 François Huaux
François Huaux- Louvain Centre for Toxicology and Applied Pharmacology (LTAP), Institut de Recherche Experimentale et Clinique (IREC), Université Catholique de Louvain, Brussels, Belgium
Immunostimulation is recognized as an important contribution in lung fibrosis in some animal models and patient subsets. With this review, we illustrate an additional scenario covering the possible implication of immunoregulation during fibrogenesis. Available animal and human data indicate that pulmonary fibrosis also includes diverse and discrete immunoregulating populations comprising regulatory lymphocytes (T and B regs) and myeloid cells (immunosuppressive macrophages and myeloid-derived suppressive cells; MDSC). They are initially recruited to limit the establishment of deleterious inflammation but participate in the development of lung fibrosis by producing immunoregulatory mediators (mainly TGF-β1 and IL-10) that directly or indirectly stimulate fibroblasts and matrix protein deposition. The existence of this silent immunoregulatory environment sustains an alternative mechanism of fibrosis that explains why in some conditions neither pro-inflammatory cytokine deficiency nor steroid and immunosuppressive therapies limit lung fibrosis. Therefore, the persistent presence of immunoregulation is an important parameter to consider for refining therapeutical strategies in lung fibrotic disorders under non-immunostimulatory conditions.
Diverse Lung Fibrotic Diseases With Diverse Mechanisms
Repair of damaged tissue is a fundamental biological process that allows the ordered replacement of dead or injured cells. However, although initially beneficial, the healing process becomes pathogenic when it is not controlled appropriately, leading to considerable tissue remodeling and the formation of permanent scar tissue and fibrosis (1). The major site of histopathological lung fibrosis is the interstitium, which consists of alveolar epithelium, pulmonary capillary endothelium, basement membrane, and perivascular and perilymphatic tissues. There are now almost 300 distinct injurious or inflammatory causes of interstitial lung disease that can result in progressive lung scarring. Many others are referred as idiopathic (i.e. idiopathic pulmonary fibrosis, or IPF) when it arises for no obvious reason (2). Silicosis is one the oldest recorded interstitial lung disease characterized by alveolitis and progressive nodular fibrosis (3). This fibroproliferative disorder can be traced back to ancient Egypt, where it was caused by inhalation of crystalline silica. It is also well known that long-term asbestos fiber inhalation causes asbestosis also comprising persistent nodular fibrosis (4). Although the incidence of silicosis and asbestosis has diminished, it continues to be a major cause of occupational lung disease in exposed workers, particularly in developing nations (5). More recently, carbon nanotubes (CNT), which present some of the physical characteristics of asbestos (long and rigid), also induce granulomatous lung disorder characterized by persistent immune responses, culminating in the development of lung fibrosis (6). Additionally, lung fibrogenic reaction may arise from other exogenous environmental stimuli such as organic dusts (bacterial, fungal and avian antigens) (7).
Suppression of chronic inflammation by immunosuppressive therapy turn off pulmonary fibrogenesis in some sub-groups of patients (e.g. non-specific interstitial pneumonia). These findings argue that inflammation represents a major pathological pathway in lung fibrosis (8, 9). A pathogenesis paradigm that did not require inflammation and immunostimulation was, however, proposed since there is little evidence of inflammation in the histopathological samples obtained from susceptible ageing individuals developing IPF undergoing surgical lung biopsy. Treatment with anti-inflammatory agents, such as steroids or anti-cytokines, seemed to have no effect on disease progression and outcome (10, 11). These last clinical observations strongly suggested that inflammation represents an important but dispensable event and that other mechanisms than immunostimulation exist or co-exist in pulmonary fibrosis (12, 13).
The Relevance of Reliable Animal Models of Lung Fibrosis
It is unanimously recognized that animal models currently available are particularly useful to discover new pro-fibrotic mediators and pathological avenues related or unrelated to the inflammatory concept. The development of experimental models producing long-lasting lesions akin to those seen in human fibrosis and defined by progressive and irreversible matrix deposition has already been the subject of many studies (14). Presently, chemical insults such as those caused by bleomycin are widely used to induce fibrotic disease. Many research studies have focused on changes in inflammatory phenomena after a single instillation of bleomycin and have yielded similar findings: the lung injuries caused by bleomycin induce acute recruitment and activation of inflammatory leukocytes which produce mediators (cytokines, chemokines, growth factors, and prostaglandins) activating fibroblast and driving fibrotic disease. While bleomycin represents the preferred molecule in this context, the lung fibrosis obtained by an unique dose of this drug is not systematically progressive and resolves itself over time after bleomycin metabolization (15).
This discrepancy led researchers to explore other models and discover new pathological avenues not apparent in the acute bleomycin-induced fibrosis model, which mainly (if not exclusively) supports that inflammation drives fibrosis. Multiple instillation of bleomycin has previously been shown to recapitulate the epithelial remodeling and fibrosis progression in the lungs of patients with IPF (16). Indeed, a repetitive alveolar epithelial injury caused by repetitive intratracheal injections of bleomycin is now proposed as the initial and major event that triggers a series of repair pathways that are in some way aberrant, leading to inappropriate fibrosis. Repeated injuries prevent epithelial cell regeneration, re-epithelialization and epithelial structure restauration and lead to a sustained disruption of alveolar epithelial morphology and fibrogenesis in injured alveoli. This experimental approach thus represents an ultimate model to address mechanistic questions on epithelial remodeling (17).
Murine models of univocal chronic lung responses and progressive fibrotic lesions using inorganic particles (crystalline silica, asbestos and CNT) are well known to cover severe fibrotic respiratory disorders in exposed human. Instillation of mineral particles into mouse and rat lungs results in the development of fibrotic nodules that resemble lesions which develop in humans. Rodents exposed to silica particles present alveolar fibrotic nodules, increased pulmonary lymphoid tissue and enhanced numbers of macrophages in the broncho-alveolar lavage fluid. Interestingly, silica, asbestos and CNT are retained in the lung and the response is characterized by a persistent and progressive fibrosis (18). Based on findings on particle-induced pulmonary fibrosis in mice, it has been recently proposed a new hypothesis that the fibrotic response may result from an exaggerated and persistent immunoregulating responses instead of or along with inflammation. This new concept have been confirmed in human silicosis but also in other animal models and patients. These results, summarized hereafter, may explain how lung fibrosis can develop in absence of immunostimulation.
History of Immunoregulating Surveillance
The quality, magnitude, and persistence of immune reactions results from the balance between immunostimulating and immunoregulating responses. The basic mechanism of immunoregulation comprises an interconnecting system involving diverse anti-inflammatory and immunosuppressive cytokines, lymphocytes and myeloid cells. There is now evidence that this sophisticated immunoregulatory systems is crucial in maintaining immune homeostasis and resolving persistent inflammation. Immunoregulatory alterations are often implicated in the pathogenesis of several inflammatory diseases such as infection, allergy, and autoimmune disorders. Conversely, uncontrolled and exacerbated immunoregulation is strategically exploited by cancer cells to survive, proliferate and escape detection by anti-tumor effector T lymphocytes (19). A well-knowledge of immunoregulation now serves for future strategies of diagnosis and treatment. The following sections are devoted to describe (i) historical aspects of immunoregulatory mechanisms (Figure 1) and (ii) matching evidence supporting a deranged immunoregulation in pulmonary fibrogenesis (Figure 2).
