 Marion Arnaud
Marion Arnaud Sara Bobisse
Sara Bobisse Johanna Chiffelle
Johanna Chiffelle Alexandre Harari
Alexandre Harari
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 July 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.701636
This article is part of the Research Topic The Clinical Application of Neoantigens View all 11 articles
Mutation-derived neoantigens are now established as attractive targets for cancer immunotherapy. The field of adoptive T cell transfer (ACT) therapy was significantly reshaped by tumor neoantigens and is now moving towards the genetic engineering of T cells with neoantigen-specific T cell receptors (TCRs). Yet, the identification of neoantigen-reactive TCRs remains challenging and the process needs to be adapted to clinical timelines. In addition, the state of recipient T cells for TCR transduction is critical and can affect TCR-ACT efficacy. Here we provide an overview of the main strategies for TCR-engineering, describe the selection and expansion of optimal carrier cells for TCR-ACT and discuss the next-generation methods for rapid identification of relevant TCR candidates for gene transfer therapy.
Pioneered by Rosenberg and colleagues, adoptive cell transfer (ACT) therapy is an immunotherapy strategy relying on the infusion of autologous tumor-infiltrating lymphocytes (TILs) to cancer patients. ACT demonstrated promising clinic outcomes in melanoma; with durable responses in 10-20% of patients (1, 2). Despite this progress, however, the majority of patients does not respond (3, 4) and the efficacy of ACT remains limited to melanoma and cervical cancer (5). One of the possible reasons for this limitation is the low frequency of antigen-specific T cells in TILs (1–3, 6–8). Furthermore, the proportion of bystander (i.e. tumor unrelated) TILs, such as viral-specific T cells, can be quite high in cellular products (9). Also, it was recently demonstrated that current cell culture conditions do not lead to a consistent clonal expansion of ex vivo TILs but rather lead to a biased immune repertoire (10). Altogether, these observations brought on the hypothesis that the proliferative potential of tumor antigen-reactive TILs is likely to be limited, therefore leading to their relative dilution in vitro by overgrowing bystander TILs.
To circumvent these issues, the field is moving towards the genetic engineering of T cells to express either chimeric antigen receptors (CARs) (11) or tumor antigen-specific T cell receptors (TCRs) (Figure 1) (12–17). CD19-targeting CAR T cells mediated complete responses in about 80% of patients with B cell acute lymphoblastic leukemia cancer (18). Yet, CARs target tissue-restricted antigens expressed on the surface of tumor cells (19), thus limiting their applications (20), while TCR-transduced cells can target any surface or intracellular antigen. In this report, we will focus on TCR-based cellular immunotherapy (Figure 1).
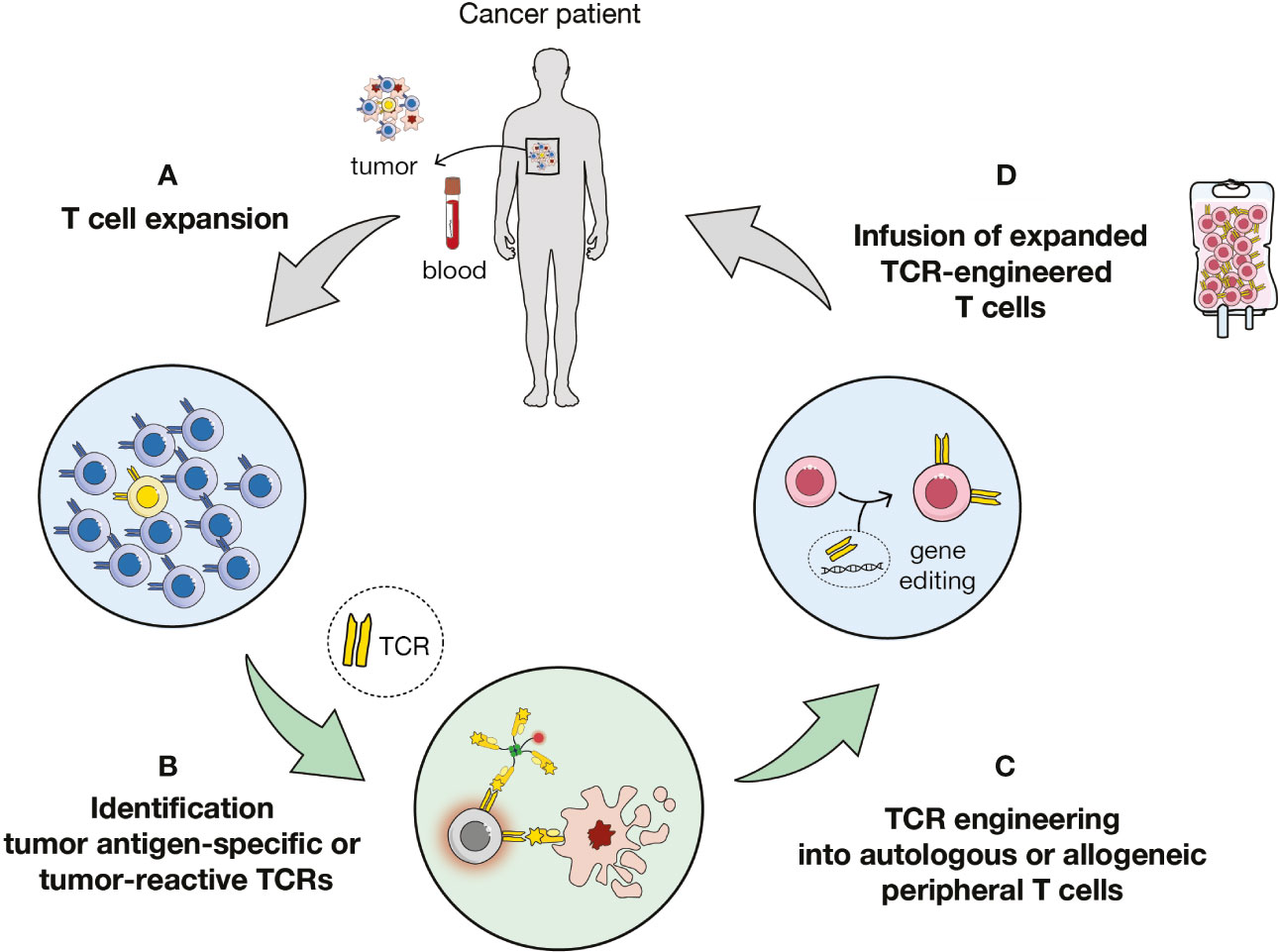
Figure 1 Overview of TCR-based immunotherapy. (A) Expansion of T cells from tumor or blood samples of patient or healthy donors. (B) Identification of antigen-specific or tumor-reactive T cells and of cognate T cell receptors (TCRs). (C) Transduction of autologous or allogenic carrier cells. (D) Adoptive cell transfer of TCR-engineered cells to the patient.
TCRs are heterodimers consisting of disulfide-linked α and β chains, each with a variable and a constant domain (21, 22). The variable regions can bind the antigen-MHC complex. The binding domain is constructed based on the recombination of multiple gene segments, leading to the great diversity of the TCR repertoire, with potentially >1015 distinct αβ TCRs (23–25). TCR gene-transfer therapy targeting tumor-associated antigens (TAAs), such as MART-1 and NY-ESO-1, achieved clinical responses ranging from about 10 to 60% of patients from different malignancies (12, 14, 26, 27). Despite these promising clinical outcomes, efficacy remained limited and important toxicities occurred (13, 27, 28). The field was then rejuvenated by the perspective of using TCRs targeting private neoantigens, which have emerged as clinically relevant targets (29, 30). In this review, we will discuss several issues including the common tools used for cells transduction, the optimal cells to transduce for ACT and the acceleration of the identification of relevant tumor-specific TCRs for gene transfer therapy.
