
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 July 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.697588
 Sajjan Rajpoot1
Sajjan Rajpoot1 Kishore K. Wary2
Kishore K. Wary2 Rachel Ibbott3
Rachel Ibbott3 Dongfang Liu4,5,6
Dongfang Liu4,5,6 Uzma Saqib7
Uzma Saqib7 Teresa L. M. Thurston3*
Teresa L. M. Thurston3* Mirza S. Baig1*
Mirza S. Baig1*The Toll-interleukin-1 Receptor (TIR) domain-containing adaptor protein (TIRAP) represents a key intracellular signalling molecule regulating diverse immune responses. Its capacity to function as an adaptor molecule has been widely investigated in relation to Toll-like Receptor (TLR)-mediated innate immune signalling. Since the discovery of TIRAP in 2001, initial studies were mainly focused on its role as an adaptor protein that couples Myeloid differentiation factor 88 (MyD88) with TLRs, to activate MyD88-dependent TLRs signalling. Subsequent studies delineated TIRAP’s role as a transducer of signalling events through its interaction with non-TLR signalling mediators. Indeed, the ability of TIRAP to interact with an array of intracellular signalling mediators suggests its central role in various immune responses. Therefore, continued studies that elucidate the molecular basis of various TIRAP-protein interactions and how they affect the signalling magnitude, should provide key information on the inflammatory disease mechanisms. This review summarizes the TIRAP recruitment to activated receptors and discusses the mechanism of interactions in relation to the signalling that precede acute and chronic inflammatory diseases. Furthermore, we highlighted the significance of TIRAP-TIR domain containing binding sites for several intracellular inflammatory signalling molecules. Collectively, we discuss the importance of the TIR domain in TIRAP as a key interface involved in protein interactions which could hence serve as a therapeutic target to dampen the extent of acute and chronic inflammatory conditions.
TIRAP, also known as MyD88-adaptor Like (MAL), is an adapter molecule associated with receptor-mediated activation of host immune signaling (1, 2). The innate immune system recognizes microbial pathogens through receptors, including Toll-like receptors (TLRs), which identify pathogen-associated molecular patterns (PAMP) (3, 4). Upon ligation of most TLRs with their respective ligands (PAMP), MyD88 is recruited directly to the cell surface receptor intracellular domain. In the case of TLR2 and TLR4, Myd88 recruitment is indirect and occurs via TIRAP. This event nucleates the formation of a large signaling complex called the “Myddosome” (5–7). The resulting downstream signaling events culminate in the activation of several transcription factors that include nuclear factor κB (NF-κB) and activated protein 1 (AP1) (6, 8–10). The activation of TLR-mediated signaling pathways is critical in driving the induction of proinflammatory cytokines by immune cells and controlling host cell survival. In this way, the cell is reprogrammed to a state that helps mitigate infection. Regulated TLR activation is required for host defense activities, yet unwanted amplification of proinflammatory cytokines is detrimental to the host. Furthermore, inappropriate receptor activation can propagate the development of autoimmune diseases in individuals harboring genetic risk factors. Therefore, it is critical to understand the TLR- and TIRAP-mediated signaling mechanisms and how these pathways are regulated.
TIRAP consists of 221 amino acids, constituting two main domains (Figure 1); a phosphatidylinositol 4,5-bisphosphate (PIP2) binding domain (PBD), which is responsible for targeting TIRAP to discrete regions of the plasma membrane upon Phosphatidylinositol 4-Phosphate 5-Kinase (PIP5Kα)-mediated production of PIP2 (11, 12) and a TIR domain, which is mainly involved in protein-protein interactions with numerous inflammatory-related proteins (6, 13, 14).
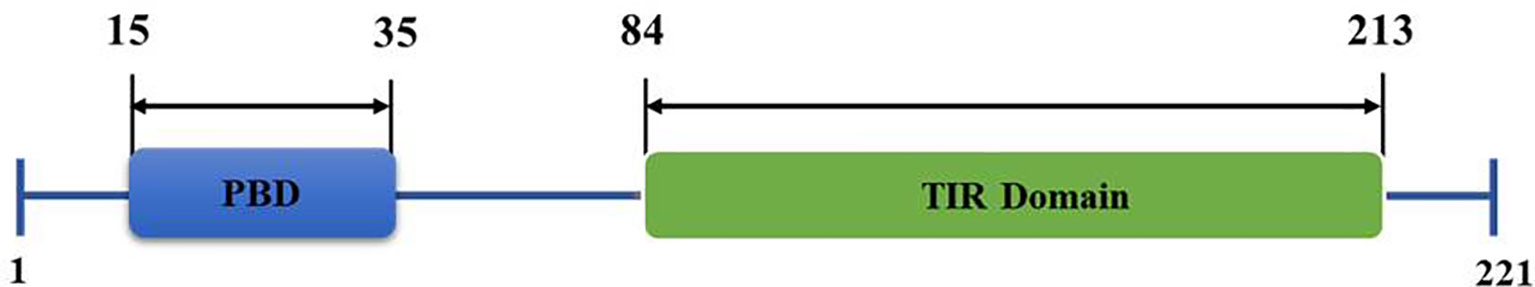
Figure 1 Structural organization of Toll/interleukin 1 receptor (TIR) domain-containing adaptor protein (TIRAP) domains. The amino acid position of an N-terminal phosphatidylinositol (PI) binding domain (PBD) and a C-terminal Toll-like receptor (TIR) domain are as shown.
This review discusses the diverse TIRAP protein interacting partners (summarized in Table 1) and the significance of these during inflammatory signaling pathways and the onset of inflammatory diseases. First, we summarized the mechanism of recruitment of TIRAP; thereafter, we discuss the downstream interactions that mediate inflammatory signaling. In that respect, the Figure 2 is mainly summarized to provide the insight of inflammatory signaling pathways mediated via multiple TIRAP interactions eventually leading to the activation of major transcription factors NF-κB and AP-1 and hence expression of proinflammatory cytokines (2, 9, 16, 20, 26, 29, 30, 32, 40, 41). On the other hand, the negative regulators of TIRAP interacting proteins are discussed and represented for their known roles (18, 22, 23, 35, 42).
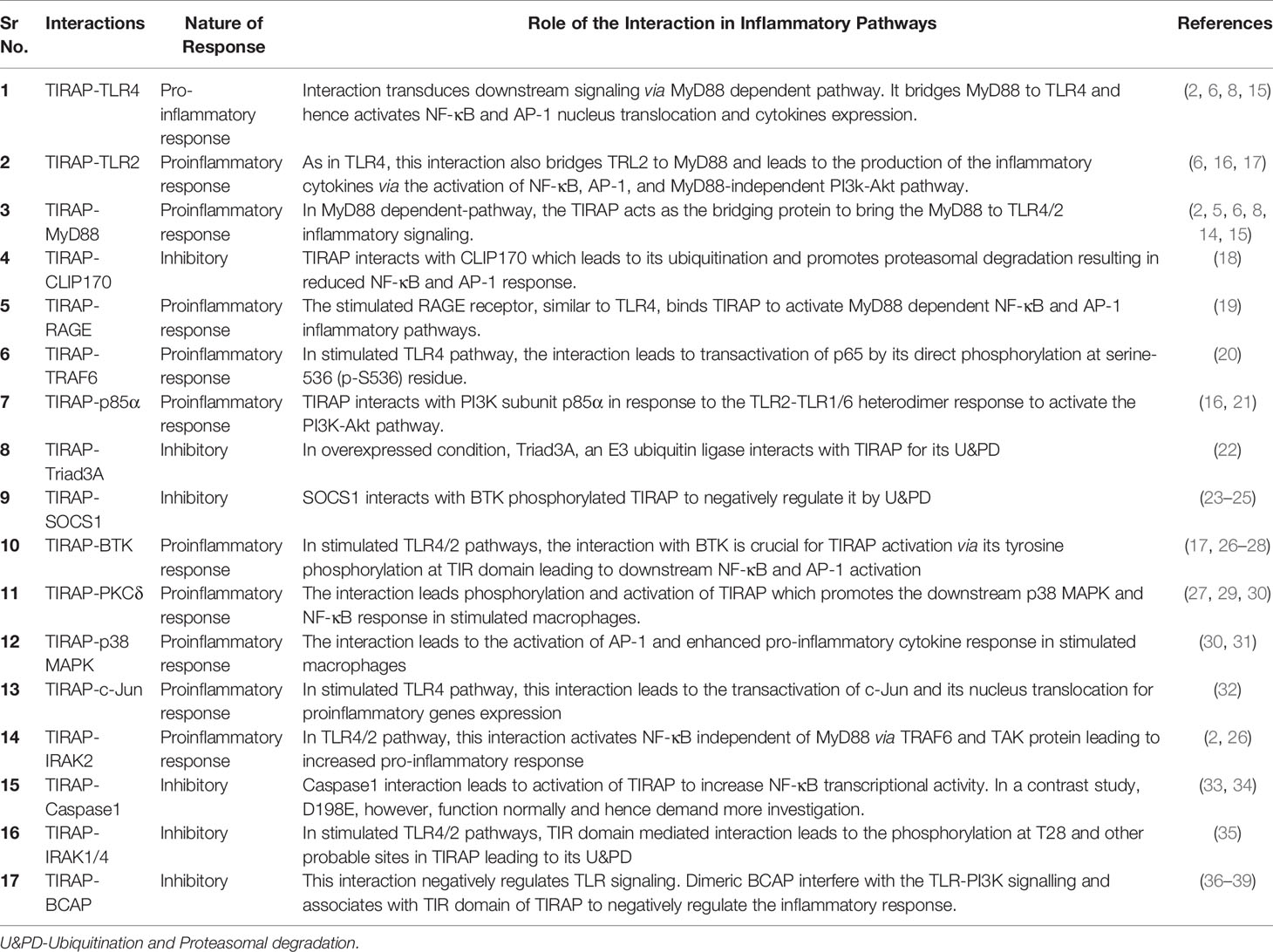
Table 1 The significance of TIRAP interaction with the partner proteins in the cellular process.
