
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 June 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.704989
This article is part of the Research Topic A Microbial View of Central Nervous System Disorders: Interplay Between Microorganisms, Neuroinflammation and Behavior View all 9 articles
 Lu Zhang1,2†
Lu Zhang1,2† Cui Jiao1,2†Lingjuan Liu1,2Aiping Wang1,2Li Tang1,2Yi Ren1,2Peng Huang1,2
Cui Jiao1,2†Lingjuan Liu1,2Aiping Wang1,2Li Tang1,2Yi Ren1,2Peng Huang1,2 Jie Xu1,2Dingan Mao1,2
Jie Xu1,2Dingan Mao1,2 Liqun Liu1,2*
Liqun Liu1,2*Nucleotide oligomerization domain-like receptors (NLRs), a class of pattern recognition receptors, participate in the host’s first line of defense against invading pathogenic microorganisms. NLR family caspase recruitment domain containing 5 (NLRC5) is the largest member of the NLR family and has been shown to play an important role in inflammatory processes, angiogenesis, immunity, and apoptosis by regulating the nuclear factor-κB, type I interferon, and inflammasome signaling pathways, as well as the expression of major histocompatibility complex I genes. Recent studies have found that NLRC5 is also associated with neuronal development and central nervous system (CNS) diseases, such as CNS infection, cerebral ischemia/reperfusion injury, glioma, multiple sclerosis, and epilepsy. This review summarizes the research progress in the structure, expression, and biological characteristics of NLRC5 and its relationship with the CNS.
The innate immune response is the first line of defense against pathogen invasion. Pattern recognition receptors (PRRs) act as pivotal sensors in these processes by recognizing specific microbial pathogens through pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PRRs induce intracellular cytokine and chemokine secretion, trigger an inflammatory response, and ultimately activate the host defense system (1). To date, biochemical studies have identified four types of PRR families: toll-like receptors (TLRs), retinoic acid-inducible gene-I-like receptors (RLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), and C-type lectin receptors (CLRs) (2). NLRs are a large protein family of PRRs located in the cytoplasm, where they are involved in the activation of the inflammatory response system and rapid removal of invasive pathogens. It was previously reported that NLRC5 [NLR family caspase recruitment domain (CARD) containing 5, also known as NOD4, NOD27, and CLR16.1] accounts for a large proportion of the NLR family, which can modulate immune responses in the context of many human diseases, such as liver disease, renal disease, rheumatoid arthritis, and heart disease, by regulating nuclear factor-κB (NF-κB), type I interferon (IFN-1), inflammasome signaling pathways and the expression of the major histocompatibility complex (MHC) I genes (3–5). However, in recent years, studies have found that NLRC5 is also associated with CNS infection (CNSI) (6–9), neuronal development (10), and neuropsychiatric disorders, such as cerebral ischemia/reperfusion (I/R) injury (11, 12), glioma (13–15), multiple sclerosis (MS) (16), epilepsy (17), schizophrenia (SCZ), and bipolar disorder (BD) (18, 19). Following recent advances in this area, we focus on a comprehensive update on the structure, expression, and biological characteristics of NLRC5, as well as its relationship with the CNS.
To date, 23 NLR genes have been identified in the human genome, whereas there are at least 34 mouse NLR genes in the mouse genome, and they are widely expressed in various cells and tissues (1, 4). It has been found that tripartite domain architecture is a common feature of the structure of this protein family: a variable N-terminal domain that interacts with downstream corresponding protein molecules, NODs that lead to the oligomerization and activation of NLRs, and a C-terminal leucine-rich repeat (LRR) domain that recognizes ligands (such as PAMPs). In terms of the N-terminal domains, NLRs can be classified into three subfamilies: NLRC containing CARD, NLRP containing pyrin domain (PYD), and NAIP containing baculovirus inhibitor domain (BIR) (1). NLRC5 was initially cloned in 2010 and has been confirmed to be located on human chromosome 16q13 (20, 21), which straddles a region of approximately 96 kbp. The encoded polypeptide contains 1,866 amino acids, with a predicted size of approximately 200 kDa (1). NLRC5 is similar to the tripartite domain of other NLRs (Figure 1). However, the N-terminal CARD of NLRC5 has been predicted to adopt a death domain fold and exhibits no obvious sequence similarity with other typical CARDs; therefore, it is called an atypical CARD (20, 22). In addition, the NOD of NLRC5 contains the Walker A and Walker B motifs, which are important for nucleoside triphosphate (NTP) binding and NTP hydrolysis, respectively (23). NLRC5 is the largest member of the NLR protein family because of the unusually long stretch of 27 LRRs in its C-terminal region (1, 20, 24). The structure of NLRC5 is highly conserved in many mammalian species; for example, the homology of human NLRC5 to mouse NLRC5 is 64% (20), which suggests that NLRC5 has similar crucial functions in various organisms.

Figure 1 Schematic representation of NLRC5 structure CARD: Caspase recruitment domain; NOD: Nucleotide oligomerization domain; LRR: Leucine-rich repeats NLS: Nuclear localization signal; Walker A: Nucleoside triphosphate (NTP)-binding site, Walker B: NTP hydrolysis site.
Previous studies have shown that NLRC5 predominantly exists in the bone marrow, human THP-1 cells, B cells, human cervical cancer cell lines, and primary myeloid and lymphoid cells (3). These locations indicate that NLRC5 plays a vital regulatory role in the occurrence and development of immune diseases. Although NLRs play a crucial role in innate immunity, there have been relatively few studies on their expression in the brain. Kuenzel et al. found that the highest expression of NLRC5 mRNA in humans was in the tissues of the brain, lung, and prostate, followed by the heart, digestive tract, and thymus (25). Subsequently, researchers found that NLRC5 is expressed in both the brain and the cells forming the blood-brain barrier (BBB), including neurons, astrocytes, microglia, oligodendrocytes, endothelial cells, and brain pericytes (Table 1) (2, 26–28). Specifically, the mRNA and protein expression of NLRC5 is relatively abundant in the hippocampus of the mouse brain. Using qPCR and western blotting analysis, Li et al. identified that there is a gradual increase in expression from 0 to 15 days after birth (10). Thus, NLRC5 may be related to the physiological and pathological states of the CNS to some extent.

Table 1 NLRC5 expression in the tissue.
Using immunofluorescence analysis, Kuenzel et al. found that NLRC5 is restricted to the cytoplasm to recognize intracellular danger signals (25). In contrast, Meissner et al. revealed that NLRC5 is located in both the cytoplasm and nuclear compartments (29). Moreover, NLRC5 has transcriptional regulation properties similar to those of the class II transactivator (CIITA), which may also be a nuclear localization protein molecule that can shuttle between the nucleus and the cytoplasm (3). NLRC5 was found to be more frequently localized to the cytoplasm of cells with high NLRC5 levels, whereas nuclear localization was more frequently observed in cells with low NLRC5 levels (1, 29). Accordingly, the nuclear localization of NLRC5 was not the result of overexpression. High expression of NLRC5 was observed in the nucleus of cells subjected to leptomycin B (an inhibitor of CRM1-dependent nuclear export) treatment (29), suggesting that the NLRC5 plasmid shuttle may be carried out in a CRM1-dependent manner. Furthermore, some studies have indicated that NLRC5 possesses a nuclear localization signal (NLS) between the CARD and NOD in duplicate, in which locus mutations within the NLS block the translocation of NLRC5 from the cytoplasm to the nucleus (29). Moreover, the CARD and NBD of NLRC5 are involved in its nuclear import because of the NLSs.
NF-κB is ubiquitously expressed in mammalian cells and plays a critical role in innate and adaptive immunity. Dysregulation of NF-κB activity leads to a variety of diseases, such as immunodeficiency, viral infections, inflammation, and cancer (1). In addition, NF-κB in the CNS can be activated by growth factors (brain-derived neurotrophic factor and nerve growth factor) and excitatory neurotransmitters, such as glutamate. It is also involved in the regulation of synaptic plasticity, learning, and memory (30), as well as the development and maintenance of addiction to a stimulus (31). The IκB kinase (IKK) complex is composed of the kinase heterodimer IKKα/IKKβ and a regulatory subunit. Cui et al. confirmed for the first time that NLRC5 is a potent negative regulator of NF-κB activation through luciferase assays (32). NLRC5 competes with NF-κB essential modulator, an NF-κB essential modulator, to directly interact with IKKα/IKKβ subunits and to block their phosphorylation and kinase activity (Figure 2), which is related to the NLRC5 LRR domain (32). NLRC5 overexpression affects the translocation of p65 NF-κB nuclear translocation and inhibits the NF-κB downstream pathway, thereby playing an anti-inflammatory role (33). Nonetheless, how NLRC5 regulates the effects of the NF-κB signaling pathway on brain function requires further study.
