 Kari Ann Shirey
Kari Ann Shirey Jorge C. G. Blanco
Jorge C. G. Blanco Stefanie N. Vogel
Stefanie N. Vogel- 1Department of Microbiology and Immunology, School of Medicine, University of Maryland, Baltimore, MD, United States
- 2Sigmovir Biosystems, Inc., Rockville, MD, United States
Respiratory viral infections have been a long-standing global burden ranging from seasonal recurrences to the unexpected pandemics. The yearly hospitalizations from seasonal viruses such as influenza can fluctuate greatly depending on the circulating strain(s) and the congruency with the predicted strains used for the yearly vaccine formulation, which often are not predicted accurately. While antiviral agents are available against influenza, efficacy is limited due to a temporal disconnect between the time of infection and symptom development and viral resistance. Uncontrolled, influenza infections can lead to a severe inflammatory response initiated by pathogen-associated molecular patterns (PAMPs) or host-derived danger-associated molecular patterns (DAMPs) that ultimately signal through pattern recognition receptors (PRRs). Overall, these pathogen-host interactions result in a local cytokine storm leading to acute lung injury (ALI) or the more severe acute respiratory distress syndrome (ARDS) with concomitant systemic involvement and more severe, life threatening consequences. In addition to traditional antiviral treatments, blocking the host’s innate immune response may provide a more viable approach to combat these infectious pathogens. The SARS-CoV-2 pandemic illustrates a critical need for novel treatments to counteract the ALI and ARDS that has caused the deaths of millions worldwide. This review will examine how antagonizing TLR4 signaling has been effective experimentally in ameliorating ALI and lethal infection in challenge models triggered not only by influenza, but also by other ALI-inducing viruses.
Summary sentence
This review focuses on the role of TLR4 in the development of virus-induced acute lung injury (ALI) and its potential for therapeutic targeting.
Introduction
Many pathogens mutate rapidly, leading to anti-microbial resistance or altered expression of immunogenic epitopes such that extant vaccines or therapeutic drugs are rendered ineffective. The ability of the host to recognize and respond to immunologic “danger” is the result of the exposure of the host to a pathogen’s unique components known as “pathogen-associated molecular patterns” (PAMP) that are readily detected by host surveillance receptors. Such “pattern recognition receptors” (PRR) (1) are expressed on cells of the innate immune system and their activation initiates intracellular signaling and transcriptional programs that lead to a rapid and strong primary response against the pathogen, e.g., Gram-negative lipopolysaccharide (LPS) interacts with Toll-like receptor 4 (TLR4) leading to the induction of thousands of genes, many of which are proinflammatory (2). Also, as part of this rapid response, host-derived molecules are often released during cellular necrosis or are secreted upon cellular stress. These molecules are collectively referred to as “danger-associated molecular patterns” (DAMPs) (1), and activate identical PRR signaling pathways, e.g., HMGB1 is a chromatin-associated protein that, when actively secreted or released from dying cells (3), engages TLR4 through its co-receptor, MD-2, and elicits a pro-inflammatory response similar to that of LPS (4).
TLR Signaling
In humans, Toll-like receptors (TLRs) are a closely related family of 10 transmembrane PRRs that sense unique microbial chemistries or PAMPs and/or host-derived DAMPs (5, 6). Ligand interaction(s) with a TLR’s N-terminal leucine-rich repeat (LRR) domain results in TLR dimerization, leading to conformational changes that cause interaction of C-terminal TLR “Toll-IL-1 receptor resistance (TIR)” domains to form a molecular scaffold (7) that facilitates recruitment of adaptor molecules (e.g., TIRAP/MyD88 or TRIF/TRAM) through TIR-TIR interactions. In turn, docking of MyD88 or TRIF to the TLR TIR domain permits further recruitment of downstream signaling proteins to form a large molecular complex called a Myddosome or Triffosome, respectively (8). The MyD88-dependent pathway is recruited by all cell surface- and endosomally-expressed TLRs except endosommaly-expressed TLR3. TLR4 recruitment of MyD88 primarily activates NF-κB, a transcription factor (TF) required for inductin of most pro-inflammatory cytokine and chemokine genes. The TRIF (MyD88-independent) pathway, that is activated only from two endosomally located TLRs (i.e., TLR4 and TLR3), activates IRF3, a TF required for induction of IFN-β and other TRIF-dependent genes. However, the TRIF-mediated pathway bifurcates and also results in delayed NF-κB activation. TLR4 is unique in that (i) it requires a non-covalently associated co-receptor, MD-2, for ligand binding, and (ii) it is the only TLR that activates both MyD88 and TRIF signaling pathways (6). That Gram-negative lipopolysaccharide (LPS) activates both MyD88 and TRIF pathways through TLR4/MD-2 is thought to explain why TLR4 signaling is associated with highly inflammatory responses as seen in Gram-negative sepsis and other diseases that are mediated by “cytokine storms”.
Influenza
New antigenic variants of influenza emerge annually, giving rise to seasonal outbreaks with significant morbidity and mortality, resulting in up to 500,000 deaths worldwide (9, 10). Hospitalization and deaths occur mainly in high-risk groups (e.g., children, elderly, chronically ill). In addition, strains to which humans have no prior immunity may appear suddenly and the resulting pandemics can be catastrophic, as illustrated by the 1918 “Spanish flu” (11, 12). In March 2009, a novel influenza A virus, pandemic H1N1, emerged that contained a previously unseen combination of genes of swine origin, and caused hospitalizations and deaths in both high-risk individuals and in healthy adults and children (12, 13). While vaccination provides significant protection, the ability to predict the influenza strains to be incorporated into the following year’s vaccine sometimes fails (14, 15). Although anti-viral therapeutics are available, patients must be treated early in infection for them to be effective. Moreover, virus resistance to neuraminidase (NA) inhibitors (i.e., oseltamivir, zanamivir) and M2 channel inhibitors (i.e., amantadine, rimantadine) is common (16, 17). Thus, a new approach to block influenza-induced, host-mediated disease therapeutically would represent a significant therapeutic treatment against infectious diseases.
A Central Role for TLR4 Signaling in Acute Lung Injury (ALI) caused by Influenza Infection
While influenza virus is recognized by multiple PRRs including TLR3, TLR7, TLR8, TLR10, retinoic acid-inducible gene I (RIG-I), and NOD-like receptor family pyrin domain containing 3 (NLRP3) (18), a central TLR4 signaling axis for the induction of ALI by multiple insults was proposed by Imai et al. (19). They proposed that diverse chemical or microbial insults to the lung trigger NADPH-dependent reactive oxygen species (ROS) that generate the formation of oxidized phospholipids such as oxidized 1-palmitoyl-2-arachidonoyl-phosphaticycholine (OxPAPC). In this study, wild-type mice were compared to mice with targeted mutations in genes involved in TLR signaling by inducing ALI by acid aspiration, instillation of UV-inactivated H5N1 influenza, infection with live bacterial pathogens, and aspiration of purified OxPAPC in vivo and confirmed in vitro. The authors concluded that a common TLR4-, TRIF-, and IL-6-dependent pathway to ALI was mediated by OxPAPC-activated lung macrophages. In support of the idea that this pathway is common to other infectious mediators of ALI, Imai et al. found that fixed lung sections of SARS-CoV-1 patients and from animals infected with anthrax, monkey pox, or Yersinia pestis also showed OxPAPC deposition (19). Shortly thereafter, Nhu et al. reported that TLR4-/- mice were highly refractory to infection with mouse-adapted influenza, A/PR/8/34 (PR8) (20). Thus, it was hypothesized that blocking TLR4 signaling might represent a viable therapeutic approach for mitigating influenza-induced ALI. Therapeutic administration of an LPS analog antagonist, Eritoran (E5564) (21, 22) that acts by competing for the LPS-binding site on MD-2 (23), resulted in a dose- and time-dependent protection of mice from lethal influenza infection when administered starting on day 2 post-infection once daily for 5 consecutive days (24). Even when Eritoran was administered to PR8-infected mice starting as late as day 6 post-infection, it still resulted in statistically significant survival, as well as improved clinical score, and inhibition of histopathology, cytokine gene and cytokine protein expression. While Eritoran was shown to decrease viral titers in the lung, it was not found to be directly antiviral (24). These findings were confirmed in the cotton rat (Sigmodon hispidus) (24, 25), a rodent species that is uniquely susceptible to non-adapted human respiratory viruses including influenza (24, 26).
Protection of mice and cotton rats from influenza-induced ALI has since been replicated with many other structurally and mechanistically distinct TLR4 antagonists including anti-TLR4 antibodies (25, 27), a cell-permeable TLR TIR decoy peptide (2R9) (28, 29), a virus-derived peptide 9R-VIPER (25, 30), a small molecule TLR4 antagonist, TAK242 (31, 32), another LPS analog antagonist (FP7) (33, 34), and the theta defensin, RC-101 (35). The “lung leak” caused by PR8 infection of mice was shown to be reversed by Eritoran administration, as well as by treatment with AT-1001 (larazotide acetate), a peptide that blocks LPS-induced tight junction disassembly in the lung (36). The blocking of TLR4 to protect against more lethal strains of influenza has also been reported. Xu et al. showed that both Eritoran (administered 2 h post-infection by intravenous inoculation), as well as another TLR4 inhibitor, epigallocatechin-3-gallate [EGCG (37)], a polyphenol naturally found in green tea (38), prolonged survival of mice infected with H9N2 influenza when administered after infection by oral gavage, and that treatment with Eritoran or EGCG diminished H9N2-induced ALI (39). While this study also showed that EGCG reduced viral titers (similar to that observed by treatment with Eritoran (24, 39), it did not assess whether EGCG was directly antiviral; however, another study reported that EGCG had greater antiviral activity against influenza B virus than influenza A virus (40). Together, these data strongly support the concept that influenza infection triggers TLR4-mediated inflammation leading to ALI.
