
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 30 July 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.705219
This article is part of the Research TopicImmune Regulations in Reproductive Organs and Organ TransplantView all 34 articles
 Maryam Shahi1†
Maryam Shahi1† Ricardo Mamber Czeresnia2†
Ricardo Mamber Czeresnia2† E. Heidi Cheek1Reade A. Quinton1Rana Chakraborty3,4
E. Heidi Cheek1Reade A. Quinton1Rana Chakraborty3,4 Elizabeth Ann L. Enninga2,3*
Elizabeth Ann L. Enninga2,3*Pregnancy is an immunological paradox whereby maternal immunity accepts a genetically unique fetus (or fetuses), while maintaining protective innate and adaptive responses to infectious pathogens. This close contact between the genetically diverse mother and fetus requires numerous mechanisms of immune tolerance initiated by trophoblast cell signals. However, in a placental condition known as villitis of unknown etiology (VUE), there appears to be a breakdown in this tolerance allowing maternal cytotoxic T-cells to traffic into the placenta to destroy fetal villi. VUE is associated with several gestational complications and an increased risk of recurrence in a subsequent pregnancy, making it a significant obstetrical diagnosis. The cause of VUE remains unclear, but dysfunctional signaling through immune checkpoint pathways, which have a critical role in blunting immune responses, may play an important role. Therefore, using placental tissue from normal pregnancy (n=8), VUE (n=8) and cytomegalovirus (CMV) infected placentae (n=4), we aimed to identify differences in programmed cell death 1 (PD-1), programmed death ligand-1 (PD-L1), LAG3 and CTLA4 expression between these etiologies by immunohistochemistry (IHC). Results demonstrated significantly lower expression of PD-L1 on trophoblast cells from VUE placentae compared to control and CMV infection. Additionally, we observed significantly higher counts of PD-1+ (>100 cells/image) and LAG3+ (0-120 cells/image) cells infiltrating into the villi during VUE compared to infection and control. Minimal CTLA4 staining was observed in all placentae, with only a few Hofbauer cells staining positive. Together, this suggests that a loss of tolerance through immune checkpoint signaling may be an important mechanism leading to the activation and trafficking of maternal cells into fetal villi during VUE. Further mechanistic studies are warranted to understand possible allograft rejection more clearly and in developing effective strategies to prevent this condition from occurring in utero.
Chronic villitis is a placental condition characterized by inflammation and lymphohistiocytic infiltration into the chorionic villi with or without necrosis, and is categorized into either infectious villitis or non-infectious villitis, also known as villitis of unknown etiology (VUE) (1). While villitis due to infectious etiologies are rare (1-4/1000 pregnancies), 6.6-33.8% of pregnancies are diagnosed with VUE following a term delivery (2–4). Infectious villitis is secondary to transplacental dissemination of microorganisms such as cytomegalovirus (CMV), Treponema pallidum, Toxoplasma gondii and Herpes simplex viruses (HSV) (5). On the other hand, the diagnosis of VUE requires exclusion of infectious etiologies and is therefore hypothesized to be an anti-fetal allograft response (6, 7). The cellular composition of VUE predominantly includes T-cells and macrophages with minor B-cell involvement (8, 9). Immunohistochemical staining and in situ hybridization demonstrate maternal origin of T-cells and both maternal and fetal origin of macrophages, further supporting a breakdown in maternal-fetal tolerance (10, 11).
Pregnancy can be likened to an allograft transplant where the fetus contains a mixture of maternal and paternal genetic material; yet maternal immune responses to the haploidentical fetus during gestation are well-regulated to support the growth and development of offspring. Therefore, the diagnosis of VUE may represent a failure of tolerance and a resulting allograft rejection response to the genetically unique fetus. This hypothesis is supported clinically in that VUE is increased in patients with autoimmune diseases such as systematic lupus erythematosus and thyroid-related disease (12, 13). Neonatal alloimmune thrombocytopenia is also associated with a VUE diagnosis (14). In addition, a higher rate of VUE has been observed in donor oocyte in vitro fertilization (IVF) pregnancies, compared to native oocyte IVF, suggesting a higher propensity of immunologic adverse effects in an entirely foreign embryo (15, 16). More recently, pregnancies resulting from IVF had a higher prevalence of VUE diagnoses compared to spontaneously conceived pregnancies (16.2% vs. 8.3%) (17). Lastly, VUE recurrence in a subsequent pregnancy is between 10-37%, which suggests that this etiology may lead to the development of a maternal anamnestic memory response, reactivated when re-exposed to the same fetal antigens (5, 18).
The instigator(s) of VUE still remain unknown. A unique feature of the placenta is the expression of non-classical major histocompatibility class I (MHCI) surface receptors, like human leukocyte antigen (HLA)-G, which is uniquely expressed on trophoblasts. These cells come in direct contact with maternal blood, and yet do not elicit immune recognition as antibodies directed at HLA-G are not detected in multigravid women (19, 20). Instead, high expression of HLA-G leads to short term tolerance by binding ILT2 and KIR receptors on numerous immune cells, and the release of soluble isoforms into the plasma throughout pregnancy (21, 22). We note that MHC I/II molecules are not expressed in normal term placentae (with the exception of HLA-C), but can be detected at high levels in placentae diagnosed with VUE (23, 24). Upregulation of MHC I/II on the trophoblast surface of placentae with VUE could promote fetal antigen presentation facilitated by activated macrophages and maternal T-cells (13). Cytokines and chemokines also play a critical role in maternal-fetal tolerance and could have an important role in maintenance of tolerance or establishment of placental inflammation. Chemokines CXCL9, CXCL10 and CXCL11 have been detected in maternal blood and placentae of cases diagnosed with VUE (23, 25). Other mechanisms to maintain tolerance include clonal deletion, FAS/FAS L interactions, indoleamine2,3-dioxygenase (IDO) expression and the presence of T-regulatory cells (26, 27).