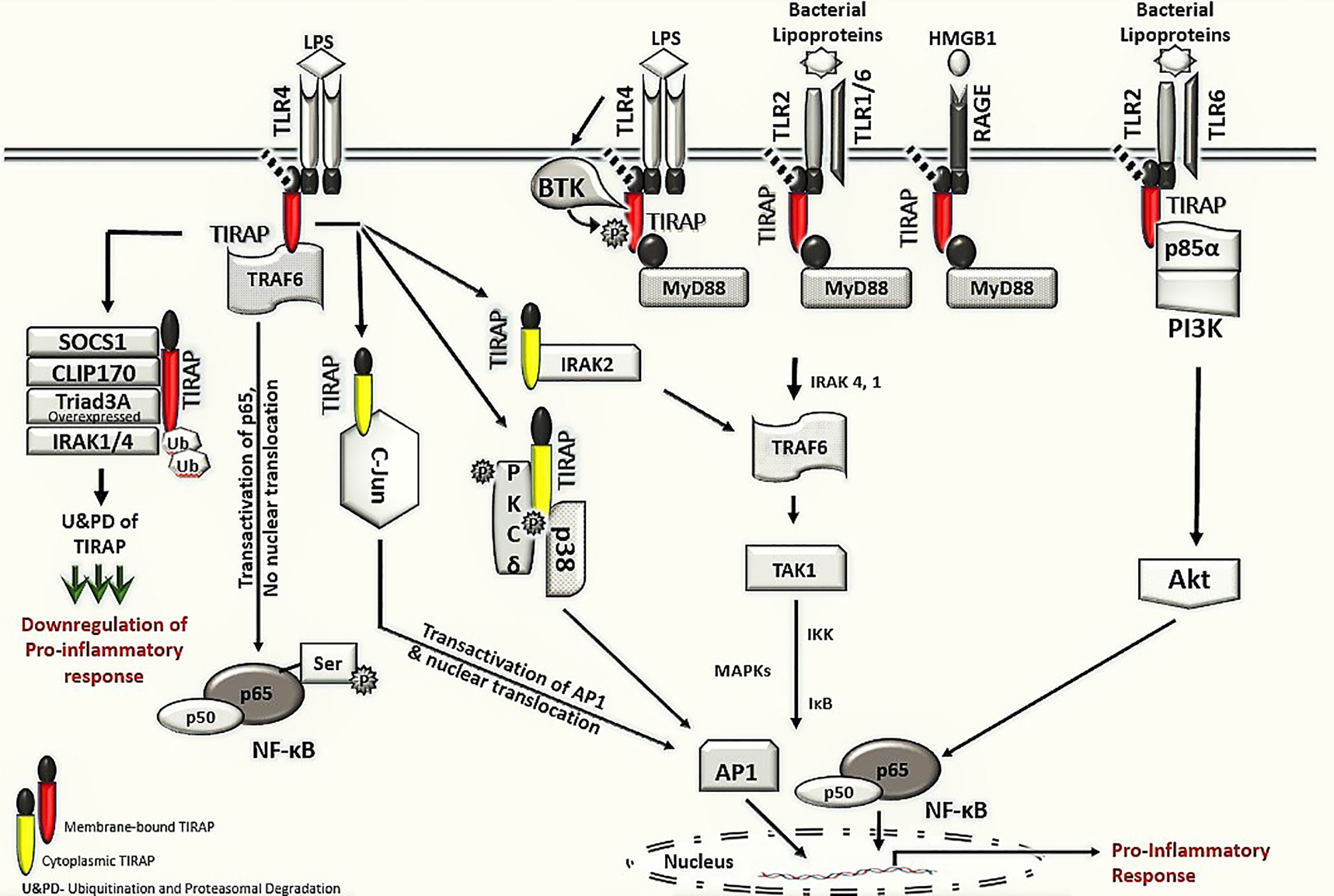
Figure 2 TIRAP interacting machinery in the activation of inflammatory signaling. The membrane localized TIRAP initiates the downstream signaling via its interaction with upstream membrane bound receptors TLR4, TLR2 and RAGE as well as with membrane localized kinases BTK, p85α subunit of PI3K and BCAP. In TLR4 mediated signaling, the membrane localized TIRAP also interacts with TRAF6 for transactivation of the p65 in NF-κB pathway. The downstream inflammatory signaling via cytoplasmic TIRAP involves an interaction with protein kinase PKCδ and p38 MAPK as well as with IRAK2 and AP-1 subunit c-Jun, respectively. The negative regulators of TIRAP protein including SOCS1, CLIP170, IRAK1/4 and Triad3A are involved in interactions with TIRAP for its ubiquitination and proteasomal degradation (U&PD) and hence downregulation in inflammatory response mediated via TIRAP. TLR- Toll -interleukin-1 receptor; TIRAP- Toll-interleukin-1 Receptor (TIR) domain-containing adaptor protein; MyD88- Myeloid differentiation factor 88; RAGE- Receptor for advanced glycation end-products; BTK- Bruton’s tyrosine kinase; PI3K- phosphoinositide 3-kinase; BCAP- B-cell adaptor for phosphoinositide 3-kinase; TRAF6, Tumor necrosis factor receptor (TNFR)-associated factor 6; NF-κB, Nuclear Factor kappa-light-chain-enhancer of activated B cells; PKCδ, protein kinase c delta; p38 MAPK, p38 mitogen-activated protein kinase; AP-1, Activator protein 1; SOCS1, Suppressor of cytokine signaling 1; CLIP170, Cytoplasmic linker protein 170; IRAK, interleukin-1 receptor-associated kinase; IKK, IκB kinase; and IκB, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor.
For TIRAP to carry out its function as a TLR adaptor protein in inflammatory signaling, it must be recruited to the membranes’ activated receptors. This occurs upon binding to PIP2 before its interaction with TLRs (12). Subsequent interaction with TLRs’ TIR domain is facilitated by tyrosine phosphorylation of selected TLRs and their adaptor molecules by several tyrosine kinases, including Bruton’s tyrosine kinase (BTK), Src, Lyn, Syk, etc (43, 44). For example, phosphorylation of the cytoplasmic tail of the TLR4 TIR domain at Y674 and Y680 and phosphorylation of TIRAP tyrosine residues at positions 86, 106, 159, and 187 by BTK are required for TLR4-TIRAP-MyD88 interaction and activation of NF-κB and MAPK (mitogen-activated protein kinase) signaling leading to proinflammatory responses (Figure 2) (17, 26, 45, 46). BTK interaction with TIRAP and its tyrosine phosphorylation in the TIR domain is crucial for TIRAP to function. Six conserved tyrosine (Y) residues in the TIR domain of human TIRAP are the potential phosphor-acceptor site. The four residues Y86, Y106, Y159 and Y187 in TIRAP are experimentally identified as the sites for BTK mediated phosphorylation. Meanwhile, the mutation of these tyrosine residues either with alanine or phenylalanine describes them as the crucial sites for BTK mediated phosphorylation, as after stimulation, the mutation impaired the BTK association with TIRAP, similar to the previously reported P125H variant of TIRAP. Also, these mutations play a dominant negative role and impair NF-κB activation (13, 17, 26). Several studies hence concluded the significance of tyrosine sites on TIRAP in the TIR domain for association with BTK immediately on LPS stimulation. However, it is important to have a detailed understanding of the mechanism of TIRAP-BTK interaction and the role of tyrosine phosphorylation regulating the downstream signaling. The prerequisite step in the TIRAP phosphorylation by BTK is its activation itself. In stimulated macrophages, src kinase is reported to activate BTK as well as PKCδ (protein kinase C delta) (27–30, 47, 48). Structural analysis of BTK and PKCδ interaction with TIRAP suggests that Y106 of TIRAP is phosphorylated by the action of both BTK and PKCδ (27). Both phosphorylation sites promote the activation of downstream p38-MAPK, NF-κB pathways, and the associated cytokine response, but due to ubiquitin-dependent degradation of phosphorylated TIRAP mediated by SOCS1 (suppressor of cytokine signaling 1), TIRAP tyrosine phosphorylation by BTK is transient (23). With the critical molecules poised to become activated at the membrane, BTK-mediated TIRAP phosphorylation and activation, followed by degradation of phospho-TIRAP, results in a rapid yet balanced inflammatory response, avoiding prolonged TLR4 or TLR2 signaling that would otherwise result in chronic inflammation and associated diseases (23). A recently concluded study on BCR-TLR interplay suggest that the high expression of BTK in TLR signaling leads to the development of pathology in a Btk-dependent model for systemic autoimmune disease (49).