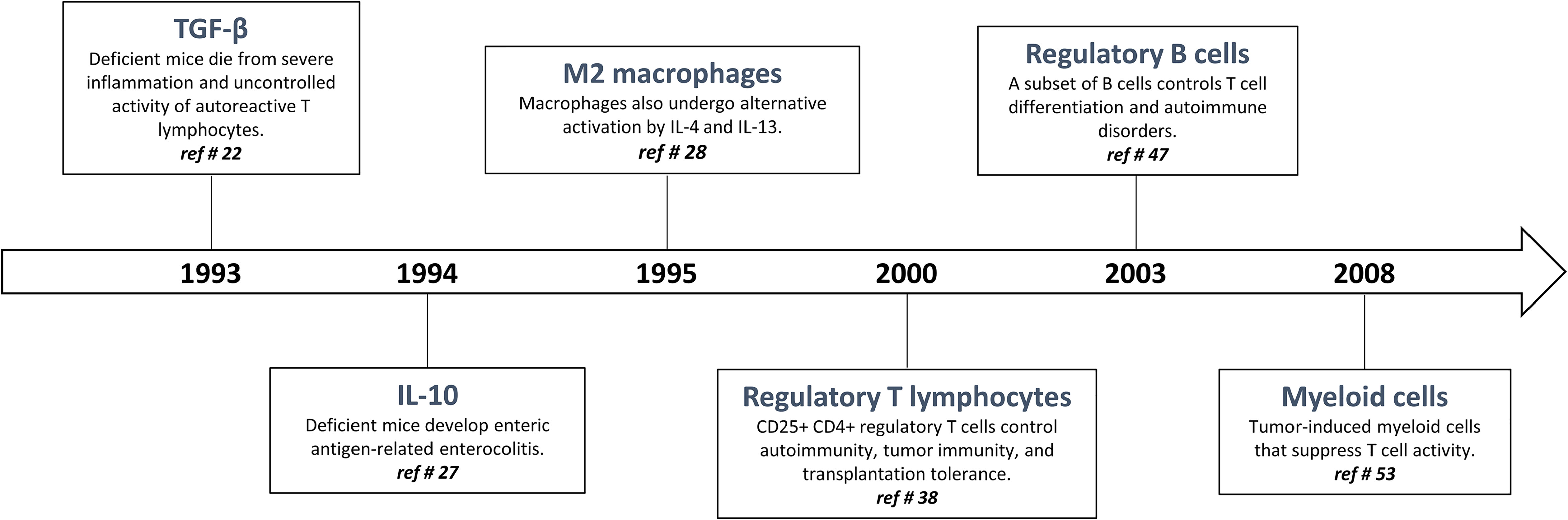
Figure 1 Timeline: historical progression of immunoregulation in the literature.
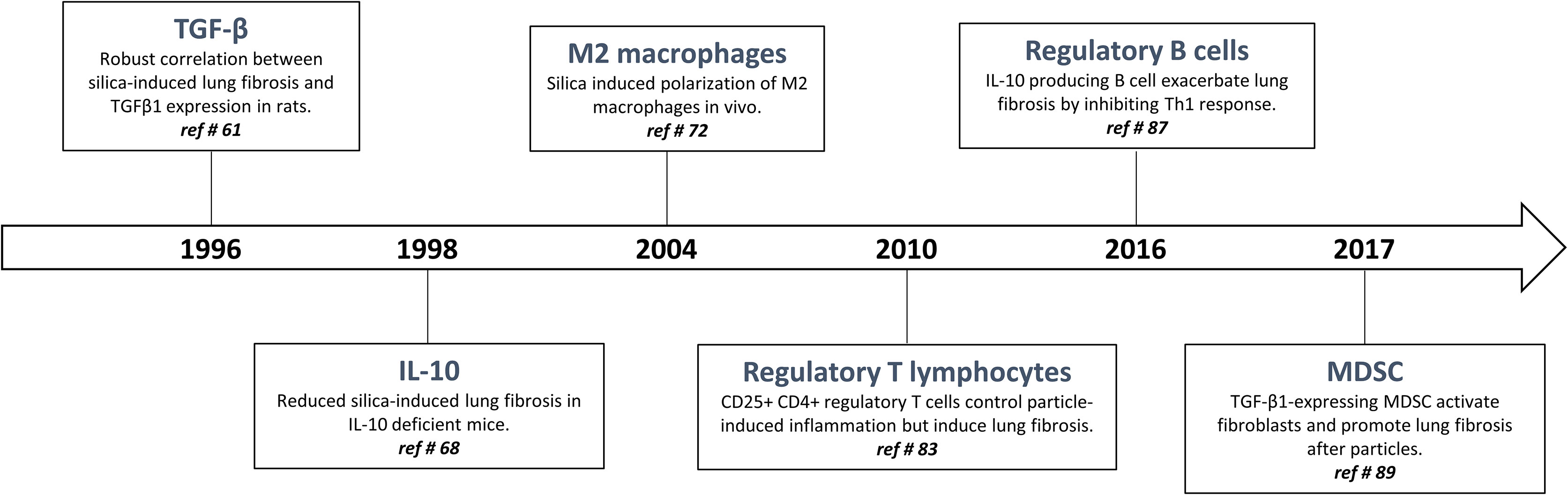
Figure 2 Timeline: historical progression of immunoregulation in particle-induced lung fibrosis.
TGF-β1 and IL-10 as Master Immunoregulating Cytokines
The most striking and non-redundant function of TGF-β1 is to regulate the immunostimulatory and inflammatory responses by orchestrating immunoregulation (20, 21). Indeed, in 1993, Kulkarni and Karlsson demonstrated that mice deficient for the gene encoding TGF-β1 die rapidly from a multi-systemic inflammatory syndrome related to deleterious T cell autoreactivity (22) (Figure 1). To become active, mature TGF-β1 must be released from the LAP (latency-associated peptide), a process referred to as TGF-β1 activation (23). The best-characterized mechanism of TGF-β1 activation imply conformational changes in the latent TGF-β1 molecule involving interaction of latent TGF-β1 with integrin αVβ8 and GARP (glycoprotein A repeats predominant protein or LRRC32) (21). The binding of the active TGF-β1 peptide to its receptors leads to a series of signaling events that are mainly mediated by SMAD2/3/4 complex that binds SMAD response elements located in the promoter regions of many genes involved in the immunoregulatory responses (21).
The cytokine interleukin-10 (IL-10) was originally described as a ‘cytokine synthesis inhibitory factor’ (CSIF), which was produced by mouse T helper 2 (Th2) clones and inhibited cytokine production by activated Th1 clones (24). IL-10 reportedly inhibits nuclear factor-κB (NF-κB) activation and expression of the most inducible cytokines and chemokines that are involved in inflammation. The anti-inflammatory activities of IL-10 also include induction of IL-1 receptor antagonist (IL-1ra) (24) and soluble p55 and p75 TNF receptor production, indicating that IL-10 induces a shift from production of pro-inflammatory to anti-inflammatory mediators (25). IL-10 strongly inhibits cytokine production and proliferation of CD4+ effector T cells via its down-regulatory effects on APC function. IL-10 also induce the differentiation of naive Th cells in a subset of regulatory T cells which are defined by their ability to produce high levels of IL-10 and TGF-β1 (26). The IL-10-related potent immunosuppressive functions and its proximity to TGF-β1 are perfectly illustrated by the work of Kuhn and colleagues demonstrating that IL-10 deficient mice suffer from chronic enterocolitis characterized by extensive inflammatory reactions and aberrant expression of major histocompatibility complex class II molecules (27) (Figure 1).
M2/Immunoregulatory Macrophages and Type 2 Like Immune Response
During the 1990s, Gordon and colleagues demonstrated that IL-4 and IL-13 induced an “alternative” form of activation in macrophages beside the “classically” polarized macrophages (28, 29) (Figure 1). In 2000, Mills and colleagues identified the fundamental M1/M2 polarization axis of macrophages and proposed that M1/inflammatory macrophages are necessary to eliminate infectious organisms and tumor cells while M2/immunoregulatory macrophages are implicated in repair process and wounds. Importantly, the uncontrolled M1 and M2 activation is associated with tissue injury and cancer progression, respectively (30, 31).
The so-called M2 phenotype express anti-inflammatory and immunosuppressive mediators such as IL-10 and TGF-β1, specific chemokines (CCL17 and CCL22, C-C motif), the subclass B scavenger receptor (CD 163), and the mannose C receptor type 1 (CD206 or MRC1) (32). The TGF-β1- and IL-10 producing M2 macrophages are thus intimately involved in the regulation of immune responses. Transcription factors such as IRF4 and STAT6 are required for M2 macrophage activation and differentiation. These different stimuli are likely to initiate during Th2-like responses (33). Th2 responses and cytokines (mainly IL-4 and IL-13) induce the differentiation of M2 macrophages.
The model categorizing macrophages (or T helper cells) in only two divergent groups M1 vs M2 (or Th1 and Th2) does not cover the intrinsic heterogeneity of macrophages (and T lymphocytes) now identified and described in diseases. A consensus now refers to M1-like or M2 like-subpopulations for avoiding oversimplified categories and highlighting the diversity and versatility of macrophages (34). M2-like macrophages possessing various functions, characteristics and deleterious activities are well known to be crucial in asthma, interstitial lung diseases and cancer (35, 36).
