 Luca Quartuccio1,2*†
Luca Quartuccio1,2*† Ginevra De Marchi1†
Ginevra De Marchi1† Simone Longhino1,2
Simone Longhino1,2 Valeria Manfrè1,2
Valeria Manfrè1,2 Maria Teresa Rizzo1,2
Maria Teresa Rizzo1,2 Saviana Gandolfo1
Saviana Gandolfo1 Alberto Tommasini3,4
Alberto Tommasini3,4 Salvatore De Vita1,2‡
Salvatore De Vita1,2‡ Robert Fox5‡
Robert Fox5‡- 1Rheumatology Clinic, ASU FC, Udine, Italy
- 2Department of Medicine, University of Udine, Udine, Italy
- 3Pediatric Immunology, IRCCS Burlo Garofolo, Trieste, Italy
- 4Department of Medical Sciences, University of Trieste, Trieste, Italy
- 5Rheumatology Clinic, Scripps Memorial Hospital and Research Foundation, La Jolla, CA, United States
Common variable immunodeficiency disorders (CVID) are a group of rare diseases of the immune system and the most common symptomatic primary antibody deficiency in adults. The “variable” aspect of CVID refers to the approximately half of the patients who develop non-infective complications, mainly autoimmune features, in particular organ specific autoimmune diseases including thyroiditis, and cytopenias. Among these associated conditions, the incidence of lymphoma, including mucosal associated lymphoid tissue (MALT) type, is increased. Although these associated autoimmune disorders in CVID are generally attributed to Systemic Lupus Erythematosus (SLE), we propose that Sjogren’s syndrome (SS) is perhaps a better candidate for the associated disease. SS is an autoimmune disorder characterized by the lymphocytic infiltrates of lacrimal and salivary glands, leading to dryness of the eyes and mouth. Thus, it is a lymphocyte aggressive disorder, in contrast to SLE where pathology is generally attributed to auto-antibody and complement activation. Although systemic lupus erythematosus (SLE) shares these features with SS, a much higher frequency of MALT lymphoma distinguishes SS from SLE. Also, the higher frequency of germ line encoded paraproteins such as the monoclonal rheumatoid factor found in SS patients would be more consistent with the failure of B-cell VDJ switching found in CVID; and in contrast to the hypermutation that characterizes SLE autoantibodies. Thus, we suggest that SS may fit as a better “autoimmune” association with CVID. Examining the common underlying biologic mechanisms that promote lymphoid infiltration by dysregulated lymphocytes and lymphoma in CVID may provide new avenues for treatment in both the diseases. Since the diagnosis of SLE or rheumatoid arthritis is usually based on specific autoantibodies, the associated autoimmune features of CVID patients may not be recognized in the absence of autoantibodies.
Introduction
Common variable immunodeficiency disorders (CVID) are a group of rare diseases of the immune system and the most common symptomatic primary antibody deficiency in adults. They comprise a group of disorders with similar antibody deficiency but a myriad of different aetiologies, most of which remain poorly understood (1–8). CVID are sometimes complicated with autoimmune features (9–11). Several biological mechanisms have been recently implicated in the development of these complications, including the decrease in the number of circulating switched memory B cells, CD21low B cell expansion, interferon (IFN) signature and B-Cell Activating Factor (BAFF) hyper-expression, and they will be addressed in the subsequent paragraphs of this review. All of these mechanisms prevent the emergence of somatic mutation among the autoantibodies in CVID patients. Thus, CVID provides an opportunity to understand processes such as neutropenia, thrombocytopenia, and lymphoproliferation in the absence of the affinity selected autoantibodies that we normally invoke as pathogenetic mechanisms.
It is worth recalling the original studies by Kunkel et al. (12) pointed out that germ line genes (encoding both heavy and light chains) were found as autoantibodies in patients with Waldenstrom’s macroglobulinemia that had not undergone significant affinity selected maturation and recombination. For example, the germ line encoded antibodies with mixed cryoglobulin or cold agglutinin activity were sequenced and found to have a limited repertoire that was defined as conserved “idiotypes” and later found to have sequence due to germ line encoded heavy and light chains. Of interest, similar limited expression of light chains was found in the rheumatoid factor (RF) of Sjögren’s syndrome (SS) patients (i.e., the 17-109 idiotype) but not in the highly variable light chains of RF in Systemic Lupus Erythematosus (SLE) patients (13). Further, the B-cell lymphomas of SS show a marked limitation of their surface immunoglobulin heavy and light chains (14). In contrast, autoantibodies with extensive somatic diversification mechanisms are the hallmark of SLE and these patients do not have the elevated frequency of B-cell Mucosa Associated Lymphoid Tissue (MALT) lymphomas (15).
In this review we look at the potential link between CVID and SS based on the high frequency of lymphoma in both groups. This contrasts with the most reviews that suggest SLE is the main “associated” systemic autoimmune disorder. This change of view is more than semantic and emphasizes that SS is a disorder of “aggressive” hyperactivated lymphocytes that infiltrate tissues in comparison to SLE that is characterized by its pathogenic antibodies that play a role through immune complexes and complement activation.
Clinical Features, Autoimmune Aspects and Heterogeneity of CVID
CVID represents the most frequent clinically expressed primary immunodeficiency (PID) in adults, accounting for more than 50% of cases of PIDs (1, 2). Worldwide geographic differences in prevalence are the consequence of discrepancies in diagnostic methods, disease awareness and data registration (3).
The term CVID was firstly coined in 1971 by the World Health Organization to express a diagnosis of exclusion from other antibody deficiency syndromes with more specific clinical and inheritance patterns (4). Since then, CVID diagnostic criteria have been revised many times (1, 5–7), matching the evolution in the clinical, immunological and genetic knowledge on the disease (7, 8).
In 2008, Chapel et al. firstly categorized CVID complications, identifying five distinct phenotypes: no complications, autoimmunity, polyclonal lymphocytic infiltration, enteropathy and lymphoid malignancy (9). Subsequently, other studies attested the classification of CVID based on the presence of complications, and the concomitance of certain features, as autoimmunity, lymphocytic interstitial lung disease and lymphoid hyperplasia, was noted (10, 11).
More recently, the 2016 International Consensus document on CVID supported further analysis on the associations between genetics, clinical presentation, disease severity and immunotype, allowing the distinction into “infection-predominant”, “inflammatory predominant” and “autoimmunity predominant” entities (1).
The latest European society of Immune Deficiency (ESID) (2019) diagnostic criteria include autoimmune and inflammatory conditions as primary clinical presentations, in addition to laboratory abnormalities (8).
In fact, it has emerged that at least 30% of patients show additional non-infectious conditions, as autoimmune, autoinflammatory, granulomatous, lymphoproliferative and/or malignant complications, especially in patients with low fraction of isotype switched memory B cells (1, 6, 11, 16).
Autoimmune diseases can be observed before CVID diagnosis in up to 17.4% of patients and as the only clinical manifestation at the time of diagnosis of CVID in 2.3% of patients (17).
Autoimmune and autoinflammatory conditions reported in CVID are summarized in Table 1.

Table 1 Autoimmune and autoinflammatory conditions reported in CVID.
Systemic autoimmune diseases, properly rheumatic diseases, were found in 5.9% of all cases in a cohort of 870 CVID patients analyzed from the USIDNET registry, accounting for almost 40% of the detected global autoimmune manifestations. One third of patients with CVID-associated rheumatologic disorders had an additional inflammatory complication or malignancy (18). Among CVID patients affected by rheumatologic conditions, a female predominance has been noted, while inflammatory arthritis has been reported as the most frequent rheumatological manifestation (3%), followed by SS (11/870 in USIDNET registry), SLE, vasculitis and Behçet’s disease and others (18–20).
Importantly, in CVID patients, the overall risk of lymphoid malignancies [e.g. extra-nodal B cell non-Hodgkin lymphoma (NHL)] ranges between 2 and 10% (11, 20), while the risk of gastric cancer was reported as 10-fold increased (21).
CVID and Autoimmunity
The pathogenesis of autoimmune complications in CVID is poorly understood, as well, and is counterintuitive because these patients are defined by their inability to make antibodies yet still mount autoimmune reactions. Some general assumptions may support this paradox:
a) the co-existence of hypo- and hyper-immune states in the same individual at the same point in time is not implausible given the complexity of the immune system;
b) both T and B cells abnormalities may contribute to the development of autoimmunity in CVID patients;
c) increased autoreactive B cells and reduced T regulatory cells may be involved in the pathogenesis of autoimmunity in CVID.
Studies on B and T cell immune dysregulation found many possible responsible factors for autoimmunity appearance, such as the expansion of CD21low/- B cells and related reduction of T regulatory cells (22); the reduction of switched memory B cells (23, 24); the low levels of naïve CD8+ (25) and CD4+ (22, 25, 26) T cells and the elevated T helper 1 and IFN gamma signature (27), related to the increase in T helper 1 and follicular T CD4+ cells (28, 29). Conflicting evidence emerged on the role of BAFF and IL-7 (10, 30–32).
Challenge of Identifying SS in CVID
Even if autoimmune clinical manifestations reported in CVID mostly resemble SLE (autoimmune cytopenias in particular), we suggest that SS may fit as a better “autoimmune” association.
SS diagnosis is not as simple as you’d think. One recent study from academic institutions with expertise in SS has shown that almost 50% of patients diagnosed as SLE with dry eye symptoms actually had SS when the patients were re-examined for the presence of anti-SSA antibody and other clinical features of SS (33). Moreover, after the patient is initially labelled as SLE, it is rare that the underlying diagnosis is re-examined. As a result, SS patients with extraglandular manifestations, that might benefit from new trials of therapy, could be never considered.
Moreover, patients affected by CVID might show SS typical manifestations even in the absence of SS related autoantibodies, determining a condition resembling seronegative SS. These clinical manifestations include both glandular (e.g. sicca symptoms) and extraglandular manifestations (e.g. constitutional manifestations, interstitial lung disease, tubular nephritis, haemolytic anemia, thrombocytopenia) (34). Thus, patients with CVID should be investigated more thoroughly for SS-related symptoms and studied in depth with functional, instrumental and histopathological tests (e.g. minor salivary gland biopsy) in addiction to laboratory parameters.