As for CAR-T cell based therapy, viral vectors were widely exploited for ex vivo TCR gene transfer into recipient T cells (Figure 2). In clinical trials, the most common viral systems used are gamma retrovirus- (RV) and lentivirus-based vectors (LV), as reviewed in (31). Both RVs and LVs allow for stable integration and efficient long-term expression of exogenous TCRs. However, safety concerns to RVs and LVs remained, that are mainly insertional mutagenesis and neoplastic transformation (32), as well as generation of replication-competent viral particles. The latter limitations boosted the development of novel vector designs, such as addition of insulator sequences (33), disruption of the long terminal repeats for self-inactivating viral vectors (34, 35) or pseudotyping (36). Of note, RV transduction requires mitotic cells for the transgene to penetrate the nucleus and integrate in the genome and thus recipient cells must be activated beforehand. Conversely, LVs allow the effective transduction of a variety of not actively dividing and terminally differentiated cells. Yet, human resting T cells are scarcely susceptible to transduction by LVs and need to be minimally stimulated to enter the G1b phase (37, 38). Importantly. several studies highlighted a positive correlation between the proliferative potential of adoptively transferred cells and their in vivo persistency (1), sustaining the use of LVs in the clinic to limit recipient cell stimulation in vitro.
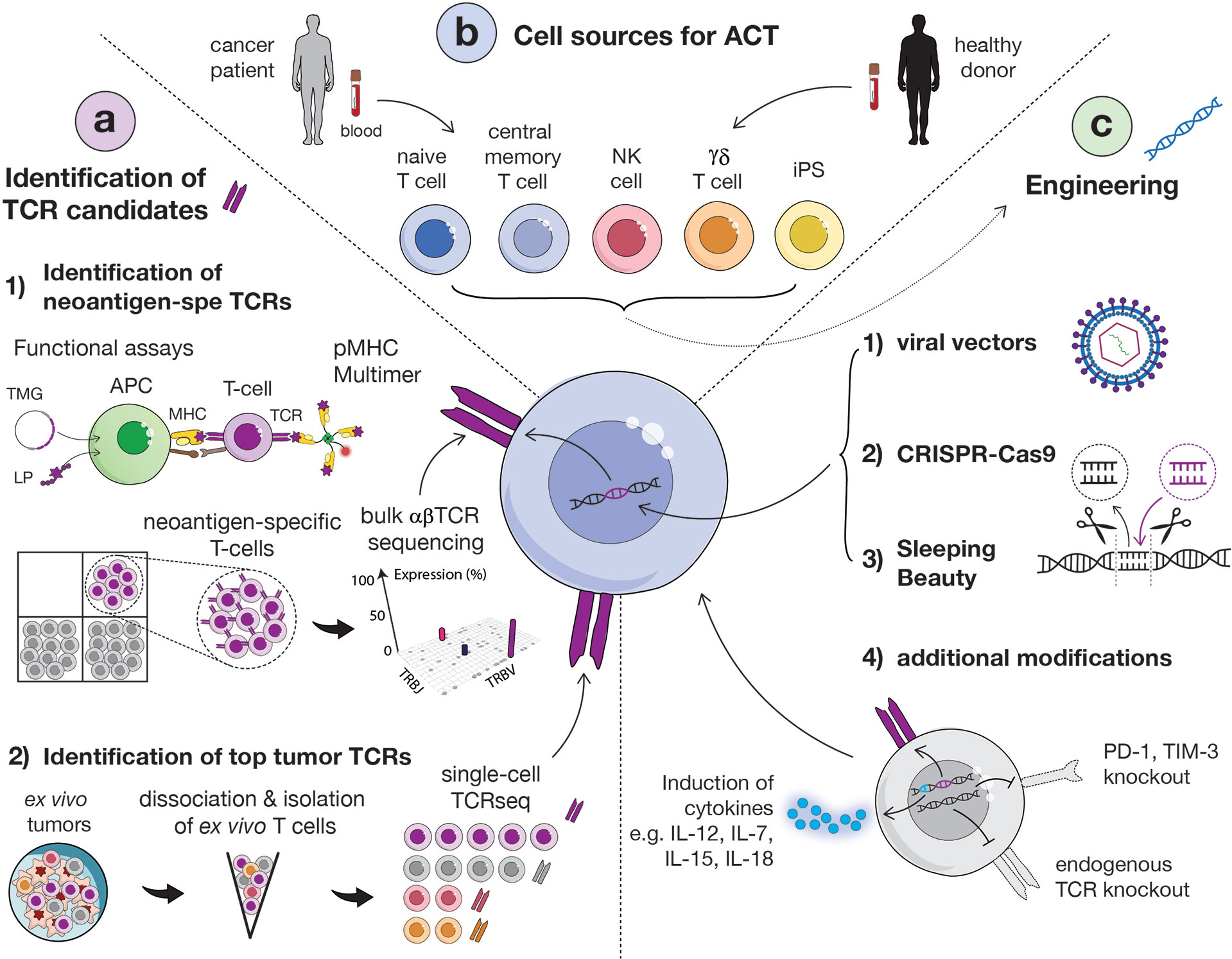
Figure 2 Overview of main strategies to identify relevant TCRs, methods for transduction and options of carrier cells. (A) Identification of TCR candidates via functional assays and sequential isolation of neoantigen-specific T cells based on pMHC multimer staining. Isolated neoantigen-reactive T cells can then be sequenced in bulk for the identification of dominant TCR clonotypes (1). Alternatively, tumor-specific TCRs can be identified directly from ex vivo fresh tumor, without intermediate culture, by sorting T cells by flow cytometry and performing single-cell TCR sequencing (2). (APC, antigen presenting cell; MHC, major histocompatibility complex; LP, long peptide; TMG, tandem minigene). (B) Several carrier cells can be used for TCR transduction, including αβ (CD4 or CD8) T cells, naïve or central memory cells, NK cells, γδ T cells and induced-pluripotent stem cells (iPSCs). Carrier cells can be derived from healthy donor or patient blood. (C) TCR engineering into carrier cells can be obtained via viral (1) or non-viral vectors [such as CRISPR-Cas9 (2) or Sleeping Beauty (3)]. Additional modifications of cells to enhance their functionality and in vivo persistency but also avoid TCR mispairing can be performed, such as the knock-out of endogenous TCR or immune inhibitor genes and the induction of cytokines (4).
As alternative to RVs and LVs, adeno-associated virus-derived vectors (AAVs) can be used for TCR-engineering. AAVs are replication-deficient systems and are not known to induce any side effects, making them ideal candidates from a safety standpoint. AAV-based cancer treatments have not yet been used in the clinic, however multiple strategies have been developed and hold great promise for the future (39–41). It is to be noted that vector manufacturing remains time consuming (over six months) and expensive (42).
Non-viral methods for stable gene transfer into T cells were investigated in CAR-T clinical trials and represent a future opportunity for TCR gene transfer clinical studies. Non-viral engineering strategies include transposon-based vector systems, such as “Sleeping Beauty”, by which a transgene of interest flanked by inverted terminal repeats is provided to target cells as discrete DNA molecule and randomly integrates in its genome when a transposase is co-supplemented in trans (i.e. mRNA, plasmid or protein) (43). Over the past decades, transposons and transposases have been extensively optimized to increase their activity and reduce toxicity (e.g. insertional mutagenesis) (44). Alternatively, exogenous TCRs can be transiently expressed in trans if transfected into T cells in the form of mRNA molecules or non-integrating vectors (45, 46). All these non-viral systems may be preferable to viral ones for the clinic because of their easier handling and cheaper production costs. Of important note, gene editing of T cells with TCRs, as opposed to CARs, has to face an extra challenge. Mispairing of exogenous and endogenous TCR chains can indeed occur and lead to off-target toxicity. Researchers have therefore developed several platforms to specifically silence the expression of the endogenous TCR, based on Sleeping Beauty, Zinc finger nucleases (47), transcription activator-like effector nucleases (TALEN) (48, 49), mega-nucleases or Clustered Regularly-Interspaced Short Palindromic Repeats (CRISPR)–Cas9 (50, 51) (Figure 2).