While the TLR4/TRIF signaling axis was suggested to be central for ALI, MyD88 has also been implicated in the host response to influenza. In survival studies, MyD88-/- mice were shown to have higher mortality rates compared to wild-type and TRIF-/- mice (41). Additionally, MyD88 deficiency resulted in significantly higher lung pathology and diminished the cytokine response to influenza after infection when compared to wildtype mice or TRIF-/- mice (41, 42). The role of IRAK4, the first enzyme recruited to MyD88 for signaling, was also examined during influenza infection. IRAK4 kinase-dead knock-in (IRAK4KDKI) mice, which have a catalytically inactive form of IRAK4 that precludes MyD88-dependent signaling, were shown to be susceptible to influenza with a slight delay in the meantime to death compared to wildtype mice. However, TRIF-/- mice infected with influenza showed less mortality compared to wildtype mice (25). Treatment with TLR4 antagonists such as Eritoran, FP7, and TAK-242 blocked both MyD88- and TRIF-dependent pathways (24, 32, 34). While MyD88-/- mice were found to be more susceptible to primary influenza infection than wildtype control mice, they were protected from a secondary influenza challenge after a low dose primary infection (41) suggesting that MyD88 was not required for protection against a secondary influenza infection. Eritoran treatment after lethal influenza challenge of wildtype mice, which inhibits both MyD88- and TRIF-dependent signaling, did not block the ability to mount an adaptive immune response to a secondary lethal influenza challenge (25). Taken together, these studies show both MyD88- and TRIF-dependent pathways contribute to influenza-induced disease.
As mentioned, influenza can induce oxidative stress by production of oxidized phospholipids (19) and reactive oxygen species (43). N-acetyl-L-cystine (NAC), a potent antioxidant (44), was also shown to protect mice against lethal H9N2 swine influenza infection. NAC, when administered prophylactically 1 h prior to infection and every 4.5 h thereafter for 5 treatments, interfered with viral replication, thereby improving survival. Along with oseltamivir treatment, NAC also reduced disease severity (45). This study also showed that rather than inhibiting TLR4 signaling, NAC inhibited expression of TLR4 (45). While this may be true, an earlier study showed that NAC also was able to inhibit influenza viral replication in vitro in A549 cells (46), suggesting multiple mechanisms by which NAC may contribute to host protection against influenza infection.
Kaempferol, a common flavonoid with known anti-inflammatory properties and anti-oxidative effects (47–50), has been shown to decrease LPS-induced TNF-α and IL-1β in macrophages (51–53). Treatment of H9N2-infected BALB/c mice with Kaempferol every 12 h starting 1 h prior to infection, resulted in improved survival, with reduced lung pathology, decreased pulmonary edema, cytokine production, and decreased viral titer in the lungs (54). While this study did not directly analyze whether Kaempferol is directly antiviral, another study showed that Kaempferol was more potent as an antiviral against influenza B virus than influenza A virus and that the mechanism was likely to work by binding to glycoproteins of the viral envelope to inhibit viral replication (40). Like NAC, Kaempferol may work both as an antiviral as well as via inhibition of TLR4 expression and thereby inhibit the TLR4 signaling pathway and inflammatory responses.
Curcumin, a polyphenol component of turmeric (55), has been widely studied for its anti-inflammatory, anti-oxidative, and other pharmacological effects (56). Due to problems with solubility and bioavailability, curcumin analogs have been developed (56, 57). Both curcumin and its analogs have been shown to block TLR4 signaling during LPS-induced sepsis and ALI through various mechanisms including binding MD-2 and inhibiting NF-κB or ERK activation (58–60). Curcumin, and a structural analog, monoacetylcurcumin (MAC), have been assessed for their effectiveness during influenza infection (61–64). Curcumin was shown to inhibit replication of multiple strains of IAV in vitro by disrupting viral attachment to cells by inhibiting hemagglutinin activity of the virus (61, 62, 64). When mice were administered curcumin twice daily for 6 days starting 24 h after influenza infection, the survival rate was significantly increased, with 150 µg/mouse of curcumin eliciting similar survival as the oseltamivir-treated control group (64). Curcumin was found to be more broad-spectrum in its effects on PRR signaling. A549 cells infected with influenza and treated with curcumin showed inhibition of influenza-induced expression of TLR2, TLR4, and TLR7, as well as inhibition of activation of MyD88- and TRIF-dependent signaling pathways (64). Taken together, curcumin can act as both a direct antiviral as well as protecting the host by inhibiting not just TLR4 signaling, but other TLRs as well.
Liu Shen Wan (LSW), a traditional Chinese medicine comprised of multiple mineral- and animal-derived components (65), has wide pharmacological effects and has been used to treat a variety of ailments for many years (66–68). LSW was shown to improve survival in a rodent model of sepsis induced by cecal ligation puncture by reducing TNF-α, but not IL-1 levels, as well as plasma malondialdehyde (MDA) contents (65). Ma et al. recently reported that LSW treatment had an anti-viral effect on influenza infection both in vitro as well as in vivo (69). LSW was found to decrease proinflammatory cytokine production in infected cells through the inhibition of the TLR4/NF-κB axis (69). When administered orally after influenza A/PR/8/34 influenza infection, LSW showed a dose-dependent increase in survival, but not as significant as treatment with oseltamivir (69). The specifics of the timing of LSW administration was not reported in this study apart from the fact that it was administered for 5 days, so the whether the LSW acts as a direct antiviral agent and/or interferes with PRR-mediated signaling remains to be determined.
Sulforaphane (SFN) is a naturally occurring compound in cruciferous vegetables such as broccoli, brussels sprouts, and cabbages (70, 71). SFN has become of interest for its use as a health supplement due to its pleiotropic effects on the immune system (71). SFN acts in an anti-inflammatory manner by suppressing TLR4 oligomerization (72), as well as by binding to MD-2 and blocking the interaction of LPS with the TLR4/MD-2 complex (70). In addition to blocking TLR4, SFN’s anti-inflammatory capabilities have been attributed to its ability to activate the anti-inflammatory TF, Nrf2, to inhibit NF-κB-induced pro-inflammatory cytokines (71). However, there are reports of cross-talk between TLRs and Nrf2 (73). Huang et al. reported that Nrf2 activation resulted in decreased TLR4-induced inflammation in the liver in an ischemia/reperfusion model (74). Several studies have examined the effects of SFN on influenza infection. In combination with β-glucan, also shown to attenuate TLR4-mediated cytokine production (75), SFN administered as an oral prophylactic supplement two weeks prior to influenza H5N1 challenge of mice, resulted in significant survival (76). However, this treatment did not decrease lung cytokine levels, but rather, increased in IFN-γ, IL-1β, and TNF-α compared to the control group (76). Importantly, the prophylactic treatment of mice with glucan-SFN also reduced viral titers in the lung (76), but this is possibly due to SFN acting on Nrf2 and blocking viral entry into and replication in epithelial cells (77, 78). To our knowledge, SFN has not been tested therapeutically.
Table I summarizes the various TLR4 antagonists and signaling inhibitors that have been shown to be efficacious against influenza infection in animal models and their mechanisms of action. The diversity of these various inhibitors of TLR4 signaling, coupled with the spectrum of mechanisms by which they act, strongly support the notion that TLR4 is key to the host inflammatory response to influenza infection.
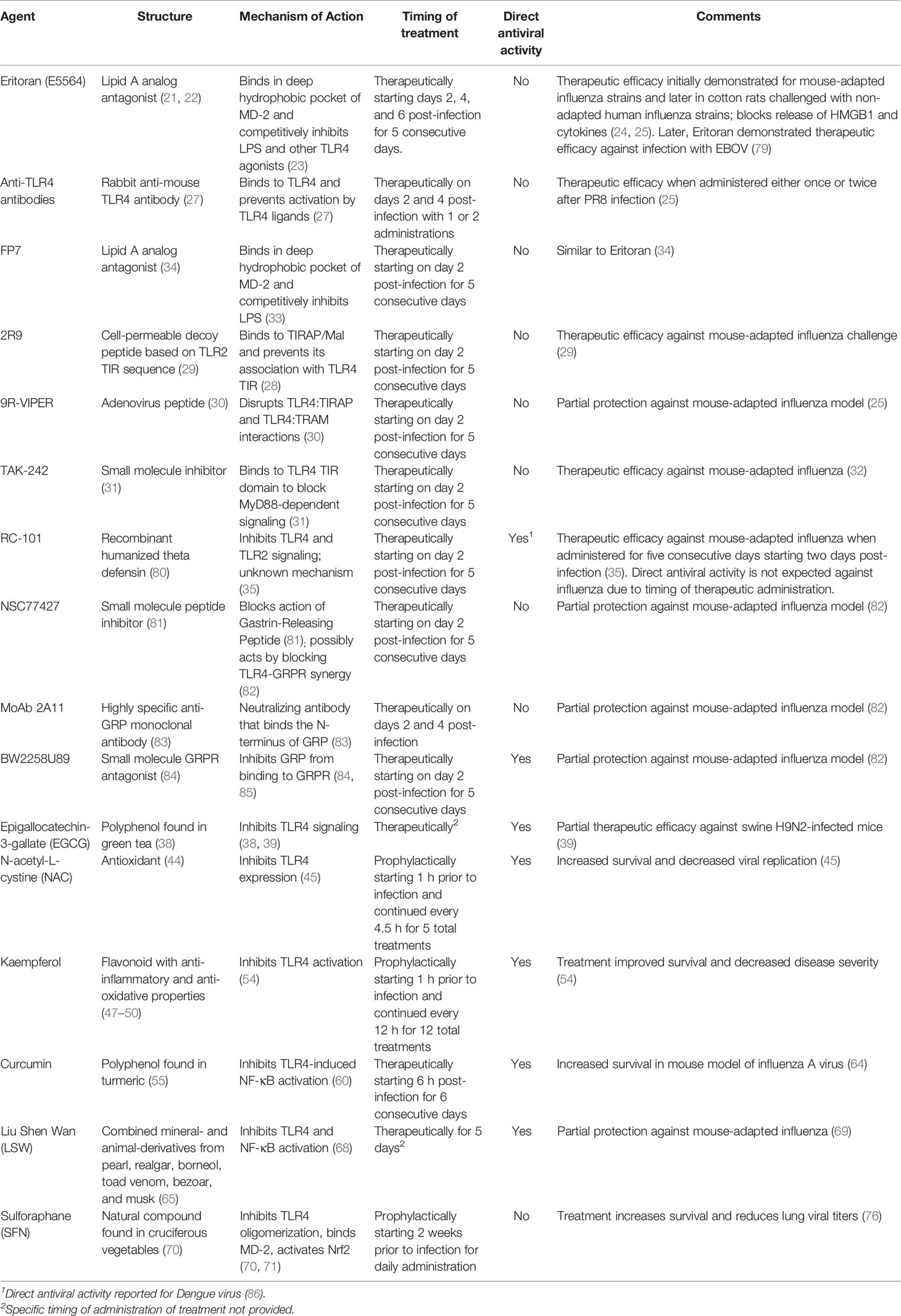
Table I Summary of agents used to treat influenza infection in rodent models.