SS-Like Features in CVID Patients Who Lack Autoantibody to SS-A
The characteristic pathologic picture in both glandular and extraglandular manifestations of SS is the “aggressive” lymphocyte that infiltrates tissues. This may be reflected in the “focus score” that counts clusters of lymphocytes in a minor salivary gland biopsy, the analogous infiltrates of the lacrimal glands, the lymphocytic clusters in the lung in lymphocytic interstitial pneumonitis (LIP) or the markedly increased frequency of lymphoma.
Although elegant models have shown that SS-A is a chaperone molecule to both single and double stranded viral nucleotides, it is the resistance of SS-A to breakdown in the apoptotic bleb that makes it an attractive candidate for perpetuating the autoimmune cycle. The binding to antibody to SS-A (whose production is closely linked to HLA-DR3) provides a mechanism for Fc internalization of the SS-A/hYRNA complex with subsequent internalization and translocation to the toll-like receptor (TLR) (35). Yet, the finding of lymphoid infiltrates and lymphoma and CVID indicate that there is more to the story.
In fact, since CVID patients lack detectable circulating autoantibodies including anti-SS-A estimation of the role of SS pathogenetic factors in CVID is likely to be grossly underestimated. For example, it has been shown that activation of TLR receptors by viral and bacterial nucleic acids plays a role in CVID by promoting IFN alpha pathway rather than TNF alpha upon stimulation (36). Also, non-coding small RNAs are important (37). Other common factors such as T-follicular type and T-helper type phenotype and B-cells expressing low levels of cellular surface CD as well as reciprocal decrease in regulatory T-cells and isotype switched memory B cells will be reviewed below.
Thus, the lesson for rheumatologists from CVID is that we have considered the cardinal feature of SS as SS-A antigen and antibody that targets. However, also in the absence of antibody to SS-A we see the lymphocyte aggressive features that characterize its dysautonomic features (dry eyes, dry mouth, dry skin, interstitial pneumonitis, interstitial nephritis, and increased frequency of lymphoma).
Relevant Clues From the Genetics of CVID
While CVID is mainly a polygenic and multifactorial disease, recent technical advances in next generation sequencing (NSG) allowed to discover a monogenic cause in up to 15-30% of cases (38, 39). Thirteen monogenic mutations associated with CVID are listed on the Online Mendelian Inheritance in Man (OMIM) database. Among them, some are specifically associated with autoimmunity, and are listed in Table 2.
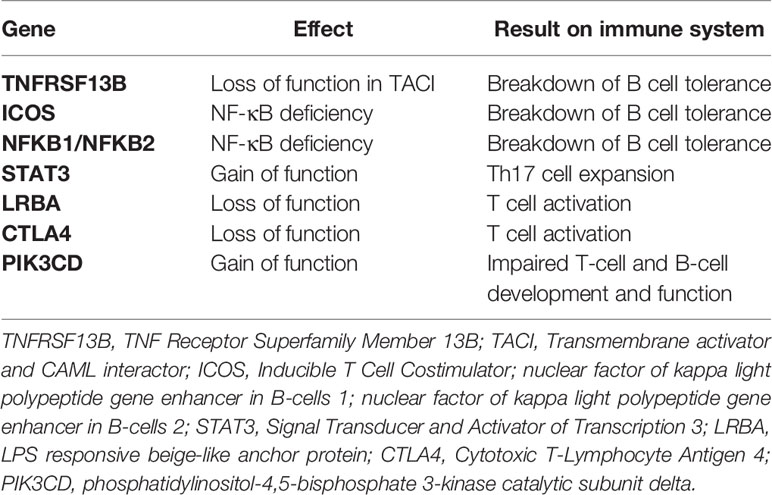
Table 2 Mutations associated with CVID and autoimmunity.
TACI may be involved in the central B cell tolerance and that reduced function results in the loss of tolerance and resultant autoimmunity (40).
ICOS and NF-κB deficiencies lead to CVID-like immunodeficiency syndromes and autoimmunity (37–39). Interestingly, dysregulation of NF-kB in glandular epithelial cells results in SS-like features (41), as well as expression of NF-κB at both the mRNA and protein level was up-regulated in SS-lymphoma-BAFF-RHis159Tyr-derived B cells, linking the innate to the adaptive immunity upregulation and lymphoma in SS (42).
STAT3 is thought to lead to autoimmunity by promoting the activation and expansion of autoimmunity-associated TH17 cells, a subtype of T cell deeply involved in the early mechanisms of autoimmunity (43–45).
LRBA and CTLA-4 are inhibitors of T cell, and their deficiencies cause excessive T cell activation and breakdown of immune tolerance, resulting in autoimmunity, as emerged even under checkpoint inhibitor therapy, namely ipilimumab (46–48).
Activated PI3Kδ syndrome (APDS) is characterized by impaired T- and B-cell development and (APDS) function, autoimmunity, and lymphoproliferation (49).
Common Features Between CVID and Sjögren’s Syndrome
Lymphoproliferation
Both CVID and primary SS are strongly related to lymphoproliferation and lymphoma, in particular B cell NHL and MALT-type lymphoma.
CVID and Lymphoproliferation
Data collected from 1091 CVID patients, showed that CVID patients with a lymphoproliferative pattern have a 2.5-fold increased risk of developing lymphoma. The most common forms of benign lymphoid hyperplasia in CVID are splenomegaly and lymphadenopathy, but lymphoid hyperplasia and polyclonal lymphoproliferative infiltrations frequently affect other organs and tissues such as lungs and gastrointestinal. The lung represents the prevalent extranodal site for lymphoproliferative disorders, which include follicular bronchiolitis, lymphoid interstitial pneumonia, and pulmonary nodular lymphoid hyperplasia (50). Chapel et al. confirmed polyclonal lymphoid infiltrate as a predictor of lymphoma in CVID, which increased by 5 times the risk of developing lymphoma (9). The main histotypes of lymphoma in CVID are represented by mature B-cell malignancies followed by Hodgkin’s lymphoma and rarely by MALT-type lymphomas (51–53).
The presence of a 2-step transformation mechanism is hypothesized, as in non-Hodgkin’s lymphoma (9). A benign lymphoproliferation is deeply linked to the immune dysregulation intrinsic to CVID patients, as observed in other primitive immunodeficiencies such as CTLA-4 haploinsufficiency and STAT 3 gain of function mutations and as do autoimmune diseases such as primary SS (43, 46, 54). In CVID, mutation of TACI, reduction of isotype-switched memory B cells, expansion of CD21low/- B cells, expression of an IFN signature, expansion of inflammatory innate lymphoid cells and retained B cell function are all linked with development of autoimmunity and lymphoproliferation (19).
SS and Lymphoproliferation
B-cell clonal expansion is a key feature of SS and progression to B-cell lymphoma occurs in about 5% of patients. The progression from polyclonal, to benign clonal lymphoproliferation, to overt lymphoma in SS is one of the few human models in which one can study B-cell lymphomagenesis and its link to immune dysregulation (55).
The pathological hallmark of SS is MALT arising in chronically inflamed tissues, mainly in salivary glands, where inflammation, autoimmunity and lymphoproliferation coexist, creating a complex biological and immunological substratum that fuels autoreactive B lymphocytes persistence and promotes their proliferation, towards a clonal selection and a possible lymphoma development (54, 56).
In SS the prevalent histological type of lymphoma is marginal zone and particularly MALT lymphoma of the salivary glands but other histotypes are described and lung is one of the prevalent organ targets of lymphoproliferation besides exocrine glands (57).
In primary SS, splenomegaly and lymphadenopathy also represent well established lymphoma risk factors, in addition to salivary gland swelling (58, 59). Moreover, in primary SS patients the typical histopathological feature of ILD is LIP (60, 61). Both lung and stomach are other sites of lymphoma development in primary SS, other than salivary and lacrimal glands (58, 59).
BAFF Hyperexpression
CVID and BAFF Hyperexpression
Knight et al. demonstrated high serum levels of BAFF, APRIL and TACI in CVID patients, however, they didn’t find a correlation with immunological or clinical phenotypes (30).
Similarly, Kreuzaler et al. showed increased BAFF serum concentration in CVID patients, without a clear correlation with clinical parameters, immunodeficiency-related inflammatory disease and B cell subsets (62).
On the contrary, Maglione et al. showed that ILD progression in CVID correlates with increased levels of IgM, particularly with the production of IgM within B cell follicles in lung parenchyma; the main stimulator of pulmonary B cell hyperplasia seems to be BAFF, which was increased both in the blood and in the lung of CVID-ILD patients (63).
SS and BAFF Hyperexpression
Among the systemic autoimmune diseases, SS showed the highest serum levels of BAFF (64–69). In mice model, the overexpression of BAFF in mice leads to hyperplasia, autoimmunity, hyperglobulinemia and splenomegaly, while the normal expression of BAFF allows B cell survival and maturation (70). In BAFF transgenic mice there’s an excessive survival signal to autoreactive B lymphocytes, probably linked to a dysregulation of tolerance at the splenic level, where they observed an enlargement of marginal zone B-cell subset. B cells with an Marginal Zone (MZ)-like phenotype infiltrate the salivary glands of BAFF transgenic mice. Parallelly, unbalanced BAFF production in the lymphoid infiltrates of the salivary glands of primary SS patients promote recruitment of a specific and potentially pathogenic subpopulation of B cells (71). Epithelial cells also produce BAFF, thus supporting the hypothesis of the crucial role of BAFF in the pathogenesis of primary SS, by an immune dysregulation through an autocrine pattern of self-stimulation (72). High levels of BAFF were correlated with the specific autoantibodies of SS, anti-SSA/SSB, and BAFF was also found mainly in local lymphoid and inflammatory microenvironments (73). In addition, BAFF upregulation correlates both with primary SS disease activity and B cell prelymphomatous and malignant lymphoproliferative disorders (74). Genetic mutations in BAFF-mediated pathway may significantly contribute to this risk of malignant evolution (42). The efficacy of belimumab, a human monoclonal antibody targeting soluble BAFF, approved for the treatment of SLE, in a phase II clinical trial of primary SS strongly supported the pathogenic role of BAFF in this autoimmune disease (75, 76), and a phase III trials of belimumab in co-administration with rituximab in primary SS is ongoing (NCT02631538).
