 Jessica N. Filderman
Jessica N. Filderman Mark Appleman
Mark Appleman Manoj Chelvanambi
Manoj Chelvanambi Jennifer L. Taylor2
Jennifer L. Taylor2 Walter J. Storkus
Walter J. Storkus
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 13 May 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.690105
This article is part of the Research Topic Tertiary Lymphoid Structures: from Basic Biology to Translational Impact in Cancer View all 15 articles
Tertiary lymphoid structures (TLS), also known as ectopic lymphoid structures (ELS) or tertiary lymphoid organs (TLO), represent a unique subset of lymphoid tissues noted for their architectural similarity to lymph nodes, but which conditionally form in peripheral tissues in a milieu of sustained inflammation. TLS serve as regional sites for induction and expansion of the host B and T cell repertoires via an operational paradigm involving mature dendritic cells (DC) and specialized endothelial cells (i.e. high endothelial venules; HEV) in a process directed by TLS-associated cytokines and chemokines. Recent clinical correlations have been reported for the presence of TLS within tumor biopsies with overall patient survival and responsiveness to interventional immunotherapy. Hence, therapeutic strategies to conditionally reinforce TLS formation within the tumor microenvironment (TME) via the targeting of DC, vascular endothelial cells (VEC) and local cytokine/chemokine profiles are actively being developed and tested in translational tumor models and early phase clinical trials. In this regard, a subset of agents that promote tumor vascular normalization (VN) have been observed to coordinately support the development of a pro-inflammatory TME, maturation of DC and VEC, local production of TLS-inducing cytokines and chemokines, and therapeutic TLS formation. This mini-review will focus on STING agonists, which were originally developed as anti-angiogenic agents, but which have recently been shown to be effective in promoting VN and TLS formation within the therapeutic TME. Future application of these drugs in combination immunotherapy approaches for greater therapeutic efficacy is further discussed.
TLS are non-encapsulated aggregates of lymphoid cells that form at sites of sustained inflammation, including tissues impacted by autoimmune disease, chronic infection, and cancer (1, 2). Recent findings suggest that the presence of TLS within tumor lesions positively correlates with favorable prognosis in most forms of solid cancer (1–4). TLS are associated with specialized vascular structures (i.e. HEV) that differentiate from CD31+ VEC or endothelial progenitor cells under pro-inflammatory, pro-angiogenic conditions (5–7). HEV express the cell surface marker peripheral node addressin (PNAd, a binding partner for CD62L expressed on lymphocytes) and produce chemokines (CCL19, CCL21, CXCL13), which facilitate the recruitment of naïve/central memory CD62L+CCR7+ T cells, naïve CD62L+CXCR5+ B cells, CXCR5+ T follicular helper (Tfh) cells and mature CCR7+ DC into the TME (1). In this context, it is believed that TLS serve as local sites for the de novo (cross)priming, expansion, and differentiation of tumor-specific T and B cells, leading to more efficient/effective anti-tumor responses within sites of active disease (2, 8–13). TLS also appear to define an operational site in which the T cell and B cell repertoires may expand their specificity against a broadened range of tumor antigenic targets, via the paradigms of epitope spreading or determinant spreading (14). Notably, TLS exhibit heterogeneity in their cellular composition and in the organization of their integrated cell subsets, which is believed to be reflective of their maturational status (2, 15). Classical (mature) TLS are characterized by the presence of i.) PNAd+ HEV surrounded by ii.) aggregates of T cells and mature DCs and iii.) distinct B cell zones containing naïve B cells around germinal center (GC)-like structures (1–3, 16, 17). Non-classical (immature) TLS contain some but not all of these three characteristics (i.e. typically lacking B cells/GC) (16). Strikingly, the presence of either classical TLS or non-classical TLS in TME portends superior prognoses in cancer patients (1–3, 10, 16–27).
An important component of TLS formation in peripheral tissues is sustained local production of homeostatic chemokines that recruit immune cells into affected tissue sites and serve as cues for establishing organized interactions between infiltrating lymphocytes and antigen presenting cells (APC). This topic has been well-described in other publications (28, 29) and elsewhere in the current volume and is therefore only briefly discussed below.
One key homeostatic chemokine associated with TLS development is CXCL13 (also known as B lymphocyte chemoattractant [BLC] or B cell-attracting chemokine 1 [BCA-1]), the ligand for CXCR5 (28). The production of CXCL13 by tumor-associated fibroblasts, Tfh cells, follicular dendritic cells (FDC) and HEV is positively-correlated with the formation of GC that contain CXCR5+ B cells (29, 30). While not yet investigated in the tumor setting, forced expression of CXCL13 in normal pancreatic β cells leads to the formation of TLS containing HEV, B cells and T cells via a process dependent on the initial infiltration of B cells into tissue and the activation of the lymphotoxin (LT)αβ-LTβR signaling cascade (31). Two additional major TLS-associated homeostatic chemokines produced by mature DC and HEV are CCL19 and CCL21, both of which serve as ligands for CCR7 (28). In normal mouse pancreatic tissue, ectopic overexpression of CCL19 or CCL21 induces the formation of TLS containing CD4+ T cells, CD11c+ DCs, and B220+ B cells surrounding HEV via a process dependent on CCL19/21-induced expression of LTα1β2 complexes on CD4+ T cells (32). Although TLS were not formally evaluated in their endpoint analyses, several studies have shown that treatment of murine tumors by injection with recombinant CCL19, viruses encoding CCL19 or CCL21, or DC engineered to express CCL21 results in robust tumor infiltration by T cells and DC in association with slowed tumor growth and extended overall survival (33–36).
In addition to these chemokines, tumor necrosis factor (TNF), interferon (IFN) and interleukin (IL)-1 superfamily cytokines also play major roles in TLS neogenesis. Lymphotoxins (LTα/TNFSF1 and LTβ/TNFSF3) and LIGHT/TNFSF14 produced by immune cells play canonical roles in the formation of TLS (29, 37). Lymphotoxins form bioactive heterotrimers (LTα3, LTα1β2, LTα2β1) that bind to LTβR/TNFRSF3, with LTα3 also binding and mediating signals through the TNFR1/TNFRSF1A, TNFR2/TNFSF1B and herpesvirus entry mediator (HVEM)/TNFRSF14 receptors (38). LIGHT also binds to HVEM/TNFRSF14 (39). TNF receptors represent important signaling receptors for endothelial cell function and proliferation and they facilitate TLS neogenesis. TNFR1/2 expression on endothelial cells has been shown to be necessary for HEV formation and T cell infiltration into murine melanoma (40). Mice lacking TNFR1/2 on the endothelium or LTα on CD8+ T cells have significantly decreased PNAd expression, demonstrating that LTα3 engagement of TNFR1 induces PNAd expression on the tumor vascular endothelium (40). In kidney and pancreatic tissues, the forced overexpression of LTα promotes lymphoid aggregate formation (containing T cells, B cells, and APCs) and tumor vascular reprograming, as indicated by increased expression of VCAM-1, ICAM-1, MAdCAM, and PNAd on VEC/HEV (41). Additionally, combined overexpression of LTα and LTβ further enhances infiltration of naïve lymphocytes and expression of homeostatic chemokines when compared to LTα overexpression alone, suggesting the synergistic action of these cytokines in TLS formation (42). In line with such findings, B16.F10 melanoma-bearing mice treated with a tumor-targeted GD2 scFv-LTα fusion protein demonstrate increased densities of intratumoral HEV and develop a diverse T cell repertoire in association with TLS neogenesis (43). Remarkably, recent reports in murine transplantable and carcinogen-induced tumor models support the operational dominance of TNFR- over LTβR-mediated signaling for HEV/TLS neogenesis in the TME (40, 44), findings which contrast with the canonical importance of LTβR-mediated signaling for HEV/TLS formation in normal tissues and in ontogenic secondary lymphoid organogenesis (1, 29, 30, 40, 44). Beyond lymphotoxins, LIGHT activation of VEC has also been shown to play a role in TLS formation in cancer models. C57BL/6 mice bearing intracranial NSCG glioblastomas treated with a fusion protein encoding LIGHT and a vascular targeting peptide (LIGHT-VTP) displayed VN and induction of classical TLS within the TME (45). In murine fibrosarcoma models, forced expression of LIGHT prompted naïve T cell infiltration and local production of homeostatic chemokines, leading to tumor rejection in the therapy setting (8, 46).
