethal allogeneic BMT experiment using total body irradiation TBI as conditioning. For recipients of lethal doses of TBI, loss of donor IFN-γ was detrimental whereas recipient of sublethal doses, the loss of IFN-γ was protective (143). Consecutive studies showed that IFN-γ deficient CD8+ T cells induce more severe GVHD in models with major and minor histocompatibility mismatch (134). These results were presumably based on the loss of apoptosis induced in activated CD8+ T cells by IFN-γ. In line with these findings, another study could prove, that the IFN-γ receptor signaling was the major pathway responsible for the migration of both conventional- but also regulatory T cells to GVHD target organs. Altered trafficking of both T cell types was mediated by expression of CXCR3 which was connected to IFN-γ receptor signaling (144). Collectively, these reports provide evidence that IFN-γ regulates the alloreactive T cell pool and can prevent excessive T cell expansion.
The role of IFN-γ in intestinal GVHD remains controversial. Multiple studies observed that IFN-γ damages intestinal epithelial cells by inducing apoptosis and production of pro-inflammatory cytokines in the intestine. On the other hand, intact IFN-γ signaling appears important for the control of alloreacitve T cell expansion, differentiation and migration. Exploring which downstream cascades are responsible for the one or the other effect might open new avenues for targeted treatment.
Type III IFNs have only recently been discovered and therefore knowledge of their role in intestinal homeostasis and inflammation is just emerging. Epithelial cells of mucosal tissues, such as the IECs, are a major target of these type of interferons (62). Both human and murine IECs show a high responsiveness to treatment with type III IFNs. Recently, mice deficient for the IFN type III receptor (IL-28 receptor alpha subunit, IL-28Rα) showed comparable thymic regeneration potential and GVHD development as wildtype mice (145). In line with these data, IL-28A protein administration did not support recovery from irradiation-induced thymus damage (145). Nevertheless, single nucleotide polymorphisms in the IFNL4 gene in donors was associated with increased risk of non-relapse mortality in humans (146). Further studies are warranted to assess the relevance of type III IFNs in GVHD.
Given the pleiotropic effects of IFNs on different cell populations involved in GVHD, it has been a challenge to develop successful clinical strategies by direct modulation of the interactions between IFN and their receptors. One indirect approach targeting IFN signaling amongst others, is the inhibition of JAK/STAT-signaling. Pre-clinical models showed, that incidence and severity of GVHD were reduced when administrating ruxolitinib, a selective inhibitor of JAK1 and -2, both being involved in the IFN-γ signaling pathway (147–149). Based on those findings, clinical trials on the potential of ruxolitinib for the treatment of glucocorticoid-refractory aGVHD showed great success and led to the approval of ruxolitinib for this indication by the Food and Drug Administration (150, 151). Another potential avenue for the use of IFN in the treatment of GVHD is related to the generation of mesenchymal stem cells (MSCs), a cell population with immunosuppressive properties. The role of IFN-γ activating MSCs has previously been described in vitro (152, 153). A first pilot study in patients suffering from severe steroid-resistant aGVHD could demonstrate MSCs as a promising treatment option (154). Nevertheless, development of a MSCs-based therapy for GVHD was impeded by factors such as a lack of standard protocol for the production of MSCs and the overall heterogeneity of MSCs derived from various donors and tissues (155–158). Regarding the role of IFN-γ in activating MSCs, it could be demonstrated, that MSCs primed with IFN-γ were able to reduce GVHD in NOD-SCID mice and to ameliorate survival rates when compared to animals receiving non-primed MSCs. The authors showed, that this effect was based on an induction of indoleamine 2,3-dioxygenase (IDO) via the IFNγ-JAK-STAT1 pathway in the MSCs, thereby enhancing their immunosuppressive properties (159). The exact mechanisms of how the various IFNs discussed in this review act in the context of GVHD remain largely unclear. It is essential to distinguish between the effects of IFNs on the hematopoietic cells of the recipient and of the donor, respectively. Furthermore, effects on the target tissues in the recipient need to be considered. Further studies are needed to elucidate the roles of IFNs in both GVHD and GVL processes after allo-HCT and to possibly make use of protective IFN administration.
In the context of genome-wide association studies, several genetic susceptibility loci for UC, CD or both were identified. These included genes which are essential key players in immunity and barrier function, amongst others. Several of those identified IBD-associated genes are involved in the type I IFN signaling pathway, for example the single nucleotide polymorphism (SNP) rs2284553, which affects the IFNAR1 gene. Other SNPs were found in the genes encoding JAK2 (rs10758669), TYK2 (rs11879191), STAT1 (rs1517352) and STAT3 (rs12942547), playing a role in several signaling pathways downstream of type I and III IFNs (28, 47, 160). Therefore, aberrations in the type I signaling network could promote an imbalanced immune response leading to induction of IBD (126). Appendicitis-appendectomy (AA) has been shown to reduce or prevent UC in adulthood, which was described in several clinical studies (161–164), and reviewed by Koutroubakis and colleagues (165). Similar observations were made regarding the prevention and ability to decrease CD severity (161, 163, 166). Cheluvappa and colleagues developed a model of AA to identify novel therapy options for colitis amelioration. In this model, mice undergoing AA were protected from experimental colitis in and age-, bacteria- and antigen- dependent manner. They found that AA led to dampened Th17 cell activity and autophagy, but most interestingly, that AA was driving the modulation of IFN-associated molecules. Significant upregulation of the ISGs IFIT1, IFIT2 and IFIT3 in the distal colon 28 days after AA could be measured. These genes are induced by IFNs, virus infections and PAMPs, mediating immunomodulatory and antiproliferative functions as well as apoptosis induction (167–169). The authors assigned the beneficial effects of AA to this mode (170). Similar results were obtained in a study where imiquimod, a virostatic agent, induced type I IFN expression in the mucosa of the GI and was able to protect against DSS-induced colitis. Notably, no systemic IFN response could be measured. Based on their findings, the authors suggested imiquimod as a potential therapeutic approach for IBD patients (171). Other studies implied that type I IFNs rather played a dual role in the context of intestinal inflammation and recovery from colitis (172). Protective actions could be seen in a study where DCs, when stimulated with TLR9 agonists, produced type I IFNs leading to the protection against experimental colitis in RAG1-deficient mice. Consistently, administration of recombinant IFN-β led to similar protection (173). In a follow-up study, the authors could show more in detail, that the type I IFN produced by DCs was able to inhibit colonic inflammation via regulation of neutrophil and monocyte trafficking into the inflamed colon (174). In a T cell-induced colitis model, the protective effect of type I IFNs was attributed to its positive influence on Tregs via increasing their cell numbers and the maintenance of Foxp3 expression (175, 176). In contrast to that, it was seen that the local delivery of IFN-β via Lactobacillus into the intestine led to an exacerbation of DSS-induced colitis accompanied by increased levels of pro-inflammatory cytokines and lower numbers of Tregs in the small intestine of mice (177). It is important to underline, that the source of IFN-β in this study was a bacterial vehicle which might have diverse and different physiological effects compared to administration of pure recombinant type I IFN. Altogether, most studies suggest that type I IFNs are protective in different preclinical models of colitis.
The type II IFN IFN-γ is one of the most highly upregulated cytokines found in IBD patients and in murine models of intestinal inflammation (41, 42, 178–180). It was demonstrated, that one aspect of the pathophysiological role of IFN-γ in IBD lied in its direct effects on the intestinal epithelium by influencing the homeostasis between cell proliferation and apoptosis via the regulation of converging of β-catenin signaling pathways. In the same study, it was observed, that TNF even increased the effects of IFN-γ, underlining a synergism between those two cytokines in the setting of intestinal inflammation (94).
Apart from that, several studies could show that IFN-γ also had significant effects on the intestinal vasculature. In vitro, it showed an overall antiangiogenic effect, including inhibition of proliferation, invasion and tube formation of endothelial cells via induction of the large GTPase guanylate binding protein‐1 (GBP‐1) (181–183). Based on these findings, Naschberger and colleagues could attribute GBP-1, resulting from IFN-γ upregulation in colorectal carcinoma (CRC), to an IFN-γ-dominated Th1-like immune reaction possessing potential angiostatic/antiangiogenic activity. They underlined that the microenvironment in GBP‐1‐positive CRC is dominated by IFN-γ, which was associated with an improved prognosis for the CRC patients (184). Interestingly, by using a neutralizing anti-IFN-γ antibody in a murine DSS-induced colitis model, it was shown, that IFN-γ exhibited an endogenous angiostatic activity in IBD and contributed to increased vascular permeability (179). In contrast to that, it was recently shown, that IFN-γ acted pathogenic in IBD by negatively impacting the vascular barrier by disruption of VE-cadherin, an adherent junction protein. By using endothelial cell-specific IFN-γ-receptor-KO mouse models, the authors of the study could show, that an endothelial-specific inhibition of the IFN-γ response led to an ameliorated outcome in DSS-induced colitis. Furthermore, IBD-associated vascular barrier dysfunction was also confirmed in human patients (185). Altogether and similar as in GVHD, IFN-γ remains a pleotropic cytokine with controversial roles in IBD pathology.