The CRISPR-Cas9 technology relies on short RNA sequences which are used to target the site of insertion instead of proteins and which are easily synthesized in vitro. As such, CRISPR-Cas9 enables the simultaneous targeting of multiple genome sites and site-specific mutagenesis, allowing the knockout (KO) of endogenous TCRs or of immune checkpoint such as PD-1 (52) and the induction of cytokine expression (e.g. IL-7, IL-12, IL-15, IL-18) (17) (Figure 2). The safety and feasibility of multiplex CRISPR-Cas9 gene editing of T cells was demonstrated in a study with advanced refractory cancer patients infused with autologous T cells KO for the three genes TRAC, TRBC and PDC1 (PD-1 loci) and transduced with a NY-ESO1 TCR (53). Of interest, the KO by CRISPR-Cas9 of cytokine-induced SH2 protein (CISH), an immune checkpoint, was found to increase the in vitro proliferation and functionality of TCR-engineered T cells and such strategy will be further explored in an upcoming clinical trial (NCT04426669) (54). Despite technical challenges which still need to be overcome, in particular regarding the delivery systems currently based on AAV and transfection (55–59), CRISPR-Cas9 will likely become the method of choice for therapeutic gene engineering in the upcoming years.
T lymphocytes are the most common source of cells used as carrier for gene transfer therapy. Following TCR engineering into recipient cells, TCR-T cells need to be expanded to reach sufficient numbers for ACT. The primary starting material is most often autologous peripheral blood (Figure 2).
Thus far, TCR gene therapy mainly focused on CD8 T cells, which represent key players of ACT. In particular, a retrospective analysis of TIL ACT infusion products and clinical efficacy from 92 patients highlighted an association between the fraction of CD8 T cells and clinical benefit (60). There is, however, an emerging clinical relevance of CD4 T cells. Early evidence was provided by Tran and colleagues who showed the antitumor potential of neoantigen-specific CD4 TILs by ACT (61). Of interest, the direct cytolytic capacity of CD4 T cells was demonstrated (62–64). In addition, CD4 T cell’s help is essential to generate efficient tumor-reactive effector CD8 T cells (65), notably during the process of epitope spreading (29, 66). Yet TCR gene transfer therapy with CD4 tumor-specific TCRs is also limited by the challenging prediction and detection of MHC class II-restricted neoantigen-specific CD4 T cells, despite major advances (67, 68).
T cell differentiation state is critical for ACT. The profile of adoptively-transferred cells is indeed likely to affect their in vivo persistency and thus treatment efficacy. T cell differentiation states range from naïve to central memory, effector memory and finally terminally differentiated (EMRA) (69). Accumulating evidence shows that the effector phenotype acquired in vitro negatively impacts the antitumor potential of T cells in vivo (70), while ACT efficacy requires a long-term persistence of transferred cells. Thus, less-differentiated T cell populations that maintain self-renewal capabilities are preferred and were associated with improved clinical benefit (71, 72). Naïve and memory T cells maintain the highest proliferative potential combined to the most potent fitness and stemness (73–76) (Figure 2). Consistently, naïve and memory subsets were found more effective than effector T cells for ACT (70, 74–77). Furthermore, Hinrich and colleagues have demonstrated that naïve CD8 T cells had a higher anti-tumor potential for ACT as compared to central memory cells (77).
The T cell state is also modulated during the expansion phase in vitro. TCR-transduced T cells are commonly expanded with anti-CD3/CD28 beads in the presence of IL-2 (12, 38, 78). It has been demonstrated that the addition to the culture medium of alternative cytokines, such as IL-7, IL-15 and/or IL-21, enabled to the generation of less-differentiated TCR-engineered T cells thus leading to increased persistency and ultimately improved efficacy (71, 79–81).
Autologous T cells are available in limited quantities and their state is likely to be affected by the multiple rounds of cancer treatments which patients undergo before ACT, complicating the manufacturing process and the feasibility of TCR gene transfer therapy. Therefore allogenic universal T cells have also been exploited for gene transfer therapy (82) (Figure 2). A few issues are to be noted, including competition with endogenous TCRs, mispairing of TCR subunits, risk of off-target toxicity due to allogenic TCR-T cell infusion and ACT product rejection by the host (83). It is thus required to KO endogenous TCRs to improve the safety and efficacy of TCR-ACT with allogenic cells (49). For all these reasons, several alternative non-T cell types were evaluated.
Mensali and colleagues provided the first proof of concept that cells other than αβ T cells could be used as recipient cells for TCR gene therapy. They transferred a TCR into NK cell line, NK-92, and demonstrated efficacy in vivo (84). More recently, Parlar and coworkers have engineered NK cells with a tyrosinase-specific TCR and highlighted their cytolytic potential in vitro (85). A potential benefit of using NK cells as a carrier is their ability to remain cytotoxic in an MHC-independent manner. This could be of interest in case of MHC loss, which is a common immune suppressive mechanism exerted by tumors (86). Alternatively, γδ T cells can also be TCR-engineered (Figure 2), thus avoiding the issue of TCR mispairing. The latter strategy was proven efficient in leukemia (87). Furthermore, as T cell exhaustion is a critical component of ACT efficacy, there is an increasing interest in using induced-pluripotent stem cells (iPSCs) for gene transfer therapy (Figure 2). Nishimura and colleagues were able to generate iPSCs from antigen-specific CD8 T cells and to re-differentiate them. In this way, they obtained rejuvenated cells with longer telomeres and a high proliferative potential, making them fitter for therapy (88). The efficacy of TCR-engineered iPSCs was shown in vivo (89).
As mentioned previously, neoantigens are attractive targets for TCR-based therapy. To date, most neoantigens originated from non-synonymous mutations. T cell reactivity to neoantigens was associated with improved clinical benefit of immunotherapy, both immune checkpoint blockade (90–92) and ACT (93–95). Early reports showed promising results in terms of safety, feasibility and efficacy with neoantigen targeting immunotherapy either in the form of vaccination (29, 96–98) or ACT (30, 99). Strikingly, complete remissions were observed following infusion of neoantigen-reactive T cells, highlighting the potency of mutanome based therapies (30, 99).
The first key challenge lies in the identification of neoantigens, which is a long and tedious process and was reviewed elsewhere (100). Upon neoantigen identification, neoepitope-specific T cells are purified and their TCR is sequenced. Candidate TCRs are then cloned to validate their antigen specificity and tumor reactivity (Figure 2).
Neoantigen-specific T cells, and hence their cognate TCR, can be identified in different samples including tumor or blood from patient or naïve T cells from healthy donors. Briefly, neoantigens can be identified from expanded TILs (7, 95, 101). However, due to low frequencies, sensitive detection sometimes requires antigen-specific in vitro stimulation (IVS). Of interest, we developed a novel strategy to improve the detection of neoantigens in TILs, based on the addition of pools of predicted neo-epitopes at the initiation of the TIL culture (7). Alternatively, TILs can be enriched prior to culture by sorting of dissociated tumor material ex vivo, based on various activation markers like PD-1 (CD279), OX40, CD137 (4-1BB), CD39 and CD103 (102–106).
Peripheral blood lymphocytes (PBLs) can also be exploited for neoantigen identification. IVS with antigen presenting cells (APC) loaded with neoantigen candidates have been extensively used to detect neoantigen reactivity (7, 30, 96, 107–110). Prior enrichment strategies have also been used by different groups including: the isolation of memory (107) or naïve T cells (108) or the sorting with PD-1 (109). Most identification processes use PBLs from autologous origin but it has been shown to be possible from allogenic sources as well (108).