The Receptor for advanced glycation end-products (RAGE) is a multi-ligand cell membrane receptor implicated in diverse chronic inflammatory states such as cardiovascular disease, cancer, neurodegeneration, and diabetes (50–52). RAGE is activated by diverse damage-associated molecular pattern molecules (DAMPs), which include advanced glycation end products (AGEs), high mobility group box-1 (HMGB1), and S100 proteins (51–53). Upon ligand binding, Protein Kinase C-ζ (PKC-ζ) phosphorylates the cytosolic domain of RAGE on Ser391, mediating interaction with the TIR domain TIRAP (19). As with TLR signaling, TIRAP acts as a bridge to MyD88. In this way, RAGE activation induces MyD88-dependent proinflammatory signaling (19) (Figure 2). The soluble RAGE is generated after either the proteolytic cleavage of its transmembrane domain to generate sRAGE or alternative splicing to give endogenous soluble RAGE (esRAGE). These proteins have distinct roles in inflammation and disease (compared to RAGE), with neither capable of inducing signaling upon binding RAGE targets. Instead, sRAGE blocks RAGE signaling effectively and appear to prevent or reduce inflammatory conditions (50, 51, 54). As mutations in the TIR domain of TIRAP result in inhibition of downstream inflammatory signaling and the role of RAGE in inducing a proinflammatory immune response during disease (19). The RAGE signalling plays a vital role in many inflammatory associated diseases (acute lung injury, sepsis, inflammatory bowl disease, atherosclerosis, cancer and other chronic infectious and noninfectious diseases). Many ligands (e.g., HMGB1, s100, etc) activate both RAGE and TLR4 leading to same inflammatory pathway via TIRAP interaction, a direct cross talk between both signalling has also been highlighted (55, 56). It should now be explored in detail whether the therapeutic intervention of TIRAP and RAGE represents a means to treat these diseased states. An earlier study on neuronal cells has been reported to disrupt this interaction through decoy RAGE peptide (RAGE-I) targeting TIRAP and abrogates the activation of cdc42, inhibiting cell migration and invasion and protecting cell death (57).
The interaction of p85α, a regulatory subunit of phosphoinositide 3-kinase (PI3K) with TIRAP, is a MyD88-independent response of the TLR2 receptor upon stimulation with bacterial lipoproteins (16, 21). This interaction is highly significant in TLR2 and TLR6 heterodimer signaling, resulting in the activation of PI3K-dependent phosphorylation of Akt, PIP3 [phosphatidylinositol (3,4,5)P3] generation, and polar shape changes of the macrophage (16). Signaling is initiated when TLR2 heterodimers with TLR6 are bound by diacylated lipoprotein ligands, or when TLR1/2 heterodimers are bound by triacylated lipoproteins (58). Ligand binding induces conformational changes within the receptor that bring their TIR domains close to downstream signal transduction (1, 59, 60). Upon diacylated lipoprotein stimulation of TLR2/6, TIRAP, but not MyD88, is essential for PI3K activity and NF-κB activation (16). Upon stimulation of macrophages, TIRAP interacts with p85α, and these proteins colocalize at the plasma membrane (16). Whereas MyD88 is not required for interaction of TIRAP with p85α, the efficiency of PI3K activity, and therefore downstream activation of Akt and NF-κB, as well as macrophage polarization, becomes delayed in MyD88 deficient cells (16). This suggests that the TIRAP interaction with p85α is direct but that MyD88 might accelerate the kinetics of Akt phosphorylation and PIP3 generation.
MALP-2 induced activation of the TLR2/6 pathway in THP-1 cells also induces the interaction of TIRAP with p85α, and this is essential for the induction of Heme Oxygenase-1 (HO-1) via Akt phosphorylation and Nrf2 activation (21). Similar to the above study, this study also reported that MyD88 deficiency resulted in decreased Akt phosphorylation but at earlier time points post-activation (60 minutes). HO-1 expression upon MALP-2 stimulation also involves c-Src and BTK, which are other binding partners of TIRAP. These proteins are likely to form a complex consisting of c-Src, BTK, TIRAP, and p85α upon stimulation, and together, they represent a potentially important target for pharmacotherapy during various chronic inflammatory diseases.
In contrast, a multimodular protein, B-cell adaptor for phosphoinositide 3-kinase (PI3K) (BCAP) has been reported to negatively regulate the TLR4/2-PI3K signalling, suggesting its association with TLR and downstream TIR domain of TIRAP, leading to its recruitment to TLR signalasome by TIR-TIR interactions (36–38). BCAP also interacts with p85α suggesting its role in regulating the downstream signalling (36). BCAP is a dimeric protein and its oligomerization depends on its ANK (ankyrin repeat) and DBB (Dof/BANK1/BCAP) domains. A recently concluded study clearly defines the importance of DBB domain in dimerization and its role in TIRAP-BCAP interaction (39). The monomeric BCAP, though fails to negatively regulate the TLR signaling suggesting that only domain dimerization drives the negative response. The TIRAP TIR domain is reported to assemble to form filament complex in vitro, an event critical for signal transduction. Such a filament could be disrupted by dimeric, and not monomeric, BCAP. Another angle suggest BCAP phosphoinositide metabolism, which cleaves PIP2 to DAG and IP3 and hence deprives TIRAP for its membrane anchor required for TLR signaling (39). Overall, the BCAP association mainly with TIRAP and p85α provides novel directions for regulatory pathways in inflammation.
As mentioned above, PKCδ represents an interacting partner of TIRAP that is required for TLR2- and TLR4-induced activation of p38 MAPK, NF-κB and proinflammatory cytokine expression (29, 30). Mice harboring PKCδ-deficiency reduces the bacterial killing function of macrophages, showing decreased NO, ROS, and H2O2, resulting in hyper-susceptible animals to infection with mycobacterium tuberculosis with increased mortality (61). Mechanistically, PKCδ constitutively interacts with and phosphorylates Y106 in the TIR domain of TIRAP as described above, and BTK phosphorylates TIRAP at Y106, as well as Y86 (27). Accordingly, Baig et al., 2017 reported a novel heterotrimeric complex consisting of TIRAP, PKCδ, and p38 MAPK required for AP1-mediated inflammatory responses in macrophages in response to LPS stimulation (30). Thus, TIRAP activity is regulated in response to receptor stimulation. In this situation, TIRAP was not required for the phosphorylation of PKCδ but was required for cytokine production (30). In summary, PKCδ activation is likely required downstream of TIRAP; however, it remains unclear how PKCδ activation is regulated in macrophages upon LPS stimulation.
Cytoplasmic linker protein 170 (CLIP170), containing two conserved cytoskeleton-associated protein Glycine rich (CAP-Gly) domains and two tandem repeats of zinc knuckle motifs (also called CCHC zinc fingers), is a multifunctional protein that binds to and regulates the dynamics of the growing plus end of microtubules (62–64). In addition, CLIP170 acts as a negative regulator of TLR4 by inducing TIRAP ubiquitination (both mono and polyubiquitination) and promoting its proteasomal degradation. Accordingly, this activity of CLIP170 decreased downstream signaling activity, including reduced NF-κB MAPK activity. Further, the activity of CLIP170 is specific to TIRAP, with MyD88 levels unaltered upon overexpression of CLIP170 (18). Analogous to many negative regulators, CLIP170 expression is induced upon stimulation with LPS. Interestingly, the pathogenic bacteria Brucella species viz. Brucella melitensis, Brucella abortus, and Brucella ovis inhibit TIRAP activity by encoding a TIR domain-containing effector protein TcpB, also called Btp1 (65). TcpB, like TIRAP, shares phosphoinositide binding properties and resemblance in its TIR domain; however, functionally, TcpB impairs TLR-4 and TLR-2 induced NF-κB activation inflammatory responses (18, 65). Mechanistically, TcpB interacts with TIRAP and promotes CLIP170-dependent polyubiquitination and subsequent proteasomal degradation of TIRAP (18). CLIP170 is also of therapeutic interest; Pregnenolone, a steroid hormone precursor, suppresses TLR4 and TLR2 mediated inflammation by promoting CLIP170-mediated ubiquitination and degradation of TIRAP (66).
Delivery of bacterial-derived pathogenic LPS to TLR4 has been shown to activate AP-1 through a series of phosphorylation events on serine/threonine residues mediated by upstream MAPKs (extracellular signal-regulated kinases; ERK, c-Jun N-terminal kinase; JNK, and p38 mitogen-activated protein kinases; p38 kinase) (67). For example, c-Jun, which contains a transactivation domain, is phosphorylated at Ser 63 and 73 by JNKs, resulting in its activation (67). c-Jun can form homo and heterodimers with other AP-1 family members, including c-Fos or ATF (Activating transcription factor) to make transcriptionally active complexes (68–70); (71). These transcriptionally active dimeric components of AP-1 now control the activation of critical proinflammatory cytokine genes such as TNF-α, IL-12, IL-23, and other proinflammatory cytokines (72–75). In recent findings, TIRAP was found to interact with the AP-1 subunit c-Jun in endotoxin-induced macrophages. This drives the transactivation of c-Jun, its translocation to the nucleus, and a proinflammatory immune response (32). Careful analysis of the molecular docking of a TIRAP-c-Jun crystal structure with immunoprecipitation experiments revealed the direct interaction of TIRAP with c-Jun (32). The pharmacological inhibitor Gefitinib, which abrogates the interaction of TIRAP with c-Jun, specifically reveals the importance of TIRAP-mediated c-Jun transactivation; treatment with Gefitinib drastically reduced the expression of several proinflammatory cytokines (IL-12, IL-23, TNF-α) in both bone-marrow-derived macrophages and in animals (32).
