 Binghan Zhou
Binghan Zhou Yuan Gao
Yuan Gao Qian Chu
Qian Chu- Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
The immune checkpoint blockade therapy has completely transformed cancer treatment modalities because of its unprecedented and durable clinical responses in various cancers. With the increasing use of immune checkpoint blockades in clinical practice, a large number of patients develop acquired resistance. However, the knowledge about acquired resistance to immune checkpoint blockades is limited and poorly summarized. In this review, we clarify the principal elements of acquired resistance to immune checkpoint blockades. The definition of acquired resistance is heterogeneous among groups or societies, but the expert consensus of The Society for Immunotherapy of Cancer can be referred. Oligo-progression is the main pattern of acquired resistance. Acquired resistance can be derived from the selection of resistant cancer cell clones that exist in the tumor mass before therapeutic intervention or gradual acquisition in the sensitive cancer cells. Specifically, tumor intrinsic mechanisms include neoantigen depletion, defects in antigen presentation machinery, aberrations of interferon signaling, tumor-induced exclusion/immunosuppression, and tumor cell plasticity. Tumor extrinsic mechanisms include upregulation of other immune checkpoints. Presently, a set of treatment modalities is applied to patients with similar clinical characteristics or resistance mechanisms for overcoming acquired resistance, and hence, further research is required.
Introduction
Since the approval of the first CTLA-4 blockade by the US Food and Drug Administration (FDA) in 2011 (1, 2), immune checkpoint blockades (ICBs) have completely changed cancer treatment modalities (3–6). Due to more extensive use of ICBs in clinical practice, several patients exhibit disease progression, although with an initial response, termed as acquired resistance (AR). However, the underlying mechanisms of AR and strategies for overcoming AR are limited and poorly summarized. Previous research (7–9) has focused on primary resistance to ICBs, providing limited description about AR, whose mechanisms and countermeasures differ from those of primary resistance. In addition, updated information on AR needs to be emphasized.
In this review, we clarify the principal elements of AR to ICBs including the (1) definition of AR; (2) clinical characteristics of AR; (3) underlying mechanisms of AR; and (4) potential strategies to overcome AR. We hope that this review will help researchers and clinicians recognize AR to ICBs more comprehensively and in detail.
Definition of AR to Immune Checkpoint Blockades
Primary resistance occurs when cancer cells do not respond to immunotherapy, whereas AR normally occurs as disease progression after an initial response. Contrary to primary resistance, the current definition of AR is highly heterogeneous among groups or societies. Different researchers define AR differently when they explore the underlying mechanisms or design clinical trials, with the main controversies being prior response (10, 11) (whether the stable disease should be regarded as a response to ICBs), treatment duration time (10, 12) (a cut-off time to distinguish from primary resistance), and treatment discontinuation (10, 13) (whether disease progression after discontinuation to ICBs should be regarded as AR). These divergent views have obstructed the integrated interpretation of each solitary study and further research in this field. To clarify the underlying resistance mechanisms to immunotherapy, Sharma et al. introduced a three-category classification of primary resistance, adaptive immune resistance, and AR (7). However, adaptive immune resistance can manifest like primary resistance or AR in the clinic, and they did not clearly define these terms (7). The International Association for the Study of Lung Cancer (IASLC) expert panel has defined primary resistance (no objective tumor radiographic response and treatment duration < 6 months) and AR (an objective tumor response and treatment duration ≥ 6 months) for lung malignancies immunotherapy, but they did not consider other clinical settings in addition to advanced disease or the issue of treatment discontinuation (14). The Society for Immunotherapy of Cancer (SITC) proposed expert consensus definitions for PD-(L)1 inhibitor monotherapy resistance in solid tumors (15) (Table 1), which can be widely applicable. However, definitions based on empirical data from clinical trials, patient registries, or previous studies investigating treatment beyond progression and/or treatment after relapse are still needed, which will fuel future research on AR to ICBs, providing guidelines for clinical practice.
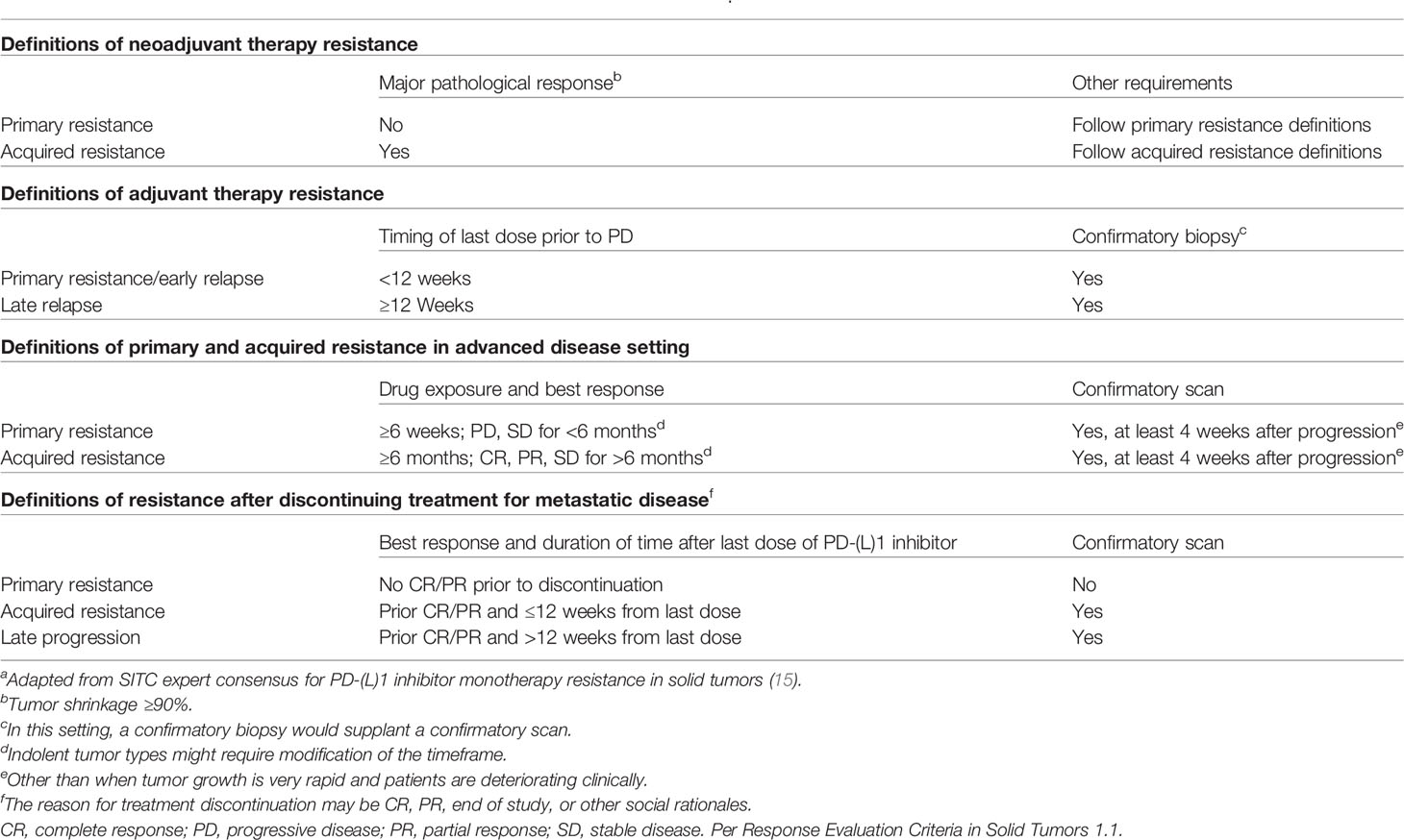
Table 1 The definitions for PD-(L)1 inhibitor monotherapy resistance in solid tumorsa.
AR is considerably different from primary resistance to ICB therapy based on three aspects— (1) immunophenotypes: AR mainly occurs in inflamed tumors, while primary resistance occurs in excluded or desert tumors, which lack sufficient immune infiltration (16); (2) underlying mechanisms: AR is attributed to the mutual evolution of tumor and tumor microenvironment (TME) under the pressure of ICB-activated immune elimination, while primary resistance can be attributed to the host, tumor, TME, and microbiome, a reflection of baseline status resisting ICBs (17–19); and (3) coping strategies: the coping strategies of AR could be specific to the resistance mechanisms, aiming at extending the patients’ survival, while the coping strategies of primary resistance are comprehensive and largely dependent on heating the cold tumor (20).
Clinical Characteristics of AR
Unlike primary resistance, the rates of AR have not been routinely reported and thus are not characterized across tumor types. Based on available data of response duration time, Schoenfeld et al. inferred the AR rate and found that AR rate ranges from 11% to 71% among different tumor types, with the median of 39%, and a tumor with a lower response rate has a higher AR rate (21). Additionally, based on clinical trial data, AR to nivolumab in melanoma at 5-year follow-up was 39% (22), while AR to nivolumab in non-small-cell lung cancer (NSCLC) at 4-year follow-up was 65% (23).
In the population of patients with response to ICBs, the median duration of response (DOR) was heterogenous among different tumor types, lines of therapies, or PD-L1 levels. Patients with melanoma had the longest DOR, with the median of more than four years. While the median DOR in other tumor types was usually less than two years, indicating that more than half responders would progress within two years (Supplementary Table 1).
In the population of patients with AR to ICBs, the clinical characteristics of these patients were not routinely reported in clinical trials. Based on the limited clinical trials and real-world studies on this topic (24–28), AR is more likely evident in patients with prior partial response (PR) instead of complete response (CR) (stable disease is not taken into consideration). The median time to resistance is inconsistent and needs further validation. Oligo-progression is the main pattern of resistance. The median survival time after AR seems heterogeneous among different patients and largely depends on the successive treatment (Table 2). However, it must be noted that the definition of AR in these studies is either not given or different from the SITC recommendations, without the requirement of 6-month drug exposure and 3-month limit for drug discontinuation.
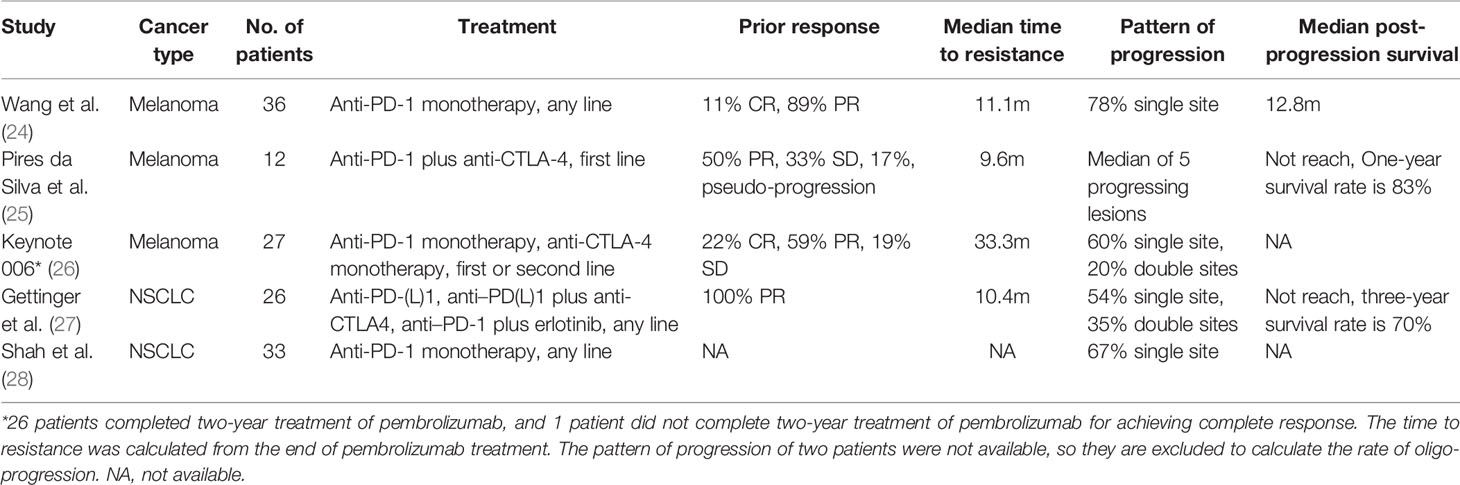
Table 2 Clinical characteristics of acquired resistance to ICBs.