Other mechanisms of immune tolerance, such as checkpoint inhibitors, have not been investigated in the setting of VUE. Immune checkpoint inhibitors on T-cells bind to their respective ligands on antigen presenting cells and abrogate immune responses, thereby preventing over-activation of immunity and the development of autoimmune disease. In the past 10 years, the critical role of the PD-1/PD-L1 interaction in tumor immune tolerance has been established, and effective treatments targeted at disrupting this interaction to reactivate an exhausted immune system against the tumor have been developed. PD-1/PD-L1 promotes immune evasion by tumor cells and may be pivotal in maternal-fetal tolerance (28, 29). PD-L1 is expressed by trophoblast cells and circulates in maternal blood during pregnancy (30, 31). Other immune checkpoint molecules include the lymphocyte activation gene protein 3 (LAG3) and cytotoxic T lymphocyte antigen-4 (CTLA4). LAG3 has been described to synergistically act with PD-1/PD-L1 to dampen immune responses by suppressing cytotoxic T-cell signaling (32). Similarly, CTLA4 competes with immune stimulating CD28 on T-cells to bind CD80 and CD86 on antigen presenting cells, inhibiting the proliferation of T-cells and the release of pro-inflammatory IL-2 (33). These semi-redundant pathways have evolved to provide multiple levels of protection against unwarranted immune responses and may have an important role in the loss of fetal-maternal tolerance that likely occurs during VUE.
Therefore, the goal of this study was to evaluate the expression of PD-1, PD-L1, LAG3 and CTLA4 in placentae diagnosed with VUE and compare it to expression in infectious villitis and normal controls. We hypothesized that the expression and abundance of these targets will significantly differ from infectious villitis and controls because of dysregulation of immune tolerance during VUE. To test this hypothesis, tissues from paraffin blocks were cut and immunohistochemistry completed to identity differences in these immune checkpoint molecules.
The Mayo Clinic Institutional Review Board approved this study (#16-006099). The aim of the study was to compare differences in checkpoint inhibitor expression in placentae with VUE, cytomegalovirus (CMV) infection, and controls by immunohistochemistry (IHC). Lymphocytes were identified by H&E and hemotoxylin-counterstained IHC slides based on their cytomorphologic features, including round small nuclei with dense chromatin pattern, inconspicuous nucleoli, and small amount of cytoplasm. Following histologic review by a pathologist, residual formalin fixed paraffin embedded placental tissues were identified. We chose 8 placentae with a diagnosis of high grade VUE per definition by Khong et al. (34) (presence of multiple foci of villitis, on more than one section, at least one of which shows inflammation affecting more than 10 contiguous villi), 4 placentae diagnosed with placental infection by cytomegalovirus (CMV) and 8 gestational age-matched controls to the VUE cases. The control cases had no identifiable pathology.
Five-micron sections were cut, and tissues underwent deparaffinization by xylene followed by antigen retrieval using citrate buffer. Tissues were then stained with the following primary antibodies overnight at 4°C: PD1 (clone NAT105, 1:200), PDL1 (clone SP142, 1:400), CTLA-4 (clone F-8, 1:50) and LAG3 (clone 11-E3, 1:200). Following secondary antibody incubation for 1 hour at room temp, slides were treated with 3,3′-Diaminobenzidine (DAB, Sigma-Aldrich) for 15 minutes followed by hemoxylin. Images were captured using a CellSens Standard (Olympus Corporation Tokyo, Japan) on a BX51 Olympus microscope (Olympus Corporation Tokyo, Japan).
Measurements of PDL-1 membrane thickness and DAB intensity were made using ImageJ Fiji, following the protocol described by Crowe et al. (35). Ten representative images from each case were gathered, blindly analyzed and compared. For PD-L1 thickness, five separate measurements of villi from each representative image were collected and averaged. Thus, for every case, the mean membrane PDL-1 deposition was obtained by fifty distinct measurements. To calculate DAB intensity, five images from each case measured at a threshold set at 131 for each case. To quantify PD-1, CTLA-4 and LAG3, the slides were reviewed by a pathologist. In scanning power (10X), the hotspot areas were identified and positive cells in 20 villi were counted in high power (400X). If ≤20 villi in the entire section had positive cells, all the positive cells in that sections were counted.
Patient demographics are reported as medians with ranges. Results from our three groups were analyzed using Kruskal-Wallis testing controlling for the false discovery rate (FDR) with the method of Benjamini, Krieger and Yekutieli. Significance was defined as a p-value ≤ 0.05. All statistical analyses and graphing were performed using GraphPad Prism software version 9.1 (GraphPad Software, San Diego, CA).
Twenty placental specimens were included in this study, 8 with a VUE diagnosis, 4 with a CMV diagnosis and 8 identified to be normal following pathological review. Demographics can be seen in Table 1, displayed as medians and ranges. There were no significant differences in our groups based on maternal age (average maternal age of 31 years in our cohort). The number of prior pregnancies was 2.5, which was similar between groups. There was a significant difference in gestational age at birth, with the CMV infected cohort having earlier births compared to controls and VUE cases. The median gestational age at birth in the VUE and control groups was also the similar. All mothers had an uncomplicated postpartum course and the infant APGARS at 1 and 5 minutes were equivalent (p=0.20 and p=0.59, respectively).
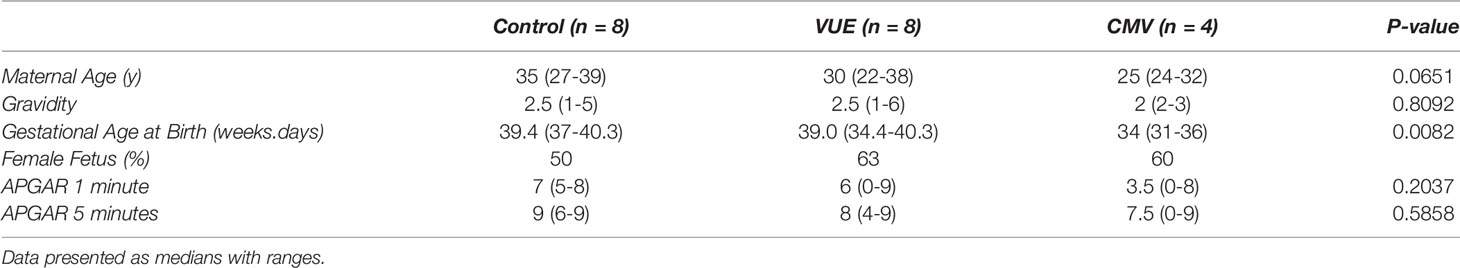
Table 1 Basic cohort demographics.