Regulatory T Cells
Regulatory T cells were initially described by Gershon et al. in the early 1970s and were called suppressive T cells because they practiced immune suppression (37). Unfortunately, despite the importance of these studies there was extensive skepticism in the immunological field about the existence of these cells, and suppressive T cells left the centre stage of immunology for decades. However, in 1996, Sakaguchi and colleagues rehabilitated the concept of “suppressive T cells”, now renamed “regulatory T cells” (38) (Figure 1). CD4+ Foxp3+ regulatory T cells (T regs) constitute a thymus-derived sub-population of CD4+ T lymphocytes that constitutively express the transcription factor Foxp3 (forkhead box P3), required for their development and their anti-inflammatory and immunosuppressive function (39, 40). T regs are developed in the thymus (naturally occurring T regs; nT regs) or are differentiated from naive T cells in the presence of TGF-β1 following T-cell receptor stimulation (induced T regs; iT regs) (41). These lymphocytes are crucial to maintain tolerance by downregulating undesired immune responses to self and non-self-antigens (42, 43). Absence or defective function of regulatory T cells is correlated with the development of immuno-pathologies such as auto-immune diseases (e.g. psoriasis and rheumatoid arthritis) and asthma. In contrast, their accumulation is associated with tolerance and implicated, for instance, in the development of cancer (44). Several mechanisms have been proposed to explain immunosuppressive functions of T regs. These include secretion by T regs of immunosuppressive cytokines, cell-contact-dependent suppression and functional modulation. Most in vivo studies indicate that T regs mediate immunosuppression by producing IL-10 and TGF-β1 (45). The strong immunosuppressive activity T regs is also related to their capacity to regulate the polarization and function of effector T lymphocytes (T eff, i.e. Th1, Th2, and Th17).
B Regs
The historical description of the new B-cell subtype named regulatory B-cells (B regs) dates back to 1974. In a mouse model of eczema, adoptive transfer of total splenocytes had a suppressive effect, whereas adoptive transfer of splenocytes from which B-lymphocytes were removed had no effect (46). In 2003, the regulatory role of B cells was shown in a mouse model of experimental autoimmune disease and this regulatory role was attributed to the ability of B cells to produce IL-10 (47) (Figure 1). B cells have been shown to restore homeostasis, possess important immunosuppressive functions and play a key role in disease control and immune tolerance (48, 49). IL‐10‐producing B cells produce anti‐inflammatory IgG4 and activate Treg cells. By promoting M2-like macrophage polarization, B regs cells also reduce auto‐reactive Th1 and Th17 cell responses and limit damaging inflammatory responses. In contrast, B regs impair cytotoxic T‐cell and NK cell responses to tumor cells and thereby promote progression of cancer (50). Beside IL-10, the exact mechanism by which B regs act in vivo remains unclear. A close contact between B-lymphocytes and the T lymphocytes, notably through the CD40-CD40L pathway are required (51, 52).
Myeloid Derived Suppressive Cells
MDSC were first identified by Van Ginderachter and his team in 2008. They detected a discrete myeloid-derived suppressor cell subpopulations with T cell-suppressive activity invading tumors. Thus, it was suggested that this MDSC population is responsible for down-regulating immune responses related to tumor progression and metastasis (53) (Figure 1). MDSC are now defined as heterogeneous and immature myeloid cells generated from the bone marrow and active in cancer development and inflammation regulation (54). Under normal physiological conditions, MDSC are rapidly differentiated into mature granulocytes, macrophages and dendritic cells. However, the differentiation of these cells into mature myeloid cells is blocked in chronic pathologies such as cancer (54). Two categories of these cells can be observed, one with granulocyte morphology and the other with monocyte morphology (55). MDSC suppress CD8, CD4 T cell activities and NK cell activity by inducing a suppressive environment (56). Several different mechanisms of action have been identified to explain the strong immunosuppressive activity of MDSC involving direct cell contacts and/or the production of several released mediators (54). MDSCs have the ability to induce differentiation and expansion of regulatory T cells by producing cytokines such as IL-10 and TGF-β1 or via CD40 (57). These cells can also deprive T cells of amino acids essential for their activity such as arginine, which is necessary for lymphocyte activation. Arginase, highly expressed by MDSCs, metabolizes arginine to urea and ornithine. Indoleamine-2.3-dioxygenase (IDO), an enzyme that catalyzes tryptophan, an essential amino acid for T cells, might also be involved in the immunosuppressive mechanisms of MDSC (54).
The Discovery of Immunoregulation in Particle-Induced Lung Fibrosis
Inorganic particle-induced lung fibrosis consists of an uncontrolled inflammation of the respiratory system tissues characterized by a chronic macrophage and neutrophil infiltration that ultimately causes fibrosis (58). Based on the widespread study of Piguet and colleagues published in 1990 in Nature, it is accepted that silica particles activate intracellular signaling pathways that culminate in the production of the pro-inflammatory mediator TNF-α, which is crucial in driving alveolitis and lung fibrosis (59). Additionally, a recent and pivotal study demonstrated that silica activate caspase-1 in a NALP3 inflammasome-dependent manner leading to the processing and secretion of the pro-inflammatory cytokine IL-1β. Evidence demonstrate that IL-1β initiates a cascade of reactions leading to inflammation and uncontrolled fibrosis (60). These observations support the concept that inflammation and sustained expression of inflammatory cell-secreted pro-fibrotic cytokines participate in the etiology of fibroproliferative diseases associated to inorganic particles.
The interconnection between inflammation and fibrosis was, however, questioned when active TGF-β1 was pointed as a key pro-fibrotic factor in response to particles. In silicosis and asbestosis, TGF-β1 expression has been found to be increased in lung tissues from patients with accelerated fibrotic disease progression (7) (Figure 2). The crucial activity of TGF-β1 during fibrogenesis has been unanimously recognized in the experimental studies using silica-, asbestos- and CNT-treated mice (6, 61–63). TGF-β1 is a major profibrogenic cytokine by delaying epithelial wound healing and expanding mesenchymal compartment (64). While the activity of TGF-β1 on fibroblasts is undisputed and not anymore debated, these observations intrigued immunologists because the presence of this powerful immunosuppressive cytokine (see above) was not in line with the IL-1/TNF-related immunostimulation axis.
Additional data challenged the inflammatory concept. IL-10, another potent immunoregulatory cytokine, has also fibrogenic activities in responses to fibrogenic particles. To examine the immune responses over the whole course of the pathological process induced by fibers and particles, validated experimental models in mice and rats with contrasting sensitivities were developed (65). By comparing these models and using deficient mice, it was newly discovered that IL-10 produced by particle-activated macrophages limit neutrophilic inflammation but is a key mediator implicated in the fibrotic lung response to silica by controlling the balance between pro- and anti-fibrotic mediator production (respectively TGF-β1 and prostaglandin E2) (65–68) (Figure 2). IL-10 is also a prevailing inducer of type-2 immune responses in particular M2-like and Th2-like pro-fibrotic cells (69, 70). The fibrogenic activity of macrophage-derived IL-10 and M2-like macrophages is not limited to silicosis (71, 72). Indeed, these macrophages are also implicated in the fibrotic lung response induced by asbestos and CNT (6, 73, 74) (Figure 2).
The little evidence of inflammation in the histopathological samples obtained from silicotic patients undergoing surgical lung biopsy (75) consolidate the pathogenesis paradigm that did not require inflammation. In contrast, high levels of IL-10 in BAL or serum were detected in patients with silicosis and asbestosis in the absence of clear inflammatory reaction (73, 76). Treatment with anti-inflammatory agents, such as corticosteroids, seemed to have no effect on outcome of these fibrotic diseases (75, 77, 78). These human observation are in accordance with several animal data indicating that steroid and numerous anti-inflammatory strategies reduce particle-induced lung inflammation but not IL-10-TGF-β1 expression and lung fibrosis in sensitive animals (79, 80). At this time, it was thus suggested that these two immunoregulatory cytokines are the major event that triggers a series of repair pathways that are aberrant and lead to inappropriate fibrosis under non-inflammatory conditions.
According to the concept that inflammation is responsible for fibrogenesis and given their ability to dampen inflammatory responses, CD4+ regulatory T lymphocytes (T regs) were first supposed to slow the progression of fibrosis (81). Indeed, it is well recognized that T regs, by restraining effector T cell responses and inducing tolerance through the production of TGF-β1 and/or IL-10, control lung inflammatory disorders (82). However, CD4+ Foxp3+ regulatory T cells are persistently recruited during long-term responses to particles (69, 83) (Figure 2). T regs purified from the lung of silica-treated mice highly express fibrogenic mediators, stimulate fibroblast proliferation in vitro and increase lung collagen deposition upon transfer into naive mice. Interestingly, the effects of T regs on fibroblast proliferation recapitulate the main function of PDGF as a primary mitogen for fibroblasts during lung fibrosis. The stimulatory effect of T regs on fibroblasts in vitro and in vivo was completely abolished by a PDGF receptor inhibitor (imatinib mesylate). It is thus likely that the role of T regs is to increase tissue fibroblast numbers, and consequently, amplify the subsequent fibroblast activation and collagen deposition (83). The pro-fibrogenic functions of T regs also comprise a stimulatory activity on Th2-like pro-fibrotic cells (69). In contrast to what it is thought, T regs thus participate in the fibrogenesis and are able to aggravate lung fibrosis induced by fibrogenic particles (i.e. silica and asbestos) in absence of inflammation (74). Finally, Xin and colleagues also observed a clear balance between inflammatory Th effector cells (Th1 and Th17) and T regs in mice treated with silica (84, 85). Neutralization of T regs-immunosuppressive activity resulted in enhanced lung inflammation and Th17 accumulation further demonstrating that T regs are initially recruited to control inflammatory responses (85, 86).