B Cell Abnormalities
Two B cell subpopulations have been shown to play a central role in both these entities: switched memory B cells and CD21low/- B cells. The formers are CD19+CD27+IgM-IgD- memory B cells which have undergone the isotypic switch; the latter are a peculiar B cell subset that under expresses CD21, a coreceptor of BCR, and at the same time expresses higher levels of IgM, CD11c, CD19 and CD95. CD21low/- B cells belong to a unique anergic B cell population which is polyclonal, pre-activated, enriched in autoreactive clones and can express highly autoreactive antibodies, including antinuclear antibody and RF (77, 78). The nature of this cell population was extensively studied also in the context of HCV related cryoglobulinemia; in both HCV related cryoglobulinemia and CVID, an anergic subset of CD21low/- B cells appears expanded and characterized by high constitutive expression of extracellular signal regulated kinase (pERK) (79–81). Moreover a BAFF hyperexpression and aberrant type I and II IFN response are thought to support CD21low/- B cell population, suggesting a profound interconnection between dysregulated innate and adaptative immunity (77, 82).
CVID and B-Cell Abnormalities
Since the early 2000s the role of switched memory B cells and CD21low/- B cells in CVID has been investigated. Warnatz et al. observed a significant decrease in class-switched B memory cells in CVID patients compared to healthy controls and identified a CVID subgroup, clinically characterized by splenomegaly and autoimmune disorders, with a high proportion of CD21low/- B cells. These findings suggested a correlation between low switched memory B cells, increased CD21low/- B cells and autoimmune and lymphoproliferative disorders in CVID (23, 83). In particular, the reduction of IgM-, IgD- CD27+ switched memory B cells represents the most common aberration in CVID, and it correlates with decrease in serum IgA and IgG levels. Sanchez Ramon et al. found that levels of switched memory B cells <0.55% had 3.3-fold higher risk to correlate with autoimmune disease (84). Many other studies confirmed these results (19, 77, 78, 85). Of note, in the large cohort of USIDNET register lower levels of switched B memory cells were observed in CVID-Rheum group (18).
SS and B-Cell Abnormalities
On the other hand, there is strong evidence of unbalance of B cell subpopulations also in SS. Many authors found that memory and switched memory B cells are reduced in primary SS compared to controls (86–88), and this unbalance appears to be related to disease duration and activity (88). Saadoun et al. found an increase of CD21low/- B cells in primary SS and in particular in primary SS with lymphoproliferative disorders, suggesting a key role of this B cell population in SS related lymphomagenesis (89, 90). As in CVID, they found that CD21low/- B cells are enriched in autoreactive clones and express highly autoreactive antibodies, such as RF, as a consequence of a chronic antigenic stimulation; this mechanism was preliminary linked to lymphoproliferation in primary SS and in HCV infection (91). The persistence of these cells can represent the initial reservoir for monoclonal expansion of a transformed clone and drive to B cell lymphoproliferation (90). Other papers support the correlation between the presence of B cell NHL in primary SS patients and the proportion of circulating CD21low/- B cells (92, 93).
Rituximab can efficiently target those subtype of B cells in CVID with autoimmune or nonmalignant lymphoproliferative manifestations, as well as other therapeutic approach aiming to specifically deplete CD21low/- B cells through an anti FcRL5 recombinant immunotoxin, originally employed in cryoglobulinemic vasculitis (94).
Interferon Signature
Type I and II IFNs are cytokines which play a central role in regulation of immunity and inflammation. Since their contribution in loss of immunotolerance, they were considered as potential therapeutic targets of drugs such as anifrolumab, an anti-type I IFN receptor monoclonal antibody [NCT 02446899], in SLE. Type II IFN (IFN gamma), instead, appears more significant in diseases characterized by a prominent lymphoproliferative component, such as primary SS (95).
This increased expression of canonical IFN stimulated genes in tissues and in circulating blood cells is defined IFN signature, which is one of the possible key items shared among primary SS and CVID with autoimmunity. In particular, an upregulated IFN signature expression distinguishes CVID patients with inflammatory complication, including autoimmunity and, at the same time, it is a hallmark of various systemic autoimmune diseases such as SLE, systemic sclerosis, myositis and primary SS (19, 96).
CVID and Interferon Signature
Regarding CVID, few papers have been published on the role of IFN signature (27, 97).
Subjects with CVID and inflammatory/autoimmune conditions displayed significantly over-expressed IFN-related transcriptional modules and pronounced downregulation of transcript related to the B cell, plasma cell and T cell modules as compared to CVID without these conditions or controls (27). Also, a significant expansion of circulating IFN gamma producing innate lymphoid cells (typically ILC3) in CVID patients with noninfectious complications compared to those without and identified these cells in the affected mucosal tissues of lung and gastrointestinal tract (97). Notably, these cells, that also correlate with inflammation and produce IL17, were detected in salivary glands of primary SS patients, although their role in this autoimmune disease is not known (98). Unger et al. demonstrated a Th1 skewed CD4 T-cell population that highly express IFN-gamma both in peripheral blood and in lymph-nodes. Notably, in the same study, IFN gamma immune environment is thought to participate in expansion of circulating CD21low/- B cells (29).
SS and Interferon Signature
On the contrary, the role of IFN signature in SS has been highlighted by many studies (99, 100). The presence of IFN-induced gene expression was demonstrated in salivary glands, peripheral blood mononuclear cells, isolated monocytes and B cells of primary SS patients and type I IFN signature was associated with higher disease activity and higher levels of autoantibodies (101–106). Type II IFN signature was also detected in salivary glands of primary SS patients (107). Two studies by Bodewes et al. confirmed the central role of overactivated innate immunity and IFN system in primary SS, particularly type II IFN (108, 109).
An aberrant activation of type I IFN response could drive autoantibody production, partly by direct activation of autoreactive B cells and partly by cytotoxic effect, accumulation of cellular debris and expression of autoantigen Ro52 (110, 111). Additionally, type I IFNs induce the expression of BAFF (109).
Interestingly, a prominent type I IFN signature was also associated with markers of B cell overactivity, such as anti-SSA antibodies, that can be attributed to type I IFN induced BAFF overproduction (106, 112). In the setting of lymphomagenesis both type I and II IFN transcript levels were considerably increased in minor salivary gland tissues from primary SS derived lymphoma, implying a direct role of these cytokines, and in particular IFN gamma, in this process (95).
Even if the main site of B cell hyperplasia and lymphoma development in CVID is the lung, also salivary glands can be involved in some cases (50–53, 113). Data suggest that IFN gamma could upregulate BAFF both in peripheral blood and in lung tissue, and locally BAFF could promote B cell survival and proliferation (63, 114).
Use of “Anti-Rheumatic Therapies” in CVID and SS
Whereas immunoglobulin replacement therapy and improved anti-microbial drugs have significantly ameliorated CVID patients survival by reducing infectious complications (16), patients with CVID affected by at least one non-infectious complication still have significant higher risk of mortality compared to the other CVID patients, since these clinical manifestations do not respond to the antibiotic and immunoglobulin replacement therapy alone (11, 20). Thus, it appears that noninfectious complications, especially gastrointestinal and pulmonary involvement, constitute the most difficult aspects of the CVID patient management (1, 20, 115–117).
Over the last 5-10 years, rituximab has been used in various non-infectious CVID complications, such as autoimmune cytopenias, granulomatous lymphocytic interstitial lung disease (GLILD) and non-malignant lymphoproliferative syndromes (118). Of note, rituximab and, more recently, belimumab, as B-cell targeted therapies, have been applied in primary SS, and they resulted effective in particular in patients with systemic features (75, 76, 119). Combination strategies with both drugs are currently under evaluation in primary SS and also in other autoimmune diseases (69).
Also, abatacept, a CTLA-4 immunoglobulin fusion protein, showed good results as a replacement therapy in patients affected by CTLA-4 and LRBA deficiency (47, 120). In addition, tocilizumab and inhibitors of Janus Kinases (JAKs) were successfully trialed in patients with STAT3 gain of function mutations, as its activation occurs downstream of both IL-6 and JAKs (43, 121).
Yet, both tocilizumab and abatacept were employed as possible new treatments of primary SS, and even JAKs inhibitors are under evaluation in primary SS (NCT04496960).
The multicenter double-blind randomized placebo-controlled trial with tocilizumab in primary SS did not improve systemic features over 24 weeks of treatment compared with placebo (122); however, tocilizumab might be effective in contrasting SS-related articular and pulmonary involvement, such as refractory organizing pneumonia (123); moreover, tocilizumab has been recently approved by FDA for pulmonary fibrosis in systemic sclerosis (124).
Two open studies have assessed abatacept in primary SS; the first demonstrated the reduction in glandular inflammation and an increase in saliva production (125), while the second one showed the decrease of ESSDAI, ESSPRI, RF, and IgG levels but salivary and glandular functions did not improved (126). Finally, the phase III trial failed to demonstrate any clinical benefit of abatacept in primary SS (127).
On the other hand, leniolisib (CDZ173), a potent and selective oral inhibitor of PI3Kdelta (128), has been successfully used in a series of patients with APDS, in which PI3Kdelta gain-of-function mutation results in lymphoproliferation of the MALT, T-cell senescence and immunodeficiency. Leniolisib normalized B cells in APDS, and improved lymphoproliferation (129). A phase II clinical trial [NCT02775916] assessing the safety, pharmacokinetics, and preliminary efficacy of leniolisib in SS has been completed in January 2021, and preliminary data were presented in 2018 (130).
Lessons From PID: Autoimmunity Is Not Just an Autoantibody!
First, SS may be probably underestimated in CVID, due to the absence of specific autoantibody, while specific symptoms may be unnoticed by specialists other than rheumatologists. On the other hand, patients with autoimmune diseases undergoing recurrent infections, or with peculiar features such as early onset or overlap syndrome should be evaluated for PID.
Second, the link between impaired B cell development, autoimmunity and lymphoma should be better elucidated based on the new growing knowledge in CVID.
Some CVID show B cell survival defects blocking the progression from transitional to naïve mature B cells, other CVID are characterized by class-switch recombination defects, impairing the evolution from follicular B cell to switched memory B cells, finally still other CVID display maturation defects into plasma cells (131). Nevertheless, the B cells in CVID are still able to produce autoantibodies, but they cannot isotype switch or affinity mature in response to new antigen challenge.
In X-linked agammaglobulinemia (XLA), BCR transcripts from peripheral blood CD19+ CD10+ IgM+ CD27−, emigrant mature naive B‐cells, represent the majority of peripheral blood B‐cells in XLA patients, and a higher proportion of which are self‐reactive and polyreactive antibodies and preferentially used VH1‐3 and VH4‐34 genes (132). Usually, the IgMkappa type encoded by the V(H)4-34 gene segment is the monoclonal immunoglobulin detected in primary chronic cold agglutinin disease (CAD), that is an autoimmune haemolytic anaemia induced by cold reactive autoantibodies (cold agglutinins) against erythrocyte surface antigens (133). Of note, unmutated VH4‐34+ B‐cells can be detected in patients with SLE memory compartment and VH4‐34‐expressing plasma cells appear to be clonally expanded during flares (134).