To address the potential role of PD-L1 expression during infectious and non-infectious etiologies, we ran IHC and took 10 representative images from each case for analysis using ImageJ. There were observable differences between the groups by microscopy (Figure 1A). As others have reported (36), PD-L1 staining was mainly localized to syncytiotrophoblasts, with moderate staining on intermediate trophoblasts and minimal expression on cytotrophoblast cells. We therefore quantified the DAB signal and found that the VUE cases had significantly lower PD-L1 staining intensity compared to control (p=0.009) and CMV placentae (p=0.0008; Figure 1B). Although we saw some evidence of increased PD-L1 expression in the CMV group compared to controls, differences between groups did not meet statistical significance (p=0.052). We then measured the thickness of staining across the membrane and note that CMV infection led to the most profuse villous staining of PD-L1 compared to controls (p=0.034) while the thickness was significantly decreased in placentae diagnosed with VUE compared to both control (p=0.004) and CMV (p=0.0001; Figure 1C). These data indicate that PD-L1 expression is decreased in placentae with a VUE diagnosis and increased during infection with CMV.
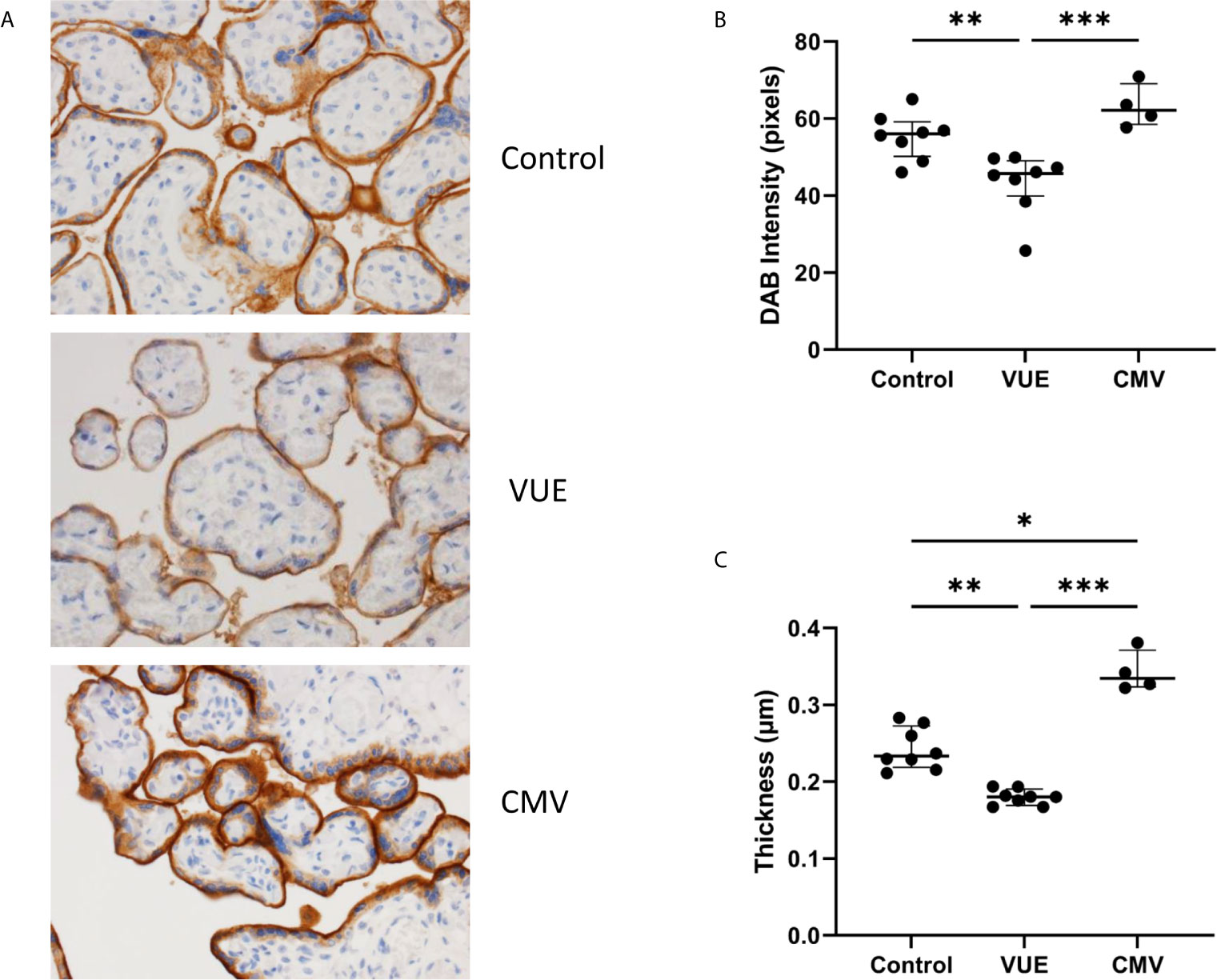
Figure 1 PD-L1 expression in placentae with infectious and non-infectious diagnoses. (A) Representative PD-L1 staining in all three groups (200X). (B) Average intensity of DAB staining between control, VUE and CMV groups. (C) Mean PD-L1 membrane thickness between groups. Graphs show median with interquartile ranges. Data was compared by Kruskal-Wallis testing with post hoc analysis using the false discovery rate method of Benjamini, Krieger and Yekutieli (n=4-8/group). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.0001.
We then quantified PD-1, the receptor to PD-L1, which is expressed on immune cells. Following IHC staining, 10 representative images from each case were captured and counted. Unsurprisingly, there were almost no PD-1+ cells that could be identified in the control placentae (Figure 2A). We then counted the number of PD-1+ cells in VUE and CMV. VUE placentae showed the highest number of PD-1+ cells (average 93 cells/image) compared to CMV (73.5 cells/image), which was not a significant difference (p=0.26; Figure 2B). Compared to control, non-infectious chronic villitis (p=0.0004) and CMV (p=0.005) had significantly more PD-1+ cells in each image. Thus, PD-1+ cells are most abundant in VUE and CMV infection compared to in normal control tissue.