Underlying Mechanisms of AR
Based on the origin of AR, AR to immunotherapy can be divided into two major categories, though the phenotype of these two categories is almost similar in the response and resistance phases (Figure 1). The first type of resistance is a special form of Darwinian natural selection that comes from the selection of genetic or epigenetic heritable traits that exist in the tumor mass before a therapeutic intervention (29). Intratumor heterogeneity can come from numerical or structural chromosomal instability, somatic mutagenesis, and epigenetic diversity (30). AR-related heterogeneity lies in various aspects of anti-tumor immunity. Additionally, the quantity and quality of resistant tumor cell clones reflect the forms of resistance. The abundant and high-quality resistant clones provide a basis for the occurrence of primary resistance, while scarce and low-quality resistant clones retain the possibility for AR.
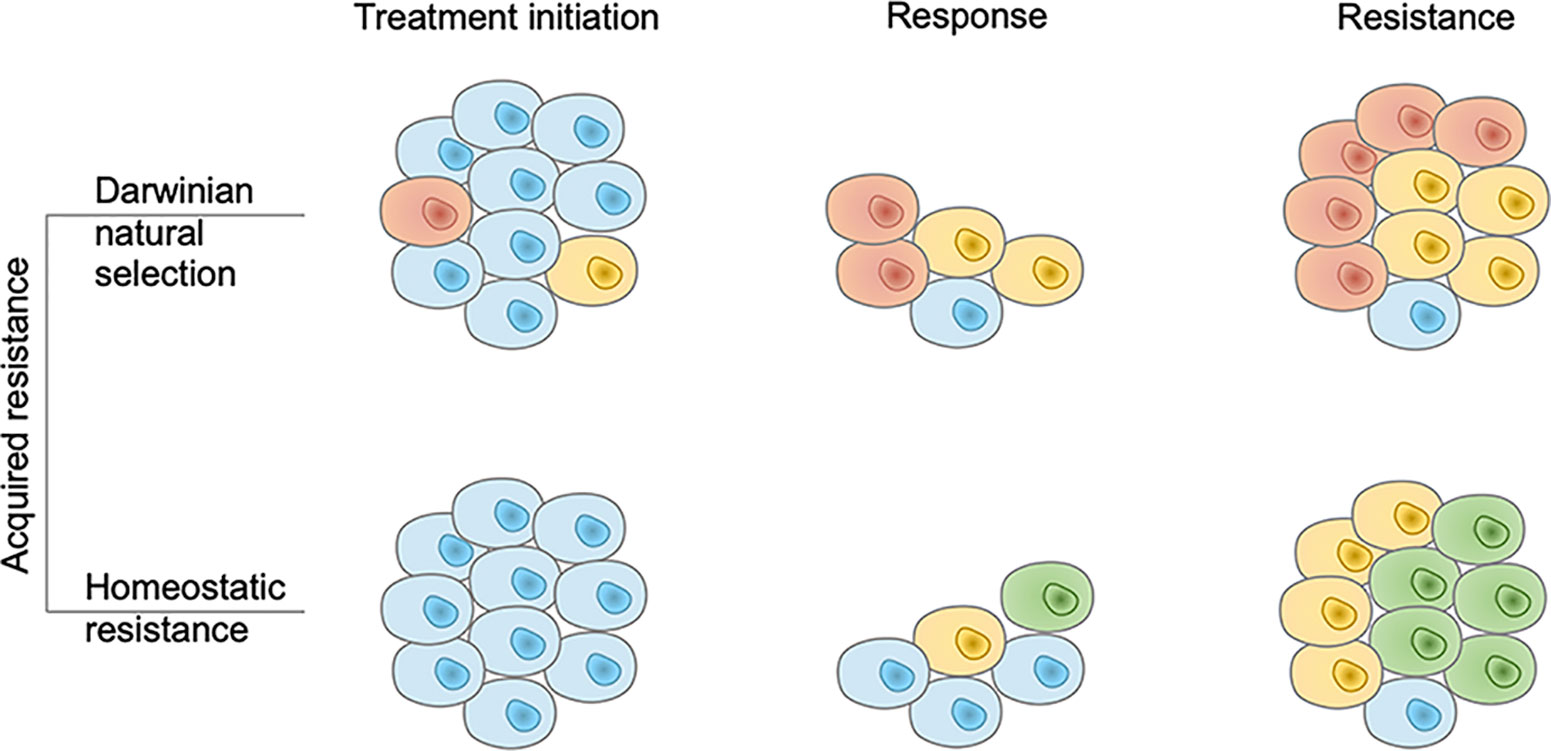
Figure 1 Two modes of acquired resistance to immunotherapy. In the Darwinian natural selection mode, immunotherapy resistant tumor cell clones pre-exist in the tumor mass. They are present at the treatment initiation phase and resist the immune response. In the homeostatic resistance mode, resistant clones are not present before the treatment initiation but emerge under the additional immune pressure it generates. Brown, yellow, and green cells denote different resistant tumor cell clones and blue cells denote sensitive tumor cell clones.
PD-L1 expression is broadly used as a biomarker for the initiation of PD-(L)1 inhibitor therapy, approved by the US FDA, although it has many shortcomings (17). However, PD-L1 expression is associated with T cell infiltration and forms a scattered but not disseminated pattern in tumor tissues (31), which can result in different responses of the individual tumor cell to PD-(L)1 inhibitor. PD-L1 expression also shows heterogeneity among different anatomic sites and decreases after ICB therapy (32). Tumor cells can express neoantigens, resulting from the instability of the genome, which can be recognized by the cytotoxic immune cells and elicit effective anti-tumor immunity (33–35). Similarly, neoantigens exhibit intratumor heterogeneity, and the clonal neoantigen load rather than the subclonal one can predict ICB-treated patient survival (36, 37). Single-cell RNA-Seq data from lung adenocarcinoma patients and cell lines revealed the intratumor heterogeneity of IFN-γ signaling pathway genes and tumor antigen expression levels (38). Proteomic analysis of the single cell-derived tumor organoids also revealed that the human leukocyte antigen (HLA) class I peptides are heterogeneous within a tumor mass (39). Rosenthal et al. reported 64 untreated early-stage NSCLC patients in the TRACERx 100 cohort with 164 tumor regions and found a great heterogeneity of the immune infiltration status among different regions of the same tumor in the same patient (40). The different immune infiltration status also leads to different immune editing levels (40).
After the initiation of ICBs, responders experienced genomic contraction, loss of the tumor cells expressing specific neoantigens, expansion of specific T cells targeting the corresponding neoantigens, and upregulation of other checkpoints (such as TIGIT, TIM-3, and VISTA), with the left cancer clones more resistant to the activated immunity (41). Gu et al. established a unique mouse system that utilized clonal tracing and mathematical modeling to monitor the growth of each cancer clone in response to ICBs, revealing that ICB-resistant cancer clones pre-exist in the tumor mass and finally result in AR (42). The smaller the tumor size, the more abundant are the ICB-resistant clones (42). One of the identified resistant clones had higher expression of genes involved in DNA replication and sterol biosynthesis and lower expression of those involved in ribosome biogenesis. The other resistant clone had a higher expression of genes involved in interferon response and lower expression of those involved in growth factor binding. Both clones had higher expression of genes indicating cytotoxic T cell dysfunction, while the MHC-I expression and the sensitivity to IFN-γ were normal (42). Additionally, Darwinian natural selection can be exemplified by the dissociated responses (43) to ICBs and the phenomenon that patients with prior PR are more likely to acquire resistance to immunotherapy compared to those with CR (Table 2).
The second type of resistance to immunotherapy is AR in sensitive tumor cells, also called “homeostatic resistance” (29). It is easy to understand that adaptive changes can occur within tumor cells to help them survive the drug-induced massacre. In the field of target therapy for lung cancer, epidermal growth factor receptor (EGFR) T790M mutation gradually emerges with the initial mutation of 19del or L858R unchanged, thus leading to AR to the first- or second-generation tyrosine kinase inhibitors (TKIs) (44, 45). One example of this type of resistance for immunotherapy is that tumor cells can upregulate PD-L1 under the pressure of the immune cell-secreted IFN-γ (46).
It is difficult to study the underlying mechanisms of AR following the two categories mentioned above, although they are easy to understand. Regarding a specific mechanism underlying AR (such as a certain gene mutation), researchers have not been able to determine whether this trait pre-exists in the tumor or is truly acquired at the individual cell level (high-resolution technologies will fuel the related research, such as single-cell RNA-seq (47) and spatial multi-omics (48)). It seems that Darwinian natural selection has a greater contribution to AR than homeostatic resistance (49, 50); however, the question remains unanswered. Furthermore, the two categories mentioned above do not completely fulfill the role of immune cells, which is fundamental for anti-tumor immunity. Thus, we classified the underlying mechanisms of AR to immunotherapy into tumor intrinsic factors and extrinsic factors (Figure 2).