Since type III IFNs are emerging as a cytokine group with specific role on epithelial barrier surfaces, several studies tested their potential role in IBD models. First data demonstrated, that IFN-λ played a protective role in a murine model of DSS-induced colitis, thereby proposing it as an anticolitogenic cytokine (81, 126). In contrast, it was found, that levels of IFN-λ were increased in inflamed ileal tissues and sera of CD patients. This was accompanied by a loss Paneth cells. Based on those findings, the authors of this study suggested, that blocking IFN-λ or reducing its concentrations in affected patients might positively affect disease outcome (186). Further studies are required to explore the therapeutic potential of IFN-λ signaling.
Studies investigating the effects of systemic administration of type IFNs to ameliorate IBD have produced controversial results. Administration of IFNs was shown to not have positive effects in the context of UC treatment (187). Overall, a Cochrane systematic literature review from 2008 investigating the efficacy and safety of type I IFN therapy (including IFN-β-1a, IFN-β-1b, IFN-α-2a, IFN-α-2b and associated PEGylated formulations) in UC showed no difference between groups of patients which were treated with type I IFNs or placebo in regards to remission achievement or symptom improvement. The authors conclude, that the data from those clinical trials do not support the use of type I IFNs to induce remission status in active UC. In accordance to the current scientific knowledge, no statistically significant benefit regarding disease amelioration could been observed in using type I IFN for the treatment of IBDs (188).
Fontolizumab, a humanized anti-IFN-γ antibody, could not induce strong clinical responses in a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study in patients suffering from moderate to severe CD. Though well tolerated, administration only led to a significant decrease in C-reactive protein levels (189). The clinical development and further investigations on Fontolizumab in the context of IBD were stopped. Also eldelumab, an anti-INF-γ-inducible protein-10 (IP-10) monoclonal antibody, could not achieve the primary endpoint in a study in patients suffering from UC (190). Interestingly, when compared to other (auto-) immune related diseases such as rheumatoid arthritis or psoriasis, it becomes apparent, that in IBD, mainly TNF antagonizing monoclonal antibodies (mAbs), including infliximab, adalimumab and golimumab, show a beneficial effect (191). IFN signaling is mediated via intracellular JAKs and TYK2. It is therefore evident, that blocking these kinases could be a promising approach to cope with the elevated signaling of proinflammatory cytokines with proposed roles in mucosal immune cells in intestinal inflammation. Examples include the successful use of tofacitinib, blocking JAK3 activation and signaling via common γ-chain containing cytokines (IL-2,-4,-7,-9,-15 and -21) in CD and UC, and the selective JAK1 inhibitor filgotinib for Crohn’s disease (192–194). This indicates that JAK inhibitors might be promising approaches for clinical therapy of IBD patients.
Regarding the therapeutic use of type III IFNs, some promising first data were collected in clinical trials for the treatment of chronic hepatitis with PEGylated forms of IFN-λ (ClinicalTrials.gov Identifier: NCT00565539). So far, there are no data available on the therapeutic potential of type III IFN administration in the context of IBD. An overview about the different IFNs and their respective role in GVHD and IBD pathogenesis can be obtained from Table 1.
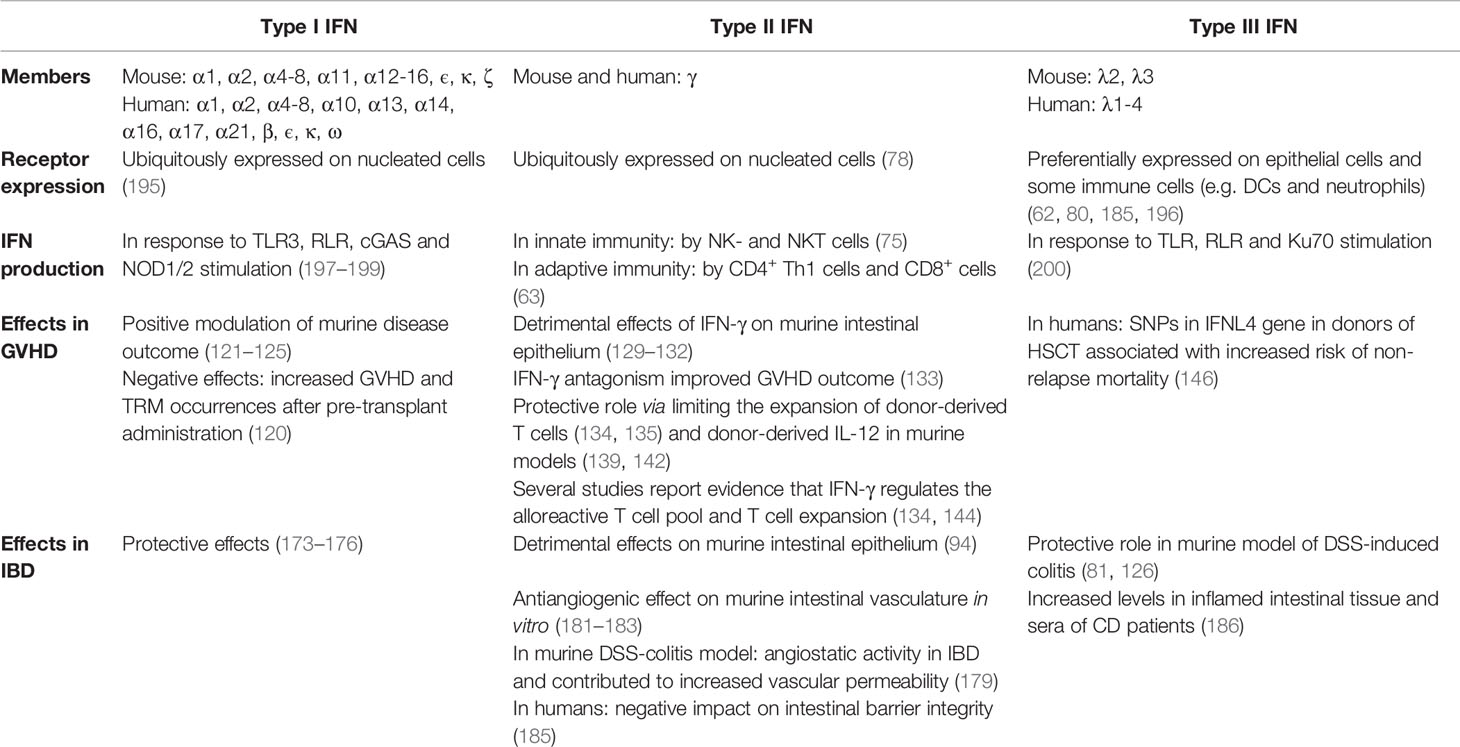
Table 1 Overview about Type I, -II and -III IFNs and their role in GVHD and IBD pathogenesis.
To date, TNF is the sole proinflammatory cytokine that has been successfully targeted in IBD. Anti-TNF therapy with various anti-TNF antibodies (including infliximab, for example) is an essential backbone for the treatment of both CD and UC patients (201). Years of research and clinical success paved the way for increased interest in other cytokines and cytokine regulatory networks regarding the pathogenesis of IBD. Unfortunately, efforts in the field of anti-IFN therapy have not yet yielded promising results, as the use of fontolizumab, an anti-IFN-γ antibody, in CD patients did not result in improved disease outcome, and further investigation and development have been discontinued (189). With regard to the therapy of GVHD, IL-6 has been the best studied and targeted cytokine in this disease. Tocilizumab, an anti-IL-6 receptor antibody, has shown efficacy in steroid-refractory intestinal aGVHD as well as cGVHD (202, 203). IFN-γ in particular has been the focus of investigation in the context of GVHD. Due to divergent and pleiotropic effects of IFN-γ blockade in preclinical mouse models, no clinical studies have yet been conducted that consider direct targeting of IFN signaling pathways in intestinal GVHD.
Overall, it is clear that a highly complex and interconnected as well as -regulated cytokine network and its imbalance plays a crucial role in the process of mucosal intestinal inflammation as well as mucosal healing. Both non-hematopoietic and hematopoietic cells of the innate and adaptive immune systems each play a central role in disease pathogenesis. In the context of GVHD, a further complication is the need to distinguish between the effect of IFNs on donor cells and, on the other hand, on recipient cells, as underscored by various preclinical models. Other factors, such as different types of MHC-mismatched BMT mouse models or even the timing of treatment in the context of IFN-cytokine network therapy, must also be considered. Further research needs to be conducted to understand why and how IFNs play such pleiotropic roles in the development and progression of both IBD and GVHD. It would be desirable to investigate the presumably positive effect of type I interferons in IBD more closely to provide the basis for eventual clinical trials. In addition, the recently discovered type III IFNs still need to be characterized in more detail, as their receptors are preferentially expressed on epithelial cells. So far, not much is known about their presumed role in signaling networks in the field of intestinal homeostasis and inflammatory processes. Ultimately, it is critical to understand better the divergent downstream signaling cascades of IFNs, and how these are connected to inflammation or tissue protection. Separating these different effects and identifying targets downstream of IFNs or their receptors might prove a promising translational approach, as seen in the example of JAK inhibition. This knowledge is essential to pave the way for more effective clinical approaches by precisely addressing the expression or functions of IFNs in intestinal inflammation.
EH and PA developed the overall concept for this review article. EH collected and reviewed literature, discussed the studies, and wrote the first draft of the manuscript. PA and RZ critically revised the manuscript. All authors contributed to the article and approved the submitted version.
