 Hao Cai
Hao Cai Yichi Zhang
Yichi Zhang Jian Wang
Jian Wang Jinyang Gu
Jinyang Gu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 June 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.690869
This article is part of the Research TopicCancer Immunotherapies: From Efficacy to Resistance MechanismsView all 42 articles
Classically activated M1 macrophages and alternatively activated M2 macrophages are two polarized subsets of macrophages at the extreme ends of a constructed continuum. In the field of cancer research, M2 macrophage reprogramming is defined as the repolarization of pro-tumoral M2 to anti-tumoral M1 macrophages. It is known that colony-stimulating factor 1 (CSF1)/CSF1 receptor (CSF1R) and CSF2/CSF2R signaling play important roles in macrophage polarization. Targeting CSF1/CSF1R for M2 macrophage reprogramming has been widely performed in clinical trials for cancer therapy. Other targets for M2 macrophage reprogramming include Toll-like receptor 7 (TLR7), TLR8, TLR9, CD40, histone deacetylase (HDAC), and PI3Kγ. Although macrophages are involved in innate and adaptive immune responses, M1 macrophages are less effective at phagocytosis and antigen presenting, which are required properties for the activation of T cells and eradication of cancer cells. Similar to T and dendritic cells, the “functionally exhausted” status might be attributed to the high expression of programmed death-ligand 1 (PD-L1) or programmed cell death protein 1 (PD-1). PD-L1 is expressed on both M1 and M2 macrophages. Macrophage reprogramming from M2 to M1 might increase the expression of PD-L1, which can be transcriptionally activated by STAT3. Macrophage reprogramming or PD-L1/PD-1 blockade alone is less effective in the treatment of most cancers. Since PD-L1/PD-1 blockade could make up for the defect in macrophage reprogramming, the combination of macrophage reprogramming and PD-L1/PD-1 blockade might be a novel treatment strategy for cancer therapy.
Macrophages exhibit a high degree of plasticity when exposed to various environmental stimuli. They are polarized to one of two opposite types in vitro, classically activated M1 macrophages that can be induced by lipopolysaccharide (LPS), interferon-γ (IFN-γ), or colony-stimulating factor 2 (CSF2, also known as granulocyte-macrophage colony-stimulating factor) or alternatively activated M2 macrophages that can be induced by interleukin 4 (IL4), IL13, or CSF1 (also known as macrophage colony-stimulating factor) (1–3). M2 macrophages are further categorized as M2a, M2b, M2c, and M2d cells upon stimulation of different M2 drivers (4). Generally, M1 macrophages exert an immune protective role via the secretion of pro-inflammatory cytokines, whereas M2 macrophages are characterized by anti-inflammatory properties, which contribute to tissue remodeling and tumor progression (3). Multiple co-stimulatory and antigen-presenting molecules are expressed on the cell membrane of antigen-presenting cells (APCs), including macrophages. When confronted by tumor antigens, macrophages engulf and present them to T cells to boost the anti-tumor immune reaction by acting synergistically with co-stimulatory molecules (1). However, the function of macrophages is more complex in the tumor microenvironment. Tumor-associated macrophages (TAMs) are thought to exhibit an M2-like phenotype, lose their antigen-presenting capacity as innate immune cells, and play a pro-tumoral role in the tumor microenvironment in a paracrine manner (5, 6). The phenotype of TAMs dynamically changes with the development and progression of tumors. At an early stage, macrophages harboring anti-tumor capacity are recruited to the tumor microenvironment; however, with tumor progression, these macrophages are “educated” by tumor-secreted cytokines to acquire an M2 phenotype (1). It is accepted that M1 and M2 are two extreme forms of polarization in vitro, and TAMs usually exhibit a mixed M1–M2 phenotype, and not a simple M1 or M2 only phenotype, in vivo (7–9).
Macrophage reprogramming, also called macrophage repolarization, is defined as the repolarization of differentiated macrophages from alternatively activated M2 phenotype to the classically activated M1 phenotype, and vice versa. Several methods have been developed to reprogram M2 macrophages, including use of targeted antibodies, small molecular inhibitors, and free or vector-delivering nucleic acids, among others. Although M2 macrophage reprogramming has been adopted in clinical trials, the treatment outcome remains uncertain. In this review, we aim to shed light on the defects in M2 macrophage reprogramming and provide better treatment strategies for cancer therapy.
Molecular targets for M2 macrophage reprogramming include Toll-like receptor 7 (TLR7), TLR8, TLR9, CD40, histone deacetylase (HDAC), PI3Kγ, CSF1, and CSF1 receptor (CSF1R) (10). TLR agonists induce M1 polarization and exert an anti-tumor effect via the increased release of pro-inflammatory mediators. CD40 agonists increase the expression of co-stimulatory and antigen-presenting molecules on macrophages and the secretion of pro-inflammatory mediators, which enhances the T cell–dependent anti-tumor effect (10). TLR signaling and CD40 are known to be activated by IFN-γ (11, 12), which is a driver of M1 polarization. Both HDAC and PI3Kγ are involved in the M2 polarization of macrophages, providing intracellular targets for macrophage reprogramming. The inhibition of HDAC or PI3Kγ exerts an anti-tumor effect via the downregulation of M2 and upregulation of M1 molecules (10). As has been reported, PI3K is present downstream of CSF1R and is epigenetically activated during M2 polarization (13).
The CSF1/CSF1R axis is the most attractive target to reprogram M2 macrophages, and multiple agents have been developed for clinical practice, including small molecule inhibitors (PLX3397, BLZ945, ARRY-382, etc.) and neutralizing antibodies against CSF1 or CSF1R (10, 14). In the tumor microenvironment, tumor cell-derived CSF1 is enriched within peri-tumoral tissues and functions as a chemoattractant to recruit circulating monocytes, subsequently resulting in increased macrophage infiltration (15). CSF1R is a transmembrane receptor for CSF1 and IL34 with tyrosine kinase activity. Binding of CSF1 or IL34 induces the homodimerization of CSF1R and the activation of downstream MEK, PI3K, and PLC-γ2 signaling pathways, which are crucial for the proliferation and differentiation of macrophages (13). It was reported that CSF1/CSF1R blockade-based anti-tumor therapy could result in loss of macrophages in the tumor either by mitigating recruitment, TAMs survival and/or differentiation from monocytes (3). Ao et al. reported that CSF1R inhibitor PLX3397 suppressed tumor growth without depletion of TAMs infiltration in a mouse model of liver cancer (16). These discrepancies might be attributed to the heterogenicity of different tumor species and different CSF1/CSF1R blockade agents. It is accepted that activation of the CSF1R signaling pathway induces the M2 polarization of macrophages (14). In contrast with CSF1/CSF1R signaling that induces M2 polarization, the CSF2/CSF2R pathway induces M1 polarization of macrophages. CSF1R and CSF2R are constitutively expressed on the cell membrane of macrophages. Both CSF1/CSF1R and CSF2/CSF2R signaling pathways play important roles in macrophage reprogramming. Infiltrating macrophages are “educated” by tumor cell-derived CSF1 or IL34 to acquire an M2 phenotype, characterized by the high expression of CD163 or CD206 (17–19). After blockade of the CSF1R signaling pathway, macrophages are repolarized instead through the CSF2/CSF2R axis to acquire an M1 phenotype (16). CSF2 enhances the antigen presentation capacity of macrophages with increased expression of major histocompatibility complex-II (MHC-II) (20). In addition to inducing M1 polarization, CSF2 is also responsible for the development of dendritic cells (DCs) and granulocytes, which are also APCs that exert a similar anti-tumor effect as M1 macrophages (21, 22).