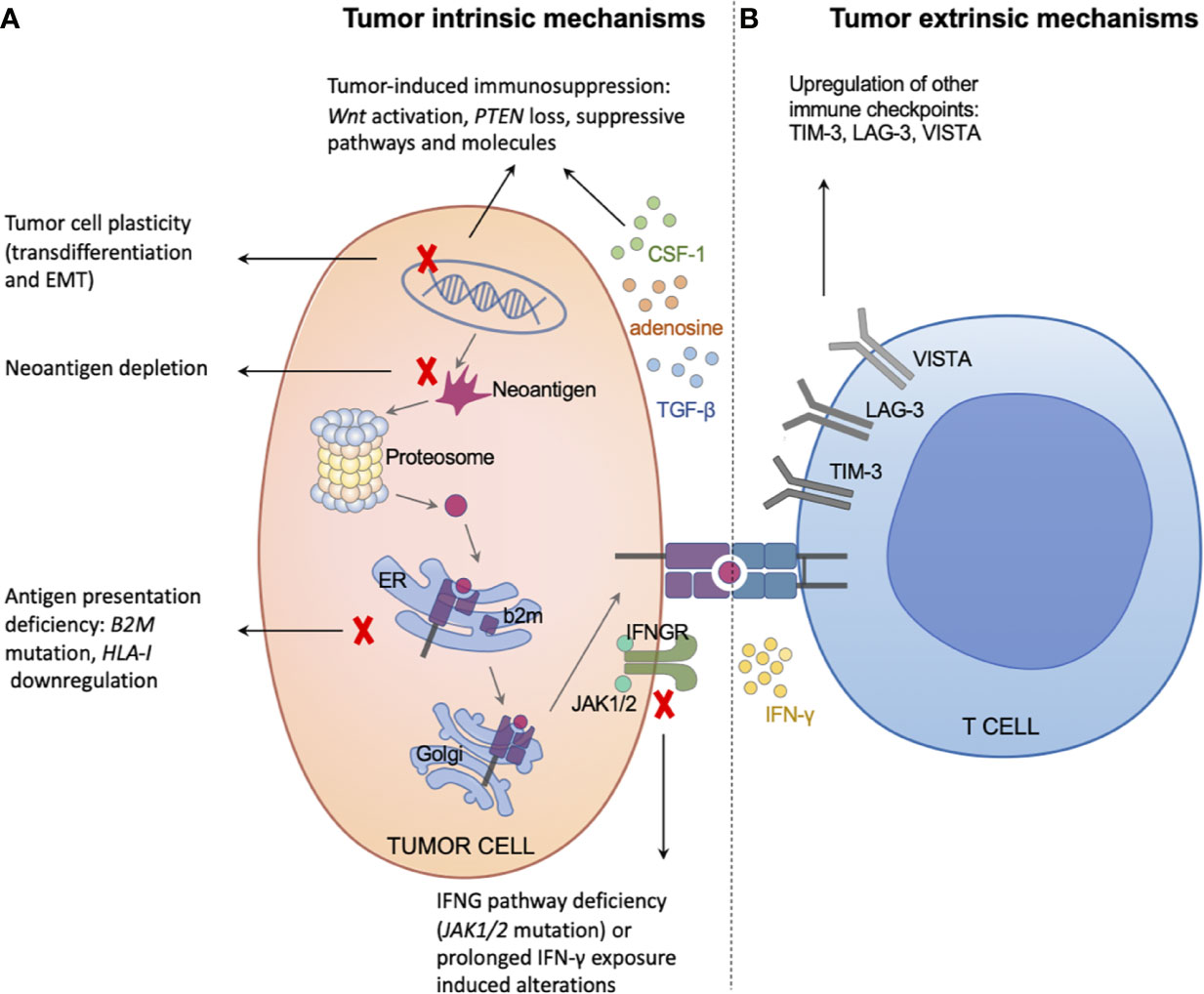
Figure 2 Tumor intrinsic and extrinsic mechanisms of acquired resistance to immunotherapy. (A) The left panel illustrates tumor intrinsic mechanisms of acquired resistance, including neoantigen depletion, defects in antigen presentation machinery, interferon signaling deficiency or prolonged exposure, tumor-induced exclusion/immunosuppression, and tumor cell plasticity. (B) The right panel illustrates tumor extrinsic mechanisms of acquired resistance, mainly through upregulating other immune checkpoints, such as TIM-3, LAG-3, and VISTA.
Tumor Intrinsic Mechanisms Underlying AR
Neoantigen Depletion
High-quality neoantigens are crucial for the efficacy of ICBs (37), and their depletion results in AR. Under the pressure of immune elimination, cancer cells can escape immune attack through the HLA loss of heterozygosity (LOH) or neoantigen depletion (40, 51). The neoantigen depletion is more pronounced in patients with intense immune infiltration or with HLA intact than HLA LOH (40). In a study (10) including four NSCLC patients with AR to ICBs, the comparison between pre-treatment and post-treatment samples revealed that some predicted neoantigens were lost in the post-treatment samples with no significant differences in the expression of PD-L1, PD-1, CTLA-4, or genetic aberrations related to immune responses. In addition, the lost neoantigens have high affinity to MHC and TCR, which can stimulate the T cells in vitro in the corresponding patients (10). Similarly, another group (52) also identified the reduced expression of high-immunoreactivity neoantigens in a metastatic uterine leiomyosarcoma patient with AR to PD-1 blockade, although the biallelic PTEN loss was also detected in the treatment-resistant tumor, indicating that the same tumor can resist immunotherapy through more than one mechanism. Notably, tumor antigen depletion is also a usual resistance mechanism in T cell transfer therapy (53, 54). However, it remains elusive whether the depletion of neoantigens is a consequence of Darwinian natural selection with the neoantigen-expressing clones eradicated or is a biological adaptation at an individual cancer cell level; the former seems inevitable for cancer immunotherapy, the strategies of which are inducing new immunogenicity, while the latter can be overcome by the modulation of certain molecular events.
Defects in Antigen Presentation Machinery
Tumor antigens can only be recognized by the cytotoxic T lymphocyte after combining with MHC-I and being presented on the cancer cell surface (55), while the defects in this process result in AR. The effective antigen presentation machinery depends on the normal functions of HLA-I, TAP, beta-2-microglobulin (B2M), and other molecules (56). Under the pressure of immune infiltration, cancer cells can evade immune attacks through aberrations of antigen-presenting genes; furthermore, the mutations of HLA genes are more likely to occur in the TCR-binding domain (57, 58). In a study (12) including four metastatic melanoma patients with AR to pembrolizumab, homozygous B2M frame-shift deletion was detected in a resistant tumor sample, with IHC validating the loss of MHC class I heavy chain at the cancer cell outer-membrane, even though diffuse intracellular staining indicated continued production of MHC class I molecules, in line with the transporting and stabilizing function of beta-2-microglobulin. B2M and the location of MHC-I were normal in the pre-treatment sample of this patient (12). The biallelic loss of B2M (homozygous mutation or heterozygous mutation combined with LOH) in the resistant tumor is also confirmed by other groups (13, 59).
However, the efficacy of ICBs is heterogeneous among patients with B2M LOH, for retaining a wide type allele of B2M. In a study by Sade-Feldman et al. (59), B2M LOH was detected in both the pre-treatment tumor and the post-treatment tumor of Pat 99, while the beta-2-microglobulin protein was lost only in the post-treatment sample, in line with the patient acquiring resistance after a prior response. In Pat 25, B2M LOH and beta-2-microglobulin protein loss were both detected in the pre-treatment tumor and post-treatment tumor, with the patient primarily resisting therapy (59). In a larger cohort, B2M LOH was also detected in patients with response to ICBs (4/36, 11.1%), although it mainly occurred in patients resistant to ICBs (20/69, 28.9%), with a significant difference (p = 0.03) (59). Additionally, B2M LOH can predict a worse survival to checkpoint blockade therapy (59). These phenomena indicate that B2M LOH can increase the possibility of beta-2-microglobulin loss, and the protein loss can result in resistance to ICBs, while B2M LOH solely cannot. The retained wide type allele of B2M may undergo epigenetic modification. Similarly, the relationship between single allelic truncating B2M mutation and AR remains elusive.
In contrast, some studies reported the inessential role of B2M in ICBs immunotherapy. Three retrospective studies of patients with colorectal carcinoma both found that a B2M mutation was more likely to happen in the microsatellite instability-high (MSI-H) patients and the B2M status was not associated with tumor immune infiltration (60–62). Furthermore, Middha et al. reported that B2M status was not associated with MHC-I expression and the MSI-H patients with inactivating mutation of B2M can still respond to ICBs, with the beta-2-microglobulin loss validated by IHC (62). Based on a study of patients with mismatch repair deficiency (d-MMR) and its supplementary information (11), the mutation of B2M can also be detected in patients with response to ICBs, though the IHC results were not available. The probable explanation of these phenomena is that the neoantigen-rich tumors (MSI-H or d-MMR) can present the antigen, independent of beta-2-microglobulin or beta-2-microglobulin, playing a different role in primary resistance compared to AR. Rizvi et al. also reported a patient with biallelic B2M mutation responding to anti-PD-1 therapy, with IHC validating the beta-2-microglobulin loss (63). However, detailed information was not available to evaluate the status of MSI or MMR (63). The relationship between beta-2-microglobulin, MHC-I, and cancer immune response needs further exploration, especially in the MSI-H or d-MMR tumors.
HLA-I is another component of antigen presentation machinery. There are limited reports about the contribution of HLA-I mutation to the efficacy of ICBs for the high polymorphism of the HLA loci (64), but it seems that there are still some recurrent mutations that are positively selected to resist immune attack (57, 64). The two studies (65, 66) contradicted each other about whether LOH of HLA-I is associated with response or resistance to ICBs, though it is widely accepted that HLA LOH is a strategy of cancer cells to evade immune attack (51) and the HLA-corrected TMB shows better predictive value than TMB (67). In a case of T cell transfer therapy targeting mutant KRAS (68), the patient progressed after the prior response to the injected HLA-C*08:02–restricted tumor-infiltrating lymphocytes. The resistant lesion was resected and found to have lost the chromosome 6 haplotype encoding the HLA-C*08:02 class I major histocompatibility complex (MHC) molecule (68). However, high-grade evidence is still needed to illustrate whether HLA-I LOH can result in AR to ICBs. Some studies reported the relationship between transcriptional downregulation of MHC class I molecule and resistance to immunotherapy. Paulson et al. (69) reported two patients with AR to combined immunotherapy (T cell transfer combined with ICBs). Comparison of the pre-treatment and post-treatment tumor from the first patient revealed the transcriptional loss of HLA-B with the injection of HLA-B restricted CD8+ T cells (69). The resistant tumor from the second patient transcriptionally downregulated HLA-A with the injection of HLA-A restricted CD8+ T cells (69). Furthermore, the transcriptional loss of HLA-I can be rescued by hypomethylating agents (69). Lee et al. (70) reported that MHC class I downregulation was a hallmark of resistance to PD-1 inhibitors and was associated with the MITFlow/AXLhigh de-differentiated phenotype and cancer-associated fibroblast signatures. In addition, the resistant phenotype was driven by TGB-β (70). Mechanically, the transcriptional loss of HLA-I can be mediated by PRC2 (71). It is worth noting that some emerging factors can also affect the antigen presentation machinery, such as HPV16 E5 (72), IL-8 (73), and autophagy (74). The relationship between these factors and AR needs to be researched in the future.
Dual Effects of Interferon Signaling
IFN-γ plays a pivotal role in ICB therapy, including directly killing cancer cells (75), upregulating MHC-I (76), upregulating PD-L1 (46), and other immune-modulating functions (77). The intact IFN-γ pathway includes IFNGR1, IFNGR2, JAK1, JAK2, STAT1, and the downstream response elements, as well as negative modulating elements such as SOCS-1 (77, 78). Gao et al. found that melanoma tumors resistant to CTLA-4 inhibitor contain genomic defects in IFN-γ signaling genes and confirmed that the knockdown of the IFNGR1 gene in B16/BL6 tumors diminished the efficacy to CTLA-4 inhibitor (79). Similarly, the biallelic JAK1/2 loss-of-function mutation leads to the defects of IFN-γ induced PD-L1 expression, resulting in primary resistance to PD-1 blockades (80). In the study of four melanoma patients with AR to pembrolizumab (12), the resistant tumors of two patients were found to harbor JAK1 or JAK2 truncating mutation combined with LOH, leading to the biallelic loss of function, while no JAK1/2 mutations were seen in the baseline tumors. On the functional level, the JAK1/2 loss of function leads to the defects of IFN-γ induced growth arrest, MHC-I expression, and PD-L1 expression (12). The findings were also validated by another group (13). Moreover, the genomic analysis revealed that the loss of tumor suppressor CDKN2A can enhance the susceptibility of JAK2 loss, increasing the rate of AR (81). However, Luo et al. reported the difference between JAK1 and JAK2, with JAK1 deficiency being able to mediate resistance to anti-PD-L1 immunotherapy while JAK2 could not (82), consistent with a recent report that metastases with JAK2 loss-of-function might be regressing under immunotherapy (83). As for the LOH or heterozygous mutations of the IFN-γ signaling genes, retaining a wide type allele, the tumors are still under effective immune attack (84, 85).