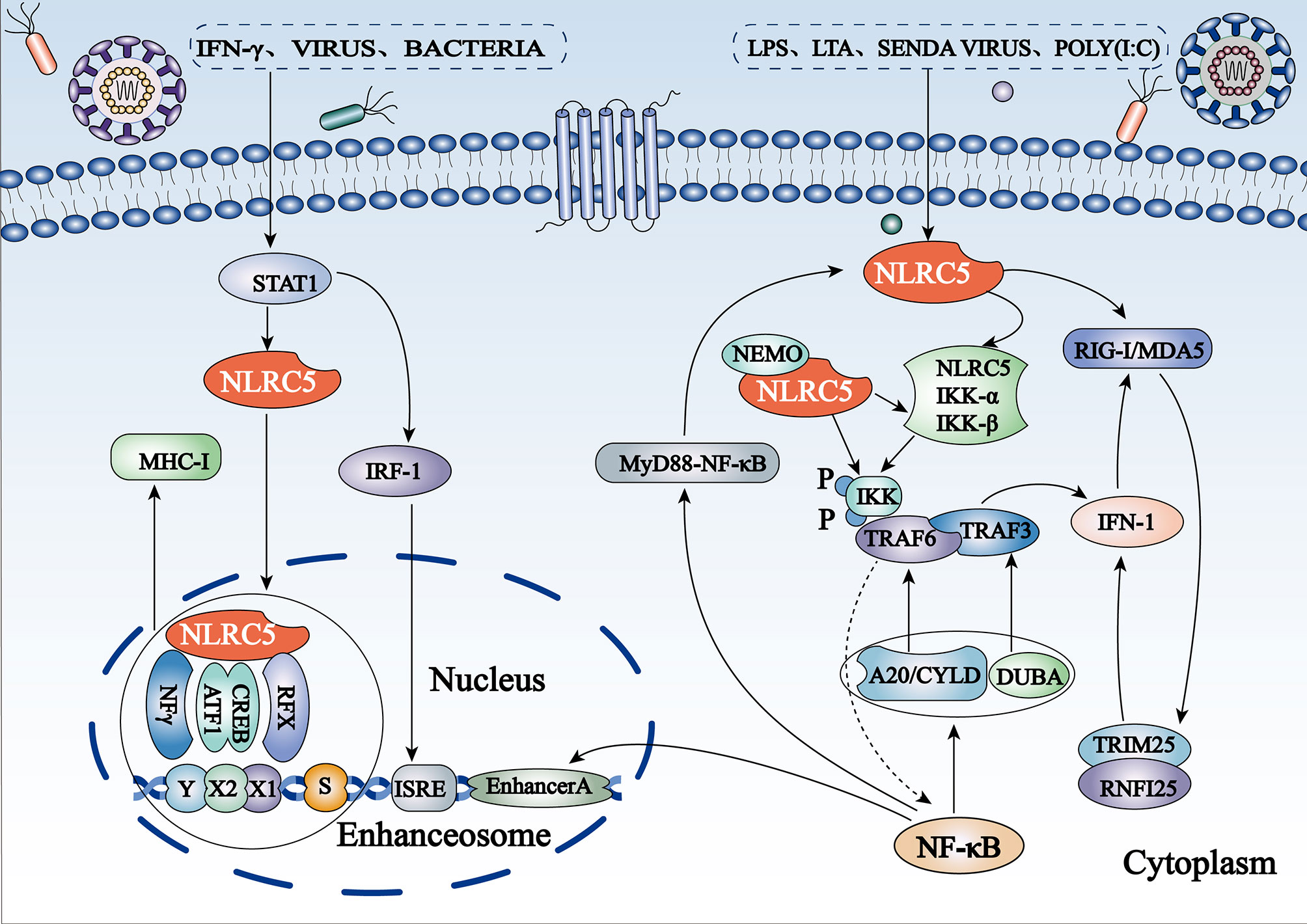
Figure 2 Schematic representation of regulation of the NF-κB and IFN-I pathways and MHC I gene expression by NLRC5 (1) NLRC5 inhibits the NF-κB pathway by binding to IKKα/IKKβ subunits and blocking their phosphorylation. The expression of NLRC5 itself is regulated by NF-κB activation to form a negative regulatory feedback loop. (2) NLRC5 can also suppress RIG-like receptor-mediated IFN-I responses by interacting with RIG-I/MDA5. (3) Stimuli such as IFN-γ, viral infection, and bacterial infection induce NLRC5 to bind directly to the GAS site in the NLRC5 promoter by activating the STAT1 homologous dimer. Subsequently, NLRC5 shuttles from the cytoplasm to the nucleus and combines with the S-X-Y motif to form a potent enhanceosome and to induce MHC I transcription.
NLRC5 was initially reported to be a positive regulator of IFN-I signaling. Vesicular stomatitis virus (VSV), polyinosinic: polycytidylic acid [poly(I:C)], and lipopolysaccharides (LPS) can induce IFN-I signal transduction (32). Endogenous NLRC5 knockdown reduced Sendai virus and poly(I:C)-mediated IFN-I pathway-dependent responses in human primary dermal fibroblasts and THP-1 cells and significantly inhibited the secretion of IFN-γ in human fibroblast cells infected with cytomegalovirus (25, 34). In contrast, growing evidence has confirmed that NLRC5 is a negative regulator of IFN-I. In vitro experiments have shown that the secretion of IFN-β is significantly increased in NLRC5-/- macrophages exposed to RIG-I/MDA5 ligands, such as VSV and poly(I:C), suggesting that NLRC5 may mainly interact with RIG-I/MDA5 to inhibit RLR-mediated IFN-I responses (32, 35) (Figure 2). In addition, NLRC5 inhibits RLR-mediated IFN-I secretion in plasmacytoid dendritic cells (36). These phenomena have also been confirmed in vivo: NLRC5-deficient mice produce higher amounts of IFN-β in sera when challenged with LPS or infected with VSV (35).
MHC I/II and co-molecules activate an adaptive immune response under many pathological conditions, such as autoimmunity, cancer, and infection, by presenting antigens to T lymphocytes. In 2010, Meissner et al. initially identified that NLRC5 may directly regulate the expression of MHC I genes (29). Gene-chip analysis showed significantly higher gene expression in wild-type and mutant Walker B cell lines, which express the active forms of NLRC5, compared to cells expressing the mutant Walker A and mutant Walker AB of NLRC5 (29). The most upregulated genes included the MHC I family (HLA-A, -B, -C, and -E) and those associated with antigen presentation and processing of MHC I genes (β2M, LMP2, and TAP1), both of which were induced by active NLRC5, which requires NTP binding instead of NTP hydrolysis (29). Given the lack of a DNA-binding domain, NLRC5-mediated MHC I transactivation requires the S-X-Y motifs in the proximal regions of MHC I genes, which are dependent on other components, called enhanceosomes, to connect to the promoter region (10). IFN-γ leads to binding of transcriptionally active signal transducers and activators of transcription-1 (STAT1) to cis-elements of the gamma interferon activation site (GAS) via stimulation of the JAK/STAT pathway to promote the upregulation of NLRC5 expression (23). NLRC5 shuttles from the cytoplasm to the nucleus and forms a potent enhanceosome with transcription factors, such as X1 box regulatory factor X (RFX), X2 box activating transcription (ATF)/cAMP response element binding protein (CREB), and nuclear factor-Y protein (NFY) complexes, thereby inducing activation of MHC I gene transcription (23) (Figure 2). Located at the enhanceosome complex, NLRC5 has been suggested to function as a scaffold to recruit transcriptional initiation and elongation factors, as well as co-activators [such as CREB binding protein (CBP), p300, GCN5, and PCAF] and chromatin modifiers (23). More intuitively, NLRC5 has been shown to be essential for the removal of the gene-silencing trimethylation of lysine 27 on histone 3 (H3K27me3) on the MHC I promoter (37), which is likely the result of specific recruitment of demethylating enzymes by NLRC5.