Does the Timing of Treatment Play a Role in Protection?
As discussed thus far, the various compounds tested were administered either prophylactically or therapeutically (summarized in Table I). This leads to two questions: First, is the timing of TLR4 inhibition critical for protection? and second, do additional mechanisms contribute to protection from influenza? To address the first question, blocking TLR4 signaling therapeutically is highly effective for protection against lethal influenza infection. Delaying the start of treatment with Eritoran as late as 6 days still provided statistically significant protection (24). However, if Eritoran was administered 3 h prior to infection, and once daily for an additional 4 days, it failed to protect from lethal influenza infection (25). Interestingly, TLR4-/- mice, which were shown to be refractory to influenza infection (20, 24, 25), failed to resist infection when Eritoran was administered 3 h prior to infection and once daily for 4 additional days. This suggests a non-TLR4 target, which is necessary for resistance early in infection, may also be targeted by Eritoran. CD14 has been found to facilitate TLR4, TLR2, and TLR3 signaling (87) and is required for cytokine and chemokine induction by influenza (88). In the case of TLR3, CD14 was reported to enhance TLR3-mediated signaling (89). Eritoran has been shown to bind to CD14 (24). It is possible that pre-treatment with Eritoran could therefore interfere with influenza-induced TLR3 activation early in influenza infection. Of note, CD14-/- mice could not be protected by Eritoran therapy after influenza infection (24). While multiple groups and studies have shown the inhibition of TLR4 is protective during influenza infection, supporting the study by Imai et al. (19), Shinya et al. (90) reported that the TLR4-TRIF pathway was protective, rather than being detrimental, in influenza infection. This study found that pre-treatment of wildtype C57BL/6J mice with LPS prior to influenza infection promoted protection and survival (90). The upregulation of TLR3 was implicated as part of the protection, perhaps supporting the notion that CD14 enhances TLR3 signaling during influenza infection.
TLR4 antagonism by other non-TLR4 specific, more complex molecules such as naturally occurring anti-inflammatory compounds suggest that other mechanisms contribute to protection during influenza infection. NAC, Kaempferol, and SFN were all administered prophylactically, but at various times prior to infection (45, 54, 76). As stated earlier, one might postulate that the protective effects observed may reflect differences in the relative abilities of these compounds to be antiviral vs. host signaling modulators. In the case of SFN, the mice were given the compound for 2 weeks, well before infection (76). SFN treatment activated host Nrf2, which blocked viral entry in epithelial cells resulting in an antiviral response (77, 78). Therapeutic treatment after influenza infection with EGCG, curcumin, and LSW (39, 64, 69) also showed protection against influenza infection, but whether these responses were the result of direct antiviral activity or altered TLR4 expression and signaling will require further investigation. Regardless of timing, the inhibition of TLR4 seems to play an important role in influenza infection.
A Central Role for DAMP-Mediated TLR4 Signaling in Acute Lung Injury (ALI) Caused by Influenza Infection
While all of the studies presented above support a key role for TLR4 in influenza-induced disease, influenza does not express any TLR4 PAMPs, suggesting that a host-derived DAMP produced during influenza infection might actually signal through TLR4. One such DAMP, HMGB1, engages TLR4 by binding to its co-receptor, MD-2 (4), and was shown to be produced during influenza infection in mice challenged with PR8 or in cotton rats challenged with non-adapted human influenza strains in a time-dependent manner (25, 91). Blocking HMGB1 therapeutically with a neutralizing anti-HMGB1 antibody or a small molecule inhibitor, P5779 (4), improved survival during influenza infection in mice to the same extent as Eritoran treatment (25, 92). Therapeutic administration of Eritoran during influenza infection inhibited HMGB1 release in mice and cotton rats (25). In addition to HMGB1, another host DAMP, Gastrin Releasing Peptide (GRP), was found to be produced during influenza infection with kinetics that paralleled that of HMGB1 (82). GRP has been implicated in lung inflammatory diseases including asthma, bronchopulmonary dysplasia, and hyperoxia-, ozone-, or radiation-induced lung injury (93). In the lung, GRP is produced by specialized epithelial cells, pulmonary neuroendocrine cells, that are activated by neuronally derived γ-aminobutyric acid (GABA) to form clusters called neuroendocrine bodies (NEB) that secrete GRP. GRP binds to its receptor, GRPR (BB2), and activates downstream signal transduction pathways (e.g., cAMP, MAPK, PI3K, Akt) (93, 94). GRP synergizes with LPS for cytokine production by macrophages (82). Blocking GRP therapeutically with a small molecule inhibitor, NSC77427 (81), an anti-GRP monoclonal antibody (2A11) (83), or a small molecule GRP receptor antagonist (BW2258U89) (84, 85), significantly reduced influenza-induced lethality, cytokine induction, and lung pathology in mice (82). Taken collectively, the host-derived DAMPs, HMGB1 and GRP, play important roles in mediating influenza-induced disease. The relationship between these two mediators of influenza-induced disease is currently under study. Figure 1 represents a hypothetical model for the involvement of TLR4 and influenza-induced DAMPs that leads to influenza pathogenesis. As has been discussed inhibiting either TLR4 expression and/or DAMP-induced TLR4 signaling mitigates ALI.
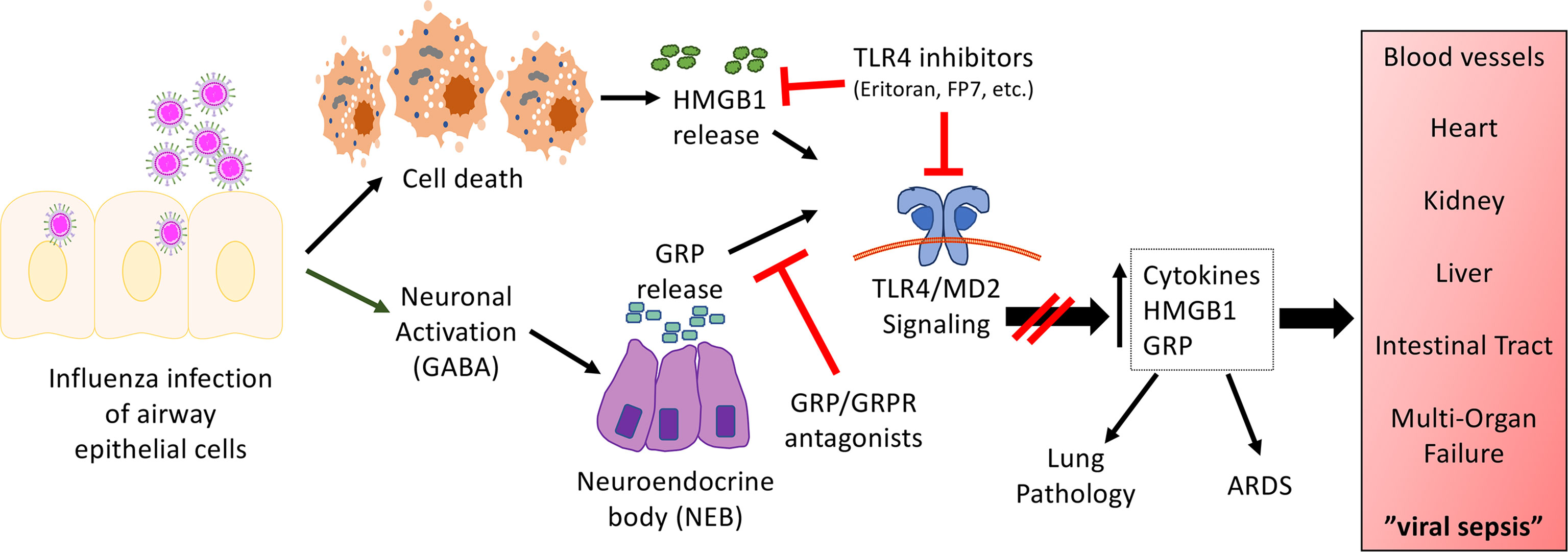
Figure 1 Hypothetical model for the involvement of TLR4 in influenza-induced disease. Our model proposes that influenza infects airway epithelial cells, leading to cell death and the release of DAMPs such as HMGB1. In turn, HMGB1 binds to the TLR4/MD2 complex to initiate signaling leading to release of cytokines and additional DAMPs. In addition, virus infection activates neurons in close proximity to NEBs, leading to the release of GRP. GRP has been shown to synergize with TLR4 agonists (82). Blocking either TLR4/MD2, HMGB1, or GRP mitigates influenza-induced ALI and disease pathogenesis. We hypothesize that this same mechanism is operative for other respiratory viral infections.
Other DAMPs have been implicated in the host response to influenza. Pulmonary surfactant proteins (SP), SP-A, SP-B, SP-C, and SP-D, were initially characterized for their ability to reduce the surface tension at the air-liquid interface of the lung (95, 96). Later, these proteins were also found to be involved in the host defense of the lung, primarily SP-A and SP-D (95, 97). SP-A and SP-D have been shown to act by binding to bacteria, viruses, and fungi allowing for microbial clearance through a variety of innate immune mechanisms (97). SP-A has been shown to stimulate NF-κB activation in both human and murine macrophages in vitro through the TLR4 signaling pathway (98). Later, it was found that SP-A was able to directly interact with the TLR4/MD2 complex (99). SP-D was found to interact with the extracellular domains of both TLR4 and TLR2 (100), while an earlier study suggested that both SP-A and SP-D could reduce inflammatory cytokines such as TNF-α by binding CD14 and, therefore, displace LPS for signaling in vitro (101). Several studies have focused on the role of SP-A and SP-D during influenza infection. Both SP-A and SP-D were found to neutralize the influenza virus through different mechanisms (102, 103). While survival of mice during influenza infection was not assessed, both SP-A-/- and SP-D-/- mice exhibit increased susceptibility to influenza compared with wildtype mice as measured by inflammatory cytokines, pathology, and viral burden (104, 105). Interestingly, the studies of SP-A and SP-D interactions with TLRs were carried out in vitro only, thus their role in influenza infection in vivo or its interplay with TLR4 has not yet been elucidated. Nonetheless, tracheal administration of exogenous SP-D to influenza-infected SP-D-/- mice at the time of infection alleviated the enhanced inflammatory response and reduced viral burden (104).