Interestingly, in ocular adnexal marginal zone B-cell lymphomas, which are observed also in SS, a strongly biased usage of V(H)4-34 in chlamydia negative patients was documented, suggesting the involvement of a particular stimulatory (auto-) antigen in their development (135). Similarly, the VH4 family usage of immunoglobulin gene rearrangement characterized also the MALT lymphomas of thyroid (136).
Yet, in Wiskott–Aldrich syndrome (WAS), a rare X‐linked PID with the classical clinical features of susceptibility to infections and autoimmune thrombocytopenia, memory B‐cells showed a low frequency of somatic hypermutation, while an increased usage of uncommon VH genes in transitional, naive and CD19highCD21low B‐cells if compared with healthy controls, some of which are enriched in autoantibodies (VH4‐34 and VH4‐61) (137). Therefore, autoimmunity in the absence of B-cell switching is possible; when B-cells are frustrated in their normal path, they can proliferate in other pathways and use germ line genes that promote autoreactivity.
Importantly, in SLE undergoing rituximab, it was reported that B-cell abnormalities resolved after effective B cell depletion and immune reconstitution, including the frequency of autoreactive VH4.34 memory B cells (138), thus, possibly explaining the efficacy of rituximab in some autoimmune manifestations of PID.
In the same way, also anti-neutrophil cytoplasmic antibodies, that may play a pathogenic role in vasculitis, showed the use of germ-line VH4 family genes (139), as well as IgG anti-platelet autoantibodies in chronic immune thrombocytopenic purpura (140).
In CVID, the available BCR repertoire data are in line with the heterogeneity of the disease and generally show lower levels of somatic hypermutation, decreased repertoire diversity and longer CDR3 segments (141). The genetic heterogeneity of CVID made it difficult to better study the sequence encoded in their B cells.
Third, lymphoproliferation over autoimmunity is the hallmark of SS rather than SLE or other systemic autoimmune diseases. Indeed, SS more than SLE shows a high frequency of monoclonal gammopathy, with germ-line gene sequences recorded in lymphomas and Waldenström’s macroglobulinemia (i.e., 17-109 crossreactive idiotype) (142). Moreover, CAD represents a spectrum of clonal lymphoproliferative disorders overlapping with Waldenström’s macroglobulinemia itself (143).
In SS, a germline and coding polymorphism of TNFAIP3 (A20), a central gatekeeper of NF-kB activation, was found associated with lymphoma, linking the impaired control of NF-kB activation in B cells to autoimmunity and to the risk of lymphoma (144).
This concept has been recently reinforced particularly in mixed cryoglobulinemia secondary to SS, where the original Waldenström RF idiotypes were shown to be used in the monoclonal RF of mixed cryoglobulinemia in SS. Interestingly, in SS, at the beginning of the cascade of the events that lead to lymphoma and cryoglobulinemia, the unmutated V(D)J germ-line combinations with the characteristics of RF antibodies of Wa and Po public idiotypes, have very weak binding to self-IgG and this low affinity for IgG autoantigen would enable these newly formed B cells to evade central and peripheral B cell tolerance checkpoints and be activated transiently by IgG forming complexes with foreign antigens. The subsequent events are affinity maturation, somatic mutation acquisition and finally soluble accumulation of particular V(D)J mutations that compromise solubility of autoantibody-antigen complexes (145). It can be argued that in CVID, the first step described above is dramatically increased by mutations that promote breakdown tolerance and infections that facilitate proliferation of the expansion of autoreactive clones, leading to autoimmunity. In this regard, in SS, circulating IgG complexed with Ro and La ribonucleoproteins represents the driving force that induces low-affinity RF B cells to proliferate. Rare immunoglobulin mutations that improve affinity for self-IgG selectively allow some clones to emerge as dominant. In CVID, the levels of autoantibodies may be rarely found in circulation because their serum levels are extremely low, as also regularly occurred in idiopathic thrombocytopenic purpura or in chronic autoimmune neutropenia, since the neutropenia and thrombocytopenia may be due either to IgG Fc-mediated clearance in the spleen or due to destruction in periphery by routine clearance. Similarly, the very low amount of IgG anti-SS-A or SS-B, as well RF, may be cleared from the circulation or the low production may be confined in the target tissue (146). Deeper molecular and tissue studies in CVID with associated SS could support this hypothesis.
Conclusions
It is paradoxical that patients with CVID have a high frequency of associated autoimmune features. Increasing pathogenetic insights allowed to reconcile the lack of B-cell maturation and autoimmunity in the wider concept of dysregulated immune system, both diseases being influenced by genetic and epigenetic factors which can lead to different clinical phenotypes (Figure 1).
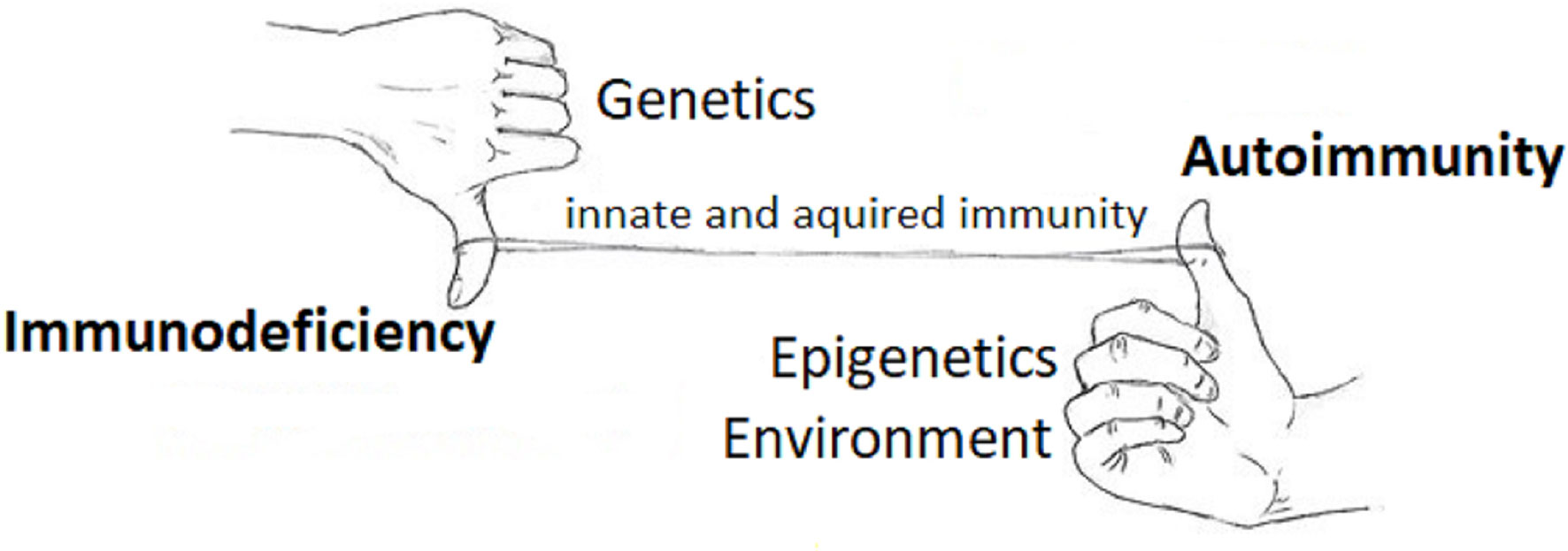
Figure 1 An integrated view of the immune system in primary immunodeficiency and autoimmune disorders. The figure illustrates the concept that immunodeficiency (downregulation of the immune system - the hand with thumb down) and autoimmunity (upregulation of the immune system - the hand with thumb up) appear different categories of human pathology, the former more related to genetics, the latter to environment and epigenetics, while the interplay between these two apparently distinct categories is guaranteed by the common line of the innate and acquired immunity, which is dysregulated in both.
The association of hypogammaglobulinemia and autoreactive B cells in CVID patients has been commonly listed as “SLE-like”. However, we propose that the autoimmunity and lymphoproliferation associated with CVID is more closely associated with a SS-like picture of immune dysregulation. In this context, CVID and SS, two conditions which can occur simultaneously and share several pathogenetic aspects, as well as targeted therapy (i.e., rituximab, abatacept), could represent a model of this immunological view. Therefore, better understanding of the underlying immunological mechanisms and specific genetic mutations that result in the immune dysregulation may lead to the development of new therapeutic targets for both the diseases.
Author Contributions
LQ, SV, RF, and GM conceived the study. All authors contributed to the literature review and interpretation of the data. The first draft of the manuscript was written by LQ, RF, SV, GM, VM, SL, and MR, and all authors commented on previous versions of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Giulia Del Frate, MD, and Dr. Donatella Colatutto, MD, for their contribution to the literature review and for making the figure, respectively.
References
1. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract (2016) 4:38–59. doi: 10.1016/j.jaip.2015.07.025
2. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
3. Modell V, Orange JS, Quinn J, Modell F. Global Report on Primary Immunodeficiencies: 2018 Update From the Jeffrey Modell Centers Network on Disease Classification, Regional Trends, Treatment Modalities, and Physician Reported Outcomes. Immunol Res (2018) 66:367–80. doi: 10.1007/s12026-018-8996-5
4. Fudenberg H, Good RA, Goodman HC, Hitzig W, Kunkel HG, Roitt IM, et al. Primary Immunodeficiencies. Report of a World Health Organization Committee. Pediatrics (1971) 47:927–46.