However, it has been reported that traditionally defined M1 macrophages with “anti-tumor properties” could also facilitate the metastasis of hepatocellular carcinoma in a paracrine manner (23). IL-1β released by M1 macrophages induces the expression of co-inhibitory molecules in tumor cells, which hampers the direct anti-tumor effect of cytotoxic T cells (24). Although these M1 macrophages are presented with high MHC-II expression, the process of phagocytosis and antigen presentation does not function as expected and needs further investigation. Therefore, after macrophage reprogramming from M2 to M1, the restoration of the phagocytic and antigen-presenting capacity of macrophages as innate immune cells should be taken into consideration to develop strategies for anti-tumor therapy.
Programmed cell death-ligand 1 (PD-L1) was reported to be expressed on a variety of cells, including tumor cells and APCs (mainly DCs and macrophages), and acts as a ligand for programmed cell death protein 1 (PD-1), which is mainly expressed on T cells. The binding of PD-L1 on macrophages to PD-1 on T cells antagonizes the co-stimulating and antigen-presenting effect of macrophages on T cells, leading to T-cell anergy and tumor cell immune escape. In addition to being a ligand for PD-1, PD-L1 inhibits the proliferation and activation of macrophages by suppressing the mechanistic target of rapamycin signaling pathway in macrophages. In addition, PD-L1 induces an immunosuppressive phenotype and inhibits the antigen-presenting capacity by reducing the expression of co-stimulatory molecules in macrophages. PD-L1 blockade increases the production of co-stimulatory molecules (CD86 and MHC-II) and pro-inflammatory cytokines (tumor necrosis factor α [TNFα] and IL12), which comprises the phenotype and expression profile of M1 macrophages (25). However, transcriptomic analysis showed that in addition to increasing the expression of pro-inflammatory genes, CCL2, a key driver of macrophage recruitment and M2 polarization is also upregulated. It was reported that PD-L1 blockade reinvigorates T cells, which is accompanied by increased IFN-γ production, further driving the M1 polarization of macrophages (26). However, the direct impact of PD-L1 on macrophage polarization is uncertain. The correlation between PD-L1 expression and macrophage polarization is ambiguous. Multiple factors involved in both M1 and M2 polarization (IFN-γ, TNFα, LPS, IL4, IL6, IL10, IL13, etc.) increase the expression of PD-L1 on macrophages (27–31). Therefore, PD-L1 is not an exclusive biomarker of M1 or M2 macrophages.
Recently, PD-1 was also found to be expressed on TAMs but not on circulating monocytes or spleen macrophages. The tumor microenvironment might play a critical role in the expression of PD-1 in macrophages. PD-1 inhibits the phagocytosis of TAMs, which is an inherent attribute of APCs in innate immunity (32). Peritoneal macrophages with high PD-1 expression are dysfunctional with reduced bactericidal capacity (33). It has been reported that macrophages with high PD-1 expression are more likely to be the pro-tumoral M2 subtype with increased CD206 expression and reduced MHC-II expression (32). Rao et al. reported that anti-PD-1 therapy induces M1 polarization in macrophages and exerts an anti-tumor effect in the absence of CD8+ T cells (34). In this study, the authors concluded that combining macrophage reprogramming with anti-PD-1 therapy is unnecessary, because anti–PD-1 alone can eliminate PD-1-expressing microglia, thus driving M2 to M1 repolarization. However, this result should be interpreted with caution. Even after PD-1-expressing macrophages have been eliminated, M1 polarization of the remaining macrophages might not occur without extra stimulation or induction. Thus, the role of CSF2 or IFN-γ should be taken into consideration.
As reported, activation of the PD-L1/PD-1 axis represents a state of “functional exhaustion” in T cells and DCs (35–37). Exhausted T cells are initially functional when exposed to antigen but gradually become silent after persistent stimulation. The exhausted T cells are characterized by increased expression of inhibitory molecules and decreased secretion of effector cytokines, including IL2, IFN-γ, and TNFα. The inhibitory receptors expressed on exhausted T cells include PD-1, cytotoxic T-lymphocyte-associated protein 4 (CTLA4), T-cell immunoreceptor with Ig and ITIM domains (TIGIT), lymphocyte-activation gene 3 (LAG-3), B and T lymphocyte attenuator (BTLA), T-cell immunoglobulin and mucin-domain-containing protein 3 (TIM3), V domain Ig suppressor of T-cell activation (VISTA), and CD96, among others (38). Similarly, these macrophages with high PD-L1 or PD-1 expression could also be called “functionally exhausted.” PD-L1 or PD-1 expression hampers the anti-tumor effect of macrophages as innate and adaptive immune cells, which could be restored after PD-L1/PD-1 blockade.
Analysis of Gene Expression Omnibus data sets (GSE95404, 71253, 66805, 95405, 69607) by comparing the transcriptional differences of M1 (induced by CSF2 or LPS/IFN-γ) and M2 (induced by CSF1 or IL4) macrophages revealed that PD-L1 expression is significantly higher in M1 macrophages than in M2 macrophages. Antonios et al. showed that the expression of PD-L1 is also significantly higher in CD163− cells than in CD163+ cells (39). A recent study identified an immune suppressor induced by CSF1, Siglec-15, which is negatively correlated with PD-L1 expression (40). CSF1R blockade increases the expression of PD-L1 in pancreatic ductal tumor cells and CD11b+Ly6Chigh monocytes (41), but the underlying mechanism remains unknown. CSF2 plays an important role in directing M1 polarization after blockade of the CSF1/CSR1R signaling pathway, which is involved in M2 polarization. Shelby et al. showed a positive correlation between PD-L1 and CSF2 concentrations in gingival crevicular fluid of patients diagnosed with periodontitis (42). It has been reported that the expression of PD-L1 on APCs is transactivated by STAT3 (43). Tumor-derived CSF2 increases the expression of PD-L1 in granulocytes by activating the JAK/STAT3 signaling pathway, which inhibits the anti-tumor effect of T cells and contributes to the progression of gastric cancer (44) and hepatocellular carcinoma (45). In liver myeloid-derived suppressor cells, activation of the CSF2/JAK2/STAT3 pathway increases the expression of PD-L1, which facilitates the intrahepatic metastasis of liver neoplasm (46). It was also reported that CSF2 increases the secretion of CXCL8 in macrophages, which further induces the expression of PD-L1 on TAMs in an autocrine manner and inhibits the anti-tumor effect of CD8+ T cells (47). However, the underlying mechanism through which CSF2 increases PD-L1 expression on macrophages has not been fully elucidated.