Role of TLR4 in Secondary Bacterial Infection After Influenza
Enhanced susceptibility to secondary bacterial infection after influenza accounts for many hospitalization and deaths, particularly during pandemics (106, 107). Co-infection with bacteria usually occurs within the first or second week of influenza infection (108). Historical records indicate that during the 1918 influenza pandemic a majority of all deaths were attributed to secondary bacterial pneumonia (106), while ~34% of the 2009 pandemic infections were associated with secondary bacterial infections (107). Studies have revealed that influenza-induced IFN-β selectively blocks induction of CXCL1/CXCL2 and neutrophil activation required to counter secondary bacterial infection with Streptococcus pneumoniae (108). We found that antagonizing TLR4 with Eritoran therapeutically during influenza virus infection, but prior to bacterial challenge, protected mice by reversing the IFN-β-mediated suppression, resulting in enhanced chemokine production (CXCL1 and CXCL2), increased myeloperoxidase activity, and reduced bacterial burden (109). Similarly, Eritoran treatment of cotton rats infected with non-adapted human pandemic H1N1, prior to infection with multi-drug resistant Staphylococcus aureus (MRSA), resulted in significant amelioration of lung pathology (109). Thus, blocking TLR4 therapeutically during influenza infection not only mitigates influenza-induced disease, but also prevents an enhanced inflammatory response to secondary bacterial infection. One of the molecular mechanisms pinpointed to explain these altered transcriptional responses are suggested by the observation that IFN-β-treatment of macrophages decreased recruitment of RNA polymerase II to the promoter of the Cxcl1 gene (109, 110). Other studies have shown similar outcomes with changes in neutrophil function after influenza that increase susceptibility to secondary bacterial infections (111, 112). Additional TFs, including STAT1, STAT2, and PPARγ, have been implicated in the host response to secondary bacterial infection after influenza infection (113–115).
Targeting TLR4 in Other Viral Respiratory Infections
The work of Imai et al. suggested that TLR4 signaling was important for induction of ALI by multiple viral and bacterial pathogens (19). While relatively few studies have been carried out to solidify this hypothesis, there are some that support it. Dengue virus (DENV) infection is endemic in many countries with approximately 400 million infections yearly (116). DENV causes a range of disease symptoms including classic dengue fever to severe hemorrhagic disease. Currently, there is no vaccine or therapy for DENV infection. The nonstructural protein 1 (NS1) of DENV can be either membrane-associated or found as a secreted, lipid-associated and soluble hexameric form (117). The soluble NS1 (sNS1) has been shown to directly activate TLR4 (as a PAMP) in both human peripheral blood mononuclear cells (PBMCs) and mouse bone marrow-derived macrophages. The induction of pro-inflammatory cytokines by DENV sNS1 was lost in TLR4-/-, but not TLR2-/- macrophages, and could be blocked by inhibiting TLR4 signaling with a TLR4 antagonist, Rhodobacter sphaeroides LPS (Rs-LPS), in human PBMCs. Capillary vascular leakage induced by NS1 in an in vivo model of DENV was also inhibited with the TLR4 antagonist Rhodobacter sphaeroides LPS (Rs-LPS) or with a neutralizing anti-TLR4 antibody (118).
Ebola virus (EBOV) made a significant impact during its re-emergence between 2013-2016 (119, 120) and continues to be of epidemiological concern for central African countries. EBOV infection fatalities are associated with significant levels of pro-inflammatory cytokines and chemokines (121). Younan et al. reported that treatment of mice with Eritoran immediately following a lethal challenge with EBOV resulted in increased protection without improving weight loss or clinical scores (79) in contrast to Eritoran treatment of mice infected with influenza (24). However, Eritoran treatment of EBOV-infected mice did significantly decrease the levels of certain cytokines and chemokines (79). Human Immunodeficiency Virus (HIV), another globally important virus, is known to dysregulate the immune system with the loss of T cell proliferation, shifting Th1 cells to Th2 cells and associated cytokines, and high levels of secreted IL-10 (122–126). The HIV Tat protein has been shown to play a role in the host immune system modulation by acting at the cell surface to stimulate cytokine secretion, particularly IL-10, on monocytes and macrophages (127, 128). The N-terminal sequence of Tat was reported to interact with TLR4 (129). Eritoran inhibited HIV-1 Tat-dependent IL-10 induction in PBMCs and the physical and functional interaction was further confirmed by using a neutralizing anti-TLR4 antibody (130).
Do Differences in Genetic Backgrounds of Mice Make a Difference?
Studies by Nhu et al. (20) and Shirey et al. (24) showed that mice with a targeted mutation in TLR4 (TLR4-/- mice; kindly provided by Shizuo Akira (131) and extensively backcrossed to C57BL/6J mice) were resistant to PR8 and other strains of human influenza. More recently, using a CRISPR/Cas9 approach, mice were engineered onto a C57BL/6J background that express two TLR4 SNPs (D298G and N397I) that are homologous to the common TLR4 SNPs found in humans (D299G and T399I) (132) and have been reported to result in LPS-hyporesponsiveness (133). As expected, the “TLR4-SNP” mice were shown to be LPS-hyporesponsive in vivo and in vitro, but were more responsive to LPS than Akira’s TLR4-/- strain backcrossed >12 times to C57BL/6J mice. When challenged with influenza PR8 (LD90), wildtype mice succumbed to infection, while TLR4-/- mice were refractory (p<0.0001) (132), as previously reported (24, 25). The PR8-infected TLR4-SNP mice, however, showed significantly reduced lethality, but were somewhat more susceptible than TLR4-/- mice. Increased resistance of the TLR4-SNP mice to PR8 infection was confirmed with respect to decreased lung pathology, as well as diminished cytokine gene and protein expression (132).
In contrast, in two other reports (134, 135), a different Tlr4 mutant mouse strain, B6.B10ScN-Tlr4lps-del/JthJ, was shown to succumb to infection with the mouse-adapted A/WSN/33 strain and also to the PR8 strain of influenza, leading to the conclusion that loss of TLR4 did not protect against influenza-induced lethality (135). It is important to emphasize that the Tlr4 mutation in the B6.B10ScN-Tlr4lps-del/JthJ strain is not the result of a targeted mutation; rather, this mouse strain was derived from a spontaneous deletion within Tlr4 that occurred in the C57BL/10ScN strain between 1947 and 1961 (136). The B6.B10ScN-Tlr4lps-del/JthJ strain originated from the C57BL/10ScN colony at NCI Frederick (NIH), was transferred to the University of Texas Southwest Medical Center where it was backcrossed for 5 generations onto a C57BL/6 background. The strain was returned to Jackson Labs in 2008 where it was backcrossed to C57BL/6J for one additional generation during re-derivation (137). Although B6.B10ScN-Tlr4lps-del/JthJ strain used for these studies was indeed LPS-unresponsive, it is entirely possible that it carries background genes derived from the C57BL/10ScN parental strain that differ from C57BL/6J inbred strain and contribute to influenza sensitivity, since minor mutations in closely related strains are known to result in significant changes in phenotypes. For example, the progenitor strain, C57BL/10ScN, and its successor, C57BL/10ScCR, both express the deletion in Tlr4; however, the latter strain possesses an additional point mutation in the gene encoding the IL-12Rβ2, causing it to express a distinct phenotype affecting IFN-γ production in response to infection (138). This has also been observed among closely related C57BL/6 substrains (139). To further evaluate the role of distinct strain backgrounds and the role of TLR4 in influenza-induced lethality, wild-type C3H/HeOuJ and C3H/HeJ mice, the latter strain shown to express a point mutation in the TIR domain of TLR4 that resulted in a loss of TLR4-mediated signaling (140, 141), were challenged with mouse-adapted influenza H1N1 PR8. As previously reported in C57BL/6J versus TLR4-/- mice (20, 24), the TLR4 signaling-deficient C3H/HeJ strain was significantly more resistant to PR8-induced lethality than the closely related, LPS-responsive C3H/OuJ strain from which it was derived when challenged with either a sublethal (p = 0.0118) or lethal dose (p = 0.0004) of influenza strain PR8. Thus, the increased resistance of TLR4-/- and C3H/HeJ mice, coupled with the ability of many distinct TLR4 antagonists to block influenza-mediated cytokine production, ALI, and lethality, strongly support a critical role for TLR4 signaling in influenza-induced disease.
Concluding Remarks
During the past year, we have all become too familiar with COVID-19 pandemic that has led to ALI, as well as the more severe acute respiratory distress syndrome (ARDS) and the deaths of millions worldwide. In support of the observation that TLR4-/- mice are extremely refractory to influenza infection (20, 24), other studies have subsequently shown that many TLR4 antagonists, that act by a variety of distinct mechanisms to prevent signaling all block influenza-induced lethality. This is independent of the influenza virus strain used and has been confirmed in other rodent models of influenza infection. More importantly, the interference with TLR4 signaling by agents like Eritoran has shown promise with other viruses, again supporting the importance of TLR4 in other virus-induced diseases. The striking similarities between influenza- and SARS-CoV-2 induced ARDS caused by a cytokine storm leading to loss of homeostasis and multiorgan failure, has been referred to as “viral sepsis” (142). By understanding the mechanisms by which the virus-induced “cytokine storm” is mitigated, we expect to discover therapeutic targets in the influenza model that will be applicable also to patients with severe SARS-CoV-2 and other virus-induced lung diseases. A recent study reported that the SARS-CoV-2 trimeric spike (S) protein directly interacted with and activated TLR4 in vitro in human and murine macrophage-like cell lines, and in bone marrow-derived macrophages from wildtype, but not TLR4-/-, mice (143). Moreover, treatment of cells with Resatorvid (TAK-242), that blocks MyD88 interaction with TLR4 (31), also blocked S protein-induced Il1b mRNA (143). However, the finding that inhibition of either MD2 or CD14 blunted gene expression suggests that the S protein may be binding not only to TLR4, as suggested by their binding data, but also to these TLR4 co-receptors. Future studies to confirm this interesting report will be warranted to define more clearly the possible interaction between S protein and the TLR4 signaling complex. Collectively, insights into key molecular interactions that underlie host-pathogen interactions support the novel concept that host-directed therapeutics that selectively target interactions between PRRs and PAMPs/DAMPs, or the signaling pathways they initiate, might serve as potentially uniquely effective therapeutic strategies that could be mobilized to target multiple infectious pathogens.
Author Contributions
KS, JB, and SV contributed to the original writing and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by AI123371 (SV), AI125215 (SV/JB), and AI159507 (KS).