Type I IFNs have also been reported to drive TLS formation in normal tissues (47, 48). In murine models, a subset of PDGFRα+ lung fibroblasts produce CXCL13 in response to infection with influenza virus or intranasal administration of IFNβ (48). IFN-I receptor (IFNAR) activation in these cells results in increased recruitment of CXCR5+ B cells and ectopic germinal center formation in the lungs, which in turn promotes the development of a broadly neutralizing repertoire of antiviral antibodies conferring cross-strain protection (48). In a hydrocarbon (TMPD)-induced model of autoimmune SLE, mice with intact IFN-I signaling had worse clinical scores and increased lupus-specific autoantibody production compared to IFNAR-deficient mice (49). It was shown that IFN-I produced by activated DCs in this model was associated with the formation of classical TLS containing B cells, CD4+ T cells, and DC along with coordinate expression of TLS homeostatic chemokines (CCL19, CCL21, CXCL13) and their receptors (CCR7, CXCR5) (50). Sustained IFN-I/IFNAR signaling in tissues has similarly been shown to promote TLS formation in additional studies via local production of pro-inflammatory CXCR3 ligand chemokines (CXCL10/11) and lymphotoxins (51, 52).
Furthermore, gene therapy delivering IL-1 family member IL-F9/IL-36γ induces HEV and TLS formation in mouse colon carcinomas in association with the development of superior T cell-mediated anti-tumor immunity and tumor growth suppression (11). Notably, the activation of IL-36R on immune and stromal cell populations has been shown to upregulate local production of pro-inflammatory, pro-TLS factors including CXCL10, LTα and IFNs (53). In humans, IL-36γ is expressed by the tumor vasculature in colorectal cancers and has been correlated with an increased density of CD20+ B cells localized in TLS in these tumors (54).
STING (STimulator of INterferon Genes) is a cytosolic DNA sensing protein that is activated upon binding to cGAMP, a catalyzed dsDNA product of cytosolic GMP/AMP synthase (cGAS) (55). Activation of STING leads to secondary activation of transcription factor IRF3 by facilitating IRF3 interaction with Tank Binding Kinase 1 (TBK1), phosphorylation of IRF3 (pIRF3), pIRF3 dimerization and translocation into the nucleus where it transactivates IFNβ and other pro-inflammatory genes (55).
As a consequence of defects in the expression/functionality of DNA repair proteins, tumors are commonly characterized by genetic instability (56, 57) and contain high concentrations of cytoplasmic DNA leading to intrinsic cGAS/STING activation and secretion of proinflammatory mediators (58–60). Progressively growing tumors have been reported to develop defects in the STING signaling pathway to avoid STING-induced apoptosis and immune surveillance (60–62). Nevertheless, dying tumor cells still release dsDNA (and/or 2’3’ cGAMP, its cGAS catalyzed product) into the TME, which may result in the activation of STING+ cells in the tumor stroma, including DC and VEC (63–65). This intrinsic inflammatory process may be therapeutically enhanced by local or systemic delivery of synthetic STING agonists (66, 67).
Activation of STING in tumor-associated VEC leads to VN (67, 68) characterized by increased vascular perfusion and upregulated expression of E-selectin/CD62E, VCAM-1 and ICAM-1 which facilitates circulating immune cell adhesion to the endothelium and consequent recruitment of tumor-infiltrating lymphocytes into the TME (63, 67, 68). This operating paradigm may underlie observations of cancers with reduced DNA repair proficiency and high comparative mutational burden presenting with brisk proinflammatory immune cell infiltrates (i.e. “hot tumors”) that are more prone to develop TLS (69, 70) and to be more responsive to interventional immunotherapy (71, 72). Notably, provision of low doses of STING agonists cGAMP and ADU-S100 (aka ML-RR-S2-CDA, MIW815) coordinately promote VN and CD8+ T cell-dependent control of tumor growth in murine models of breast carcinoma, lung carcinoma and melanoma (67, 68, 73). Yang et al. (68) further confirmed the importance of STING agonist-induced Type-I IFN produced by tumor VEC with the therapeutic benefits associated with this treatment approach. Most recently (Figure 1), Chelvanambi et al. has demonstrated that VN induced by intratumoral administration of low doses of the STING agonist ADU-S100 results in sustained inflammation within the TME of B16 melanomas and local production of homeostatic cytokines/chemokines (LTα, LTβ, LIGHT, CCL19 and CCL21, but remarkably not CXCL13) and pro-inflammatory/pro-TLS mediators (CXCL10, IL-36β, IFNβ) (67). These therapy-associated changes were associated with coordinate neogenesis of non-classical TLS and the development of a unique tumor-infiltrating T cell receptor (TCR) repertoire in the TLS+ TME that was not detectable in the peripheral immune cell compartment (67). Parallel studies using STING-KO mice confirmed the strict requirement for STING expression in host but not tumor cells for therapeutic response to intratumoral administration of ADU-S100, including TLS formation and slowed tumor growth (67).
