
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 23 August 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.692263
This article is part of the Research TopicType III Interferons: Emerging Roles Beyond Antiviral Barrier DefenseView all 8 articles
 Olusegun O. Onabajo1‡
Olusegun O. Onabajo1‡ Fang Wang1†‡
Fang Wang1†‡ Mei-Hsuan Lee2‡
Mei-Hsuan Lee2‡ Oscar Florez-Vargas1
Oscar Florez-Vargas1 Adeola Obajemu1Chizu Tanikawa3
Adeola Obajemu1Chizu Tanikawa3 Joselin M. Vargas1
Joselin M. Vargas1 Shu-Fen Liao2,4
Shu-Fen Liao2,4 Ci Song5Yu-Han Huang2Chen-Yang Shen6
Ci Song5Yu-Han Huang2Chen-Yang Shen6 A. Rouf Banday1
A. Rouf Banday1 Thomas R. O’Brien7
Thomas R. O’Brien7 Zhibin Hu5Koichi Matsuda8
Zhibin Hu5Koichi Matsuda8 Ludmila Prokunina-Olsson1*
Ludmila Prokunina-Olsson1*IFNL3/IFNL4 polymorphisms are inversely associated with the risk of chronic hepatitis C virus (HCV) infection and cirrhosis, two major risk factors for developing hepatocellular carcinoma (HCC). To further explore these inverse associations and their molecular underpinnings, we analyzed IFNL3/IFNL4 polymorphisms represented by the IFNL4 genotype (presence of rs368234815-dG or rs12979860-T alleles) in HCV patients: 2969 from Japan and 2931 from Taiwan. IFNL4 genotype was associated with an increased risk of HCV-related HCC (OR=1.28, 95%CI=1.07-1.52, P=0.0058) in the general population of Japanese patients, but not in Taiwanese patients who achieved treatment-induced viral clearance. IFNL4 genotype was also associated with a decreased risk of cirrhosis (OR=0.66, 95%CI=0.46-0.93, P=0.018, in Taiwanese patients). We then engineered HepG2 cells to inducibly express IFN-λ4 in the presence or absence of interferon lambda receptor 1 (IFNLR1). Induction of IFN-λ4 resulted in its intracellular accumulation, mainly in lysosomes and late endosomes, and increased ER stress, leading to apoptosis and reduced proliferation. We identified the very-low-density lipoprotein receptor (VLDLR), which facilitates HCV entry into hepatocytes, as a transcript induced by IFN-λ4 but not IFN-λ3. Our results suggest that the molecular mechanisms underlying the anti-cirrhotic but pro-HCV associations observed for IFNL3/IFNL4 polymorphisms are, at least in part, contributed by intracellular accumulation of IFN-λ4 causing ER stress in hepatic cells.
Infection with hepatitis C virus (HCV) persists in a chronic stage in 75-85% of infected individuals; 10-20% of these patients progress to liver cirrhosis and then to hepatocellular carcinoma (HCC), the main type of primary liver cancer, with a rate of 1-4% per year (1). Genetic polymorphisms within the IFNL3/IFNL4 genomic region have been identified as the strongest predictors of spontaneous and treatment-induced clearance of HCV infection (2–5).
Due to strong linkage disequilibrium (LD) in the IFNL3/IFNL4 region (6), many markers provide similar genetic associations, including rs368234815 that controls IFN-λ4 production (7), rs4803217 that was suggested to affect IFN-λ3 stability (8), and rs12979860, a non-functional polymorphism in the first intron of IFNL4 (3). Among these, a dinucleotide polymorphism, rs368234815-TT/dG, is the most informative marker for predicting HCV clearance in all populations (6, 7). Individuals with the rs368234815-dG allele have a frameshift in the first exon of the IFNL4 gene, which creates an open reading frame for interferon lambda 4 (IFN-λ4), a type III IFN. Homozygous carriers of the rs368234815-TT allele, including most Asians (~90%) and Europeans (~50%), but only a minority of individuals of African ancestry (~10%), are natural IFN-λ4 knockouts (6).
Carriers of the IFNL4-dG allele are more likely to develop chronic HCV infection and fail to respond to HCV treatment. Given that chronic HCV infection is a strong risk factor for HCC, the IFNL4-dG allele would be expected to be associated with the risk of HCC as well. However, associations between IFNL3/IFNL4 polymorphisms and HCC risk have been inconsistent and reported by some studies (9–13), but not others (14, 15). Surprisingly, the alleles associated with poor HCV clearance were also associated with reduced risk of liver fibrosis (16, 17), which is an aberrant wound-healing response to chronic liver injury and a pre-stage of liver cirrhosis (18). Thus, the interplay between the IFNL3/IFNL4 polymorphisms, HCV infection, and the risk of liver cirrhosis and HCC remains unclear. Here, we addressed these questions using genetic, genomic, and functional tools.
A total of 2969 individuals were from BioBank Japan, which between 2003 and 2007 recruited ~200,000 patients with 47 common diseases from 12 Japanese medical institutes representing 67 hospitals (19). From the BioBank Japan database, we selected all the patients who were HCV-RNA positive at the time of recruitment and ascertained for primary HCC status (yes/no) based on histology, imaging, and laboratory tests. No information about HCV treatment and outcomes was available. The project was approved by the ethical committees of the University of Tokyo, and all participants provided written informed consent. Genomic DNA from peripheral blood was genotyped with an Illumina HumanOmniExpressExome BeadChip or a combination of the Illumina HumanOmniExpress and HumanExome BeadChips (20). The genotypes were prephased with MACH (21) and imputed with Minimac using the 1000 Genomes Project Phase 1 (version 3) East Asian reference panel (22). IFNL4-rs12979860 was imputed with high confidence, and association analysis was conducted using a logistic regression model using imputed gene dosage, adjusting for age, sex, and the top 2 principal components (PCs).
The REVEAL-II (Risk Evaluation of Viral Load Elevation and Associated Liver Diseases) clinical cohort has been described (23). Briefly, treatment-naive HCV+ patients without HCC were enrolled in the prospective study during 2004–2014. The patients were treated with Peg-IFN and ribavirin (RBV) for 48 or 24 weeks (for patients with HCV genotype 1 and other genotypes, respectively), and sustained virologic response (SVR) was determined from the serum HCV RNA test results 24 weeks post-treatment. Patients who didn’t achieve an SVR were re-treated with the same regimen, and the combined SVR was estimated based on the treatment and retreatment.
Demographic and clinical data were obtained via chart reviews with standardized forms. Cirrhosis at baseline (before treatment) was determined by abdominal ultrasound (in 81.6% of patients) or liver biopsy (in 18.4%) and categorized as yes/no. DNA samples were genotyped for IFNL4-rs368234815 by a custom TaqMan genotyping assay, as previously described (7). Only individuals with available information about baseline cirrhosis and IFNL4-rs368234815 genotypes were included in the analysis (n=2,931). Incident HCC diagnoses after treatment completion were verified by medical records and electronic linkage with the Taiwan National Cancer Registration and the National Death Certification databases.
The person-years of follow-up for each patient were calculated from the enrolment date to either the date of HCC identification, the date of death, or December 31, 2014, whichever came first. The incidence rates of HCC per 1,000 person-years were calculated by dividing the number of newly developed HCC cases by person-years of follow-up. Multivariable Cox proportional hazards models were used to examine the outcomes in relation to IFNL4 genotypes (as 0 and 1), controlling for several clinical predictors including age, sex, baseline liver cirrhosis, serum alanine aminotransferase (ALT), HCV genotype (genotype 1 vs. other), and SVR. Hazard ratios (HRs) with 95% confidence intervals (CIs) and statistical significance levels were determined by a two-sided p-value of 0.05. The proportionality assumption of Cox models was examined, and the assumption was not violated. All analyses were performed using the SAS statistical software package (version 9.1; SAS Institute Inc., Cary, NC, USA).
HBV-positive HCC patients (n=1,300) were consecutively recruited between 2006 and 2010 in Nantong and Nanjing, China (24). Age and sex-matched controls were recruited from the same geographical areas and included 1,344 HBV persistent carriers that were positive for both HBsAg and antibody against hepatitis B core antigen (anti-HBc), as well as 1,344 subjects with natural clearance of HBV-negative for HBsAg but positive for antibodies against hepatitis B surface antigen (anti-HBs) and anti-HBc. All the cases and controls were negative for HCV antibody (anti-HCV). No information about HBV treatment and outcomes was available. Genomic DNA was extracted from blood leukocytes with phenol-chloroform. DNA samples were genotyped for IFNL4-rs368234815 by a custom TaqMan genotyping assay, as previously described (7). Association analysis was conducted using a logistic regression model adjusting for relevant covariates as indicated.
Human cell lines: HepG2 (hepatoma) and 293T (embryonal kidney) were acquired from the American Type Culture Collection (ATCC), while LX-2 (hepatic stellate) was purchased from Millipore-Sigma. Stable HepG2 cell lines expressing doxycycline (dox)-inducible GFP-tagged IFN-λ3 and IFN-λ4 (IFN-λ3-GFP and IFN-λ4-GFP cells, respectively), have been described (25). All cell lines were maintained in DMEM supplemented with 10% FBS (Invitrogen), penicillin, and streptomycin; 5 μg/ml blasticidin and 1 mg/ml neomycin were additionally used for the stable cell lines. Expression of IFN-λ3–GFP and IFN-λ4–GFP was induced by 0.5 μg/ml or specified concentrations of dox for indicated time points. LX-2 cells were maintained in DMEM supplemented with 2% FBS, 100 units/ml penicillin, 100 µg/ml streptomycin, and 2 nM glutamine media. Primary human hepatic stellate cells (HSCs) were purchased from ScienCell Research Laboratories and maintained in stellate cell media with 10% FBS and stellate cell growth supplement (ScienCell). All cell lines were tested bi-monthly for mycoplasma contamination using the MycoAlert Mycoplasma Detection kit (Lonza) and were authenticated annually using the AmpFLSTR Identifiler Plus Kit (ThermoFisher Scientific) by the Cancer Genomics Research Laboratory (CGR, NCI). Fresh primary human hepatocytes (PHHs) from 12 anonymous donors purchased from BioreclamationIVT and genotyped for IFNL4-rs368234815 were previously described (25).