5. Conley ME, Notarangelo LD, Etzioni A. Diagnostic Criteria for Primary Immunodeficiencies. Clin Immunol (1999) 93:190–7. doi: 10.1006/clim.1999.4799
6. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass Trial: Defining Subgroups in Common Variable Immunodeficiency. Blood (2008) 111:76–85. doi: 10.1182/blood-2007-06-091744
7. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New Diagnostic Criteria for CVID. Expert Rev Clin Immunol (2014) 10:183–6. doi: 10.1586/1744666X.2014.875274
8. Seidel MG, Kindle G, Gathmann B, Quinti I, Buckland M, van Montfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J Allergy Clin Immunol Pract (2019) 7:1763–70. doi: 10.1016/j.jaip.2019.02.004
9. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common Variable Immunodeficiency Disorders: Division Into Distinct Clinical Phenotypes. Blood (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
10. Maglione PJ, Overbey JR, Radigan L, Bagiella E, Cunningham-Rundles C. Pulmonary Radiologic Findings in Common Variable Immunodeficiency: Clinical and Immunological Correlations. Ann Allergy Asthma Immunol (2014) 113:452–9. doi: 10.1016/j.anai.2014.04.024
11. Gathmann B, Mahlaoui N, Gérard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical Picture and Treatment of 2212 Patients With Common Variable Immunodeficiency. J Allergy Clin Immunol (2014) 134:116–26. doi: 10.1016/j.jaci.2013.12.1077
12. Kunkel HG, Allen JC, Grey HM, Martensson L, Grubb R. A Relationship Between the H Chain Groups of 7S Γ-Globulin and the Gm System [23]. Nature (1964) 203:413–4. doi: 10.1038/203413a0
13. Crowley JJ, Goldfien RD, Schrohenloher RE, Spiegelberg HL, Silverman GJ, Mageed RA, et al. Incidence of Three Cross-Reactive Idiotypes on Human Rheumatoid Factor Paraproteins. J Immunol (1988) 140:3411–8.
14. Hansen A, Reiter K, Pruss A, Loddenkemper C, Kaufmann O, Jacobi AM, et al. Dissemination of a Sjögren’s Syndrome-Associated Extranodal Marginal-Zone B Cell Lymphoma: Circulating Lymphoma Cells and Invariant Mutation Pattern of Nodal Ig Heavy- and Light-Chain Variable-Region Gene Rearrangements. Arthritis Rheum (2006) 54:127–37. doi: 10.1002/art.21558
15. Ladouceur A, Tessier-Cloutier B, Clarke AE, Ramsey-Goldman R, Gordon C, Hansen JE, et al. Cancer and Systemic Lupus Erythematosus. Rheum Dis Clin North Am (2020) 46:533–50. doi: 10.1016/j.rdc.2020.05.005
16. Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al. The Burden of Common Variable Immunodeficiency Disorders : A Retrospective Analysis of the European Society for Immunodeficiency (ESID) Registry Data. Orphanet J Rare Dis (2018) 13:201. doi: 10.1186/s13023-018-0941-0
17. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-Term Follow-Up and Outcome of a Large Cohort of Patients With Common Variable Immunodeficiency. J Clin Immunol (2007) 27:308–16. doi: 10.1007/s10875-007-9075-1
18. Gutierrez MJ, Sullivan KE, Fuleihan R, Bingham CO. Phenotypic Characterization of Patients With Rheumatologic Manifestations of Common Variable Immunodeficiency. Semin Arthritis Rheum (2018) 48:318–26. doi: 10.1016/j.semarthrit.2018.02.013
19. Maglione PJ. Autoimmune and Lymphoproliferative Complications of Common Variable Immunodeficiency. Curr Allergy Asthma Rep (2016) 16:19. doi: 10.1007/s11882-016-0597-6
20. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and Mortality in Common Variable Immune Deficiency Over 4 Decades. Blood (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
21. Dhalla F, da Silva SP, Lucas M, Travis S, Chapel H. Review of Gastric Cancer Risk Factors in Patients With Common Variable Immunodeficiency Disorders, Resulting in a Proposal for a Surveillance Programme. Clin Exp Immunol (2011) 165:1–7. doi: 10.1111/j.1365-2249.2011.04384.x
22. Arumugakani G, Wood PMD, Carter CRD. Frequency of Treg Cells Is Reduced in CVID Patients With Autoimmunity and Splenomegaly and Is Associated With Expanded CD21lo B Lymphocytes. J Clin Immunol (2010) 30:292–300. doi: 10.1007/s10875-009-9351-3
23. Warnatz K, Denz A, Dräger R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe Deficiency of Switched Memory B Cells (CD27+IgM-IgD-) in Subgroups of Patients With Common Variable Immunodeficiency: A New Approach to Classify a Heterogeneous Disease. Blood (2002) 99:1544–51. doi: 10.1182/blood.V99.5.1544
24. Boileau J, Mouillot G, Gérard L, Carmagnat M, Rabian C, Oksenhendler E, et al. Autoimmunity in Common Variable Immunodeficiency: Correlation With Lymphocyte Phenotype in the French DEFI Study. J Autoimmun (2011) 36:25–32. doi: 10.1016/j.jaut.2010.10.002
25. Bateman EAL, Ayers L, Sadler R, Lucas M, Roberts C, Woods A, et al. T Cell Phenotypes in Patients With Common Variable Immunodeficiency Disorders: Associations With Clinical Phenotypes in Comparison With Other Groups With Recurrent Infections. Clin Exp Immunol (2012) 170:202–11. doi: 10.1111/j.1365-2249.2012.04643.x
26. Mouillot G, Carmagnat M, Gérard L, Garnier JL, Fieschi C, Vince N, et al. B-Cell and T-Cell Phenotypes in CVID Patients Correlate With the Clinical Phenotype of the Disease. J Clin Immunol (2010) 30:746–55. doi: 10.1007/s10875-010-9424-3
27. Park J, Munagala I, Xu H, Blankenship D, Maffucci P, Chaussabel D, et al. Interferon Signature in the Blood in Inflammatory Common Variable Immune Deficiency. PloS One (2013) 8:e74893. doi: 10.1371/journal.pone.0074893
28. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
29. Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The TH1 Phenotype of Follicular Helper T Cells Indicates an IFN-γ–Associated Immune Dysregulation in Patients With CD21low Common Variable Immunodeficiency. J Allergy Clin Immunol (2018) 141:730–40. doi: 10.1016/j.jaci.2017.04.041
30. Knight AK, Radigan L, Marron T, Langs A, Zhang L, Cunningham-Rundles C. High Serum Levels of BAFF, APRIL, and TACI in Common Variable Immunodeficiency. Clin Immunol (2007) 124:182–9. doi: 10.1016/j.clim.2007.04.012
31. Varzaneh FN, Keller B, Unger S, Aghamohammadi A, Warnatz K, Rezaei N. Cytokines in Common Variable Immunodeficiency as Signs of Immune Dysregulation and Potential Therapeutic Targets - A Review of the Current Knowledge. J Clin Immunol (2014) 34:524–43. doi: 10.1007/s10875-014-0053-0
32. Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors Beyond Lack of Antibody Govern Pulmonary Complications in Primary Antibody Deficiency. J Clin Immunol (2019) 39:440–7. doi: 10.1007/s10875-019-00640-5
33. Rasmussen A, Radfar L, Lewis D, Grundahl K, Stone DU, Kaufman CE, et al. Previous Diagnosis of Sjögren’s Syndrome as Rheumatoid Arthritis or Systemic Lupus Erythematosus. Rheumatol (United Kingdom) (2016) 55:1195–201. doi: 10.1093/rheumatology/kew023
34. Ramos-Casals M, Brito-Zerón P, Seror R, Bootsma H, Bowman SJ, Dörner T, et al. Characterization of Systemic Disease in Primary Sjögren’s Syndrome: EULAR-SS Task Force Recommendations for Articular, Cutaneous, Pulmonary and Renal Involvements. Rheumatol (United Kingdom) (2015) 54:2230–8. doi: 10.1093/rheumatology/kev200
35. Konttinen YT, Stegajev V, Al-Samadi A, Porola P, Hietanen J, Ainola M. Sjögren’s Syndome and Extragonadal Sex Steroid Formation: A Clue to a Better Disease Control? J Steroid Biochem Mol Biol (2015) 145:237–44. doi: 10.1016/j.jsbmb.2014.08.014
36. Tavasolian P, Sharifi L, Aghamohammadi A, Noorbakhsh F, Sanaei R, Shabani M, et al. Toll-Like Receptors Pathway in Common Variable Immune Deficiency (CVID) and X-Linked Agammaglobulinemia (XLA). Eur Cytokine Netw (2018) 29:153–8. doi: 10.1684/ecn.2018.0420
37. Babaha F, Yazdani R, Shahkarami S, Esfahani ZH, Abolhahassani H, Sadr M, et al. Evaluation of miR-210 Expression in Common Variable Immunodeficiency: Patients With Unsolved Genetic Defect. Allergol Immunopathol (Madr) (2021) 49:84–93. doi: 10.15586/aei.v49i2.39
38. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic Diagnosis Using Whole Exomesequencing in Common Variable Immunodeficiency. Front Immunol (2016) 7:220. doi: 10.3389/fimmu.2016.00220
39. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, Azucena González-Navarro E, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
40. Salzer U, Bacchelli C, Buckridge S, Pan-Hammarström Q, Jennings S, Lougaris V, et al. Relevance of Biallelic Versus Monoallelic TNFRSF13B Mutations in Distinguishing Disease-Causing From Risk-Increasing TNFRSF13B Variants in Antibody Deficiency Syndromes. Blood (2009) 113:1967–76. doi: 10.1182/blood-2008-02-141937
41. Wang X, Shaalan A, Liefers S, Coudenys J, Elewaut D, Proctor GB, et al. Dysregulation of NF-kB in Glandular Epithelial Cells Results in Sjögren’s-Like Features. PloS One (2018) 13:e0200212. doi: 10.1371/journal.pone.0200212
42. Papageorgiou A, Mavragani CP, Nezos A, Zintzaras E, Quartuccio L, De Vita S, et al. A BAFF Receptor His159Tyr Mutation in Sjögren’s Syndrome-Related Lymphoproliferation. Arthritis Rheumatol (Hoboken NJ) (2015) 67:2732–41. doi: 10.1002/art.39231
43. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-Onset Lymphoproliferation and Autoimmunity Caused by Germline STAT3 Gain-of-Function Mutations. Blood (2015) 125:591–9. doi: 10.1182/blood-2014-09-602763
44. Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing Regulation of the Locus Encoding IL-17 Through Direct, Reciprocal Actions of STAT3 and STAT5. Nat Immunol (2011) 12:247–54. doi: 10.1038/ni.1995
45. Haapaniemi EM, Kaustio M, Rajala HLM, Van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, Hypogammaglobulinemia, Lymphoproliferation, and Mycobacterial Disease in Patients With Activating Mutations in STAT3. Blood (2015) 125:639–48. doi: 10.1182/blood-2014-04-570101
46. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
47. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients With LRBA Deficiency Show CTLA4 Loss and Immune Dysregulation Responsive to Abatacept Therapy. Sci (80-) (2015) 349:436–40. doi: 10.1126/science.aaa1663
48. Chan KK, Bass AR. Autoimmune Complications of Immunotherapy: Pathophysiology and Management. BMJ (2020) 369:m736. doi: 10.1136/bmj.m736
49. Nunes-Santos CJ, Uzel G, Rosenzweig SD. PI3K Pathway Defects Leading to Immunodeficiency and Immune Dysregulation. J Allergy Clin Immunol (2019) 143:1676–87. doi: 10.1016/j.jaci.2019.03.017
50. Yakaboski E, Fuleihan RL, Sullivan KE, Cunningham-Rundles C, Feuille E. Lymphoproliferative Disease in CVID: A Report of Types and Frequencies From a US Patient Registry. J Clin Immunol (2020) 40:524–30. doi: 10.1007/s10875-020-00769-8
51. Desar IME, Keuter M, Raemaekers JMM, Jansen JBMJ, van Krieken JHJ, van der Meer JWM. Extranodal Marginal Zone (MALT) Lymphoma in Common Variable Immunodeficiency. Neth J Med (2006) 64:136–40.