PA is supported by the German Cancer Consortium (DKTK) (FR-01-375). The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding programme Open Access Publishing. RZ was supported by the SFB 1479 (P01) OncoEscape.
RZ received honoraria from Novartis, Incyte and Mallinckrodt.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Copelan EA. Hematopoietic Stem-Cell Transplantation. N Engl J Med (2006) 354(17):1813–26. doi: 10.1056/NEJMra052638
2. Welniak LA, Blazar BR, Murphy WJ. Immunobiology of Allogeneic Hematopoietic Stem Cell Transplantation. Annu Rev Immunol (2007) 25:139–70. doi: 10.1146/annurev.immunol.25.022106.141606
3. Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-Versus-Leukemia Reactions in Allogeneic Chimeras. Blood (2004) 103(3):767–76. doi: 10.1182/blood-2003-02-0342
4. Zeiser R, Blazar BR. Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med (2017) 377(22):2167–79. doi: 10.1056/NEJMra1609337
5. Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group Report. Biol Blood Marrow Transplant (2015) 21(3):389–401.e1. doi: 10.1016/j.bbmt.2014.12.001
6. Ferrara JL, Cooke KR, Teshima T. The Pathophysiology of Acute Graft-Versus-Host Disease. Int J Hematol (2003) 78(3):181–7. doi: 10.1007/BF02983793
7. Ball LM, Egeler RM, Party EPW. Acute GvHD: Pathogenesis and Classification. Bone Marrow Transplant (2008) 41(Suppl 2):S58–64. doi: 10.1038/bmt.2008.56
8. Zeiser R. Advances in Understanding the Pathogenesis of Graft-Versus-Host Disease. Br J Haematol (2019) 187(5):563–72. doi: 10.1111/bjh.16190
9. Zeiser R, Socie G, Blazar BR. Pathogenesis of Acute Graft-Versus-Host Disease: From Intestinal Microbiota Alterations to Donor T Cell Activation. Br J Haematol (2016) 175(2):191–207. doi: 10.1111/bjh.14295
10. MacMillan ML, DeFor TE, Weisdorf DJ. What Predicts High Risk Acute Graft-Versus-Host Disease (GVHD) at Onset?: Identification of Those at Highest Risk by a Novel Acute GVHD Risk Score. Br J Haematol (2012) 157(6):732–41. doi: 10.1111/j.1365-2141.2012.09114.x
11. Johansson JE, Ekman T. Gut Toxicity During Hemopoietic Stem Cell Transplantation may Predict Acute Graft-Versus-Host Disease Severity in Patients. Dig Dis Sci (2007) 52(9):2340–5. doi: 10.1007/s10620-006-9404-x
12. Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total Body Irradiation and Acute Graft-Versus-Host Disease: The Role of Gastrointestinal Damage and Inflammatory Cytokines. Blood (1997) 90(8):3204–13. doi: 10.1182/blood.V90.8.3204
13. Takashima S, Kadowaki M, Aoyama K, Koyama M, Oshima T, Tomizuka K, et al. The Wnt Agonist R-spondin1 Regulates Systemic Graft-Versus-Host Disease by Protecting Intestinal Stem Cells. J Exp Med (2011) 208(2):285–94. doi: 10.1084/jem.20101559
14. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 Promotes Intestinal-Stem-Cell-Mediated Epithelial Regeneration. Nature (2015) 528(7583):560–4. doi: 10.1038/nature16460
15. Haring E, Uhl FM, Andrieux G, Proietti M, Bulashevska A, Sauer B, et al. Bile Acids Regulate Intestinal Antigen Presentation and Reduce Graft-Versus-Host Disease Without Impairing the Graft-Versus-Leukemia Effect. Haematologica (2020). doi: 10.3324/haematol.2019.242990
16. Norona J, Apostolova P, Schmidt D, Ihlemann R, Reischmann N, Taylor G, et al. Glucagon-Like Peptide 2 for Intestinal Stem Cell and Paneth Cell Repair During Graft-Versus-Host Disease in Mice and Humans. Blood (2020) 136(12):1442–55. doi: 10.1182/blood.2020005957
17. Eriguchi Y, Takashima S, Oka H, Shimoji S, Nakamura K, Uryu H, et al. Graft-Versus-Host Disease Disrupts Intestinal Microbial Ecology by Inhibiting Paneth Cell Production of Alpha-Defensins. Blood (2012) 120(1):223–31. doi: 10.1182/blood-2011-12-401166
18. Eriguchi Y, Nakamura K, Hashimoto D, Shimoda S, Shimono N, Akashi K, et al. Decreased Secretion of Paneth Cell Alpha-Defensins in Graft-Versus-Host Disease. Transpl Infect Dis (2015) 17(5):702–6. doi: 10.1111/tid.12423
19. Levine JE, Huber E, Hammer ST, Harris AC, Greenson JK, Braun TM, et al. Low Paneth Cell Numbers at Onset of Gastrointestinal Graft-Versus-Host Disease Identify Patients at High Risk for Nonrelapse Mortality. Blood (2013) 122(8):1505–9. doi: 10.1182/blood-2013-02-485813
20. Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, et al. Regulation of Intestinal Inflammation by Microbiota Following Allogeneic Bone Marrow Transplantation. J Exp Med (2012) 209(5):903–11. doi: 10.1084/jem.20112408
21. Holler E, Butzhammer P, Schmid K, Hundsrucker C, Koestler J, Peter K, et al. Metagenomic Analysis of the Stool Microbiome in Patients Receiving Allogeneic Stem Cell Transplantation: Loss of Diversity is Associated With Use of Systemic Antibiotics and More Pronounced in Gastrointestinal Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2014) 20(5):640–5. doi: 10.1016/j.bbmt.2014.01.030
22. Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, et al. Intestinal Blautia is Associated With Reduced Death From Graft-versus-Host Disease. Biol Blood Marrow Transplant (2015) 21(8):1373–83. doi: 10.1016/j.bbmt.2015.04.016
23. Kakihana K, Fujioka Y, Suda W, Najima Y, Kuwata G, Sasajima S, et al. Fecal Microbiota Transplantation for Patients With Steroid-Resistant Acute Graft-Versus-Host Disease of the Gut. Blood (2016) 128(16):2083–8. doi: 10.1182/blood-2016-05-717652
24. Spindelboeck W, Schulz E, Uhl B, Kashofer K, Aigelsreiter A, Zinke-Cerwenka W, et al. Repeated Fecal Microbiota Transplantations Attenuate Diarrhea and Lead to Sustained Changes in the Fecal Microbiota in Acute, Refractory Gastrointestinal Graft-Versus-Host-Disease. Haematologica (2017) 102(5):e210–e3. doi: 10.3324/haematol.2016.154351
25. DeFilipp Z, Peled JU, Li S, Mahabamunuge J, Dagher Z, Slingerland AE, et al. Third-Party Fecal Microbiota Transplantation Following allo-HCT Reconstitutes Microbiome Diversity. Blood Adv (2018) 2(7):745–53. doi: 10.1182/bloodadvances.2018017731
26. van Lier YF, Davids M, Haverkate NJE, de Groot PF, Donker ML, Meijer E, et al. Donor Fecal Microbiota Transplantation Ameliorates Intestinal Graft-Versus-Host Disease in Allogeneic Hematopoietic Cell Transplant Recipients. Sci Transl Med (2020) 12(556):3668–77. doi: 10.1126/scitranslmed.aaz8926
27. Neuman MG. Immune Dysfunction in Inflammatory Bowel Disease. Transl Res (2007) 149(4):173–86. doi: 10.1016/j.trsl.2006.11.009
28. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491(7422):119–24. doi: 10.1038/nature11582
29. Kaplan GG. Ibd: Global Variations in Environmental Risk Factors for IBD. Nat Rev Gastroenterol Hepatol (2014) 11(12):708–9. doi: 10.1038/nrgastro.2014.182
30. Baumgart DC, Carding SR. Inflammatory Bowel Disease: Cause and Immunobiology. Lancet (2007) 369(9573):1627–40. doi: 10.1016/S0140-6736(07)60750-8
31. Yan JB, Luo MM, Chen ZY, He BH. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J Immunol Res (2020) 2020:8813558. doi: 10.1155/2020/8813558
32. Kim KU, Kim J, Kim WH, Min H, Choi CH. Treatments of Inflammatory Bowel Disease Toward Personalized Medicine. Arch Pharm Res (2021) 44(3):293–309. doi: 10.1007/s12272-021-01318-6
33. Danese S, Fiocchi C. Ulcerative Colitis. N Engl J Med (2011) 365(18):1713–25. doi: 10.1056/NEJMra1102942
34. Baumgart DC, Sandborn WJ. Crohn’s Disease. Lancet (2012) 380(9853):1590–605. doi: 10.1016/S0140-6736(12)60026-9
35. Odenwald MA, Turner JR. Intestinal Permeability Defects: Is it Time to Treat? Clin Gastroenterol Hepatol (2013) 11(9):1075–83. doi: 10.1016/j.cgh.2013.07.001
36. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
37. Nalle SC, Turner JR. Intestinal Barrier Loss as a Critical Pathogenic Link Between Inflammatory Bowel Disease and Graft-Versus-Host Disease. Mucosal Immunol (2015) 8(4):720–30. doi: 10.1038/mi.2015.40
38. Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, et al. IL23 Differentially Regulates the Th1/Th17 Balance in Ulcerative Colitis and Crohn’s Disease. Gut (2008) 57(12):1682–9. doi: 10.1136/gut.2007.135053