Based on the immunosuppressive profile of silica-treated mice, other immunosuppressive populations among lymphocytes have been investigated. Regulatory B lymphocytes (B regs), another immunosuppressive cell population (see above), also accumulate and participate in granuloma formation and fibrosis development by producing lung IL-10 in mice treated with silica. A heightened accumulation of inflammatory T effector cells (Th1 and Th17) but limited pulmonary fibrosis were observed in B reg-depleted mice treated with silica (87) (Figure 2). IL-10-producing B regs were also noted in silicotic patients in absence of inflammatory reaction (76). B regs exacerbate fibrogenesis by stimulating T regs functions and polarization via the release of IL-10 (88). These findings indicated that the accumulation and polarization of immunoregulatory lymphocytes is a central event during particle-induced pulmonary fibrosis with limited immunostimulation.
Finally, M-MDSC are also progressively and specifically accumulated during the development of pulmonary fibrosis. Indeed, a close relationship between the accumulation of MDSC, pulmonary immunosuppression and lung fibrosis was clearly found in mice treated with silica or CNT (89, 90) (Figure 2). Beside M2-like macrophages and T regs, immunosuppressive MDSC also expressed TGF-β1 conferring to these myeloid cells the capacity to down regulate T effector cell activity (91). In order to define their role in fibrosis, lung MDSC were purified from silica-treated mice and co-cultured with naive lung fibroblasts. MDSC stimulates lung fibroblasts to release tissue inhibitor of metalloproteinase and collagenolytic activity by expressing TGF-β1. They contribute to lung fibrogenesis by inducing a non-degrading collagen microenvironment (89).
The persistent accumulation of immunoregulatory macrophages, lymphocytes and myeloid cells in the lung during the progressive establishment of experimental silicosis is consistent with studies on tuberculosis (92) and lung cancer (93) that often affect the silicotic patients. Indeed, these cells control neutrophilic inflammation and anti-tumor T effector lymphocytes dedicated to microorganism and tumoral cell elimination. However, authors found that human silicosis is accompanied by a reduced number of blood regulatory T cells and speculated that the absence of these regulatory cells may explain the occurrence of autoimmune diseases (e.g. systemic scleroderma, rheumatoid arthritis and systemic lupus erythematosus) (94, 95). These conflicting results highlight the possible limitations of the mouse models. Injection of silica in mice does not fulfill all conditions encountered in patients with silicosis (infection, cancer and autoimmune diseases). Moreover, experimental models used to study the effects of silica are relatively short compared to human silicosis. These contradictory results also suggest that the fibrogenic activity of immunoregulation is only effective in a non-inflammatory environment.
Inclusive Implication of Immunoregulatory Mediators And Cells in Lung Fibrotic Diseases
Immunoregulation is operative in different fibrotic context and not specifically concomitant to particle-induced fibrogenesis. Indeed, recent investigations using complementary mouse models of lung fibrosis also reported that inflammation is not an absolute prerequisite for fibrogenesis and that the fibrotic pathological process can develop through immunoregulation.
IL-10 induce lung collagen deposition and fibrosis when overexpressed in transgenic mice (96, 97). These observations correspond to those noted when TGF-β1 is overexpressed in murine lungs (98). Intratracheal transfer of adenoviral recombinant IL-10 or TGF-β1 to murine lung has been shown to dramatically increase fibroblast accumulation and expression of type I and type III collagen around airways as well as in the pulmonary interstitium (70, 99, 100). The pro-fibrotic function of IL-10 is associated to infiltration of fibrocytes and M2-like macrophages (97). In addition; IL-10 and TGF-β1 contribute to lung injury and fibrosis by sensitizing epithelial cells to apoptosis (98, 101). TIM-3+ M2-like macrophages that possess strong immunoregulatory functions is now considered as an important pro-fibrotic population by being a key source of TGF-β1 and IL-10. Adoptive transfer of this immunoregulatory population promoted bleomycin-induced lung fibrosis by highly secreting TGF-β1 and IL-10 (102). Recent data at single cell level suggest that pro-fibrotic macrophages did not demonstrate a shaped and clear M2-like polarization but resemble to alveolar macrophages deriving from monocytes (103). Altogether, these studies indicated that the long-term overexpression of M2 like-related immunoregulating cytokines that suppress inflammation such as TGF-β1 and IL-10 are also profibrotic factors in the lung.
In vivo expansion of lung CD4+CD25+Foxp3+ T regs cells during bleomycin-induced lung fibrosis unexpectedly led to an increase of fibrogenesis. More important, this pro-fibrotic effect was a lymphocyte-dependent process. A marked down-regulation of type 1 and an increase of type 2 immune responses in the lungs were proposed to explain T reg fibrogenic activity (104, 105). These observations were corroborated by Chakraborthy and colleagues. Depletion of T regs ameliorate bleomycin-induced acute lung fibrosis by modulating Th effector cell balance. In addition, adoptive transfer of Sema 7a1 T regs induces fibrosis in the TGF-β1–exposed murine lung by altering the production of T-cell mediators (106).
Accumulation of MDSC with functional immunosuppressive activity was also noted in bleomycin-induced experimental pulmonary fibrosis and their potential role in fibroblast activation investigated (107, 108). Purified MDSC differentiate into lung fibroblasts as manifested by significantly elevated α-smooth muscle actin and TGF-β1 expression. Differentiation of MDSC into fibrocytes could also be possible during tissue repair processes. Indeed, there is some evidence that peripheral monocytes and MDSCs differentiate into fibrocytes (109, 110). These cells have the ability to promote fibroblast proliferation, migration, and collagen production but also differentiate into myofibroblasts (111, 112). Altogether, these previous experimental studies are well in accordance with the immunoregulation concept elaborated from findings reported after particle exposure and supporting that persitent immunoregulatory environment is profibrotic in absence of immunostimulation (Figure 3).
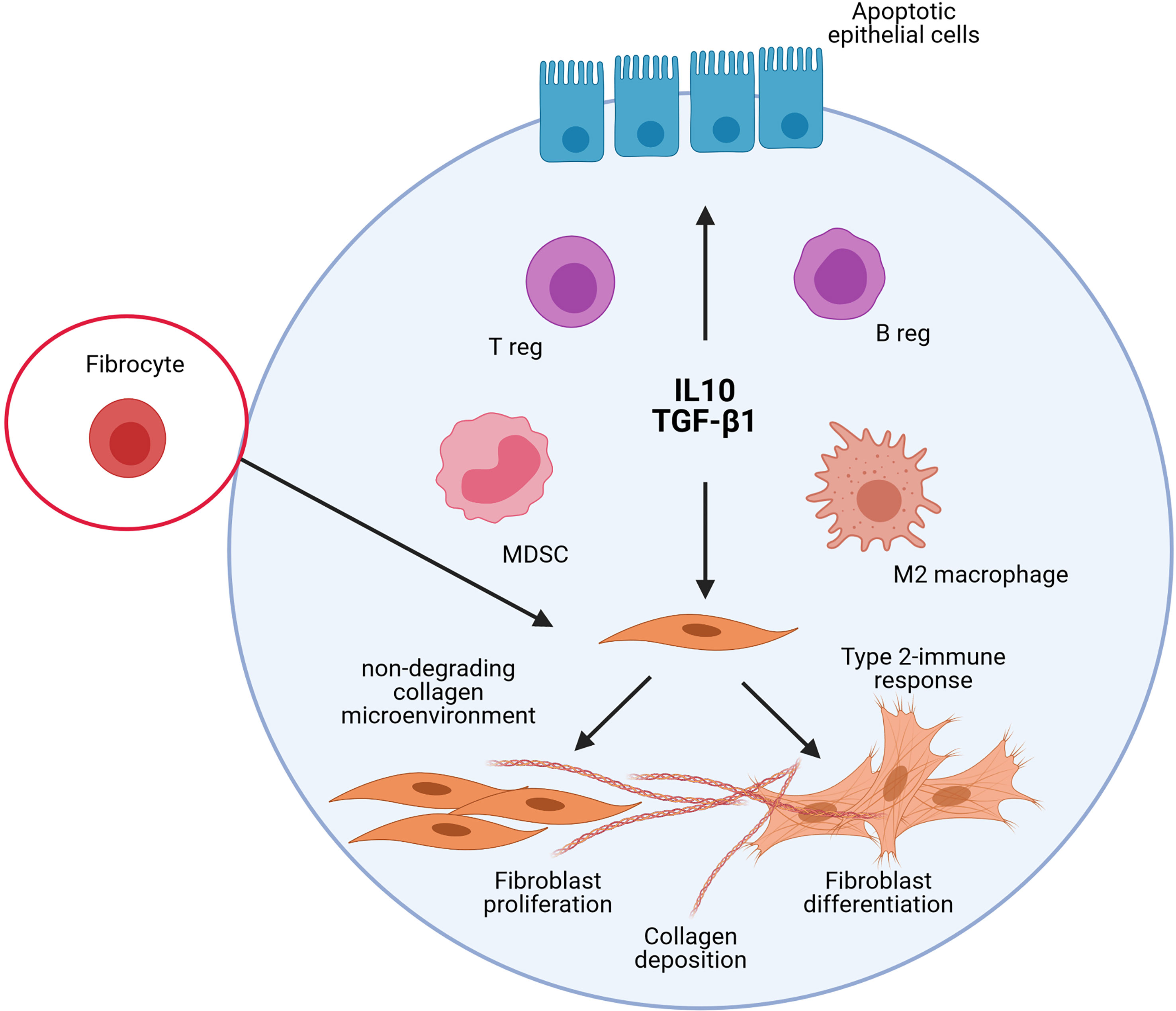
Figure 3 Schematic representation of the implication of the different immunoregulatory cells and mediators in lung fibrosis. The studies on experimental lung fibrosis have highlighted a new pathological pathway, which suggests that pulmonary fibrosis is orchestrated by an immunoregulatory response characterized by a persistent accumulation of pulmonary immunoregulatory cells (regulatory T and B lymphocytes, i.e. T and B regs; regulatory myeloid cells, i.e. M2/immunosuppressive macrophages and Myeloid Derived Suppressive Cells, MDSC) and a sustained production of IL-10 and TGF-β1. The persistent accumulation of these elements to control immunostimulatory responses in the lungs contribute, however, to pulmonary fibrosis. This sustains the view that immunoregulation is important pro-fibrotic environment that could markedly explain the development of the lung fibrotic response under non-inflammatory conditions.