Inflammasomes are multiprotein complexes that are assembled by PRRs in the cytoplasm. These complexes are a significant part of the innate immune system and are known for their ability to recognize PAMPs or DAMPs and activate the proteolytic enzyme caspase-1. Activated caspase-1 promotes proteolysis and maturation of the pro-inflammatory cytokines IL-1β and IL-18, stimulating the occurrence of pyroptosis (38). It has been reported that inflammasome activation is associated with a variety of brain diseases, including ischemic and traumatic brain injury and neurodegenerative diseases (12, 39, 40). Co-immunoprecipitation experiments revealed that NLRC5 directly binds to the NLR family PYD containing 3 (NLRP3) and apoptosis-associated speck-like protein containing CARD (ASC), which is considered to be an activator and synergetic component of the inflammasome (41). In vitro studies have shown that the overexpression of NLRC5 promotes IL-1β production by stimulating caspase-1 in HEK293 cells (42). Candidate pathogens such as Escherichia coli, Shigella flexneri, and Staphylococcus aureus require NLRP3 and ASC to activate the inflammasome, and RNA-mediated suppression of NLRC5 was shown to significantly block IL-1β secretion in THP-1 cells compared to the control group (41). In vivo, NLRC5 was shown to significantly promote IL-1β secretion in Listeria monocytogenes, and the recruitment of neutrophils and bacterial clearance were significantly reduced in the liver and spleen of Nlrc5-deficient mice (43). When attacked by pathogenic microorganisms or stimulated by endogenous danger signals, host cells produce caspase-1-dependent pyroptosis. Different from chromatin pyknosis, DNA fragmentation, cell shrinkage and apoptotic body formation in the process of apoptosis, pyroptosis will cause the infected cells to swell and rupture the cell membrane in a short period of time, thus releasing inflammatory factors such as IL-1 β and IL-18 into the interstitial space, activating the corresponding receptors in the adjacent cells and triggering a broader immune response (44, 45). These findings suggest that NLRC5 regulates inflammasome activation and participates in the formation of IL-1β in response to multiple bacterial pathogens. In macrophages, NLRC5 overexpression promotes caspase-1-dependent IL-1β production. However, NLRC5 deficiency does not inhibit caspase-1 activation induced by LPS plus ATP or nigericin, MSU, curdlan, poly-IC and poly (dA:dT), which is dependent on NLRP3, AIM2, NLRC4 or NLRP3 inflammasomes. Therefore, NLRC5-mediated inflammasomes activation may be induced by unknown pathogenic factors or endogenous activators, and its activation may be cell-type specific (42). Overall, it remains to be seen whether NLRC5 is an activating receptor of inflammasomes.
DNA methylation usually occurs on cytosine residues of cytosine-guanosine (CpG) dinucleotides (46). Changes in CpG methylation induce changes in gene expression that can lead to changes in protein and metabolite levels, which are associated with many complex disorders (47). CpG sites in NLRC5 (cg07839457, cg16411857, cg08159663, cg00218406, and cg08099136), associated with one or several of the seven proteins of the immune system (CD48, CD163, CXCL10, CXCL11, LAG3, FCGR3B, and B2M), form a network involving the methylation of NLRC5 (47). NLRC5 increases the expression of the B2M protein of MHC class I; indirect links have been found between the five CpG sites and the associated proteins through various interleukins (IL-6 and IL-10), major histocompatibility complexes [HLA-A, HLA-ABC, MHC class I, and interferon regulatory factor 3 (IRF3)], and NF-κB (47). Moreover, it has been found that the methylation of NLRC5 is linked to numerous diseases. For example, NLRC5 methylation at cg07839457 is associated with BMI and obesity in Africans (48), whereas cg07839457 and cg16411857 methylation are associated with HIV infection (49). Moreover, NLRC5 methylation has been associated with lupus (46) and rheumatoid arthritis (50). In melanoma and bladder cancer, Low methylation of the NLRC5 promoter is associated with higher survival rate (51).Therefore, the NLRC5 methylation network plays a role in diseases and immunity, and its relationship with CNS diseases and specific disease mechanisms needs to be further elucidated.
NLRC5 exerts different effects in different diseases by promoting and/or inhibiting the PI3K/AKT signaling pathway. Activation of the PI3K/AKT signaling pathway is a marker of cancer progression (52). NLRC5 promotes AN3CA cell migration and invasion by activating the PI3K/AKT signaling pathway in endometrial cancer; however, it can be inhibited by LY294002, a specific inhibitor of the PI3K/AKT signaling pathway (53). Moreover, integrin alpha x (ITGAX) stimulates cancer angiogenesis through PI3K/AKT signaling-mediated VEGFR2/VEGF-A overexpression in blood vessel endothelial cells in hepatocellular carcinoma (HCC) (54). There is a positive correlation between NLRC5 and VEGF-A expression, which also coordinates the activation of the PI3K/AKT signaling pathway (55), demonstrating that NLRC5 promotes HCC progression via the AKT/VEGF-A signaling pathway (Figure 3). Furthermore, Feng et al. found that knockdown of NLRC5 efficiently inhibited H/R-induced oxidative stress and apoptosis in HK-2 cells and attenuated renal I/R injury in vitro through the activation of the PI3K/AKT signaling pathway (56).
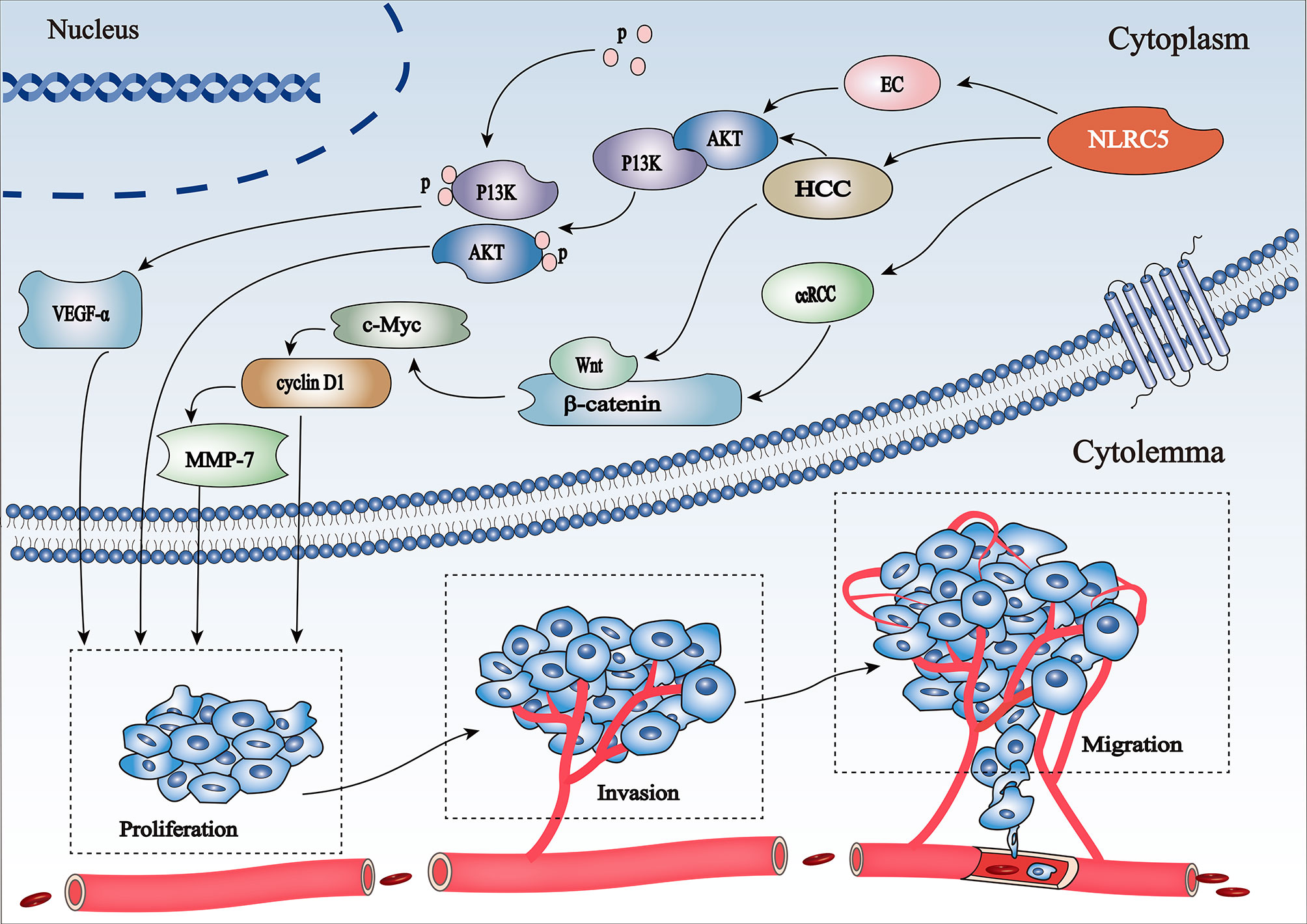
Figure 3 Schematic representation of regulation of the PI3K/Akt and Wnt/β-catenin pathways involved in cancer progression by NLRC5 (1) NLRC5 promotes HCC progression via the AKT/VEGF-A signaling pathway. (2) NLRC5 mediates cell proliferation, invasion, and migration in HCC and ccRCC by activating c-Myc and cyclin D1, the key target genes of the Wnt/β-catenin signaling pathway.