Conflict of Interest
JB was employed by Sigmovir Biosystems, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALI, Acute lung injury; ARDS, Acute respiratory distress syndrome; DAMP, Danger-associated molecular pattern; DENV, Dengue virus; EGCG, Epigallocatechin-3-gallate; EBOV, Ebola virus; GABA, γ-amibobutric acid; GRP, Gastrin releasing peptide; HMGB1, High-mobility group box 1; HIV, Human Immunodeficiency virus; IAV, Influenza A virus; LRR, Leucine-rich repeats; LPS, Lipopolysaccharide; LSW, Liu Shen Wan; MDA, Malondialdehyde; MAC, Monoacetylcurcumin; MRSA, Multi-drug resistant Staphylococcus aureus; NAC, N-acetyl-L-cystine; NA, Neuraminidase; NEB, Neuroendocrine bodies; OxPAPC, Oxidized 1-palmitoyl-2-arachidonoyl-phosphatidycholine; PAMP, Pathogen-associated molecular pattern; PRR, Pattern recognition receptors; PBMC, Peripheral blood mononuclear cells; ROS, Reactive oxygen species; Rs-LPS, Rhodobacter sphaeroides LPS; SNP, Single nucleotide polymorphisms; (S) protein, Spike protein; SFN, Sulforaphane; SP, Surfactant protein; TIR, Toll-IL-1 receptor resistance; TLR, Toll-like receptor; TF, Transcription factor.
References
1. Tolle LB, Standifor T. Danger-Associated Molecular Patterns (DAMPs) in Acute Lung Injury. J Pathol (2013) 229:145–56. doi: 10.1002/path.4124
2. Gao JJ, Diesl V, Wittmann T, Morrison DC, Ryan JL, Vogel SN, et al. Regulation of Gene Expression in Mouse Macrophages Stimulated With Bacterial CpG-DNA and Lipopolysaccharide. J Leukoc. Biol (2002) 72:1234–45. doi: 10.1189/jlb.72.6.1234
3. Andersson U, Yang H, Harris H. High-Mobility Group Box 1 Protein (HMGB1) Operates as an Alarmin Outside as Well as Inside Cells. Semin Immunol (2018) 38:40–8. doi: 10.1016/j.smim.2018.02.011
4. Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W, et al. MD-2 Is Required for Disulfide HMGB1-Dependent TLR4 Signaling. J Exp Med (2015) 212:5–14. doi: 10.1084/jem.20141318
5. Iwasaki I. A Virological View of Innate Immune Recognition. Annu Rev Microbiol (2012) 66:177–96. doi: 10.1146/annurev-micro-092611-150203
6. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
7. Toshchakov VY, Vogel SN. Cell-Penetrating TIR BB Loop Decoy Peptides a Novel Class of TLR Signal Inhibitors and a Tool to Study Topology of TIR-TIR Interactions. Exp Opin Biol Ther (2007) 7:1035–50. doi: 10.1517/14712598.7.7.1035
8. Bryant CE, Gay NJ, Heymans S, Sacre S, Schaefer L, Midwood KS. Advances in Toll-Like Receptor Biology: Modes of Activation by Diverse Stimuli. Crit Rev Biochem Mol Biol (2015) 50:359–79. doi: 10.3109/10409238.2015.1033511
9. Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, et al. Influenza-Associated Hospitalizations in the United States. Jama (2004) 292:1333–40. doi: 10.1001/jama.292.11.1333
10. Reid AH, Taubenberger JK, Fanning TG. The 1918 Spanish Influenza: Integrating History and Biology. Microbes Infect (2001) 3:81–7. doi: 10.1016/S1286-4579(00)01351-4
11. Taubenberger JK, Reid AH, Janczewski TA, Fanning TG. Integrating Historical, Clinical and Molecular Genetic Data in Order to Explain the Origin and Virulence of the 1918 Spanish Influenza Virus. Philos Trans R Soc Lond B Biol Sci (2001) 356:1829–39. doi: 10.1098/rstb.2001.1020
12. Brockwell-Staats C, Webster RG, Webby RJ. Diversity of Influenza Viruses in Swine and the Emergence of a Novel Human Pandemic Influenza A (H1n1). Influenza Other Respi Viruses (2009) 3:207–13. doi: 10.1111/j.1750-2659.2009.00096.x
13. Osterholm MT, Kelley NS, Sommer A, Belongia EA. Efficacy and Effectiveness of Influenza Vaccines: A Systematic Review and Meta-Analysis. Lancet Infect Dis (2012) 12:36–44. doi: 10.1016/S1473-3099(11)70295-X
14. Centers for Disease Control and Prevention (CDC)Interim Adjusted Estimates of Seasonal Influenza Vaccine Effectiveness - United States, February 2013. MMWR Morb Mortal Wkly Rep (2013) 62(7):119–23.
15. Centers for Disease Control and Prevention (CDC) Emergence of Avian Influenza A (H7N9) Virus Causing Severe Human Illness – China, February – April 2013. MMWR Morb Mortal Wkly Rep (2013) 62:366–71.
16. Baranovich T, Webster RG, Govorkova EA. Fitness of Neuraminidase Inhibitor-Resistant Influenza A Viruses. Curr Opin Virol (2011) 1:574–81. doi: 10.1016/j.coviro.2011.08.006
17. Leang SK, Deng YM, Shaw R, Caldwell N, Iannello P, Komadina N, et al. Influenza Antiviral Resistance in the Asia-Pacific Region During 2011. Antiviral Res (2013) 97:206–10. doi: 10.1016/j.antiviral.2012.12.016
18. Iwasaki A, Pillai PS. Innate Immunity to Influenza Virus Infection. Nat Rev Immunol (2014) 14:315–28. doi: 10.1038/nri3665
19. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo, et al. Identification of Oxidative Stress and Toll-Like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell (2008) 133:235–49. doi: 10.1016/j.cell.2008.02.043
20. Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, et al. Novel Signaling Interactions Between Proteinase-Activated Receptor 2 and Toll-Like Receptors In Vitro and In Vivo. Mucosal Immunol (2010) 3:29–39. doi: 10.1038/mi.2009.120
21. Mullarkey M, Rose JR, Bristol J, Kuwata T, Kimura A, Kobayashi S, et al. Inhibition of Endotoxin Response by E5564, a Novel Toll-Like Receptor 4-Directed Endotoxin Antagonist. J Pharmacol Exp Ther (2003) 304:1093–102. doi: 10.1124/jpet.102.044487
22. Kalil AC, LaRosa SP, Jagadish G, Melvyn L, Opal SM. The Eritoran Sepsis Study Grou. Influence of Severity of Illness on the Effects of Eritoran Tetrasodium (E5564) and on Other Therapies for Severe Sepsis. Shock (2011) 36:327–31. doi: 10.1097/SHK.0b013e318227980e
23. Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The Structural Basis of Lipopolysaccharide Recognition by the TLR4-MD-2 Complex. Nature (2009) 458:1191–5. doi: 10.1038/nature07830
24. Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM. The TLR4 Antagonist Eritoran Protects Mice From Lethal Influenza Infection. Nature (2013) 497:498–502. doi: 10.1038/nature12118
25. Shirey KA, Lai W, Patel MC, Pletneva LM, Pang C, Kurt-Jones E, et al. Novel Strtegies for Targeting Innate Immune Responses to Influenza. Mucosal Immunol (2016) 9:1173–82. doi: 10.1038/mi.2015.141
26. Blanco JC, Boukhvalova MS, Perez DR, Vogel SN, Kajon A. Modeling Human Respiratory Viral Infections in the Cotton Rat (Sigmodon Hispidus). J Antivir Antiretrovir (2014) 6:40–2. doi: 10.4172/jaa.1000093
27. Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson A-L, Mauri D, et al. Protection From Lethal Gram-Negative Bacterial Sepsis by Targeting Toll-Like Receptor 4. Proc Natl Acad Sci USA (2009) 106:2348–52. doi: 10.1073/pnas.0808146106
28. Couture LA, Piao W, Ru LW, Vogel SN, Toshchakov VY. Targeting Toll-Like Receptor (TLR) Signaling by Toll/Interleukine-1 Receptor (TIR) Domain-Containing Adapter Protein/MyD88 Adapter-Like (TIRAP/Mal)-Derived Decoy Peptides. J Biol Chem (2012) 287:24641–8. doi: 10.1074/jbc.M112.360925
29. Piao W, Shirey KA, Ru LW, Lai W, Szmacinski H, Snyder GA, et al. A Decoy Peptide That Disrupts TIRAP Recruitment to TLRs Protects Mice in a Murine Model of Influenza. Cell Rep (2015) 11:1941–52. doi: 10.1016/j.celrep.2015.05.035
30. Lysakova-Devine T, Keogh B, Harrington B, Nagpal K, Halle A, Golenbock DT, et al. Viral Inhibitory Peptide of TLR4, A Peptide Derived From Vaccinia Protein A46, Specifically Inhibits TLR4 by Directly Targeting MyD88 Adaptor-Like and TRIF-Related Adaptor Molecule. J Immunol (2010) 185:4261–71. doi: 10.4049/jimmunol.1002013
31. Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, et al. A Novel Cyclohexene Derivative, Ethyl (6R)-60[N-(2-Chloro-4-fluorophenyl0sulfamoyl]cyclohex-1-ene-1-carboxylate (Tak-242), Selectively Inhibits Toll-Like Receptor 4-Mediated Cytokine Production Through Suppression of Intracellular Signaling. Mol Pharmacol (2006) 69:1288–95. doi: 10.1124/mol.105.019695
32. Shirey KA, Lai W, Brown LJ, Blanco JCG, Beadenkopf R, Wang Y, et al. Select Targeting of Intracellular Toll-Interleukin-1 Receptor Resistance Domains for Protection Against Influenza-Induced Disease. Innate Immun (2020) 26:26–34. doi: 10.1177/1753425919846281