As mentioned above, p38 MAPK is one of three members of the MAPK protein family, that in response to LPS, induces the expression of proinflammatory cytokines (IL-12, IL-23, TNF-α, IL-6, and IL-1β) through activation of the downstream transcription factor AP-1 (31). In addition, the MyD88-dependent TLR4/TLR2 induced activation of MAPKs and NF-κB and subsequent proinflammatory response is well characterized (3, 5, 15, 31, 76). Interestingly, this inflammatory response is also regulated through the direct interaction of TIRAP with p38 MAPK and PKCδ in LPS-stimulated macrophages (30) and suggests that TIRAP has multiple functions in the induction of MAPK signaling.
Two reports on the interaction of TIRAP with Caspase-1 have highlighted the importance of this interaction to macrophage signaling events (33, 34). Caspases are evolutionarily conserved enzymes with aspartate-specific, cysteine dependent proteolytic activity, which are involved in inflammation and apoptosis (77, 78). Caspase-1, formerly called IL-1beta converting enzyme (ICE), is synthesized as a zymogen precursor and is cleaved into its p20 (20kDa) and p10 (10kDa) active catalytic subunits and a non-catalytic Caspase Activation and Recruitment Domain (CARD) (79, 80). Active caspase-1, often found in a multi-protein complex called the inflammasome, cleaves the inactive pro-forms of IL-1β and IL-18 to generate active cytokines. This function of caspase-1, as well as activation of gasdermin-D (GSDMD), is critical for the inflammatory immune response of LPS-primed macrophages (80, 81). TIRAP first found to interact with caspase-1 by yeast two-hybrid, is not required for the activation of Caspase-1 (33). Instead, caspase-1 appears to cleave TIRAP (but not MyD88), and this is required for TLR4, and TLR2 mediated NF-κB, p38 MAPK activation, and cytokine production but not IL-1 and TLR7 signaling, which is TIRAP independent (33, 34). After LPS stimulation of macrophages, caspase-1 was reported to cleave after D198 in the C-terminal region of TIRAP. However, the significance of this cleavage was questioned by another study, which found that cleavage was not required for NF-κB activity (34). Instead, it appears that mutation of D198A disrupts TLR4 mediated signaling due to the loss in the acidic amino acid at this site and not due to loss of caspase-1 cleavage. Indeed, TIRAP-D198E remained functional, suggesting that TIRAP is functional in its full-length form (34). Therefore, whether the caspase-1-TIRAP interaction is of biological relevance requires further study.
A novel role for TIRAP in NF-κB p65 trans-activation was described after the identification that TIRAP interacts with TRAF-6 (20). The interaction following activation of TLR2 or TLR4 is direct in nature and independent of membrane localization (Figure 2). The TRAF family of adaptor proteins and E3 ubiquitin ligases comprises of 6 members, viz., TRAF1, TRAF2, TRAF3, TRAF4, TRAF5, and TRAF6, with each member having distinct functions in the regulation of immune signaling (82). A short motif, Pro-X-Glu-X-X-Z (X: any amino acid; Z: aromatic/acidic residue), acts as a TRAF6 binding motif that is found in various receptors and adaptor proteins (82). TIRAP contains a TRAF6 binding domain, with mutation of this motif (TIRAP-E190A), abolishing interaction with TRAF6 and preventing signal transduction (20). Functionally, the interaction of TIRAP with TRAF6 promotes serine-536 phosphorylation of the p65 subunit of NF-κB, which regulates its transcriptional activation, rather than nuclear translocation (20). In contrast, a recent study based on a mathematical data-model suggests that TIRAP-independent MyD88 activation and Myddosome complex formation in TLR4 signaling does not require TRAF6 (83).
Triad3A is a RING finger-type E3 ubiquitin-protein ligase that recognizes and interacts with the TIR domain of TLRs, promoting their proteolytic degradation (22, 42). Specifically, Triad3A causes K48-linked ubiquitination and degradation of TLR4 and TLR9. In addition, Fearns et al. (2006) demonstrated that overexpressed Triad3A directly interacts with and results in degradation of the TLR4 adaptor proteins TIRAP, TRIF, and RIP1 but not other adaptor proteins such as MyD88 and TRAM (22). This suggests that TIRAP is post-translationally regulated via different E3 ligases (CLIP170 and Triad3A). Future work should determine the relative contributions and functions to proinflammatory immune signaling.
The interleukin-1 receptor-associated kinase (IRAK) family protein kinase consists of four members, IRAK-1, IRAK-2, IRAK-3/M, and IRAK-4 (84). The sequential activation and recruitment of IRAKs, excluding IRAK-M, in MyD88 dependent signaling is well studied and reported in TLR signaling (76, 85–87). In the TLR-4-TIRAP-MyD88 mydossome complex, active MyD88 interacts with the N- terminal death domains of IRAK4, triggering a series of phosphorylation events and recruitment of IRAK-1 and IRAK-2, which are required for TRAF-6 ubiquitination, activation, and downstream activation of NF-κB (88, 89). In addition, IRAK-2, but not IRAK-1, directly interacts with TIRAP, and this results in NF-κB activation (2, 26). Further evidence for the critical role of IRAK-2 comes from the observation that a dominant-negative variant blocks TIRAP-induced NF-κB activation. The precise residues that mediate the TIRAP-IRAK-2 interaction remain unknown, but the critical tyrosine residues of TIRAP (Y86, Y106, and Y159) are not required (26). Further research is required to understand the precise physiological role of the IRAK-2-TIRAP signaling axis and test whether it represents a means for developing targeted therapeutics that control inflammation.
Interestingly, the interaction of TIRAP with IRAK-4 and IRAK-1 can result in TIRAP degradation following its phosphorylation and ubiquitination, and therefore inhibition of signaling. In this way, IRAK-1/-4 negatively regulates TLR-4/2 signaling by expressing an auto-active IRAK4 inducing TIRAP degradation (35). The interaction appears to be mediated by the TIR domain of TIRAP, with mutation of proline at position 125 (P125H) in the BB loop abolishing the interaction. Both IRAK-4 and IRAK-1 phosphorylate multiple sites, including T28 on TIRAP, that are required for subsequent ubiquitination and degradation (35). Additional research is required to dissect the additional phosphorylation sites within TIRAP, as well as the ubiquitination sites that control TIRAP degradation. Furthermore, the selective role of IRAK-4 and IRAK-1 both as positive and negative regulators is an important consideration in the investigation of inflammation. It is interesting to speculate that over time IRAK-1 and IRAK-4 switch from a positive role in MyD88 dependent inflammatory signaling to a negative one, helping to dampen signaling and prevent too much inflammation. What controls this switch should be investigated using a dose-dependent endotoxin challenge over time, with careful monitoring of TIRAP complex composition.
The suppressor of cytokine signaling (SOCS1) serves as a key physiological regulator of both innate and adaptive immunity acting through macrophages, dendritic cells, B-cells, and T-cells (90). Structurally, the eight intracellular members of the SOCS family, SOCS1-SOCS7 and CIS, have a central SH2 domain, N-terminal extended SH2 subdomain and a variable region, and C-terminal 40 amino acid SOCS box. The SOCS box recruits factors for E3 ubiquitin ligase mediated target protein ubiquitination followed by proteasomal degradation (90, 91). Previous studies described SOCS1 as a negative intracellular regulator in the cytokine-induced JAK-STAT pathway; however, subsequent studies have addressed the role of SOCS1 that is associated with TLR4 and TLR2 signaling as an E3 ligase that polyubiquitinates TIRAP along with other proteins such as p65 and IRAK1, causing their 26S proteasomal degradation (23–25). Analogous to PEST (Proline, Glutamic acid, Serine, and Threonine) motif-containing proteins such as IκB, the PEST region of TIRAP mediates its phosphorylation, lysine polyubiquitination, and degradation, after LPS and Pam3Cys induced TLR4 and TLR2 signaling, but not following TLR7 and TLR9. Unlike TIRAP, MyD88 does not contain a PEST domain. This degradation appears dependent on SOCS1; a mutant SOCS1 (SH2 and SOCS Box) variant, as well as SOCS1 deficient macrophages, are unable to target and degrade TIRAP in response to LPS ligation, whereas wild type SOCS1 interacted and degraded TIRAP as early as 15 to 30 minutes post LPS stimulation (23). As tyrosine phosphorylation of TIRAP is required for SOCS1-mediated degradation, it appears that TIRAP is degraded only after BTK has been activated and has phosphorylated TIRAP (23, 24). Further, the tyrosine phosphorylated TIRAP generates a binding site for SH2-domain of SOCS1 (24). Accordingly, SOCS1-mediates the negative regulation of TIRAP to prevent sustained NF-κB and MAPK activities in macrophages (23). In addition to TIRAP phosphorylation, SOCS1 functional activity also depends on NOS1-derived NO production (74, 92). In response to LPS in TLR4 signaling, NOS1 derived NO production resulted in degradation of wild-type SOCS1 post-S-nitrosation, whereas pharmacological inhibition of NOS1 resulted in increased SOCS1 expression and a concomitant increase in TIRAP degradation (74). Therefore, it will be a rewarding effort to elucidate how the NOS1 axis, propagating sustained TIRAP activation, balances with TIRAP degradation mediated via BTK and SOCS1 during different inflammatory events (Figure 2).