The disconnection between fibrosis and inflammation is not limited to patients developing silicosis or asbestosis. Clinical measurements of inflammation in IPF patients developing fibrosis fail to correlate scar formation with inflammation and immunostimulation. Corticosteroids have never conclusively been shown to significantly alter the course of pulmonary fibrosis in patients and have, at best, limited efficacy in the treatment of scarring disease (113).
Interestingly, the expression TGF-β1 and IL-10 mainly by macrophages was increased in lung biopsies from patients with IPF compared with controls, suggesting the presence of M2-like macrophages (114, 115). Single-cell multi-omics approaches characterizing macrophage populations in health and lung fibrotic disease at high resolution tempered a clear accumulation of M2-like polarized macrophages in fibrotic tissue. Fibrotic macrophages in IPF patients are now identified as proliferating SSP1-positive macrophages (116, 117). However, it has been suggested that Th2-related cytokines such IL-4 and IL-3 as well as M-CSF activate this subset of proliferating macrophages (117). Clinical reports showed increased number and function of CD4+CD25+FoxP3+ T regs in the lungs and blood of patients with IPF associated with a more progressive clinical course (106, 116, 118–120). More recently, the importance of MDSC was also suggested by the observation of MDSC accumulation in IPF lungs (108, 121).
In conclusion, the presence of immunoregulatory microenvironment may be relevant to human pathology. Based on these recent findings, it is important to consider the possibility that regulatory lymphocytes and myeloid cells may also drive fibroproliferative wound healing. Consequently, these cells and their cytokine products could become therapeutic targets in patients developing fibrotic diseases. Particularly, these studies identified as potentially important targets the production of TGF-β1 and IL-10 by immunoregulatory cells in non-immunostimulatory conditions. The clinical separation of patients reaches from immunoregulatory or immunostimulatory scar formation could offer novel markers for the pathological assessment as well as novel regulators and drug targets to treat pulmonary fibroproliferative diseases.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was funded by the Actions de Recherche Concertées, Fédération Wallonie-Bruxelles (ARC 19/24-098, CYTAID), Fondation Contre le Cancer (2019-219), Fonds de la Recherche Scientifique (FNRS, PDR T.0119.13), ANSES (Agence nationale française de sécurité sanitaire de l’alimentation, de l’environnement et du travail, MacFibOsis) and European Commission under H2020 project (Contract no. 874707, Eximious). FH is a Senior Research Associate with the FNRS, Belgium.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Henderson NC, Rieder F, Wynn TA. Fibrosis: From Mechanisms to Medicines. Nature (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
2. du Bois RM. Strategies for Treating Idiopathic Pulmonary Fibrosis. Nat Rev Drug Discov (2010) 9:129–40. doi: 10.1038/nrd2958
3. Hoy RF, Chambers DC. Silica-Related Diseases in the Modern World. Allergy (2020) 75:2805–17. doi: 10.1111/all.14202
4. Gulati M, Redlich CA. Asbestosis and Environmental Causes of Usual Interstitial Pneumonia. Curr Opin Pulm Med (2015) 21:193–200. doi: 10.1097/MCP.0000000000000144
6. Dong J, Ma Q. Type 2 Immune Mechanisms in Carbon Nanotube-Induced Lung Fibrosis. Front Immunol (2018) 9:1120. doi: 10.3389/fimmu.2018.01120
7. Araya J, Nishimura SL. Fibrogenic Reactions in Lung Disease. Annu Rev Pathol (2010) 5:77–98. doi: 10.1146/annurev.pathol.4.110807.092217
8. Kondoh Y, Taniguchi H, Yokoi T, Nishiyama O, Ohishi T, Kato T, et al. Cyclophosphamide and Low-Dose Prednisolone in Idiopathic Pulmonary Fibrosis and Fibrosing Nonspecific Interstitial Pneumonia. Eur Respir J (2005) 25:528–33. doi: 10.1183/09031936.05.00071004
9. Lee SH, Park MS, Kim SY, Kim DS, Kim YW, Chung MP, et al. Factors Affecting Treatment Outcome in Patients With Idiopathic Nonspecific Interstitial Pneumonia: A Nationwide Cohort Study. Respir Res (2017) 18:204. doi: 10.1186/s12931-017-0686-7
10. Raghu G. Idiopathic Pulmonary Fibrosis: Lessons From Clinical Trials Over the Past 25 Years. Eur Respir J (2017) 50:1701209. doi: 10.1183/13993003.01209-2017
11. Parker JM, Glaspole IN, Lancaster LH, Haddad TJ, She D, Roseti SL, et al. A Phase 2 Randomized Controlled Study of Tralokinumab in Subjects With Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2018) 197:94–103. doi: 10.1164/rccm.201704-0784OC
12. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic Pulmonary Fibrosis. Nat Rev Dis Primers (2017) 3:17074. doi: 10.1038/nrdp.2017.74
13. Mora AL, Rojas M, Pardo A, Selman M. Emerging Therapies for Idiopathic Pulmonary Fibrosis, A Progressive Age-Related Disease. Nat Rev Drug Discov (2017) 16:755–72. doi: 10.1038/nrd.2017.170
14. Moore BB, Hogaboam CM. Murine Models of Pulmonary Fibrosis. Am J Physiol Lung Cell Mol Physiol (2008) 294:L152–60. doi: 10.1152/ajplung.00313.2007
15. O’Dwyer DN, Moore BB. Animal Models of Pulmonary Fibrosis. Methods Mol Biol (2018) 1809:363–78. doi: 10.1007/978-1-4939-8570-8_24
16. Cao Z, Lis R, Ginsberg M, Chavez D, Shido K, Rabbany SY, et al. Targeting of the Pulmonary Capillary Vascular Niche Promotes Lung Alveolar Repair and Ameliorates Fibrosis. Nat Med (2016) 22:154–62. doi: 10.1038/nm.4035
17. Redente EF, Black BP, Backos DS, Bahadur AN, Humphries SM, Lynch DA, et al. Persistent, Progressive Pulmonary Fibrosis and Epithelial Remodeling in Mice. Am J Respir Cell Mol Biol (2021) 64:669–76. doi: 10.1165/rcmb.2020-0542MA
18. Dong J, Ma Q. Integration of Inflammation, Fibrosis, and Cancer Induced by Carbon Nanotubes. Nanotoxicology (2019) 13:1244–74. doi: 10.1080/17435390.2019.1651920
19. Chen W, Ten Dijke P. Immunoregulation by Members of the TGFbeta Superfamily. Nat Rev Immunol (2016) 16:723–40. doi: 10.1038/nri.2016.112
20. Theron AJ, Anderson R, Rossouw TM, Steel HC. The Role of Transforming Growth Factor Beta-1 in the Progression of HIV/AIDS and Development of Non-AIDS-Defining Fibrotic Disorders. Front Immunol (2017) 8:1461. doi: 10.3389/fimmu.2017.01461