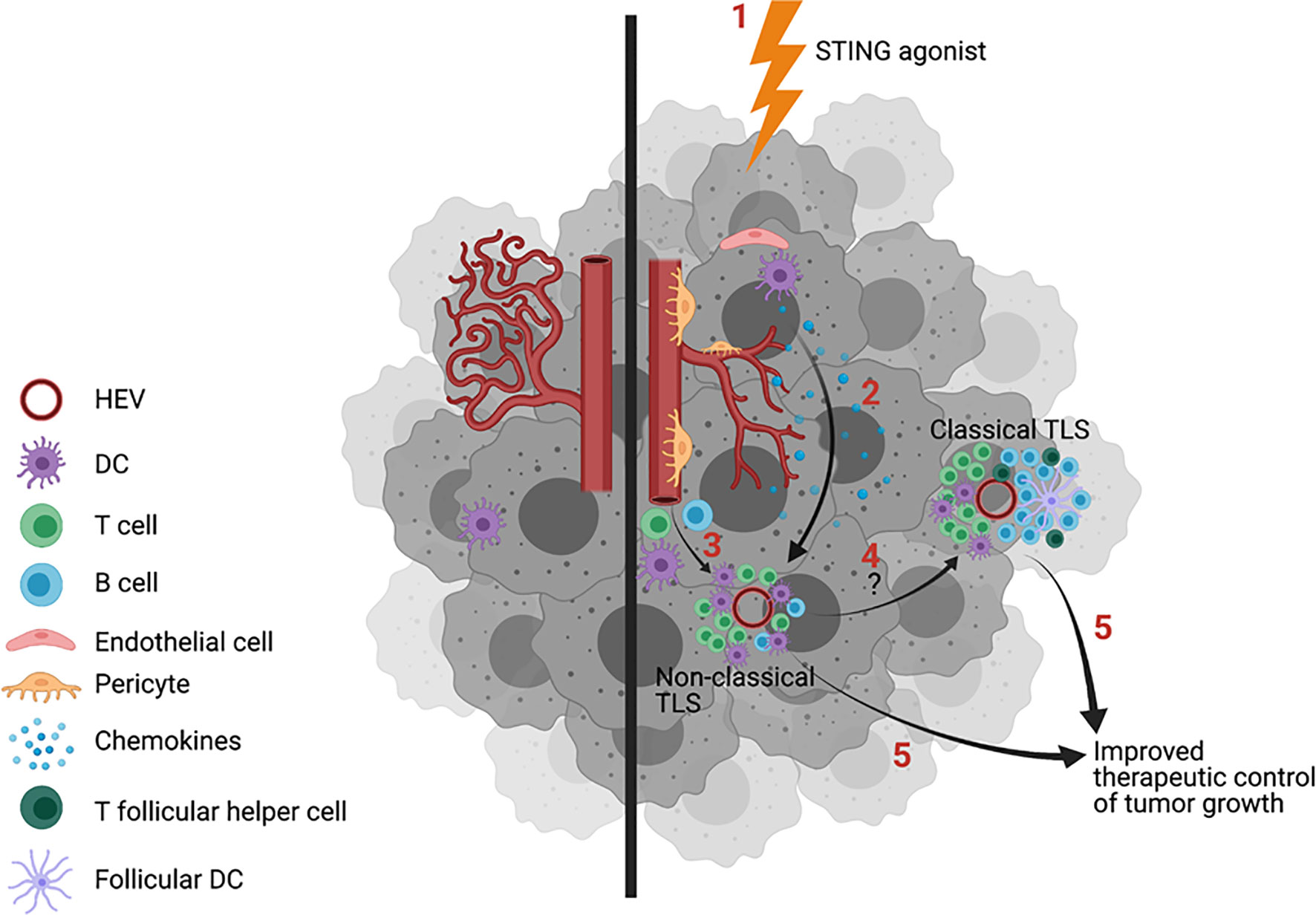
Figure 1 Treatment of tumors with STING agonists induces vascular normalization (VN), increased inflammatory immune cell infiltration and TLS formation. Untreated tumors exhibit dysfunctional blood vessels that limit immune cell entry into the tumor microenvironment (TME) in support of tumor progression (left). Provision of STING agonists (1, right) into the TME leads to the activation of STING+ stromal cells (2), including dendritic cells (DC) and vascular endothelial cells (VEC), leading to enhanced endothelial cell expression of adhesion molecules, improved vascular integrity/perfusion, and pro-inflammatory immune cell infiltration. A subset of therapeutically-conditioned VEC may differentiate into PNAd+ high endothelial venules (HEV). STING activated DC and HEV produce TLS-promoting cytokines/chemokines CCL19, CCL21, lymphotoxins, CXCL10 and IFNβ, which serve to recruit T cells and DC into the TME in support of non-classic, immature TLS formation proximal to HEV (3). CXCL13, required for optimal B cell recruitment into the TME and for germinal center (GC) formation within TLS, is only poorly produced in the STING agonist-conditioned tumors, precluding formation of classical, mature TLS. Combination protocols will likely be required for conversion of immature TLS into B cell/GC-rich classical, mature TLS (4) and/or to improve the therapeutic benefits associated with treatment-induced TLS formation (5). Image created with BioRender.com.
The finding that STING agonists promote the formation of non-classical/immature TLS is consistent with the inability of these agents to augment production of CXCL13 within the treated TME, a prerequisite for CXCR5+ B cell/Tfh cell recruitment and the formation of GC within classical/mature TLS (67). While the exact mechanism underlying this deficiency in CXCL13 production remains unclear, it could relate to the known regulatory action of STING signaling in B cells. For instance, the activation of STING in B cells via genetic engineering to express a constitutively activated form of STING or by treatment of B cells with STING agonists results in endoplasmic reticulum (ER)-associated degradation of membrane-bound immunoglobulin, muted BCR signaling via enhanced SHP1 phosphatase activity and increased rates of B cell apoptosis (74–76).
Since STING agonist-associated treatment benefits occur in association with non-classical/immature TLS formation, with seemingly minimal input from B cells, these findings also reenergize discussions related to the operational importance of B cells in therapeutic anti-tumor immune responses. Despite several recent reports citing the association of B cells and GC within tumor-associated TLS and positive clinical prognosis and response to interventional immunotherapy (19, 77–80), the literature is balanced by observations for an immunosuppressive influence for intratumoral B cells in promoting tumor progression, poor patient prognosis and immune-related adverse events (irAEs) in response to immunotherapy (80–82). Translational modeling in murine tumor models has similarly provided equivocal findings. Hence, B cell deficiency (muMT) or B cell depletion (using anti-CD20 mAbs) has resulted in either decreased (83–86) or increased (87) tumor growth. In the former cases, B cells are hypothesized to serve as intrinsic immunoregulatory cells or facilitators of Treg recruitment/development/function (82–85), while in the latter situation, B cells are believed to serve as supportive antigen-presenting cells and/or producers of pro-TLS cytokines (i.e. LIGHT) and therapeutic anti-tumor antibodies (19, 78, 88–93).
In recent years, more attention has been devoted to discerning the impact of B cells in TLS that form in patients’ tumors with a generally beneficial role for B cells emerging. In both melanoma and renal cell carcinoma, B cell gene signatures are enriched in the tumors of patients who respond to immune checkpoint blockade with positive correlations observed at baseline and on-treatment (79). When tumor samples were histologically analyzed, TLS containing B cells were more commonly identified in tumor biopsies obtained from clinical responders vs. non-responders, and these mature TLS appeared more secondary-follicle-like and contained CD21+ follicular DC and CD23+ germinal center B cells (79). Furthermore, the presence of B cells in classical, mature TLS was associated with T cells exhibiting more activated, functional phenotypes and expanded repertoires (79, 94–96).
If these more recent observations can be generalized, they suggest that optimal benefits from interventional immunotherapies may require treatment-associated development of classic, mature TLS containing B cells. As such, STING agonist-based regimens should be combined (synchronously or potentially after STING agonists) with co-treatments that coordinately induce the entry of therapeutic B cells, as well as FDC and Tfh cells, into the TME to improve TLS-associated anti-tumor immune responses. Candidate co-therapies include a range of toll-receptor agonists (97–104), agonist anti-TNFR1 antibodies (105) and DNA methylase inhibitors (106) which have each been reported to augment production of CXCL13 by stromal cell populations. This augmentation would be expected to improve tumor infiltrating B cell content and GC formation within the TLS+ TME, conceivably improving immune-mediated control of tumor growth. Indeed, several recent reports support therapeutic synergy using treatment regimens combining STING agonists and TLR1/2 agonist Pam3Csk (97), TLR4 agonist monophosphoryl lipid A (102), TLR7/8 agonist MEDI9197 (103) or TLR9 agonist CpG (104). Although the impact of these interventional protocols on TLS formation within the TME and the evolving anti-tumor immune response remains unknown, these aspects are expected to be actively pursued in future studies.
Given the ability of STING agonists to promote robust pro-inflammatory responses in tumor-associated stromal cells, it is perhaps not surprising that these agents are competent to initiate the development of non-classical, immature TLS within the TME (67). And even though treatment of tumor-bearing mice with STING agonists leads to reduced levels of tumor-associated myeloid-derived suppressor cells (MDSC) and Treg cells (107, 108), these regimens promote compensatory activation of immune regulatory pathways by augmenting expression of arginase-2 (ARG2), cyclooxygenase-2 (COX2/PTGS2), indoleamine 2,3 dioxygenase (IDO), programmed death-1 (PD-1), programmed death ligand-1 (PD-L1) and prostaglandin E synthase (PTGES) within the TME (67, 109–111). Hence, combined treatment protocols that include STING agonists and antagonists of these regulatory pathways would be anticipated to enhance/sustain inflammation within the TME in support of TLS formation/maintenance and improved host control of tumor growth. While the formation of TLS has yet to be investigated as a therapeutic endpoint in translational models of such combination treatment protocols, therapeutic synergy has been observed for regimens combining STING agonists with inhibitors of COX2 (Celecoxib) or IDO (BMS-986205), or antagonist anti-PD1 and/or anti-PD-L1 antibodies (107, 109, 112).