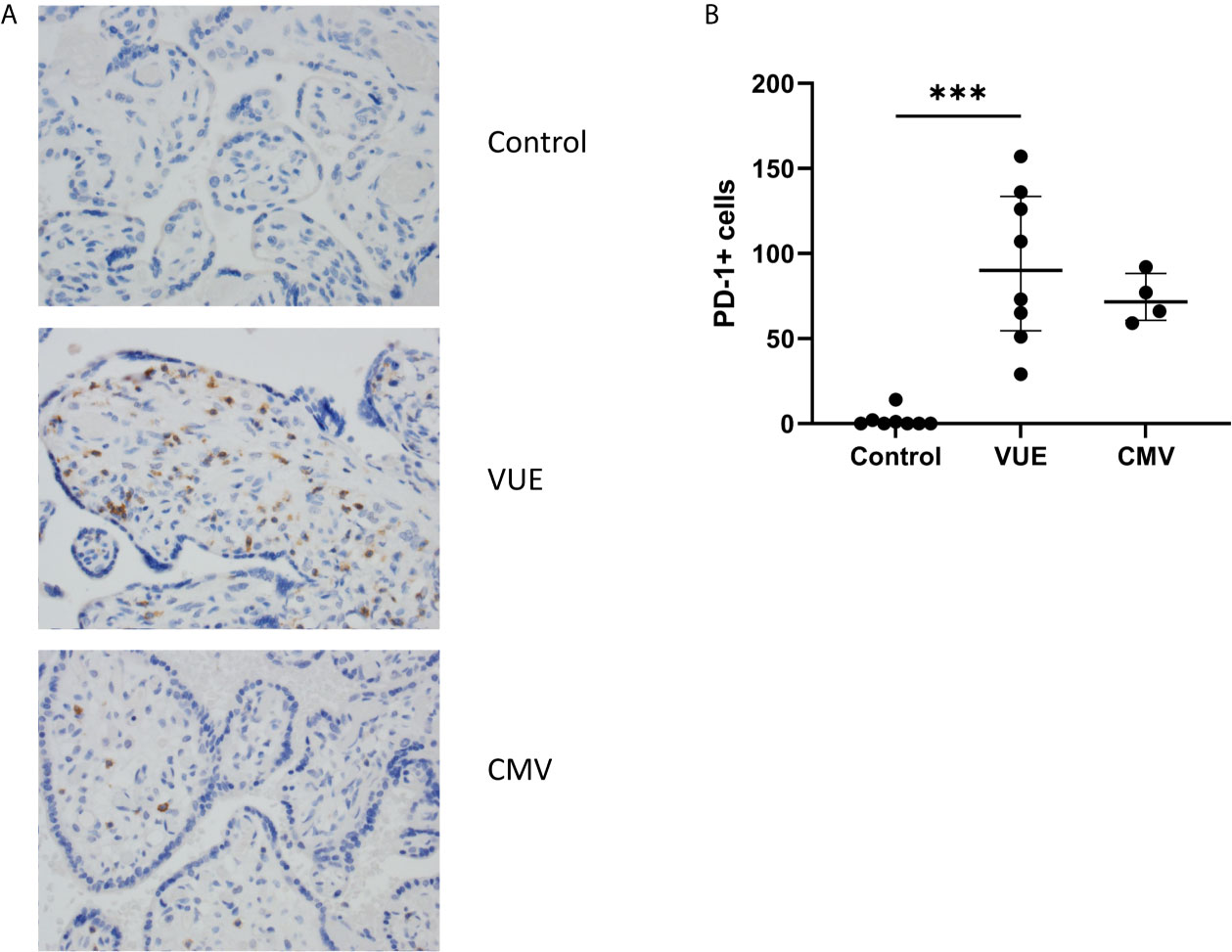
Figure 2 PD-1+ cells in placentae with CMV, VUE and controls. (A) Representative PD-1 staining in all three groups (200X). (B) Average count of PD-1+ cells per 20 villi in control, VUE and CMV placentae. Graphs show median with interquartile ranges and significance was determined by Kruskal-Wallis testing with false discovery rate correction by Benjamini, Krieger and Yekutieli (n=4-8/group). ***p ≤ 0.0001.
Lastly, we stained for LAG3 and CTLA4, other known checkpoint molecules, in our three groups. Positive cells were counted and averaged from a total of 10 images taken from each case. Figure 3A staining demonstrated significant infiltration of LAG3+ cells in VUE compared to controls (49 vs. 0.2 cells/image; p=0.05). However, there was a high level of variability noted across the VUE cases (0-116 cells/image; Figure 3B). We did not see evidence of a difference in LAG3+ cells between VUE and CMV infection (49 vs. 7 cells/image; p=0.399), and CMV infection and control cases (7 vs. 0.2 cell/image; p=0.40). In contrast, overall expression of CTLA4+ cells in the placental villi was negative in all three groups, with only a few fetal macrophages staining positive (Figure 4). Together, VUE appears to be associated with greater infiltration of LAG3+ cells compared to infection and controls, but cells expressing CTLA4 are generally absent in the placenta from our three groups.
Figure 3 Placental abundance of LAG3+ cells during CMV, VUE and in controls. (A) Representative LAG3 staining in all three groups (200X). (B) Average count of LAG3+ cells image in each group. Median with interquartile ranges are represented in the graph. Significance was determined by Kruskal-Wallis with false discovery rate correction by Benjamini, Krieger and Yekutieli (n=4-8/group). *p ≤ 0.05.