Although the defects of IFN-γ signaling can drive AR, the prolonged interferon signaling can also elicit resistance to ICBs through multiple inhibitory pathways, such as upregulation of IDO and other immune checkpoint ligands (86). IFN-γ serves as an essential element for cytotoxic T cell-dependent cancer genome immunoediting (87). In the preclinical study (88), IFN-β can increase the tumor infiltration of regulatory T cells via NOS2, resulting in immunosuppression and AR. Besides, IFN-β can coordinate with all-trans retinoic acid to upregulate CD38, resulting in resistance via the adenosine receptor signaling (89).
Tumor-Induced Exclusion/Immunosuppression
The immune desert tumors are well-known to resist immunotherapy due to the lack of pre-existing immune elements (90). Spranger et al. first reported that the tumor-intrinsic Wnt/β-catenin activation is the main cause of immune desert (91). Besides, Peng et al. reported that loss of PTEN can decrease the immune infiltration, thus promoting the formation of immune desert (92). PTEN loss of function occurs via the PI3K-Akt pathway (93). In the study of Peng et al. (92), PTEN loss resulted in primary resistance in mice receiving adoptive cell therapy, and PTEN loss positively correlated with primary resistance in a patient cohort receiving anti-PD-1 therapy. As for AR, George et al. reported a patient with metastatic uterine leiomyosarcoma receiving anti-PD-1 monotherapy, the resistant sample of which was detected with biallelic PTEN loss (52). Trujillo et al. also reported biallelic PTEN loss as an AR mechanism for a patient with melanoma receiving combined immunotherapy of anti-PD-1 and anti-CTLA-4 (94). They also reported a patient with melanoma receiving melanoma-peptide/interleukin-12 vaccine, the resistant sample of which was detected with β-catenin activation (94). In the cohort of melanoma patients with AR to ICBs (95), PTEN loss was detected in four patients and β-catenin activation was detected in two patients, without overlap.
In addition to genetic changes, tumor cells can also induce immunosuppression to resist immunotherapy via multiple approaches. Under the pressure of ICBs-induced immune activation, tumor cells can increase the infiltration of regulatory T cells via IFN-β/NOS2 (88), recruit myeloid-derived suppressor cells (MDSCs) via PD-L1-NLRP3-Wnt5a-CXCR2 (96), and secrete CSF-1 to increase the level of tumor-associated macrophages (TAMs) (97). However, it remains elusive whether CSF-1 induced TAMs are immunoreactive or immunosuppressive (16). The clinical trial of anti-CSF-1R antibody largely failed (98). Intriguingly, TAMs can also steal anti-PD-1 mAbs to alleviate efficacy (99). Kim et al. identified two immune subtypes of triple-negative breast cancer, including neutrophil-enriched (NES) and macrophage-enriched subtypes (MES), and the MES-to-NES conversion can mediate acquired ICBs resistance (100). Furthermore, tumor-derived bone morphogenetic protein 7 (BMP7), a member of the TGF-β superfamily, promotes resistance to immunotherapy (101). Adenosine is considered an important immunosuppressive cytokine (102). During the treatment of PD-1 inhibitors, tumor cells can upregulate CD73 to increase adenosine production, leading to AR (103). Moreover, the upregulation of CD73 was validated in the resistant tumor from a melanoma patient with initial CR to pembrolizumab, while CD73 was at a relatively low level in the baseline tumor (103). Similarly, tumor cells can also upregulate CD38 to promote adenosine production, leading to AR (89). Indoleamine 2,3 dioxygenase (IDO) is the rate-limiting enzyme that catabolizes tryptophan (Trp) into kynurenine (Kyn), controlling the function of T cells (104). The upregulation of IDO in tumor cells can also induce immunosuppression, resulting in resistance (105, 106).
Tumor Cell Plasticity Drives Acquired Resistance
Tumor cell plasticity represents the ability of tumor cells to undergo phenotypic changes in response to environmental stimuli without modifying their genome. The contribution of tumor cell plasticity to the acquired anti-tumor therapy resistance has been recognized recently (107, 108).
One important issue is transdifferentiation. The transdifferentiation of non-small-cell lung cancer (NSCLC) to small-cell lung cancer (SCLC) is considered to be one of the AR mechanisms to target therapy (109). It is thought that this transdifferentiation depends on the inactivation of RB1, without the dependency of EGFR status or tyrosine kinase inhibitors use (109). There are indeed several case reports about the ICBs induced transdifferentiation of NSCLC to SCLC (110–112). There were five patients in total. Four of them were treated with nivolumab and one with pembrolizumab. After progression on ICB, three of them received EC chemotherapy (etoposide-carboplatin) and all responded. However, the ICBs induced transdifferentiation and the underlying mechanisms data from large cohorts is yet to be confirmed.
Another issue is the epithelial−mesenchymal transition (EMT). EMT is thought to be associated with extensive anti-tumor therapy resistance (107, 108, 113). Sehgal et al. found that immunotherapy persister cells, which can resist CD8 T-cell mediated killing, expressed Snai1 and stem cell antigen-1 (Sca-1), and exhibited hybrid epithelial-mesenchymal features (114). SOX2, another transcription factor associated with EMT, was reported to promote resistance to ICBs (115). Additionally, Dongre et al. found that quasi-mesenchymal cells can not only resist anti-CTLA-4 therapy but also protect epithelial cells from immune attack (116). Tumor cell plasticity is a concept that easy to understand but hard to research. The high-resolution technologies may help the related research, such as single-cell RNA-seq (47) and spatial multi-omics (48).
Tumor Extrinsic Mechanisms Underlying AR
The main tumor extrinsic mechanism for AR is the upregulation of other immune checkpoints, such as TIM-3, LAG-3, and VISTA (117). With tumor progression, other immune checkpoints are sequentially upregulated, with PD-1 upregulated as an early event, while LAG-3 and BTLA upregulated as a late event (118). BTLA has two classical inhibitory motifs, ITIM and ITSM, and an immune activating motif Grb2 (119, 120). Ritthipichai et al. reported that CD8+BTLA+ tumor- infiltrating lymphocytes exerted better anti-tumor immunity than the BTLA- counterpart (120). Therefore, the specific contribution of BTLA to AR needs further exploration. Moreover, T cells with PD-1 high expression are more severely exhausted and show poorer response to PD-1 inhibitors, while T cells with PD-1 moderate expression can be saved by PD-1 inhibitors (118). Riaz et al. found that multiple immune checkpoints were upregulated during nivolumab treatment (41). Through mouse models with AR to PD-1 inhibitors, Koyama et al. found that PD-1 inhibitors were still bound to the T cells in drug-resistant specimens, but TIM-3 upregulation resulted in drug resistance (121). In addition, the anti-TIM-3 antibody was able to overcome the drug resistance, but CTLA-4 and LAG-3 were upregulated sequentially (121). Furthermore, they also reported two cases of AR to PD-1 inhibitors, in which TIM-3 was upregulated (121). Shayan et al. found that the PD-1 inhibitors induced TIM-3 upregulation was achieved through the PI3K-Akt pathway (122). Recently, the TIM-3/Galectin-9 pathway has been shown to induce primary resistance and AR to ICBs (123). In the preclinical study, Huang et al. found that when one of these three immune checkpoints, PD-1, LAG-3, and CTLA-4, was inhibited, the other two were upregulated, thus limiting the efficacy of the single inhibitor (124). In the cohort of NSCLC patients with AR to ICBs (13), eight patients were able to obtain matched samples before and after treatment, of which three patients were detected with TIM-3 upregulation, five with LAG-3 upregulation. In addition, the three patients with TIM-3 upregulation also demonstrated a co-upregulation with LAG-3 (13). MHC-II is a ligand of LAG-3 (125). For the MHC-II positive tumors, not only LAG-3 was upregulated in the resistant specimens, but also FCRL6, another receptor of MHC-II, was upregulated and suppressed the immune function (126). FCRL6 might be an emerging immune checkpoint (126). Furthermore, their group also found that the upregulation of TIM-3 and LAG-3 was not associated with primary resistance, but only with AR (126).
Gao et al. first reported the upregulation of VISTA in prostate cancer patients after CTLA-4 inhibitor treatment (127). In the cohort of melanoma patients with AR to ICBs (95), twelve patients were able to obtain matched samples before and after treatment, eight of which were detected with VISTA upregulation.
TIGIT was first identified by Yu et al. in 2009 as an immune checkpoint rheostat that suppresses the activation of T cells (128). TIGIT is mainly expressed on T cell subsets (including Tregs and memory T cells) and NK cells (128, 129). Indeed, a preclinical study showed that TIGIT blockade can prevent NK cell exhaustion and elicit potent anti-tumor immunity (130). A preliminary phase II clinical trial showed that the combination of TIGIT blockade and anti-PD-L1 antibody has better objective response rate (ORR) and median Progression Free Survival (mPFS) than anti-PD-L1 antibody alone in the first line setting (131). However, the specific contribution of TIGIT to AR is still not clear. It is unknown whether the superiority of the combination with TIGIT blockade can be translated into later line settings, which means conquering the real AR to prior immune checkpoints.
Although the upregulation of other immune checkpoints has been considered the main tumor extrinsic mechanism to AR, some other tumor extrinsic factors also potentially result in AR and need further research, including tumor vasculature, systemic and tumoral metabolic status, microbiome, and multiple cytokines.
Potential strategies to Overcome AR
Understanding the clinical characteristics and underlying mechanisms of AR is ultimately required for overcoming them. As mentioned above, the mechanisms of AR are highly diversified, including neoantigen depletion, defects in antigen presentation machinery, aberrations of interferon signaling, tumor-induced exclusion/immunosuppression, tumor cell plasticity, and upregulation of other immune checkpoints. Patients may resist ICBs through one of these mechanisms or a combo of them. So, it is hard to find one strategy to fit all the situations. The more executable strategy is to apply a set of treatment modalities to patients with the same clinical characteristics or resistance mechanisms (Table 3).