Wnt/β-catenin is associated with the proliferation, invasion, and migration of tumor cells (57). NLRC5 activates β-catenin transcription and translation by stimulating the Wnt/β-catenin signaling pathway, thereby promoting tumor development and progression. The oncogene c-Myc (58) and cell cycle regulator cyclin D1 (59) are key target genes of the Wnt/β-catenin signaling pathway. NLRC5 mediates cell proliferation, migration, and invasion by regulating the Wnt/β-catenin signaling pathway in clear cell renal cell carcinoma (ccRCC) (60) (Figure 3). However, knockdown of NLRC5 can weaken the expression of c-Myc and cyclin D1, thereby inhibiting the growth of tumor cells (60). Moreover, upregulation of NLRC5 not only positively correlates with the increase in β-catenin, but also coordinates the activation of the downstream Wnt/β-catenin signaling pathway in HCC (61).
In addition to the aforementioned pathways, NLRC5 is involved in the regulation of the TLR4/MyD88/NF-κB, JAK2/STAT3, and TGF-β1/Smad signaling pathways, and plays a role in hepatic fibrosis, malignant tumors, ethanol-induced hepatic injury and other diseases, as shown in Table 2.
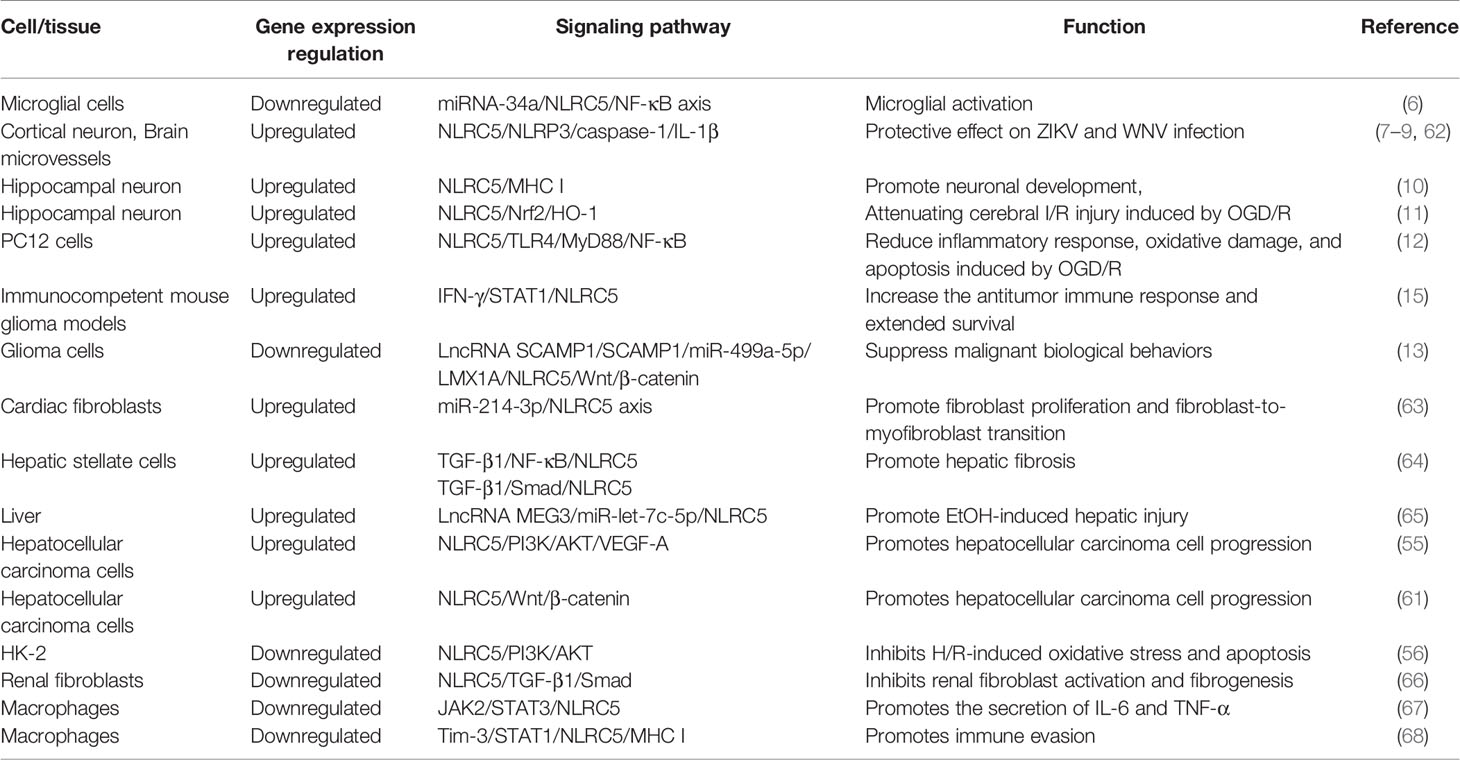
Table 2 Role of NLRC5 in common signaling pathways.
Previous studies have shown that MHC I is involved in regulating synaptic plasticity in the lateral geniculate nucleus during development, neurite outgrowth, and polarization in young cultured hippocampal neurons. Moreover, it is critical for hippocampal-dependent memory by inhibiting NMDAR function (69–71), which is of great significance to the development of neurons. Li et al. suggested that the expression of endogenous NLRC5 mRNA and MHC I is correlated with hippocampal development (10). MHC I transcription may be regulated by NLRC5 in hippocampal neurons. The specific regulatory mechanism involves transport of NLRC5 from the cytoplasm to the nucleus and induction of the activation of MHC I transcription by binding to RFX5 and RFXANK of X1 box, CREB of X2 box, and NFY complexes (10). According to double immunostaining analysis, NLRC5 and MHC I are co-localized in hippocampal neurons, and Nlrc5-/- mouse hippocampal MHC I levels are significantly decreased (10), further suggesting that NLRC5 may play a vital role in regulating neuron development by upregulation of MHC I in hippocampal neurons. However, no relevant functional experiments have revealed how NLRC5 affects hippocampal neuronal function changes.
The BBB is the interface that separates neural tissue from circulating blood and maintains a safe and homeostatic milieu for proper neuronal function and synaptic transmission (72). As a key part of the BBB, the brain endothelial cells can directly contact the blood and elicit bacteria–host interactions that assist bacterial pathogens, such as E. coli, Streptococcus pneumoniae, Meningitis neisseria, and S. aureus, in invading the brain (73). NLRC5 is widely expressed in cerebral endothelial cells and brain pericytes and is regulated by inflammatory mediators (2, 26–28); for example, IFN-γ, TNF-α, and IL-1β upregulate the expression of NLRC5 in brain pericytes (27).However, the role of NLRC5 in the subsequent inflammatory response and its mechanism needs to be further studied.
CNS complications in people infected with HIV are collectively termed as neuroHIV, and its pathological mechanism involves aggravation of cellular oxidative stress, disturbance of energy metabolism, immune activation, inflammation, and neuronal damage (6, 74). Many reports have confirmed both protein and mRNA expression of HIV-1 Tat in the brains of HIV-1-infected individuals. HIV-1 Tat can induce neuroinflammation by activating glial cells and modulating various signaling pathways to impact microglial function, thus exerting neurotoxic effects on many CNS cells (6). miRNA-34a is abundant in the brain and is upregulated in numerous neurological diseases and aging-related diseases (75). Periyasamy et al. showed that HIV-1 Tat-mediated upregulation of miRNA-34a targets the 3’-UTR of NLRC5 in mouse primary microglial cells and that the downregulated NLRC5 inflammasome negatively regulates the NF-κB p65 signaling axis via modulation of the phosphorylated IKK complex, leading to increased expression of pro-inflammatory cytokines such as IL-1β and IL-6 and the consequent microglial activation (6). In summary, the miRNA-34a/NLRC5/NF-κB signaling pathway may be an entry point for the treatment of HIV-1 Tat-mediated microglial inflammation.
Neurotropic viruses can invade the CNS via various routes and irreversibly disrupt the complex structural and functional architecture of the CNS, including retrograde axonal transport along motor and olfactory neurons and hematogenous spread across the BBB via direct infection of endothelial cells (76). Zika virus (ZIKV) and West Nile virus (WNV) have significant neuroinvasive characteristics and are regarded as neurotropic (7). High ZIKV replication was observed as early as 12 h and peaked at 48 h after infection, similar to the expression of NLRC5 mRNA in primary cortical neurons of mice (8). Leda et al. evaluated the impact of ZIKV infection on brain microvasculature and the BBB by evaluating the transcriptomic profile of brain microvessels via high-throughput RNA sequencing, and the analysis revealed upregulation of NLRC5 (7). Moreover, NLRC5 expression was upregulated in the mouse brain after WNV NY99 infection (9), suggesting that neurotropic virus invading the CNS could induce the expression of NLRC5 in the brain of mice. Previous studies have confirmed that NLRP3 activates caspase-1 by recruiting ASC to form inflammasomes, resulting in the proteolytic cleavage of pro-IL-1β into mature IL-1β. The NLRP3 inflammasome pathway and IL-1β signaling are key factors controlling WNV infection and immunity in the CNS (62). However, the biological function of NLRC5 in the CNS against viral infection and whether it cooperates with activation of the NLRP3 inflammasome remain unclear and need to be further studied.