21. Dedobbeleer O, Stockis J, van der Woning B, Coulie PG, Lucas S. Cutting Edge: Active TGF-Beta1 Released From GARP/TGF-Beta1 Complexes on the Surface of Stimulated Human B Lymphocytes Increases Class-Switch Recombination and Production of IgA. J Immunol (2017) 199:391–6. doi: 10.4049/jimmunol.1601882
22. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming Growth Factor Beta 1 Null Mutation in Mice Causes Excessive Inflammatory Response and Early Death. Proc Natl Acad Sci USA (1993) 90:770–4. doi: 10.1073/pnas.90.2.770
23. Taylor AW. Review of the Activation of TGF-Beta in Immunity. J Leukoc Biol (2009) 85:29–33. doi: 10.1189/jlb.0708415
24. Fiorentino DF, Bond MW, Mosmann TR. Two Types of Mouse T Helper Cell. IV. Th2 Clones Secrete a Factor That Inhibits Cytokine Production by Th1 Clones. J Exp Med (1989) 170:2081–95. doi: 10.1084/jem.170.6.2081
25. Walter MR. The Molecular Basis of IL-10 Function: From Receptor Structure to the Onset of Signaling. Curr Top Microbiol Immunol (2014) 380:191–212. doi: 10.1007/978-3-662-43492-5_9
26. Saraiva M, O’Garra A. The Regulation of IL-10 Production by Immune Cells. Nat Rev Immunol (2010) 10:170–81. doi: 10.1038/nri2711
27. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-Deficient Mice Develop Chronic Enterocolitis. Cell (1993) 75:263–74. doi: 10.1016/0092-8674(93)80068-P
28. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J Exp Med (1992) 176:287–92. doi: 10.1084/jem.176.1.287
29. Doyle AG, Herbein G, Montaner LJ, Minty AJ, Caput D, Ferrara P, et al. Interleukin-13 Alters the Activation State of Murine Macrophages In Vitro: Comparison With Interleukin-4 and Interferon-Gamma. Eur J Immunol (1994) 24:1441–5. doi: 10.1002/eji.1830240630
30. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
31. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
32. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
33. Murray PJ, Wynn TA. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol (2011) 11:723–37. doi: 10.1038/nri3073
34. Mantovani A, Marchesi F, Jaillon S, Garlanda C, Allavena P. Tumor-Associated Myeloid Cells: Diversity and Therapeutic Targeting. Cell Mol Immunol (2021) 18:566–78. doi: 10.1038/s41423-020-00613-4
35. Ogger PP, Byrne AJ. Macrophage Metabolic Reprogramming During Chronic Lung Disease. Mucosal Immunol (2021) 14:282–95. doi: 10.1038/s41385-020-00356-5
36. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
38. Sakaguchi S, Toda M, Asano M, Itoh M, Morse SS, Sakaguchi N. T Cell-Mediated Maintenance of Natural Self-Tolerance: Its Breakdown as a Possible Cause of Various Autoimmune Diseases. J Autoimmun (1996) 9:211–20. doi: 10.1006/jaut.1996.0026
39. Sakaguchi S. Regulatory T Cells: Key Controllers of Immunologic Self-Tolerance. Cell (2000) 101:455–8. doi: 10.1016/S0092-8674(00)80856-9
40. Ohkura N, Sakaguchi S. A Novel Modifier of Regulatory T Cells. Nat Immunol (2009) 10:685–6. doi: 10.1038/ni0709-685
41. Wan YY. Regulatory T Cells: Immune Suppression and Beyond. Cell Mol Immunol (2010) 7:204–10. doi: 10.1038/cmi.2010.20
42. Fehervari Z, Sakaguchi S. CD4+ Tregs and Immune Control. J Clin Invest (2004) 114:1209–17. doi: 10.1172/JCI200423395
43. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity (2019) 50:302–16. doi: 10.1016/j.immuni.2019.01.020
44. Munoz-Rojas AR, Mathis D. Tissue Regulatory T Cells: Regulatory Chameleons. Nat Rev Immunol (2021) Online ahead of print. doi: 10.1038/s41577-021-00519-w
45. Rocamora-Reverte L, Melzer FL, Wurzner R, Weinberger B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front Immunol (2020) 11:616949. doi: 10.3389/fimmu.2020.616949
46. Mauri C, Ehrenstein MR. The ‘Short’ History of Regulatory B Cells. Trends Immunol (2008) 29:34–40. doi: 10.1016/j.it.2007.10.004
47. Mauri C, Gray D, Mushtaq N, Londei M. Prevention of Arthritis by Interleukin 10-Producing B Cells. J Exp Med (2003) 197:489–501. doi: 10.1084/jem.20021293
48. Rosser EC, Mauri C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
49. Menon M, Hussell T, Ali Shuwa H. Regulatory B Cells in Respiratory Health and Diseases. Immunol Rev (2021) 299:61–73. doi: 10.1111/imr.12941
50. Jansen K, Cevhertas L, Ma S, Satitsuksanoa P, Akdis M, van de Veen W. Regulatory B Cells, A to Z. Allergy (2021) Online ahead of print. doi: 10.1111/all.14763
51. Mauri C, Bosma A. Immune Regulatory Function of B Cells. Annu Rev Immunol (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
52. Mauri C. Regulation of Immunity and Autoimmunity by B Cells. Curr Opin Immunol (2010) 22:761–7. doi: 10.1016/j.coi.2010.10.009
53. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of Discrete Tumor-Induced Myeloid-Derived Suppressor Cell Subpopulations With Distinct T Cell-Suppressive Activity. Blood (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
54. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat Rev Immunol (2021) 21:485–498. doi: 10.1038/s41577-020-00490-y
55. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
56. Talmadge JE, Gabrilovich DI. History of Myeloid-Derived Suppressor Cells. Nat Rev Cancer (2013) 13:739–52. doi: 10.1038/nrc3581
57. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune Stimulatory Receptor CD40 Is Required for T-Cell Suppression and T Regulatory Cell Activation Mediated by Myeloid-Derived Suppressor Cells in Cancer. Cancer Res (2010) 70:99–108. doi: 10.1158/0008-5472.CAN-09-1882
58. Sayan M, Mossman BT. The NLRP3 Inflammasome in Pathogenic Particle and Fibre-Associated Lung Inflammation and Diseases. Part Fibre Toxicol (2016) 13:51. doi: 10.1186/s12989-016-0162-4
59. Piguet PF, Collart MA, Grau GE, Sappino AP, Vassalli P. Requirement of Tumour Necrosis Factor for Development of Silica-Induced Pulmonary Fibrosis. Nature (1990) 344:245–7. doi: 10.1038/344245a0
60. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome Through Phagosomal Destabilization. Nat Immunol (2008) 9:847–56. doi: 10.1038/ni.1631
61. Mariani TJ, Roby JD, Mecham RP, Parks WC, Crouch E, Pierce RA. Localization of Type I Procollagen Gene Expression in Silica-Induced Granulomatous Lung Disease and Implication of Transforming Growth Factor-Beta as a Mediator of Fibrosis. Am J Pathol (1996) 148:151–64.