52. Reichenberger F, Wyser C, Gonon M, Cathomas G, Tamm M. Pulmonary Mucosa-Associated Lymphoid Tissue Lymphoma in a Patient With Common Variable Immunodeficiency Syndrome. Respiration (2001) 68:109–12. doi: 10.1159/000050475
53. Cunningham-Rundles C, Cooper DL, Duffy TP, Strauchen J. Lymphomas of Mucosal-Associated Lymphoid Tissue in Common Variable Immunodeficiency. Am J Hematol (2002) 69:171–8. doi: 10.1002/ajh.10050
54. De Vita S, De Marchi G, Sacco S, Gremese E, Fabris M, Ferraccioli G. Preliminary Classification of Nonmalignant B Cell Proliferation in Sjögren’s Syndrome: Perspectives on Pathobiology and Treatment Based on an Integrated Clinico-Pathologic and Molecular Study Approach. Blood Cells Mol Dis (2001) 27:757–66. doi: 10.1006/bcmd.2001.0446
55. De Vita S, Boiocchi M, Sorrentino D, Carbone A, Avellini C, Dolcetti R, et al. Characterization of Prelymphomatous Stages of B Cell Lymphoproliferation in Sjogren’s Syndrome. Arthritis Rheum (1997) 40:318–31. doi: 10.1002/art.1780400217
56. Anderson LG, Talal N. The Spectrum of Benign to Malignant Lymphoproliferation in Sjögren’s Syndrome. Clin Exp Immunol (1972) 10:199–221.
57. Nocturne G, Mariette X. Sjögren Syndrome-Associated Lymphomas: An Update on Pathogenesis and Management. Br J Haematol (2015) 168:317–27. doi: 10.1111/bjh.13192
58. Retamozo S, Brito-Zerón P, Ramos-Casals M. Prognostic Markers of Lymphoma Development in Primary Sjögren Syndrome. Lupus (2019) 28:923–36. doi: 10.1177/0961203319857132
59. Papageorgiou A, Voulgarelis M, Tzioufas AG. Clinical Picture, Outcome and Predictive Factors of Lymphoma in Sjögren Syndrome. Autoimmun Rev (2015) 14:641–9. doi: 10.1016/j.autrev.2015.03.004
60. Sambataro G, Ferro F, Orlandi M, Sambataro D, Torrisi SE, Quartuccio L, et al. Clinical, Morphological Features and Prognostic Factors Associated With Interstitial Lung Disease in Primary Sjögren’s Syndrome: A Systematic Review From the Italian Society of Rheumatology. Autoimmun Rev (2020) 19:102447. doi: 10.1016/j.autrev.2019.102447
61. Dong X, Zhou J, Guo X, Li Y, Xu Y, Fu Q, et al. A Retrospective Analysis of Distinguishing Features of Chest HRCT and Clinical Manifestation in Primary Sjögren’s Syndrome-Related Interstitial Lung Disease in a Chinese Population. Clin Rheumatol (2018) 37:2981–8. doi: 10.1007/s10067-018-4289-6
62. Kreuzaler M, Rauch M, Salzer U, Birmelin J, Rizzi M, Grimbacher B, et al. Soluble BAFF Levels Inversely Correlate With Peripheral B Cell Numbers and the Expression of BAFF Receptors. J Immunol (2012) 188:497–503. doi: 10.4049/jimmunol.1102321
63. Maglione PJ, Gyimesi G, Cols M, Radigan L, Ko HM, Weinberger T, et al. BAFF-Driven B Cell Hyperplasia Underlies Lung Disease in Common Variable Immunodeficiency. JCI Insight (2019) 4:e122728. doi: 10.1172/jci.insight.122728
64. Nakayamada S, Tanaka Y. BAFF- and APRIL-Targeted Therapy in Systemic Autoimmune Diseases. Inflamm Regener (2016) 36:6. doi: 10.1186/s41232-016-0015-4
65. Magliozzi R, Marastoni D, Calabrese M. The BAFF / APRIL System as Therapeutic Target in Multiple Sclerosis. Expert Opin Ther Targets (2020) 24:1135–45. doi: 10.1080/14728222.2020.1821647
66. Szodoray P, Jonsson R. The BAFF/APRIL System in Systemic Autoimmune Diseases With a Special Emphasis on Sjögren’s Syndrome. Scand J Immunol (2005) 62:421–8. doi: 10.1111/j.1365-3083.2005.01688.x
67. Möckel T, Basta F, Weinmann-Menke J, Schwarting A. B Cell Activating Factor (BAFF): Structure, Functions, Autoimmunity and Clinical Implications in Systemic Lupus Erythematosus (SLE). Autoimmun Rev (2021) 20:102736. doi: 10.1016/j.autrev.2020.102736
68. Fabris M, Quartuccio L, Sacco S, De Marchi G, Pozzato G, Mazzaro C, et al. B-Lymphocyte Stimulator (BLyS) Up-Regulation in Mixed Cryoglobulinaemia Syndrome and Hepatitis-C Virus Infection. Rheumatology (2007) 46:37–43. doi: 10.1093/rheumatology/kel174
69. De Vita S, Quartuccio L, Salvin S, Picco L, Scott CA, Rupolo M, et al. Sequential Therapy With Belimumab Followed by Rituximab in Sjögren’s Syndrome Associated With B-Cell Lymphoproliferation and Overexpression of BAFF: Evidence for Long-Term Efficacy. Clin Exp Rheumatol (2014) 32:490–4.
70. Stohl W, Xu D, Kim KS, Koss MN, Jorgensen TN, Deocharan B, et al. BAFF Overexpression and Accelerated Glomerular Disease in Mice With an Incomplete Genetic Predisposition to Systemic Lupus Erythematosus. Arthritis Rheum (2005) 52:2080–91. doi: 10.1002/art.21138
71. Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al. Association of BAFF/BLyS Overexpression and Altered B Cell Differentiation With Sjögren’s Syndrome. J Clin Invest (2002) 109:59–68. doi: 10.1172/jci14121
72. Daridon C, Devauchelle V, Hutin P, Le Berre R, Martins-Carvalho C, Bendaoud B, et al. Aberrant Expression of BAFF by B Lymphocytes Infiltrating the Salivary Glands of Patients With Primary Sjögren’s Syndrome. Arthritis Rheum (2007) 56:1134–44. doi: 10.1002/art.22458
73. Mariette X, Roux S, Zhang J, Bengoufa D, Lavie F, Zhou T, et al. The Level of BLyS (BAFF) Correlates With the Titre of Autoantibodies in Human Sjögren’s Syndrome. Ann Rheum Dis (2003) 62:168–71. doi: 10.1136/ard.62.2.168
74. Quartuccio L, Salvin S, Fabris M, Maset M, Pontarini E, Isola M, et al. Blys Upregulation in Sjögren’s Syndrome Associated With Lymphoproliferative Disorders, Higher ESSDAI Score and B-Cell Clonal Expansion in the Salivary Glands. Rheumatol (United Kingdom) (2013) 52:276–81. doi: 10.1093/rheumatology/kes180
75. Mariette X, Seror R, Quartuccio L, Baron G, Salvin S, Fabris M, et al. Efficacy and Safety of Belimumab in Primary Sjögren’s Syndrome: Results of the BELISS Open-Label Phase II Study. Ann Rheum Dis (2015) 74:526–31. doi: 10.1136/annrheumdis-2013-203991
76. De Vita S, Quartuccio L, Seror R, Salvin S, Ravaud P, Fabris M, et al. Efficacy and Safety of Belimumab Given for 12 Months in Primary Sjögren’s Syndrome: The BELISS Open-Label Phase II Study. Rheumatol (United Kingdom) (2015) 54:2249–56. doi: 10.1093/rheumatology/kev257
77. Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement Receptor 2/CD21- Human Naive B Cells Contain Mostly Autoreactive Unresponsive Clones. Blood (2010) 115:5026–36. doi: 10.1182/blood-2009-09-243071
78. Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low B Cells in Common Variable Immunodeficiency Resemble Tissue Homing, Innate-Like B Cells. Proc Natl Acad Sci USA (2009) 106:13451–6. doi: 10.1073/pnas.0901984106
79. Gandolfo S, De Vita S. Double Anti-B Cell and Anti-BAFF Targeting for the Treatment of Primary Sjögren’s Syndrome. Clin Exp Rheumatol (2019) 37:S199–208.
80. Charles ED, Brunetti C, Marukian S, Ritola KD, Talal AH, Marks K, et al. Clonal B Cells in Patients With Hepatitis C Virus-Associated Mixed Cryoglobulinemia Contain an Expanded Anergic CD21low B-Cell Subset. Blood (2011) 117:5425–37. doi: 10.1182/blood-2010-10-312942
81. Visentini M, Marrapodi R, Conti V, Mitrevski M, Camponeschi A, Lazzeri C, et al. Dysregulated Extracellular Signal-Regulated Kinase Signaling Associated With Impaired B-Cell Receptor Endocytosis in Patients With Common Variable Immunodeficiency. J Allergy Clin Immunol (2014) 134:401–10. doi: 10.1016/j.jaci.2014.03.017
82. Karnell JL, Kumar V, Wang J, Wang S, Voynova E, Ettinger R. Role of CD11c+ T-Bet+ B Cells in Human Health and Disease. Cell Immunol (2017) 321:40–5. doi: 10.1016/j.cellimm.2017.05.008
83. Warnatz K, Wehr C, Dräger R, Schmidt S, Eibel H, Schlesier M, et al. Expansion of CD19hiCD21lo/neg B Cells in Common Variable Immunodeficiency (CVID) Patients With Autoimmune Cytopenia. Immunobiology (2002) 206:502–13. doi: 10.1078/0171-2985-00198
84. Sánchez-Ramón S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B Cells in Common Variable Immunodeficiency: Clinical Associations and Sex Differences. Clin Immunol (2008) 128:314–21. doi: 10.1016/j.clim.2008.02.013
85. Vodjgani M, Aghamohammadi A, Samadi M, Moin M, Hadjati J, Mirahmadian M, et al. Analysis of Class-Switched Memory B Cells in Patients With Common Variable Immunodeficiency and Its Clinical Implications. J Investig Allergol Clin Immunol (2007) 17:321–8.