TLS are increasingly viewed as important operational components supporting the development and maintenance of protective immune responses that impact patient prognosis and response to interventional immunotherapy. The ability to predictably and reproducibly promote or augment TLS formation in a patient’s tumor(s) via the administration of therapeutic agents may dramatically improve objective response rates over those currently observed for standard of care treatments, including immune checkpoint blockade antibodies. Although previous animal modeling of gene therapy and targeted antibody approaches to deliver individual TLS homeostatic cytokines/chemokines have proven successful in controlling tumor growth and promoting TLS formation in mice (113–115), these strategies have yet to be effectively translated into the clinic, and they rely on the biologic dominance of a single agent to initiate the complex biologic process of TLS formation. In this regard, STING agonism provides the opportunity to coordinately activate a range of tumor-associated stromal cell populations, including vascular endothelial cells and immune cells, leading to VN, enhanced immune cell infiltration, and the establishment of a pro-inflammatory TME in which TLS-associated homeostatic chemokines and cytokines are produced and TLS formation is facilitated. While provision of STING agonist ADU-S100 into B16 melanomas resulted in the development of a T cell repertoire unique to the therapeutic TLS+ TME and to some abscopal benefit in regulating the growth of distal, untreated tumor lesions, the current approach has several limitations.
First, the approach involves direct injection of a second-generation STING agonist (which can only be administered locally) into an accessible lesion, with the intent to treat disseminated disease. In this regard, while local injection of STING agonists [i.e. ADU-S100/MIW815 (NCT02675439), MK-1454 (NCT03010176)] as monotherapies has provided some evidence for pro-inflammatory changes in the TME or patient sera, therapeutic benefits have been minimal (i.e. < 5% objective response rate) in early phase clinical trials treating advanced-stage cancer patients (as described in greater detail in a series of recent outstanding reviews) (55, 116, 117). This deficiency may be circumvented by the provision of next-generation, systemic STING agonists for more effective treatment of patients with multifocal, disseminated disease in visceral tissue sites. Several of these agents (i.e. E7766 [NCT04109092], GSK-3745417 [NCT03843359], MSA-2, SB-11285 [NCT04096638], TAK-676 [NCT04420884]) (55, 118, 119) are planned for, or are currently being evaluated in, phase I clinical trials. Given the pro-apoptotic impact of high-doses of STING agonists on VEC (i.e. vasoablative) and immune cell populations (67, 73–76, 120, 121), but the ability of low-dose regimens to promote VN and enhanced pro-inflammatory immune function in pre-clinical models, it might be anticipated that low-dose protocols will provide optimal immunotherapeutic benefits in these trials. While it is not clear that the formation of TLS represents an exploratory endpoint in these ongoing trial designs, one might expect that low-dose regimens of these next-generation STING agonists will prove effective in inducing de novo development of TLS or expansion of existing TLS within the tumor of treated patients. It is also possible given enhanced autoimmune manifestations in older (cancer) patients (122), many of which have known associations with the formation of TLS in affected tissues (123), that treatment with systemic STING agonists may exacerbate the incidence and severity of irAEs.
Second, the TLS promoted by ADU-S100 in the TME appear rich in CD8+ T cells, DC and HEV, but they are poor in CXCL13 production and infiltrating B cell/GC content (67) [i.e. representative of non-classical, immature TLS (2, 16)]. If B cells are indeed crucial to superior therapeutic benefits associated with TLS formation in tumors, additional co-therapies that i.) reinforce local CXCL13 production, ii.) B cell, Tfh and FDC infiltration and iii.) GC formation, may need to be combined with STING agonists to achieve maximal interventional benefit.
Third, the natural checks and balances in evolving immune responses must be considered in conditionally optimizing STING agonist-based immunotherapies. The robust pro-inflammatory responses evoked by these agents result in an upregulation in immune regulatory pathways within the TME, including but not limited to prostaglandin E production and immune suppression mediated by arginase, IDO and co-inhibitory receptors (67, 68). These regulatory pathways may be antagonized (individually or collectively) in combination STING agonist protocols using available, in-clinic targeted inhibitors. Such approaches would be expected to augment and prolong inflammation within the TME in support of TLS formation and the mobilization of broadly-reactive anti-tumor immune responses in the therapy setting. However, as suggested above, such deregulated reinforcement of TLS formation in tumors and normal tissues carries increased risk for the evolution of severe (autoimmune) irAEs.
Finally, previous findings suggest that in certain cases, STING activation and the presence of HEV/TLS may be associated with tumor progression. Hence, in murine lung carcinoma models (109, 111), provision of STING agonists (i.e. CDA) initially slowed primary tumor growth but ultimately resulted in disease progression and metastasis due to treatment-associated enhancement in immune regulatory/tolerogenic pathways (COX2, IDO, PD-1), which could be mitigated using targeted inhibitors in combination protocols (109, 111). Furthermore, tumors with pronounced chromosomal instability and intrinsic STING signaling competency have been reported to exhibit STING-dependent metastatic potential (124), which might be envisioned to be further exacerbated by treatment with STING agonists. Additional reports caution that TLS enriched in Treg cells or immature (thin-walled) HEV may be associated with poor immune infiltration of tumors, poor patient prognosis and increased tumor metastasis (125). Therefore, baseline tumor STING signaling competency and the quality and cell composition of STING-agonized TLS should be carefully monitored for correlative impact on cancer patient outcome.
All authors contributed to the article and approved the submitted version.