Increasing evidence shows that NLR proteins are involved in the pathogenesis of cerebral I/R injury (77, 78). The production of reactive oxygen species (ROS) and subsequent oxidative stress, inflammation, and apoptosis are the core processes of cerebral I/R injury (79–81). Previous studies have confirmed that NLRC5 may play a key role in alleviating liver and renal I/R injury (56, 82). Li et al. showed that the mRNA and protein levels of NLRC5 are significantly decreased in oxygen-glucose deprivation (OGD)/reoxygenation (R)-induced neurons (11). Overexpression of NLRC5 has been shown to cause a significant increase in cell viability and increased expression of bcl-2, nuclear factor erythroid 2-related factor 2 (Nrf2), heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO-1), glutathione peroxidase 3 (GPx-3), and downstream genes of the Nrf2/HO-1 pathway, as well as decreased expression of Bax and ROS (11). In conclusion, these results suggest that the protective effects of NLRC5 are mediated by the Nrf2/HO-1 pathway. Thus, NLRC5 may serve as an effective target for the treatment of cerebral I/R injury. Furthermore, Zhang et al. found that the expression of NLRC5 in neonates with cerebral ischemia and OGD/R-induced PC12 cells was significantly reduced (12). However, NLRC5 overexpression was found to suppress the levels of inflammatory cytokines such as TNF-α, IL-6, IL-1β, ROS, malondialdehyde (MDA), Bax, and caspase-3, as well as apoptosis. Moreover, it inhibits the levels of TLR4, MyD88, and NF-κB p-p65 and upregulates the expression of superoxide dismutase (SOD) and bcl-2 (12). Therefore, NLRC5 alleviates inflammation, oxidative damage, and apoptosis in PC12 cells under OGD/R conditions by suppressing the activation of the TLR4/MyD88/NF-κB pathway. Collectively, these results indicate that NLRC5 plays a neuroprotective role in cerebral I/R injury through different approaches, and it is expected to be an effective target for the treatment of cerebral I/R injury.
Glioblastoma (GBM) is the most common and lethal type of tumor in the CNS. NLRC5, an IFN-related gene, has been shown to be associated with overall survival of GBM patients (14). IFN-γ is mainly produced by T and NK cells It can induce a positive feedback in the STAT1 pathway that triggers the expression and activation of STAT1 and other downstream genes, such as NLRC5, CIITA, and TAP1, and increases the immune response in the tumor microenvironment (5, 15). In vivo genetic knockdown of the proto-oncogene Fyn can induce changes in IFN-γ secretion, promote the overexpression of STAT1 and its downstream genes, and further increase the antitumor immune response in immunocompetent mouse glioma models, which significantly extends survival (15). In addition, upregulation of the lncRNA secretory carrier membrane protein 1 (SCAMP1), which functions as an oncogene in glioma cells, significantly promotes glioma cell proliferation, migration, and invasion and inhibits apoptosis (13). In contrast, miR-499a-5p exerts a tumor-suppressive function in glioma cells (13). SCAMP1 can increase LIM homeobox transcription factor 1 alpha (LMX1A) levels by negatively regulating miR-499a-5p expression, following LMX1A-mediated transcriptional activation of NLRC5, and can promote malignant biological behavior of glioma cells by attenuating the activity of the Wnt/β-catenin signaling pathway (13). This suggests that NLRC5 plays a regulatory role in the development of glioma and is related to IFN signaling and the SCAMP1/miR-499a-5p/LMX1A/NLRC5 pathway.
Multiple sclerosis (MS) is an autoimmune disease of the CNS, associated with inappropriate activation of lymphocytes, hyperinflammatory responses, demyelination, and neuronal damage (83). NLRs, as inflammatory regulators expressed in various cells in the CNS, mediate a variety of signaling pathways and play a role in the pathogenesis of MS (83). Falcão et al. performed single-cell transcriptomic analysis of oligodendrocyte lineage cells from the spinal cord of mice with experimental autoimmune encephalomyelitis (EAE) and showed that EAE-specific oligodendrocyte lineage populations express genes involved in antigen processing and presentation via MHC I and MHC II and genes involved in immunoprotection (16). Transcription factors (such as NLRC5, a transactivator of MHC I) are expressed in all oligodendrocyte lineage cells. These factors are induced by the IFN-γ pathway and can be used as MS-specific markers (16). Overall, as the transactivator of MHC I, the role of NLRC5 in MS and its mechanism are worthy of further exploration.
Epilepsy is a group of diseases characterized by periodic seizures and unpredictable occurrences caused by abnormal synchronous activity of neurons. Theiler’s murine encephalomyelitis virus (TMEV) infection induces a well-characterized experimental model of epilepsy, resulting in increased expression of NLRC5 and MHC I (H2-Kb) mRNAs and significantly decreased expression of IFN-I and pro-inflammatory mediators in normal mouse brains (17). It was found that infected Nlrc5-/- mice had significantly fewer seizures, lower sickness scores, weight loss, and neuroinflammation than wild-type mice (17). NLRC5 deficiency can reduce epileptic seizures caused by immune abnormalities in TMEV-infected mice, suggesting that NLRC5 can enhance the inflammatory response, regulate antiviral immunity, and promote the occurrence of epileptic seizures. Thus, inhibiting the acute elevation of NLRC5 levels or its downstream signaling molecules, such as IL-1β, is a promising therapeutic strategy for epilepsy. In addition, other members of the NLR family, such as NLRP1, have previously been implicated in epilepsy and were found to contribute to neuronal death and chronic seizure activity in the amygdala kindling-induced rat model of epilepsy (84). Since NLRC5 can interact with other NLRs (such as NLRP3) and form inflammasomes, further study of the synergistic effect of NLRC5 and other inflammasomes on epileptic seizures is also necessary.
SCZ and BD are highly heritable psychiatric disorders with overlapping susceptibility loci and symptomatology, such as impulsive and risk-taking behaviors (18, 19). NLRC5 is upregulated in SCZ and is located in the genetic signaling regions of BD and SCZ (18). Furthermore, Pacifico et al. reported the first transcriptome sequencing of the postmortem human dorsal striatum, their data showed that NLRC5 was differentially expressed between bipolar (n = 18) and control (n = 17) subjects (19). However, whether the highly expressed nlrc5 is involved in the development of SCZ and BD remains to be elucidated.
MHC I antigen presentation, MHC I-regulated of neuronal development, activation of the NF-κB signaling pathway, and activation of inflammasomes are important components of the immune response in nervous system development and nervous system diseases. NLRC5, the largest member of the NLR family, participates in the pathophysiological process of the CNS by regulating multiple pathways such as NF-κB, IFN-I, and inflammatory signaling pathways and MHC I gene expression. On the one hand, it can promote the development of neurons, has a neuroprotective effect against HIV and other viral encephalitis and brain I/R injury, and can also enhance the immune response against cerebral tumors. However, it may also play a negative role in the occurrence of neuropsychiatric diseases such as epilepsy, SCZ, and BD and may promote the malignant biological behavior of brain tumor cells. In summary, the findings from studies on NLRC5 provide new insights into the pathogenesis and treatment of various neurological diseases.
Although progress has been made in the research into the effect of NLRC5 in the CNS, its role and application still need to be further explored and expanded. For example, CNSI by bacteria is the most common and severe infectious disease in children, which can lead to serious neurological sequelae. However, there are few studies on NLRC5 and bacterial infections of the CNS. Based on its unique role in innate immunity, we can further focus on the immune signal regulation pathway in bacterial CNSI, which is conducive to providing a new theoretical basis for the targeted therapy of clinically relevant diseases. In addition, NLRC5 upregulates MHC I in neurons, and its downstream target, NF-κB, is closely related to synaptic plasticity. Therefore, in addition to the molecular mechanism, the effects of NLRC5 on synaptic function and neural information transmission can be studied in combination with electrophysiological and other functional experiments to explore its mechanism in neural development and neuropsychiatric diseases.