86. Szabó K, Papp G, Szántó A, Tarr T, Zeher M. A Comprehensive Investigation on the Distribution of Circulating Follicular T Helper Cells and B Cell Subsets in Primary Sjögren’s Syndrome and Systemic Lupus Erythematosus. Clin Exp Immunol (2016) 183:76–89. doi: 10.1111/cei.12703
87. Bohnhorst JØ, Bjørgan MB, Thoen JE, Natvig JB, Thompson KM. Bm1–Bm5 Classification of Peripheral Blood B Cells Reveals Circulating Germinal Center Founder Cells in Healthy Individuals and Disturbance in the B Cell Subpopulations in Patients With Primary Sjögren’s Syndrome. J Immunol (2001) 167:3610–8. doi: 10.4049/jimmunol.167.7.3610
88. Barcelos F, Martins C, Papoila A, Geraldes C, Cardigos J, Nunes G, et al. Association Between Memory B-Cells and Clinical and Immunological Features of Primary Sjögren’s Syndrome and Sicca Patients. Rheumatol Int (2018) 38:1063–73. doi: 10.1007/s00296-018-4018-0
89. Saadoun D, Terrier B, Bannock J, Vazquez T, Massad C, Kang I, et al. Expansion of Autoreactive Unresponsive CD21-/Low B Cells in Sjögren’s Syndrome-Associated Lymphoproliferation. Arthritis Rheum (2013) 65:1085–96. doi: 10.1002/art.37828
90. Glauzy S, Boccitto M, Bannock JM, Delmotte FR, Saadoun D, Cacoub P, et al. Accumulation of Antigen-Driven Lymphoproliferations In Complement Receptor 2/CD21–/Low B Cells From Patients With Sjögren’s Syndrome. Arthritis Rheumatol (2018) 70:298–307. doi: 10.1002/art.40352
91. De Re V, De Vita S, Gasparotto D, Marzotto A, Carbone A, Ferraccioli G, et al. Salivary Gland B Cell Lymphoproliferative Disorders in Sjögren’s Syndrome Present a Restricted Use of Antigen Receptor Gene Segments Similar to Those Used by Hepatitis C Virus-Associated Non-Hodgkin’s Lymphomas. Eur J Immunol (2002) 32:903–10. doi: 10.1002/1521-4141(200203)32:3<903::AID-IMMU903>3.0.CO;2-D
92. Theander E, Henriksson G, Ljungberg O, Mandl T, Manthorpe R, Jacobsson LTH. Lymphoma and Other Malignancies in Primary Sjögren’s Syndrome: A Cohort Study on Cancer Incidence and Lymphoma Predictors. Ann Rheum Dis (2006) 65:796–803. doi: 10.1136/ard.2005.041186
93. Zintzaras E, Voulgarelis M, Moutsopoulos HM. The Risk of Lymphoma Development in Autoimmune Diseases: A Meta-Analysis. Arch Intern Med (2005) 165:2337–44. doi: 10.1001/archinte.165.20.2337
94. Terrier B, Nagata S, Ise T, Rosenzwajg M, Pastan I, Klatzmann D, et al. CD21-/Low Marginal Zone B Cells Highly Express Fc Receptor-Like 5 Protein and are Killed by Anti-Fc Receptor-Like 5 Immunotoxins in Hepatitis C Virus-Associated Mixed Cryoglobulinemia Vasculitis. Arthritis Rheumatol (2014) 66:433–43. doi: 10.1002/art.38222
95. Nezos A, Gravani F, Tassidou A, Kapsogeorgou EK, Voulgarelis M, Koutsilieris M, et al. Type I and II Interferon Signatures in Sjogren’s Syndrome Pathogenesis: Contributions in Distinct Clinical Phenotypes and Sjogren’s Related Lymphomagenesis. J Autoimmun (2015) 63:47–58. doi: 10.1016/j.jaut.2015.07.002
96. Rönnblom L, Eloranta ML. The Interferon Signature in Autoimmune Diseases. Curr Opin Rheumatol (2013) 25:248–53. doi: 10.1097/BOR.0b013e32835c7e32
97. Cols M, Rahman A, Maglione PJ, Garcia-Carmona Y, Simchoni N, Ko HBM, et al. Expansion of Inflammatory Innate Lymphoid Cells in Patients With Common Variable Immune Deficiency. J Allergy Clin Immunol (2016) 137:1206–15. doi: 10.1016/j.jaci.2015.09.013
98. Ciccia F, Guggino G, Rizzo A, Ferrante A, Raimondo S, Giardina AR, et al. Potential Involvement of IL-22 and IL-22-Producing Cells in the Inflamed Salivary Glands of Patients With Sjögren’s Syndrome. Ann Rheum Dis (2012) 71:295–301. doi: 10.1136/ard.2011.154013
99. Kiripolsky J, McCabe LG, Kramer JM. Innate Immunity in Sjögren’s Syndrome. Clin Immunol (2017) 182:4–13. doi: 10.1016/j.clim.2017.04.003
100. Low HZ, Witte T. Aspects of Innate Immunity in Sjögren’s Syndrome. Arthritis Res Ther (2011) 13:218. doi: 10.1186/ar3318
101. Wildenberg ME, van Helden-Meeuwsen CG, van de Merwe JP, Drexhage HA, Versnel MA. Systemic Increase in Type I Interferon Activity in Sjögren’s Syndrome: A Putative Role for Plasmacytoid Dendritic Cells. Eur J Immunol (2008) 38:2024–33. doi: 10.1002/eji.200738008
102. Hjelmervik TOR, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene Expression Profiling of Minor Salivary Glands Clearly Distinguishes Primary Sjögren’s Syndrome Patients From Healthy Control Subjects. Arthritis Rheum (2005) 52:1534–44. doi: 10.1002/art.21006
103. Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, et al. Peripheral Blood Gene Expression Profiling in Sjögren’s Syndrome. Genes Immun (2009) 10:285–96. doi: 10.1038/gene.2009.20
104. Imgenberg-Kreuz J, Sandling JK, Almlöf JC, Nordlund J, Signér L, Norheim KB, et al. Genome-Wide DNA Methylation Analysis in Multiple Tissues in Primary Sjögren’s Syndrome Reveals Regulatory Effects at Interferon-Induced Genes. Ann Rheum Dis (2016) 75:2029–36. doi: 10.1136/annrheumdis-2015-208659
105. Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN Pathways and Plasmacytoid Dendritic Cell Recruitment in Target Organs of Primary Sjögren’s Syndrome. Proc Natl Acad Sci USA (2006) 103:2770–5. doi: 10.1073/pnas.0510837103
106. Brkic Z, Maria NI, Van Helden-Meeuwsen CG, Van De Merwe JP, Van Daele PL, Dalm VA, et al. Prevalence of Interferon Type I Signature in CD14 Monocytes of Patients With Sjögren’s Syndrome and Association With Disease Activity and BAFF Gene Expression. Ann Rheum Dis (2013) 72:728–35. doi: 10.1136/annrheumdis-2012-201381
107. Hall JC, Baer AN, Shah AA, Criswell LA, Shiboski CH, Rosen A, et al. Molecular Subsetting of Interferon Pathways in Sjögren’s Syndrome. Arthritis Rheumatol (2015) 67:2437–46. doi: 10.1002/art.39204
108. Bodewes ILA, Al-Ali S, van Helden-Meeuwsen CG, Maria NI, Tarn J, Lendrem DW, et al. Systemic Interferon Type I and Type II Signatures in Primary Sjögren’s Syndrome Reveal Differences in Biological Disease Activity. Rheumatol (United Kingdom) (2018) 57:921–30. doi: 10.1093/RHEUMATOLOGY/KEX490
109. Bodewes ILA, Björk A, Versnel MA, Wahren-Herlenius M. Innate Immunity and Interferons in the Pathogenesis of Sjögren’s Syndrome. Rheumatology (2019) 60:2561–73. doi: 10.1093/rheumatology/key360
110. Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A. Role of Type I Interferons in the Activation of Autoreactive B Cells. Immunol Cell Biol (2012) 90:498–504. doi: 10.1038/icb.2012.10
111. Strandberg L, Ambrosi A, Espinosa A, Ottosson L, Eloranta ML, Zhou W, et al. Interferon-α Induces Up-Regulation and Nuclear Translocation of the Ro52 Autoantigen as Detected by a Panel of Novel Ro52-Specific Monoclonal Antibodies. J Clin Immunol (2008) 28:220–31. doi: 10.1007/s10875-007-9157-0
112. Mavragani CP, Niewold TB, Moutsopoulos NM, Pillemer SR, Wahl SM, Crow MK. Augmented Interferon-α Pathway Activation in Patients With Sjögren’s Syndrome Treated With Etanercept. Arthritis Rheum (2007) 56:3995–4004. doi: 10.1002/art.23062
113. Gangemi S, Allegra A, Musolino C. Lymphoproliferative Disease and Cancer Among Patients With Common Variable Immunodeficiency. Leuk Res (2015) 39:389–96. doi: 10.1016/j.leukres.2015.02.002
114. Matson EM, Abyazi ML, Bell KA, Hayes KM, Maglione PJ. B Cell Dysregulation in Common Variable Immunodeficiency Interstitial Lung Disease. Front Immunol (2021) 11:622114. doi: 10.3389/fimmu.2020.622114
115. Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection Outcomes in Patients With Common Variable Immunodeficiency Disorders: Relationship to Immunoglobulin Therapy Over 22 Years. J Allergy Clin Immunol (2010) 125:1354–60. doi: 10.1016/j.jaci.2010.02.040
116. Agarwal S, Cunningham-Rundles C. Autoimmunity in Common Variable Immunodeficiency. Ann Allergy Asthma Immunol (2019) 123:454–60. doi: 10.1016/j.anai.2019.07.014
117. Yazdani R, Habibi S, Sharifi L, Azizi G, Abolhassani H, Olbrich P, et al. Common Variable Immunodeficiency: Epidemiology, Pathogenesis, Clinical Manifestations, Diagnosis, Classification, and Management. J Investig Allergol Clin Immunol (2020) 30:14–34. doi: 10.18176/jiaci.0388