Upon their identification, neoantigen-specific T cells can be purified based on pMHC multimer staining or based on the up-regulation of activation markers following specific-activation, such as 4-1BB and OX40 (7, 107, 108, 111, 112). Isolated cells then undergo bulk α and β TCR sequencing in order to select the dominant α and β TCR clonotypes. Of note, TCR repertoire analysis strategies are challenging due to the high diversity of TCR repertoires (113). Advances in next-generation sequencing has improved the interpretation of TCR repertoires. Using RNA as a source of material allows allelic exclusion thereby avoiding to overestimate repertoires diversity. RNA is also more sensitive than DNA despite being less quantitative due to variation in expression levels (114). Among the different sequencing methods, multiplex polymerase chain reaction (PCR) (115) remains the most commonly used strategy, despite misrepresentation of clonotypes proportion introduced by heterogeneity in primers efficiency (116). Other approaches rely on the addition of adaptors prior to PCR amplification (117, 118), such as the 5’ RACE PCR or TCR amplification following gene capture (119). For each method, the bias in quantification reduces the ability for easy pairing of α and β chains, which is required for therapeutic applications. A concept based on multiple sequencing and combinatorial analysis was developed to pair αβ TCR chains, yet this strategy is limited to high-frequency clonotypes and requires large cell numbers (120). To avoid the above-mentioned limitations, both T cell cloning or single-cell sequencing can be used.
Interrogation of neoantigen-specificity and antitumor-reactivity of candidate TCRs can be assessed following the expression of TCR candidates into recipient cells, a strategy hereafter referred as TCR cloning. Antigen-specificity can be challenged by transducing TCRs into activated PBLs or Jurkat T cells which are then co-cultured with APCs loaded with neoantigens (29, 86, 94, 95, 99, 101, 102, 107, 121–123). Next, tumor-reactivity can be measured by co-culture of TCR-engineered T cells with autologous tumor cells or APCs pulsed with tumor lysate (86, 94, 101, 102, 121, 124, 125). To enable a rapid identification of neoantigen-reactive TCRs, Paria and colleagues have developed a TCR cloning methodology using Jurkat T cells electroporated with RNAs encoding TCR α and β chain, respectively (126). The benefit of this approach lies in the use of RNA electroporation, which is faster and more efficient than TCR transduction by genetic engineering. TCR expression is transient but sufficient for TCR interrogation. Interestingly, they used the Jurkat luciferase system (under NFAT promotor) which is a rapid and easy read-out.
Of important note, the reactivity of validated neoantigen-specific TCRs to the wild-type peptide should be evaluated to avoid autoimmunity and thus ensure patient safety. This can done by performing a peptide dose response with APCs (86, 94, 95, 99, 102, 107). Another method consists of using a high-throughput genetic platform (127).
As an alternative to the aforementioned time-consuming strategies (based on TCR isolation and downstream TCR validation), new developments enable the direct identification of tumor-specific TCRs by pre-selecting the most frequent clonotypes from fresh tumor samples (121) (Figure 2). Selected αβ TCR pairs are then challenged against tumor cells by TCR cloning. Others have however found that the tumor reactivity of intra-tumoral TCR repertoire was low (125), highlighting a potential limitation of using this strategy for gene transfer therapy. Identified tumor-reactive TCRs can then be examined retrospectively for antigen specificity using ligand discovery approaches, based for instance on trogocytosis, a process in which T cells exchange membrane proteins with APC presenting candidate antigens (128). Other possibilities for TCR ligand discovery include the use of chimeric signaling and antigen presenting bifunctional receptors (SABRs) (129). It is to be noted that the development of microfluidic-based platforms will accelerate and automate the isolation of relevant TCRs for TCR-ACT in the near future (130–132).
Instead of direct TCR identification, combined single-cell transcriptomics and TCR sequencing (133) could be exploited to define intra-tumoral signatures of neoantigen-specific or tumor-reactive TCRs (134). Old-generation single-cell methodologies are tedious and allow the sequencing of a limited number of cells (135, 136). Single-cell TCR sequencing and profiling of T cells was recently eased by the development of a commercially available strategy by 10x Genomics (137–139). Briefly, the latter microfluidic-based technology is able to generate an emulsion containing one trapped cell with a uniquely barcoded bead and, upon cell lysis polyadenylated mRNA is captured. The process results in barcoded libraries, which undergo both downstream single-cell TCR sequencing and transcriptomic profiling. Overall, 15’000-20’000 cells can be covered. The 10x Genomics strategy allows the analysis of clones in a timely manner and was applied for the identification of a signature of clone persistency in the circulation after ACT (140). In the near future, intra-tumoral signatures of neoantigen and/or tumor-reactive TCRs may be defined based on a comprehensive database of TCRs combined to their ex vivo transcriptomic profiling. We foresee that these signatures will enable the direct identification of relevant TCR pairs from ex vivo tumor, which will considerably facilitate and accelerate the selection of TCR candidates for gene transfer therapy.
In this review, we provided an overview of the available therapeutic engineering systems, but also of the optimal carrier cells for TCR gene transfer therapy, and finally we described strategies to identify new TCR candidates. The field is constantly evolving and future advances are likely to reshape the landscape of TCR-ACT. Multiple clinical trials are currently ongoing in different types of cancer (17) and the results are awaited with much anticipation. In the future, we could take advantage of unique intra-tumoral transcriptomic signatures from identified neoantigen-specific or tumor-reactive TCRs. These signatures may then be used to directly and rapidly fish out TCRs of interest for gene transfer therapy. Challenges of TCR-ACT remain, in particular immune-editing following therapy, ultimately leading to immune evasion and progression (86). To increase the efficacy of TCR-ACT therapy and counter potential immune-editing, multiple TCRs should be targeted, which is currently possible with the CRISPR-Cas9 technology. Future strategies may also focus on the simultaneous targeting of MHC-restricted epitopes together with membrane antigens. Importantly, to extend the reach of therapy and benefit more cancer patients, ‘shared’ neoantigens (141, 142) arising from ‘hotspots’ mutations shared between unrelated individuals should be preferentially targeted and these approaches will be further explored in upcoming clinical trials (e.g. NCT03190941). Additionally, it is likely that next-generation improvement of carrier cells will further potentiate TCR-ACT efficacy. Given the plethora of existing technologies and their constant amelioration, together with the many ongoing clinical trials, we expect the immune-oncology field to be fundamentally modified by TCR gene transfer therapy in the upcoming years.
All authors contributed to the article and approved the submitted version.
This research was supported by the Ludwig Institute for Cancer Research and grant 310030_182384 from the Swiss National Science Foundation (A.H.).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Kaat de Jonge for her kind support editing the manuscript.
1. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable Complete Responses in Heavily Pretreated Patients With Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
2. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-Lasting Complete Responses in Patients With Metastatic Melanoma After Adoptive Cell Therapy With Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin Cancer Res (2016) 22:3734–45. doi: 10.1158/1078-0432.CCR-15-1879
3. Andersen R, Westergaard MCW, Kjeldsen JW, Muller A, Pedersen NW, Hadrup SR, et al. T-Cell Responses in the Microenvironment of Primary Renal Cell Carcinoma-Implications for Adoptive Cell Therapy. Cancer Immunol Res (2018) 6:222–35. doi: 10.1158/2326-6066.CIR-17-0467
4. Dafni U, Michielin O, Lluesma SM, Tsourti Z, Polydoropoulou V, Karlis D, et al. Efficacy of Adoptive Therapy With Tumor-Infiltrating Lymphocytes and Recombinant Interleukin-2 in Advanced Cutaneous Melanoma: A Systematic Review and Meta-Analysis. Ann Oncol (2019) 30:1902–13. doi: 10.1093/annonc/mdz398
5. Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete Regression of Metastatic Cervical Cancer After Treatment With Human Papillomavirus-Targeted Tumor-Infiltrating T Cells. J Clin Oncol (2015) 33:1543–50. doi: 10.1200/JCO.2014.58.9093
6. Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebæk E, et al. Dissection of T-Cell Antigen Specificity in Human Melanoma. Cancer Res (2012) 72:1642–50. doi: 10.1158/0008-5472.CAN-11-2614
7. Bobisse S, Genolet R, Roberti A, Tanyi JL, Racle J, Stevenson BJ, et al. Sensitive and Frequent Identification of High Avidity Neo-Epitope Specific CD8+ T Cells in Immunotherapy-Naive Ovarian Cancer. Nat Commun (2018) 9:1092. doi: 10.1038/s41467-018-03301-0
8. Gokuldass A, Draghi A, Papp K, Borch TH, Nielsen M, Westergaard MCW, et al. Qualitative Analysis of Tumor-Infiltrating Lymphocytes Across Human Tumor Types Reveals a Higher Proportion of Bystander CD8+ T Cells in Non-Melanoma Cancers Compared to Melanoma. Cancers (Basel) (2020) 12:3344. doi: 10.3390/cancers12113344
9. Simoni Y, Becht E, Fehlings M, Loh CY, Koo S, Wei K, et al. Bystander CD8+ T Cells Are Abundant and Phenotypically Distinct in Human Tumour Infiltrates. Nature (2018) 557:575–80. doi: 10.1038/s41586-018-0130-2
10. Poschke IC, Hassel JC, Rodriguez-Ehrenfried A, Lindner KAM, Heras-Murillo I, Appel LM, et al. The Outcome of Ex Vivo TIL Expansion Is Highly Influenced by Spatial Heterogeneity of the Tumor T-Cell Repertoire and Differences in Intrinsic In Vitro Growth Capacity Between T-Cell Clones. Clin Cancer Res (2020) 26:4289–301. doi: 10.1158/1078-0432.CCR-19-3845
11. Crowther MD, Svane IM, Met Ö. T-Cell Gene Therapy in Cancer Immunotherapy: Why It Is No Longer Just CARs on The Road. MDPI (2020) 9:1–18. doi: 10.3390/cells9071588
12. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science (2006) 314:126–9. doi: 10.1126/science.1129003
13. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J Immunother (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
14. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A Pilot Trial Using Lymphocytes Genetically Engineered With an NY-ESO-1-Reactive T-Cell Receptor: Long-Term Follow-Up and Correlates With Response. Clin Cancer Res (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
15. Rosenberg SA, Restifo NP. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science (2015) 348:62–8. doi: 10.1126/science.aaa4967
16. Sadelain M, Rivière I, Riddell S. Therapeutic T Cell Engineering. Nature (2017) 545:423–31. doi: 10.1038/nature22395
17. Gaissmaier L, Elshiaty M, Christopoulos P. Breaking Bottlenecks for the TCR Therapy of Cancer. Cells (2020) 9:1–21. doi: 10.3390/cells9092095
18. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-Up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
19. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
20. Sterner RC, Sterner RM. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
21. Bäckström BT, Milia E, Peter A, Jaureguiberry B, Baldari CT, Palmer E. A Motif Within the T Cell Receptor Alpha Chain Constant Region Connecting Peptide Domain Controls Antigen Responsiveness. Immunity (1996) 5:437–47. doi: 10.1016/s1074-7613(00)80500-2
22. Dong D, Zheng L, Lin J, Zhang B, Zhu Y, Li N, et al. Structural Basis of Assembly of the Human T Cell Receptor-CD3 Complex. Nature (2019) 573:546–52. doi: 10.1038/s41586-019-1537-0
23. Bassing CH, Swat W, Alt FW. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell (2002) 109 Suppl:S45–55. doi: 10.1016/s0092-8674(02)00675-x
24. Lythe G, Callard RE, Hoare RL, Molina-París C. How Many TCR Clonotypes Does a Body Maintain? J Theor Biol (2016) 389:214–24. doi: 10.1016/j.jtbi.2015.10.016
25. Sewell AK. Why Must T Cells be Cross-Reactive? Nat Rev Immunol (2012) 12:669–77. doi: 10.1038/nri3279
26. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene Therapy With Human and Mouse T-Cell Receptors Mediates Cancer Regression and Targets Normal Tissues Expressing Cognate Antigen. Blood Adv (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
27. Bethune MT, Joglekar AV. Personalized T Cell-Mediated Cancer Immunotherapy: Progress and Challenges. Curr Opin Biotechnol (2017) 48:142–52. doi: 10.1016/j.copbio.2017.03.024
28. Van Den Berg JH, Gomez-eerland R, Van De Wiel B, Hulshoff L, Van Den Broek D, Bins A, et al. Case Report of a Fatal Serious Adverse Event Upon Administration of T Cells Transduced With a MART-1-Specific T-Cell Receptor. Am Soc Gene Cell Ther (2015) 23:1541–50. doi: 10.1038/mt.2015.60
29. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature (2017) 547:222–6. doi: 10.1038/nature23003
30. Chen F, Wei J, Liu B, Chen F, Zou Z, Du J, et al. Neoantigen Identification Strategies Enable Personalized Immunotherapy in Refractory Solid Tumors. JCI (2019) 129:2056–70. doi: 10.1172/JCI99538
31. Manfredi F, Cianciotti BC, Potenza A, Tassi E, Noviello M, Biondi A, et al. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front Immunol (2020) 11:1689. doi: 10.3389/fimmu.2020.01689
32. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional Mutagenesis Combined With Acquired Somatic Mutations Causes Leukemogenesis Following Gene Therapy of SCID-X1 Patients. J Clin Invest (2008) 118:3143–50. doi: 10.1172/JCI35798
33. Ellis J. Silencing and Variegation of Gammaretrovirus and Lentivirus Vectors. Hum Gene Ther (2005) 16:1241–6. doi: 10.1089/hum.2005.16.1241
34. Loew R, Meyer Y, Kuehlcke K, Gama-Norton L, Wirth D, Hauser H, et al. A New PG13-Based Packaging Cell Line for Stable Production of Clinical-Grade Self-Inactivating Gamma-Retroviral Vectors Using Targeted Integration. Gene Ther (2010) 17:272–80. doi: 10.1038/gt.2009.134
35. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A Third-Generation Lentivirus Vector With a Conditional Packaging System. J Virol (1998) 72:8463–71. doi: 10.1128/JVI.72.11.8463-8471.1998
36. Pistello M, Vannucci L, Ravani A, Bonci F, Chiuppesi F, del Santo B, et al. Streamlined Design of a Self-Inactivating Feline Immunodeficiency Virus Vector for Transducing Ex Vivo Dendritic Cells and T Lymphocytes. Genet Vaccines Ther (2007) 5:8. doi: 10.1186/1479-0556-5-8
37. Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science (1996) 272:263–7. doi: 10.1126/science.272.5259.263
38. Bobisse S, Rondina M, Merlo A, Tisato V, Mandruzzato S, Amendola M, et al. Reprogramming T Lymphocytes for Melanoma Adoptive Immunotherapy by T-Cell Receptor Gene Transfer With Lentiviral Vectors. Cancer Res (2009) 69:9385–94. doi: 10.1158/0008-5472.CAN-09-0494