PD-L1 expression is known to increase in M1 macrophages when induced with CSF2, LPS, or IFN-γ. An in vitro study showed that phorbol-12-myristate 13-acetate-activated THP-1 macrophages exhibit higher PD-L1 expression after LPS stimulation (48). IFN-γ released by tumor-infiltrating T lymphocytes increases PD-L1 expression in macrophages, which in turn inhibits the anti-tumor effect of T cells, resulting in a state of “adaptive immune tolerance” (49, 50). Both IFN-γ- and LPS-induced PD-L1 expression on tumor-infiltrating macrophages is dependent on the activation of STAT3 (51, 52). Moreover, activation of the TLR4/ERK axis is responsible for LPS-induced PD-L1 expression (53). In addition to CSF1/CSF1R blockade, macrophage reprogramming using HDAC inhibitors or agonistic anti-CD40 antibodies increases the expression of PD-L1 on macrophages, which limits the anti-tumor effect of reprogrammed TAMs (54, 55). Although the impact of macrophage reprogramming targeting PI3Kγ on PD-L1 expression is unknown, it has been reported that PI3Kγ inhibition increases the expression of PD-L1 on myeloid-derived suppressor cells (56). This evidence shows that all of these macrophage reprogramming strategies have the same side effect of increased PD-L1 expression on macrophages (Figure 1). Although PD-1 is also reportedly found on macrophages, the impact of macrophage reprogramming on PD-1 expression has not yet been reported.
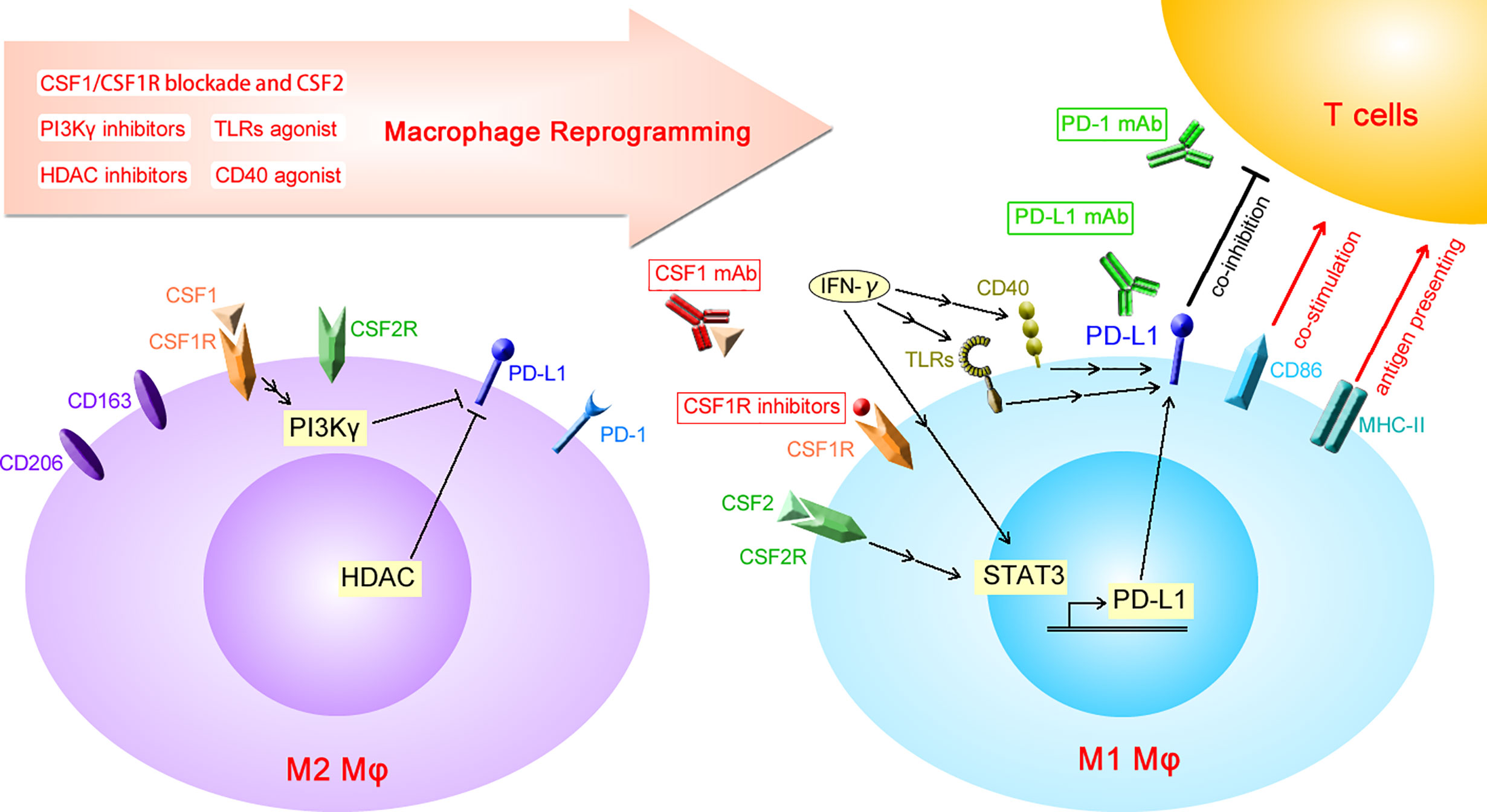
Figure 1 Diagrammatic sketch of macrophage reprogramming increasing the expression of PD-L1. PD-L1, programmed death-ligand 1; PD-1, programmed cell death protein 1; CSF1, colony-stimulating factor 1; CSF1R, CSF1 receptor; CSF2, colony-stimulating factor 2; CSF2R, CSF2 receptor; HDAC, histone deacetylase; IFN-γ, interferon-γ; TLRs, toll-like receptors; MHC-II, major histocompatibility complex-II; mAb, monoclonal antibody; Mφ, macrophages.
In recent years, PD-L1/PD-1 monoclonal antibodies have gained widespread attention for cancer treatment. They show a strong anti-tumor effect in certain cancers such as Hodgkin’s disease and desmoplastic melanoma, among others. However, in most cancers, including non-small-cell lung carcinoma, gastroesophageal cancer, urinary neoplasm, and hepatocellular carcinoma, PD-L1/PD-1 blockade alone is only effective in a small proportion of patients (with objective response rates ranging from 15% to 25%) (57). Resistance to PD-L1/PD-1 blockade might be attributed to a lack of pre-existing T-cell infiltration in certain tumors. Another mechanism for ineffective anti-PD-L1/PD-1 monotherapy is the increased macrophage infiltration in the tumor microenvironment (58). Tumor-derived CSF1 induces the expression of granulin in macrophages, which impedes the infiltration of cytotoxic CD8+ T cells at the site of the tumor lesion, resulting in resistance to immune checkpoint therapy (59, 60). Therefore, therapies combining anti-PD-L1/PD-1 monoclonal antibodies have been adopted as a priority for clinical practice for selected cancers. For advanced hepatocellular carcinoma and clear cell renal cell carcinoma, anti-PD-L1/PD-1 single-agent therapy is only recommended as a second-line treatment option when first-line treatment fails (61, 62).