LQL, LZ, CJ, and DM conceived the idea of this review. CJ and LZ performed literature searching and drafted the manuscript. LJL, AW, LT, YR, PH, and JX created the figures. All authors critically reviewed and edited the content of this manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (No. 81873762 to LQL and No. 81501039 to LJL), and the Program of Hunan Provincial Department of Science and Technology, China (No. 2018SK2069 to LQL), and the Natural Science Foundation of Hunan Province, China (No. 2019JJ50876 to JX), and the Program from Health Commission of Hunan Province, China (No. B2018-0311 to LQL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ASC, Apoptosis-associated speck-like protein containing CARD; ATF, Activating transcription; BBB, blood-brain barrier; BD, bipolar disorder; BIR, Baculovirus inhibitor domain; CARD, Containing caspase recruitment domain; CBP, CREB binding protein; ccRCC, Clear cell renal cell carcinoma; CIITA, Class II transactivator; CLRs, C-type lectin receptors; CNS, Central nervous system; CNSI, Central nervous system infection; CpG, Cytosine-guanosine dinucleotides; CREB, cAMP response element binding protein; DAMPs, Damage-associated molecular patterns; EAE, Experimental autoimmune encephalomyelitis; GAS, Gamma interferon activation site; GBM, Glioblastoma; GPx-3, Glutathione peroxidase 3; HCC, Hepatocellular carcinoma; HO-1, Heme oxygenase-1; IFN-1, Type I interferon; I/R, Ischemia/reperfusion; ITGAX, Integrin alpha x; LMX1A, LIM homeobox transcription factor 1, alpha; LRRs, Leucine-rich repeats; MDA, Malondialdehyde; MS, Multiple sclerosis; MHC, major histocompatibility complex; NF-κB, Nuclear factor-κB; NFY, Nuclear factor-Y protein; NLRs, NOD-like receptors; NLRC5, NLR family, CARD containing 5; NLRP3, NLR family pyrin domain containing 3; NLS, Nuclear localization signal; NODs, Nucleotide oligomerization domain; NQO-1, NAD(P)H,quinone oxidoreductase 1; Nrf2, Nuclear factor erythroid 2-related factor 2; NTP, nucleoside triphosphate; OGD, Oxygen-glucose deprivation; PAMPs, Pathogen-associated molecular patterns; PRRs, Pattern recognition receptors; PYD, Pyrin domain; RFX, Regulatory factor X; RLRs, Retinoic acid-inducible gene-I-like receptors; ROS, oxygen species; TLRs, Toll-like receptors; SCAMP1, Secretory carrier membrane protein 1; SCZ, Schizophrenia; TMEV, Theiler’s murine encephalomyelitis virus; VSV, Vesicular stomatitis virus; SOD, Superoxide dismutase; STAT1, Signal transducers and activators of transcription-1; WNV, West Nile virus; ZIKV, Zika virus.
1. Wu Y, Shi T, Li J. NLRC5: A Paradigm for NLRs in Immunological and Inflammatory Reaction. Cancer Lett (2019) 451:92–9. doi: 10.1016/j.canlet.2019.03.005
2. Kong X, Yuan Z, Cheng J. The Function of NOD-like Receptors in Central Nervous System Diseases. J Neurosci Res (2017) 95(8):1565–73. doi: 10.1002/jnr.24004
3. Wang JQ, Liu YR, Xia Q, Chen RN, Liang J, Xia QR, et al. Emerging Roles for NLRC5 in Immune Diseases. Front Pharmacol (2019) 10:1352. doi: 10.3389/fphar.2019.01352
4. Cao L, Wu XM, Hu YW, Xue NN, Nie P, Chang MX. The Discrepancy Function of NLRC5 Isoforms in Antiviral and Antibacterial Immune Responses. Dev Comp Immunol (2018) 84:153–63. doi: 10.1016/j.dci.2018.02.013
5. Qiu L, Ma T, Chang G, Liu X, Guo X, Xu L, et al. Expression Patterns of NLRC5 and Key Genes in the STAT1 Pathway Following Infection With Salmonella pullorum. Gene (2017) 597:23–9. doi: 10.1016/j.gene.2016.10.026
6. Periyasamy P, Thangaraj A, Bendi VS, Buch S. HIV-1 Tat-mediated Microglial Inflammation Involves a Novel miRNA-34a-NLRC5-NfκB Signaling Axis. Brain Behav Immun (2019) 80:227–37. doi: 10.1016/j.bbi.2019.03.011
7. Leda AR, Bertrand L, Andras IE, El-Hage N, Nair M, Toborek M. Selective Disruption of the Blood-Brain Barrier by Zika Virus. Front Microbiol (2019) 10:2158. doi: 10.3389/fmicb.2019.02158
8. Azouz F, Arora K, Krause K, Nerurkar VR, Kumar M. Integrated MicroRNA and Mrna Profiling in Zika Virus-Infected Neurons. Viruses (2019) 11(2):162. doi: 10.3390/v11020162
9. Kumar M, Belcaid M, Nerurkar VR. Identification of Host Genes Leading to West Nile Virus Encephalitis in Mice Brain Using RNA-seq Analysis. Sci Rep (2016) 6:26350. doi: 10.1038/srep26350
10. Li P, Shen Y, Cui P, Hu Y, Zhang Y, Miao F, et al. Neuronal NLRC5 Regulates MHC Class I Expression in Neuro-2a Cells and Also During Hippocampal Development. J Neurochem (2020) 152(2):182–94. doi: 10.1111/jnc.14876
11. Li L, Yu M, Pang H, Chen L, Liu J, Hou S. NLRC5 Protects Neurons From Oxygen-Glucose Deprivation-Induced Injury Through Activating the Nrf2/HO-1 Pathway. J Recept Signal Transduct Res (2021) 41(1):53–8. doi: 10.1080/10799893.2020.1786840
12. Zhang Z, Sun Y, Chen X. NLRC5 Alleviated OGD/R-Induced PC12-Cell Injury by Inhibiting Activation of the TLR4/Myd88/NF-κb Pathway. J Int Med Res (2020) 48(8):300060520940455. doi: 10.1177/0300060520940455
13. Zong Z, Song Y, Xue Y, Ruan X, Liu X, Yang C, et al. Knockdown of LncRNA SCAMP1 Suppressed Malignant Biological Behaviours of Glioma Cells via Modulating miR-499a-5p/LMX1A/NLRC5 Pathway. J Cell Mol Med (2019) 23(8):5048–62. doi: 10.1111/jcmm.14362
14. Zhu C, Zou C, Guan G, Guo Q, Yan Z, Liu T, et al. Development and Validation of an Interferon Signature Predicting Prognosis and Treatment Response for Glioblastoma. Oncoimmunology (2019) 8(9):e1621677. doi: 10.1080/2162402X.2019.1621677
15. Comba A, Dunn PJ, Argento AE, Kadiyala P, Ventosa M, Zamler DB, et al. The Proto-Oncogene Fyn Inhibits the Anti-Glioblastoma Immune Response [Preprint] (2019). doi: 10.1101/608505
16. Falcão AM, van Bruggen D, Marques S, Meijer M, Jäkel S, Agirre E, et al. Disease-Specific Oligodendrocyte Lineage Cells Arise in Multiple Sclerosis. Nat Med (2018) 24(12):1837–44. doi: 10.1038/s41591-018-0236-y
17. Bijalwan M. Pathogenesis of Theiler’s Murine Encephalomyelitis Virus (TMEV) in an Experimental Model of Epilepsy. College Station, Texas, USA: Texas A&M University (2017).