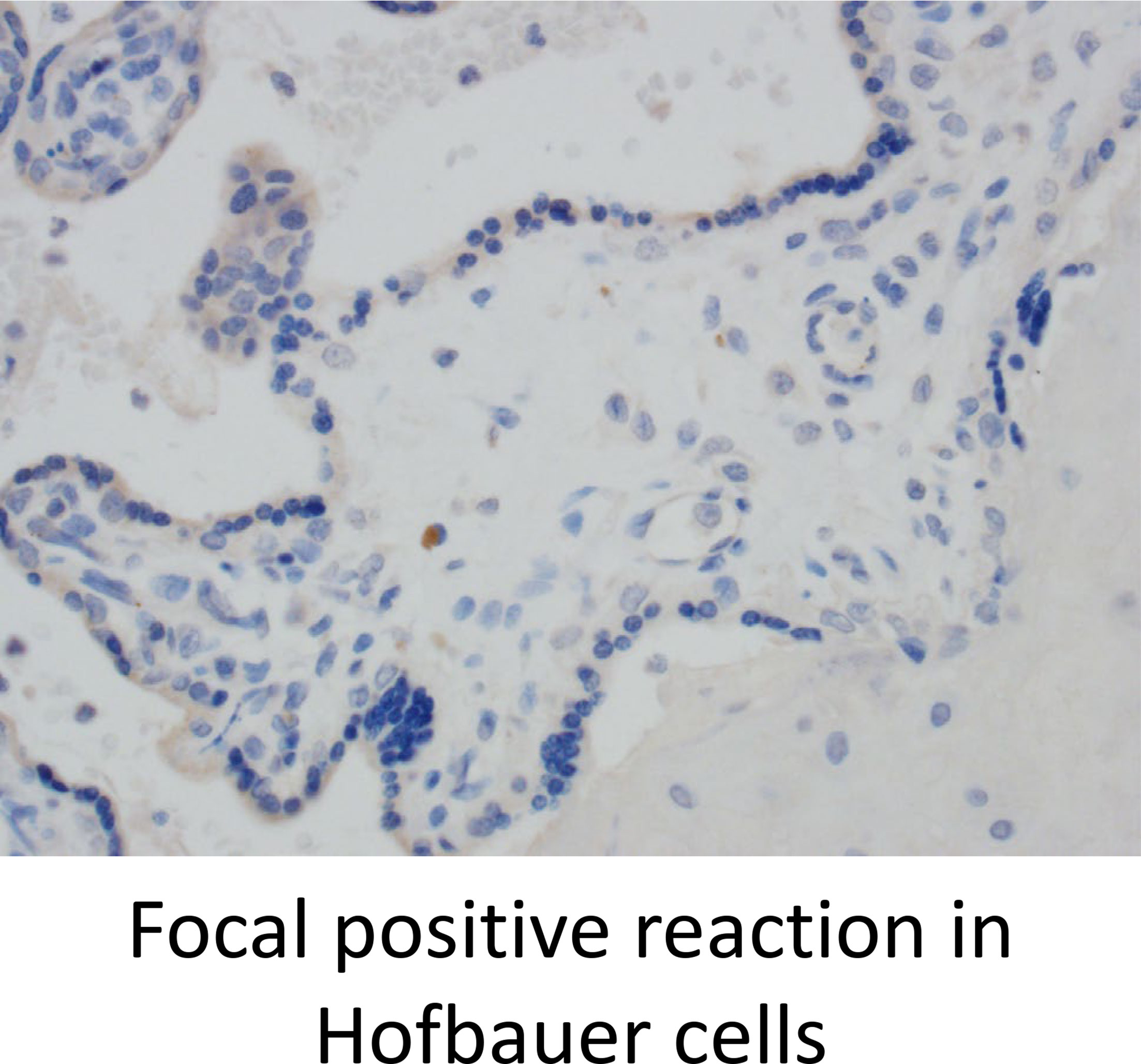
Figure 4 CTLA4 is weakly expressed in the placenta but is found on a few Hofbauer cells. Representative CTLA4 staining in all three groups (200X).
VUE, a diagnosis characterized by the infiltration of maternal CD8+ T-cells into villous tissue of the placenta without infectious cause, is hypothesized to be the result of an immune rejection response targeted against the haploidentical fetus during pregnancy. While the cause remains unknown, we hypothesized that this could be reflect dysregulation of checkpoint molecules which play a critical role in maintaining maternal-fetal tolerance. Our results demonstrate that VUE leads to downregulation of PD-L1 and upregulation of receptors PD-1 and LAG3 on infiltrating cells. This expression pattern is unique from placentae infected with CMV, and the changes in these checkpoint interactions, which ultimately dampen immune responses, may have a significant role in initiation of VUE pathogenesis.
The discovery of proteins that turn off activated lymphocytes, termed checkpoint receptors, has revolutionized the field of tumor immunology through the development of therapeutic strategies that block these interactions, thereby maintaining immune cell activation and tumor cell killing. The first was CTLA4, which binds CD80/CD86 to negatively regulate T-cell activation and blockade leads to anti-tumor immune responses (37, 38). Next, the interaction between PD-1 and PD-L1 was also found to cause cell death in activated T-cells, which tumors expressing high levels of PD-L1 use to evade the immune system (39, 40). Recent focus has turned to disrupting LAG3 signaling through MHC class II to promote immune activation against tumors (41, 42). Like the tumor microenvironment, trophoblasts interact with decidual immune cells at the maternal-fetal interface through checkpoint receptors, which are critical for pregnancy success. Guleria et al. reported increased allogeneic, but not syngeneic, fetal loss in murine models treated with PD-L1 blockade, which also increased proinflammatory cytokine signaling (28). However, other groups have shown that deletion of PD-1 or PD-L1 has no impact on pregnancy outcomes (7). T regulatory cells (T regs) in the normal decidua have been observed to express high levels of CTLA4 and, in specimens from spontaneous abortion cases, the proportion of CTLA4+ T reg cells were found to be significantly lower (43, 44). More recently, upregulation of PD-1 and LAG3 has been found on CD8+ maternal T-cells that recognize fetal antigen from a first pregnancy and are subsequently exposed to the same fetal antigens during a second pregnancy (45). This data proposes a priming mechanism occurs between pregnancies which promotes fetal antigen-specific immune tolerance responses in the mother. Together, these studies indicate a critical role for immune checkpoint receptors in mediating maternal immune tolerance to the allogenic fetus.
Expression of checkpoint receptors in VUE have not been examined, but as this etiology has been hypothesized to be an allograft rejection response, presence or absence of these suppressive regulatory signals are pertinent. In tissue transplant, allograft rejection is a common complication, and occurs when lymphocytes from the recipient infiltrate into donor tissue leading to organ failure (46). Antibody-mediated organ failure, as measured by C4d expression, is used clinically to assess transplant rejection (47). Placentae diagnosed with VUE have been found to have increased staining for C4d in the syncytiotrophoblast layer (48). ICAM-1, a surface glycoprotein that is upregulated upon cytokine stimulation resulting in the migration of immune cells into tissue, is upregulated on trophoblast cells and leukocytes in chronic villitis (49). Interestingly, anti-ICAM-1 antibodies showed preclinical promise for improving allograft survival, randomized clinical trials demonstrated no benefit (50, 51). Demise of a fetus after 20 weeks gestation could be considered an extreme rejection response. In a cohort of 40 fetal demise cases, placental examination showed that approximately 58% had a chronic inflammatory lesion defined as chorioamnionitis, VUE or chronic deciduitis (52). Importantly, chronic inflammation did not correlate with the detection of microorganisms.
Despite the recent advancements in our understanding of VUE as a fetal rejection response, it is not yet possible to rule out the presence of a subclinical infectious etiology. At least in a subset of VUE cases, undiagnosed pathogens were later detected (5, 53). While a bacterial etiology could not be confirmed by PCR for universal bacterial 16S rRNA, at least one herpesvirus strain has been detected in half of the VUE cases tested (54, 55). However, differences in histological and clinical manifestations of VUE from the known infectious etiologies suggest distinct inflammatory processes. Morphologically, VUE tends to involve the placental parenchyma in a patchy pattern with a higher rate of distal villi distribution, whereas in infectious villitis, the distribution is more diffuse with concurrent involvement of umbilical cord, chorionic plate, and membranes (5). While the presence of plasma cells does not exclude a VUE diagnosis, marked plasmacytic infiltration is more often associated with an infectious processes like CMV or other viruses (56).