39. Marcucci KT, Jadlowsky JK, Hwang W, Suhoski-davis M, Gonzalez VE, Kulikovskaya I, et al. Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused HIV and Oncology Patients. Mol Ther (2018) 26:269–79. doi: 10.1016/j.ymthe.2017.10.012
40. Li C, Samulski RJ. Engineering Adeno-Associated Virus Vectors for Gene Therapy. Nat Rev Genet (2020) 21:255–72. doi: 10.1038/s41576-019-0205-4
41. Hacker UT, Bentler M, Kaniowska D, Morgan M, Büning H. Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives Ulrich. MDPI (2020) 12:1889. doi: 10.3390/cancers12071889
42. Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, et al. High-Throughput Sequencing of T-Cell Receptors Reveals a Homogeneous Repertoire of Tumour-Infiltrating Lymphocytes in Ovarian Cancer. J Pathol (2013) 231:433–40. doi: 10.1002/path.4260
43. Hackett PB, Largaespada DA, Cooper LJN. A Transposon and Transposase System for Human Application. Mol Ther (2010) 18:674–83. doi: 10.1038/mt.2010.2
44. Cain AK, Barquist L, Goodman AL, Paulsen IT, Parkhill J, van Opijnen T. A Decade of Advances in Transposon-Insertion Sequencing. Nat Rev Genet (2020) 21:526–40. doi: 10.1038/s41576-020-0244-x
45. Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple Injections of Electroporated Autologous T Cells Expressing a Chimeric Antigen Receptor Mediate Regression of Human Disseminated Tumor. Cancer Res (2010) 70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880
46. Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-Efficiency Transfection of Primary Human and Mouse T Lymphocytes Using RNA Electroporation. Mol Ther (2006) 13:151–9. doi: 10.1016/j.ymthe.2005.07.688
47. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T Cell Specificity Towards Leukemia by Zinc Finger Nucleases and Lentiviral Gene Transfer. Nat Med (2012) 18:807–15. doi: 10.1038/nm.2700
48. Wood AJ, Lo T-W, Zeitler B, Pickle CS, Ralston EJ, Lee AH, et al. Targeted Genome Editing Across Species Using ZFNs and TALENs. Science (2011) 333:307. doi: 10.1126/science.1207773
49. Berdien B, Mock U, Atanackovic D, Fehse B. TALEN-Mediated Editing of Endogenous T-Cell Receptors Facilitates Efficient Reprogramming of T Lymphocytes by Lentiviral Gene Transfer. Gene Ther (2014) 21:539–48. doi: 10.1038/gt.2014.26
50. Doudna JA, Charpentier E. Genome Editing. The New Frontier of Genome Engineering With CRISPR-Cas9. Science (2014) 346:1258096. doi: 10.1126/science.1258096
51. Pickar-Oliver A, Gersbach CA. The Next Generation of CRISPR-Cas Technologies and Applications. Nat Rev Mol Cell Biol (2019) 20:490–507. doi: 10.1038/s41580-019-0131-5
52. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients With Refractory Non-Small-Cell Lung Cancer. Nat Med (2020) 26:732–40. doi: 10.1038/s41591-020-0840-5
53. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-Engineered T Cells in Patients With Refractory Cancer. Science (2020) 367:eaba7365. doi: 10.1126/science.aba7365
54. Palmer DC, Webber BR, Patel Y, Johnson MJ, Kariya CM, Walker S, et al. Internal Checkpoint Regulates T Cell Neoantigen Reactivity and Susceptibility to PD1 Blockade. bioRxiv (2020). doi: 10.1101/2020.09.24.306571
55. Od Z, Condori J, Peterson N, Zhou S, Krenciute G. Integration and Expression in T Cells: Protocol and Application for T-Cell Therapy. MDPI (2020) 12:1704. doi: 10.3390/cancers12061704
56. Lino CA, Harper JC, Carney JP, Timlin JA, Lino CA, Harper JC, et al. Delivering CRISPR: A Review of the Challenges and Approaches. Drug Delivery (2018) 25:1234–57. doi: 10.1080/10717544.2018.1474964
57. Sharma G, Sharma AR, Bhattacharya M, Lee S, Chakraborty C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol Ther (2021) 29:571–86. doi: 10.1016/j.ymthe.2020.09.028
58. Hirakawa MP, Krishnakumar R, Timlin JA, Carney JP, Butler KS. Gene Editing and CRISPR in the Clinic: Current and Future Perspectives. Biosci Rep (2020) 40:BSR20200127. doi: 10.1042/BSR20200127
59. Rui Y, Wilson DR, Green JJ. Non-Viral Delivery To Enable Genome Editing. Trends Biotechnol (2018) 37:1–13. doi: 10.1016/j.tibtech.2018.08.010
60. Prieto PA, Durflinger KH, Wunderlich JR, Rosenberg SA, Dudley ME. Enrichment of CD8+ Cells From Melanoma Tumor-Infiltrating Lymphocyte Cultures Reveals Tumor Reactivity for Use in Adoptive Cell Therapy. J Immunother (2010) 33:547–56. doi: 10.1097/CJI.0b013e3181d367bd
61. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient With Epithelial Cancer. Science (2014) 344:642–4. doi: 10.1126/science.1251102
62. Cachot A, Bilous M, Liu Y, Li X, Saillard M, Cenerenti M, et al. Tumor-Specific Cytolytic CD4 T Cells Mediate Immunity Against Human Cancer. Sci Adv (2021) 7:eabe3348. doi: 10.1126/sciadv.abe3348
63. Merlo A, Turrini R, Bobisse S, Zamarchi R, Alaggio R, Dolcetti R, et al. Virus-Specific Cytotoxic CD4 T Cells for the Treatment of EBV-Related Tumors. J Immunol (2010) 184:5895– 902. doi: 10.4049/jimmunol.0902850
64. Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4+ T Cells Mediate Anti-Tumor Cytotoxicity in Human Bladder Cancer. Cell (2020) 181:1612–25.e13. doi: 10.1016/j.cell.2020.05.017
65. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, Dupage M, Luoma AM, et al. MHC-II Neoantigens Shape Tumour Immunity and Response to Immunotherapy. Nature (2019) 574:696–701. doi: 10.1038/s41586-019-1671-8
66. Diekmann J, Kreiter S, Vormehr M, Van De Roemer N, Diken M, Lo M, et al. Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature (2015) 250:692–6. doi: 10.1038/nature14426
67. Racle J, Michaux J, Rockinger GA, Arnaud M, Bobisse S, Chong C, et al. Robust Prediction of HLA Class II Epitopes by Deep Motif Deconvolution of Immunopeptidomes. Nat Biotechnol (2019) 37:1283–6. doi: 10.1038/s41587-019-0289-6
68. Rockinger GA, Guillaume P, Cachot A, Saillard M, Speiser DE, Coukos G, et al. Optimized Combinatorial pMHC Class II Multimer Labeling for Precision Immune Specific CD4 T Monitoring of Tumor-Speciifc CD4 T Cells in Patients. J Immunother Cancer (2020) 8:1–14. doi: 10.1136/jitc-2019-000435
69. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function Throughout Life. Immunity (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
70. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of Full Effector Function In Vitro Paradoxically Impairs the In Vivo Antitumor Efficacy of Adoptively Transferred CD8+ T Cells. J Clin Invest (2005) 115:1616–26. doi: 10.1172/JCI24480
71. Mclellan AD, Ali SM, Rad H. Chimeric Antigen Receptor T Cell Persistence and Memory Cell Formation. Immunol Cell Biol (2019) 97:664–74. doi: 10.1111/imcb.12254
72. Krishna S, Lowery FJ, Copeland AR, Bahadiroglu E, Mukherjee R, Jia L, et al. Stem-Like CD8 T Cells Mediate Response of Adoptive Cell Immunotherapy Against Human Cancer. Science (2020) 370:1328–34. doi: 10.1126/science.abb9847
73. Busch DH, Fräßle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of Memory T Cell Subsets for Adoptive Immunotherapy. Semin Immunol (2016) 28:28–34. doi: 10.1016/j.smim.2016.02.001
74. Gattinoni L, Klebanoff CA, Restifo NP. Paths to Stemness: Building the Ultimate Antitumour T Cell. Nat Rev Cancer (2012) 12:671–84. doi: 10.1038/nrc3322
75. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive Transfer of Effector CD8+ T Cells Derived From Central Memory Cells Establishes Persistent T Cell Memory in Primates. J Clin Invest (2008) 118:294–305. doi: 10.1172/JCI32103
76. Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial Transfer of Single-Cell-Derived Immunocompetence Reveals Stemness of CD8(+) Central Memory T Cells. Immunity (2014) 41:116–26. doi: 10.1016/j.immuni.2014.05.018
77. Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human Effector CD8+ T Cells Derived From Naive Rather Than Memory Subsets Possess Superior Traits for Adoptive Immunotherapy. Blood (2011) 117:808–14. doi: 10.1182/blood-2010-05-286286
78. D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor Activity Associated With Prolonged Persistence of Adoptively Transferred NY-ESO-1 (C259)T Cells in Synovial Sarcoma. Cancer Discov (2018) 8:944–57. doi: 10.1158/2159-8290.CD-17-1417
79. Zhou J, Jin L, Wang F, Zhang Y, Liu B, Zhao T. Chimeric Antigen Receptor T (CAR-T) Cells Expanded With IL-7/IL-15 Mediate Superior Antitumor Effects. Protein Cell (2019) 10:764–9. doi: 10.1007/s13238-019-0643-y
80. Kaneko S, Mastaglio S, Bondanza A, Ponzoni M, Sanvito F, Aldrighetti L, et al. IL-7 and IL-15 Allow the Generation of Suicide Gene Modified Alloreactive Self-Renewing Central Memory Human T Lymphocytes. Blood (2009) 113:1006–15. doi: 10.1182/blood-2008-05-156059
81. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely Related T-Memory Stem Cells Correlate With In Vivo Expansion of CAR. CD19-T Cells and Are Preserved by IL-7 and IL-15. Gene Ther (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174.The
82. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular Remission of Infant B-ALL After Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci Transl Med (2017) 2013:1–9. doi: 10.1126/scitranslmed.aaj2013
83. Bunse M, Bendle GM, Linnemann C, Bies L, Schulz S, Schumacher TN, et al. RNAi-Mediated TCR Knockdown Prevents Autoimmunity in Mice Caused by Mixed TCR Dimers Following TCR Gene Transfer. Mol Ther (2014) 22:1983–91. doi: 10.1038/mt.2014.142
84. Mensali N, Dillard P, Hebeisen M, Lorenz S, Theodossiou T, Renée M, et al. NK Cells Specifically TCR-Dressed to Kill Cancer Cells. EBioMedicine (2019) 40:106–17. doi: 10.1016/j.ebiom.2019.01.031
85. Parlar A, Canan Sayitoglu E, Ozkazank D, Georgoudaki A-M, Pamukcu CP, Aras M, et al. Engineering Antigen-Specific NK Cell Lines Against the Melanoma-Associated Antigen Tyrosinase via TCR Gene Transfer. Eur J Immunol (2019) 49:1278–90. doi: 10.1002/eji.201948140
86. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
87. Van Der Veken LT, Hagedoorn RS, Van Loenen MM, Willemze R, Falkenburg JHF, Heemskerk MHM. Alphabeta T-Cell Receptor Engineered; Gamma Delta T-Cells Mediate Effective Antileukemic Reactivity. Cancer Res (2006) 66:3331–8. doi: 10.1158/0008-5472.CAN-05-4190
88. Nishimura T, Kaneko S, Kawana-tachikawa A, Tajima Y, Goto H, Zhu D, et al. Short Article Generation of Rejuvenated Antigen-Specific T Cells by Reprogramming to Pluripotency and Redifferentiation. Stem Cell (2011) 12:114–26. doi: 10.1016/j.stem.2012.11.002
89. Minagawa A, Yoshikawa T, Yasukawa M, Hotta A, Kunitomo M, Iriguchi S. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Short Article Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell (2018) 23:850–58.e4. doi: 10.1016/j.stem.2018.10.005
90. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science (2016) 351:1463–9. doi: 10.1126/science.aaf1490
91. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
92. Wu D, Liu Y, Li X, Liu Y, Yang Q, Liu Y, et al. Identification of Clonal Neoantigens Derived From Driver Mutations in an EGFR-Mutated Lung Cancer Patient Benefitting From Anti-PD-1. Front Immunol (2020) 11:1366. doi: 10.3389/fimmu.2020.01366
93. Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and Putative Neoantigen Load Predict Clinical Benefit of Adoptive T Cell Therapy in Melanoma. Nat Commun (2017) 8:1–10. doi: 10.1038/s41467-017-01460-0
94. Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, et al. Durable Complete Response From Metastatic Melanoma After Transfer of Autologous T Cells Recognizing 10 Mutated Tumor Antigens. Cancer Immunol Res (2016) 4:669–79. doi: 10.1158/2326-6066.CIR-15-0215
95. Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, et al. Unique Neoantigens Arise From Somatic Mutations in Patients With Gastrointestinal Cancers. Cancer Discov (2019) 9:1022–35. doi: 10.1158/2159-8290.cd-18-1494
96. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C, et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature (2019) 565:240–5. doi: 10.1038/s41586-018-0810-y
97. Linette GP, Carreno BM. Neoantigen Vaccines Pass the Immunogenicity Test. Trends Mol Med (2017) 23:869–71. doi: 10.1016/j.molmed.2017.08.007
98. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An Immunogenic Personal Neoantigen Vaccine for Patients With Melanoma. Nature (2017) 547:217–21. doi: 10.1038/nature22991
99. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y, Zheng Z, et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat Med (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
100. Arnaud M, Duchamp M, Bobisse S, Renaud P, Coukos G, Harari A. Biotechnologies to Tackle the Challenge of Neoantigen Identification. Curr Opin Biotechnol (2020) 65:52–9. doi: 10.1016/j.copbio.2019.12.014
101. Lo W, Parkhurst M, Robbins PF, Tran E, Lu YC, Jia L, et al. Immunologic Recognition of a Shared P53 Mutated Neoantigen in a Patient With Metastatic Colorectal Cancer. Cancer Immunol Res (2019) 7:534–43. doi: 10.1158/2326-6066.CIR-18-0686
102. Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced Detection of Neoantigen-Reactive T Cells Targeting Unique and Shared Oncogenes for Personalized Cancer Immunotherapy. JCI Insight (2018) 3:1–16. doi: 10.1172/JCI.INSIGHT.122467
103. Seliktar-Ofir S, Merhavi-Shoham E, Itzhaki O, Yunger S, Markel G, Schachter J, et al. Selection of Shared and Neoantigen-Reactive T Cells for Adoptive Cell Therapy Based on CD137 Separation. Front Immunol (2017) 8:1211. doi: 10.3389/fimmu.2017.01211
104. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, De Miranda NF, et al. Co-Expression of CD39 and CD103 Identifies Tumor-Reactive CD8 T Cells in Human Solid Tumors. Nat Commun (2018) 9:2724. doi: 10.1038/s41467-018-05072-0
105. Parkhurst M, Gros A, Pasetto A, Prickett T, Crystal JS, Robbins P, et al. Isolation of T-Cell Receptors Specifically Reactive With Mutated Tumor-Associated Antigens From Tumor-Infiltrating Lymphocytes Based on CD137 Expression. Clin Cancer Res (2017) 23:2491–505. doi: 10.1158/1078-0432.CCR-16-2680
106. Bianchi V, Harari A, Coukos G. Neoantigen-Specific Adoptive Cell Therapies for Cancer: Making T-Cell Products More Personal. Front Immunol (2020) 11:1215. doi: 10.3389/fimmu.2020.01215
107. Cafri G, Yossef R, Pasetto A, Deniger DC, Lu Y-C, Parkhurst M, et al. Memory T Cells Targeting Oncogenic Mutations Detected in Peripheral Blood of Epithelial Cancer Patients. Nat Commun (2019) 10:449. doi: 10.1038/s41467-019-08304-z
108. Ali M, Giannakopoulou E, Böschen M, Strønen E, Yang W, Toebes M, et al. Induction of Neoantigen-Reactive T Cells From Healthy Donors. Nat Protoc (2019) 14:1926–43. doi: 10.1038/s41596-019-0170-6
109. Gros A, Tran E, Parkhurst MR, Ilyas S, Pasetto A, Groh EM, et al. Recognition of Human Gastrointestinal Cancer Neoantigens by Circulating PD-1+ Lymphocytes. J Clin Invest (2019) 129:1–13. doi: 10.1172/JCI127967