62. Ma Q. Polarization of Immune Cells in the Pathologic Response to Inhaled Particulates. Front Immunol (2020) 11:1060. doi: 10.3389/fimmu.2020.01060
63. Huaux F. Emerging Role of Immunosuppression in Diseases Induced by Micro- and Nano-Particles: Time to Revisit the Exclusive Inflammatory Scenario. Front Immunol (2018) 9:2364. doi: 10.3389/fimmu.2018.02364
64. Gharaee-Kermani M, Hu B, Phan SH, Gyetko MR. Recent Advances in Molecular Targets and Treatment of Idiopathic Pulmonary Fibrosis: Focus on TGFbeta Signaling and the Myofibroblast. Curr Med Chem (2009) 16:1400–17. doi: 10.2174/092986709787846497
65. Barbarin V, Nihoul A, Misson P, Arras M, Delos M, Leclercq I, et al. The Role of Pro- and Anti-Inflammatory Responses in Silica-Induced Lung Fibrosis. Respir Res (2005) 6:112. doi: 10.1186/1465-9921-6-112
66. Barbarin V, Arras M, Misson P, Delos M, McGarry B, Phan SH, et al. Characterization of the Effect of Interleukin-10 on Silica-Induced Lung Fibrosis in Mice. Am J Respir Cell Mol Biol (2004) 31:78–85. doi: 10.1165/rcmb.2003-0299OC
67. Huaux F, Arras M, Vink A, Renauld JC, Lison D. Soluble Tumor Necrosis Factor (TNF) Receptors P55 and P75 and Interleukin-10 Downregulate TNF-Alpha Activity During the Lung Response to Silica Particles in NMRI Mice. Am J Respir Cell Mol Biol (1999) 21:137–45. doi: 10.1165/ajrcmb.21.1.3570
68. Huaux F, Louahed J, Hudspith B, Meredith C, Delos M, Renauld JC, et al. Role of Interleukin-10 in the Lung Response to Silica in Mice. Am J Respir Cell Mol Biol (1998) 18:51–9. doi: 10.1165/ajrcmb.18.1.2911
69. Liu F, Liu J, Weng D, Chen Y, Song L, He Q, et al. CD4+CD25+Foxp3+ Regulatory T Cells Depletion May Attenuate the Development of Silica-Induced Lung Fibrosis in Mice. PloS One (2010) 5:e15404. doi: 10.1371/journal.pone.0015404
70. Barbarin V, Xing Z, Delos M, Lison D, Huaux F. Pulmonary Overexpression of IL-10 Augments Lung Fibrosis and Th2 Responses Induced by Silica Particles. Am J Physiol Lung Cell Mol Physiol (2005) 288:L841–8. doi: 10.1152/ajplung.00329.2004
71. Zhao Y, Hao C, Bao L, Wang D, Li Y, Qu Y, et al. Silica Particles Disorganize the Polarization of Pulmonary Macrophages in Mice. Ecotoxicol Environ Saf (2020) 193:110364. doi: 10.1016/j.ecoenv.2020.110364
72. Misson P, van den Brule S, Barbarin V, Lison D, Huaux F. Markers of Macrophage Differentiation in Experimental Silicosis. J Leukoc Biol (2004) 76:926–32. doi: 10.1189/jlb.0104019
73. Murthy S, Larson-Casey JL, Ryan AJ, He C, Kobzik L, Carter AB. Alternative Activation of Macrophages and Pulmonary Fibrosis Are Modulated by Scavenger Receptor, Macrophage Receptor With Collagenous Structure. FASEB J (2015) 29:3527–36. doi: 10.1096/fj.15-271304
74. Kumagai-Takei N, Lee S, Srinivas B, Shimizu Y, Sada N, Yoshitome K, et al. The Effects of Asbestos Fibers on Human T Cells. Int J Mol Sci (2020) 21:6987. doi: 10.3390/ijms21196987
75. Greenberg MI, Waksman J, Curtis J. Silicosis: A Review. Dis Mon (2007) 53:394–416. doi: 10.1016/j.disamonth.2007.09.020
76. Chen Y, Li C, Lu Y, Zhuang H, Gu W, Liu B, et al. IL-10-Producing CD1d(hi)CD5(+) Regulatory B Cells May Play a Critical Role in Modulating Immune Homeostasis in Silicosis Patients. Front Immunol (2017) 8:110. doi: 10.3389/fimmu.2017.00110
77. Pardo A, Selman M. Idiopathic Pulmonary Fibrosis: New Insights in Its Pathogenesis. Int J Biochem Cell Biol (2002) 34:1534–8. doi: 10.1016/S1357-2725(02)00091-2
78. Selman M, Rojas M, Mora AL, Pardo A. Aging and Interstitial Lung Diseases: Unraveling an Old Forgotten Player in the Pathogenesis of Lung Fibrosis. Semin Respir Crit Care Med (2010) 31:607–17. doi: 10.1055/s-0030-1265901
79. Rabolli V, Lo Re S, Uwambayinema F, Yakoub Y, Lison D, Huaux F. Lung Fibrosis Induced by Crystalline Silica Particles Is Uncoupled From Lung Inflammation in NMRI Mice. Toxicol Lett (2011) 203:127–34. doi: 10.1016/j.toxlet.2011.03.009
80. Re SL, Giordano G, Yakoub Y, Devosse R, Uwambayinema F, Couillin I, et al. Uncoupling Between Inflammatory and Fibrotic Responses to Silica: Evidence From MyD88 Knockout Mice. PloS One (2014) 9:e99383. doi: 10.1371/journal.pone.0099383
81. Wilson MS, Wynn TA. Pulmonary Fibrosis: Pathogenesis, Etiology and Regulation. Mucosal Immunol (2009) 2:103–21. doi: 10.1038/mi.2008.85
82. Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, et al. Expansion and Function of Foxp3-Expressing T Regulatory Cells During Tuberculosis. J Exp Med (2007) 204:2159–69. doi: 10.1084/jem.20062105
83. Lo Re S, Lecocq M, Uwambayinema F, Yakoub Y, Delos M, Demoulin JB, et al. Platelet-Derived Growth Factor-Producing CD4+ Foxp3+ Regulatory T Lymphocytes Promote Lung Fibrosis. Am J Respir Crit Care Med (2011) 184:1270–81. doi: 10.1164/rccm.201103-0516OC
84. Tang W, Liu F, Chen Y, Song L, Dai W, Li C, et al. Reduction of IL-17A Might Suppress the Th1 Response and Promote the Th2 Response by Boosting the Function of Treg Cells During Silica-Induced Inflammatory Response In Vitro. Mediators Inflamm (2014) 2014:570894. doi: 10.1155/2014/570894
85. Dai W, Liu F, Li C, Lu Y, Lu X, Du S, et al. Blockade of Wnt/beta-Catenin Pathway Aggravated Silica-Induced Lung Inflammation Through Tregs Regulation on Th Immune Responses. Mediators Inflamm (2016) 2016:6235614. doi: 10.1155/2016/6235614
86. Thakur C, Wolfarth M, Sun J, Zhang Y, Lu Y, Battelli L, et al. Oncoprotein Mdig Contributes to Silica-Induced Pulmonary Fibrosis by Altering Balance Between Th17 and Treg T Cells. Oncotarget (2015) 6:3722–36. doi: 10.18632/oncotarget.2914
87. Liu F, Dai W, Li C, Lu X, Chen Y, Weng D, et al. Role of IL-10-Producing Regulatory B Cells in Modulating T-Helper Cell Immune Responses During Silica-Induced Lung Inflammation and Fibrosis. Sci Rep (2016) 6:28911. doi: 10.1038/srep28911
88. Lu Y, Liu F, Li C, Chen Y, Weng D, Chen J. IL-10-Producing B Cells Suppress Effector T Cells Activation and Promote Regulatory T Cells in Crystalline Silica-Induced Inflammatory Response In Vitro. Mediators Inflamm (2017) 2017:8415094. doi: 10.1155/2017/8415094
89. Lebrun A, Lo Re S, Chantry M, Izquierdo Carerra X, Uwambayinema F, Ricci D, et al. CCR2(+) Monocytic Myeloid-Derived Suppressor Cells (M-MDSCs) Inhibit Collagen Degradation and Promote Lung Fibrosis by Producing Transforming Growth Factor-Beta1. J Pathol (2017) 243:320–30. doi: 10.1002/path.4956
90. Shvedova AA, Tkach AV, Kisin ER, Khaliullin T, Stanley S, Gutkin DW, et al. Carbon Nanotubes Enhance Metastatic Growth of Lung Carcinoma Via Up-Regulation of Myeloid-Derived Suppressor Cells. Small (2013) 9:1691–5. doi: 10.1002/smll.201201470
91. Shvedova AA, Kisin ER, Yanamala N, Tkach AV, Gutkin DW, Star A, et al. MDSC and TGFbeta Are Required for Facilitation of Tumor Growth in the Lungs of Mice Exposed to Carbon Nanotubes. Cancer Res (2015) 75:1615–23. doi: 10.1158/0008-5472.CAN-14-2376
92. Konecny P, Ehrlich R, Gulumian M, Jacobs M. Immunity to the Dual Threat of Silica Exposure and Mycobacterium Tuberculosis. Front Immunol (2018) 9:3069. doi: 10.3389/fimmu.2018.03069
93. Sato T, Shimosato T, Klinman DM. Silicosis and Lung Cancer: Current Perspectives. Lung Cancer (Auckl) (2018) 9:91–101. doi: 10.2147/LCTT.S156376
94. Otsuki T, Maeda M, Murakami S, Hayashi H, Miura Y, Kusaka M, et al. Immunological Effects of Silica and Asbestos. Cell Mol Immunol (2007) 4:261–8.