39. Seiderer J, Elben I, Diegelmann J, Glas J, Stallhofer J, Tillack C, et al. Role of the Novel Th17 Cytokine IL-17F in Inflammatory Bowel Disease (IBD): Upregulated Colonic IL-17F Expression in Active Crohn’s Disease and Analysis of the IL17F p.His161Arg Polymorphism in IBD. Inflammation Bowel Dis (2008) 14(4):437–45. doi: 10.1002/ibd.20339
40. Huang Y, Chen Z. Inflammatory Bowel Disease Related Innate Immunity and Adaptive Immunity. Am J Transl Res (2016) 8(6):2490–7. doi: 10.1038/nri3661
41. Verma R, Verma N, Paul J. Expression of Inflammatory Genes in the Colon of Ulcerative Colitis Patients Varies With Activity Both at the mRNA and Protein Level. Eur Cytokine Netw (2013) 24(3):130–8. doi: 10.1684/ecn.2013.0343
42. Neurath MF. Cytokines in Inflammatory Bowel Disease. Nat Rev Immunol (2014) 14(5):329–42. doi: 10.1038/nri3661
43. Bleich A, Mahler M. Environment as a Critical Factor for the Pathogenesis and Outcome of Gastrointestinal Disease: Experimental and Human Inflammatory Bowel Disease and Helicobacter-Induced Gastritis. Pathobiology (2005) 72(6):293–307. doi: 10.1159/000091327
44. Ferrara JL. Novel Strategies for the Treatment and Diagnosis of Graft-Versus-Host-Disease. Best Pract Res Clin Haematol (2007) 20(1):91–7. doi: 10.1016/j.beha.2006.11.004
45. Ng SC, Kamm MA. Therapeutic Strategies for the Management of Ulcerative Colitis. Inflammation Bowel Dis (2009) 15(6):935–50. doi: 10.1002/ibd.20797
46. Thomas A, Lodhia N. Advanced Therapy for Inflammatory Bowel Disease: A Guide for the Primary Care Physician. J Am Board Fam Med (2014) 27(3):411–20. doi: 10.3122/jabfm.2014.03.130224
47. Andreou NP, Legaki E, Gazouli M. Inflammatory Bowel Disease Pathobiology: The Role of the Interferon Signature. Ann Gastroenterol (2020) 33(2):125–33. doi: 10.20524/aog.2020.0457
48. Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and Their Actions. Annu Rev Biochem (1987) 56:727–77. doi: 10.1146/annurev.bi.56.070187.003455
49. Pestka S, Kotenko SV, Muthukumaran G, Izotova LS, Cook JR, Garotta G. The Interferon Gamma (IFN-Gamma) Receptor: A Paradigm for the Multichain Cytokine Receptor. Cytokine Growth Factor Rev (1997) 8(3):189–206. doi: 10.1016/S1359-6101(97)00009-9
50. Aaronson DS, Horvath CM. A Road Map for Those Who Don’t Know JAK-STAT. Science (2002) 296(5573):1653–5. doi: 10.1126/science.1071545
51. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated Signalling. Nat Rev Immunol (2005) 5(5):375–86. doi: 10.1038/nri1604
52. Der SD, Zhou A, Williams BR, Silverman RH. Identification of Genes Differentially Regulated by Interferon Alpha, Beta, or Gamma Using Oligonucleotide Arrays. Proc Natl Acad Sci USA (1998) 95(26):15623–8. doi: 10.1073/pnas.95.26.15623
53. Platanias LC, Fish EN. Signaling Pathways Activated by Interferons. Exp Hematol (1999) 27(11):1583–92. doi: 10.1016/S0301-472X(99)00109-5
54. Bach EA, Aguet M, Schreiber RD. The IFN Gamma Receptor: A Paradigm for Cytokine Receptor Signaling. Annu Rev Immunol (1997) 15:563–91. doi: 10.1146/annurev.immunol.15.1.563
55. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Science (1994) 264(5164):1415–21. doi: 10.1126/science.8197455
56. Decker T, Kovarik P, Meinke A. GAS Elements: A Few Nucleotides With a Major Impact on Cytokine-Induced Gene Expression. J Interferon Cytokine Res (1997) 17(3):121–34. doi: 10.1089/jir.1997.17.121
57. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-Gamma: An Overview of Signals, Mechanisms and Functions. J Leukoc Biol (2004) 75(2):163–89. doi: 10.1189/jlb.0603252
58. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-Lambdas Mediate Antiviral Protection Through a Distinct Class II Cytokine Receptor Complex. Nat Immunol (2003) 4(1):69–77. doi: 10.1038/ni875
59. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. Il-28, IL-29 and Their Class II Cytokine Receptor IL-28R. Nat Immunol (2003) 4(1):63–8. doi: 10.1038/ni873
60. Stanifer ML, Pervolaraki K, Boulant S. Differential Regulation of Type I and Type Iii Interferon Signaling. Int J Mol Sci (2019) 20(6). doi: 10.3390/ijms20061445
61. Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, et al. An Important Role for Type III Interferon (IFN-Lambda/IL-28) in TLR-induced Antiviral Activity. J Immunol (2008) 180(4):2474–85. doi: 10.4049/jimmunol.180.4.2474
62. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-Lambda (IFN-Lambda) is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PloS Pathog (2008) 4(3):e1000017. doi: 10.1371/journal.ppat.1000017
63. Schoggins JW. Interferon-Stimulated Genes: What Do They All do? Annu Rev Virol (2019) 6(1):567–84. doi: 10.1146/annurev-virology-092818-015756
64. Takaoka A, Yanai H. Interferon Signalling Network in Innate Defence. Cell Microbiol (2006) 8(6):907–22. doi: 10.1111/j.1462-5822.2006.00716.x
65. Crouse J, Kalinke U, Oxenius A. Regulation of Antiviral T Cell Responses by Type I Interferons. Nat Rev Immunol (2015) 15(4):231–42. doi: 10.1038/nri3806
66. Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, et al. Comparative Analysis of IRF and IFN-alpha Expression in Human Plasmacytoid and Monocyte-Derived Dendritic Cells. J Leukoc Biol (2003) 74(6):1125–38. doi: 10.1189/jlb.0603255
67. Gilliet M, Cao W, Liu YJ. Plasmacytoid Dendritic Cells: Sensing Nucleic Acids in Viral Infection and Autoimmune Diseases. Nat Rev Immunol (2008) 8(8):594–606. doi: 10.1038/nri2358
68. Blasius AL, Beutler B. Intracellular Toll-Like Receptors. Immunity (2010) 32(3):305–15. doi: 10.1016/j.immuni.2010.03.012
69. Radvanyi LG, Banerjee A, Weir M, Messner H. Low Levels of Interferon-Alpha Induce CD86 (B7.2) Expression and Accelerates Dendritic Cell Maturation From Human Peripheral Blood Mononuclear Cells. Scand J Immunol (1999) 50(5):499–509. doi: 10.1046/j.1365-3083.1999.00625.x
70. Parlato S, Santini SM, Lapenta C, Di Pucchio T, Logozzi M, Spada M, et al. Expression of CCR-7, Mip-3beta, and Th-1 Chemokines in Type I IFN-Induced Monocyte-Derived Dendritic Cells: Importance for the Rapid Acquisition of Potent Migratory and Functional Activities. Blood (2001) 98(10):3022–9. doi: 10.1182/blood.V98.10.3022
71. Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I Interferons Produced by Dendritic Cells Promote Their Phenotypic and Functional Activation. Blood (2002) 99(9):3263–71. doi: 10.1182/blood.V99.9.3263
72. Padovan E, Spagnoli GC, Ferrantini M, Heberer M. Ifn-alpha2a Induces IP-10/CXCL10 and MIG/CXCL9 Production in Monocyte-Derived Dendritic Cells and Enhances Their Capacity to Attract and Stimulate CD8+ Effector T Cells. J Leukoc Biol (2002) 71(4):669–76. doi: 10.1189/jlb.71.4.669
73. Rouzaut A, Garasa S, Teijeira A, Gonzalez I, Martinez-Forero I, Suarez N, et al. Dendritic Cells Adhere to and Transmigrate Across Lymphatic Endothelium in Response to IFN-Alpha. Eur J Immunol (2010) 40(11):3054–63. doi: 10.1002/eji.201040523
74. Simmons DP, Wearsch PA, Canaday DH, Meyerson HJ, Liu YC, Wang Y, et al. Type I IFN Drives a Distinctive Dendritic Cell Maturation Phenotype That Allows Continued Class II MHC Synthesis and Antigen Processing. J Immunol (2012) 188(7):3116–26. doi: 10.4049/jimmunol.1101313
75. Boehm U, Klamp T, Groot M, Howard JC. Cellular Responses to Interferon-Gamma. Annu Rev Immunol (1997) 15:749–95. doi: 10.1146/annurev.immunol.15.1.749
76. Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced In Vivo Growth and Resistance to Rejection of Tumor Cells Expressing Dominant Negative IFN Gamma Receptors. Immunity (1994) 1(6):447–56. doi: 10.1016/1074-7613(94)90087-6
77. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. Ifngamma and Lymphocytes Prevent Primary Tumour Development and Shape Tumour Immunogenicity. Nature (2001) 410(6832):1107–11. doi: 10.1038/35074122
78. Alspach E, Lussier DM, Schreiber RD. Interferon Gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol (2019) 11(3). doi: 10.1101/cshperspect.a028480
80. Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, et al. Lambda Interferon Renders Epithelial Cells of the Respiratory and Gastrointestinal Tracts Resistant to Viral Infections. J Virol (2010) 84(11):5670–7. doi: 10.1128/JVI.00272-10
81. Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, et al. IFN-Lambda Determines the Intestinal Epithelial Antiviral Host Defense. Proc Natl Acad Sci USA (2011) 108(19):7944–9. doi: 10.1073/pnas.1100552108
82. Stanifer ML, Guo C, Doldan P, Boulant S. Importance of Type I and III Interferons at Respiratory and Intestinal Barrier Surfaces. Front Immunol (2020) 11:608645. doi: 10.3389/fimmu.2020.608645
83. Chirdo FG, Millington OR, Beacock-Sharp H, Mowat AM. Immunomodulatory Dendritic Cells in Intestinal Lamina Propria. Eur J Immunol (2005) 35(6):1831–40. doi: 10.1002/eji.200425882
84. Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, et al. Interferon-Lambda and Interleukin 22 Act Synergistically for the Induction of Interferon-Stimulated Genes and Control of Rotavirus Infection. Nat Immunol (2015) 16(7):698–707. doi: 10.1038/ni.3180
85. Mahlakoiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. Leukocyte-Derived IFN-alpha/beta and Epithelial IFN-lambda Constitute a Compartmentalized Mucosal Defense System That Restricts Enteric Virus Infections. PloS Pathog (2015) 11(4):e1004782. doi: 10.1371/journal.ppat.1004782
86. Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, et al. Expression of Ifnlr1 on Intestinal Epithelial Cells is Critical to the Antiviral Effects of Interferon Lambda Against Norovirus and Reovirus. J Virol (2017) 91(7). doi: 10.1128/JVI.02079-16
87. Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, et al. Interferon-Lambda Cures Persistent Murine Norovirus Infection in the Absence of Adaptive Immunity. Science (2015) 347(6219):269–73. doi: 10.1126/science.1258100
88. Broggi A, Granucci F, Zanoni I. Type III Interferons: Balancing Tissue Tolerance and Resistance to Pathogen Invasion. J Exp Med (2020) 217(1). doi: 10.1084/jem.20190295
89. Schnoor M, Betanzos A, Weber DA, Parkos CA. Guanylate-Binding Protein-1 is Expressed at Tight Junctions of Intestinal Epithelial Cells in Response to Interferon-Gamma and Regulates Barrier Function Through Effects on Apoptosis. Mucosal Immunol (2009) 2(1):33–42. doi: 10.1038/mi.2008.62
90. Mirpuri J, Brazil JC, Berardinelli AJ, Nasr TR, Cooper K, Schnoor M, et al. Commensal Escherichia Coli Reduces Epithelial Apoptosis Through IFN-alphaA-mediated Induction of Guanylate Binding Protein-1 in Human and Murine Models of Developing Intestine. J Immunol (2010) 184(12):7186–95. doi: 10.4049/jimmunol.0903116
91. Pervolaraki K, Stanifer ML, Munchau S, Renn LA, Albrecht D, Kurzhals S, et al. Type I and Type Iii Interferons Display Different Dependency on Mitogen-Activated Protein Kinases to Mount an Antiviral State in the Human Gut. Front Immunol (2017) 8:459. doi: 10.3389/fimmu.2017.00459
92. Saxena K, Simon LM, Zeng XL, Blutt SE, Crawford SE, Sastri NP, et al. A Paradox of Transcriptional and Functional Innate Interferon Responses of Human Intestinal Enteroids to Enteric Virus Infection. Proc Natl Acad Sci USA (2017) 114(4):E570–9. doi: 10.1073/pnas.1615422114
93. Stanifer ML, Kee C, Cortese M, Zumaran CM, Triana S, Mukenhirn M, et al. Critical Role of Type Iii Interferon in Controlling SARS-Cov-2 Infection in Human Intestinal Epithelial Cells. Cell Rep (2020) 32(1):107863. doi: 10.1016/j.celrep.2020.107863
94. Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, et al. Interferon-Gamma Regulates Intestinal Epithelial Homeostasis Through Converging Beta-Catenin Signaling Pathways. Immunity (2010) 32(3):392–402. doi: 10.1016/j.immuni.2010.03.001
95. Capaldo CT, Beeman N, Hilgarth RS, Nava P, Louis NA, Naschberger E, et al. IFN-Gamma and TNF-alpha-induced GBP-1 Inhibits Epithelial Cell Proliferation Through Suppression of Beta-Catenin/TCF Signaling. Mucosal Immunol (2012) 5(6):681–90. doi: 10.1038/mi.2012.41
96. Nava P, Kamekura R, Quiros M, Medina-Contreras O, Hamilton RW, Kolegraff KN, et al. Ifngamma-Induced Suppression of Beta-Catenin Signaling: Evidence for Roles of Akt and 14.3.3zeta. Mol Biol Cell (2014) 25(19):2894–904. doi: 10.1091/mbc.e13-09-0512
97. Raetz M, Hwang SH, Wilhelm CL, Kirkland D, Benson A, Sturge CR, et al. Parasite-Induced TH1 Cells and Intestinal Dysbiosis Cooperate in IFN-gamma-dependent Elimination of Paneth Cells. Nat Immunol (2013) 14(2):136–42. doi: 10.1038/ni.2508
98. Farin HF, Karthaus WR, Kujala P, Rakhshandehroo M, Schwank G, Vries RG, et al. Paneth Cell Extrusion and Release of Antimicrobial Products is Directly Controlled by Immune Cell-Derived IFN-Gamma. J Exp Med (2014) 211(7):1393–405. doi: 10.1084/jem.20130753
99. Yitbarek A, Alkie T, Taha-Abdelaziz K, Astill J, Rodriguez-Lecompte JC, Parkinson J, et al. Gut Microbiota Modulates Type I Interferon and Antibody-Mediated Immune Responses in Chickens Infected With Influenza Virus Subtype H9N2. Benef Microbes (2018) 9(3):417–27. doi: 10.3920/BM2017.0088
100. Bradley KC, Finsterbusch K, Schnepf D, Crotta S, Llorian M, Davidson S, et al. Microbiota-Driven Tonic Interferon Signals in Lung Stromal Cells Protect From Influenza Virus Infection. Cell Rep (2019) 28(1):245–56.e4. doi: 10.1016/j.celrep.2019.05.105
101. Winkler ES, Shrihari S, Hykes BL Jr., Handley SA, Andhey PS, Huang YS, et al. The Intestinal Microbiome Restricts Alphavirus Infection and Dissemination Through a Bile Acid-Type I Ifn Signaling Axis. Cell (2020) 182(4):901–18 e18. doi: 10.1016/j.cell.2020.06.029
102. Bird L. Gut Bacteria Support Antiviral Immunity. Nat Rev Immunol (2020) 20(9):520–1. doi: 10.1038/s41577-020-00412-y
103. Sivaprakasam S, Bhutia YD, Ramachandran S, Ganapathy V. Cell-Surface and Nuclear Receptors in the Colon as Targets for Bacterial Metabolites and Its Relevance to Colon Health. Nutrients (2017) 9(8). doi: 10.3390/nu9080856
104. Zeng MY, Inohara N, Nunez G. Mechanisms of Inflammation-Driven Bacterial Dysbiosis in the Gut. Mucosal Immunol (2017) 10(1):18–26. doi: 10.1038/mi.2016.75
105. Correa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA. Regulation of Immune Cell Function by Short-Chain Fatty Acids. Clin Transl Immunol (2016) 5(4):e73. doi: 10.1038/cti.2016.17
106. Butzner JD, Parmar R, Bell CJ, Dalal V. Butyrate Enema Therapy Stimulates Mucosal Repair in Experimental Colitis in the Rat. Gut (1996) 38(4):568–73. doi: 10.1136/gut.38.4.568
107. Scheppach W, Sommer H, Kirchner T, Paganelli GM, Bartram P, Christl S, et al. Effect of Butyrate Enemas on the Colonic Mucosa in Distal Ulcerative Colitis. Gastroenterology (1992) 103(1):51–6. doi: 10.1016/0016-5085(92)91094-K
108. Vernia P, Marcheggiano A, Caprilli R, Frieri G, Corrao G, Valpiani D, et al. Short-Chain Fatty Acid Topical Treatment in Distal Ulcerative Colitis. Aliment Pharmacol Ther (1995) 9(3):309–13. doi: 10.1111/j.1365-2036.1995.tb00386.x
109. Steinhart AH, Hiruki T, Brzezinski A, Baker JP. Treatment of Left-Sided Ulcerative Colitis With Butyrate Enemas: A Controlled Trial. Aliment Pharmacol Ther (1996) 10(5):729–36. doi: 10.1046/j.1365-2036.1996.d01-509.x
110. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review Article: The Role of Butyrate on Colonic Function. Aliment Pharmacol Ther (2008) 27(2):104–19. doi: 10.1111/j.1365-2036.2007.03562.x
111. Vidali G, Boffa LC, Bradbury EM, Allfrey VG. Butyrate Suppression of Histone Deacetylation Leads to Accumulation of Multiacetylated Forms of Histones H3 and H4 and Increased Dnase I Sensitivity of the Associated DNA Sequences. Proc Natl Acad Sci USA (1978) 75(5):2239–43. doi: 10.1073/pnas.75.5.2239
112. Davie JR. Inhibition of Histone Deacetylase Activity by Butyrate. J Nutr (2003) 133(7 Suppl):2485S–93S. doi: 10.1093/jn/133.7.2485S
113. Klampfer L, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Inhibition of Interferon Gamma Signaling by the Short Chain Fatty Acid Butyrate. Mol Cancer Res (2003) 1(11):855–62.