Studies have described the role of TIRAP in modifying the innate immune response elicited largely by endotoxin (4, 5, 9, 15, 17, 93). However, the increasing number of TIRAP partner proteins and their role in the activation of inflammatory response (Figures 2 and 3) pose great potential as therapeutic targets, demanding further structural elucidation. Here we have attempted to give a brief insight into the basic architecture of TIRAP and its protein-binding domain. The human TIRAP gene encodes a 221 amino acid protein (10). Segregating TIRAP protein sequence gives us an N-terminal region spanning from the 1st to 83rd amino acid containing the PBD (Phosphatidylinositol 4,5-bisphosphate (PIP2) Binding Domain) from the 15th to 35th amino acid sequence that is rich in lysine residues. A C-terminal region mainly encodes the TIR Domain, spanning for 130 amino acid long from 84th to 213th (10, 13, 94). Also, we used the Motif search tool at www.genome.jp (https://www.genome.jp/tools/motif/) to obtain the domain details from different databases as produced by the tool search results.
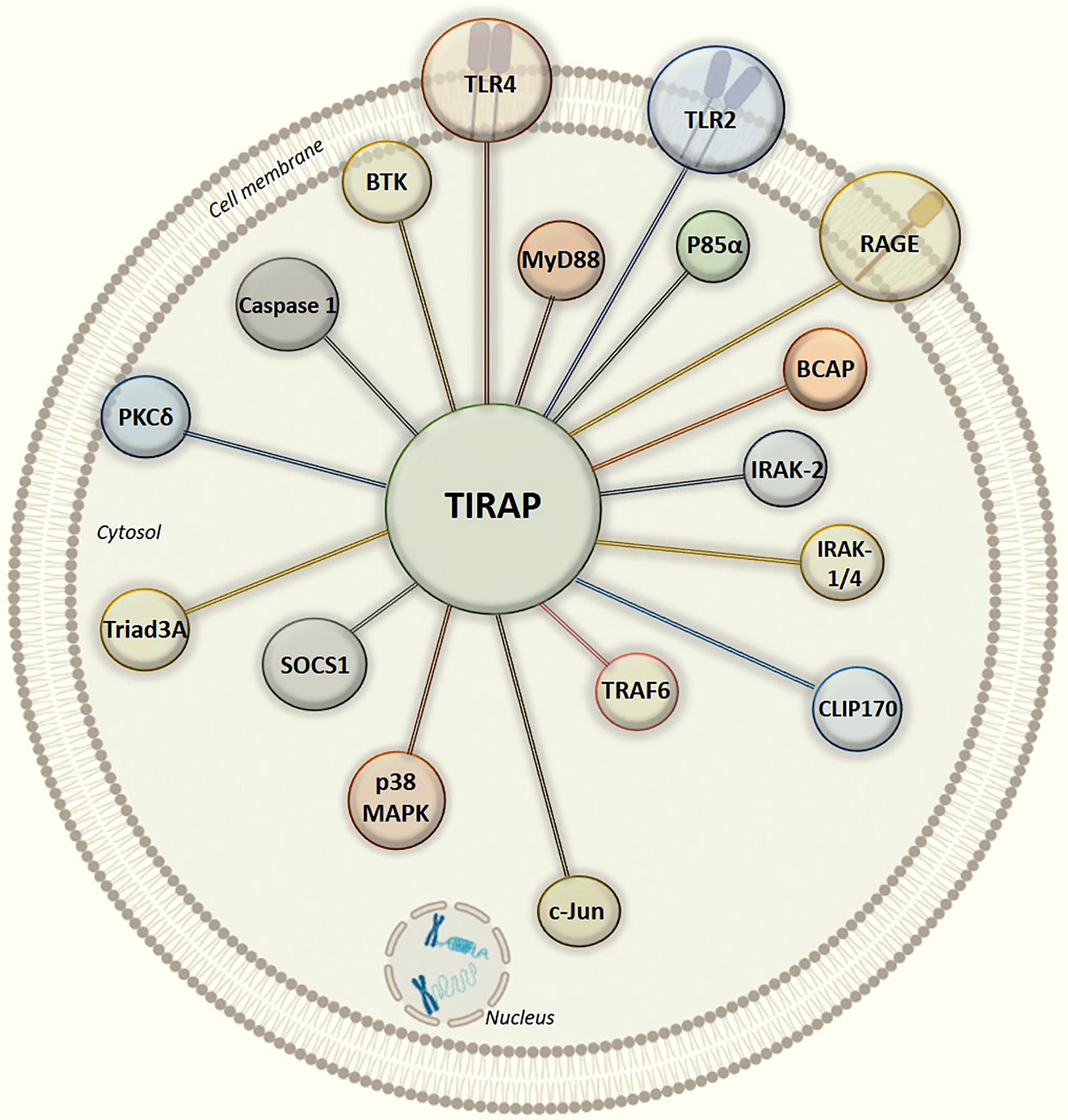
Figure 3 Graphical illustration of intracellular protein-protein interaction network of TIRAP with experimentally defined partner proteins.
In all the interactions discussed above (Figure 3), it is interesting to note that the TIR domain is centrally involved in making interactions with the TIRAP partner proteins. Though, there may be numerous post-translational modifications which can regulate TIRAP interactions, however, the experimental evidence of tyrosine phosphorylation in the TIR domain is one of the most common and validated. Besides, the macrophages largely produce NOS (Nitric oxide synthase) mediated NO (nitric oxide) which has a crucial role in regulating inflammatory responses, for example, NOS1 protects TIRAP degradation from SOCS1 (74, 92). Unlike phosphorylation, there is no experimental evidence of NO mediated nitrosylation sites yet in TIRAP, however, we may presume that the presence of Cystine S-nitrosylation site in TIR domain may be another key factor in regulating the interaction and signaling events. However, S-glutathionylation of cysteine at C91 in TIR domain has been shown to increase the interaction of TIRAP with MyD88 (95).
Besides the known phosphorylation sites, TIR domain might also encompass other sites which might be crucial for a string of TIRAP mediated interactions. Hence, in the current review, we also attempted to computationally predict possible Serine/threonine/tyrosine phosphorylation and S-nitrosylation sites on TIRAP. Briefly, the human TIRAP TIR domain sequence retrieved from UniProt (UniProtKB- P58753) was used for the prediction and study. We searched the protein phosphorylation database PhosphoSitePlus server (https://www.phosphosite.org/homeAction) and the same sites were retrieved for human TIRAP TIR domain. Further, the predictions of possible phosphorylation sites in TIR domain were obtained from kinase-based phosphorylation prediction tools; GPS-phosphorylation (http://gps.biocuckoo.cn/) (96) and NetPhos 3.1 (http://www.cbs.dtu.dk/services/NetPhos/) (97). Including the experimentally proved sites, both the tools predicted same number of a total 21 probable phosphorylation sites (10 serine sites, 05 threonine sites, and 06 tyrosine sites) (Supplementary Figure 1). Similarly, the S-Nitrosylation (SNO) sites in TIR domain were predicted from three different computational tools [GPS-SNO (http://sno.biocuckoo.org) (98), DeepNitro (www.deepnitro.renlab.org) (99) and SNOSite (http://csb.cse.yzu.edu.tw/SNOSite/predict.php)] (100). Interestingly, all the SNO sites predicted from these three independent tools were found similar (Supplementary Figure 1). The graphical representation of TIRAP and its domains, as presented in Supplementary Figure 1, was further depicted using the DOG Domain illustrator tool (http://dog.biocuckoo.org/). In support of these predictions, the tyrosine residues Y86, Y106, Y159, and Y187 have been reported to be the experimentally confirmed phosphorylation sites which modulate the interaction of TIRAP with BTK protein and downstream transcription factors activation (13, 17, 26, 27).
Since the discovery of TIRAP, it has been shown to have an elaborate role in inflammatory signalling. As summarized in this review, many investigators have identified its diverse functions other than that of its role as an adaptor protein. The dynamic nature of TIRAP interaction with several upstream and downstream signaling proteins in varied inflammatory pathways draws immense curiosity towards the post-translational modifications in its TIR domain. Interestingly, a recent study described the interaction of TIRAP and MyD88 with IL1R1 (interleukin-1 receptor like-1) receptor in response to a Helicobacter pylori released stimuli (101). Though this study focuses on experimentally validated phosphorylation sites of TIRAP TIR domain, it would be interesting to experimentally identify novel modifications sites on TIRAP which may reveal other crucial interactions involved in signaling pathways.
The report that TIRAP acts as a second adaptor protein after MyD88 in the year 2001 was marked as a major discovery in the mechanism of TLR4-dependent inflammatory signaling. Subsequently, two more adaptor proteins, TRIF and TRAM, mostly responsible for IRF3 activation, were added to the family of TIR domain-containing adaptor proteins. More recent studies show that TIRAP not only acts as a bridging protein between TLR4/2 and MyD88, but also propagates transduction of downstream signaling events, sometimes in a MyD88-independent manner. Clearly, the ability of TIRAP to interact and collaborate with several signaling molecules in a context-dependent manner means this protein is a major regulator of cell signaling.
Conceptualization: MB. Investigation: MB. Writing (Original Draft): MB and SR. Reviewing and editing TT, RI, KW, US, and DL. Supervision: TT. All authors contributed to the article and approved the submitted version.