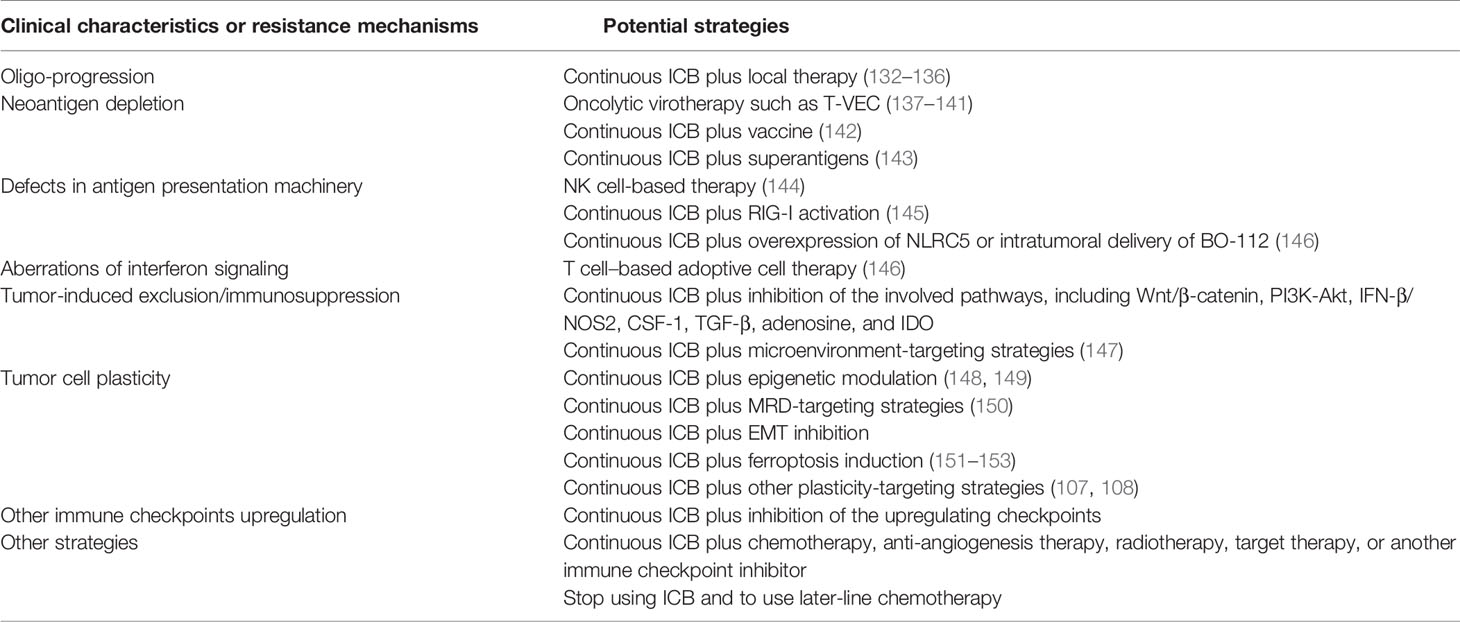
Table 3 The potential strategies to overcome acquired resistance.
Strategies for Oligo-Progression
Since most patients with AR to ICBs develop oligo-progression (Table 2), local therapy becomes the most available way to overcome resistance. Radiotherapy can not only control the local disease but also shows a synergistic effect on immunotherapy (132, 133), thus being a preferred choice. Local resection is another choice worthy of consideration that has been proved safe for patients with advanced NSCLC after ICB therapy (134, 135). Cryotherapy also seems feasible under certain conditions (136). In the Keynote 006 study, three patients received local resection and a second-course pembrolizumab therapy after the single-site progression with initial complete response, two of whom achieved a second-course complete response while one achieved stable disease (26). In a retrospective cohort of 26 patients with NSCLC, 15 patients who received local therapy achieved better overall survival than the total population (27).
Strategies for Neoantigen Depletion
Oncolytic virotherapy is an effective way to elicit new immunogenicity for tumors lacking in neoantigen (137–140). In the case reports of talimogene laherparepvec (T-VEC) treatment to overcome AR to ICBs in melanoma patients, three of six patients were evaluated as PR (141). Simultaneous anti-PD-1 and vaccine therapy can also reverse resistance (142). Furthermore, superantigens (SAgs) treatment emerges as a promising strategy to elicit immunogenicity (143). The detailed approaches to heat the cold tumor can be referred to in another review (20).
Strategies for Defects in Antigen Presentation Machinery
As for the defects in antigen presentation machinery, the coping strategy is either repairing the defects or activating the anti-tumor immunity independent of antigen presentation. However, the current knowledge about both approaches is very lacking. NK cell-based therapy might be an effective way to overcome this type of resistance, for the MHC-I loss is an activating signal for NK cell toxicity (144). RIG-I activation is also a promising strategy (145). Kalbasi et al. reported that overexpression of NLRC5 (nucleotide-binding oligomerization domain-like receptor family caspase recruitment domain containing 5) and intratumoral delivery of BO-112, a potent nanoplexed version of polyinosinic: polycytidylic acid (poly I: C), each restored MHC-I expression (146).
Strategies for Aberrations of Interferon Signaling
Defects in tumor-intrinsic interferon (IFN) signaling fail ICBs against cancer, but these tumors still maintain sensitivity to the T cell-based adoptive cell therapy (ACT). Kalbasi et al. reported that ACT was effective against tumors with JAK2 loss (146). As tumors with JAK1 loss show significant downregulation of MHC-I, ACT combined with overexpression of NLRC5 or intratumoral delivery of BO-112 was an effective approach (146). As for the IFN-β induced resistance, IFN-β inhibition might be an effective strategy.
Strategies for Tumor-Induced Exclusion/Immunosuppression
Since tumor-induced exclusion/immunosuppression involves the activation of multiple signaling pathways, pharmacological inhibition would be effective in this regard, including pathways of Wnt/β-catenin, PI3K-Akt, IFN-β/NOS2, CSF-1, TGF-β, adenosine, and IDO. Additionally, microenvironment-targeting combinations are also the future directions of cancer immunotherapy and can be referred to in another review (147).
Strategies for Tumor Cell Plasticity
Epigenetic modulation is an effective approach to restrict tumor cell plasticity, but concrete strategies need to be explored (148, 149). Targeting minimal residual disease (MRD) is worthy of consideration for decreasing the tumor load to restrict plasticity (150). Selective inhibition of the EMT program might also help overcome AR to ICBs. Tsoi et al. found that melanoma can be categorized into four subtypes following a differentiation trajectory with subtype-specific sensitivity to ferroptosis induction. This presents a therapeutic approach to target the differentiation plasticity to increase the efficacy of immunotherapy (151). Besides, induction of ferroptosis has other immune-activating functions (152, 153). The detailed strategies to target tumor cell plasticity can be referred to in the other reviews (107, 108).
Strategies for Upregulation of Other Immune Checkpoints
Theoretically, the sequential inhibition of other immune checkpoint receptors such as TIM-3, LAG-3, and VISTA can help overcome AR. Indeed, dozens of early-phase clinical trials have been initiated to explore the clinical efficacy of these checkpoint blockades with or without another checkpoint blockade, which can be referred to in other reviews (154–156). TIGIT is expressed on both T cells and NK cells and TIGIT blockade can elicit NK cell-based anti-tumor immunity (128–130), which might have specific significance for patients with AR to T cell-based ICB therapy. Besides, a phase II clinical trial has showed that TIGIT blockade can synergize with anti-PD-L1 antibody (131). The TIGIT blockade combinations are also promising candidates to overcome AR. It should be noted that the line of the designed therapy is important with regard to overcoming AR. Even if a combination strategy shows superior efficacy in the first-line setting, whether it can overcome AR to prior immune checkpoint blockades (in the second-line setting after prior ICB failure) is still unanswered. Above all, the evidence from these clinical trials and real-world studies is awaited.
Other Strategies
Since the guidelines or expert opinions about the treatment after ICB failure are scarce, there are various approaches in clinical practice to treat patients with AR to ICBs. Continuation of prior ICB combined with other therapies, such as chemotherapy, anti-angiogenesis therapy, radiotherapy, target therapy, and another immune checkpoint inhibitor, is frequently used. Another approach usually used is by changing immunotherapy into classical later-line modalities of chemotherapy. The evidence to support these attempts is at its infancy and arises a core question as to whether to stop using the prior ICBs. The INSIGNA study (https://clinicaltrials.gov, NCT03793179) will preliminarily answer the question about which is superior between prior ICB plus chemotherapy and chemotherapy alone for the patients with AR to ICBs.
Discussion
With the increasing use of ICBs in clinical practice, a large number of patients develop AR. However, the knowledge about AR to ICBs is limited and poorly summarized. Through reviewing the published papers associated with this term, we clarified the principal elements of it. The definition of AR to ICBs is inconsistent at present, but expert opinions are available for this question. Oligo-progression is the main resistance pattern. The underlying mechanisms of AR involve tumor intrinsic factors and extrinsic factors. Neoantigen depletion can protect tumor cells from immune attack, however, it is unclear whether the depletion of neoantigens is a consequence of Darwinian natural selection with the neoantigen-expressing clones being eradicated or is a biological adaptation at an individual cancer cell level. Antigen presentation deficiency can also protect tumor cells from being recognized and killed by the immune cells, which includes biallelic B2M mutation and HLA-I down-regulation. IFN-γ is a crucial mediator of anti-tumor immunity. The defects in the IFNG pathway can lead to AR, while the prolonged IFN-γ exposure can also elicit resistance by upregulating IDO and other immune checkpoint ligands. Under the pressure of immune elimination, tumor cells can induce an immunosuppressive microenvironment through the genetic approaches (Wnt/β-catenin activation and PTEN loss) and the non-genetic approaches (recruiting Treg, TAM, and MDSC and upregulating IDO, adenosine, and TGF-β pathway). Tumor cell plasticity preserves the great potential for AR. One important issue is transdifferentiation from NSCLC cells to SCLC cells, the other is the EMT program. The tumor extrinsic mechanisms of AR are mainly through the upregulation of other immune checkpoints, such as TIM-3, LAG-3, and VISTA. The potential strategy to overcome AR is to apply a set of modalities to patients with the same clinical characteristics or resistance mechanisms. Although not discussed in this review, monitoring resistance is an important issue, and the circulating tumor DNA could be an effective tool.
One of the future directions in this field is to explore the mechanisms underlying AR to ICBs more comprehensively. New tools, such as CRISPER screen, single-cell RNA sequencing, and spatial multi-omics, will fuel the related research. It is worthy of noting that many mechanisms of primary resistance to ICBs have not been completely elucidated in the field of AR. For example, gut microbiota and tumor metabolism have been acknowledged to play important roles in primary resistance to ICBs (157–159), while their contribution to AR is largely unknown and thus needs future exploration. The other issue in this field is that translational research is urgently needed to extend patients’ survival. Through efforts to overcome AR, immunotherapy can be a promising therapeutic modality for curing cancer.