114. Chen L, Sun M, Wu W, Yang W, Huang X, Xiao Y, et al. Microbiota Metabolite Butyrate Differentially Regulates Th1 and Th17 Cells’ Differentiation and Function in Induction of Colitis. Inflamm Bowel Dis (2019) 25(9):1450–61. doi: 10.1093/ibd/izz046
115. Zhai R, Xue X, Zhang L, Yang X, Zhao L, Zhang C. Strain-Specific Anti-inflammatory Properties of Two Akkermansia Muciniphila Strains on Chronic Colitis in Mice. Front Cell Infect Microbiol (2019) 9:239. doi: 10.3389/fcimb.2019.00239
116. Yang K, Hou Y, Zhang Y, Liang H, Sharma A, Zheng W, et al. Suppression of Local Type I Interferon by Gut Microbiota-Derived Butyrate Impairs Antitumor Effects of Ionizing Radiation. J Exp Med (2021) 218(3). doi: 10.1084/jem.20201915
117. Gutierrez-Merino J, Isla B, Combes T, Martinez-Estrada F, Maluquer De Motes C. Beneficial Bacteria Activate Type-I Interferon Production Via the Intracellular Cytosolic Sensors STING and MAVS. Gut Microbes (2020) 11(4):771–88. doi: 10.1080/19490976.2019.1707015
118. Antunes KH, Fachi JL, de Paula R, da Silva EF, Pral LP, Dos Santos AA, et al. Microbiota-Derived Acetate Protects Against Respiratory Syncytial Virus Infection Through a GPR43-type 1 Interferon Response. Nat Commun (2019) 10(1):3273. doi: 10.1038/s41467-019-11152-6
119. Reyes VE, Klimpel GR. Interferon Alpha/Beta Synthesis During Acute Graft-Versus-Host Disease. Transplantation (1987) 43(3):412–6. doi: 10.1097/00007890-198703000-00018
120. Morton AJ, Gooley T, Hansen JA, Appelbaum FR, Bruemmer B, Bjerke JW, et al. Association Between Pretransplant Interferon-Alpha and Outcome After Unrelated Donor Marrow Transplantation for Chronic Myelogenous Leukemia in Chronic Phase. Blood (1998) 92(2):394–401. doi: 10.1182/blood.V92.2.394.414k42_394_401
121. Hehlmann R, Hochhaus A, Kolb HJ, Hasford J, Gratwohl A, Heimpel H, et al. Interferon-Alpha Before Allogeneic Bone Marrow Transplantation in Chronic Myelogenous Leukemia Does Not Affect Outcome Adversely, Provided it is Discontinued at Least 90 Days Before the Procedure. Blood (1999) 94(11):3668–77. doi: 10.1182/blood.V94.11.3668
122. Robb RJ, Kreijveld E, Kuns RD, Wilson YA, Olver SD, Don AL, et al. Type I-IFNs Control GVHD and GVL Responses After Transplantation. Blood (2011) 118(12):3399–409. doi: 10.1182/blood-2010-12-325746
123. Fischer JC, Bscheider M, Eisenkolb G, Lin CC, Wintges A, Otten V, et al. Rig-I/MAVS and STING Signaling Promote Gut Integrity During Irradiation- and Immune-Mediated Tissue Injury. Sci Transl Med (2017) 9(386). doi: 10.1126/scitranslmed.aag2513
124. Swimm A, Giver CR, DeFilipp Z, Rangaraju S, Sharma A, Ulezko Antonova A, et al. Indoles Derived From Intestinal Microbiota Act Via Type I Interferon Signaling to Limit Graft-Versus-Host Disease. Blood (2018) 132(23):2506–19. doi: 10.1182/blood-2018-03-838193
125. Fischer JC, Bscheider M, Gottert S, Thiele Orberg E, Combs SE, Bassermann F, et al. Type I Interferon Signaling Before Hematopoietic Stem Cell Transplantation Lowers Donor T Cell Activation Via Reduced Allogenicity of Recipient Cells. Sci Rep (2019) 9(1):14955. doi: 10.1038/s41598-019-51431-2
126. Pott J, Stockinger S. Type I and III Interferon in the Gut: Tight Balance Between Host Protection and Immunopathology. Front Immunol (2017) 8:258. doi: 10.3389/fimmu.2017.00258
127. Burman AC, Banovic T, Kuns RD, Clouston AD, Stanley AC, Morris ES, et al. Ifngamma Differentially Controls the Development of Idiopathic Pneumonia Syndrome and GVHD of the Gastrointestinal Tract. Blood (2007) 110(3):1064–72. doi: 10.1182/blood-2006-12-063982
128. Krenger W, Ferrara JL. Graft-Versus-Host Disease and the Th1/Th2 Paradigm. Immunol Res (1996) 15(1):50–73. doi: 10.1007/BF02918284
129. Ellison CA, Fischer JM, HayGlass KT, Gartner JG. Murine Graft-Versus-Host Disease in an F1-hybrid Model Using IFN-gamma Gene Knockout Donors. J Immunol (1998) 161(2):631–40.
130. Ellison CA, Natuik SA, McIntosh AR, Scully SA, Danilenko DM, Gartner JG. The Role of Interferon-Gamma, Nitric Oxide and Lipopolysaccharide in Intestinal Graft-Versus-Host Disease Developing in F1-hybrid Mice. Immunology (2003) 109(3):440–9. doi: 10.1046/j.1365-2567.2003.01663.x
131. Eriguchi Y, Nakamura K, Yokoi Y, Sugimoto R, Takahashi S, Hashimoto D, et al. Essential Role of IFN-gamma in T Cell-Associated Intestinal Inflammation. JCI Insight (2018) 3(18). doi: 10.1172/jci.insight.121886
132. Takashima S, Martin ML, Jansen SA, Fu Y, Bos J, Chandra D, et al. T Cell-Derived Interferon-Gamma Programs Stem Cell Death in Immune-Mediated Intestinal Damage. Sci Immunol (2019) 4(42). doi: 10.1126/sciimmunol.aay8556
133. Mowat AM. Antibodies to IFN-gamma Prevent Immunologically Mediated Intestinal Damage in Murine Graft-Versus-Host Reaction. Immunology (1989) 68(1):18–23.