Exon 3 of the NM_170173.3 transcript, which is the first exon common for all four alternative isoforms of IFNLR1, was selected as a target region for designing six candidate gRNAs with a CRISPR design tool (http://crispr.mit.edu/). Double-stranded oligonucleotides were cloned into the linearized SmartNuclease vector (EF1-T7-hspCas9-T2A-RFP-H1-gRNA) with a red fluorescent protein (RFP, System Biosciences) and IFNLR1-guide RNA (IFNLR1-gRNA) plasmids were validated by DNA sequencing. 293T cells were transiently transfected in 6-well plates with six individual IFNLR1-gRNA plasmids using Lipofectamine 3000 (Life Technologies). The cells were harvested 4 days post-transfection and subjected to fluorescence-activated cell sorting (FACS) for RFP with a FACS Aria III (BD Biosciences). Genomic DNA from sorted RFP-positive cells was extracted using DNeasy Blood & Tissue Kit (Qiagen) and used for sequencing the exon 3 target region. One out of 6 IFNLR1-gRNA plasmids was selected as the most optimal (Figure S1A) and was transfected into IFN-λ4-GFP cells in a 12-well plate, together with linearized screening puromycin marker (Clontech). Two days post-transfection, cells were plated in a 10-cm plate, and fresh media containing puromycin (2 µg/ml) was added to the plate two days later. After three weeks of selection, visible clones were picked and transferred into 24-well plates with fresh media without puromycin. DNA was extracted from each of the clones and sequenced as described above. Final clones represented the stable IFN-λ4-GFP-IFNLR1KO cells. Potential off-target sites were predicted with the online tool CCTop (http://crispr.cos.uni-heidelberg.de) (26). The top ten predicted sites were tested by sequencing the genomic DNA of IFN-λ4-GFP-IFNLR1KO cells, and no off-target mutations were detected (data not shown).
Cells were seeded in 96-well plates (2 × 104 cells/well) and reverse-transfected with 100 ng/well of Cignal ISRE-Luc reporter (Qiagen) using Lipofectamine 3000. After 24 hours of transfection, cells were treated for 8 hours in triplicates with recombinant IFNα (0.5 ng/ml, PBL), custom IFN-λ3 (20 ng/ml) and IFN-λ4 (50 ng/ml) (25), or were induced for 12 or 24 hours with dox (0.5 µg/ml), with untreated cells used as a negative control. Induction of ISRE-Luc reporter was evaluated by measuring Firefly and Renilla luciferase activity with dual-luciferase reporter assays on Glomax-Multi Detection System (Promega). The results were presented as relative light units (RLU), corresponding to Firefly normalized by Renilla luciferase activity.
Cells were seeded in 12-well plates and treated for 8 hours in biological quadruplicates with IFNα (0.5 ng/ml, R&D Systems), IFNβ (0.5 ng/ml, GenScript), IFNγ (1 ng/ml, R&D Systems), IFN-λ1 (5 ng/ml, R&D Systems), IFN-λ2 (60 ng/ml, R&D Systems), and custom IFN-λ3 (20 ng/ml) and IFN-λ4 (50 ng/ml) (25), with no treatment used as a negative control.
Cells were lysed in RIPA buffer (Sigma) supplemented with protease inhibitor cocktail (Promega) and PhosSTOP (Roche). Cell lysates were resolved on Blot 4-12% Bis-Tris gel (ThermoFisher) and transferred to nitrocellulose membranes with iBlot 2 Dry Blotting System (ThermoFisher). The membranes were probed with primary antibodies against IFNLR1 (#NBP1-84381, Novus Biologicals), STAT1 (#9172, Cell Signaling Technology), phospho-STAT1 (Tyr701, #58D6, Cell Signaling Technology), IFN-λ4 (ab196984; Abcam), GAPDH (ab37168, Abcam) and HRP-linked secondary antibody, goat anti-rabbit IgG (#7074; Cell Signaling Technology) or goat anti-mouse IgG (San Cruz, sc-2031). Signal detection was done with HyGLO™ quick spray chemiluminescent HRP antibody detection reagent (Denville Scientific) and Chemidoc Touch Imaging System (BioRad) and quantified by ImageJ software.
HepG2 cells were transfected for 24 hours with corresponding Halo-tagged constructs in 4-well chambered slides for fixed cells or in 4-well coverslip slides for live cells (2 × 105 cells/well, LabTek). For fixed cells, a BacMam system (ThermoFisher) was used to deliver baculoviruses expressing GFP-tagged proteins targeted to specific organelles (N-acetylgalactosaminyltransferase for Golgi, Rab5a for early endosomes, Rab7a for late endosomes, and LAMP1 for lysosomes), 6 hours prior to transfection. Cells were incubated with cell-permeant TMR red Halo-tag ligand (1:2,000 for 15 min, Promega) 24 hours post-transfection, fixed with 4% paraformaldehyde, mounted with Prolong Gold antifade mounting media with DAPI (ThermoFisher) and coverslip.
For live cells, media in chambered slides was replaced with live-cell imaging solution (Life Technologies), supplemented with 20 mM glucose, 24 hours post-transfection. Cell-permeant TMR red Halo-tag ligand was added to cells (1:2,000 for 15 min). Imaging was done on an LSM700 confocal laser scanning microscope (Carl Zeiss) using an inverted oil lens at 40x magnification; live imaging was done using a temperature and CO2-controlled chamber, with 6 sec scans every minute for 12 hours.
HepG2 cells were treated with or without tunicamycin (20 µg/ml, Sigma) for 24 hours. For SeV infection, PHHs and HSCs were infected with SeV (7.5 × 105 chicken embryo ID50 [CEID50] per milliliter, Charles River Laboratories) and collected at the indicated time points. All experiments were done in triplicates.
Total RNA was extracted using an RNeasy Mini Kit with on-column DNase digestion (Qiagen). RNA quantity and quality were evaluated by NanoDrop 8000 (Thermo Scientific) and Bioanalyzer 2100 (Agilent Technologies). RNA integrity numbers (RIN) of all RNA samples were > 9.5. cDNA was synthesized from the total RNA with the RT2 First Strand Kit (Qiagen), with an additional DNase I treatment step. qRT-PCR assays were performed in technical quadruplicates in 384-well plates on QuantStudio 7 instrument (Life Technologies), with RT² SYBR Green (Qiagen) or TaqMan (Thermo Fisher) expression assays). The expression of target genes was normalized by geometric means of endogenous controls (GAPDH and ACTB), presented as ΔCt values. The 2-ΔΔCT method, in which ΔΔCt = ΔCt (experiment) – ΔCt (control), and fold = 2-ΔΔCt was used to calculate the relative abundance of target mRNA expression and fold change. Data are shown as mean ± SEM based on biological replicates.
For proliferation assays, HepG2 cells were induced with 0.5µg/ml dox and treated with 10 μM 5-bromo-2′-deoxyuridine (BrdU) for 3 h. Dead cells were detected with a near-IR Live/Dead kit (ThermoFisher) and stained using a PE BrdU flow kit (BD Biosciences). For coculture experiments and proliferation experiments, HepG2 cells were labeled with Far Red dye (ThermoFisher). Cells were analyzed with multiparametric flow cytometry on a FACS Aria III (BD Biosciences) and FlowJo.v10 software (BD Biosciences).
Cells seeded in 96-well plates (8000 cells/well) were mock- or dox-induced (0.5 µg/ml) in quadruplicates for 24, 48, and 72 hours, then subjected to cell viability assays using CellTiter-Glo assays (Promega) and cell apoptosis assays using ApoTox-Glo Triplex assay (Promega). Luminescence was measured on the GloMax-multi detection system (Promega). For the colony formation assays, cells (2,000 cells/well) were mock- or dox-induced (0.5 µg/ml) in triplicates in 6-well plates for two weeks. The colonies were stained with 0.5% crystal violet, photographed, and counted using ImageJ software. Cell cycle was evaluated using PI staining and analyzed on FlowJo.10 software (BD Biosciences). Seeded cells were fixed with 70% ethanol on ice for 2 hours, washed twice with PBS, and treated with RNAse A (100µg/ml, Qiagen). Cells were then stained with PI (20µg/ml) for 10 minutes and were immediately analyzed on an Accuri C6 (BD Biosciences).
Total RNA was extracted from cells using an RNeasy Mini Kit with an on-column DNase digestion (Qiagen) and quantitated using Qubit 4 Fluorometer (ThermoFisher). Ribosomal RNA was depleted with Ribo-Zero treatment, then 60 ng was used for library preparation using KAPA Stranded RNA-Seq Library Preparation Kit (Illumina, KR0934, v-1.13). The samples were barcoded and pooled in sets of eight per run to generate paired 150-bp reads with a NextSeq 550 Sequencing System (Illumina), generating 21.2 - 118.8 million reads per sample. Quality control analysis of RNA-seq data was carried out with the FastQC pipeline. Paired RNA-seq reads were aligned to the ENSEMBL human reference genome GRCh37.75 (hg19) with STAR v 2.5.3a (27) using default parameters, and BAM files were generated with SAMtools v 1.5 (28). Data normalization and analysis of differential gene expression were done with DESeq2 R package v 1.22.2 (29, 30). Thresholds of fold change ≥ 1.5 (log2 fold change = 0.58) and false-discovery rate (FDR) adjusted P-value < 0.05 were used to identify differentially expressed genes (DEG) between experimental conditions. Identified DEGs were analyzed with Ingenuity Pathway Analysis (IPA) software (Qiagen). The generated RNA-seq dataset for 108 samples (54 individual samples in duplicates) has been deposited to NCBI Gene Expression Omnibus (GEO) with an accession number GSE145038.
Analysis of experimental data: Experimental data were analyzed using a two-sided, unpaired Student’s t-test, and P-values <0.05 were considered statistically significant. Statistical significance of the RNA-seq differential expression data was determined using Wald negative binomial test with Benjamini–Hochberg adjustment for multiple testing (30). Unless otherwise specified, data plotting and statistical analyses were performed with Prism 7 (GraphPad), SPSS v.25 (IBM), or R packages. Means are presented with standard errors (SEM) based on biological replicates.
First, we evaluated progression to HCC in 2969 HCV-infected patients from the BioBank Japan (19), using an intronic IFNL4 marker rs12979860, which is completely linked with rs368234815 in Asians (r2 = 1.0). The IFNL4-rs12979860-T allele (a proxy for IFNL4-dG, which supports the production of IFN-λ4) was associated with an increased risk of HCC (per-allele OR=1.28, p=0.0058, Table 1). Because HCV-positive patients were selected from the general population, this association likely represents patients who have never been treated as well as those who failed to clear HCV after treatment, but these questions were not explored due to a lack of information on treatments and outcomes.

Table 1 IFNL4 genotype is associated with increased risk of HCC in HCV-infected patients in Japan.