Even though macrophage reprogramming using CSF1R inhibitors or CSF1 antibodies has been achieved for anti-tumor therapy, their effectiveness is still uncertain in most cancers. The CSF1R inhibitor PLX3397 has shown significant anti-tumor effects in animal models (63–67), but its treatment efficacy in clinical settings remains unknown. PLX3397 is effective for tenosynovial giant cell tumors (68, 69) with a 39% overall response rate in a phase III clinical trial (69). However, the treatment outcome was rather disappointing for glioma (70). A poor objective response rate was reported for the CSF1R inhibitor ARRY-382 and CSF1R monoclonal antibody emactuzumab in treating solid tumors. Data on tumor control are unavailable for the CSF1 monoclonal antibody lacnotuzumab and CSF1R monoclonal antibodies cabiralizumab and LY3022855 (14). Similar to CSF1/CSF1R blockade, CSF2 induces M1 polarization by activating the CSF2R signaling pathway. Moreover, CSF2 is used to augment the recruitment and maturation of DCs in clinical trials for cancer therapy. However, CSF2 is typically not used as a single agent for macrophage reprogramming in clinical settings. Except for CSF1/CSF1R blockade and CSF2, other macrophage reprogramming agents including CD40 agonists, TLR agonists, HDAC inhibitors, and PI3Kγ inhibitors have been approved in clinical practice for the treatment of selected types of tumors (71–74). All of these macrophage reprogramming strategies are only effective in a small number of patients. As has been detailed in this review, the side effect of increased PD-L1 expression might explain the relatively poor anti-tumor effect of macrophage reprogramming. The anti-tumor nature of macrophages is inhibited by PD-L1, which could be rescued by PD-L1/PD-1 blockade.
Macrophages play a critical role in the activation of T cells (75). Activated CD8+ T cells mediate tumor cell killing directly, whereas activated CD4+ T cells exert anti-tumor effects indirectly by enhancing the cytotoxic effect of CD8+ T cells. The T cell–dependent anti-tumor response requires not only the blockade of co-inhibitory signals on T cells and APCs but also the restoration of co-stimulatory and antigen-presenting molecules on APCs. Macrophage reprogramming-induced PD-L1 expression provides a therapeutic target for PD-L1/PD-1 monoclonal antibodies. Moreover, PD-L1/PD-1 monoclonal antibodies could make up for the defect in macrophage reprogramming, thus improving the anti-tumor effectiveness of macrophage reprogramming. Roemer et al. reported that the expression of both MHC-II and PD-L1 is associated with a favorable outcome with PD-1 blockade (76). Bioinformatics analysis has shown that M1 macrophages are required for the efficacy of anti-PD-L1/PD-1 therapy (77), which implies better treatment outcomes by combining macrophage reprogramming and anti-PD-L1/PD-1 therapy compared to those of single-agent anti-tumor strategies. In this review, CSF1/CSF1R and CSF2/CSF2R-based macrophage reprogramming and its combination with PD-L1/PD-1 blockade is emphasized. The completed and ongoing clinical trials combining macrophage reprogramming and PD-L1/PD-1 blockade in cancer therapy are summarized in Table 1.
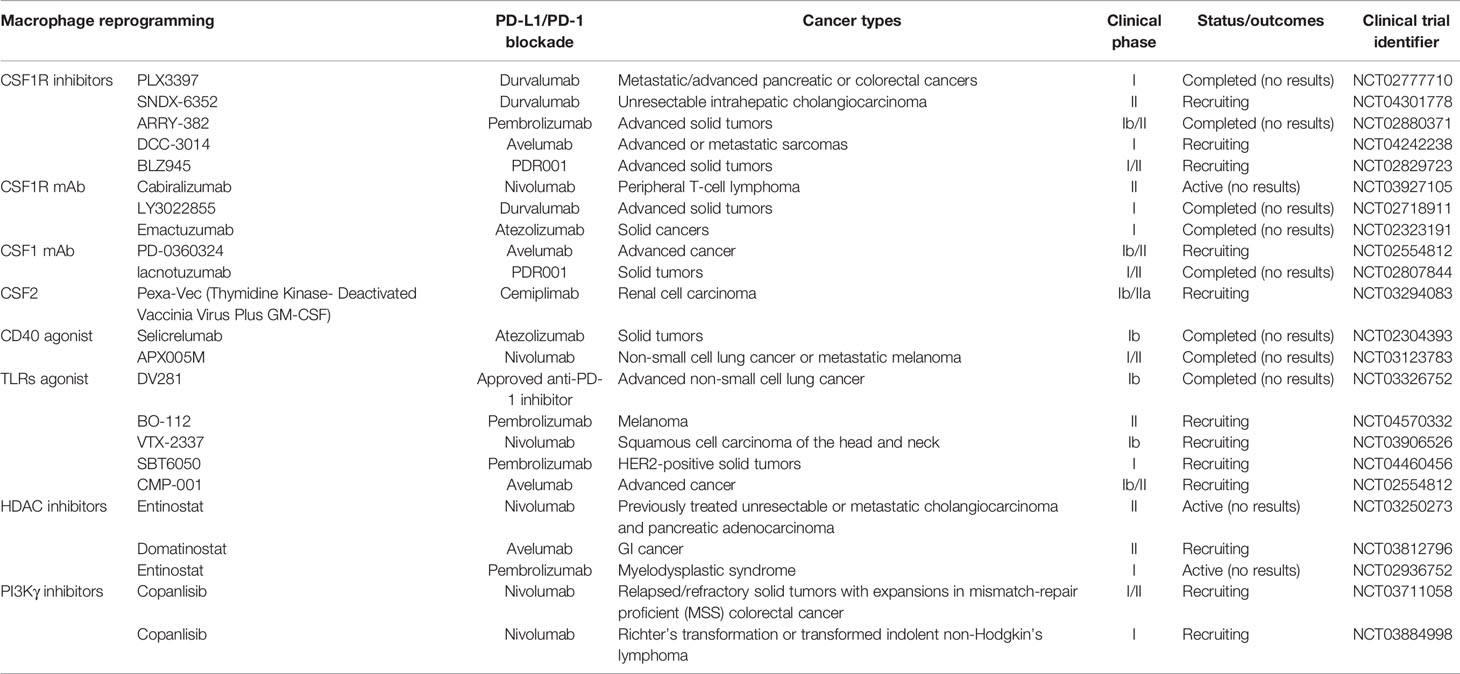
Table 1 Completed and ongoing clinical trials combining macrophage reprogramming and PD-L1/PD-1 blockade in cancer therapy.