This work was supported by NIH grants R01 CA204419 and P01 CA234212 (both to WS) and NIH T32 CA082084 (to JF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, Slingluff CL. Immune Cell Infiltration and Tertiary Lymphoid Structures as Determinants of Antitumor Immunity. J Immunol (2018) 200:432–42. doi: 10.4049/jimmunol.1701269
2. Weinstein AM, Storkus WJ. Therapeutic Lymphoid Organogenesis in the Tumor Microenvironment. Adv Cancer Res (2015) 128:197–233. doi: 10.1016/bs.acr.2015.04.003
3. Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front Immunol (2016) 7:407. doi: 10.3389/fimmu.2016.00407
4. Dieu-Nosjean M-C, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautès-Fridman C. Tertiary Lymphoid Structures, Drivers of the Anti-Tumor Responses in Human Cancers. Immunol Rev (2016) 271:260–75. doi: 10.1111/imr.12405
5. Weinstein AM, Storkus WJ. Biosynthesis and Functional Significance of Peripheral Node Addressin in Cancer-Associated TLO. Front Immunol (2016) 7:301. doi: 10.3389/fimmu.2016.00301
6. Jones E, Gallimore A, Ager A. Defining High Endothelial Venules and Tertiary Lymphoid Structures in Cancer. Methods Mol Biol (2018) 1845:99–118. doi: 10.1007/978-1-4939-8709-2_7
7. Ager A, May MJ. Understanding High Endothelial Venules: Lessons for Cancer Immunology. Oncoimmunology (2015) 4:e1008791. doi: 10.1080/2162402X.2015.1008791
8. Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of Naive T Cells Inside Tumors Leads to Eradication of Established Tumors. Nat Immunol (2004) 5:141–9. doi: 10.1038/ni1029
9. Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor Masses Support Naive T Cell Infiltration, Activation, and Differentiation Into Effectors. J Exp Med (2010) 207:1791–804. doi: 10.1084/jem.20092454
10. Teillaud J-L, Dieu-Nosjean M-C. Tertiary Lymphoid Structures: An Anti-Tumor School for Adaptive Immune Cells and an Antibody Factory to Fight Cancer? Front Immunol (2017) 8:830. doi: 10.3389/fimmu.2017.00830
11. Weinstein AM, Chen L, Brzana EA, Patil PR, Taylor JL, Fabian KL, et al. Tbet and IL-36γ Cooperate in Therapeutic DC-mediated Promotion of Ectopic Lymphoid Organogenesis in the Tumor Microenvironment. OncoImmunology (2017) 6:e1322238. doi: 10.1080/2162402X.2017.1322238
12. Goc J, Germain C, Vo-Bourgais TKD, Lupo A, Klein C, Knockaert S, et al. Dendritic Cells in Tumor-Associated Tertiary Lymphoid Structures Signal a Th1 Cytotoxic Immune Contexture and License the Positive Prognostic Value of Infiltrating CD8+ T Cells. Cancer Res (2014) 74:705–15. doi: 10.1158/0008-5472.CAN-13-1342
13. Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, et al. Induced Bronchus-Associated Lymphoid Tissue Serves as a General Priming Site for T Cells and is Maintained by Dendritic Cells. J Exp Med (2009) 206:2593–601. doi: 10.1084/jem.20091472
14. Kuerten S, Schickel A, Kerkloh C, Recks MS, Addicks K, Ruddle NH, et al. Tertiary Lymphoid Organ Development Coincides With Determinant Spreading of the Myelin-Specific T Cell Response. Acta Neuropathol (2012) 124:861–73. doi: 10.1007/s00401-012-1023-3
15. Posch F, Silina K, Leibl S, Mündlein A, Moch H, Siebenhüner A, et al. Maturation of Tertiary Lymphoid Structures and Recurrence of Stage II and III Colorectal Cancer. Oncoimmunology (2017) 7:e1378844. doi: 10.1080/2162402X.2017.1378844
16. Hiraoka N, Ino Y, Yamazaki-Itoh R. Tertiary Lymphoid Organs in Cancer Tissues. Front Immunol (2016) 7:244. doi: 10.3389/fimmu.2016.00244
17. Stowman AM, Hickman AW, Mauldin IS, Mahmutovic A, Gru AA, Slingluff CL Jr. Lymphoid Aggregates in Desmoplastic Melanoma Have Features of Tertiary Lymphoid Structures. Melanoma Res (2018) 28:237–45. doi: 10.1097/CMR.0000000000000439
18. Wirsing AM, Rikardsen OG, Steigen SE, Uhlin-Hansen L, Hadler-Olsen E. Characterisation and Prognostic Value of Tertiary Lymphoid Structures in Oral Squamous Cell Carcinoma. BMC Clin Pathol (2014) 14:38. doi: 10.1186/1472-6890-14-38
19. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B Cells in Tertiary Lymphoid Structures is Associated With a Protective Immunity in Patients With Lung Cancer. Am J Respir Crit Care Med (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
20. Kroeger DR, Milne K, Nelson BH. Tumor Infiltrating Plasma Cells are Associated With Tertiary Lymphoid Structures, Cytolytic T-cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res (2016) 22:3005–15. doi: 10.1158/1078-0432.CCR-15-2762
21. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-Term Survival for Patients With Non- Small-Cell Lung Cancer With Intratumoral Lymphoid Structures. J Clin Oncol (2008) 26:4410–7. doi: 10.1200/JCO.2007.15.0284
22. Siliņa K, Soltermann A, Attar FM, Casanova R, Uckeley ZM, Thut H, et al. Germinal Centers Determine the Prognostic Relevance of Tertiary Lymphoid Structures and are Impaired by Corticosteroids in Lung Squamous Cell Carcinoma. Cancer Res (2018) 78:1308–20. doi: 10.1158/0008-5472.CAN-17-1987
23. Castino GF, Cortese N, Capretti G, Serio S, Di Caro G, Mineri R, et al. Spatial Distribution of B Cells Predicts Prognosis in Human Pancreatic Adenocarcinoma. Oncoimmunology (2016) 5:e1085147. doi: 10.1080/2162402X.2015.1085147
24. Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, et al. Occurrence of Tertiary Lymphoid Tissue is Associated With T Cell Infiltration and Predicts Better Prognosis in Early- Stage Colorectal Cancers. Clin Cancer Res (2014) 20:2147–58. doi: 10.1158/1078-0432.CCR-13-2590
25. Lee HJ, Kim JY, Park IA, Song IH, Yu JH, Ahn JH, et al. Prognostic Significance of Tumor Infiltrating Lymphocytes and the Tertiary Lymphoid Structures in HER2-positive Breast Cancer Treated With Adjuvant Trastuzumab. Am J Clin Pathol (2015) 144:278–88. doi: 10.1309/AJCPIXUYDVZ0RZ3G
26. Savas P, Salgado R, Sotiriou C, Denkert C, Darcy PK, Smyth MJ, et al. Clinical Relevance of Host Immunity in Breast Cancer: From TILs to the Clinic. Nat Rev Clin Oncol (2016) 13:228–41. doi: 10.1038/nrclinonc.2015.215
27. Martinet L, Filleron T, Le Guellec S, Rochaix P, Garrido I, Girardet JP. High Endothelial Venule Blood Vessels for Tumor-Infiltrating Lymphocytes are Associated With Lymphotoxin β-Producing Dendritic Cells in Human Breast Cancer. J Immunol (2013) 191:2001–8. doi: 10.4049/jimmunol.1300872
28. Nerviani A, Pitzalis C. Role of Chemokines in Ectopic Lymphoid Structures Formation in Autoimmunity and Cancer. J Leukoc Biol (2018) 104:333–41. doi: 10.1002/JLB.3MR0218-062R
29. Ruddle NH. Lymphoid Neo-Organogenesis: Lymphotoxin’s Role in Inflammation and Development. Immunol Res (1999) 19:119–25. doi: 10.1007/BF02786481
30. Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, et al. Lymphotoxin Beta Receptor Signaling is Required for Inflammatory Lymphangiogenesis in the Thyroid. Proc Natl Acad Sci USA (2007) 104:5026–31. doi: 10.1073/pnas.0606697104
31. Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC Expression in Pancreatic Islets Causes B Cell Recruitment and Lymphotoxin-Dependent Lymphoid Neogenesis. Immunity (2000) 12:471–81. doi: 10.1016/s1074-7613(00)80199-5
32. Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, et al. Differing Activities of Homeostatic Chemokines CCL19, CCL21, and CXCL12 in Lymphocyte and Dendritic Cell Recruitment and Lymphoid Neogenesis. J Immunol (2002) 169:424–33. doi: 10.4049/jimmunol.169.1.424
33. Sharma S, Stolina M, Luo J, Strieter RM, Burdick M, Li X, et al. Secondary Lymphoid Tissue Chemokine Mediates T Cell-Dependent Antitumor Responses In Vivo. J Immunol (2000) 164:4558–63. doi: 10.4049/jimmunol.164.9.4558
34. Li J, O’Malley M, Sampath P, Kalinski P, Bartlett DL, Thorne SH. Expression of CCL19 From Oncolytic Vaccinia Enhances Immunotherapeutic Potential While Maintaining Oncolytic Activity. Neoplasia (2012) 14:1115–21. doi: 10.1593/neo.121272
35. Chen P, Luo S, Wen YJ, Li YH, Li J, Wang YS, et al. Low-Dose Paclitaxel Improves the Therapeutic Efficacy of Recombinant Adenovirus Encoding CCL21 Chemokine Against Murine Cancer. Cancer Sci (2014) 105:1393–401. doi: 10.1111/cas.12537
36. Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase I Trial of Intratumoral Injection of CCL21 Gene-Modified Dendritic Cells in Lung Cancer Elicits Tumor-Specific Immune Responses and CD8+ T-Cell Infiltration. Clin Cancer Res (2017) 23:4556–68. doi: 10.1158/1078-0432
37. Tang H, Zhu M, Qiao J, Fu YX. Lymphotoxin Signalling in Tertiary Lymphoid Structures and Immunotherapy. Cell Mol Immunol (2017) 14:809–18. doi: 10.1038/cmi.2017.13
38. Upadhyay V, Fu YX. Lymphotoxin Signalling in Immune Homeostasis and the Control of Microorganisms. Nat Rev Immunol (2013) 13:270–9. doi: 10.1038/nri3406
39. Ware CF. Targeting Lymphocyte Activation Through the Lymphotoxin and LIGHT Pathways. Immunol Rev (2008) 223:86–201. doi: 10.1111/j.1600-065X.2008.00629.x
40. Peske JD, Thompson ED, Gemta L, Baylis RA, Fu Y-X, Engelhard VH. Effector Lymphocyte-Induced Lymph Node-Like Vasculature Enables Naive T-Cell Entry Into Tumours and Enhanced Anti-Tumour Immunity. Nat Commun (2015) 6:7114. doi: 10.1038/ncomms8114
41. Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic Inflammation Caused by Lymphotoxin is Lymphoid Neogenesis. J Exp Med (1996) 183:1461–72. doi: 10.1084/jem.183.4.1461
42. Drayton DL, Ying X, Lee J, Lesslauer W, Ruddle NH. Ectopic LT Alpha Beta Directs Lymphoid Organ Neogenesis With Concomitant Expression of Peripheral Node Addressin and a HEV-restricted Sulfotransferase. J Exp Med (2003) 197:1153–63. doi: 10.1084/jem.20021761
43. Schrama D, thor Straten P, Fischer WH, McLellan AD, Brocker EB, Reisfeld RA, et al. Targeting of Lymphotoxin-Alpha to the Tumor Elicits an Efficient Immune Response Associated With Induction of Peripheral Lymphoid-Like Tissue. Immunity (2001) 14:111–21. doi: 10.1016/s1074-7613(01)00094-2
44. Colbeck EJ, Jones E, Hindley JP, Smart K, Schulz R, Browne M, et al. Treg Depletion Licenses T Cell-Driven HEV Neogenesis and Promotes Tumor Destruction. Cancer Immunol Res (2017) 5:1005–15. doi: 10.1158/2326-6066.CIR-17-0131
45. He B, Jabouille A, Steri V, Johansson-Percival A, Michael IP, Kotamraju VR, et al. Vascular Targeting of LIGHT Normalizes Blood Vessels in Primary Brain Cancer and Induces Intratumoural High Endothelial Venules. J Pathol (2018) 245:209–21. doi: 10.1002/path.5080
46. Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. NK-Cell Activation by LIGHT Triggers Tumor-Specific CD8+ T-Cell Immunity to Reject Established Tumors. Blood (2006) 107:1342–51. doi: 10.1182/blood-2005-08-3485
47. Mourik BC, Lubberts E, de Steenwinkel JEM, Ottenhoff THM, Leenen PJM. Interactions Between Type 1 Interferons and the Th17 Response in Tuberculosis: Lessons Learned From Autoimmune Diseases. Front Immunol (2017) 8:294. doi: 10.3389/fimmu.2017.00294
48. Denton AE, Innocentin S, Carr EJ, Bradford BM, Lafouresse F, Mabbott NA, et al. Type I Interferon Induces CXCL13 to Support Ectopic Germinal Center Formation. J Exp Med (2019) 216:621–37. doi: 10.1084/jem.20181216
49. Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E, et al. Deficiency of the Type I Interferon Receptor Protects Mice From Experimental Lupus. Arthritis Rheumatol (2007) 56:3770–83. doi: 10.1002/art.23023
50. Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, et al. Type I Interferon Production by Tertiary Lymphoid Tissue Developing in Response to 2,6,10,14-Tetramethyl-Pentadecane (Pristane). Am J Pathol (2006) 168:1227–40. doi: 10.2353/ajpath.2006.050125
51. Ogasawara K, Hida S, Weng Y, Saiura A, Sato K, Takayanagi H, et al. Requirement of the IFN-alpha/beta-induced CXCR3 Chemokine Signalling for CD8+ T Cell Activation. Genes Cells (2002) 7:309–20. doi: 10.1046/j.1365-2443.2002.00515.x
52. Banks TA, Rickert S, Benedict CA, Ma L, Ko M, Meier J, et al. A lymphotoxin-IFN-beta Axis Essential for Lymphocyte Survival Revealed During Cytomegalovirus Infection. J Immunol (2005) 174:7217–25. doi: 10.4049/jimmunol.174.11.7217
53. Chelvanambi M, Weinstein AM, Storkus WJ. Il-36 Signaling in the Tumor Microenvironment. Adv Exp Med Biol (2020) 1240:95–110. doi: 10.1007/978-3-030-38315-2_8
54. Weinstein AM, Giraldo NA, Petitprez F, Julie C, Lacroix L, Peschaud F, et al. Association of IL-36γ With Tertiary Lymphoid Structures and Inflammatory Immune Infiltrates in Human Colorectal Cancer. Cancer Immunol Immunother (2019) 68:109–20. doi: 10.1007/s00262-018-2259-0
55. Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING Pathway Agonism as a Cancer Therapeutic. Immunol Rev (2019) 290:24–38. doi: 10.1111/imr.12765
56. Kiwerska K, Szyfter K. DNA Repair in Cancer Initiation, Progression, and Therapy-a Double-Edged Sword. J Appl Genet (2019) 60:329–34. doi: 10.1007/s13353-019-00516-9
57. Hoeijmakers JH. Genome Maintenance Mechanisms for Preventing Cancer. Nature (2001) 411:366–74. doi: 10.1038/35077232
58. Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, et al. RAD51 Interconnects Between DNA Replication, DNA Repair and Immunity. Nucleic Acids Res (2017) 45:4590–605. doi: 10.1093/nar/gkx126
59. Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, et al. Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell (2021) 39:109–21. doi: 10.1016/j.ccell.2020.11.004
60. Talens F, Van Vugt MATM. Inflammatory Signaling in Genomically Instable Cancers. Cell Cycle (2019) 18:1830–48. doi: 10.1080/15384101.2019.1638192