18. Bergen SE, O’Dushlaine CT, Ripke S, Lee PH, Ruderfer DM, Akterin S, et al. Genome-Wide Association Study in a Swedish Population Yields Support for Greater CNV and MHC Involvement in Schizophrenia Compared With Bipolar Disorder. Mol Psychiatry (2012) 17(9):880–6. doi: 10.1038/mp.2012.73
19. Pacifico R, Davis RL. Transcriptome Sequencing Implicates Dorsal Striatum-Specific Gene Network, Immune Response and Energy Metabolism Pathways in Bipolar Disorder. Mol Psychiatry (2017) 22(3):441–9. doi: 10.1038/mp.2016.94
20. Yao Y, Qian Y. Expression Regulation and Function of NLRC5. Protein Cell (2013) 4(3):168–75. doi: 10.1007/s13238-012-2109-3
21. Tang F, Xu Y, Zhao B. NLRC5: New Cancer Buster? Mol Biol Rep (2020) 47(3):2265–77. doi: 10.1007/s11033-020-05253-5
22. Gutte PG, Jurt S, Grütter MG, Zerbe O. Unusual Structural Features Revealed by the Solution NMR Structure of the NLRC5 Caspase Recruitment Domain. Biochemistry (2014) 53(19):3106–17. doi: 10.1021/bi500177x
23. Jongsma MLM, Guarda G, Spaapen RM. The Regulatory Network Behind MHC Class I Expression. Mol Immunol (2019) 113:16–21. doi: 10.1016/j.molimm.2017.12.005
24. Mótyán JA, Bagossi P, Benkő S, Tőzsér J. A Molecular Model of the Full-Length Human NOD-like Receptor Family CARD Domain Containing 5 (NLRC5) Protein. BMC Bioinf (2013) 14:275. doi: 10.1186/1471-2105-14-275
25. Kuenzel S, Till A, Winkler M, Häsler R, Lipinski S, Jung S, et al. The Nucleotide-Binding Oligomerization Domain-Like Receptor NLRC5 is Involved in IFN-Dependent Antiviral Immune Responses. J Immunol (2010) 184(4):1990–2000. doi: 10.4049/jimmunol.0900557
26. Lénárt N, Brough D, Dénes Á. Inflammasomes Link Vascular Disease With Neuroinflammation and Brain Disorders. J Cereb Blood Flow Metab (2016) 36(10):1668–85. doi: 10.1177/0271678X16662043
27. Nyúl-Tóth Á, Kozma M, Nagyőszi P, Nagy K, Fazakas C, Haskó J, et al. Expression of Pattern Recognition Receptors and Activation of the Non-Canonical Inflammasome Pathway in Brain Pericytes. Brain Behav Immun (2017) 64:220–31. doi: 10.1016/j.bbi.2017.04.010
28. Nagyőszi P, Nyúl-Tóth Á, Fazakas C, Wilhelm I, Kozma M, Molnár J, et al. Regulation of NOD-Like Receptors and Inflammasome Activation in Cerebral Endothelial Cells. J Neurochem (2015) 135(3):551–64. doi: 10.1111/jnc.13197
29. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR Family Member NLRC5 Is a Transcriptional Regulator of MHC Class I Genes. Proc Natl Acad Sci U S A (2010) 107(31):13794–9. doi: 10.1073/pnas.1008684107
30. Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B Functions in Synaptic Signaling and Behavior. Nat Neurosci (2003) 6(10):1072–8. doi: 10.1038/nn1110
31. Robison AJ, Nestler EJ. Transcriptional and Epigenetic Mechanisms of Addiction. Nat Rev Neurosci (2011) 12(11):623–37. doi: 10.1038/nrn3111
32. Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, et al. NLRC5 Negatively Regulates the NF-kappaB and Type I Interferon Signaling Pathways. Cell (2010) 141(3):483–96. doi: 10.1016/j.cell.2010.03.040
33. Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 Limits the Activation of Inflammatory Pathways. J Immunol (2010) 185(3):1681–91. doi: 10.4049/jimmunol.0903900
34. Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hösel M, et al. A Role for the Human Nucleotide-Binding Domain, Leucine-Rich Repeat-Containing Family Member NLRC5 in Antiviral Responses. J Biol Chem (2010) 285(34):26223–32. doi: 10.1074/jbc.M110.109736
35. Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF. Enhanced TLR-induced Nf-κb Signaling and Type I Interferon Responses in NLRC5 Deficient Mice. Cell Res (2012) 22(5):822–35. doi: 10.1038/cr.2012.53
36. Fekete T, Bencze D, Szabo A, Csoma E, Biro T, Bacsi A, et al. Regulatory NLRs Control the RLR-Mediated Type I Interferon and Inflammatory Responses in Human Dendritic Cells. Front Immunol (2018) 9:2314. doi: 10.3389/fimmu.2018.02314
37. Robbins GR, Truax AD, Davis BK, Zhang L, Brickey WJ, Ting JP. Regulation of Class I Major Histocompatibility Complex (MHC) by Nucleotide-Binding Domain, Leucine-Rich Repeat-Containing (NLR) Proteins. J Biol Chem (2012) 287(29):24294–303. doi: 10.1074/jbc.M112.364604
38. Hu Z, Chai J. Structural Mechanisms in NLR Inflammasome Assembly and Signaling. Curr Top Microbiol Immunol (2016) 397:23–42. doi: 10.1007/978-3-319-41171-2_2
39. Yu JW, Lee MS. Mitochondria and the NLRP3 Inflammasome: Physiological and Pathological Relevance. Arch Pharm Res (2016) 39(11):1503–18. doi: 10.1007/s12272-016-0827-4
40. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome Inhibition Prevents α-Synuclein Pathology and Dopaminergic Neurodegeneration in Mice. Sci Transl Med (2018) 10(465):eaah4066. doi: 10.1126/scitranslmed.aah4066
41. Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, et al. Cutting Edge: NLRC5-Dependent Activation of the Inflammasome. J Immunol (2011) 186(3):1333–7. doi: 10.4049/jimmunol.1003111
42. Kumar H, Pandey S, Zou J, Kumagai Y, Takahashi K, Akira S, et al. NLRC5 Deficiency Does Not Influence Cytokine Induction by Virus and Bacteria Infections. J Immunol (2011) 186(2):994–1000. doi: 10.4049/jimmunol.1002094
43. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 Regulates MHC Class I Antigen Presentation in Host Defense Against Intracellular Pathogens. Cell Res (2012) 22(5):836–47. doi: 10.1038/cr.2012.56
44. Labbé K, Saleh M. Cell Death in the Host Response to Infection. Cell Death Differ (2008) 15(9):1339–49. doi: 10.1038/cdd.2008.91
45. Fink SL, Bergsbaken T, Cookson BT. Anthrax Lethal Toxin and Salmonella Elicit the Common Cell Death Pathway of Caspase-1-Dependent Pyroptosis via Distinct Mechanisms. Proc Natl Acad Sci U S A (2008) 105(11):4312–7. doi: 10.1073/pnas.0707370105
46. Yeung KS, Chung BH, Choufani S, Mok MY, Wong WL, Mak CC, et al. Genome-Wide DNA Methylation Analysis of Chinese Patients With Systemic Lupus Erythematosus Identified Hypomethylation in Genes Related to the Type I Interferon Pathway. PLoS One (2017) 12(1):e0169553. doi: 10.1371/journal.pone.0169553
47. Zaghlool SB, Kühnel B, Elhadad MA, Kader S, Halama A, Thareja G, et al. Epigenetics Meets Proteomics in an Epigenome-Wide Association Study With Circulating Blood Plasma Protein Traits. Nat Commun (2020) 11(1):15. doi: 10.1038/s41467-019-13831-w
48. Meeks KAC, Henneman P, Venema A, Burr T, Galbete C, Danquah I, et al. An Epigenome-Wide Association Study in Whole Blood of Measures of Adiposity Among Ghanaians: The RODAM Study. Clin Epigenet (2017) 9:103. doi: 10.1186/s13148-017-0403-x
49. Zhang X, Justice AC, Hu Y, Wang Z, Zhao H, Wang G, et al. Epigenome-Wide Differential DNA Methylation Between HIV-Infected and Uninfected Individuals. Epigenetics (2016) 11(10):750–60. doi: 10.1080/15592294.2016.1221569
50. Zou J, Lippert C, Heckerman D, Aryee M, Listgarten J. Epigenome-Wide Association Studies Without the Need for Cell-Type Composition. Nat Methods (2014) 11(3):309–11. doi: 10.1038/nmeth.2815
51. Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC Class I Transactivator is a Target for Immune Evasion in Cancer. Proc Natl Acad Sci U S A (2016) 113(21):5999–6004. doi: 10.1073/pnas.1602069113
52. Song M, Bode AM, Dong Z, Lee MH. AKT as a Therapeutic Target for Cancer. Cancer Res (2019) 79(6):1019–31. doi: 10.1158/0008-5472.CAN-18-2738
53. Fan Y, Dong Z, Shi Y, Sun S, Wei B, Zhan L. NLRC5 Promotes Cell Migration and Invasion by Activating the PI3K/AKT Signaling Pathway in Endometrial Cancer. J Int Med Res (2020) 48(5):300060520925352. doi: 10.1177/0300060520925352