A preclinical study showed that PLX3397 combined with a PD-1 monoclonal antibody enhances the anti-tumor effect of a DC vaccine in a mouse glioma model (39). Another CSF1R inhibitor, BLZ945, was also reported to synergize with PD-1/PD-L1–blocking antibodies for the treatment of murine neuroblastoma (78). However, in clinical practice, a lack of evidence on tumor control has limited the use of the combination of CSF1/CSF1R blockade and PD-L1/PD-1 monoclonal antibody to treat solid tumors. A phase II clinical trial using the combination of a CSF1R antibody (AMG820) and pembrolizumab has revealed an acceptable safety profile. However, the anti-tumor effect was insufficient for further evaluation, which might be because most recruited patients were resistant to the PD-1 antibody (79). Recently, mannose-modified macrophage-derived microparticles loaded with metformin have been developed to reprogram M2 to M1 macrophages, which can synergistically enhance anti-PD-1 therapy (80).
A preclinical animal study has shown that anti-PD-1 therapy increases the anti-tumor effect of CSF2 (81). CSF2 is used as a single agent for treating melanoma and was shown to provide no survival benefits in a phase III clinical trial (82). Holi et al. reported that treatment with immune checkpoint inhibitor ipilimumab plus sargramostim (CSF2) showed longer survival (1-year overall survival: 68.9% vs 52.9%) and lower toxicity (grades 3–5 toxicity: 44.9% vs 58.3%) than ipilimumab alone in treating metastatic melanoma (83). In most cases, CSF2 is usually anchored to the tumor vaccine as an adjuvant. PD-L1/PD-1 blockade was shown to increase the anti-tumor effect of the anchored-CSF2 tumor vaccine (84–86). Tian et al. constructed a new type of tumor vaccine that produces both PD-1 antibody and CSF2, which has shown a promising anti-tumor effect (87). In gallbladder cancer, conventional chemotherapy supplemented with CSF2 and PD-L1 blockade was shown to decrease local cancer recurrence after surgery (88). Therefore, CSF2, in combination with a PD-L1/PD-1 monoclonal antibody, is superior to single-agent therapy.
Macrophage reprogramming has been adopted in clinical trials for cancer therapy. Several reprogramming strategies have been developed by targeting TLR7, TLR8, TLR9, CD40, histone deacetylase (HDAC), PI3Kγ, CSF1, and CSF1R. The CSF1/CSF1R axis is the most attractive target to reprogram M2 macrophages in clinical trials. However, traditionally defined M1 macrophages with “anti-tumor properties” could also facilitate cancer progression, even with high expression of co-stimulatory and antigen-presenting molecules. The side effect of increased PD-L1 expression results in a “functionally exhausted” status in macrophages, which limits the anti-tumor effect of reprogrammed macrophages. PD-L1/PD-1 blockade could make up for the defect in macrophage reprogramming, providing a potentially promising treatment strategy by combining macrophage reprogramming with PD-L1/PD-1 monoclonal antibodies.
JG, HC, and YZ conceived the topic of this review article. HC, YZ, and JW searched reference articles and extracted key information for this review article. HC and YZ wrote this manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (81772507, 82072646 to JG), Key Program of Development Center for Medical Science & Technology National Health Commission of China (NHC2018RWS01007 to JG), Shanghai Municipal Education Commission---Gaofeng Clinical Medicine Grant Support (20191910 to JG) and Clinical Research Plan of SHDC (no. SHDC2020CR3005A to JG).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the contributions of Hui-Chuan Sun from Zhongshan Hospital Fudan University that aided the efforts of the authors.
CSF1, colony-stimulating factor 1; CSF1R, CSF1 receptor; DCs, dendritic cells; PD-1, programmed cell death protein 1; PD-L1, programmed cell death-ligand 1; APCs, antigen-presenting cells; IFN-γ, interferon-γ; IL, interleukin; LPS, lipopolysaccharide; TAMs, tumor-associated macrophages; HDAC, histone deacetylase; TLR7, Toll-like receptor 7; MHC-II, major histocompatibility complex-II.
1. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage Plasticity, Polarization, and Function in Health and Disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
2. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J Immunol (2006) 177(10):7303–11. doi: 10.4049/jimmunol.177.10.7303
3. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res (2021) 81(5):1201–8. doi: 10.1158/0008-5472.CAN-20-2990
4. Abdelaziz MH, Abdelwahab SF, Wan J, Cai W, Huixuan W, Jianjun C, et al. Alternatively Activated Macrophages; a Double-Edged Sword in Allergic Asthma. J Trans Med (2020) 18(1):58. doi: 10.1186/s12967-020-02251-w
5. Belgiovine C, D’Incalci M, Allavena P, Frapolli R. Tumor-Associated Macrophages and Anti-Tumor Therapies: Complex Links. Cell Mol Life Sci: CMLS (2016) 73(13):2411–24. doi: 10.1007/s00018-016-2166-5
6. Biswas SK, Mantovani A. Macrophage Plasticity and Interaction With Lymphocyte Subsets: Cancer as a Paradigm. Nat Immunol (2010) 11(10):889–96. doi: 10.1038/ni.1937
7. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
8. Helm O, Mennrich R, Petrick D, Goebel L, Freitag-Wolf S, Roder C, et al. Comparative Characterization of Stroma Cells and Ductal Epithelium in Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma. PloS One (2014) 9(5):e94357. doi: 10.1371/journal.pone.0094357
9. Wu MF, Lin CA, Yuan TH, Yeh HY, Su SF, Guo CL, et al. The M1/M2 Spectrum and Plasticity of Malignant Pleural Effusion-Macrophage in Advanced Lung Cancer. Cancer Immunol Immunother (2021) 70(5):1435–50. doi: 10.1007/s00262-020-02781-8
10. Bart VMT, Pickering RJ, Taylor PR, Ipseiz N. Macrophage Reprogramming for Therapy. Immunology (2021) 163(2):128–44. doi: 10.1111/imm.13300