134. Yang YG, Qi J, Wang MG, Sykes M. Donor-Derived Interferon Gamma Separates Graft-Versus-Leukemia Effects and Graft-Versus-Host Disease Induced by Donor CD8 T Cells. Blood (2002) 99(11):4207–15. doi: 10.1182/blood.V99.11.4207
135. Asavaroengchai W, Wang H, Wang S, Wang L, Bronson R, Sykes M, et al. An Essential Role for IFN-gamma in Regulation of Alloreactive CD8 T Cells Following Allogeneic Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant (2007) 13(1):46–55. doi: 10.1016/j.bbmt.2006.09.014
136. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 Cd4+ T Cells Through IL-12 Produced by Listeria-induced Macrophages. Science (1993) 260(5107):547–9. doi: 10.1126/science.8097338
137. Zundler S, Neurath MF. Interleukin-12: Functional Activities and Implications for Disease. Cytokine Growth Factor Rev (2015) 26(5):559–68. doi: 10.1016/j.cytogfr.2015.07.003
138. Sykes M, Szot GL, Nguyen PL, Pearson DA. Interleukin-12 Inhibits Murine Graft-Versus-Host Disease. Blood (1995) 86(6):2429–38. doi: 10.1182/blood.V86.6.2429.bloodjournal8662429
139. Dey BR, Yang YG, Szot GL, Pearson DA, Sykes M. Interleukin-12 Inhibits Graft-Versus-Host Disease Through an Fas-mediated Mechanism Associated With Alterations in Donor T-cell Activation and Expansion. Blood (1998) 91(9):3315–22. doi: 10.1182/blood.V91.9.3315
140. Yang YG, Sykes M. The Role of interleukin-12 in Preserving the Graft-Versus-Leukemia Effect of Allogeneic CD8 T Cells Independently of GVHD. Leuk Lymphoma (1999) 33(5-6):409–20. doi: 10.3109/10428199909058446
141. Sykes M, Pearson DA, Taylor PA, Szot GL, Goldman SJ, Blazar BR. Dose and Timing of Interleukin (IL)-12 and Timing and Type of Total-Body Irradiation: Effects on Graft-vs.-Host Disease Inhibition and Toxicity of Exogenous IL-12 in Murine Bone Marrow Transplant Recipients. Biol Blood Marrow Transplant (1999) 5(5):277–84. doi: 10.1016/S1083-8791(99)70002-9
142. Wang H, Yang YG. The Complex and Central Role of Interferon-Gamma in Graft-Versus-Host Disease and Graft-Versus-Tumor Activity. Immunol Rev (2014) 258(1):30–44. doi: 10.1111/imr.12151
143. Welniak LA, Blazar BR, Anver MR, Wiltrout RH, Murphy WJ. Opposing Roles of Interferon-Gamma on CD4+ T Cell-Mediated Graft-Versus-Host Disease: Effects of Conditioning. Biol Blood Marrow Transplant (2000) 6(6):604–12. doi: 10.1016/S1083-8791(00)70025-5
144. Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, et al. Ifngammar Signaling Mediates Alloreactive T-cell Trafficking and GVHD. Blood (2012) 120(19):4093–103. doi: 10.1182/blood-2012-01-403196
145. Fischer JC, Lin CC, Heidegger S, Wintges A, Schlapschy M, Beudert M, et al. Regeneration After Radiation- and Immune-Mediated Tissue Injury is Not Enhanced by Type Iii Interferon Signaling. Int J Radiat Oncol Biol Phys (2019) 103(4):970–6. doi: 10.1016/j.ijrobp.2018.11.038
146. Gadalla SM, Wang Y, Wang T, Onabajo OO, Banday AR, Obajemu A, et al. Association of Donor IFNL4 Genotype and non-Relapse Mortality After Unrelated Donor Myeloablative Haematopoietic Stem-Cell Transplantation for Acute Leukaemia: A Retrospective Cohort Study. Lancet Haematol (2020) 7(10):e715–23. doi: 10.1016/S2352-3026(20)30294-5
147. Choi J, Cooper ML, Alahmari B, Ritchey J, Collins L, Holt M, et al. Pharmacologic Blockade of JAK1/JAK2 Reduces GvHD and Preserves the Graft-Versus-Leukemia Effect. PloS One (2014) 9(10):e109799. doi: 10.1371/journal.pone.0109799
148. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of Therapeutic JAK 1/2 Blockade in Graft-Versus-Host Disease. Blood (2014) 123(24):3832–42. doi: 10.1182/blood-2013-12-543736
149. Carniti C, Gimondi S, Vendramin A, Recordati C, Confalonieri D, Bermema A, et al. Pharmacologic Inhibition of JAK1/JAK2 Signaling Reduces Experimental Murine Acute GVHD While Preserving Gvt Effects. Clin Cancer Res (2015) 21(16):3740–9. doi: 10.1158/1078-0432.CCR-14-2758
150. Jagasia M, Perales MA, Schroeder MA, Ali H, Shah NN, Chen YB, et al. Ruxolitinib for the Treatment of Steroid-Refractory Acute GVHD (REACH1): A Multicenter, Open-Label Phase 2 Trial. Blood (2020) 135(20):1739–49. doi: 10.1182/blood.2020004823
151. Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N Engl J Med (2020) 382(19):1800–10. doi: 10.1056/NEJMoa1917635
152. Ryan JM, Barry F, Murphy JM, Mahon BP. Interferon-Gamma Does Not Break, But Promotes the Immunosuppressive Capacity of Adult Human Mesenchymal Stem Cells. Clin Exp Immunol (2007) 149(2):353–63. doi: 10.1111/j.1365-2249.2007.03422.x
153. Polchert D, Sobinsky J, Douglas G, Kidd M, Moadsiri A, Reina E, et al. IFN-Gamma Activation of Mesenchymal Stem Cells for Treatment and Prevention of Graft Versus Host Disease. Eur J Immunol (2008) 38(6):1745–55. doi: 10.1002/eji.200738129
154. Ringden O, Uzunel M, Rasmusson I, Remberger M, Sundberg B, Lonnies H, et al. Mesenchymal Stem Cells for Treatment of Therapy-Resistant Graft-Versus-Host Disease. Transplantation (2006) 81(10):1390–7. doi: 10.1097/01.tp.0000214462.63943.14
155. Sekiya I, Larson BL, Smith JR, Pochampally R, Cui JG, Prockop DJ. Expansion of Human Adult Stem Cells From Bone Marrow Stroma: Conditions That Maximize the Yields of Early Progenitors and Evaluate Their Quality. Stem Cells (2002) 20(6):530–41. doi: 10.1634/stemcells.20-6-530
156. Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M. Characterization of the Optimal Culture Conditions for Clinical Scale Production of Human Mesenchymal Stem Cells. Stem Cells (2006) 24(2):462–71. doi: 10.1634/stemcells.2004-0331
157. Phinney DG. Functional Heterogeneity of Mesenchymal Stem Cells: Implications for Cell Therapy. J Cell Biochem (2012) 113(9):2806–12. doi: 10.1002/jcb.24166
158. Phinney DG. Biochemical Heterogeneity of Mesenchymal Stem Cell Populations: Clues to Their Therapeutic Efficacy. Cell Cycle (2007) 6(23):2884–9. doi: 10.4161/cc.6.23.5095
159. Kim DS, Jang IK, Lee MW, Ko YJ, Lee DH, Lee JW, et al. Enhanced Immunosuppressive Properties of Human Mesenchymal Stem Cells Primed by Interferon-Gamma. EBioMedicine (2018) 28:261–73. doi: 10.1016/j.ebiom.2018.01.002
160. Liu TC, Stappenbeck TS. Genetics and Pathogenesis of Inflammatory Bowel Disease. Annu Rev Pathol (2016) 11:127–48. doi: 10.1146/annurev-pathol-012615-044152
161. Russel MG, Dorant E, Brummer RJ, van de Kruijs MA, Muris JW, Bergers JM, et al. Appendectomy and the Risk of Developing Ulcerative Colitis or Crohn’s Disease: Results of a Large Case-Control Study. South Limburg Inflammatory Bowel Disease Study Group. Gastroenterology (1997) 113(2):377–82. doi: 10.1053/gast.1997.v113.pm9247453
162. Derby LE, Jick H. Appendectomy Protects Against Ulcerative Colitis. Epidemiology (1998) 9(2):205–7. doi: 10.1097/00001648-199803000-00018
163. Duggan AE, Usmani I, Neal KR, Logan RF. Appendicectomy, Childhood Hygiene, Helicobacter Pylori Status, and Risk of Inflammatory Bowel Disease: A Case Control Study. Gut (1998) 43(4):494–8. doi: 10.1136/gut.43.4.494
164. Andersson RE, Olaison G, Tysk C, Ekbom A. Appendectomy and Protection Against Ulcerative Colitis. N Engl J Med (2001) 344(11):808–14. doi: 10.1056/NEJM200103153441104
165. Koutroubakis IE, Vlachonikolis IG, Kouroumalis EA. Role of Appendicitis and Appendectomy in the Pathogenesis of Ulcerative Colitis: A Critical Review. Inflamm Bowel Dis (2002) 8(4):277–86. doi: 10.1097/00054725-200207000-00007
166. Radford-Smith GL, Edwards JE, Purdie DM, Pandeya N, Watson M, Martin NG, et al. Protective Role of Appendicectomy on Onset and Severity of Ulcerative Colitis and Crohn’s Disease. Gut (2002) 51(6):808–13. doi: 10.1136/gut.51.6.808
167. Guo J, Peters KL, Sen GC. Induction of the Human Protein P56 by Interferon, Double-Stranded RNA, or Virus Infection. Virology (2000) 267(2):209–19. doi: 10.1006/viro.1999.0135
168. Fensterl V, Sen GC. The ISG56/IFIT1 Gene Family. J Interferon Cytokine Res (2011) 31(1):71–8. doi: 10.1089/jir.2010.0101
169. Reich NC. A Death-Promoting Role for ISG54/IFIT2. J Interferon Cytokine Res (2013) 33(4):199–205. doi: 10.1089/jir.2012.0159
170. Cheluvappa R. Identification of New Potential Therapies for Colitis Amelioration Using an Appendicitis-Appendectomy Model. Inflammation Bowel Dis (2019) 25(3):436–44. doi: 10.1093/ibd/izy332
171. Sainathan SK, Bishnupuri KS, Aden K, Luo Q, Houchen CW, Anant S, et al. Toll-Like Receptor-7 Ligand Imiquimod Induces Type I Interferon and Antimicrobial Peptides to Ameliorate Dextran Sodium Sulfate-Induced Acute Colitis. Inflamm Bowel Dis (2012) 18(5):955–67. doi: 10.1002/ibd.21867