The results of our study show increased expression of PD-L1 in CMV infectious villitis. CMV infected dendritic cells have been reported to express higher levels of PDL-1 (57). Thus, PD-L1 overexpression by CMV-infected cells could be a mechanism to support viral replication. Having only studied CMV related infectious villitis, we cannot infer that other agents induce similar PD-L1 expression. While much of the focus is on T-cells, immunologic activation in the Hofbauer cells during VUE has also been observed, and studies have demonstrated that CMV has a tropism for Hofbauer cells of the placenta (58, 59). Therefore, Hofbauer cells have an important role in the pathophysiology of both conditions, which requires further characterization. In VUE, our results show increased PD-1 expression in infiltrating lymphocytes and decreased PD-L1 expression in trophoblasts, implying a possible loss suppressive signals leading to the propagation of T-cell activation signals and resulting inflammation. This has been observed in autoimmune disease, where depleting PD-1+ autoreactive T cells in mice with type I diabetes or autoimmune encephalomyelitis led to delayed disease onset and improvement of symptoms (60). Though our small sample size is small, the data demonstrates differences in immune checkpoint receptor expression in infectious versus non-infectious villitis which should be explored further.
To conclude, differential expression of PD-L1 and abundance of PD-1 and LAG3 is seen in placentae diagnosed with VUE compared to CMV infection. These data suggest that disruption of maternal-fetal tolerance through checkpoint receptor signaling may be an important mechanism in the development of VUE. Further understanding of the precise etiology and pathophysiology of VUE will require a multidisciplinary and systems biology approach to effectively address this sometimes-devastating placental condition.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Mayo Clinic Institutional Review Board 200 First St. SW Rochester, MN 55905. The patients/participants provided their written informed consent to participate in this study.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This work was supported by HD065987 (EALE), the Marcia T. Kreyling Career Development Award in Pediatric and Neonatal Research (EALE), the Mayo Clinic Division of Anatomical Pathology (MS) and AI131566, HD097843, HD103498 (RC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Altshuler G, Russell P. The Human Placental Villitides: A Review of Chronic Intrauterine Infection. Curr Top Pathol (1975) 60:64–112. doi: 10.1007/978-3-642-66215-7_3
2. Knox WF, Fox H. Villitis of Unknown Aetiology: Its Incidence and Significance in Placentae From a British Population. Placenta (1984) 5(5):395–402. doi: 10.1016/S0143-4004(84)80019-3
3. Russell P. Inflammatory Lesions of the Human Placenta. III. The Histopathology of Villitis of Unknown Aetiology. Placenta (1980) 1(3):227–44. doi: 10.1016/S0143-4004(80)80005-1
4. Chan JS. Villitis of Unknown Etiology and Massive Chronic Intervillositis. Surg Pathol Clin (2013) 6(1):115–26. doi: 10.1016/j.path.2012.11.004
5. Redline RW. Villitis of Unknown Etiology: Noninfectious Chronic Villitis in the Placenta. Hum Pathol (2007) 38(10):1439–46. doi: 10.1016/j.humpath.2007.05.025
6. Redline RW, Patterson P. Villitis of Unknown Etiology Is Associated With Major Infiltration of Fetal Tissue by Maternal Inflammatory Cells. Am J Pathol (1993) 143(2):473–9.
7. Taglauer ES, Adams Waldorf KM, Petroff MG. The Hidden Maternal-Fetal Interface: Events Involving the Lymphoid Organs in Maternal-Fetal Tolerance. Int J Dev Biol (2010) 54(2-3):421–30. doi: 10.1387/ijdb.082800et
8. Kim JS, Romero R, Kim MR, Kim YM, Friel L, Espinoza J, et al. Involvement of Hofbauer Cells and Maternal T Cells in Villitis of Unknown Aetiology. Histopathology (2008) 52(4):457–64. doi: 10.1111/j.1365-2559.2008.02964.x
9. Kapur P, Rakheja D, Gomez AM, Sheffield J, Sanchez P, Rogers BB. Characterization of Inflammation in Syphilitic Villitis and in Villitis of Unknown Etiology. Pediatr Dev Pathol (2004) 7(5):453–8; discussion 21. doi: 10.1007/s10024-004-2124-3
10. Enninga EAL, Raber P, Quinton RA, Ruano R, Ikumi N, Gray CM, et al. Maternal T Cells in the Human Placental Villi Support an Allograft Response During Noninfectious Villitis. J Immunol (2020) 204(11):2931–9. doi: 10.4049/jimmunol.1901297
11. Myerson D, Parkin RK, Benirschke K, Tschetter CN, Hyde SR. The Pathogenesis of Villitis of Unknown Etiology: Analysis With a New Conjoint Immunohistochemistry-in Situ Hybridization Procedure to Identify Specific Maternal and Fetal Cells. Pediatr Dev Pathol (2006) 9(4):257–65. doi: 10.2350/08-05-0103.1
12. Magid MS, Kaplan C, Sammaritano LR, Peterson M, Druzin ML, Lockshin MD. Placental Pathology in Systemic Lupus Erythematosus: A Prospective Study. Am J Obstet Gynecol (1998) 179(1):226–34. doi: 10.1016/S0002-9378(98)70277-7
13. Labarrere CA, Catoggio LJ, Mullen EG, Althabe OH. Placental Lesions in Maternal Autoimmune Diseases. Am J Reprod Immunol Microbiol (1986) 12(3):78–86. doi: 10.1111/j.1600-0897.1986.tb00068.x