A Biotechnology and Biological Sciences Research Council (BBSRC) David Phillips Fellowship (BB/R011834/1) funds TLMT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge the Indian Institute of Technology Indore (IITI) for providing facilities and other support. This work was supported by Cumulative Professional Development Allowance (CPDA) from IITI to MB. The authors also acknowledge American Heart Association's (AHA) support to KW and National Institutes of Health's (NIH) support to DL.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.697588/full#supplementary-material
Supplementary Figure 1 | Representation of TIRAP TIR (Toll/Interleukin-1 Receptor) domain and computational prediction of serine (S), threonine (T) and tyrosine (Y) phosphorylation and cysteine (C) S-nitrosylation (SNO) positions. The TIRAP protein containing an N-terminal PBD and C-terminal TIR domain are highlighted. The predicted phosphorylation sites on top and SNO sites at bottom of domain map are indicated in blue and light brown colors, respectively. The experimentally proved phosphorylation sites (Y86, Y106, Y159, and Y187) are marked with asterisk. The predicted phosphorylation sites were obtained from kinase-based computational tools GPS-Phosphorylation (http://gps.biocuckoo.cn/) and NetPhos 3.1 (http://www.cbs.dtu.dk/services/NetPhos/). For cysteine (C) SNO sites, computational tools GPS-SNO (http://sno.biocuckoo.org/index.php), DeepNitro (www.deepnitro.renlab.org), and SNOSite (http://csb.cse.yzu.edu.tw/SNOSite/predict.php) were used.
1. O’Neill LA, Bowie AG. The Family of Five: TIR-Domain-Containing Adaptors in Toll-Like Receptor Signalling. Nat Rev Immunol (2007) 7(5):353–64. doi: 10.1038/nri2079
2. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-Adapter-Like) Is Required for Toll-Like Receptor-4 Signal Transduction. Nature (2001) 413(6851):78–83. doi: 10.1038/35092578
3. Kawai T, Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
4. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
5. Balka KR, De Nardo D. Understanding Early TLR Signaling Through the Myddosome. J Leukoc Biol (2019) 105(2):339–51. doi: 10.1002/JLB.MR0318-096R
6. Bernard NJ, O’Neill LA. Mal, More Than a Bridge to Myd88. IUBMB Life (2013) 65(9):777–86. doi: 10.1002/iub.1201
7. Latty SL, Sakai J, Hopkins L, Verstak B, Paramo T, Berglund NA, et al. Activation of Toll-Like Receptors Nucleates Assembly of the MyDDosome Signaling Hub. Elife (2018) 7:e31377. doi: 10.7554/eLife.31377
8. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 Signal Transduction Pathway. Cytokine (2008) 42(2):145–51. doi: 10.1016/j.cyto.2008.01.006
9. Palsson-McDermott EM, O’Neill LA. Signal Transduction by the Lipopolysaccharide Receptor, Toll-Like Receptor-4. Immunology (2004) 113(2):153–62. doi: 10.1111/j.1365-2567.2004.01976.x
10. Belhaouane I, Hoffmann E, Chamaillard M, Brodin P, Machelart A. Paradoxical Roles of the MAL/Tirap Adaptor in Pathologies. Front Immunol (2020) 11:569127. doi: 10.3389/fimmu.2020.569127
11. Kagan JC, Medzhitov R. Phosphoinositide-Mediated Adaptor Recruitment Controls Toll-Like Receptor Signaling. Cell (2006) 125(5):943–55. doi: 10.1016/j.cell.2006.03.047
12. Patra MC, Choi S. Insight Into Phosphatidylinositol-Dependent Membrane Localization of the Innate Immune Adaptor Protein Toll/Interleukin 1 Receptor Domain-Containing Adaptor Protein. Front Immunol (2018) 9:75. doi: 10.3389/fimmu.2018.00075
13. Lin Z, Lu J, Zhou W, Shen Y. Structural Insights Into TIR Domain Specificity of the Bridging Adaptor Mal in TLR4 Signaling. PloS One (2012) 7(4):e34202. doi: 10.1371/journal.pone.0034202
14. Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, et al. Structural Basis for the Multiple Interactions of the MyD88 TIR Domain in TLR4 Signaling. Proc Natl Acad Sci USA (2009) 106(25):10260–5. doi: 10.1073/pnas.0812956106
15. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-Like Receptor-4 Mediates Lipopolysaccharide-Induced Signal Transduction. J Biol Chem (1999) 274(16):10689–92. doi: 10.1074/jbc.274.16.10689
16. Santos-Sierra S, Deshmukh SD, Kalnitski J, Kuenzi P, Wymann MP, Golenbock DT, et al. Mal Connects TLR2 to PI3Kinase Activation and Phagocyte Polarization. EMBO J (2009) 28(14):2018–27. doi: 10.1038/emboj.2009.158
17. Gray P, Dunne A, Brikos C, Jefferies CA, Doyle SL, O’Neill LA. MyD88 Adapter-Like (Mal) is Phosphorylated by Bruton’s Tyrosine Kinase During TLR2 and TLR4 Signal Transduction. JJoBC (2006) 281(15):10489–95. doi: 10.1074/jbc.M508892200
18. Jakka P, Bhargavi B, Namani S, Murugan S, Splitter G, Radhakrishnan G. Cytoplasmic Linker Protein CLIP170 Negatively Regulates TLR4 Signaling by Targeting the TLR Adaptor Protein TIRAP. J Immunol (2018) 200(2):704–14. doi: 10.4049/jimmunol.1601559
19. Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, et al. TIRAP, an Adaptor Protein for TLR2/4, Transduces a Signal From RAGE Phosphorylated Upon Ligand Binding. PloS One (2011) 6(8):e23132. doi: 10.1371/journal.pone.0023132
20. Verstak B, Nagpal K, Bottomley SP, Golenbock DT, Hertzog PJ, Mansell A. MyD88 Adapter-Like (Mal)/TIRAP Interaction With TRAF6 is Critical for TLR2- and TLR4-Mediated NF-kappaB Proinflammatory Responses. J Biol Chem (2009) 284(36):24192–203. doi: 10.1074/jbc.M109.023044
21. You X, Liu L, Zeng Y, Li R, He J, Ma X, et al. Macrophage-Activating Lipopeptide-2 Requires Mal and PI3K for Efficient Induction of Heme Oxygenase-1. PloS One (2014) 9(7):e103433. doi: 10.1371/journal.pone.0103433
22. Fearns C, Pan Q, Mathison JC, Chuang TH. Triad3A Regulates Ubiquitination and Proteasomal Degradation of RIP1 Following Disruption of Hsp90 Binding. J Biol Chem (2006) 281(45):34592–600. doi: 10.1074/jbc.M604019200
23. Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, et al. Suppressor of Cytokine Signaling 1 Negatively Regulates Toll-Like Receptor Signaling by Mediating Mal Degradation. Nat Immunol (2006) 7(2):148–55. doi: 10.1038/ni1299
24. Fujimoto M, Naka T. SOCS1, a Negative Regulator of Cytokine Signals and TLR Responses, in Human Liver Diseases. Gastroenterol Res Pract (2010) 2010:470468. doi: 10.1155/2010/470468
25. Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a Negative Regulator of LPS-Induced Macrophage Activation. Immunity (2002) 17(5):583–91. doi: 10.1016/S1074-7613(02)00446-6
26. Piao W, Song C, Chen H, Wahl LM, Fitzgerald KA, O’Neill LA, et al. Tyrosine Phosphorylation of MyD88 Adapter-Like (Mal) is Critical for Signal Transduction and Blocked in Endotoxin Tolerance. J Biol Chem (2008) 283(6):3109–19. doi: 10.1074/jbc.M707400200
27. Paracha RZ, Ali A, Ahmad J, Hussain R, Niazi U, Muhammad SA. Structural Evaluation of BTK and PKCdelta Mediated Phosphorylation of MAL at Positions Tyr86 and Tyr106. Comput Biol Chem (2014) 51:22–35. doi: 10.1016/j.compbiolchem.2014.04.001
28. Jefferies CA, O’Neill LA. Bruton’s Tyrosine Kinase (Btk)-The Critical Tyrosine Kinase in LPS Signalling? Immunol Lett (2004) 92(1-2):15–22. doi: 10.1016/j.imlet.2003.11.017
29. Kubo-Murai M, Hazeki K, Sukenobu N, Yoshikawa K, Nigorikawa K, Inoue K, et al. Protein Kinase Cdelta Binds TIRAP/Mal to Participate in TLR Signaling. Mol Immunol (2007) 44(9):2257–64. doi: 10.1016/j.molimm.2006.11.005
30. Baig MS, Liu D, Muthu K, Roy A, Saqib U, Naim A, et al. Heterotrimeric Complex of P38 MAPK, PKCdelta, and TIRAP is Required for AP1 Mediated Inflammatory Response. Int Immunopharmacol (2017) 48:211–8. doi: 10.1016/j.intimp.2017.04.028
31. Yang Y, Kim SC, Yu T, Yi YS, Rhee MH, Sung GH, et al. Functional Roles of P38 Mitogen-Activated Protein Kinase in Macrophage-Mediated Inflammatory Responses. Mediators Inflammation (2014) 2014:352371. doi: 10.1155/2014/352371