Author Contributions
QC conceptualized this review. BZ searched the papers and drafted the manuscript. YG, PZ, and QC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of China (Grant No. 82072597 and No. 81974483).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.693609/full#supplementary-material
References
1. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
2. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab Plus Dacarbazine for Previously Untreated Metastatic Melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
3. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab Versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med (2016) 375(19):1823–33. doi: 10.1056/NEJMoa1606774
4. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373(2):123–35. doi: 10.1056/NEJMoa1504627
5. Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab Plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med (2018) 379(23):2220–9. doi: 10.1056/NEJMoa1809064
6. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab Plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
7. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
8. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br J Cancer (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
9. Syn NL, Teng MWL, Mok TSK, Soo RA. De-Novo and Acquired Resistance to Immune Checkpoint Targeting. Lancet Oncol (2017) 18(12):e731–41. doi: 10.1016/s1470-2045(17)30607-1
10. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape During Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov (2017) 7(3):264–76. doi: 10.1158/2159-8290.Cd-16-0828
11. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
12. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated With Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
13. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov (2017) 7(12):1420–35. doi: 10.1158/2159-8290.Cd-17-0593
14. Remon J, Passiglia F, Ahn MJ, Barlesi F, Forde PM, Garon EB, et al. Immune Checkpoint Inhibitors in Thoracic Malignancies: Review of the Existing Evidence by an IASLC Expert Panel and Recommendations. J Thorac Oncol (2020) 15(6):914–47. doi: 10.1016/j.jtho.2020.03.006
15. Kluger HM, Tawbi HA, Ascierto ML, Bowden M, Callahan MK, Cha E, et al. Defining Tumor Resistance to PD-1 Pathway Blockade: Recommendations From the First Meeting of the SITC Immunotherapy Resistance Taskforce. J Immunother Cancer (2020) 8(1):e000398. doi: 10.1136/jitc-2019-000398
16. Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity (2020) 52(1):17–35. doi: 10.1016/j.immuni.2019.12.011
17. Havel JJ, Chowell D, Chan TA. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat Rev Cancer (2019) 19(3):133–50. doi: 10.1038/s41568-019-0116-x
18. O’Donnell JS, Teng MWL, Smyth MJ. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
19. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ Book (2019) 39:147–64. doi: 10.1200/edbk_240837
20. Ochoa de Olza M, Navarro Rodrigo B, Zimmermann S, Coukos G. Turning Up the Heat on non-Immunoreactive Tumours: Opportunities for Clinical Development. Lancet Oncol (2020) 21(9):e419–30. doi: 10.1016/s1470-2045(20)30234-5
21. Schoenfeld AJ, Hellmann MD. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell (2020) 37(4):443–55. doi: 10.1016/j.ccell.2020.03.017
22. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
23. Antonia SJ, Borghaei H, Ramalingam SS, Horn L, De Castro Carpeño J, Pluzanski A, et al. Four-Year Survival With Nivolumab in Patients With Previously Treated Advanced non-Small-Cell Lung Cancer: A Pooled Analysis. Lancet Oncol (2019) 20(10):1395–408. doi: 10.1016/s1470-2045(19)30407-3
24. Wang DY, Eroglu Z, Ozgun A, Leger PD, Zhao S, Ye F, et al. Clinical Features of Acquired Resistance to Anti-PD-1 Therapy in Advanced Melanoma. Cancer Immunol Res (2017) 5(5):357–62. doi: 10.1158/2326-6066.Cir-16-0287
25. Pires da Silva I, Lo S, Quek C, Gonzalez M, Carlino MS, Long GV, et al. Site-Specific Response Patterns, Pseudoprogression, and Acquired Resistance in Patients With Melanoma Treated With Ipilimumab Combined With Anti-PD-1 Therapy. Cancer (2020) 126(1):86–97. doi: 10.1002/cncr.32522
26. Robert C, Ribas A, Schachter J, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab Versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results From an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol (2019) 20(9):1239–51. doi: 10.1016/s1470-2045(19)30388-2
27. Gettinger SN, Wurtz A, Goldberg SB, Rimm D, Schalper K, Kaech S, et al. Clinical Features and Management of Acquired Resistance to PD-1 Axis Inhibitors in 26 Patients With Advanced Non-Small Cell Lung Cancer. J Thorac Oncol (2018) 13(6):831–9. doi: 10.1016/j.jtho.2018.03.008
28. Shah S, Wood K, Labadie B, Won B, Brisson R, Karrison T, et al. Clinical and Molecular Features of Innate and Acquired Resistance to Anti-PD-1/PD-L1 Therapy in Lung Cancer. Oncotarget (2018) 9(4):4375–84. doi: 10.18632/oncotarget.23315
29. Restifo NP, Smyth MJ, Snyder A. Acquired Resistance to Immunotherapy and Future Challenges. Nat Rev Cancer (2016) 16(2):121–6. doi: 10.1038/nrc.2016.2
30. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell (2017) 168(4):613–28. doi: 10.1016/j.cell.2017.01.018
31. Kim TK, Herbst RS, Chen L. Defining and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends Immunol (2018) 39(8):624–31. doi: 10.1016/j.it.2018.05.001
32. Hong L, Negrao MV, Dibaj SS, Chen R, Reuben A, Bohac JM, et al. Programmed Death-Ligand 1 Heterogeneity and Its Impact on Benefit From Immune Checkpoint Inhibitors in NSCLC. J Thorac Oncol (2020) 15(9):1449–59. doi: 10.1016/j.jtho.2020.04.026
33. Riaz N, Morris L, Havel JJ, Makarov V, Desrichard A, Chan TA. The Role of Neoantigens in Response to Immune Checkpoint Blockade. Int Immunol (2016) 28(8):411–9. doi: 10.1093/intimm/dxw019
34. Chen DS, Mellman I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
35. Srivastava RM, Purohit TA, Chan TA. Diverse Neoantigens and the Development of Cancer Therapies. Semin Radiat Oncol (2020) 30(2):113–28. doi: 10.1016/j.semradonc.2019.12.001
36. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
37. McGranahan N, Swanton C. Neoantigen Quality, Not Quantity. Sci Transl Med (2019) 11(506):eaax7918. doi: 10.1126/scitranslmed.aax7918
38. Ma KY, Schonnesen AA, Brock A, Van Den Berg C, Eckhardt SG, Liu Z, et al. Single-Cell RNA Sequencing of Lung Adenocarcinoma Reveals Heterogeneity of Immune Response-Related Genes. JCI Insight (2019) 4(4):e121387. doi: 10.1172/jci.insight.121387
39. Demmers LC, Kretzschmar K, Van Hoeck A, Bar-Epraïm YE, van den Toorn HWP, Koomen M, et al. Single-Cell Derived Tumor Organoids Display Diversity in HLA Class I Peptide Presentation. Nat Commun (2020) 11(1):5338. doi: 10.1038/s41467-020-19142-9
40. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-Directed Immune Escape in Lung Cancer Evolution. Nature (2019) 567(7749):479–85. doi: 10.1038/s41586-019-1032-7
41. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution During Immunotherapy With Nivolumab. Cell (2017) 171(4):934–49. doi: 10.1016/j.cell.2017.09.028
42. Gu SS, Wang X, Hu X, Jiang P, Li Z, Traugh N, et al. Clonal Tracing Reveals Diverse Patterns of Response to Immune Checkpoint Blockade. Genome Biol (2020) 21(1):263. doi: 10.1186/s13059-020-02166-1
43. Borcoman E, Kanjanapan Y, Champiat S, Kato S, Servois V, Kurzrock R, et al. Novel Patterns of Response Under Immunotherapy. Ann Oncol (2019) 30(3):385–96. doi: 10.1093/annonc/mdz003
44. Gainor JF, Shaw AT. Emerging Paradigms in the Development of Resistance to Tyrosine Kinase Inhibitors in Lung Cancer. J Clin Oncol (2013) 31(31):3987–96. doi: 10.1200/jco.2012.45.2029
45. Chong CR, Jänne PA. The Quest to Overcome Resistance to EGFR-Targeted Therapies in Cancer. Nat Med (2013) 19(11):1389–400. doi: 10.1038/nm.3388
46. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of Inflammatory Response With B7-H1 Expression in Human Melanocytic Lesions Supports an Adaptive Resistance Mechanism of Immune Escape. Sci Transl Med (2012) 4(127):127ra37. doi: 10.1126/scitranslmed.3003689
47. Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, Livak KJ. Applying High-Dimensional Single-Cell Technologies to the Analysis of Cancer Immunotherapy. Nat Rev Clin Oncol (2021) 18(4):244–56. doi: 10.1038/s41571-020-00449-x
48. Liu Y, Yang M, Deng Y, Su G, Enninful A, Guo CC, et al. High-Spatial-Resolution Multi-Omics Sequencing Via Deterministic Barcoding in Tissue. Cell (2020) 183(6):1665–81.e18. doi: 10.1016/j.cell.2020.10.026
49. Riva L, Pandiri AR, Li YR, Droop A, Hewinson J, Quail MA, et al. The Mutational Signature Profile of Known and Suspected Human Carcinogens in Mice. Nat Genet (2020) 52(11):1189–97. doi: 10.1038/s41588-020-0692-4
50. Lopez-Bigas N, Gonzalez-Perez A. Are Carcinogens Direct Mutagens? Nat Genet (2020) 52(11):1137–8. doi: 10.1038/s41588-020-00730-w
51. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell (2017) 171(6):1259–71. doi: 10.1016/j.cell.2017.10.001
52. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated With Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity (2017) 46(2):197–204. doi: 10.1016/j.immuni.2017.02.001
53. Mehta A, Kim YJ, Robert L, Tsoi J, Comin-Anduix B, Berent-Maoz B, et al. Immunotherapy Resistance by Inflammation-Induced Dedifferentiation. Cancer Discov (2018) 8(8):935–43. doi: 10.1158/2159-8290.Cd-17-1178
54. Wylie B, Chee J, Forbes CA, Booth M, Stone SR, Buzzai A, et al. Acquired Resistance During Adoptive Cell Therapy by Transcriptional Silencing of Immunogenic Antigens. Oncoimmunology (2019) 8(8):1609874. doi: 10.1080/2162402x.2019.1609874
55. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
56. Springer S. Transport and Quality Control of MHC Class I Molecules in the Early Secretory Pathway. Curr Opin Immunol (2015) 34:83–90. doi: 10.1016/j.coi.2015.02.009
57. Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep (2016) 15(4):857–65. doi: 10.1016/j.celrep.2016.03.075
58. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and Genetic Properties of Tumors Associated With Local Immune Cytolytic Activity. Cell (2015) 160(1-2):48–61. doi: 10.1016/j.cell.2014.12.033
59. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to Checkpoint Blockade Therapy Through Inactivation of Antigen Presentation. Nat Commun (2017) 8(1):1136. doi: 10.1038/s41467-017-01062-w
60. Yeon Yeon S, Jung SH, Jo YS, Choi EJ, Kim MS, Chung YJ, et al. Immune Checkpoint Blockade Resistance-Related B2M Hotspot Mutations in Microsatellite-Unstable Colorectal Carcinoma. Pathol Res Pract (2019) 215(1):209–14. doi: 10.1016/j.prp.2018.11.014