33. Cighetti R, Ciaramelli C, Enza S, Zanoni I, Kubik L, Ardá-Freire A, et al. Modulation of CD14 and TLR4-MD2 Activities by a Synthetic Lipid A Mimetic. ChemBioChem (2014) 15:250–8. doi: 10.1002/cbic.201300588
34. Perrin-Cocon L, Aublin-Gex A, Sestito SE, Shirey KA, Patel MC, André P, et al. The Synthetic TLR4 Antagonist FP7 Inhibits LPS-Induced Cytokine Production and Metabolic Reprogramming in Dendritic Cells, and Protects Mice From Lethal Influenza Infection. Sci Rep (2017) 7:40791. doi: 10.1038/srep40791
35. Prantner D, Shirey KA, Lai W, Lu W, Cole AM, Vogel SN, et al. The θ-Defensin Retrocyclin 101 Inhibits TLR4- and TLR2-Dependent Signaling and Protects Mice Against Influenza Infection. J Leuk Biol (2017) 102:1103–13. doi: 10.1189/jlb.2A1215-567RR
36. Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Labg MS, Grailer JJ, et al. Zonulin as Prehaptoglobin2 Regulates Lung Permeability and Activates the Complement System. Am J Physiol Lung Cell Mol Physiol (2013) 304:L863–72. doi: 10.1152/ajplung.00196.2012
37. Seong KJ, Lee HG, Kook MS, Ko Jung JY, Kim WJ. Epigallocatechin-3-Gallate Rescues LPS-Impaired Adult Hippocampal Neurogenesis Through Suppressing the TLR4-NF- B Signaling Pathway in Mice. Korean J Physiol Pharmacol (2016) 20:41–51. doi: 10.4196/kjpp.2016.20.1.41
38. Hong BE, Fujimura Y, Yamada K, Tachibana H. TLR4 Signaling Inhibitory Pathway Induced by Green Tea Polyphenol Epigallocatechin-3-Gallate Through 67-kDa Laminin Receptor. J Immunol (2010) 185:33–45. doi: 10.4049/jimmunol.0903742
39. Xu M-J, Liu B-J, Wang C-L, Wang G-H, Tian Y, Wang S-H, et al. Epigallocatechin-3-Gallate Inhibits TLR4 Signaling Through the 67-kDa Laminin Receptor and Effectively Alleviates Acute Lung Injury Induced by H9N2 Swine Influenza Virus. Int Immunopharmacol (2017) 52:24–33. doi: 10.1016/j.intimp.2017.08.023
40. Yang Z-F, Bai L-P, Huang W-B, Li X-S, Zhao S-S, Zhong N-S, et al. Comparison of In Vitro Antiviral Activity of Tea Polyphenols Against Influenza A and B Viruses and Structure-Activity Relationship Analysis. Fitoterapia (2014) 93:47–53. doi: 10.1016/j.fitote.2013.12.011
41. Seo S-U, Kwon H-J, Song J-H, Byun Y-H, Seong BL, Kawai T, et al. MyD88 Signaling Is Indispensable for Primary Influenza A Virus Infection, But Dispensable for Secondary Infection. J Virol (2010) 84:12713–22. doi: 10.1128/JVI.01675-10
42. Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MBA. Mapping the Innate Signaling Cascade Essential for Cytokine Storm During Influenza Virus Infection. Proc Natl Acad Sci USA (2014) 111:3799–804. doi: 10.1073/pnas.1400593111
43. He G, Dong C, Luan Z, McAllan BM, Xu T, Zhao L, et al. Oxygen Free Radical Involvement in Acute Lung Injury Induced by H5N1 Virus in Mice. Influenza Other Resp Viruses (2013) 6:945–53. doi: 10.1111/irv.12067
44. Dekhuijzen PNR. Antioxidant Properties of N-Acetylcysteine: Their Relevance in Relation to Chronic Obstructive Pulmonary Disease. Eur Respir J (2004) 23:629–36. doi: 10.1183/09031936.04.00016804
45. Zhang R-H, Li C-H, Wang C-L, Xu M-J, Xu T, Wei D, et al. N-acetyl-L-cystine (NAC) Protects Against H9N2 Swine Influenza Virus-Induced Acute Lung Injury. Int Immunopharmacol (2014) 22:1–8. doi: 10.1016/j.intimp.2014.06.013
46. Geiler J, Michaelis M, Naczk P, Leutz A, Langer K, Doer HW, et al. N-Acetyl-L-Cystein (NAC) Inhibits Virus Replication and Expression of Pro-Inflammatory Molecules in A549 Cells Infected With Highly Pathogenic H5N1 Influenza A Virus. Biochem Pharmacol (2010) 79:413–20. doi: 10.1016/j.bcp.2009.08.025
47. Byun MR, Jeong H, Bae SJ, Kim AR, Hwang ES, Hong JH. TAZ Is Required for the Osteogenic and Anti-Adipogenic Activities of Kaempferol. Bone (2012) 50:364–72. doi: 10.1016/j.bone.2011.10.035
48. Xiao HB, Lu XY, Sun ZL, Zhang HB. Kaempferol Regulates OPN-CD44 Pathway to Inhibit the Atherogenesis of Apolipoprotein E Deficient Mice. Toxicol Appl Pharmacol (2011) 257:405–11. doi: 10.1016/j.taap.2011.09.024
49. Crespo I, García-Mediavilla MV, Gutiérrez B, Sánchez-Campos S, Tuñón MJ, González-Gallego J. A Comparison of the Effects of Kaempferol and Quercetin on Cytokine-Induced Pro-Inflammatory Status of Cultured Human Endothelial Cells. Br J Nutr (2008) 100:968–76. doi: 10.1017/S0007114508966083
50. Olszewska M. Separation of Quercetin, Sexangularetin, Kaempferol and Isorhamnetin for Simultaneous HPLC Determination of Flavonoid Aglycones in Inflorescences, Leaves and Fruits of Three Sorbus Species. J Pharm Biomed Anal (2008) 48:629–35. doi: 10.1016/j.jpba.2008.06.004
51. Kowalski J, Samojedny A, Paul M, Pietsz G, Wilczok T. Effect of Apigenin, Kaempferol, and Resveratrol on the Expression of interleukin-1β and Tumor Necrosis Factor-α Genes In J774.2 Macrophages. Pharmacol Rep (2005) 57:390–4.
52. Khajuria V, Gupt S, Sharma N, Tiwari H, Bhardwaj S, Dutt P, et al. Kaempferol-3-O-β-D-Glucuronate Exhibit Potential Anti-Inflammatory Effect in LPS Stimulated RAW 264.7 Cells and Mice Model. Int Immunopharmacol (2018) 57:62–71. doi: 10.1016/j.intimp.2018.01.041
53. Lee S-B, Shin J-S, Han H-S, Lee H-H, Park JC, Lee K-T. Kaempferol 7-O-β-D-Glucoside Isolated From the Leave of Cudrania Tricuspidata Inhibits LPS-Induced Expression of Pro-Inflammatory Mediators Through Inactivation of NF-kB, Ap-1, and JAK-STAT in RAW 264.7 Macrophages. Chemico-Biological Interact (2018) 284:101–11. doi: 10.1016/j.cbi.2018.02.022
54. Zhang R, Ai X, Duan Y, Xue M, He W, Wang C, et al. Kaempferol Ameliorates H9N2 Swine Influenza Virus-Induced Acute Lung Injury by Inactivation of TLR4/MyD88-Mediated NF-Kb and MAPK Signaling Pathways. Biomedicine Pharmacotherapy (2017) 89:660–72. doi: 10.1016/j.biopha.2017.02.081
55. Hewling SJ, Kalman DS. Curcumin: A Review of It’s Effects on Human Health. Foods (2017) 6:92. doi: 10.3390/foods6100092
56. Prasad S, Tyagi AK, Aggarwal BB. Recent Developments in Delivery, Bioavailability, Absorption and Metabolism of Curcumin: The Golden Pigment From Golden Spice. Cancer Res Treat (2014) 46:2–18. doi: 10.4143/crt.2014.46.1.2
57. Liang G, Yang S, Zhou H, Shao L, Huang K, Xiao J, et al. Synthesis, Crystal Structure and Anti-Inflammatory Properties of Curcumin Analogues. Eur J Med Chem (2009) 44:915–9. doi: 10.1016/j.ejmech.2008.01.031
58. Wang Y, Shan X, Dai Y, Jiang L, Chen G, Zhang Y, et al. Curcumin Analog L48H37 Prevents Lipopolysaccharide-Induced TLR4 Signaling Pathway Activation and Sepsis Via Targeting MD2. J Pharmacol Exp Ther (2015) 353:539–50. doi: 10.1124/jpet.115.222570
59. Zhang Y, Liang D, Dong L, Ge X, Xu F, Chen W, et al. Anti-Inflammatory Effects of Novel Curcumin Analogs in Experimental Acute Lung Injury. Respir Res (2015) 16:4. doi: 10.1186/s12931-015-0199-1
60. Jurenka JS. Anti-Inflammatory Properties of Curcumin, a Major Constituent of Curcuma Longa: A Review of Preclinical and Clinical Research Altern. Med Rev: A J Clin Ther (2009) 14(2):141–53.
61. Chen D-Y, Shien J-H, Tiley L, Chiou S-S, Wang S-Y, Chang T-J, et al. Curcumin Inhibits Influenza Virus Infection and Haemagglutination Activity. Food Chem (2010) 119:1346–51. doi: 10.1016/j.foodchem.2009.09.011
62. Chen T-Y, Chen D-Y, Wen H-W, Ou J-L, Chiou S-S, Chen J-M, et al. Inhibition of Enveloped Viruses Infectivity by Curcumin. PloS One (2013) 8:e62482. doi: 10.1371/journalpone.0062482
63. Richart SM, Li Y-L, Mizushina Y, Chang Y-Y, Chung T-Y, Chen G-H, et al. Synergic Effect of Curcumin and Its Structural Analogue (Monoacetylcurcumin) on Anti-Influenza Virus Infection. Drug Anal (2018) 26:1015–23. doi: 10.1016/j.jfda.2017.12.006
64. Dai J, Gu L, Su Y, Wang Q, Zhao Y, Chen X, et al. Inhibition of Curcumin on Influenza A Virus Infection and Influenzal Pneumonia Via Oxidative Stress, TLR2/4, P38/JNK MAPK and NF-kB Pathways. Int Immunopharmacol (2018) 54:177–87. doi: 10.1016/j.intimp.2017.11.009
65. Ma H, Kou J, Zhu D, Yan Y, Yu B. Liu-Shen-Wan, a Traditional Chinese Medicine, Improves Survival in Sepsis Induced by Cecal Ligation and Puncture Via Reducing TNF-α Levels, MDA Content and Enhancing Macrophage Phagocytosis. Int Immunopharmacol (2006) 6:1355–62. doi: 10.1016/j.intimp.2006.03.003
66. Zheng GH, Wang GH. Progress of Clinical Applications and Pharmacological Studies of Liu-Shen-Wan. Hubei J Tradit Chin Med (1996) 18:56–7.