95. Brilland B, Beauvillain C, Mazurkiewicz G, Rucay P, Roquelaure Y, Tabiasco J, et al. T Cell Dysregulation in Non-Silicotic Silica Exposed Workers: A Step Toward Immune Tolerance Breakdown. Front Immunol (2019) 10:2743. doi: 10.3389/fimmu.2019.02743
96. Lee CG, Homer RJ, Cohn L, Link H, Jung S, Craft JE, et al. Transgenic Overexpression of Interleukin (IL)-10 in the Lung Causes Mucus Metaplasia, Tissue Inflammation, and Airway Remodeling Via IL-13-Dependent and -Independent Pathways. J Biol Chem (2002) 277:35466–74. doi: 10.1074/jbc.M206395200
97. Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, et al. New Concepts of IL-10-Induced Lung Fibrosis: Fibrocyte Recruitment and M2 Activation in a CCL2/CCR2 Axis. Am J Physiol Lung Cell Mol Physiol (2011) 300:L341–53. doi: 10.1152/ajplung.00122.2010
98. Lee CG, Kang HR, Homer RJ, Chupp G, Elias JA. Transgenic Modeling of Transforming Growth Factor-Beta(1): Role of Apoptosis in Fibrosis and Alveolar Remodeling. Proc Am Thorac Soc (2006) 3:418–23. doi: 10.1513/pats.200602-017AW
99. Ask K, Bonniaud P, Maass K, Eickelberg O, Margetts PJ, Warburton D, et al. Progressive Pulmonary Fibrosis Is Mediated by TGF-Beta Isoform 1 But Not TGF-Beta3. Int J Biochem Cell Biol (2008) 40:484–95. doi: 10.1016/j.biocel.2007.08.016
100. Decologne N, Kolb M, Margetts PJ, Menetrier F, Artur Y, Garrido C, et al. TGF-Beta1 Induces Progressive Pleural Scarring and Subpleural Fibrosis. J Immunol (2007) 179:6043–51. doi: 10.4049/jimmunol.179.9.6043
101. Sun L, Hult EM, Cornell TT, Kim KK, Shanley TP, Wilke CA, et al. Loss of Myeloid-Specific Protein Phosphatase 2A Enhances Lung Injury and Fibrosis and Results in IL-10-Dependent Sensitization of Epithelial Cell Apoptosis. Am J Physiol Lung Cell Mol Physiol (2019) 316:L1035–48. doi: 10.1152/ajplung.00299.2018
102. Wang Y, Kuai Q, Gao F, Wang Y, He M, Zhou H, et al. Overexpression of TIM-3 in Macrophages Aggravates Pathogenesis of Pulmonary Fibrosis in Mice. Am J Respir Cell Mol Biol (2019) 61:727–36. doi: 10.1165/rcmb.2019-0070OC
103. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-Derived Alveolar Macrophages Drive Lung Fibrosis and Persist in the Lung Over the Life Span. J Exp Med (2017) 214:2387–404. doi: 10.1084/jem.20162152
104. Birjandi SZ, Palchevskiy V, Xue YY, Nunez S, Kern R, Weigt SS, et al. CD4(+)CD25(hi)Foxp3(+) Cells Exacerbate Bleomycin-Induced Pulmonary Fibrosis. Am J Pathol (2016) 186:2008–20. doi: 10.1016/j.ajpath.2016.03.020
105. Moore MW, Herzog EL. Regulatory T Cells in Idiopathic Pulmonary Fibrosis: Too Much of a Good Thing? Am J Pathol (2016) 186:1978–81. doi: 10.1016/j.ajpath.2016.06.002
106. Reilkoff RA, Peng H, Murray LA, Peng X, Russell T, Montgomery R, et al. Semaphorin 7a+ Regulatory T Cells Are Associated With Progressive Idiopathic Pulmonary Fibrosis and Are Implicated in Transforming Growth Factor-Beta1-Induced Pulmonary Fibrosis. Am J Respir Crit Care Med (2013) 187:180–8. doi: 10.1164/rccm.201206-1109OC
107. Bryant AJ, Shenoy V, Fu C, Marek G, Lorentsen KJ, Herzog EL, et al. Myeloid-Derived Suppressor Cells Are Necessary for Development of Pulmonary Hypertension. Am J Respir Cell Mol Biol (2018) 58:170–80. doi: 10.1165/rcmb.2017-0214OC
108. Oliveira AC, Fu C, Lu Y, Williams MA, Pi L, Brantly ML, et al. Chemokine Signaling Axis Between Endothelial and Myeloid Cells Regulates Development of Pulmonary Hypertension Associated With Pulmonary Fibrosis and Hypoxia. Am J Physiol Lung Cell Mol Physiol (2019) 317:L434–44. doi: 10.1152/ajplung.00156.2019
109. Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes Represent a Novel MDSC Subset Circulating in Patients With Metastatic Cancer. Blood (2013) 122:1105–13. doi: 10.1182/blood-2012-08-449413
110. Wright AK, Newby C, Hartley RA, Mistry V, Gupta S, Berair R, et al. Myeloid-Derived Suppressor Cell-Like Fibrocytes Are Increased and Associated With Preserved Lung Function in Chronic Obstructive Pulmonary Disease. Allergy (2017) 72:645–55. doi: 10.1111/all.13061
111. Pilling D, Zheng Z, Vakil V, Gomer RH. Fibroblasts Secrete Slit2 to Inhibit Fibrocyte Differentiation and Fibrosis. Proc Natl Acad Sci USA (2014) 111:18291–6. doi: 10.1073/pnas.1417426112
112. Verstovsek S, Manshouri T, Pilling D, Bueso-Ramos CE, Newberry KJ, Prijic S, et al. Role of Neoplastic Monocyte-Derived Fibrocytes in Primary Myelofibrosis. J Exp Med (2016) 213:1723–40. doi: 10.1084/jem.20160283
113. Selman M, Pardo A. Idiopathic Pulmonary Fibrosis: An Epithelial/Fibroblastic Cross-Talk Disorder. Respir Res (2002) 3:3. doi: 10.1186/rr198
114. Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, et al. Cytokine Profiles in Idiopathic Pulmonary Fibrosis Suggest an Important Role for TGF-Beta and IL-10. Eur Respir J (2003) 22:69–76. doi: 10.1183/09031936.03.00014703
115. Freeburn RW, Armstrong L, Millar AB. Cultured Alveolar Macrophages From Patients With Idiopathic Pulmonary Fibrosis (IPF) Show Dysregulation of Lipopolysaccharide-Induced Tumor Necrosis Factor-Alpha (TNF-Alpha) and Interleukin-10 (IL-10) Inductions. Eur Cytokine Netw (2005) 16:5–16.
116. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-Cell RNA-Seq Reveals Ectopic and Aberrant Lung-Resident Cell Populations in Idiopathic Pulmonary Fibrosis. Sci Adv (2020) 6:eaba1983. doi: 10.1126/sciadv.aba1983
117. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-Expressing Macrophages in Idiopathic Pulmonary Fibrosis. Eur Respir J (2019) 54:1802441. doi: 10.1183/13993003.02441-2018
118. Galati D, De Martino M, Trotta A, Rea G, Bruzzese D, Cicchitto G, et al. Peripheral Depletion of NK Cells and Imbalance of the Treg/Th17 Axis in Idiopathic Pulmonary Fibrosis Patients. Cytokine (2014) 66:119–26. doi: 10.1016/j.cyto.2013.12.003
119. Hou Z, Ye Q, Qiu M, Hao Y, Han J, Zeng H. Increased Activated Regulatory T Cells Proportion Correlate With the Severity of Idiopathic Pulmonary Fibrosis. Respir Res (2017) 18:170. doi: 10.1186/s12931-017-0653-3
120. Wang F, Xia H, Yao S. Regulatory T Cells Are a Double-Edged Sword in Pulmonary Fibrosis. Int Immunopharmacol (2020) 84:106443. doi: 10.1016/j.intimp.2020.106443
Keywords: immunosuppression, inflammation, regulatory lymphocytes and myeloid cells, carbon nanotubes, silica and asbestos
Citation: Huaux F (2021) Interpreting Immunoregulation in Lung Fibrosis: A New Branch of the Immune Model. Front. Immunol. 12:690375. doi: 10.3389/fimmu.2021.690375
Received: 02 April 2021; Accepted: 06 July 2021;
Published: 20 August 2021.
Edited by:
Enrico Conte, University of Catania, ItalyReviewed by:
Athol Wells, Royal Brompton Hospital, United KingdomXiaofeng Zhou, University of Michigan, United States
Copyright © 2021 Huaux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: François Huaux, ZnJhbmNvaXMuaHVhdXhAdWNsb3V2YWluLmJl