11. Schroder K, Sweet MJ, Hume DA. Signal Integration Between Ifngamma and TLR Signalling Pathways in Macrophages. Immunobiology (2006) 211(6-8):511–24. doi: 10.1016/j.imbio.2006.05.007
12. Lee SJ, Qin H, Benveniste EN. Simvastatin Inhibits IFN-Gamma-Induced CD40 Gene Expression by Suppressing STAT-1alpha. J Leukocyte Biol (2007) 82(2):436–47. doi: 10.1189/jlb.1206739
13. Stanley ER, Chitu V. CSF-1 Receptor Signaling in Myeloid Cells. Cold Spring Harbor Perspect Biol (2014) 6(6):a021857. doi: 10.1101/cshperspect.a021857
14. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors in Cancer Therapy. J Immunother Cancer (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
15. Sossey-Alaoui K, Pluskota E, Bialkowska K, Szpak D, Parker Y, Morrison CD, et al. Kindlin-2 Regulates the Growth of Breast Cancer Tumors by Activating CSF-1-Mediated Macrophage Infiltration. Cancer Res (2017) 77(18):5129–41. doi: 10.1158/0008-5472.CAN-16-2337
16. Ao JY, Zhu XD, Chai ZT, Cai H, Zhang YY, Zhang KZ, et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol Cancer Ther (2017) 16(8):1544–54. doi: 10.1158/1535-7163.MCT-16-0866
17. Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Monkkonen J, et al. Human Breast Cancer Cells Educate Macrophages Toward the M2 Activation Status. Breast Cancer Res: BCR (2015) 17:101. doi: 10.1186/s13058-015-0621-0
18. Raggi C, Correnti M, Sica A, Andersen JB, Cardinale V, Alvaro D, et al. Cholangiocarcinoma Stem-Like Subset Shapes Tumor-Initiating Niche by Educating Associated Macrophages. J Hepatol (2017) 66(1):102–15. doi: 10.1016/j.jhep.2016.08.012
19. Cai H, Zhu XD, Ao JY, Ye BG, Zhang YY, Chai ZT, et al. Colony-Stimulating Factor-1-Induced AIF1 Expression in Tumor-Associated Macrophages Enhances the Progression of Hepatocellular Carcinoma. Oncoimmunology (2017) 6(9):e1333213. doi: 10.1080/2162402X.2017.1333213
20. Ushach I, Zlotnik A. Biological Role of Granulocyte Macrophage Colony-Stimulating Factor (GM-CSF) and Macrophage Colony-Stimulating Factor (M-CSF) on Cells of the Myeloid Lineage. J Leukocyte Biol (2016) 100(3):481–9. doi: 10.1189/jlb.3RU0316-144R
21. Yan WL, Shen KY, Tien CY, Chen YA, Liu SJ. Recent Progress in GM-CSF-Based Cancer Immunotherapy. Immunotherapy (2017) 9(4):347–60. doi: 10.2217/imt-2016-0141
22. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity (2015) 42(6):1197–211. doi: 10.1016/j.immuni.2015.05.018
23. Wang H, Wang X, Li X, Fan Y, Li G, Guo C, et al. CD68(+)HLA-DR(+) M1-Like Macrophages Promote Motility of HCC Cells Via NF-Kappab/FAK Pathway. Cancer Lett (2014) 345(1):91–9. doi: 10.1016/j.canlet.2013.11.013
24. Zong Z, Zou J, Mao R, Ma C, Li N, Wang J, et al. M1 Macrophages Induce PD-L1 Expression in Hepatocellular Carcinoma Cells Through IL-1beta Signaling. Front Immunol (2019) 10:1643. doi: 10.3389/fimmu.2019.01643
25. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol Res (2018) 6(10):1260–73. doi: 10.1158/2326-6066.CIR-17-0537
26. Xiong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, et al. Anti-PD-L1 Treatment Results in Functional Remodeling of the Macrophage Compartment. Cancer Res (2019) 79(7):1493–506. doi: 10.1158/0008-5472.CAN-18-3208
27. Galbraith NJ, Manek S, Walker S, Bishop C, Carter JV, Cahill M, et al. The Effect of Ikappak-16 on Lipopolysaccharide-Induced Impaired Monocytes. Immunobiology (2018) 223(4-5):365–73. doi: 10.1016/j.imbio.2017.10.045
28. Hartley G, Regan D, Guth A, Dow S. Regulation of PD-L1 Expression on Murine Tumor-Associated Monocytes and Macrophages by Locally Produced TNF-Alpha. Cancer Immunol Immunother: CII (2017) 66(4):523–35. doi: 10.1007/s00262-017-1955-5
29. Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front Immunol (2017) 8:1300. doi: 10.3389/fimmu.2017.01300
30. Rodriguez-Garcia M, Porichis F, de Jong OG, Levi K, Diefenbach TJ, Lifson JD, et al. Expression of PD-L1 and PD-L2 on Human Macrophages Is Up-Regulated by HIV-1 and Differentially Modulated by IL-10. J Leukocyte Biol (2011) 89(4):507–15. doi: 10.1189/jlb.0610327
31. Zhang J, Cao D, Yu S, Chen L, Wei D, Shen C, et al. Amphotericin B Suppresses M2 Phenotypes and B7-H1 Expression in Macrophages to Prevent Raji Cell Proliferation. BMC Cancer (2018) 18(1):467. doi: 10.1186/s12885-018-4266-0
32. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
33. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. PD-1 Expression by Macrophages Plays a Pathologic Role in Altering Microbial Clearance and the Innate Inflammatory Response to Sepsis. Proc Natl Acad Sci USA (2009) 106(15):6303–8. doi: 10.1073/pnas.0809422106
34. Rao G, Latha K, Ott M, Sabbagh A, Marisetty A, Ling X, et al. Anti-PD-1 Induces M1 Polarization in the Glioma Microenvironment and Exerts Therapeutic Efficacy in the Absence of CD8 Cytotoxic T Cells. Clin Cancer Res (2020) 26(17):4699–712. doi: 10.1158/1078-0432.CCR-19-4110
35. Ma J, Zheng B, Goswami S, Meng L, Zhang D, Cao C, et al. PD1(Hi) CD8(+) T Cells Correlate With Exhausted Signature and Poor Clinical Outcome in Hepatocellular Carcinoma. J Immunother Cancer (2019) 7(1):331. doi: 10.1186/s40425-019-0814-7
36. Wang X, Zhang Z, Zhang S, Fu J, Yao J, Jiao Y, et al. B7-H1 Up-Regulation Impairs Myeloid DC and Correlates With Disease Progression in Chronic HIV-1 Infection. Eur J Immunol (2008) 38(11):3226–36. doi: 10.1002/eji.200838285
37. Sachdeva M, Sharma A, Arora SK. Increased Expression of Negative Regulators of Cytokine Signaling During Chronic HIV Disease Cause Functionally Exhausted State of Dendritic Cells. Cytokine (2017) 91:118–23. doi: 10.1016/j.cyto.2016.08.010
38. Catakovic K, Klieser E, Neureiter D, Geisberger R. T Cell Exhaustion: From Pathophysiological Basics to Tumor Immunotherapy. Cell Commun Signal: CCS (2017) 15(1):1. doi: 10.1186/s12964-016-0160-z
39. Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, et al. Immunosuppressive Tumor-Infiltrating Myeloid Cells Mediate Adaptive Immune Resistance Via a PD-1/PD-L1 Mechanism in Glioblastoma. Neuro-oncology (2017) 19(6):796–807. doi: 10.1093/neuonc/now287
40. Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an Immune Suppressor and Potential Target for Normalization Cancer Immunotherapy. Nat Med (2019) 25(4):656–66. doi: 10.1038/s41591-019-0374-x
41. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-Cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723
42. Shelby A, Pendleton C, Thayer E, Johnson GK, Xie XJ, Brogden KA. PD-L1 Correlates With Chemokines and Cytokines in Gingival Crevicular Fluid From Healthy and Diseased Sites in Subjects With Periodontitis. BMC Res Notes (2020) 13(1):532. doi: 10.1186/s13104-020-05376-9
43. Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, et al. PD-L1 Expression on Tolerogenic Apcs Is Controlled by STAT-3. Eur J Immunol (2011) 41(2):413–24. doi: 10.1002/eji.201040979
44. Zhang X, Xu W. Neutrophils Diminish T-Cell Immunity to Foster Gastric Cancer Progression: The Role of GM-CSF/PD-L1/PD-1 Signalling Pathway. Gut (2017) 66(11):1878–80. doi: 10.1136/gutjnl-2017-313923
45. He G, Zhang H, Zhou J, Wang B, Chen Y, Kong Y, et al. Peritumoural Neutrophils Negatively Regulate Adaptive Immunity Via the PD-L1/PD-1 Signalling Pathway in Hepatocellular Carcinoma. J Exp Clin Cancer Res: CR (2015) 34:141. doi: 10.1186/s13046-015-0256-0
46. Thorn M, Guha P, Cunetta M, Espat NJ, Miller G, Junghans RP, et al. Tumor-Associated GM-CSF Overexpression Induces Immunoinhibitory Molecules Via STAT3 in Myeloid-Suppressor Cells Infiltrating Liver Metastases. Cancer Gene Ther (2016) 23(6):188–98. doi: 10.1038/cgt.2016.19
47. Lin C, He H, Liu H, Li R, Chen Y, Qi Y, et al. Tumour-Associated Macrophages-Derived CXCL8 Determines Immune Evasion Through Autonomous PD-L1 Expression in Gastric Cancer. Gut (2019) 68(10):1764–73. doi: 10.1136/gutjnl-2018-316324
48. Narayanapillai SC, Han YH, Song JM, Kebede ME, Upadhyaya P, Kassie F. Modulation of the PD-1/PD-L1 Immune Checkpoint Axis During Inflammation-Associated Lung Tumorigenesis. Carcinogenesis (2020) 41(11):1518–28. doi: 10.1093/carcin/bgaa059
49. Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. New Engl J Med (2016) 375(18):1767–78. doi: 10.1056/NEJMra1514296
50. Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, et al. The IFN-Gamma/PD-L1 Axis Between T Cells and Tumor Microenvironment: Hints for Glioma Anti-PD-1/PD-L1 Therapy. J Neuroinflamm (2018) 15(1):290. doi: 10.1186/s12974-018-1330-2
51. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep (2019) 29(11):3766. doi: 10.1016/j.celrep.2019.11.113
52. Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y, et al. Macrophage-Derived CCL5 Facilitates Immune Escape of Colorectal Cancer Cells Via the P65/STAT3-CSN5-PD-L1 Pathway. Cell Death Different (2020) 27(6):1765–81. doi: 10.1038/s41418-019-0460-0
53. Kang X, Li P, Zhang C, Zhao Y, Hu H, Wen G. The TLR4/ERK/PDL1 Axis May Contribute to NSCLC Initiation. Int J Oncol (2020) 57(2):456–65. doi: 10.3892/ijo.2020.5068
54. Li X, Su X, Liu R, Pan Y, Fang J, Cao L, et al. HDAC Inhibition Potentiates Anti-Tumor Activity of Macrophages and Enhances Anti-PD-L1-Mediated Tumor Suppression. Oncogene (2021) 40(10):1836–50. doi: 10.1038/s41388-020-01636-x
55. Zippelius A, Schreiner J, Herzig P, Muller P. Induced PD-L1 Expression Mediates Acquired Resistance to Agonistic Anti-CD40 Treatment. Cancer Immunol Res (2015) 3(3):236–44. doi: 10.1158/2326-6066.CIR-14-0226
56. Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 Efficacy can be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells With a Selective Inhibitor of PI3Kdelta/Gamma. Cancer Res (2017) 77(10):2607–19. doi: 10.1158/0008-5472.CAN-16-2534
57. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
58. Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol (2018) 4(1):93–7. doi: 10.1001/jamaoncol.2017.1617
59. Quaranta V, Rainer C, Nielsen SR, Raymant ML, Ahmed MS, Engle DD, et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res (2018) 78(15):4253–69. doi: 10.1158/0008-5472.CAN-17-3876
60. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages Impede CD8 T Cells From Reaching Tumor Cells and Limit the Efficacy of Anti-PD-1 Treatment. Proc Natl Acad Sci USA (2018) 115(17):E4041–E50. doi: 10.1073/pnas.1720948115
61. Chen LT, Martinelli E, Cheng AL, Pentheroudakis G, Qin S, Bhattacharyya GS, et al. Pan-Asian Adapted ESMO Clinical Practice Guidelines for the Management of Patients With Intermediate and Advanced/Relapsed Hepatocellular Carcinoma: A TOS-ESMO Initiative Endorsed by CSCO, ISMPO, JSMO, KSMO, MOS and SSO. Ann Oncol (2020) 31(3):334–51. doi: 10.1016/j.annonc.2019.12.001
62. Rini BI, Battle D, Figlin RA, George DJ, Hammers H, Hutson T, et al. The Society for Immunotherapy of Cancer Consensus Statement on Immunotherapy for the Treatment of Advanced Renal Cell Carcinoma (RCC). J Immunother Cancer (2019) 7(1):354. doi: 10.1186/s40425-019-0813-8
63. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 Receptor Improves the Antitumor Efficacy of Adoptive Cell Transfer Immunotherapy. Cancer Res (2014) 74(1):153–61. doi: 10.1158/0008-5472.CAN-13-1816
64. Patwardhan PP, Surriga O, Beckman MJ, de Stanchina E, Dematteo RP, Tap WD, et al. Sustained Inhibition of Receptor Tyrosine Kinases and Macrophage Depletion by PLX3397 and Rapamycin as a Potential New Approach for the Treatment of Mpnsts. Clin Cancer Res (2014) 20(12):3146–58. doi: 10.1158/1078-0432.CCR-13-2576
65. Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony Stimulating Factor 1 Receptor Inhibition Delays Recurrence of Glioblastoma After Radiation by Altering Myeloid Cell Recruitment and Polarization. Neuro-oncology (2016) 18(6):797–806. doi: 10.1093/neuonc/nov272
66. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, et al. Inhibition of Colony Stimulating Factor-1 Receptor Abrogates Microenvironment-Mediated Therapeutic Resistance in Gliomas. Oncogene (2017) 36(43):6049–58. doi: 10.1038/onc.2017.261