118. Pecoraro A, Crescenzi L, Galdiero MR, Marone G, Rivellese F, Rossi FW, et al. Immunosuppressive Therapy With Rituximab in Common Variable Immunodeficiency. Clin Mol Allergy (2019) 17:9. doi: 10.1186/s12948-019-0113-3
119. Devauchelle-Pensec V, Mariette X, Jousse-Joulin S, Berthelot J-M, Perdriger A, Puéchal X, et al. Treatment of Primary Sjögren Syndrome With Rituximab. Ann Intern Med (2014) 160:233–42. doi: 10.7326/m13-1085
120. Verma N, Burns SO, Walker LSK, Sansom DM. Immune Deficiency and Autoimmunity in Patients With CTLA-4 (CD152) Mutations. Clin Exp Immunol (2017) 190:1–7. doi: 10.1111/cei.12997
121. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the Treatment of Immune Dysregulation in Patients With Gain-of-Function Signal Transducer and Activator of Transcription 1 (STAT1) or STAT3 Mutations. J Allergy Clin Immunol (2018) 142:1665–9. doi: 10.1016/j.jaci.2018.07.020
122. Felten R, Devauchelle-Pensec V, Seror R, Duffau P, Saadoun D, Hachulla E, et al. Interleukin 6 Receptor Inhibition in Primary Sjögren Syndrome: A Multicentre Double-Blind Randomised Placebo-Controlled Trial. Ann Rheum Dis (2020) 80:329–38. doi: 10.1136/annrheumdis-2020-218467
123. Justet A, Ottaviani S, Dieudé P, Taillé C. Tocilizumab for Refractory Organising Pneumonia Associated With Sjögren’s Disease. BMJ Case Rep (2015) 2015:bcr2014209076. doi: 10.1136/bcr-2014-209076
124. Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in Systemic Sclerosis: A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Respir Med (2020) 8:963–74. doi: 10.1016/S2213-2600(20)30318-0
125. Adler S, Körner M, Förger F, Huscher D, Caversaccio MD, Villiger PM. Evaluation of Histologic, Serologic, and Clinical Changes in Response to Abatacept Treatment of Primary Sjögren’s Syndrome: A Pilot Study. Arthritis Care Res (2013) 65:1862–8. doi: 10.1002/acr.22052
126. Meiners PM, Vissink A, Kroese FGM, Spijkervet FKL, Smitt-Kamminga NS, Abdulahad WH, et al. Abatacept Treatment Reduces Disease Activity in Early Primary Sjögren’s Syndrome (Open-Label Proof of Concept ASAP Study). Ann Rheum Dis (2014) 73:1393–6. doi: 10.1136/annrheumdis-2013-204653
127. Baer AN, Gottenberg JE, St Clair EW, Sumida T, Takeuchi T, Seror R, et al. Efficacy and Safety of Abatacept in Active Primary Sjögren’s Syndrome: Results of a Phase III, Randomised, Placebo-Controlled Trial. Ann Rheum Dis (2021) 80:339–48. doi: 10.1136/annrheumdis-2020-218599
128. Hoegenauer K, Soldermann N, Zécri F, Strang RS, Graveleau N, Wolf RM, et al. Discovery of CDZ173 (Leniolisib), Representing a Structurally Novel Class of PI3K Delta-Selective Inhibitors. ACS Med Chem Lett (2017) 8:975–80. doi: 10.1021/acsmedchemlett.7b00293
129. Rao VK, Webster S, Dalm VASH, Sediva A, van Hagen PM, Do HM, et al. Safety and Efficacy of Long Term Suppression of PI3Kinase Pathway by Small Molecule PI3K-Delta Inhibitor, Leniolisib in Apds (Activated Pi3kδ Syndrome). Blood (2018) 132:3706. doi: 10.1182/blood-2018-99-113426
130. Dörner T, Zeher M, Laessing U, Chaperon F, De Buck S, Hasselberg A, et al. OP0250 A Randomised, Double-Blind Study to Assess the Safety, Tolerability and Preliminary Efficacy of Leniolisib (CDZ173) in Patients With Primary Sjögren’s Syndrome. Ann Rheum Dis (2018) 77:174. doi: 10.1136/annrheumdis-2018-eular.3111
131. Ghraichy M, Galson JD, Kelly DF, Trück J. B-Cell Receptor Repertoire Sequencing in Patients With Primary Immunodeficiency: A Review. Immunology (2018) 153:145–60. doi: 10.1111/imm.12865
132. Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, Meffre E. Bruton’s Tyrosine Kinase Is Essential for Human B Cell Tolerance. J Exp Med (2004) 200:927–34. doi: 10.1084/jem.20040920
133. Berentsen S, Ulvestad E, Tjonnfjord G. B-Lymphocytes as Targets for Therapy in Chronic Cold Agglutinin Disease. Cardiovasc Hematol Disord Targets (2008) 7:219–27. doi: 10.2174/187152907781745279
134. Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, Cellular Origin and Autoreactivity of Antibody-Secreting Cell Population Expansions in Acute Systemic Lupus Erythematosus. Nat Immunol (2015) 16:755–65. doi: 10.1038/ni.3175
135. Van Maldegem F, Wormhoudt TAM, Mulder MMS, Oud MECM, Schilder-Tol E, Musler AR, et al. Chlamydia Psittaci-Negative Ocular Adnexal Marginal Zone B-Cell Lymphomas Have Biased V H 4-34 Immunoglobulin Gene Expression and Proliferate in a Distinct Inflammatory Environment. Leukemia (2012) 26:1647–53. doi: 10.1038/leu.2012.28
136. Sato Y, Nakamura N, Nakamura S, Sakugawa S, Ichimura K, Tanaka T, et al. Deviated VH4 Immunoglobulin Gene Usage Is Found Among Thyroid Mucosa-Associated Lymphoid Tissue Lymphomas, Similar to the Usage at Other Sites, But Is Not Found in Thyroid Diffuse Large B-Cell Lymphomas. Mod Pathol (2006) 19:1578–84. doi: 10.1038/modpathol.3800692
137. Simon KL, Anderson SM, Garabedian EK, Moratto D, Sokolic RA, Candotti F. Molecular and Phenotypic Abnormalities of B Lymphocytes in Patients With Wiskott-Aldrich Syndrome. J Allergy Clin Immunol (2014) 133:896–9. doi: 10.1016/j.jaci.2013.08.050
138. Anolik JH, Barnard J, Cappione A, Pugh-Bernard AE, Felgar RE, Looney RJ, et al. Rituximab Improves Peripheral B Cell Abnormalities in Human Systemic Lupus Erythematosus. Arthritis Rheum (2004) 50:3580–90. doi: 10.1002/art.20592
139. Longhurst C, Ehrenstein MR, Leaker B, Stevenson FK, Spellerberg M, Chapman C, et al. Analysis of Immunoglobulin Variable Region Genes of a Human IgM Anti-Myeloperoxidase Antibody Derived From a Patient With Vasculitis. Immunology (1996) 87:334–8. doi: 10.1046/j.1365-2567.1996.463529.x
140. Olee T, En J, Lai CJ, Mo L, Cho CS, Wei X, et al. Generation and Analysis of an IgG Anti-Platelet Autoantibody Reveals Unusual Molecular Features. Br J Haematol (1997) 96:836–45. doi: 10.1046/j.1365-2141.1997.d01-2112.x
141. Roskin KM, Simchoni N, Liu Y, Lee JY, Seo K, Hoh RA, et al. IgH Sequences in Common Variable Immune Deficiency Reveal Altered B Cell Development and Selection. Sci Transl Med (2015) 7:302. doi: 10.1126/scitranslmed.aab1216
142. Fox RI, Fong S, Chen PP, Kipps TJ. Autoantibody Production in Sjogren’s Syndrome: A Hypothesis Regarding Defects in Somatic Diversification of Germ Line Encoded Genes. In Vivo (Brooklyn) (1988) 2:47–55.
143. Berentsen S. Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia in Waldenström’s Macroglobulinemia. Clin Lymphoma Myeloma (2009) 9:110–2. doi: 10.3816/CLM.2009.n.030
144. Nocturne G, Tarn J, Boudaoud S, Locke J, Miceli-Richard C, Hachulla E, et al. Germline Variation of TNFAIP3 in Primary Sjögren’s Syndrome-Associated Lymphoma. Ann Rheum Dis (2016) 75:780–3. doi: 10.1136/annrheumdis-2015-207731
145. Singh M, Jackson KJL, Wang JJ, Schofield P, Field MA, Koppstein D, et al. Lymphoma Driver Mutations in the Pathogenic Evolution of an Iconic Human Autoantibody. Cell (2020) 180:878–94. doi: 10.1016/j.cell.2020.01.029
Keywords: immunodeficiency, Sjögren’s syndrome, autoimmunity, lymphoproliferation, B cells
Citation: Quartuccio L, De Marchi G, Longhino S, Manfrè V, Rizzo MT, Gandolfo S, Tommasini A, De Vita S and Fox R (2021) Shared Pathogenetic Features Between Common Variable Immunodeficiency and Sjögren’s Syndrome: Clues for a Personalized Medicine. Front. Immunol. 12:703780. doi: 10.3389/fimmu.2021.703780
Received: 30 April 2021; Accepted: 22 June 2021;
Published: 12 July 2021.
Edited by:
Alan Baer, Johns Hopkins University, United StatesReviewed by:
Gaetane Nocturne, INSERM U1184 Centre de recherche en Immunologie des Infections virales et des maladies auto-immunes, FranceJoanne Reed, Garvan Institute of Medical Research, Australia
Copyright © 2021 Quartuccio, De Marchi, Longhino, Manfrè, Rizzo, Gandolfo, Tommasini, De Vita and Fox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Quartuccio, bHVjYS5xdWFydHVjY2lvQHVuaXVkLml0
†These authors share first authorship
‡These authors share senior authorship