61. Snahnicanova Z, Kasubova I, Kalman M, Grendar M, Mikolajcik P, Gabonova E, et al. Genetic and Epigenetic Analysis of the Beta-2-Microglobulin Gene in Microsatellite Instable Colorectal Cancer. Clin Exp Med (2020) 20(1):87–95. doi: 10.1007/s10238-019-00601-7
62. Middha S, Yaeger R, Shia J, Stadler ZK, King S, Guercio S, et al. Majority of B2M-Mutant and -Deficient Colorectal Carcinomas Achieve Clinical Benefit From Immune Checkpoint Inhibitor Therapy and are Microsatellite Instability-High. JCO Precis Oncol (2019) 3:PO.18.00321. doi: 10.1200/po.18.00321
63. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J Clin Oncol (2018) 36(7):633–41. doi: 10.1200/jco.2017.75.3384
64. Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive Analysis of Cancer-Associated Somatic Mutations in Class I HLA Genes. Nat Biotechnol (2015) 33(11):1152–8. doi: 10.1038/nbt.3344
65. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science (2018) 359(6375):582–7. doi: 10.1126/science.aao4572
66. Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, et al. Integrative Molecular and Clinical Modeling of Clinical Outcomes to PD1 Blockade in Patients With Metastatic Melanoma. Nat Med (2019) 25(12):1916–27. doi: 10.1038/s41591-019-0654-5
67. Shim JH, Kim HS, Cha H, Kim S, Kim TM, Anagnostou V, et al. HLA-Corrected Tumor Mutation Burden and Homologous Recombination Deficiency for the Prediction of Response to PD-(L)1 Blockade in Advanced Non-Small-Cell Lung Cancer Patients. Ann Oncol (2020) 31(7):902–11. doi: 10.1016/j.annonc.2020.04.004
68. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375(23):2255–62. doi: 10.1056/NEJMoa1609279
69. Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, et al. Acquired Cancer Resistance to Combination Immunotherapy From Transcriptional Loss of Class I HLA. Nat Commun (2018) 9(1):3868. doi: 10.1038/s41467-018-06300-3
70. Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A, et al. Transcriptional Downregulation of MHC Class I and Melanoma De- Differentiation in Resistance to PD-1 Inhibition. Nat Commun (2020) 11(1):1897. doi: 10.1038/s41467-020-15726-7
71. Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell (2019) 36(4):385–401. doi: 10.1016/j.ccell.2019.08.008
72. Miyauchi S, Sanders PD, Guram K, Kim SS, Paolini F, Venuti A, et al. HPV16 E5 Mediates Resistance to PD-L1 Blockade and Can Be Targeted With Rimantadine in Head and Neck Cancer. Cancer Res (2020) 80(4):732–46. doi: 10.1158/0008-5472.Can-19-1771
73. Yuen KC, Liu LF, Gupta V, Madireddi S, Keerthivasan S, Li C, et al. High Systemic and Tumor-Associated IL-8 Correlates With Reduced Clinical Benefit of PD-L1 Blockade. Nat Med (2020) 26(5):693–8. doi: 10.1038/s41591-020-0860-1
74. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature (2020) 581(7806):100–5. doi: 10.1038/s41586-020-2229-5
75. Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced In Vivo Growth and Resistance to Rejection of Tumor Cells Expressing Dominant Negative IFN Gamma Receptors. Immunity (1994) 1(6):447–56. doi: 10.1016/1074-7613(94)90087-6
76. Shirayoshi Y, Burke PA, Appella E, Ozato K. Interferon-Induced Transcription of a Major Histocompatibility Class I Gene Accompanies Binding of Inducible Nuclear Factors to the Interferon Consensus Sequence. Proc Natl Acad Sci USA (1988) 85(16):5884–8. doi: 10.1073/pnas.85.16.5884
77. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
78. Chow KT, Gale M Jr. Snapshot: Interferon Signaling. Cell (2015) 163(7):1808–. doi: 10.1016/j.cell.2015.12.008
79. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell (2016) 167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069
80. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov (2017) 7(2):188–201. doi: 10.1158/2159-8290.Cd-16-1223
81. Horn S, Leonardelli S, Sucker A, Schadendorf D, Griewank KG, Paschen A. Tumor CDKN2A-Associated JAK2 Loss and Susceptibility to Immunotherapy Resistance. J Natl Cancer Inst (2018) 110(6):677–81. doi: 10.1093/jnci/djx271
82. Luo N, Formisano L, Gonzalez-Ericsson PI, Sanchez V, Dean PT, Opalenik SR, et al. Melanoma Response to Anti-PD-L1 Immunotherapy Requires JAK1 Signaling, But Not JAK2. Oncoimmunology (2018) 7(6):e1438106. doi: 10.1080/2162402x.2018.1438106
83. Makohon-Moore AP, Lipson EJ, Hooper JE, Zucker A, Hong J, Bielski CM, et al. The Genetic Evolution of Treatment-Resistant Cutaneous, Acral, and Uveal Melanomas. Clin Cancer Res (2021) 27(5):1516–25. doi: 10.1158/1078-0432.Ccr-20-2984
84. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired Ifnγ Resistance Impairs Anti-Tumor Immunity and Gives Rise to T-Cell-Resistant Melanoma Lesions. Nat Commun (2017) 8:15440. doi: 10.1038/ncomms15440
85. Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic Features of Response to Combination Immunotherapy in Patients With Advanced Non-Small-Cell Lung Cancer. Cancer Cell (2018) 33(5):843–52. doi: 10.1016/j.ccell.2018.03.018
86. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell (2016) 167(6):1540–54. doi: 10.1016/j.cell.2016.11.022
87. Takeda K, Nakayama M, Hayakawa Y, Kojima Y, Ikeda H, Imai N, et al. IFN-γ Is Required for Cytotoxic T Cell-Dependent Cancer Genome Immunoediting. Nat Commun (2017) 8:14607. doi: 10.1038/ncomms14607
88. Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, et al. Sustained Type I Interferon Signaling as a Mechanism of Resistance to PD-1 Blockade. Cell Res (2019) 29(10):846–61. doi: 10.1038/s41422-019-0224-x
89. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape From PD-1/PD-L1 Blockade. Cancer Discov (2018) 8(9):1156–75. doi: 10.1158/2159-8290.Cd-17-1033
90. Hegde PS, Karanikas V, Evers S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res (2016) 22(8):1865–74. doi: 10.1158/1078-0432.Ccr-15-1507
91. Spranger S, Bao R, Gajewski TF. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
92. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov (2016) 6(2):202–16. doi: 10.1158/2159-8290.Cd-15-0283
93. Song MS, Salmena L, Pandolfi PP. The Functions and Regulation of the PTEN Tumour Suppressor. Nat Rev Mol Cell Biol (2012) 13(5):283–96. doi: 10.1038/nrm3330
94. Trujillo JA, Luke JJ, Zha Y, Segal JP, Ritterhouse LL, Spranger S, et al. Secondary Resistance to Immunotherapy Associated With β-Catenin Pathway Activation or PTEN Loss in Metastatic Melanoma. J Immunother Cancer (2019) 7(1):295. doi: 10.1186/s40425-019-0780-0
95. Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, et al. Negative Immune Checkpoint Regulation by VISTA: A Mechanism of Acquired Resistance to Anti-PD-1 Therapy in Metastatic Melanoma Patients. Mod Pathol (2017) 30(12):1666–76. doi: 10.1038/modpathol.2017.89
96. Theivanthiran B, Evans KS, DeVito NC, Plebanek M, Sturdivant M, Wachsmuth LP, et al. A Tumor-Intrinsic PD-L1/NLRP3 Inflammasome Signaling Pathway Drives Resistance to Anti-PD-1 Immunotherapy. J Clin Invest (2020) 130(5):2570–86. doi: 10.1172/jci133055
97. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T Cell-Induced CSF1 Promotes Melanoma Resistance to PD1 Blockade. Sci Transl Med (2018) 10(436):eaan3311. doi: 10.1126/scitranslmed.aan3311
98. Pradel LP, Ooi CH, Romagnoli S, Cannarile MA, Sade H, Rüttinger D, et al. Macrophage Susceptibility to Emactuzumab (RG7155) Treatment. Mol Cancer Ther (2016) 15(12):3077–86. doi: 10.1158/1535-7163.Mct-16-0157
99. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage-Mediated Resistance Pathway in Anti-PD-1 Therapy. Sci Transl Med (2017) 9(389):eaal3604. doi: 10.1126/scitranslmed.aal3604
100. Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-Subtyping of Breast Cancer Reveals Distinct Myeloid Cell Profiles and Immunotherapy Resistance Mechanisms. Nat Cell Biol (2019) 21(9):1113–26. doi: 10.1038/s41556-019-0373-7
101. Cortez MA, Masrorpour F, Ivan C, Zhang J, Younes AI, Lu Y, et al. Bone Morphogenetic Protein 7 Promotes Resistance to Immunotherapy. Nat Commun (2020) 11(1):4840. doi: 10.1038/s41467-020-18617-z
102. Leone RD, Emens LA. Targeting Adenosine for Cancer Immunotherapy. J Immunother Cancer (2018) 6(1):57. doi: 10.1186/s40425-018-0360-8
103. Reinhardt J, Landsberg J, Schmid-Burgk JL, Ramis BB, Bald T, Glodde N, et al. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 During Immunotherapy. Cancer Res (2017) 77(17):4697–709. doi: 10.1158/0008-5472.Can-17-0395
104. Munn DH, Mellor AL. Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends Immunol (2013) 34(3):137–43. doi: 10.1016/j.it.2012.10.001
105. Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bossé D, et al. Metabolomic Adaptations and Correlates of Survival to Immune Checkpoint Blockade. Nat Commun (2019) 10(1):4346. doi: 10.1038/s41467-019-12361-9
106. Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M, et al. Indoleamine 2,3-Dioxygenase Provides Adaptive Resistance to Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Cancer Immunol Immunother (2018) 67(8):1305–15. doi: 10.1007/s00262-018-2190-4
107. Marine JC, Dawson SJ, Dawson MA. Non-Genetic Mechanisms of Therapeutic Resistance in Cancer. Nat Rev Cancer (2020) 20(12):743–56. doi: 10.1038/s41568-020-00302-4
108. Boumahdi S, de Sauvage FJ. The Great Escape: Tumour Cell Plasticity in Resistance to Targeted Therapy. Nat Rev Drug Discov (2020) 19(1):39–56. doi: 10.1038/s41573-019-0044-1
109. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation From Non-Small-Cell Lung Cancer to Small-Cell Lung Cancer: Molecular Drivers and Cells of Origin. Lancet Oncol (2015) 16(4):e165–72. doi: 10.1016/s1470-2045(14)71180-5
110. Abdallah N, Nagasaka M, Abdulfatah E, Shi D, Wozniak AJ, Sukari A. Non-Small Cell to Small Cell Lung Cancer on PD-1 Inhibitors: Two Cases on Potential Histologic Transformation. Lung Cancer (Auckl) (2018) 9:85–90. doi: 10.2147/lctt.S173724
111. Imakita T, Fujita K, Kanai O, Terashima T, Mio T. Small Cell Lung Cancer Transformation During Immunotherapy With Nivolumab: A Case Report. Respir Med Case Rep (2017) 21:52–5. doi: 10.1016/j.rmcr.2017.03.019
112. Bar J, Ofek E, Barshack I, Gottfried T, Zadok O, Kamer I, et al. Transformation to Small Cell Lung Cancer as a Mechanism of Resistance to Immunotherapy in Non-Small Cell Lung Cancer. Lung Cancer (2019) 138:109–15. doi: 10.1016/j.lungcan.2019.09.025
113. Shibue T, Weinberg RA. EMT. Cscs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat Rev Clin Oncol (2017) 14(10):611–29. doi: 10.1038/nrclinonc.2017.44