14. Althaus J, Weir EG, Askin F, Kickler TS, Blakemore K. Chronic Villitis in Untreated Neonatal Alloimmune Thrombocytopenia: An Etiology for Severe Early Intrauterine Growth Restriction and the Effect of Intravenous Immunoglobulin Therapy. Am J Obstet Gynecol (2005) 193(3 Pt 2):1100–4. doi: 10.1016/j.ajog.2005.06.043
15. Styer AK, Parker HJ, Roberts DJ, Palmer-Toy D, Toth TL, Ecker JL. Placental Villitis of Unclear Etiology During Ovum Donor In Vitro Fertilization Pregnancy. Am J Obstet Gynecol (2003) 189(4):1184–6. doi: 10.1067/S0002-9378(03)00577-5
16. Perni SC, Predanic M, Predanik M, Cho JE, Baergen RN. Placental Pathology and Pregnancy Outcomes in Donor and Non-Donor Oocyte In Vitro Fertilization Pregnancies. J Perinat Med (2005) 33(1):27–32. doi: 10.1515/JPM.2005.004
17. Herman HG, Tamayev L, Feldstein O, Bustan M, Rachmiel Z, Schreiber L, et al. Placental-Related Disorders of Pregnancy and IVF: Does Placental Histological Examination Explain the Excess Risk? Reprod BioMed Online (2020) 41(1):81–7. doi: 10.1016/j.rbmo.2020.04.001
18. Feeley L, Mooney EE. Villitis of Unknown Aetiology: Correlation of Recurrence With Clinical Outcome. J Obstet Gynaecol (2010) 30(5):476–9. doi: 10.3109/01443611003802339
19. Hunt JS, Pace JL, Morales PJ, Ober C. Immunogenicity of the Soluble Isoforms of HLA-G. Mol Hum Reprod (2003) 9(11):729–35. doi: 10.1093/molehr/gag087
20. Hunt JS, Petroff MG, McIntire RH, Ober C. HLA-G and Immune Tolerance in Pregnancy. FASEB J (2005) 19(7):681–93. doi: 10.1096/fj.04-2078rev
21. Hunt JS, Jadhav L, Chu W, Geraghty DE, Ober C. Soluble HLA-G Circulates in Maternal Blood During Pregnancy. Am J Obstet Gynecol (2000) 183:682–8. doi: 10.1067/mob.2000.106762
22. Alegre E, Rizzo R, Bortolotti D, Fernandez-Landázuri S, Fainardi E, González A. Some Basic Aspects of HLA-G Biology. J Immunol Res (2014) 2014:657625. doi: 10.1155/2014/657625
23. Enninga EAL, Leontovich AA, Fedyshyn B, Wakefield L, Gandhi M, Markovic SN, et al. Upregulation of HLA-Class I and II in Placentas Diagnosed With Villitis of Unknown Etiology. Reprod Sci (2020) 27(5):1129–38. doi: 10.1007/s43032-019-00101-9
24. Papúchová H, Meissner TB, Li Q, Strominger JL, Tilburgs T. The Dual Role of HLA-C in Tolerance and Immunity at the Maternal-Fetal Interface. Front Immunol (2019) 10:2730. doi: 10.3389/fimmu.2019.02730
25. Kim MJ, Romero R, Kim CJ, Tarca AL, Chhauy S, LaJeunesse C, et al. Villitis of Unknown Etiology Is Associated With a Distinct Pattern of Chemokine Up-Regulation in the Feto-Maternal and Placental Compartments: Implications for Conjoint Maternal Allograft Rejection and Maternal Anti-Fetal Graft-Versus-Host Disease. J Immunol (2009) 182(6):3919–27. doi: 10.4049/jimmunol.0803834
26. Moldenhauer LM, Hayball JD, Robertson SA. Utilising T Cell Receptor Transgenic Mice to Define Mechanisms of Maternal T Cell Tolerance in Pregnancy. J Reprod Immunol (2010) 87(1-2):1–13. doi: 10.1016/j.jri.2010.05.007
27. Erlebacher A, Vencato D, Price KA, Zhang D, Glimcher LH. Constraints in Antigen Presentation Severely Restrict T Cell Recognition of the Allogeneic Fetus. J Clin Invest (2007) 117(5):1399–411. doi: 10.1172/JCI28214
28. Guleria I, Khosroshahi A, Ansari MJ, Habicht A, Azuma M, Yagita H, et al. A Critical Role for the Programmed Death Ligand 1 in Fetomaternal Tolerance. J Exp Med (2005) 202(2):231–7. doi: 10.1084/jem.20050019
29. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on Tumor Cells Is Sufficient for Immune Evasion in Immunogenic Tumors and Inhibits CD8 T Cell Cytotoxicity. J Exp Med (2017) 214(4):895–904. doi: 10.1084/jem.20160801
30. Enninga EAL, Harrington SM, Creedon DJ, Ruano R, Markovic SN, Dong H, et al. Immune Checkpoint Molecules Soluble Program Death Ligand 1 and Galectin-9 Are Increased in Pregnancy. Am J Reprod Immunol (2018) 79(2):1–6. doi: 10.1111/aji.12795
31. Petroff MG, Chen L, Phillips TA, Azzola D, Sedlmayr P, Hunt JS. B7 Family Molecules are Favorably Positioned at the Human Maternal-Fetal Interface. Biol Reprod (2003) 68(5):1496–504. doi: 10.1095/biolreprod.102.010058
32. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 Co-Inhibitory Molecules Collaborate to Limit CD8+ T Cell Signaling and Dampen Antitumor Immunity in a Murine Ovarian Cancer Model. Oncotarget (2015) 6(29):27359–77. doi: 10.18632/oncotarget.4751
33. Krummel MF, Allison JP. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J Exp Med (1995) 182(2):459–65. doi: 10.1084/jem.182.2.459
34. Khong TY, Mooney EE, Ariel I, Balmus NC, Boyd TK, Brundler MA, et al. Sampling and Definitions of Placental Lesions: Amsterdam Placental Workshop Group Consensus Statement. Arch Pathol Lab Med (2016) 140(7):698–713. doi: 10.5858/arpa.2015-0225-CC
35. Crowe AR, Yue W. Semi-Quantitative Determination of Protein Expression Using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc (2019) 9(24):1–11. doi: 10.21769/BioProtoc.3465
36. Veras E, Kurman RJ, Wang TL, Shih IM. PD-L1 Expression in Human Placentas and Gestational Trophoblastic Diseases. Int J Gynecol Pathol (2017) 36(2):146–53. doi: 10.1097/PGP.0000000000000305
37. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 Can Function as a Negative Regulator of T Cell Activation. Immunity (1994) 1:405–13. doi: 10.1016/1074-7613(94)90071-X
38. Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734
39. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced Expression of PD-1, A Novel Member of the Immunoglobulin Gene Superfamily, Upon Programmed Cell Death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
40. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat Med (2002) 8(8):793–800. doi: 10.1038/nm730
41. Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, et al. Characterization of the Major Histocompatibility Complex Class II Binding Site on LAG-3 Protein. Proc Natl Acad Sci USA (1997) 94(11):5744–9. doi: 10.1073/pnas.94.11.5744
42. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, A Novel Lymphocyte Activation Gene Closely Related to CD4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/jem.171.5.1393
43. Sasaki Y, Sakai M, Miyazaki S, Higuma S, Shiozaki A, Saito S. Decidual and Peripheral Blood CD4+CD25+ Regulatory T Cells in Early Pregnancy Subjects and Spontaneous Abortion Cases. Mol Hum Reprod (2004) 10(5):347–53. doi: 10.1093/molehr/gah044
44. Heikkinen J, Möttönen M, Alanen A, Lassila O. Phenotypic Characterization of Regulatory T Cells in the Human Decidua. Clin Exp Immunol (2004) 136(2):373–8. doi: 10.1111/j.1365-2249.2004.02441.x
45. Kinder JM, Turner LH, Stelzer IA, Miller-Handley H, Burg A, Shao TY, et al. CD8+ T Cell Functional Exhaustion Overrides Pregnancy-Induced Fetal Antigen Alloimmunization. Cell Rep (2020) 31(12):107784. doi: 10.1016/j.celrep.2020.107784
46. Valujskikh A, Lantz O, Celli S, Matzinger P, Heeger PS. Cross-Primed CD8(+) T Cells Mediate Graft Rejection via a Distinct Effector Pathway. Nat Immunol (2002) 3(9):844–51. doi: 10.1038/ni831
47. Troxell ML, Weintraub LA, Higgins JP, Kambham N. Comparison of C4d Immunostaining Methods in Renal Allograft Biopsies. Clin J Am Soc Nephrol (2006) 1(3):583–91. doi: 10.2215/CJN.00900805
48. Rudzinski E, Gilroy M, Newbill C, Morgan T. Positive C4d Immunostaining of Placental Villous Syncytiotrophoblasts Supports Host-Versus-Graft Rejection in Villitis of Unknown Etiology. Pediatr Dev Pathol (2013) 16(1):7–13. doi: 10.2350/12-05-1195-OA.1
49. Egal ES, Mariano FV, Blotta MH, Piña AR, Montalli VA, Almeida OP, et al. ICAM-1 Expression on Immune Cells in Chronic Villitis. Placenta (2014) 35(12):1021–6. doi: 10.1016/j.placenta.2014.10.004
50. Poritz LS, Page MJ, Tilberg AF, Koltun WA. Amelioration of Graft Versus Host Disease With Anti-ICAM-1 Therapy. J Surg Res (1998) 80(2):280–6. doi: 10.1006/jsre.1998.5422
51. Salmela K, Wramner L, Ekberg H, Hauser I, Bentdal O, Lins LE, et al. A Randomized Multicenter Trial of the Anti-ICAM-1 Monoclonal Antibody (Enlimomab) for the Prevention of Acute Rejection and Delayed Onset of Graft Function in Cadaveric Renal Transplantation: A Report of the European Anti-ICAM-1 Renal Transplant Study Group. Transplantation (1999) 67(5):729–36. doi: 10.1097/00007890-199903150-00015
52. Lannaman K, Romero R, Chaiworapongsa T, Kim YM, Korzeniewski SJ, Maymon E, et al. Fetal Death: An Extreme Manifestation of Maternal Anti-Fetal Rejection. J Perinat Med (2017) 45(7):851–68. doi: 10.1515/jpm-2017-0073
53. Benirschke K, Coen R, Patterson B, Key T. Villitis of Known Origin: Varicella and Toxoplasma. Placenta (1999) 20(5-6):395–9. doi: 10.1053/plac.1999.0405
54. Ernst LM, Bockoven C, Freedman A, Wang V, Pellerite M, Wylie TN, et al. Chronic Villitis of Unknown Etiology: Investigations Into Viral Pathogenesis. Placenta (2021) 107:24–30. doi: 10.1016/j.placenta.2021.02.020
55. Ernst LM, Crouch J, Rinder H, Howe JG. Bacterial Etiology for Chronic Villitis Is Not Supported by Polymerase Chain Reaction for 16S rRNA DNA. Pediatr Dev Pathol (2005) 8(6):647–53. doi: 10.1007/s10024-005-0412-1
56. Mostoufi-zadeh M, Driscoll SG, Biano SA, Kundsin RB. Placental Evidence of Cytomegalovirus Infection of the Fetus and Neonate. Arch Pathol Lab Med (1984) 108(5):403–6.
57. Benedict CA, Loewendorf A, Garcia Z, Blazar BR, Janssen EM. Dendritic Cell Programming by Cytomegalovirus Stunts Naive T Cell Responses via the PD-L1/PD-1 Pathway. J Immunol (2008) 180(7):4836–47. doi: 10.4049/jimmunol.180.7.4836
58. Johnson EL, Boggavarapu S, Johnson ES, Lal AA, Agrawal P, Bhaumik SK, et al. Human Cytomegalovirus Enhances Placental Susceptibility and Replication of Human Immunodeficiency Virus Type 1 (HIV-1), Which May Facilitate In Utero HIV-1 Transmission. J Infect Dis (2018) 218(9):1464–73. doi: 10.1093/infdis/jiy327
59. Johnson EL. Human Cytomegalovirus Preferentially Infects Early Gestation Placental Macrophages and Evades Their Antiviral Immunogenicity Through Evasion of the Type I IFN Response. J Immunol (2020) 204:204.
Keywords: villitis, cytomegalovirus, placenta, immune tolerance, PD-1, PD-L1, LAG3
Citation: Shahi M, Mamber Czeresnia R, Cheek EH, Quinton RA, Chakraborty R and Enninga EAL (2021) Expression of Immune Checkpoint Receptors in Placentae With Infectious and Non-Infectious Chronic Villitis. Front. Immunol. 12:705219. doi: 10.3389/fimmu.2021.705219
Received: 04 May 2021; Accepted: 16 July 2021;
Published: 30 July 2021.
Edited by:
Andrew L. Mellor, Newcastle University, United KingdomReviewed by:
Frans H. J. Claas, Leiden University, NetherlandsCopyright © 2021 Shahi, Mamber Czeresnia, Cheek, Quinton, Chakraborty and Enninga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth Ann L. Enninga, ZW5uaW5nYS5lbGl6YWJldGhhbm5AbWF5by5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.