54. Wang J, Yang L, Liang F, Chen Y, Yang G. Integrin Alpha X Stimulates Cancer Angiogenesis Through PI3K/Akt Signaling-Mediated VEGFR2/VEGF-A Overexpression in Blood Vessel Endothelial Cells. J Cell Biochem (2019) 120(2):1807–18. doi: 10.1002/jcb.27480
55. He YH, Li MF, Zhang XY, Meng XM, Huang C, Li J. NLRC5 Promotes Cell Proliferation Via Regulating the AKT/VEGF-A Signaling Pathway in Hepatocellular Carcinoma. Toxicology (2016) 359-360:47–57. doi: 10.1016/j.tox.2016.06.012
56. Han F, Gao Y, Ding CG, Xia XX, Wang YX, Xue WJ, et al. Knockdown of NLRC5 Attenuates Renal I/R Injury In Vitro Through the Activation of PI3K/Akt Signaling Pathway. BioMed Pharmacother (2018) 103:222–7. doi: 10.1016/j.biopha.2018.04.040
57. Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell (2017) 169(6):985–99. doi: 10.1016/j.cell.2017.05.016
58. Wang Q, Zhou Y, Rychahou P, Harris JW, Zaytseva YY, Liu J, et al. Deptor Is a Novel Target of Wnt/β-Catenin/c-Myc and Contributes to Colorectal Cancer Cell Growth. Cancer Res (2018) 78(12):3163–75. doi: 10.1158/0008-5472.CAN-17-3107
59. Wang Z, Li B, Zhou L, Yu S, Su Z, Song J, et al. Prodigiosin Inhibits Wnt/β-Catenin Signaling and Exerts Anticancer Activity in Breast Cancer Cells. Proc Natl Acad Sci U S A (2016) 113(46):13150–5. doi: 10.1073/pnas.1616336113
60. Wang Q, Ding H, He Y, Li X, Cheng Y, Xu Q, et al. NLRC5 Mediates Cell Proliferation, Migration, and Invasion by Regulating the Wnt/β-Catenin Signalling Pathway in Clear Cell Renal Cell Carcinoma. Cancer Lett (2019) 444:9–19. doi: 10.1016/j.canlet.2018.11.024
61. Peng YY, He YH, Chen C, Xu T, Li L, Ni MM, et al. NLRC5 Regulates Cell Proliferation, Migration and Invasion in Hepatocellular Carcinoma by Targeting the Wnt/β-Catenin Signaling Pathway. Cancer Lett (2016) 376(1):10–21. doi: 10.1016/j.canlet.2016.03.006
62. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1β Signaling Promotes CNS-Intrinsic Immune Control of West Nile Virus Infection. PLoS Pathog (2012) 8(11):e1003039. doi: 10.1371/journal.ppat.1003039
63. Yang K, Shi J, Hu Z, Hu X. The Deficiency of miR-214-3p Exacerbates Cardiac Fibrosis via miR-214-3p/NLRC5 Axis. Clin Sci (Lond) (2019) 133(17):1845–56. doi: 10.1042/CS20190203
64. Xu T, Ni MM, Xing L, Li XF, Meng XM, Huang C, et al. NLRC5 Regulates TGF-beta1-induced Proliferation and Activation of Hepatic Stellate Cells During Hepatic Fibrosis. Int J Biochem Cell Biol (2016) 70:92–104. doi: 10.1016/j.biocel.2015.11.010
65. Wang Q, Li M, Shen Z, Bu F, Yu H, Pan X, et al. The Long Non-Coding RNA MEG3/Mir-Let-7c-5p Axis Regulates Ethanol-Induced Hepatic Steatosis and Apoptosis by Targeting NLRC5. Front Pharmacol (2018) 9:302. doi: 10.3389/fphar.2018.00302
66. Wang S, Zhao X, Yang S, Chen B, Shi J. Knockdown of NLRC5 Inhibits Renal Fibroblast Activation via Modulating TGF-Beta1/Smad Signaling Pathway. Eur J Pharmacol (2018) 829:38–43. doi: 10.1016/j.ejphar.2018.03.045
67. Li L, Xu T, Huang C, Peng Y, Li J. NLRC5 Mediates Cytokine Secretion in RAW264.7 Macrophages and Modulated by the JAK2/STAT3 Pathway. Inflammation (2014) 37(3):835–47. doi: 10.1007/s10753-013-9804-y
68. Wang Z, Li G, Dou S, Zhang Y, Liu Y, Zhang J, et al. Tim-3 Promotes Listeria Monocytogenes Immune Evasion by Suppressing Major Histocompatibility Complex Class I. J Infect Dis (2020) 221(5):830–40. doi: 10.1093/infdis/jiz512
69. Lee H, Brott BK, Kirkby LA, Adelson JD, Cheng S, Feller MB, et al. Synapse Elimination and Learning Rules Co-Regulated by MHC Class I H2-Db. Nature (2014) 509(7499):195–200. doi: 10.1038/nature13154
70. Shen Y, Zhao H, Li P, Peng Y, Cui P, Miao F, et al. MHC Class I Molecules and PirB Shape Neuronal Morphology by Affecting the Dendritic Arborization of Cortical Neurons. Neurochem Res (2019) 44(2):312–22. doi: 10.1007/s11064-018-2676-7
71. Fourgeaud L, Davenport CM, Tyler CM, Cheng TT, Spencer MB, Boulanger LM. MHC Class I Modulates NMDA Receptor Function and AMPA Receptor Trafficking. Proc Natl Acad Sci U S A (2010) 107(51):22278–83. doi: 10.1073/pnas.0914064107
72. Langen UH, Ayloo S, Gu C. Development and Cell Biology of the Blood-Brain Barrier. Annu Rev Cell Dev Biol (2019) 35:591–613. doi: 10.1146/annurev-cellbio-100617-062608
73. Al-Obaidi MMJ, Desa MNM. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial-Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell Mol Neurobiol (2018) 38(7):1349–68. doi: 10.1007/s10571-018-0609-2
74. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-Associated Neurocognitive Disorder–Pathogenesis and Prospects for Treatment. Nat Rev Neurol (2016) 12(4):234–48. doi: 10.1038/nrneurol.2016.27
75. Jauhari A, Singh T, Singh P, Parmar D, Yadav S. Regulation of miR-34 Family in Neuronal Development. Mol Neurobiol (2018) 55(2):936–45. doi: 10.1007/s12035-016-0359-4
76. Ludlow M, Kortekaas J, Herden C, Hoffmann B, Tappe D, Trebst C, et al. Neurotropic Virus Infections as the Cause of Immediate and Delayed Neuropathology. Acta Neuropathol (2016) 131(2):159–84. doi: 10.1007/s00401-015-1511-3
77. Wang H, Zhong D, Chen H, Jin J, Liu Q, Li G. NLRP3 Inflammasome Activates Interleukin-23/interleukin-17 Axis During Ischaemia-Reperfusion Injury in Cerebral Ischaemia in Mice. Life Sci (2019) 227:101–13. doi: 10.1016/j.lfs.2019.04.031
78. Meng C, Zhang J, Zhang L, Wang Y, Li Z, Zhao J. Effects of NLRP6 in Cerebral Ischemia/Reperfusion (I/R) Injury in Rats. J Mol Neurosci (2019) 69(3):411–8. doi: 10.1007/s12031-019-01370-4
79. Huang Q, Lou T, Wang M, Xue L, Lu J, Zhang H, et al. Compound K Inhibits Autophagy-Mediated Apoptosis Induced by Oxygen and Glucose Deprivation/Reperfusion via Regulating AMPK-mTOR Pathway in Neurons. Life Sci (2020) 254:117793. doi: 10.1016/j.lfs.2020.117793
80. Deng Y, Ma G, Dong Q, Sun X, Liu L, Miao Z, et al. Overexpression of miR-224-3p Alleviates Apoptosis From Cerebral Ischemia Reperfusion Injury by Targeting FIP200. J Cell Biochem (2019) 120(10):17151–8. doi: 10.1002/jcb.28975
81. Shi YS, Zhang Y, Liu B, Li CB, Wu J, Li Y. Nomilin Protects Against Cerebral Ischemia-Reperfusion Induced Neurological Deficits and Blood-Brain Barrier Disruption via the Nrf2 Pathway. Food Funct (2019) 10(9):5323–32. doi: 10.1039/C9FO01481K
82. Chen Z, Ding T, Ma CG. Dexmedetomidine (DEX) Protects Against Hepatic Ischemia/Reperfusion (I/R) Injury by Suppressing Inflammation and Oxidative Stress in NLRC5 Deficient Mice. Biochem Biophys Res Commun (2017) 493(2):1143–50. doi: 10.1016/j.bbrc.2017.08.017
83. Gharagozloo M, Gris KV, Mahvelati T, Amrani A, Lukens JR, Gris D. NLR-Dependent Regulation of Inflammation in Multiple Sclerosis. Front Immunol (2017) 8:2012. doi: 10.3389/fimmu.2017.02012
Keywords: pattern recognition receptor, NLRC5, central nervous system, disease, development
Citation: Zhang L, Jiao C, Liu L, Wang A, Tang L, Ren Y, Huang P, Xu J, Mao D and Liu L (2021) NLRC5: A Potential Target for Central Nervous System Disorders. Front. Immunol. 12:704989. doi: 10.3389/fimmu.2021.704989
Received: 04 May 2021; Accepted: 07 June 2021;
Published: 18 June 2021.
Edited by:
Robert O. Watson, Texas A&M University, United StatesReviewed by:
Juan Pablo de Rivero Vaccari, University of Miami, United StatesCopyright © 2021 Zhang, Jiao, Liu, Wang, Tang, Ren, Huang, Xu, Mao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liqun Liu, bGl1bGlxdW5AY3N1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.