We then analyzed the clinical REVEAL II cohort from Taiwan (23), which included 2,931 HCV-infected patients monitored for progression to HCC after treatment with peg-IFNα/RBV (Table 2). We genotyped IFNL4-rs368234815, which has a direct functional effect on IFN-λ4 production (7, 25). Since the rs368234815-dG allele is uncommon in Asian populations (<7% in 1000 Genomes Project), we used a dominant genetic model, combining all carriers of the dG allele (TT/dG and dG/dG) in one group, designated as the IFNL4 genotype group. In the REVEAL II cohort, carriers of IFNL4 genotype, as expected (2, 4), had reduced sustained virologic response (SVR) after treatment or retreatment with IFNα/RBV (OR=0.30, p=1.63E-18, Table 3). Among all REVEAL II patients, IFNL4 genotype was associated with progression to HCC (OR=1.79, p=0.011, Table 4), but this association was not significant after accounting for SVR after treatment or retreatment (Tables 4, 5). A context-dependent variant, rs117648444 G->A, alters amino acid 70 within exon 2 from Serine to Proline only in the presence of rs368234815-dG allele (IFNL4 transcript), and results in functionally weak IFNλ4-S70 instead of strong IFNλ4-P70 protein (6, 7, 31). The rs117648444-A allele is relatively common in European (12%) and African (7.5%) ancestries, where it has a statistically significant modifying effect on associations of rs368234815-dG allele (31–33). However, this allele has <0.5% frequency in East-Asian populations (based on five reference populations from the 1000 Genomes Project), precluding further analyses based on the combination of rs368234815 (rs12979860) and rs117648444 in patients from Japan and Taiwan.
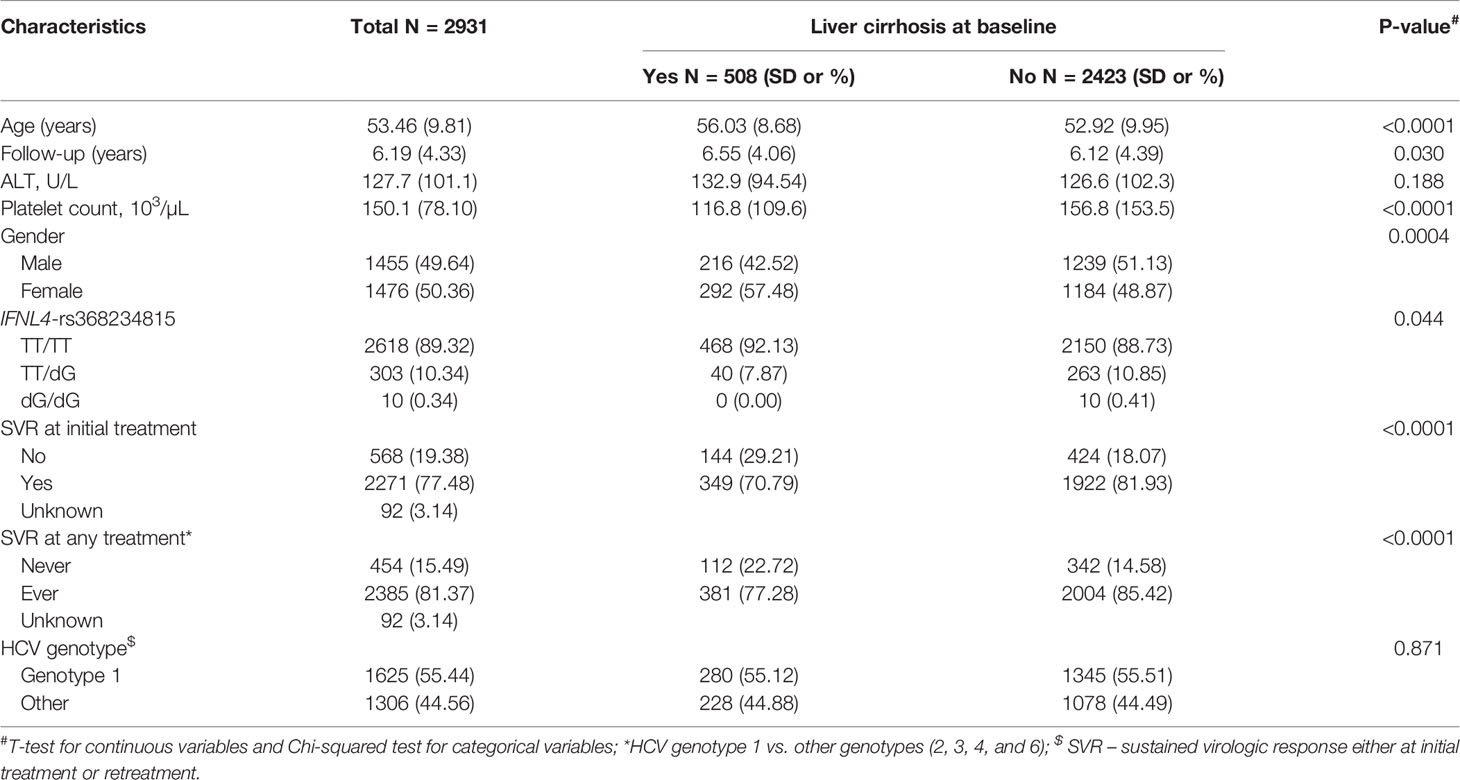
Table 2 Baseline characteristics of HCV-infected patients in the REVEAL II cohort, Taiwan.
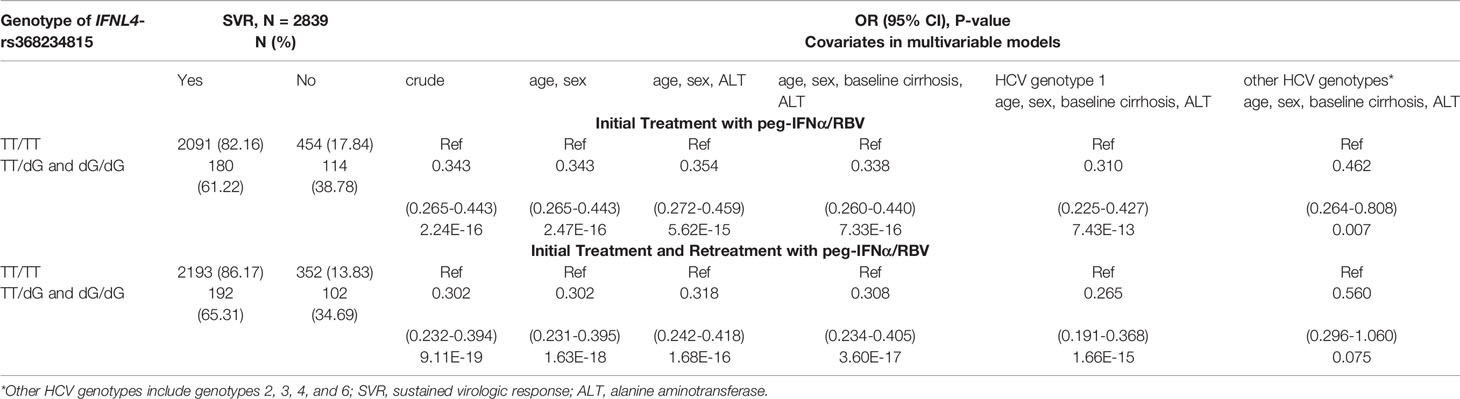
Table 3 Association of the IFNL4 genotype with reduced SVR in HCV-infected patients in the REVEAL II cohort, Taiwan.
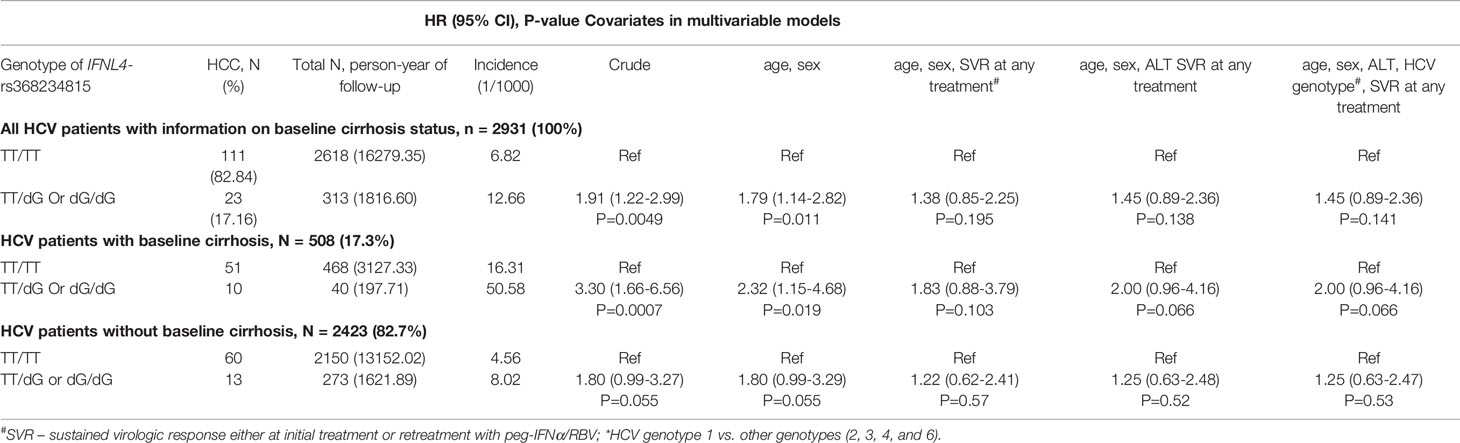
Table 4 Progression to HCC in HCV-infected patients in the REVEAL II cohort, Taiwan in relation to IFNL4 genotype and cirrhosis.

Table 5 Progression to HCC in HCV-infected patients in the REVEAL II cohort, Taiwan in relation to IFNL4 genotype and SVR after treatment/retreatment with peg-IFNα/RBV.
IFNL4 genotype and other markers in the IFNL3/IFNL4 region have been linked with reduced hepatic inflammation and fibrosis, a pre-stage for cirrhosis (16, 17, 34), potentially mitigating progression to HCC. In the REVEAL II cohort, we also observed a lower prevalence of cirrhosis at baseline in the presence of IFNL4 genotype (OR=0.66, p=0.018, Table 6). Analyses stratified according to baseline cirrhosis status showed an association of IFNL4 genotype with HCC progression in patients with cirrhosis (OR=2.32, p=0.019, Table 4) but not without cirrhosis (OR=1.80, p=0.055, Table 4), although this association was eliminated after adjusting for SVR. IFNL4 genotype was not associated with progression to HCC in Chinese patients with HBV (Table 7), in line with the absence of association between IFNL4 genotype and HBV risk (35, 36). Thus, our results indicate that IFNL4 genotype is moderately associated with protection from cirrhosis but not with progression to HCC after accounting for viral clearance.