32. Srivastava M, Saqib U, Banerjee S, Wary K, Kizil B, Muthu K, et al. Inhibition of the TIRAP-C-Jun Interaction as a Therapeutic Strategy for AP1-Mediated Inflammatory Responses. Int Immunopharmacol (2019) 71:188–97. doi: 10.1016/j.intimp.2019.03.031
33. Miggin SM, Pålsson-McDermott E, Dunne A, Jefferies C, Pinteaux E, Banahan K, et al. NF-κb Activation by the Toll-IL-1 Receptor Domain Protein MyD88 Adapter-Like is Regulated by Caspase-1. Proc Natl Acad Sci USA (2007) 104(9):3372–7. doi: 10.1073/pnas.0608100104
34. Ulrichts P, Bovijn C, Lievens S, Beyaert R, Tavernier J, Peelman F. Caspase-1 Targets the TLR Adaptor Mal at a Crucial TIR-Domain Interaction Site. J Cell Sci (2010) 123(Pt 2):256–65. doi: 10.1242/jcs.056002
35. Dunne A, Carpenter S, Brikos C, Gray P, Strelow A, Wesche H, et al. IRAK1 and IRAK4 Promote Phosphorylation, Ubiquitination, and Degradation of MyD88 Adaptor-Like (Mal). J Biol Chem (2016) 291(47):24802. doi: 10.1074/jbc.A109.098137
36. Halabi S, Sekine E, Verstak B, Gay NJ, Moncrieffe MC. Structure of the Toll/interleukin-1 Receptor (TIR) Domain of the B-Cell Adaptor That Links Phosphoinositide Metabolism With the Negative Regulation of the Toll-Like Receptor (TLR) Signalosome. JJoBC (2017) 292(2):652–60. doi: 10.1074/jbc.M116.761528
37. Ni M, MacFarlane AW, Toft M, Lowell CA, Campbell KS, Hamerman JA. B-Cell Adaptor for PI3K (BCAP) Negatively Regulates Toll-Like Receptor Signaling Through Activation of PI3K. JPotNAoS (2012) 109(1):267–72. doi: 10.1073/pnas.1111957108
38. Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, et al. Role for B-Cell Adapter for PI3K (BCAP) as a Signaling Adapter Linking Toll-Like Receptors (TLRs) to Serine/Threonine Kinases PI3K/Akt. Proc Natl Acad Sci USA (2012) 109(1):273–8. doi: 10.1073/pnas.1118579109
39. Lauenstein JU, Scherm MJ, Udgata A, Moncrieffe MC, Fisher DI, Gay NJ. Negative Regulation of TLR Signaling by BCAP Requires Dimerization of Its DBB Domain. JTJoI (2020) 204(8):2269–76. doi: 10.4049/jimmunol.1901210
40. Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, et al. Essential Role for TIRAP in Activation of the Signalling Cascade Shared by TLR2 and TLR4. Nature (2002) 420(6913):324–9. doi: 10.1038/nature01182
41. Sakaguchi M, Kinoshita R, Putranto EW, Ruma IMW, Sumardika IW, Youyi C, et al. Signal Diversity of Receptor for Advanced Glycation End Products. Acta Med Okayama (2017) 71(6):459–65. doi: 10.18926/AMO/55582
42. Chuang TH, Ulevitch RJ. Triad3A, an E3 Ubiquitin-Protein Ligase Regulating Toll-Like Receptors. Nat Immunol (2004) 5(5):495–502. doi: 10.1038/ni1066
43. Leifer CA, Medvedev AE. Molecular Mechanisms of Regulation of Toll-Like Receptor Signaling. JJolb (2016) 100(5):927–41. doi: 10.1189/jlb.2MR0316-117RR
44. Lannoy V, Côté-Biron A, Asselin C, Rivard NJCC. Signaling. Phosphatases in Toll-Like Receptors Signaling: The Unfairly-Forgotten. Cell Commun Signal (2021) 19(1):1–15. doi: 10.1186/s12964-020-00693-9
45. Chattopadhyay S, Sen GC. Tyrosine Phosphorylation in Toll-Like Receptor Signaling. Cytokine Growth Factor Rev (2014) 25(5):533–41. doi: 10.1016/j.cytogfr.2014.06.002
46. Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, et al. Bruton’s Tyrosine Kinase Is a Toll/interleukin-1 Receptor Domain-Binding Protein That Participates in Nuclear Factor kappaB Activation by Toll-Like Receptor 4. J Biol Chem (2003) 278(28):26258–64. doi: 10.1074/jbc.M301484200
47. Steinberg SF. Distinctive Activation Mechanisms and Functions for Protein Kinase Cdelta. Biochem J (2004) 384(Pt 3):449–59. doi: 10.1042/BJ20040704
48. Gschwendt M, Kielbassa K, Kittstein W, Marks F. Tyrosine Phosphorylation and Stimulation of Protein Kinase C Delta From Porcine Spleen by Src In Vitro. Dependence on the Activated State of Protein Kinase C Delta. FEBS Lett (1994) 347(1):85–9. doi: 10.1016/0014-5793(94)00514-1
49. Rip J, De Bruijn MJ, Appelman MK, Pal Singh S, Hendriks RW, Corneth O. Toll-Like Receptor Signaling Drives Btk-Mediated Autoimmune Disease. Front Immunol (2019) 10:95. doi: 10.3389/fimmu.2019.00095
50. Chavakis T, Bierhaus A, Nawroth PP. RAGE (Receptor for Advanced Glycation End Products): A Central Player in the Inflammatory Response. Microbes Infect (2004) 6(13):1219–25. doi: 10.1016/j.micinf.2004.08.004
51. Hudson BI, Lippman ME. Targeting RAGE Signaling in Inflammatory Disease. Annu Rev Med (2018) 69:349–64. doi: 10.1146/annurev-med-041316-085215
52. Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligandsAnd Their Role in Cancer and Inflammation. J Transl Med (2009) 7:17. doi: 10.1186/1479-5876-7-17
53. Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, The Receptor for Advanced Glycation End Products. J Mol Med (Berl) (2005) 83(11):876–86. doi: 10.1007/s00109-005-0688-7
54. Kierdorf K, Fritz G. RAGE Regulation and Signaling in Inflammation and Beyond. J Leukoc Biol (2013) 94(1):55–68. doi: 10.1189/jlb.1012519
55. Prantner D, Nallar S, Vogel SNJTFJ. The Role of RAGE in Host Pathology and Crosstalk Between RAGE and TLR4 in Innate Immune Signal Transduction Pathways. FASEB J (2020) 34(12):15659–74. doi: 10.1096/fj.202002136R
56. Watanabe M, Toyomura T, Wake H, Liu K, Teshigawara K, Takahashi H, et al. Differential Contribution of Possible Pattern-Recognition Receptors to Advanced Glycation End Product–Induced Cellular Responses in Macrophage-Like Raw264. 7 cells. Biotechnol Appl Biochem (2020) 67: (2):265–72. doi: 10.1002/bab.1843
57. Putranto EW, Murata H, Yamamoto KI, Kataoka K, Yamada H, Futami JI, et al. Inhibition of RAGE Signaling Through the Intracellular Delivery of Inhibitor Peptides by PEI Cationization. Int J Mol Med (2013) 32(4):938–44. doi: 10.3892/ijmm.2013.1467
58. Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host Innate Immune Receptors and Beyond: Making Sense of Microbial Infections. Cell Host Microbe (2008) 3(6):352–63. doi: 10.1016/j.chom.2008.05.003
59. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, et al. Crystal Structure of the TLR1-TLR2 Heterodimer Induced by Binding of a Tri-Acylated Lipopeptide. Cell (2007) 130(6):1071–82. doi: 10.1016/j.cell.2007.09.008
60. Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, et al. Lipoproteins are Critical TLR2 Activating Toxins in Group B Streptococcal Sepsis. J Immunol (2008) 180(9):6149–58. doi: 10.4049/jimmunol.180.9.6149
61. Parihar SP, Ozturk M, Marakalala MJ, Loots DT, Hurdayal R, Beukes D, et al. Protein Kinase C-Delta (PKCdelta), a Marker of Inflammation and Tuberculosis Disease Progression in Humans, is Important for Optimal Macrophage Killing Effector Functions and Survival in Mice. Mucosal Immunol (2018) 11(2):579–80. doi: 10.1038/mi.2017.108
62. Dixit R, Barnett B, Lazarus JE, Tokito M, Goldman YE, Holzbaur EL. Microtubule Plus-End Tracking by CLIP-170 Requires Eb1. Proc Natl Acad Sci USA (2009) 106(2):492–7. doi: 10.1073/pnas.0807614106
63. Maekawa H, Schiebel E. CLIP-170 Family Members: A Motor-Driven Ride to Microtubule Plus Ends. Dev Cell (2004) 6(6):746–8. doi: 10.1016/j.devcel.2004.05.017
64. Pierre P, Scheel J, Rickard JE, Kreis TE. CLIP-170 Links Endocytic Vesicles to Microtubules. Cell (1992) 70(6):887–900. doi: 10.1016/0092-8674(92)90240-D
65. Sengupta D, Koblansky A, Gaines J, Brown T, West AP, Zhang D, et al. Subversion of Innate Immune Responses by Brucella Through the Targeted Degradation of the TLR Signaling Adapter, MAL. J Immunol (2010) 184(2):956–64. doi: 10.4049/jimmunol.0902008
66. Murugan S, Jakka P, Namani S, Mujumdar V, Radhakrishnan G. The Neurosteroid Pregnenolone Promotes Degradation of Key Proteins in the Innate Immune Signaling to Suppress Inflammation. J Biol Chem (2019) 294(12):4596–607. doi: 10.1074/jbc.RA118.005543