67. Han SJ, Pei GS. Clinical Application of Liu-Shen-Wan. Chin J Ethnomed Ethnopharmacy (2000) 10:57–8.
68. Ma H-Y, Kou J-P, Wang J-R, Yu B-Y. Evaluation of the Anti-Inflammatory and Analgesic Activities of Liu-Shen-Wan and Its Individual Fractions. J Ethnopharmacol (2007) 112:108–14. doi: 10.1016/j.jep.2007.02.008
69. Ma Q, Huang W, Zhao J, Yang Z. Liu Shen Wan Inhibits Influenza a Virus and Excessive Virus-Induced Inflammatory Response Via Suppression of TLR4/NF-kB Signaling Pathway In Vitro and In Vivo. J Ethnopharmacology (2020) 252:112584. doi: 10.1016/j.jep.2020.112584
70. Molteni M, Bosi A, Rossetti C. Natural Products With Toll-Like Receptor 4 Antagonist Activity. Int J Inflammation (2018) 2018:2859135. doi: 10.1155/2018/2859135
71. Mahn A, Castillo A. Potential of Sulforaphane as a Natural Immune System Enhancer: A Review. Molecules (2021) 26:752. doi: 10.3390/molecules26030752
72. Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM, et al. Sulforaphane Suppresses Oligomerization of TLR4 in a Thiol-Dependent Manner. J Immunol (2010) 184:411–9. doi: 10.4049/jimmunol.0803988
73. Mohan S, Gupta D. Crosstalk of Toll-Like Receptors Signaling and Nrf2 Pathway for Regulation of Inflammation. Biomedicine Pharmacotherapy (2018) 108:1866–78. doi: 10.1016/j.biopha.2018.10.019
74. Huang S, Yue B, Ke J, Zhu XD, Shen Y, Zhai M, et al. Nuclear Factor Erythroid 2-Related Factor 2 Regulates Toll-Like Receptor 4 Innate Responses in Mouse Liver Ischemia-Reperfusion Injury Through Akt-forkhead Box Protein O1 Signaling Network. Transplantation (2014) 98:721–8. doi: 10.1097/TP.0000000000000316
75. Shah VB, Williams DL, Keshvara L. β-Glucan Attenuates TLR2- and TLR4-Mediated Cytokine Production by Microglia. Neurscience Lett (2009) 458:111–5. doi: 10.1016/j.neulet.2009.04.039
76. Vaclav V, Jana VA. Novel Glucan-Sulforaphane Combination Stimulates Immune Response to Influenza in Mouse Model. Am J Immunol (2016) 12:20–8. doi: 10.3844/ajisp.2016.20.28
77. Kesic MJ, Simmons SO, Bauer R. Nrf2 Expression Modifies Influenza A Entry and Replication in Nasal Epithelial Cells. Free Radic Biol Med (2011) 51:444–53. doi: 10.1016/j.freeradbiomed.2011.04.027
78. Li Z, Liu Y, Fang Z, Yang L, Zhuang M, Zhang Y, et al. Natural Sulforaphane From Broccoli Seeds Against Influenza A Virus Replication in MDCK Cells. Nat Prod Commun (2019) 14:1–8. doi: 10.1177/1934578X19858221
79. Younan P, Ramanathan P, Graber J, Gusovsky F, Bukreyev A. The Toll-Like Receptor 4 Antagonist Eritoran Protects Mice From Lethal Filovirus Challenge. mBio (2017) 8:e00226–17. doi: 10.1128/mBio.00226-17
80. Wang W, Cole AM, Hong T, Waring AJ, Lehrer RI. Retrocyclin, an Antiretroviral θ-Defensin, Is a Lectin. J Immunol (2003) 170:4708–16. doi: 10.4049/jimmunol.170.9.4708
81. Martínez A, Zudaire E, Julián M, Moody TW, Cuttitta F. Gastrin-Releasing Peptide (GRP) Induces Angiogenesis and the Specific GRP Blocker 77427 Inhibits Tumor Growth In Vitro and In Vivo. Oncogene (2005) 24:4106–13. doi: 10.1038/sj.onc.1208581
82. Shirey KA, Sunday ME, Lai W, Patel MC, Blanco JCG, Cuttitta F, et al. Novel Role of Gastrin Releasing Peptide-Mediated Signaling in the Host Response to Influenza Infection. Mucosal Immunol (2019) 12:223–31. doi: 10.1038/s41385-018-0081-9
83. Cuttitta F, Desmond CN, Mulshine J, Moody TW, Fedorko J, Fischler A, et al. Bombesin-Like Peptides can Function as Autocrine Growth Factors in Human Small-Cell Lung Cancer. Nature (1985) 316:823–6. doi: 10.1038/316823a0
84. Moody TW, Venugopal R, Zia F, Patierno S, Leban JJ, McDermed J. BW2258U89: A GRP Receptor Antagonist Which Inhibits Small Cell Lung Cancer Growth. Life Sci (1995) 56:521–9. doi: 10.1016/0024-3205(94)00481-7
85. Marquez C, Treston A, Moody E, Jakowlew S, Moody TW. The Metabolism of BW2258U89, a GRP Receptor Antagonist. Neuropeptides (2000) 34:108–15. doi: 10.1054/npep.2000.0798
86. Rothan HA, Han HC, Ramasamy TS, Othman S, Rahman NA, Yusof R. Inhibition of Dengue NS2B-NS3 Protease and Viral Replication in Vero Cells by Recombinant Retrocyclin-1. BMC Infect Dis (2012) 21:314. doi: 10.1186/1471-2334-12-314
87. Anas A, van der Poll T, de Vos AF. Role of CD14 in Lung Inflammation and Infection. In: Vincent JL, editor. Yearbook of Intensive Care and Emergency Medicine. Berlin, Heidelberg: Springer (2010). p. 129–40.
88. Pauligk C, Nain M, Reiling N, Gemsa D, Kaufmann D. CD14 Is Required for Influenza A Virus-Induced Cytokine and Chemokine Production. Immunobiology (2004) 209:3–10. doi: 10.1016/j.imbio.2004.04.002
89. Lee H-K, Dunzendorfer S, Soldau K, Tobias PS. Double-Stranded RNA-Mediated TLR3 Activation Is Enhanced by CD14. Immunity (2006) 24:153–63. doi: 10.1016/j.immuni.2005.12.012
90. Shinya K, Ito M, Makino A, Tanaka M, Miyake K, Eisfeld AJ, et al. The TLR4-TRIF Pathway Protects Against H5N1 Influenza Virus Infection. J Virol (2012) 19:19–24. doi: 10.1128/JVI.06168-11
91. Patel MC, Shirey KA, Boukhvalova MS, Vogel SN, Blanco JCG. Serum High-Mobility-Group Box 1 as a Biomarker and a Therapeutic Target During Respiratory Virus Infections. mBio (2018) 9:e00246–18. doi: 10.1128/mBio.00246-18
92. Nosaka N, Yashiro M, Yamada M, Fujii Y, Tsukahara H, Liu K, et al. Anti-High Mobility Group Box-1 Monoclonal Antibody Treatment Provides Protection Against Influenza A Virus (H1N1)-Induced Pneumonia in Mice. Crit Care (2015) 19:249. doi: 10.1186/s13054-015-0983-9
93. Sunday MW. Oxygen, Gastrin-Releasing Peptide, and Pediatric Lung Disease: Life in the Balance. Front Pediatr (2014) 18:72. doi: 10.3389/fped.2014.00072
94. Jaeger N, Czepielewski RS, Bagatini M, Porto BN, Bonorino C. Neuropeptide Gastrin-Releasing Peptide Induces PI3K/Reactive Oxygen Species-Dependent Migration in Lung Adenocarcinoma Cells. Tumor Biol (2017) 39:1–11. doi: 10.1177/1010428317694321
95. Wright J. Immunoregulatory Functions of Surfactant Proteins. Nat Rev Immunol (2005) 5:58–68. doi: 10.1038/nri1528
96. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. Surfactant Proteins SP-A and SP-D: Structure, Function and Receptors. Mol Immun (2006) 43:1293–315. doi: 10.1016/j.molimm.2005.08.004
97. Nayak A, Dodagatta-Marri E, Tsolaki AG, Kishore U. An Insight Into the Diverse Roles of Surfactant Proteins, SP-A and SP-D in Innate and Adaptive Immunity. Front Immun (2012) 3:131. doi: 10.3389/fimmu.2012.00131
98. Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting Edge: The Immunostimulatory Activity of the Lung Surfactant Protein-a Involves Toll-Like Receptor 4. J Immunol (2002) 168:5989–92. doi: 10.4049/jimmunol.168.12.5989
99. Yamada C, Sano H, Shimizu T, Mitsuzawa H, Nishitani C, Himi T. Surfactant Protein A Directly Interacts With TLR4 and MD-2 and Regulates Inflammatory Cellular Response. J Biol Chem (2006) 28:21771–80. doi: 10.1074/jbc.M513041200
100. Ohya M, Nishitani C, Sano H, Yamada C, Mitsuzawa H, Shimizu T, et al. Human Pulmonary Surfactant Protein D Binds the Extracellular Domains of Toll-Like Receptors 2 and 4 Through the Carbohydrate Recognition Domain by a Mechanism Different From Its Binding to Phosphatidylinositol and Lipopolysaccharide. Biochemistry (2006) 45:8657–64. doi: 10.1021/bi060176z
101. Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. Surfactant Proteins A and D Bind CD14 by Different Mechanisms. J Bio. Chem (2000) 275:22442–51. doi: 10.1074/jbc.M001107200
102. Benne C, Kraaijeveld C, Van Strijp J, Brouwer E, Harmen M, Verhoef J, et al. Interactions of Surfactant Protein A With Influena A Viruses: Binding and Neutralization. J Infect Dis (1995) 171:335–41. doi: 10.1093/infdis/171.2.335
103. Hartshorn KL, Webby R, White MR, Tecle T, Pan C, Boucher S, et al. Role of Viral Hemagglutinin Glycosylation in Anti-Influenza Activities of Recombinant Surfactant Protein D. Respir Res (2008) 9(65). doi: 10.1186/1465-9921-9-65
104. LeVine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. Surfactant Protein D Enhances Clearance of Influenza A Virus From the Lung In Vivo. J Immunol (2001) 167:5868–73. doi: 10.4049/jimmunol.167.10.5868
105. LeVine AM, Hartshorn K, Elliot J, Whitsett J, Korfhagen T. Absensence of SP-A Modulates Innate and Adaptive Defense Responses to Pulmonary Influenza Infection. Am J Physiol Lung Cell Mol Physiol (2002) 282:L563–72. doi: 10.1152/ajplung.00280.2001
106. Morens DM, Taubenberger JK, Fauci AS. Predominant Role of Bacterial Pneumonia as a Cause of Death in Pandemic Influenza: Implications for Pandemic Influenza Preparedness. J Infect Dis (2008) 198:962–70. doi: 10.1086/591708