172. Rauch I, Hainzl E, Rosebrock F, Heider S, Schwab C, Berry D, et al. Type I Interferons Have Opposing Effects During the Emergence and Recovery Phases of Colitis. Eur J Immunol (2014) 44(9):2749–60. doi: 10.1002/eji.201344401
173. Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-Like Receptor 9-Induced Type I IFN Protects Mice From Experimental Colitis. J Clin Invest (2005) 115(3):695–702. doi: 10.1172/JCI22996
174. Abe K, Nguyen KP, Fine SD, Mo JH, Shen C, Shenouda S, et al. Conventional Dendritic Cells Regulate the Outcome of Colonic Inflammation Independently of T Cells. Proc Natl Acad Sci USA (2007) 104(43):17022–7. doi: 10.1073/pnas.0708469104
175. Lee SE, Li X, Kim JC, Lee J, Gonzalez-Navajas JM, Hong SH, et al. Type I Interferons Maintain Foxp3 Expression and T-regulatory Cell Functions Under Inflammatory Conditions in Mice. Gastroenterology (2012) 143(1):145–54. doi: 10.1053/j.gastro.2012.03.042
176. Kole A, He J, Rivollier A, Silveira DD, Kitamura K, Maloy KJ, et al. Type I Ifns Regulate Effector and Regulatory T Cell Accumulation and Anti-Inflammatory Cytokine Production During T Cell-Mediated Colitis. J Immunol (2013) 191(5):2771–9. doi: 10.4049/jimmunol.1301093
177. McFarland AP, Savan R, Wagage S, Addison A, Ramakrishnan K, Karwan M, et al. Localized Delivery of Interferon-Beta by Lactobacillus Exacerbates Experimental Colitis. PloS One (2011) 6(2):e16967. doi: 10.1371/journal.pone.0016967
178. Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, et al. Interferon-Gamma is Causatively Involved in Experimental Inflammatory Bowel Disease in Mice. Clin Exp Immunol (2006) 146(2):330–8. doi: 10.1111/j.1365-2249.2006.03214.x
179. Haep L, Britzen-Laurent N, Weber TG, Naschberger E, Schaefer A, Kremmer E, et al. Interferon Gamma Counteracts the Angiogenic Switch and Induces Vascular Permeability in Dextran Sulfate Sodium Colitis in Mice. Inflamm Bowel Dis (2015) 21(10):2360–71. doi: 10.1097/MIB.0000000000000490
180. Singh UP, Singh NP, Murphy EA, Price RL, Fayad R, Nagarkatti M, et al. Chemokine and Cytokine Levels in Inflammatory Bowel Disease Patients. Cytokine (2016) 77:44–9. doi: 10.1016/j.cyto.2015.10.008
181. Guenzi E, Topolt K, Cornali E, Lubeseder-Martellato C, Jorg A, Matzen K, et al. The Helical Domain of GBP-1 Mediates the Inhibition of Endothelial Cell Proliferation by Inflammatory Cytokines. EMBO J (2001) 20(20):5568–77. doi: 10.1093/emboj/20.20.5568
182. Guenzi E, Topolt K, Lubeseder-Martellato C, Jorg A, Naschberger E, Benelli R, et al. The Guanylate Binding Protein-1 GTPase Controls the Invasive and Angiogenic Capability of Endothelial Cells Through Inhibition of MMP-1 Expression. EMBO J (2003) 22(15):3772–82. doi: 10.1093/emboj/cdg382
183. Weinlander K, Naschberger E, Lehmann MH, Tripal P, Paster W, Stockinger H, et al. Guanylate Binding Protein-1 Inhibits Spreading and Migration of Endothelial Cells Through Induction of Integrin Alpha4 Expression. FASEB J (2008) 22(12):4168–78. doi: 10.1096/fj.08-107524
184. Naschberger E, Croner RS, Merkel S, Dimmler A, Tripal P, Amann KU, et al. Angiostatic Immune Reaction in Colorectal Carcinoma: Impact on Survival and Perspectives for Antiangiogenic Therapy. Int J Cancer (2008) 123(9):2120–9. doi: 10.1002/ijc.23764
185. Mennechet FJ, Uze G. Interferon-Lambda-Treated Dendritic Cells Specifically Induce Proliferation of FOXP3-expressing Suppressor T Cells. Blood (2006) 107(11):4417–23. doi: 10.1182/blood-2005-10-4129
186. Gunther C, Ruder B, Stolzer I, Dorner H, He GW, Chiriac MT, et al. Interferon Lambda Promotes Paneth Cell Death Via STAT1 Signaling in Mice and Is Increased in Inflamed Ileal Tissues of Patients With Crohn’s Disease. Gastroenterology (2019) 157(5):1310–22 e13. doi: 10.1053/j.gastro.2019.07.031
187. Danese S, Angelucci E, Malesci A, Caprilli R. Biological Agents for Ulcerative Colitis: Hypes and Hopes. Med Res Rev (2008) 28(2):201–18. doi: 10.1002/med.20103
188. Wang Y, MacDonald JK, Benchimol EI, Griffiths AM, Steinhart AH, Panaccione R, et al. Type I Interferons for Induction of Remission in Ulcerative Colitis. Cochrane Database Syst Rev (2015) 2015(9):CD006790. doi: 10.1002/14651858.CD006790.pub3
189. Reinisch W, de Villiers W, Bene L, Simon L, Racz I, Katz S, et al. Fontolizumab in Moderate to Severe Crohn’s Disease: A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Multiple-Dose Study. Inflamm Bowel Dis (2010) 16(2):233–42. doi: 10.1002/ibd.21038
190. Sandborn WJ, Colombel JF, Ghosh S, Sands BE, Dryden G, Hebuterne X, et al. Eldelumab [Anti-Ip-10] Induction Therapy for Ulcerative Colitis: A Randomised, Placebo-Controlled, Phase 2b Study. J Crohns Colitis (2016) 10(4):418–28. doi: 10.1093/ecco-jcc/jjv224
191. Kaser A. Not All Monoclonals are Created Equal - Lessons From Failed Drug Trials in Crohn’s Disease. Best Pract Res Clin Gastroenterol (2014) 28(3):437–49. doi: 10.1016/j.bpg.2014.04.005
192. Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, et al. Tofacitinib, an Oral Janus Kinase Inhibitor, in Active Ulcerative Colitis. N Engl J Med (2012) 367(7):616–24. doi: 10.1056/NEJMoa1112168
193. Sandborn WJ, Ghosh S, Panes J, Vranic I, Wang W, Niezychowski W, et al. A Phase 2 Study of Tofacitinib, an Oral Janus Kinase Inhibitor, in Patients With Crohn’s Disease. Clin Gastroenterol Hepatol (2014) 12(9):1485–93.e2. doi: 10.1016/j.cgh.2014.01.029
194. Vermeire S, Schreiber S, Petryka R, Kuehbacher T, Hebuterne X, Roblin X, et al. Clinical Remission in Patients With Moderate-to-Severe Crohn’s Disease Treated With Filgotinib (the FITZROY Study): Results From a Phase 2, Double-Blind, Randomised, Placebo-Controlled Trial. Lancet (2017) 389(10066):266–75. doi: 10.1016/S0140-6736(16)32537-5
195. Novick D, Cohen B, Rubinstein M. The Human Interferon Alpha/Beta Receptor: Characterization and Molecular Cloning. Cell (1994) 77(3):391–400. doi: 10.1016/0092-8674(94)90154-6
196. Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, et al. Interferon-Lambda Mediates non-Redundant Front-Line Antiviral Protection Against Influenza Virus Infection Without Compromising Host Fitness. Immunity (2017) 46(5):875–90.e6. doi: 10.1016/j.immuni.2017.04.025
197. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
198. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic Sensing of Viruses. Immunity (2013) 38(5):855–69. doi: 10.1016/j.immuni.2013.05.007
199. Paludan SR, Bowie AG. Immune Sensing of DNA. Immunity (2013) 38(5):870–80. doi: 10.1016/j.immuni.2013.05.004
200. Syedbasha M, Egli A. Interferon Lambda: Modulating Immunity in Infectious Diseases. Front Immunol (2017) 8:119. doi: 10.3389/fimmu.2017.00119
201. van Dullemen HM, van Deventer SJ, Hommes DW, Bijl HA, Jansen J, Tytgat GN, et al. Treatment of Crohn’s Disease With Anti-Tumor Necrosis Factor Chimeric Monoclonal Antibody (Ca2). Gastroenterology (1995) 109(1):129–35. doi: 10.1016/0016-5085(95)90277-5
202. Ganetsky A, Frey NV, Hexner EO, Loren AW, Gill SI, Luger SM, et al. Tocilizumab for the Treatment of Severe Steroid-Refractory Acute Graft-Versus-Host Disease of the Lower Gastrointestinal Tract. Bone Marrow Transplant (2019) 54(2):212–7. doi: 10.1038/s41409-018-0236-z
203. Kattner AS, Holler E, Holler B, Klobuch S, Weber D, Martinovic D, et al. IL6-Receptor Antibody Tocilizumab as Salvage Therapy in Severe Chronic Graft-Versus-Host Disease After Allogeneic Hematopoietic Stem Cell Transplantation: A Retrospective Analysis. Ann Hematol (2020) 99(4):847–53. doi: 10.1007/s00277-020-03968-w
Keywords: graft-versus-host disease, inflammatory bowel disease, interferon, intestine, ulcerative colitis, Crohn’s disease
Citation: Haring E, Zeiser R and Apostolova P (2021) Interfering With Inflammation: Heterogeneous Effects of Interferons in Graft-Versus-Host Disease of the Gastrointestinal Tract and Inflammatory Bowel Disease. Front. Immunol. 12:705342. doi: 10.3389/fimmu.2021.705342
Received: 05 May 2021; Accepted: 10 June 2021;
Published: 24 June 2021.
Edited by:
Jaebok Choi, Washington University School of Medicine in St. Louis, United StatesReviewed by:
Senthilnathan Palaniyandi, University of Kentucky, United StatesCopyright © 2021 Haring, Zeiser and Apostolova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petya Apostolova, cGV0eWEuYXBvc3RvbG92YUB1bmlrbGluaWstZnJlaWJ1cmcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.