Table 6 Prevalence of cirrhosis in HCV-infected patients in relation to IFNL4 genotype in the REVEAL II cohort, Taiwan.
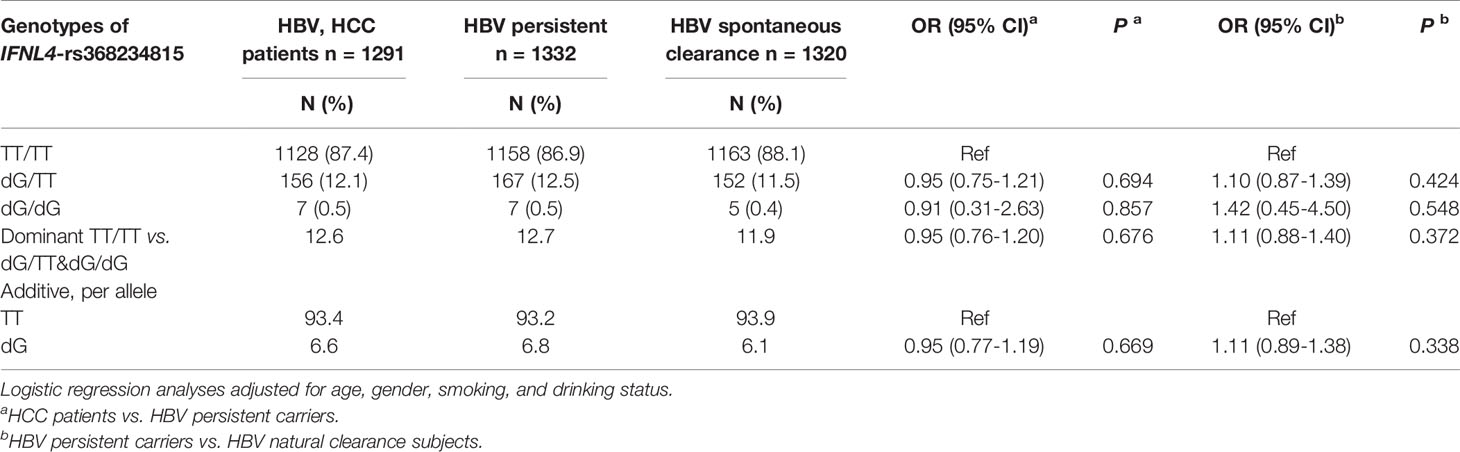
Table 7 IFNL4 genotype is not associated with an increased risk of HCC in HBV-infected patients in China.
Based on previous reports linking IFNL3/IFNL4 genotypes and fibrosis risk (16, 17, 34) and our current results on association with cirrhosis risk in HCV patients, we explored molecular mechanisms that could explain these associations. Genetic variants might affect the expression and function of both IFN-λ3 and IFN-λ4, making it hard to delineate the individual contribution of these IFNs by genetic analysis alone. Thus, we explored functional effects of both IFN-λ3 and IFN-λ4 in a panel of hepatoma HepG2 cell lines, in which we inducibly expressed IFN-λ3-GFP or IFN-λ4-GFP and used CRISPR-Cas9 gene editing to eliminate IFNLR1, the receptor used by all type III IFNs (Figure S1). After doxycycline (dox)-induction of these cell lines for 8, 24, and 72 hrs, we analyzed their global transcriptome by RNA-seq.
IFN-λ4 induction for 72 hrs resulted in the most extensive set of differentially expressed genes (DEGs, P-FDR < 0.05 and dox+/dox-fold change +/-≥ 1.5, n=2880); this set was used for further comparative analyses (Figure S2 and Table S1). Of these genes, 2735 and 145 were classified as IFNLR1-dependent and -independent, respectively, based on their differential expression in IFN-λ4-GFP and IFN-λ4-GFP-IFNLR1KO cells (Figure 1A).
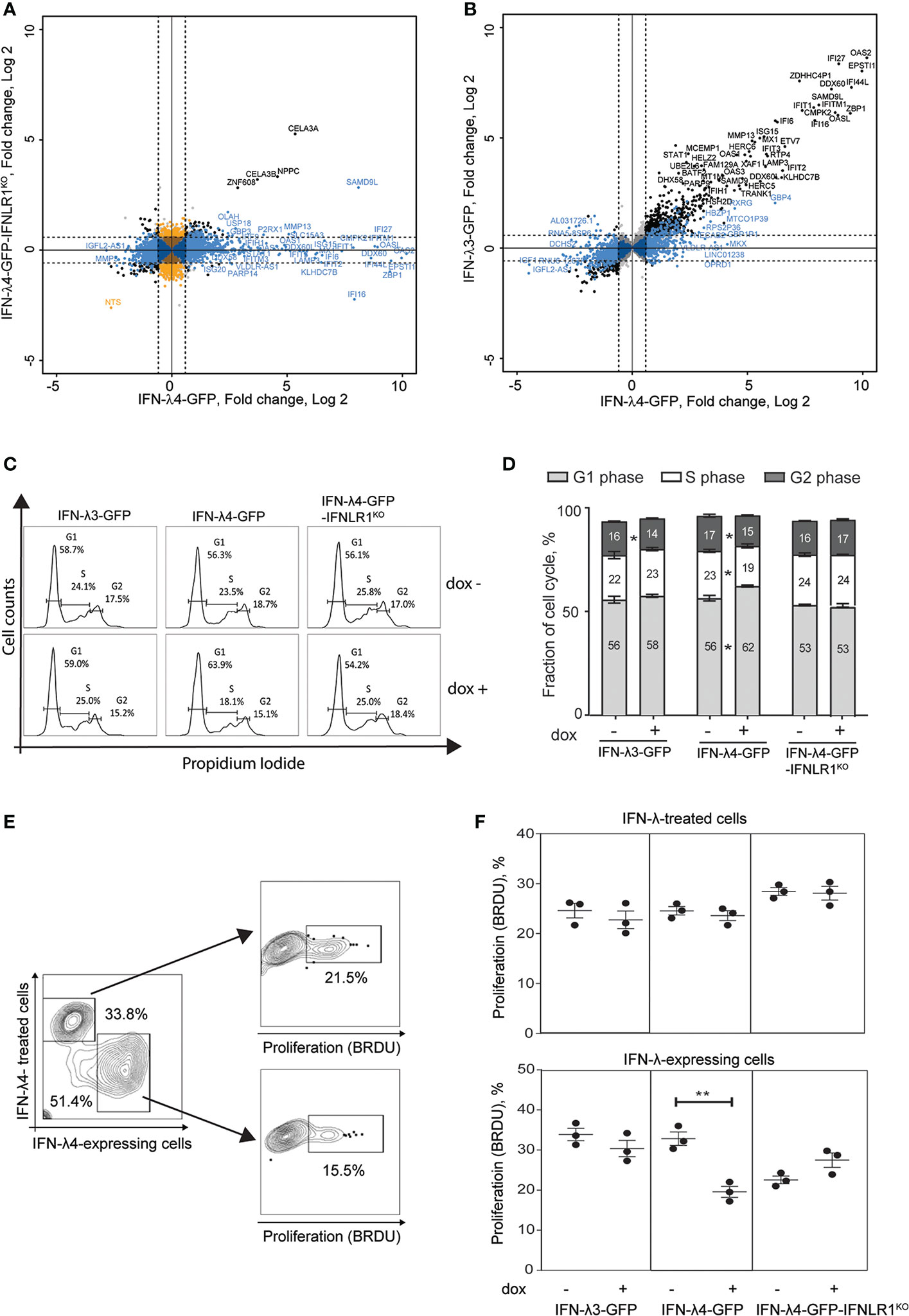
Figure 1 IFN-λ4 inhibits the proliferation of HepG2 cells. RNA-sequencing analysis was used to identify differentially expressed genes (DEGs, P-FDR < 0.05) in IFN-λ3-GFP, IFN-λ4-GFP and IFN-λ4-GFP-IFNLR1KO HepG2 cells after 72 hrs of induction by dox, compared to controls (dox- conditions). The cutoff threshold (fold change > +/-1.5) is indicated by dotted lines. (A) Analysis of all DEGs (n = 3251) detected for IFN-λ4-GFP or IFN-λ4-GFP-IFNLR1KO cells. In blue - DEGs (n = 2,735) specific to IFN-λ4-GFP and considered IFNLR1-dependent. In black - DEGs (n = 145) shared between both groups and considered IFNLR1-independent. In orange - DEGs (n = 371) specific to IFN-λ4-GFP-IFNLR1KO. (B) DEGs of IFN-λ4-GFP analyzed in IFN-λ3-GFP transcriptome. In black - DEGs (n = 1,506) shared in IFN-λ4-GFP and IFN-λ3-GFP and in blue - IFN-λ4-signature DEGs (n = 1,229) detected in IFN-λ4-GFP but not in IFN-λ3-GFP producing cells. Additional details are provided in Figure S2 and Table S1. (C, D) Cell cycle analysis of cells synchronized by 24 hrs of serum starvation, treated with or without dox (0.5 µg/ml) for 72 hrs, and analyzed by flow cytometry after PI staining. The plot shows a representative picture and the percentage of cells in each phase of the cell cycle. All data are shown as mean± SEM from triplicate experiments. *P < 0.05, Student’s T-test comparing no dox to dox+ for each cell cycle stage. (E, F) Bromodeoxyuridine (BrdU, %) incorporation indicating cell proliferation in HepG2 cells expressing IFN-λ3-GFP, IFN-λ4-GFP and IFN-λ4-GFP-IFNLR1KO. Cells were cocultured with HepG2 cells labeled with Far Red proliferation dye, dox-induced for 72 hrs, and treated with BrdU for 3 hrs before analysis. Gates show HepG2 cells exposed to IFN-λs (IFN-λ treated cells) and HepG2 expressing IFN-λs. P-values compare corresponding dox+ vs. dox- HepG2 cells, **p < 0.01, Student’s T-test. Graphs represent one of three independent experiments, each in biological triplicates.