110. Yang W, Lee K, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic Neoantigens Derived From Gene Fusions Stimulate T Cell Responses. Nat Med (2019) 25:767–75. doi: 10.1038/s41591-019-0434-2
111. Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, et al. Large-Scale Detection of Antigen-Specific T Cells Using Peptide-MHC-I Multimers Labeled With DNA Barcodes. Nat Biotechnol (2016) 34:1037–45. doi: 10.1038/nbt.3662
112. Bentzen AK, Hadrup SR. Evolution of MHC-Based Technologies Used for Detection of Antigen-Responsive T Cells. Cancer Immunol Immunother (2017) 66:657–66. doi: 10.1007/s00262-017-1971-5
113. Krangel MS. Mechanics of T Cell Receptor Gene Rearrangement. Curr Opin Immunol (2010) 21:133–9. doi: 10.1016/j.coi.2009.03.009.Mechanics
114. Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, Franke A. Overview of Methodologies for T-Cell Receptor Repertoire Analysis. BMC Biotechnol (2017) 17:61. doi: 10.1186/s12896-017-0379-9
115. Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive Assessment of T-Cell Receptor Beta-Chain Diversity in Alpha/Beta T Cells. Immunobiology (2009) 114:4099–107. doi: 10.1182/blood-2009-04-217604
116. Polz MF, Cavanaugh CM. Bias in Template-to-Product Ratios in Multitemplate PCR. Appl Environ Microbiol (1998) 64:3724–30. doi: 10.1128/AEM.64.10.3724-3730.1998
117. Freeman D, Warren RL, Webb JR, Nelson BH, Freeman JD, Holt RA. Profiling the T-Cell Receptor Beta-Chain Repertoire by Massively Parallel Sequencing. Genome Res (2009) 19:1817–24. doi: 10.1101/gr.092924.109.19
118. Ruggiero E, Nicolay JP, Fronza R, Arens A, Paruzynski A, Nowrouzi A, et al. High-Resolution Analysis of the Human T-Cell Receptor Repertoire. Nat Commun (2015) 6:1–7. doi: 10.1038/ncomms9081
119. Linnemann C, Heemskerk B, Kvistborg P, Kluin RJC, Bolotin DA, Chen X, et al. High-Throughput Identification of Antigen-Specific TCRs by TCR Gene Capture. Nat Med (2013) 19:1534–41. doi: 10.1038/nm.3359
120. Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, et al. High-Throughput Pairing of T Cell Receptor Alpha and Beta Sequences. Sci Transl Med (2015) 7:301ra131. doi: 10.1126/scitranslmed.aac5624
121. Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, et al. Tumor- and Neoantigen-Reactive T-Cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res (2016) 4:734–43. doi: 10.1158/2326-6066.CIR-16-0001
122. Sarkar S, Motwani V, Sabhachandani P, Cohen N, Konry T. T Cell Dynamic Activation and Functional Analysis in Nanoliter Droplet Microarray Saheli. J Clin Cell Immunol (2015) 176:139–48. doi: 10.1016/j.physbeh.2017.03.040
123. Poran A, Scherer J, Bushway ME, Besada R, Balogh KN, Wanamaker A, et al. Combined TCR Repertoire Profiles and Blood Cell Phenotypes Predict Melanoma Patient Response to Personalized Neoantigen Therapy Plus Anti-PD-1. Cell Rep Med (2020) 1:100141. doi: 10.1016/j.xcrm.2020.100141
124. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of Neoantigen-Specific T Cells From Tumor and Peripheral Lymphocytes. J Clin Invest (2015) 125:3981–91. doi: 10.1172/JCI82416
125. Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, De RMAJ, et al. Low and Variable Tumor Reactivity of the Intratumoral TCR Repertoire in Human Cancers. Nat Med (2019) 25:89–94. doi: 10.1038/s41591-018-0266-5
126. Paria BC, Levin N, Lowery FJ, Pasetto A, Deniger DC, Parkhurst MR, et al. Rapid Identification and Evaluation of Neoantigen-Reactive T-Cell Receptors From Single Cells. J Immunother (2020) 44:1–8. doi: 10.1097/CJI.0000000000000342
127. Gejman RS, Jones HF, Klatt MG, Chang AY, Oh CY, Chandran SS, et al. Identification of the Targets of T-Cell Receptor Therapeutic Agents and Cells by Use of a High-Throughput Genetic Platform. Cancer Immunol Res (2020) 8:1–13. doi: 10.1158/2326-6066.CIR-19-0745
128. Li G, Bethune MT, Wong S, Joglekar AV, Leonard MT, Wang JK, et al. T Cell Antigen Discovery via Trogocytosis. Nat Methods (2019) 16:183–90. doi: 10.1038/s41592-018-0305-7
129. Joglekar AV, Leonard MT, Jeppson JD, Swift M, Li G, Wong S, et al. T Cell Antigen Discovery via Signaling and Antigen-Presenting Bifunctional Receptors. Nat Methods (2019) 16:191–8. doi: 10.1038/s41592-018-0304-8
130. Segaliny AI, Li G, Kong L, Ren C, Chen X, Wang JK, et al. Functional TCR T Cell Screening Using Single-Cell Droplet Microfluidics. Lab Chip (2018) 18:3733–49. doi: 10.1039/c8lc00818c
131. Peng S, Zaretsky JM, Ng AHC, Baltimore D, Ribas A, Heath JR, et al. Sensitive Detection and Analysis of Neoantigen- Specific T Cell Populations From Tumors and Blood. Cell (2019) 28:2728–38. doi: 10.1016/j.celrep.2019.07.106
132. Ng AHC, Peng S, Xu AM, Noh WJ, Guo K, Bethune MT, et al. MATE-Seq: Microfluidic Antigen-TCR Engagement Sequencing. Lab Chip (2019) 19:3011–21. doi: 10.1039/c9lc00538b
133. Friedensohn S, Khan TA, Reddy ST. Advanced Methodologies in High-Throughput Sequencing of Immune Repertoires. Trends Biotechnol (2016) 35:203–14. doi: 10.1016/j.tibtech.2016.09.010
134. van der Leun AM, Thommen DS, Schumacher TN. CD8+ T Cell States in Human Cancer: Insights From Single-Cell Analysis. Nat Rev Cancer (2020) 20:218–32. doi: 10.1038/s41568-019-0235-4
135. Kim S, Bhonsle L, Besgen P, Nickel J, Backes A, Held K, et al. Analysis of the Paired TCR a - and B -Chains of Single Human T Cells. PloS One (2012) 7:e37338. doi: 10.1371/journal.pone.0037338
136. Han A, Glanville J, Hansmann L, Davis MM. Linking T-Cell Receptor Sequence to Functional Phenotype at the Single-Cell Level. Nat Biotechnol (2015) 32:684–92. doi: 10.1038/nbt.2938
137. Azizi E, Carr AJ, Plitas G, Mazutis L, Rudensky AY, Pe D, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment Resource Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell (2018) 174:1293–308.e36. doi: 10.1016/j.cell.2018.05.060
138. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal Replacement of Tumor-Specific T Cells Following PD-1 Blockade. Nat Med (2019) 25:1251–9. doi: 10.1038/s41591-019-0522-3
139. Sheih A, Voillet V, Hanafi LA, DeBerg HA, Yajima M, Hawkins R, et al. Clonal Kinetics and Single-Cell Transcriptional Profiling of CAR-T Cells in Patients Undergoing CD19 CAR-T Immunotherapy. Nat Commun (2020) 11:1–13. doi: 10.1038/s41467-019-13880-1
140. Lu YC, Jia L, Zheng Z, Tran E, Robbins PF, Rosenberg SA. Single-Cell Transcriptome Analysis Reveals Gene Signatures Associated With T-Cell Persistence Following Adoptive Cell Therapy. Cancer Immunol Res (2019) 7:1824–36. doi: 10.1158/2326-6066.CIR-19-0299
141. Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, et al. T-Cell Responses to TP53 Hotspot Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin Cancer Res (2018) 24:5562–73. doi: 10.1158/1078-0432.CCR-18-0573
Keywords: cancer immunotherapy, T cell receptor, gene transfer therapy, adoptive cell transfer, cell engineering, T cells
Citation: Arnaud M, Bobisse S, Chiffelle J and Harari A (2021) The Promise of Personalized TCR-Based Cellular Immunotherapy for Cancer Patients. Front. Immunol. 12:701636. doi: 10.3389/fimmu.2021.701636
Received: 28 April 2021; Accepted: 01 July 2021;
Published: 30 July 2021.
Edited by:
Min Cheng, Weifang Medical University, ChinaReviewed by:
Esther Giehl, University Hospital Carl Gustav Carus, GermanyCopyright © 2021 Arnaud, Bobisse, Chiffelle and Harari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marion Arnaud, bWFyaW9uLmFybmF1ZEB1bmlsLmNo; Alexandre Harari, YWxleGFuZHJlLmhhcmFyaUBjaHV2LmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.