107. Chertow DS, Memoli MJ. Bacterial Coinfection in Influenza: A Grand Rounds Review. JAMA (2013) 309:275–82. doi: 10.1001/jama.2012.194139
108. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and Bacterial Superinfection: Illuminating the Immunologic Mechanisms of Disease. Infect Immun (2015) 83:3764–70. doi: 10.1128/IAI.00298-15
109. Shirey KA, Perkins DJ, Lai W, Zhang W, Fernando LR, Gusovsky F, et al. Influenza “Trains” the Host for Enhanced Susceptibility to Secondary Bacterial Infection. mBio (2019) 10:e00810–19. doi: 10.1128/mBio.00810-19
110. Perkins DJ, Rajaiah R, Tennant SM, Ramachandran G, Higginson EE, Dyson TN, et al. Salmonella Typhimurium Co-Opts the Host Type I IFN System to Restrict Macrophage Innate Immune Transcriptional Responses Selectively. J Immunol (2015) 195:2461–71. doi: 10.4049/jimmunol.1500105
111. Ishikawa H, Fukui T, Ino S, Sasaki H, Awano N, Kohda C, et al. Influenza Virus Infection Causes Neutrophil Dysfunction Through Reduced G-CSF Production and an Increased Risk of Secondary Bacteria Infection in the Lung. Virology (2016) 499:23–9. doi: 10.1016/j.virol.2016.08.025
112. Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, et al. Influenza A Virus Exacerbates Staphylococcus Aureus Pneumonia in Mice by Attenuating Antimicrobial Peptide Production. J Infect Dis (2014) 209:865–75. doi: 10.1093/infdis/jit527
113. Lee B, Gopal R, Manni ML, McHugh KJ, Mandalapu S, Robinson KM, et al. STAT1 Is Required for Suppression of Type 17 Immunity During Influenza and Bacterial Superinfection. Immunohorizons (2017) 1:81–91. doi: 10.4049/immunohorizons.1700030
114. Gopal R, Lee B, McHugh KJ, Rich HE, Ramanan K, Mandalapu S, et al. STAT2 Signaling Regulates Macrophage Phenotype During Influenza and Bacterial Super-Infection. Front Immunol (2018) 9:2018.02151. doi: 10.3389/fimmu.2018.02151
115. Gopal R, Mendy A, Marinelli MA, Richwalls LJ, Seger PJ, Pate LS, et al. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance During Influenza-Bacterial Superinfection. Viruses (2019) 11:505. doi: 10.3390/v11060505
116. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The Global Distribution and Burden of Dengue. Nature (2013) 496:504–7. doi: 10.1038/nature12060
117. Jacobs MG, Robinson PJ, Bletchly C, Mackenzie JM, Young PR. Dengue Virus Non-Structural Protein 1 Is Expressed in a Glycosyl-Phosphatidylinositol-Linked Form That Is Capable of Signal Transduction. FASEB J (2000) 14:1603–10. doi: 10.1096/fj.99-0829com
118. Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, et al. Dengue Virus NS1 Protein Activates Cells Via Toll-Like Receptor 4 and Disrupts Endothelial Cell Monolayer Integrity. Sci Trans Med (2015) 7:304ra142. doi: 10.1126/scitranslmed.aaa3863
119. Centers for Disease Control and Prevention. 2014-2016 Ebola Outbreak in West Africa (2016). Available at: https://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/index.html (Accessed April 27, 2021).
120. Kilgore PE, Grabenstein JD, Salim AM, Rybak M. Treatment of Ebola Virus Disease. Pharmacotherapy (2015) 35:43–53. doi: 10.1002/phar.1545
121. Falasca L, Agrati C, Perosillo N, DiCaro A, Capobianchi MR, Ippolito G, et al. Molecular Mechanisms of Ebola Virus Pathogenesis: Focus on Cell Death. Cell Death Differ (2015) 22:1250–9. doi: 10.1038/cdd.2015.67
122. Clerici M, Shearer GMA. TH1–>TH2 Switch Is a Critical Step in the Etiology of HIV Infection. Immunol Today (1993) 14:107–11. doi: 10.1016/0167-5699(93)90208-3
123. Clerici M, Sarin A, Coffman RL, Wynn TA, Blatt SP, Hendrix CW, et al. Type 1/Type 2 Cytokine Modulation of T-Cell Programmed Cell Death as a Model for Human Immunodeficiency Virus Pathogenesis. Proc Natl Acad Sci USA (1994) 91:11811–5. doi: 10.1073/pnas.91.25.11811
124. Emilie D, Ftor R, Llorente L, Marfaing-Koka A, Peuchmaur M, Devergne O, et al. Cytokines From Lymphoid Organs of HIV-Infected Patients: Production and Role in the Immune Disequilibrium of the Disease and in the Development of B Lymphomas. Immunol Rev (1994) 140:5–34. doi: 10.1111/j.1600-065X.1994.tb00863.x
125. Graziosi C, Pantaleo G, Fauci AS. Comparative Analysis of Constitutive Cytokine Expression in Peripheral Blood and Lymph Nodes of HIV-Infected Individuals. Res Immunol (1994) 145:602–7. doi: 10.1016/S0923-2494(05)80040-9
126. Alfano M, Poli G. The Cytokine Network in HIV Infection. Curr Mol Med (2002) 2:677–89. doi: 10.2174/1566524023361925
127. Bennasser Y, Badou A, Tkaczuk J, Bahraoui E. Signaling Pathways Triggered by HIV-1 Tat in Human Monocytes to Induce TNF-Alpha. Virology (2002) 303:174–80. doi: 10.1006/viro.2002.1676
128. Leghmari K, Contreras X, Moureau C, Bahraoui E. Hiv-1 Tat Protein Induces TNF-Alpha and IL-10 Production by Human Macrophages: Differential Implication of PKC-BetaII and -Delta Isozymes and MAP Kinases ERK1/2 and P38. Cell Immunol (2008) 254:46–55. doi: 10.1016/j.cellimm.2008.06.011
129. Haij NB, Leghmari K, Planes R, Thieblemont N, Bahraoui E. Hiv-1 Tat Protein Binds to TLR4-MD2 and Signals to Induce TNF-Alpha and IL-10. Retrovirology (2013) 10:123. doi: 10.1186/1742-4690-10-123
130. Bahraoui E, Briant L, Chazal N. E5564 Inhibits Immunosuppressive Cytokine IL-10 Induction Promoted by HIV-1 Tat Protein. Virol J (2014) 11:214. doi: 10.1186/s12985-014-0214-z
131. Hoshino K, Takeuchi O, Kawi T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting Edge: Toll-Like Receptor 4 (TLR4)-Deficient Mice Are Hyporesponsive to Lipopolysaccharide: Evidence for TLR4 as the Lps Gene Product. J Immunol (1999) 162:3749–52.
132. Richard K, Piepenbrink KH, Shirey KA, Gopalakrishnan A, Nallar S, Prantner DJ, et al. A Mosue Model of Human TLR4 D299g/T399i SNPs Reveals Mechanisms of Altered LPS and Pathogen Responses. J Exp Med (2021) 218:e20200675. doi: 10.1084/jem.20200675
133. Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, et al. TLR4 Mutations Are Associated With Endotoxin Hyporesponsiveness in Humans. Nat Genet (2000) 25:187–91. doi: 10.1038/76048
134. Abdul-Careem MF, Firoz Mian M, Gillgrass AE, Chenoweth MJ, Barra NG, Chan T, et al. FimH, A TLR4 Ligand, Induces Innate Antiviral Responses in the Lung Leading to Protection Against Lethal Influenza Infection in Mice. Antiviral Res (2011) 292:346–55. doi: 10.1016/j.antiviral.2011.09.004
135. Morales-Nebreda L, Mutlu GM, Scott Budinger GR, Radigan KA. Loss of TLR4 Does Not Prevent Influenza A-Induced Mortality. Am J Respir Crit Care Med (2014) 180:1280–1. doi: 10.1164/rccm.201401-0193LE
136. Vogel SN, Hansen CT, Rosenstreich DL. Characterization of a Congenitally LPS-Resistant, Athymic Mouse Strain. J Immunol (1979) 122:619–22.
137. The Jackson Laboratory. B6.B10ScN-Tlr4lps-del/JthJ (2020). Available at: http://jaxmice.jax.org/strain/007227.html (Accessed April 27, 2021).
138. Poltorak A, Merlin T, Nielsen PJ, Sandra O, Smirnova I, Schupp I, et al. A Point Mutation in the IL-12Rβ2 Gene Underlies the IL-12 Unresponsiveness of Lps-Defective C57bl/10scCr Mice. J Immunol (2001) 167:2106–11. doi: 10.4049/jimmunol.167.4.2106
139. Bourdi M, Davies JS, Pohl LR. Mispairing C57BL/6 Substrains of Genetically Engineered Mice and Wild-Type Controls can Lead to Confounding Results as It Did in Studies of JNK2 in Acetaminophen and Concanavalin A Liver Injury. Chem Res Toxicol (2011) 24:794–6. doi: 10.1021/tx200143x
140. Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel C, Du X, et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCR Mice: Mutations in TLR4 Gene. Science (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
141. Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-Tolerant Mice Have Mutations in Toll-Like Receptor 4 (Tlr4). J Exp Med (1999) 189:615–25. doi: 10.1084/jem.189.4.615
142. Li H, Liu L, Zhang D, Xu J, Dai H, Tang N, et al. Sars-CoV-2 and Viral Sepsis: Observations and Hypotheses. Lancet (2020) 395:1517–20. doi: 10.1016/S0140-6736(20)30920-X
Keywords: TLR4, influenza, ALI, viruses, HMGB1
Citation: Shirey KA, Blanco JCG and Vogel SN (2021) Targeting TLR4 Signaling to Blunt Viral-Mediated Acute Lung Injury. Front. Immunol. 12:705080. doi: 10.3389/fimmu.2021.705080
Received: 04 May 2021; Accepted: 10 June 2021;
Published: 02 July 2021.
Edited by:
Michael H. Lehmann, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Holger Heine, Research Center Borstel (LG), GermanyLoic Guillot, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Jay Evans, University of Montana, United States
Copyright © 2021 Shirey, Blanco and Vogel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kari Ann Shirey, S1NoaXJleUBzb20udW1hcnlsYW5kLmVkdQ==