The set of IFN-λ4-induced and IFNLR1-dependent genes (n=2735) was then compared with the expression of the same gene set in IFN-λ3-producing cells. IFNL3 and IFNL4 transcripts in these cell lines were induced to a similar magnitude (Figure S3A), although actual protein levels may differ. A set of 1506 genes was induced by IFN-λ3 or IFN-λ4 (P-FDR < 0.05), with 766 genes induced by ≥1.5 fold in both groups. As expected, ISGs were the top-induced genes activated stronger by IFN-λ4 compared to IFN-λ3 (Figure S3B and Figure 1B). The remaining 1229 (of 2735) genes were considered as IFN-λ4-specific DEGs (Figure S2G and Table S1) and enriched with genes related to cell cycle inhibition (Figure S3C and Table S1).
In line with our previous results (37), we observed G1/G0 cell cycle arrest (Figures 1C, D) and reduced proliferation (Figure S4) in IFN-λ4-expressing HepG2 cells. However, another study did not demonstrate the antiproliferative effect of recombinant IFN-λ4 in hepatic cells (38). To address this discrepancy, we examined proliferation in HepG2 cells expressing IFN-λ4 (and thus exposed to this recombinant protein both endogenously and exogenously), and bystander cells not producing but only exposed to secreted IFN-λ4 exogenously. We used cell-permeable dyes to discriminate IFN-λ4-expressing from IFN-λ4-exposed HepG2 cells and estimated proliferation in both populations using a flow cytometry-based proliferation assay. We observed that in contrast with IFN-λ4-expressing cells there was no inhibition of proliferation in bystander cells exposed to recombinant IFN-λ4 (Figures 1E, F). Interestingly, the antiproliferative effect of intracellular IFN-λ4 was also attenuated in IFN-λ4-GFP-IFNLR1KO cells (Figures 1E, F). These results indicate that IFN-λ4 causes cell cycle arrest and reduced proliferation of hepatic cells through a mechanism that is IFNLR1-dependent and requires intracellular expression of IFN-λ4.
The absence of antiproliferative effects in HepG2 cells exposed to IFN-λ4 (Figures 1E, F) suggests that paracrine IFN signaling was insufficient to inhibit cell proliferation, prompting us to search for additional intrinsic factors associated with IFN-λ4 expression. Unfolded protein response (UPR) was one of the main pathways we identified by Ingenuity Pathway Analysis (IPA) as significantly activated in IFN-λ4-expressing HepG2 cells (Figure S5). UPR due to endoplasmic reticulum (ER) stress has been shown to inhibit proliferation and induce apoptosis in some conditions (39, 40). UPR is induced to relieve ER stress by increasing the expression of several chaperones that aid protein folding (39, 41), and we found a more robust induction of select UPR effectors, curated from IPA, in IFN-λ4-expressing compared to IFN-λ3-expressing and IFN-λ4-IFNLR1KO HepG2 cells (Figures S6A–C). We previously showed that infection of primary human hepatocytes (PHH) with Sendai virus (SeV) resulted in intracellular accumulation of IFN-λ4 (25), possibly causing ER stress. We observed that the expression of an ER stress maker, DDIT3, was significantly upregulated in PHH from donors with vs. without IFNL4 genotype (Figure S6D), while for other ER stress markers, we observed a trend, which did not reach statistical significance, possibly due to low sample size (Figure S6E).
The induction of ER stress (Figures S6A–C) and intracellular accumulation of IFN-λ4 (Figures 1E, F) (25, 37) suggested that IFN-λ4 is an inefficiently folded protein that induces the misfolded protein ER stress response (40, 42). ER stress is mitigated by targeting misfolded proteins to lysosomes for degradation (39, 42). Live imaging in HepG2 cells showed that IFN-λ4 was accumulated in lysosomes (Figure 2A) via late endosome trafficking (Figure 2B and Video S1) but was excluded from early recycling endosomes (Figure 2A). Continued accumulation of IFN-λ4 led to lysosomal enlargement (Figure 2C), followed by membrane blebbing and cell death, implicating apoptosis (Figure 2D and Video S2). We confirmed apoptosis in IFN-λ4-expressing cells using a biochemical assay for caspase activity (Figure 2E) and cell viability (Figure 2F). Prolonged ER stress causes apoptosis via the activity of the UPR-induced transcription factor DDIT3 (43), downregulating the expression of anti-apoptotic factors, including BCL2 (44), which we also observed (Figure S6A). These results suggest that prolonged ER stress due to the continued endogenous production of misfolded IFN-λ4 could be contributing to apoptosis of hepatic cells.
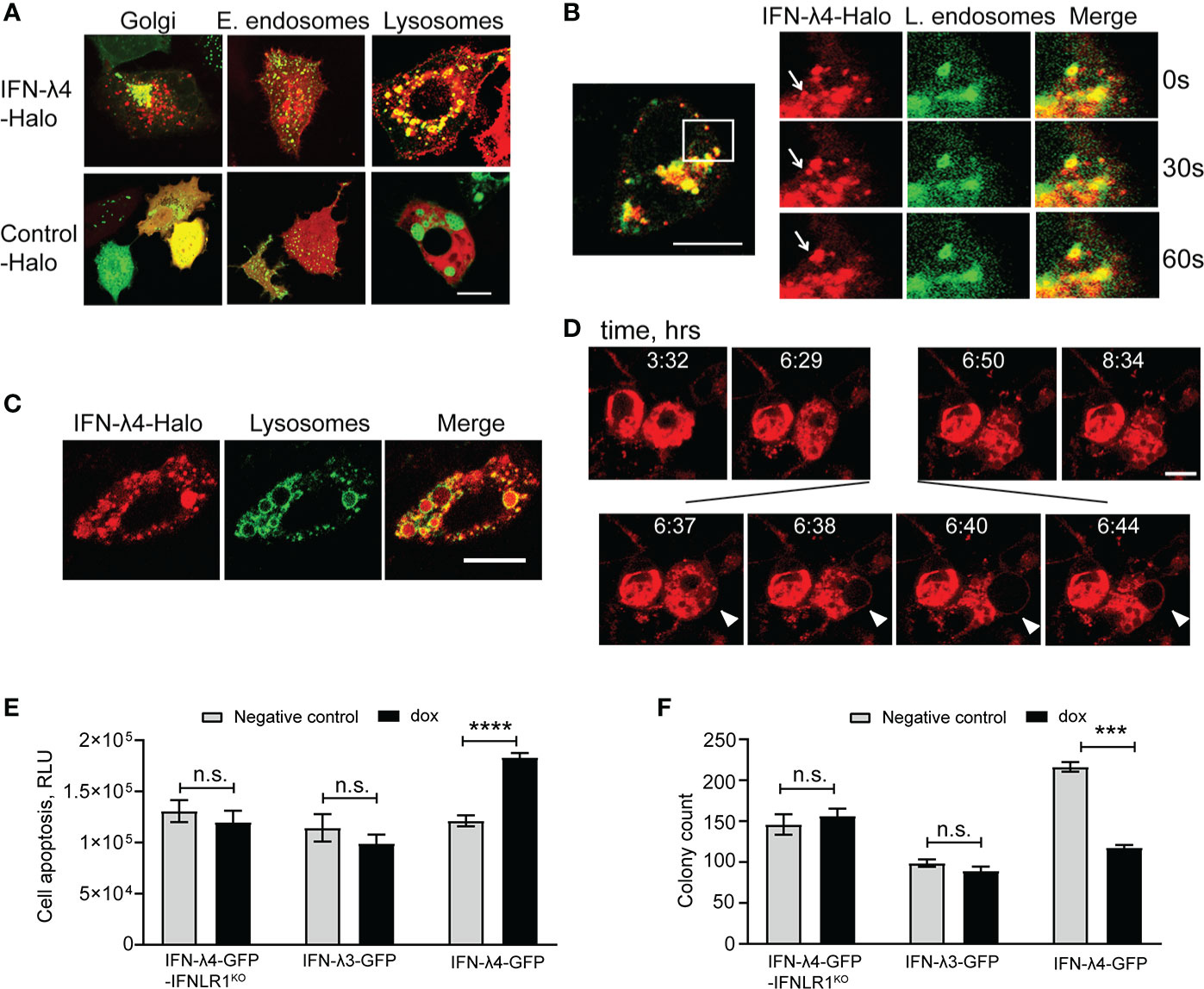
Figure 2 IFN-λ4 is a misfolded protein that induces ER stress. Representative confocal images of HepG2 cells transduced with a mammalian baculovirus delivery system (BacMam) of GFP-tagged proteins targeting specific organelles - lysosomes, Golgi, early and late endosomes. After transduction for 6 hrs, cells were transiently transfected with Halo-tagged constructs for IFN-λ4 or control for indicated times, stained with cell-permeant Halo-tag ligand TMR (red), and imaged. (A) Confocal images showing IFN-λ4 accumulation in lysosomes but not in early endosomes. (B) Late endosomal trafficking of IFN-λ4, with the inset showing larger magnification. (C) Unfolded protein response (UPR) is represented by lysosomal enlargement after protein accumulation. (D) Live images of IFN-λ4-expressing HepG2 cells undergoing apoptosis, characterized by membrane blebbing and cell death. Images were scanned every minute for 12 hrs. Scale bars – 10 um. (E) Apoptosis detection with ApoTox-Glo assays in corresponding untreated and dox-induced cells for indicated time points. RLU, relative luminescence units. (F) Graph showing counts from colony formation assay for HepG2 cells expressing IFN-λ3, IFN-λ4 or IFNLR1 KO grown in 6-well plates with or without dox for 13 days. Cell colonies were stained with crystal violet and counted with ImageJ software. The graph represents the number of colonies as a percentage of initial plated counts. n.s., not significant, ***p < 0.001, ****p < 0.0001, Student’s T-test.
Activated hepatic stellate cells (HSCs) are key drivers of liver fibrosis during HCV infection (45). While HCV is not known to replicate in HSCs, type-III IFNs can be produced in HSCs in response to poly I:C, while another study showed that HCV binds HSCs via CD81 where it upregulates MMP2 (46). Thus, we examined whether IFN-λ4 can induce ER stress not only in hepatocytes but also in HSCs. We infected primary HSCs with SeV to induce IFNs and observed significant induction both of IFNL4 (Figure 3A) and ER stress genes (Figure 3B). The induction of OAS1, an ISG, in response to IFN-λ4 treatment indicated the presence of the functional type III signaling machinery, including IFNLR1, in primary HSCs (Figure 3C). When we overexpressed IFN-λ4 in LX2 (a human stellate cell line), similarly to hepatocytes, we observed upregulation of ER stress genes and inhibition of cell proliferation (Figures 3D–G). These results suggest that decreased proliferation of activated HSCs, which has been shown to contribute to reduced cirrhosis (47), could partly explain the association of IFNL4 genotype with reduced cirrhosis in patients with HCV.