114. Sehgal K, Portell A, Ivanova EV, Lizotte PH, Mahadevan NR, Greene JR, et al. Dynamic Single-Cell RNA Sequencing Identifies Immunotherapy Persister Cells Following PD-1 Blockade. J Clin Invest (2021) 131(2):e135038. doi: 10.1172/jci135038
115. Wu R, Wang C, Li Z, Xiao J, Li C, Wang X, et al. SOX2 Promotes Resistance of Melanoma With PD-L1 High Expression to T-Cell-Mediated Cytotoxicity That Can Be Reversed by SAHA. J Immunother Cancer (2020) 8(2):e001037. doi: 10.1136/jitc-2020-001037
116. Dongre A, Rashidian M, Eaton EN, Reinhardt F, Thiru P, Zagorulya M, et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition (EMT)-Mediated Immunosuppression in Breast Carcinomas. Cancer Discov (2020) 11(5):1286–305. doi: 10.1158/2159-8290.Cd-20-0603
117. Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
118. Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of Lung Cancer is Associated With Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol Res (2015) 3(12):1344–55. doi: 10.1158/2326-6066.Cir-15-0097
119. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not All Immune Checkpoints are Created Equal. Front Immunol (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
120. Ritthipichai K, Haymaker CL, Martinez M, Aschenbrenner A, Yi X, Zhang M, et al. Multifaceted Role of BTLA in the Control of CD8(+) T-Cell Fate After Antigen Encounter. Clin Cancer Res (2017) 23(20):6151–64. doi: 10.1158/1078-0432.Ccr-16-1217
121. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive Resistance to Therapeutic PD-1 Blockade Is Associated With Upregulation of Alternative Immune Checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
122. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive Resistance to Anti-PD1 Therapy by Tim-3 Upregulation is Mediated by the PI3K-Akt Pathway in Head and Neck Cancer. Oncoimmunology (2017) 6(1):e1261779. doi: 10.1080/2162402x.2016.1261779
123. Limagne E, Richard C, Thibaudin M, Fumet JD, Truntzer C, Lagrange A, et al. Tim-3/Galectin-9 Pathway and Mmdsc Control Primary and Secondary Resistances to PD-1 Blockade in Lung Cancer Patients. Oncoimmunology (2019) 8(4):e1564505. doi: 10.1080/2162402x.2018.1564505
124. Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory Upregulation of PD-1, LAG-3, and CTLA-4 Limits the Efficacy of Single-Agent Checkpoint Blockade in Metastatic Ovarian Cancer. Oncoimmunology (2017) 6(1):e1249561. doi: 10.1080/2162402x.2016.1249561
125. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol Rev (2017) 276(1):80–96. doi: 10.1111/imr.12519
126. Johnson DB, Nixon MJ, Wang Y, Wang DY, Castellanos E, Estrada MV, et al. Tumor-Specific MHC-II Expression Drives a Unique Pattern of Resistance to Immunotherapy Via LAG-3/FCRL6 Engagement. JCI Insight (2018) 3(24):e120360. doi: 10.1172/jci.insight.120360
127. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an Inhibitory Immune Checkpoint That Is Increased After Ipilimumab Therapy in Patients With Prostate Cancer. Nat Med (2017) 23(5):551–5. doi: 10.1038/nm.4308
128. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
129. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. the Interaction of TIGIT With PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc Natl Acad Sci USA (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
130. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
131. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary Analysis of a Randomized, Double-Blind, Phase II Study of the Anti-TIGIT Antibody Tiragolumab (Tira) Plus Atezolizumab (Atezo) Versus Placebo Plus Atezo as First-Line (1L) Treatment in Patients With PD-L1-Selected NSCLC (CITYSCAPE). J Clin Oncol (2020)38(15_suppl):9503. doi: 10.1200/JCO.2020.38.15_suppl.9503
132. Hwang WL, Pike LRG, Royce TJ, Mahal BA, Loeffler JS. Safety of Combining Radiotherapy With Immune-Checkpoint Inhibition. Nat Rev Clin Oncol (2018) 15(8):477–94. doi: 10.1038/s41571-018-0046-7
133. Theelen W, Chen D, Verma V, Hobbs BP, Peulen HMU, Aerts J, et al. Pembrolizumab With or Without Radiotherapy for Metastatic non-Small-Cell Lung Cancer: A Pooled Analysis of Two Randomised Trials. Lancet Respir Med (2020) 9(5):467–75. doi: 10.1016/s2213-2600(20)30391-x
134. Chaft JE, Hellmann MD, Velez MJ, Travis WD, Rusch VW. Initial Experience With Lung Cancer Resection After Treatment With T-Cell Checkpoint Inhibitors. Ann Thorac Surg (2017) 104(3):e217–8. doi: 10.1016/j.athoracsur.2017.03.038
135. Hu ZI, Hellmann MD, Wolchok JD, Vyas M, Shia J, Stadler ZK, et al. Acquired Resistance to Immunotherapy in MMR-D Pancreatic Cancer. J Immunother Cancer (2018) 6(1):127. doi: 10.1186/s40425-018-0448-1
136. Adam LC, Raja J, Ludwig JM, Adeniran A, Gettinger SN, Kim HS. Cryotherapy for Nodal Metastasis in NSCLC With Acquired Resistance to Immunotherapy. J Immunother Cancer (2018) 6(1):147. doi: 10.1186/s40425-018-0468-x
137. Guo ZS, Liu Z, Bartlett DL. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front Oncol (2014) 4:74. doi: 10.3389/fonc.2014.00074
138. Chouljenko DV, Ding J, Lee IF, Murad YM, Bu X, Liu G, et al. Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes. Biomedicines (2020) 8(11):484. doi: 10.3390/biomedicines8110484
139. Liu Y, Cai J, Liu W, Lin Y, Guo L, Liu X, et al. Intravenous Injection of the Oncolytic Virus M1 Awakens Antitumor T Cells and Overcomes Resistance to Checkpoint Blockade. Cell Death Dis (2020) 11(12):1062. doi: 10.1038/s41419-020-03285-0
140. Groeneveldt C, Kinderman P, van den Wollenberg DJM, van den Oever RL, Middelburg J, Mustafa DAM, et al. Preconditioning of the Tumor Microenvironment With Oncolytic Reovirus Converts CD3-Bispecific Antibody Treatment Into Effective Immunotherapy. J Immunother Cancer (2020) 8(2):e001191. doi: 10.1136/jitc-2020-001191
141. Fröhlich A, Niebel D, Fietz S, Egger E, Buchner A, Sirokay J, et al. Talimogene Laherparepvec Treatment to Overcome Loco-Regional Acquired Resistance to Immune Checkpoint Blockade in Tumor Stage IIIB-IV M1c Melanoma Patients. Cancer Immunol Immunother (2020) 69(5):759–69. doi: 10.1007/s00262-020-02487-x
142. Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, et al. PD-1 Blockade in Subprimed CD8 Cells Induces Dysfunctional PD-1(+)CD38(Hi) Cells and Anti-PD-1 Resistance. Nat Immunol (2019) 20(9):1231–43. doi: 10.1038/s41590-019-0441-y
143. Knopick P, Terman D, Riha N, Alvine T, Larson R, Badiou C, et al. Endogenous HLA-DQ8αβ Programs Superantigens (SEG/SEI) to Silence Toxicity and Unleash a Tumoricidal Network With Long-Term Melanoma Survival. J Immunother Cancer (2020) 8(2):e001493. doi: 10.1136/jitc-2020-001493
144. Shimasaki N, Jain A, Campana D. NK Cells for Cancer Immunotherapy. Nat Rev Drug Discov (2020) 19(3):200–18. doi: 10.1038/s41573-019-0052-1
145. Such L, Zhao F, Liu D, Thier B, Le-Trilling VTK, Sucker A, et al. Targeting the Innate Immunoreceptor RIG-I Overcomes Melanoma-Intrinsic Resistance to T Cell Immunotherapy. J Clin Invest (2020) 130(8):4266–81. doi: 10.1172/jci131572
146. Kalbasi A, Tariveranmoshabad M, Hakimi K, Kremer S, Campbell KM, Funes JM, et al. Uncoupling Interferon Signaling and Antigen Presentation to Overcome Immunotherapy Resistance Due to JAK1 Loss in Melanoma. Sci Transl Med (2020) 12(565):eabb0152. doi: 10.1126/scitranslmed.abb0152
147. Murciano-Goroff YR, Warner AB, Wolchok JD. The Future of Cancer Immunotherapy: Microenvironment-Targeting Combinations. Cell Res (2020) 30(6):507–19. doi: 10.1038/s41422-020-0337-2
148. Gonda TA, Fang J, Salas M, Do C, Hsu E, Zhukovskaya A, et al. A DNA Hypomethylating Drug Alters the Tumor Microenvironment and Improves the Effectiveness of Immune Checkpoint Inhibitors in a Mouse Model of Pancreatic Cancer. Cancer Res (2020) 80(21):4754–67. doi: 10.1158/0008-5472.Can-20-0285
149. Hellmann MD, Jänne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, et al. Entinostat Plus Pembrolizumab in Patients With Metastatic NSCLC Previously Treated With Anti-PD-(L)1 Therapy. Clin Cancer Res (2021) 27(4):1019–28. doi: 10.1158/1078-0432.Ccr-20-3305
150. Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell (2018) 174(4):843–55. doi: 10.1016/j.cell.2018.06.025
151. Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-Stage Differentiation Defines Melanoma Subtypes With Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell (2018) 33(5):890–904. doi: 10.1016/j.ccell.2018.03.017
152. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the Crossroads of Cancer-Acquired Drug Resistance and Immune Evasion. Nat Rev Cancer (2019) 19(7):405–14. doi: 10.1038/s41568-019-0149-1
153. Efimova I, Catanzaro E, van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination With Early Ferroptotic Cancer Cells Induces Efficient Antitumor Immunity. J Immunother Cancer (2020) 8(2):e001369. doi: 10.1136/jitc-2020-001369
154. Wolf Y, Anderson AC, Kuchroo VK. TIM3 Comes of Age as an Inhibitory Receptor. Nat Rev Immunol (2020) 20(3):173–85. doi: 10.1038/s41577-019-0224-6
155. Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. Lymphocyte-Activation Gene 3 (LAG3): The Next Immune Checkpoint Receptor. Semin Immunol (2019) 42:101305. doi: 10.1016/j.smim.2019.101305
156. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel Immune Checkpoint Targets: Moving Beyond PD-1 and CTLA-4. Mol Cancer (2019) 18(1):155. doi: 10.1186/s12943-019-1091-2
157. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The Human Tumor Microbiome is Composed of Tumor Type-Specific Intracellular Bacteria. Science (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
158. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-Programmed Nutrient Partitioning in the Tumour Microenvironment. Nature (2021) 593(7858):282–8. doi: 10.1038/s41586-021-03442-1
Keywords: immune checkpoint blockade therapy, acquired resistance, neoantigen depletion, interferon signaling aberration, tumor-induced exclusion, immunosuppression, tumor cell plasticity, treatment modalities.
Citation: Zhou B, Gao Y, Zhang P and Chu Q (2021) Acquired Resistance to Immune Checkpoint Blockades: The Underlying Mechanisms and Potential Strategies. Front. Immunol. 12:693609. doi: 10.3389/fimmu.2021.693609
Received: 11 April 2021; Accepted: 28 May 2021;
Published: 14 June 2021.
Edited by:
Lazar Vujanovic, University of Pittsburgh, United StatesReviewed by:
Joe-Marc Chauvin, University of Pittsburgh Medical Center, United StatesDayana Rivadeneira, University of Pittsburgh, United States
Copyright © 2021 Zhou, Gao, Zhang and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Chu, cWlhbmNodUB0amgudGptdS5lZHUuY24=