61. He L, Xiao X, Yang X, Zhang Z, Wu L, Liu Z. STING Signaling in Tumorigenesis and Cancer Therapy: A Friend or Foe? Cancer Lett (2017) 402:203–12. doi: 10.1016/j.canlet.2017.05.026
62. Reisländer T, Groelly FJ, Tarsounas M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell (2020) 80:21–8. doi: 10.1016/j.molcel.2020.07.026
63. Campisi M, Sundararaman SK, Shelton SE, Knelson EH, Mahadevan NR, Yoshida R, et al. Tumor-Derived cGAMP Regulates Activation of the Vasculature. Front Immunol (2020) 11:2090. doi: 10.3389/fimmu.2020.02090
64. Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, et al. Cancer-Cell-Intrinsic Cgas Expression Mediates Tumor Immunogenicity. Cell Rep (2019) 29:1236–48. doi: 10.1016/j.celrep.2019.09.065
65. Andzinski L, Spanier J, Kasnitz N, Kröger A, Jin L, Brinkmann MM, et al. Growing Tumors Induce a Local STING Dependent Type I IFN Response in Dendritic Cells. Int J Cancer (2016) 139:1350–7. doi: 10.1002/ijc.30159
66. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep (2015) 11:1018–30. doi: 10.1016/j.celrep.2015.04.031
67. Chelvanambi M, Fecek RJ, Taylor JL, Storkus WJ. STING Agonist-Based Treatment Promotes Vascular Normalization and Tertiary Lymphoid Structure Formation in the Therapeutic Melanoma Microenvironment. J Immunother Cancer (2021) 9:e001906. doi: 10.1136/jitc-2020-001906
68. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING Activation Reprograms Tumor Vasculatures and Synergizes With VEGFR2 Blockade. J Clin Invest (2019) 129:4350–64. doi: 10.1172/JCI125413
69. Lin Z, Huang L, Li S, Gu J, Cui X, Zhou Y. Pan-Cancer Analysis of Genomic Properties and Clinical Outcome Associated With Tumor Tertiary Lymphoid Structure. Sci Rep (2020) 10:21530. doi: 10.1038/s41598-020-78560-3
70. Salem D, Chelvanambi M, Storkus WJ, Fecek RJ. Cutaneous Melanoma: Mutational Status and Potential Links to Tertiary Lymphoid Structure Formation. Front Immunol (2021) 12:629519. doi: 10.3389/fimmu.2021.629519
71. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor Mutational Load Predicts Survival After Immunotherapy Across Multiple Cancer Types. Nat Genet (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
72. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther (2017) 16:2598–608. doi: 10.1158/1535-7163.MCT-17-0386
73. Farshchi Adli AD, Jahanban-Esfahlan R, Seidi K, Samandari-Rad S, Zarghami N. An Overview on Vadimezan (Dmxaa): The Vascular Disrupting Agent. Chem Biol Drug Des (2018) 91:996–1006. doi: 10.1111/cbdd.13166
74. Tang CHA, Lee AC, Chang S, Xu Q, Shao A, Lo Y, et al. STING Regulates BCR Signaling in Normal and Malignant B Cells. Cell Mol Immunol (2021) 18:1016–31. doi: 10.1038/s41423-020-00552-0.Online ahead of print
75. Tang CHA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res (2016) 76:2137–52. doi: 10.1158/0008-5472.CAN-15-1885
76. Jing Y, Dai X, Yang L, Kang D, Jiang P, Li N, et al. STING Couples With PI3K to Regulate Actin Reorganization During BCR Activation. Sci Adv (2020) 6:eaax9455. doi: 10.1126/sciadv.aax9455
77. Sakimura C, Tanaka H, Okuno T, Hiramatsu S, Muguruma K, Hirakawa K, et al. B Cells in Tertiary Lymphoid Structures are Associated With Favorable Prognosis in Gastric Cancer. J Surg Res (2017) 215:74–82. doi: 10.1016/j.jss.2017.03.033
78. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature (2020) 577:561–5. doi: 10.1038/s41586-019-1914-8
79. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B Cells and Tertiary Lymphoid Structures Promote Immunotherapy Response. Nature (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
80. Willsmore ZN, Harris RJ, Crescioli S, Hussein K, Kakkassery H, Thapa D, et al. B Cells in Patients With Melanoma: Implications for Treatment With Checkpoint Inhibitor Antibodies. Front Immunol (2021) 11:622442. doi: 10.3389/fimmu.2020.622442
81. Corsiero E, Delvecchio FR, Bombardieri M, Pitzalis C. B Cells in the Formation of Tertiary Lymphoid Organs in Autoimmunity, Transplantation and Tumorigenesis. Curr Opin Immunol (2019) 57:46–52. doi: 10.1016/j.coi.2019.01.004
82. Zhang Y, Gallastegui N, Rosenblatt JD. Regulatory B Cells in Anti-Tumor Immunity. Int Immunol (2015) 27:521–30. doi: 10.1093/intimm/dxv034
83. Zhang Y, Eliav Y, Shin SU, Schreiber TH, Podack ER, Tadmor T, et al. B Lymphocyte Inhibition of Anti-Tumor Response Depends on Expansion of Treg But is Independent of B-cell Il-10 Secretion. Cancer Immunol Immunother (2013) 62:87–99. doi: 10.1007/s00262-012-1313-6
84. Shah S, Divekar AA, Hilchey SP, Cho HM, Newman CL, Shin SU, et al. Increased Rejection of Primary Tumors in Mice Lacking B Cells: Inhibition of Anti-Tumor CTL and TH1 Cytokine Responses by B Cells. Int J Cancer (2005) 117:574–86. doi: 10.1002/ijc.21177
85. Zhang Y, Morgan R, Chen C, Cai Y, Clark E, Noor Khan W, et al. Mammary-Tumor-Educated B Cells Acquire LAP/TGF-β and PD-L1 Expression and Suppress Anti-Tumor Immune Responses. Int Immunol (2016) 28:423–33. doi: 10.1093/intimm/dxw007
86. Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. PD-1hi Identifies a Novel Regulatory B Cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discovery (2016) 6:546–59. doi: 10.1158/2159-8290.CD-15-1408
87. DiLillo DJ, Yanaba K, Tedder TF. B Cells are Required for Optimal CD4+ and CD8+ T Cell Tumor Immunity: Therapeutic B Cell Depletion Enhances B16 Melanoma Growth in Mice. J Immunol (2010) 184:4006–16. doi: 10.4049/jimmunol.0903009
88. Fridman WH, Petitprez F, Meylan M, Chen TWW, Sun CM, Roumenina LT, et al. B Cells and Cancer: to B or Not to B? J Exp Med (2021) 218:e20200851. doi: 10.1084/jem.20200851
89. Nelson BH. CD20+ B Cells: The Other Tumor-Infiltrating Lymphocytes. J Immunol (2010) 185:4977–82. doi: 10.4049/jimmunol.1001323
90. Kang YM, Kim SY, Kang JH, Han SW, Nam EJ, Kyung HS, et al. LIGHT Up-Regulated on B Lymphocytes and Monocytes in Rheumatoid Arthritis Mediates Cellular Adhesion and Metalloproteinase Production by Synoviocytes. Arthritis Rheumatol (2007) 56:1106–17. doi: 10.1002/art.22493
91. Schlößer HA, Thelen M, Lechner A, Wennhold K, Garcia-Marquez MA, Rothschild SI, et al. B Cells in Esophago-Gastric Adenocarcinoma are Highly Differentiated, Organize in Tertiary Lymphoid Structures and Produce Tumor Specific Antibodies. Oncoimmunology (2019) 8:e1512458. doi: 10.1080/2162402X.2018.1512458
92. Coronella JA, Spier C, Welch M, Trevor KT, Stopeck AT, Villar H, et al. Antigen-Driven Oligoclonal Expansion of Tumor- Infiltrating B Cells in Infiltrating Ductal Carcinoma of the Breast. J Immunol (2002) 169:1829–36. doi: 10.4049/jimmunol.169.4.1829
93. Nzula S, Going JJ, Stott DI. Antigen- Driven Clonal Proliferation, Somatic Hypermutation, and Selection of B Lymphocytes Infiltrating Human Ductal Breast Carcinomas. Cancer Res (2003) 63:3275–80.