Figure 3 IFN-λ4 inhibits the proliferation of hepatic stellate cells (HSCs). qRT-PCR expression analysis showing (A) IFNL4 expression after SeV infection of primary HSCs and (B) induction of UPR markers. (C) qRT-PCR showing induction of OAS1 after treatment of primary HSCs with recombinant IFN-λ4 (50ng/ml). (D) qRT-PCR showing induction of UPR markers in LX-2 hepatic stellate cell line transiently transfected for 24 hrs with Halo-tagged constructs for IFN-λ4 or control. (E–G) Proliferation analysis in LX-2 cells transiently transfected with Halo-tagged constructs for IFN-λ4 or control, labeled with Far Red proliferation dye and analyzed by flow cytometry after 72 hrs. (E) Gating for Halo-tag expression. (F) Histogram representing the proliferation of gated cells, comparing cells expressing Halo-tagged IFN-λ4 vs. control. (G) Graph representing the geometric mean expression of Far Red proliferation dye with higher values indicating reduced proliferation. **p < 0.01, ***p < 0.001, ****p < 0.0001, Student’s T-test.
Primary receptors for HCV include occludin (OCLN) (48), CD81, scavenger receptor class B type 1 (SCARB1), low-density lipoprotein receptor (LDLR), and surface glycosaminoglycans (49). Very-low-density lipoprotein receptor (VLDLR) was recently identified as an additional receptor for HCV (50). Since VLDLR is induced in response to ER stress in some conditions (51), we tested if IFN-λ4-induced ER stress could be contributing to the upregulation of VLDLR expression. RNA-seq data showed significant upregulation of VLDLR, while other receptors (OCLN, CD81 and SCARB1) were not induced in response to IFN-λ4 expression in HepG2 cells (Figure 4A). Analysis with gene-specific expression assays using independent biological replicates of HepG2 cells validated the induction of VLDLR by IFN-λ4 (Figure 4B). We then tested whether ER stress could lead to upregulation of VLDLR. Indeed, in normal HepG2 cells, tunicamycin treatment led to significant upregulation of VLDLR (by 3-fold, Figure 4C). These results suggest that IFN-λ4 could be increasing the expression of VLDLR in hepatic cells by inducing ER stress, thus, promoting and sustaining HCV entry.
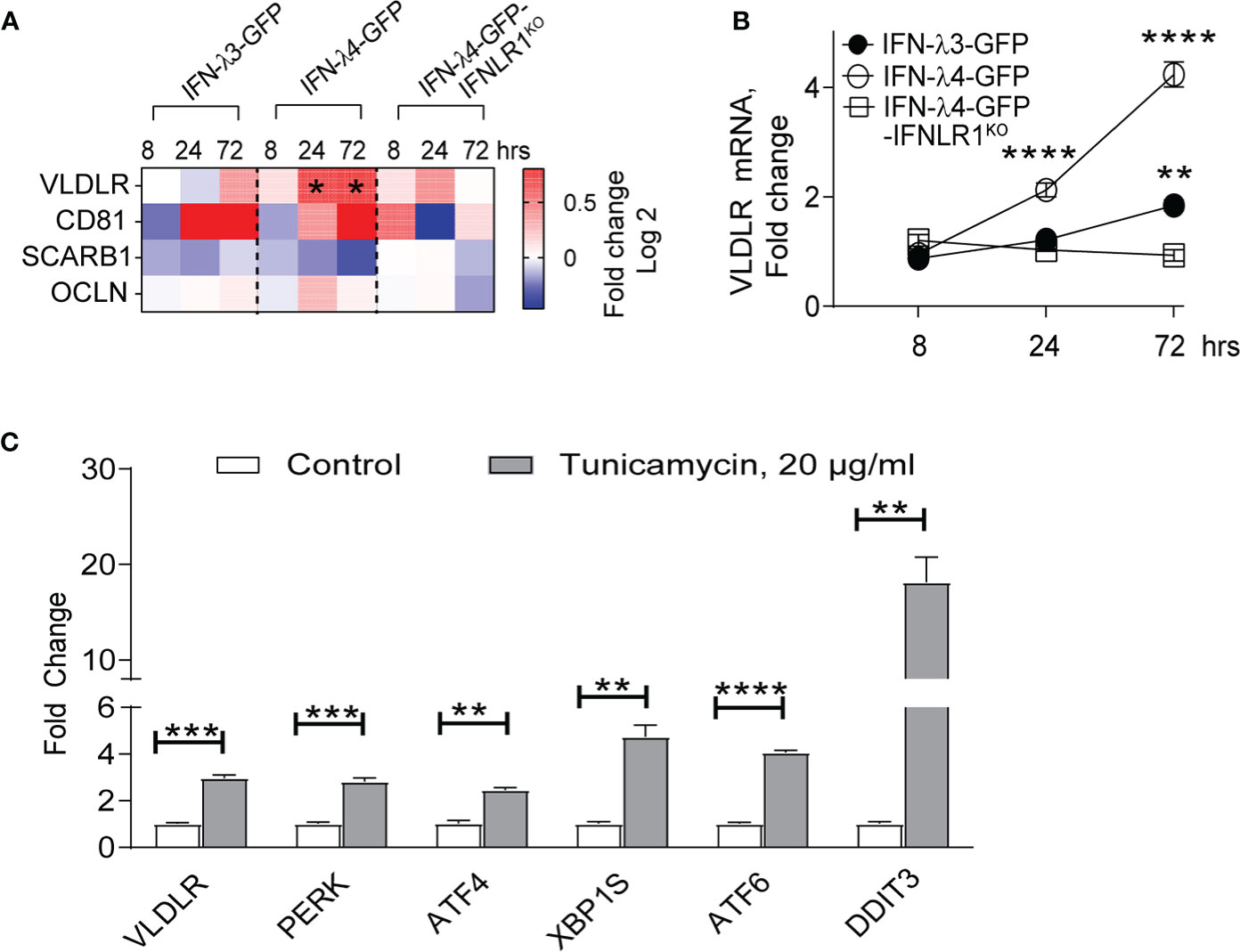
Figure 4 IFN-λ4 upregulates VLDLR expression. (A) Differential expression of HCV receptors identified by RNA-seq analysis (Table S1) comparing corresponding dox+ and dox- HepG2 cells induced for indicated times points and presented as fold change (Log2). * represents significant (p<0.05) induction above 1.5 fold. (B) qRT-PCR analysis of VLDLR after IFN-λ4 induction in corresponding HepG2 cells. Mean +/- SEM (C) qRT-PCR analysis of VLDLR, as well as select UPR-associated genes in HepG2 cells following tunicamycin treatment for 24 hours. Each gene expression was normalized by the expression of endogenous controls (GAPDH and ACTB) and by expression in corresponding untreated groups. **P < 0.01, ***P < 0.001, ****P < 0.0001, Student’s T-test.
Genetic variants within the IFNL3/IFNL4 region have been associated with increased risk of chronic HCV but reduced fibrosis. Reconciling these opposing phenotypes has been further complicated by strong linkage disequilibrium in this locus (6), nominating several IFNL3 and IFNL4 variants as potentially contributing to these associations. We used IFNL4 genotype as a representative marker in this region to explore genetic associations in large sets of HCV patients from Japan and Taiwan. We observed a moderately increased risk of progression to HCC in HCV patients with IFNL4 genotype in the general population, but this risk was eliminated in patients achieving viral clearance, explaining inconsistencies in previous reports on HCC risk (10–15). Thus, our results support the role of IFN-λ4 in HCV risk but not in progression to HCC once HCV is cleared.
We also observed that HCV patients with IFNL4 genotype were less likely to manifest liver cirrhosis, in line with the previous studies (16, 17, 34). Our data suggest that reduced fibrosis/cirrhosis could be due to ER stress induced by intracellular accumulation of IFN-λ4, but not IFN-λ3. Specifically, we observed that inducible expression of IFN-λ4 was associated with cell cycle arrest, apoptosis, and decreased cell proliferation. In addition to primary hepatocytes, HSCs could be critical for antiproliferative effects of IFN-λ4. Viral hepatitis is characterized by repeated cycles of hepatic cell death and regeneration. Breakdown in the formation of the extracellular matrix in combination with continuous proliferative pressure leads to the development of fibrotic tissue (18, 52–54). This mechanism is mediated by HSCs, which differentiate into myofibroblasts and accumulate in fibrotic tissue (54, 55). As a result, signaling pathways that lead to cell cycle arrest in HSCs inhibit the development of fibrosis and cirrhosis (47, 54). Indeed, we observed that IFN-λ4-induced ER stress coupled with IFN signaling reduced proliferation in HSCs. Although the role of ER stress in liver homeostasis is complex, with conflicting findings emerging from mouse knockout studies (53), induction of ER stress as an adaptive process could be beneficial in other inflammatory conditions of the liver, such as non-alcohol fatty liver disease (NAFLD) (53).
We were somewhat surprised that IFNLR1 knockout attenuated the ER-stress response, suggesting a role for type-III IFN signaling in the antiproliferative effect, although IFN-λ3-GFP expression did not induce ER-stress. We propose that IFN-λ4 associated UPR coupled with type-III IFN signaling leads to significant ER-stress and antiproliferative effects. Some studies have shown that type-I IFNs can induce ER-stress in certain cancer cells (56–58). Although acting through different receptors, type-I and type-III IFNs induce a similar set of ISGs, supporting the idea that in the right context, type-III IFNs could induce ER-stress. This may be the case for IFN-λ4, where ER-stress could be induced by type-III IFN signaling coupled with UPR, while neither mechanism may be sufficient to induce ER stress on its own.
Our experimental model utilized hepatic cell lines designed to examine the specific effects of IFN-λ4 or the closely related IFN-λ3 in mediating antiviral effects. This approach enabled us to identify novel phenotypes associated with IFN-λ4 expression. Importantly, in virally infected PHH, where we previously demonstrated intracellular accumulation of IFN-λ4 (25), IFNL4 genotype was also associated with increased ER stress, supporting the role of endogenously expressed IFN-λ4 in contributing to increased ER stress during viral infections.