67. Dammeijer F, Lievense LA, Kaijen-Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP, et al. Depletion of Tumor-Associated Macrophages With a CSF-1R Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol Res (2017) 5(7):535–46. doi: 10.1158/2326-6066.CIR-16-0309
68. Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. New Engl J Med (2015) 373(5):428–37. doi: 10.1056/NEJMoa1411366
69. Tap WD, Gelderblom H, Palmerini E, Desai J, Bauer S, Blay JY, et al. Pexidartinib Versus Placebo for Advanced Tenosynovial Giant Cell Tumour (ENLIVEN): A Randomised Phase 3 Trial. Lancet (2019) 394(10197):478–87. doi: 10.1016/S0140-6736(19)30764-0
70. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally Administered Colony Stimulating Factor 1 Receptor Inhibitor PLX3397 in Recurrent Glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium Phase II Study. Neuro-oncology (2016) 18(4):557–64. doi: 10.1093/neuonc/nov245
71. Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med (2020) 71:47–58. doi: 10.1146/annurev-med-062518-045435
72. Qiu X, Tian Y, Liang Z, Sun Y, Li Z, Bian J. Recent Discovery of Phosphoinositide 3-Kinase Gamma Inhibitors for the Treatment of Immune Diseases and Cancers. Future Med Chem (2019) 11(16):2151–69. doi: 10.4155/fmc-2019-0010
73. McClure JJ, Li X, Chou CJ. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv Cancer Res (2018) 138:183–211. doi: 10.1016/bs.acr.2018.02.006
74. Keshavarz A, Pourbagheri-Sigaroodi A, Zafari P, Bagheri N, Ghaffari SH, Bashash D. Toll-Like Receptors (TLRs) in Cancer; With an Extensive Focus on TLR Agonists and Antagonists. IUBMB Life (2021) 73(1):10–25. doi: 10.1002/iub.2412
75. Guerriero JL. Macrophages: Their Untold Story in T Cell Activation and Function. Int Rev Cell Mol Biol (2019) 342:73–93. doi: 10.1016/bs.ircmb.2018.07.001
76. Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, Ouyang J, et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J Clin Oncol (2018) 36(10):942–50. doi: 10.1200/JCO.2017.77.3994
77. Zhao R, Wan Q, Wang Y, Wu Y, Xiao S, Li Q, et al. M1-Like Tams Are Required for the Efficacy of PD-L1/PD-1 Blockades in Gastric Cancer. Oncoimmunology (2020) 10(1):1862520. doi: 10.1080/2162402X.2020.1862520
78. Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin Cancer Res (2016) 22(15):3849–59. doi: 10.1158/1078-0432.CCR-15-1912
79. Razak AR, Cleary JM, Moreno V, Boyer M, Calvo Aller E, Edenfield W, et al. Safety and Efficacy of AMG 820, An Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Combination With Pembrolizumab in Adults With Advanced Solid Tumors. J Immunother Cancer (2020) 8(2):e001006. doi: 10.1136/jitc-2020-001006
80. Wei Z, Zhang X, Yong T, Bie N, Zhan G, Li X, et al. Boosting Anti-PD-1 Therapy With Metformin-Loaded Macrophage-Derived Microparticles. Nat Commun (2021) 12(1):440. doi: 10.1038/s41467-020-20723-x
81. Urdinguio RG, Fernandez AF, Moncada-Pazos A, Huidobro C, Rodriguez RM, Ferrero C, et al. Immune-Dependent and Independent Antitumor Activity of GM-CSF Aberrantly Expressed by Mouse and Human Colorectal Tumors. Cancer Res (2013) 73(1):395–405. doi: 10.1158/0008-5472.CAN-12-0806
82. Lawson DH, Lee S, Zhao F, Tarhini AA, Margolin KA, Ernstoff MS, et al. Randomized, Placebo-Controlled, Phase III Trial of Yeast-Derived Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Versus Peptide Vaccination Versus GM-CSF Plus Peptide Vaccination Versus Placebo in Patients With No Evidence of Disease After Complete Surgical Resection of Locally Advanced and/or Stage IV Melanoma: A Trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). J Clin Oncol (2015) 33(34):4066–76. doi: 10.1200/JCO.2015.62.0500
83. Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, et al. Ipilimumab Plus Sargramostim vs Ipilimumab Alone for Treatment of Metastatic Melanoma: A Randomized Clinical Trial. Jama (2014) 312(17):1744–53. doi: 10.1001/jama.2014.13943
84. Zhang X, Shi X, Li J, Mo L, Hu Z, Gao J, et al. PD-1 Blockade Overcomes Adaptive Immune Resistance in Treatment With Anchored-GM-CSF Bladder Cancer Cells Vaccine. J Cancer (2018) 9(23):4374–81. doi: 10.7150/jca.25423
85. Shi X, Zhang X, Li J, Zhao H, Mo L, Shi X, et al. PD-1/PD-L1 Blockade Enhances the Efficacy of SA-GM-CSF Surface-Modified Tumor Vaccine in Prostate Cancer. Cancer Lett (2017) 406:27–35. doi: 10.1016/j.canlet.2017.07.029
86. Shi X, Zhang X, Li J, Mo L, Zhao H, Zhu Y, et al. PD-1 Blockade Enhances the Antitumor Efficacy of GM-CSF Surface-Modified Bladder Cancer Stem Cells Vaccine. Int J Cancer (2018) 142(10):2106–17. doi: 10.1002/ijc.31219
87. Tian H, Shi G, Wang Q, Li Y, Yang Q, Li C, et al. A Novel Cancer Vaccine With the Ability to Simultaneously Produce Anti-PD-1 Antibody and GM-CSF in Cancer Cells and Enhance Th1-Biased Antitumor Immunity. Signal Transduct Target Ther (2016) 1:16025. doi: 10.1038/sigtrans.2016.25
88. Miyake M, Hori S, Ohnishi S, Toritsuka M, Fujii T, Shimizu T, et al. Supplementary Granulocyte Macrophage Colony-Stimulating Factor to Chemotherapy and Programmed Death-Ligand 1 Blockade Decreases Local Recurrence After Surgery in Bladder Cancer. Cancer Sci (2019) 110(10):3315–27. doi: 10.1111/cas.14158
Keywords: macrophage reprogramming, colony stimulating factors, programmed cell death ligand 1/programmed cell death 1, functional exhaustion, combination therapy
Citation: Cai H, Zhang Y, Wang J and Gu J (2021) Defects in Macrophage Reprogramming in Cancer Therapy: The Negative Impact of PD-L1/PD-1. Front. Immunol. 12:690869. doi: 10.3389/fimmu.2021.690869
Received: 04 April 2021; Accepted: 02 June 2021;
Published: 23 June 2021.
Edited by:
Lazar Vujanovic, University of Pittsburgh, United StatesReviewed by:
Adam C. Soloff, University of Pittsburgh, United StatesCopyright © 2021 Cai, Zhang, Wang and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinyang Gu, Z2p5bnlkQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.