94. Zhu W, Germain C, Liu Z, Sebastian Y, Devi P, Knockaert S, et al. A High Density of Tertiary Lymphoid Structure B Cells in Lung Tumors is Associated With Increased CD4+ T Cell Receptor Repertoire Clonality. Oncoimmunology (2015) 4:e1051922. doi: 10.1080/2162402X.2015.1051922
95. Gnjatic S, Atanackovic D, Jäger E, Matsuo M, Selvakumar A, Altorki NK, et al. Survey of Naturally Occurring CD4+ T Cell Responses Against NY- ESO-1 in Cancer Patients: Correlation With Antibody Responses. Proc Natl Acad Sci USA (2003) 100:8862–7. doi: 10.1073/pnas.1133324100
96. Montfort A, Pearce O, Maniati E, Vincent BG, Bixby L, Böhmet S, et al. A Strong B Cell Response is Part of the Immune Landscape in Human High-Grade Serous Ovarian Metastases. Clin Cancer Res (2017) 23:250–62. doi: 10.1158/1078-0432.CCR-16-0081
97. Hu HG, Wu J, Zhang BD, Li WH, Li YM. Pam 3 CSK 4-Cdg SF Augments Antitumor Immunotherapy by Synergistically Activating TLR1/2 and STING. Bioconjug Chem (2020) 31:2499–503. doi: 10.1021/acs.bioconjchem.0c00522
98. Vanpouille-Box C, Hoffmann JA, Galluzzi L. Pharmacological Modulation of Nucleic Acid Sensors - Therapeutic Potential and Persisting Obstacles. Nat Rev Drug Discovery (2019) 18:845–67. doi: 10.1038/s41573-019-0043-2
99. Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, et al. The Proteoglycan Biglycan Regulates Expression of the B Cell Chemoattractant CXCL13 and Aggravates Murine Lupus Nephritis. J Clin Invest (2010) 120:4251–72. doi: 10.1172/JCI42213
100. Bellamri N, Viel R, Morzadec C, Lecureur V, Joannes A, de Latour B, et al. TNF-Alpha and IL-10 Control CXCL13 Expression in Human Macrophages. J Immunol (2020) 204:2492–502. doi: 10.4049/jimmunol.1900790
101. Robinet M, Villeret B, Maillard S, Cron MA, Berrih-Aknin S, Le Panse R. Use of Toll-Like Receptor Agonists to Induce Ectopic Lymphoid Structures in Myasthenia Gravis Mouse Models. Front Immunol (2017) 8:1029. doi: 10.3389/fimmu.2017.01029
102. Lorkowski ME, Atukorale PU, Bielecki PA, Tong KH, Covarrubias G, Zhang Y, et al. Immunostimulatory Nanoparticle Incorporating Two Immune Agonists for the Treatment of Pancreatic Tumors. J Control Release (2021) 330:1095–105. doi: 10.1016/j.jconrel.2020.11.014
103. Mullins SR, Vasilakos JP, Deschler K, Grigsby I, Gillis P, John J, et al. Intratumoral Immunotherapy With TLR7/8 Agonist MEDI9197 Modulates the Tumor Microenvironment Leading to Enhanced Activity When Combined With Other Immunotherapies. J Immunother Cancer (2019) 7:244. doi: 10.1186/s40425-019-0724-8.(B16)
104. Temizoz B, Kuroda E, Ohata K, Jounai N, Ozasa K, Kobiyama K, et al. TLR9 and STING Agonists Synergistically Induce Innate and Adaptive type-II Ifn. Eur J Immunol (2015) 45:1159–69. doi: 10.1002/eji.201445132
105. Mandik-Nayak L, Huang G, Sheehan KC, Erikson J, Chaplin DD. Signaling Through TNF Receptor p55 in TNF-Alpha-Deficient Mice Alters the CXCL13/CCL19/CCL21 Ratio in the Spleen and Induces Maturation and Migration of Anergic B Cells Into the B Cell Follicle. J Immunol (2001) 167:1920–8. doi: 10.4049/jimmunol.167.4.1920
106. Ma D, Fan SB, Hua N, Li GH, Chang Q, Liu X. Hypermethylation of Single CpG Dinucleotides At the Promoter of CXCL13 Gene Promote Cell Migration in Cervical Cancer. Curr Cancer Drug Targets (2020) 20:355–63. doi: 10.2174/1568009620666200102123635
107. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING Activation With T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol Res (2017) 5:676–84. doi: 10.1158/2326-6066.CIR-17-0049
108. Zhang CX, Ye SB, Ni JJ, Cai TT, Liu YN, Huang DJ, et al. STING Signaling Remodels the Tumor Microenvironment by Antagonizing Myeloid-Derived Suppressor Cell Expansion. Cell Death Differ (2019) 26:2314–28. doi: 10.1038/s41418-019-0302-0
109. Lemos H, Ou R, McCardle C, Lin Y, Calver J, Minett J, et al. Overcoming Resistance to STING Agonist Therapy to Incite Durable Protective Antitumor Immunity. J Immunother Cancer (2020) 8:e001182. doi: 10.1136/jitc-2020-001182
110. Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, et al. Antitumor Activity of a Systemic STING-activating non-Nucleotide cGAMP Mimetic. Science (2020) 369:993–9. doi: 10.1126/science.abb4255
111. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. Sting Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res (2016) 76:2076–81. doi: 10.1158/0008-5472.CAN-15-1456
112. Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, et al. A Nanoparticle-Incorporated STING Activator Enhances Antitumor Immunity in PD-L1-insensitive Models of Triple-Negative Breast Cancer. JCI Insight (2018) 3:e120638. doi: 10.1172/jci.insight.120638
113. Sharma S, Kadam P, Dubinett S. CCL21 Programs Immune Activity in Tumor Microenvironment. Adv Exp Med Biol (2020) 1231:67–78. doi: 10.1007/978-3-030-36667-4_7
114. Hisada M, Yoshimoto T, Kamiya S, Magami Y, Miyaji H, Yoneto T, et al. Synergistic Antitumor Effect by Coexpression of Chemokine CCL21/SLC and Costimulatory Molecule LIGHT. Cancer Gene Ther (2004) 11:280–8. doi: 10.1038/sj.cgt.7700676
115. Skeate JG, Otsmaa ME, Prins R, Fernandez DJ, Da Silva DM, Kast WM. TNFSF14: LIGHTing the Way for Effective Cancer Immunotherapy. Front Immunol (2020) 11:922. doi: 10.3389/fimmu.2020.00922
116. Le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial Watch: STING Agonists in Cancer Therapy. Oncoimmunology (2020) 9:1777624. doi: 10.1080/2162402X.2020.1777624
117. Gogoi H, Mansouri S, Jin L. The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines (2020) 8:453. doi: 10.3390/vaccines8030453
118. Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An Orally Available non-Nucleotide STING Agonist With Antitumor Activity. Science (2020) 369:eaba6098. doi: 10.1126/science.aba6098
119. Aval LM, Pease JE, Sharma R, Pinato DJ. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J Clin Med (2020) 9:3323. doi: 10.3390/jcm9103323
120. Baguley BC. Antivascular Therapy of Cancer: DMXAA. Lancet Oncol (2003) 4:141–8. doi: 10.1016/s1470-2045(03)01018-0
121. Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-Mediated Disruption of Calcium Homeostasis Chronically Activates ER Stress and Primes T Cell Death. J Exp Med (2019) 216:867–83. doi: 10.1084/jem.20182192
122. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S. Franceschi, CChronic Inflammation in the Etiology of Disease Across the Life Span. Nat Med (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
123. Pipi E, Nayar S, Gardner DH, Colafrancesco S, Smith C, Barone F. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front Immunol (2018) 9:1952. doi: 10.3389/fimmu.2018.01952
124. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal Instability Drives Metastasis Through a Cytosolic DNA Response. Nature (2018) 553:467–72. doi: 10.1038/nature25432
Keywords: dendritic cells, immunotherapy, STING agonists, tertiary lymphoid structures, T cells, tumor, vaccine, vascular normalization
Citation: Filderman JN, Appleman M, Chelvanambi M, Taylor JL and Storkus WJ (2021) STINGing the Tumor Microenvironment to Promote Therapeutic Tertiary Lymphoid Structure Development. Front. Immunol. 12:690105. doi: 10.3389/fimmu.2021.690105
Received: 02 April 2021; Accepted: 30 April 2021;
Published: 13 May 2021.
Edited by:
Catherine Sautes-Fridman, U1138 Centre de Recherche des Cordeliers (INSERM), FranceReviewed by:
Itziar Otano, Research Institute Hospital 12 de Octubre, SpainCopyright © 2021 Filderman, Appleman, Chelvanambi, Taylor and Storkus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter J. Storkus, c3Rvcmt1c3dqQHVwbWMuZWR1
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.