Several mechanisms have been proposed for the association of IFNL4 genotype with HCV persistence. We and others have demonstrated an association of IFNL4 genotype with increased ISG induction and enhanced negative regulation of IFN responses (7, 25). Here, we describe a potentially novel mechanism contributing to the association of IFNL4 with chronic HCV via the upregulation of VLDLR, a putative entry receptor for HCV (50). LDLs and VLDLs can shield HCV from antibody neutralization (59), and their circulating levels can be reduced by increased expression of VLDLR in the liver (60). Interestingly, IFNL4 genotype has been associated with lower circulating levels of LDL in HCV (61), which may be linked to increased VLDLR expression. ER stress-induced VLDLR expression appears to be dependent on protein kinase RNA‐like ER kinase–activating transcription factor 4 (PERK) signaling pathway (51), which was also induced by IFN-λ4 overexpression in our models.
In conclusion, we present genetic and functional results suggesting a role for IFN-λ4 in antiproliferative mechanisms via intracellular accumulation and induction of ER stress, with implications for the development of HCV, cirrhosis, and HCC. We acknowledge the limitations of our study, including the lack of animal models complicated by the absence of IFNL4 in the mouse genome. Further dissecting the role of IFN-λ4 in the context of HCV infection and developing clinically relevant applications will require replicating these findings in comparable cohorts of patients and controlling for HCV treatments and outcomes.
The RNA-seq dataset for 108 samples (54 individual samples in duplicates) has been deposited to NCBI Gene Expression Omnibus (GEO) with an accession number GSE145038 and is accessible through the link https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145038.
The study was conducted in accordance with the ethical principles stated in the Declaration of Helsinki and was approved by the Ethics Committee of the National Taiwan University Hospital, Kaohsiung Medical University Hospital, China Medical University Hospital, Chang Gung Memorial Hospital in Kaohsiung and Linkou, and Academia Sinica. The project was approved by the ethical committees of the University of Tokyo. The patients/participants provided their written informed consent to participate in this study.
OO and LP-O: Study concept and design. OO, FW, M-HL, AO, CT, JV, S-FL, CS, Y-HH, C-YS, AB, ZH, and KM: Acquisition of data. OO, FW, M-HL, OF-V, AO, CT, JV, SF-L, CS, Y-HH, C-YS, AB, TO’B, ZH, KM, and LP-O: Analysis and interpretation of data. OO and LP-O: Drafting of the manuscript. OO, FW, M-HL, OF-V, TO’B, and LP-O: Critical revision of the manuscript for important intellectual content. OO, FW, M-HL, OF-V, and LP-O: Statistical analysis. LP-O, M-HL: Obtained funding, technical, or material support. LP-O: Study supervision. All authors contributed to the article and approved the submitted version.
This study was supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, US National Cancer Institute, research grants from the Ministry of Science and Technology, Taipei, Taiwan (105-2628-B-010-003-MY4 and 107-2314-B-010-004-MY2). Funders had no role in study design; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript, and decision to submit the manuscript for publication.
TO’B and LP-O are co-inventors on IFN-λ4-related patents issued to NCI/NIH and receive royalties for antibodies for IFN-λ4 detection.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Cancer Genomics Research Laboratory (DCEG/NCI) for help with RNA-seq and cell line authentication. We acknowledge all of the collaborators for REVEAL-II cohort: Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan: Chung-Feng Huang, Chia-Yen Dai, Jee-Fu Huang, Ming-Lun Yeh, Ching-I Huang, Ming-Lung Yu, Wan-Long Chuang; Division of Hepatogastroenterology, Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan: Hsueh-Chou Lai, Wen-Pang Su, Jung-Ta Kao, Sheng-Hung Chen, Po-Heng Chuang, Cheng-Yuan Peng; Department of Gastroenterology and Hepatology, Linkou Medical Center, Chang Gung Memorial Hospital, Kweishan, Taoyuan, Taiwan: Chun-Yen Lin, Wen-Juei Jeng, I-Shyan Sheen; Department of Internal Medicine, National Taiwan University Hospital and National Taiwan University College of Medicine, Taipei, Taiwan: Chen-Hua Liu, Chun-Jen Liu, Hung-Chih Yang, Chieh-Chang Chen, Shih-Jer Hsu, Jia-Horng Kao; Division of Hepato-Gastroenterology, Department of Internal Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan: Jing-Houng Wang, Kwong-Ming Kee, Sheng-Nan Lu; Academia Sinica, Taipei, Taiwan: Hwai-I Yang, Chien-Jen Chen; Global Health Economics and Outcomes Research, Bristol Myers-Squibb, Princeton, NJ, USA: Yong Yuan.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.692263/full#supplementary-material
1. Chen SL, Morgan TR. The Natural History of Hepatitis C Virus (HCV) Infection. Int J Med Sci (2006) 3(2):47–52. doi: 10.7150/ijms.3.47
2. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic Variation in IL28B Predicts Hepatitis C Treatment-Induced Viral Clearance. Nature (2009) 461(7262):399–401. doi: 10.1038/nature08309
3. Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic Variation in IL28B and Spontaneous Clearance of Hepatitis C Virus. Nature (2009) 461(7265):798–801. doi: 10.1038/nature08463
4. Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, et al. IL28B Is Associated With Response to Chronic Hepatitis C Interferon-Alpha and Ribavirin Therapy. Nat Genet (2009) 41(10):1100–4. doi: 10.1038/ng.447
5. Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, et al. Genome-Wide Association of IL28B With Response to Pegylated Interferon-Alpha and Ribavirin Therapy for Chronic Hepatitis C. Nat Genet (2009) 41(10):1105–9. doi: 10.1038/ng.449
6. Prokunina-Olsson L. Genetics of the Human Interferon Lambda Region. J Interferon Cytokine Res (2019) 39(10):599–608.
7. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A Variant Upstream of IFNL3 (IL28B) Creating a New Interferon Gene IFNL4 Is Associated With Impaired Clearance of Hepatitis C Virus. Nat Genet (2013) 45(2):164–71. doi: 10.1038/ng.2521
8. McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, Shapiro BA, et al. The Favorable IFNL3 Genotype Escapes mRNA Decay Mediated by AU-Rich Elements and Hepatitis C Virus-Induced MicroRNAs. Nat Immunol (2014) 15(1):72–9. doi: 10.1038/ni.2758
9. Moreira JP, Malta Fde M, Diniz MA, Kikuchi L, Chagas AL, Lima Lde S, et al. Interferon Lambda and Hepatitis C Virus Core Protein Polymorphisms Associated With Liver Cancer. Virology (2016) 493:136–41. doi: 10.1016/j.virol.2016.03.008
10. Chang KC, Tseng PL, Wu YY, Hung HC, Huang CM, Lu SN, et al. A Polymorphism in Interferon L3 Is an Independent Risk Factor for Development of Hepatocellular Carcinoma After Treatment of Hepatitis C Virus Infection. Clin Gastroenterol Hepatol (2015) 13(5):1017–24. doi: 10.1016/j.cgh.2014.10.035
11. Asahina Y, Tsuchiya K, Nishimura T, Muraoka M, Suzuki Y, Tamaki N, et al. Genetic Variation Near Interleukin 28B and the Risk of Hepatocellular Carcinoma in Patients With Chronic Hepatitis C. J Gastroenterol (2014) 49(7):1152–62. doi: 10.1007/s00535-013-0858-2
12. Hodo Y, Honda M, Tanaka A, Nomura Y, Arai K, Yamashita T, et al. Association of Interleukin-28B Genotype and Hepatocellular Carcinoma Recurrence in Patients With Chronic Hepatitis C. Clin Cancer Res (2013) 19(7):1827–37. doi: 10.1158/1078-0432.CCR-12-1641
13. Lee MH, Yang HI, Lu SN, Lin YJ, Jen CL, Wong KH, et al. Polymorphisms Near the IFNL3 Gene Associated With HCV RNA Spontaneous Clearance and Hepatocellular Carcinoma Risk. Sci Rep (2015) 5:17030. doi: 10.1038/srep17030
14. Joshita S, Umemura T, Katsuyama Y, Ichikawa Y, Kimura T, Morita S, et al. Association of IL28B Gene Polymorphism With Development of Hepatocellular Carcinoma in Japanese Patients With Chronic Hepatitis C Virus Infection. Hum Immunol (2012) 73(3):298–300. doi: 10.1016/j.humimm.2011.12.021
15. Agundez JA, Garcia-Martin E, Maestro ML, Cuenca F, Martinez C, Ortega L, et al. Relation of IL28B Gene Polymorphism With Biochemical and Histological Features in Hepatitis C Virus-Induced Liver Disease. PloS One (2012) 7(5):e37998. doi: 10.1371/journal.pone.0037998
16. Eslam M, McLeod D, Kelaeng KS, Mangia A, Berg T, Thabet K, et al. IFN-Lambda3, Not IFN-Lambda4, Likely Mediates IFNL3-IFNL4 Haplotype-Dependent Hepatic Inflammation and Fibrosis. Nat Genet (2017) 49(5):795–800. doi: 10.1038/ng.3836
17. Eslam M, Hashem AM, Leung R, Romero-Gomez M, Berg T, Dore GJ, et al. Interferon-Lambda Rs12979860 Genotype and Liver Fibrosis in Viral and Non-Viral Chronic Liver Disease. Nat Commun (2015) 6:6422. doi: 10.1038/ncomms7422
18. Moreira RK. Hepatic Stellate Cells and Liver Fibrosis. Arch Pathol Lab Med (2007) 131(11):1728–34. doi: 10.5858/2007-131-1728-HSCALF
19. Hirata M, Kamatani Y, Nagai A, Kiyohara Y, Ninomiya T, Tamakoshi A, et al. Cross-Sectional Analysis of Biobank Japan Clinical Data: A Large Cohort of 200,000 Patients With 47 Common Diseases. J Epidemiol (2017) 27(3s):S9–s21. doi: 10.1016/j.je.2016.12.003
20. Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, et al. Genome-Wide Association Study Identifies 112 New Loci for Body Mass Index in the Japanese Population. Nat Genet (2017) 49(10):1458–67. doi: 10.1038/ng.3951
21. Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. Mach: Using Sequence and Genotype Data to Estimate Haplotypes and Unobserved Genotypes. Genet Epidemiol (2010) 34(8):816–34. doi: 10.1002/gepi.20533
22. Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A Global Reference for Human Genetic Variation. Nature (2015) 526(7571):68–74. doi: 10.1038/nature15393
23. Lee MH, Huang CF, Lai HC, Lin CY, Dai CY, Liu CJ, et al. Clinical Efficacy and Post-Treatment Seromarkers Associated With the Risk of Hepatocellular Carcinoma Among Chronic Hepatitis C Patients. Sci Rep (2017) 7(1):3718. doi: 10.1038/s41598-017-02313-y
24. Hu L, Zhai X, Liu J, Chu M, Pan S, Jiang J, et al. Genetic Variants in Human Leukocyte Antigen/DP-DQ Influence Both Hepatitis B Virus Clearance and Hepatocellular Carcinoma Development. Hepatology (2012) 55(5):1426–31. doi: 10.1002/hep.24799
25. Obajemu AA, Rao N, Dilley KA, Vargas JM, Sheikh F, Donnelly RP, et al. IFN-Lambda4 Attenuates Antiviral Responses by Enhancing Negative Regulation of IFN Signaling. J Immunol (2017) 199(11):3808–20. doi: 10.4049/jimmunol.1700807
26. Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, Mateo JL. Cctop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PloS One (2015) 10(4):e0124633. doi: 10.1371/journal.pone.0124633
27. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics (2013) 29(1):15–21. doi: 10.1093/bioinformatics/bts635
28. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map Format and Samtools. Bioinformatics (2009) 25(16):2078–9. doi: 10.1093/bioinformatics/btp352
29. Love MI, Huber W, Anders S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data With Deseq2. Genome Biol (2014) 15(12):550. doi: 10.1186/s13059-014-0550-8
30. Anders S, Huber W. Differential Expression Analysis for Sequence Count Data. Genome Biol (2010) 11(10):1–12. doi: 10.1186/gb-2010-11-10-r106
31. Terczynska-Dyla E, Bibert S, Duong FH, Krol I, Jorgensen S, Collinet E, et al. Reduced Ifnlambda4 Activity Is Associated With Improved HCV Clearance and Reduced Expression of Interferon-Stimulated Genes. Nat Commun (2014) 5:5699. doi: 10.1038/ncomms6699
32. Gadalla SM, Wang Y, Wang T, Onabajo OO, Banday AR, Obajemu A, et al. Association of Donor IFNL4 Genotype and Non-Relapse Mortality After Unrelated Donor Myeloablative Haematopoietic Stem-Cell Transplantation for Acute Leukaemia: A Retrospective Cohort Study. Lancet Haematol (2020) 7(10):e715–23. doi: 10.1016/S2352-3026(20)30294-5
33. Prokunina-Olsson L, Morrison RD, Obajemu A, Mahamar A, Kim S, Attaher O, et al. IFN-Lambda4 Is Associated With Increased Risk and Earlier Occurrence of Several Common Infections in African Children. Genes Immun (2021) 22(1):44–55. doi: 10.1038/s41435-021-00127-7
34. Bochud PY, Bibert S, Kutalik Z, Patin E, Guergnon J, Nalpas B, et al. IL28B Alleles Associated With Poor Hepatitis C Virus (HCV) Clearance Protect Against Inflammation and Fibrosis in Patients Infected With Non-1 HCV Genotypes. Hepatology (2012) 55(2):384–94. doi: 10.1002/hep.24678
35. Zhao J, Zhang X, Fang L, Pan H, Shi J. Association Between IL28B Polymorphisms and Outcomes of Hepatitis B Virus Infection: A Meta-Analysis. BMC Med Genet (2020) 21(1):88. doi: 10.1186/s12881-020-01026-w
36. Martin MP, Qi Y, Goedert JJ, Hussain SK, Kirk GD, Hoots WK, et al. IL28B Polymorphism Does Not Determine Outcomes of Hepatitis B Virus or HIV Infection. J Infect Dis (2010) 202(11):1749–53. doi: 10.1086/657146
37. Onabajo OO, Porter-Gill P, Paquin A, Rao N, Liu L, Tang W, et al. Expression of Interferon Lambda 4 Is Associated With Reduced Proliferation and Increased Cell Death in Human Hepatic Cells. J Interferon Cytokine Res (2015) 35(11):888–900. doi: 10.1089/jir.2014.0161
38. Hong M, Schwerk J, Lim C, Kell A, Jarret A, Pangallo J, et al. Interferon Lambda 4 Expression Is Suppressed by the Host During Viral Infection. J Exp Med (2016) 213(12):2539–52. doi: 10.1084/jem.20160437
39. Hetz C. The Unfolded Protein Response: Controlling Cell Fate Decisions Under ER Stress and Beyond. Nat Rev Mol Cell Biol (2012) 13(2):89–102. doi: 10.1038/nrm3270
40. Pluquet O, Pourtier A, Abbadie C. The Unfolded Protein Response and Cellular Senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am J Physiol Cell Physiol (2015) 308(6):C415–25. doi: 10.1152/ajpcell.00334.2014
41. Ron D, Walter P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat Rev Mol Cell Biol (2007) 8(7):519–29. doi: 10.1038/nrm2199
42. MacGurn JA. Garbage on, Garbage Off: New Insights Into Plasma Membrane Protein Quality Control. Curr Opin Cell Biol (2014) 29:92–8. doi: 10.1016/j.ceb.2014.05.001
43. Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S. Ectopic Expression of CHOP (GADD153) Induces Apoptosis in M1 Myeloblastic Leukemia Cells. FEBS Lett (1996) 395(2-3):143–7. doi: 10.1016/0014-5793(96)01016-2
44. Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, et al. Identification of Novel Stress-Induced Genes Downstream of Chop. EMBO J (1998) 17(13):3619–30. doi: 10.1093/emboj/17.13.3619
45. Wang Y, Li J, Wang X, Sang M, Ho W. Hepatic Stellate Cells, Liver Innate Immunity, and Hepatitis C Virus. J Gastroenterol Hepatol (2013) 28 Suppl 1:112–5. doi: 10.1111/jgh.12023
46. Mazzocca A, Sciammetta SC, Carloni V, Cosmi L, Annunziato F, Harada T, et al. Binding of Hepatitis C Virus Envelope Protein E2 to CD81 Up-Regulates Matrix Metalloproteinase-2 in Human Hepatic Stellate Cells. J Biol Chem (2005) 280(12):11329–39. doi: 10.1074/jbc.M410161200
47. Liu X, Xu J, Rosenthal S, Zhang LJ, McCubbin R, Meshgin N, et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology (2020) 158(6):1728–44. doi: 10.1053/j.gastro.2020.01.027
48. Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, et al. Human Occludin Is a Hepatitis C Virus Entry Factor Required for Infection of Mouse Cells. Nature (2009) 457(7231):882–6. doi: 10.1038/nature07684
49. Cormier EG, Tsamis F, Kajumo F, Durso RJ, Gardner JP, Dragic T. CD81 Is an Entry Coreceptor for Hepatitis C Virus. Proc Natl Acad Sci USA (2004) 101(19):7270–4. doi: 10.1073/pnas.0402253101
50. Ujino S, Nishitsuji H, Hishiki T, Sugiyama K, Takaku H, Shimotohno K. Hepatitis C Virus Utilizes VLDLR as a Novel Entry Pathway. Proc Natl Acad Sci USA (2016) 113(1):188–93. doi: 10.1073/pnas.1506524113
51. Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, et al. Endoplasmic Reticulum Stress Induces Hepatic Steatosis via Increased Expression of the Hepatic Very Low-Density Lipoprotein Receptor. Hepatology (2013) 57(4):1366–77. doi: 10.1002/hep.26126
52. Chakraborty JB, Oakley F, Walsh MJ. Mechanisms and Biomarkers of Apoptosis in Liver Disease and Fibrosis. Int J Hepatol (2012) 2012:648915. doi: 10.1155/2012/648915
53. Maiers JL, Malhi H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin Liver Dis (2019) 39(2):235–48. doi: 10.1055/s-0039-1681032
54. Liu X, Xu J, Brenner DA, Kisseleva T. Reversibility of Liver Fibrosis and Inactivation of Fibrogenic Myofibroblasts. Curr Pathobiol Rep (2013) 1(3):209–14. doi: 10.1007/s40139-013-0018-7
55. Mirabello L, Yeager M, Yu K, Clifford GM, Xiao Y, Zhu B, et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell (2017) 170(6):1164–74.e6. doi: 10.1016/j.cell.2017.08.001
56. Mihailidou C, Papavassiliou AG, Kiaris H. Cell-Autonomous Cytotoxicity of Type I Interferon Response via Induction of Endoplasmic Reticulum Stress. FASEB J (2017) 31(12):5432–9. doi: 10.1096/fj.201700152R
57. Shi WY, Cao C, Liu L. Interferon Alpha Induces the Apoptosis of Cervical Cancer Hela Cells by Activating Both the Intrinsic Mitochondrial Pathway and Endoplasmic Reticulum Stress-Induced Pathway. Int J Mol Sci (2016) 17(11):312–24. doi: 10.3390/ijms17111832
58. Lombardi A, Tomer Y. Interferon Alpha Impairs Insulin Production in Human Beta Cells via Endoplasmic Reticulum Stress. J Autoimmun (2017) 80:48–55. doi: 10.1016/j.jaut.2017.02.002
59. Scheel TK, Rice CM. Understanding the Hepatitis C Virus Life Cycle Paves the Way for Highly Effective Therapies. Nat Med (2013) 19(7):837–49. doi: 10.1038/nm.3248
60. Goldstein JL, Brown MS. The LDL Receptor. Arterioscler Thromb Vasc Biol (2009) 29(4):431–8. doi: 10.1161/ATVBAHA.108.179564
Keywords: IFNL4, HCC (hepatocellular carcinoma), liver cirrhosis, ER stress, HCV (Hepatitis C)
Citation: Onabajo OO, Wang F, Lee M-H, Florez-Vargas O, Obajemu A, Tanikawa C, Vargas JM, Liao S-F, Song C, Huang Y-H, Shen C-Y, Banday AR, O’Brien TR, Hu Z, Matsuda K and Prokunina-Olsson L (2021) Intracellular Accumulation of IFN-λ4 Induces ER Stress and Results in Anti-Cirrhotic but Pro-HCV Effects. Front. Immunol. 12:692263. doi: 10.3389/fimmu.2021.692263
Received: 08 April 2021; Accepted: 02 August 2021;
Published: 23 August 2021.
Edited by:
Achille Broggi, Boston Children’s Hospital and Harvard Medical School, United StatesReviewed by:
Amariliz Rivera, The State University of New Jersey, United StatesCopyright © 2021 Onabajo, Wang, Lee, Florez-Vargas, Obajemu, Tanikawa, Vargas, Liao, Song, Huang, Shen, Banday, O’Brien, Hu, Matsuda and Prokunina-Olsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludmila Prokunina-Olsson, cHJva3VuaW5hbEBtYWlsLm5paC5nb3Y=
†Present address: Fang Wang, Department of Pathogen Biology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin, China
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.