67. Atsaves V, Leventaki V, Rassidakis GZ, Claret FXJC. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers (Basel) (2019) 11(7):1037. doi: 10.3390/cancers11071037
68. Eferl R, Wagner EF. AP-1: A Double-Edged Sword in Tumorigenesis. Nat Rev Cancer (2003) 3(11):859–68. doi: 10.1038/nrc1209
69. Kerppola TK, Curran T. Fos-Jun Heterodimers and Jun Homodimers Bend DNA in Opposite Orientations: Implications for Transcription Factor Cooperativity. Cell (1991) 66(2):317–26. doi: 10.1016/0092-8674(91)90621-5
70. Wisdom R. AP-1: One Switch for Many Signals. Exp Cell Res (1999) 253(1):180–5. doi: 10.1006/excr.1999.4685
71. Chida K, Nagamori S, Kuroki T. Nuclear Translocation of Fos is Stimulated by Interaction With Jun Through the Leucine Zipper. Cell Mol Life Sci (1999) 55(2):297–302. doi: 10.1007/s000180050291
72. Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, et al. AP-1 Activated by Toll-Like Receptors Regulates Expression of IL-23 P19. J Biol Chem (2009) 284(36):24006–16. doi: 10.1074/jbc.M109.025528
73. Liu X, Yin S, Chen Y, Wu Y, Zheng W, Dong H, et al. LPSinduced Proinflammatory Cytokine Expression in Human Airway Epithelial Cells and Macrophages via NFkappaB, STAT3 or AP1 Activation. Mol Med Rep (2018) 17(4):5484–91. doi: 10.3892/mmr.2018.8542
74. Srivastava M, Baig MS. NOS1 Mediates AP1 Nuclear Translocation and Inflammatory Response. BioMed Pharmacother (2018) 102:839–47. doi: 10.1016/j.biopha.2018.03.069
75. Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting Inflammation by Modulating the Jun/AP-1 Pathway. Ann Rheum Dis (2011) 70 Suppl 1:i109–12. doi: 10.1136/ard.2010.140533
76. Abraham C, Vogel SN, Perkins DJ. Signaling Mechanisms Regulating Innate Immune Responses. In: Mucosal Immunology. Cambridge, Massachusetts, United States: Academic Press (2015). p. 605–22. Academic Press
77. Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, et al. A Unified Model for Apical Caspase Activation. Mol Cell (2003) 11(2):529–41. doi: 10.1016/S1097-2765(03)00051-0
78. Lamkanfi M, Kalai M, Saelens X, Declercq W, Vandenabeele P. Caspase-1 Activates Nuclear Factor of the Kappa-Enhancer in B Cells Independently of its Enzymatic Activity. J Biol Chem (2004) 279(23):24785–93. doi: 10.1074/jbc.M400985200
79. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential Requirement for Caspase-1 Autoproteolysis in Pathogen-Induced Cell Death and Cytokine Processing. Cell Host Microbe (2010) 8(6):471–83. doi: 10.1016/j.chom.2010.11.007
80. Martinon F, Mayor A, Tschopp J. The Inflammasomes: Guardians of the Body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
81. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A Novel Heterodimeric Cysteine Protease Is Required for Interleukin-1βprocessing in Monocytes. Nature (1992) 356(6372):768–74. doi: 10.1038/356768a0
82. Xie P. TRAF Molecules in Cell Signaling and in Human Diseases. J Mol Signal (2013) 8(1):7. doi: 10.1186/1750-2187-8-7
83. Li X, Zhong C-Q, Yin Z, Qi H, Xu F, He Q, et al. Data-Driven Modeling Identifies TIRAP-Independent MyD88 Activation Complex and Myddosome Assembly Strategy in LPS/TLR4 Signaling. Int J Mol Sci (2020) 21(9):3061. doi: 10.3390/ijms21093061
84. Janssens S, Beyaert R. Functional Diversity and Regulation of Different Interleukin-1 Receptor-Associated Kinase (IRAK) Family Members. Mol Cell (2003) 11(2):293–302. doi: 10.1016/S1097-2765(03)00053-4
85. Flo TH, Aderem A. Pathogen Recognition by Toll-Like Receptors. In: Bertók L, Chow DA, editors. NeuroImmune Biology, vol. 5p . Amsterdam, Netherlands: Elsevier (2005). p. 167–82. Elsevier
86. de Diego RP, Rodríguez-Gallego C. Chapter 34 - Other TLR Pathway Defects. In: Sullivan KE, Stiehm ER, editors. Stiehm’s Immune Deficiencies. Amsterdam: Academic Press (2014). p. 687–710.
87. Kumar H, Takeuchi O, Akira S. Toll-Like Receptors. In: Lennarz WJ, Lane MD, editors. Encyclopedia of Biological Chemistry, 2nd ed. Waltham: Academic Press (2013). p. 396–401.
88. Mandraju R, Troutman TD, Pasare C. Toll-Like Receptor Function and Signaling. In: Reference Module in Biomedical Sciences. Amsterdam, Netherlands: Elsevier (2014).
89. Biswas C. Chapter 18 - Inflammation in Systemic Immune Diseases: Role of TLR9 Signaling and the Resultant Oxidative Stress in Pathology of Lupus. In: Chatterjee S, Jungraithmayr W, Bagchi D, editors. Immunity and Inflammation in Health and Disease. Cambridge, Massachusetts, United States: Academic Press (2018). p. 223–37.
90. Yoshimura A, Naka T, Kubo M. SOCS Proteins, Cytokine Signalling and Immune Regulation. Nat Rev Immunol (2007) 7(6):454–65. doi: 10.1038/nri2093
91. Sharma J, Larkin J 3rd. Therapeutic Implication of SOCS1 Modulation in the Treatment of Autoimmunity and Cancer. Front Pharmacol (2019) 10:324. doi: 10.3389/fphar.2019.00324
92. Baig MS, Zaichick SV, Mao M, de Abreu AL, Bakhshi FR, Hart PC, et al. NOS1-Derived Nitric Oxide Promotes NF-kappaB Transcriptional Activity Through Inhibition of Suppressor of Cytokine Signaling-1. J Exp Med (2015) 212(10):1725–38. doi: 10.1084/jem.20140654
93. Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, et al. Role of TLR4 Tyrosine Phosphorylation in Signal Transduction and Endotoxin Tolerance. J Biol Chem (2007) 282(22):16042–53. doi: 10.1074/jbc.M606781200
94. Zhao X, Xiong W, Xiao S, Tang TX, Ellena JF, Armstrong GS, et al. Membrane Targeting of TIRAP is Negatively Regulated by Phosphorylation in its Phosphoinositide-Binding Motif. Sci Rep (2017) 7:43043. doi: 10.1038/srep43043
95. Hughes MM, Lavrencic P, Coll RC, Ve T, Ryan DG, Williams NC, et al. Solution Structure of the TLR Adaptor MAL/TIRAP Reveals an Intact BB Loop and Supports MAL Cys91 Glutathionylation for Signaling. Proc Natl Acad Sci USA (2017) 114(32):E6480–E9. doi: 10.1073/pnas.1701868114
96. Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, et al. GPS 2.1: Enhanced Prediction of Kinase-Specific Phosphorylation Sites With an Algorithm of Motif Length Selection. Protein Eng Des Sel (2011) 24(3):255–60. doi: 10.1093/protein/gzq094
97. Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak SJP. Prediction of Post-Translational Glycosylation and Phosphorylation of Proteins From the Amino Acid Sequence. Proteomics (2004) 4(6):1633–49. doi: 10.1002/pmic.200300771
98. Xue Y, Liu Z, Gao X, Jin C, Wen L, Yao X, et al. GPS-SNO: Computational Prediction of Protein S-Nitrosylation Sites With a Modified GPS Algorithm. PloS One (2010) 5(6):e11290. doi: 10.1371/journal.pone.0011290
99. Xie Y, Luo X, Li Y, Chen L, Ma W, Huang J, et al. DeepNitro: Prediction of Protein Nitration and Nitrosylation Sites by Deep Learning. Genomics Proteomics Bioinformatics (2018) 16(4):294–306. doi: 10.1016/j.gpb.2018.04.007
100. Lee T-Y, Chen Y-J, Lu T-C, Huang H-D, Chen Y-J. SNOSite: Exploiting Maximal Dependence Decomposition to Identify Cysteine S-Nitrosylation With Substrate Site Specificity. PLoS One (2011) 6(7):e21849. doi: 10.1371/journal.pone.0021849
Keywords: inflammation, TLR signaling, inflammatory disease, TIRAP (TIR domain-containing adaptor protein), protein-protein interaction (PPI)
Citation: Rajpoot S, Wary KK, Ibbott R, Liu D, Saqib U, Thurston TLM and Baig MS (2021) TIRAP in the Mechanism of Inflammation. Front. Immunol. 12:697588. doi: 10.3389/fimmu.2021.697588
Received: 19 April 2021; Accepted: 23 June 2021;
Published: 08 July 2021.
Edited by:
You-Me Kim, Korea Advanced Institute of Science and Technology, South KoreaReviewed by:
Hiroyuki Oshiumi, Kumamoto University, JapanCopyright © 2021 Rajpoot, Wary, Ibbott, Liu, Saqib, Thurston and Baig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa L. M. Thurston, dC50aHVyc3RvbkBpbXBlcmlhbC5hYy51aw==; Mirza S. Baig, bXNiLmlpdEBpaXRpLmFjLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.